
Three-cohort targeted gene screening reveals a non-synonymous TRKA polymorphism associated with...
127
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Three-cohort targeted gene screening reveals a non-synonymous TRKA polymorphism associated with...


Towards candidate genes for schizophrenia: the APO-SUS/APO-UNSUS rat model
and human association studies

Towards candidate genes for schizophrenia: the APO-SUS/APO-UNSUS rat model and human association studies Jessica Elisabeth Lathouwers - van Schijndel ISBN: 978-90-9025821-8 Printed by Ipskamp Drukkers B.V. Enschede The studies described in this thesis were performed at the Department of Molecular Animal Physiology, Donders Institute for Brain, Cognition and Behaviour, Centre for Neuroscience and Nijmegen Centre for Molecular Life Sciences (NCMLS), Faculty of Science, Radboud University Nijmegen, Nijmegen, The Netherlands.

Towards candidate genes for schizophrenia:
the APO-SUS/APO-UNSUS rat model and human association studies
Een wetenschappelijke proeve op het gebied van de Natuurwetenschappen, Wiskunde en Informatica
Proefschrift
ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen
op gezag van de rector magnificus prof. mr. S.C.J.J Kortmann, volgens besluit van het college van decanen
in het openbaar te verdedigen op vrijdag 17 december 2010 om 13.00 uur precies
door
Jessica Elisabeth van Schijndel
geboren 12 november 1980 te Heeswijk-Dinther

Promotor: Prof. dr. G.J.M. Martens Manuscriptcommissie: Prof. dr. E.W. Roubos Prof. dr. J.K. Buitelaar Dr. B.A. Ellenbroek (Evotec AG, Hamburg, Duitsland)

Contents Chapter 1 General Introduction 7 Chapter 2 Gene expression profiling in brain regions of a rat model displaying schizophrenia-related features 35 Chapter 3 Dopamine susceptibility of APO-SUS rats is not per se coupled to HPA-axis activity 47 Chapter 4 Three-cohort targeted gene screening reveals a non-synonymous TRKA polymorphism associated with schizophrenia 61 Chapter 5 Dual association of a TRKA polymorphism with schizophrenia 85 Chapter 6 General Discussion 97 Summary / Samenvatting 113 Dankwoord 123 Curriculum vitae 124 List of publications 125


Chapter 1 General Introduction
Part of this chapter has been published as: Van Schijndel JE and Martens GJM.
Gene expression profiling in rodent models for schizophrenia. Current Neuropharmacology, 2010, 8:382-393.
.


Chapter 1 - General Introduction
9
Individuals are afflicted worldwide with psychiatric disorders. A survey in developed and less-developed countries (in the Americas, Europe, the Middle East, Africa and Asia) has shown a 12-month prevalence of having a mental disorder described in the Diagnostic and Statistical Manual of Mental Disorders, Fourth
Edition (DSM-IV) from 4.3% in Shanghai to 26.4% in the United States (Demyttenaere et al., 2004). In The Netherlands, the 12-month prevalence of mental disorders is between 14.9% and 23.3% (Bijl et al., 1998; Demyttenaere et al., 2004), whereas the lifetime prevalence is 41.2% (Bijl et al., 1998). The mental disorder schizophrenia has a worldwide lifetime risk of ~1%, with an enormous impact on society in terms of expenditure and human suffering (Van Os et al., 2009). The first symptoms of this complex psychiatric disorder usually manifest during late adolescence or early adulthood, but the origin of the disorder is thought to be neurodevelopmental (Rehn et al., 2005; Weinberger, 1995). In this thesis, studies on a rat model for schizophrenia as well as human association studies on schizophrenia case-control cohorts are presented. In this introductory chapter, various topics dealt with in this thesis will be described, including human aetiology studies, mRNA expression studies on various animal models for schizophrenia and the rat model that is central in this thesis. Human aetiology studies on schizophrenia Schizophrenia is characterized by delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behaviour, and negative symptoms (American Psychiatric Association, 1994). Negative symptoms concern the absence of or reduction in basic emotional and behavioural processes and include flat or blunted emotions, poverty of speech, the inability to experience pleasure, and lack of motivation. Although at present not considered a diagnostic criterion, many patients suffer from cognitive impairments such as problems with attention, concentration, learning, memory and abstract thinking (Keefe et al., 2007; Mueser et al., 2004). Environmental factors in combination with predisposing genes appear to be important for the aetiology of schizophrenia. Environmental factors include prenatal infections, maternal malnutrition during pregnancy, obstetric complications, poverty, child abuse and neglect, war trauma, loss of parent and migration (Maki et al., 2005; Mueser et al., 2004; Read et al., 2008; Read et al., 2005). Such factors may affect gene expression by epigenetic mechanisms (Read et al., 2009; Weaver et al., 2004). Family, twin and adoption studies have shown that schizophrenia is a disorder with a substantial genetic contribution (Cardno et al., 2000; McGuffin, 2004). On the basis of linkage analyses, loci on 21 of the 23 chromosomes have been linked to the disorder. A meta-analysis has revealed that in various populations chromosomal region 2q, but also 1q, 8p, 22q and other loci, are associated with susceptibility to schizophrenia (Lewis et al., 2003). Follow-up studies on the regions implicated by the genome scans have resulted in positional candidate genes

Chapter 1 - General Introduction
10
such as neuregulin-1 (NRG1; 8p12) and catechol-O-methyltransferase (COMT; 22q11.21; Riley et al., 2006). In multiple case-control cohorts, a number of other candidate genes have been tested for association with schizophrenia, including brain-derived neurotrophic factor (BDNF; Kanazawa et al., 2007), dysbindin, disrupted in schizophrenia 1 (DISC1), D-amino acid oxidase activator (DAOA/G72) and proline dehydrogenase (PRODH; Karayiorgou et al., 2006). Yet, variations in these genes account for the aetiology of only a small percentage of schizophrenia patients. New technologies have resulted in platforms that allow association studies for hundreds of thousands of single-nucleotide polymorphisms (SNPs) to search for polymorphisms associated with schizophrenia and other complex diseases. In the last few years, a genome-wide association (GWA) scan has been reported for schizophrenia. For this study, genomic DNAs from 178 Caucasian schizophrenia patients were hybridized to two chips containing ~262000 and ~238000 SNPs (Affymetrix). Compared with 144 healthy controls, one SNP demonstrated association beyond the significance threshold, namely rs4129148, neighboring colony stimulating factor 2 receptor alpha chain (CSF2RA) in the pseudoautosomal region. Sequencing of CSF2RA and the neighboring gene interleukin 3 receptor alpha (IL3RA) revealed several novel, rare missense variants associated with schizophrenia (Lencz et al., 2007). The association of SNPs in cytokine receptor genes may explain the relation of schizophrenia and prenatal infection and/or the altered immune system observed in schizophrenia patients (Brown et al., 2010; Leucht et al., 2007). SNP rs6603272 in IL3RA has been associated with schizophrenia in two Chinese studies (Sun et al., 2008; Sun et al., 2009). However, replication studies are necessary to confirm these findings. However, the final outcome of genetic research on schizophrenia is at present unclear, the search for vulnerability genes for schizophrenia has not resulted in consistent findings, and many genes associated with psychiatric disorders have only a small effect and are mostly linked to more than one disorder (Bentall et al., 2008). The inconsistent results may arise from the clinical heterogeneity displayed by these disorders (Ruggeri et al., 2009). The debate continues on whether in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) schizophrenia should be considered as a single disorder or as a group of several disease entities (Gaebel et al., 2008).
Animal models for schizophrenia In order to get a better insight into the function of candidate genes and the aetiology of schizophrenia, multiple animal models have been developed. A problem encountered here is that the most prominent symptoms of schizophrenia - delusions, hallucinations and thought disorder - can not be reproduced in rodents and even not in non-human primates. Furthermore, a well-established genotype, cellular phenotype or other biological marker that is characteristic for the disorder is not

Chapter 1 - General Introduction
11
available to validate a model. Nevertheless, a number of symptoms such as e.g. behaviours related to increased dopaminergic transmission (dopamimetric-induced hyperlocomotion), social withdrawal (reduced contact with unfamiliar partners), loss of prepulse inhibition (PPI, a symptom at the interface of psychosis and cognition) and cognitive deficits (impaired performance in a spatial memory test) have been observed in animal models (Lipska et al., 2000; Van den Buuse, 2010 2010). It is therefore generally accepted that animal models may provide new insights into the pathophysiology and aetiology of schizophrenia (Lipska et al., 2000). In general and based on the way they have been created, animal models can be divided into three groups: (1) neurodevelopmental models, (2) pharmacological models and (3) genetic models. Such animal models for schizophrenia and their characteristics have been described in many excellent papers and reviews (Desbonnet et al., 2009; Marcotte et al., 2001; Tordjman et al., 2007). In this chapter, mRNA expression profiling studies on these animal models will be described (table 1). mRNA expression profiling in neurodevelopmental animal models To generate a neurodevelopmental animal model for schizophrenia, environmental factors such as stress and viral infection are employed and since they should affect brain development the factors are applied to animals prenatally (or at a young age). Epidemiologic evidence has suggested that maternal infection by a virus may lead to the genesis of schizophrenia (Dalman et al., 2008; Yudofsky, 2009). In mice, prenatal influenza infection causes effects on brain structure and function such as increased serotonin levels (Winter et al., 2008). To establish a schizophrenia animal model, C57BL6J mice have been infected on pregnancy day 18 with H1N1 influenza virus. This resulted in a moderate, sublethal infection with a mean virus titer of 105.25 cell culture infectious dose (CCID)50/ml (Fatemi et al., 2008). Brain tissues from the offspring of these mice were collected for mRNA expression profiling at post natal day (PND) 0, 14 (childhood) and 56 (adulthood). Analysis with the mouse genome 430 2.0 array (Affymetrix) showed that in the brains of the prenatally exposed mice compared to the brains of control offspring the highest number of genes were up- or downregulated at PND 0 (72, 175 and 157 genes in frontal, hippocampal and cerebellar brain areas, respectively) and PND 56 (110, 62 and 96 genes, respectively), relative to PND 14 (33, 21 and 16, respectively). A number of genes was validated by quantitative polymerase chain reaction (qPCR), including v-erb-B2 avian erythroblastic leukaemia viral oncogene (Erbb4), semaphorin 3A (Sema3a), very-low density lipoprotein receptor (Vldlr), and ATPase,Na+/K+ transporting, beta 2 polypeptide (Atp1b2; Fatemi et al., 2008). These genes had been implicated previously in the aetiopathology of schizophrenia (Birchmeier, 2009; Eastwood et al., 2003; Suzuki et al., 2008).

Chapter 1 - General Introduction
12
Table 1. An overview of schizophrenia animal models used in mRNA expression profiling.
Animal model
Approach
Number of gene(s) with affected mRNA expression
Reference
C57BL6J mice infected with H1N1 influenza virus on pregnancy day 18
Mouse genome 430 2.0 array (Affymetrix) covering >39000 transcripts
Hippocampus: 175 (PND0), 21 (PND14) and 62 (PND56) Cerebellum: 157 (PND0), 16 (PND14) and 96 (PND56) Prefrontal cortex: 72 (PND0), 33 (PND14) and 110 (PND56)
(Fatemi et al., 2008)
Balb/c mice infected with H1N1 influenza virus on pregnancy day 9
Murine 430 high-density oligonucleotide array (Affymetrix) covering ~20000 mouse genes and ESTs
Whole brain: 39 genes (Fatemi et al., 2005)
Lewis rat pups inoculated with NBD within 12 hours of birth, sacrificed at 4 weeks of age
3DNA Array 350 Expression Array (Genisphere) covering 8064 genes
Cerebellum: MT-I/-II (Mt1a) 9.09 fold increase Hippocampus: MT-I/-II (Mt1a) 2.95 fold increase
(Williams et al., 2006)
Pregnant Sprague-Dawley rats subjected to a repeated variable stress paradigm from ED14 to ED22, male pups were subjected to acute stress at PND56 and sacrificed
Affymetrix RG U34A microarray covering 8799 genes
Frontal pole: 35 genes (Kinnunen et al., 2003)
VH-lesioned Fisher 344 (F344) rats, daily i.p. haloperidol treatment for 14 days starting at PND55
GeneFilter® DNA microarray blots (Research Genetics Inc., Huntsville) covering 37575 cDNA clones
VH-lesioned F344 versus sham-lesioned F344 rats - Frontal tissue: 828 genes - Temporal tissue: 768 genes Haloperidol treated versus control VH-lesioned F344 - Frontal tissue: 404 genes - Temporal tissue: 595 genes
(Wong et al., 2005)
New born F344 rats separated from their dams for 8 hours every other day (PND2 to 10)
Affymetrix rat U34A microarray
Hippocampal slices: 24 genes including Ttr (4.85 fold decrease)
(Kohda et al., 2006)
Institute for Cancer Research (ICR) Mice subjected to isolated rearing for 4 weeks
GeneChip mouse genome 430 2.0 Array (Affymetrix) covering 45101 probe sets
Dentate gyrus: 22 genes including Nr4a2/Nurr1 and Npas4
(Ibi et al., 2008)
ICR mice, daily i.p. PCP for 24 days
Clontech Atlas glass array mouse
Cerebral cortical tissue: 73 genes
(Toyooka et al., 2002)
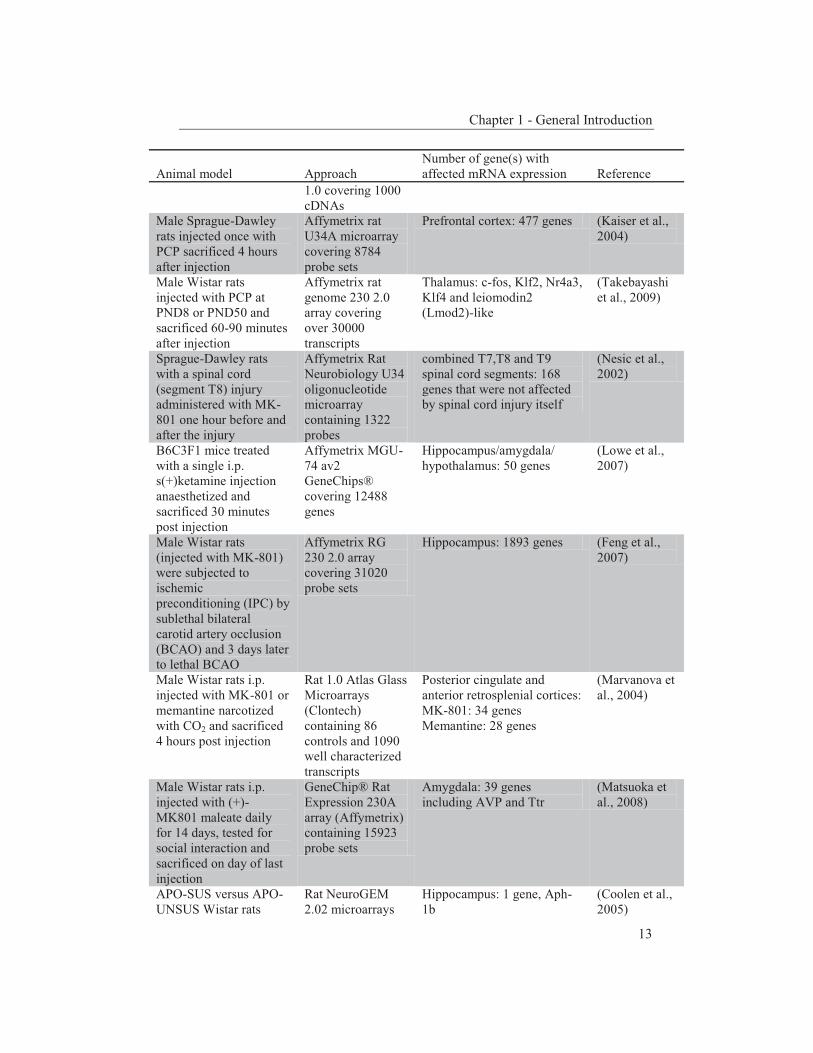
Chapter 1 - General Introduction
13
Animal model
Approach
Number of gene(s) with affected mRNA expression
Reference
1.0 covering 1000 cDNAs
Male Sprague-Dawley rats injected once with PCP sacrificed 4 hours after injection
Affymetrix rat U34A microarray covering 8784 probe sets
Prefrontal cortex: 477 genes (Kaiser et al., 2004)
Male Wistar rats injected with PCP at PND8 or PND50 and sacrificed 60-90 minutes after injection
Affymetrix rat genome 230 2.0 array covering over 30000 transcripts
Thalamus: c-fos, Klf2, Nr4a3, Klf4 and leiomodin2 (Lmod2)-like
(Takebayashi et al., 2009)
Sprague-Dawley rats with a spinal cord (segment T8) injury administered with MK-801 one hour before and after the injury
Affymetrix Rat Neurobiology U34 oligonucleotide microarray containing 1322 probes
combined T7,T8 and T9 spinal cord segments: 168 genes that were not affected by spinal cord injury itself
(Nesic et al., 2002)
B6C3F1 mice treated with a single i.p. s(+)ketamine injection anaesthetized and sacrificed 30 minutes post injection
Affymetrix MGU-74 av2 GeneChips® covering 12488 genes
Hippocampus/amygdala/ hypothalamus: 50 genes
(Lowe et al., 2007)
Male Wistar rats (injected with MK-801) were subjected to ischemic preconditioning (IPC) by sublethal bilateral carotid artery occlusion (BCAO) and 3 days later to lethal BCAO
Affymetrix RG 230 2.0 array covering 31020 probe sets
Hippocampus: 1893 genes (Feng et al., 2007)
Male Wistar rats i.p. injected with MK-801 or memantine narcotized with CO2 and sacrificed 4 hours post injection
Rat 1.0 Atlas Glass Microarrays (Clontech) containing 86 controls and 1090 well characterized transcripts
Posterior cingulate and anterior retrosplenial cortices: MK-801: 34 genes Memantine: 28 genes
(Marvanova et al., 2004)
Male Wistar rats i.p. injected with (+)-MK801 maleate daily for 14 days, tested for social interaction and sacrificed on day of last injection
GeneChip® Rat Expression 230A array (Affymetrix) containing 15923 probe sets
Amygdala: 39 genes including AVP and Ttr
(Matsuoka et al., 2008)
APO-SUS versus APO-UNSUS Wistar rats
Rat NeuroGEM 2.02 microarrays
Hippocampus: 1 gene, Aph-1b
(Coolen et al., 2005)

Chapter 1 - General Introduction
14
Animal model
Approach
Number of gene(s) with affected mRNA expression
Reference
containing 8478 Sprague Dawley rat nervous system cDNAs
Male Sprague–Dawley albino rats administered i.p. daily with olanzapine and sacrificed 24 h after the last injection under deep anaesthesia
Affymetrix rat genome 230 2.0 array set containing ~28000 rat genes and ESTs
Frontal cortex: 69 genes (Fatemi et al., 2006)
Male C57BL/6 mice i.p. injected with haloperidol, olanzapine, or clozapine daily for 7 days and sacrificed under 4% halothane anaesthesia five hours post injection
Affymetrix GeneChip Mouse Genome 430 2.0 microarrays containing over 39000 transcripts
Whole brain:11941 transcripts by one or more drugs, 249 transcripts by all 3 drugs
(Duncan et al., 2008)
Male Wistar rats injected i.p. with clozapine and sacrificed 1, 6 or 24 hrs post injection
Atlas Rat 1.2 Array filter (Clontech) covering 1176 sequence-verified cDNAs
Anterior cingulate and prefrontal cortices: 217 genes including 22 genes with a presynaptic function
(Kontkanen et al., 2002)
ICR mice injected subcutaneous with clozapine daily for 20 days and sacrificed 20 hours post injection
Atlas Rat 1.2 Array filter (Clontech) covering 1176 sequence-verified cDNAs
Cerebral cortical tissues: undefined number of genes including potassium channel, prostaglandin D2 synthase, serotonin receptor type 2 and myelin-associated oligodendrocytic basic protein
(Takahashi et al., 2004)
C57BL6 mice received daily for 31 days haloperidol or clozapine or phenylpropanolamine via food pellets
Murine Genome U74A Array (Affymetrix) containing ~12000 genes
Cortex: 1156 transcripts in one or more models
(Mehler-Wex et al., 2006)
Sprague–Dawley rats injected i.p. daily with Haloperidol or Risperidone and sacrificed 96 hours (acute treatment) or 4 weeks (chronic treatment) after initiation of drug treatment
PXM oligo-nucleotide slides (Full Moon Biosystems) containing 8000 rat gene-specific, amino-modified oligonucleotides
Prefrontal/fronto-parieto-temporal cortices: Haloperidol: - acute: 28 genes - chronic: 9 genes Risperidone: - acute: 89 genes - chronic: 17 genes
(Feher et al., 2005)
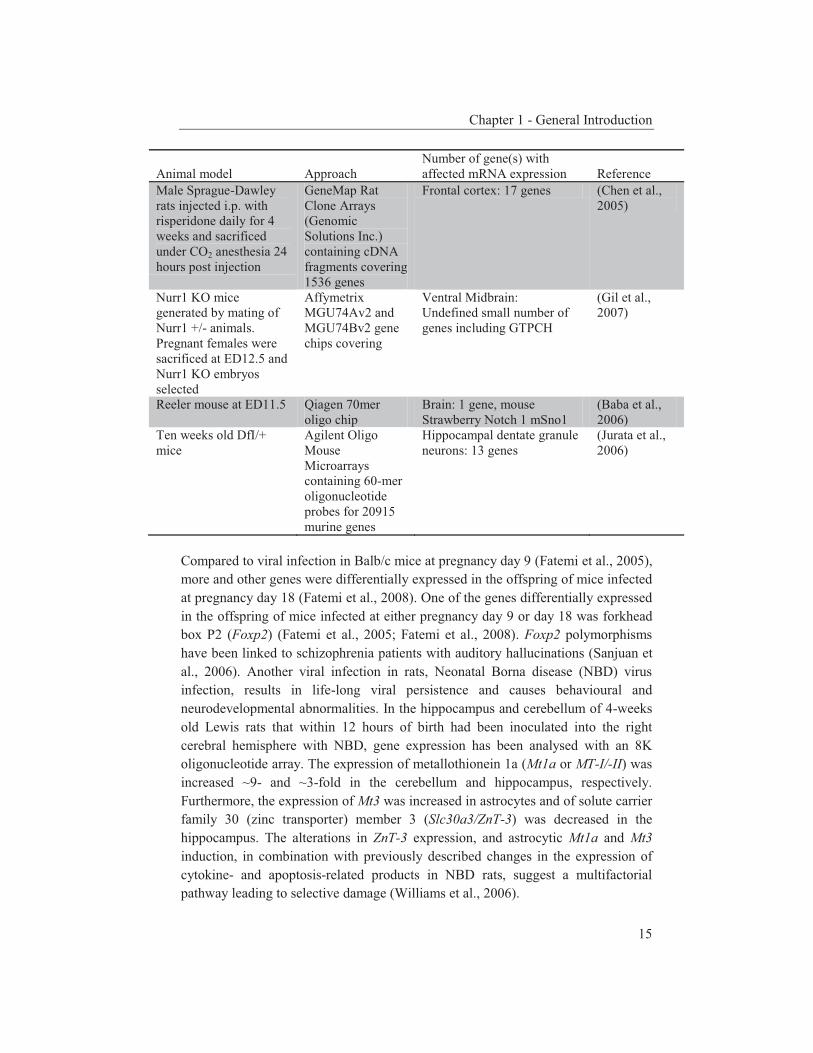
Chapter 1 - General Introduction
15
Animal model
Approach
Number of gene(s) with affected mRNA expression
Reference
Male Sprague-Dawley rats injected i.p. with risperidone daily for 4 weeks and sacrificed under CO2 anesthesia 24 hours post injection
GeneMap Rat Clone Arrays (Genomic Solutions Inc.) containing cDNA fragments covering 1536 genes
Frontal cortex: 17 genes (Chen et al., 2005)
Nurr1 KO mice generated by mating of Nurr1 +/- animals. Pregnant females were sacrificed at ED12.5 and Nurr1 KO embryos selected
Affymetrix MGU74Av2 and MGU74Bv2 gene chips covering
Ventral Midbrain: Undefined small number of genes including GTPCH
(Gil et al., 2007)
Reeler mouse at ED11.5 Qiagen 70mer oligo chip
Brain: 1 gene, mouse Strawberry Notch 1 mSno1
(Baba et al., 2006)
Ten weeks old DfI/+ mice
Agilent Oligo Mouse Microarrays containing 60-mer oligonucleotide probes for 20915 murine genes
Hippocampal dentate granule neurons: 13 genes
(Jurata et al., 2006)
Compared to viral infection in Balb/c mice at pregnancy day 9 (Fatemi et al., 2005), more and other genes were differentially expressed in the offspring of mice infected at pregnancy day 18 (Fatemi et al., 2008). One of the genes differentially expressed in the offspring of mice infected at either pregnancy day 9 or day 18 was forkhead box P2 (Foxp2) (Fatemi et al., 2005; Fatemi et al., 2008). Foxp2 polymorphisms have been linked to schizophrenia patients with auditory hallucinations (Sanjuan et al., 2006). Another viral infection in rats, Neonatal Borna disease (NBD) virus infection, results in life-long viral persistence and causes behavioural and neurodevelopmental abnormalities. In the hippocampus and cerebellum of 4-weeks old Lewis rats that within 12 hours of birth had been inoculated into the right cerebral hemisphere with NBD, gene expression has been analysed with an 8K oligonucleotide array. The expression of metallothionein 1a (Mt1a or MT-I/-II) was increased ~9- and ~3-fold in the cerebellum and hippocampus, respectively. Furthermore, the expression of Mt3 was increased in astrocytes and of solute carrier family 30 (zinc transporter) member 3 (Slc30a3/ZnT-3) was decreased in the hippocampus. The alterations in ZnT-3 expression, and astrocytic Mt1a and Mt3 induction, in combination with previously described changes in the expression of cytokine- and apoptosis-related products in NBD rats, suggest a multifactorial pathway leading to selective damage (Williams et al., 2006).

Chapter 1 - General Introduction
16
Another environmental factor that has been implicated in the aetiology of schizophrenia is prenatal stress (Koenig et al., 2002). To create an animal model, repeated variable stress paradigms have been applied to pregnant Sprague–Dawley rats during the last week of gestation. A microarray analysis of the frontal pole brain area of the prenatally stressed adult offspring and non-stressed adult controls revealed significant changes in 35 genes. Three differentially expressed genes encoded neurotransmitter receptor subunits or associated proteins, two were involved in calcium/calmodulin signaling, two were associated with neurotransmitter vesicles or vesicle recycling and two were Na+/K+-transporting ATPases (Kinnunen et al., 2003). Both the neurodevelopmental animal model based on prenatal influenza infection (Fatemi et al., 2008) and the model generated by prenatal stress (Kinnunen et al., 2003) displayed differential expression of Na+/K+-ATPase genes and potassium voltage-gated channel genes. However, the majority of the differentially expressed genes was differentially expressed in only a single animal model. A third neurodevelopmental animal model constitutes the neonatal ventral-hippocampal (VH) lesion rat model that exhibits many of the behavioural features of pharmacological schizophrenia models (Lipska et al., 1993; Lipska et al., 2000; , 2002). The locomotor abnormalities induced by the VH lesions differ between rat strains and therefore transcript expression was compared between saline- or haloperidol-treated adult VH-lesioned Fischer-344 and Lewis rats (Wong et al., 2005). Three variables were compared, namely with respect to strain (Fischer-344 versus Lewis), to age (day 6 versus day 70) and to treatment (saline versus haloperidol), and suppression subtraction hybridization was used to identify changes in transcript levels. Genes differentially expressed in all three cases included mitochondrial cytochrome oxidase (complex IV) subunits including COX1 and 2, mitochondrial tRNA, g and z isoforms of 14-3-3, adenosine monophosphate deaminase 3 (Ampd3), and a 25-kD synaptosomal associated protein (SNAP-25; Wong et al., 2005). A further neurodevelopmental animal model concerns the maternal deprivation rat model. Maltreatment, such as abuse and neglect, is a susceptibility factor for psychiatric disorders (Palomo et al., 2002). Fisher 344 rat pups were separated for 8 hours from their mother every other day between PND 2 and 10. At the age of 13 weeks, the rats were sacrificed and gene expression in hippocampal slices was analysed with an U34A (Affymetrix) array. The expression of fifteen genes was upregulated and of nine genes downregulated. The most prominent change in expression was found for the transthyretin (Ttr) gene. In the brain the expression of this gene is nearly restricted to the choroid plexus and therefore the authors speculated about a possible function for the choroid plexus in stress (Kohda et al., 2006). However, in other studies the differential expression of Ttr was considered

Chapter 1 - General Introduction
17
to be the result of a tissue-isolation artefact (contaminating choroid plexus) rather than being a differentially expressed gene of interest (Coolen et al., 2005). Yet another example of a neurodevelopmental animal model concerns three-weeks old, weaned Institute for Cancer Research mice that were isolated for 3 days, 2 weeks or 4 weeks, a treatment that may also affect brain development (Whitaker-Azmitia et al., 2000). In these mice, mRNA expression in the dentate gyrus was analysed with a GeneChip mouse genome 430 2.0 array (Affymetrix), but no obvious changes were found. However, using a general linear model incorporating the feeding period the downregulation of two genes was observed, namely of nuclear receptor subfamily 4, group A, member 2 (Nr4a2/Nurr1) and neuronal PAS domain protein 4 (Npas4; Ibi et al., 2008). mRNA expression profiling in pharmacological animal models The neuropharmacological animal models for schizophrenia are based on specific neurotransmitter systems (typically dopamine (DA) and glutamate). Psychotomimetic substances (DA agonists, N-methyl-D-aspartate (NMDA) antagonists) have been used to generate schizophrenia-like symptoms in animals. In rodents, phencyclidine (PCP) and other antagonists of the NMDA-type glutamate receptor produce positive and negative symptoms, and cognitive disturbances remarkably similar to those observed in schizophrenia patients (Bubenikova-Valesova et al., 2008). Animals in which these symptoms were induced by PCP or related compounds have therefore been considered to represent useful pharmacological models for this complex disorder. A single administration of PCP during the late foetal or early postnatal period in rats (corresponding to the third trimester of human pregnancy) produced behavioral symptoms indicative of NMDA receptor blockade lasting for 8 to 10 hours and resulted in increased neuronal death by apoptosis (Ikonomidou et al., 1999). Acute and chronic prenatal or neonatal administration of PCP to rats led to changes in behaviour of the adult animal, such as hyperlocomotion, impairments in information processing and increased stereotypic behaviour, and changes in the glutamatergic, dopaminergic and GABAergic systems (reviewed in Bubenikova-Valesova et al., 2008). The influence of PCP administration on mRNA expression levels has been reported already in 2002. Applying a Clontech Atlas glass mouse 1.0 arrays, 28 genes have been found to be upregulated (including tyrosine 3-hydroxylase, Th) and 45 genes were downregulated (including voltage-gated potassium channel, subfamily H (eag-related), member 1, Kcnh1) in mice injected with PCP once a day for 24 days (Toyooka et al., 2002). Using newer microarrays representing more genes and following acute PCP treatment, mRNA expression changes in 477 genes have been observed in adult rat prefrontal cortex (PFC), a brain area associated with cognitive dysfunction in schizophrenia. One of the downregulated genes was voltage-gated K+ channel Kcna4 (Kaiser et al., 2004), yet another potassium voltage-gated

Chapter 1 - General Introduction
18
channel gene differentially expressed in an animal model for schizophrenia. The observed upregulation of c-fos, Klf2, Nr4a3, Klf4 has been confirmed in an independent study that also reported the upregulation of Leiomodin 2 (Lmod2) in the thalamus of male rats following acute PCP administration at PND 50 but not at PND 8, suggesting that Lmod2 may be involved in the age-dependent onset of drug-induced schizophrenia-like psychosis (Takebayashi et al., 2009). Another antagonist of the NMDA receptor is ketamine, a PCP derivative (Annetta et al., 2005). In the to our knowledge only report on the genome-wide analysis of mRNA expression following ketamine administration, 50 genes have been found to be differentially expressed in the combined hippocampus, amygdala and hypothalamus regions of singly injected mice (Lowe et al., 2007). A number of the differentially expressed genes is associated with tissue/organ/system development, such as gastrulation brain homeobox 2 (GBX2) and early growth response 2 (EGR2) (Chen et al., 2009; De et al., 2005), or cancer/apoptosis, such as protein kinase C delta (PRKCD), secreted phosphoprotein 1 (SPP1/OPN), prostaglandin-endoperoxide synthase 2 (Ptgs2; also named cyclo-oxygenase 2, COX-2) and growth arrest and DNA-damage-inducible 45 gamma (GADD45g) (Basu, 2003; de Moraes et al., 2007; Sheikh et al., 2000; Wai et al., 2004). The investigators followed up on Troponin T1 (Tnnt1) since this gene showed consistently strongly (2- to 4-fold) elevated expression in the brains of treated mice and was found to be exclusively regulated by FoxO1, a gene thought to be involved in metabolic pathways and energy homeostasis (Lowe et al., 2007). The activation of FoxO1 in the hypothalamus of normal mice increases food intake and body weight, whereas its inhibition decreases both processes, suggesting that FoxO1 indeed plays a role in central energy homeostasis and hunger/appetite behaviours (Kim et al., 2006). Weight gain and abdominal adiposity may be central to the pathophysiology of the metabolic syndrome observed among neuropsychiatric patients (Holt et al., 2004), and Tnnt1 may be the link between ketamine and weight gain (Lowe et al., 2007). A further antagonist of the NMDA receptor is MK-801 or dizocilpine (Woodruff et al., 1987). The effect of MK-801 on mRNA expression levels following spinal cord injury (SCI) has already been analysed in 2002 (Nesic et al., 2002). Although SCI is not linked to schizophrenia, it is still of interest to compare the effects of the NMDA receptor antagonists PCP, ketamine, and MK-801 following SCI. One hour after traumatizing adult male Sprague-Dawley rats, the mRNA levels of 165 genes and expressed sequence tags (ESTs) were changed on rat neurobiology U34 microarrays (Affymetrix). The affected mRNA levels were mostly of genes that are involved in transcription, inflammation, cell survival and membrane excitability. Intrathecal administration of MK-801 directly to the injury site at 1 hour before the injury and at 1 hour after the injury reversed the effect of SCI on the expression levels of about half of the SCI-affected mRNAs. Additionally, MK-801 treatment changed the mRNA levels of 168 genes and ESTs that had not been affected by SCI

Chapter 1 - General Introduction
19
(Nesic et al., 2002). In another study, ischemic insults were induced in fasted male Wistar rats and hippocampal mRNA levels were analysed with an Affymetrix Rat Genome 230 2.0 array (31020 probe sets). Directly following the lethal ischemic insult 967 genes were upregulated in vehicle/IPC versus MK-801/IPC rat brains, while 926 genes were downregulated. The investigators further investigated the down regulation of COX-2(Ptgs2) and confirmed by Western blot analysis that at 24 h following lethal ischemia the expression of COX-2/Ptgs2 was substantially reduced in the vehicle/IPC group in comparison to the vehicle/sham (non-preconditioned) and MK801/IPC groups. Thus, in this study MK-801 treatment increased the expression of COX-2/Ptgs2 while in the study of Lowe et al (2007) ketamine treatment reduced the expression of this gene (Feng et al., 2007; Lowe et al., 2007). Since the two studies were published simultaneously, this discrepancy was not discussed by the authors. The use different conditions (3 mg/kg MK-801 prior to IPC, followed by lethal ischemia after 72 hours versus a single i.p. injection of 80 mg/kg ketamine) may explain the discrepancy. In another study, the influences of MK-801 and the uncompetitive NMDA receptor antagonist memantine (1-amino-3,5-dimethyladamantane) on brain mRNA expression were compared (Marvanova et al., 2004). Male adult Wistar rats were i.p. injected with 5-50 mg/kg memantine or 1 mg/kg MK-801 and the posterior cingulate and anterior retrosplenial cortices were dissected for RNA isolation. Microarray analyses (1090 genes) revealed that 34 genes were regulated by the MK-801 treatment and that 28 genes were differentially expressed by the memantine treatment. Of these, 13 genes were regulated by both treatments, including nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (Nfkbia), neurotrophic tyrosine kinase, receptor, type 2 (Trk2), heat shock protein 4 (Hspa4/Hsp70), heat shock protein 5 (Hspa5/GRP78), GDP dissociation inhibitor Gdi11 (), hippocalcin-like 4 (Hpcal4/NVP-2), glutamate receptor, ionotropic, N-methyl D-aspartate 2B (Grin2b/NMDAR2B), Neuromedin B transporter (Nmbr), neuropeptide Y (Npy), corticotropin releasing hormone (Crh/CRF), Na+/K+ transporting ATPase 2β (Atp1b2), Ca2+ transporting ATPase-isoform 2 (Atp2b2) and protein phosphatase 2 (formerly 2A), catalytic subunit, beta isoform (Ppp2cb; Marvanova et al., 2004). In a related study, male Wistar rats were repeatedly i.p. injected with 0.13 mg/kg MK-801 once daily for 14 days. Gene expression in the bilateral amygdalae was analysed with a Rat Expression Array 230A Array spotted with 15923 probe sets (Affymetrix). Twenty-three genes were downregulated and sixteen genes were upregulated of which arginine vasopressin (Avp), prostaglandin D2 synthase (Ptgds), myelin-associated oligodendrocyte basic protein (Mobp; all downregulated) and insulin-like growth factor binding protein 2 (Igfbp2), somatostatin receptor 2 (Sstr2), ectonucleotide pyrophosphatase/phosphodiesterase 2 (Enpp2) and Ttr (upregulated) were confirmed by qPCR (Matsuoka et al., 2008). In the four studies applying MK-801 treatment (Feng et al., 2007; Marvanova et al.,

Chapter 1 - General Introduction
20
2004; Matsuoka et al., 2008; Nesic et al., 2002), four different sets of affected genes have been identified, presumably due to the differences in the use of animals, injection methods, brain tissues and arrays. The significance of these four gene sets remains to be established. The most common hypothesis concerning the aetiology of schizophrenia is the dopaminergic hypothesis which was built on the finding that the clinical effectiveness of antipsychotic drugs is related to their affinity for DA receptors. Since then advances in neurochemical imaging and a further understanding of the genetic aetiology of schizophrenia have led to the hypothesis that multiple (genetic, environmental) factors interact, resulting in DA dysregulation in schizophrenia patients (Howes et al., 2009). It is therefore not surprising that DA receptor agonists are useful in creating animal models to explore this hypothesis. An example of a DA receptor agonist is apomorphine (Depatie et al., 2001). Apomorphine-susceptible rats (so-called APO-SUS rats) constitute a model for schizophrenia. These rats have been pharmacogenetically selected based on their susceptibility for apomorphine as measured by their gnawing behaviour (>500 gnawing counts in 45 minutes). Extensive characterization of the APO-SUS rats and their phenotypic counterparts apomorphine-unsusceptible (APO-UNSUS) rats has shown that both genetic and environmental factors influence the complex phenotype of these rats (Ellenbroek et al., 2000). Microarray analysis of hippocampal mRNA using Rat NeuroGEM 2.02 microarrays containing 8478 Sprague Dawley rat nervous system cDNAs revealed that only one gene was differentially expressed between the two rat lines, namely Aph-1b. Further analysis showed that the expression difference of this γ-secretase component was caused by a gene-dosage imbalance, namely one or two copies of Aph-1b in APO-SUS rats and three copies in APO-UNSUS rats (Coolen et al., 2005). Pharmacological models can also be generated by the treatment of animals with schizophrenia drugs such as olanzapine or clozapine (Geyer, 1998). Olanzapine is a second-generation antipsychotic agent that exhibits greater 5HT2A than D2 receptor antagonism and has high affinity for a number of other receptors, such as
-adrenergic, histamine-H1, 5HT2A, 5HT2C, 5HT1A, 5HT6, 5HT7, and 5HT3 receptors (Bymaster et al., 1997). Additionally, olanzapine facilitates NMDA neurotransmission via its effects on glutamatergic NMDA and AMPA receptors (Tarazi et al., 2003). To study the influence of chronic olanzapine treatment, adult male Sprague–Dawley albino rats were administered daily for 21 days with 1 ml olanzapine (2 mg/kg/day, i.p.) and sacrificed 24 h after the last injection under deep anaesthesia. Gene expression in the frontal cortex was analysed using an Affymetrix rat genome 230 2.0 array set. The results showed down regulation of 31 genes and upregulation of 38 genes in the drug-treated versus control brains. The application of real-time qPCR verified the magnitude and direction of the changes for a number of significantly affected genes, including

Chapter 1 - General Introduction
21
calbindin 3 (S100g), reelin, RGS2, and insulin 2 (Fatemi et al., 2006). Recently, Duncan et al. (2008) compared the effects of the conventional antipsychotic drug haloperidol on gene expression with the effects of the atypical drugs olanzapine and clozapine (Duncan et al., 2008). Haloperidol (1 mg/kg), olanzapine (10 mg/kg), or clozapine (10 mg/kg) was administered via daily i.p. injection for 7 days to male C57BL/6 mice. Five hours after the last injection, the mice were sacrificed under 4% halothane anaesthesia. Whole-brain gene expression profiles were analysed using the Affymetrix GeneChip Mouse Genome 430 2.0 microarrays. Data analysis revealed that ~12,000 out of over 39,000 transcripts showed > 1.5-fold change by at least one drug compared with saline-treated controls and 249 transcripts were coregulated by all three antipsychotic drugs. Twenty candidate genes were chosen for further expression analysis based on their regulation by multiple drugs, the chromosomal location of their human orthologs, pattern of expression, or relevant neurobiological function (Duncan et al., 2008). Some of these genes have been genetically associated with schizophrenia, namely neuroplastin (Nptn/Sdfr1), numb homolog (Drosophila)-like (Numbl), proteolipid protein 1 (Plp1), gamma-aminobutyric acid (GABA) A receptor, α1 (Gabra1), and reticulon 4 (Rtn4/NogoA) (Novak et al., 2002; Passos Gregorio et al., 2006; Petryshen et al., 2005; Qin et al., 2005; Saito et al., 2007). Thirteen out of the twenty genes showed a statistically significant change in mRNA expression as determined by qPCR. To examine whether the changes in mRNA expression had led to changes into protein expression, whole-brain expression of three of the proteins was analysed with Western blots. The Kv channel interacting protein 3 (Kcnip3) was downregulated in brain tissues of the haloperidol- and olanzapine-treated rats, Kv1.1 was upregulated in the haloperidol-treated rats and NEDD4 was downregulated in the olanzapine-treated rats. The authors hypothesised that the specific effects of multiple antipsychotic drugs on the expression of four genes regulating voltage-gated ion channels –potassium voltage-gated channel, Shaker-related subfamily, member 1 (Kcna1), Potassium voltage-gated channel, Shaker-related subfamily, beta member 1 (Kcnab1), Kcnip3 and Neural precursor cell expressed, developmentally down-regulated gene 4 (Nedd4)– may indicate a role for these neuronal regulators in antipsychotic drug action (Duncan et al., 2008). In another study on clozapine-treated rats, no genes encoding a potassium voltage-gated ion channel have been found to be deregulated (Kontkanen et al., 2002). Male Wistar rats (~200 g) were given a single clozapine (25 mg/kg) i.p. injection and sacrificed 1, 6 or 24 hrs after the drug administration. Gene expression in the prefrontal and anterior cingulate cortices was analyzed with an Atlas Rat 1.2 Array filter (Clontech) of 1176 sequence-verified cDNAs. In situ hybridization was used to validate the regulation of 35 genes with the most pronounced changes in the filter array experiments or their close functional relatives. These genes belong to many different functional classes. A modest but consistent increase (~15%) of chromogranin A (Chga)

Chapter 1 - General Introduction
22
mRNA was observed in the outer and inner layers of the prefrontal cortex at 6 hrs after a single clozapine injection. Twenty four hrs after clozapine treatment synaptotagmin V mRNA was down regulated in the inner layers of the frontal cortex (~30%), and in the outer and inner layers of the parietal cortex (~15–20%). These results were consistent with the filter array results. In situ hybridization confirmed the upregulation (~20%) of calcineurin A mRNA in the inner layers of the infralimbic cortex 6 hrs after clozapine treatment and a more pronounced upregulation (~70%) in the inner layers of the frontal cortex 24 hrs after the treatment. Following a chronic treatment (daily injection for 17 days) with haloperidol or clozapine, six of the 35 genes were differentially expressed as shown by in situ hybridization. The chronic clozapine treatment changed the expression of Chga, Son of sevenless (Sos) and Syntaxin binding protein 1 (Stxbp1/Sec1) (Kontkanen et al., 2002). An effect of such chronic clozapine treatment has been reported previously for Chga mRNA expression in striatum, nucleus accumbens, dorsal raphe nucleus and prefrontal cortex (Bauer et al., 2000; Kroesen et al., 1995). The expression of Inhibitor of DNA-binding 2 (Id2), RAB12, member RAS oncogene family (Rab12) and Visinin-like protein 2 was changed by chronic treatment with haloperidol. Interestingly, yisinin-like protein 1 has been found previously to be down regulated 1 and 24 hrs after a single PCP injection (8 mg/kg) in the entorhinal cortex and nucleus accumbens of male Sprague-Dawley rats, respectively (Kajimoto et al., 1995). Investigators who studied previously the effect of PCP on gene expression in mice (Toyooka et al., 2002) also studied gene expression in male Institute of Cancer Research mice (Charles River Laboratories) that received daily subcutaneous injections of clozapine (7.5 mg/kg) for 20 days. Mice were sacrificed 20 hrs after the last injection and mRNA expression in the cerebral cortical tissues was analysed with a Clontech Atlas Mouse 1.2 Array membrane. Of the 1176 genes on this membrane, the expression of potassium channel Kcnab1 and Ptgds was decreased, while the expression of serotonin receptor type 2 and myelin-associated oligodendrocytic basic protein was increased (Takahashi et al., 2004). In a more recent study, the chronic effects of haloperidol, clozapine and phenylpropanolamine (an anorexiant drug used to encourage weight loss) on 10-12 week old male C57BL6 mice were examined (Mehler-Wex et al., 2006). Mice were administered with the drugs orally via food pellets daily for 31 days and gene expression in the cortices was analysed using the Murine Genome U74A Array (Affymetrix) containing ~12000 genes. The authors focused on differentially expressed genes involved in body weight regulation since antipsychotic drugs such as clozapine are associated with clinically significant weight gain (Taylor et al., 2000). However, although the mice treated with clozapine had the highest food intake they weighed significantly less than the control mice, presumably because the induced weight gain in rodents treated with antipsychotic drugs is influenced by a variety of factors (Cooper et al., 2008). The

Chapter 1 - General Introduction
23
genes aldolase 3 (Aldo3) and phospholipase C beta 1 (Plcb1), involved in glycolysis and lipid metabolism, respectively, were differentially expressed in the clozapine-treated but not in the haloperidol-treated mice (Mehler-Wex et al., 2006), suggesting that these genes are involved in the regulation of body weight rather than in schizophrenia. Although drugs such as haloperidol and the atypical antipsychotic risperidone may have different receptor affinities, they interfere with neural plasticity and signal transduction in a similar manner (Feher et al., 2005). Adult Sprague-Dawley rats were daily injected i.p. with haloperidol or risperidone and following acute or chronic (4 weeks) treatment gene expression in the prefrontal/fronto-parieto-temporal cortices was analysed with arrays containing 8000 rat genes. The acute treatment with risperidone affected the expression of more genes than the acute haloperidol treatment (89 and 28 genes, respectively). This could be due to the fact that the high-potential D2-, 5HT2- and NA-antagonist risperidone binds to diverse receptors, while haloperidol acts only as a D2-antagonist. Of the 28 genes differentially expressed upon haloperidol treatment, 18 were also differentially expressed following risperidone treatment. In general, each of the drugs affected genes with specific functions in signal transduction, transcriptional and translational regulation, protein turnover, and cellular metabolism (Feher et al., 2005). The effect of chronic risperidone treatment of rats on mRNA expression levels in the frontal cortex was also studied by Chen and Chen (2005). The use of GeneMap Rat Clone Arrays containing cDNA fragments of 1536 genes revealed 17 genes to be differentially expressed, eight of which were validated by qPCR. None of these eight genes were found to be differentially expressed in the acute and chronic risperidone study of Fehér et al. (Feher et al., 2005). mRNA expression profiling in genetic animal models In most cases, a genetic animal model for schizophrenia carries a mutation in a gene involved in brain development (Chen et al., 2006). The mutation is usually in a dopaminergic or a glutamatergic gene, in line with the use of DA agonists and NMDA antagonists in pharmacological models for schizophrenia (Gainetdinov et al., 2001). Examples of dopaminergic and glutamatergic mutant animals include the DA transporter (DAT), Nurr1 and glutamate receptor, ionotropic, NMDA2A (Grin2a/NR2A) knockout (KO) mice, and glutamate receptor, ionotropic, NMDA1 (Grin1/NR1) knock down mice (Miyamoto et al., 2001; Mohn et al., 1999; Spielewoy et al., 2000). To our knowledge, gene expression has not been analyzed in any of these nor in other dopaminergic or glutamatergic mutant mice, except in the Nurr1 KO mice. Nurr1, an orphan member of the steroid/thyroid hormone receptor family, is an absolute requirement for the proper development of midbrain DA neurons. Homozygous Nurr1 KO mice fail to generate midbrain DA neurons, are hypoactive and die soon after birth (Zetterstrom et al., 1997). Gene expression

Chapter 1 - General Introduction
24
has been analyzed in the ventral midbrain of homozygous Nurr1 KO mice at embryonic day 12.5 and PND0 using Affymetrix MGU74Av2 and MGU74Bv2 gene chips. Among the differentially expressed genes were DA-related genes such as tyrosine hydroxylase (TH) and DAT, and also guanosine 5’-triphospate (GTP) cyclohydrolase 1 (Gch1/GTPCH), a rate-determining enzyme in the synthesis of tetrahydrobiopterin (BH4), a cofactor essential for TH (Gil et al., 2007). Genetic animal models have also been developed on the basis of genes thought to play an important role in the aetiology of schizophrenia. One of such genes is Reelin (RELN) (Grayson et al., 2006; Impagnatiello et al., 1998; Kahler et al., 2008) and therefore the heterozygous reeler mouse, with a reelin insufficiency, has become of interest as an animal model (Tueting et al., 1999). In the reeler mouse brain, the expression of the mouse homologue of Strawberry Notch (Sno) was reduced. In vitro assays have shown that reelin treatment indeed enhanced the expression of Sno. Being a component of the notch signaling pathway in Drosophila (Majumdar et al., 1997), Sno thus links the reelin signal to the regulation of neurogenesis (Baba et al., 2006). A further schizophrenia animal model is represented by the Df1/+ mouse because it carries a hemizygous deletion from Es2 to Ufd1l (referred to as Df1 (Lindsay et al., 1999)), a region syntenic with a human 22q11 deletion causing DiGeorge syndrome; 35% of the DiGeorge syndrome patients develop schizophrenia (Jurata et al., 2006). In hippocampal dentate granule neurons of 10-weeks-old Df1/+ mice, two genes with a ribosomal protein function (Rps2 and Arbp) have been found to be upregulated. Eleven genes were downregulated, ten of which were located in the 22q11 region, including the gene encoding COMT. The only down-regulated gene not located in the deleted region was B-cell CLL/lymphoma 7a (Bcl7a), a gene with an unknown function (Jurata et al., 2006). Other genetic animal models for schizophrenia concern mice lacking the protein kinase C interacting protein/histidine triad nucleotide binding protein 1 (PKCI/HINT1) (Barbier et al., 2007; Vawter et al., 2004), brain-derived neurotrophic factor (BDNF) (Gorski et al., 2003; Kernie et al., 2000; Lyons et al., 1999), neural cell adhesion molecule (Ncam) 180 (Wood et al., 1998), cyclin-dependent kinase 5 (Cdk5) (Ohshima et al., 1996), neural PAS domain protein 3 (NPAS3) (Erbel-Sieler et al., 2004) or LIM homeobox protein 5 (Lhx5) (Paylor et al., 2001), mice carrying a mutation in proline dehydrogenase 2 (PRODH2) (Gogos et al., 1999), Disrupted-in-Schizophrenia-1 (DISC1) (Li et al., 2007; Pletnikov et al., 2008) or neuregulin-1 (Karl et al., 2007) as well as the Chakragati (Ckr) mouse (Torres et al., 2004). No gene expression analyses have been performed on any of these genetic animal models. This overview of the microarray analyses performed in animal models for schizophrenia illustrates that we still can not provide a conclusive answer to the question which genes play a role in the aetiology of this complex disorder. The

Chapter 1 - General Introduction
25
numerous mRNA expression studies have resulted in even more genes that could play a role in the onset of schizophrenia than was previously thought. APO-SUS and APO-UNSUS rat model In this thesis, the APO-SUS/APO-UNSUS rat model mentioned above is examined. This model has been generated from Wistar rats on the basis of their susceptibility to the DA receptor agonist apomorphine and is characterized by dopaminergic dysregulation. The APO-SUS rats display a high susceptibility to apomorphine, an increased susceptibility to dopamimetric drugs, an increased expression of TH (arcuate nucleus and substantia nigra pars compacta) and DA receptor D1 (lateral caudate putamen) mRNAs, and an increased DA receptor D2 binding in the lateral and medial caudate putamen when compared to APO-UNSUS rats (Cools et al., 1990; Ellenbroek et al., 2000; Rots et al., 1996a). The pharmacogenetic selection has been performed twice, independently and with a decade difference in time, resulting in the so-called original and replicate APO-SUS and APO-UNSUS rat lines (Ellenbroek et al., 2000). The genetic and epigenetic background of both the original and replicate rat lines have been studied (Coolen et al., 2006a; Coolen et al., 2006b; Coolen et al., 2005; Van Loo et al., 2007). The APO-SUS and APO-UNSUS rats differ not only in their dopaminergic pathway, but also in a number of other aspects. For example, APO-SUS rats display a reduced prepulse inhibition (PPI) (Ellenbroek et al., 1995), a relative dominance of type 2 helper T cells (Kavelaars et al., 1997), an increased occurrence of periodontitis (Breivik et al., 2000), and a reduced sensitivity for (lung) cancer/metastasis (Teunis et al., 2002) and rheumatoid arthritis (Van de Langerijt et al., 1994) relative to APO-UNSUS rats. Interestingly, schizophrenia patients also display abnormalities in their brain information processing and immune system (Braff et al., 1990; Leucht et al., 2007). The rats also differ in their neuroendocrine systems. The APO-SUS rats from the original line have a hyperactive hypothalamic-pituitary-adrenal (HPA) axis compared to APO-UNSUS rats. In the plasma of APO-SUS rats the basal and stress-induced levels of adrenocorticotropic hormone (ACTH) are higher and this difference in the HPA-axis seems to occur earlier in the development of the rats than the difference in the dopaminergic system (Rots et al., 1995; Rots et al., 1996b; Rots et al., 1996c). Interestingly, in schizophrenia patients higher basal levels of ACTH and an hyperactive HPA-axis have also been described (Ryan et al., 2004; Walker et al., 2008; Walsh et al., 2005). On the basis of the above, the APO-SUS rats have been proposed to represent a model with features reminiscent of characteristics displayed by schizophrenia patients (Ellenbroek et al., 1995). Scope and outline of this thesis The aim of this thesis was to identify candidate genes associated with schizophrenia. To this end, two research lines were pursued. The APO-SUS rats

Chapter 1 - General Introduction
26
were studied to get an insight into the molecular pathways underlying their dopaminergic dysregulation and to get more information on the connection between the HPA-axis and the dopaminergic pathway. Furthermore, human genetic association studies were performed to identify non-synonymous SNPs in neurodevelopmental genes associated with schizophrenia. Chapter 2 describes mRNA expression profiling in various APO-SUS and APO-UNSUS brain regions. The analysis using an Affymetrix GeneChip® Rat Exon 1.0 ST Array harbouring 27,342 transcripts reveals that besides the previously reported Aph-1b only one other gene, encoding the potassium channel-interacting protein KCnIP1, is significantly differentially expressed with a similar regulation (upregulated) in the brain tissues from both the replicate and original APO-SUS rats. We discuss the possible implication of this finding for the APO-SUS/APO-UNSUS rat model and for the complex disorder schizophrenia. Chapter 3 reports a difference between the original and replicate APO-SUS/APO-UNSUS rat lines with respect to the activities of their HPA-axes. It has been reported previously that the APO-SUS rats from the original lines display a hyperactive HPA-axis (Rots et al., 1995) and we here measure ACTH and free corticosterone to replicate these findings. However, we find that APO-SUS rats from the replicate line do not display a hyperactive HPA-axis. We therefore challenge the previously hypothesized synergistic relationship between the HPA-axis and the dopaminergic pathway. Furthermore, we describe the role of a non-synonymous SNP in corticosteroid-binding globulin (CBG), a SNP that has previously been associated with affected free-corticosterone levels (Smith et al., 1991). Chapter 4 communicates an association study on 396 selected non-synonymous SNPs in three independent Caucasian schizophrenia case-control cohorts. A meta-analysis shows ten SNPs that are associated with schizophrenia. Correction for multiple testing reveals SNP rs6336 in neurotrophic tyrosine kinase receptor (NTRK1, better known as TRKA) as the most attractive SNP for further study. We discuss the potential role of this SNP and of TRKA in schizophrenia. Chapter 5 examines TRKA SNP rs6336 in, besides the three cohorts reported in Chapter 4, an additional five independent Caucasian schizophrenia case-control cohorts. In this study, the SNP is found as a risk allele in five cohorts, whereas it acts as a protective allele in three cohorts. We discuss the dual character of this SNP and speculate on possible causes for this duality.

Chapter 1 - General Introduction
27
In the general discussion (Chapter 6), the results described in chapters two to five are discussed and placed in a broader context. References AmericanPsychiatricAssociation. Diagnostic and statistical manual of mental disorders (4th ed.).
Arlington: American Psychiatric Publishing, Inc. 1994. Annetta MG, Iemma D, Garisto C, Tafani C, Proietti R. Ketamine: new indications for an old
drug. Curr Drug Targets 2005;6:789-794. Baba K, Dekimoto H, Muraoka D, Agata K, Terashima T, Katsuyama Y. A mouse homologue of
Strawberry Notch is transcriptionally regulated by Reelin signal. Biochem Biophys Res Commun 2006;350:842-849.
Barbier E, Zapata A, Oh E, Liu Q, Zhu F, Undie A, et al. Supersensitivity to amphetamine in protein kinase-C interacting protein/HINT1 knockout mice. Neuropsychopharmacology 2007;32:1774-1782.
Basu A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med 2003;7:341-350.
Bauer R, Mayr A, Lederer W, Needham PL, Kilpatrick IC, Fleischhacker WW, et al. Further evidence that behavioral tests and neuropeptide mRNA and tissue level alterations can differentiate between typical and atypical antipsychotic drugs. Neuropsychopharmacology 2000;23:46-55.
Bentall RP, Fernyhough C. Social predictors of psychotic experiences: specificity and psychological mechanisms. Schizophr Bull 2008;34:1012-1020.
Bijl RV, Ravelli A, van Zessen G. Prevalence of psychiatric disorder in the general population: results of The Netherlands Mental Health Survey and Incidence Study (NEMESIS). Soc Psychiatry Psychiatr Epidemiol 1998;33:587-595.
Birchmeier C. ErbB receptors and the development of the nervous system. Exp Cell Res 2009;315:611-618.
Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry 1990;47:181-188.
Breivik T, Sluyter F, Hof M, Cools A. Differential susceptibility to periodontitis in genetically selected Wistar rat lines that differ in their behavioral and endocrinological response to stressors. Behav Genet 2000;30:123-130.
Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010;167:261-280.
Bubenikova-Valesova V, Horacek J, Vrajova M, Hoschl C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev 2008;32:1014-1023.
Bymaster FP, Rasmussen K, Calligaro DO, Nelson DL, DeLapp NW, Wong DT, et al. In vitro and in vivo biochemistry of olanzapine: a novel, atypical antipsychotic drug. J Clin Psychiatry 1997;58 Suppl 10:28-36.
Cardno AG, Gottesman, II. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet 2000;97:12-17.
Chen J, Lipska BK, Weinberger DR. Genetic mouse models of schizophrenia: from hypothesis-based to susceptibility gene-based models. Biol Psychiatry 2006;59:1180-1188.
Chen L, Guo Q, Li JY. Transcription factor Gbx2 acts cell-nonautonomously to regulate the formation of lineage-restriction boundaries of the thalamus. Development 2009;136:1317-1326.
Chen ML, Chen CH. Microarray analysis of differentially expressed genes in rat frontal cortex under chronic risperidone treatment. Neuropsychopharmacology 2005;30:268-277.
Coolen MW, van Loo KM, Ellenbroek BA, Cools AR, Martens GJ. Ontogenic reduction of Aph-1b mRNA and gamma-secretase activity in rats with a complex neurodevelopmental phenotype. Mol Psychiatry 2006a;11:787-793.

Chapter 1 - General Introduction
28
Coolen MW, van Loo KM, van Bakel NN, Ellenbroek BA, Cools AR, Martens GJ. Reduced Aph-1b expression causes tissue- and substrate-specific changes in gamma-secretase activity in rats with a complex phenotype. Faseb J 2006b;20:175-177.
Coolen MW, Van Loo KM, Van Bakel NN, Pulford DJ, Serneels L, De Strooper B, et al. Gene dosage effect on gamma-secretase component Aph-1b in a rat model for neurodevelopmental disorders. Neuron 2005;45:497-503.
Cools AR, Brachten R, Heeren D, Willemen A, Ellenbroek B. Search after neurobiological profile of individual-specific features of Wistar rats. Brain Res Bull 1990;24:49-69.
Cooper GD, Harrold JA, Halford JC, Goudie AJ. Chronic clozapine treatment in female rats does not induce weight gain or metabolic abnormalities but enhances adiposity: implications for animal models of antipsychotic-induced weight gain. Prog Neuropsychopharmacol Biol Psychiatry 2008;32:428-436.
Dalman C, Allebeck P, Gunnell D, Harrison G, Kristensson K, Lewis G, et al. Infections in the CNS during childhood and the risk of subsequent psychotic illness: a cohort study of more than one million Swedish subjects. Am J Psychiatry 2008;165:59-65.
de Moraes E, Dar NA, de Moura Gallo CV, Hainaut P. Cross-talks between cyclooxygenase-2 and tumor suppressor protein p53: Balancing life and death during inflammatory stress and carcinogenesis. Int J Cancer 2007;121:929-937.
De S, Turman JE, Jr. Krox-20 gene expression: influencing hindbrain-craniofacial developmental interactions. Arch Histol Cytol 2005;68:227-234.
Demyttenaere K, Bruffaerts R, Posada-Villa J, Gasquet I, Kovess V, Lepine JP, et al. Prevalence, severity, and unmet need for treatment of mental disorders in the World Health Organization World Mental Health Surveys. Jama 2004;291:2581-2590.
Depatie L, Lal S. Apomorphine and the dopamine hypothesis of schizophrenia: a dilemma? J Psychiatry Neurosci 2001;26:203-220.
Desbonnet L, Waddington JL, O'Tuathaigh CM. Mutant models for genes associated with schizophrenia. Biochem Soc Trans 2009;37:308-312.
Duncan CE, Chetcuti AF, Schofield PR. Coregulation of genes in the mouse brain following treatment with clozapine, haloperidol, or olanzapine implicates altered potassium channel subunit expression in the mechanism of antipsychotic drug action. Psychiatr Genet 2008;18:226-239.
Eastwood SL, Law AJ, Everall IP, Harrison PJ. The axonal chemorepellant semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol Psychiatry 2003;8:148-155.
Ellenbroek BA, Geyer MA, Cools AR. The behavior of APO-SUS rats in animal models with construct validity for schizophrenia. J Neurosci 1995;15:7604-7611.
Ellenbroek BA, Sluyter F, Cools AR. The role of genetic and early environmental factors in determining apomorphine susceptibility. Psychopharmacology (Berl) 2000;148:124-131.
Erbel-Sieler C, Dudley C, Zhou Y, Wu X, Estill SJ, Han T, et al. Behavioral and regulatory abnormalities in mice deficient in the NPAS1 and NPAS3 transcription factors. Proc Natl Acad Sci U S A 2004;101:13648-13653.
Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse 2005;57:91-99.
Fatemi SH, Reutiman TJ, Folsom TD, Bell C, Nos L, Fried P, et al. Chronic olanzapine treatment causes differential expression of genes in frontal cortex of rats as revealed by DNA microarray technique. Neuropsychopharmacology 2006;31:1888-1899.
Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, et al. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res 2008;99:56-70.
Feher LZ, Kalman J, Puskas LG, Gyulveszi G, Kitajka K, Penke B, et al. Impact of haloperidol and risperidone on gene expression profile in the rat cortex. Neurochem Int 2005;47:271-280.

Chapter 1 - General Introduction
29
Feng Z, Davis DP, Sasik R, Patel HH, Drummond JC, Patel PM. Pathway and gene ontology based analysis of gene expression in a rat model of cerebral ischemic tolerance. Brain Res 2007;1177:103-123.
Gaebel W, Zielasek J. The DSM-V initiative "deconstructing psychosis" in the context of Kraepelin's concept on nosology. Eur Arch Psychiatry Clin Neurosci 2008;258 Suppl 2:41-47.
Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci 2001;24:527-533.
Geyer MA. Behavioral studies of hallucinogenic drugs in animals: implications for schizophrenia research. Pharmacopsychiatry 1998;31 Suppl 2:73-79.
Gil M, McKinney C, Lee MK, Eells JB, Phyillaier MA, Nikodem VM. Regulation of GTP cyclohydrolase I expression by orphan receptor Nurr1 in cell culture and in vivo. J Neurochem 2007;101:142-150.
Gogos JA, Santha M, Takacs Z, Beck KD, Luine V, Lucas LR, et al. The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nat Genet 1999;21:434-439.
Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience 2003;121:341-354.
Grayson DR, Chen Y, Costa E, Dong E, Guidotti A, Kundakovic M, et al. The human reelin gene: transcription factors (+), repressors (-) and the methylation switch (+/-) in schizophrenia. Pharmacol Ther 2006;111:272-286.
Holt RI, Peveler RC, Byrne CD. Schizophrenia, the metabolic syndrome and diabetes. Diabet Med 2004;21:515-523.
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull 2009;35:549-562.
Ibi D, Takuma K, Koike H, Mizoguchi H, Tsuritani K, Kuwahara Y, et al. Social isolation rearing-induced impairment of the hippocampal neurogenesis is associated with deficits in spatial memory and emotion-related behaviors in juvenile mice. J Neurochem 2008;105:921-932.
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999;283:70-74.
Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A 1998;95:15718-15723.
Jurata LW, Gallagher P, Lemire AL, Charles V, Brockman JA, Illingworth EL, et al. Altered expression of hippocampal dentate granule neuron genes in a mouse model of human 22q11 deletion syndrome. Schizophr Res 2006;88:251-259.
Kahler AK, Djurovic S, Kulle B, Jonsson EG, Agartz I, Hall H, et al. Association analysis of schizophrenia on 18 genes involved in neuronal migration: MDGA1 as a new susceptibility gene. Am J Med Genet B Neuropsychiatr Genet 2008;147B:1089-1100.
Kaiser S, Foltz LA, George CA, Kirkwood SC, Bemis KG, Lin X, et al. Phencyclidine-induced changes in rat cortical gene expression identified by microarray analysis: implications for schizophrenia. Neurobiol Dis 2004;16:220-235.
Kajimoto Y, Shirakawa O, Kuno T, Nishino N, Nakai H. Delayed changes in neural visinin-like calcium-binding protein gene expression caused by acute phencyclidine administration. J Neural Transm Gen Sect 1995;100:257-262.
Kanazawa T, Glatt SJ, Kia-Keating B, Yoneda H, Tsuang MT. Meta-analysis reveals no association of the Val66Met polymorphism of brain-derived neurotrophic factor with either schizophrenia or bipolar disorder. Psychiatr Genet 2007;17:165-170.
Karayiorgou M, Gogos JA. Schizophrenia genetics: uncovering positional candidate genes. Eur J Hum Genet 2006;14:512-519.
Karl T, Duffy L, Scimone A, Harvey RP, Schofield PR. Altered motor activity, exploration and anxiety in heterozygous neuregulin 1 mutant mice: implications for understanding schizophrenia. Genes Brain Behav 2007;6:677-687.
Kavelaars A, Heijnen CJ, Ellenbroek B, van Loveren H, Cools A. Apomorphine-susceptible and apomorphine-unsusceptible Wistar rats differ in their susceptibility to inflammatory and

Chapter 1 - General Introduction
30
infectious diseases: a study on rats with group-specific differences in structure and reactivity of hypothalamic-pituitary-adrenal axis. J Neurosci 1997;17:2580-2584.
Keefe RS, Fenton WS. How should DSM-V criteria for schizophrenia include cognitive impairment? Schizophr Bull 2007;33:912-920.
Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. Embo J 2000;19:1290-1300.
Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci 2006;9:901-906.
Kinnunen AK, Koenig JI, Bilbe G. Repeated variable prenatal stress alters pre- and postsynaptic gene expression in the rat frontal pole. J Neurochem 2003;86:736-748.
Koenig JI, Kirkpatrick B, Lee P. Glucocorticoid hormones and early brain development in schizophrenia. Neuropsychopharmacology 2002;27:309-318.
Kohda K, Jinde S, Iwamoto K, Bundo M, Kato N, Kato T. Maternal separation stress drastically decreases expression of transthyretin in the brains of adult rat offspring. Int J Neuropsychopharmacol 2006;9:201-208.
Kontkanen O, Toronen P, Lakso M, Wong G, Castren E. Antipsychotic drug treatment induces differential gene expression in the rat cortex. J Neurochem 2002;83:1043-1053.
Kroesen S, Marksteiner J, Mahata SK, Mahata M, Fischer-Colbrie R, Saria A, et al. Effects of haloperidol, clozapine and citalopram on messenger RNA levels of chromogranins A and B and secretogranin II in various regions of rat brain. Neuroscience 1995;69:881-891.
Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry 2007;12:572-580.
Leucht S, Burkard T, Henderson J, Maj M, Sartorius N. Physical illness and schizophrenia: a review of the literature. Acta Psychiatr Scand 2007;116:317-333.
Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet 2003;73:34-48.
Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci U S A 2007;104:18280-18285.
Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 1999;401:379-383.
Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology 1993;9:67-75.
Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology 2000;23:223-239.
Lipska BK, Weinberger DR. A neurodevelopmental model of schizophrenia: neonatal disconnection of the hippocampus. Neurotox Res 2002;4:469-475.
Lowe XR, Lu X, Marchetti F, Wyrobek AJ. The expression of Troponin T1 gene is induced by ketamine in adult mouse brain. Brain Res 2007;1174:7-17.
Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A 1999;96:15239-15244.
Majumdar A, Nagaraj R, Banerjee U. strawberry notch encodes a conserved nuclear protein that functions downstream of Notch and regulates gene expression along the developing wing margin of Drosophila. Genes Dev 1997;11:1341-1353.
Maki P, Veijola J, Jones PB, Murray GK, Koponen H, Tienari P, et al. Predictors of schizophrenia--a review. Br Med Bull 2005;73-74:1-15.
Marcotte ER, Pearson DM, Srivastava LK. Animal models of schizophrenia: a critical review. J Psychiatry Neurosci 2001;26:395-410.

Chapter 1 - General Introduction
31
Marvanova M, Lakso M, Wong G. Identification of genes regulated by memantine and MK-801 in adult rat brain by cDNA microarray analysis. Neuropsychopharmacology 2004;29:1070-1079.
Matsuoka T, Tsunoda M, Sumiyoshi T, Takasaki I, Tabuchi Y, Seo T, et al. Effect of MK-801 on gene expressions in the amygdala of rats. Synapse 2008;62:1-7.
McGuffin P. Nature and nurture interplay: schizophrenia. Psychiatr Prax 2004;31 Suppl 2:S189-193.
Mehler-Wex C, Grunblatt E, Zeiske S, Gille G, Rausch D, Warnke A, et al. Microarray analysis reveals distinct gene expression patterns in the mouse cortex following chronic neuroleptic and stimulant treatment: implications for body weight changes. J Neural Transm 2006;113:1383-1393.
Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T. Hyperfunction of dopaminergic and serotonergic neuronal systems in mice lacking the NMDA receptor epsilon1 subunit. J Neurosci 2001;21:750-757.
Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999;98:427-436.
Mueser KT, McGurk SR. Schizophrenia. Lancet 2004;363:2063-2072. Nesic O, Svrakic NM, Xu GY, McAdoo D, Westlund KN, Hulsebosch CE, et al. DNA microarray
analysis of the contused spinal cord: effect of NMDA receptor inhibition. J Neurosci Res 2002;68:406-423.
Novak G, Kim D, Seeman P, Tallerico T. Schizophrenia and Nogo: elevated mRNA in cortex, and high prevalence of a homozygous CAA insert. Brain Res Mol Brain Res 2002;107:183-189.
Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A 1996;93:11173-11178.
Palomo T, Kostrzewa RM, Archer T, Beninger RJ. Neurodevelopmental liabilities in schizophrenia and affective disorders. Neurotox Res 2002;4:397-408.
Passos Gregorio S, Gattaz WF, Tavares H, Kieling C, Timm S, Wang AG, et al. Analysis of coding-polymorphisms in NOTCH-related genes reveals NUMBL poly-glutamine repeat to be associated with schizophrenia in Brazilian and Danish subjects. Schizophr Res 2006;88:275-282.
Paylor R, Zhao Y, Libbey M, Westphal H, Crawley JN. Learning impairments and motor dysfunctions in adult Lhx5-deficient mice displaying hippocampal disorganization. Physiol Behav 2001;73:781-792.
Petryshen TL, Middleton FA, Tahl AR, Rockwell GN, Purcell S, Aldinger KA, et al. Genetic investigation of chromosome 5q GABAA receptor subunit genes in schizophrenia. Mol Psychiatry 2005;10:1074-1088, 1057.
Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 2008;13:173-186, 115.
Qin W, Gao J, Xing Q, Yang J, Qian X, Li X, et al. A family-based association study of PLP1 and schizophrenia. Neurosci Lett 2005;375:207-210.
Read J, Bentall RP, Fosse R. Time to abandon the bio-bio-bio model of psychosis: Exploring the epigenetic and psychological mechanisms by which adverse life events lead to psychotic symptoms. Epidemiol Psichiatr Soc 2009;18:299-310.
Read J, Fink PJ, Rudegeair T, Felitti V, Whitfield CL. Child Maltreatment and Psychosis: A Return to a Genuinely Integrated Bio-Psycho-Social Model. Clinical Schizophrenia & Related Psychoses 2008;7:235-254.
Read J, van Os J, Morrison AP, Ross CA. Childhood trauma, psychosis and schizophrenia: a literature review with theoretical and clinical implications. Acta Psychiatr Scand 2005;112:330-350.
Rehn AE, Rees SM. Investigating the neurodevelopmental hypothesis of schizophrenia. Clin Exp Pharmacol Physiol 2005;32:687-696.
Riley B, Kendler KS. Molecular genetic studies of schizophrenia. Eur J Hum Genet 2006;14:669-680.

Chapter 1 - General Introduction
32
Rots NY, Cools AR, Berod A, Voorn P, Rostene W, de Kloet ER. Rats bred for enhanced apomorphine susceptibility have elevated tyrosine hydroxylase mRNA and dopamine D2-receptor binding sites in nigrostriatal and tuberoinfundibular dopamine systems. Brain Res 1996a;710:189-196.
Rots NY, Cools AR, de Jong J, De Kloet ER. Corticosteroid feedback resistance in rats genetically selected for increased dopamine responsiveness. J Neuroendocrinol 1995;7:153-161.
Rots NY, Cools AR, Oitzl MS, de Jong J, Sutanto W, de Kloet ER. Divergent prolactin and pituitary-adrenal activity in rats selectively bred for different dopamine responsiveness. Endocrinology 1996b;137:1678-1686.
Rots NY, Workel J, Oitzl MS, Berod A, Rostene W, Cools AR, et al. Development of divergence in dopamine responsiveness in genetically selected rat lines is preceded by changes in pituitary-adrenal activity. Brain Res Dev Brain Res 1996c;92:164-171.
Ruggeri M, Tansella M. The interaction between genetics and epidemiology: the puzzle and its pieces. Epidemiol Psichiatr Soc 2009;18:77-80.
Ryan MC, Sharifi N, Condren R, Thakore JH. Evidence of basal pituitary-adrenal overactivity in first episode, drug naive patients with schizophrenia. Psychoneuroendocrinology 2004;29:1065-1070.
Saito A, Fujikura-Ouchi Y, Kuramasu A, Shimoda K, Akiyama K, Matsuoka H, et al. Association study of putative promoter polymorphisms in the neuroplastin gene and schizophrenia. Neurosci Lett 2007;411:168-173.
Sanjuan J, Tolosa A, Gonzalez JC, Aguilar EJ, Perez-Tur J, Najera C, et al. Association between FOXP2 polymorphisms and schizophrenia with auditory hallucinations. Psychiatr Genet 2006;16:67-72.
Sheikh MS, Hollander MC, Fornance AJ, Jr. Role of Gadd45 in apoptosis. Biochem Pharmacol 2000;59:43-45.
Smith CL, Hammond GL. An amino acid substitution in biobreeding rat corticosteroid binding globulin results in reduced steroid binding affinity. J Biol Chem 1991;266:18555-18559.
Spielewoy C, Roubert C, Hamon M, Nosten-Bertrand M, Betancur C, Giros B. Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice. Behav Pharmacol 2000;11:279-290.
Sun S, Wang F, Wei J, Cao LY, Wu GY, Lu L, et al. Association between interleukin-3 receptor alpha polymorphism and schizophrenia in the Chinese population. Neurosci Lett 2008;440:35-37.
Sun S, Wei J, Li H, Jin S, Li P, Ju G, et al. A family-based study of the IL3RA gene on susceptibility to schizophrenia in a Chinese Han population. Brain Res 2009;1268:13-16.
Suzuki K, Nakamura K, Iwata Y, Sekine Y, Kawai M, Sugihara G, et al. Decreased expression of reelin receptor VLDLR in peripheral lymphocytes of drug-naive schizophrenic patients. Schizophr Res 2008;98:148-156.
Takahashi Y, Kumanishi T, Hayashi S. Using a DNA microarray method to examine gene expression in brain from clozapine-injected mice. Ann N Y Acad Sci 2004;1025:561-569.
Takebayashi H, Yamamoto N, Umino A, Nishikawa T. Developmentally regulated and thalamus-selective induction of leiomodin2 gene by a schizophrenomimetic, phencyclidine, in the rat. Int J Neuropsychopharmacol 2009;1-16.
Tarazi FI, Baldessarini RJ, Kula NS, Zhang K. Long-term effects of olanzapine, risperidone, and quetiapine on ionotropic glutamate receptor types: implications for antipsychotic drug treatment. J Pharmacol Exp Ther 2003;306:1145-1151.
Taylor DM, McAskill R. Atypical antipsychotics and weight gain--a systematic review. Acta Psychiatr Scand 2000;101:416-432.
Teunis MA, Kavelaars A, Voest E, Bakker JM, Ellenbroek BA, Cools AR, et al. Reduced tumor growth, experimental metastasis formation, and angiogenesis in rats with a hyperreactive dopaminergic system. Faseb J 2002;16:1465-1467.

Chapter 1 - General Introduction
33
Tordjman S, Drapier D, Bonnot O, Graignic R, Fortes S, Cohen D, et al. Animal models relevant to schizophrenia and autism: validity and limitations. Behav Genet 2007;37:61-78.
Torres G, Hallas BH, Vernace VA, Jones C, Gross KW, Horowitz JM. A neurobehavioral screening of the ckr mouse mutant: implications for an animal model of schizophrenia. Brain Res Bull 2004;62:315-326.
Toyooka K, Usui M, Washiyama K, Kumanishi T, Takahashi Y. Gene expression profiles in the brain from phencyclidine-treated mouse by using DNA microarray. Ann N Y Acad Sci 2002;965:10-20.
Tueting P, Costa E, Dwivedi Y, Guidotti A, Impagnatiello F, Manev R, et al. The phenotypic characteristics of heterozygous reeler mouse. Neuroreport 1999;10:1329-1334.
Van de Langerijt AG, Van Lent PL, Hermus AR, Sweep CG, Cools AR, Van den Berg WB. Susceptibility to adjuvant arthritis: relative importance of adrenal activity and bacterial flora. Clin Exp Immunol 1994;97:33-38.
Van den Buuse M. Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr Bull 2010;36:246-270.
Van Loo KM, Martens GJ. Identification of genetic and epigenetic variations in a rat model for neurodevelopmental disorders. Behav Genet 2007;37:697-705.
Van Os J, Kapur S. Schizophrenia. Lancet 2009;374:635-645. Vawter MP, Shannon Weickert C, Ferran E, Matsumoto M, Overman K, Hyde TM, et al. Gene
expression of metabolic enzymes and a protease inhibitor in the prefrontal cortex are decreased in schizophrenia. Neurochem Res 2004;29:1245-1255.
Wai PY, Kuo PC. The role of Osteopontin in tumor metastasis. J Surg Res 2004;121:228-241. Walker E, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the
developmental course of schizophrenia. Annu Rev Clin Psychol 2008;4:189-216. Walsh P, Spelman L, Sharifi N, Thakore JH. Male patients with paranoid schizophrenia have
greater ACTH and cortisol secretion in response to metoclopramide-induced AVP release. Psychoneuroendocrinology 2005;30:431-437.
Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004;
Weinberger DR. From neuropathology to neurodevelopment. Lancet 1995;346:552-557. Whitaker-Azmitia P, Zhou F, Hobin J, Borella A. Isolation-rearing of rats produces deficits as
adults in the serotonergic innervation of hippocampus. Peptides 2000;21:1755-1759. Williams BL, Yaddanapudi K, Kirk CM, Soman A, Hornig M, Lipkin WI. Metallothioneins and
zinc dysregulation contribute to neurodevelopmental damage in a model of perinatal viral infection. Brain Pathol 2006;16:1-14.
Winter C, Reutiman TJ, Folsom TD, Sohr R, Wolf RJ, Juckel G, et al. Dopamine and serotonin levels following prenatal viral infection in mouse--implications for psychiatric disorders such as schizophrenia and autism. Eur Neuropsychopharmacol 2008;18:712-716.
Wong AH, Lipska BK, Likhodi O, Boffa E, Weinberger DR, Kennedy JL, et al. Cortical gene expression in the neonatal ventral-hippocampal lesion rat model. Schizophr Res 2005;77:261-270.
Wood GK, Tomasiewicz H, Rutishauser U, Magnuson T, Quirion R, Rochford J, et al. NCAM-180 knockout mice display increased lateral ventricle size and reduced prepulse inhibition of startle. Neuroreport 1998;9:461-466.
Woodruff GN, Foster AC, Gill R, Kemp JA, Wong EH, Iversen LL. The interaction between MK-801 and receptors for N-methyl-D-aspartate: functional consequences. Neuropharmacology 1987;26:903-909.
Yudofsky SC. Contracting schizophrenia: lessons from the influenza epidemic of 1918-1919. Jama 2009;301:324-326.
Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science 1997;276:248-250.


Chapter 2 Gene expression profiling in brain regions of a rat model displaying schizophrenia-related features
Published as: Van Schijndel JE, Van Zweeden M, Van Loo KMJ, Martens GJM.
Gene expression profiling in brain regions of a rat model displaying schizophrenia-related features.
Behavioural Brain Research, 2010, 207:476-479.

Chapter 2 - Gene expression in a rat model for complex phenotype
36
Abstract Animal models allow insights into complex neurodevelopmental disorders. Apomorphine-susceptible rats (so-called APO-SUS rats) provide a model that displays a complex phenotype with schizophrenia-related features and together with its phenotypic counterpart (APO-UNSUS rats) has been independently generated twice (original and replicate rat lines). To understand the molecular basis underlying this phenotype, we here performed mRNA expression profiling in various APO-SUS and APO-UNSUS rat brain regions. The expression of only the previously reported Aph-1b and the newly discovered KCnIP1 (a member of the potassium channel-interacting protein family that is known to modulate neuronal channel activity) was significantly different in the APO-SUS and APO-UNSUS rats from both the original and replicate rat lines. Thus, KCnIP1 may constitute a novel candidate gene playing a role in the complex phenotype of the APO-SUS/APO-UNSUS rat model and further studies on this gene are warranted.

Chapter 2 - Gene expression in a rat model for complex phenotype
37
Introduction In order to get a better insight into the aetiology of complex neuropsychiatric disorders such as schizophrenia, in the last decades a number of animal models have been developed. Animal models have drawbacks in that some symptoms of neuropsychiatric disorders (e.g. delusions and hallucinations in schizophrenia) can not be reproduced in rodents and even not in non-human primates. Furthermore, to validate animal models a well-established genotype and a cellular phenotype or another biological marker is often lacking. Nevertheless, animal models provide new insights into the pathophysiology and aetiology of complex neurodevelopmental disorders (Lipska et al., 2000). For instance, animal models allow studies on the role of genetic and environmental factors in the aetiology of the disorders. In addition, the models are being used to identify molecular pathways and neuronal circuits that are responsible for specific neural abnormalities and their associated behavioral impairment. For our research, we use rats selected for a high susceptibility to the dopamine (DA) receptor agonist apomorphine (so-called APO-SUS rats). These rats display also an increased susceptibility to dopamimetic drugs, an increased expression of both tyrosine hydroxylase and DA receptor D1 mRNA, and an increased DA receptor D2 binding when compared to rats with a low susceptibility to apomorphine (APO-UNSUS rats) (Cools et al., 1990; Ellenbroek et al., 2000; Rots et al., 1996a). Dopaminergic dysregulation has been observed in schizophrenic patients as well (Corripio et al., 2006; Muller-Spahn et al., 1998). Furthermore, as in schizophrenia the disturbances in the dopaminergic system occur from adolescence onwards. For instance, in the lateral caudate putamen of 10- and 18-days old APO-SUS and APO-UNSUS rats dopamine D1 receptor expression was not different, while in this brain area an increase in expression was detected at the age of ~60 days (Rots et al., 1996a; Rots et al., 1996c). The APO-SUS and APO-UNSUS rats differ not only in their dopaminergic pathway, but also in their information processing, neuroendocrine and immune systems. For example, APO-SUS rats display a reduced prepulse inhibition (PPI) (Ellenbroek et al., 1995), an increased hypothalamus-pituitary-adrenal axis response to stress (Rots et al., 1996b), a relative dominance of type 2 helper T cells (Kavelaars et al., 1997), an increased occurrence of periodontitis (Breivik et al., 2000), and a reduced sensitivity for (lung) cancer/metastasis (Teunis et al., 2002) and rheumatoid arthritis (Van de Langerijt et al., 1994) relative to APO-UNSUS rats. Interestingly, epidemiological studies have shown that schizophrenic patients have a reduced PPI (Braff et al., 1990), a higher prevalence of bacterial (tuberculosis) and viral (HIV, hepatitis B/C) infections and dental problems, and a lower risk of cancer and rheumatoid arthritis (Leucht et al., 2007). Thus, the APO-SUS characteristics other than its dopaminergic susceptibility are also reminiscent

Chapter 2 - Gene expression in a rat model for complex phenotype
38
of features observed in schizophrenia. A number of the APO-SUS characteristics (and indeed of the schizophrenia characteristics) have not been found in other neuropsychiatric or apomorphine-related disorders. For example, no link has been found between bipolar disorder and cancer risk (Hippisley-Cox et al., 2007) or between Parkinson’s disease and rheumatoid arthritis (Rugbjerg et al., 2009). Crossbreeding experiments have shown that, like in schizophrenia, genetic factors contribute to the phenotype of the APO-SUS/APO-UNSUS rats (Abel, 2004; Ellenbroek et al., 2000). Moreover, cross-fostering of APO-SUS pups (by an APO-UNSUS mother) significantly reduced apomorphine-induced gnawing in the adult APO-SUS rats, whereas maternal deprivation of APO-UNSUS pups resulted in increased apomorphine-induced gnawing of these rats later in life (Ellenbroek et al., 2000). The two phenotypes can therefore be permanently reversed by experiences during critical periods in early life (environmental factors). Based on the above-mentioned findings, the APO-SUS rats have been proposed to represent a model displaying features reminiscent of schizophrenia (Ellenbroek et al., 1995). The pharmacogenetic selection for apomorphine susceptibility has been performed twice, independently and with a decade difference in time, resulting in the so-called original and replicate APO-SUS and APO-UNSUS rat lines (Ellenbroek et al., 2000). Hippocampal mRNA expression profiling using the Rat NeuroGEM 2.02 microarray (8,478 transcripts), which represents less than a third of the genes known to date, has shown that in both the original and replicate rat lines only one gene was differentially expressed, namely anterior pharynx defective 1 homolog B (Aph1b) (Coolen et al., 2005); the Aph1b protein is a component of the γ-secretase complex that cleaves many type I transmembrane proteins (Shirotani et al., 2004). In order to identify differentially expressed genes other than Aph1b and to get a more comprehensive insight into the pathways underlying rats with a dysregulated dopaminergic system, we now performed mRNA expression profiling in a number of brain regions of the APO-SUS and APO-UNSUS rats with an Affymetrix GeneChip® Rat Exon 1.0 ST Array harbouring 27,342 transcripts and thus covering most of the rat transcriptome. Materials and methods Animals The generation of the APO-SUS and APO-UNSUS Wistar rat lines with a high or low susceptibility for apomorphine, respectively, has been described previously for the replicate and original lines (Cools et al., 1990; Ellenbroek et al., 2000). Rats were bred and reared in the Central Animal Facility of the Radboud University Nijmegen under approved animal protocols and in accordance with institutional guidelines. Rats were tested for their apomorphine susceptibility two weeks before decapitation. Adult (~ 250 g) male APO-SUS and APO-UNSUS rats were injected

Chapter 2 - Gene expression in a rat model for complex phenotype
39
with apomorphine (1.5 mg/kg subcutaneously) and gnawing scores were measured in a gnawing box for 45 minutes, as described previously (Cools et al., 1990). After decapitation, the hippocampus, striatum, cortex, and mid- and interbrain (hereafter referred to as midbrain) were dissected, and the tissues were quick-frozen immediately following dissection in liquid nitrogen and stored at -80°C. Microarray experiment For mRNA expression profiling, brain tissue samples from APO-SUS and APO-UNSUS rats (cortex, striatum and midbrain; each n = 2) were homogenized with the Tissue Lyser (Qiagen) and total RNA was prepared with TRIzol Reagent (Invitrogen) and purified and DNase treated on NucleoSpin columns (Bioke). Array experiments were performed at ServiceXS (Leiden, The Netherlands). Quality control of the samples was performed with the Bioanalyzer (Agilent). The Affymetrix Transcript (WT) Single-Stranded Target Labeling Kit and Control Reagents were used to synthesize Biotin-labeled Single-Stranded cDNA. The Affymetrix protocols were strictly followed for hybridizations, washing, staining and scanning of the Affymetrix Rat Gene-1.0ST Genechips. All data were within the quality control specifications, according to Affymetrix Command Console specifications. Microarray data analysis The Affymetrix Command Console CEL output files were analysed using GeneSpring GX software (Agilent Technologies). Intensity values were normalized and summarized using robust multi-array analysis (RMA) for each brain tissue. Transcripts were filtered at the level of expression (20.0 - 100.0th Percentile). For cortex, striatum and midbrain samples this resulted in 22481, 22478, and 22351 transcripts, respectively. Normalized intensity values were used to determine the fold-change ratio for each transcript as an average normalized expression ratio of intensity for APO-SUS versus APO-UNSUS. Expression levels were statistically compared using a two-tailed Student’s t-Test with equal variance. Statistical significance was set at a p-value of less than 0.05. p-Values were not corrected for multiple testing since the differences in expression of all interesting genes found on the microarrays were replicated and validated by quantitative PCR. Real-time quantitative reverse transcriptase-PCR Three gene transcripts found on the microarrays to be differentially expressed (RGD1303232, Ke2, and LOC692042) were validated by quantitative PCR (qPCR; n = 4), showing the validity of the microarray results. Furthermore, the differential mRNA expression detected by microarray analysis was replicated using qPCR (n = 8). For this purpose, total RNA from hippocampus was prepared with TRIzol Reagent (Invitrogen). Genomic DNA was removed from the RNA using DNaseI (1

Chapter 2 - Gene expression in a rat model for complex phenotype
40
U/μl; Fermentas) following manufacturer’s instructions. The reverse transcription reaction was performed using the RevertAidTM H Minus First Strand cDNA Synthesis Kit (Fermentas) according to manufacturer’s instructions. The 10 μl qPCR reaction mixture contained 3 μl of 13.3-fold diluted cDNA, 1x Absolute SYBR Green mix (Thermo Scientific) and 4 μM forward and reverse primer (Biolegio). Pipeting was performed with a CAS-1200 robotic workstation (Corbett Robotics). Primers were designed using Primer Express 2.0 (sequences available upon request). Amplification was performed on a Corbett Life Science Rotor-Gene 6000 using the following protocol: denaturation for 15 min at 95°C and 40 cycles of 15 s 95°C, 15 s 60°C, 45 s 72°C. SYBR Green fluorescence was measured at the end of the extension step (72°C). After amplification, amplified DNA was dissociated by a melt from 72°C to 95°C. SYBR Green fluorescence was measured during this step to confirm single amplicon amplification. Serial dilutions of cDNA standards were used to determine the efficiency of each primer set. Critical cycle threshold (Ct) values were determined using Rotor-Gene 6000 Series Software (Corbett Research) and the relative expression of the gene of interest (GOI) was expressed as a ratio of the GOI to β-actin using the formula according to Pfaffl (2001). Results In an attempt to identify the genes that are differentially expressed in the brains of rats with a dysregulated dopaminergic pathway, we determined the mRNA expression profiles in cortex, striatum and midbrain of adult rats from the replicate APO-SUS and APO-UNSUS lines. In cortex, striatum and midbrain, 73, 93 and 42 transcripts, respectively, were significantly (p<0.05) differentially expressed with a fold change over 1.3 (fc > 1.3). Seven transcripts, including Aph1b, were significantly differentially expressed in all of the three tissues (fc > 1.3; table 1a). Fifteen transcripts tended to be differentially expressed (fc > 1.3) in all of the three tissues, although not significantly (table 1b). Thirteen transcripts were nearly significantly differentially expressed (p<0.1) in the three tissues with an >1.3-fold change in at least one tissue and an >1.2-fold change in the other two tissues (table 1c). Of these thirteen transcripts six represented ribosomal protein L38 (Rpl38), a gene displaying nucleotide sequence similarity with regions on nearly all rat chromosomes. To examine whether the thirty-five transcripts that were differentially expressed in the three brain tissues would be differentially expressed in other tissues as well, we analyzed the mRNA expression levels of twenty-one of these genes by qPCR on hippocampal mRNA of APO-SUS and APO-UNSUS rats from the replicate lines. The expression of seventeen genes (81%) was indeed differentially regulated in the hippocampus similar to that in the other three tissues with the difference being significant in eleven cases (52%; p<0.05; table 1). To
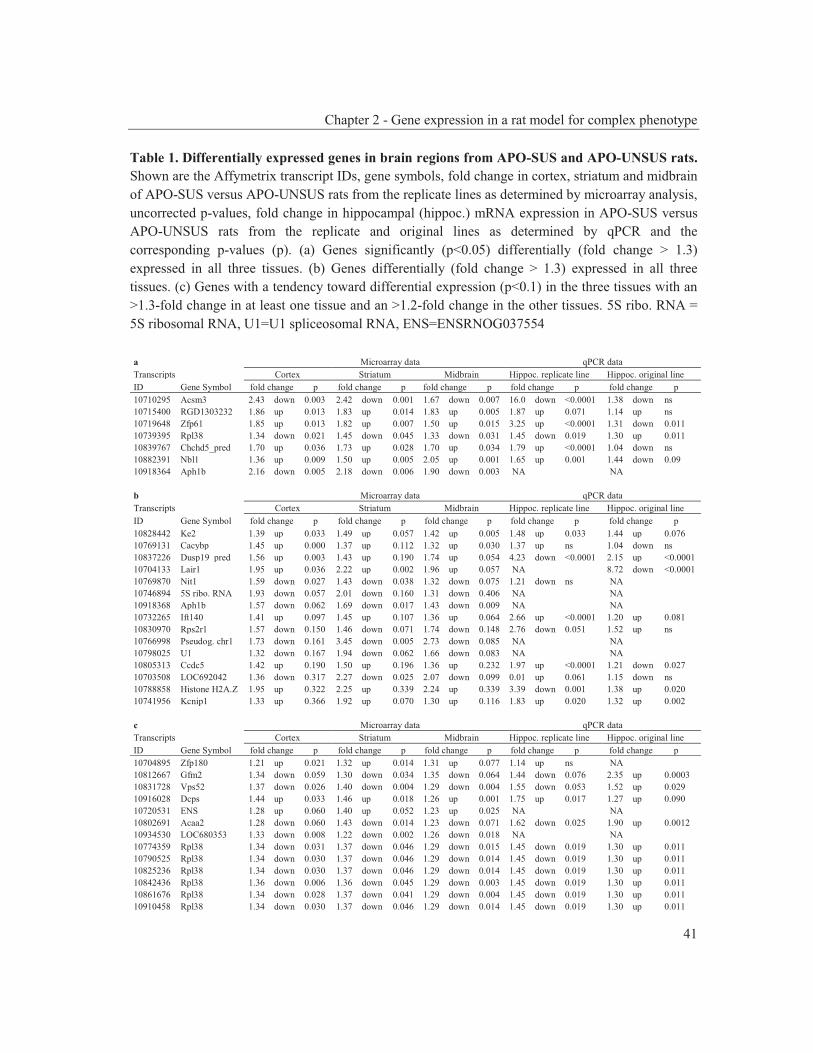
Chapter 2 - Gene expression in a rat model for complex phenotype
41
Table 1. Differentially expressed genes in brain regions from APO-SUS and APO-UNSUS rats. Shown are the Affymetrix transcript IDs, gene symbols, fold change in cortex, striatum and midbrain of APO-SUS versus APO-UNSUS rats from the replicate lines as determined by microarray analysis, uncorrected p-values, fold change in hippocampal (hippoc.) mRNA expression in APO-SUS versus APO-UNSUS rats from the replicate and original lines as determined by qPCR and the corresponding p-values (p). (a) Genes significantly (p<0.05) differentially (fold change > 1.3) expressed in all three tissues. (b) Genes differentially (fold change > 1.3) expressed in all three tissues. (c) Genes with a tendency toward differential expression (p<0.1) in the three tissues with an >1.3-fold change in at least one tissue and an >1.2-fold change in the other tissues. 5S ribo. RNA = 5S ribosomal RNA, U1=U1 spliceosomal RNA, ENS=ENSRNOG037554 a Microarray data qPCR data Transcripts Cortex Striatum Midbrain Hippoc. replicate line Hippoc. original line ID Gene Symbol fold change p fold change p fold change p fold change p fold change p 10710295 Acsm3 2.43 down 0.003 2.42 down 0.001 1.67 down 0.007 16.0 down <0.0001 1.38 down ns 10715400 RGD1303232 1.86 up 0.013 1.83 up 0.014 1.83 up 0.005 1.87 up 0.071 1.14 up ns 10719648 Zfp61 1.85 up 0.013 1.82 up 0.007 1.50 up 0.015 3.25 up <0.0001 1.31 down 0.011 10739395 Rpl38 1.34 down 0.021 1.45 down 0.045 1.33 down 0.031 1.45 down 0.019 1.30 up 0.011 10839767 Chchd5_pred 1.70 up 0.036 1.73 up 0.028 1.70 up 0.034 1.79 up <0.0001 1.04 down ns 10882391 Nbl1 1.36 up 0.009 1.50 up 0.005 2.05 up 0.001 1.65 up 0.001 1.44 down 0.09 10918364 Aph1b 2.16 down 0.005 2.18 down 0.006 1.90 down 0.003 NA NA b Microarray data qPCR data Transcripts Cortex Striatum Midbrain Hippoc. replicate line Hippoc. original line ID Gene Symbol fold change p fold change p fold change p fold change p fold change p 10828442 Ke2 1.39 up 0.033 1.49 up 0.057 1.42 up 0.005 1.48 up 0.033 1.44 up 0.076 10769131 Cacybp 1.45 up 0.000 1.37 up 0.112 1.32 up 0.030 1.37 up ns 1.04 down ns 10837226 Dusp19_pred 1.56 up 0.003 1.43 up 0.190 1.74 up 0.054 4.23 down <0.0001 2.15 up <0.0001 10704133 Lair1 1.95 up 0.036 2.22 up 0.002 1.96 up 0.057 NA 8.72 down <0.0001 10769870 Nit1 1.59 down 0.027 1.43 down 0.038 1.32 down 0.075 1.21 down ns NA 10746894 5S ribo. RNA 1.93 down 0.057 2.01 down 0.160 1.31 down 0.406 NA NA 10918368 Aph1b 1.57 down 0.062 1.69 down 0.017 1.43 down 0.009 NA NA 10732265 Ift140 1.41 up 0.097 1.45 up 0.107 1.36 up 0.064 2.66 up <0.0001 1.20 up 0.081 10830970 Rps2r1 1.57 down 0.150 1.46 down 0.071 1.74 down 0.148 2.76 down 0.051 1.52 up ns 10766998 Pseudog. chr1 1.73 down 0.161 3.45 down 0.005 2.73 down 0.085 NA NA 10798025 U1 1.32 down 0.167 1.94 down 0.062 1.66 down 0.083 NA NA 10805313 Ccdc5 1.42 up 0.190 1.50 up 0.196 1.36 up 0.232 1.97 up <0.0001 1.21 down 0.027 10703508 LOC692042 1.36 down 0.317 2.27 down 0.025 2.07 down 0.099 0.01 up 0.061 1.15 down ns 10788858 Histone H2A.Z 1.95 up 0.322 2.25 up 0.339 2.24 up 0.339 3.39 down 0.001 1.38 up 0.020 10741956 Kcnip1 1.33 up 0.366 1.92 up 0.070 1.30 up 0.116 1.83 up 0.020 1.32 up 0.002 c Microarray data qPCR data Transcripts Cortex Striatum Midbrain Hippoc. replicate line Hippoc. original line ID Gene Symbol fold change p fold change p fold change p fold change p fold change p 10704895 Zfp180 1.21 up 0.021 1.32 up 0.014 1.31 up 0.077 1.14 up ns NA 10812667 Gfm2 1.34 down 0.059 1.30 down 0.034 1.35 down 0.064 1.44 down 0.076 2.35 up 0.0003 10831728 Vps52 1.37 down 0.026 1.40 down 0.004 1.29 down 0.004 1.55 down 0.053 1.52 up 0.029 10916028 Dcps 1.44 up 0.033 1.46 up 0.018 1.26 up 0.001 1.75 up 0.017 1.27 up 0.090 10720531 ENS 1.28 up 0.060 1.40 up 0.052 1.23 up 0.025 NA NA 10802691 Acaa2 1.28 down 0.060 1.43 down 0.014 1.23 down 0.071 1.62 down 0.025 1.90 up 0.0012 10934530 LOC680353 1.33 down 0.008 1.22 down 0.002 1.26 down 0.018 NA NA 10774359 Rpl38 1.34 down 0.031 1.37 down 0.046 1.29 down 0.015 1.45 down 0.019 1.30 up 0.011 10790525 Rpl38 1.34 down 0.030 1.37 down 0.046 1.29 down 0.014 1.45 down 0.019 1.30 up 0.011 10825236 Rpl38 1.34 down 0.030 1.37 down 0.046 1.29 down 0.014 1.45 down 0.019 1.30 up 0.011 10842436 Rpl38 1.36 down 0.006 1.36 down 0.045 1.29 down 0.003 1.45 down 0.019 1.30 up 0.011 10861676 Rpl38 1.34 down 0.028 1.37 down 0.041 1.29 down 0.004 1.45 down 0.019 1.30 up 0.011 10910458 Rpl38 1.34 down 0.030 1.37 down 0.046 1.29 down 0.014 1.45 down 0.019 1.30 up 0.011

Chapter 2 - Gene expression in a rat model for complex phenotype
42
examine which of the genes that were differentially expressed in the APO-SUS and APO-UNSUS rats from the replicate lines were also differentially expressed in rats from the original lines, we analyzed hippocampal mRNA expression in the original APO-SUS and APO-UNSUS rats. The regulation of mRNA expression of three genes, namely of Decapping enzyme, scavenger (Dcps), intraflagellar transport 140 homolog (Ift140) and Ke2 (also named prefoldin 6, Pfdn6), showed the same trend in the replicate and original lines, but their regulation was not significantly different in the APO-SUS and APO-UNSUS rats from the original lines. Only one gene, potassium channel-interacting protein KCnIP1, was significantly differentially expressed with the same regulation (upregulated) in the brain tissues from both the replicate and original APO-SUS rats (table 1). The other three members of the voltage-gated potassium (Kv) channel-interacting protein family (KCnIP2, -3 and -4) were not differentially expressed (data not shown). Discussion This study focussed on the differences in mRNA expression profiles in various brain regions of APO-SUS and APO-UNSUS rats from the original and replicate lines. The APO-SUS rats constitute a model for certain aspects of schizophrenia (Ellenbroek et al., 2002; Ellenbroek et al., 1995; Teunis et al., 2002). Most of the genes that were differentially expressed in the APO-SUS and APO-UNSUS rats from the replicate lines were not differentially expressed in the original rat lines. This remarkable finding suggests that these genes are not directly responsible for the dopaminergic dysregulation in the APO-SUS rats, a phenotype that is well established in both the replicate and original rat lines. Seven transcripts were differentially expressed in three brain tissues from the replicate APO-SUS/APO-UNSUS rats (fc > 1.3). Since Dcps, Ift140 and Ke2 were differentially expressed in both the original and the replicate lines, albeit not significant in the original line, they may be linked to one or more aspects of the complex phenotype of APO-SUS rats. Aside from the previously described gene encoding the γ-secretase component Aph-1b (Coolen et al., 2006; Coolen et al., 2005), the only other gene significantly higher expressed in APO-SUS than APO–UNSUS rats from both the original and the replicate lines was KCnIP1, a member of the potassium channel-interacting protein family (also named KChIPs). The results of a number of studies suggest that KCnIP1 may well represent a candidate gene linked to the dopaminergic dysregulation of the APO-SUS rats and the APO-SUS phenotype in general. First, KCnIP3 knockout mice show complex neurological phenotypes (Cheng et al., 2002; Lilliehook et al., 2003; Pruunsild et al., 2005), suggesting a role for KCnIPs in brain development and function; KCnIP1 knockout mice have not been described. Second, in mice treated with conventional and atypical antipsychotic drugs, four of the observed 249 differentially expressed

Chapter 2 - Gene expression in a rat model for complex phenotype
43
genes were linked to voltage-gated potassium (Kv) channels, including KCnIP3. The fact that in these drug-treated mice KCnIP3 was downregulated in the hippocampus, prefrontal cortex and ventral tegmental area suggests that this KCnIP family member may play a role in mesocortical dopamine neuronal firing (Duncan et al., 2008). Such firing is believed to be dysfunctional in schizophrenia and may underlie cognitive deficits (Abi-Dargham, 2004). Other aspects of the APO-SUS phenotype may be linked to KCnIP1 as well. KCnIPs have been shown to modulate neuronal and cardiac A-type Kv4 channels (An et al., 2000; Rhodes et al., 2004) that have been implicated in multiple disorders, including immune disorders, cancer and epilepsy (Hong et al., 2003; Strauss et al., 2000; Wang, 2004). Interestingly, APO-SUS rats show a different immune system (Kavelaars et al., 1997), a lower risk of developing (lung) cancer (Teunis et al., 2002) and more spike wave discharges associated with absence epilepsy when compared to APO-UNSUS rats (de Bruin et al., 2000). Thus, multiple aspects of the complex phenotype of APO-SUS rats may result from an affected expression of KCnIP1. Intriguingly, genes encoding the potassium channels themselves have been implicated in the aaetiology of schizophrenia as well, such as KCNN3 (Tomita et al., 2003) and very recently KCNH2 (Huffaker et al., 2009). This holds in particular for the latter gene since the presence of disease-associated SNPs together with compelling, converging evidence from (functional) brain imaging, mRNA expression and electrophysiological studies suggests that KCNH2 is a strong contributor to the schizophrenia phenotype (Horvath et al., 2009). Since gene × environment interactions are known to play a role in the APO-SUS/APO-UNSUS rat model as well as in schizophrenia, it is of interest to analyse in future studies the expression of KCnIP1 under various experimental conditions, such as maternal deprivation and cross-fostering. Of further interest will be to explore the KCnIP1 expression pattern in brain areas of developing APO-SUS and APO-UNSUS rats, as well as of the female rat lines. In conclusion, in this study we revealed a gene other than Aph1b, namely the gene encoding the potassium channel-interacting protein KCnIP1, which is significantly differentially expressed in a number of brain tissues of APO-SUS and APO-UNSUS rats from both the original and the replicate lines. Together with its characteristics, our findings make KCnIP1 an attractive novel candidate gene that may play a role in the complex phenotype of the APO-SUS rats. Acknowledgements We thank Luuk Lubbers for animal breeding and assistance. This work was supported by grant T5-209 from the Dutch Top Institute Pharma.

Chapter 2 - Gene expression in a rat model for complex phenotype
44
References Abel KM. Foetal origins of schizophrenia: testable hypotheses of genetic and environmental
influences. Br J Psychiatry 2004;184:383-385. Abi-Dargham A. Do we still believe in the dopamine hypothesis? New data bring new evidence.
Int J Neuropsychopharmacol 2004;7 Suppl 1:S1-5. An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, et al. Modulation of A-type
potassium channels by a family of calcium sensors. Nature 2000;403:553-556. Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies.
Arch Gen Psychiatry 1990;47:181-188. Breivik T, Sluyter F, Hof M, Cools A. Differential susceptibility to periodontitis in genetically
selected Wistar rat lines that differ in their behavioral and endocrinological response to stressors. Behav Genet 2000;30:123-130.
Cheng HY, Pitcher GM, Laviolette SR, Whishaw IQ, Tong KI, Kockeritz LK, et al. DREAM is a critical transcriptional repressor for pain modulation. Cell 2002;108:31-43.
Coolen MW, van Loo KM, van Bakel NN, Ellenbroek BA, Cools AR, Martens GJ. Reduced Aph-1b expression causes tissue- and substrate-specific changes in gamma-secretase activity in rats with a complex phenotype. Faseb J 2006;20:175-177.
Coolen MW, Van Loo KM, Van Bakel NN, Pulford DJ, Serneels L, De Strooper B, et al. Gene dosage effect on gamma-secretase component Aph-1b in a rat model for neurodevelopmental disorders. Neuron 2005;45:497-503.
Cools AR, Brachten R, Heeren D, Willemen A, Ellenbroek B. Search after neurobiological profile of individual-specific features of Wistar rats. Brain Res Bull 1990;24:49-69.
Corripio I, Perez V, Catafau AM, Mena E, Carrio I, Alvarez E. Striatal D2 receptor binding as a marker of prognosis and outcome in untreated first-episode psychosis. Neuroimage 2006;29:662-666.
de Bruin NM, van Luijtelaar EL, Jansen SJ, Cools AR, Ellenbroek BA. Dopamine characteristics in different rat genotypes: the relation to absence epilepsy. Neurosci Res 2000;38:165-173.
Duncan CE, Chetcuti AF, Schofield PR. Coregulation of genes in the mouse brain following treatment with clozapine, haloperidol, or olanzapine implicates altered potassium channel subunit expression in the mechanism of antipsychotic drug action. Psychiatr Genet 2008;18:226-239.
Ellenbroek BA, Cools AR. Apomorphine susceptibility and animal models for psychopathology: genes and environment. Behav Genet 2002;32:349-361.
Ellenbroek BA, Geyer MA, Cools AR. The behavior of APO-SUS rats in animal models with construct validity for schizophrenia. J Neurosci 1995;15:7604-7611.
Ellenbroek BA, Sluyter F, Cools AR. The role of genetic and early environmental factors in determining apomorphine susceptibility. Psychopharmacology (Berl) 2000;148:124-131.
Hippisley-Cox J, Vinogradova Y, Coupland C, Parker C. Risk of malignancy in patients with schizophrenia or bipolar disorder: nested case-control study. Arch Gen Psychiatry 2007;64:1368-1376.
Hong YM, Jo DG, Lee MC, Kim SY, Jung YK. Reduced expression of calsenilin/DREAM/KChIP3 in the brains of kainic acid-induced seizure and epilepsy patients. Neurosci Lett 2003;340:33-36.
Horvath S, Mirnics K. Breaking the gene barrier in schizophrenia. Nat Med 2009;15:488-490. Huffaker SJ, Chen J, Nicodemus KK, Sambataro F, Yang F, Mattay V, et al. A primate-specific,
brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med 2009;15:509-518.
Kavelaars A, Heijnen CJ, Ellenbroek B, van Loveren H, Cools A. Apomorphine-susceptible and apomorphine-unsusceptible Wistar rats differ in their susceptibility to inflammatory and infectious diseases: a study on rats with group-specific differences in structure and reactivity of hypothalamic-pituitary-adrenal axis. J Neurosci 1997;17:2580-2584.

Chapter 2 - Gene expression in a rat model for complex phenotype
45
Leucht S, Burkard T, Henderson J, Maj M, Sartorius N. Physical illness and schizophrenia: a review of the literature. Acta Psychiatr Scand 2007;116:317-333.
Lilliehook C, Bozdagi O, Yao J, Gomez-Ramirez M, Zaidi NF, Wasco W, et al. Altered Abeta formation and long-term potentiation in a calsenilin knock-out. J Neurosci 2003;23:9097-9106.
Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology 2000;23:223-239.
Muller-Spahn F, Modell S, Ackenheil M, Brachner A, Kurtz G. Elevated response of growth hormone to graded doses of apomorphine in schizophrenic patients. J Psychiatr Res 1998;32:265-271.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001;29:e45.
Pruunsild P, Timmusk T. Structure, alternative splicing, and expression of the human and mouse KCNIP gene family. Genomics 2005;86:581-593.
Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, et al. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J Neurosci 2004;24:7903-7915.
Rots NY, Cools AR, Berod A, Voorn P, Rostene W, de Kloet ER. Rats bred for enhanced apomorphine susceptibility have elevated tyrosine hydroxylase mRNA and dopamine D2-receptor binding sites in nigrostriatal and tuberoinfundibular dopamine systems. Brain Res 1996a;710:189-196.
Rots NY, Cools AR, Oitzl MS, de Jong J, Sutanto W, de Kloet ER. Divergent prolactin and pituitary-adrenal activity in rats selectively bred for different dopamine responsiveness. Endocrinology 1996b;137:1678-1686.
Rots NY, Workel J, Oitzl MS, Berod A, Rostene W, Cools AR, et al. Development of divergence in dopamine responsiveness in genetically selected rat lines is preceded by changes in pituitary-adrenal activity. Brain Res Dev Brain Res 1996c;92:164-171.
Rugbjerg K, Friis S, Ritz B, Schernhammer ES, Korbo L, Olsen JH. Autoimmune disease and risk for Parkinson disease. A population-based case-control study. Neurology 2009;
Shirotani K, Edbauer D, Prokop S, Haass C, Steiner H. Identification of distinct gamma-secretase complexes with different APH-1 variants. J Biol Chem 2004;279:41340-41345.
Strauss U, Wissel K, Jung S, Wulff H, Hansel W, Zhu J, et al. K(+) channel-blocking alkoxypsoralens inhibit the immune response of encephalitogenic T line cells and lymphocytes from Lewis rats challenged for experimental autoimmune encephalomyelitis. Immunopharmacology 2000;48:51-63.
Teunis MA, Kavelaars A, Voest E, Bakker JM, Ellenbroek BA, Cools AR, et al. Reduced tumor growth, experimental metastasis formation, and angiogenesis in rats with a hyperreactive dopaminergic system. Faseb J 2002;16:1465-1467.
Tomita H, Shakkottai VG, Gutman GA, Sun G, Bunney WE, Cahalan MD, et al. Novel truncated isoform of SK3 potassium channel is a potent dominant-negative regulator of SK currents: implications in schizophrenia. Mol Psychiatry 2003;8:524-535, 460.
Van de Langerijt AG, Van Lent PL, Hermus AR, Sweep CG, Cools AR, Van den Berg WB. Susceptibility to adjuvant arthritis: relative importance of adrenal activity and bacterial flora. Clin Exp Immunol 1994;97:33-38.
Wang Z. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch 2004;448:274-286.


Chapter 3 Dopamine susceptibility of APO-SUS
rats is not per se coupled to HPA-axis activity
Published as: Van Schijndel JE, Van Zweeden M, Van Loo KMJ,
Lubbers LJ, Pesman GJ, Sweep FCGJ, Martens GJM. Dopamine susceptibility of APO-SUS rats is not per se coupled to HPA-axis activity.
Physiology & Behavior, in press

Chapter 3 - Dopamine sensitivity and HPA-axis
48
Abstract A synergistic relationship is thought to exist between hypothalamic-pituitary-adrenal (HPA) axis activity and dopamine neurotransmission. To test whether a high response to dopamine indeed implies a hyperactive HPA-axis, we here used Wistar rats that were selected twice independently (original and replicate lines) for a high or low susceptibility to the dopamine receptor agonist apomorphine (so-called APO-SUS and APO-UNSUS rats, respectively). The APO-SUS rats from the original line displayed a hyperactive HPA-axis in that higher basal and stress-induced adrenocorticotropic hormone (ACTH) levels, and lower basal free-corticosterone levels were observed than those found in the original APO-UNSUS rats. In contrast, the activity of the HPA-axis in the APO-SUS rats from the replicate line did not differ from that in the replicate APO-UNSUS rats. Thus, in the APO-SUS/APO-UNSUS rat model the level of HPA-axis activity is not necessarily causally linked to dopamine responsiveness, implying that a hyperactive HPA-axis is not a prerequisite for a high dopaminergic response.

Chapter 3 - Dopamine sensitivity and HPA-axis
49
Introduction The hypothalamic-pituitary-adrenal (HPA) axis governs a hormonal cascade that has important effects on brain function. The HPA-axis involves the release of corticotrophin-releasing hormone from the paraventricular nucleus of the hypothalamus causing the secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland, which in turn leads to the release of glucocorticoids (GCs; cortisol in primates and corticosterone in rodents) from the adrenals. Although the exact mechanisms are not yet understood, ACTH can directly elevate gene expression for catecholamine biosynthesis (Serova et al., 2008) and the release of GC influences dopamine activity in the central nervous system (reviewed in Czyrak et al., 2003). It has been previously postulated that a synergistic relationship exists between the HPA-axis and the dopaminergic system (Walker et al., 2008). For instance, in healthy human subjects a psychosocial stress caused a salivary cortisol response that was positively correlated with the level of dopamine release in the ventral striatum (Pruessner et al., 2004). Moreover, in rats GC suppression by adrenalectomy resulted in lower levels of extracellular dopamine in the nucleus accumbens in basal conditions which could be reversed by corticosterone administration (Piazza et al., 1996). An interesting animal model to study the previously postulated synergistic HPA-axis/dopaminergic relationship (Walker et al., 2008) involves Wistar rats that were selected for a high susceptibility to the dopamine receptor agonist apomorphine (APO-SUS rats) and rats with a low susceptibility to apomorphine (APO-UNSUS rats; Cools et al., 1990; Ellenbroek et al., 1995). The neuropharmacological selection from the Wistar rats was performed twice and independently with a time span difference of a decade, resulting in the so-called original and replicate lines (Cools et al., 1990; Ellenbroek et al., 2000). Most studies have been performed with the original APO-SUS and APO-UNSUS rats, and these rat lines differ not only in their apomorphine susceptibility and therefore their dopaminergic system (Cools et al., 1993; Ellenbroek et al., 1995), but also in their activity of the HPA-axis. APO-SUS rats from the original line have a hyperactive HPA-axis compared to APO-UNSUS rats and this difference in the HPA-axis seems to occur earlier in the development of the rats than the difference in the dopaminergic system (Rots et al., 1995; Rots et al., 1996a; Rots et al., 1996b). In view of the above results, we wondered whether a high HPA-axis activity is a prerequisite for the apomorphine susceptibility of the APO-SUS rats. The availability of the two separate, independent APO-SUS and APO-UNSUS rat lines offers a unique opportunity to explore the relationship between dopamine responsiveness and HPA-axis activity. We therefore here tested the activity of the HPA-axis (ACTH and corticosterone levels) in APO-SUS and APO-UNSUS rats from the replicate lines as well as from the original lines. Furthermore, since the

Chapter 3 - Dopamine sensitivity and HPA-axis
50
non-synonymous single-nucleotide polymorphism (SNP) Met276Ile in the gene encoding corticosteroid-binding globulin (CBG/Serpina6) is thought to influence corticosterone binding of the protein (Smith et al., 1991) we screened the APO-SUS and APO-UNSUS rat genomes for CBG SNPs. Materials and methods Animals The generation of the APO-SUS and APO-UNSUS rat lines with a high or low susceptibility for apomorphine, respectively, has been described previously for the replicate and original lines (Cools et al., 1990; Ellenbroek et al., 2000). Rats were bred and reared in the Central Animal Facility of the Radboud University Nijmegen under approved animal protocols and in accordance with institutional guidelines. All rats were reared and housed in macrolon cages (42 × 26 × 15 cm; n = 2-3 per cage) under a fixed 12/12 h light/dark cycle (lights on: 7.00 a.m.) in a temperature-controlled room (21 ± 1.7 °C). Water and food pellets were available ad libitum. For each group, APO-SUS and APO-UNSUS rats (n = 10 and n = 8 per group for the original rat lines and replicate rat lines, respectively) were tested for their apomorphine susceptibility at least two weeks before decapitation. Male APO-SUS and APO-UNSUS rats at the age of 94±1.5 days (replicate lines) and 132±7.4 days (original lines) were injected with apomorphine (1.5 mg/kg subcutaneously) and gnawing scores were measured in a gnawing box for 45 minutes, as described previously (Cools et al., 1990). Open-field stress induction APO-SUS and APO-UNSUS rats were individually housed and for habituation they were handled twice a day starting four days prior to the open-field test (that took place between 7.30 and 13.00 h) and decapitation. Rats were placed on a black square table (160 x 160 cm; 95 cm elevated above the floor, surrounded by a white natural background, illuminated by white light of 170 Lux at the middle of the open field) for a period of 30 minutes. As described by Cools et al. (1990), behaviour was recorded with a video camera and analysed with a computerised tracking system (Cools et al., 1990). Rats (n = 4 per group for both the original and replicate rat lines) were decapitated immediately after the 30-minutes stress induction in a separate room with no other rats or cadavers present. Decapitation of control APO-SUS and APO-UNSUS rats was performed either alternately with the stress-induced rats (n = 6 and n = 4 per group for the original and replicate rat lines, respectively) or on a separate day alternately with replicate APO-SUS and APO-UNSUS rats (n = 7 and n = 8 rats, respectively). The trunk blood of the decapitated animals was collected in ethylenediaminetetraacetic acid (EDTA)-coated tubes for plasma and in uncoated tubes for serum. The blood was stored on ice (0-4°C) until

Chapter 3 - Dopamine sensitivity and HPA-axis
51
further processing. After centrifugation the samples were aliquoted and stored at -20 °C. The tails of the rats were collected for the isolation of genomic deoxyribonucleic acid (DNA). The tails were quickly frozen in liquid nitrogen and stored at -80°C until DNA isolation. Determination of ACTH ACTH was measured in EDTA plasma by a two-step immunoradiometric assay (IRMA, Dynotest BRAHMS, Berlin, Germany) based on two monoclonal mouse antibodies directed against different antigenic determinants on the human ACTH-(1-39) molecule. The tube was coated with anti-human ACTH-(15-21) and a C-terminal 125Iodine labeled anti-human ACTH-(34-39) was used as a detection signal. Addition of ACTH-fragments 1-17, 1-24 and 7-38 at concentrations up to 4.4 nmol/L did not result in any inhibition of intact-ACTH binding. Within-assay coefficients of variation (CV) were 7.9, 3.5 and 1.3 % (at 1.0, 9.3 and 173.0 pmol/L, respectively). Between-assay CVs were 16.2, 4.7 and 4.4 % (at 1.1, 2.7 and 5.3 pmol/L, respectively). A functional detection limit of 0.5 pmol/L was established. Determination of free corticosterone In basal conditions the levels of free corticosterone have been found to be different between APO-SUS and APO-UNSUS rats, while total corticosterone levels were comparable between the two rat lines (Rots et al., 1996a; Rots et al., 1996b). In this study, we therefore determined free corticosterone levels. Serum corticosterone was measured by radioimmunoassy using an antiserum raised against corticosterone-21-hemisuccinate bovine serum albumin, as described previously (Sweep et al., 1992). The percent free corticosterone was assessed by addition of chromatographically prepurified tritiated corticosterone to 250 μl plasma with subsequent ultrafiltration of a 150 μl aliquot in a Millipore Microcon centrifugal ultrafiltration device (Millipore Corporation, Billerica, MA, U.S.A) equipped with a YM-10 membrane (10000 M.W. cut-off). Centrifugation took place at 37o C in a preheated Sorvall RC24 centrifuge (SS34 fixed-angle rotor, Goffin-Meyvis, Etten-Leur, The Netherlands) at 2500 g. Since variations in the total corticosterone levels would gloss over the variations in free corticosterone levels, we calculated the percentage of free corticosterone from the ratio of measured count rates in 50 μl aliquots from filtrate and original serum. High-resolution melting (HRM) Genomic DNA was isolated from the tail following the standard protocol of proteinase K and phenol extraction. For amplifying the region encompassing the Met276Ile SNP in CBG, forward (5’-ggtgaacctatacatcccgaaatt-3’) and reverse (5’- tctttggtgttgcctgagaaatc-3’) primers were used. Polymerase chain reaction (PCR)

Chapter 3 - Dopamine sensitivity and HPA-axis
52
amplifications were performed in a total volume of 10 μl on a Rotor-GeneTM 6000 real-time rotary analyzer (Corbett Research, Sydney, Australia). The reaction mixture contained 10 ng genomic DNA, 1x TITANIUMTM Taq PCR Buffer (Clontech), 0.25 mM dNTP, 375 nM forward and 375 nM reverse primers, 1.5 μM SYTO® 9 green fluorescent nucleic acid stain (Invitrogen), and 0.25x TITANIUMTM Taq polymerase (Clontech). To amplify the amplicon, the cycling conditions were as follows: one cycle of 95 °C for 3 minutes; 45 cycles of 95 °C for 10 seconds, 60 °C for 15 seconds and 72 °C for 20 seconds. Before the HRM one cycle of 90 °C for 1 minute and 40 °C for 1 minute was applied to form random DNA duplexes. The HRM protocol was a 90-seconds pre-melt at 65 °C followed by a gradient rise in temperature from 65 °C to 95 °C, 0.1 °C and 1 second per step. HRM was analyzed with the Rotor-Gene 6000 Series Software using the HRM (normalized and difference graphs) and melt analysis tools. DNA sequencing The PCR fragment generated with the HRM primers and encompassing SNP Met276Ile in rat CBG (see above) was purified by agarose gel electrophoresis and sequenced using the BigDye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) and the ABI310 machine. Statistics Results are presented as means ± standard deviations. An F-test was used to determine the homogeneity/heterogeneity of the variances. Significance of the variables was determined with the one-tailed Student’s t-test or a t-test with Welch's correction for unequal variances (depending on the results of the F-test). A p-value < 0.05 was considered statistically significant (GraphPad Software Inc, San Diego, USA). Statistical results are presented as the t-value (t), degrees of freedom of the t-test (df) and p-value (p) for the t-test, and F, df and p for the one-way analysis of variance (ANOVA). The nonparametric (Spearman) correlation was used to calculate correlations (GraphPad Software Inc, San Diego, USA). Statistical results of this test are presented as correlation coefficient (r) and two-tailed p. Results To test whether the level of activity of the HPA-axis is directly associated with the response to the dopamine receptor agonist apomorphine, APO-SUS and APO-UNSUS rats were first tested for their apomorphine susceptibility. As expected, APO-SUS rats had a high gnawing score (1170 ± 323 and 1212 ± 337 gnaws/45 minutes for the original and replicate rat lines, respectively) and APO-UNSUS rats had a low gnawing score (23 ± 57 and 4 ± 3 gnaws/45 minutes for the original and replicate rat lines, respectively). The differences in gnawing scores were highly
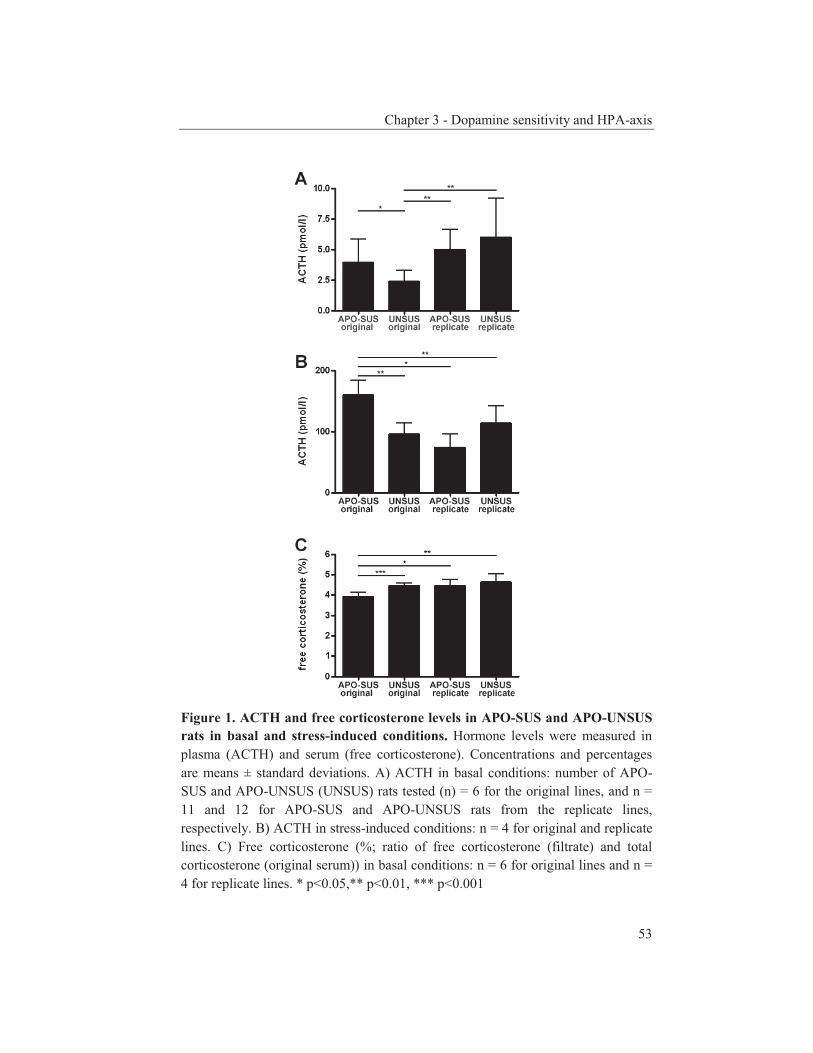
Chapter 3 - Dopamine sensitivity and HPA-axis
53
Figure 1. ACTH and free corticosterone levels in APO-SUS and APO-UNSUS rats in basal and stress-induced conditions. Hormone levels were measured in plasma (ACTH) and serum (free corticosterone). Concentrations and percentages are means ± standard deviations. A) ACTH in basal conditions: number of APO-SUS and APO-UNSUS (UNSUS) rats tested (n) = 6 for the original lines, and n = 11 and 12 for APO-SUS and APO-UNSUS rats from the replicate lines, respectively. B) ACTH in stress-induced conditions: n = 4 for original and replicate lines. C) Free corticosterone (%; ratio of free corticosterone (filtrate) and total corticosterone (original serum)) in basal conditions: n = 6 for original lines and n = 4 for replicate lines. * p<0.05,** p<0.01, *** p<0.001

Chapter 3 - Dopamine sensitivity and HPA-axis
54
significant between APO-SUS and APO-UNSUS rats (original lines: t = 12.15, df = 9, p < 0.0001 and replicate lines: t = 10.15, df = 7, p < 0.0001). Gnawing scores were comparable between APO-SUS rats from the original and replicate rat lines (t = 0.05, df = 16, two-tailed p = 0.96) and between APO-UNSUS rats from the two rat lines (t = 1.07, df = 9, two-tailed p = 0.31). APO-SUS and APO-UNSUS rats from both the replicate and the original lines were then exposed to an environmental stressor (thirty minutes in a new environment on the open field). The distance walked (step size 16 cm) by rats from the original lines was 4514 ± 1396 cm (n = 3) and 2842 ± 1818 cm (n = 4) for APO-SUS and APO-UNSUS rats, respectively (t = 1.24, df = 5, p = 0.12). Rats from the replicate lines walked 6858 ± 950 cm (n = 3) and 3998 ± 2619 cm (n = 4) for APO-SUS and APO-UNSUS rats, respectively (t = 1.77, df = 5, p = 0.07). The environmental challenge evoked a significant ACTH response in both APO-SUS and APO-UNSUS rats from the replicate lines. Average plasma ACTH levels were elevated from 5.00 ± 1.62 pmol/l to 74.0 ± 22.1 pmol/l and from 6.00 ± 3.16 pmol/l to 113.3 ± 28.1 pmol/l in the APO-SUS and APO-UNSUS rats, respectively (figure 1). The average ACTH levels were comparable between the replicate APO-SUS and APO-UNSUS rats in both basal (t = 0.97, df = 16, p = 0.17) and stress-induced conditions (t = 2.20, df = 6, one-tailed p = 1.00, two-tailed p = 0.07). The percentage of free corticosterone in basal conditions was not different between the APO-SUS rats (4.45 ± 0.3%) and APO-UNSUS rats (4.65 ± 0.4%; figure 1) from the replicate lines (t = 0.78, df = 6, p = 0.23). The ACTH levels in basal or stress-induced conditions did not correlate with the gnawing scores (APO-SUS basal: r = -0.40, p = 0.75; APO-SUS stress: r = -0.50, p = 0.42; APO-UNSUS basal: r = -0.60, p = 0.42; APO-UNSUS stress: r = -0.63, p = 0.42). The percentage of free corticosterone did not correlate with the gnawing scores (APO-SUS: r = -0.80, p = 0.33; APO-UNSUS: r = -0.32, p = 0.75). We next determined the basal and stress-induced ACTH levels, and basal free-corticosterone levels in challenged APO-SUS and APO-UNSUS rats from the original lines. Exposure to the open field evoked a significant ACTH response in the original APO-SUS rats as well as in the original APO-UNSUS rats. The average plasma ACTH levels were elevated from 3.93 ± 1.90 pmol/l to 159.5 ± 24.0 pmol/l and from 2.36 ± 0.90 pmol/l to 95.4 ± 18.1 pmol/l in the APO-SUS and APO-UNSUS rats, respectively (figure 1). The average ACTH levels were significantly higher in the APO-SUS rats compared to the APO-UNSUS rats in both basal (t = 1.82, df =10, p = 0.05) and stress-induced (t = 4.27, df = 6, p = 0.003) conditions. The percentage of free corticosterone in basal conditions was significantly (t = 5.31, df = 10, p = 0.0002) lower in the APO-SUS rats (3.91 ± 0.2%) than in the APO-UNSUS rats (4.46 ± 0.1%; figure 1) from the original lines. The ACTH levels in basal or stress-induced conditions did not correlate with the gnawing scores (APO-SUS basal: r = 0.14, p = 0.82; APO-SUS stress: r = 0.00, p = 1.08; APO-UNSUS basal: r = -0.29, p = 0.56; APO-UNSUS stress: r = 0.00, p = 1.08). The percentage
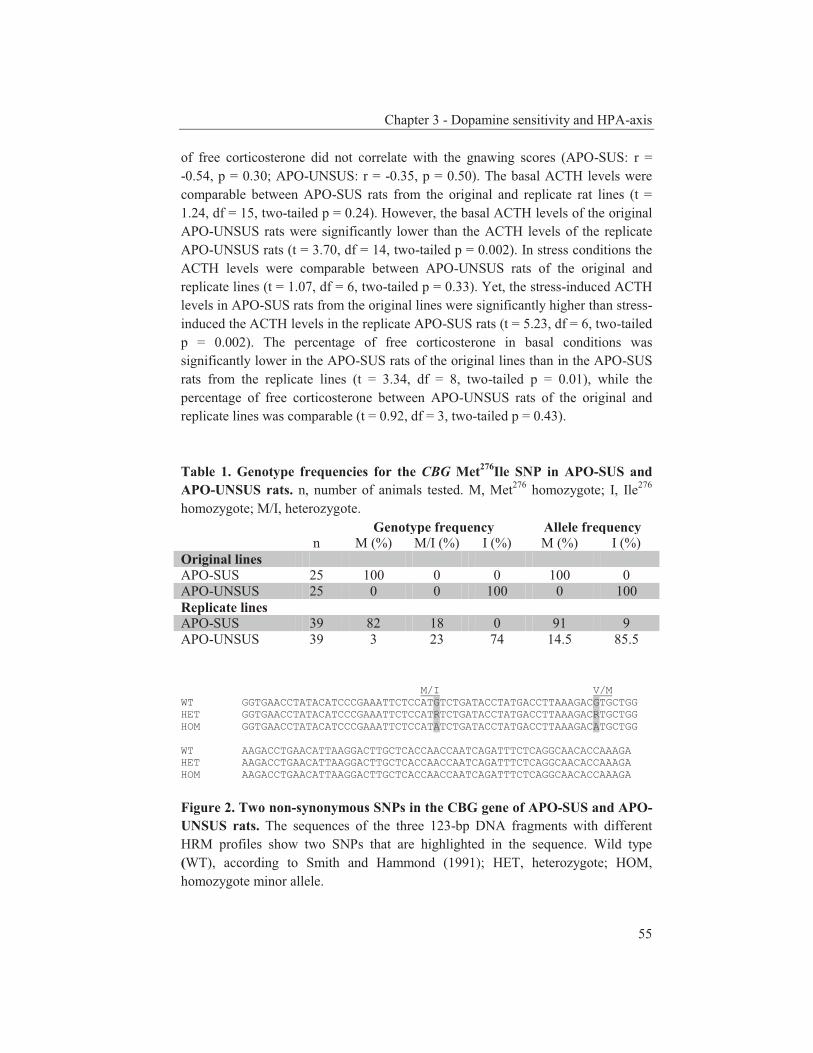
Chapter 3 - Dopamine sensitivity and HPA-axis
55
of free corticosterone did not correlate with the gnawing scores (APO-SUS: r = -0.54, p = 0.30; APO-UNSUS: r = -0.35, p = 0.50). The basal ACTH levels were comparable between APO-SUS rats from the original and replicate rat lines (t = 1.24, df = 15, two-tailed p = 0.24). However, the basal ACTH levels of the original APO-UNSUS rats were significantly lower than the ACTH levels of the replicate APO-UNSUS rats (t = 3.70, df = 14, two-tailed p = 0.002). In stress conditions the ACTH levels were comparable between APO-UNSUS rats of the original and replicate lines (t = 1.07, df = 6, two-tailed p = 0.33). Yet, the stress-induced ACTH levels in APO-SUS rats from the original lines were significantly higher than stress-induced the ACTH levels in the replicate APO-SUS rats (t = 5.23, df = 6, two-tailed p = 0.002). The percentage of free corticosterone in basal conditions was significantly lower in the APO-SUS rats of the original lines than in the APO-SUS rats from the replicate lines (t = 3.34, df = 8, two-tailed p = 0.01), while the percentage of free corticosterone between APO-UNSUS rats of the original and replicate lines was comparable (t = 0.92, df = 3, two-tailed p = 0.43). Table 1. Genotype frequencies for the CBG Met276Ile SNP in APO-SUS and APO-UNSUS rats. n, number of animals tested. M, Met276 homozygote; I, Ile276 homozygote; M/I, heterozygote.
Genotype frequency Allele frequency n M (%) M/I (%) I (%) M (%) I (%) Original lines APO-SUS 25 100 0 0 100 0 APO-UNSUS 25 0 0 100 0 100 Replicate lines APO-SUS 39 82 18 0 91 9 APO-UNSUS 39 3 23 74 14.5 85.5 M/I V/M WT GGTGAACCTATACATCCCGAAATTCTCCATGTCTGATACCTATGACCTTAAAGACGTGCTGG HET GGTGAACCTATACATCCCGAAATTCTCCATRTCTGATACCTATGACCTTAAAGACRTGCTGG HOM GGTGAACCTATACATCCCGAAATTCTCCATATCTGATACCTATGACCTTAAAGACATGCTGG WT AAGACCTGAACATTAAGGACTTGCTCACCAACCAATCAGATTTCTCAGGCAACACCAAAGA HET AAGACCTGAACATTAAGGACTTGCTCACCAACCAATCAGATTTCTCAGGCAACACCAAAGA HOM AAGACCTGAACATTAAGGACTTGCTCACCAACCAATCAGATTTCTCAGGCAACACCAAAGA
Figure 2. Two non-synonymous SNPs in the CBG gene of APO-SUS and APO-UNSUS rats. The sequences of the three 123-bp DNA fragments with different HRM profiles show two SNPs that are highlighted in the sequence. Wild type (WT), according to Smith and Hammond (1991); HET, heterozygote; HOM, homozygote minor allele.

Chapter 3 - Dopamine sensitivity and HPA-axis
56
Since SNP Met276Ile in rat CBG has been reported to give rise to affected free-corticosterone levels (Smith et al., 1991), we explored whether the observed difference in free-corticosterone levels in the APO-SUS and APO-UNSUS rats from the original lines may be explained by the presence or absence of this SNP. DNA sequence analysis and HRM profiling showed that the Met276 allele is present in all (original line) or most (91%; replicate line) of the APO-SUS rats, and not (original line) or much less (14.5%; replicate line) in the APO-UNSUS rats (table 1). In the replicate rat lines, the CBG genotype was not related with the percentage of free corticosterone (one-way ANOVA; F = 1.18, df = 7, p = 0.38). Furthermore, DNA sequence analysis revealed a novel SNP in CBG in perfect linkage disequilibrium (LD) with Met276Ile, namely Val285Met (figure 2). Discussion To get a better insight into the postulated causal relationship between the activity of the HPA-axis and the dopaminergic system (Walker et al., 2008), we investigated the HPA-axis activity in rats with a known difference in their dopaminergic system, namely the APO-SUS and APO-UNSUS rats. The APO-SUS and APO-UNSUS rat lines were selected twice, independently from each other and with a decade difference in time, resulting in the original and replicate lines. The availability of these rat lines thus provided us with the unique opportunity to examine twice the hypothesized relationship between the stress and dopamine pathways. As a stressor we used exposure to a novel environment. Plasma ACTH levels were elevated in APO-SUS and APO-UNSUS rats from both the replicate and original lines indicating that the open field is indeed a genuine stressor. In contrast to APO-SUS and APO-UNSUS rats from the replicate lines, in basal conditions the APO-SUS rats from the original line showed higher ACTH levels and lower free-corticosterone levels than the APO-UNSUS rats. These results concerning the original rat lines are consistent with previous studies (Rots et al., 1995). The lower free-corticosterone levels in the original APO-SUS rats may indicate a lower threshold or enhanced sensitivity for stress-induced activation of the HPA-axis in these animals, which would lead to higher levels of ACTH (Rots et al., 1995). However, one has to realize that, although highly significant, the difference in free-corticosterone levels found between the original rat lines is small. Also, in the APO-UNSUS rats of the original line the free-corticosterone levels are similar to those in both replicate lines, whereas the basal ACTH levels in the original APO-UNSUS rats are lower than those in the three other groups. Yet, in the original APO-SUS rats the lower free-corticosterone level is accompanied by a higher basal ACTH level and in the replicate lines comparable free-corticosterone levels occur with similar basal ACTH levels. Taking these observations concerning the free-

Chapter 3 - Dopamine sensitivity and HPA-axis
57
corticosterone and basal ACTH levels together, we have to conclude that their physiological significance is at present unclear. We found that, in contrast to what holds for the rats from the original lines, the HPA-axis activity of the APO-SUS rats from the replicate lines did not differ from the HPA-axis activity of the replicate APO-UNSUS rats. Nevertheless, the replicate APO-SUS/-UNSUS rats display differential apomorphine susceptibility. A number of studies have shown the causal relation between the HPA-axis and the dopaminergic system (Czyrak et al., 2003; Piazza et al., 1996; Pruessner et al., 2004; Serova et al., 2008; Sullivan et al., 1998). Nevertheless, in schizophrenia patients an altered dopaminergic system may occur without an affected HPA-axis (Mokrani et al., 1995; Risch et al., 1992). Interestingly, our present results show that, as in the schizophrenia patients, in the replicate APO-SUS rat model the dopaminergic system is altered, whereas no differences occur in the HPA-axis. Thus, although this study does not exclude the possibility of a causal relationship between the activity of the HPA-axis and the dopaminergic system, we conclude that a hyperactive HPA-axis is not indispensable for an altered dopaminergic system. Other factors may as well play a role in the development of a dysfunctioning dopaminergic system, e.g. the functioning of the dopamine receptor systems or neuromodulators such as neurotensin (Liang et al., 2010). The fact that various parameters, one of them being a hyperactive HPA-axis, may lead to an altered dopaminergic response complicates research on disorders associated with a dysfunctioning dopaminergic system. A non-synonymous SNP (Met276Ile) in CBG has been proposed to influence CBG binding of corticosterone resulting in affected free corticosterone levels. The Met to Ile substitution results in a reduced steroid-binding affinity of rat CBG (Smith et al., 1991). The higher amount of free corticosterone in the APO-UNSUS rats of the original rat lines could therefore be due to the reduced steroid-binding affinity of the (276Ile) CBG protein of APO-UNSUS rats. Besides the methionine to isoleucine substitution, we discovered a valine to methionine substitution of residue 285 in CBG which was in complete LD with this SNP. The Met276 and Val285 alleles were mostly absent in the APO-UNSUS rats and present in the APO-SUS rats from not only the original but also from the replicate lines. These findings imply that the two SNPs do not segregate with the levels of free corticosterone because in the replicate APO-SUS and APO-UNSUS rats these levels were not different and therefore do not correlate with their CBG genotypes. The one-way ANOVA-analysis of the results from the replicate lines also indicated that there is no relation between the CBG genotype and the levels of free corticosterone. Thus, the combination of the Met276 and Val285 alleles is likely not causing a higher steroid-binding affinity of rat CBG. To elucidate the effect of the SNPs on steroid-binding activity, the free-corticosterone levels could be determined in the serum of animals in which the

Chapter 3 - Dopamine sensitivity and HPA-axis
58
SNPs are not in complete LD or the binding affinity of recombinant CBG proteins could be studied. In conclusion, we found that the APO-SUS/-UNSUS rats from the replicate lines do not display the same HPA-axis activity as the APO-SUS/-UNSUS rats from the original lines. Therefore, our study suggests that, in contrast to what has been hypothesized previously (Rots et al., 1996b), a hyperactive HPA-axis is not a prerequisite for a high apomorphine response. Acknowledgements We thank I. Poyck for technical assistance. This work was supported by grant T5-209 from the Dutch Top Institute Pharma. References Cools AR, Brachten R, Heeren D, Willemen A, Ellenbroek B. Search after neurobiological
profile of individual-specific features of Wistar rats. Brain Res Bull 1990;24:49-69. Cools AR, Dierx J, Coenders C, Heeren D, Ried S, Jenks BG, et al. Apomorphine-susceptible and
apomorphine-unsusceptible Wistar rats differ in novelty-induced changes in hippocampal dynorphin B expression and two-way active avoidance: a new key in the search for the role of the hippocampal-accumbens axis. Behav Brain Res 1993;55:213-221.
Czyrak A, Mackowiak M, Chocyk A, Fijal K, Wedzony K. Role of glucocorticoids in the regulation of dopaminergic neurotransmission. Pol J Pharmacol 2003;55:667-674.
Ellenbroek BA, Geyer MA, Cools AR. The behavior of APO-SUS rats in animal models with construct validity for schizophrenia. J Neurosci 1995;15:7604-7611.
Ellenbroek BA, Sluyter F, Cools AR. The role of genetic and early environmental factors in determining apomorphine susceptibility. Psychopharmacology (Berl) 2000;148:124-131.
Liang Y, Boules M, Li Z, Williams K, Miura T, Oliveros A, et al. Hyperactivity of the dopaminergic system in NTS1 and NTS2 null mice. Neuropharmacology 2010;58:1199-1205.
Mokrani MC, Duval F, Crocq MA, Bailey PE, Macher JP. Multihormonal responses to apomorphine in mental illness. Psychoneuroendocrinology 1995;20:365-375.
Piazza PV, Barrot M, Rouge-Pont F, Marinelli M, Maccari S, Abrous DN, et al. Suppression of glucocorticoid secretion and antipsychotic drugs have similar effects on the mesolimbic dopaminergic transmission. Proc Natl Acad Sci U S A 1996;93:15445-15450.
Pruessner JC, Champagne F, Meaney MJ, Dagher A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci 2004;24:2825-2831.
Risch SC, Lewine RJ, Kalin NH, Jewart RD, Risby ED, Caudle JM, et al. Limbic-hypothalamic-pituitary-adrenal axis activity and ventricular-to-brain ratio studies in affective illness and schizophrenia. Neuropsychopharmacology 1992;6:95-100.
Rots NY, Cools AR, de Jong J, De Kloet ER. Corticosteroid feedback resistance in rats genetically selected for increased dopamine responsiveness. J Neuroendocrinol 1995;7:153-161.
Rots NY, Cools AR, Oitzl MS, de Jong J, Sutanto W, de Kloet ER. Divergent prolactin and pituitary-adrenal activity in rats selectively bred for different dopamine responsiveness. Endocrinology 1996a;137:1678-1686.

Chapter 3 - Dopamine sensitivity and HPA-axis
59
Rots NY, Workel J, Oitzl MS, Berod A, Rostene W, Cools AR, et al. Development of divergence in dopamine responsiveness in genetically selected rat lines is preceded by changes in pituitary-adrenal activity. Brain Res Dev Brain Res 1996b;92:164-171.
Serova LI, Gueorguiev V, Cheng SY, Sabban EL. Adrenocorticotropic hormone elevates gene expression for catecholamine biosynthesis in rat superior cervical ganglia and locus coeruleus by an adrenal independent mechanism. Neuroscience 2008;153:1380-1389.
Smith CL, Hammond GL. An amino acid substitution in biobreeding rat corticosteroid binding globulin results in reduced steroid binding affinity. J Biol Chem 1991;266:18555-18559.
Sullivan RM, Gratton A. Relationships between stress-induced increases in medial prefrontal cortical dopamine and plasma corticosterone levels in rats: role of cerebral laterality. Neuroscience 1998;83:81-91.
Sweep CG, van der Meer MJ, Hermus AR, Smals AG, van der Meer JW, Pesman GJ, et al. Chronic stimulation of the pituitary-adrenal axis in rats by interleukin-1 beta infusion: in vivo and in vitro studies. Endocrinology 1992;130:1153-1164.
Walker E, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol 2008;4:189-216.


Chapter 4 Three-cohort targeted gene screening
reveals a non-synonymous TRKA polymorphism associated
with schizophrenia
Published as: Van Schijndel JE, Van Loo KMJ, Van Zweeden M, Djurovic S,
Andreassen OA, Hansen T, Werge T, Kallunki P, Pedersen JT, Martens GJM. Three-cohort targeted gene screening reveals a non-synonymous
TRKA polymorphism associated with schizophrenia. Journal of Psychiatric Research, 2009, 43:1195-1199.

Chapter 4 - An association study in three schizophrenia case-control cohorts
62
Abstract Schizophrenia is a complex neurodevelopmental disorder that is thought to be induced by an interaction between predisposing genes and environmental stressors. To identify predisposing genetic factors, we performed a targeted (mostly neurodevelopmental) gene approach involving the screening of 396 selected non-synonymous single-nucleotide polymorphisms (SNPs) in three independent Caucasian schizophrenia case-control cohorts (USA, Denmark and Norway). A meta-analysis revealed ten non-synonymous SNPs that were nominally associated with schizophrenia, nine of which have not been previously linked to the disorder. Risk alleles are in TRKA (rs6336), BARD1 (rs28997576), LAMA5 (rs3810548), DKK2 (rs7037102), NOD2 (rs2066844) and RELN (rs2229860), whereas protective alleles are in NOD2 (rs2066845), NRG1 (rs10503929), ADAM7 (rs13259668) and TNR (rs859427). Following correction for multiple testing, the most attractive candidate for further study concerns SNP rs6336 (q = 0.12) that causes the substitution of an evolutionarily highly conserved amino acid residue in the kinase domain of the neurodevelopmentally important receptor TRKA. Thus, TRKA signaling may represent a novel susceptibility pathway for schizophrenia.

Chapter 4 - An association study in three schizophrenia case-control cohorts
63
Introduction Schizophrenia is a complex disorder with a lifetime risk of ~1%. The first symptoms usually manifest during late adolescence or early adulthood, but the origin of the disorder is thought to be neurodevelopmental (Weinberger, 1987). Predisposing genes in combination with environmental factors (including prenatal infections, maternal malnutrition during pregnancy, obstetric complications and migration) appear to be of importance in the aetiology of schizophrenia (Maki et al., 2005). Twin studies have shown that schizophrenia is a genetic disorder with a heritability risk of up to ~80%. On the basis of linkage analyses loci on 21 of the 23 chromosomes have been linked to schizophrenia. A meta-analysis has revealed that chromosomal region 2q, but also 1q, 8p, 22q and other loci, increase susceptibility to schizophrenia in diverse populations (Lewis et al., 2003). Follow-up studies on the regions implicated by the genome scans have resulted in positional candidate genes. A haplotype in the 5’-region of one of these genes, neuregulin 1 (NRG1; 8p12), shows significant association with schizophrenia, but the functional consequence of this gene variant is unclear. The Val158Met single-nucleotide polymorphism (SNP) in the catechol O-methyltransferase (COMT; 22q11.21) gene has been studied in many case-control cohorts and is functional, but association with schizophrenia was not supported by meta-analysis. A number of other candidate genes have been tested for association with schizophrenia in multiple case-control cohorts. Among these genes are brain-derived neurotrophic factor (BDNF; Kanazawa et al., 2007), dysbindin, disrupted in schizophrenia 1 (DISC1), D-amino acid oxidase activator (DAOA/G72) and proline dehydrogenase (PRODH), reviewed by Karayiorgou et al. (2006). Nevertheless, variations in these genes would account for the aetiology of only a small percentage of schizophrenic patients and it is likely that more polymorphisms in other genes are involved. Technological advances have resulted in platforms that allow association studies for hundreds of thousands of SNPs to search for polymorphisms associated with schizophrenia and other complex diseases. In the last few years, such genome-wide association (GWA) scans have been reported for complex diseases like bipolar disorder (Baum et al., 2008) and schizophrenia (Lencz et al., 2007). Associated SNPs found with such scans often occur with a relatively high minor allele frequency (MAF) and the functionality of the SNPs is usually not directly apparent. We therefore decided to perform a screening of non-synonymous SNPs, with a focus on SNPs that have a relatively low occurrence in the Caucasian population (MAF <10%). In view of the neurodevelopmental hypothesis of schizophrenia (Weinberger, 1987), we selected most of the SNPs from coding regions of genes that are involved in neurodevelopmental processes or otherwise potentially associated with schizophrenia. The SNPs were tested in three independent

Chapter 4 - An association study in three schizophrenia case-control cohorts
64
Caucasian schizophrenia-control cohorts. Here, we report ten SNPs that were found to be nominally associated with schizophrenia, with a SNP in the neurotrophic tyrosine kinase receptor NTRK1 (better known and hereafter referred to as TRKA) at a nearly significant confidence level following correction for multiple testing. Materials and methods Cohorts USA The USA cohort consisted of 491 Caucasian schizophrenic patients and 298 unrelated controls. Samples were derived from the PrecisionMed Inc. Human Biological Sample Bank collection (San Diego, California, USA). Patients (age 50.9 ± 9.8; 333 males, age 50.1 ± 9.9 and 158 females, age 52.6 ± 9.4) were diagnosed according the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV). Controls (age 72.1 ± 14.1; 113 males, age 73.5 ± 13.4 and 185 females, age 71.2 ± 14.5) had no (family) history of neuropsychiatric disorder and were recruited from the same geographic region as the schizophrenic patients. All patients and controls were Caucasian; both parents and all four grandparents had to be Caucasian for the subject to be enrolled as a Caucasian subject. All subjects had given written informed consent of participation. Denmark The Danish cohort consisted of 383 Caucasian schizophrenic patients and 400 unrelated Caucasian controls. Patients (age 44.6 ± 12.2, age of onset 27.1 ± 8.9; 229 males, age 43.7 ± 11.7 and 154 females, age 45.9 ± 12.8) were clinically diagnosed with schizophrenia or schizoaffective disorder according to ICD-10. The vast majority (96%) of the patients were also diagnosed with schizophrenia according to the DSM-IV standards (Hansen et al., 2007). The majority (87%) of the patients were ethnical Danish, i.e. the patients and both parents were born in Denmark, while in a minor fraction of the cases (13%) one parent was born in another north-western European country. The healthy control subjects (age 44.4 ± 12.1; 238 males, age 43.6 ± 11.6 and 162 females, age 45.7 ± 12.6) were recruited among 15 000 blood-donors from the Danish Blood Donor Corps in the Copenhagen area. Apparent behavioural abnormality was an exclusion criterion and all individuals stated that they felt completely healthy with a possibility to discuss any health-related issues with a physician. The study was approved by the Danish Scientific-Ethical Committees and the Danish Protection Agency. All patients had given written informed consent before inclusion into the project. For more detailed sample description, see Hansen et al (2007).

Chapter 4 - An association study in three schizophrenia case-control cohorts
65
Norway 134 schizophrenic patients (age 37.8 ± 10.8; 75 males, age 36.9 ± 9.8 and 59 females, age 38.8 ± 11.6) were recruited from the Oslo TOP (Thematic Organized Psychoses Research) Study and were selected according to DSM-IV. 195 Controls were randomly selected from statistical records of persons from the same catchments area as the patient groups. The control subjects (age 37.0 ± 10.3; 90 males, age 37.5 ± 10.2 and 105 females, age 36.7 ± 10.5) were screened by clinical interviews and with the Primary Care Evaluation of Mental Disorders (PRIME-MD). The majority of the patients (90%) and controls (85%) were ethnical Norwegian, i.e. the patient and both parents were born in Norway, while in rest of the cases one parent was born outside Norway in another north-western European country. All subjects had given written informed consent of participation and the study has been approved by The Norwegian Scientific-Ethical Committees and the Norwegian Data Protection Agency. For more detailed sample description, see Hansen et al (2007). SNP selection For genotyping, we selected 396 SNPs located in 216 genes that are either involved in neurodevelopmental processes, or have been reported to be associated with schizophrenia or another disease (see Supplementary Table 1 for details) and thus presumably represent functional SNPs. All selected SNPs were non-synonymous, except for the NRG1 haplotype SNPs. None of the SNPs could be predicted by another selected SNP (r2 < 0.80) and the majority of the SNPs has been genotyped in a database cohort of European descent (NCBI dbSNP Build 126). The number of SNPs fulfilling the criteria used by us (non-synonymous SNPs and with a MAF below 10% in HapMap-CEU) is surprisingly limited (in ~3000 brain genes ~500 SNPs). Furthermore, we selected more SNPs (> 40% of the total) with a MAF below 10% in HapMap-CEU than what generally holds for SNPs selected for genome-wide analyses (<15% of the total).
Genotyping SNPs were genotyped using iPLEXTM chemistry on a MALDI-TOF Mass Spectrometer by Sequenom Inc. (San Diego, CA, USA, http://www.sequenom.com). SNP-containing genomic regions were amplified in a multiplex PCR followed by deactivation of the remaining nucleotides by SAP treatment. A single-base primer extension step was performed and the primer extension products were analyzed using MALDI TOF MS. An assay was successful when over 90% of the samples were genotyped. In the first round of genotyping (USA cohort) 50 out of the 396 SNP assays failed, with 24 of these having a call rate between 80 and 90%. In the second round of genotyping (Danish and Norwegian cohorts) ten out of the 189 SNP assays failed, with two assays having a

Chapter 4 - An association study in three schizophrenia case-control cohorts
66
call rate between 80 and 90%. Call rates below 90% were equally divided among cases and controls (table S1). Three SNPs were genotyped in duplicate and the duplicates were > 99% in agreement. The genotyping results for SNP rs6336 were confirmed by applying an allele-specific PCR (primers and conditions available on request; data not shown). Statistical analysis Differences between cases and controls were analyzed by standard contingency table analysis using two-tailed χ2 test probabilities. Genotype frequencies of the control samples were tested for the Hardy-Weinberg equilibrium (p<0.05). Heterogeneity between studies was assessed using the Q Chochran test, and meta-analysis was performed using fixed-effects models. Meta-analysis calculations were performed using EasyMA2001 (Cucherat et al., 1997). To take multiple testing into account, false discovery rate (FDR) q-values were calculated using the bootstrap method within the QVALUE software package. The q-value of a test measures the minimum FDR that is incurred when calling that test significant. This method is superior to traditional methods, such as Bonferroni, since it provides a better balance between the purposes to control false discoveries and to find true positives (Storey, 2003). Like in other studies using a targeted candidate gene approach (e.g. Sullivan et al., 2008), in the present study the q-value threshold for declaring significance was 0.10. To evaluate the putative haplotype block in NOD2, the D’ value was calculated using Haploview 3.32 (Barrett et al., 2005). Results For our targeted SNP screening of three independent Caucasian schizophrenia case-control cohorts (USA, Denmark, and Norway), we selected non-synonymous SNPs in neurodevelopmental genes and in genes associated with schizophrenia or non-synonymous SNPs presumably functional since they were linked to diseases such as diabetes mellitus or cancer. A total of 396 SNPs (table S1) was initially tested for association in the USA cohort. In this cohort, 23 SNPs were nominally associated with schizophrenia (uncorrected p-value <0.05; table S2). Of the 396 SNPs, 189 SNPs were selected for further analysis based on a trend for association with schizophrenia in the USA cohort, their reported association with schizophrenia or their MAF (1<MAF<20%). The 189 SNPs were analyzed in two additional schizophrenia-control cohorts from Europe (Denmark and Norway). In both the Danish and Norwegian cohort, nine SNPs were nominally associated with schizophrenia (uncorrected p-value <0.05; table S2). A meta-analysis revealed that in the three Caucasian cohorts ten SNPs were significantly (p<0.05) associated with schizophrenia (table 1) and did not deviate from the Hardy Weinberg equilibrium in the control groups (table S1). The top list
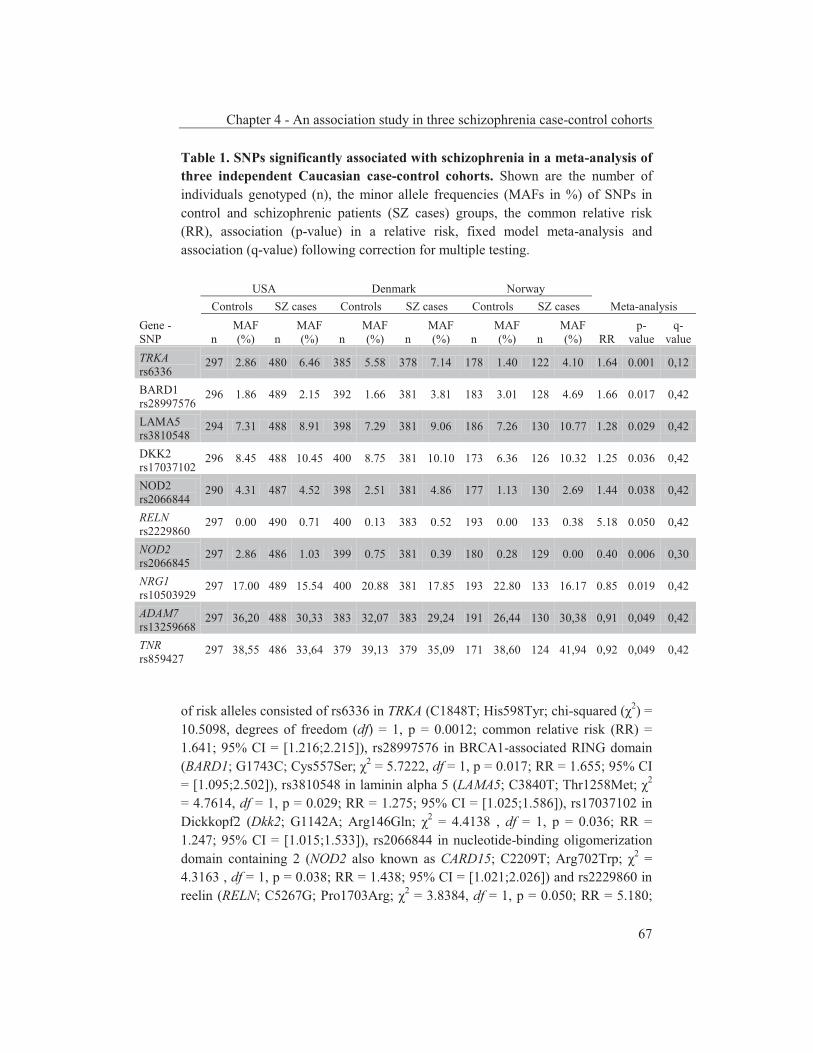
Chapter 4 - An association study in three schizophrenia case-control cohorts
67
Table 1. SNPs significantly associated with schizophrenia in a meta-analysis of three independent Caucasian case-control cohorts. Shown are the number of individuals genotyped (n), the minor allele frequencies (MAFs in %) of SNPs in control and schizophrenic patients (SZ cases) groups, the common relative risk (RR), association (p-value) in a relative risk, fixed model meta-analysis and association (q-value) following correction for multiple testing.
USA Denmark Norway Controls SZ cases Controls SZ cases Controls SZ cases Meta-analysis
Gene - SNP n
MAF (%) n
MAF (%) n
MAF (%) n
MAF (%) n
MAF (%) n
MAF (%) RR
p-value
q-value
TRKA rs6336
297 2.86 480 6.46 385 5.58 378 7.14 178 1.40 122 4.10 1.64 0.001 0,12
BARD1 rs28997576
296 1.86 489 2.15 392 1.66 381 3.81 183 3.01 128 4.69 1.66 0.017 0,42
LAMA5 rs3810548
294 7.31 488 8.91 398 7.29 381 9.06 186 7.26 130 10.77 1.28 0.029 0,42
DKK2 rs17037102
296 8.45 488 10.45 400 8.75 381 10.10 173 6.36 126 10.32 1.25 0.036 0,42
NOD2 rs2066844
290 4.31 487 4.52 398 2.51 381 4.86 177 1.13 130 2.69 1.44 0.038 0,42
RELN rs2229860
297 0.00 490 0.71 400 0.13 383 0.52 193 0.00 133 0.38 5.18 0.050 0,42
NOD2 rs2066845
297 2.86 486 1.03 399 0.75 381 0.39 180 0.28 129 0.00 0.40 0.006 0,30
NRG1 rs10503929
297 17.00 489 15.54 400 20.88 381 17.85 193 22.80 133 16.17 0.85 0.019 0,42
ADAM7 rs13259668
297 36,20 488 30,33 383 32,07 383 29,24 191 26,44 130 30,38 0,91 0,049 0,42
TNR rs859427
297 38,55 486 33,64 379 39,13 379 35,09 171 38,60 124 41,94 0,92 0,049 0,42
of risk alleles consisted of rs6336 in TRKA (C1848T; His598Tyr; chi-squared (χ2) = 10.5098, degrees of freedom (df) = 1, p = 0.0012; common relative risk (RR) = 1.641; 95% CI = [1.216;2.215]), rs28997576 in BRCA1-associated RING domain (BARD1; G1743C; Cys557Ser; χ2 = 5.7222, df = 1, p = 0.017; RR = 1.655; 95% CI = [1.095;2.502]), rs3810548 in laminin alpha 5 (LAMA5; C3840T; Thr1258Met; χ2 = 4.7614, df = 1, p = 0.029; RR = 1.275; 95% CI = [1.025;1.586]), rs17037102 in Dickkopf2 (Dkk2; G1142A; Arg146Gln; χ2 = 4.4138 , df = 1, p = 0.036; RR = 1.247; 95% CI = [1.015;1.533]), rs2066844 in nucleotide-binding oligomerization domain containing 2 (NOD2 also known as CARD15; C2209T; Arg702Trp; χ2 = 4.3163 , df = 1, p = 0.038; RR = 1.438; 95% CI = [1.021;2.026]) and rs2229860 in reelin (RELN; C5267G; Pro1703Arg; χ2 = 3.8384, df = 1, p = 0.050; RR = 5.180;

Chapter 4 - An association study in three schizophrenia case-control cohorts
68
95% CI = [0.999;26.862]). Significantly associated as protective alleles were rs2066845 in NOD2 (G2827C; Gly908Arg; χ2 = 7.5526, df = 1, p = 0.0060; RR = 0.398; 95% CI = [0.206;0.768]), rs10503929 in neuregulin1 (NRG1; T973C; Met294Thr; χ2 = 5.4978, df = 1, p = 0.019; RR = 0.847; 95% CI = [0.738;0.973]), rs13259668 in ADAM metallopeptidase domain 7 (ADAM7; A2025C; Asn638His; χ2 = 3.8763, df = 1, p = 0.049; RR = 0.909; 95% CI = [0.826;1.000]) and rs859427 in TenascinR (TNR; A2483G; Lys643Arg; χ2 = 3.8669, df = 1, p = 0.049; RR = 0.919; 95% CI = [0.844;1.000]). We then used the bootstrap method to calculate FDR q-values in order to correct for multiple testing of samples. For the ten associated SNPs, the q-values ranged from 0.42 for SNP rs2229860 to 0.12 for SNP rs6336 (table 1). Applying the q<0.10 significance threshold for candidate gene studies (Sullivan et al., 2008), in the three independent cohorts SNP rs6336 in TRKA was nearly significantly (q = 0.12) associated with schizophrenia. Removal of a single cohort from the meta-analysis confirmed that none of the cohorts was decisive per se (table S3). Discussion In this study, we found ten SNPs to be nominally associated with schizophrenia in a meta-analysis of three independent case-control cohorts of European descent. Of the ten SNPs, only the NRG1 SNP has been associated before with schizophrenia (Rosa et al., 2007) and thus the other nine are novel. Furthermore, it is noteworthy that, except for those in NRG1 and RELN, none of the SNPs that have been repeatedly reported to be associated with schizophrenia or were selected from schizophrenia genes, such as the Val158Met SNP in COMT (p = 0.45) and the Val66Met SNP in BDNF (p = 0.40), represented risk or protective alleles in our analysis. Of the ten polymorphisms, six SNPs represented risk alleles. The most strongly associated SNP rs6336 results in a histidine to tyrosine change at position 598 within the kinase domain of TRKA. BARD1 Cys557Ser SNP rs28997576 lowers levels of the tumor suppressor p53 protein and suppresses apoptosis in vitro (Sauer et al., 2005). The fact that the neurodevelopmentally important p53 protein has been associated with schizophrenia (Ni et al., 2005) and that the 557Ser allele causes reduced p53 levels supports a role for this genetic variation in BARD1 (best known for its interaction with the Breast Cancer 1 protein BRCA1) in neurodevelopmental disorders such as schizophrenia. The Thr1258Met SNP (rs3810548) is located in the proline-rich domain (1222-1277) of the extracellular matrix protein LAMA5 and deletes one of its seven predicted O-glycosylation sites (Julenius et al., 2005). SNP rs17037102 results in a change of the basic and evolutionarily highly conserved arginine to a glutamine at position 146 of the Dkk2 protein. Dkk2 is a member of the Dickkopf family and directly activates Wnt/β-catenin signaling (Wu et al., 2000) that is critical during (neuro)development (Cotter et al., 1998). SNP

Chapter 4 - An association study in three schizophrenia case-control cohorts
69
rs2066844 in NOD2, encoding an intracellular bacterial receptor activating nuclear factor-κB (NF-κB), causes an arginine to thryptophan change (Arg702Trp). In vitro assays have shown that the NF-κB response of the 702Trp variant to bacterial glycan components is reduced compared to that of wild-type NOD2 (Bonen et al., 2003). Thus, NOD2 SNP rs2066844 may influence the immune response and as such provide a link between schizophrenia and infection. The rare SNP rs2229860 results in a proline to arginine change at position 1703 of reelin, a protein important for the positioning of neurons during brain development (Tissir et al., 2003). Pro1703 is well conserved from mammals to invertebrates, indicating an important role for this residue. A reduced expression of reelin mRNA and protein in postmortem brains of schizophrenia patients (Impagnatiello et al., 1998), epigenetic abnormalities (Grayson et al., 2006) and an association study have linked reelin to schizophrenia (Kahler et al., 2008). In our analysis, four SNPs were associated with schizophrenia as protective alleles. Protective SNP rs2066845 is located in NOD2 and results in a glycine to arginine change at position 908 in the leucine-rich repeat of the protein. Two SNPs in NOD2 were thus found to be associated with schizophrenia, one protective (Gly908Arg) and one risk (Arg702Trp). Although the r2 between the two NOD2 SNPs is 0.0001, the fact that D’=1 indicates that the two SNPs are in LD. The second protective SNP, rs10503929 in NRG1, leads to a methionine to threonine change and a predicted extra N-glycosylation site (http://www.cbs.dtu.dk/services/ NetNGlyc). The threonine allele has recently been identified as a protective allele in a Spanish schizophrenia family study as well (144 families, p=0.03; Rosa et al., 2007). Haplotypes and SNPs in NRG1 have previously been associated with schizophrenia as risk factors (Tosato et al., 2005), whereas our as well as the Spanish study suggests that minor allele rs10503929 acts as a protective factor. ADAM7 SNP rs13259668 results in a change of the asparagine at position 638 to a histidine located in the cysteine-rich domain that complements the binding capacity of the disintegrin domain (Smith et al., 2002). Finally, TNR SNP rs875326 involves a lysine to an arginine substitution at position 643 in the fibronectin type-III domain of this extracellular matrix protein that is expressed primarily in the central nervous system. In contrast to TNR polymorphisms, SNPs in TNXB, another tenascin gene family member, have been previously associated with schizophrenia (Tochigi et al., 2007). The ten SNPs with a p<0.05 displayed a FDR q-value (for correction of multiple sample testing) between 0.12 and 0.42. The q-value of SNP rs6336 (His598Tyr) in TRKA (q = 0.12) was just above the 0.10 q-value threshold used in association studies (Sullivan et al., 2008). Like its directly surrounding region within the kinase domain, His598 is conserved among all known vertebrate TRKA sequences as well as in a snail TRKA-related protein sequence (U61728; Van Kesteren et al., 1998), the only known invertebrate TRKA-like receptor. Furthermore, at the corresponding

Chapter 4 - An association study in three schizophrenia case-control cohorts
70
position in the kinase domain the histidine is conserved in all known vertebrate sequences of the two additional NTRK family members TRKB and TRKC, but not in other tyrosine receptor kinases. The His598Tyr substitution is predicted to have a deleterious effect on TRKA because of the loss of a hydrogen bond and overpacking (www.SNPs3D.org; Yue et al., 2006). TRKA is the high-affinity receptor for nerve growth factor (NGF) that plays a crucial role in neurodevelopment. Some components of neurotrophin signaling cascades, such as BDNF, have been linked to schizophrenia through genetic and expression studies (Kanazawa et al., 2007). An altered expression of TRKA and NGF has been found in animal models for schizophrenia (Becker et al., 2008; Terry et al., 2007) and of NGF in (post-mortem) studies on schizophrenic patients (Shoval et al., 2005), but TRKA has not been genetically associated before with the disorder. In conclusion, we have found in a meta-analysis over three independent Caucasian case-control cohorts ten non-synonymous SNPs that are associated with schizophrenia, nine of which have not been described in relation to the disorder before. Additional screenings of these SNPs in other schizophrenia case-control cohorts are needed for validation. TRKA SNP rs6336 is of special interest and advances a novel susceptibility pathway for the development of schizophrenia. Acknowledgements Authors wish to thank patients who participated and made this study possible. Authors also acknowledge expert assistance by numerous mental health professionals in the various clinical departments. This work was supported by grant T5-209 from the Dutch Top Institute Pharma. References Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype
maps. Bioinformatics 2005;21:263-265. Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, et al. A genome-wide
association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry 2008;13:197-207.
Becker A, Grecksch G, Schwegler H, Roskoden T. Expression of mRNA of neurotrophic factors and their receptors are significantly altered after subchronic ketamine treatment. Med Chem 2008;4:256-263.
Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 2003;124:140-146.
Cotter D, Kerwin R, al-Sarraji S, Brion JP, Chadwich A, Lovestone S, et al. Abnormalities of Wnt signalling in schizophrenia--evidence for neurodevelopmental abnormality. Neuroreport 1998;9:1379-1383.
Cucherat M, Boissel JP, Leizorovicz A, Haugh MC. EasyMA: a program for the meta-analysis of clinical trials. Comput Methods Programs Biomed 1997;53:187-190.

Chapter 4 - An association study in three schizophrenia case-control cohorts
71
Grayson DR, Chen Y, Costa E, Dong E, Guidotti A, Kundakovic M, et al. The human reelin gene: transcription factors (+), repressors (-) and the methylation switch (+/-) in schizophrenia. Pharmacol Ther 2006;111:272-286.
Hansen T, Olsen L, Lindow M, Jakobsen KD, Ullum H, Jonsson E, et al. Brain expressed microRNAs implicated in schizophrenia etiology. PLoS ONE 2007;2:e873.
Impagnatiello F, Guidotti AR, Pesold C, Dwivedi Y, Caruncho H, Pisu MG, et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A 1998;95:15718-15723.
Julenius K, Molgaard A, Gupta R, Brunak S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 2005;15:153-164.
Kahler AK, Djurovic S, Kulle B, Jonsson EG, Agartz I, Hall H, et al. Association analysis of schizophrenia on 18 genes involved in neuronal migration: MDGA1 as a new susceptibility gene. Am J Med Genet B Neuropsychiatr Genet 2008;147B:1089-1100.
Kanazawa T, Glatt SJ, Kia-Keating B, Yoneda H, Tsuang MT. Meta-analysis reveals no association of the Val66Met polymorphism of brain-derived neurotrophic factor with either schizophrenia or bipolar disorder. Psychiatr Genet 2007;17:165-170.
Karayiorgou M, Gogos JA. Schizophrenia genetics: uncovering positional candidate genes. Eur J Hum Genet 2006;14:512-519.
Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry 2007;12:572-580.
Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet 2003;73:34-48.
Maki P, Veijola J, Jones PB, Murray GK, Koponen H, Tienari P, et al. Predictors of schizophrenia--a review. Br Med Bull 2005;73-74:1-15.
Ni X, Trakalo J, Valente J, Azevedo MH, Pato MT, Pato CN, et al. Human p53 tumor suppressor gene (TP53) and schizophrenia: case-control and family studies. Neurosci Lett 2005;388:173-178.
Rosa A, Gardner M, Cuesta MJ, Peralta V, Fatjo-Vilas M, Miret S, et al. Family-based association study of neuregulin-1 gene and psychosis in a Spanish sample. Am J Med Genet B Neuropsychiatr Genet 2007;144:954-957.
Sauer MK, Andrulis IL. Identification and characterization of missense alterations in the BRCA1 associated RING domain (BARD1) gene in breast and ovarian cancer. J Med Genet 2005;42:633-638.
Shoval G, Weizman A. The possible role of neurotrophins in the pathogenesis and therapy of schizophrenia. Eur Neuropsychopharmacol 2005;15:319-329.
Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW. The cysteine-rich domain regulates ADAM protease function in vivo. J Cell Biol 2002;159:893-902.
Storey JD. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann Stat 2003;31:2013-2035.
Sullivan PF, Lin D, Tzeng JY, van den Oord E, Perkins D, Stroup TS, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry 2008;13:570-584.
Terry AV, Jr., Gearhart DA, Warner SE, Zhang G, Bartlett MG, Middlemore ML, et al. Oral haloperidol or risperidone treatment in rats: temporal effects on nerve growth factor receptors, cholinergic neurons, and memory performance. Neuroscience 2007;146:1316-1332.
Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci 2003;4:496-505. Tochigi M, Zhang X, Ohashi J, Hibino H, Otowa T, Rogers M, et al. Association study between
the TNXB locus and schizophrenia in a Japanese population. Am J Med Genet B Neuropsychiatr Genet 2007;144B:305-309.
Tosato S, Dazzan P, Collier D. Association between the neuregulin 1 gene and schizophrenia: a systematic review. Schizophr Bull 2005;31:613-617.

Chapter 4 - An association study in three schizophrenia case-control cohorts
72
Van Kesteren RE, Fainzilber M, Hauser G, van Minnen J, Vreugdenhil E, Smit AB, et al. Early evolutionary origin of the neurotrophin receptor family. Embo J 1998;17:2534-2542.
Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 1987;44:660-669.
Wu W, Glinka A, Delius H, Niehrs C. Mutual antagonism between dickkopf1 and dickkopf2 regulates Wnt/beta-catenin signalling. Curr Biol 2000;10:1611-1614.
Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics 2006;7:166.
Chapter 4 - An association study in three schizophrenia case-control cohorts
73
Supplementary table 1. SNPs tested for association with schizophrenia in three independent Caucasian case-control cohorts. Shown are the minor allele frequencies (MAFs in %) of SNPs in control and schizophrenic patient (SZ) groups and allelic association per cohort (p-value) , association in a relative risk, fixed model meta-analysis (p-value) and association following correction for multiple testing (q-value). The genes and SNPs are divided into "neurodevelopmental", "linked to schizophrenia" and "linked to a disease". The diseases are indicated in brackets behind the SNP ID. AD = Alzheimer's Disease, AMD = Age-related Macular Degeneration, As = Asthma, BC = Breast Cancer, BMD = Bone Mineral Density, CAD = Coronary Artery Disease, CC = Colon Cancer, CD = Crohn’s disease, CP = Cancer Progression, CrC = Colorectal Cancer, DM = Diabetes Mellitus, DMc = DM complications, LC = Lung Cancer, MI = Myocardial Infarction, NTD = Neural tube defect, PC = Prostate Cancer.
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
Neurodevelopmental genes ADAM12 rs3740199 44,26 42,63 0,53 ADAM15 rs6427128 12,96 13,64 0,71 10,33 10,42 0,95 11,93 12,30 0,89 0,73 0,51 ADAM22 rs17255978 Assay Failure (0.0%) ADAM22 rs2279542 47,30 48,36 0,68 ADAM28 rs17736699 Assay Failure (86.2) ADAM28 rs7829965 1,87 2,78 0,26 3,26 3,40 0,87 3,63 1,56 0,12 0,83 0,53 ADAM30 rs2641348 9,69 10,06 0,81 9,23 12,50 0,04 10,44 11,81 0,59 0,083 0,42 ADAM33 rs2280090 15,57 12,37 0,08 ADAM33 rs3918392 2,70 3,89 0,21 3,14 3,03 0,90 2,59 3,76 0,40 0,29 0,45 ADAM7 rs13259668 36,20 30,33 0,02 32,07 29,24 0,23 26,44 30,38 0,27 0,049 0,42 ADAM7 rs3736281 1,35 2,06 0,30 Assay Failure (0.0%) ADAM8 rs12257830 0,17 0,00 0,19 ADAM8 rs2275725 10,65 9,65 0,52 9,52 8,40 0,44 8,12 5,38 0,18 0,17 0,44 ADAMTS1 rs428785 19,69 22,92 0,14 ADAMTS13 rs2301612 42,71 40,27 0,34 ADAMTS13 rs685523 10,30 11,93 0,32 Assay Failure (0.0%) ADAMTS14 rs10823607 11,28 12,99 0,32 13,38 12,43 0,58 10,96 11,60 0,80 0,73 0,51 ADAMTS14 rs12774070 Assay Failure (62.9%) ADAMTS14 rs4747096 14,07 17,15 0,11 14,41 16,71 0,21 18,23 17,83 0,90 0,081 0,42 ADAMTS16 rs16875054 3,87 4,08 0,84 3,58 3,41 0,86 2,75 3,52 0,58 0,82 0,53 ADAMTS16 rs1863968 38,68 39,69 0,69 ADAMTS16 rs2086310 15,40 19,32 0,06 23,12 23,10 0,99 20,73 20,31 0,90 0,33 0,47 ADAMTS17 rs2573652 Assay Failure (89.7%) ADAMTS17 rs28567966 16,84 14,57 0,23 15,23 14,79 0,81 13,16 15,89 0,33 0,58 0,50 ADAMTS17 rs7496668 Assay Failure (86.8%) ADAMTS18 rs11640912 34,68 36,75 0,41 ADAMTS18 rs11643211 25,35 24,69 0,77 ADAMTS18 rs12935394 11,99 10,86 0,49 11,90 11,97 0,97 12,69 19,23 0,02 0,50 0,50 ADAMTS18 rs9930984 36,94 38,15 0,64 ADAMTS19 rs11749126 19,02 17,08 0,33 18,38 21,07 0,18 21,47 17,72 0,25 0,85 0,53 ADAMTS19 rs6595908 0,17 0,21 0,87
Chapter 4 - An association study in three schizophrenia case-control cohorts
74
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
ADAMTS2 rs1054480 25,00 27,51 0,28 ADAMTS2 rs11750821 (a) 11,69 12,65 0,58 ADAMTS2 rs17667857 0,00 0,00 1,00 ADAMTS3 rs788908 38,18 37,30 0,73 ADAMTS4 rs4233367 39,86 40,57 0,78 ADAMTS5 rs226794 11,62 12,14 0,75 11,88 9,55 0,14 11,46 14,02 0,33 0,77 0,53 ADAMTS5 rs2830585 18,35 15,10 0,09 15,83 15,67 0,93 14,92 12,12 0,31 0,12 0,42 ADAMTS5 rs457947 13,64 16,70 0,10 12,06 14,25 0,20 12,18 9,23 0,24 0,13 0,42 ADAMTS7 rs3825807 42,74 44,18 0,58 45,58 43,08 0,32 40,05 44,27 0,29 0,90 0,54 ADAMTS9 rs6787633 0,68 1,23 0,29 ALDH1A1 rs8187929 0,00 0,00 1,00 ALK rs17694720 0,00 0,00 1,00 0,00 0,00 1,00 0.00 c 0,41 0,24 0,60 0,50 ALK rs1881420 20,20 23,87 0,09 24,06 22,82 0,56 25,13 21,32 0,27 0,85 0,53 ALK rs1881421 31,14 37,03 0,02 37,59 36,22 0,57 39,47 35,55 0,32 0,56 0,50 APLP2 rs3740881 0,17 0,31 0,59 ARHGEF10 rs17683288 7,51 5,38 0,09 7,05 9,97 0,04 10,44 6,87 0,12 0,84 0,53 ARHGEF10 rs2294039 2,87 4,40 0,13 3,39 3,40 0,99 3,91 3,79 0,94 0,36 0,47 ARHGEF10 rs9657362 14,97 15,83 0,65 13,28 14,27 0,57 10,70 13,46 0,29 0,29 0,45 ARHGEF11 rs945508 47,79 45,76 0,44 ARHGEF12 rs2305013 4,04 4,50 0,66 6,77 4,33 0,04 7,25 5,43 0,36 0,12 0,42 ARHGEF15 rs3744647 35,25 37,71 0,33 ARHGEF15 rs871841 49,15 49,38 0,93 ARHGEF16 rs2185639 9,09 13,24 0,02 13,66 11,94 0,31 14.29 c 13,93 0,90 0,52 0,50 ARHGEF18 rs2287918 21,89 25,67 0,09 27,50 27,94 0,85 30,63 29,77 0,82 0,31 0,47 ARHGEF19 rs221057 26,02 26,67 0,78 ARHGEF19 rs221058 Assay Failure (84.4%) ARHGEF3 rs3772219 32,29 30,15 0,38 ATF4 rs4894 0,00 0,00 1,00 Axin2 rs2240308 44,76 47,23 0,34 BMP4 rs17563 (a) 43.26 c 44,73 0,59 BTC rs11938093 23,78 26,56 0,23 BTC rs28549760 13,27 12,53 0,68 10,35 13,32 0,07 14,32 13,26 0,70 0,48 0,50 CASP6 rs11574697 0,00 0,00 1,00 CASP6 rs5030593 0,00 0,00 1,00 CASP6 rs5030674 0,00 0,00 1,00 CASP8 rs1045485 (a) 10,31 11,55 0,45 CD44 rs1467558 15,71 19,76 0,05 19,80 16,49 0,09 16,06 19,08 0,32 0,60 0,50 CD44 rs9666607 29,97 33,74 0,12 31,19 29,71 0,53 27,86 32,33 0,22 0,28 0,44 CD82 rs1139971 Assay Failure (88.6%) CDH1 rs9282655 0,00 0,00 1,00 CDH2 rs17445840 0,00 0,41 0,12 0,00 0,00 1,00 0,00 0,00 1,00 0,39 0,49 CDH2 rs2289664 2,36 2,56 0,81 2,38 2,75 0,65 2,53 4,80 0,13 0,26 0,44 CER1 rs10115703 7,74 8,47 0,61 9,15 8,53 0,67 8,03 10,77 0,24 0,59 0,50 CER1 rs3747532 30,47 32,35 0,44 CHRD rs16858780 15,17 17,50 0,24 Assay Failure (79.9%) CLSTN2 rs17348572 6,23 6,22 1,00 7,13 6,27 0,50 4,71 4,58 0,94 0,62 0,50 CLSTN2 rs7632885 39,23 40,31 0,67

Chapter 4 - An association study in three schizophrenia case-control cohorts
75
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
CLSTN3 rs7302230 7,74 6,34 0,29 5,90 6,30 0,74 5,21 6,02 0,66 0,73 0,51 CNTN2 rs2229867 0,00 0,00 1,00 CNTN2 rs2229866 Assay Failure (65.0%) CNTN2 rs2275697 15,49 16,42 0,63 15,97 17,96 0,31 16,94 18,22 0,68 0,25 0,44 CNTN3 rs626578 5,24 5,99 0,53 3,25 4,31 0,27 3,37 3,44 0,96 0,26 0,44 CNTN5 rs11223168 10,00 8,08 0,20 8,35 7,83 0,71 11,92 9,40 0,31 0,13 0,42 CNTN5 rs1216183 9,60 12,81 0,05 9,60 10,39 0,60 11.43 c 10,71 0,78 0,15 0,42 CNTN5 rs1944169 0,17 0,00 0,21 CSPG5 rs3732530 36,36 34,59 0,48 CTNNA3 rs4548513 41,16 35,59 0,03 32,45 32,38 0,98 39,06 39,69 0,87 0,16 0,42 CUL3 rs3738952 8,64 10,58 0,22 9,00 10,44 0,33 7,81 10,61 0,22 0,056 0,42 CUL4A rs2302757 23,57 23,52 0,98 DDX11 rs2272522 18,92 20,89 0,35 DDX11 rs6442827 0,00 0,00 1,00 DKK2 rs17037102 8,45 10,45 0,19 8,75 10,10 0,36 6.36 c 10,32 0,08 0,036 0,42 DKK3 rs3206824 20,88 21,56 0,75 DKKL1 rs2303759 (a) 7,83 8,14 0,83 DKKL1 rs2288481 24,41 22,54 0,39 DKKL1 rs919364 Assay Failure (86.3%) DNMT1 rs16999593 0,00 0,22 0,26 DNMT1 rs8111085 7,77 8,61 0,56 5,88 6,14 0,83 4,66 4,55 0,94 0,60 0,50 DNMT2 rs11254408 (a) 13,18 16,36 0,09 15,19 14,36 0,64 9,84 14,29 0,08 0,15 0,42 DNMT2 rs11254413 Assay Failure (84.5%) DNMT3B rs17123590 0,00 0,00 1,00 DNMT3L rs7354779 25,34 25,97 0,78 EFNA1 rs4745 46,46 45,59 0,74 EFNA3 rs17723260 12,67 12,61 0,97 12,91 14,14 0,48 17,89 16,54 0,65 0,82 0,53 EFNB1 rs16989105 0,00 0,20 0,27 ELP4 rs2295748 22,53 24,28 0,43 EN2 rs3735653 Assay Failure (79.7%) EPHA1 rs11768549 2,08 2,27 0,80 1,25 2,23 0,14 0,79 0,78 0,99 0,26 0,44 EPHA1 rs4725617 7,07 7,24 0,90 10,05 11,15 0,48 11,78 9,92 0,46 0,81 0,53 EPHA1 rs6967117 Assay Failure (88.6%) EPHA10 rs12405650 (b) 7,68 8,07 0,78 6,50 7,22 0,57 5,83 5,04 0,67 0,69 0,51 EPHA10 rs17511304 (a) 1,01 0,82 0,70 EPHA10 rs6670599 Assay Failure (72.2%) EPHA10 rs6671088 15,33 16,36 0,60 16,33 18,23 0,32 19,25 13,28 0,05 0,82 0,53 EPHA3 rs17801309 10,47 9,90 0,71 7,54 10,89 0,02 7,81 9,54 0,44 0,13 0,42 EPHA7 rs2278107 3,56 1,99 0,06 2,47 3,55 0,22 1,91 5,47 0,02 0,53 0,50 EPHB6 rs8177146 2,70 1,84 0,26 0,88 0,39 0,23 1,12 0,78 0,67 0,11 0,42 EPHB6 rs8177173 1,18 1,32 0,82 1,56 1,31 0,67 0,53 0,76 0,71 0,97 0,56 EPHB6 rs8177175 0,00 0,00 1,00 FBXO2 rs9614 (a,b) 19,59 20,92 0,53 19,75 21,58 0,37 19,57 18,08 0,64 0,42 0,50 FEZ1 rs597570 19,09 18,00 0,59 17,63 17,41 0,91 13,74 20,54 0,02 0,64 0,50 FN1 rs1250259 25,34 24,11 0,59 FN1 rs17449032 1,86 1,95 0,90 2,81 2,24 0,48 2,50 3,88 0,33 0,96 0,56 FZD6 rs3808553 46,46 49,18 0,30
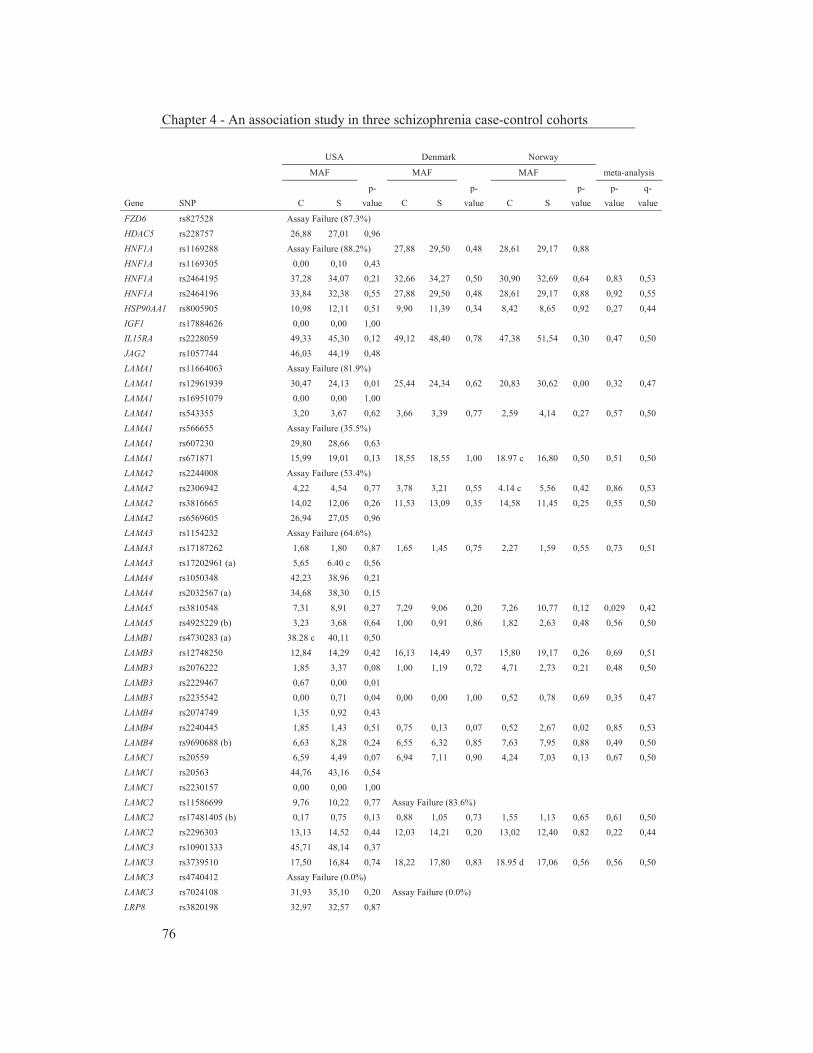
Chapter 4 - An association study in three schizophrenia case-control cohorts
76
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
FZD6 rs827528 Assay Failure (87.3%) HDAC5 rs228757 26,88 27,01 0,96 HNF1A rs1169288 Assay Failure (88.2%) 27,88 29,50 0,48 28,61 29,17 0,88 HNF1A rs1169305 0,00 0,10 0,43 HNF1A rs2464195 37,28 34,07 0,21 32,66 34,27 0,50 30,90 32,69 0,64 0,83 0,53 HNF1A rs2464196 33,84 32,38 0,55 27,88 29,50 0,48 28,61 29,17 0,88 0,92 0,55 HSP90AA1 rs8005905 10,98 12,11 0,51 9,90 11,39 0,34 8,42 8,65 0,92 0,27 0,44 IGF1 rs17884626 0,00 0,00 1,00 IL15RA rs2228059 49,33 45,30 0,12 49,12 48,40 0,78 47,38 51,54 0,30 0,47 0,50 JAG2 rs1057744 46,03 44,19 0,48 LAMA1 rs11664063 Assay Failure (81.9%) LAMA1 rs12961939 30,47 24,13 0,01 25,44 24,34 0,62 20,83 30,62 0,00 0,32 0,47 LAMA1 rs16951079 0,00 0,00 1,00 LAMA1 rs543355 3,20 3,67 0,62 3,66 3,39 0,77 2,59 4,14 0,27 0,57 0,50 LAMA1 rs566655 Assay Failure (35.5%) LAMA1 rs607230 29,80 28,66 0,63 LAMA1 rs671871 15,99 19,01 0,13 18,55 18,55 1,00 18.97 c 16,80 0,50 0,51 0,50 LAMA2 rs2244008 Assay Failure (53.4%) LAMA2 rs2306942 4,22 4,54 0,77 3,78 3,21 0,55 4.14 c 5,56 0,42 0,86 0,53 LAMA2 rs3816665 14,02 12,06 0,26 11,53 13,09 0,35 14,58 11,45 0,25 0,55 0,50 LAMA2 rs6569605 26,94 27,05 0,96 LAMA3 rs1154232 Assay Failure (64.6%) LAMA3 rs17187262 1,68 1,80 0,87 1,65 1,45 0,75 2,27 1,59 0,55 0,73 0,51 LAMA3 rs17202961 (a) 5,65 6.40 c 0,56 LAMA4 rs1050348 42,23 38,96 0,21 LAMA4 rs2032567 (a) 34,68 38,30 0,15 LAMA5 rs3810548 7,31 8,91 0,27 7,29 9,06 0,20 7,26 10,77 0,12 0,029 0,42 LAMA5 rs4925229 (b) 3,23 3,68 0,64 1,00 0,91 0,86 1,82 2,63 0,48 0,56 0,50 LAMB1 rs4730283 (a) 38.28 c 40,11 0,50 LAMB3 rs12748250 12,84 14,29 0,42 16,13 14,49 0,37 15,80 19,17 0,26 0,69 0,51 LAMB3 rs2076222 1,85 3,37 0,08 1,00 1,19 0,72 4,71 2,73 0,21 0,48 0,50 LAMB3 rs2229467 0,67 0,00 0,01 LAMB3 rs2235542 0,00 0,71 0,04 0,00 0,00 1,00 0,52 0,78 0,69 0,35 0,47 LAMB4 rs2074749 1,35 0,92 0,43 LAMB4 rs2240445 1,85 1,43 0,51 0,75 0,13 0,07 0,52 2,67 0,02 0,85 0,53 LAMB4 rs9690688 (b) 6,63 8,28 0,24 6,55 6,32 0,85 7,63 7,95 0,88 0,49 0,50 LAMC1 rs20559 6,59 4,49 0,07 6,94 7,11 0,90 4,24 7,03 0,13 0,67 0,50 LAMC1 rs20563 44,76 43,16 0,54 LAMC1 rs2230157 0,00 0,00 1,00 LAMC2 rs11586699 9,76 10,22 0,77 Assay Failure (83.6%) LAMC2 rs17481405 (b) 0,17 0,75 0,13 0,88 1,05 0,73 1,55 1,13 0,65 0,61 0,50 LAMC2 rs2296303 13,13 14,52 0,44 12,03 14,21 0,20 13,02 12,40 0,82 0,22 0,44 LAMC3 rs10901333 45,71 48,14 0,37 LAMC3 rs3739510 17,50 16,84 0,74 18,22 17,80 0,83 18.95 d 17,06 0,56 0,56 0,50 LAMC3 rs4740412 Assay Failure (0.0%) LAMC3 rs7024108 31,93 35,10 0,20 Assay Failure (0.0%) LRP8 rs3820198 32,97 32,57 0,87
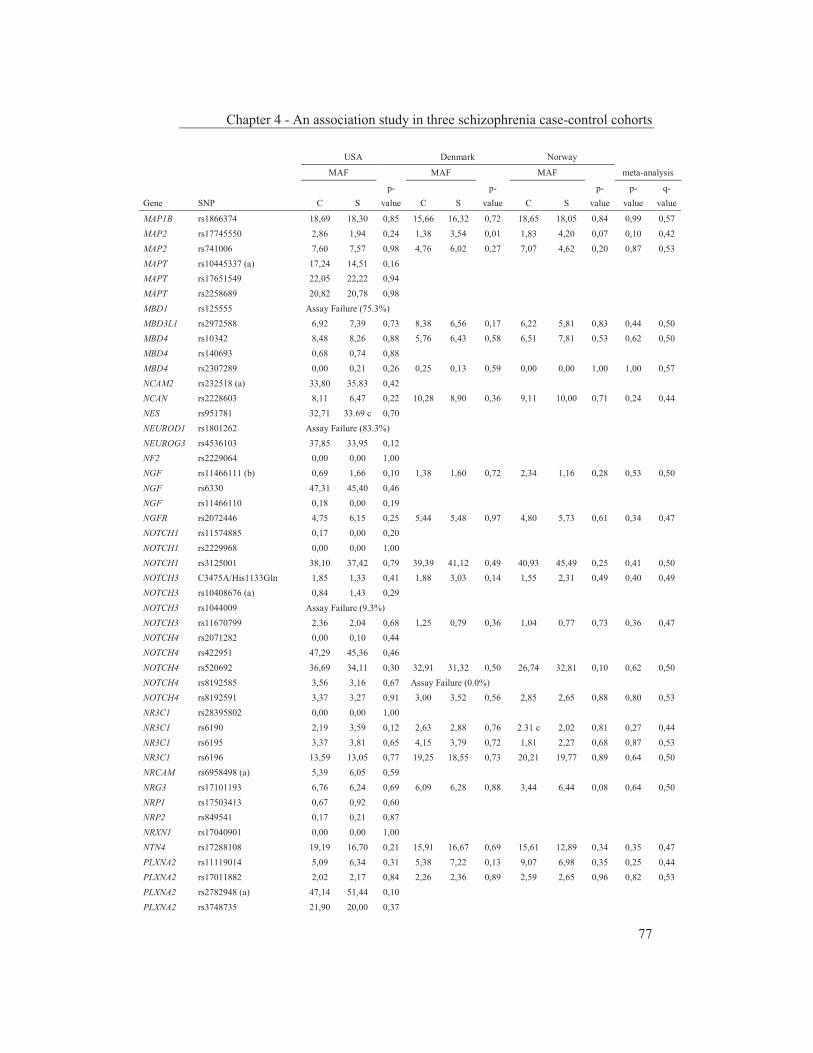
Chapter 4 - An association study in three schizophrenia case-control cohorts
77
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
MAP1B rs1866374 18,69 18,30 0,85 15,66 16,32 0,72 18,65 18,05 0,84 0,99 0,57 MAP2 rs17745550 2,86 1,94 0,24 1,38 3,54 0,01 1,83 4,20 0,07 0,10 0,42 MAP2 rs741006 7,60 7,57 0,98 4,76 6,02 0,27 7,07 4,62 0,20 0,87 0,53 MAPT rs10445337 (a) 17,24 14,51 0,16 MAPT rs17651549 22,05 22,22 0,94 MAPT rs2258689 20,82 20,78 0,98 MBD1 rs125555 Assay Failure (75.3%) MBD3L1 rs2972588 6,92 7,39 0,73 8,38 6,56 0,17 6,22 5,81 0,83 0,44 0,50 MBD4 rs10342 8,48 8,26 0,88 5,76 6,43 0,58 6,51 7,81 0,53 0,62 0,50 MBD4 rs140693 0,68 0,74 0,88 MBD4 rs2307289 0,00 0,21 0,26 0,25 0,13 0,59 0,00 0,00 1,00 1,00 0,57 NCAM2 rs232518 (a) 33,80 35,83 0,42 NCAN rs2228603 8,11 6,47 0,22 10,28 8,90 0,36 9,11 10,00 0,71 0,24 0,44 NES rs951781 32,71 33.69 c 0,70 NEUROD1 rs1801262 Assay Failure (83.3%) NEUROG3 rs4536103 37,85 33,95 0,12 NF2 rs2229064 0,00 0,00 1,00 NGF rs11466111 (b) 0,69 1,66 0,10 1,38 1,60 0,72 2,34 1,16 0,28 0,53 0,50 NGF rs6330 47,31 45,40 0,46 NGF rs11466110 0,18 0,00 0,19 NGFR rs2072446 4,75 6,15 0,25 5,44 5,48 0,97 4,80 5,73 0,61 0,34 0,47 NOTCH1 rs11574885 0,17 0,00 0,20 NOTCH1 rs2229968 0,00 0,00 1,00 NOTCH1 rs3125001 38,10 37,42 0,79 39,39 41,12 0,49 40,93 45,49 0,25 0,41 0,50 NOTCH3 C3475A/His1133Gln 1,85 1,33 0,41 1,88 3,03 0,14 1,55 2,31 0,49 0,40 0,49 NOTCH3 rs10408676 (a) 0,84 1,43 0,29 NOTCH3 rs1044009 Assay Failure (9.3%) NOTCH3 rs11670799 2,36 2,04 0,68 1,25 0,79 0,36 1,04 0,77 0,73 0,36 0,47 NOTCH4 rs2071282 0,00 0,10 0,44 NOTCH4 rs422951 47,29 45,36 0,46 NOTCH4 rs520692 36,69 34,11 0,30 32,91 31,32 0,50 26,74 32,81 0,10 0,62 0,50 NOTCH4 rs8192585 3,56 3,16 0,67 Assay Failure (0.0%) NOTCH4 rs8192591 3,37 3,27 0,91 3,00 3,52 0,56 2,85 2,65 0,88 0,80 0,53 NR3C1 rs28395802 0,00 0,00 1,00 NR3C1 rs6190 2,19 3,59 0,12 2,63 2,88 0,76 2.31 c 2,02 0,81 0,27 0,44 NR3C1 rs6195 3,37 3,81 0,65 4,15 3,79 0,72 1,81 2,27 0,68 0,87 0,53 NR3C1 rs6196 13,59 13,05 0,77 19,25 18,55 0,73 20,21 19,77 0,89 0,64 0,50 NRCAM rs6958498 (a) 5,39 6,05 0,59 NRG3 rs17101193 6,76 6,24 0,69 6,09 6,28 0,88 3,44 6,44 0,08 0,64 0,50 NRP1 rs17503413 0,67 0,92 0,60 NRP2 rs849541 0,17 0,21 0,87 NRXN1 rs17040901 0,00 0,00 1,00 NTN4 rs17288108 19,19 16,70 0,21 15,91 16,67 0,69 15,61 12,89 0,34 0,35 0,47 PLXNA2 rs11119014 5,09 6,34 0,31 5,38 7,22 0,13 9,07 6,98 0,35 0,25 0,44 PLXNA2 rs17011882 2,02 2,17 0,84 2,26 2,36 0,89 2,59 2,65 0,96 0,82 0,53 PLXNA2 rs2782948 (a) 47,14 51,44 0,10 PLXNA2 rs3748735 21,90 20,00 0,37
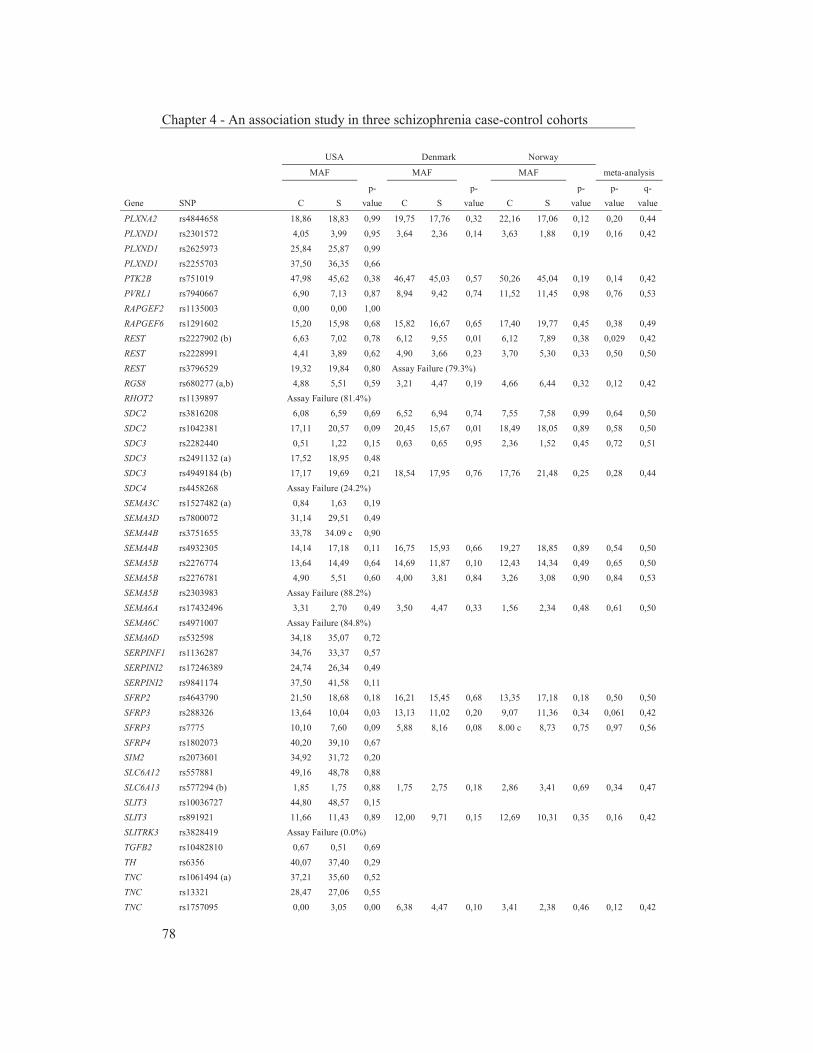
Chapter 4 - An association study in three schizophrenia case-control cohorts
78
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
PLXNA2 rs4844658 18,86 18,83 0,99 19,75 17,76 0,32 22,16 17,06 0,12 0,20 0,44 PLXND1 rs2301572 4,05 3,99 0,95 3,64 2,36 0,14 3,63 1,88 0,19 0,16 0,42 PLXND1 rs2625973 25,84 25,87 0,99 PLXND1 rs2255703 37,50 36,35 0,66 PTK2B rs751019 47,98 45,62 0,38 46,47 45,03 0,57 50,26 45,04 0,19 0,14 0,42 PVRL1 rs7940667 6,90 7,13 0,87 8,94 9,42 0,74 11,52 11,45 0,98 0,76 0,53 RAPGEF2 rs1135003 0,00 0,00 1,00 RAPGEF6 rs1291602 15,20 15,98 0,68 15,82 16,67 0,65 17,40 19,77 0,45 0,38 0,49 REST rs2227902 (b) 6,63 7,02 0,78 6,12 9,55 0,01 6,12 7,89 0,38 0,029 0,42 REST rs2228991 4,41 3,89 0,62 4,90 3,66 0,23 3,70 5,30 0,33 0,50 0,50 REST rs3796529 19,32 19,84 0,80 Assay Failure (79.3%) RGS8 rs680277 (a,b) 4,88 5,51 0,59 3,21 4,47 0,19 4,66 6,44 0,32 0,12 0,42 RHOT2 rs1139897 Assay Failure (81.4%) SDC2 rs3816208 6,08 6,59 0,69 6,52 6,94 0,74 7,55 7,58 0,99 0,64 0,50 SDC2 rs1042381 17,11 20,57 0,09 20,45 15,67 0,01 18,49 18,05 0,89 0,58 0,50 SDC3 rs2282440 0,51 1,22 0,15 0,63 0,65 0,95 2,36 1,52 0,45 0,72 0,51 SDC3 rs2491132 (a) 17,52 18,95 0,48 SDC3 rs4949184 (b) 17,17 19,69 0,21 18,54 17,95 0,76 17,76 21,48 0,25 0,28 0,44 SDC4 rs4458268 Assay Failure (24.2%) SEMA3C rs1527482 (a) 0,84 1,63 0,19 SEMA3D rs7800072 31,14 29,51 0,49 SEMA4B rs3751655 33,78 34.09 c 0,90 SEMA4B rs4932305 14,14 17,18 0,11 16,75 15,93 0,66 19,27 18,85 0,89 0,54 0,50 SEMA5B rs2276774 13,64 14,49 0,64 14,69 11,87 0,10 12,43 14,34 0,49 0,65 0,50 SEMA5B rs2276781 4,90 5,51 0,60 4,00 3,81 0,84 3,26 3,08 0,90 0,84 0,53 SEMA5B rs2303983 Assay Failure (88.2%) SEMA6A rs17432496 3,31 2,70 0,49 3,50 4,47 0,33 1,56 2,34 0,48 0,61 0,50 SEMA6C rs4971007 Assay Failure (84.8%) SEMA6D rs532598 34,18 35,07 0,72 SERPINF1 rs1136287 34,76 33,37 0,57 SERPINI2 rs17246389 24,74 26,34 0,49 SERPINI2 rs9841174 37,50 41,58 0,11 SFRP2 rs4643790 21,50 18,68 0,18 16,21 15,45 0,68 13,35 17,18 0,18 0,50 0,50 SFRP3 rs288326 13,64 10,04 0,03 13,13 11,02 0,20 9,07 11,36 0,34 0,061 0,42 SFRP3 rs7775 10,10 7,60 0,09 5,88 8,16 0,08 8.00 c 8,73 0,75 0,97 0,56 SFRP4 rs1802073 40,20 39,10 0,67 SIM2 rs2073601 34,92 31,72 0,20 SLC6A12 rs557881 49,16 48,78 0,88 SLC6A13 rs577294 (b) 1,85 1,75 0,88 1,75 2,75 0,18 2,86 3,41 0,69 0,34 0,47 SLIT3 rs10036727 44,80 48,57 0,15 SLIT3 rs891921 11,66 11,43 0,89 12,00 9,71 0,15 12,69 10,31 0,35 0,16 0,42 SLITRK3 rs3828419 Assay Failure (0.0%) TGFB2 rs10482810 0,67 0,51 0,69 TH rs6356 40,07 37,40 0,29 TNC rs1061494 (a) 37,21 35,60 0,52 TNC rs13321 28,47 27,06 0,55 TNC rs1757095 0,00 3,05 0,00 6,38 4,47 0,10 3,41 2,38 0,46 0,12 0,42

Chapter 4 - An association study in three schizophrenia case-control cohorts
79
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
TNC rs2104772 42,26 46,02 0,15 42,61 42,39 0,93 38,22 38,76 0,89 0,35 0,47 TNN rs10798333 38,98 40,62 0,52 TNN rs16847812 Assay Failure (64.8%) TNN rs17374761 0,00 0,00 1,00 TNN rs2072032 (a) 47,30 47,22 0,98 47,61 52,36 0,06 49,47 48,85 0,88 0,24 0,44 TNN rs2285215 40,97 38,99 0,45 TNR rs2239819 25,93 27,04 0,63 TNR rs859427 38,55 33,64 0,05 39,13 35,09 0,10 38.60 c 41,94 0,41 0,049 0,42 TRKA rs6336 2,86 6,46 0,00 5,58 7,14 0,21 1,40 4,10 0,04 0,0012 0,12 VCAN rs160278 48,14 48,00 0,96 VCAN rs188703 39,39 39,08 0,90 VCAN rs2287926 13,80 12,45 0,44 10,87 11,94 0,51 7,38 10,94 0,12 0,66 0,50 VCAN rs2652098 (a) 49,65 49,89 0,93 VCAN rs309559 Assay Failure (88.7%) WNT16 rs2707466 44,75 45,81 0,68 WNT2B rs351371 0,00 0,00 1,00 WNT8B rs3793771 24,07 21,22 0,19 25,62 25,47 0,95 26.49 c 22.22 c 0,25 0,19 0,44 WNT9B rs4968281 27,05 22,53 0,05 26,75 29,63 0,21 23,30 28,24 0,16 0,79 0,53 Genes and SNPs linked to schizophrenia BDNF rs6265 19,02 19,80 0,71 22,47 18,54 0,05 15,89 17,56 0,57 0,40 0,49 COMT rs4680 47,46 50,65 0,23 45,74 43,06 0,29 45,31 51,13 0,14 0,45 0,50 COMT rs6267 0,00 0,31 0,18 0,13 0,00 0,33 0,52 0,78 0,68 0,62 0,50 DISC1 rs3738401 Assay Failure (81.9%) DISC1 rs6675281 12,84 13,63 0,66 17,27 15,91 0,48 16,39 13,78 0,38 0,56 0,50 DISC1 rs821616 27,95 28,44 0,83 30,78 29,00 0,44 33,06 30,15 0,44 0,48 0,50 DRD2 rs1800496 0,67 0,00 0,01 0,13 0,00 0,33 0,00 0,00 1,00 0,16 0,42 DRD2 rs1801028 Assay Failure (90.0%) 2,63 1,33 0,07 1,82 1,53 0,78 DRD3 rs6280 (a,b) 29,06 26,11 0,22 29,11 29,40 0,90 27.98 c 32,52 0,24 0,86 0,53 DTNBP1 rs16876589 0,00 0,00 1,00 DTNBP1 rs17470454 5,22 5,89 0,58 7,52 8,03 0,71 6.97 d 7,66 0,75 0,48 0,50 GRID1 rs2306265 1,52 1,43 0,89 1,75 1,96 0,75 2.29 c 3,23 0,48 0,64 0,50 GRM3 rs17161026 1,40 1,67 0,68 0,63 0,39 0,52 1,30 0,00 0,07 0,83 0,53 HTR2A rs1805055 1,52 0,76 0,16 1,55 1,20 0,57 3,11 3,60 0,74 0,38 0,49 HTR2A rs6308 0,17 0,44 0,37 0,13 0,79 0,05 0,52 0,38 0,79 0,16 0,42 HTR2A rs6314 Assay Failure (36.2%) IL2RB rs228942 Assay Failure (37.1%) IL2RB rs3218273 0,00 0,00 1,00 NRG1 rs10503929 17,00 15,54 0,44 20,88 17,85 0,13 22,80 16,17 0,04 0,019 0,42 NRG1 rs3735774 Assay Failure (82.1%) NRG1 rs3924999 39,15 40,46 0,61 35,68 35,47 0,93 35,68 37,02 0,73 0,67 0,50 NRG1 SNP8NRG221132 11,99 11,15 0,62 11,38 10,86 0,75 11,14 7,58 0,13 0,28 0,44 NRG1 SNP8NRG221533 33,33 39.86 c 0,01 35,35 35,19 0,94 35,94 34,47 0,70 0,18 0,44 NRG1 SNP8NRG241930 33,90 33,88 1,00 34,71 33,29 0,55 36,79 29,17 0,04 0,24 0,44 NRG1 SNP8NRG433E1006 Assay Failure (0.1%) RELN rs2229860 0,00 0,71 0,04 0,13 0,52 0,16 0,00 0,38 0,23 0,050 0,42 RELN rs362691 10,68 10,48 0,90 11,11 10,24 0,58 13,28 9,09 0,10 0,27 0,44 SLC6A4 rs6355 (b) 1,35 1,64 0,65 2,88 1,98 0,25 1,12 0,78 0,67 0,47 0,50

Chapter 4 - An association study in three schizophrenia case-control cohorts
80
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
SLC6A5 rs1443547 (a) 45,90 42,65 0,21 SLC6A5 rs1443548 23,65 24,72 0,64 SLC6A5 rs16906628 0,51 0,93 0,36 SLC6A5 rs3740870 16,61 16,81 0,92 16,54 16,40 0,94 17,97 17,44 0,86 0,95 0,56 TNXB rs1135809 37,27 40.25 c 0,26 TNXB rs1150752 (a) 4,38 6,99 0,04 10,48 9,16 0,38 12,69 11,28 0,59 0,95 0,56 TNXB rs12211410 7,88 9,71 0,22 6,38 6,99 0,63 4,23 6,75 0,17 0,10 0,42 TNXB rs17201602 4,22 4,74 0,64 Assay Failure (0.0%) TNXB rs17421133 Assay Failure (88.2%) TNXB rs185819 (a) 49,82 44,32 0,04 51,04 50,91 0,96 55,65 53,03 0,51 0,12 0,42 TNXB rs204896 Assay Failure (61.9%) TNXB rs204900 Assay Failure (77.4%) 10,90 9,08 0,23 10,29 8,40 0,44 TNXB rs2856453 Assay Failure (71.6%) Genes and SNPs linked to a disease A2M rs669 (AD) 35,86 33,37 0,31 APC rs28933380 (Cancer) Assay Failure (17.0%) APOE rs429358 (AD; a) 1,21 1,47 0,67 Assay Failure (86.9%) APOE rs7412 (AD; a) 8,62 5,72 0,03 Assay Failure (0.0%) APOE rs769456 (AD) 0,00 0,00 1,00 ATM rs3092857 (Leukemia) 0,00 0,00 1,00 AURKA rs2273535 (CC; a) 19,87 22,41 0,24 BARD1 rs28997576 (BC) 1,86 2,15 0,69 1,66 3,81 0,01 3,01 4,69 0,27 0,017 0,42 BLMH rs1050565 (AD) 30,38 30,21 0,95 BRCA1 rs1800709 (BC) 0,34 0,21 0,62 BRCA1 rs4986852 (BC) 2,36 2,16 0,80 2,14 1,18 0,14 1,55 1,89 0,74 0,36 0,47 BRCA2 rs144848 (BC) 27,29 30,02 0,25 CFH rs1061170 (AMD) Assay Failure (13.2%) CHEK2 rs17879961 (PC) 0,17 0,21 0,87 CHEK2 rs17883862 (LC) 0,00 0,00 1,00 CTLA4 rs231775 (DM) 36,15 37,30 0,65 DCC rs2229080 (CrC) 36,87 39,49 0,30 DCC rs984274 (CrC) 0,00 0,31 0,18 DCC rs9951523 (CrC) 2,70 0,97 0,01 1,25 1,18 0,89 2,33 1,88 0,70 0,062 0,42 DCDC2 rs2274305 (Dyslexia) 34,51 33,44 0,66 DCDC2 rs9460973 (Dyslexia; b) 1,21 2,63 0,06 2,38 2,36 0,98 3,37 1,94 0,28 0,61 0,50 ELAC2 rs4792311 (PC; a) 29,44 25,85 0,13 28,25 26,33 0,41 28,13 28,68 0,88 0,15 0,42 ENPP1 rs1044498 (DM) 13,22 15,60 0,20 14,45 14,79 0,85 11,20 15,15 0,14 0,13 0,42 EXO1 rs4149963 (Cancer) 9,18 7,88 0,37 8,08 6,28 0,17 6,74 8,65 0,36 0,28 0,44 EXO1 rs9350 (Cancer) 14,81 15,73 0,62 16,17 16,49 0,86 14,84 15,12 0,92 0,65 0,50 FGFR4 rs351855 (CP) Assay Failure (82.9%) HFE rs1799945 (AD) 17,23 16,09 0,55 12,03 11,84 0,91 11,20 9,09 0,39 0,43 0,50 HFE rs1800562 (AD) 5,93 5,14 0,51 6,82 5,94 0,48 7,11 7,63 0,80 0,45 0,50 LEPR rs1137100 (Obesity) Assay Failure (0.1%) LEPR rs1137101 (Obesity) 44,63 48,02 0,19 LEPR rs8179183 (Obesity) 18,86 16.86 c 0,33 15,99 15,39 0,75 13,21 17,42 0,14 0,80 0,53 LRP1 rs2229278 (AD) 0,00 0,00 1,00 LRP1 rs2229279 (AD) 0,00 0,00 1,00
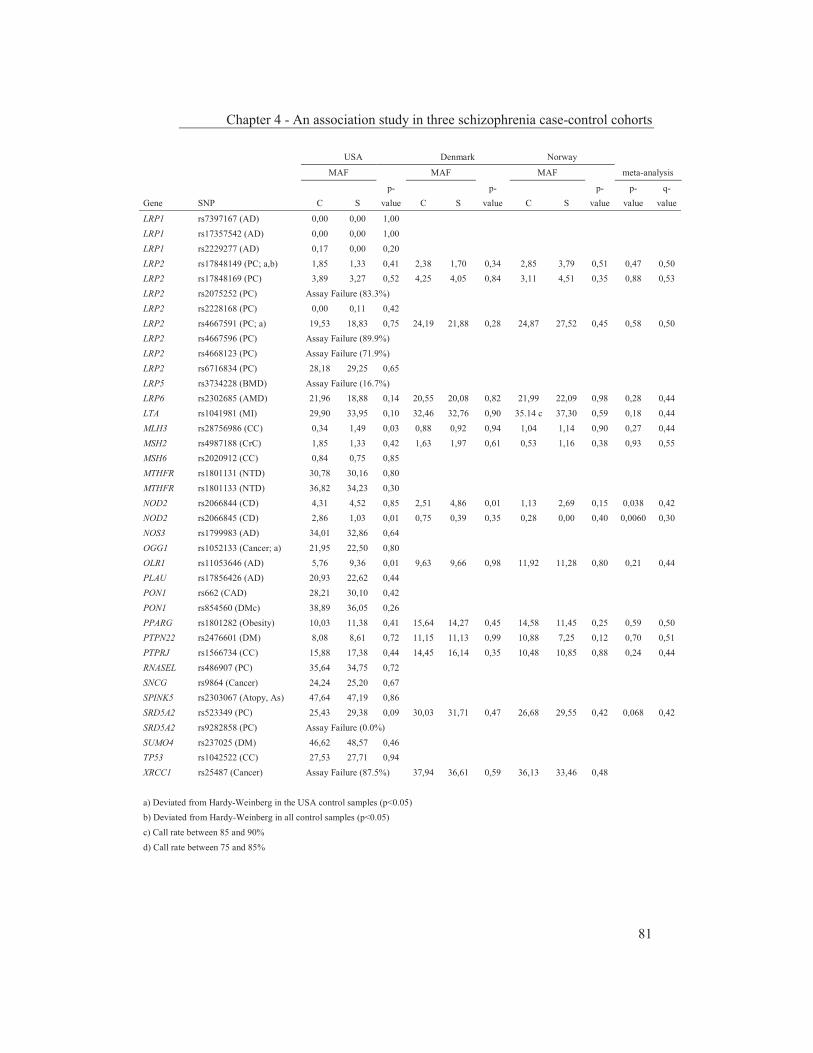
Chapter 4 - An association study in three schizophrenia case-control cohorts
81
USA Denmark Norway
MAF MAF MAF meta-analysis
p- p- p- p- q- Gene SNP C S value C S value C S value value value
LRP1 rs7397167 (AD) 0,00 0,00 1,00 LRP1 rs17357542 (AD) 0,00 0,00 1,00 LRP1 rs2229277 (AD) 0,17 0,00 0,20 LRP2 rs17848149 (PC; a,b) 1,85 1,33 0,41 2,38 1,70 0,34 2,85 3,79 0,51 0,47 0,50 LRP2 rs17848169 (PC) 3,89 3,27 0,52 4,25 4,05 0,84 3,11 4,51 0,35 0,88 0,53 LRP2 rs2075252 (PC) Assay Failure (83.3%) LRP2 rs2228168 (PC) 0,00 0,11 0,42 LRP2 rs4667591 (PC; a) 19,53 18,83 0,75 24,19 21,88 0,28 24,87 27,52 0,45 0,58 0,50 LRP2 rs4667596 (PC) Assay Failure (89.9%) LRP2 rs4668123 (PC) Assay Failure (71.9%) LRP2 rs6716834 (PC) 28,18 29,25 0,65 LRP5 rs3734228 (BMD) Assay Failure (16.7%) LRP6 rs2302685 (AMD) 21,96 18,88 0,14 20,55 20,08 0,82 21,99 22,09 0,98 0,28 0,44 LTA rs1041981 (MI) 29,90 33,95 0,10 32,46 32,76 0,90 35.14 c 37,30 0,59 0,18 0,44 MLH3 rs28756986 (CC) 0,34 1,49 0,03 0,88 0,92 0,94 1,04 1,14 0,90 0,27 0,44 MSH2 rs4987188 (CrC) 1,85 1,33 0,42 1,63 1,97 0,61 0,53 1,16 0,38 0,93 0,55 MSH6 rs2020912 (CC) 0,84 0,75 0,85 MTHFR rs1801131 (NTD) 30,78 30,16 0,80 MTHFR rs1801133 (NTD) 36,82 34,23 0,30 NOD2 rs2066844 (CD) 4,31 4,52 0,85 2,51 4,86 0,01 1,13 2,69 0,15 0,038 0,42 NOD2 rs2066845 (CD) 2,86 1,03 0,01 0,75 0,39 0,35 0,28 0,00 0,40 0,0060 0,30 NOS3 rs1799983 (AD) 34,01 32,86 0,64 OGG1 rs1052133 (Cancer; a) 21,95 22,50 0,80 OLR1 rs11053646 (AD) 5,76 9,36 0,01 9,63 9,66 0,98 11,92 11,28 0,80 0,21 0,44 PLAU rs17856426 (AD) 20,93 22,62 0,44 PON1 rs662 (CAD) 28,21 30,10 0,42 PON1 rs854560 (DMc) 38,89 36,05 0,26 PPARG rs1801282 (Obesity) 10,03 11,38 0,41 15,64 14,27 0,45 14,58 11,45 0,25 0,59 0,50 PTPN22 rs2476601 (DM) 8,08 8,61 0,72 11,15 11,13 0,99 10,88 7,25 0,12 0,70 0,51 PTPRJ rs1566734 (CC) 15,88 17,38 0,44 14,45 16,14 0,35 10,48 10,85 0,88 0,24 0,44 RNASEL rs486907 (PC) 35,64 34,75 0,72 SNCG rs9864 (Cancer) 24,24 25,20 0,67 SPINK5 rs2303067 (Atopy, As) 47,64 47,19 0,86 SRD5A2 rs523349 (PC) 25,43 29,38 0,09 30,03 31,71 0,47 26,68 29,55 0,42 0,068 0,42 SRD5A2 rs9282858 (PC) Assay Failure (0.0%) SUMO4 rs237025 (DM) 46,62 48,57 0,46 TP53 rs1042522 (CC) 27,53 27,71 0,94 XRCC1 rs25487 (Cancer) Assay Failure (87.5%) 37,94 36,61 0,59 36,13 33,46 0,48
a) Deviated from Hardy-Weinberg in the USA control samples (p<0.05) b) Deviated from Hardy-Weinberg in all control samples (p<0.05) c) Call rate between 85 and 90% d) Call rate between 75 and 85%
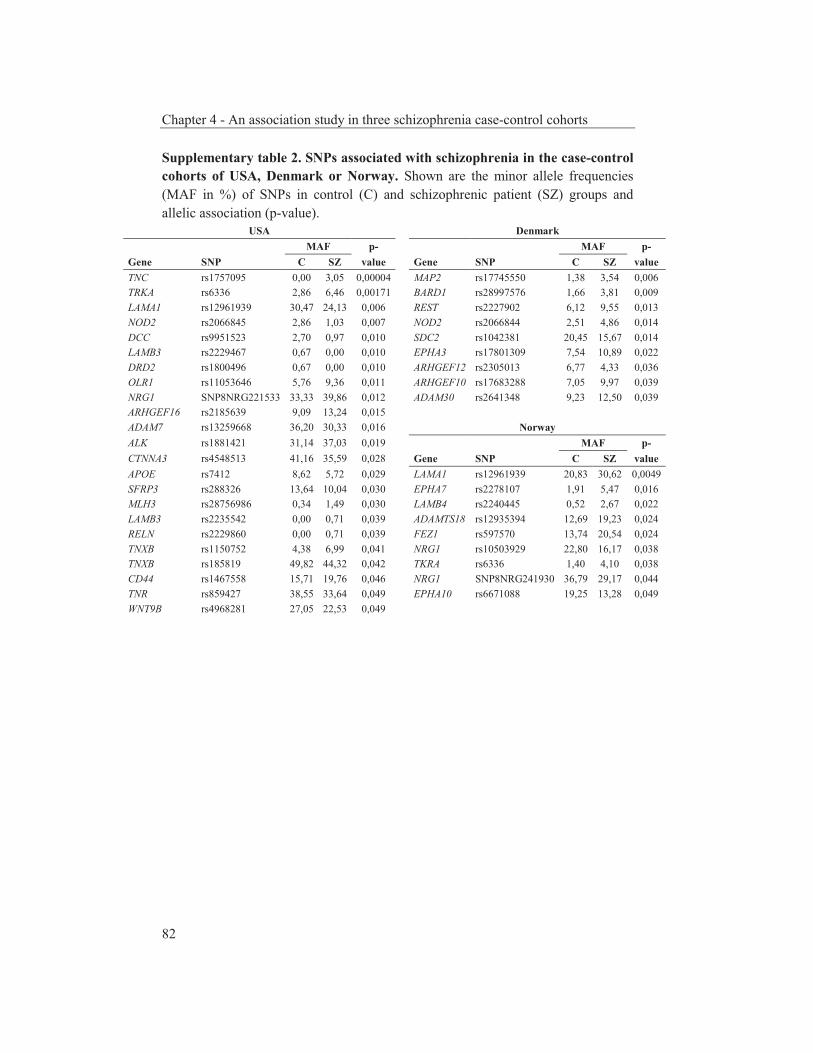
Chapter 4 - An association study in three schizophrenia case-control cohorts
82
Supplementary table 2. SNPs associated with schizophrenia in the case-control cohorts of USA, Denmark or Norway. Shown are the minor allele frequencies (MAF in %) of SNPs in control (C) and schizophrenic patient (SZ) groups and allelic association (p-value).
USA Denmark MAF p- MAF p-
Gene SNP C SZ value Gene SNP C SZ value TNC rs1757095 0,00 3,05 0,00004 MAP2 rs17745550 1,38 3,54 0,006 TRKA rs6336 2,86 6,46 0,00171 BARD1 rs28997576 1,66 3,81 0,009 LAMA1 rs12961939 30,47 24,13 0,006 REST rs2227902 6,12 9,55 0,013 NOD2 rs2066845 2,86 1,03 0,007 NOD2 rs2066844 2,51 4,86 0,014 DCC rs9951523 2,70 0,97 0,010 SDC2 rs1042381 20,45 15,67 0,014 LAMB3 rs2229467 0,67 0,00 0,010 EPHA3 rs17801309 7,54 10,89 0,022 DRD2 rs1800496 0,67 0,00 0,010 ARHGEF12 rs2305013 6,77 4,33 0,036 OLR1 rs11053646 5,76 9,36 0,011 ARHGEF10 rs17683288 7,05 9,97 0,039 NRG1 SNP8NRG221533 33,33 39,86 0,012 ADAM30 rs2641348 9,23 12,50 0,039 ARHGEF16 rs2185639 9,09 13,24 0,015 ADAM7 rs13259668 36,20 30,33 0,016 Norway ALK rs1881421 31,14 37,03 0,019 MAF p- CTNNA3 rs4548513 41,16 35,59 0,028 Gene SNP C SZ value APOE rs7412 8,62 5,72 0,029 LAMA1 rs12961939 20,83 30,62 0,0049 SFRP3 rs288326 13,64 10,04 0,030 EPHA7 rs2278107 1,91 5,47 0,016 MLH3 rs28756986 0,34 1,49 0,030 LAMB4 rs2240445 0,52 2,67 0,022 LAMB3 rs2235542 0,00 0,71 0,039 ADAMTS18 rs12935394 12,69 19,23 0,024 RELN rs2229860 0,00 0,71 0,039 FEZ1 rs597570 13,74 20,54 0,024 TNXB rs1150752 4,38 6,99 0,041 NRG1 rs10503929 22,80 16,17 0,038 TNXB rs185819 49,82 44,32 0,042 TKRA rs6336 1,40 4,10 0,038 CD44 rs1467558 15,71 19,76 0,046 NRG1 SNP8NRG241930 36,79 29,17 0,044 TNR rs859427 38,55 33,64 0,049 EPHA10 rs6671088 19,25 13,28 0,049 WNT9B rs4968281 27,05 22,53 0,049
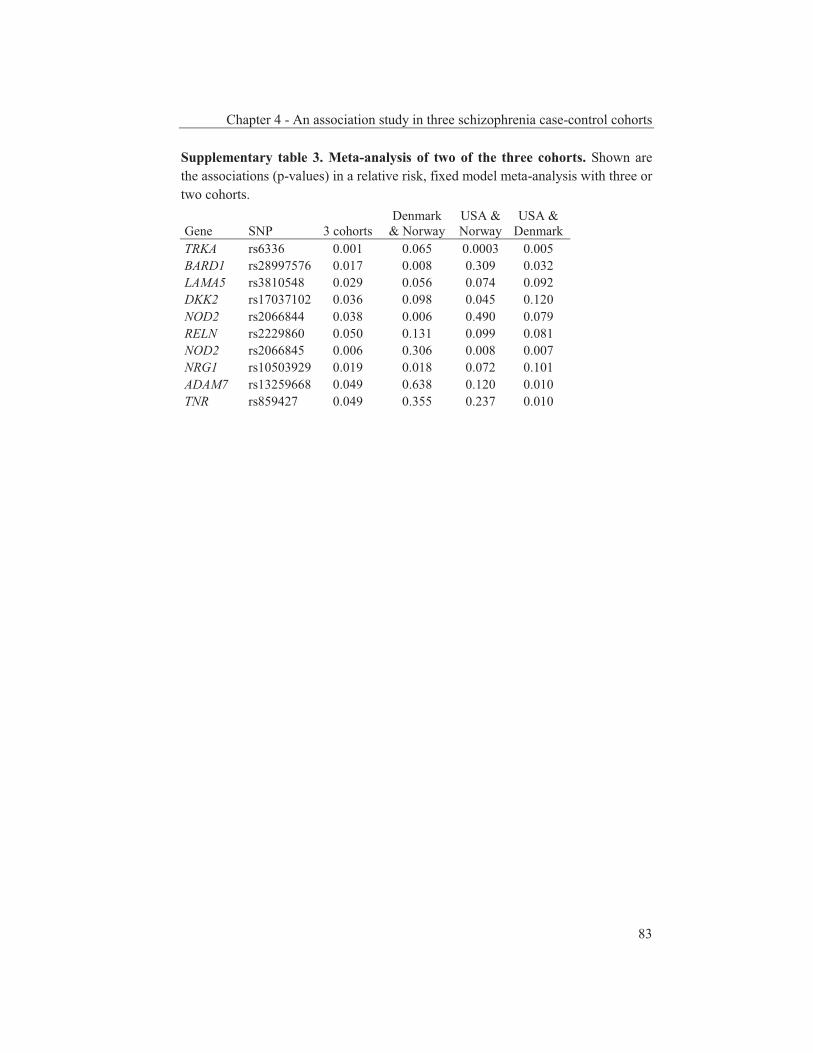
Chapter 4 - An association study in three schizophrenia case-control cohorts
83
Supplementary table 3. Meta-analysis of two of the three cohorts. Shown are the associations (p-values) in a relative risk, fixed model meta-analysis with three or two cohorts.
Gene SNP 3 cohorts Denmark
& Norway USA & Norway
USA & Denmark
TRKA rs6336 0.001 0.065 0.0003 0.005 BARD1 rs28997576 0.017 0.008 0.309 0.032 LAMA5 rs3810548 0.029 0.056 0.074 0.092 DKK2 rs17037102 0.036 0.098 0.045 0.120 NOD2 rs2066844 0.038 0.006 0.490 0.079 RELN rs2229860 0.050 0.131 0.099 0.081 NOD2 rs2066845 0.006 0.306 0.008 0.007 NRG1 rs10503929 0.019 0.018 0.072 0.101 ADAM7 rs13259668 0.049 0.638 0.120 0.010 TNR rs859427 0.049 0.355 0.237 0.010


Chapter 5 Dual association of a TRKA
polymorphism with schizophrenia
A modified version of this chapter has been submitted as: Van Schijndel JE, Van Zweeden M, Van Loo KMJ, Djurovic S, Andreassen OA, Hansen T, Werge T, Nyegaard M, Sørensen KM, Nordentoft M, Mortensen PB,
Mors O, Børglum AD, Del-Favero J, Norrback KF, Adolfsson R, De Hert M, Claes S, Cichon S, Rietschel M, Nöthen MM, Kallunki P, Pedersen JT, Martens GJM.
Dual association of a TRKA polymorphism with schizophrenia.

Chapter 5 - Association study on TRKA SNP in schizophrenia
86
Abstract An interaction between predisposing genes and environmental stressors is thought to underlie the neurodevelopmental disorder schizophrenia. In a targeted gene screening, we previously found that the minor allele of the single-nucleotide polymorphism (SNP) rs6336 in the neurotrophic tyrosine kinase receptor 1 gene (NTRK1/TRKA) is associated with schizophrenia as a risk factor. We now investigated the TRKA SNP in a total of eight independent Caucasian schizophrenia case-control groups. Remarkably, whereas in five of the groups a higher frequency of the risk allele was indeed found in the patients compared to the controls, in the three other groups the SNP acted as a protective factor. An intriguing possibility is that this dual character of the TRKA SNP is caused by its interaction with endophenotypic and/or epistatic factors.

Chapter 5 - Association study on TRKA SNP in schizophrenia
87
Introduction Schizophrenia is a complex disorder characterized by psychotic symptoms, negative symptoms and cognitive impairment. Although these symptoms are usually displayed during late adolescence or early adulthood, the aetiology of the disorder is thought to be neurodevelopmental (Weinberger, 1995). Family and adoption studies have shown that both genetic and environmental factors play a role in the aetiology of schizophrenia. The genetic factors result in a concordance rate of 40-65% in monozygotic twins (Cardno et al., 2000). Linkage analyses have revealed multiple susceptibility chromosomal regions (Lewis et al., 2003) that contain various candidate genes. Meta-analyses based on studies on a number of single-nucleotide polymorphisms (SNPs) in these genes have elucidated nominally significant SNPs but with low average summary odds ratios (ORs; Allen et al., 2008). Recently, genome-wide association studies have revealed yet additional candidate SNPs and genes (O'Donovan et al., 2008) and copy number variations (Rujescu et al., 2008; Stefansson et al., 2008). Among the multiple SNPs and genes genetically associated with schizophrenia a substantial number is involved in neurodevelopment, including a neurotrophin gene, namely the gene encoding the brain-derived neurotrophic factor BDNF (Jonsson et al., 2006). On the basis of mRNA and protein expression profiles other neurotrophins and neurotrophic tyrosine kinase receptors (NTRKs) have been linked to schizophrenia (Buckley et al., 2007; Shoval et al., 2005). For example, compared to controls lower plasma levels of nerve growth factor (NGF) have been detected in both first-episode psychotic and medicated chronic schizophrenia patients (Parikh et al., 2003). Furthermore, the mRNA levels of NGF and its high-affinity receptor NTRK1 (better known and hereafter referred to as TRKA) have been found to be reduced in the hippocampus, striatum and (hypo)thalamus of rats treated with subchronic levels of ketamine, which in healthy humans induces psychotic and negative symptoms and cognitive impairment (Becker et al., 2008). At the genetic level, SNPs in TRKA have been studied in genome-wide association studies on schizophrenia case-control cohorts, but no association has been found (affymetrix 500K array set; Lencz et al., 2007). Recently, however, we performed a targeted gene screening involving non-synonymous SNPs in mostly neurodevelopmental genes and found that only TRKA SNP rs6336 was associated with schizophrenia (OR = 1.73; chapter 4). Yet, schizophrenia is a disorder with a high level of heterogeneity and therefore reliable association data are difficult to obtain. Consequently, screenings of additional cohorts are necessary to establish the validity of an association. We therefore decided to examine TRKA SNP rs6336 in five additional independent Caucasian schizophrenia case-control groups.

Chapter 5 - Association study on TRKA SNP in schizophrenia
88
Materials and methods Cohorts Since we genotyped SNP rs6336 previously in three independent Caucasian schizophrenia case-control groups (from USA, Norway and Denmark I; chapter 4), below these three groups are described only briefly, whereas the five additional, independent Caucasian schizophrenia case-control groups genotyped here are defined in more detail.
USA, Norway and Denmark I The USA groups consisted of 488 Caucasian schizophrenic patients (age 50.9 ± 9.8; 332 males, age 50.1 ± 9.9, and 156 females, age 52.7 ± 9.4), diagnosed according the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV), and 297 unrelated Caucasian controls (age 72.1 ± 14.1; 113 males, age 73.5 ± 13.4, and 184 females, age 71.2 ± 14.5). The Norwegian groups involved 133 schizophrenic patients (age 37.8 ± 10.8; 74 males, age 36.9 ± 9.9, and 59 females, age 38.8 ± 11.6), selected according to DSM-IV, and 195 unrelated Caucasian controls (age 37.0 ± 10.3; 90 males, age 37.5 ± 10.2, and 105 females, age 36.7 ± 10.5). The Danish I groups consisted of 451 Caucasian schizophrenic patients (age 44.9 ± 12.2, age of onset 27.3 ± 8.9; 264 males, age 43.8 ± 11.9, and 187 females, age 46.3 ± 12.5), clinically diagnosed with schizophrenia or schizoaffective disorder according to the International Classification of Disease version 10 (ICD-10) and 96% of the patients also to the DSM-IV standards, and 1032 unrelated Caucasian controls (age 44.5± 12.2; 607 males, age 43.7 ± 11.7, and 425 females, age 45.7 ± 12.7). For detailed sample description of the USA, Norway and Denmark I case-control groups, see chapter 4. Denmark II The second Danish sample (Denmark II) consisted of 248 incident schizophrenia cases (133 males and 114 females) and 286 medical student volunteers as controls (76 males and 210 females). The cases were diagnosed according to ICD 10-DCR and DSM IV using SCAN interviews and a best estimate procedure. The cases and controls were of Danish parentage for three generations. The study was approved by the Danish Data Protection Agency and the Ethical Committees in Denmark.
Denmark III The third Danish sample (Denmark III) consisted of 363 cases with schizophrenia and 430 control individuals. The 363 cases (209 males and 154 females) were obtained from the Danish Newborn Screening Biobank (Norgaard-Pedersen et al., 2007). The cases, all born in Denmark, were clinically diagnosed with schizophrenia according to ICD 10. The cases were matched on gender and date of

Chapter 5 - Association study on TRKA SNP in schizophrenia
89
birth to at least one control individual from the Biobank. In total 430 controls (232 males and 198 females) with no record of mental illness were included. The study was approved by the Danish Data Protection Agency and the Ethical Committees in Denmark. Sweden The patient sample was comprised of 486 unrelated Caucasian individuals (age 53.1 ± 15.1 years; 180 females, 306 males; age at onset 24.8 ± 7.3), fulfilling the DSM-IV criteria of schizophrenia (American Psychiatric Association, 1994). The patients were initially identified through the in-patient hospital registers in the north part of Sweden and had at least on one occasion received a discharge diagnosis of schizophrenia. The ascertainment of the patients was performed during 1992-2005 (for details, see Ekholm et al., 2005). The control sample of 512 Caucasians (age 58.0 ± 13.0; 273 females, 238 males) were randomly selected from the same geographic area as the patients (Nilsson et al., 1997). A diagnosis of schizophrenia or other psychotic disorders were excluded after nurses' health interview, a self-report questionnaire and studies of psychiatric case records. All subjects participated after giving written informed consent. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethical Review Board of the universities of Umeå and Antwerp. Belgium The Belgium sample consisted of 518 Caucasian schizophrenic patients (age 38.7 ± 12.1; 343 males, age 36.3 ± 11.2 and 175 females, age 43.4 ± 12.5) and 246 randomly selected Caucasian controls (age 37.8 ± 13.1; 153 males, age 41.9 ± 14.6 and 93 females, age 31.0 ± 5.6) from the same geographic region (Leuven, Belgium). Patients were selected according the DSM-IV criteria (schizophrenia: disorganized type (295.1): 10 patients; paranoid type (295.3): 216 patients; schizophreniform disorder (295.4): 58 patients; residual type (295.6): 2 patients; schizoaffective disorder (295.7): 106 patients; undifferentiated type (295.9): 126 patients). All patients and controls had given informed consent. Germany The German schizophrenia sample was recruited from consecutive admissions to the inpatient unit of the Department of Psychiatry and Psychotherapy of the University of Bonn and of the Central Institute of Mental Health in Mannheim. The sample comprised 866 patients with a DSM-IV lifetime diagnosis of schizophrenia, made by a consensus best-estimate procedure (Leckman et al., 1982) based on all available information, including a structured interview (Spitzer et al., 1992), medical records, and the family history method. We also used the OPCRIT (Farmer et al., 1992) system to obtain detailed polydiagnostic documentation of symptoms.

Chapter 5 - Association study on TRKA SNP in schizophrenia
90
360 (41.57%) of the patients were female, 506 (58.43%) were male. The mean age was 36.9 ± 11.9 years. All individuals and their parents were of German descent. The German control sample consisted of 1,000 population-based individuals, 52.2% were females, 47.8% were males. The mean age was 48.8 ± 15.6 years. All individuals were German. For all patient and control individuals, written informed consent was obtained prior to study participation. Protocols and procedures were approved by the ethics committees of the Faculties of Medicine at the Universities of Bonn and Mannheim/Heidelberg. High-Resolution Melting (HRM) The exons of TRKA were analysed by HRM in 71 samples from Caucasian schizophrenia patients. Primers surrounding the 16 exons were developed (sequences available on request). Despite various attempts involving a number of primer sets, we did not succeed in amplifying exon 1. PCR amplifications were performed in a total volume of 10 μl on a Rotor-GeneTM 6000 real-time rotary analyzer (Corbett Research, Sydney, Australia). The reaction mixture contained 10 ng human genomic DNA, 1x TITANIUMTM Taq PCR Buffer (Clontech), 0.25 mM dNTP, 375 nM forward and reverse primers, 1.5 μM SYTO® 9 green fluorescent nucleic acid stain (Invitrogen), and 0.25x TITANIUMTM Taq polymerase (Clontech). To amplify the amplicon the cycling conditions were as follows: one cycle of 95°C for 3 minutes; 45 cycles of 95°C for 10 seconds, 60°C for 15 seconds and 72°C for 20 seconds. Before the HRM one cycle of 90°C for 1 minute and 40°C for 1 minute was applied to form random DNA duplexes. The HRM protocol was a 90 seconds pre-melt wait at 65°C followed by a grandient rise in temperature from 65°C to 95°C, 0.1°C and 1 second per step. HRM was analysed with the Rotor-Gene 6000 Series Software using the HRM (normalised and difference graphs) and melt analysis tools. SNP Selection NCBI database (build 129) searches for SNPs located in the TRKA gene revealed fifteen non-synonymous SNPs. Nine SNPs are not validated and do not have frequency data. Two have frequency data but are not present in 60 HAPMAP samples, indicating low minor allele frequencies (MAFs). Only four (including SNPs rs6336 and rs6339) have MAFs above zero in the HAPMAP or another European sample. In a search for unknown polymorphisms, we scanned the exons of TRKA with HRM in 71 schizophrenia genomic DNA samples, but did not detect new non-synonymous SNPs. In exon 15, we detected the variations corresponding to SNPs rs6336 and rs6339. DNA sequencing and PCR experiments together with information derived from the Genome Variation Server (GVS) of the SeattleSNPs Program for Genomic Applications revealed that these two SNPs are in complete linkage disequilibrium (LD; r2 between 0.82 and 1.00).

Chapter 5 - Association study on TRKA SNP in schizophrenia
91
Genotyping The samples of the cohorts of USA, Denmark I, Norway, Denmark II, Denmark III and Germany were genotyped for rs6336 using iPLEXTM chemistry on a MALDI-TOF Mass Spectrometer by Sequenom Inc. (San Diego, CA, USA, http://www.sequenom.com). SNP-containing genomic regions were amplified in a multiplex PCR followed by deactivation of the remaining nucleotides by SAP treatment. A single-base primer extension step was performed and the primer extension (iPLEX) products were desalted using Resin and spotted on a SpectroCHIP (Sequenom) using a nanodispenser. The samples were analyzed using a Bruker MALDI TOF MS and the genotypes were determined using the MassARRAY Typer 3.4 software (Sequenom). Assay conditions and primer sequences are available upon request. Confirmation of those genotypes and genotyping of the Belgium cohort was done by allele-specific PCR using primers specific for SNP rs6336 (General primers: forward (fw): 5’-GGCAAGGGCTGAGTCTG-3’ and reverse (rv): 5’-TCCATCTGGGATAGCGAAGG-3’. Specific rv primers: for the “T-allele”: 5’-CTTGGCATCAGGTCCATA-3’ and for the “C-allele”: 5’-CTTGGCATCAGGTCCATG-3’) or HRM using fw primer 5’-GGAGTTCTATCCTCCCAGCCT-3’ and rv primer 5’-CCTGGAGCCACATCCTCCC-3’. Genotyping of the Swedish cohort was performed by pyrosequencing on a PSQ HS96 pyrosequencer (http://www.biotage.com/). Biotinylated PCR products were immobilized onto streptavidin-coated sepharose beads (Amersham Biosciences, Sweden). Biotinylated ssDNA was obtained by incubating the immobilized PCR products in 0.5 M NaOH, followed by two sequential washes in 10 mM Tris-Acetate, pH 7.6. Primer annealing was performed by incubation at 80°C for 2 min and then at room temperature for 5 min. Pyrosequencing was performed on the PSQ96 pyrosequencer (Biotage, Sweden). Standard PCR was performed with fw primer 5’- ATCCTCCCAGCCTATCCCCTCT-3’ and 5’ biotine labeled rv primer 5’- CCAGCAGCTTGGCATCAGG-3’ at an annealing temperature of 68°C. Genotyping was performed using primer 5-CTTTTCTTGTTCACAGATCC-3’. Statistical Analysis Differences in allele frequencies between cases and controls were analyzed by standard contingency table analysis using two-tailed χ2 test probabilities. The genotype distribution was tested in all of the control samples and no deviation was found from Hardy-Weinberg equilibrium. Heterogeneity between studies was assessed using the Q Cochran test and the Cochran Mantel Haenszel test was used to calculate the overall association across all samples (EasyMA2001; Cucherat et al., 1997). Power calculations were estimated using Quanto v1.2 (Gauderman, 2002) and showed that power was sufficient to detect a risk effect in the 2475 new

Chapter 5 - Association study on TRKA SNP in schizophrenia
92
cases (99.9%, assuming an OR of 1.73, a disease allele frequency of 5% and a disease prevalence of 1%). Results We first performed HRM to scan TRKA for the presence of any SNP other than rs6336. This analysis revealed only one additional non-synonymous SNP (rs6339), but this polymorphism is in LD with rs6336 (0.82 < r2 < 1.00; see Materials and Methods for details). We therefore decided to examine in more detail TRKA SNP rs6336. Further to our previous screening of three independent Caucasian schizophrenia case-control groups (chapter 4), we now genotyped SNP rs6336 in five additional case-control groups from Denmark (two groups), Sweden, Belgium and Germany. In these additional screenings, only in the Belgium and Denmark II groups SNP rs6336 also showed a higher frequency of the risk allele in the schizophrenic patients compared to the healthy controls, although not significantly different (Belgium: OR = 1.30; 95% confidence interval (CI) = [0.79;2.12]; p = 0.30, Denmark II: OR = 1.16; 95% CI = [0.69;1.97]; p=0.58). Surprisingly, in the three other groups the SNP displayed a lower frequency of the risk allele in the patients than in the controls (Sweden OR = 0.68, Denmark III OR = 0.71, Germany OR = 0.74; table I). The Cochran Mantel Haenszel test revealed that SNP rs6336 was not associated with schizophrenia when analyzing across all eight samples (OR = 1.00; 95% CI = [0.87;1.16]; p = 0.98). However, to exclude the hypothesis-generating data from the original three-sample screenings, we performed an analysis of the five new case-control groups. The Cochran Mantel Haenszel meta-analysis of the five new samples revealed that SNP rs6336 was significantly associated with schizophrenia as a protective factor (OR = 0.820; 95% CI = [0.687;0.978]; p = 0.032). This result is opposite to the hypothesis-generating data. Discussion In our previous targeted gene screening approach in three independent Caucasian schizophrenia case-control groups, TRKA SNP rs6336 was significantly associated with schizophrenia as a risk factor (chapter 4). However, the present study involving the screening of this SNP in five additional Caucasian schizophrenia case-control groups showed that the SNP was a risk factor in two of the extra groups, whereas in the three other groups it was found as a protective factor. Another remarkable observation in our studies was that the MAFs for SNP rs6336 greatly varied among the groups, in particular among the control cohorts (MAFs ranging from 1.8 to 7.6%). Such a variation in MAFs is not a feature unique for TRKA SNP rs6336 since in schizophrenia association studies reported by others similar observations have been made as well. For example, in a screening of Table
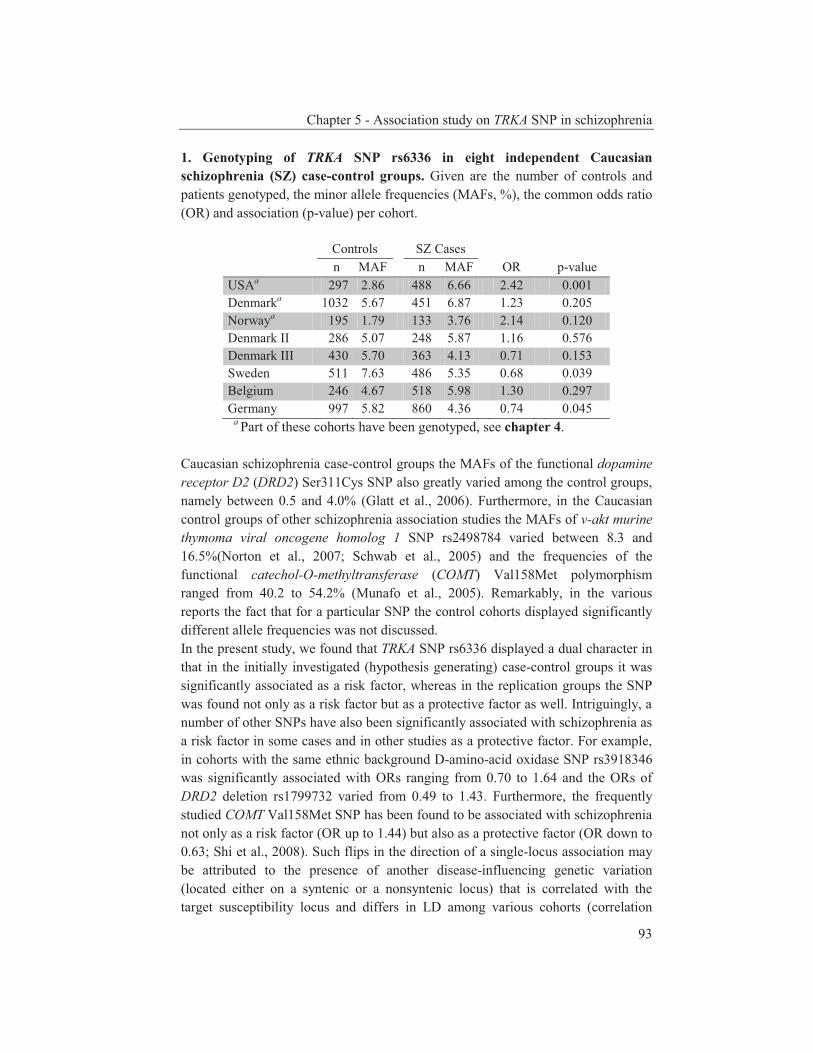
Chapter 5 - Association study on TRKA SNP in schizophrenia
93
1. Genotyping of TRKA SNP rs6336 in eight independent Caucasian schizophrenia (SZ) case-control groups. Given are the number of controls and patients genotyped, the minor allele frequencies (MAFs, %), the common odds ratio (OR) and association (p-value) per cohort.
Controls SZ Cases n MAF n MAF OR p-value USAa 297 2.86 488 6.66 2.42 0.001 Denmarka 1032 5.67 451 6.87 1.23 0.205 Norwaya 195 1.79 133 3.76 2.14 0.120 Denmark II 286 5.07 248 5.87 1.16 0.576 Denmark III 430 5.70 363 4.13 0.71 0.153 Sweden 511 7.63 486 5.35 0.68 0.039 Belgium 246 4.67 518 5.98 1.30 0.297 Germany 997 5.82 860 4.36 0.74 0.045
a Part of these cohorts have been genotyped, see chapter 4. Caucasian schizophrenia case-control groups the MAFs of the functional dopamine receptor D2 (DRD2) Ser311Cys SNP also greatly varied among the control groups, namely between 0.5 and 4.0% (Glatt et al., 2006). Furthermore, in the Caucasian control groups of other schizophrenia association studies the MAFs of v-akt murine thymoma viral oncogene homolog 1 SNP rs2498784 varied between 8.3 and 16.5%(Norton et al., 2007; Schwab et al., 2005) and the frequencies of the functional catechol-O-methyltransferase (COMT) Val158Met polymorphism ranged from 40.2 to 54.2% (Munafo et al., 2005). Remarkably, in the various reports the fact that for a particular SNP the control cohorts displayed significantly different allele frequencies was not discussed. In the present study, we found that TRKA SNP rs6336 displayed a dual character in that in the initially investigated (hypothesis generating) case-control groups it was significantly associated as a risk factor, whereas in the replication groups the SNP was found not only as a risk factor but as a protective factor as well. Intriguingly, a number of other SNPs have also been significantly associated with schizophrenia as a risk factor in some cases and in other studies as a protective factor. For example, in cohorts with the same ethnic background D-amino-acid oxidase SNP rs3918346 was significantly associated with ORs ranging from 0.70 to 1.64 and the ORs of DRD2 deletion rs1799732 varied from 0.49 to 1.43. Furthermore, the frequently studied COMT Val158Met SNP has been found to be associated with schizophrenia not only as a risk factor (OR up to 1.44) but also as a protective factor (OR down to 0.63; Shi et al., 2008). Such flips in the direction of a single-locus association may be attributed to the presence of another disease-influencing genetic variation (located either on a syntenic or a nonsyntenic locus) that is correlated with the target susceptibility locus and differs in LD among various cohorts (correlation

Chapter 5 - Association study on TRKA SNP in schizophrenia
94
coefficient; Lin et al., 2007). In a further attempt to try to understand the dual character of a SNP, the contribution of schizophrenia endophenotypes has been considered. For instance, the COMT polymorphism is associated with lifetime symptomatology and sensorimotor gating (Goghari et al., 2008; Quednow et al., 2008). Yet, a meta-analysis including the endophenotypes should be performed to confirm the initial data. Furthermore, the issue whether schizophrenia should be considered as a single neurodevelopmental disorder or represents a collection of endophenotypes is currently being discussed in connection with the preparation of the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V; Gaebel et al., 2008). Thus, in the future a better classification of the patients may well lead to the identification of TRKA SNP rs6336 as a risk or protective factor for a certain endophenotype. In addition to gene-environment interactions, gene-gene interactions may influence the final outcome of a phenotype and may thus affect the eventual result of an association study. Since susceptibility genes can often be grouped into a family with roles in the same pathways or processes (Carter, 2006), SNPs within members of such a gene family may interact (pair-wise interacting SNPs). For schizophrenia, examples may include interactions between SNPs in the ERBB and neuregulin gene families (Benzel et al., 2007) or between an intronic tryptophan hydroxylase SNP and the short allele of the serotonin transporter gene (Chotai et al., 2005). Thus, SNPs in components of the TRKA pathway may interact with TRKA SNP rs6336 and as such contribute to the observed dual character of rs6336. Another explanation for the dual character may depend on the parental origin of the genetic variation. Recently, parental-origin-specific SNP associations with breast cancer, basal-cell carcinoma, and type 2 diabetes have been reported (Kong et al., 2009). Thus, TRKA SNP rs6336 may be a risk allele when inherited from the father and a protective allele when inherited from the mother or vise versa. In conclusion, we find TRKA SNP rs6336 either as a risk allele or a protective factor for schizophrenia. However, further research on large, well-characterized cohorts is necessary to explore whether this TRKA SNP is linked to a certain endophenotype and/or interacts with another SNP(s) in the TRKA signaling pathway to explain its apparently dual association with schizophrenia. Acknowledgements Authors wish to thank patients who participated and made this study possible. Authors also acknowledge expert assistance by numerous mental health professionals in the various clinical departments. This work was supported by the Dutch Top Institute Pharma (grant T5-209).

Chapter 5 - Association study on TRKA SNP in schizophrenia
95
References Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic
meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 2008;40:827-834.
Becker A, Grecksch G, Schwegler H, Roskoden T. Expression of mRNA of neurotrophic factors and their receptors are significantly altered after subchronic ketamine treatment. Med Chem 2008;4:256-263.
Benzel I, Bansal A, Browning BL, Galwey NW, Maycox PR, McGinnis R, et al. Interactions among genes in the ErbB-Neuregulin signalling network are associated with increased susceptibility to schizophrenia. Behav Brain Funct 2007;3:31.
Buckley PF, Mahadik S, Pillai A, Terry A, Jr. Neurotrophins and schizophrenia. Schizophr Res 2007;94:1-11.
Cardno AG, Gottesman, II. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet 2000;97:12-17.
Carter CJ. Schizophrenia susceptibility genes converge on interlinked pathways related to glutamatergic transmission and long-term potentiation, oxidative stress and oligodendrocyte viability. Schizophr Res 2006;86:1-14.
Chotai J, Serretti A, Lorenzi C. Interaction between the tryptophan hydroxylase gene and the serotonin transporter gene in schizophrenia but not in bipolar or unipolar affective disorders. Neuropsychobiology 2005;51:3-9.
Cucherat M, Boissel JP, Leizorovicz A, Haugh MC. EasyMA: a program for the meta-analysis of clinical trials. Comput Methods Programs Biomed 1997;53:187-190.
Ekholm B, Ekholm A, Adolfsson R, Vares M, Osby U, Sedvall GC, et al. Evaluation of diagnostic procedures in Swedish patients with schizophrenia and related psychoses. Nord J Psychiatry 2005;59:457-464.
Farmer AE, Wessely S, Castle D, McGuffin P. Methodological issues in using a polydiagnostic approach to define psychotic illness. Br J Psychiatry 1992;161:824-830.
Gaebel W, Zielasek J. The DSM-V initiative "deconstructing psychosis" in the context of Kraepelin's concept on nosology. Eur Arch Psychiatry Clin Neurosci 2008;258 Suppl 2:41-47.
Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol 2002;155:478-484.
Glatt SJ, Jonsson EG. The Cys allele of the DRD2 Ser311Cys polymorphism has a dominant effect on risk for schizophrenia: evidence from fixed- and random-effects meta-analyses. Am J Med Genet B Neuropsychiatr Genet 2006;141B:149-154.
Goghari VM, Sponheim SR. Differential association of the COMT Val158Met polymorphism with clinical phenotypes in schizophrenia and bipolar disorder. Schizophr Res 2008;103:186-191.
Jonsson EG, Edman-Ahlbom B, Sillen A, Gunnar A, Kulle B, Frigessi A, et al. Brain-derived neurotrophic factor gene (BDNF) variants and schizophrenia: an association study. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:924-933.
Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental origin of sequence variants associated with complex diseases. Nature 2009;462:868-874.
Leckman JF, Sholomskas D, Thompson WD, Belanger A, Weissman MM. Best estimate of lifetime psychiatric diagnosis: a methodological study. Arch Gen Psychiatry 1982;39:879-883.
Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry 2007;12:572-580.
Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet 2003;73:34-48.

Chapter 5 - Association study on TRKA SNP in schizophrenia
96
Lin PI, Vance JM, Pericak-Vance MA, Martin ER. No gene is an island: the flip-flop phenomenon. Am J Hum Genet 2007;80:531-538.
Munafo MR, Bowes L, Clark TG, Flint J. Lack of association of the COMT (Val158/108 Met) gene and schizophrenia: a meta-analysis of case-control studies. Mol Psychiatry 2005;10:765-770.
Nilsson LG, Bäckman L, Erngrund K, Nyberg L, Adolfsson R, G. B, et al. The Betula prospective cohort study: memory, health and aging. Aging Neuropsychol. Cognit. 1997;4:1-32.
Norgaard-Pedersen B, Hougaard DM. Storage policies and use of the Danish Newborn Screening Biobank. J Inherit Metab Dis 2007;30:530-536.
Norton N, Williams HJ, Dwyer S, Carroll L, Peirce T, Moskvina V, et al. Association analysis of AKT1 and schizophrenia in a UK case control sample. Schizophr Res 2007;93:58-65.
O'Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet 2008;
Parikh V, Evans DR, Khan MM, Mahadik SP. Nerve growth factor in never-medicated first-episode psychotic and medicated chronic schizophrenic patients: possible implications for treatment outcome. Schizophr Res 2003;60:117-123.
Quednow BB, Wagner M, Mossner R, Maier W, Kuhn KU. Sensorimotor Gating of Schizophrenia Patients Depends on Catechol O-Methyltransferase Val158Met Polymorphism. Schizophr Bull 2008;
Rujescu D, Ingason A, Cichon S, Pietilainen OP, Barnes MR, Toulopoulou T, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet 2008;
Schwab SG, Hoefgen B, Hanses C, Hassenbach MB, Albus M, Lerer B, et al. Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol Psychiatry 2005;58:446-450.
Shi J, Gershon ES, Liu C. Genetic associations with schizophrenia: Meta-analyses of 12 candidate genes. Schizophr Res 2008;104:96-107.
Shoval G, Weizman A. The possible role of neurotrophins in the pathogenesis and therapy of schizophrenia. Eur Neuropsychopharmacol 2005;15:319-329.
Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry 1992;49:624-629.
Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008;455:232-236.
Weinberger DR. From neuropathology to neurodevelopment. Lancet 1995;346:552-557.

Chapter 6 General Discussion


Chapter 6 - General Discussion
99
In order to get more insight into the aetiology of the complex neuropsychiatric disorder schizophrenia, in the last four decades a myriad of studies on animal models as well as human association studies have been performed. These studies have highlighted many genes and pathways that may be involved in the development of schizophrenia, but unfortunately they have raised more questions than answers. The aim of this thesis was to identify candidate genes associated with schizophrenia. In this final chapter the results obtained from the studies on the APO-SUS/APO-UNSUS rat model and the human association studies are discussed and placed in a broader context. mRNA expression profiling in animal models for schizophrenia In the last ten years, a number of microarray analyses have been performed in animal models for schizophrenia. An overview of the results of these analyses (chapter 1) illustrates that we still can not provide a conclusive answer to the question which genes play a role in the aetiology of this complex disorder. The numerous mRNA expression studies have resulted in even more genes that could play a role in the onset of schizophrenia than was previously thought. Most of these genes have been found in only a single study and their significance is at present unclear. The three kinds of animal models for schizophrenia (neurodevelopmental, pharmacological and genetic) have been created in different ways. This circumstance may explain the near lack of overlap between the genes found to be differentially expressed in the various animal models. Moreover, heterogeneity such as the ages of the animals and the ways of treating or sacrificing the animals occurs even within one group of animal models (either neurodevelopmental, pharmacological or genetic). For instance, in the various studies on the pharmacological models the animals were treated differently in that substances were administered at different concentrations and at different time intervals, and the time points at which the animals were sacrificed following drug administration were not the same. Furthermore, before being sacrificed in some of these studies the animals were anaesthetized, while it is known that anaesthesia may well affect gene expression levels (Culley et al., 2006; Dampney et al., 1995; Nakazato et al., 2009; Vahl et al., 2005). In addition, the methods of dissecting the brain areas differed and a number of brain regions were analysed, with each region having its specific mRNA expression pattern. For the gene expression analyses, a variety of microarrays and platforms have been used, ranging in their coverage from a couple of thousand genes to the nearly complete genome. The variation among the various experiments was further increased by the use of different statistical methods and cut-off values. The studies performed with the early microarrays and dealing with only a limited number of genes should therefore be repeated with the newest microarrays or deep-sequencing approaches and using the latest knowledge, e.g. regarding the effect of anaesthesia on gene expression. More overlap in the results

Chapter 6 - General Discussion
100
of the future genome-wide mRNA expression studies may then occur, possibly leading to the identification of common molecular pathways that may well play a role in the aetiology of schizophrenia. At present however the studies performed thus far do not provide decisive insights. Clearly, more molecular analyses of the various animal models for schizophrenia are necessary. It would be of interest to include in such studies new genome-wide technologies to explore, besides mRNA expression profiles, epigenetic (e.g. DNA methylation or histone acetylation), and noncoding RNA and (phospho)protein expression patterns as well. The family of sodium and potassium voltage-gated channels and schizophrenia As mentioned, most genes found to be differentially expressed in mRNA expression studies on schizophrenia animal models have been reported only once. However, an exception may constitute the family of genes encoding sodium and potassium voltage-gated channels (consisting of ~200 members).. In seven out of the twenty-six microarray studies discussed in chapter 1 at least one member of this family has been found to be differentially expressed (Duncan et al., 2008; Fatemi et al., 2006; Kaiser et al., 2004; Kinnunen et al., 2003; Marvanova et al., 2004; Takahashi et al., 2004; Toyooka et al., 2002). In chapter 2, we describe the differential expression of the potassium channel-interacting protein KCnIP1 in the hippocampus of both the original and the replicate APO-SUS/APO-UNSUS rat lines. Intriguingly, genes encoding potassium channels have already been implicated in the aetiology of schizophrenia, such as KCNN3 (Tomita et al., 2003) and recently KCNH2 (Huffaker et al., 2009). In particular KCNH2 (also known as the human ether-a-go-go-related gene (HERG)) appears to be a strong contributor to the schizophrenia phenotype because of the presence of disease-associated single-nucleotide polymorphisms (SNPs) in its gene together with compelling, converging evidence from (functional) brain imaging, mRNA expression and electrophysiological studies (Horvath et al., 2009). High-affinity antagonists of the KCNH2 channel are the antipsychotic drugs sertindole, pimozide and thioridazine. In fact, these drugs displayed little or no selectivity for dopamine D(2) or 5-HT(2A) receptors relative to their KCNH2 channel affinities (Kongsamut et al., 2002; Rampe et al., 1998). Recent studies have shown that the openers of potassium voltage-gated KQT-like (KCNQ) channels represent a class of antipsychotics as well (Sotty et al., 2009). KCNQ channels suppress basal activity of dopaminergic neurons and dopaminergic firing and activation of KCNQ attenuates the central stimulating effects of dopamine, cocaine, methylphenidate, and PCP (Dalby-Brown et al., 2006). Interestingly, the non-synonymous SNP rs10828317 (N251S) in phosphatidylinositol-5-phosphate 4-kinase, type II, alpha (PIP5K2A), a gene that controls the function of KCQN channels, has been associated with schizophrenia (Bakker et al., 2007). Use of the Xenopus oocyte expression system has shown that wild-type PIP5K2A but not the N251S mutant activated heteromeric KCNQ2/KCNQ3 and KCNQ3/KCNQ5

Chapter 6 - General Discussion
101
channels, indicating that this associated SNP is indeed functional (Fedorenko et al., 2008). Differential expression of KCnIP1 in the APO-SUS/-UNSUS rat model KCnIP1 is differentially expressed in both the original and replicate APO-SUS and APO-UNSUS hippocampus (chapter 2), but the reason for this is at present unclear. Possibilities for an explanation include the presence of a copy number variation (CNV), SNP, or differential expression caused by DNA methylation and/or histone modification. CNVs may be involved in the aetiology of complex disorders (Lee et al., 2006). To test whether CNVs play a role in the development of the APO-SUS/APO-UNSUS rat model, genomic DNAs of rats from the replicate lines were applied to a rat Agilent 244A array containing 235,000 probes (~60-nucleotides coding and non coding rat sequences with an average spatial resolution of 6.0 kb). A total of 367 probes were identified to represent a difference in copy number, but none of these was located in KCnIP1 (K.M.J. Van Loo, personal communication). It is therefore unlikely that the differential expression of KCnIP1 is directly caused by the presence of a CNV in the gene. The differential expression of KCnIP1 might also be due to SNPs that affect the stability or splicing of the mRNA. The promoter region, splice sites and exons of KCnIP1 should be analyzed for SNPs, which can be done with high-resolution melting (HRM). In the last decade the involvement of DNA methylation and histone modification (so-called epigenetics) in psychiatric disorders has become more acknowledged (Bredy et al., 2010). Methylation occurs on cytosines at CpG dinucleotides and affects the expression of genes by triggering histone deacetylation, chromatin condensation and gene silencing. Epigenetics including DNA methylation is defined as heritable changes in gene expression patterns (Wolffe et al., 1999), but can also be influenced and changed by environmental factors such as maternal care (Weaver et al., 2004). Therefore, differences in DNA methylation may well represent the mechanism underlying the differential gene expression in APO-SUS and APO-UNSUS rats since the apomorphine susceptibility is heritable but is also influenced by maternal deprivation and cross fostering (Ellenbroek et al., 2000). Tissue-specific epigenetic differences do indeed exist between the APO-SUS and APO-UNSUS rats (Van Loo et al., 2007). Sequence analysis reveals two small CpG islands (~105 nucleotides) in the 1-kb region upstream of the transcription initiation site of KCnIP1 (http://emboss.bioinformatics.nl; parameters: observed/expected ratio >0.60, Percent C + Percent G > 50.00, and Length > 100). Methylation of these CgG islands may affect the expression of KCnip1 and therefore it would be of interest to examine the methylation status of these CpG islands in the APO-SUS and APO-UNSUS rat genomes.

Chapter 6 - General Discussion
102
The potassium channel-interacting protein KCnIP1 and schizophrenia KCnIP (or KChIP) proteins coassemble with the α subunits of the pore-forming voltage-gated potassium channels 4 (Kv4) and regulate the surface density and gating properties of these channels (Birnbaum et al., 2004; Rhodes et al., 2004). As mentioned, potassium channels have been implicated in the aetiology of schizophrenia (Bakker et al., 2007; Rampe et al., 1998; Sotty et al., 2009). Differential KCnIP1 expression or a change in the mode of action of KCnIP1 due to e.g. a SNP might therefore be associated with schizophrenia as well. KCnIP proteins belong to the neuronal calcium sensors (NCS) together with gyanylyl cyclase-activating proteins (GCAPs), recoverins, NCS-1 (or frequenin), and visinin-like proteins (VILIPs). Since neuronal changes in cytosolic Ca2+ concentration regulate many different events (Berridge, 1997), NCS proteins are involved in the regulation of a wide range of neuronal functions, including the regulation of receptors, ion channels, membrane traffic and cell survival (reviewed by Burgoyne et al., 2004). The human nucleotide sequence of KCnIP1 displays one non-synonymous SNP (E206K) which is located in the C-terminal region of its well-conserved, functional EF-hand Ca2+-binding domain (Liao et al., 2009). This SNP therefore represents a candidate genetic variation that is worthwhile to be examined in a human association study. No correlation between a high dopamine susceptibility and a hyperactive HPA-axis Over a decade ago it was demonstrated that besides their apomorphine susceptibility APO-SUS rats of the original line displayed a hyperactive hypothalamus-pituitary-adrenal (HPA) axis (Rots et al., 1995; Rots et al., 1996a). Upon a conditioned emotional response higher levels of adenocorticotropic hormone (ACTH) and a prolonged elevation of ACTH levels were measured in plasma of the APO-SUS rats when compared to those in the APO-UNSUS rats. Under basal conditions the levels of ACTH were higher and the levels of free corticosterone were lower in the APO-SUS than the APO-UNSUS rats (Rots et al., 1996a). This difference in basal ACTH levels was already present at postnatal day 18, while at that age no differences were detected in the levels of dopamine D1 and D2 receptor, and tyrosine hydroxylase (TH) mRNAs. Based on these results it was hypothesized that the divergence in HPA-axis characteristics, such as basal and stress-induced ACTH levels, precedes the difference in dopamine responsiveness and may be causal to the differences observed within the dopaminergic system (Rots et al., 1996b). The causal correlation between a hyperactive HPA-axis and a high dopamine susceptibility has been postulated before since ACTH can directly elevate gene expression for catecholamine biosynthesis and the release of glucocorticoids (e.g. corticosterone in rodents) influences dopamine activity in the central nervous system (Czyrak et al., 2003; Serova et al., 2008). Since the

Chapter 6 - General Discussion
103
experiments performed by Rots and colleagues were all with rats of the original lines, we decided to replicate and validate these experiments using rats of the original and replicate lines. As described in chapter 3, APO-SUS rats of the original lines indeed showed elevated basal and stress-induced ACTH levels and lower free-corticosterone levels under basal conditions. However, in the rats from the replicate lines both the ACTH levels as well as the free-corticosterone levels did not differ. Although not proven, these results suggest that the hyperactive HPA-axis in the APO-SUS rats is likely not causally linked to the responsiveness to the dopamine D1/D2 receptor antagonist apomorphine. Differences between the APO-SUS / APO-UNSUS original and replicate rat lines: Consequences for the rat model As shown in chapter 3, APO-SUS rats from the replicate line do not display all aspects of the original APO-SUS phenotype. The differences found in the activity of the HPA-axis of the rats from the original lines were not observed in the replicate rat lines. Besides consequences for the hypothesized correlation between HPA-axis and dopamine responsiveness, this might also have consequences for other phenotypes that have been observed in APO-SUS rats of the original lines such as the immunological differences. For instance, a correlation between the activity of the HPA-axis and the Th1/Th2 ratio has been reported (reviewed by Elenkov, 2004). Kavelaars and colleagues found in rats from the original lines a difference in T helper (Th) cell response. The APO-SUS rats, but not APO-UNSUS rats, generated a T helper (Th) 2 response upon a Trichinella spiralis infection, whereas APO-UNSUS rats, but not APO-SUS rats, were susceptible for a Th1-mediated experimental autoimmune encephalomyelitis. Furthermore, the ratio of Th1-derived cytokine interferon γ and Th2-derived cytokine interleukin 4 was significantly higher in APO-UNSUS rats than in APO-SUS rats form the original lines (Kavelaars et al., 1997). The Th1/Th2 balances in APO-SUS and APO-UNSUS rats from the replicate lines remain to be established. Recent studies on the replicate rat lines have shown that the weights of APO-SUS and APO-UNSUS thymuses are different (V.D. Eijsink, R.W.M. Van Vugt and G.J.M. Martens, personal communication), which might indicate a disturbance in the immune system and in the Th1/Th2 balance since the thymus is an important organ in the cytokine network (Yarilin et al., 2004). Knowledge of the Th1/Th2 balance in the rat model is of interest since this balance has also shifted towards the Th2 system in schizophrenia patients (Muller et al., 1999); it represents one of the phenotypic similarities between the original APO-SUS rats and schizophrenia patients. Besides parameters of the immune system also other phenotypes described for the APO-SUS rats from the original line are worthwhile retesting in APO-SUS rats from both the original and the replicate lines. The APO-SUS/APO-UNSUS selection during breeding was solely based on apomorphine susceptibility and other parameters such as behaviour

Chapter 6 - General Discussion
104
on the open field, prepulse inhibition (PPI) and the occurrence of lung metastasis have hardly been studied in the replicate lines. Therefore, some of the differences reported in the past may not be represented anymore in the current original and/or replicate rat lines. Recent studies have shown that the reduced PPI and the higher activity on the open field are still present in the replicate APO-SUS rats. However, in rats from the original lines the difference in the behaviour on the open field could not be observed anymore (V.D. Eijsink, M.M.M. Verheij and G.J.M. Martens, personal communication). Extensive phenotypic analysis will establish whether (one of) the APO-SUS rat lines is/are still a good rat model for schizophrenia or rather represent(s) a rat model to study only the dopaminergic pathway. TRKA and schizophrenia The aim of this thesis was to identify genes associated with schizophrenia. To this end, we used, besides the APO-SUS rat model, human association studies. In chapter 4, the associations of a number of non-synonymous SNPs were examined. The 396 selected SNPs were located in genes important in neurodevelopment or associated with schizophrenia or another disease. After testing in USA Caucasian schizophrenia case-control groups, 189 SNPs were further tested in Caucasian groups from Denmark and Norway. The most promising SNP, with a corrected p-value of 0.12, was rs6336 in TRKA (NTRK1). The follow-up was an association study of this SNP in five other Caucasian schizophrenia case-control groups (chapter 5). Remarkably, whereas in two of these groups a higher frequency of the risk allele was indeed found in the patients compared to the controls, in the three other groups the SNP acted as a protective factor. In a ninth case-control group, the SNP also acted as a protective factor (p = 0.001, data not shown; in collaboration with R.A. Ophoff, Department of Medical Genetics, UMC Utrecht). A number of other SNPs have also been significantly associated with schizophrenia as a risk factor in some cases and as a protective factor in other groups (Shi et al., 2008). In an attempt to understand these remarkable results, various hypotheses have been postulated. The flips in the direction of a single-locus association may be attributed to the presence of another disease-influencing genetic variation (located either on a syntenic or a nonsyntenic locus) that is correlated with the target susceptibility locus and differs in linkage disequilibrium (LD) among various cohorts (correlation coefficient; Lin et al., 2007). To explore this hypothesis for rs6336, the associations of this SNP and other SNPs that from cohort to cohort vary in LD with rs6336 have to be studied in a large case-control group. Another hypothesis comprises the contribution of different phenotypes. For instance, the COMT Val158Met polymorphism has been associated with lifetime symptomatology and sensorimotor gating (Goghari et al., 2008; Quednow et al., 2008), while it failed association in a meta-analysis (Shi et al., 2008). Yet, a meta-analysis considering these clinical phenotypes should be performed to confirm the initial data. To explore whether

Chapter 6 - General Discussion
105
rs6336 is associated with one or more clinical schizophrenia phenotypes, more clinical data on the eight groups are needed. This is extremely difficult since the patient groups were collected by different psychiatrists and thus different clinical phenotypes have been recorded for each group. Another hypothesis concerns the parental origin of alleles. In 2009, parental-origin-specific associations with breast cancer, basal-cell carcinoma and type 2 diabetes have been reported, illustrating that SNPs in imprinted genes can be a risk allele when inherited from the father and a protective allele when inherited from the mother or vise versa (Kong et al., 2009). Kong et al. (2009) have reported 48 imprinted genes, but this group does not include TRKA. However, the group is not complete, e.g. the imprinting of the gene encoding GABA(A) receptor beta(2) subunit (GABRB2) has recently been reported (Pun et al., 2010). Interestingly, transmission disequilibrium tests with family trios have shown significant differences between paternal and maternal transmissions of the schizophrenia-associated SNP rs6556547 in the imprinted GABRB2 gene (Pun et al., 2010). Thus, the TRKA flip in direction of association might be parent specific. However, to investigate this possibility, genomic DNAs from parents and/or relatives are needed, but these are difficult to obtain. In conclusion, from the meta-analysis of the genotyping of the eight Caucasian cohorts we have to infer that TRKA SNP rs6336 is not associated with schizophrenia. However, further research on large, well-characterized cohorts could reveal whether this TRKA SNP interacts with another SNP(s), is linked to a certain clinical phenotype and/or is parental-origin-specifically associated with schizophrenia. If rs6336 and TRKA are indeed associated with schizophrenia, functional studies have to demonstrate whether the SNP is functional. The effect of rs6336, representing a H598Y substitution, on autophosphorylation of TRKA has been studied by Mardy and colleagues. They concluded that the H598Y (rs6336) substitution had no significant effect on the autophosphorylation of TRKA transfected into COS-1 and SH-SY5Y cells. Also SNP rs6339, a SNP in complete LD with rs6336 and representing a G607V substitution, had no effect on TRKA autophosphorylation (Mardy et al., 2001). However, the effect of both substitutions together on TRKA autophosphorylation was not tested. This knowledge would be of interest as the fact that rs6336 and rs6339 are in complete LD implies that the substitutions will always occur simultaneously. The phosphorylation event is essential since phosphorylated TRKA represents the activated state of the receptor. Recently, it was shown that the antipsychotics risperidone, haloperidol and olanzapine significantly decrease TRKA phosphorylation in rat hippocampus (Terry et al., 2010). In other schizophrenia animal models using risperidone and haloperidol, the expression level of TRKA was altered (Becker et al., 2008; Terry et al., 2007). Therefore, SNPs that affect the phosphorylation and thus the activity of TRKA might well be associated with the aetiology of schizophrenia and TRKA might represent a drug target.

Chapter 6 - General Discussion
106
Candidate genes other than TRKA and schizophrenia The association study reported in chapter 4 revealed ten SNPs that in a meta-analysis over three Caucasian schizophrenia case-control groups were associated with schizophrenia. Eight of the ten SNPs were also tested in some of the Caucasian schizophrenia case-control groups described in chapter 5 (data not shown). Seven? of these more broadly tested SNPs had a flip in the direction of association, likely causing the fact that they were not significantly associated with schizophrenia when tested in the meta-analysis. This observation underlines the necessity of correcting association data for multiple testing, i.e. with a p-value of 0.05 testing 100 SNPs implies that 5 SNPs will be falsely associated. The only SNP not showing a flip in association direction is rs2066844 in NOD2 (CARD15); this SNP was tested in a total of six cohorts and when tested in the meta-analysis it was significantly associated with schizophrenia. SNP rs2066844 causes an arginine to thryptophan change (Arg702Trp) in NOD2, a protein that activates the intracellular bacterial receptor activating nuclear factor-κB (NF-κB). In vitro assays have shown that the NF-κB response of the 702Trp variant to bacterial glycan components is reduced compared to that of wild-type NOD2 (Bonen et al., 2003). Therefore, NOD2 SNP rs2066844 may influence the immune response and as such provide a link between schizophrenia and infection. NOD2 has not been associated with schizophrenia before, neither in SNP association studies nor in animal or human expression studies. It would be interesting to analyze SNP rs2066844 in the additional three case-control groups that were used for analyzing rs6336 to examine whether the NOD2 SNP is genuinely associated with schizophrenia. Future perspectives The data discussed in this thesis raise more questions than give answers, but do provide leads for further research on both the APO-SUS / APO-UNSUS animal model and human association studies. For instance, the microarray data described in chapter 2 can be interpreted in several ways. With the purpose of finding genetic differences between the two rat lines, genes were considered to be of interest if their expression levels were different in three brain tissues. However, the most interesting gene KCnIP1 was expressed at a low level in the pituitary and this expression was not significantly different between the APO-SUS and APO-UNSUS rats (data not shown). Therefore, it may well be that other genes are equally interesting but just not or not high enough expressed in one of the three tissues. To study the genetic background of the APO-SUS and APO-UNSUS rats, extensive genome-wide SNP screenings with a rat SNP microarray and complete sequencing of the two genomes would be of interest. Given the fact that the genomes of the APO-SUS and APO-UNSUS rats are expected to contain at least 400.000 SNPs (K.M.J. Van Loo, personal communication), many rats should be sequenced to identify the most interesting and genuinely associated SNPs. Furthermore, these rats

Chapter 6 - General Discussion
107
have been bred for so many years that it is likely that SNPs can be 100% associated with APO-SUS rats and not with the APO-UNSUS rats without being functionally involved in the APO-SUS phenotype. To circumvent this problem, inter-/backcross lines of APO-SUS and APO-UNSUS rats can be generated by crossbreeding procedures. The generated F2 rat lines should be tested for the desired phenotypes (apomorphine susceptibility, open field, PPI, etc) and then be subjected to SNP testing. Also, further differential gene expression microarray studies involving more animals could be performed. When differentially expressed genes are discovered, their association with one or more of the phenotypes should be tested, which also holds for KCnIP1. In addition, to establish why a gene is differentially expressed one has to analyze whether its expression is differential in every tissue where the gene is expressed. A genomic cause will lead to differential expression in each tissue where the gene is expressed, whereas an epigenetic cause may lead to differential gene expression in only a few tissues (Van Loo et al., 2007). Examining neurodevelopmental expression in various tissues may provide more insight into the role of the gene during development. Furthermore, expression studies following cross-fostering and maternal deprivation, which are known to influence the phenotype (Ellenbroek et al., 2000), should be performed. Gene expression could also be studied after a pharmacological challenge (e.g. with apomorphine) or an environmental stressor (e.g. the open field or a forced swimming test). Since the APO-SUS and APO-UNSUS rats will behave differently in these challenges, it is likely that also certain molecular pathways will be induced to a different extent and some of the pathways induced in one line may not be induced in the other line. Chapter 3 showed that at least one phenotype that is present in the original APO-SUS / APO-UNSUS rat lines is not present in the replicate rat lines and therefore not yet tested phenotypes should be retested in both rat lines. Although this may result in the finding that the (replicate) APO-SUS rats are not a genuine model for schizophrenia, the affected dopaminergic pathway (since the apomorphine response is still differential) can provide new leads useful in schizophrenia research. Knowledge of genes responsible for this affected dopaminergic pathway is thus still very interesting. Therefore, genetic variations in KCnIP1 and in other genes resulting from further genomic and expression studies should be studied in human to determine their association with schizophrenia and/or other complex neurodevelopmental disorders. Yet, for future human association research a number of aspects have to be considered. First of all, the cohorts need to be of sufficient sizes to demonstrate association, in particular when the odds ratios (OR) and the risk allele frequencies are low. Over ten thousand cases and an equal number of controls are needed to detect significant association (power = 90%, two-sided α = 0.001) of a risk allele with an allele frequency in controls of 1% and an OR of 1.5. To detect such an association with an OR of 1.3, one would even need over twenty-seven thousand cases (Hattersley et al., 2005). Secondly, the rationale behind the

Chapter 6 - General Discussion
108
SNPs that are tested for association should be evident. Non-synonymous SNPs can be studied with for example the Affymetrix Targeted Genotyping GeneChip Human (Panels 1 & 2) 20K which consists of two panels containing twenty thousand SNPs changing amino acids and representing more than ten thousand genes. However, since interesting SNPs might have a regulatory function (e.g. in splice sites) rather than changing an amino acid residue, one could also argue to study as many SNPs as possible. For this purpose an array such as the Affymetrix Genome-Wide Human SNP Array 6.0, which contains over 900K SNPs and 900K probes for the detection of CNVs, can be used. Last but not least the case-control groups should be well defined. Schizophrenia is a complex disorder that comprises more than one symptom. It might well be that in patients that are more affected by negative symptoms, such as the reduction in emotional processes, other genes are associated with the complex disorder than in patients that are affected by cognitive impairment. Therefore, it is important that as much patient information as possible is collected. Furthermore, the debate continues on whether in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) schizophrenia should be considered as a single disorder or as a group of several disease entities (Gaebel et al., 2008). The genes reported in chapter 4 might be associated with one or more schizophrenia-related phenotypes and symptoms. In future research the functionality of the SNPs in e.g. TRKA and NOD2/CARD15 can be tested in in vitro tests and ultimately in an animal model. Animals carrying the risk alleles should be examined in various tests that correspond to one or more phenotypes of schizophrenia. In the last decades many genetic animal models have been developed (see chapter 1 for a number of examples) and the mutant models for e.g. disrupted in schizophrenia 1 (DISC1) and neuregulin 1 (NRG1) have been well characterized, which has led to new insights (Desbonnet et al., 2009). Furthermore, mice with a disruption in the region equivalent to human chromosomal region 22q11.2, a region associated with schizophrenia, have disruptions in synchronous firing between prefrontal cortex and hippocampal neurons, a phenomenon that has been associated with disruptions in learning and memory. Disruption of communication between these brain regions may indeed underlie schizophrenia (Sigurdsson et al., 2010). In conclusion, this thesis describes a number of genes, such as KCnIP1, TRKA, and NOD2/CARD15, that may well be involved in the aetiology of schizophrenia. Although clearly more research is necessary to establish the exact roles of these genes in this complex disorder, they do provide new leads in the continuing search to understand the molecular mechanisms underlying schizophrenia.

Chapter 6 - General Discussion
109
References Bakker SC, Hoogendoorn ML, Hendriks J, Verzijlbergen K, Caron S, Verduijn W, et al. The
PIP5K2A and RGS4 genes are differentially associated with deficit and non-deficit schizophrenia. Genes Brain Behav 2007;6:113-119.
Becker A, Grecksch G, Schwegler H, Roskoden T. Expression of mRNA of neurotrophic factors and their receptors are significantly altered after subchronic ketamine treatment. Med Chem 2008;4:256-263.
Berridge MJ. Elementary and global aspects of calcium signalling. J Physiol 1997;499 ( Pt 2):291-306.
Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev 2004;84:803-833.
Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 2003;124:140-146.
Bredy TW, Sun YE, Kobor MS. How the epigenome contributes to the development of psychiatric disorders. Dev Psychobiol 2010;
Burgoyne RD, O'Callaghan DW, Hasdemir B, Haynes LP, Tepikin AV. Neuronal Ca2+-sensor proteins: multitalented regulators of neuronal function. Trends Neurosci 2004;27:203-209.
Culley DJ, Yukhananov RY, Xie Z, Gali RR, Tanzi RE, Crosby G. Altered hippocampal gene expression 2 days after general anesthesia in rats. Eur J Pharmacol 2006;549:71-78.
Czyrak A, Mackowiak M, Chocyk A, Fijal K, Wedzony K. Role of glucocorticoids in the regulation of dopaminergic neurotransmission. Pol J Pharmacol 2003;55:667-674.
Dalby-Brown W, Hansen HH, Korsgaard MP, Mirza N, Olesen SP. K(v)7 channels: function, pharmacology and channel modulators. Curr Top Med Chem 2006;6:999-1023.
Dampney RA, Li YW, Hirooka Y, Potts P, Polson JW. Use of c-fos functional mapping to identify the central baroreceptor reflex pathway: advantages and limitations. Clin Exp Hypertens 1995;17:197-208.
Desbonnet L, Waddington JL, O'Tuathaigh CM. Mutant models for genes associated with schizophrenia. Biochem Soc Trans 2009;37:308-312.
Duncan CE, Chetcuti AF, Schofield PR. Coregulation of genes in the mouse brain following treatment with clozapine, haloperidol, or olanzapine implicates altered potassium channel subunit expression in the mechanism of antipsychotic drug action. Psychiatr Genet 2008;18:226-239.
Elenkov IJ. Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci 2004;1024:138-146. Ellenbroek BA, Sluyter F, Cools AR. The role of genetic and early environmental factors in
determining apomorphine susceptibility. Psychopharmacology (Berl) 2000;148:124-131.
Fatemi SH, Reutiman TJ, Folsom TD, Bell C, Nos L, Fried P, et al. Chronic olanzapine treatment causes differential expression of genes in frontal cortex of rats as revealed by DNA microarray technique. Neuropsychopharmacology 2006;31:1888-1899.
Fedorenko O, Strutz-Seebohm N, Henrion U, Ureche ON, Lang F, Seebohm G, et al. A schizophrenia-linked mutation in PIP5K2A fails to activate neuronal M channels. Psychopharmacology (Berl) 2008;199:47-54.
Gaebel W, Zielasek J. The DSM-V initiative "deconstructing psychosis" in the context of Kraepelin's concept on nosology. Eur Arch Psychiatry Clin Neurosci 2008;258 Suppl 2:41-47.
Goghari VM, Sponheim SR. Differential association of the COMT Val158Met polymorphism with clinical phenotypes in schizophrenia and bipolar disorder. Schizophr Res 2008;103:186-191.
Hattersley AT, McCarthy MI. What makes a good genetic association study? Lancet 2005;366:1315-1323.
Horvath S, Mirnics K. Breaking the gene barrier in schizophrenia. Nat Med 2009;15:488-490.

Chapter 6 - General Discussion
110
Huffaker SJ, Chen J, Nicodemus KK, Sambataro F, Yang F, Mattay V, et al. A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med 2009;15:509-518.
Kaiser S, Foltz LA, George CA, Kirkwood SC, Bemis KG, Lin X, et al. Phencyclidine-induced changes in rat cortical gene expression identified by microarray analysis: implications for schizophrenia. Neurobiol Dis 2004;16:220-235.
Kavelaars A, Heijnen CJ, Ellenbroek B, van Loveren H, Cools A. Apomorphine-susceptible and apomorphine-unsusceptible Wistar rats differ in their susceptibility to inflammatory and infectious diseases: a study on rats with group-specific differences in structure and reactivity of hypothalamic-pituitary-adrenal axis. J Neurosci 1997;17:2580-2584.
Kinnunen AK, Koenig JI, Bilbe G. Repeated variable prenatal stress alters pre- and postsynaptic gene expression in the rat frontal pole. J Neurochem 2003;86:736-748.
Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, et al. Parental origin of sequence variants associated with complex diseases. Nature 2009;462:868-874.
Kongsamut S, Kang J, Chen XL, Roehr J, Rampe D. A comparison of the receptor binding and HERG channel affinities for a series of antipsychotic drugs. Eur J Pharmacol 2002;450:37-41.
Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron 2006;52:103-121.
Liao YS, Chen KC, Chang LS. Functional role of EF-hands 3 and 4 in membrane-binding of KChIP1. J Biosci 2009;34:203-211.
Lin PI, Vance JM, Pericak-Vance MA, Martin ER. No gene is an island: the flip-flop phenomenon. Am J Hum Genet 2007;80:531-538.
Mardy S, Miura Y, Endo F, Matsuda I, Indo Y. Congenital insensitivity to pain with anhidrosis (CIPA): effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Hum Mol Genet 2001;10:179-188.
Marvanova M, Lakso M, Wong G. Identification of genes regulated by memantine and MK-801 in adult rat brain by cDNA microarray analysis. Neuropsychopharmacology 2004;29:1070-1079.
Muller N, Riedel M, Ackenheil M, Schwarz MJ. The role of immune function in schizophrenia: an overview. Eur Arch Psychiatry Clin Neurosci 1999;249 Suppl 4:62-68.
Nakazato K, Yoshida Y, Takemori K, Kobayashi K, Sakamoto A. Expressions of genes encoding drug-metabolizing enzymes are altered after sevoflurane, isoflurane, propofol or dexmedetomidine anesthesia. Biomed Res 2009;30:17-24.
Pun FW, Zhao C, Lo WS, Ng SK, Tsang SY, Nimgaonkar V, et al. Imprinting in the schizophrenia candidate gene GABRB2 encoding GABA(A) receptor beta(2) subunit. Mol Psychiatry 2010;
Quednow BB, Wagner M, Mossner R, Maier W, Kuhn KU. Sensorimotor Gating of Schizophrenia Patients Depends on Catechol O-Methyltransferase Val158Met Polymorphism. Schizophr Bull 2008;
Rampe D, Murawsky MK, Grau J, Lewis EW. The antipsychotic agent sertindole is a high affinity antagonist of the human cardiac potassium channel HERG. J Pharmacol Exp Ther 1998;286:788-793.
Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, et al. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J Neurosci 2004;24:7903-7915.
Rots NY, Cools AR, de Jong J, De Kloet ER. Corticosteroid feedback resistance in rats genetically selected for increased dopamine responsiveness. J Neuroendocrinol 1995;7:153-161.
Rots NY, Cools AR, Oitzl MS, de Jong J, Sutanto W, de Kloet ER. Divergent prolactin and pituitary-adrenal activity in rats selectively bred for different dopamine responsiveness. Endocrinology 1996a;137:1678-1686.
Rots NY, Workel J, Oitzl MS, Berod A, Rostene W, Cools AR, et al. Development of divergence in dopamine responsiveness in genetically selected rat lines is preceded by changes in pituitary-adrenal activity. Brain Res Dev Brain Res 1996b;92:164-171.

Chapter 6 - General Discussion
111
Serova LI, Gueorguiev V, Cheng SY, Sabban EL. Adrenocorticotropic hormone elevates gene expression for catecholamine biosynthesis in rat superior cervical ganglia and locus coeruleus by an adrenal independent mechanism. Neuroscience 2008;153:1380-1389.
Shi J, Gershon ES, Liu C. Genetic associations with schizophrenia: Meta-analyses of 12 candidate genes. Schizophr Res 2008;104:96-107.
Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature 2010;464:763-767.
Sotty F, Damgaard T, Montezinho LP, Mork A, Olsen CK, Bundgaard C, et al. Antipsychotic-like effect of retigabine [N-(2-Amino-4-(fluorobenzylamino)-phenyl)carbamic acid ester], a KCNQ potassium channel opener, via modulation of mesolimbic dopaminergic neurotransmission. J Pharmacol Exp Ther 2009;328:951-962.
Takahashi Y, Kumanishi T, Hayashi S. Using a DNA microarray method to examine gene expression in brain from clozapine-injected mice. Ann N Y Acad Sci 2004;1025:561-569.
Terry AV, Gearhart DA, Pillai A, Zhang G, Bartlett MG. Chronic antipsychotic treatment: protracted decreases in phospho-TrkA levels in the rat hippocampus. Int J Neuropsychopharmacol 2010;1-7.
Terry AV, Jr., Gearhart DA, Warner SE, Zhang G, Bartlett MG, Middlemore ML, et al. Oral haloperidol or risperidone treatment in rats: temporal effects on nerve growth factor receptors, cholinergic neurons, and memory performance. Neuroscience 2007;146:1316-1332.
Tomita H, Shakkottai VG, Gutman GA, Sun G, Bunney WE, Cahalan MD, et al. Novel truncated isoform of SK3 potassium channel is a potent dominant-negative regulator of SK currents: implications in schizophrenia. Mol Psychiatry 2003;8:524-535, 460.
Toyooka K, Usui M, Washiyama K, Kumanishi T, Takahashi Y. Gene expression profiles in the brain from phencyclidine-treated mouse by using DNA microarray. Ann N Y Acad Sci 2002;965:10-20.
Vahl TP, Ulrich-Lai YM, Ostrander MM, Dolgas CM, Elfers EE, Seeley RJ, et al. Comparative analysis of ACTH and corticosterone sampling methods in rats. Am J Physiol Endocrinol Metab 2005;289:E823-828.
Van Loo KM, Martens GJ. Identification of genetic and epigenetic variations in a rat model for neurodevelopmental disorders. Behav Genet 2007;37:697-705.
Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004;
Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science 1999;286:481-486. Yarilin AA, Belyakov IM. Cytokines in the thymus: production and biological effects. Curr Med
Chem 2004;11:447-464.


Summary Samenvatting

Summary/Samenvatting
114
Summary Schizophrenia is a complex psychiatric disorder with a lifetime risk of ~1% and an enormous impact on society. The disorder is characterized by psychotic and negative symptoms, and most patients also suffer from cognitive impairments. The first symptoms usually manifest during late adolescence or early adulthood, but the origin of the disorder is thought to be neurodevelopmental. Environmental factors in combination with predisposing genes appear to be important for the aetiology of schizophrenia. The aim of this thesis was to identify candidate genes associated with schizophrenia. To this end, studies on a rat model for schizophrenia as well as human association studies on schizophrenia case-control cohorts are presented. The identification of predisposing genes for schizophrenia is however not simple. For example, loci on 21 of the 23 chromosomes have been reported to be associated with the disorder on the basis of linkage analyses. Furthermore, a number of candidate genes have been tested in multiple case-control cohorts for association with schizophrenia. Yet, variations in these genes account for the aetiology of only a small percentage of schizophrenia patients. In order to get a better insight into the aetiology of schizophrenia and the function of candidate genes, a variety of animal models have been developed. In chapter 1 mRNA expression profiling studies on animal models for schizophrenia are reviewed. From this overview it becomes clear that the various studies have found many candidate genes. However, most of these genes have been reported in only one expression study and thus the question which genes really play a role in the aetiology of schizophrenia remains unanswered. Chapter 1 also introduces the schizophrenia animal model central in this thesis, namely the apomorphine-susceptible and apomorphine-unsusceptible (APO-SUS and APO-UNSUS, respectively) rats. This model has been generated from Wistar rats on the basis of their susceptibility to the dopamine (DA) receptor agonist apomorphine and is characterized by dopaminergic dysregulation. The pharmacogenetic selection has been performed twice, independently and with a decade difference in time, resulting in the so-called original and replicate APO-SUS and APO-UNSUS rat lines. The APO-SUS and APO-UNSUS rats differ not only in their dopaminergic pathway, but also in a number of other aspects such as their brain information processing, and immune and neuroendocrine systems. An mRNA expression profiling study on various APO-SUS and APO-UNSUS brain regions is described in chapter 2. The analysis revealed that besides the previously reported Aph-1b only one other gene, encoding the potassium channel-interacting protein KCnIP1, was significantly differentially expressed with a similar regulation (upregulated) in brain tissues from both the replicate and original APO-SUS rats. Although the mechanism underlying the differential expression of KCnIP1 in the APO-SUS/APO-UNSUS rat model is not known yet, this gene is of interest for further research as many members of the family of sodium and potassium voltage-

Summary/Samenvatting
115
gated channels have been found to be associated with schizophrenia in both human and animal studies. Seven out of the twenty-six mRNA expression studies reviewed in chapter 1 have also reported the differential expression of a member of this family. Therefore, chapter 6 concludes that the family of genes encoding sodium and potassium voltage-gated channels deserves further study in relation to schizophrenia. In chapter 3 it is demonstrated that the replicate and original APO-SUS/APO-UNSUS rat lines differ in at least one phenotype. It has been previously demonstrated that original APO-SUS rats have a hyperactive HPA-axis. As expected, we found that APO-SUS rats from the original line indeed showed significantly elevated adenocorticotropic hormone (ACTH) levels in basal and stress conditions and the basal free-corticosterone levels were lower than in the original APO-UNSUS rats. However, the basal and stress-induced ACTH levels and basal free-corticosterone levels were comparable in APO-SUS and APO-UNSUS rats from the replicate lines. Furthermore, in chapter 3 we studied in the gene encoding corticosteroid-binding globulin (CBG/Serpina6) the Met276Ile SNP that is hypothesized to affect the corticosterone binding of the CBG protein. This SNP was in complete linkage disequilibrium (LD) with a Val285Met SNP in the same gene. The Met276 and Val285 alleles were associated with the APO-SUS rats of both the replicate and original lines, indicating that these SNPs likely do not segregate with the levels of free corticosterone. The phenotypes of the APO-SUS rats of the original and replicate rat lines are thus not identical and it is worthwhile to retest other phenotypes described in APO-SUS rats from both the original and the replicate lines. The selection during breeding was solely based on apomorphine susceptibility and other parameters have hardly been studied. Therefore, some of the differences reported in the past may not be present in the original and/or replicate rat lines anymore. Extensive phenotypic analysis will establish whether (one of) the APO-SUS rat lines is/are still a good rat model for schizophrenia or rather represent(s) a rat model to study the dopaminergic pathway. Besides the animal studies, human association studies aimed at identifying schizophrenia candidate genes were performed. An association study on 396 selected non-synonymous SNPs in three independent Caucasian schizophrenia case-control cohorts is reported in chapter 4. The meta-analysis showed ten SNPs that were associated with schizophrenia. Correction for multiple testing revealed SNP rs6336 in neurotrophic tyrosine kinase receptor (NTRK1, better known as TRKA) as the most attractive SNP for further study. Therefore, this SNP was tested for association in five additional independent Caucasian schizophrenia case-control cohorts in chapter 5. In this study, the SNP was found as a risk allele in five cohorts, whereas it acted as a protective allele in three cohorts. This flip in direction of association has been seen before for other SNPs and various hypotheses have been postulated to explain this phenomenon. We discuss the dual character of the

Summary/Samenvatting
116
TRKA SNP and speculate on potential causes for this duality. The possible effect of this SNP on the phosphorylation of TRKA has been discussed in chapters 4 and 6. In chapter 4, nine other SNPs (in BARD1 (rs28997576), LAMA5 (rs3810548), DKK2 (rs7037102), NOD2 (rs2066844), RELN (rs2229860), NOD2 (rs2066845), NRG1 (rs10503929), ADAM7 (rs13259668), and TNR (rs859427)) are shown to be associated with schizophrenia. Additional results concerning these SNPs and the implications are discussed in chapter 6. Especially SNP rs2066844 in NOD2 (CARD15), which causes an arginine to thryptophan change (Arg702Trp), may be of interest to study in more detail in additional case-control groups and with cellular or animal models. Suggestions for further research are given in chapter 6, including a discussion on how additional microarray studies may provide more leads to the genetic background of the APO-SUS/APO-UNSUS rat model. Furthermore, the impact of the differences between the replicate and original APO-SUS rat lines, the complex interpretation of the results from the human association studies and suggestions to optimize these studies are discussed. In conclusion, this thesis postulates that KCnIP1, TRKA, and NOD2/CARD15 may well be involved in the aetiology of schizophrenia and provides new leads in the continuing search to understand the molecular mechanisms underlying this devastating complex disorder.

Summary/Samenvatting
117
Samenvatting Schizofrenie is een complexe psychiatrische stoornis met een enorme impact op de maatschappij. De stoornis wordt gekarakteriseerd door psychotische symptomen (de zogenoemde "positieve" symptomen) en het ontbreken van basale emoties ("negatieve" symptomen), en de meeste patiënten leiden ook aan cognitieve stoornissen. De eerste symptomen openbaren zich gewoonlijk tijdens de late adolescentie of tijdens vroege volwassenheid, maar men denkt dat de stoornis ontstaat tijdens de vroege ontwikkeling van de hersenen. Omgevingsfactoren in combinatie met genetische factoren lijken belangrijk te zijn bij het ontstaan van schizofrenie. Het doel van het onderzoek beschreven in dit proefschrift was om genen te identificeren die geassocieerd zouden kunnen zijn met schizofrenie. Om dit te bewerkstellingen, werden zowel studies aan een rattenmodel als genetische associatie studies uitgevoerd. Het identificeren van de genetische factoren die betrokken zijn bij het ontstaan van schizofrenie is niet gemakkelijk. Zo zijn op basis van koppelingsanalyse locaties op 21 van de 23 chromosomen gerapporteerd als zijnde geassocieerd met de stoornis. Vele kandidaatgenen zijn getest in meerdere casus - controle groepen, maar deze genen dragen slechts bij een klein percentage van de schizofreniepatiënten bij aan het ontstaan van de stoornis. Om meer inzicht te krijgen in het ontstaan van schizofrenie en de functie van de kandidaatgenen zijn er verschillende diermodellen ontwikkeld. In hoofdstuk 1 wordt een overzicht gegeven van mRNA expressiestudies die zijn uitgevoerd aan diermodellen voor schizofrenie. Uit dit overzicht blijkt dat door de diverse studies vele kandidaatgenen zijn gevonden. De meeste van deze genen worden echter slechts genoemd in één expressiestudie en de vraag welke genen nu echt een rol spelen bij het ontstaan van schizofrenie blijft dus onbeantwoord. Het diermodel voor schizofrenie dat centraal staat in dit proefschrift wordt ook geïntroduceerd in hoofdstuk 1, namelijk apomorfine-gevoelige en apomorfine-ongevoelige ratten (in het Engels: apomorphine-susceptible, APO-SUS, en apomorphine-unsusceptible, APO-UNSUS). Dit rattenmodel werd gegenereerd vanuit Wistar laboratoriumratten op basis van hun gevoeligheid voor de dopamine receptor agonist apomorfine en wordt gekarakteriseerd door een verschil in regulatie van de neurotransmitter dopamine. Deze farmacogenetische selectie is twee keer uitgevoerd, onafhankelijk van elkaar en met een decennium tijdsverschil, wat geresulteerd heeft in de zogenoemde originele en replica APO-SUS en APO-UNSUS ratten. De APO-SUS en APO-UNSUS ratten verschillen niet alleen in hun dopaminerge systeem, maar ook op andere gebieden, zoals informatieverwerking in de hersenen, het immuunsysteem en het neuro-endocriene systeem. Een mRNA expressiestudie van verschillende hersengebieden van de APO-SUS en APO-UNSUS ratten staat beschreven in hoofdstuk 2. Deze studie bracht, naast het eerder gerapporteerde Aph-1b gen, slecht één ander gen aan het licht, namelijk het gen dat

Summary/Samenvatting
118
codeert voor het kaliumkanaal-interactieve eiwit KCnIP1. Dit gen kwam significant hoger tot expressie in breinweefsel van zowel de originele als de replica APO-SUS ratten. Alhoewel het waarom van deze differentiële expressie nog niet bekend is, is KCnIP1 wel een interessant gen voor toekomstig onderzoek aangezien vele leden van de genfamilie coderend voor calcium- of kaliumkanalen geassocieerd zijn met schizofrenie in zowel mens als dierstudies. Zeven van de zesentwintig mRNA expressiestudies beschreven in hoofdstuk 1 rapporteerden een lid van deze familie. Daarom wordt in hoofdstuk 6 geconcludeerd dat met betrekking tot schizofrenie de familie van genen coderend voor calcium- of kaliumkanalen verdere bestudering verdient. In hoofdstuk 3 wordt aangetoond dat de originele en replica ratten wat betreft tenminste één fenotype verschillen. In het verleden is aangetoond dat ratten van de originele APO-SUS lijn een hyperactieve hypothalamus-hypofyse-bijnieras (beter bekend onder de Engelse afkorting HPA-as; Hypothalamic-pituitary-adrenal, HPA-axis) hebben. Zoals eerder gerapporteerd, konden we aantonen dat in zowel basale als stressomstandigheden de APO-SUS ratten van de originele lijn inderdaad significant hogere waarden hadden van het adrenocorticotroop hormoon (ACTH) en dat de vrije corticosteronwaardes onder basale condities lager waren dan die van de APO-UNSUS ratten van de originele lijn. De ACTH- en vrije corticosteronwaardes waren echter vergelijkbaar tussen de APO-SUS en APO-UNSUS ratten van de replica lijn. Ook bestudeerden we in hoofdstuk 3 het gen coderend voor corticosteroïde-bindend globuline (CBG/Serpina6). In dit gen bevindt zich een polymorfisme van een enkel nucleotide (in het Engels single-nucleotide polymorphism, SNP) dat zorgt voor de aminozuurverandering Met276Ile. De hypothese is dat deze SNP de binding van het CBG-eiwit aan corticosteron beïnvloedt. Deze SNP is 100% gekoppeld aan een andere SNP in hetzelfde gen, namelijk de Val285Met SNP. De Met276 en Val285 allelen waren geassocieerd met de APO-SUS ratten van zowel de originele als de replica lijn, wat impliceert dat deze SNPs niet segregeren met de vrije corticosteronwaardes (deze waren immers verschillend tussen de originele en replica lijnen). De fenotypes van de originele en replica rattenlijnen zijn dus niet identiek en het is de moeite waard om andere fenotypes die voorheen beschreven zijn voor APO-SUS ratten te hertesten in ratten van zowel de originele als de replica rattenlijnen. De selectie tijdens het fokken van deze ratten was enkel gebaseerd op de gevoeligheid voor apomorfine en andere fenotypes zijn nauwelijks bestudeerd in de replica lijnen. Het is daarom mogelijk dat verschillen die in het verleden beschreven zijn, nu niet meer aanwezig zijn in ratten van de originele en/of replica rattenlijnen. Een uitgebreide fenotypische analyse zal antwoord geven op de vraag of (een van) de APO-SUS rattenlijn op dit moment nog een goed rattenmodel voor schizofrenie is of dat ze slechts een rattenmodel vormen voor de bestudering van het dopaminerge systeem.

Summary/Samenvatting
119
Naast dierstudies, werden ook associatiestudies bij de mens uitgevoerd om kandidaatgenen voor schizofrenie te ontdekken. Een associatiestudie van 396 geselecteerde SNPs (die leiden tot een aminozuurverandering) in drie onafhankelijke Caucasische schizofrenie casus - controle groepen is beschreven in hoofdstuk 4. De meta-analyse toonde aan dat tien SNPs geassocieerd konden worden met schizofrenie. Na een correctie voor het testen van meerdere variabelen (multiple testing) bleef één SNP over als de meest aantrekkelijke SNP voor verdere studie. Dit is SNP rs6336 in het gen coderend voor de neurotrofische tyrosine kinase receptor (TRKA). Daarom werd deze SNP verder getest voor associatie in vijf andere, onafhankelijke Caucasische schizofrenie casus - controle groepen (hoofdstuk 5). In deze studie werd gevonden dat in twee groepen de SNP geassocieerd is met schizofrenie als risicofactor, maar in drie andere groepen als beschermende factor. Deze tegenstelling in associatie is in het verleden beschreven voor andere SNPs en er zijn verschillende hypotheses die dit fenomeen proberen te verklaren. We bediscussiëren het duale karakter van de SNP in TRKA en speculeren over de potentiële oorzaken van deze dualiteit. Het eventuele effect van de SNP op de fosforylering van TRKA wordt bediscussieerd in de hoofdstukken 4 en 6. In hoofdstuk 4 worden nog negen andere SNPs (in de genen BARD1 (rs28997576), LAMA5 (rs3810548), DKK2 (rs7037102), NOD2 (rs2066844 en rs2066845), RELN (rs2229860), NRG1 (rs10503929), ADAM7 (rs13259668) and TNR (rs859427)) beschreven die geassocieerd zijn met schizofrenie. Resultaten van meerdere associatiestudies met deze SNPs en de implicaties hiervan staan beschreven in hoofdstuk 6. Vooral SNP rs2066844 in NOD2 (CARD15), welke een aminozuurverandering (Arg702Trp) veroorzaakt, kan interessant zijn voor verdere studie in meerdere schizofrenie casus - controle groepen en in cellulaire modellen of diermodellen. Suggesties voor verder onderzoek worden gegeven in hoofdstuk 6, inclusief een discussie over hoe toekomstige microarraystudies meer inzicht kunnen geven in de genetische achtergrond van het APO-SUS/APO-UNSUS rattenmodel. Tevens worden de impact van de verschillen tussen de originele en replica rattenlijnen, de moeizame interpretaties van de associatiestudies bij mensen en suggesties om deze studies te optimaliseren in dit hoofdstuk bediscussieerd. Concluderend kan gesteld worden dat de resultaten van dit proefschrift laten zien dat KCnIP1, TRKA, en NOD2/CARD15 betrokken kunnen zijn bij het ontstaan van schizofrenie en ze bieden nieuwe aanknopingspunten voor de voortschrijdende pogingen om de moleculaire mechanismen die ten grondslag liggen aan deze destructieve complexe stoornis beter te begrijpen.


Dankwoord Curriculum vitae
List of publications

Dankwoord
122

Curriculum vitae
123
Curriculum vitae Jessica Elisabeth Lathouwers - van Schijndel werd op 12 november 1980 geboren te Heeswijk-Dinther. Na het behalen van haar gymnasium diploma aan Gymnasium Bernrode te Bernheze begon ze in 1998 met de studie bioprocestechnologie aan de Wageningen Universiteit (toen nog Landbouwuniversiteit Wageningen geheten). Ze koos voor de specialisatie cellulair-moleculair, met afstudeervakken bij het Laboratorium voor Virologie onder leiding van dr. ir. Gorben Pijlman en de leerstoelgroep Celbiologie en Immunologie onder leiding van dr. ir. Mark Huising, en een stage bij the Department of Microbiology and Immunology of the Melbourne University onder leiding van dr. Barbara Coulson. In het voorjaar van 2004 werd het doctoraaldiploma behaald. Aansluitend begon ze als junior onderzoeker aan haar promotie-onderzoek op de afdeling Moleculaire Dierfysiologie (Radboud Universiteit Nijmegen), onder leiding van prof. dr. Gerard Martens. Sinds februari 2009 is ze werkzaam als junior projectleider bij de afdeling Viral Bioprocess Technology & Support (BTS-V) van Intervet/Schering Plough Animal Health te Boxmeer.

List of publications
124
List of publications van Schijndel JE, van Zweeden M, van Loo KMJ, Djurovic S, Andreassen OA, Hansen T, Werge T, Nyegaard M, Sørensen KM, Nordentoft M, Mortensen PB, Mors O, Børglum AD, Del-Favero J, Norrback KF, Adolfsson R, De Hert M, Claes S, Cichon S, Rietschel M, Nöthen MM, Kallunki P, Pedersen JT, Martens GJM.
Dual association of a TRKA polymorphism with schizophrenia. Submitted. van Schijndel JE, van Zweeden M,van Loo KMJ, Lubbers LJ, Pesman GJ, Sweep FCGJ, Martens GJM. Dopamine susceptibility of APO-SUS rats is not per se coupled to HPA-axis activity. Physiol Behav. In press. van Schijndel JE, Martens GJM. Gene expression profiling in rodent models for schizophrenia. Curr Neuropharmacol. 2010; 8(4):382-393. van Schijndel JE, van Zweeden M, van Loo KMJ, Martens GJM. Gene expression profiling in brain regions of a rat model displaying schizophrenia-related features. Behav Brain Res. 2010 Mar; 207(2):476-479. van Schijndel JE*, van Loo KMJ*, van Zweeden M, Djurovic S, Andreassen OA, Hansen T, Werge T, Kallunki P, Pedersen JT, Martens GJM. Three-cohort targeted gene screening reveals a non-synonymous TRKA polymorphism associated with schizophrenia. J Psychiatr Res. 2009 Oct; 43(15):1195-1199. *equal contribution Moons T, Claes S, Martens GJM, Peuskens J, van Loo KMJ, van Schijndel JE, De Hert MA, van Winkel R. Clock genes and body composition in patients with schizophrenia under treatment with antipsychotic drugs. Schizophr Res. In press. van Loo KMJ, van Schijndel JE, van Zweeden M, van Manen D, Trip MD, Petersen DC, Schuitemaker H, Hayes VM, Martens GJM. Correlation between HIV-1 seropositivity and prevalence of a gamma-secretase polymorphism in two distinct ethnic populations. J Med Virol. 2009 Nov; 81(11):1847-1851. van Loo KMJ, Dejaegere T, van Zweeden M, van Schijndel JE, Wijmenga C, Trip MD, Martens GJM. Male-specific association between a gamma-secretase polymorphism and premature coronary atherosclerosis. PLoS One. 2008; 3(11):e3662.

List of publications
125
Huising MO, van Schijndel JE, Kruiswijk CP, Nabuurs SB, Savelkoul HF, Flik G, Verburg-van Kemenade BM. The presence of multiple and differentially regulated interleukin-12p40 genes in bony fishes signifies an expansion of the vertebrate heterodimeric cytokine family. Mol Immunol. 2006 Apr; 43(10):1519-1533. Huising MO, Kruiswijk CP, van Schijndel JE, Savelkoul HF, Flik G, Verburg-van Kemenade BM. Multiple and highly divergent IL-11 genes in teleost fish. Immunogenetics. 2005 Jul; 57(6):432-443. Pijlman GP, van Schijndel JE, Vlak JM. Spontaneous excision of BAC vector sequences from bacmid-derived baculovirus expression vectors upon passage in insect cells. J Gen Virol. 2003 Oct; 84(Pt 10):2669-2678.





















