
Multiscale modeling of the plasticity in an aluminum single crystal

Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533
Contents lists available at ScienceDirect
Spectrochimica Acta Part A: Molecular andBiomolecular Spectroscopy
journal homepage: www.elsevier .com/locate /saa
Thermally induced single crystal to single crystal transformation leadingto polymorphism
http://dx.doi.org/10.1016/j.saa.2014.04.0321386-1425/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding authors. Tel.: +91 9831115427; fax: +91 33 2414 6584.E-mail addresses: [email protected] (K. Dey), [email protected] (A. Ghosh), [email protected] (S. Kumar).
Rajat Saha a, Susobhan Biswas a, Sanjoy Kumar Dey a,f, Arijit Sen a, Madhusudan Roy b, Ian M. Steele c,Kamalendu Dey d,⇑, Ashutosh Ghosh e,⇑, Sanjay Kumar a,⇑a Department of Physics, Jadavpur University, Kolkata 700032, Indiab Applied Material Science Division, SINP, Kolkata 700064, Indiac Department of the Geophysical Sciences, The University of Chicago, USAd Department of Chemistry, University of Kalyani, Kalyani, Nadia, Indiae Department of Chemistry, University of Calcutta, Kolkata 700009, Indiaf Department of Physics, NITMAS, 24-paragana(S) 743368, India
h i g h l i g h t s
� Single crystal to single crystaltransformation.� Polymorphism.� 2D homochiral and achiral
supramolecular sheets.� DSC analysis.� Photoluminescence study.
g r a p h i c a l a b s t r a c t
[La(1,10-phen)2(NO3)3] undergoes ‘thermally induced reversible single crystal to single crystal transfor-mation’ between its two polymorphic phases at 100 K and 293 K. Structural analysis shows that the com-plex forms 2D achiral sheets at 100 K whereas at 293 K forms two different homochiral 2D sheets. DSCanalysis indicates that this structural transformation occurs at 246 K. This structural transformation pro-ceeds through coherent movement of ligands.
a r t i c l e i n f o
Article history:Received 2 December 2013Received in revised form 10 March 2014Accepted 6 April 2014Available online 18 April 2014
Keywords:Single crystal to single crystaltransformationPolymorphismSupramolecular interactionsHomochirality and achiralityPhotoluminescence
a b s t r a c t
The robust complex [La(1,10-phen)2(NO3)3] (1,10-phen = 1,10-phenanthroline) exhibits thermally inducedsingle crystal to single crystal transformation from one polymorphic phase to another. The complex crystal-lizes in monoclinic C2/c space group with C2 molecular symmetry at 293 K while at 100 K it shows P21/c spacegroup with C1 molecular symmetry. Supramolecular investigation shows that at 100 K the complex forms 2Dachiral sheets whereas at 293 K forms two different homochiral 2D sheets. Low temperature DSC analysisindicates that this structural transformation occurs at 246 K and also this transformation is reversible in nat-ure. We have shown that thermally induced coherent movement of ligands changes the molecular symmetryof the complex and leads to polymorphism. Photoluminescence property of complex has been studied inboth solid state and in methanolic solution at room temperature. The effect of the presence low-lying LUMOorbital of p-character in the complex is elucidated by theoretical calculation using DFT method.
� 2014 Elsevier B.V. All rights reserved.

R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533 527
Introduction
In the last decade, escalating attention has been devoted to thestudy of solid-state structural transformation due to its paramountimportance in the field of solid state chemistry and material sci-ence [1–4]. A special case of solid-state structural transformationis ‘single crystal to single crystal’ (SC–SC) transformation in whichsignificant changes within the crystal structure take place withoutappreciable loss of crystal mosiacity [2]. The challenge before us isto understand why atoms in solid move cooperatively during SC–SC transformation and how a crystal can exhibit such surprisingfluidity.
Generally, solid state structural transformation occurs inresponse to several physical factors such as thermal perturbations,photochemical excitations etc. and also due to some chemical fac-tors. In literature, there are several examples of such structuraltransformations of flexible host frameworks through removal andexchange of solvent molecules [5–12]. In contrast, such structuraltransformation for robust frameworks, where the complex doesnot contain any solvent molecule, is a rare phenomenon [13,14].For robust complexes SC–SC transformations are manly triggeredby thermal and photochemical excitations. During structural trans-formations of robust frameworks, all the atoms move collectivelyand cooperatively in response to external stimulation. Such atomicmovement leads to (i) bond breaking-bond making, (ii) movementof the ligands, or both and in turn the supramolecular interactionsof the complex will also change. There are only scattered examplesin literature on such structural transformations of robust frame-works [13,14]. It may be noted that Hu et al. have established thatone-dimensional polymer [ZnCl2(l-bipy)]n is converted to two-dimensional network [Zn(l-Cl)2(l-bipy)]n upon cooling below130 K [13].
Single crystal to single crystal transformation of robust frame-work leads to polymorphism as both the initial and final phaseshave the identical composition with different geometry [15,16].Polymorphism is of major industrial importance today [17]. Whilea lot of effort is being given in the study of polymorphic (organic)modifications of drugs [18], in the literature [19–22], there are onlyscattered examples on the systematic investigation of inorganicpolymorphism available. Metal–organic complexes which containflexible ligands can exhibit polymorphism [23–27]. Based uponvarious conformations of flexible ligand, such complex can achievedifferent type of structures and consequently polymorphism arises.But, for robust metal–organic complexes containing rigid ligands,polymorphism is rare to find [28–32].
In this endeavor, we are going to report that the robust complex[La(1,10-phen)2(NO3)3] containing rigid 1,10-phenanthroline andnitrate ligands undergoes thermally induced structural transfor-mation between its two polymorphic phases. Single crystal struc-ture analyses indicate the complex changes its space group fromP21/c to C2/c upon heating from 100 K to 293 K. Low temperatureDSC analysis indicates that this reversible structural conversionoccurs at �248 K.
Experimental
Materials and method
All the chemicals used were of A.R. grade. The solvents andchemicals were purified and dried before use by standardprocedures. La(NO3)3, 6H2O (Merck) and 1,l0-phenanthroline(1,10-phen) (Merck) were used as received. Elemental analysis(C, H, N) were carried out using a Perkin–Elmer 240C elementalanalyzer. The thermal analyses were carried out using a MettlerToledo TGA–DTA 85 thermal analyzer under a flow of N2 (30 ml/
min). The sample was heated at a rate of 10 �C min�1 with inertalumina as a reference. The electronic spectra were recorded byShimadzu UV-2401PC spectrophotometers and IR spectra weremeasured on Nicolet Impact 410 spectrometer between 400 and4000 cm�1, using the KBr pellet method. Low temperature andhigh temperature DSC analyses were carried out using DSC 204F1, NETZSCH at nitrogen atmosphere. The sample was first cooledfrom room temperature to �80 �C and followed by heating to roomtemperature at a rate of 10 �C min�1.
Synthesis of complex [La(1,10-phen)2(NO3)3]
5 ml aqueous solution of La(NO3)3�6H2O (1.08 g, 0.0025 mol)was added drop-wise to 5 ml methanolic solution of 1,10-phen(0.90 g, 0.005 mol). The resulting solution was filtered off. Colorlesssingle crystals started to appear and were collected by filtrationand dried. Fig. S1 depicts image of the crystal recorded by polariz-ing microscope. Anal. calc. (%) for C24H16LaN7O9: C: 42.06; H: 2.35;N: 14.31. Found (%): C: 42.02; H: 2.39; N: 14.36. IR (KBr, cm�1):33507br, 3062vw, 2836vw, 1625s, 1592s, 1575s, 1470m, 1385s,1292m, 1102m, 1027w, 843m, 730w, 637w.
Crystallographic data collection and refinement
Suitable single crystal of the complex was mounted on a BrukerSMART diffractometer equipped with a graphite monochromatorand Mo Ka (k = 0.71073 ÅA
0
) radiation. The single crystal data ofthe complex have been recorded at 100 K and 293 K. The structureswere solved by Patterson method using the SHELXS97 software.Subsequent difference Fourier synthesis and least-squarerefinement revealed the positions of the non-hydrogen atoms.Non-hydrogen atoms were refined with independent anisotropicdisplacement parameters. Hydrogen atoms were placed in ideal-ized positions and their displacement parameters were fixed tobe 1.2 times larger than those of the attached non-hydrogen atom.Successful convergence was indicated by the maximum shift/errorof 0.001 for the last cycle of the least squares refinement. All calcu-lations were carried out using SHELXS 97 [33], SHELXL 97 [34],PLATON 99 [35], ORTEP-32 [36] and WinGX software Ver-1.64[37]. Data collection, structure refinement parameters and crystal-lographic data are given in Table 1. Selected coordination bondlengths, bond angles and non-covalent interaction parameters arepresented in Tables S1–S6.
Computational details
Full geometry optimization of the structures obtained at 100 Kand 293 K were carried out in singlet state using Gaussian 09 soft-ware package [38]. For this particular purpose the analytical gradi-ent methods of DFT with Becke’s three parameters (B3) exchangefunctional together with the Lee–Yang–Parr (LYP) non-local corre-lation functional (symbolized as B3LYP) was employed. We havechosen LANL2DZ basis set with effective core potential and theHOMO–LUMO energy gap has been determined at the same level.
Result and discussion
Crystal structure description of the complex at 100 K
According to the results of X-ray single crystal structural study,the complex has monoclinic structure with P21/c space group at100 K. Each asymmetric unit consists of one La3+ ion, three nitrateanions and two 1,10-phenanthroline moieties, as shown in Fig. 1.Each lanthanide ion is 10 coordinated. For La1(III) ion, four N donoratoms (N1, N2, N3 and N4) of two different phen ligands, six

Table 1Comparative crystal data and details of the structure determination of the complex at100 and 293 K.
100 K 293 K
Crystal dataFormula C24H16LaN7O9 C24H16LaN7O9
Color Colorless ColorlessFormula weight 685.35 685.35Crystal system Monoclinic MonoclinicSpace group P21/c C2/ca (Å) 11.125(4) 11.2219(3)b (Å) 17.816(6) 18.1758(4)c (Å) 15.461(4) 13.1333(4)b (�) 124.719(17) 100.394(1)V (Å3) 2518.8(15) 2634.80(12)Z 4 4D(calc) (g/cm3) 1.807 1.728l(Mo Ka) (/mm) 1.765 1.688F(000) 1352 1352
Data collectionTemperature (K) 100 293Radiation (Å) Mo Ka 0.71073 Mo Ka 0.71073Theta min–max (�) 2.0, 28.5 2.2, 26.5Dataset �14:8; �22:23;
�18:20�14:14; �22:22;�16:16
Tot. 15854 10362Uniq. data 6107 2749R (int) 0.062 0.070Observed data [I > 2.0
sigma(I)]3972 2689
RefinementNref 6107 2749Npar 370 187R 0.0482 0.0624wR2 0.0929 0.1535S 0.82 1.09Max. and av. shift/error 0.00, 0.00 0.00, 0.00Min. and max. resd. dens.
(e/Å3)�0.99, 1.75 �1.96, 2.22
528 R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533
oxygen atoms (O1, O2, O4, O5, O7 and O9) of three different nitrateions (two from each nitrate) fulfill the coordination environment.All the three nitrates show l�2 chelating mode through two donoroxygen atoms while one oxygen atom of each nitrate remainsuncoordinated. Some selected coordination bond lengths and bondangles are given in Table S1. La–O and La–N bond lengths vary inthe range of 2.562(4)–2.605(5) Å and 2.630(4)–2.682(4) Å respec-tively. These values agree very well with the 10-coordinated La–O and La–N distances of the earlier reported complexes [39,40].Monomeric units are further connected by supramolecular interac-tions to form 3D supramolecular structure.
Supramolecular structure of the complex at 100 K
The packing analysis of the complex at 100 K indicates that 3Dsupramolecular structure has been formed by p� � �p, lone pair� � �pand C–H� � �O hydrogen bonding interactions among the differentmonomeric units. The pyridyl ring R1 (N1–C1–C2–C3–C4–C5)interacts (p� � �p) with another pyridyl ring R2 (N4–C21–C20–C22–C23–C24) at �1 + x, 3/2 � y, �1/2 + z and in this way mono-meric units are connected to form 1D supramolecular chain alongthe crystallographic c-axis, Fig. S2. The distance between centroidsis 3.608(4) Å and the dihedral angle is 1.2(3)�. Hydrogen bondinginteractions C19–H19� � �O7 and C22–H22� � �O7 further stabilizethe 1D supramolecular chains along c-axis. These 1D chains arefurther connected by O3� � �R2 and O9� � �R1 lone pair� � �p interac-tions. Oxygen atom O3 interacts with R2 pyridyl ring at x, 3/2 � y, �1/2 + z and oxygen atom O9 interacts with R1 pyridyl ringat x, 3/2 � y, 1/2 + z to form 2D supramolecular sheets in the ac-plane, Fig. 2. The hydrogen bonding interactions C15–H15� � �O2
and C7–H7� � �O9 along b-axis augment the 2D sheets into a 3Dsupramolecular structure, Fig. S3. Along the crystallographic b-axis, two supramolecular helices are formed. All p� � �p, lonepair� � �p and hydrogen bonding interactions parameters are pre-sented in Tables S2 and S3.
Crystal structure description of the complex at 293 K
Single crystal structure of the complex at 293 K was previouslydescribed in detail in the article of Frechette et al. [40]. At 293 K,the complex crystallizes in C2/c space group. Each asymmetric unitconsists of 0.5 La3+ ions, 1.5 nitrate ions and one 1,10-phenanthro-line moiety. It is noteworthy that the asymmetric unit of the com-plex at 293 K is half of the corresponding asymmetric unit at 100 K.The dihedral angle between two 1,10-phenanthroline rings in a[La(1,10-phen)2(NO3)3] molecule is 39.68� at 100 K and 41.80� at293 K. And also at 293 K, the nitrate groups are in a perfect sym-metric arrangement while at 100 K one nitrate group is tiltedand this nitrate group is responsible for breaking of symmetry at100 K structure. Thus, the molecular symmetry of the complex ischanged and consequently the supramolecular structure becomesdifferent. Some selected coordination bond lengths and bondangles of the complex at 293 K are given in Table S4.
In the previously reported complex [39], La–O bond distancesare 2.580(3), 2.611(3) and 2.599(3) Å and La–N bond distances are2.701(3) and 2.646(3) Å but in our case La–O bond distances are2.602(6), 2.586(6) and 2.554(8) Å and La–N bond distancesare 2.645(5) and 2.718(6) Å. So there is a little change in bonddistance parameters.
Supramolecular structure of the complex at 293 K
The packing analysis of the complex at 293 K shows that 3Dsupramolecular structure has been formed by p� � �p, lone pair� � �pand C–H� � �O hydrogen bonding interactions among differentmonomeric units. The pyridyl ring R1 (N3–C1–C2–C3–C4–C5)interacts (p� � �p) with another pyridyl ring R1 (N3–C1–C2–C3–C4–C5) at 1/2 � x, 1/2 � y, 1�z and in this way monomeric unitsare connected to form 1D supramolecular chain along a-axis,Fig. S4. The distance between centroids is 3.724(4) Å and the dihe-dral angle is 0�. These 1D chains are now connected by O3� � �R2lone pair� � �p interactions. Oxygen atom O3 interacts with R2(N4–C6–C7–C8–C9–C10) pyridyl rings at position to form 2Dsupramolecular sheets in the ac-plane, Fig. 3. The hydrogen bond-ing interactions C2–H2� � �O5 and C8–H8� � �O2 augment the 2Dsheets into 3D supramolecular structure, Fig. S2. Along the crystal-lographic b-axis, two supramolecular helices are formed. All p� � �p,lone pair� � �p and hydrogen bonding interactions parameters arelisted in Tables S5 and S6.
Helicity with chirality/achirality and polymorphism
Different crystalline forms containing identical compositionsare called polymorphs. Structural analyses of the complex at differ-ent temperatures show that the complex [La(1,10-phen)2(NO3)3]undergoes thermally induced structural transformation from onesingle crystalline phase to another phase without any change inchemical composition. Thus, it can be said that the complex under-goes structural transformation between its two polymorphicphases i.e. both forms are polymorphic in nature.
Here, we describe the supramolecular structure of these twopolymorphic phases in another way. PLATON study indicates thatat 100 K the complex has no molecular symmetry (C1) whereasat 293 K it has C2 molecular symmetry. This difference arises dueto dynamic movement of the ligands in response to externalstimuli (thermal energy). So, due to this transformation,
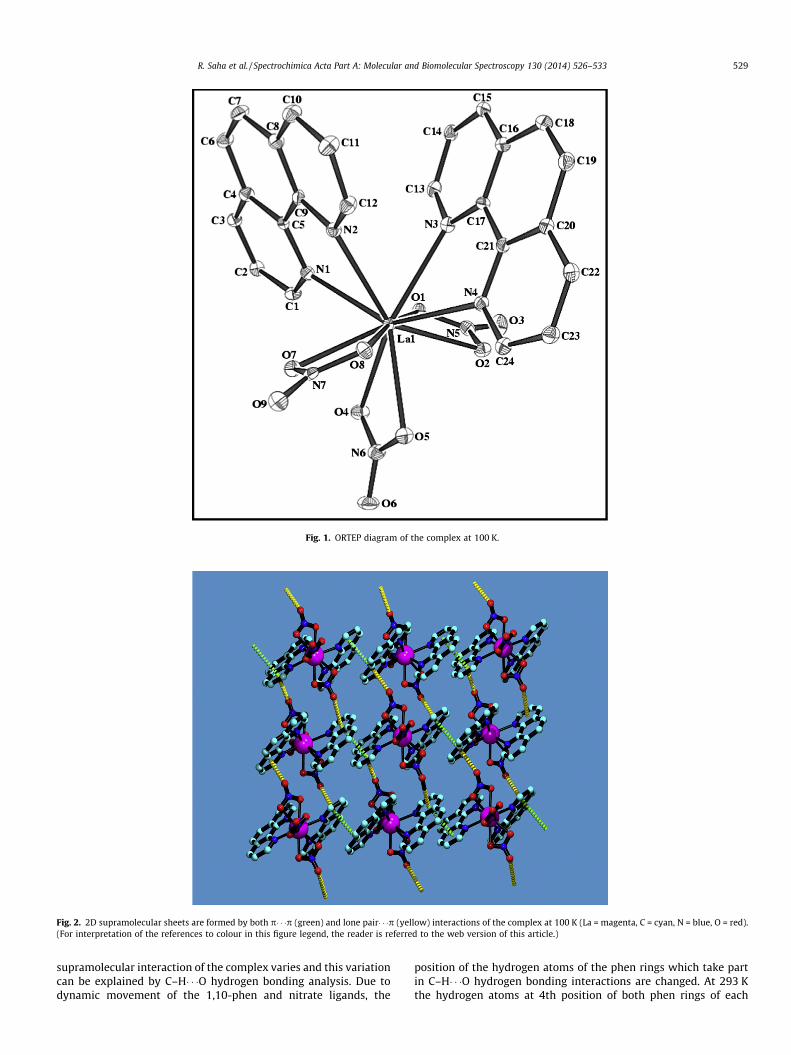
Fig. 1. ORTEP diagram of the complex at 100 K.
Fig. 2. 2D supramolecular sheets are formed by both p� � �p (green) and lone pair� � �p (yellow) interactions of the complex at 100 K (La = magenta, C = cyan, N = blue, O = red).(For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533 529
supramolecular interaction of the complex varies and this variationcan be explained by C–H� � �O hydrogen bonding analysis. Due todynamic movement of the 1,10-phen and nitrate ligands, the
position of the hydrogen atoms of the phen rings which take partin C–H� � �O hydrogen bonding interactions are changed. At 293 Kthe hydrogen atoms at 4th position of both phen rings of each

Fig. 3. The supramolecular 2D sheet formed by p� � �p (magenta) and lone pair� � �p (yellow) interactions within the ac-plane of the complex at 293 K (La = orange, C = cyan,N = blue, O = red). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
530 R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533
molecular unit take part in hydrogen bonding interactions while at100 K (for the lower symmetric structure) hydrogen atoms at dif-ferent positions of two different phen rings (4th position of onephen ring and 5th position of another phen ring) of each molecularunit take part in hydrogen bonding interactions, Fig. 4. Thesehydrogen bonding interactions take part in supramolecular helixformation in both cases. However, the respective distance of thehydrogen atom at 4th position of the phen ring is too long(>2.6 Å) for C–H� � �O hydrogen bonding at 100 K.
The optimized molecular confirmations of the complexes asestablished by DFT/B3LYP method agree well with those obtainedfrom X-ray structure analyses. Selected optimized bond lengthsand angles are given in Tables S7–S10. The small amount of devia-tion is observed between the calculated and experimentallyobserved bond lengths and bond angles. These small changes ofgeometrical parameters are expected because of the fact that thetheoretical calculations have been carried out with isolated mole-cules in the gaseous phase whereas the experimental values corre-spond to molecules in the crystalline state.
Fig. 4. Hydrogen bonding SBU of both forms, A = SBU of the complex at 100 K, B = SBUO = red). (For interpretation of the references to colour in this figure legend, the reader
The highest occupied molecular orbital (HOMO) is concentratedon the metal and the nitrate ligands whereas the lowest unoccu-pied molecular orbital (LUMO) is concentrated on 1,10-phenligands which have p-bonding character. The contour plots ofHOMO and LUMO are shown in Fig. S14. The orbital energy calcu-lations reveal that EHOMO and ELUMO for the complex at 100 K are�7.1088 eV and �2.7256 eV while EHOMO and ELUMO for the com-plex at 293 K are �7.1097 eV and �2.7258 eV respectively. Thecontour plots of the frontier orbital show overlapping of p-electrondistribution functions of two phenanthroline ligands attached withlanthanum coordination center and due to such overlapping newlow lying energy level will appear. Thus in the complex low lyingorbital (LUMO acceptor-) of p-character is present in the interli-gand pockets of 1,10-phenanthroline ligands coordinated withthe metal center. Therefore the system acts not only as a p-electrondonor, but also as a p-electron acceptor towards both the coordina-tion centers and other molecules [41,42]. Thus, because of thecombined effect of hydrogen bonding and p-electron distributionfunctions of the whole system, the complex [La(1,10-phen)2
of the complex at 293 K, C = 1,10-phenanthroline (La = orange, C = cyan, N = blue,is referred to the web version of this article.)
R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533 531
(NO3)3] undergoes thermally induced structural transformationfrom one single crystalline phase to another phase without anychange in chemical composition.
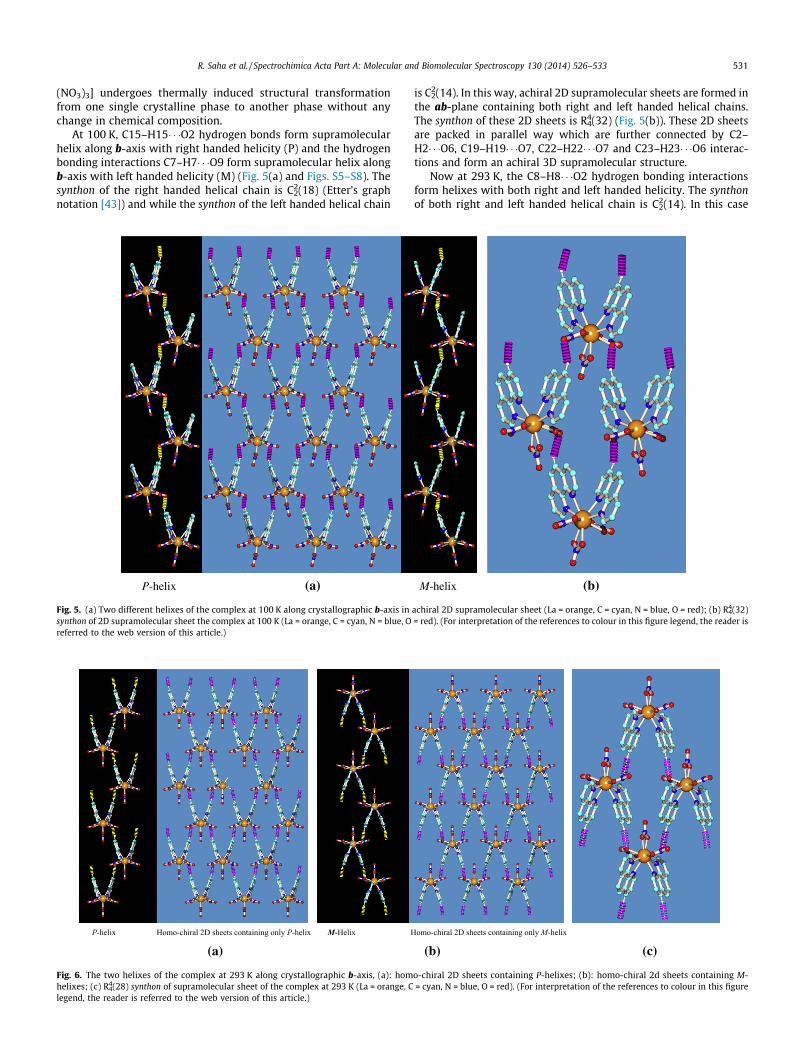
At 100 K, C15–H15� � �O2 hydrogen bonds form supramolecularhelix along b-axis with right handed helicity (P) and the hydrogenbonding interactions C7–H7� � �O9 form supramolecular helix alongb-axis with left handed helicity (M) (Fig. 5(a) and Figs. S5–S8). Thesynthon of the right handed helical chain is C2
2(18) (Etter’s graphnotation [43]) and while the synthon of the left handed helical chain
(a)
Fig. 5. (a) Two different helixes of the complex at 100 K along crystallographic b-axis insynthon of 2D supramolecular sheet the complex at 100 K (La = orange, C = cyan, N = blue, Oreferred to the web version of this article.)
(a)
Fig. 6. The two helixes of the complex at 293 K along crystallographic b-axis, (a): homhelixes; (c) R4
4(28) synthon of supramolecular sheet of the complex at 293 K (La = orange, Clegend, the reader is referred to the web version of this article.)
is C22(14). In this way, achiral 2D supramolecular sheets are formed in
the ab-plane containing both right and left handed helical chains.The synthon of these 2D sheets is R4
4(32) (Fig. 5(b)). These 2D sheetsare packed in parallel way which are further connected by C2–H2� � �O6, C19–H19� � �O7, C22–H22� � �O7 and C23–H23� � �O6 interac-tions and form an achiral 3D supramolecular structure.
Now at 293 K, the C8–H8� � �O2 hydrogen bonding interactionsform helixes with both right and left handed helicity. The synthonof both right and left handed helical chain is C2
2(14). In this case
(b)
achiral 2D supramolecular sheet (La = orange, C = cyan, N = blue, O = red); (b) R44(32)
= red). (For interpretation of the references to colour in this figure legend, the reader is
(b) (c)
o-chiral 2D sheets containing P-helixes; (b): homo-chiral 2d sheets containing M-= cyan, N = blue, O = red). (For interpretation of the references to colour in this figure

Fig. 8. Photoluminescence spectra of the complex in (a) solid state and (b) solutionstate.
532 R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533
homo-chiral 2D sheets are formed alternatively; one type of 2Dsheets contain only right handed helixes while the other type con-tains only left handed helixes (Fig. 6(a) and (b) and Figs. S9 andS10) and the 2D homochiral sheets are packed in anti-parallel way.The synthon of these 2D sheets is R4
4(28), Fig. 6(c). These homo-chiral2D supramolecular sheets with opposite chirality are connected byC2–H2� � �O5 hydrogen bonding interactions and develop an achiral3D supramolecular structure.
PXRD, DSC and thermal analysis
Phase purity of the complex was determined by collecting thePXRD data at room temperature, Fig. S11. In order to determinethe phase transition temperature of the complex, low temperatureDSC analysis was carried out (Fig. 7). During cooling, the curveshows an exothermic peak at 243 K and the correspondingenthalpy change is �0.2634 J/g. The heating cycle shows an endo-thermic peak at 248 K and the associated enthalpy change is0.2651 J/g. Therefore, the changes in enthalpy of the complex inboth heating and cooling cycles are nearly equal to each other.So, the transformation occurs at �246 K and it is reversible in nat-ure. Now, high temperature DSC analysis shows that any furtherstructural transformation does not occur at higher temperaturerange and the complex melts at 318 �C, Fig. S12.
Thermogravimetric analysis shows that the complex is stableup to 340 �C, Fig. S13. A large mass loss took place between 340–390 �C. It is clear from thermal plot that the mass loss up to390 �C is 53.17% (52.57% calculated) corresponds to two 1,10-phe-nanthroline moieties. And weight loss up to 520 �C corresponds todecomposition of nitrate ions leaving La2O3 as residue.
Electronic and luminescence property of both complexes
The absorption spectrum of the complex was taken in both solidand solution (methanol). The absorption band of free phen(267 nm) remains unchanged in both solid and in methanolicsolution.
The emission spectrum of the compound was investigated inboth solid and solution at room-temperature. As the absence of4f states, no ligand–lanthanide ion transition can occur in this caseand emission from the ligand triplet states is detected as verybroad bands in the near UV or visible region with maxima at about373 nm in the complex in solid state (Fig. 8(a)) whereas in solutionthe compound shows broad bands in the near UV or visible regionwith maxima at 368 nm (kex = 323 nm) with a new peak at 413 nm
Fig. 7. Low temperature DSC analysis of the complex.
(Fig. 8(b)). The emission maxima at 373 nm in solid state and368 nm in solution may due to p–p* transition between the twointermolecular aromatic p rings. The small peak around 413 nmin solution may be caused by the interaction of solvent. The solventmolecules associate with the complex and increase in the sizewhich causes the shift of the peak.
Conclusion
Here, we have reported that the complex [La(1,10-phen)2
(NO3)3] undergoes thermally induced SC–SC transformationbetween its two polymorphic phases due cooperative movementof the ligands. At 293 K, the complex shows C2/c space group withC2 molecular symmetry while it transforms into P21/c space groupwith C1 molecular symmetry at 100 K. The change in the molecularsymmetry differentiates the overall supramolecular structure. Thecomplex forms 2D achiral supramolecular sheets containing bothright and left handed helical chains at 100 K while at 293 K it formstwo different types of homochiral 2D supramolecular sheets, onetype contains only right handed helical chains and the other typecontains only left handed helical chains. The overall 3D supramo-lecular structures are achiral in both temperatures. Low tempera-ture DSC analysis corroborates with the finding and indicatesthat this transformation is reversible in nature. Presence of p-ligand allows the complex to be a fluorescence material.
Finally, we have presented a distinct case of thermally inducedSC–SC transformation of a robust inorganic complex containingrigid ligands leading to polymorphism. The complex exhibit ther-mally induced structural transformation from one single crystal-line phase to another phase due to the combined effect of

R. Saha et al. / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 526–533 533
hydrogen bonding and p-electron distribution functions of thewhole system. Further, it has been established that molecular sym-metry plays the key role in determining the overall supramolecularstructure of the individual phases. So, it can be concluded thatthermally triggered collective and cooperative movement ofligands changes the molecular symmetry as well as the supramo-lecular interactions that results into polymorphism in this robustcomplex.
Acknowledgment
This paper is dedicated to late Dr. Golam Mostafa, Dept. of Phys-ics, Jadavpur University. R.S. acknowledges financial support fromthe Council of Scientific and Industrial Research (CSIR), New Delhiunder the SRF program (09/096(0565)2008-EMR-I). We gratefullyacknowledge the financial support granted by Department of Sci-ence and Technology (DST), Govt. of India through PURSE and FISTprogram to Physics department, JU.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.saa.2014.04.032.
References
[1] M. Albrecht, M. Lutz, A.L. Spek, G. van Koten, Nature 406 (2000) 970–974.[2] L.J. Barbour, Aust. J. Chem. 59 (2006) 595–596.[3] K. Nagayoshi, M.K. Kabir, H. Tobita, K. Honda, M. Kawahara, M. Katada, K.
Adachi, H. Nishikawa, I. Ikemoto, H. Kumagai, Y. Hosokoshi, K. Inoue, S.Kitagawa, S. Kawata, J. Am. Chem. Soc. 125 (2003) 221–232.
[4] K. Uemura, S. Kitagawa, K. Fukui, K. Saito, J. Am. Chem. Soc. 126 (2004) 3817–3828.
[5] C. Massera, M. Melegari, E. Kalenius, F. Ugozzoli, E. Dalcanale, Chem. Eur. J. 17(2011) 3064–3068.
[6] X. Cheng, W. Zhang, X. Chen, J. Am. Chem. Soc. 129 (2007) 15738–15739.[7] A. Aslani, A. Morsali, M. Zeller, Dalton Trans. (2008) 5173–5177.[8] A.M.P. Peedikakkal, J.J. Vittal, Cryst. Growth Des. 11 (2011) 4696–4703.[9] S.M. Mobin, A.K. Srivastava, P. Mathur, G.K. Lahiri, Inorg. Chem. 48 (2009)
4652–4654.[10] H. Sadeghzadeh, A. Morsali, Inorg. Chem. 48 (2009) 10871–10873.[11] Y. Zhang, T. Liu, S. Kanegawa, O. Sato, J. Am. Chem. Soc. 131 (2009) 7942–7943.[12] S.K. Ghosh, W. Kaneko, D. Kiriya, M. Ohba, S. Kitagawa, Angew. Chem. Int. Ed.
47 (2008) 8843–8847.[13] Q. Chu, D.C. Swenson, L.R. MacGillivray, Angew. Chem. Int. Ed. 44 (2005) 3569–
3572.
[14] C. Hu, U. Englert, Angew. Chem. Int. Ed. 44 (2005) 2281–2283.[15] J. Dunitz, J. Bernstein, Acc. Chem. Res. 28 (1995) 193–200.[16] D. Braga, F. Grepioni, G.R. Desiraju, Chem. Rev. 98 (1998) 1375–1405.[17] S.L. Price, Acc. Chem. Res. 42 (1) (2009) 117–126.[18] A. Kalliokoski, P.J. Neuvonen, M. Niemi, Basic Clin. Pharm. Toxicol. 107 (4)
(2010) 775–781.[19] B.M. Foxman, P.L. Goldberg, H. Mazurek, Inorg. Chem. 20 (1981) 4368–4375.[20] B.L. Bennett, D.M. Roddick, Inorg. Chem. 35 (1996) 4703–4707.[21] T.L. Hennigar, D.C. MacQuarrie, P. Losier, R.D. Rogers, M.J. Zaworotko, Angew.
Chem. Int. Ed. 36 (1997) 972–973.[22] D. O’Connell, J.C. Patterson, T.R. Spalding, G. Ferguson, J.F. Gallagher, Y.-W. Li,
J.D. Kennedy, R. Macías, M. Thornton-Pett, J. Holub, J. Chem. Soc., Dalton Trans.(1996) 3323–3333.
[23] F. Dai, H. He, D. Sun, Inorg. Chem. 48 (11) (2009) 4613–4615.[24] P. Maniam, N. Stock, Inorg. Chem. 50 (11) (2011) 5085–5097.[25] A. Gunay, W.W. Brennessel, W.D. Jones, Acta, Cryst. E64 (2008) m454.[26] S. Grimme, J.-P. Djukic, Inorg. Chem. 50 (6) (2011) 2619–2628.[27] D. Braga, F. Grepioni, Chem. Soc. Rev. 29 (2000) 229–238.[28] N. Masciocchi, S. Bruni, E. Cariati, F. Cariati, S. Galli, A. Siron, Inorg. Chem. 40
(23) (2001) 5897–5905.[29] D.V. Soldatov, A.S. Zanina, G.D. Enright, C.I. Ratcliffe, J.A. Ripmeester, Cryst.
Growth Des. 3 (6) (2003) 1005–1013.[30] D.V. Soldatov, G.D. Enright, J.A. Ripmeester, Cryst. Growth Des. 4 (6) (2004)
1185–1194.[31] D.M. Shin, I.S. Lee, Y.K. Chung, M.S. Lah, Chem. Commun. (2003) 1036–1037.[32] A.M. Goforth, K. Gerth, M.D. Smith, S. Shotwell, U.H.F. Bunz, H-C.Z. Loye, Solid
State Sci. 7 (9) (2005) 1083–1095.[33] G.M Sheldrick, SHELXS 97, Program for Structure Solution, University of
Göttingen, Germany, 1997.[34] G.M Sheldrick, SHELXL 97, Program for Crystal Structure Refinement,
University of Göttingen, Germany, 1997.[35] A.L. Spek, PLATON, molecular geometry program, J. Appl. Crystallogr. 36 (2003)
7–13.[36] L.J. Farrugia, J. Appl. Crystallogr. 30 (1997) 565.[37] L.J. Farrugia, J. Appl. Crystallogr. 32 (1999) 837–838.[38] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato,X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M.Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro,M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, T. Keith, R.Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J.Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken,C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R.Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski,G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B.Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision C.01,Gaussian Inc, Wallingford CT, 2010.
[39] Z.P. Ji, R.D. Rogers, J. Chem. Cryst. 26 (1996) 573–577.[40] M. Frechette, I.R. Butler, R. Hynes, C. Detellier, Inorg. Chem. 31 (1992) 1650–
1656.[41] W. Linert, V. Gutmann, Z. Phys. Chem. N.F. 142 (1984) 221–238.[42] W. Linert, V. Gutmann, Coord. Chem. Rev. 117 (1992) 159–183.[43] M.C. Etter, J.C. MacDonald, J. Bernstein, Acta Cryst. B 46 (1990) 256–262.
Copyright © 2022 FDOKUMEN




















