
Thermal Treatment of Flame‐Synthesized Amorphous Tricalcium Phosphate Nanoparticles
9
Thermal Treatment of Flame-Synthesized Amorphous Tricalcium Phosphate Nanoparticles Nicola Do¨ belin, w,z Tobias J. Brunner, y,ww Wendelin J. Stark, y Martin Fisch, z Egle Conforto, J and Marc Bohner z z RMS Foundation, CH-2544 Bettlach, Switzerland y Department of Chemistry and Applied Biosciences, Institute for Chemical and Bioengineering, ETH Zu¨ rich, CH-8093 Zu¨ rich, Switzerland z Institut fu¨ r Geologie, Universita¨ t Bern, CH-3012 Bern, Switzerland J Fe´ de´ ration de Recherche en Environnement pour le De´ veloppement Durable (FR-EDD), FR CNRS 3097, Centre Commun d’Analyses, F-17071 La Rochelle Cedex 9, France Flame-spray synthesis was used to produce nanoparticulate X-ray amorphous tricalcium phosphate. Upon heating, the ma- terial crystallized at temperatures between 5251 and 6001C, yielding a-tricalcium phosphate (TCP) with minor amounts of b-TCP and hydroxyapatite (HA). Further heating induced a gradual transformation of a- to b-TCP accompanied by crystal- lite growth and particle fusion. a-TCP was completely trans- formed into b-TCP at 9501C. The high-temperature polymorph a 0 -TCP was not observed. In transmission electron microscopy and selected area electron diffraction analyses, the raw material appeared completely amorphous and samples heated previously to 6001 and 7001C comprised single-crystalline particles of a-, b-TCP, and HA. The results of this study demonstrate that nanoparticulate a-TCP can be obtained from flame-spray-syn- thesized amorphous nanoparticles at temperatures where sinte- ring effects such as particle growth and fusion are moderate, if not negligible. I. Introduction S INCE several decades calcium phosphate (CaP) ceramics have been a popular synthetic alternative to bone autografts, allo- grafts, and xenografts from natural sources in the reconstruction of large bone defects. 1–4 Some of these synthetic ceramics show excellent biocompatibility due to their similarity to the mineral phase of bone. The degradation rate after implantation depends on the solubility of the CaP phases in the human body, 5 and on the physical properties of the implant such as the degree of po- rosity, pore diameter, pore interconnectivity, specific surface area (SSA), and particle size. 6 The biological performance and resorption characteristics of bone substituting CaP products can thus be tailored by optimizing these parameters, 7 and by com- bining phases with favorable properties. CaP materials are obtained from various processes including: (a) precipitation from aqueous solutions, (b) cement reactions of solid phases with an aqueous component, and (c) solid-state de- hydration reactions or phase transformations at high tempera- ture. 8 Several physical properties of the synthesis products depend on the formation conditions: phases synthesized by low-temperature processes (a) and (b) are less crystalline and possess a higher SSA than phases obtained by high-temperature processes (c). In case of high-temperature formation, on the other hand, sinter effects lead to a reduction of the SSA, an in- creased particle growth, a high degree of crystallinity, and dens- ification and shrinkage by particle fusion. These sinter processes often result in hard and brittle solid materials. b-tricalcium phosphate (b-TCP; Ca 3 (PO 4 ) 2 ) is one of the most prominent CaP phases in modern implants. It is classically obtained from various precursor materials with a Ca:P ratio of 1.50 by thermal treatment above 7501C. 9,10 Above 11201–11501C, it transforms into the high-temperature poly- morph a-TCP 11–14 by undergoing a reconstructive reversible phase transformation. 15 The reverse transformation to b-TCP can be suppressed by rapid cooling to room temperature, which maintains a-TCP as a metastable phase. In fact, it was shown that even at moderate cooling rates ( 21C/min) no reverse transformation from a- to b-TCP occurs. 11,14,15 Another less known high-temperature polymorph of a- and b-TCP was ob- served by optical microscopy 16 and neutron diffraction 17 at temperatures above 14301C. The latter authors refined the crys- tal structure; however, to our best knowledge no atomic coor- dinates have been published to date. The phase has been called ‘‘super-a-TCP,’’ 16 a-TCP, 12 or more recently a 0 -TCP. 17 This phase could not be maintained in a metastable form at room temperature by quenching. Metastable a-TCP is soluble and reacts with water to give calcium-deficient hydroxyapatite (CDHA) at low tempera- tures. 18–20 Several bone cements have been developed based on this setting reaction. 21–23 Besides solubility-controlling chemical parameters such as pH and chemical composition of the liquid component, the kinetics of the hydration reaction depend on physical parameters such as the SSA and particle size of the solid component. 24–26 Being able to control these parameters allows to control the setting and handling properties of the cement without having to add retarding or accelerating agents. How- ever, due to the pronounced sintering effects (crystallite growth, particle fusion, reduction of the SSA) in the stability field of a-TCP above 11501C, it is not possible to obtain nanoparticu- late a-TCP with a high SSA by conventional high-temperature preparation route. Milling of larger particles is possible, but it further increases the enormous cost of a-TCP production and may require further thermal treatment. 27 A theory known as the Ostwald step rule 28 provides a solu- tion to this problem. It states that in the crystallization from a solution or of an amorphous solid phase, the least stable phase often occurs first, followed by transformations to thermody- namically more stable phases. Indeed, it was shown by Somrani et al. 29 that the crystalline phases of a precipitated amorphous TCP (ATCP) powder obtained after calcination in the range J. Ferreira—contributing editor This work was supported by the Gebert Ru¨ f Foundation under grant no. GRS-048/04. w Author to whom correspondence should be addressed. e-mail: nicola.doebelin@ rms-foundation.ch ww Present address: Biotronik AG, CH-8180 Bu¨ lach, Switzerland Manuscript No. 26512. Received July 21, 2009; approved April 17, 2010. J ournal J. Am. Ceram. Soc., 93 [10] 3455–3463 (2010) DOI: 10.1111/j.1551-2916.2010.03856.x r 2010 The American Ceramic Society 3455
-
Upload
rms-foundation -
Category
Documents
-
view
1 -
download
0
Transcript of Thermal Treatment of Flame‐Synthesized Amorphous Tricalcium Phosphate Nanoparticles

Thermal Treatment of Flame-Synthesized AmorphousTricalcium Phosphate Nanoparticles
Nicola Dobelin,w,z Tobias J. Brunner,y,wwWendelin J. Stark,yMartin Fisch,z Egle Conforto,J andMarc Bohnerz
zRMS Foundation, CH-2544 Bettlach, Switzerland
yDepartment of Chemistry and Applied Biosciences, Institute for Chemical and Bioengineering, ETH Zurich,CH-8093 Zurich, Switzerland
zInstitut fur Geologie, Universitat Bern, CH-3012 Bern, Switzerland
JFederation de Recherche en Environnement pour le Developpement Durable (FR-EDD), FR CNRS 3097,Centre Commun d’Analyses, F-17071 La Rochelle Cedex 9, France
Flame-spray synthesis was used to produce nanoparticulateX-ray amorphous tricalcium phosphate. Upon heating, the ma-terial crystallized at temperatures between 5251 and 6001C,yielding a-tricalcium phosphate (TCP) with minor amounts ofb-TCP and hydroxyapatite (HA). Further heating induced agradual transformation of a- to b-TCP accompanied by crystal-lite growth and particle fusion. a-TCP was completely trans-formed into b-TCP at 9501C. The high-temperature polymorpha0-TCP was not observed. In transmission electron microscopyand selected area electron diffraction analyses, the raw materialappeared completely amorphous and samples heated previouslyto 6001 and 7001C comprised single-crystalline particles of a- ,b-TCP, and HA. The results of this study demonstrate thatnanoparticulate a-TCP can be obtained from flame-spray-syn-thesized amorphous nanoparticles at temperatures where sinte-ring effects such as particle growth and fusion are moderate, ifnot negligible.
I. Introduction
SINCE several decades calcium phosphate (CaP) ceramics havebeen a popular synthetic alternative to bone autografts, allo-
grafts, and xenografts from natural sources in the reconstructionof large bone defects.1–4 Some of these synthetic ceramics showexcellent biocompatibility due to their similarity to the mineralphase of bone. The degradation rate after implantation dependson the solubility of the CaP phases in the human body,5 and onthe physical properties of the implant such as the degree of po-rosity, pore diameter, pore interconnectivity, specific surfacearea (SSA), and particle size.6 The biological performance andresorption characteristics of bone substituting CaP products canthus be tailored by optimizing these parameters,7 and by com-bining phases with favorable properties.
CaP materials are obtained from various processes including:(a) precipitation from aqueous solutions, (b) cement reactions ofsolid phases with an aqueous component, and (c) solid-state de-hydration reactions or phase transformations at high tempera-ture.8 Several physical properties of the synthesis productsdepend on the formation conditions: phases synthesized bylow-temperature processes (a) and (b) are less crystalline and
possess a higher SSA than phases obtained by high-temperatureprocesses (c). In case of high-temperature formation, on theother hand, sinter effects lead to a reduction of the SSA, an in-creased particle growth, a high degree of crystallinity, and dens-ification and shrinkage by particle fusion. These sinter processesoften result in hard and brittle solid materials.
b-tricalcium phosphate (b-TCP; Ca3(PO4)2) is one of the mostprominent CaP phases in modern implants. It is classicallyobtained from various precursor materials with a Ca:P ratioof 1.50 by thermal treatment above 7501C.9,10 Above11201–11501C, it transforms into the high-temperature poly-morph a-TCP11–14 by undergoing a reconstructive reversiblephase transformation.15 The reverse transformation to b-TCPcan be suppressed by rapid cooling to room temperature, whichmaintains a-TCP as a metastable phase. In fact, it was shownthat even at moderate cooling rates (� 21C/min) no reversetransformation from a- to b-TCP occurs.11,14,15 Another lessknown high-temperature polymorph of a- and b-TCP was ob-served by optical microscopy16 and neutron diffraction17 attemperatures above 14301C. The latter authors refined the crys-tal structure; however, to our best knowledge no atomic coor-dinates have been published to date. The phase has been called‘‘super-a-TCP,’’16 �a-TCP,12 or more recently a0-TCP.17 Thisphase could not be maintained in a metastable form at roomtemperature by quenching.
Metastable a-TCP is soluble and reacts with water to givecalcium-deficient hydroxyapatite (CDHA) at low tempera-tures.18–20 Several bone cements have been developed based onthis setting reaction.21–23 Besides solubility-controlling chemicalparameters such as pH and chemical composition of the liquidcomponent, the kinetics of the hydration reaction depend onphysical parameters such as the SSA and particle size of the solidcomponent.24–26 Being able to control these parameters allowsto control the setting and handling properties of the cementwithout having to add retarding or accelerating agents. How-ever, due to the pronounced sintering effects (crystallite growth,particle fusion, reduction of the SSA) in the stability field ofa-TCP above 11501C, it is not possible to obtain nanoparticu-late a-TCP with a high SSA by conventional high-temperaturepreparation route. Milling of larger particles is possible, but itfurther increases the enormous cost of a-TCP production andmay require further thermal treatment.27
A theory known as the Ostwald step rule28 provides a solu-tion to this problem. It states that in the crystallization from asolution or of an amorphous solid phase, the least stable phaseoften occurs first, followed by transformations to thermody-namically more stable phases. Indeed, it was shown by Somraniet al.29 that the crystalline phases of a precipitated amorphousTCP (ATCP) powder obtained after calcination in the range
J. Ferreira—contributing editor
This work was supported by the Gebert Ruf Foundation under grant no. GRS-048/04.wAuthor to whom correspondence should be addressed. e-mail: nicola.doebelin@
rms-foundation.chwwPresent address: Biotronik AG, CH-8180 Bulach, Switzerland
Manuscript No. 26512. Received July 21, 2009; approved April 17, 2010.
Journal
J. Am. Ceram. Soc., 93 [10] 3455–3463 (2010)
DOI: 10.1111/j.1551-2916.2010.03856.x
r 2010 The American Ceramic Society
3455

from 6001 to 8001C comprise more than 90 mol% a-TCP, eventhough the maximum temperature of the formation process wasfar below the stability field of a-TCP. However, Li et al.30
showed that the crystalline phase obtained after heating ofprecipitated amorphous TCP at 8001C depended on synthesisparameters, namely on the pH of the mother solution, and onthe aging time of the precipitate in the mother solution. Purea-TCP phase was obtained by heating precipitated amorphousTCP prepared at highly alkaline pH, and/or aged for a shorttime. Mildly alkaline pH of the mother solution, as well aslonger aging times, favored the direct formation of b-TCP in-stead. Their results demonstrate that short-range order and syn-thesis parameters of X-ray amorphous TCP affect the phaseevolution occurring at early stages of temperature-induced crys-tallization. The assumption that any amorphous TCP is boundto form metastable a-TCP as a first crystalline phase at lowtemperature appears to be not valid. According to Li et al.,30
amorphous TCP materials may possess different degrees andtypes of short-range structures, which predetermine the crystal-line phase occurring first after heat-induced crystallization. Thistheory could also explain why a0-TCP was so far never observedbelow 14501C, despite the prediction of the Ostwald step rule.
In the present study, in situ high-temperature X-ray powderdiffraction (XRD) was used to monitor the phase compositionand crystallite growth induced by thermal treatment up to10001C of a nanoparticulate amorphous TCP powder preparedby flame-spray synthesis.31 The preparation method of the ma-terial was vastly different from the precipitation methods fre-quently described in the literature. It was thus of interest toinvestigate how an amorphous TCP material formed at a veryhigh temperature in a water-free environment would crystallize,and whether a0-TCP could be obtained. With increasing particlegrowth and fusion at higher temperatures, particularly inthe stability field of a-TCP above 11501C, the material startsto lose its unique and outstanding features of nanosized weaklyagglomerated particles. The temperature range analyzed in thepresent study was thus restricted to temperatures up to 10001Cand we concentrated on the temperature of the initial crystalli-zation. Preliminary results of the study focusing on the occur-rence of a0-TCP,32 on variations of the Ca:P ratio,33 and on thereactivity of as-prepared phases34 have been published before. Inthis follow-up study, we provide a more in-depth discussion ofthe results. We also used scanning and transmission electronmicroscope techniques to provide a thorough characterizationof the material, in particular the phase composition, particlemorphology, and crystal domain size, in its amorphous andcrystalline states.
II. Experimental Procedure
ATCP were prepared by flame-spray synthesis as described byLoher et al.31 and Brunner et al.35 A liquid precursor solutionwas prepared from calcium hydroxide (Ph. Eur., Riedel-deHaen,Seelze, Germany) dissolved in 2-ethylhexanoic acid (technicalgrade, Soctech, Bucharest, Romania) and tributyl phosphate(Acros Organics, Geel, Belgium). The liquid mixture was fedthrough a capillary (0.4 mm in diameter) into a burning methane(1.13 L/min)/oxygen (2.4 L/min, both technical grade, Pan Gas,Dagmersellen, Switzerland) supporting flame at a rate of 10 mL/min using a gear-ring pump (HNP Mikrosysteme, Parchim,Germany). Oxygen (10 L/min) was used to disperse the liquidleaving the capillary. During flame-spray synthesis, the nano-particles form in a high-temperature environment (core part ofthe flame: 15001–20001C; residence time: 1–3 ms) and are rapidlyquenched by air entrainment from the surrounding. This syn-thesis procedure favors the formation of kinetically controlledphase distributions. The particles were separated from the gasphase using PTFE filters and subsequently sieved with 250 mmmesh sieves.
In situXRD data sets were collected using a powder diffracto-meter (X’Pert Pro MPD, Panalytical, Almelo, the Netherlands)
in y/y configuration using Ni-filtered CuKa radiation and a fixeddivergence slit of 1/81 opening. A heating chamber (HTK-1200,Anton Paar, Ostfildern, Germany) was attached to the gonio-meter. The samples were prepared in a ceramic sample holderspinning at approximately 1 rps. Data sets were collected from91–651 2y with a step size of 0.0161 2y and a counting time of 1 sper step at the following temperatures: 251, 5001, 5251, 5501,5751, 6001, 6501, 7001, 7501, 8001, 8501, 9001, 9501, 10001C, andagain at 251C. The heating rate was set to 11C/min for all heat-ing sequences, which was slow enough to prevent overshootingwithin the accuracy of the furnace control unit (711C). Duringdata collection, the temperature was held constant, and for thelast data acquisition the sample was cooled from 10001 to 251Cat 601C/min.
XRD patterns were analyzed by Rietveld refinement usingthe software FullProf.2k.36 Starting models for the crystal struc-tures were taken from Mathew et al.37 for a-TCP, Dickenset al.38 for b-TCP, and Sudarsanan and Young39 for hydrox-yapatite (HA). No additional crystalline phases were observed inthe diffraction patterns. The refinements were performed inthree steps, starting with vertical sample displacement, scale fac-tors for all phases, and sampled background points with linearinterpolation until convergence was reached. In the second step,the cell parameters of all phases were released for refinement,and in the last step isotropic peak broadening of all phases andone common overall isotropic displacement parameter (Boverall)were optimized. The instrument resolution function and peakasymmetry were determined with an ‘‘NIST SRM 660a—LineProfile LaB6’’ standard before the refinements of the sampledata sets. Peak shapes were modeled with a pseudo-Voigt func-tion accounting for peak asymmetry due to axial divergence.40
Average crystallite sizes were calculated from isotropic Gaussianpeak broadening using the Scherrer equation.
The most common method to determine X-ray amorphousfractions in a specimen is to add a known quantity of a crystal-line internal standard. However, at temperatures up to 10001C,solid-state reactions of the specimen with the standard powderare to be expected. Relative weight fractions WP of the crystal-line phase P, on the other hand, can be calculated from refinedparameters and chemical information of all phases as follows41:
WP ¼SPðZMVÞPPi SiðZMVÞi
(1)
where S is the Rietveld scale factor, Z the number of formulaunits per unit cell, M the molecular mass of the formula unit, Vthe unit cell volume, and i iterates over all crystalline phases. Theabsolute mass mP of each crystalline component in the specimenis proportional to
mP / SPðZMVÞP (2)
By making the reasonable assumptions that (i) the intensity ofthe primary beam and the device configuration were identicalfor all data sets, (ii) the total amount of powder in the specimenremained constant during the experiment, and (iii) all phasesinvolved, including the amorphous powder, have similar particlesizes and absorption coefficients, the dependency of SP could bereduced to the phase abundance and the overall atomic dis-placement parameter. The latter, as well as the unit cell volume,are temperature dependent. The value mP calculated from Eq.(2) for a constant amount of a phase P will thus show a slightlinear decrease with the increasing temperature.
In order to determine the amount of X-ray amorphous ma-terial in the specimen, a 100% crystalline reference was needed.As the sum of mP values of all crystalline phases remained con-stant between 8001 and 10001C, except for a steady decreasecaused by the temperature-dependent parameters, it was as-sumed that all amorphous material was crystallized below8001C. A linear regression through the sums of mP values
3456 Journal of the American Ceramic Society—Dobelin et al. Vol. 93, No. 10

between 8001 and 10001C thus served as references for 100%crystallinity for the calculation of absolute phase quantities.
Scanning electron micrographs (SEM) were recorded using aLEO 1530 Gemini (Zeiss/LEO, Oberkochen, Germany) aftersputtering the samples with B4 nm of platinum. Particle sizeswere measured manually by determining the diameter of 50 par-ticles per sample using the software Image Access Premium 7(Imagic, Glattbrugg, Switzerland).
For transmission electron microscopy (TEM) and selectedarea electron diffraction (SAED), one sample was used withoutthermal treatment, a second one was heated for 1 h at 6001C(11C/min), and a third one was heated for 1 h at 7001C (11C/min). Figures 5–7 were recorded using a TEM (JEM-2011,JEOL Ltd., Tokyo, Japan) operated at 200 kV in the bright-field mode with a resolution of 0.25 nm. The instrument wasequipped with a Link ISIS EDS system (Oxford Instruments,Abingdon, U.K.) and a Dualview CDD camera (Gatan France,Grandchamp, France). A double-tilt specimen holder was usedto spatially orient crystals in the SAED mode. This procedureallowed the identification of crystalline phases though the in-dexation of mono- and polycrystal diffractograms. CaP particlessuspended in ultra pure ethanol were submitted to ultra soundagitation for 15 min to separate particles forming agglomeratesand to obtain homogeneous dispersion. The suspensions werethen dropped on TEM grids covered with carbon films. Figure 8was recorded using a CM30 ST (LaB6 cathode, operatedat 300 kV, point resolution 0.20 nm), transmission electronmicroscope (Philips Electron Optics, Eindhoven, the Nether-lands). Particles were deposited onto a carbon foil supported ona copper grid.
III. Results
In XRD patterns, the material appeared X-ray amorphous up to5001C. First diffraction peaks were observed at 5251C (Fig. 1),and at 5501C the first refinable diffraction pattern was obtainedfor which the phase quantities and crystallite sizes could be de-termined despite considerable peak broadening. The dominantphase at early stages of the crystallization was identified asa-TCP, and the minor phases as b-TCP and HA (Fig. 2). Thehighest absolute amount of a-TCP was 69 wt% found at 6001C.Further heating induced a gradual transformation of a- intob-TCP and narrowing of the peak shape due to crystal domaingrowth. Above 9001C, no a-TCP could be detected. HA con-centrations converged to 7–8 wt% above 7501C, indicating aslight mismatch of the Ca:P ratio in the bulk material. Calcu-lation of absolute phase quantities showed that the degree of
crystallinity reached 52 wt% at 5501C, and 88 wt% at 5751C. At6001C and above, the material was 100% crystalline and noamorphous content was detectable.
Average crystallite sizes shown in Fig. 3 were calculated fromisotropic peak broadening. The broad peak shapes observed inthe 5501C X-ray diffraction pattern corresponded to an averagespherical crystallite size of 30–40 nm. This was slightly largerthan the values measured from SEM images before thermaltreatment (Fig. 4). With the increasing temperature, the crystal-lites grew, reaching 150 nm at 10001C and thus exceeding thenanorange (o100 nm). Particle growth was faster than crystallitegrowth, which became particularly pronounced above 8001C. Athigh temperatures, some reflections of the b-TCP pattern lostintensity due to excessive thermal vibration of some atomic sites.It was not attempted to refine individual atomic displacementparameters, instead higher RBragg values were accepted, as peakshape and phase quantity refinements did not appear to be neg-atively influenced.
TEM observations of the amorphous raw material confirmedthat the particles were originally amorphous (Fig. 5). However,they were very sensitive to the electron beam irradiation andstarted to crystallize after exposure of some seconds up to 1 min.
Fig. 1. In situ X-ray diffraction patterns and theoretical peak positionsfor a-tricalcium phosphate (TCP), b-TCP, and hydroxyapatite (HA).Just after crystallization, the main phase was a-TCP, followed by agradual transformation to b-TCP. The peak shift of the top pattern wascaused by the thermal contraction of the unit cell when cooled from10001C to room temperature.
Fig. 2. Absolute concentrations of crystalline phases as refined fromthe X-ray powder diffraction patterns. The pattern observed at 5251Ccould not be quantified. The degree of crystallinity reached 52 wt% at5501C and 100 wt% at 6001C. The standard deviation of the sum ofcrystalline phases is 1.5 wt% for three-phase compositions, and 1.0 wt%for two-phase compositions.
Fig. 3. Average crystallite sizes were refined from X-ray powderdiffraction (XRD) data by measuring isotropic peak broadening.Particle sizes were measured from scanning electron microscopy(SEM) images by averaging the diameters of 50 particles.
October 2010 Thermal Treatment of Flame-Synthesized ATCP Nanoparticles 3457

Bright-field images showed morphological transformationsfrom the amorphous spherical particles to the structured ones.In Fig. 6, we observed the corresponding electron diffractionpattern evolution from amorphous particles before irradiationto a high degree of crystallinity after irradiation for severalminutes.
The sample calcined at 6001C was crystalline, but tended tobecome amorphous under the electron beam. Electron diffrac-tion patterns of a- , b-TCP, and HA were observed (Fig. 7) forparticle ensembles, but no individual particles containing morethan one phase simultaneously were found.
IV. Discussion
(1) Phase Evolution
Despite the difference in the preparation route, the temperature-dependent phase evolution observed in our experiment generallyagrees with the results published in previous studies.29,31,34 Thefirst phases observed were a- , b-TCP, and HA. The a-TCPcontent was relatively high with an a:b-TCP ratio between 76:24
Fig. 4. Scanning electron microscopy images of the amorphous powderbefore thermal treatment showed spherical particles with an averagediameter of 28 nm (standard deviation5 4 nm, determined from 50particles).
Fig. 5. Bright-field transmission electron microscopy images of the amorphous sample: not irradiated (A), after irradiation during 1 s (B), 20 s (C), 40 s(D), and 1 min (E).
3458 Journal of the American Ceramic Society—Dobelin et al. Vol. 93, No. 10

and 85:15 from the beginning of crystallization up to 6501C(Table I). a0-TCP, which is less stable than a-TCP and shouldthus, according to the Ostwald step rule,28 occur first in thecrystallization sequence could not be observed in the presentstudy. The gradual transformation of a-TCP into b-TCP afterthe initial crystallization was expected according to thermody-namics and the Ostwald step rule. It represents the transitionfrom a metastable to a thermodynamically stable phase.
The high-temperature XRD setup used in this study, eventhough allowing in situ data collection at high temperatures, wasnot a real-time analysis technique. It showed the static phasecomposition at a given temperature, but no real-time informa-tion about ongoing reactions. In fact, the design of the exper-iment was chosen in a way that phase transformations primarilyoccurred between data acquisitions when the temperature wasincreased rather than during data collections when the temper-ature was kept constant. From the XRD data, we learned thata0-TCP does not occur as a metastable or stable phase in thecrystallization sequence of the amorphous flame-synthesizedTCP. But we cannot exclude that it occurred temporarily at
early stages of the crystallization, and if it does, a certain size ofcoherent crystal domains would still be required in order togenerate a diffraction pattern.
(2) Particle Growth
Before thermal treatment, the raw material was composed ofweakly connected spherical particles with a mean particle diam-eter of approximately 28 nm (Fig. 4). XRD data show that thepowder was transformed from a purely X-ray amorphous stateat 5001C into purely crystalline material with crystallite sizes ofapproximately 50 nm at 6001C (Fig. 3). At temperatures up to7001C, the crystallite sizes determined from isotropic peakbroadening were slightly larger than the particle sizes measuredfrom SEM images. Because this constellation has no physicalmeaning, it is rather an indication of a calibration error of thediffractometer resolution function, or an interpretation error ofthe SEM images. Considering the large number of parametersthat can influence the interpretation of isotropic peak broaden-ing (strain, anisotropic crystallite shapes), it is known and no
Fig. 6. Selected area electron diffraction patterns of the amorphous sample: not irradiated (A), after some seconds of irradiation (B), and particlesexposed to the electron beam for different (increasing) times until almost complete crystallization (C–F).
October 2010 Thermal Treatment of Flame-Synthesized ATCP Nanoparticles 3459

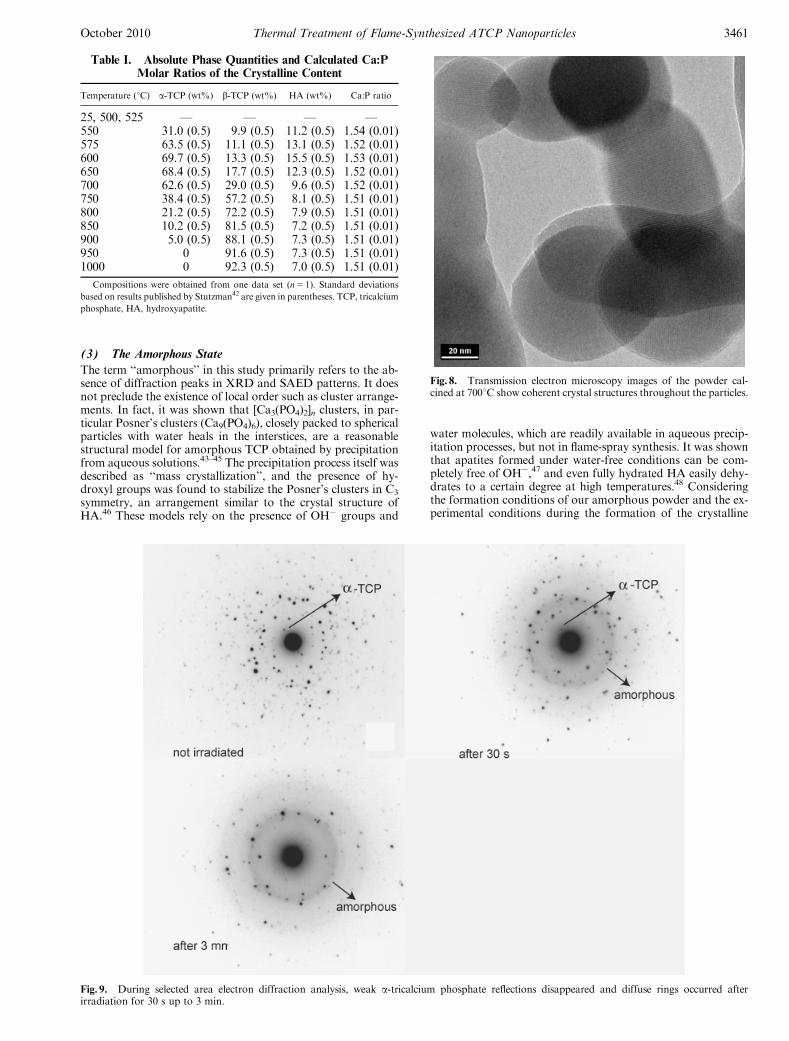
surprise that the calculated values obtained by the Scherrerequation do not reach the accuracy of optical measurements.The data, however, show that crystallite and particle sizes are inthe same range. In other words, just after crystallization theparticles were single crystals with a coherent crystal structure.TEM images of the 7001C data set (Fig. 8) confirmed this in-terpretation by showing coherent crystal domains throughoutthe particles, and SAED results supported the data by provingthe single-phase composition of the particles. With the increas-ing temperature, it became increasingly difficult to define the sizeof the particles in SEM images due to the formation of necksand particle intergrowth. As the particles agglomerated, the dis-crepancy between particle size and crystallite size increased, andso did the particle size distribution. The XRD data showed nosign of anisotropic crystal growth such as needles or platelets.
In a gradual phase transformation as it was observed in thisstudy, one would expect to find partially transformed particles.The fact that a- and b-TCP coexisted and transformations froma- to b-TCP occurred but no multiphase particles were found isremarkable, but it can be explained by new insights on theb-2a-TCP phase transformation recently published byCarrodeguas et al.15 These authors suggested that the transitionis reconstructive and requires a considerable amount of activa-tion energy to proceed in both directions. Our observationsconfirm that once the activation barrier is surpassed, the trans-formation proceeds throughout the particle too quickly to ap-pear as crystal domains of sub-particle size in XRD patterns orto be ‘‘frozen’’ in the samples used for ex situ TEM analysis.Hence, we only observed untransformed a-TCP or fully trans-formed b-TCP particles.
Fig. 7. Six examples of crystalline phases identified through selected area electron diffraction patterns in the sample calcined at 6001C: particleensembles containing a-TCP and hydroxyapatite HA (A, B), particles containing only a-TCP (C, D), and particles containing only b-TCP (E, F).
3460 Journal of the American Ceramic Society—Dobelin et al. Vol. 93, No. 10
(3) The Amorphous State
The term ‘‘amorphous’’ in this study primarily refers to the ab-sence of diffraction peaks in XRD and SAED patterns. It doesnot preclude the existence of local order such as cluster arrange-ments. In fact, it was shown that [Ca3(PO4)2]n clusters, in par-ticular Posner’s clusters (Ca9(PO4)6), closely packed to sphericalparticles with water heals in the interstices, are a reasonablestructural model for amorphous TCP obtained by precipitationfrom aqueous solutions.43–45 The precipitation process itself wasdescribed as ‘‘mass crystallization’’, and the presence of hy-droxyl groups was found to stabilize the Posner’s clusters in C3
symmetry, an arrangement similar to the crystal structure ofHA.46 These models rely on the presence of OH� groups and
water molecules, which are readily available in aqueous precip-itation processes, but not in flame-spray synthesis. It was shownthat apatites formed under water-free conditions can be com-pletely free of OH�,47 and even fully hydrated HA easily dehy-drates to a certain degree at high temperatures.48 Consideringthe formation conditions of our amorphous powder and the ex-perimental conditions during the formation of the crystalline
Table I. Absolute Phase Quantities and Calculated Ca:PMolar Ratios of the Crystalline Content
Temperature (1C) a-TCP (wt%) b-TCP (wt%) HA (wt%) Ca:P ratio
25, 500, 525 — — — —550 31.0 (0.5) 9.9 (0.5) 11.2 (0.5) 1.54 (0.01)575 63.5 (0.5) 11.1 (0.5) 13.1 (0.5) 1.52 (0.01)600 69.7 (0.5) 13.3 (0.5) 15.5 (0.5) 1.53 (0.01)650 68.4 (0.5) 17.7 (0.5) 12.3 (0.5) 1.52 (0.01)700 62.6 (0.5) 29.0 (0.5) 9.6 (0.5) 1.52 (0.01)750 38.4 (0.5) 57.2 (0.5) 8.1 (0.5) 1.51 (0.01)800 21.2 (0.5) 72.2 (0.5) 7.9 (0.5) 1.51 (0.01)850 10.2 (0.5) 81.5 (0.5) 7.2 (0.5) 1.51 (0.01)900 5.0 (0.5) 88.1 (0.5) 7.3 (0.5) 1.51 (0.01)950 0 91.6 (0.5) 7.3 (0.5) 1.51 (0.01)1000 0 92.3 (0.5) 7.0 (0.5) 1.51 (0.01)
Compositions were obtained from one data set (n5 1). Standard deviations
based on results published by Stutzman42 are given in parentheses. TCP, tricalcium
phosphate, HA, hydroxyapatite.
Fig. 8. Transmission electron microscopy images of the powder cal-cined at 7001C show coherent crystal structures throughout the particles.
Fig. 9. During selected area electron diffraction analysis, weak a-tricalcium phosphate reflections disappeared and diffuse rings occurred afterirradiation for 30 s up to 3 min.
October 2010 Thermal Treatment of Flame-Synthesized ATCP Nanoparticles 3461

phases, it is to be expected that the material, including the minorHA phase, is in fact completely dehydrated. This is a majordifference between our starting material and the one used inother studies, e.g. by Somrani et al.29 and Li et al.,30 who usedamorphous TCP precipitated from an aqueous solution thusbeing fully hydrated before thermal treatment. These consider-ations, as well as the fact that no signs of order were observed inXRD and SAED patterns, suggest the absence of cluster ar-rangements in the amorphous flame-spray-synthesized material.
In order to understand the phase changes occurring under theTEM electron beam, it is important to observe that: first, bothtypes of transformation are induced by the interaction of a low-intensity (pA–nA), high-energy (200 keV) electron probe withthe matter under vacuum (10�5 Pa). These transformations arenot general but local, and induced by the kinetic energy trans-ferred from electrons to the sample atoms. Second, the degree ofcrystalline order of samples, as well as the crystalline phases in-volved in the amorphization and crystallization processes isdifferent. In the crystalline sample calcined at 6001C, a complextransformation occurs: The crystalline order of the a-TCP phasedecreases under the beam irradiation, and simultaneously, a newamorphous phase appears. This is highlighted by the fact that,on one hand, a-TCP diffraction spots become less visible, andon the other, a diffuse diffraction ring is observed (Fig. 9). HAand b-TCP phases remain unchanged. In the uncalcined amor-phous sample, however, the crystalline phase obtained after ir-radiation was identified (Fig. 6) as being mainly HA.
(4) Ca:P Ratio
The Ca:P ratios of the bulk material calculated from the refinedphase compositions remained constant at 7501C and above, buta higher ratio was determined for lower temperatures (Table I).For the calculations, it was assumed that all crystalline phaseswere stoichiometric, i.e. the Ca:P ratios were assumed to be 1.50for the TCP phases, and 1.667 for HA. However, in the observedchemical system, no known reaction could explain the apparentchange in the bulk Ca:P ratio. Obviously, the assumption thatall phases were stoichiometric was not correct. There are twotendencies coinciding with the calculated change in the bulkCa:P ratio: (i) a slight reduction of the HA content, and (ii) amassive reduction of the a-TCP content (Fig. 2). If one of thesephases was deficient in Ca, the calculation assuming stoichio-metric composition would have over estimated the bulk Cacontent, but the error would diminish as the Ca-deficient phasediminishes. Ca deficiency in HA is a common phenomenon. Itwas shown by Raynaud et al.49 that nonstoichiometric HA witha Ca:P ratio below 1.667 decomposes into b-TCP and stoichio-metric HA between 7001 and 7501C. This stays in agreementwith the observed decrease of HA between 6001 and 7501C andthe over estimated bulk Ca:P ratios calculated below 7501C.However, the charge balance in CDHA (Ca10�x(HPO4)x(PO4)6�x(OH)2�x) is maintained by the incorporation ofHPO4 substituting for PO4, and the phase is typically formedby hydration or precipitation reactions such as the hydration ofa-TCP at low temperatures.20 It is thus questionable whetherCDHA forms in a heat-induced crystallization of a fired rawmaterial.
On the other hand, HA is not the only phase in the observedsystem known for a variable Ca content; a certain degree of Cadeficiency has also been reported for both a- and b-TCP,12,50 butthe data available in this study are not sufficient to determinethe precise Ca:P ratios of the individual phases and to revealthe origin of the Ca:P over-estimation was not the scope of thepresent study.
The slight excess of Ca as compared with the stoichiometricTCP composition may have had an effect on the crystallinephase composition of the thermally treated material, in partic-ular on the formation of a-TCP. It was shown by Lemaıtreet al.51 that Ca:P ratios slightly above 1.50 promoted theformation of a-TCP when heat treated at 13001C, whereasCa-deficient compositions tended to form b-TCP instead. The
opposite trend was observed in a previous study analyzing thereactivity of flame-spray-synthesized nanoparticles produced inthe same reactor as the material used in the present study,34
where compositions with small amounts of HA showed slightlyless a-TCP and more b-TCP than HA-free samples after heattreatment below 10001C. Ca excess appears to have a differentinfluence on the formation of a- and b-TCP depending onwhether it crystallized below 10001C or at the phase boundaryat 11501C.
V. Conclusion
Previous studies have shown that flame-spray synthesis is a suit-able method for the production of X-ray amorphous CaP nano-particles, and that the process can be scaled up to produce largerquantities. Our study demonstrates that the as-preparedpowders can be transformed into a nanoparticulate crystallinepowder with high a-TCP content by calcination at relatively lowtemperatures. The process forms single-crystalline a-, b-TCP,and HA particles with minimum particle fusion and inter-growth. Nanoparticles sometimes allow to modify the reactionkinetics and flow properties of hydraulic cement pastes. Thematerial characterized in the present study could thus be an in-teresting reactant for hydraulic bone cements to optimize thesetting reaction and injectability.
Acknowledgments
The authors would like to thank Dr. Urs Eggenberger from the University ofBern for his support with the XRD data collection, and Norman Luchinger fromthe ETH Zurich for SEM images.
References
1H. Albee and S. J. Morrison, ‘‘Studies in Bone Growth Triple Calcium Phos-phate as a Stimulus to Osteogenesis,’’ Ann. Surg., 71 [1] 32–9 (1920).
2T. Driskell, ‘‘Calcium Phosphate Resorbable Ceramics: A Potential Alternativeto Bone Grafting,’’ J. Dent. Res., 52, 123 (1973).
3J. J. Klawitter and S. F. Hulbert, ‘‘Application of Porous Ceramics for theAttachment of Load Bearing Internal Orthopedic Applications,’’ J. Biomed.Mater. Res. Symp., 2 [1] 161–229 (1971).
4S. F. Hulbert, F. A. Young, R. S. Mathews, J. J. Klawitter, C. D. Talbert, andF. H. Stelling, ‘‘Potential of Ceramic Materials as Permanently Implantable Skel-etal Prostheses,’’ J. Biomed. Mater. Res., 4 [3] 433–56 (1970).
5F. C. M. Driessens and R. M. Verbeeck, ‘‘Relation Between Physico-ChemicalSolubility and Biodegradability of Calcium Phosphates’’; pp. 105–11 in Advancesin Biomaterials, Vol. 8, Edited by C. de Putter, G. L. de Lange, K. De Groot, andA. J. C. Lee. Elsevier Science Publishers, Amsterdam, 1988.
6R. Z. LeGeros, ‘‘Biodegradation and Bioresorption of Calcium PhosphateCeramics,’’ Clin. Mater., 14 [1] 65–88 (1993).
7M. C. von Doernberg, B. von Rechenberg, M. Bohner, S. Grunenfelder, G. H.van Lenthe, R. Muller, B. Gasser, R. Mathys, G. Baroud, and J. Auer, ‘‘In VivoBehavior of Calcium Phosphate Scaffolds with Four Different Pore Sizes,’’Biomaterials, 27 [30] 5186–98 (2006).
8M. Bohner, ‘‘Calcium Orthophosphates in Medicine: From Ceramics toCalcium Phosphate Cements,’’ Injury, 31 [Suppl. 4] 37–47 (2000).
9A. Tampieri, G. Celotti, F. Szontagh, and E. Landi, ‘‘Sintering and Charac-terization of HA and TCP Bioceramics with Control of their Strength and PhasePurity,’’ J. Mater. Sci. Mater. Med., 8 [1] 29–37 (1997).
10I. R. Gibson, I. Rehman, S. M. Best, and W. Bonfield, ‘‘Characterization ofthe Transformation from Calcium-Deficient Apatite to Beta-TricalciumPhosphate,’’ J. Mater. Sci. Mater. Med., 11 [9] 533–9 (2000).
11H. Monma and M. Goto, ‘‘Behavior of the a–b Phase Transformation inTricalcium Phosphate,’’ Yogyo-Kyokai-Shi, 91 [10] 473–5 (1983).
12J. H. Welch and W. Gutt, ‘‘High-Temperature Studies of the System CalciumOxide–Phosphorus Pentoxide,’’ J. Chem. Soc., 874, 4442–4 (1961).
13W. Fix, H. Heymann, and R. Heinke, ‘‘Subsolidus Relations in the System2CaO �SiO2–3CaO �P2O5,’’ J. Am. Ceram. Soc., 52 [6] 346–7 (1969).
14R. Enderle, F. Gotz-Neunhoeffer, M. Gobbels, F. A. Muller, and P. Greil,‘‘Influence of Magnesium Doping on the Phase Transformation Temperature ofBeta-TCP Ceramics Examined by Rietveld Refinement,’’ Biomaterials, 26 [17]3379–84 (2005).
15R. G. Carrodeguas, A. H. De Aza, X. Turrillas, P. Pena, and S. De Aza,‘‘New Approach to the Beta-Alpha Polymorphic Transformation in Magnesium-Substituted Tricalcium Phosphate and its Practical Implications,’’ J. Am. Ceram.Soc., 91 [4] 1281–6 (2008).
16R. W. Nurse, J. H. Welch, and W. Gutt, ‘‘A New Form of TricalciumPhosphate,’’ Nature, 182 [1] 1230 (1958).
17M. Yashima and A. Sakai, ‘‘High-Temperature Neutron Powder DiffractionStudy of the Structural Phase Transition between a and a0 Phases in TricalciumPhosphate Ca3(PO4)2,’’ Chem. Phys. Lett., 372 [5–6] 779–83 (2003).
3462 Journal of the American Ceramic Society—Dobelin et al. Vol. 93, No. 10

18H. Monma and T. Kanazawa, ‘‘The Hydration of a-Tricalcium Phosphate,’’Yogyo-Kyokai-Shi, 84 [4] 209–13 (1976).
19C. Durucan and P. W. Brown, ‘‘Alpha-Tricalcium Phosphate Hydrolysis toHydroxyapatite at and Near Physiological Temperature,’’ J. Mater. Sci. Mater.Med., 11 [6] 365–71 (2000).
20C. Durucan and P. W. Brown, ‘‘Kinetic Model for Alpha-Tricalcium Phos-phate Hydrolysis,’’ J. Am. Ceram. Soc., 85 [8] 2013–8 (2002).
21F. C.M. Driessens, M. G. Boltong, O. Bermudez, J. A. Planell, M. P. Ginebra,and E. Fernandez, ‘‘Effective Formulations for the Preparation of CalciumPhosphate Bone Cements,’’ J. Mater. Sci. Mater. Med., 5, 164–70 (1994).
22F. C. Driessens, J. A. Planell, M. G. Boltong, I. Khairoun, andM. P. Ginebra,‘‘Osteotransductive Bone Cements,’’ Proc. Inst. Mech. Eng. [H], 212 [6] 427–35(1998).
23M. Bohner, ‘‘Physical and Chemical Aspects of Calcium Phosphates Used inSpinal Surgery,’’ Eur. Spine J., 10 [Suppl. 2] S114–21 (2001).
24M. P. Ginebra, E. Fernandez, F. C. M. Driessens, and J. A. Planell,‘‘Modeling of the Hydrolysis of Alpha-Tricalcium Phosphate,’’ J. Am. Ceram.Soc., 82 [10] 2808–12 (1999).
25C. Durucan and P. W. Brown, ‘‘Reactivity of Alpha-Tricalcium Phosphate,’’J. Mater. Sci., 37 [5] 963–9 (1999).
26M. Bohner, A. K. Malsy, C. L. Camire, and U. Gbureck, ‘‘Combining ParticleSize Distribution and Isothermal Calorimetry Data to Determine the ReactionKinetics of a-Tricalcium Phosphate–Water Mixtures,’’ Acta Biomater., 2 [3] 343–8(2006).
27M. Bohner, R. Luginbuhl, C. Reber, N. Doebelin, G. Baroud, and E. Con-forto, ‘‘A Physical Approach to Modify the Hydraulic Reactivity of Alpha-Tricalcium Phosphate Powder,’’ Acta Biomater., 5 [9] 3524–35 (2009).
28R. A. Van Santen, ‘‘The Ostwald Step Rule,’’ J. Phys. Chem., 88 [24] 5768–9(1984).
29S. Somrani, C. Rey, and M. Jemal, ‘‘Thermal Evolution of AmorphousTricalcium Phosphate,’’ J. Mater. Chem., 13 [4] 888–92 (2003).
30Y. Li, W. Weng, and K. C. Tam, ‘‘Novel Highly Biodegradable BiphasicTricalcium Phosphates Composed of a-Tricalcium Phosphate and b-TricalciumPhosphate,’’ Acta Biomater., 3 [2] 251–4 (2007).
31S. Loher, W. J. Stark, M. Maciejewski, A. Baiker, S. E. Pratsinis, D.Reichardt, F. Maspero, F. Krumeich, and D. Gunther, ‘‘Fluoro-Apatite andCalcium Phosphate Nanoparticles by Flame Synthesis,’’ Chem. Mater., 17, 36–42(2005).
32N. Dobelin, T. J. Brunner, W. J. Stark, M. Eggimann, M. Fisch, and M.Bohner, ‘‘Phase Evolution of Thermally Treated Amorphous TricalciumPhosphate Nanoparticles,’’ Key Eng. Mater., 396–398, 595–8 (2009).
33M. Maciejewski, T. J. Brunner, S. F. Loher, W. J. Stark, and A. Baiker,‘‘Phase Transitions in Amorphous Calcium Phosphates with Different Ca/PRatios,’’ Thermochim. Acta, 468 [1–2] 75–80 (2008).
34M. Bohner, T. J. Brunner, N. Doebelin, R. Tang, and W. J. Stark, ‘‘Effectof Thermal Treatments on the Reactivity of Nanosized Tricalcium PhosphatePowders,’’ J. Mater. Chem., 18 [37] 4460–7 (2008).
35T. J. Brunner, M. Bohner, C. Dora, C. Gerber, and W. J. Stark, ‘‘Comparisonof Amorphous TCP Nanoparticles to Micron-Sized alpha-TCP as StartingMaterials for Calcium Phosphate Cements,’’ J. Biomed. Mater. Res. B Appl.Biomater., 83 [2] 400–7 (2007).
36J. Rodriguez-Carvajal, ‘‘Recent Developments of the ProgramFULLPROF,’’ Commission on Powder Diffraction (IUCr),’’ Newsletter, 26,12–9 (2001).
37M. Mathew, L. W. Schroeder, B. Dickens, and W. E. Brown, ‘‘The CrystalStructure of a-Ca3(PO4)2,’’ Acta Crystallogr., B33, 1325–33 (1977).
38B. Dickens, L. W. Schroeder, and W. E. Brown, ‘‘Crystallographic Studies onthe Role ofMg as a Stabilizing Impurity in b-Ca3(PO4)2 I. The Crystal Structure ofPure b-Ca3(PO4)2,’’ J. Solid State Chem., 10, 232–48 (1974).
39K. Sudarsanan and R. A. Young, ‘‘Significant Precision in Crystal StructureDetails: Holly Springs Hydroxyapatite,’’ Acta Crystallogr., B25, 1534–43 (1969).
40L. W. Finger, D. E. Cox, and A. P. Jephcoat, ‘‘A Correction for PowderDiffraction Peak Asymmetry Due to Axial Divergence,’’ J. Appl. Cryst., 27,892–900 (1994).
41R. J. Hill and C. J. Howard, ‘‘Quantitative Phase Analysis from NeutronPowder Diffraction Data using the Rietveld Method,’’ J. Appl. Cryst., 20, 467–74(1987).
42P. Stutzman, ‘‘Powder Diffraction Analysis of Hydraulic Cements:ASTM Rietveld Round-Robin Results on Precision,’’ Powder Diffr., 20 [2] 97–100 (2005).
43G. Treboux, P. Layrolle, N. Kanzaki, K. Onuma, and A. Ito, ‘‘Existence ofPosner’s Cluster in Vacuum,’’ J. Phys. Chem. A, 104 [21] 5111–4 (2000).
44N. Kanzaki, G. Treboux, K. Onuma, S. Tsutsumi, and A. Ito, ‘‘CalciumPhosphate Clusters,’’ Biomaterials, 22 [21] 2921–9 (2001).
45A. S. Posner and F. Betts, ‘‘Synthetic Amorphous Calcium Phosphate and itsRelation to Bone Mineral Structure,’’ Acc. Chem. Res., 8 [8] 273–81 (1975).
46E. I. Suvorova and P. A. Buffat, ‘‘Size Effect in X-Ray and Electron Diffrac-tion Patterns from Hydroxyapatite Particles,’’ Crystallog. Rep., 46 [5] 722–9(2001).
47J. C. Trombe and G. Montel, ‘‘Some Features of the Incorporation of Oxygenin Different Oxidation States in the Apatitic Lattice—I,’’ J. Inorg. Nucl. Chem., 40,15–21 (1978).
48P. V. Riboud, ‘‘Composition et Stabilite des Phases a Structure d’ ApatiteDans le Systeme CaO–P2O5–Oxyde de Fer–H2O a Haute Temperature,’’ Ann.Chim., 8, 381–90 (1973).
49S. Raynaud, E. Champion, D. Bernache-Assollant, and P. Thomas, ‘‘CalciumPhosphate Apatites with Variable Ca/P Atomic Ratio I. Synthesis, Characterisat-ion and Thermal Stability of Powders,’’ Biomaterials, 23 [4] 1065–72 (2002).
50J. R. VanWazer, ‘‘Orthophosphoric Acid, its Salts and Esters’’; pp. 479–599 inPhosphorus and its Compounds, Vol. 1: Chemistry, Edited by J. R. Van Wazer.Interscience Publishers, New York, 1958.
51J. Lemaıtre, H. Andrianjatovo, C. Biourge, K. Ohura, and P. Harodouin, ‘‘ANew High Strength, Resorbable Biocement,’’ Oberflachen Werkstoffe, 9, 13–8(1994). &
October 2010 Thermal Treatment of Flame-Synthesized ATCP Nanoparticles 3463




















