
The RNA Ontology (RNAO): An ontology for integrating RNA sequence and structure data
37
Applied Ontology 6 (2011) 53–89 53 DOI 10.3233/AO-2011-0082 IOS Press The RNA Ontology (RNAO): An ontology for integrating RNA sequence and structure data Robert Hoehndorf a , Colin Batchelor b , Thomas Bittner c , Michel Dumontier d , Karen Eilbeck e , Rob Knight f,l , Chris J. Mungall g , Jane S. Richardson h , Jesse Stombaugh f , Eric Westhof i , Craig L. Zirbel j and Neocles B. Leontis k,∗ a Department of Genetics, University of Cambridge, Cambridge, UK b Royal Society of Chemistry, Cambridge, UK c Department of Philosophy, University at Buffalo, Buffalo, NY, USA d Department of Biology School of Computer Science, Carleton University, Ottawa, ON, Canada e Department of Human Genetics, Eccles Institute of Human Genetics, University of Utah, Salt Lake City, UT, USA f Department of Chemistry and Biochemistry, University of Colorado at Boulder, Boulder, CO,USA g Life Sciences Division, Lawrence Berkeley National Lab, CA, USA h Department of Biochemistry, Duke University Medical School, Durham, NC, USA i Architecture et réactivité de l’ARN, Université de Strasbourg, Institut de Biologie Moléculaire et Cellulaire du CNRS, Strasbourg, France j Department of Mathematics and Statistics, Bowling Green State University, Bowling Green, OH, USA k Department of Chemistry, Bowling Green State University, Bowling Green, OH, USA l Howard Hughes Medical Institute, Boulder, CO, USA Abstract. Biomedical Ontologies integrate diverse biomedical data and enable intelligent data-mining and help translate basic research into useful clinical knowledge. We present the RNA Ontology (RNAO), an ontology for integrating diverse RNA data, including RNA sequences and sequence alignments, three-dimensional structures, and biochemical and functional data. For example, individual atomic resolution RNA structures have broader significance as representatives of classes of homologous molecules, which can differ significantly in sequence while sharing core structural features and common roles or functions. Thus, structural data gain value by being linked to homologous sequences in genomic data and databases of sequence align- ments. Likewise, the value of genomic data is enhanced by annotation of shared structural features, especially when these can be linked to specific functions. Moreover, the significance of biochemical, functional and mutational analyses of RNA mole- cules are most fully understood when linked to molecular structures and phylogenies. To achieve these goals, RNAO provides logically rigorous definitions of the components of RNA primary, secondary and tertiary structure and the relations between these entities. RNAO is being developed to comply with the developing standards of the Open Biomedical Ontologies (OBO) Consortium. The RNAO can be accessed at http://code.google.com/p/rnao/. Keywords: Ontology of molecules, RNA ontology, covalent bonding relation, non-covalent bonding relation, base pairing relation, base stacking relation, RNA motif, RNA sequence alignment * Corresponding author: Neocles B. Leontis, Department of Chemistry, Bowling Green State University, Bowling Green, OH 43402, USA. E-mail: [email protected]. 1570-5838/11/$27.50 © 2011 – IOS Press and the authors. All rights reserved
-
Upload
bensakovich -
Category
Documents
-
view
5 -
download
0
Transcript of The RNA Ontology (RNAO): An ontology for integrating RNA sequence and structure data

Applied Ontology 6 (2011) 53–89 53DOI 10.3233/AO-2011-0082IOS Press
The RNA Ontology (RNAO): An ontologyfor integrating RNA sequence andstructure data
Robert Hoehndorf a, Colin Batchelor b, Thomas Bittner c, Michel Dumontier d, Karen Eilbeck e,Rob Knight f,l, Chris J. Mungall g, Jane S. Richardson h, Jesse Stombaugh f, Eric Westhof i,Craig L. Zirbel j and Neocles B. Leontis k,∗
a Department of Genetics, University of Cambridge, Cambridge, UKb Royal Society of Chemistry, Cambridge, UKc Department of Philosophy, University at Buffalo, Buffalo, NY, USAd Department of Biology School of Computer Science, Carleton University, Ottawa, ON, Canadae Department of Human Genetics, Eccles Institute of Human Genetics, University of Utah,Salt Lake City, UT, USAf Department of Chemistry and Biochemistry, University of Colorado at Boulder, Boulder, CO, USAg Life Sciences Division, Lawrence Berkeley National Lab, CA, USAh Department of Biochemistry, Duke University Medical School, Durham, NC, USAi Architecture et réactivité de l’ARN, Université de Strasbourg, Institut de Biologie Moléculaire etCellulaire du CNRS, Strasbourg, Francej Department of Mathematics and Statistics, Bowling Green State University, Bowling Green, OH, USAk Department of Chemistry, Bowling Green State University, Bowling Green, OH, USAl Howard Hughes Medical Institute, Boulder, CO, USA
Abstract. Biomedical Ontologies integrate diverse biomedical data and enable intelligent data-mining and help translate basicresearch into useful clinical knowledge. We present the RNA Ontology (RNAO), an ontology for integrating diverse RNA data,including RNA sequences and sequence alignments, three-dimensional structures, and biochemical and functional data. Forexample, individual atomic resolution RNA structures have broader significance as representatives of classes of homologousmolecules, which can differ significantly in sequence while sharing core structural features and common roles or functions.Thus, structural data gain value by being linked to homologous sequences in genomic data and databases of sequence align-ments. Likewise, the value of genomic data is enhanced by annotation of shared structural features, especially when these canbe linked to specific functions. Moreover, the significance of biochemical, functional and mutational analyses of RNA mole-cules are most fully understood when linked to molecular structures and phylogenies. To achieve these goals, RNAO provideslogically rigorous definitions of the components of RNA primary, secondary and tertiary structure and the relations betweenthese entities. RNAO is being developed to comply with the developing standards of the Open Biomedical Ontologies (OBO)Consortium. The RNAO can be accessed at http://code.google.com/p/rnao/.
Keywords: Ontology of molecules, RNA ontology, covalent bonding relation, non-covalent bonding relation, base pairingrelation, base stacking relation, RNA motif, RNA sequence alignment
*Corresponding author: Neocles B. Leontis, Department of Chemistry, Bowling Green State University, Bowling Green, OH43402, USA. E-mail: [email protected].
1570-5838/11/$27.50 © 2011 – IOS Press and the authors. All rights reserved

54 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
1. Introduction
The Ribonucleic acid (RNA) Ontology (RNAO) is being developed under the auspices of the RNAOntology Consortium (ROC). The goal of the ROC is to create an integrated conceptual frameworkdefining a common, controlled and structured vocabulary to describe, characterize and annotate RNAsequences, secondary structures, three-dimensional (3D) structure dynamics and functions (Leontis etal., 2006). Previous work in this field includes the RiboWeb ontology, which was part of a knowledgebasefor studying the bacterial ribosome (Altman et al., 1999; Chen et al., 1997), the Multiple AlignmentOntology (Thompson et al., 2005) for nucleic acid and protein sequences, and RNAML, which is anXML schema for exchanging information about RNA secondary structures, tertiary structures, sequencesand sequence alignments (Waugh et al., 2002).
The immediate context of RNAO is the Open Biomedical Ontologies (OBO) project, which seeks tocoordinate the development of biological ontologies (Smith et al., 2007). The relationships among someof these ontologies are as follows. Small molecules, parts of small molecules, and the roles and disposi-tions of small molecules are the responsibility of ChEBI, the ontology of Chemical Entities of BiologicalInterest (Degtyarenko et al., 2008). Genes and their functions are covered by the Gene Ontology (GO),macromolecular sequences (DNA, RNA and polypeptide) are covered by the Sequence Ontology (SO),and proteins by the Protein Ontology (Ashburner et al., 2000; Eilbeck et al., 2005; Reeves et al., 2008;Smith et al., 2007; Thompson et al., 2005). RNAO is distinct from its neighbors, but shares relationshipsand refers to terms from the other ontologies where necessary. RNAO defines RNA-specific terms thatcan be used by other ontologies, including SO and GO. RNAO terms fill a specific niche in annotatingRNA sequences, secondary and 3D structures, structure databases such as PDB and NDB, and sequencedatabases and multiple sequence alignments.
The paper is organized as follows: in Section 2 we informally introduce the chemical structure andfolding of RNA molecules and how RNA structure relates to RNA function. In Section 3 we adopta formal approach to represent the chemical entities that compose RNA molecules and the chemicaland spatial relations that hold between those entities when RNA chains fold into biologically activeshapes. We conclude Section 3 with a preliminary analysis of RNAO relation composition. In Section 4we provide an example of how the formal relations can be used to annotate recurrent 3D motifs inRNA structure. This exemplifies an important use case of RNAO. In Section 5 we give a frameworkfor describing the backbone conformations found in folded RNA molecules. In Section 6, we presentan outline of relations needed to annotate RNA sequence alignments with conserved structural relationsidentified in 3D structures of RNA homologues. This exemplifies a second important use case of RNAO,and shows how RNAO facilitates the integration of RNA structural and sequence data. We describethe relationship of RNAO to top-level ontologies in Section 7 and to the Sequence Ontology (SO) inSection 8. Finally, in Section 9 we discuss the implementations of RNAO in OWL, OBO and SPASS.The implementations are available from http://code.google.com/p/rnao.
2. Overview of RNA structural biology
2.1. RNA components
RNA molecules are chains of repeating units called nucleotide residues (nt) that are linked by cova-lent chemical bonds. Branched or cyclic RNA molecules occur and can be represented by the proposedformalism. The nucleotides themselves consist of three covalently linked components: the nucleobases,

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 55
Fig. 1. Chemical structures of representative purine (left) and pyrimidine (right) nucleotides. Nucleotide residues (left) purine(Adenosine) and (right) pyrimidine (Uridine). Atoms are labeled with letters, for the element type (N – nitrogen, O – oxygen,C – carbon, H – hydrogen, P – phosphorus), and numbers, for the position in the molecule. Ribose atoms are numbered withprime symbols (e.g., C1′, O2′, etc.). Hydrogen atoms attached to ribose carbon atoms are omitted for clarity. Covalent bondsare represented as single or double lines connecting two atoms. The three base “edges”, the Hoogsteen, Watson–Crick andSugar edges, are labeled for each base. The 3′-“face” of each base is toward the reader (see Section 3.2.2).
ribose sugars and phosphate groups (Fig. 1). There are well-established conventions for naming andnumbering the atoms within the nucleotides; these are used to describe structural data in atomic detail.RNA nucleobases are planar one- or two-ring heterocyclic moieties that come in four main varieties(A – adenine, G – guanine, C – cytosine and U – uracil), as well as a large number of modified varieties(Dunin-Horkawicz et al., 2006) that fulfill specialized functions in living cells, The representation ofmodified nucleobases will be detailed elsewhere. As shown in Fig. 1, the one-ring pyrimidine bases(exemplified by U) are each covalently linked at their N1 positions by glycosidic bonds to the C1′ posi-tions of ribose rings, while the two-ring purine bases (exemplified by A) form glycosidic bonds at theirN9 positions. The atoms comprising the nucleobases are numbered sequentially by their positions in thering(s). Hydrogen atoms are numbered according to the number of the heavy atom (N, C or O) to whichthey are attached.
Ribose sugars are aldopentoses that exist as five-membered furanose rings when joined to nucle-obases. They consist of the atoms C1′, C2′, O2′, C3′, O3′, C4′, O4′, C5′ and O5′ (Fig. 1) and theirattached hydrogen atoms (not shown except for the RNA-defining 2′ OH). A nucleobase joined to asugar ring is called a nucleoside. Phosphate groups consist of a central phosphorus atom (“P”) in a +5valence state covalently bonded to four oxygen atoms, labeled OP1, OP2, O3′ and O5′ in Fig. 1. O5′ alsobelongs to the ribose of the same nucleotide while O3′ belongs to the ribose of the preceding nucleotidein the chain, so that O3′ and O5′ are parts shared by a ribose and a phosphate, while the phosphate groupoverlaps adjacent nucleotide residues. In RNA at cellular pH, each phosphate group has a −1 electriccharge.
2.2. The RNA chain
Most RNA molecules are linear-chain polymers. The basic repeating unit of RNA chains is the ribonu-cleotide residue (called a “nucleotide” in RNAO), a covalent assembly comprising a nucleobase, a riboseand a phosphate group; a total of two OH groups and two H atoms are lost from the starting materials in

56 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
Fig. 2. Partition of RNA chain into nucleotide residues or suites. Diagram of a three-nucleotide-long segment (CUC) of anRNA chain. The 5′-to-3′ chain direction runs from top to bottom. Black horizontal lines indicate the partitioning of the chaininto disjoint nucleotide residues, from phosphate to phosphate. Green lines indicate the alternative partitioning into overlapping“suites” from ribose to ribose, used for classifying RNA backbone chain conformations (Richardson et al., 2008). Suites arecentered on a phospho-diester linkage and span the adjacent nucleotides. For example suite i + 1 spans the bases and ribosesof nucleotides i and i + 1, so that adjacent suites overlap. While covalent bond lengths and bond angles are relatively rigid, thetorsion (dihedral) angles of single bonds along the chain can vary significantly giving rise to different backbone conformations(see Section 5). The Greek letters label torsion angles for the glycosidic bonds and for the successive covalent bonds along thebackbone. Non-carbon atoms are shown as color-coded spheres: N – blue, O – red and P – yellow; H atoms are omitted forclarity.
each residue’s worth of that assembly process. Successive ribonucleotide units are linked in a directionalmanner by phospho-diester linkages between the 3′-carbon of the ribose ring of one nucleotide and the5′-carbon of the ribose of the next nucleotide to form RNA chains (Fig. 2). The phospho-diester linkagesconsist of a series of covalent bonds through the O3′, P and O5′ of the connecting phosphate (covalentconnection relations are described in Section 3.1). By convention, each phosphate group, excepting itsO3′ atom, is associated with the following nucleotide, to which it is connected via the 5′-carbon of thecorresponding ribose moiety. Thus, each RNA chain is considered to be directed (and numbered) fromits 5′- to its 3′-end, which is the direction in which RNA molecules are synthesized in the cell, in aDNA-templated, enzymatically catalyzed process called “transcription”.
Each RNA molecule begins with a nucleotide that has a free 5′-end (meaning that its O5′ is notattached to another nucleotide) and ends with a nucleotide that has a free 3′-end. Nucleotides on the 5′-side of a given nucleotide are said to be “upstream” and those on the 3′-side are said to be “downstream”along the chain.
Linear RNA chain segments are collections of nucleotides, each of which is attached via phosphategroups at the 3′- and 5′-positions to exactly two other nucleotides belonging to the collection, except

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 57
for the nucleotides on the 5′- and 3′-ends of the segment. Since the RNAO is primarily concerned withbiopolymers rather than the chemical units from which they are formed, we will adopt much of theshortcut terminology standard in the field, such as using “nucleotide” to mean “ribonucleotide residue”,but will provide explicit definitions of which atoms constitute each such unit (see Section 3.2). Whenreferring to a free nucleotide such as AMP, we will keep the nomenclature explicit. As part of theRNA Ontology, we will provide a dictionary of synonyms that translate between those shortcuts andtheir verbose and precise equivalents in chemical terminology (e.g., the adenosine nucleoside is 9-β-D-ribofuranosyladenine).
2.3. Folding of the RNA chain to form biologically active three-dimensional structure
Cells synthesize many kinds of RNA molecules to carry out a wide range of functions: some partici-pate as carriers of information while others perform catalytic or regulatory roles. RNA molecules partic-ipate in nearly all stages of gene expression, from remodeling of chromosome structure, to transcriptionand maturation of messenger RNA (mRNA), to mRNA-templated protein synthesis and protein local-ization. Some RNAs are produced at specific times and places, i.e., in specific cells and/or sub-cellularcompartments. Unlike DNA, most RNA molecules are single-stranded and range in size from <20 nt totens of thousands of nt. Under physiological conditions, most RNA molecules fold in part or in whole toachieve specific, biologically active three-dimensional (3D) structures; these structures are dynamic andcan change in response to environmental cues or regulatory signals, including changes of temperature orof the concentrations of molecules that interact with the RNA. Thus, the functions of a given RNA de-pend on achieving the appropriate architecture at the appropriate time and place in the cell or organism.Therefore, representing RNA secondary structure (2◦) and 3-dimensional (3D) structure in a manner thatcan be reasoned over in automated ways is highly desirable to advance RNA science. The hierarchicalnature of RNA structure, outlined in the next paragraphs, provides a starting point for constructing usefulrepresentations.
2.4. From sequence to RNA secondary structure
The flexibility of the RNA chain allows it to double back on itself so that pairs of short RNA chainsegments of appropriate sequence can interact with each other, nucleobase by nucleobase, to form anti-parallel double helical structures akin to those formed by DNA. RNA double helices also consist ofWatson–Crick basepairs, stacked on each other, like rungs of a ladder. The backbones of the chainsegments (or “strands”) wrap around each other in a right-handed manner and run in opposite directions(“anti-parallel strand orientation”). Thus, if one of the two Watson–Crick basepaired strands is oriented5′-to-3′ then the other strand is oriented 3′-to-5′. In other words, since the numbering of nucleotides inthe chain increases in the 5′-to-3′ direction, then starting with the same initial basepair, the numberingof nucleotides increases for one strand while it decreases for the other strand in moving along the helix.Anti-parallel double helices define RNA secondary structure and are also the simplest and most regularkind of RNA 3D motif, as they consist of a single type of base-pairing interaction, a repeated and regularpattern of base-stacking interactions, and a repeated backbone conformation (A-form, as opposed to theB-form typical in DNA). Watson–Crick basepairs are only one of many possible kinds of basepairs, aswill be explained in Section 3.4.2. They belong to the cis Watson–Crick/Watson–Crick basepair family,and include the “isosteric” (i.e., occupying the same space) base combinations AU, UA, GC and CG, withoccasional GU or UG combinations, which can form near isosteric basepairs that do not significantlyperturb the regular double helix geometry. Double-helical segments in RNA molecules tend to be quite

58 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
short, typically between two and twelve Watson–Crick basepairs. The set of Watson–Crick base-pairedhelices comprise the secondary structure of each RNA. Some RNA molecules, or their parts, can foldinto more than one secondary structure, producing more than one 3D structure, and can be inducedby appropriate perturbations to switch between these possible structures to carry out their biologicalfunctions.
2.5. Other local motifs
The strand segments at which RNA chains double back on themselves form structures called “stem-loops”, which consist of a double helix capped by a “hairpin loop” (Fig. 3). There are many kinds ofhairpin loops, many of which have specific 3D shapes, structured and stabilized by characteristic sets ofinteractions between the nucleotides belonging to the hairpin loop. In Section 4, we use the “GNRA”hairpin loop motif to illustrate the use of RNAO to define and annotate a recurrent 3D motif. Manyhelices of a folded RNA are capped on one end by hairpin loop motifs. For example, tRNAs (RNAsof length ∼80 nt that deliver amino acids to ribosomes for protein synthesis) have three hairpin loops,including the critical anticodon loop that recognizes cognate codons in mRNA. A pair of sequence seg-ments joining two helices forms a so-called “internal loop” – as illustrated in Fig. 3. Internal loops mayalso form distinct 3D motifs, structured by multiple interactions between the component nucleotides.“Junction” (or “multi-helix”) loops result when three or more helical segments are joined together di-rectly or by strand segments that do not form Watson–Crick basepairs. The simplest junctions are three-way junctions (Fig. 3), but up to 7-way junctions have been observed. Junctions are frequently importantfunctional sites of RNA molecules. For example, a 3-way junction is the site of catalytic activity in thehammerhead ribozyme and a complex multi-helix junction in Domain 5 of 23S rRNA is the catalyticallyactive peptidyl-transferase center of the ribosome. Junctions, with their property of increasing the RNAstructure complexity by branching, combine with other local 3D motifs to define the characteristic 3Darchitectures and folds of functional RNA molecules.
The 3D motifs formed by hairpin, internal and junction “loops” are thus critical elements of RNA 3Dstructure. Many of them involve well-defined geometric arrangements of their component nucleotides
Fig. 3. Schematic depiction of the secondary structure of a hypothetical RNA molecule illustrating different kinds of localmotifs. The Sugar–phosphate backbone is indicated by the continuous heavy line, beginning at the 5′-end and extending to the3′-end. The chain folds back on itself to form four short anti-parallel double helical regions, labeled H1 to H4. Watson–Crickbasepairs are indicated by thin lines. The folding also creates two hairpin loops, an internal loop and a junction loop, in thiscase a 3-way junction.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 59
and recur in different RNA molecules. The structures of 3D motifs can be strongly conserved during evo-lution, even when the sequences that form them have diverged. Although non-helical RNA 3D motifsare largely devoid of Watson–Crick basepairs, they contain other stabilizing inter-nucleotide interactionsand thus require for their descriptions comprehensive representations of all such interactions. Advancesin structural biology have revealed that these interactions include non-Watson–Crick basepairs, base-stacking, base–backbone (phosphate or ribose) and backbone–backbone interactions, and specific back-bone conformations, in addition to the covalent bonds that link nucleotides to each other to form theRNA chain. A major focus of the RNAO is therefore the formal description of appropriate relations andtheir logical constraints to represent each type of interaction. Once defined, these relations can be usedto annotate RNA 3D structures and alignments of homologous RNA sequences.
3. RNA Ontology
Above, we laid out the basic structural biology of RNA and described the components of RNA struc-ture. Next we define the ontological terms that we need to formalize our understanding of RNA. Webegin with the components of RNA molecules – atoms, molecules and sub-molecules – and the con-nection relation that holds between atoms that are covalently bonded. Then we propose a second kindof connection relation to represent the weaker, non-covalent interactions, including hydrogen bondingand van der Waals interactions, which hold between parts of RNA molecules that associate to form thefolded 3D structure. This provides the basis for the ontological description of relations representing non-covalent interactions responsible for base-pairing, base-stacking, base–phosphate and other interactionsinvolving the backbone, all of which stabilize 3D structure. We conclude Section 3 with a discussion ofthe composition of these relations.
3.1. Atoms and molecules
The topics in this subsection are more the province of ChEBI than RNAO, but they have not yet beenaddressed systematically by ChEBI. ChEBI has committed itself to an approach similar to that previouslyoutlined (Batchelor, 2008). We develop them here for completeness and are actively discussing thesematters with the ChEBI team with the aim that ChEBI will cover these topics in the near future.
Molecules are objects composed of atoms connected to each other by covalent bonds in a semi-permanent way. Atoms in turn are composed of positively charged, massive nuclei and negativelycharged, much lighter electrons. Atoms, molecules, and the sub-atomic particles of which they arecomposed require treatments based on quantum mechanics and quantum chemistry to account for theirproperties. For example, molecules are not simple mereological sums of free atoms. Free atoms, in theabsence of a perturbing field, are spherically symmetric and the volume occupied by the electron(s)associated with each atom only contains one nucleus. In a molecule, however, the electrons responsi-ble for covalent bonding, the “valence electrons”, are “shared” between atoms, which is to say that thevolume where there is a significant probability of finding each valence electron contains more than onenucleus. Although electrons are point-like particles, the regions that electrons occupy are much larger.In relation to the nuclei, electrons are distributed over a comparatively huge volume. It is a quantummechanical principle that their exact positions in 3D space at any time cannot be specified precisely. Themost complete specification possible is a complex valued function, the “wave function”, the square ofwhich is interpreted as a probability distribution for finding the electron in 3D space. When a covalentbond forms between two atoms, the wave function describing the probability distribution for the two

60 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
electrons forming the bond extends over both atoms. Thus, what we think of as atoms within moleculesare fiat divisions of the molecule and in no sense objects in their own right.
The mereotopology described by the upper-level ontology for the OBO Foundry, the “Basic FormalOntology” (BFO, see http://www.ifomis.org/bfo), and the OBO Relation Ontology (RO), is based onthe topology of macroscopic objects. It is not yet possible to represent quantum mechanical probabilitydistributions in this framework. Therefore, as a first approximation, we will take atoms as primitiveentities in RNAO and will define molecules and sub-molecules as collections of specific atoms joinedin specific patterns of covalent connection, using the relation of covalent bonding between atoms asa primitive relation, without reference to electrons. We adopt the theory of collections and partitionsoutlined by Bittner et al. (2009) and re-use the member_of relation as primitive in RNAO. In Section 7we discuss the relationship of RNAO to alternative upper level ontologies.
BFO distinguishes between instances (individuals), collections and classes (universals). The in-stance_of relation assigns individuals to classes. Its domain is individuals and its range is classes. Theclasses of individuals treated in this version of RNAO include atoms, molecules and sub-molecules, butnot electrons or nuclei. Collections are finite sets of individuals having at least one member. Universalsof RNAO will be discussed below (Section 3.3). When applying relations to continuants, the BFO andthe RO include a temporal argument in the relations. For the purpose of formalizing RNAO, we ignorethis temporal argument. It is a subject for future work to extend the RNAO with temporally qualifiedrelations.
We begin our formal ontology by taking the part_of relation as a primitive instance-level relation anduse it to define the instance-level overlaps_with relation (D1). Then we define the mereological sum ofmembers of a collection (D2) and the concept of being mereologically maximal (D3).
Definitions and axioms will be numbered as they appear and designated with an intial “D” (for defin-itions) or “A” (for axioms). In definitions and axioms we will display relations in bold face and uncap-italized (“part_of”), and classes of entities capitalized and italicized (“Atom”). We will use lower caseletters (“x”) to denote variables that refer to individuals and upper case letters (“P ”) for variables thatrefer to collections.
D1. Overlaps_with. x overlaps_with y iff there exists a z such that z part_of x and also z part_of y.
Using the notion of collection and the overlaps_with relation we define the mereological sum of acollection (sum_of_collection), a relation whose domain is a collection and range is an individual. Wealso use the primitive relation member_of, which is the relation between individuals and a collection towhich they belong.
D2. Sum_of_collection. x is the sum_of_collection P iff for all z, z overlaps_with x iff (there is a ysuch that y member_of P and y overlaps_with z).
D3. Mereologically_maximal. x is mereologically_maximal iff for all y, if y overlaps_with x, theny part_of of x.
In this framework, we will define a molecule x as the mereological sum of a collection P of co-valently bonded atoms and require x to be mereologically maximal (D5 and D6). Therefore, in ad-dition to parthood relations, we require relations that represent covalent and non-covalent bonding,as they are fundamental to the ontology of molecules. We will call these covalently_bonded_to andweakly_interacting_with (non-covalently bonded to). Note that “non-covalently bonded to” is not the

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 61
logical negation of “covalently bonded to”, but means a kind of bonding that does not involve sharing ofelectrons between atoms.
We declare covalently_bonded_to as a primitive relation in RNAO. Its domain and range are in-stances of Atom and it is a symmetric relation. Like the spatial connectedness relation of the RO, cova-lently_bonded_to is not transitive, but unlike spatial connectedness, it is irreflexive. We state this withAxiom A1.
A1. For all x, x is not covalently_bonded_to x.
Finally, covalently_bonded_to is generally not a functional relation. (A functional relation assigns asingle entity in its range to each entity in its domain.) That is, depending on the kind of atom and thebonds a particular atom forms (single, double, triple), atom x may be able to covalently bond to morethan one distinct atom (Axiom A2).
A2. There exists x, y, z instance_of Atom such that [x covalently_bonded_to y and x cova-lently_bonded_to z and y not equal_to z].
Next, we define the relation covalently_bonded_to*. Covalently_bonded_to* is the transitive clo-sure of the relation covalently_bonded_to, and its formalization requires second-order logic.
D4a. Covalently_bonded_to*. Covalently_bonded_to* is the smallest transitive relation includingcovalently_bonded_to.
We will also need a ternary version of this relation, which we define in D4b, to define the cova-lently_bonded_sum_of relation (D5).
D4b. Covalently_bonded_to_in*. x Covalently_bonded_to_in* y in P is the smallest transitive rela-tion including covalently_bonded_to ranging over members of the collection P .
Based on these relations, we can define covalently bonded sums as follows.
D5. Covalently_bonded_sum_of: x is the covalently_bonded_sum_of P iff [P instance_of Collec-tion_of_atoms and x is the sum_of_collection P and (for all y and z in P , y covalently_bonded_to* zin P )].
This definition says that if a path of covalent bonds connects each atom in P to every other atom in Pthen together the members of P form a new whole, the covalently connected sum. We use this definitionto define the class Molecule.
D6. Molecule. x instance_of Molecule iff there exists P such that P instance_of Collection_of_atomsand x is the covalently_bonded_sum_of P and x is mereologically_maximal.
We note that additional restrictions are needed in order to ensure that every Molecule, in the sense ofD6, is an instance of some universal such as RNA, DNA, water and so on, in accordance with the lawsof chemistry and physics.

62 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
We can define the relation sub_molecule_of to identify the parts of a molecule larger than individualatoms that form distinct entities such as the constituant nucleotides of an RNA molecule or the compo-nent parts (such as the nucleobase) of a nucleotide, as shown in Figs 1 and 2. We first define the relationsub_collection_of.
D7. Sub_collection_of. x sub_collection_of y iff for all z (if z member_of x then z member_of y).
D8. Sub_molecule_of. y sub_molecule_of x iff there exist P , R such that P , R instances_of Collec-tions_of_atoms and y is a covalently_bonded_sum_of R and x is a covalently_bonded_sum_of P andR is a sub_collection_of P .
Based on this definition, each nucleotide residue is a sub-molecule of the RNA to which it belongs andeach ribose and nucleobase is a sub-molecule of the nucleotide to which it belongs. We note that eachinternal phosphate group in an RNA molecule includes four oxygen atoms, one of which, the O3′ atom,is taken by convention to belong to a different nucleotide residue from the others. Therefore, a phosphategroup is not a sub-molecule of any nucleotide residue, but is a sub-molecule of the entire RNA molecule.In spite of some evident disadvantages, it is essential that RNAO follow the convention of placing theO3′ atom in the preceding nucleotide, because the most fundamental units of exchange among databasesand software are the individual atoms, which are in universal practice identified by element type, positionname, and residue number, and therefore must be defined as belonging to that residue.
Next we introduce the primitive relation weakly_interacting_with to represent non-covalent interac-tions, which are also fundamental to RNA Ontology. The domain and range of weakly_interacting_withare Atoms, Sub_molecules, Molecules or Non-covalent_boundaries, which we introduce in Axiom A6.
Like the covalently_bonded_to relation, the weakly_interacting_with relation is symmetric butnot transitive. However, as weakly_interacting_with can be applied to entities larger than individualatoms, one cannot say it is irreflexive, that is, there are large molecules that have sub-molecules that areweakly_interacting_with one another. We express this fact with Axiom A3.
A3. There exists x such that [(x instance_of Sub-molecule or x instance_of Molecule) and xweakly_interacting_with x].
Much of the paper will deal with specific non-covalent interactions, which are sub-relations of theweakly_interacting_with relation. To lay the groundwork for defining these relations, we need to con-sider the boundaries of molecules.
3.2. Boundaries
Spatial boundaries are important ontological concepts as they demarcate the spatial region occupiedby an entity, the distinct spatial regions occupied by different parts of a larger entity, or the possiblesites of interaction between two distinct entities. The boundaries of macroscopic n-dimensional entitiesare entities of lower dimension. Thus 3-dimensional entities such as molecules can have one- or two-dimensional boundaries, corresponding to lines or surfaces. Examples of 2-dimensional boundaries arethe external surfaces forming the boundary between the region occupied by a 3-dimensional object andthe rest of space or the 2-dimensional surface defining the interior boundary separating distinct, non-overlapping, yet adjacent proper parts of a larger entity. Examples of one-dimensional boundaries are

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 63
lines dividing the exterior surface of a 3-dimensional object, such as the equator of the Earth, which sepa-rates the northern and southern hemispheres of the Earth’s surface. In macroscopic ontology, boundariescan be bona fide (physical) or fiat (human demarcated) (Smith & Varzi, 1997).
As discussed above, in dealing with atoms and molecules, we think in terms of quantum mechan-ical spatial probability distributions for the electrons as defining most of the volume occupied by themolecule. The electron probability density at the effective external boundary of an individual atom ormolecule only asymptotically approaches zero. Likewise, defining the boundary between two covalentlybonded atoms requires quantum chemistry. Unless the atoms are identical, it generally does not corre-spond to the midpoint of the line connecting the two nuclei. Thus the external and internal boundariesof RNA (and other) molecules are intrinsically fuzzy and not well defined. Nonetheless, external bound-aries for molecules can be defined approximately as the collection of points in 3D space at which theelectron probability density diminishes to a small enough value to permit molecules to approach andexert attractive forces on one another, without experiencing overwhelming electrostatic repulsion. Theseare bona fide boundaries as they correspond to an underlying physical reality, although, in detail, theirposition differs for interaction with different partner molecules.
We do not attempt to represent these aspects of RNA molecules directly and we know of no attemptsso far to do so. For our purposes it is sufficient to simply assert that there exists a Covalent_boundarybetween two atoms whenever they are covalently_bonded_to each other and similarly to assert thatthere exists a Non-covalent_boundary between two entities (atoms, sub-molecules or molecules) when-ever they are weakly_interacting_with each other. The latter boundary corresponds roughly to whatchemists call the van der Waals surface of the entity, with closer boundaries between atoms that happento be hydrogen bonded. We assume that boundaries of entities are parts of these entities as well. We assertthe existence of these boundaries with two Axioms A4 and A5, which introduce the primitive ternary re-lations covalent_boundary_between and non-covalent_boundary_between. We also define the rela-tion external_boundary_of and the classes of entities Covalent_boundary and Non_covalent_boundary(D9 and D11).
A4. If x covalently_bonded_to y, then, there exists exactly one Boundary B1 such that B1 is the cova-lent_boundary_between x and y.
D9. Covalent_boundary. B is an instance_of Covalent_boundary iff there exists an x and y such thatB is the covalent_boundary_between x and y.
A5. If x weakly_interacting_with y, then, there exists a Boundary B2 such that B2 is a non-covalent_boundary_between x and y.
D10. External_boundary_of. B is external_boundary_of x iff (B instance_of Boundary) and[x instance_of (Atom or Sub_molecule or Molecule)] and (B part_of x) and [there is no y such that(B is the covalent_boundary_between x and y) or (B is the non-covalent_boundary_between xand y)].
D11. External_boundary. B is an instance_of External_boundary iff there exists an x such that B isexternal_boundary_of x.
D12. Non-covalent_boundary. B is an instance_of Non-covalent_boundary iff (B is the exter-nal_boundary_of some y and y instance_of (Atom or Sub_molecule or Molecule)) or B is non-covalent_boundary_between x and y and (x and y instance_of Atom or Sub_molecule or Molecule).

64 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
With these definitions and axioms we have the means to restrict the domain and range ofthe weakly_interacting_with relation to instances of Atoms, Sub_molecules, Molecules and Non-covalent_boundaries in Axiom A6.
A6. If x weakly_interacting_with y then (x and y are instances_of (Atoms or Sub_molecules or Mole-cules or Non-covalent_boundaries, in any combination)).
We note parenthetically that chemists will often choose the position of a non-covalent boundary tocoincide with a surface of minimum electron density where x and y meet; however there are otherpossible choices.
3.2.1. Boundaries within nucleotides and between adjacent nucleotides in the same chainTo describe the characteristic modes of interaction between the different parts of each RNA molecule
when it folds into its biologically active shape, we need to define fiat boundaries along the RNA chainto partition the RNA chain in useful ways. Covalent_boundaries serve to define such boundaries. Aseach RNA molecule is a continuous covalent chain synthesized by successively linking together the fournucleotides in a particular order characteristic of that type of RNA, it is useful to partition the RNA chainby defining fiat boundaries between the nucleotides. By convention each phosphate group is primarilyassociated with the ribose to which it is attached at the 5′-position of the ribose ring and each nucleobaseis associated with the ribose to which it is attached. The formal boundary between nucleotide residues isa Covalent_boundary associated with the covalent bond between the phosphorus atom of one nucleotideresidue and the O3′ oxygen of the preceding nucleotide residue. This is the convention used by majormacromolecular databases such as the Protein Data Bank (PDB; Berman et al., 2000) and the NucleicAcid Database (NDB; Berman et al., 1992), adopted to reflect the known chemical origin of the O3′
atom. An alternative partition of the RNA chain, defining the formal boundary between nucleotides asthe Covalent_boundary between the O3′ atom and the C3′ atom to which it is bonded would have thevirtue of grouping together all four oxygen atoms of each phosphate moiety, but that would disruptlong-established community standards of information exchange.
The boundary between the nucleobase and the ribose is the covalent boundary associated with the gly-cosidic bond, which is the covalent bond between C1′ of the ribose ring and the N1 atom of pyrimidinenucleobases or the N9 atom of purine nucleobases (see Figs 1 and 2). Since the phosphate is not a properpart of any one nucleotide, we will not define fiat boundaries between the ribose and the phosphate,since that could not in any case complete a partition of the nucleotide. The phosphate and ribose aremore usefully considered as units that overlap by a shared oxygen atom on each side.
An alternative partitioning of an RNA molecule into overlapping sub-molecules, that is useful forclassifying the backbone configurations of RNA molecules, is the partition into suites (see Fig. 2 andSection 5).
3.2.2. External boundaries of nucleotides and their regionsOur chief interest, however, is in the non-covalent boundaries of nucleotides, because it is at these
boundaries that base-pairing, base-stacking and other non-covalent interactions take place between dif-ferent parts of an RNA molecule when it folds into its biologically active states.
We partition the non-covalent boundary of each nucleotide in a way that is useful for describingand classifying the characteristic interactions between nucleotides that form and stabilize the native3D shapes of RNA molecules. The focus remains the nucleobase, the characteristic chemical part ofeach nucleotide. Given that the nucleobases have approximately the shape of right triangular prisms,

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 65
Fig. 4. Abstraction of an RNA nucleobase as a flattened triangular prism connected to the backbone by a glycosidic covalentbond in the default anti configuration. Left: top view looking down on the 3′-face of the nucleobase from the 3′-end of the chain.Right: perspective view of the nucleobase revealing the two triangular faces and three elongated rectangular base “edges”, theHoogsteen edge (“H”), the Watson–Crick edge (“W”) and the Sugar edge (“S”) as shown. Note that the 5′ face is definedrelative to the 5′ strand direction in the canonical double helix, not relative to the actual local direction. Operationally, if thenumbering of atoms around the base ring that includes N1/9 increases clockwise, you are looking at the 5′, or “up” face; ifcounterclockwise (as in Fig. 1), you are looking at the 3′ or “down” face.
they present two types of surface boundaries – two triangular “faces” and three roughly rectangular andelongated “edges” (see Fig. 4). Because of the inherent asymmetry of nucleosides, the two triangularfaces are not equivalent; see Section 3.4.3 for their central role in defining base-stacking relations.
The three base edges are also distinct and are central to describing basepair relations. They are namedthe Watson–Crick, the Hoogsteen and the Sugar edges (Fig. 4). The boundaries between the faces andthe edges are one-dimensional (fiat) boundaries. The boundaries between the three edges are demarcatedby chemical functional groups: the glycosidic bond connecting the ribose and nucleobase demarcatesthe boundary between the Hoogsteen and Sugar edges in both pyrimidines and purines; the exocyclicfunctional groups at the pyrimidine 4-position (–NH2 for C and =O for U) and the purine 6-position(–NH2 for A and =O for G) demarcate the boundary between the Hoogsteen and Watson–Crick edges;and the exocyclic functional groups at the pyrimidine and purine 2-positions (=O for C and U, –NH2
for G, and –H for adenine) demarcate the boundary between the Watson–Crick and Sugar edges (seeFig. 1). The 2′-OH functional group of the ribose, which is a primary distinguishing feature of RNAcompared to DNA, frequently participates in the base-pairing interactions involving the Sugar edgeof the nucleobase and is considered part of the Sugar edge. These definitions of the fiat boundaries thatpartition the bona fide external boundary of each nucleoside will be used below to enumerate and classifythe possible types of base-pairing and base-stacking interactions. Given the inherent asymmetry of thenucleosides and the importance of the glycosidic bond in connecting the nucleobase and ribose moieties,it will be seen that the relative positions of the glycosidic bonds of two interacting nucleosides will playa crucial role in this classification.
3.3. RNA classes
The most important classes (or “universals” or “types”) in RNA science are RNA molecules. In gen-eral, each organism makes many copies of any given RNA encoded in its genome. It is now acknowl-edged that organisms of a given species do present variations in their genomes. It is important to notethat most of the RNAs discussed in practice (e.g., most RNA families in the Rfam database) are families

66 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
of functional RNA molecules, which may be homologous, i.e., molecules related by evolution. Con-sequently, families of RNA molecules cannot be neatly defined by sequence or structure. Nonetheless,generally one can define a consensus sequence (and structure) that embeds the main functional and struc-tural characteristics of a RNA family and that can accommodate the minor sequence variations occurringwith a given species (or bacterial strain) or even several species.
On the other hand, these classes of molecules can also be considered to be individuals in the senseof collectives of molecules related through evolutionary relations. These collectives are made up ofindividual molecules, each of which has a particular sequence and structure. This is consistent with thephilosophical view that species are best understood to be individuals, that is, to have the same ontologicalstatus as individual organisms (Crane, 2004; Hull, 1978). In this view, individual organisms are spatio-temporal parts of species rather than instances, just as organs are parts of organisms.
Within RNAO, designations such as “E. coli 16S rRNA molecule” represent a “consensus” classwhose extension includes every member of the 16S rRNA collective individual encoded in the genome ofsome E. coli strain. Thus, classes of molecules in RNAO are at present defined based on the molecularsequence and structure, not necessarily through their evolutionary lineage. The application of RNAOto elucidate the relationship of RNA sequence to structure and function, which are more conservedthan sequence is intended to provide tools for systematically studying evolutionary processes in RNA,and thus to elucidate evolutionary relations between RNA molecules (see Section 6). Important issuesthat need to be addressed have been delineated in previous work (Brown et al., 2009). As discussedin Section 6, RNAO constitutes the first steps towards the formalization of the concepts described byBrown et al. (2009).
Some other classes fundamental to RNA science are those that are instantiated by the chemical compo-nents of RNA molecules. One such class is “Nucleotide”. The class Nucleotide consists of two primarysub-classes, Purines and Pyrimidines, each of which includes a large number of sub-classes, the mostimportant of which are the purine sub-classes Adenosine (A) and Guanosine (G), and the pyrimidinesub-classes Cytidine (C) and Uridine (U). These classes are related to Nucleotide by the is_a relation.Thus, “Purine is_a Nucleotide”, “Adenosine is_a Purine” and “Adenosine is_a Nucleotide”. Each ofthese classes, in turn, are super-classes that include nucleotides that are derived from the parent bychemical modification. Thus, “1-Methyl-Adenosine is_a Adenosine”. Because such classes are alreadyhandled by ChEBI, we need not duplicate them in RNAO.
Other classes are instantiated by parts of nucleotides (here meaning “ribonucleotide residues”), whichinclude the class of nucleobases and the class of ribose rings. Each instance of these classes is a sub-molecule of the corresponding nucleotide instance.
In future work we will define class level relations for all RNA relations. For simplicity, this paperfocuses on defining the underlying instance-level relations needed by RNAO.
3.4. Inter-nucleotide relations
Inspired by Villanueva-Rosales and Dumontier (2007), we formally treat all interactions, covalentand non-covalent, as connection relations. We note that covalent bond connections, generally cannotbe established or broken without affecting the type (universal) the RNA molecule instantiates, at leastwhen this involves adding or deleting nucleotides. By contrast, non-covalent connections, which includehydrogen-bonding, van der Waals or dispersion and electrostatic interactions, can generally be estab-lished or disrupted without affecting the type the molecule instantiates. Physically, wholesale disruptionof these interactions results in processes biochemists call “denaturation” of the folded form of the RNA

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 67
chain. In principle, under the right conditions such disruptions are reversible. The logical properties ofthe covalent and non-covalent connection relations were described above. We now use these relations todefine more specific interactions, needed to describe RNA structures.
3.4.1. Covalent connection between sub-moleculesWe use the parent relation covalently_bonded_to, that models covalent bonding between two atoms,
to define a new relation, covalently_connected_to (abbreviated “cov_conn_to”) that has submoleculesas both domain and range. Its intended meaning is that two sub-molecules m and n (for example, twonucleotides) are covalently connected if some atom in m forms a covalent bond to some atom of n.
D13. Cov_conn_to. m cov_conn_to n iff [(m and n instances_of Sub_molecules) and not (m equal n)and there exist x and y such that (x and y instances_of Atoms and x part_of m and y part_of n and xcovalently_bonded_to y)].
This relation is irreflexive, symmetric and non-transitive. “Non-transitive” means that if one atom inA is bonded to some atom in B, and another atom in B is bonded to some atom in C, there will not, ingeneral, exist an atom in A which is bonded to some atom in C, although this is also not excluded.
To formally describe the covalent bonding between nucleotides to form RNA chains, we specialize thecov_conn_to relation for phospho-diester linkages in RNA. The domain and range for these relations isNucleotides (abbreviated “nt”).
D14. Cov_conn_3′_5′. nt1 cov_conn_3′_5′ nt2 iff nt1, nt2 instances_of Nucleotides and there exist x,y such that [x instance_of Ribose_O3′_atom and y instance_of Phosphorus_atom and x part_of nt1
and y part_of nt2 and x covalently_bonded_to y].
Note that cov_conn_3′_5′ is the relation between successive nucleotides in the normal 5′-to-3′ direc-tion. In a similar manner we can define cov_conn_5′_3′ and, for completeness, relations cov_conn_5′_5′
and cov_conn_3′_3′, although these are rare in natural RNAs. To describe lariat structures formed dur-ing intron splicing or present in certain cyclic nucleotides, we can also define relations that involve the2′-position of the ribose, for example cov_conn_3′_2′.
D15. Cov_conn_5′_3′. nt1 cov_conn_5′_3′ nt2 iff nt1, nt2 instances_of Nucleotides and there exist x,y such that [x instance_of Phosphorus_atom and y instance_of Ribose_O3′_atom and x Part_of nt1
and y Part_of nt2 and x covalently_bonded_to y].
For example, in Fig. 2, the relations (nt1 cov_conn_3′_5′ nt2) and (nt2 cov_conn_5′_3′ nt1) hold,showing that these are inverse relations. Neither of these relations is reflexive, symmetric or transitive.However, each of these relations is functional.
A7. If (nt1 cov_conn_3′_5′ nt2 and nt1 cov_conn_3′_5′ nt3) then nt2 equals nt3.
A8. If (nt1 cov_conn_5′_3′ nt2 and nt1 cov_conn_5′_3′ nt3) then nt2 equals nt3.
Other properties of these relations are discussed below under Relation Composition (Section 3.5).Similar to the relation covalently_bonded_to, we introduce transitive versions R* for each con-nectedness relation R as the smallest transitive relations including R. For example, the relationcov_conn_5′_3′* is the smallest transitive relation including cov_conn_5′_3′.

68 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
It is also useful to define the relation same_chain for nucleotides that belong to the same RNA chain(i.e., RNA molecule). “Nucleotide” is the domain and range of this relation. We restrict this relation toRNA chains in which all nucleotides are linked to each other by cov_conn_5′_3′ or cov_conn_3′_5′
relations.
D16. Same_chain. nt1 same_chain nt2 iff [(nt1 equals nt2) or (nt1 cov_conn_3′_5′* nt2) or (nt1
cov_conn_5′_3′* nt2)].
As a consequence of this definition, the binary relation same_chain is reflexive, symmetric and tran-sitive.
We define the relations 5′_to and 3′_to that correspond to the notions “upstream” and “downstream”that are used in molecular biology. These relations are also inverses of each other (cf. the upstream anddownstream relations as applied to sequences in Eilbeck and Mungall (2009)). They are transitive, butnot reflexive or symmetric.
D17. 5′_to: nt1 5′_to nt2 iff (nt1 same_chain nt2) and (nt1 cov_conn_3′_5′* nt2).
In a similar way we define 3′_to.
D18. 3′_to. nt1 3′_to nt2 iff (nt1 same_chain nt2) and (nt1 cov_conn_5′_3′* nt2).
These two relations are inverses of each other. They are neither reflexive nor symmetric but they aretransitive:
If (x 5′_to y and y 5′_to z) then x 5′_to z.
If (x 3′_to y and y 3′_to z) then x 3′_to z.
These statements follow from the definitions and do not require axioms.We next define the class of continuous RNA strand segments consisting of standard phosphate link-
ages, “5′_to_3′_strand_segment”.
D19. 5′_to_3′_strand_segment. x instance_of 5′_to_3′_strand_segment iff there exists P such that[P collection_of Nucleotides and x is the sum_of_collection of P and for all y, z in P , (y 5′_to zOR y 3′_to z) and {for all y, w, and z if (y, z in P and y 5′_to z and w 3′_to y and w 5′_to z) then wpart_of x}].
Finally, we define the class of canonical RNA molecules, meaning those molecules in which all pairsof covalently bonded nucleotides are connected 5′-to-3′ or 3′-to-5′. We call these molecules instances of5′_to_3′_RNA_molecule.
D20. 5′_to_3′_RNA_molecule. x instance_of 5′_to_3′_RNA_molecule iff (x instance_of 5′_to_3′_strand_segment and x instance_of Molecule).

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 69
3.4.2. RNA base-pairingAs discussed above, the shape of an RNA nucleobase, whether purine or pyrimidine, can be approxi-
mated as a relatively flat triangular prism. This abstraction is supported by exhaustive analysis of RNA3D structure data showing that RNA nucleobases form two main kinds of interactions – face-to-face“base-stacking” on the one hand, and edge-to-edge “base-pairing” on the other. Evidence for the central-ity of these interactions to RNA structure is that most bases in 3D structures form one or more basepairsas well as one or more base-stacking interactions while only a small number of bases form only stackingor pairing interactions or none at all. Detailed structural analysis indicates the usefulness of partitioningthe bona fide non-covalent boundary of each nucleobase into upper and lower triangle-shaped “faces”,at which stacking interactions take place, and elongated rectangular “edges” around the base perimeter,at which base-pairing interactions take place. Consistent with this partitioning into three edges, eachnucleobase can basepair edge-to-edge with up to three other nucleobases, one at each edge (Nasalean etal., 2009). The definitions of the edges were shown for adenosine (A), representing purines, and uridine(U), representing pyrimidines, in Fig. 1.
In this subsection, we discuss the edge-to-edge pairing interactions, which we capture as sub-relationsof the weakly_interacting_with relation. In the next section we discuss base-stacking relations, whichare also represented as sub-relations of weakly_interacting_with.
RNA basepairs can form between two RNA nucleobases when they lie approximately in the sameplane and approach each other at their edges sufficiently close to form non-covalent bonds. For this to bepossible, the two bases must present complementary edges. This means that in the chosen orientation,hydrogen bond donors on the interacting edge of the first base must be juxtaposed with hydrogen bondacceptors on the edge of the second base and vice versa. Generally two or more hydrogen bonds areneeded to form a basepair. Hydrogen bonds are attractive interactions between electropositive hydrogenatoms covalently attached to “donor” atoms (N or O in RNA) and electronegative “acceptor” groups (Nor O atoms deficient in hydrogens).
Basepairs can form between any pair of edges having complementary arrangements of donor andacceptor groups, and this depends on the nature of the bases (A, C, G or U), on the edges involved, aswell as their relative orientation. The relative orientation is defined with respect to the glycosidic bonds(see Fig. 5). With six different combinations of edge interaction (WC–WC, H–H, S–S, WC–H, WC–Sand H–S), and two relative orientations (cis and trans) for the interaction of the nucleosides, there resulttwelve geometric base-pairing classes or families in the Leontis–Westhof classification scheme (Leontis& Westhof, 2001). Each class is named descriptively by indicating the interacting edges of the twobases, Watson–Crick (W), Hoogsteen (H) or Sugar (S), and whether the interaction is cis (c) or trans (t)
Fig. 5. Cis vs trans base-pairing. For each combination of interacting base edges (here Watson–Crick edge of A with Wat-son–Crick edge of U), two kinds of basepair can form which differ in the relative orientation of the glycosidic bonds. Left panelshows cis WW base-pairing and right panel shows trans WW base-pairing.

70 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
(see Table 1). Thus cWH (cis Watson–Crick/Hoogsteen) GA is a different basepair from cWW GAor cWH AG. The resulting base-pairing classes have the logical property of being mutually disjoint.Moreover, they largely exhaust the types of edge-to-edge pairing interactions observed in 3D atomic-resolution structures (Leontis et al., 2002; Stombaugh et al., 2009). In this scheme, the “Watson–Crick”basepairs, which are defined as those pairings that form regular anti-parallel helices, are the “canonical”AU or GC cis Watson–Crick/Watson–Crick (cWW) basepairs. The cWW GU (“wobble”) basepair alsooccurs in RNA helices, but is not strictly speaking considered a canonical Watson–Crick pair. Thus theWatson–Crick pairs are restricted to certain base-combinations (AU, UA, GC and CG) in the cWWfamily. All other basepairs, including the other cWW base-combinations as well as all tWW (transWatson–Crick/Watson–Crick) are called “non-Watson–Crick basepairs”. A comprehensive catalog ofRNA basepairs, organized by geometric family, is provided at http://rna.bgsu.edu/FR3D/basepairs/. Thiscompilation indicates which base combinations are possible for each basepair family and provides 3Dexemplars calculated from instances extracted from the PDB database of atomic resolution RNA 3Dstructures (Stombaugh et al., 2009). Information regarding which base combinations are allowed foreach basepair family is included in RNAO.
To formalize these ideas we introduce the relations edge_of and face_of and their specializations sowe can use these to define base-pairing and base-stacking relations. Then we define classes of specificedges and faces.
A9. If n instance_of Nucleotide then there exists exactly one x, y, z such that (x Watson–Crick_edge_of n and y Hoogsteen_edge_of n and z Sugar_edge_of n).
D21. Edge_of. e edge_of n iff (e Watson–Crick_edge_of n or e Hoogsteen_edge_of n or esugar_edge_of n).
D22. Watson–Crick_edge. e instance_of Watson–Crick_edge iff there exists x such that (x instance_ofNucleotide and e Watson–Crick_edge_of x).
In a similar manner we can define the classes of Hoogsteen and Sugar edges. We also define the inverserelation has_edge (D23) and its specializations (not shown):
D23. Has_edge. n has_edge e iff e edge_of n.
Similarly for faces:
A10. If n instance_of Nucleotide then there exists exactly one f , g such that (f 5′-face_of n and g3′-face_of n).
D24. f face_of n iff (f 5′-face_of n or f 3′-face_of n).
D25. 5′-face. f instance_of 5′-face iff there exists n such that (n instance_of Nucleotide and f5′-face_of n).
In a similar manner we can define the class of 3′-faces. We also define the inverse relation has_faceand its specializations (not shown):
D26. Has_face. n has_face f iff f face_of n.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 71
Edge_of and face_of are functional relations, meaning each edge or face belongs to one and only onenucleotide. These facts are stated as axioms.
A11. If (e edge_of x and e edge_of y) then x equals y.
A12. If (f face_of x and f face_of y) then x equals y.
Although the inverse relations, has_edge and has_face are not functional, their specializations are.Therefore we have a set of axioms for each edge and face relation exemplified by Axiom A13.
A13. If (n has_Watson–Crick_edge e1 and n has_Watson–Crick_edge e2) then e1 equals e2.
With these relations we can define the general base-pairing relation “pairs_with”.
D27. Pairs_with. nt1 pairs_with nt2 iff [nt1 and nt2 instances_of Nucleotide and (nt1 has_edge e1 andnt2 has_edge e2 and e1 weakly_interacting_with e2)].
We next specialize the pairs_with relation by specifying interacting edges. We give one example, asa template for defining the other edge-to-edge base-pairing relations.
D28. Pairs_with_WH. nt1 pairs_with_WH nt2 iff [nt1 pairs_with nt2 and nt1 has_Watson_Crick_edge f1 and nt2 has_Hoogsteen_edge f2 and f1 weakly_interacting_with f2.]
Finally we specialize each of these relations by specifying the relative orientation of the glycosidicbond angles of the two nucleotides forming the basepair (see Fig. 5). For these definitions we requiretwo new binary relations, cis_Glycosidic_bond_orientation and trans_Glycosidic_bond_orientation,which indicate which of the two orientations of the glycosidic bonds hold for the basepair formed by nt1
and nt2 (see Fig. 5).
D29. Pairs_with_cWH. nt1 pairs_with_cWH nt2 iff [(nt1 pairs_with_WH nt2) and (nt1 cis_Glycosidic_bond_orientation nt2)].
In summary, we capture the parent base-pairing relation pairs_with as non-covalent connection be-tween edges of two nucleotides. This relation is symmetric but not reflexive or transitive. It is not func-tional as a base can pair with more than one other base (up to three). We specialize the pairs_with rela-tion according to the edges involved and then according to whether the interacting bases are cis or trans.In this way we obtain, pairs_with_WW, pairs_with_WH, . . . , pairs_with_SS. From pairs_with_WWwe obtain pairs_with_cWW and pairs_with_tWW by specifying whether the pair is cis or trans andlikewise for the other families. Note that relations corresponding to asymmetric basepair classes, forexample, pairs_with_cWH and pairs_with_cHW, are distinct, inverse relations, while relations thatinvolve the same edge on each base are self-inverse. Thus, there are 18 specific base-pairing relations,but only 12 basepair classes, as shown in Table 1. We define the general basepair class (D30) and providea sample definition for a specific class of basepairs (D31).
D30. Base-pair. bp instance_of Base-pair iff there exist nt1 and nt2 such that (nt1 pairs_with nt2 andfor all p with p part_of bp, p overlaps_with nt1 or p overlaps_with nt2).

72R
.Hoehndorfetal./R
NA
O:
An
ontologyfor
integratingR
NA
sequenceand
structuredata
Table 1
Base-pairing relations in RNAO
Classes Relations Symbol Triangle abstraction
Base-pairing relations Inverse relation Prevents pairingswith third base
family_1_base_pair pairs_with_WW pairs_with_cWW Self-inverse W-first pairingsW-second pairings
family_2_base_pair pairs_with_tWW Self-inverse W-first pairingsW-second pairings
family_3_base_pair pairs_with_WH pairs_with_cWH pairs_with_cHW W-first pairingsH-second pairings
pairs_with_HW pairs_with_cHW pairs_with_cWH H-first pairingsW-second pairings
family_4_base_pair pairs_with_WH pairs_with_tWH pairs_with_tHW W-first pairingsH-second pairings
pairs_with_HW pairs_with_tHW pairs_with_tWH H-first pairingsW-second pairings
family_5_base_pair pairs_with_WS pairs_with_cWS pairs_with_cSW W-first pairingsS-second pairings
pairs_with_SW pairs_with_cSW pairs_with_cWS S-first pairingsW-second pairings
family_6_base_pair pairs_with_WS pairs_with_tWS pairs_with_tSW W-first pairingsS-second pairings
pairs_with_SW pairs_with_tSW pairs_with_tWS S-first pairingsW-second pairings
family_7_base_pair pairs_with_HH pairs_with_cHH Self-inverse H-first pairingsH-second pairings

R.H
oehndorfetal./RN
AO
:A
nontology
forintegrating
RN
Asequence
andstructure
data73
Table 1
(Continued)
Classes Relations Symbol Triangle abstraction
Base-pairing relations Inverse relation Prevents pairingswith third base
family_8_base_pair pairs_with_tHH Self-inverse H-first pairingsH-second pairings
family_9_base_pair pairs_with_HS pairs_with_cHS pairs_with_cSH H-first pairingsS-second pairings
pairs_with_SH pairs_with_cSH pairs_with_cHS S-first pairingsH-second pairings
family_10_base_pair pairs_with_HS pairs_with_tHS pairs_with_tSH H-first pairingsS-second pairings
pairs_with_SH pairs_with_tSH pairs_with_tHS S-first pairingsH-second pairings
family_11_base_pair pairs_with_SS pairs_with_cSS Self-inverse S-first pairingsS-second pairings(this will change in v2)
family_12_base_pair pairs_with_tSS Self-inverse S-first pairingsS-second pairings(this will change in v2)
Notes: Sub-relations of the pairs_with relation, listed according to interacting edges and glycosidic bond orientations. Each relation is assigned a unique symbolusing circles to represent Watson–Crick (W) edges, squares to represent Hoogsteen (H) edges and triangles to represent Sugar edges (S). Filled symbols designatecis basepairs and open symbols trans pairs.

74 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
D31. cWH_Base-pair. bp instance_of cWH_Base-pair iff there exist nt1 and nt2 such that (nt1
pairs_with_cWH nt2 and for all p with p part_of bp, p overlaps_with nt1 or p overlaps_with nt2).
The fact that there are only 12 basepair classes follows logically from the definitions. For example:
If bp instance_of cWH_Base-pair then bp instance_of cHW_Base-pair.
None of the base-pairing relations is transitive or reflexive. However, these relations are mutuallyexclusive. This means that if nt1 pairs_with_Ei_Ej nt2, where Ei and Ej are specific edges, W, H or S,then nt1 cannot simultaneously form a different kind of basepair with nt2.
To a first approximation, each edge of a nucleoside may interact only with a single edge of a differentnucleoside. This is generally true, but an oversimplification as there are special cases in which a singleedge of a base can interact simultaneously with two different nucleobases. One case involves the Sugaredge of a purine (usually A) interacting with the sugar edges of two other bases which themselves forma cWW basepair. In the usual case, these interactions define a base triple with cWW(GC) and tSS(GA)and cSS(CA). A preliminary discussion of relation composition using base-pairing relations is givenbelow.
3.4.3. Base-stacking relationsBase-stacking interactions are non-covalent interactions between the faces of two approximately par-
allel nucleobases, as shown in Fig. 4. We begin by defining the parent stacking relation stack.
D32. Stack. nt1 stack nt2 iff [nt1 and nt2 instances_of Nucleotide and (nt1 has_face f1 and nt2 has_facef2 and f1 weakly_interacting_with f2)].
The parent relationship is irreflexive, symmetric, and neither transitive nor functional.Because the nucleobase faces are not equivalent (see Fig. 4), four distinct, basic types of stacking re-
lations are possible between RNA nucleobases. The base faces have been labeled by some authors as the“Up” or “Down” faces and by other authors as the “5′-” or “3′-faces”, by reference to the strand directionthey face in the canonical double helix. We will use the designations 5′-face and 3′-face, but the ontologyitself declares both designations as synonyms. Stacking can occur between two 5′-faces, between two3′-faces or between the 5′- and 3′-faces of two bases. We formally define the most frequently observedrelation of this type.
D33. Stack_3′_5′. nt1 stack_3′_5′ nt2 iff [nt1 stack nt2 and nt1 has_3′-face f1 and nt2 has_5′-face f2
and f1 weakly_interacting_with f2].
The other three stacking relations are defined in similar fashion. These relations are not reflexive ortransitive. stack_5′_3′ is the inverse of stack_3′_5′ whereas stack_5′_5′ and stack_3′_3′ are symmetricand self-inverse:
nt1 stack_5′_3′ nt2 iff nt2 stack_3′_5′ nt1 (Inverse relations),
nt1 stack_3′_3′ nt2 iff nt2 stack_3′_3′ nt1 (Self-Inverse),
nt1 stack_5′_5′ nt2 iff nt2 stack_5′_5′ nt1 (Self-Inverse).

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 75
All these statements follow from the definitions. Importantly, stacking and base-pairing relations aredisjoint. We state this fact with an Axiom A14 and a Corollary C15 that follows logically from it, usingthe parent relations, as these apply to all the sub-relations:
A14. If nt1 pairs_with nt2, then not (nt1 stack nt2).
C15. If nt1 stack nt2, then not (nt1 pairs_with nt2).
In Section 3.5.2 we describe relation composition for stacking relations.
3.5. Relation composition
In the previous section we have attempted to ground the spatial and topological relations relating thechemical constituents of RNA molecules in fundamental ontological relations. We have laid out the basiclogical properties of the relations used to characterize RNA structures. Here we ask the general question,what logical constraints come into play when relations are composed? That is, for three RNA entities, e1,e2 and e3, if R1 holds between e1 and e2 and R2 holds between e2 and e3, what constraints are there on therelation R3 between e1 and e3? Typically, the possible relations R3 are presented in a composition tablein which the rows are labeled by “e1 R1 e2”, the columns are labeled by “e2 R2 e3” and the entries of thetable display all possible R3 such that e1 R3 e3. This is the standard way to display relation composition.An equivalent approach, that helps clarify the possible relations that e1 can form simultaneously, is todisplay possible relations R3, where e2 R3 e3, given e1 R1 e2 and e1 R2 e3. Replacing e1 R1 e2 by itsinverse relation, R1-inv, and swapping the designation of e1 and e2 shows these are equivalent. We willuse the former approach in the composition tables for covalent bonding and base-stacking relations. Wecan also ask the question, what are the constraints of e1 R1 e2 on possible R2 such that e1 R2 e2 or e2 R2
e3? This approach will be used to explore the properties of base-pairing relations.
3.5.1. Composition of covalent connection relationsWe begin with a self-contained composition table (Table 2) involving covalent relations between RNA
nucleotides (labeled nt1, nt2, nt3). As discussed above, cov_conn_5′_3′ and cov_conn_3′_5′ are thestandard, overwhelmingly most frequent covalent connection relations in biological RNA molecules.“nt1 cov_conn_3′_5′ nt2” means that the 3′-end of nt1 is covalently attached via a phosphate moiety to
Table 2
Composition table for covalent relations
Notes: Rows and columns correspond to different covalent connectivity relations between nt1 and nt2 and between nt2 and nt3,respectively. Possible relations between nt1 and nt3 are shown in the cells. “Not allowed” means that the nt1 R1 nt2 and nt2 R2nt3 cannot hold simultaneously. “=” is the identity relation. *The same_chain relation must hold between nt1 and nt3, but it isnot necessarily the only relationship that can hold between them. It is not possible a priori to predict other relations.

76 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
the 5′-end of nt2, where nt1 and nt2 are nucleotides. These two relations are inverses of each other, asfollows from the definitions:
nt1 cov_conn_3′_5′ nt2 iff nt2 cov_conn_5_3′ nt1.
Furthermore, each of the covalently connected relations is functional. Each nucleotide can becov_conn_5′_3′ or cov_conn_3′_5′ to at most one other nucleotide at a time. These facts require axiomsand we provide Axiom A16 as an example.
A16. If (nt1 cov_conn_3′_5′ nt2 and nt1 cov_conn_3′_5′ nt3), then nt2 equals nt3.
As the inverse relations are also functional it follows that:
If (nt1 cov_conn_3′_5′ nt2 and nt2 cov_conn_5′_3′ nt3), then nt1 equals nt3.
Combining these logical properties together results in the following composition table for four possi-ble covalent relations between nucleotides.
3.5.2. Composition of base-stacking relationsUnlike the covalent connection relations, the stacking relations are not functional; two different bases
can have the same stacking relation to another base. We provide an example axiom.
A17. There exists nt1, nt2, nt3 such that (nt1 stack_5′_5′ nt2) and (nt2 stack_5′_5′ nt3) and not (nt1
equal nt3).
For four of the eight ways of composing stacking relation, including the four in which a stackingrelation is composed with its inverse relation, if nt1 �= nt3 and both nt1 and nt3 stack with good overlapon nt2 and lie approximately in the same plane to each other, then it is probable, but not necessary, thatnt1 and nt3 form some kind of basepair. Moreover, in this case it is highly unlikely that nt1 and nt3 willthemselves form a stacking relation.
In other cases, nt1 and nt3 are necessarily different nucleotides because they stack on nt2 using differentfaces. Thus it follows from the definitions that:
If (nt1 stack_5′_5′ nt2) and (nt2 stack_5′_3′ nt3) then not (nt1 equal nt3).
Again, if it is also the case that nt1 and nt3 stack optimally on nt2, then it is very likely that nt1 and nt3
also form a basepair of some kind. This is because they are stacked on the same face of nt2 (the 5′-facein this case).
In the following case, nt1 and nt3 are also necessarily different nucleotides, but they cannot form abasepair because they are stacked on different faces of nt2. This also follows from the definitions:
If (nt1 stack_3′_5′ nt2) and (nt2 stack_3′_5′ nt3)
then [not (nt1 equal nt3) and not (nt1 pairs_with nt3) and not (nt1 stack nt3)].
These are the different possibilities that can occur when composing stacking relations. They are sum-marized in Table 3a, with logical statements, and in Table 3b, in a graphical manner.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 77
Table 3a
Composition table for base-stacking relations
Notes: Rows and columns correspond to different stacking relation between nt1 and nt2 and between nt2 and nt3, respectively.Possible relations between nt1 and nt3 are shown in the cells. These relations are illustrated graphically in Table 3b.
Table 3b
Base-stacking relation composition illustrated graphically
Notes: Each rectangle represents a nucleotide. The red colored region corresponds to the 3′-face and the blue region correspondsto the 5′-face. As in Table 3a, rows and columns correspond to different stacking relation between nt1 and nt2 and between nt2and nt3, respectively. Possible relations between nt1 and nt3 are shown in the cells. When nt1 and nt3 are stacked on the sameside of nt2, they usually, but not necessarily, form a basepair.
3.5.3. Composition of base-pairing relationsWhen base-pairing relations are composed, base-triples are formed, in which three bases interact
through hydrogen bonding. Such RNA arrangements are frequent and will be treated exhaustively ina forthcoming work (Abu Almakarem et al., 2010). The first issue is to identify which combinationsof basepair relations, R1 and R2, are compatible with each other under the general rule (which hasvery few exceptions) that a given edge of a nucleotide can interact only with one nucleotide at a timeto make a full edge-to-edge basepair interaction. We illustrate in Table 4 for the basepair relationsR1 = pairs_with_cWW, pairs_with_cWH and pairs_with_cHW holding in turn between nt1 andnt2. The second column of Table 4 shows which relations R2 can hold between nt2 and a third base nt3 �=nt1, given that R1 holds between nt1 and nt2. The third column lists relations which cannot hold betweennt2 and nt3. A more detailed analysis must consider each possible base combination for each R1 and
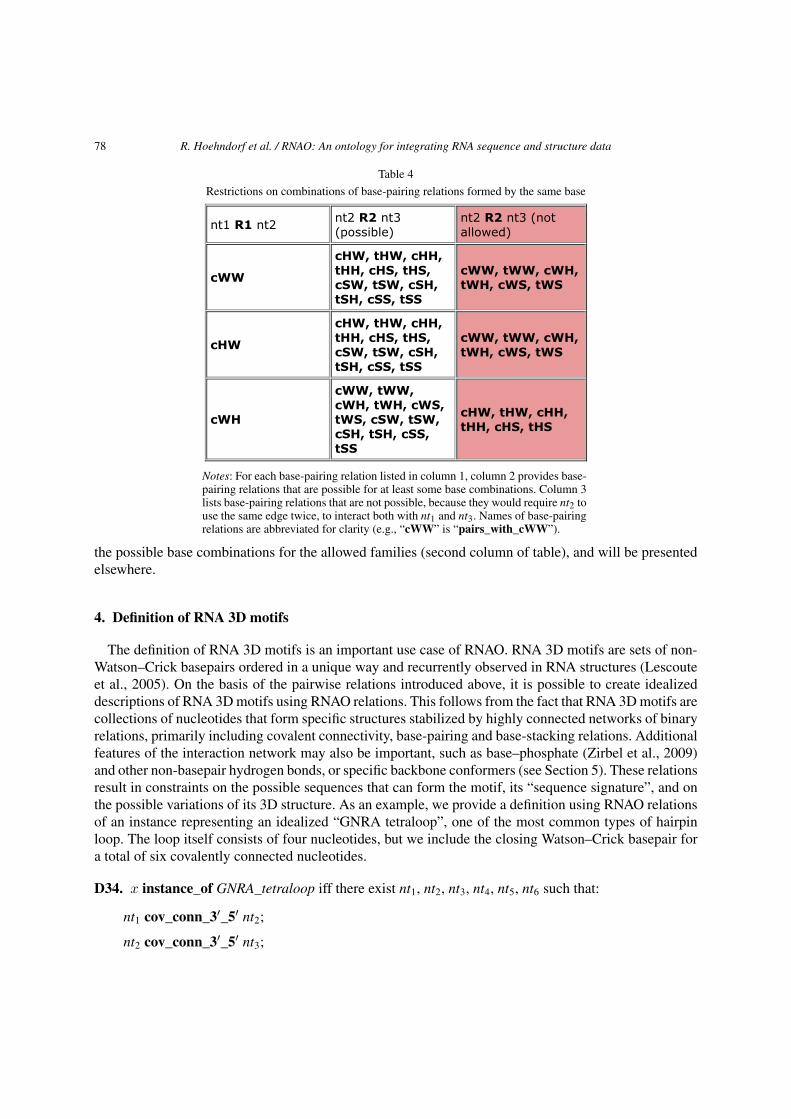
78 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
Table 4
Restrictions on combinations of base-pairing relations formed by the same base
Notes: For each base-pairing relation listed in column 1, column 2 provides base-pairing relations that are possible for at least some base combinations. Column 3lists base-pairing relations that are not possible, because they would require nt2 touse the same edge twice, to interact both with nt1 and nt3. Names of base-pairingrelations are abbreviated for clarity (e.g., “cWW” is “pairs_with_cWW”).
the possible base combinations for the allowed families (second column of table), and will be presentedelsewhere.
4. Definition of RNA 3D motifs
The definition of RNA 3D motifs is an important use case of RNAO. RNA 3D motifs are sets of non-Watson–Crick basepairs ordered in a unique way and recurrently observed in RNA structures (Lescouteet al., 2005). On the basis of the pairwise relations introduced above, it is possible to create idealizeddescriptions of RNA 3D motifs using RNAO relations. This follows from the fact that RNA 3D motifs arecollections of nucleotides that form specific structures stabilized by highly connected networks of binaryrelations, primarily including covalent connectivity, base-pairing and base-stacking relations. Additionalfeatures of the interaction network may also be important, such as base–phosphate (Zirbel et al., 2009)and other non-basepair hydrogen bonds, or specific backbone conformers (see Section 5). These relationsresult in constraints on the possible sequences that can form the motif, its “sequence signature”, and onthe possible variations of its 3D structure. As an example, we provide a definition using RNAO relationsof an instance representing an idealized “GNRA tetraloop”, one of the most common types of hairpinloop. The loop itself consists of four nucleotides, but we include the closing Watson–Crick basepair fora total of six covalently connected nucleotides.
D34. x instance_of GNRA_tetraloop iff there exist nt1, nt2, nt3, nt4, nt5, nt6 such that:
nt1 cov_conn_3′_5′ nt2;
nt2 cov_conn_3′_5′ nt3;

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 79
nt3 cov_conn_3′_5′ nt4;
nt4 cov_conn_3′_5′ nt5;
nt5 cov_conn_3′_5′ nt6;
nt1 pairs_with_cWW nt6;
nt2 pairs_with_tSH nt5;
nt1 stack_3′_5′ nt2;
nt3 stack_3′_5′ nt4;
nt4 stack_3′_5′ nt5;
nt2 instance_of Guanine;
nt4 instance_of Purine;
nt5 instance_of Adenosine;
x sum_of_collection (nt1, nt2, nt3, nt4, nt5, nt6).
Three representations of the 3D structure of an instance of a GNRA hairpin motif that conforms inall respects to this definition are shown in Fig. 6. In the left panel of Fig. 6, the covalent bonds betweenphosphate and ribose atoms are colored black and the bases are colored according to the scheme usedin the right panel. The hydrogen bonds of the basepaired nucleotides are indicated by dashed lines inthe left and center panels. The center panel indicates the stacking relationships, where the red shadedregion corresponds to the 3′-face and the blue shaded region corresponds to the 5′-face. The right panelshows the base-pairing and base–phosphate relations. Finally, the instance_of relations in the motifdescription above reflect the fact that the vast majority of hairpin loops having this geometry followthe pattern GNRA, where N stands for any base (A, C, G, U) and R stands for purines, A or G. Thisparticular instance, GAGA, conforms to this pattern.
Fig. 6. Three representations of a classic GNRA hairpin loop motif. Left: 3D structure with covalent bonds drawn as solid linesand hydrogen bonds as dashed lines. Middle: Bases represented as rectangles with 5′-faces colored blue and 3′-faces coloredred to show stacking relations. Right: Annotated 2D structure using the Leontis–Westhof symbols for basepairs (see Table 1).The curved arrow indicates that the chain changes direction between G2659 and A2660. A base–phosphate relation, found inmany GNRA instances, is also shown involving G2659. The specific instance shown is from the sarcin/ricin loop of E. coli 23SrRNA (PDB file: 1q9a at 1.04 Å resolution).

80 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
For the GNRA hairpin loop, there are three other features nearly always present:
(1) the N7(G)-to-OP2(A) base–phosphate hydrogen bond, shown at right in Fig. 6;(2) the 2′OH(G)-to-N7(R) base-ribose hydrogen bond, which explains the sequence preference for a
purine in the R position;(3) the 1g backbone conformer that provides the stack switch and change in chain direction between
G and N.
Like all physical molecules, RNA molecules are in constant molecular motion, even in a crystal; theconformation of a local motif is affected by the specific base sequence and by the structural context;moreover the experimental 3D models contain occasional errors, especially in base–base interactions,ribose pucker and backbone torsions. Thus, particular instances of a given motif from experimental 3Dstructures show some variability; not all instances will be found to have all of the pairwise interactionsidentified in the classic case. Many GNRA tetraloops have other pairwise relations that appear lessconserved than the ones listed in the definition, but which may, in fact, represent the idealized stateabout which the physical GNRA molecule fluctuates over time. This shows the need, in a future versionof the RNAO, to introduce relations that can account for experimental uncertainty, for local distortions,and for structural fluctuations and perturbations in response to environmental changes. In defining theGNRA motif we have chosen the set of relations least sensitive to variation or error and presumed to bemost conserved across real variations in the molecule. In summary, RNAO provides a powerful set oftools for annotating instances of 3D motifs and abstracting the most characteristic features to constructdefinitions of motif classes. These tools should enable the search within structural and sequence databases, as well as facilitate the transfer of information content between different levels of complexity.
5. Backbone conformers
To fully describe RNA 3D structure, we also need to specify and classify the conformations of thehighly flexible RNA backbone. Specific backbone conformations are characteristic of RNA sites that(1) bind ions, small molecules, proteins, or other nucleic acids segments or (2) that catalyze reactions.Furthermore, recurrent RNA motifs exhibit characteristic backbone conformations in high-resolutionstructures, knowledge of which is useful for generating theoretical models or evaluating less accuratestructures.
In RNAO we need both a generic definition of the backbone that includes all the atoms in the RNAmolecule that do not belong to the nucleobase (in common usage, the “Sugar–phosphate backbone”) anda more restrictive definition referring to the shortest-path linear chain of covalently bonded atoms thatspans the length of the molecule. The generic definition will be needed to specify atoms that take partin non-covalent backbone–backbone or base–backbone interactions, while the restrictive linear chainserves to define torsion angles sufficient for specifying backbone conformations. We therefore focushere on the restrictive definition, to which we refer as “the linear backbone”.
The linear backbone of an RNA molecule comprises a continuous chain of covalently bonded atomsthat runs the length of the molecule. Some of the atoms of the backbone are parts of the phosphate sub-molecules (O5′, P, O3′) and others are parts of the ribose rings (C3′–C4′–C5′). The atoms of the linearbackbone are linked to each other by single bonds that are subject to torsional rotation with 3-fold,2-fold or more complex preference patterns (Richardson et al., 2008). The torsion angles are named α toζ successively along the chain, starting at the phosphate with α, the O3′–P–O5′–C5′ torsion angle (see

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 81
Fig. 2). Thus, the RNA backbone has many degrees of freedom and each set of adjacent torsion anglesdefines a particular backbone conformation and trajectory of the RNA chain through 3D space.
As described earlier and illustrated in Fig. 2, the RNA molecule can be partitioned in two differentways. The first, more traditional partition is in non-overlapping nucleotide residues, separated by thecovalent boundary between the O3′ of one nucleotide and the P atom of the subsequent nucleotide. Thesecond partition is into suites, which consist of two consecutive nucleobases and ribose rings, plus thebackbone and phospho-diester linkage between them (Murray et al., 2003; Richardson et al., 2008).Successive suites overlap, and a given suite starts at C4′ of one nucleotide and ends at C3′ of the nextnucleotide; a suite can be designated either using both residue numbers or more simply by just givingthe number of the second residue, which contributes the P atom and four of seven torsion angles of thesuite. The backbone conformation of a suite depends on the values of the 7 linear backbone torsion anglesincluded within the suite, starting with δ, ε and ζ of Nti−1, and continuing with α, β, γ and δ of Nti. Suitesare especially useful for classifying backbone conformations because they relate the positions of twoconsecutive nucleotides, and because the values of their torsion angles are much more strongly correlatedthan are values of the six torsions, α to ζ, within a single nucleotide. In the high-dimensional space ofsuite backbone torsion angles, there is a small number of energetically favorable local minima, knownas RNA “backbone conformers”. A suite backbone conformer is a distinct, restricted, and recurrentlyobserved region in the 7-dimensional conformation space, representing a local energy minimum, inanalogy with side-chain rotamers in proteins (Lovell et al., 2000).
To formalize these ideas, we first define the suite_of relation (D35) and use this definition to defineSuite (D36). This allows us to define the relation linear_RNA_backbone_of in a general way, so it canapply to an RNA molecule, to a nucleotide, or to a suite.
D35. Suite_of: x suite_of m iff m instance_of 5′_to_3′_RNA_molecule and x part_of m and thereexist nti, ntj , such that: {nti, ntj instance_of Nucleotide and nti part_of m and nti part_of m andnti cov_conn_3′_5′ ntj and there exists R such that: [R instance_of Collection_of_atoms and for all zsuch that: (if (z part_of nti and z instance_of either O2′ or O3′ or O4′ or C1′ or C2′ or C3′ or C4′ orNucleobase) or (z part_of ntj and z instance_of either P or OP1 or OP2 or O2′ or O4′ or O5′ or C2′ orC1′ or C3′ or C4′ or C5′ or Nucleobase) then z member_of R and x covalently_bonded_sum_of R)]}.
D36. Suite. x instance-of Suite iff there exists some y, such that: x suite_of y.
We define the relation linear_RNA_backbone_of so that the linear RNA backbone of an entity is thecovalently_bonded_sum_of all phosphate atoms O5′, P and O3′ and all ribose atoms C3′, C4′ and C5′
that belong to that entity.
D37. Linear_RNA_backbone_of. x linear_RNA_backbone_of m iff {(m instance_of 5′_to_3′_RNA_molecule or m instance_of Nucleotide or m instance_of Suite) and there exists R such that[R instance_of Collection_of_atoms and for all z, if z part_of m and z instance_of (P or O3′ or O5′ orC3′ or C4′ or C5′) then z member_of R] and x covalently_bonded_sum_of R}.
The RNA backbone working group of the ROC has defined a community-consensus nomenclature andidentified, to date, 54 recurrent RNA backbone conformers (Richardson et al., 2008), each of which isa region in 7-dimensional torsion space around a specific combination of energetically favorable valuesfor the 7 torsion angles in a suite. Each suite conformer is assigned a 2-character number–letter name,such as 1a for the standard A-form conformation and 5z for the suite that makes the initial S-curve of the

82 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
S-motif. The number codes the approximate δ–ε–ζ torsion values for the first half of the suite (odd if thefirst ribose ring has the conformation C3′-endo pucker, even if C2′-endo pucker), and the letter codes theapproximate α–β–γ–δ torsion values for the second half of the suite (early alphabet half if the secondribose has C3′-endo pucker, and late alphabet if C2′-endo). Because the conformers were developed fromempirical study of the data-point clusters in the 7-dimensional torsion-angle space, suite conformersare defined only for the case of normal cov_conn_3′_5′ connectivity. The rules for assigning suite-conformer identities for a given RNA structure have been described in previous work and implementedin the Suitename program (Richardson et al., 2008). Suite-conformer identities can also be obtained on-line by running a structure through the MolProbity web service at http://molprobity.biochem.duke.edu(Chen et al., 2010).
Initially, we considered representing backbone conformers using sub-relations of covalent connectionrelations, for example, specializing cov_conn_3′_5′ into cov_conn_3′_5′_1g or cov_conn_3′_5′_5z.However, it subsequently became clear that conformers are better represented as time-varying quali-ties of covalent structures rather than sub-types. Therefore, we plan in future work to add conformerqualities to RNAO to represent suite backbone conformers. We anticipate this approach will also beapplicable to ribose ring pucker conformers, which are currently not represented in RNAO. We furtheranticipate that this change will facilitate the integration of representations of backbone conformers andtheir composition rules with the various aspects of local structure description in previous sections of thispaper.
Because suites overlap at torsion angle δ (cf. Fig. 2), there are strict restrictions on consecutive suiteconformers as shown in Table 5 (where the * in a two-character name acts as a wild card and possiblealternative characters are provided in parentheses). For instance, *a 1* is a valid consecutive pair, be-cause the shared ribose ring is in the conformation C3′-endo, whereas *a 2* is prohibited because thiswould require the shared ribose ring to simultaneously have a C2′-endo and a C3′-endo conformation,which is not possible.
The two-character suite-conformer names can be alternated with base names to provide a concise,string description that encodes both sequence and 3D backbone conformation for a stretch of RNA.Thus, each structural motif can be described by its consensus string. For instance, if nt1 and nt6 arecWW basepaired then the following conformer string represents a GNRA tetraloop having the relations
Table 5
Restrictions on two consecutive suite conformers
Suite 2 (1, 3, 5, 7, 9, &) * (2, 4, 6, 8, 0, #) *Suite 1 C3′-endo C2′-endo* (a, c–n) required prohibitedC3′-endo* (b, o–z,[ ) prohibited requiredC2′-endo
Notes: Backbone conformer names are composed of a number followed by a letter. The numbercodes the approximate δ–ε–ζ values for residue i − 1 of the suite, which includes the i −1 ribose pucker; the letter codes the approximate α–β–γ–δ values for residue i of the suite,which includes the i ribose pucker. Consecutive suites overlap at the ribose ring, which thereforerestricts the possible combinations of consecutive suite conformers, as shown in the table. Theasterisk (“*”) acts as a wildcard for the first part of the name of suite 1 and the second part ofthe name of suite 2. Possible alternative charactes are given in parentheses.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 83
listed in D34:
N 1a G 1g N 1a R 1a A 1c N 1a.
Geometrically, the 1g suite swings the chain up and over into the NRA base stack, and the 1c suitemakes a small adjustment back into alignment with the stem underneath the tetraloop. We anticipatebeing able to express backbone interaction sites in a similar way. For example, N 1[ N defines a con-former that provides a site where a third base can intercalate. In only 12 of the 54 suite conformersis it possible to position the two consecutive bases to allow for intercalation of another base or smallmolecule between them; the commonest intercalation sites are 1[ and 1m. Certain suites imply specificinteractions. For example, if suite i has conformer 5z, then there is a backbone hydrogen bond betweenOP2(i − 1) and O2′(i). Other suites exclude certain interactions. Thus, if suite i has conformer 1e, thennt(i−1) cannot stack on nt(i1). In future work, relevant axioms and theorems will be included expressingsuch restrictions.
6. RNA alignments
The integration of 3D structural data with RNA sequence data is a central reason for creating RNAO.Like other biopolymers, RNA molecules constitute families related by common descent or by commonstructure and function. Usually these groupings coincide. Thus, all 16S-like “Small-subunit ribosomalRNAs” (“SSRs”) share descent from a common ancestor as well as a common core 3D structure. Theyalso share common functions in their respective ribosomes. By contrast, “hammerhead ribozymes” havebeen isolated from unrelated organisms and at least twice by in vitro selection from random-sequencepools. Thus hammerhead ribozymes share a common structure and function but not a common ancestor(Salehi-Ashtiani & Szostak, 2001; Tang & Breaker, 2000). For a discussion, the reader can consultBrown et al. (2009) and Hammann and Westhof (2007).
Biopolymers that belong to the same family share a common 3D structure more conserved than thenucleotide sequence. Thus, many sequences can fold into a similar 3D core architecture. The purpose ofaligning the nucleotide sequences of related RNA molecules is to place parts of each RNA molecule intocorrespondence with equivalent parts of other RNA molecules of the same family to make the commonstructural features explicit. If the structures are sufficiently conserved, the common structural featurescan include specific pairing, stacking, or other binary spatial relations. Well-constructed sequence align-ments have multiple uses in biological research and are powerful tools for inferring biological function,establishing phylogenetic relationships and modeling 3D structures. High-quality sequence alignmentsare characterized by the accuracy with which correspondences are defined. Here, our concern is to definethe correspondence relations pertaining to RNA molecules. This is crucial for accurate transfer of anno-tations of structural relations (for example base-pairing or base-stacking relations) from 3D structures tocorresponding parts of aligned RNA sequences.
In its most general form, the correspondence relation (“corresponds_to”) has as domain a collec-tion of nucleotides, all of which are parts of one RNA molecule, and has as its range a collection ofnucleotides that are part of a second RNA molecule and related to the first molecule by evolution orstructure or usually, both.
In some cases, it is possible to identify individual nucleotides or basepairs in each RNA moleculethat can be placed into correspondence, either because they can be shown to share common descent or

84 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
because they coincide in 3D space when the structures of the RNA molecules are superposed. The mostreliable correspondences at the nucleotide level are made on the basis of both evolutionary and structuralevidence. We take the general correspondence relation between arbitrary collections of nucleotides asprimitive and introduce it with an Axiom A18. Its domain and range are collections of nucleotides.
A18. If x corresponds_to y then [x and y instances_of Collection and there exist an M1, M2 such that(M1, M2 instance_of 5′_to_3′_RNA_Molecule and for all z ((z member_of x implies z part_of M1)and (z member_of y implies z part_of M2)))].
We specialize this relation to the nucleotide level with D36. The specialized relation, corre-sponds_to_nt_nt, has as its domain individual nucleotides in one RNA molecule and for its rangeindividual nucleotides in a second RNA molecule.
D38. Corresponds_to_nt_nt. x corresponds_to_nt_nt y iff x corresponds_to y and there exist ex-actly one a and one b such that (a member_of x and b member_of y and a, b instance_of Nucleotide).
Using this new relation, we can define a correspondence relation for basepairs, “corresponds_to_bp_bp”, for which the domain is a basepair in one RNA molecule and the range is a basepair in a secondRNA molecule. We denote a basepair as an ordered pair of nucleotides, for example “(x1, x2)”.
D39. Corresponds_to_bp_bp. (x1, x2) corresponds_to_bp_bp (y1, y2) iff (x1, x2, y1) and y2 in-stances_of Nucleotide and there exists (X and Y ) such that (X and Y ) instances_of 5′_to_3′_RNA_Molecule and (x1 and x2) part_of X and (y1 and y2) part_of Y and x1 corresponds_to_nt_nt y1 andx2 corresponds_to_nt_nt y2 and x1 pairs_with x2 and y1 pairs_with y2.
We note that when corresponds_to_bp_bp holds, the pairs (x1, x2) and (y1, y2) almost always formthe same type of basepair, e.g., tWH, cSH, or cWW (Stombaugh et al., 2009). Thus, an important usecase of RNAO is to evaluate the quality of multiple sequence alignments, by checking whether the corre-sponding basepairs in each sequence of an alignment can form the annotated basepair. For example, if thecWW relation is assigned to a pair of columns of the alignment for which the corresponds_to_bp_bprelation is also assigned and one of the sequences contains two Gs in these columns, this will be flaggedas not consistent with the annotations because the GG base combination does not occur in the cWWbasepair family. This information can be used to re-evaluate the alignment of that sequence or the as-signment of the correspondence relation.
Very often, when aligning RNA molecules, it is neither possible (due to lack of information) normeaningful (due to sequence divergence) to establish nucleotide or basepair level correspondences forall parts of each RNA. The lack of information may be due, for example, to a lack of a sufficient numberof suitably divergent RNA sequences or 3D structures. Further, RNA molecules may have diverged somuch in the course of evolution, by insertion, deletion or rearrangement of sequence elements, that it isimpossible to assign correspondences at the nucleotide or basepair level. Even so, it may still be possibleto assign correspondence at the level of larger elements or domains of structure. Possible cases of thistype were identified and examples from real RNA molecules provided in a recent ROC publication(Brown et al., 2009). Suggestions were made there for additional correspondence relations. Future workwill formalize these sub-relations of the corresponds_to relation.
We conclude by noting that corresponds_to and its sub-relations are reflexive, symmetric and tran-sitive. Additional relations will be developed in future work to record the kinds of evidence supportingcorrespondence relations between parts of RNA molecule belonging to the same family.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 85
7. Foundation in top-level ontology
The axioms of the RNA Ontology are self-contained and do not rely on other ontologies. However,to facilitate interoperability between the RNAO and other ontologies, as well as to provide additionalconstraints on primitive classes and relations, a foundation in a top-level ontology is beneficial. The BFO(http://www.ifomis.org/bfo) and the RO (Smith et al., 2005) are two complementary top-level ontologiesof particular relevance in the biomedical domain. To interoperate with ontologies based on either theBFO or the RO, we re-used several classes and relations from these ontologies. However, we provideadditional axioms for these classes and relations which may not be generally applicable in other domains.
In particular, we use the axioms of extensional mereology (Simons, 1987) for parthood relations, andprovide a weak characterization of boundaries within RNAO. For this purpose, we treat boundaries asdependent continuants and not subclasses of BFO’s Object boundary. In RNAO, we re-use the continuantclasses from BFO, in particular Material Object. We ignore the distinctions that BFO makes betweenthe subclasses of Material Object, because several classes in RNAO can become subclasses of eitherObject or Object Aggregate, which are disjoint in BFO. The assumptions we make about these classesare explicit in the axiom system, and we believe that they are compatible with other top-level ontologiessuch as the Descriptive Ontology for Linguistic and Cognitive Engineering (DOLCE) (Masolo et al.,2003) or the General Formal Ontology (GFO) (Herre et al., 2006). Both DOLCE and GFO containclasses for material objects. DOLCE does not contain a theory of boundaries, and the basic axiomswe provide can be used to introduce boundaries. The GFO provides a theory of boundaries (Baumann,2009) that is distinct from our assumption that boundaries are parts of objects. To integrate the RNAOand GFO, we have to remove the assertion that boundaries are parts of objects, and GFO’s axioms forboundaries can compensate for the loss of inference power obtained by removing the axiom.
8. RNAO and the Sequence Ontology
A close neighbor of the RNAO in the OBO library is the Sequence Ontology (SO) (Eilbeck et al.,2005). SO is a structured controlled vocabulary for the description of biological sequences. It is primarilyused for the annotation of genomic sequences in DNA, or in RNA in the case of RNA viruses, butcan equally be used for the annotation of sequences derived from genomic sequences such as thosein RNA and in protein sequence (Reeves et al., 2008). The main branch of SO provides annotationsfor sequence features. A sequence feature is an extent of biological sequence that has start and endcoordinates with respect to a reference, such as one end of a chromosome. The SO is used by manymodel organism genome databases to type the kinds of features located within the sequence (Eilbeck etal., 2005; Mungall & Emmert, 2007). SO is the basis of the GFF3 file format, one of the major formatsfor exchanging genomic data.
Like RNAO, SO is a member of the OBO Library (Smith et al., 2007). To facilitate interoperabilitybetween ontologies, SO entities are classified according to the Basic Formal Ontology (BFO), and the re-lations used by the ontology are taken, where possible, from RO. Earlier versions of SO described the re-lations between features with a mereotopology built from the part_of relation. To better describe therelationships between SO features, over 20 more specific spatial and temporal relations were recentlyintroduced (Mungall et al., 2010). In SO, sequence features are defined to be generically dependentcontinuants. This means that sequence features inhere within the bearer molecules (the independentcontinuants), by virtue of the shapes (the specifically dependent continuants) of the individual mole-cules. Sequence features are themselves transferable through copying. Therefore, SO itself does not

86 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
represent molecules. Equally, molecules encoded by the genome are explicitly excluded from the scopeof Chemical Entities of Biological Interest (ChEBI), the OBO ontology for small molecules. This gapin the OBO ontologies is being filled by a new project called Sequence Ontology: Molecules (SOM)(Mungall et al., 2010) which does represent the polymeric molecules in which the sequence featuresinhere. SOM is tied closely to SO and equivalent terms share the numerical part of their ID, for exampleintron as a feature has the ID SO:0000188 and intron as a segment of an RNA molecule has the IDSOM:0000188. The development of SOM facilitates automatic ontology development by cross productsbetween OBO ontologies. The monomeric residues of which the polymeric molecules are composed are,however, within the scope of ChEBI, and therefore where a logical definition that relies on a residue isneeded then SOM uses a class from ChEBI. SOM will be of use to RNAO development by providing thetypes of polymeric molecules not currently present in existing ontologies such as ChEBI, with which toassociate RNA annotations. Thus, RNAO, SO and SOM are closely associated. SO represents sequencefeatures for which SOM represents the molecule and RNAO represents the two-dimensional or three-dimensional structure. For example, SO represents the many kinds of functional RNA transcribed bythe genome, whereas RNAO provides the means to describe and catalogue their secondary and tertiarystructures. The new relations in SO that relate features of sequences as they are copied from one kindof molecule to another through biological stages and inhere within different kinds of molecule are rele-vant to the integration of SO and RNAO. In response to the development of the RNAO there are termsin SO which describe structural RNA elements, that are being deprecated from and transitioned intoRNAO, including base_pair and its descendents, and structural motifs such as stem_loop, pseudoknotand g_quartet. RNA sequence features will be submitted to the Sequence Ontology Term tracker foraddition into SO. Orthogonality will be maintained through this division of labor and by communication,such as workshops, between the two groups.
9. Ontology implementation and administration
The RNA Ontology is available in three versions, an implementation in first-order logic, in OWL andin the OBO Flatfile Format (http://www.geneontology.org/GO.format.obo-1_2.shtml). The axioms anddefinitions we provide in this paper are implemented using the SPASS theorem prover (Weidenbach etal., 2009). SPASS is a theorem prover for first-order logic with equality. Some axioms in this manuscriptrequire second-order logic, in particular the transitive closure of relations, and could not be implementedcompletely using SPASS. Instead, we approximate these by introducing a transitive relation that includesthe original relations. We do not state that this relation is the smallest relation satisfying the axioms.
The axioms in first-order logic serve as a reference for the definitions of the classes in RNAO, whichcan be used by both humans and machine as documentation of the basic distinctions that RNAO provides.We used the SPASS theorem prover to verify the theorems we state in the text. The natural languagestatements of the theorems are included as comments in the SPASS input file. In addition to the axiomsin first-order logic, we are also making available an implementation in OWL and the OBO Flat fileFormat. The implementation of the OWL version of the RNA Ontology was guided by the axioms andinferences using the SPASS reasoner, and the top-level distinctions made by RNAO are present both inthe SPASS and OWL version. The OWL version contains several specific domain classes that are notincluded in the SPASS version of RNAO. The OBO version of the RNAO was automatically generatedusing a transformation from OWL to OBO. In the OWL and OBO versions. The implementation isavailable from http://code.google.com/p/rnao. This site includes an issue tracker and we welcome inputfrom readers and users of the ontology.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 87
10. Conclusions
In this paper we have presented a description of RNA structure in formal language that is easilytranslated into first-order logic, or, when necessary, second-order logic. We have described the com-ponents from which RNA molecules are built up through covalent bonding, the local conformationsof those polynucleotide chains, and the dominant forms of internal interactions through edge-to-edgebase-pairing and face-to-face stacking contacts. This is built on a foundation of a description of cova-lent and non-covalent interactions that should be useful to other molecular level ontologies, for exampleChEBI. Future extensions will address non-basepair hydrogen bonds, modified bases, and unusual con-nectivities (such as lariat loops involving 2′-5′ and 3′-5′ phospho-diester linkages). We anticipate thatthe RNAO edge-to-edge, stacking, and other interactions will be useful for future ontological work de-scribing protein–RNA, protein–DNA and RNA–DNA interactions.
We have also outlined a set of relations describing sequence and structure alignments that will befurther elaborated elsewhere. While useful for RNA, this new approach should be equally applicable toprotein sequence alignments.
We have described how RNAO builds upon and interacts with existing domain ontologies in neigh-boring fields such as ChEBI and SO, and also how it relates to foundational work in BFO, RO andGFO. Very important future work will focus on investigating tractable methods for spatial reasoning,based on the RNA Ontology, over experimental RNA structures as recorded in, for example, the NDB.Spatial reasoning is known in general to be poorly handled by existing OWL reasoners. Expressing realstructural variability, and perhaps even allowing for experimental error, would be extremely valuablebut may remain outside the feasible scope of the ontology. We also plan to investigate methods for tem-poral reasoning over RNA structures, extending RNAO to accommodate time dependence of molecularstructures, including interactions with other molecules and structural changes arising from them. Forthis purpose, we will use temporally indexed relations together with an ontological representation ofmolecular processes. The RNAO in its current form serves as a foundation for these efforts by providingthe means to specify RNA molecules at time points.
Acknowledgements
This paper is the result of collaborative meetings sponsored by the RNA Ontology Consortium (ROC).We thank all individuals who have taken part in ROC meetings and contributed their comments to im-prove the paper. We especially thank Fabian Neuhaus and the anonymous reviewers of this manuscriptfor their carefully reading and constructive and thoughtful comments. The ROC is supported by a Re-search Coordination Network (RCN) grant from the National Science Foundation (Grant no. 0443508).
References
Abu Almakarem, A., Petrov, A., Stombaugh, J., Zirbel, C.L. & Leontis, N.B. (2011). Comprehensive survey and geometricclassification of base triples in RNA structures. Submitted.
Altman, R.B., Michael, B., Chai, X.J., Michelle Whirl, C., Chen, R.O. & Abernethy, N.F. (1999). RiboWeb: an ontology-basedsystem for collaborative molecular biology. IEEE Intell. Syst., 14(5), 68–76.
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M. et al. (2000). Gene ontology: tool for the unificationof biology. Nat. Genet., 25(1), 25–29.
Batchelor, C. (2008). An upper-level ontology for chemistry. In Proceeding of the 2008 Conference on Formal Ontology inInformation Systems: Proceedings of the Fifth International Conference (FOIS 2008).

88 R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data
Baumann, R. (2009). The mereotopological structure of the brentanoraum B(3) and material entities. Unpublished MSc thesis,Department of Mathematics, Universität Leipzig, Leipzig, Germany.
Berman, H.M., Olson, W.K., Beveridge, D.L., Westbrook, J., Gelbin, A., Demeny, T. et al. (1992). The nucleic acid database.A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J., 63(3), 751–759.
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H. et al. (2000). The protein data bank. Nucleic AcidsRes., 28(1), 235–242.
Bittner, T., Donnelly, M. & Smith, B. (2009). A spatio-temporal ontology for geographic information integration. Int. J. Geogr.Inf. Sci., 23(6), 765–798.
Bittner, T. & Smith, B. (2003). Vague reference and approximating judgments. Spatial Cognit. Comput., 3(2), 137–156.Brown, J.W., Birmingham, A., Griffiths, P.E., Jossinet, F., Kachouri-Lafond, R., Knight, R. et al. (2009). The RNA structure
alignment ontology. RNA, 15(9), 1623–1631.Chen, R.O., Felciano, R. & Altman, R.B. (1997). RIBOWEB: linking structural computations to a knowledge base of published
experimental data. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology (Vol. 5,pp. 84–87).
Chen, V.B., Arendall 3rd, W.B., Headd, J.J., Keedy, D.A., Immormino, R.M., Kapral, G.J. et al. (2010). MolProbity: all-atomstructure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr., 66(Pt 1), 12–21.
Crane, J. (2004). On the metaphysics of species. Philosophy of Science, 71(2), 156–173.Degtyarenko, K., de Matos, P., Ennis, M., Hastings, J., Zbinden, M., McNaught, A. et al. (2008). ChEBI: a database and
ontology for chemical entities of biological interest. Nucleic Acids Res., 36(Database issue), D344–D350.Dunin-Horkawicz, S., Czerwoniec, A., Gajda, M.J., Feder, M., Grosjean, H. & Bujnicki, J.M. (2006). MODOMICS: a database
of RNA modification pathways. Nucleic Acids Res., 34(Database issue), D145–D149.Eilbeck, K., Lewis, S.E., Mungall, C.J., Yandell, M., Stein, L., Durbin, R. et al. (2005). The Sequence Ontology: a tool for the
unification of genome annotations. Genome Biol., 6(5), R44.Eilbeck, K. & Mungall, C.J. (2009). Evolution of the sequence ontology terms and relationships. Nature Precedings. Available
at: http://dx.doi.org/10.1038/npre.2009.3495.1. Posted 28 July 2009.Hammann, C. & Westhof, E. (2007). Searching genomes for ribozymes and riboswitches. Genome Biol., 8(4), 210.Herre, H., Heller, B., Burek, P., Hoehndorf, R., Loebe, F. & Michalek, H. (2006). General Formal Ontology (GFO): A Foun-
dational Ontology Integrating Objects and Processes. Part I: Basic Principles (Version 1.0), Research Group Ontologies inMedicine (Onto-Med), University of Leipzig.
Hull, D.L. (1978). A matter of individuality. Philosophy of Science, 45, 335–360.Leontis, N.B., Altman, R.B., Berman, H.M., Brenner, S.E., Brown, J.W., Engelke, D.R. et al. (2006). The RNA Ontology
Consortium: an open invitation to the RNA community. RNA, 12(4), 533–541.Leontis, N.B., Stombaugh, J. & Westhof, E. (2002). The non-Watson–Crick base pairs and their associated isostericity matrices.
Nucleic Acids Res., 30(16), 3497–3531.Leontis, N.B. & Westhof, E. (2001). Geometric nomenclature and classification of RNA base pairs. RNA, 7(4), 499–512.Lescoute, A., Leontis, N.B., Massire, C. & Westhof, E. (2005). Recurrent structural RNA motifs, isostericity matrices and
sequence alignments. Nucleic Acids Res., 33(8), 2395–2409.Lovell, S.C., Word, J.M., Richardson, J.S. & Richardson, D.C. (2000). The penultimate rotamer library. Proteins: Struct. Func.
Genet., 40, 389–408.Masolo, C., Borgo, S., Gangemi, A., Guarino, N. & Oltramari, A. (2003). WonderWeb deliverable D18 ontology library (final).
Available at: wonderweb.semanticweb.org/deliverables/documents/D18.pdf.Mungall, C.J., Batchelor, C. & Eilbeck, K. (2010). Evolution of the Sequence Ontology terms and relationships. J. Biomed.
Inform., available at: doi: 10.1016/j.jbi.2010.03.002.Mungall, C.J. & Emmert, D.B. (2007). A Chado case study: an ontology-based modular schema for representing genome-
associated biological information. Bioinformatics, 23(13), i337–i346.Murray, L.J., Arendall 3rd, W.B., Richardson, D.C. & Richardson, J.S. (2003). RNA backbone is rotameric. Proc. Natl. Acad.
Sci. USA, 100(24), 13904–13909.Nasalean, L., Stombaugh, J., Zirbel, C.L. & Leontis, N.B. (2009). RNA 3D structural motifs: definition, identification, anno-
tation, and database searching. In N.G. Walter, S.A. Woodson & R.T. Batey (Eds.), Non-Protein Coding RNAs (pp. 1–26).Berlin–Heidelberg: Springer.
Reeves, G.A., Eilbeck, K., Magrane, M., O’Donovan, C., Montecchi-Palazzi, L., Harris, M.A. et al. (2008). The Protein FeatureOntology: a tool for the unification of protein feature annotations. Bioinformatics, 24(23), 2767–2772.
Richardson, J.S., Schneider, B., Murray, L.W., Kapral, G.J., Immormino, R.M., Headd, J.J. et al. (2008). RNA backbone:consensus all-angle conformers and modular string nomenclature (an RNA Ontology Consortium contribution). RNA, 14(3),465–481.
Salehi-Ashtiani, K. & Szostak, J.W. (2001). In vitro evolution suggests multiple origins for the hammerhead ribozyme. Nature,414(6859), 82–84.
Simons, P.M. (1987). Parts: A Study in Ontology/Peter Simons. Oxford: Clarendon Press.

R. Hoehndorf et al. / RNAO: An ontology for integrating RNA sequence and structure data 89
Smith, B., Ashburner, M., Rosse, C., Bard, J., Bug, W., Ceusters, W. et al. (2007). The OBO Foundry: coordinated evolution ofontologies to support biomedical data integration. Nat. Biotechnol., 25(11), 1251–1255.
Smith, B., Ceusters, W., Klagges, B., Kohler, J., Kumar, A., Lomax, J. et al. (2005). Relations in biomedical ontologies. GenomeBiol., 6(5), R46.
Smith, B. & Varzi, A.C. (1997). Fiat and bona fide boundaries: Towards on ontology of spatially extended objects. In SpatialInformation Theory (pp. 103–119).
Smith, B. & Varzi, A.C. (2000). Fiat and bona fide boundaries. Philosophy and Phenomenological Research, 60(2), 401–420.Stombaugh, J., Zirbel, C.L., Westhof, E. & Leontis, N.B. (2009). Frequency and isostericity of RNA base pairs. Nucleic Acids
Res., 37(7), 2294–2312.Tang, J. & Breaker, R.R. (2000). Structural diversity of self-cleaving ribozymes. Proc. Natl. Acad. Sci. USA, 97(11), 5784–5789.Thompson, J.D., Holbrook, S.R., Katoh, K., Koehl, P., Moras, D., Westhof, E. et al. (2005). MAO: a Multiple Alignment
Ontology for nucleic acid and protein sequences. Nucleic Acids Res., 33(13), 4164–4171.Villanueva-Rosales, N. & Dumontier, M. (2007). Describing chemical functional groups in OWL-DL for the classification of
chemical compounds. In OWL: Experiences and Directions, Innsbruck, Austria.Waugh, A., Gendron, P., Altman, R., Brown, J.W., Case, D., Gautheret, D. et al. (2002). RNAML: a standard syntax for
exchanging RNA information. RNA, 8(6), 707–717.Weidenbach, C., Dimova, D., Fietzke, A., Kumar, R., Suda, M. & Wischnewski, P. (2009). SPASS version 3.5. In Proceedings
of the 22nd International Conference on Automated Deduction.Zirbel, C.L., Sponer, J.E., Sponer, J., Stombaugh, J. & Leontis, N.B. (2009). Classification and energetics of the base–phosphate
interactions in RNA. Nucleic Acids Res., 37(15), 4898–4918.




















