
The pJan25 vector series: An enhancement of the gateway-compatible vector pGWB533 for broader...
8
Short Communication The pJan25 vector series: An enhancement of the Gateway-compatible vector pGWB533 for broader promoter testing applications Ahmed Mansour Alzohairy a,b , Margaret H. MacDonald b , Benjamin F. Matthews b,⇑ a Genetics Department, Faculty of Agriculture, Zagazig University, Egypt b USDA-ARS Soybean Genomics and Improvement Laboratory, PSI, Beltsville, MD 20705, United States article info Article history: Received 31 August 2012 Accepted 14 January 2013 Available online 23 January 2013 Communicated by Dr. G.J. Phillips Keywords: Plasmid pJan25 Gateway eGFP GUS Promoter abstract Agrobacterium-mediated transformation of plants has enhanced our ability to progress more rapidly in plant genetic engineering. Development of binary vectors for Agrobacte- rium has played a major role in advancing plant biology. Here, we report new features added to the Gateway-compatible vector pGWB533 for promoter testing with the reporter gene encoding b-glucuronidase (GUS). The original vector contains the spectinomycin/ streptomycin adenylyltransferase (aadA) gene for bacterial selection and the hygromycin phosphotransferase gene (hpt) for transformed plant selection. However, some bacterial strains used to transform plants, such as Agrobacterium rhizogenes strain K599, have ele- vated tolerance to spectinomycin and streptomycin, thus making bacterial selection of pGWB533 inefficient. Although pGWB533 confers chemical selection for transgenic plants using hygromycin resistance, the plasmid has no visual marker that enables visual selec- tion of transformed plants or transgenic tissue. In this regard, adding a gene to constitu- tively express green fluorescent protein (eGFP) makes it easier to visually select the transformed tissue and trim out the non-transformed. In this report we describe a series of vectors, pJan25S (NCBI: KC416200), pJan25T (NCBI: KC416201) and pJan25X (NCBI: KC416202), that are enhancements of pGWB533 for pro- moter testing. All three vectors contain the gene encoding eGFP as a visual marker for transformed tissue. However, in pJan25S and pJan25T, eGFP is controlled by the rolD pro- moter for root-specific expression, while in pJan25X it is controlled by the CaMV35S pro- moter for constitutive expression in all plant tissues. Spectinomycin and streptomycin resistance remains in pJan25S for bacterial selection; however, pJan25T and pJan25X con- tain the gene encoding tetracycline resistance (tet) for bacterial selection. These changes resulted in enhanced vectors with better visual and chemical selection that should have broad application in promoter studies. Published by Elsevier Inc. 1. Introduction Many applications in biotechnology require promoters that provide specific temporal and spatial gene expression at a certain level. This can be achieved through the use of specific promoters to express a gene of interest in the cor- rect amount at the specific place and time where it is needed. In this regard, testing promoters to evaluate their temporal and spatial specificity and level of expression is an important area of study in genomics. Many expression vectors with different resistance and reporter genes were developed by Nakagawa et al. (2007) using the Gateway Ò recombinase cloning system (Invitrogen, Carlsbad, CA). Most of these vectors were derived from pPZP221 0147-619X/$ - see front matter Published by Elsevier Inc. http://dx.doi.org/10.1016/j.plasmid.2013.01.005 ⇑ Corresponding author. Address: USDA-ARS PSI, Soybean Genomics and Improvement Laboratory, 10300 Beltsville Ave., Bldg., 006, Beltsville, MD 20705, United States. Tel.: +1 (301) 504 5730; fax: +1 (301) 504 5728. E-mail addresses: [email protected], [email protected] (B.F. Matthews). Plasmid 69 (2013) 249–256 Contents lists available at SciVerse ScienceDirect Plasmid journal homepage: www.elsevier.com/locate/yplas
Transcript of The pJan25 vector series: An enhancement of the gateway-compatible vector pGWB533 for broader...

Plasmid 69 (2013) 249–256
Contents lists availabl e at SciVerse ScienceDi rect
Plasmid
journal homepage: www.elsevier .com/ locate/yplas
Short Communication
The pJan25 vector series: An enhancement of the Gateway-compatible vector pGWB533 for broader promoter testing applications
0147-619X/$ - see front matter Published by Elsevier Inc.http://dx.doi.org/10.1016/j.plasmid.2013.01.005
⇑ Corresponding author. Address: USDA-ARS PSI, Soybean Genomics and Improvement Laboratory, 10300 Beltsville Ave., Bldg., 006, Beltsville,MD 20705, United States. Tel.: +1 (301) 504 5730; fax: +1 (301) 504 5728.
E-mail addresses: [email protected], [email protected](B.F. Matthews).
Ahmed Mansour Alzohairy a,b, Margaret H. MacDonald b, Benjamin F. Matthews b,⇑a Genetics Department, Faculty of Agriculture, Zagazig University, Egypt b USDA-ARS Soybean Genomics and Improvement Laboratory, PSI, Beltsville, MD 20705, United States
a r t i c l e i n f o a b s t r a c t
Article history:Received 31 August 2012 Accepted 14 January 2013 Available online 23 January 2013 Communicated by Dr. G.J. Phillips
Keywords:PlasmidpJan25GatewayeGFPGUSPromoter
Agrobacterium-mediated transformat ion of plants has enhanced our ability to progress more rapidly in plant genetic engineering. Development of binary vectors for Agrobacte-rium has played a major role in advancing plant biology. Here, we report new features added to the Gateway-compa tible vector pGWB533 for promoter testing with the reporter gene encoding b-glu curonidase (GUS). The original vector contains the spectino mycin/ streptomycin adenylyltransferase (aadA) gene for bacterial selection and the hygromycin phosphotransferase gene (hpt) for transformed plant selection. However, some bacter ial strains used to transform plants, such as Agrobacterium rhizogenes strain K599, have ele- vated tolerance to spectinomycin and streptomycin, thus making bacterial selection ofpGWB533 inefficient. Although pGWB533 confers chemical selection for transgenic plants using hygromycin resistance, the plasmid has no visual marker that enables visual selec- tion of transformed plants or transgenic tissue. In this regard, adding a gene to constitu- tively express green fluorescent protein (eGFP) makes it easier to visually select the transformed tissue and trim out the non-transformed.
In this report we describe a series of vectors, pJan25S (NCBI: KC416200), pJan25T (NCBI:KC416201) and pJan25X (NCBI: KC416202), that are enhancements of pGWB533 for pro- moter testing. All three vectors contain the gene encoding eGFP as a visual marker for transformed tissue. However, in pJan25S and pJan25T, eGFP is controlled by the rolD pro-moter for root-specific expression, while in pJan25X it is controlled by the CaMV35S pro- moter for constitutive expression in all plant tissues. Spectinomycin and streptomycin resistance remains in pJan25S for bacterial selection; however, pJan25T and pJan25X con- tain the gene encoding tetracycline resistance (tet) for bacterial selection. These chan ges resulted in enhanced vectors with better visual and chemical selection that should have broad application in promoter studies.
Published by Elsevier Inc.
1. Introduction at a certain level. This can be achieved through the use of
Many applications in biotechnolo gy require promoters that provide specific temporal and spatial gene expression
specific promote rs to express a gene of interest in the cor- rect amount at the specific place and time where it isneeded. In this regard, testing promoters to evaluate their temporal and spatial specificity and level of expression isan important area of study in genomics. Many expression vectors with different resistance and reporter genes were developed by Nakagawa et al. (2007) using the Gateway �
recombinas e cloning system (Invitrogen, Carlsbad, CA).Most of these vectors were derived from pPZP221

250 A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256
(Hajdukiewicz et al., 1994 ), which contains aadA, the gene encoding adenylyltran sferase. This enzyme confers bacte- rial resistance to spectinomycin and streptomyci n.Although the Gateway � cloning system has been used toconstruct numerous plant expression vectors, these vec- tors cannot be used for all applications that are required for in-depth promoter studies due to certain limitatio ns(Traore and Zhao, 2011 ). For instance, most of the Gate- way�-compatible vectors use only chemical selection for transformed plants, e.g., kanamyc in or hygromycin resis- tance (Nakamura et al., 2009 ). This requires growing the transformed plants on tissue culture media with the right concentratio n of chemical and requires sterile technique with all the costs and risks of microbial contaminat ion. Inthis regard, visual selection of transformed plant tissue ismuch less expensive and easier. For instance, when using Agrobacteriu m rhizogene s-mediated root transformat ion,some root tissue could be transformed while other tissue is not. However, if the transforming vector contains the gene encoding enhanced green fluorescent protein (eGFP;Haseloff et al., 1997 ) to transform roots, the transformed roots can be readily distinguished from non-transform edroots by eGFP fluorescence and thus untransform ed roots can be easily excised and discarded.
Several research studies have used eGFP fluorescence for distinguishing transformed from non-transfor med plant material. For example, gene silencing constructs were examined to determine if they conferred resistance to soy- bean cyst nematodes if expresse d in soybean roots of com- posite plants (Klink et al., 2009 ; Ibrahim et al., 2011 ).Transforme d roots were visualized using eGFP and non- transformed roots were excised before adding nematodes to the transformed roots. Mature female nematodes were counted 35 days later. In this case, A.rhizogen es was used for root transformat ion and the vector contained the gene encoding eGFP, controlled by the root-speci fic rolD pro-moter from A. rhizogene s (White et al., 1985; Elmayan and Tepfer, 1995 ). Sterile technique and chemical selection was not required to identify and select transformed roots.
In this study, the pGWB533 vector (Fig. 1a) was used asthe DNA backbone to develop the pJan25S, pJan25T and pJan25X vectors to be used with A. rhizogenes and A. tum- efaciens-mediated root and whole plant transformat ion,respectively (Tepfer, 1984; Ko et al., 2006 ). The Gateway �
(Invitrogen, Carlsbad, CA) compatible pJan25 destination vectors were engineered specifically for Agrobacteri um-based plant transformat ion and visualizati on of trans- formed tissue and plants.
2. Material and methods
2.1. Addition of tetracycline resistance and eGFP
The genes encoding tetracycline resistance (tet) and green fluorescent protein (eGFP) were obtained from the pRAP17 vector (Klink et al., 2009 ). Antibiotic selection ofbacteria transformed with the vector is provided by tet,while the eGFP gene provides a visual marker for identify- ing transformed plant tissue. The gene encoding eGFP pro- tein was controlle d by the rolD promoter and the T35s
terminator. The rolD-eGFP-T35s cassette was amplifiedusing Platinum � Taq DNA Polymerase High Fidelity (Invit-rogen, Carlsbad, CA) from pRAP17 as template (Klink et al.,2009) with cassette-specific primers including HindIII sites for cloning into pGWB533 (Table 1). Amplification reac- tions were performed according to Williams et al. (1990).Briefly, each reaction mixture containe d l� of 10� HighFidelity PCR Buffer (600 mM Tris–SO4 (pH 8.9), 180 mMammonium sulfate), 0.2 mM dNTPs (Pharmacia), 2 mMMgSO4, 0.2 lM of each primer, 50 ng of pRAP17 DNA,and 1.0 unit of Platinum � Taq DNA Polymerase High Fidel- ity in a final volume of 50 ll. The PCR program had an ini- tial denaturation step at 94 �C for 2 min followed by 35cycles of: denature at 94 �C for 30 s, anneal at 55 �C for 30 s, and extend at 68 �C for 2 min, followed by a finalextension step at 68 �C for 10 min. Ten microlite rs of PCR product stained with 2 ll E-Z Vision � One DNA Loading Buffer (Amresco, USA) was separated by agarose (1) gel electrophor esis run at 100 V in 1X SB (10 mM NaOH solu- tion with boric acid, pH 8.5) and photographed on a UVtransilluminat or (Pharmacia) by a Canon S5 digital camera with a UV filter adaptor. A negative control that containe dall the necessary PCR components except template DNA was included for PCRs. The 40 ll remainder of the rolD-eGFP-T35s cassette amplicon was digested with HindIIIand purified using the MinElute PCR Purification Kit (Qia-gen, Santa Clarita, CA) following the manufac turer’s instructions. The rolD-eGFP-T35s HindIII cassette was li- gated to the HindIII site of pGWB533 following the manu- facturer’s instructions, resulting in pJan25S. The HindIIIsite lies inside the left and right T-DNA borders that deli- mit the region that is transferred into plant tissue upon transformat ion (Fig. 1a and b). The 50-phosphate from the pGWB53 3 linear plasmid was removed using shrimp alkaline phosphat ase (SAP) (Novagen, Madison, WI) at37 �C for 1 h to prevent self-ligat ion of the plasmid. The SAP was inactivated by incubation at 65 �C for 15 min.The rolD-eGFP-T35s cassette was then ligated to the pGWB53 3-phosphatase d vector with T4 DNA ligase (Invit-rogen, Carlsbad, CA) overnight at 14 �C. One microlite r ofthe ligation was then transformed into One Shot 2T1R ccdBSurvival Cells (Invitrogen), plated on LB plates supple- mented with 100 mg L�1 spectinomy cin and incubate dovernight at 37 �C.
The cassette containing tet was isolated from pRAP17 by digesting with BstEII (Invitogen, Carlsbad, CA) fol- lowed by gel purification using a QIAquick � GelExtraction Kit (Qiagen, Santa Clarita, CA) accordin g tothe manufactur er’s instructions. The quality and concen- tration of the purified fragments were measured using aNanoDrop (Thermo Fisher, Wilmington , DE) spectropho- tometer. The tet cassette was ligated into the BstEII site of the Gateway �-compatible vector pJan25S, which was digested by the same restriction enzyme and phosphatas ed before ligation. The BstEII site lies outside the left and right T-DNA borders and so is not trans- ferred to plant tissue (Fig. 2b). When placed at this site,tet also interrupts the addA gene, rendering the spectinomyci n and streptomycin resistance nonfunc- tional. Adding the tet cassette to pJan25S resulted inthe formation of pJan25T. Replacing the rolD-eGFP-T35s
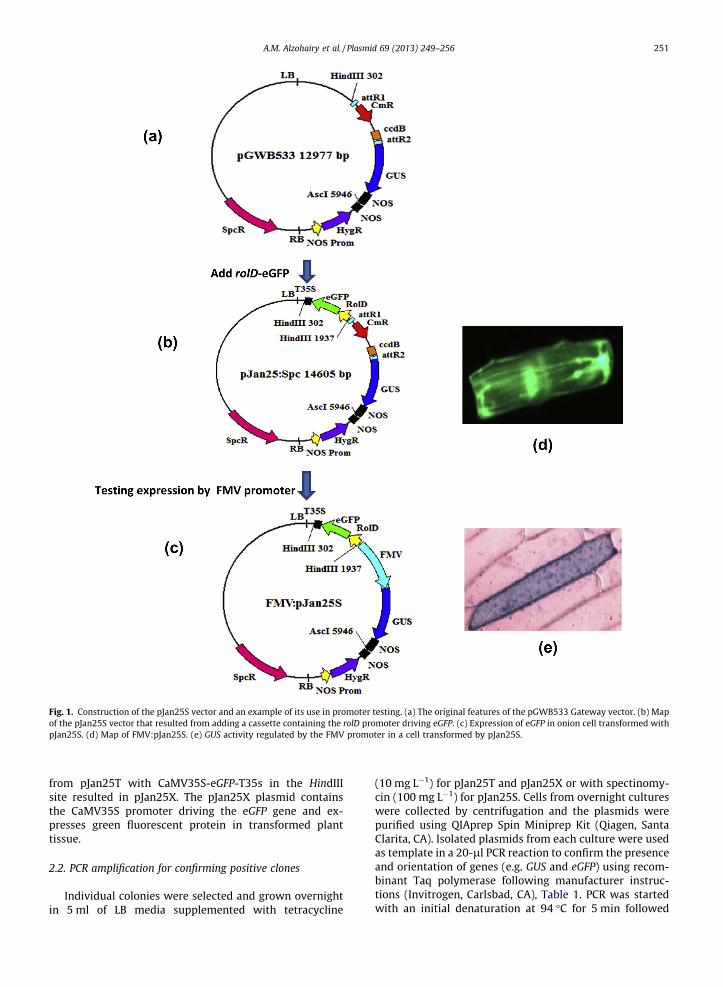
Fig. 1. Construction of the pJan25S vector and an example of its use in promoter testing. (a) The original features of the pGWB533 Gateway vector. (b) Map of the pJan25S vector that resulted from adding a cassette containing the rolD promoter driving eGFP. (c) Expression of eGFP in onion cell transformed with pJan25S. (d) Map of FMV:pJan25S. (e) GUS activity regulated by the FMV promoter in a cell transformed by pJan25S.
A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256 251
from pJan25T with CaMV35S-e GFP-T35s in the HindIIIsite resulted in pJan25X. The pJan25X plasmid contains the CaMV35S promote r driving the eGFP gene and ex- presses green fluorescent protein in transformed plant tissue.
2.2. PCR amplification for confirming positive clones
Individual colonies were selected and grown overnight in 5 ml of LB media supplemented with tetracycline
(10 mg L�1) for pJan25T and pJan25X or with spectinomy- cin (100 mg L�1) for pJan25S. Cells from overnight cultures were collected by centrifugation and the plasmids were purified using QIAprep Spin Miniprep Kit (Qiagen, Santa Clarita, CA). Isolated plasmids from each culture were used as template in a 20- ll PCR reaction to confirm the presence and orientation of genes (e.g. GUS and eGFP) using recom- binant Taq polymerase following manufactur er instruc- tions (Invitrogen, Carlsbad, CA), Table 1. PCR was started with an initial denaturation at 94 �C for 5 min followed

Table 1Primer pairs used in the isolation of genes.
Primer name Sequence
rolD Hin dIII forward ATTTTAAGCTTGCATGCCTGCAGGTTAG T35s HindIII reverse GCCCCAAGCTTGCAGGTCACTGGATTTTGG CAMV35S HindIII
forward CGTCAAGCTTAACATGGTGGAGCACGACAC
FMV forward plus CACC
CACCTTTACAGTAAGAACTGATAAC
FMV reverse CACTCCCCCTCTCTAAAAAA eGfp complete F TGGTGAGCAAGGGCGAGGAGC eGfp complete R TCGTCCATGCCGAGAGAGATCCCG tet-BF (BstE II) TCGTGGTCACCATCAGCGATCGGCTCGTT tet-BR (BstE II) AGCGGTGACCATCAATCGTCACCCAAA GUS forward GGA CTT TGC AAG TGG TGA ATC CGUS reverse CGA ATC CTT TGC CAC GTA AGT C
252 A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256
by 25 cycles of denaturation at 94 �C for 35 s, annealing atthe primer Tm (Table 1) for 35 s, and extension at 72 �C for 2 min per kb of PCR product. Final extension of the ampli- fication was for 10 min at 72 �C. All PCR reactions were performed using a DNA Engine Tetrad 2 Peltier Thermal Cycler (Bio-Rad, USA).
2.3. Transfer of the FMV promoter from the pENTR plasmid tothe pJan25 series
To test the new vectors and ensure that the selection markers and promote r site worked properly together, the FMV-sgt promote r fragment, �270 to +31 from the tran- scription start site (Bhattachar yya et al., 2002 ), was trans- ferred from a FMV: pENTR clone to the pJan25T, pJan25S and pJan25X plasmids using the Gateway � LR Clonase™II Enzyme Mix kit following the manufacturer’s instruc- tions (Invitrogen, Carlsbad, CA). The LR clonase reaction re- placed the ccdB gene between attR1 and attR2 sites with the FMV promoter, thus allowing the Escherichia coli cellsto grow. The plasmid was transformed into E. coli (OneShot� Mach1™-T1R chemically competent cells, Invitro- gen, Carlsbad, CA). clones, The transformed cells were grown overnight at 37 �C on LB selective media containing 10 mg L�1 tetracycl ine for pJan25T and pJan25X, and 100 mg L�1 spectinomyci n for pJan25S. Plasmids were iso- lated from overnight cultures using the QiaPrep Spin Mini- prep Kit (Qiagen, Santa Clarita, CA). Their sequences were confirmed by DNA sequencing.
2.4. Microprojecti le bombardmen t
Microprojecti le bombardment (Sanford, 1990 ) was conducted using the Biolistic Particle Delivery System (PDS-1000/He, BioRad, Hercules, CA) according to the manufactur er’s instructions. Gold microprojec tiles coated with 6 ll of plasmid DNA (1 lg/ll) were bombarded into onion cells at 1100 PSI. Each construct was delivered into three replicates.
Fluorescenc e was monitored using a Nikon SMZ 1500 stereomicros cope with an eGFP filter (Nikon Corporat ion;Tokyo, Japan). Stereomicro scope images were captured with an Optronics MagnaFire model S99802 CCD camera
(Optronics; Goleta, CA). The functiona lity of the pJan25 series plasmids was tested using the FMV promote r driving expression of the GUS reporter gene. Bombarded onion tis- sue was incubate d with GUS staining solution to determine b-glucuronidase activity after 24 h. GUS staining solution consisted of 2 mM 5-bromo -4-chloro-3-in dolyl glucuro- nide, 100 mM potassium phosphate buffer pH 7.0, 10 mMEDTA, 0.5 mM potassium ferricyanide , 0.5 mM potassium ferrocyanide, 0.1% Triton X-100 (Jefferson et al., 1987 ).The colorimetric reaction proceeded by immersion inGUS stain and subsequent vacuum infiltration with 500 ll of GUS stain for 10 min. Tissue was subsequent lyincubated at 37 �C overnight to promote developmen t ofthe GUS stain.
2.5. Soybean root transformati on with A. rhizogenes
Plasmids in the pJan25 series harborin g the FMV pro- moter were used to transform chemically competent A.rhizogenes (strain K599) cells. The transformed cells were grown overnight at 26 �C on LB selective medium contain- ing 5 mg L�1 tetracycline for pJan25T and pJan25X, and 100 mg L�1 spectinomyci n for pJan25S. For soybean trans- formation, an A. rhizogenes clone containing a pJan25 plas- mid was grown overnight at 26 �C in TB media supplemented with 5 mg L�1 tetracycline or 100 mg L�1
spectinomyci n. Cells were resuspended in MS medium with 3% sucrose, pH 5.7, to yield a suspension with anOD600 of approximat ely 0.5. Soybean seedlings 7–9 days old were cut at the soil line and transferred to 50-ml beak- ers (25 plants/be aker) containing 40 ml of the A. rhizogene ssuspension. The cell suspension was vacuum infiltratedinto the base of the stem of the cut plants for 30 min, after which the plants were cocultivated in the same solution at23 �C for 16 h on a rotating platform moving at 65 rpm. The plants were washed with reverse osmosis water and kept in water in the 50-ml beakers for 2 days before being planted in Promix peat-bas ed growing medium in a green- house. Four weeks later, eGFP fluorescence of transformed roots was observed using a Dark Reader Spot lamp (ClareChemical Research, Dolores, CO). Approximatel y 65–70%of the seedlings had transformed roots.
3. Results and discussion
3.1. Overview of pJan25S, pJan25T and pJan25X
The pJan25 vectors were developed from the pGWB533 vector (12,977 bp), which contains the GATEWAY � attR1and attR2 sites for easy cloning of promoters of interest.The attR2 site is adjacent to the gene encoding GUS whichserves as a reporter gene for promoter activity (Fig. 1a).
3.2. Construction of pJan25S
The pJan25S plasmid was designed to add root-specificeGFP expression to the pGWB533 vector (Fig. 1a), thus allowing visual selection of transformed root material via eGFP fluorescence. Consequently, the rolD promoter, which is highly active in roots, was used to regulate eGFP gene

Fig. 2. Construction of the pJan25T vector and an example of its use in promoter testing. Maps of pJan25S before (a) and after (b) adding the tetracycline resistance (tet) gene, which resulted in pJan25T. (c) Onion cell transformed with pJan25T exhibiting eGFP fluorescence. (d) Map of pJan25T with FMV promoter. (e) FMV driving GUS activity in onion cell.
A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256 253
expression. A rolD-eGFP-T35s cassette was added topGWB533, resulting in the new vector pJan25S (Fig. 1b).Presence of the eGFP gene in the plasmid confirmed byPCR. As in pGWB533, the pJan25S plasmid provides specti- nomycin resistance for bacterial selection. The functional- ity of pJan25S for promoter studies was tested byinserting the figwort mosaic virus subgenomic transcript (FMV) promoter (Bhattachary ya et al., 2002 ) into the attR1-attR2 cloning site upstream from the GUS gene(Fig. 1 c) and using this construct to transiently transform onion cells via microprojec tile bombardme nt. Expression of both eGFP and GUS proteins in transformed onion cells was confirmed visually (Fig. 1d, e).
3.3. Construction of pJan25T
A.rhizogenes (strain K599) has been used for plant trans- formation and in the production of composite plants with transformed roots (Klink et al., 2009 ; Ibrahim et al.,2011). However , strain K599 has an elevated tolerance tospectinomyci n but is very sensitive to tetracycline .Thus, addition of the tetracycline resistance gene (tet) topJan25S allows more efficient selection of transformed A.rhizogenes cells. Therefore, tetracycline resistance was engineered into pJan25S (Fig. 2a), outside the left and right borders, resulting in the new vector pJan25T (Fig. 2b). Pres- ence of the tet gene was confirmed by PCR. The FMV
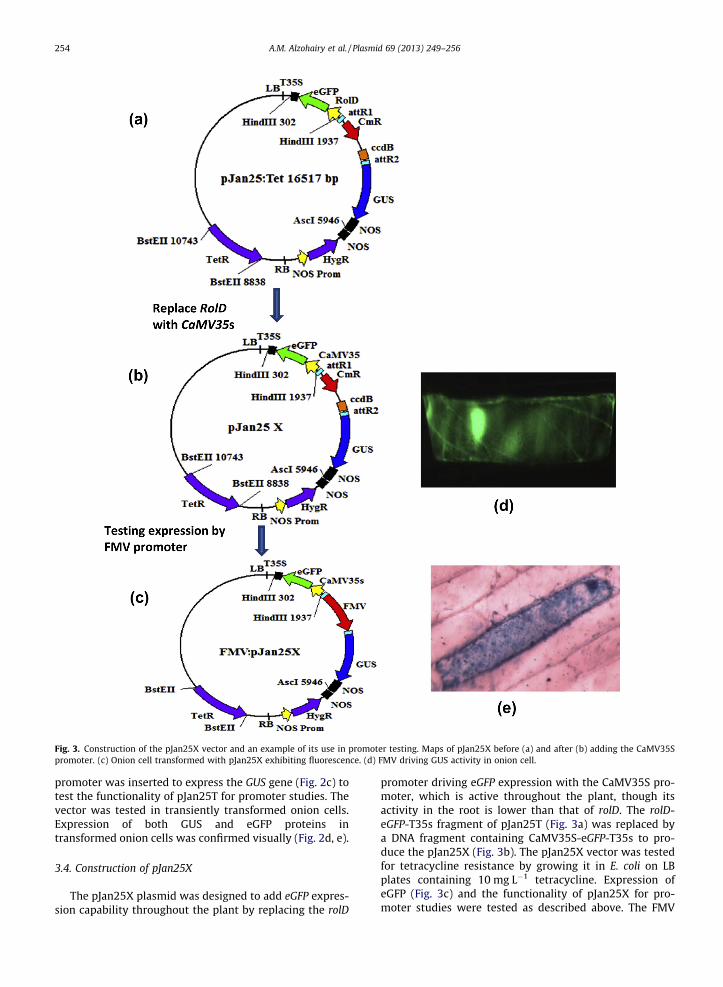
Fig. 3. Construction of the pJan25X vector and an example of its use in promoter testing. Maps of pJan25X before (a) and after (b) adding the CaMV35S promoter. (c) Onion cell transformed with pJan25X exhibiting fluorescence. (d) FMV driving GUS activity in onion cell.
254 A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256
promoter was inserted to express the GUS gene (Fig. 2c) totest the functionality of pJan25T for promoter studies. The vector was tested in transiently transformed onion cells.Expression of both GUS and eGFP proteins intransformed onion cells was confirmed visually (Fig. 2d, e).
3.4. Construction of pJan25X
The pJan25X plasmid was designed to add eGFP expres-sion capability througho ut the plant by replacing the rolD
promoter driving eGFP expression with the CaMV35S pro- moter, which is active throughout the plant, though its activity in the root is lower than that of rolD. The rolD-eGFP-T35s fragment of pJan25T (Fig. 3a) was replaced bya DNA fragment containing CaMV35S-e GFP-T35s to pro- duce the pJan25X (Fig. 3b). The pJan25X vector was tested for tetracycline resistance by growing it in E. coli on LBplates containing 10 mg L�1 tetracycline. Expression ofeGFP (Fig. 3c) and the functiona lity of pJan25X for pro- moter studies were tested as described above. The FMV
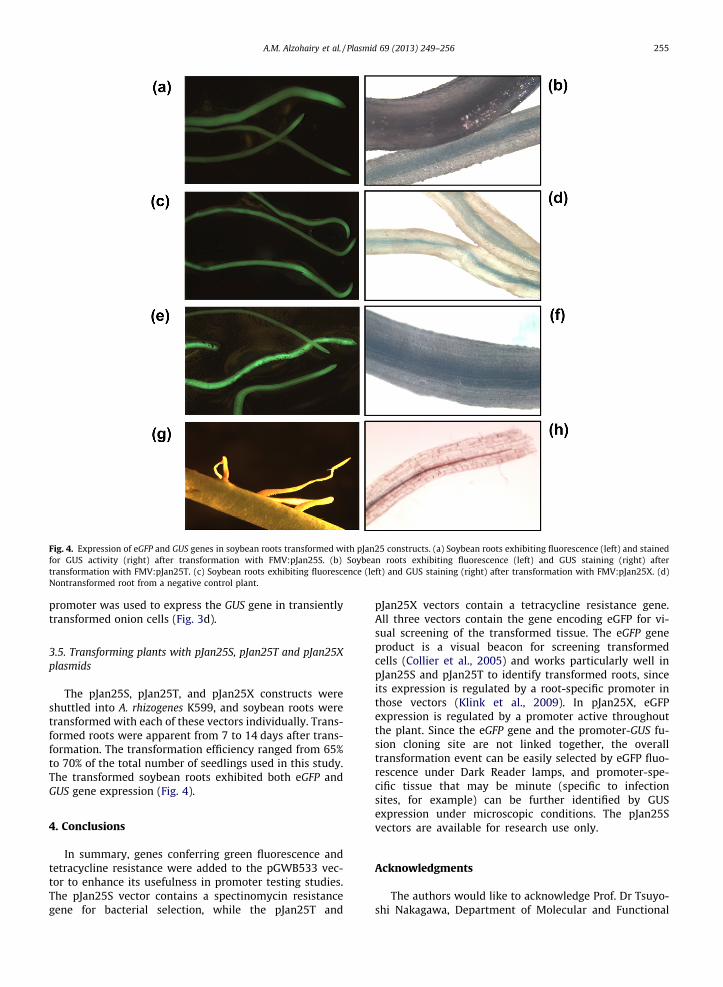
Fig. 4. Expression of eGFP and GUS genes in soybean roots transformed with pJan25 constructs. (a) Soybean roots exhibiting fluorescence (left) and stained for GUS activity (right) after transformation with FMV:pJan25S. (b) Soybean roots exhibiting fluorescence (left) and GUS staining (right) after transformation with FMV:pJan25T. (c) Soybean roots exhibiting fluorescence (left) and GUS staining (right) after transformation with FMV:pJan25X. (d)Nontransformed root from a negative control plant.
A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256 255
promoter was used to express the GUS gene in transiently transformed onion cells (Fig. 3d).
3.5. Transformi ng plants with pJan25S, pJan25T and pJan25X plasmids
The pJan25S, pJan25T, and pJan25X constructs were shuttled into A. rhizogenes K599, and soybean roots were transformed with each of these vectors individually. Trans- formed roots were apparent from 7 to 14 days after trans- formation. The transformat ion efficiency ranged from 65%to 70% of the total number of seedlings used in this study.The transformed soybean roots exhibited both eGFP andGUS gene expression (Fig. 4).
4. Conclusions
In summary, genes conferring green fluorescence and tetracycline resistance were added to the pGWB533 vec- tor to enhance its usefulness in promoter testing studies.The pJan25S vector contains a spectinomycin resistance gene for bacterial selection , while the pJan25T and
pJan25X vectors contain a tetracycline resistance gene.All three vectors contain the gene encoding eGFP for vi- sual screening of the transformed tissue. The eGFP geneproduct is a visual beacon for screening transformed cells (Collier et al., 2005 ) and works particular ly well inpJan25S and pJan25T to identify transformed roots, since its expression is regulated by a root-specific promoter inthose vectors (Klink et al., 2009 ). In pJan25X, eGFP expression is regulated by a promoter active throughout the plant. Since the eGFP gene and the promoter- GUS fu-sion cloning site are not linked together, the overall transformat ion event can be easily selected by eGFP fluo-rescence under Dark Reader lamps, and promote r-spe- cific tissue that may be minute (specific to infection sites, for example) can be further identified by GUS expression under microscop ic conditions. The pJan25S vectors are available for research use only.
Acknowled gments
The authors would like to acknowled ge Prof. Dr Tsuyo- shi Nakagawa, Department of Molecular and Functional

256 A.M. Alzohairy et al. / Plasmid 69 (2013) 249–256
Genomics, Center for Integrate d Research in Science, Shi- mane University, for providing the Gateway vector pGWB533 for research purposes. We also thank Dr. I. Maiti,University of Kentucky, for providing the figwort mosaic virus subgenomic transcript (FMV) promoter. The authors thank Eric Brewer for critical review of the manuscript,and gratefully acknowled ge support from United Soybean Board and from the Fulbright and Arab Fund fellowship programs. Mention of trade name, proprieta ry product, orvendor does not constitute a guarante e or warranty ofthe product by the U.S. Department of Agriculture or imply its approval to the exclusion of other products or vendors that also may be suitable. The names of these new plas- mids were inspired by the Egyptian revolution in January 25th 2011 (AMA).
References
Bhattacharyya, S., Dey, N., Maiti, I.B., 2002. Analysis of cis-sequence ofsubgenomic transcript promoter from the figwort mosaic virus and comparison of promoter activity with the cauliflower mosaic virus promoters in monocot and dicot cells. Virus Res. 90, 47–62.
Collier, R., Fuchs, B., Walter, N., Kevin Lutke, W., Taylor, C.G., 2005. Ex vitro composite plants: an inexpensive, rapid method for root biology.Plant J. 43, 449–457.
Elmayan, T., Tepfer, M., 1995. Evaluation in tobacco of the organ specificity and strength of the rolD promoter, domain A of the 35S promoter and the 35S2 promoter. Transgenic Res. 4, 388–396.
Haseloff, J., Siemering, K.R., Prasher, D.C., Hodge, S., 1997. Removal of acryptic intron and subcellular localization of green fluorescentprotein are required to mark transgenic arabidopsis plants brightly.Proc. Natl. Acad. Sci. USA 94, 2122–2127.
Hajdukiewicz, P., Svab, Z., Maliga, P., 1994. The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol. Biol. 25 (6), 989–994.
Ibrahim, H.M.M., Alkharouf, N.W., Meyer, S.L.F., Aly, M.A.M., El-Din,A.E.K.G., Hussein, E.H.A., Matthews, B.F., 2011. Post-transcriptional gene silencing of root knot-nematode in transformed soybean roots.Exp. Parasitol. 127, 90–99.
Jefferson, R.A., Kavanagh, T.A., Bevan, M.W., 1987. GUS fusions: b-glucuronidase as a sensitive and versatile gene fusion marker inhigher plants. EMBO J. 6, 3901–3907.
Klink, V.P., Kim, K.H., Martins, V.E., MacDonald, M.H., Beard, H.S.,Alkharouf, N.W., Park, S.C., Matthews, B.F., 2009. A correlation between host-mediated expression of parasite genes as tandem inverted repeats and abrogation of the formation of female Heterodera glycines cysts during infection of Glycine max. Planta 230, 53–71.
Ko, T.S., Korban, S.S., Somers, D.A., 2006. Soybean (glycine max)transformation using immature cotyledon explants. Methods Mol.Biol. (Clifton, NJ) 343, 397–405.
Nakagawa, T., Suzuki, T., Murata, S., Nakamura, S., Hino, T., Maeo, K.,Tabata, R., Kawai, T., Tanaka, K., Niwa, Y., Watanabe, Y., Nakamura, K.,Kimura, T., Ishiguro, S., 2007. Improved gateway binary vectors: high- performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci. Biotechnol. Biochem. 71 (8), 2095–2100.
Nakamura, S., Nakao, A., Kawamukai, M., Kimura, T., Ishiguro, S.,Nakagawa, T., 2009. Development of gateway binary vectors,R4L1pGWBs, for promoter analysis in higher plants. Biosci.Biotechnol. Biochem. 73 (11), 2556–2559.
Sanford, J.C., 1990. Biolistic plant transformation. Physiol. Plant. 79, 206–209.
Tepfer, D., 1984. Transformation of several species of higher plants byAgrobacterium rhizogenes : sexual transmission of the transformed genotype and phenotype. Cell 37, 959–967.
Traore, S.M., Zhao, B., 2011. A novel Gateway �-compatible binary vector allows direct selection of recombinant clones in Agrobacteriumtumefaciens. Plant Methods 7 (1), 42.
Williams, J.G.K., Kubelik, A.R., Livak, K.J., Rafalski, J.A., Tingey, S.V., 1990.DNA polymorphisns amplified by arbitrary primers are useful asgenetic markers. Nucleic Acids Res. 18, 6531–6535.
White, F.F., Taylor, B.H., Huffman, G.A., Gordon, M.P., Nester, E.W., 1985.Molecular and genetic analysis of the transferred DNA regions of the root-inducing plasmid of Agrobacterium rhizogenes . J. Bacteriol. 164,33–44.




















