
Multiple Sulfatase Deficiency: Clinical, Neuropathological, Ultrastructural and Biochemical Studies

Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435
Design, synthesis and biochemical evaluation of AC ring mimics as novelinhibitors of the enzyme estrone sulfatase (ES)
Sabbir Ahmeda,∗, Karen Jamesb, Caroline P. Owenaa School of Chemical and Pharmaceutical Sciences, Kingston University, Penrhyn Road, Kingston upon Thames, Surrey, KT1 2EE, UK
b Institute of Cancer Research, Cotswold Road, Sutton, UK
Received 27 May 2002; accepted 1 October 2002
Abstract
We report the results of our study into a series of 4′-O-sulfamoyl-4-biphenyl based compounds as novel inhibitors of the enzyme estronesulfatase (ES). From the results of the molecular modeling design process, it was suggested that these compounds would be able to mimicboth the A and C rings of the steroid backbone, and thus possess inhibitory activity against ES. The results of the biochemical evaluationstudy show that these compounds are indeed good inhibitors, possessing greater inhibitory activity than COUMATE, but weaker inhibitoryactivity than EMATE or the tricyclic derivative of COUMATE, namely 667-COUMATE. Furthermore, the compounds are observed to beirreversible inhibitors.© 2002 Elsevier Science Ltd. All rights reserved.
Keywords: Estrone sulfatase; EMATE; COUMATE
1. Introduction
In the treatment of hormone-dependent breast cancer, ex-tensive research has been undertaken to produce compoundswhich are both potent and selective inhibitors of the cy-tochrome P-450 enzyme aromatase (AR), which converts an-drogens to estrogens[1]. However, the use of AR inhibitorsdoes not result in the inhibition of all of the biosynthetic pro-cesses that lead to estrogen formation. That is, the enzymeestrone sulfatase (ES) converts the stored (sulfated) formof the estrogens to the active (non-sulfated) forms (Fig. 1),thereby allowing the stimulation of tumors via a non-ARpathway which, in general, is not blocked by AR inhibitors.
A number of steroidal and non-steroidal sulfamatecontaining compounds[2,3] have been investigated aspotent inhibitors of this enzyme (Fig. 2), includingestrone-3-O-sulfamate (EMATE; IC50 = 65 pM; a time-and concentration-dependent irreversible steroidal inhibitor)and 4-methylcoumarin-7-O-sulfamate (COUMATE; an ir-reversible non-steroidal inhibitor;Fig. 2). COUMATE hasbeen further derivatised since, in contrast to EMATE, it hasbeen shown to lack significant levels of estrogenicity, anda series of tricyclic compounds such as 667- (Fig. 2), 668-
∗ Corresponding author. Tel.:+44-20-8547-2000;fax: +44-20-8547-7562.
E-mail address: [email protected] (S. Ahmed).
and 669-COUMATE[3] has resulted. In general, the sulfa-mate moiety is believed to be involved in the irreversibleinhibition of ES.
In an effort to overcome the lack of detailed informa-tion regarding the active site of ES, we initiated a series ofstructure-activity relationship (SAR) determination studies[4–6]. From the results of our initial molecular modellingstudy and a review of potential mechanisms for ES, we hy-pothesized that compounds which were able to mimic thesteroidal backbone would allow us to probe the active siteof ES. We therefore undertook a design process, incorporat-ing the results of our SAR and molecular modelling studies,which included the derivation of a potential transition-statefor the reaction catalyzed by ES. From these studies, weconcluded that the sulfamated biphenyl compounds wouldallow us to mimic the A and C rings of the steroid backbone,whilst the functional group R (Fig. 3) would undergo hydro-gen bonding with any potential H-bonding group(s) withinthe active site of ES (the usefulness of the biphenyl moietyas a steroid mimic has been extensively studied[7]).
Here, we report the results of our study to probe theactive site of ES using mimics of the A and C rings ofestrone sulfate and therefore describe the design processinvolving the derivation of the transition-state for the reac-tion catalyzed by ES; synthesis of a number of derivativesof 4′-O-sulfamoyl-4-biphenyl; in vitro biochemical evalu-ation of the synthesized compounds, and the determination
0960-0760/02/$ – see front matter © 2002 Elsevier Science Ltd. All rights reserved.PII: S0960-0760(02)00228-5

426 S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435
Fig. 1. Action of the enzyme ES on estrone sulfate.
Fig. 2. Potent inhibitors of ES.
S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435 427
Fig. 3. Diagram to show the rationale for the synthesis of biphenylsulfamates.
of the mode of action of the synthesized compounds, i.e.reversible or irreversible.
2. Experimental
2.1. Transition-state determination and thedesign process
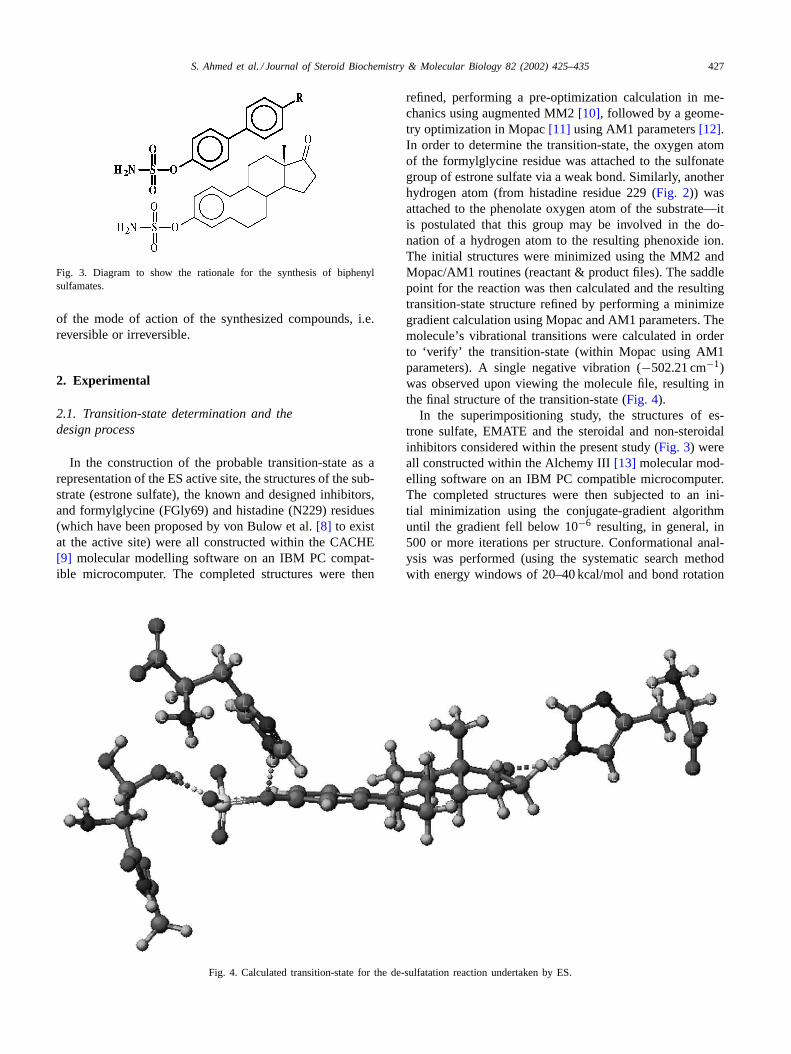
In the construction of the probable transition-state as arepresentation of the ES active site, the structures of the sub-strate (estrone sulfate), the known and designed inhibitors,and formylglycine (FGly69) and histadine (N229) residues(which have been proposed by von Bulow et al.[8] to existat the active site) were all constructed within the CACHE[9] molecular modelling software on an IBM PC compat-ible microcomputer. The completed structures were then
Fig. 4. Calculated transition-state for the de-sulfatation reaction undertaken by ES.
refined, performing a pre-optimization calculation in me-chanics using augmented MM2[10], followed by a geome-try optimization in Mopac[11] using AM1 parameters[12].In order to determine the transition-state, the oxygen atomof the formylglycine residue was attached to the sulfonategroup of estrone sulfate via a weak bond. Similarly, anotherhydrogen atom (from histadine residue 229 (Fig. 2)) wasattached to the phenolate oxygen atom of the substrate—itis postulated that this group may be involved in the do-nation of a hydrogen atom to the resulting phenoxide ion.The initial structures were minimized using the MM2 andMopac/AM1 routines (reactant & product files). The saddlepoint for the reaction was then calculated and the resultingtransition-state structure refined by performing a minimizegradient calculation using Mopac and AM1 parameters. Themolecule’s vibrational transitions were calculated in orderto ‘verify’ the transition-state (within Mopac using AM1parameters). A single negative vibration (−502.21 cm−1)was observed upon viewing the molecule file, resulting inthe final structure of the transition-state (Fig. 4).
In the superimpositioning study, the structures of es-trone sulfate, EMATE and the steroidal and non-steroidalinhibitors considered within the present study (Fig. 3) wereall constructed within the Alchemy III[13] molecular mod-elling software on an IBM PC compatible microcomputer.The completed structures were then subjected to an ini-tial minimization using the conjugate-gradient algorithmuntil the gradient fell below 10−6 resulting, in general, in500 or more iterations per structure. Conformational anal-ysis was performed (using the systematic search methodwith energy windows of 20–40 kcal/mol and bond rotation

428 S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435
between 20 and 50◦) on flexible parts of the inhibitors usingPowersearch[13] in order to determine the low energy con-formers. The derived transition-state was converted to theAlchemy file format prior to the superimpositioning study.
On the assumption that the shape of estrone sulfate wouldreflect the nature of the binding site of ES, the lowest energyconformers of the inhibitors were superimposed by specifi-cation of three or more points on the sulfonate group of thesubstrate (Fig. 1) on the transition-state in the fitting pro-cess (an energy window of�E = 5 kcal/mol was used indetermining the conformers to be used). The superimposi-tioning points were chosen since the sulfamate group wascommon to most of the potent inhibitors, as such, we postu-lated that this group was important in mimicking the naturalsubstrate’s binding to the active site.
2.2. Chemistry
2.2.1. Methods and materialsChemicals were purchased from Sigma–Aldrich (Poole,
Dorset, England), and checked for purity by1H and13CNMR (Brucker 300 and 75.5 MHz respectively) usingeither CDCl3, or d6-acetone as the solvent. Infrared spectrawere obtained on a Perkin-Elmer Fourier transform-Paragon1000 IR. High-resolution masses of the synthesized com-pounds were obtained from the EPSRC Mass SpectrometryService Centre at the University of Wales College Swansea,UK, using a VG ZAB-E instrument. Gas chromatography-mass spectrometry was carried out on a Hewlett 5890Packard series II GC-MS at a flow rate of 0.58 ml/min, and atemperature range increasing from 120 to 270◦C at the rateof 10◦C/min. Melting points are uncorrected and were ob-tained on a BUCHI 512 or a Gallenkamp Instrument. Purityof the compounds was checked using three different mobilephases on a HPLC system with a reverse phase C18 column.
1,1′Biphenyl-4-yl sulfamate (10): sodium hydride (NaH;60% dispersion in mineral oil, 0.15 g, 3.75 mmol) was addedto a stirred solution of 4-phenylphenol (0.5 g, 2.94 mmol) indimethyl formamide (DMF; 20 ml) under an atmosphere ofnitrogen gas at 0◦C. After evolution of hydrogen had ceased,aminosulfonyl chloride in toluene (20 ml,∼20 mmol) wasadded after 30 min in one portion and the reaction allowed tostir for 10 h. The reaction was then quenched with saturatedsodium bicarbonate (NaHCO3) solution (50 ml), extractedinto dichloromethane (DCM; 2 ml× 50 ml), washed withwater (3 ml× 30 ml) and dried over anhydrous magnesiumsulfate (MgSO4). The mixture was filtered and the solventremoved under vacuum to give a yellow oil, which was puri-fied using flash chromatography to give10 (0.12 g, 16.0%) asa pure white solid [mp 146–149◦C (literature 165◦C, [14]);Rf 0.31 diethyl ether/petroleum ether 40–60◦C (50/50)].
νmax (film) cm−1: 3421.3, 3301.9 (NH), 1382.0, 1177.9(S=O). δH (d6-acetone) 7.45 (9H, m, ArH), 7.12 (2H,s, NH2). δC (d6-acetone): 129.603, 128.734, 127.529,123.402 (CAr). MS m/z found: MNH4+ 267.0798,(C12H11NO3S)NH4
+ requires 267.0803.
4′-Cyano-1,1′-biphenyl-4-yl sulfamate (11): compound11 was synthesized following the same procedure as forcompound10 except that NaH (60% dispersion in min-eral oil, 0.13 g, 3.25 mmol) was added to a stirred solutionof 4-hydroxy-4-biphenyl carbonitrile (0.5 g, 2.56 mmol) inDMF (10 ml). Aminosulfonyl chloride in toluene (20 ml,∼20 mmol) was added after 30 min. Removal of the solventunder vacuum produced a yellow oil, which was purifiedusing flash chromatography to give11 (0.17 g, 24.8%)as a pure white solid [mp 132–136◦C; Rf 0.33 diethylether/petroleum ether 40–60◦C (70/30)].
νmax (film) cm−1: 3366.3, 3300.2 (NH2), 1383.9, 1157.9(S=O). δH (d6-acetone): 7.61 (8H, m, ArH), 7.11 (2H, s,NH2). δC (CDCl3): 133.4, 129.2, 128.4, 123.6 (CAr). MSm/z 274 (M+).
Methyl-(4′-hydroxy)-1,1′-biphenyl-4-carboxylate (12):concentration H2SO4 (1 ml) was added to a suspensionof 4-hydroxybiphenyl carboxylic acid (0.6 g, 2.8 mmol) inmethanol (20 ml) and the solution refluxed for 1 h. Aftercooling to room temperature, NaOH (∼15 ml) was added toneutralize the solution. The resulting mixture was allowedto stand for 15 min, before being poured into a cool beaker,and made up to 500 ml with water. The white precipitatewas filtered and dried (80◦C), to give 12 (0.58 g, 90.2%)as a white solid [mp 220–222◦C (literature 224–225◦C[10]); Rf 0.55 diethyl ether/petroleum ether 40–60◦C(70/30)].
νmax (film) cm−1: 3408.4 (OH), 2926.5 (CH aliphatic),1696.0 (C=O). The 90 MHzδH (acetone): 7.5 (8H, m, ArH),3.9 (3H, s, CH3), 2.8 (1H, s, OH). GCMStR 18.208 m/z 228(M+).
Methyl-4′-[(aminosulfonyl)oxy]-1,1′-biphenyl-4-carboxy-late (13): compound 13 was synthesized following thesame procedure as for compound10 except that NaH (60%dispersion in mineral oil, 0.1 g, 2.5 mmol) was added to astirred solution of12 (0.4 g, 1.75 mmol) in DMF (10 ml).Aminosulfonyl chloride in toluene (20 ml,∼20 mmol) wasadded after 30 min. Removal of the solvent under vacuumproduced a yellow oil, which was purified using flash chro-matography to give13 (0.2 g, 37.6%) as a pure white solid[mp 162–165◦C; Rf 0.52 diethyl ether/petroleum ether40–60◦C (70/30)].
νmax (film) cm−1: 3363.6, 3234.7 (NH2), 1708.0 (C=O),1388.6, 1161.1 (S=O). δH (d6-acetone): 8.11 (2H, d,J =9 Hz, ArH), 7.82 (2H, d,J = 9 Hz, ArH), 7.81 (2H, d,J =9 Hz, ArH), 7.45 (2H, d,J = 9 Hz, ArH), 7.23 (2H, s, NH2),3.91 (3H, s, OCH3). δC (d6-acetone): 151.5, 144.8, 138.8,130.6, 130.0, 129.1, 127.7, 123.6 (CAr), 52.2 (OCH3).MS m/z found: MNH4
+ 325.0854, (C14H13NO5S)NH4+
requires 325.0858.Ethyl-(4′-hydroxy)-1,1′-biphenyl-4-carboxylate (14): com-
pound14 was synthesized following the same proceduresas for compound12, except that conc. H2SO4 (1 ml) wasadded to a suspension of 4-hydroxybiphenyl carboxylicacid (0.5 g, 2.34 mmol) in ethanol (20 ml). The white pre-cipitate was filtered and dried (80◦C), to give 14 (0.46 g,

S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435 429
81.23%) as a white solid [mp 141–143◦C; Rf 0.63 diethylether/petroleum ether 40–60◦C (70/30)].
νmax (film) cm−1: 3335.9 (OH), 1681.4 (C=O). δH(CDCl3): 8.00 (2H, d, J = 8 Hz, ArH), 7.60 (2H, d,J = 8 Hz, ArH), 7.53 (2H, d,J = 8 Hz, ArH), 6.95 (2H, d,J = 8 Hz, ArH), 5.81 (1H, s, OH), 4.41 (2H, q,J = 7 Hz,CH2CH3), 1.42 (3H, t,J = 7 Hz, CH2CH3). δC (CDCl3):167.2 (C=O), 157.3, 145.6, 132.5, 130.1, 128.6, 128.4,126.4, 115.9 (CAr), 61.1 (OCH2CH3), 14.3 (OCH2CH3).GCMS tR 19.223 m/z 242 (M+).
Ethyl-4′-[(aminosulfonyl)oxy]-1,1′-biphenyl-4 - carboxy-late (15): compound15 was synthesized following the sameprocedure as for compound10 except that NaH (60% dis-persion in mineral oil, 0.05 g, 1.25 mmol) was added to astirred solution of14 (0.2 g, 0.83 mmol) in DMF (10 ml).Aminosulfonyl chloride in toluene (10 ml,∼10 mmol) wasadded after 30 min. Removal of the solvent under vacuumproduced a solid, which was purified using flash chromatog-raphy to give15 (0.08 g, 30.0%) as a pure white solid [mp171.2–173.5◦C; Rf 0.30 petroleum ether 40–60◦C: ethylacetate (65/35)].
νmax (film) cm−1: 3343.7, 3222.2 (NH), 1693.9 (C=O)1401.2, 1162.5 (S=O). δH (CDCl3): 8.12 (2H, d,J = 9 Hz,ArH), 7.80 (2H, dJ = 9 Hz, ArH), 7.79 (2H, d,J = 9 Hz,ArH), 7.44 (2H, d,J = 9 Hz, ArH), 5.03 (2H, s, NH2), 4.41(2H, q,J = 7 Hz, CH2–), 1.42 (3H, t,J = 7 Hz, CH2CH3).δc (CDCl3): 199.7 (C=O), 154.2, 135.0, 131.4, 130.3, 122.7,115.2 (C–Ar), 32.0 (OCH2), 8.2 (CH3). MS m/z 321 (M+).
Propyl-(4′-hydroxy)-1,1′-biphenyl-4-carboxylate (16): co-mpound16 was synthesized following the same proceduresas for compound12, except that conc. H2SO4 (1 ml) wasadded to a suspension of 4-hydroxybiphenyl carboxylic acid(0.6 g, 2.8 mmol) in propanol (20 ml). The white precipitatewas filtered and dried (80◦C), to give16 (0.65 g, 88.9%) as awhite solid [mp 117–121◦C; Rf 0.67 diethyl ether/petroleumether 40–60◦C (70/30)].
νmax (film) cm−1: 3317.2 (OH), 2964.1 (CH aliphatic),1714.1 (C=O). δH (CDCl3): 8.10 (2H, d,J = 9 Hz, ArH),7.61 (2H, dJ = 9 Hz, ArH), 7.46 (2H, d,J = 9 Hz, ArH),6.96 (2H, d,J = 9 Hz, ArH), 5.70 (1H, s, OH), 4.32 (2H,t, J = 7 Hz, OCH2–), 1.81 (2H, m, OCH2CH2–), 1.06(3H, t, J = 7 Hz, –CH3). δC(CDCl3): 167.0 (C=O), 156.1,145.3, 132.5, 130.1, 128.6, 128.5, 126.5, 115.9 (C–Ar), 66.7(OCH2), 22.1 (CH2), 10.5 (CH3). GCMStR 20.170 m/z 256(M+).
Propyl-4′-[(aminosulfonyl)oxy]-1,1′-biphenyl-4-carboxy-late (17): compound 17 was synthesized following thesame procedure as for compound10 except that NaH (60%dispersion in mineral oil, 0.1 g, 2.5 mmol) was added to astirred solution of16 (0.48 g, 1.86 mmol) in DMF (10 ml).Aminosulfonyl chloride in toluene (20 ml,∼20 mmol) wasadded after 30 min. Removal of the solvent under vacuumproduced a yellow oil, which was purified using flash chro-matography to give17 (0.19 g, 30.5%) as a pure white solid[mp 136–139◦C; Rf 0.55 diethyl ether/petroleum ether40–60◦C (70/30)].
νmax (film) cm−1: 3324.6, 3197.1 (NH), 1687.8 (C=O)1369.7, 1193.3 (S=O). δH (CDCl3): 8.13 (2H, d, J =9 Hz, ArH), 7.65 (2H, d,J = 9 Hz, ArH), 7.63 (2H, d,J = 9 Hz, ArH), 7.44 (2H, d,J = 9 Hz, ArH), 5.01 (2H,s, NH2), 4.32 (2H, t,J = 7 Hz, OCH2–), 1.83 (2H, m,OCH2CH2–), 1.06 (3H, t,J = 7 Hz, –CH3). δH (CDCl3):166.2 (C=O), 151.3, 144.6, 138.7, 130.5, 130.1, 129.0,127.5, 123.5 (C–Ar), 66.7 (OCH2), 22.4 (CH2), 10.4 (CH3).MS m/z found: MH+ 336.0900, (C16H17NO5S)H+ requires336.0905.
Butyl-(4′-hydroxy)-1,1′-biphenyl-4-carboxylate (18): co-mpound18 was synthesized following the same procedureas for compound12, except that conc. H2SO4 (1 ml) wasadded to a suspension of 4-hydroxybiphenyl carboxylic acid(0.6 g, 2.8 mmol) in butanol (20 ml). After quenching in icewater, the product was extracted into ether (3 ml× 30 ml).Removal of the solvent under vacuum, followed by distilla-tion to remove excess butanol, gave crude18 (0.35 g, 46.1%)as a pale brown solid [Rf 0.71 diethyl ether/petroleumether 40–60◦C (70:30)]. GCMS tR 21.585 m/z 270(M+). The product from this reaction was used directlyin the synthesis of the aminosulfonated derivative of18.
Butyl-4′-[(aminosulfonyl)oxy]-1,1′- biphenyl-4- carboxy-late (19): compound 19 was synthesized following thesame procedure as for compound10 except that NaH (60%dispersion in mineral oil, 0.1 g, 2.5 mmol) was added to astirred solution of18 (0.3 g, 1.11 mmol) in DMF (10 ml).Aminosulfonyl chloride in toluene (20 ml,∼20 mmol) wasadded after 30 min. Removal of the solvent under vacuumproduced a yellow oil, which was purified using flash chro-matography to give19 (0.11 g, 28.4%) as a pure white solid[mp 100–104◦C; Rf 0.57 diethyl ether/petroleum ether40–60◦C (70/30)].
νmax (film) cm−1: 3415.0, 3300.3 (NH), 1712.5 (C=O)1378.9, 1179.0 (S=O). δH (CDCl3): 8.11 (2H, d, J =9 Hz, ArH), 7.61 (2H, d,J = 9 Hz, ArH), 7.59 (2H, d,J = 9 Hz, ArH), 7.43 (2H, d,J = 9 Hz, ArH), 5.01 (2H,s, NH2), 4.35 (2H, t,J = 7 Hz, OCH2–), 1.77 (2H, m,OCH2CH2–), 1.50 (2H, m, OCH2CH2CH2–) 1.00 (3H,t, J = 7 Hz, –CH3). δC (CDCl3): 167.2 (C=O), 150.0,130.1, 128.8, 127.0, 122.6 (CAr), 65.0 (OCH2), 30.8(OCH2CH2), 19.3 (OCH2CH2CH2), 13.8 (CH3). MS m/z349 (M+).
2.3. Biochemical evaluation of synthesized inhibitors
The synthesized compounds were evaluated for in-hibitory activity against ES using standard literature method[15] in order to determine the initial screening inhibitionand IC50 values, whilst the mode of action (reversible orirreversible inhibition) was determined using a methodinvolving dialysis of bound/unbound inhibitor[16]. Theresults of the biochemical evaluation are shown inTable 1,together with the calculated log P of the parent phenolicstructure.

430 S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435
Table 1Showing the inhibition data for the synthesized compounds, EMATE andCOUMATE (log P was calculated for the parent phenol) (a= [I] of 5 �M;b = [I] of 10 �M; c = [I] of 0.01 �M)
Compound (R) Compoundnumber
Inhibition(%)
IC50 (�M/Tube) Log P
H 10 5.5 a 76.1± 0.9 3.447CN 11 44.2 a 6.7± 0.01 3.483COOMe 13 44.2 a 5.2± 0.07 3.177COOEt 15 53.4 a 4.2± 0.01 3.519COOPr 17 60.5 a 3.5± 0.01 3.988COOBu 19 48.4 a 5.8± 0.02 4.384COUMATE – 47.6 b 10± 0.3 1.698667-COUMATE – 98.2 a 0.2± 0.01 2.651EMATE – 16.4 c 0.5± 0.01 3.870
2.3.1. ES assayIn the biochemical evaluation, the standard literature
method was used[17]. The total assay volume was 1 ml.3H-estrone sulfate (25�l, 50�M; 750,000 dpm/tube) andthe inhibitors (50�M) dissolved in ethanol were added toa 10 ml assay tube, and the ethanol removed with a streamof nitrogen. Tris–HCl buffer (0.05 M, pH 7.2, 0.2 ml) wasadded to each tube. Placental microsomes were then di-luted with Tris–HCl buffer. The microsomes and assaytubes were pre-incubated for 5 min at 37◦C in a shakingwater bath prior to the addition of the microsomes (0.8 ml)to the tubes. After 20 min incubation (at 37◦C), toluene(4 ml) was added to quench the assay, and the tubes placedon ice. The quenched samples were vortexed for 45 s andcentrifuged (3000 rpm, 10 min). The 1 ml of toluene wasremoved and added to 5 ml scintillation cocktail (CocktailT). The efficiency of the extraction process was monitoredusing 14C-estrone. The aliquots were counted for 3 min.All samples were run in triplicate. Control samples withno inhibitor were incubated simultaneously. Blank sampleswere obtained by incubating with boiled microsomes. Itshould also be noted that the basal sulfatase activity for theassay was found to be 85,000 dpm, i.e. approximately 45%conversion.
2.3.2. Irreversible ES assayThe irreversible inhibition was determined using the
procedure described by Purohit et al.[17] using EMATE(10�M), COUMATE (100�M) and compound 15(100�M). Placental microsomes (18 mg/ml, 55�l) wereincubated with each of the inhibitors (25�l in ethanol,removed with a stream of nitrogen) in Tris–HCl buffer(50 mM, pH 7.2, 945�l) at 37◦C for 10 min. A controltube with no inhibitor was incubated simultaneously (100%tubes). An aliquot (100�l) in triplicate, was taken fromeach sample and tested for ES activity using the procedureabove, except that 900�l of Tris–HCl buffer was added tothe assay tubes. A second aliquot (100�l) in triplicate, wassubjected to dialysis at 4◦C for 16 h, with regular changes ofTris–HCl buffer. The microsomes were then removed from
the dialysis tubing and tested for ES activity as describedabove.
2.3.3. Time and concentration dependency assayAn experiment, modified from Purohit et al.[17] was con-
ducted to determine whether the active compounds were in-hibiting ES in a time and concentration dependent manner.Dilutions of each compound (in ethanol) were placed in 5 mlassay tubes and the ethanol removed with a stream of ni-trogen. Tris–HCl buffer (50 mM, pH 7.2, 0.2 ml) was addedand the tubes warmed in a shaking water bath (37◦C, 5 min).At the appointed time the microsomes were added and theassay tubes incubated for between 0 and 60 min. After in-cubation, dextran coated charcoal (1% (w/v) in Tris–HClbuffer, 500�l) was added and the tubes allowed to shake at37◦C for a further 10 min. After this time the tubes were im-mediately centrifuged (1,000 rpm, 3 min), and a portion ofthe supernatant (750�l) added to pre-incubated 10 ml assaytubes containing3H-estrone sulfate (25�l (ethanol removed)to give final concentration 50�M, 750,000 dpm/tube) andTris–HCl buffer (50 mM, pH 7.2, 250�l). After 30 min ofincubation at 37◦C, the assay tubes were quenched by theaddition of toluene (4 ml) and placed on ice. Each tube wasvortexed for 45 s, and centrifuged (3,000 rpm) for 10 min.Aliquots (1 ml) of each toluene layer were added to CocktailT (5 ml) and counted by liquid scintillation.
3. Results and discussion
3.1. Molecular modelling
Consideration of the derived transition-state shows thatthe amino acid residues postulated to be involved in thereaction mechanism are positioned close to the sulfategroup of estrone sulfate and that the C(2) position of thesteroidal backbone is hindered (the nearest amino acid atomto steroid C(2) is 2.1 Å (Fig. 4)). However, consideration ofthe C(4) position shows that it is slightly less hindered thanthe C(2), the nearest amino acid atom to steroid C(4) being3.5 Å. This observation is therefore consistent with exper-imental data[18] which shows that the 2-nitro derivative ofEMATE is a poorer inhibitor than the 4-nitro derivative. Wetherefore propose that these groups are involved in stericinteraction with the amino acid residues leading to desta-bilization of the enzyme-inhibitor complex.Fig. 5 showsthe two derivatives superimposed onto the transition-state,with 2-nitro EMATE resulting in a C(2)-NO2 to the near-est amino acid atom distance of 1.7 Å and 4-nitro EMATEresulting in a C(2)-NO2 to the nearest amino acid atomdistance of 3.7 Å. The hypothesis that steric interaction isresponsible for lowered activity within the 2-substitutedderivatives of EMATE is further supported by compoundssuch as the 2-alkyl derivatives of EMATE which are foundto be some 36,000 times less active than 4-nitro EMATE.The superimpositioning of the 2-alkyl derivative shows that

S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435 431
Fig. 5. Superimpositioning of the 2-nitro EMATE (red) and 4-nitro EMATE (green) derivatives onto the transition state.
high levels of steric interaction is possible between the alkylgroup and the enzyme active site residues.
When alternative steroidal inhibitors were superimposedusing the sulfonate group onto estrone sulfate within thetransition-state, it was discovered that the positions of thesteroidal backbone of the different steroidal inhibitors didnot correspond well. For example, when EMATE (IC50 =65 pM, Fig. 6) was superimposed using only the sulfamategroup onto the sulfate moiety within estrone sulfate, it wasdiscovered that a poor fit was obtained (with root meansquare fit value of 0.17) and a distance of 1.6 Å was ob-served between the EMATE C(17)=O and the estrone sulfateC(17)=O group. Consideration of alternative estrogen-basedinhibitors shows similar results. That is, the low energy con-formers of the weaker inhibitors based upon estradiol (i.e.compounds containing a�-hydroxy group instead of theC(17)=O, compounds2 and 3 (Fig. 2)), show that the in-
Fig. 6. Low energy conformer of EMATE (green) superimposed onto the transition-state.
hibitors superimpose in a similar manner to EMATE, how-ever, resulting in an increased substrate C(17)=O to inhibitorC(17)-�OH group distance of 3.7 Å for compound2. Fromthe consideration of these steroidal inhibitors, in particu-lar, the positioning of the C(17) hydrogen-bonding groupof these compounds with respect to the C(17)=O of estronesulfate within the transition-state, we conclude that the mostrelevant part of the steroid-derived structure is the posses-sion of a C(3) sulfonate/sulfamate group. We therefore pos-tulated that the C(17) position may be involved in polar-polarinteraction with the active site, however, this interaction isdependent upon the functional group at the C(17) position,thus the hydroxy moiety may result in weaker interactionthan the carbonyl group.
The consideration of the steroidal compounds suggeststhe existence of hydrogen-bonding groups at the active sitecorresponding to the C(17) area of the steroidal backbone.

432 S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435
Fig. 7. Low energy conformer of 667-COUMATE superimposed onto the transition-state.
In particular, the inhibitory activity suggests the existenceof groups able to interact with a C=O since the posses-sion of a hydroxy group leads to reduced inhibitory activity.However, in the case of the non-steroidal inhibitors (and inparticular the potent inhibition exhibited by some of thesecompounds) the above hypothesis is contradicted since mostof the non-steroidal compounds contain no clear functionalgroup which is able to mimic the C(17) hydrogen-bondinggroup in estrone sulfate or the steroidal inhibitors.
For example, compounds such as4 (Fig. 2) are potentinhibitors (IC50 values= 1�M), however, this compounddoes not possess any polar groups that can mimic thesteroidal C(17) region of the steroid backbone. Compoundssuch as COUMATE (IC50 = 380 nM), 667-COUMATE(IC50 = 8 nM) and 5 (IC50 = 55.8 nM) possess groups(the carboxy groups of the lactone and amide respectively)potentially capable of undergoing hydrogen-bonding withthe region of the active site corresponding to the steroidC(17) position. Superimpositioning of these compounds inan attempt to allow such interactions to take place results inthe groups being some distance away from the appropriatehydrogen-bonding group at the active site. Furthermore,667-COUMATE possesses a heptyl cyclic moiety whichdoes not mimic the C(17)=O in any way, however, it isthe most potent compound knowntodate and has enteredphase II clinical trials. Furthermore, superimpositioning of667-COUMATE onto the transition-state shows that thelowest energy conformer possessed an inhibitor carbonyl
Fig. 8. Low energy conformer of6 superimposed onto the transition-state.
group to active site C(17) hydrogen bonding group distanceof 6.4 Å (Fig. 7), a distance which is considered to be toolarge for strong interaction.
The compounds considered thus far contain the less hin-dered sulfamate moiety (OSO2NH2). However, there aresulfamate derivatives, such as inhibitor6 (Fig. 2), which pos-sess a bulky group attached to the sulfonate group. Thesetypes of compound have not been considered previouslyas it was unclear as to how they could be accommodatedwithin the active site, indeed, they were initially presumedto be weak inhibitors. However, the results of the biochemi-cal evaluation showed these compounds to possess good in-hibitory activity. We believe that with our representation ofthe active site, the activity of these bulky group-containingcompounds can now be rationalized. That is, we discoveredthat on undertaking conformational analysis of the rotatablebonds within the sulfamate group, conformers were found(which were within a�E ∼ 5 kcal/mol range) which al-lowed the bulky group to be accommodated within the ac-tive site without undergoing close interaction between thecomponents of the active site and the inhibitors (Fig. 8).
In summary, the results of the molecular modellingof known steroidal inhibitors suggest that groups able tomimic the sulfate group of estrone sulfate is the most im-portant factor in the inhibition of ES, however, within thearea of the active site occupied by the sulfate group, thereappear to be ‘pockets’ able to accommodate phenyl groupsrather than aliphatic based moieties. The use of interactions
S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435 433
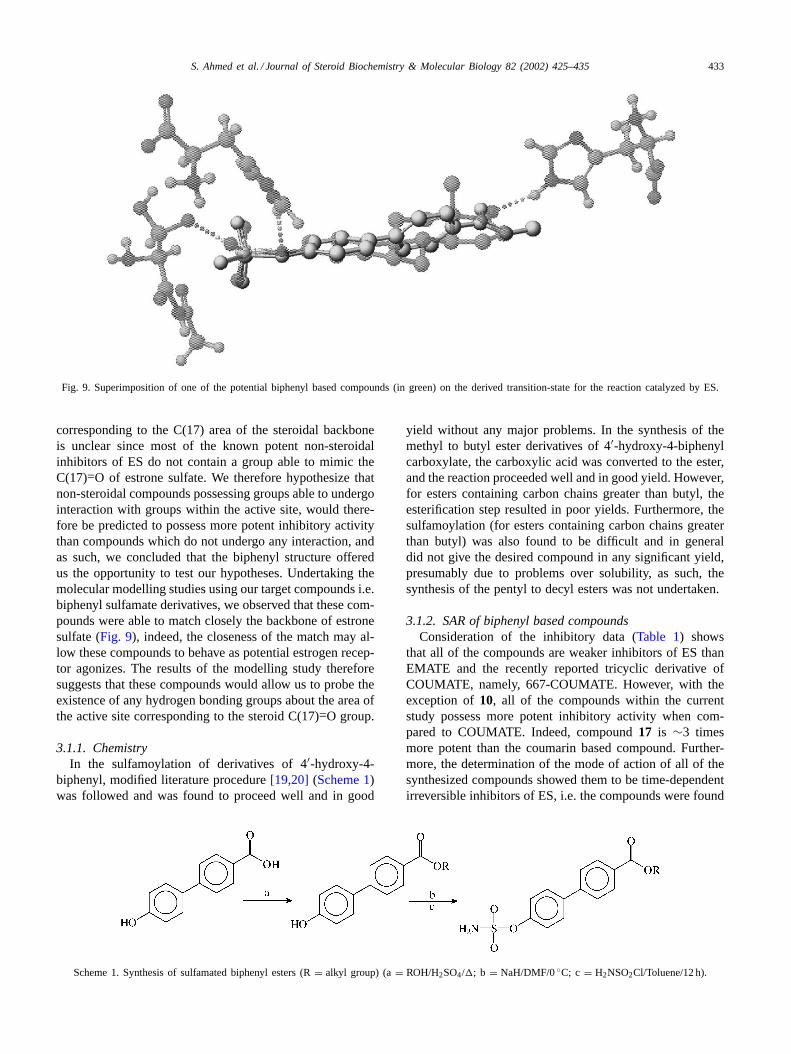
Fig. 9. Superimposition of one of the potential biphenyl based compounds (in green) on the derived transition-state for the reaction catalyzed by ES.
corresponding to the C(17) area of the steroidal backboneis unclear since most of the known potent non-steroidalinhibitors of ES do not contain a group able to mimic theC(17)=O of estrone sulfate. We therefore hypothesize thatnon-steroidal compounds possessing groups able to undergointeraction with groups within the active site, would there-fore be predicted to possess more potent inhibitory activitythan compounds which do not undergo any interaction, andas such, we concluded that the biphenyl structure offeredus the opportunity to test our hypotheses. Undertaking themolecular modelling studies using our target compounds i.e.biphenyl sulfamate derivatives, we observed that these com-pounds were able to match closely the backbone of estronesulfate (Fig. 9), indeed, the closeness of the match may al-low these compounds to behave as potential estrogen recep-tor agonizes. The results of the modelling study thereforesuggests that these compounds would allow us to probe theexistence of any hydrogen bonding groups about the area ofthe active site corresponding to the steroid C(17)=O group.
3.1.1. ChemistryIn the sulfamoylation of derivatives of 4′-hydroxy-4-
biphenyl, modified literature procedure[19,20] (Scheme 1)was followed and was found to proceed well and in good
Scheme 1. Synthesis of sulfamated biphenyl esters (R= alkyl group) (a= ROH/H2SO4/�; b = NaH/DMF/0◦C; c = H2NSO2Cl/Toluene/12 h).
yield without any major problems. In the synthesis of themethyl to butyl ester derivatives of 4′-hydroxy-4-biphenylcarboxylate, the carboxylic acid was converted to the ester,and the reaction proceeded well and in good yield. However,for esters containing carbon chains greater than butyl, theesterification step resulted in poor yields. Furthermore, thesulfamoylation (for esters containing carbon chains greaterthan butyl) was also found to be difficult and in generaldid not give the desired compound in any significant yield,presumably due to problems over solubility, as such, thesynthesis of the pentyl to decyl esters was not undertaken.
3.1.2. SAR of biphenyl based compoundsConsideration of the inhibitory data (Table 1) shows
that all of the compounds are weaker inhibitors of ES thanEMATE and the recently reported tricyclic derivative ofCOUMATE, namely, 667-COUMATE. However, with theexception of10, all of the compounds within the currentstudy possess more potent inhibitory activity when com-pared to COUMATE. Indeed, compound17 is ∼3 timesmore potent than the coumarin based compound. Further-more, the determination of the mode of action of all of thesynthesized compounds showed them to be time-dependentirreversible inhibitors of ES, i.e. the compounds were found

434 S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435
Fig. 10. Plot of IC50 vs. log P to show the relationship between the two parameters for the synthesized compounds studied within this report.
to bind irreversibly to the ES active site and could not be di-alyzed with time. For example, a solution of15 (at 100�Mconcentration) was incubated with the ES enzyme and fol-lowing dialysis of the compound, the enzyme was foundto show some 14% conversion compared to the known re-versible inhibitor, danazol, which showed in excess of 90%conversion after dialysis of the inhibitor. In comparison, theincubation of ES with the standard compound EMATE (at1�M concentration) resulted in the enzyme recovering itscatalytic activity by some 23% after dialysis of the inhibitor.
From the initial consideration of the inhibition data of thebiphenyl-based compounds considered within the presentstudy, we are able to conclude that these compounds may beable to undergo polar-polar or hydrogen bonding interactionswith the active site. That is, where the R group contains apotential hydrogen-bonding moiety (Fig. 3) the compoundsare able to undergo favorable interaction that results in anincrease in the binding ability of these compounds comparedto COUMATE, resulting in good inhibitory activity. Sincethese compounds are closer mimics of the steroidal backbonecompared to 667-COUMATE, we propose that an alternativefactor may be responsible for the potent inhibition observedwithin this compound which are not observed within thebiphenyl substituted compounds. The reduction in inhibitoryactivity observed with increasing alkyl chain length withinthe ester moiety is proposed to be due to steric interactionbetween the inhibitor and components within the active siteof ES (corresponding to the C(17) area of the steroid back-bone). As such, the current study adds further support tothe observations from the superimpositioning studies of thesteroidal inhibitors, i.e. hydrogen-bonding group(s) may ex-ist at the active site which are able to interact with the C(17)area of the steroid backbone. Since the compounds were de-signed using the derived transition-state of the desulfatationreaction, the results of our study add further support to theaccuracy of the derived transition-state.
A detailed consideration of the physicochemical prop-erties of these compounds, in particular, the alkyl esters,shows that within the ester derivatives, there appears to be agood correlation between log P and inhibitory activity (IC50value) (Fig. 10). Further detailed consideration ofFig. 5shows that the ‘optimum’ log P appears to be approximately3.7. We have previously rationalized[15] the hydrophobicrequirement as being an important factor in the determi-
nation of the inhibitory activity of compounds against ES.Through the consideration of the SAR determination studyof a large number of alkyl and phenyl sulfamate based com-pounds, we proposed that the hydrophobic nature of the car-bon backbone of the inhibitors was necessary to destabilizethe phenoxide ion resulting from the desulfatation reaction,which resulted in the loss of the carbon backbone of the in-hibitor (phenoxide ion containing moiety) from the activesite [15]. From our studies, we have also hypothesized thatcompounds which mimicked the estrone backbone with re-spect to hydrophobicity (calculated log P of estrone is 3.8)would possess good inhibition (assuming the pKa factor wasin the range of 7.7–8.2). As can be observed, the optimumlog P within the present study is similar to that of estrone, assuch, the results of this study therefore add further supportto our previous work.
4. Conclusion
In conclusion, we have successfully designed and syn-thesized a range of compounds so as to mimic the steroidbackbone of estrone. Furthermore, from the considerationof the inhibitory data, we have been able to propose the ex-istence of a potential hydrogen bonding group at an areacorresponding to the C(17) area of the steroid backbone andhave added further support to the accuracy of the derivedtransition-state for the desulfatation reaction catalyzed byES. These compounds therefore are good lead compoundsin the mimicking of the steroid backbone and thus allow usthe opportunity to derivatise the biphenyl structure so as toincrease potency.
Acknowledgements
The high resolution mass spectra were undertaken by theEPSRC National Mass Spectrometry centre at the Universityof Wales College Swansea.
References
[1] A.M.H. Brodie, V.C.O. Njar, Aromatase inhibitors and their appli-cation in breast cancer treatment, Steroids 65 (2000) 171–179.

S. Ahmed et al. / Journal of Steroid Biochemistry & Molecular Biology 82 (2002) 425–435 435
[2] A. Purohit, G.J. Williams, N.M. Howarth, B.V.L. Potter, M.J. Reed,Inactivation of steroid sulfatase by an active-site-directed inhibitor,estrone-3-O-sulfamate, Biochemistry 34 (1995) 11508–11514.
[3] L.W.L. Woo, A. Purohit, B. Malini, M.J. Reed, B.V.L. Potter,Potent active site-directed inhibition of steroid sulphatase by tricycliccoumarin-based sulphamates, Chem. Biol. 7 (2000) 773–791.
[4] S. Ahmed, K. James, L. Sampson, C. Mastri, Structure-activityrelationship study of steroidal and non-steroidal inhibitors of theenzyme estrone sulfatase, Biochem. Biophys. Res. Commun. 254(1999) 811–815.
[5] S. Ahmed, K. James, C.P. Owen, C.K. Patel, M. Patel, Determinationand use of a transition state for the enzyme estrone sulfatase (ES)from a proposed reaction mechanism, Bioorg. Med. Chem. Lett. 11(2001) 3001–3005.
[6] S. Ahmed, K. James, C.P. Owen, C.K. Patel, Acid dissociation cons-tant, a potential physicochemical factor in the inhibition of theenzyme estrone sulfatase (ES), Bioorg. Med. Chem. Lett. 12 (2001)899–902.
[7] A.D. Abell, B.R. Henderson, Steroidal and non-steroidal inhibitorsof steroid 5-alpha-reductase, Curr. Med. Chem. 2 (1995) 583–597.
[8] R. von Bulow, B. Schmidt, T. Dierks, K. von Figura, I. Uson, Crystalstructure of an enzyme-substrate complex provides insight into theinteraction between human arylsulfatase A and its substrates duringcatalysis, J. Mol. Biol. 305 (2001) 269–277.
[9] CACHE (Trademark) Oxford Molecular Ltd., Oxford Science Park,Oxford, UK.
[10] N.L. Allinger, Conformational analysis. 130. MM2. A hydrocarbonforce field utilizing V1 and V2 torsional terms, J. Am. Chem. Soc.99 (1977) 8127–8134.
[11] Mopac is a Copyright of the US Air Force Academy, Frank J. SeilerResearch Laboratory, US Air Force Academy, Colorado Springs, CO80840-6528.
[12] J.J.P. Stewart, Optimization of parameters for semiempirical methods.1. Method, J. Comput. Chem. 10 (1989) 209–220.
[13] Alchemy IIITM, PowersearchTM (Trademarks) Tripos Associates Inc.,1699 South Hanley Road, Suite 303, St. Louis, Missouri 63144,USA.
[14] G. Hedayatullah, Synthese et reduction d’azidosulfates d’aryle, TetraLetts. 22 (1975) 2455–2456.
[15] A. Purohit, K.A. Vernon, A.E.W. Hummelinck, L.W.L. Woo, H.A.M.Hejaz, B.V.L. Potter, M.J. Reed, The development of a-ring modifiedanalogues of oestrone-3-O-sulphamate as potent steroid sulphataseinhibitors with reduced oestrogenicity, J. Steroid Biochem. Mol. Biol.64 (1998) 269–275.
[16] A. Purohit, L.W.L. Woo, B.V.L. Potter, M.J. Reed, In vivo inhibitionof estrone sulfatase activity and growth of nitrosomethylurea-inducedmammary tumors by 667 COUMATE, Cancer Res. 60 (2000) 3394–3396.
[17] A. Purohit, G.J. Williams, N.M. Howarth, B.V.L. Potter, M.J.Reed, Inactivation of steroid sulfatase by an active-site-directedinhibitor, estrone-3-O-sulfamate, Biochemistry 34 (1995) 11508–11514.
[18] L.W.L. Woo, N.M. Howarth, A. Purohit, H.A.M. Hejaz, M.J. Reed,B.V.L. Potter, Steroidal and non-steroidal sulfamates as potentinhibitors of steroid sulfatase, J. Med. Chem. 41 (1998) 1068–1083.
[19] S. Ahmed, K. James, C.P. Owen, C.K. Patel, M. Patel, Novelinhibitors of the enzyme estrone sulfatase (ES), Bioorg. Med. Chem.Lett. 11 (2001) 841–844.
[20] S. Ahmed, K. James, C.P. Owen, C.K. Patel, L. Sampson, Synthesisand biochemical evaluation of novel and potent inhibitors of theenzyme oestrone sulphatase (ES), J. Steroid Biochem. Mol. Biol. 80(2002) 419–427.
Copyright © 2022 FDOKUMEN


![EP2306 [2-(4-biphenyl)-4-methyl-octahydro-1,4-benzoxazin-2-ol, hydrobromide], a novel squalene synthase inhibitor, reduces atherosclerosis in the cholesterol-fed rabbit](https://static.fdokumen.com/doc/165x107/6337a6386f78ac31240eae17/ep2306-2-4-biphenyl-4-methyl-octahydro-14-benzoxazin-2-ol-hydrobromide-a.jpg)




![The photophysics of singlet, triplet, and degradation trap states in 4,4-N,N[sup ʹ]-dicarbazolyl-1,1[sup ʹ]-biphenyl](https://static.fdokumen.com/doc/165x107/634397aac405478ed30633d9/the-photophysics-of-singlet-triplet-and-degradation-trap-states-in-44-nnsup.jpg)







![Condensed bridgehead nitrogen heterocyclic system: Synthesis and pharmacological activities of 1,2,4-triazolo-[3,4- b]-1,3,4-thiadiazole derivatives of ibuprofen and biphenyl-4-yloxy](https://static.fdokumen.com/doc/165x107/632834412089eb31f609dd2b/condensed-bridgehead-nitrogen-heterocyclic-system-synthesis-and-pharmacological.jpg)




![Intrinsically Conductive Organo-Silver Linear Chain Polymers [– S–Ag–S–biphenyl–]n assembled on Roughened Elemental Silver](https://static.fdokumen.com/doc/165x107/632805dd6989153a060bbeb3/intrinsically-conductive-organo-silver-linear-chain-polymers-sagsbiphenyln.jpg)