
Reactivity of Pd/Fe bimetallic nanotubes in dechlorination of coplanar polychlorinated biphenyls
Upload
independentCategory
view
1download
0
Identification of the metabolically active members of abacterial community in a polychlorinated biphenyl-polluted moorland soil
Balbina Nogales, * Edward R. B. Moore,Wolf-Rainer Abraham and Kenneth N. TimmisDivision of Microbiology, GBF-National Research Centrefor Biotechnology, Mascheroder Weg 1, D-38124Braunschweig, Germany.
Summary
The presumptive metabolically active members of abacterial community in a moorland soil in Germany,highly polluted with polychlorinated biphenyls (PCBs),were identified by sequencing of cloned reversetranscription–polymerase chain reaction (RT–PCR)amplification products of 16S rRNA generated fromtotal RNA extracts. Analysis of the 16S rRNA clonelibrary revealed a considerable diversity of metaboli-cally active bacteria in the soil, despite the acidic pHand high concentrations of PCBs. Cloned sequencetypes clustered within the Proteobacteria (34% alpha-,33% beta- and 7% gamma-subclasses), the Holo-phaga –Acidobacterium phylum (14%), the Actinobac-teria (6.5%) and the Planctomycetales (2%). Threecloned sequence types were not affiliated to anydescribed phylogenetic group. An unusual feature ofthis soil was the abundance of sequence types withinthe beta-subclass of the Proteobacteria , most ofwhich were similar to the 16S rRNA gene sequencesof species from only two genera, Burkholderia andVariovorax . Three other numerous 16S rRNA sequencetypes were similar to the sequences of Sphingomonasspecies, members of the Rhodopila globiformisgroup and Acidobacterium capsulatum . Some of thesequence types retrieved were similar to the 16SrRNA sequences of bacterial isolates able to degradea variety of organic pollutants, including PCBs. As thePCB contamination is the major source of measurablecarbon in this soil, some of the 16S rRNA sequencetypes detected and presumed to represent the meta-bolically active members of the community indicatethe organisms likely to be involved, directly or
indirectly, in the utilization of the PCBs as carbonand energy sources.
Introduction
Microorganisms are important agents in the naturalcleansing of the biosphere of organic wastes of biologicalorigin and play significant roles in the detoxification ofenvironmental pollutants of non-biological origin resulting,for example, from industrial activities. As human popula-tions and industrial activities increased and the rate of pol-lution accelerated, so it became necessary to acceleratenatural detoxification activities by developing efficient bio-technological processes, such as waste treatment plants.This has not, however, eliminated industrial pollution of theenvironment, and the number of contaminated sites thatrequire to be cleaned up is substantial. Some of thesesites may be treated by thermic or chemical processes,but others, particularly those involving large areas ofunderground and aquifer material, must be treated biologi-cally in situ (Bouwer et al., 1994).
In situ bioremediation is a growing technology, still in itsinfancy and lacking a knowledge base on the underlyingmicrobiology and ecology. Knowledge of the microorgan-isms that are the most effective pollutant metabolizersunder different conditions, as well as the nature, popula-tion densities and activities of such organisms in pollutedsites, will promote the development of optimal bioremedia-tion processes (Liu and Suflita, 1993). Thus far, our knowl-edge about pollutant degradative capabilities is limited tothose demonstrated by cultivated microorganisms, althoughit is generally recognized that these may not be the mostimportant microorganisms in natural microbial communities.
In order to gain information on the nature of the bacteriathat may play important roles in pollutant degradation, wehave exploited a culture-independent method for examin-ing the diversity and composition of the metabolicallyactive members of a microbial community present in asoil highly polluted with polychlorinated biphenyls (PCBs).The approach taken was the identification of the individual16S ribosomal RNA sequences (16S rRNA) in clonelibraries generated from reverse transcription–polymer-ase chain reaction (RT–PCR) amplification productsobtained from total RNA.
Environmental Microbiology (1999) 1(3), 199–212
Q 1999 Blackwell Science Ltd
Received 28 September, 1998; revised 6 December, 1998; accepted21 December, 1998. *For correspondence. E-mail [email protected]; Tel.(þ49) 531 6181 405; Fax (þ49) 531 6181 411.
Results
The Wittenberg site
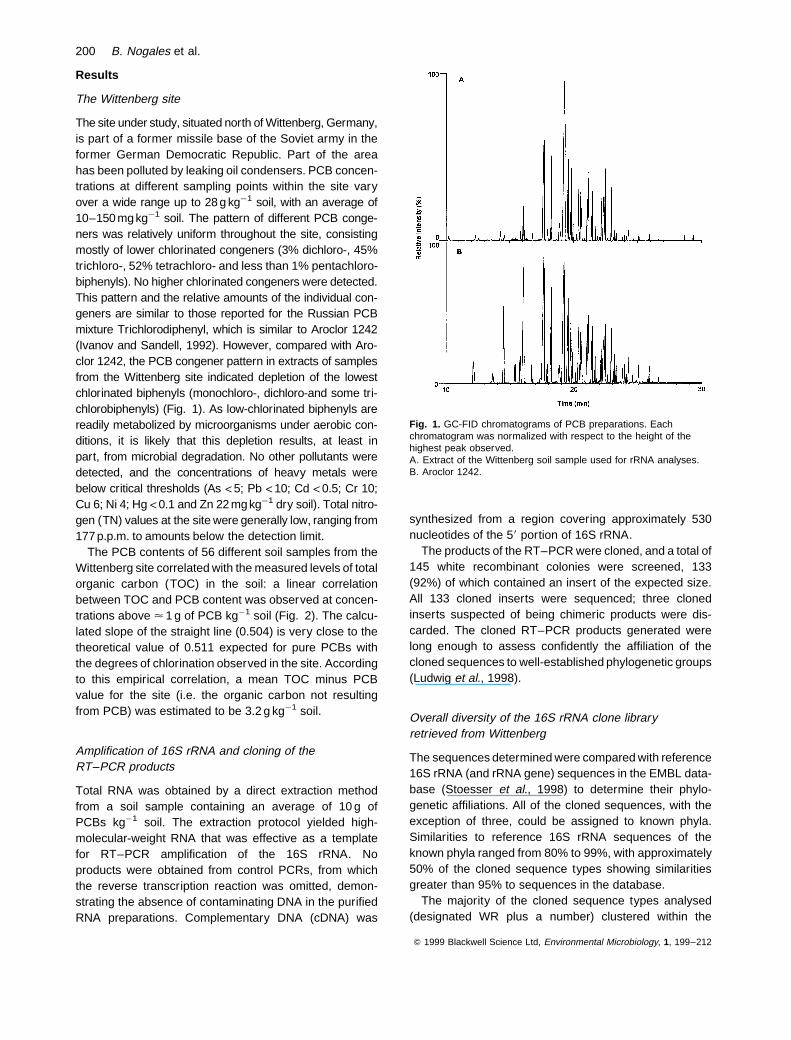
The site under study, situated north of Wittenberg, Germany,is part of a former missile base of the Soviet army in theformer German Democratic Republic. Part of the areahas been polluted by leaking oil condensers. PCB concen-trations at different sampling points within the site varyover a wide range up to 28 g kg¹1 soil, with an average of10–150 mgkg¹1 soil. The pattern of different PCB conge-ners was relatively uniform throughout the site, consistingmostly of lower chlorinated congeners (3% dichloro-, 45%trichloro-, 52% tetrachloro- and less than 1% pentachloro-biphenyls). No higher chlorinated congeners were detected.This pattern and the relative amounts of the individual con-geners are similar to those reported for the Russian PCBmixture Trichlorodiphenyl, which is similar to Aroclor 1242(Ivanov and Sandell, 1992). However, compared with Aro-clor 1242, the PCB congener pattern in extracts of samplesfrom the Wittenberg site indicated depletion of the lowestchlorinated biphenyls (monochloro-, dichloro-and some tri-chlorobiphenyls) (Fig. 1). As low-chlorinated biphenyls arereadily metabolized by microorganisms under aerobic con-ditions, it is likely that this depletion results, at least inpart, from microbial degradation. No other pollutants weredetected, and the concentrations of heavy metals werebelow critical thresholds (As <5; Pb <10; Cd <0.5; Cr 10;Cu 6; Ni 4; Hg <0.1 and Zn 22 mgkg¹1 dry soil). Total nitro-gen (TN) values at the site were generally low, ranging from177 p.p.m. to amounts below the detection limit.
The PCB contents of 56 different soil samples from theWittenberg site correlated with the measured levels of totalorganic carbon (TOC) in the soil: a linear correlationbetween TOC and PCB content was observed at concen-trations above < 1 g of PCB kg¹1 soil (Fig. 2). The calcu-lated slope of the straight line (0.504) is very close to thetheoretical value of 0.511 expected for pure PCBs withthe degrees of chlorination observed in the site. Accordingto this empirical correlation, a mean TOC minus PCBvalue for the site (i.e. the organic carbon not resultingfrom PCB) was estimated to be 3.2 g kg¹1 soil.
Amplification of 16S rRNA and cloning of theRT–PCR products
Total RNA was obtained by a direct extraction methodfrom a soil sample containing an average of 10 g ofPCBs kg¹1 soil. The extraction protocol yielded high-molecular-weight RNA that was effective as a templatefor RT–PCR amplification of the 16S rRNA. Noproducts were obtained from control PCRs, from whichthe reverse transcription reaction was omitted, demon-strating the absence of contaminating DNA in the purifiedRNA preparations. Complementary DNA (cDNA) was
synthesized from a region covering approximately 530nucleotides of the 58 portion of 16S rRNA.
The products of the RT–PCR were cloned, and a total of145 white recombinant colonies were screened, 133(92%) of which contained an insert of the expected size.All 133 cloned inserts were sequenced; three clonedinserts suspected of being chimeric products were dis-carded. The cloned RT–PCR products generated werelong enough to assess confidently the affiliation of thecloned sequences to well-established phylogenetic groups(Ludwig et al., 1998).
Overall diversity of the 16S rRNA clone libraryretrieved from Wittenberg
The sequences determined were compared with reference16S rRNA (and rRNA gene) sequences in the EMBL data-base (Stoesser et al., 1998) to determine their phylo-genetic affiliations. All of the cloned sequences, with theexception of three, could be assigned to known phyla.Similarities to reference 16S rRNA sequences of theknown phyla ranged from 80% to 99%, with approximately50% of the cloned sequence types showing similaritiesgreater than 95% to sequences in the database.
The majority of the cloned sequence types analysed(designated WR plus a number) clustered within the
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Fig. 1. GC-FID chromatograms of PCB preparations. Eachchromatogram was normalized with respect to the height of thehighest peak observed.A. Extract of the Wittenberg soil sample used for rRNA analyses.B. Aroclor 1242.
200 B. Nogales et al.

alpha (34.1% of the total number of sequence types)and beta (33.3%) subdivisions of the Proteobacteria(Stackebrandt et al., 1988). A smaller number of clonedsequence types clustered within the gamma subdivision(7.3%). Sequence types related to the Holophaga–Acidobacterium phylum (Ludwig et al., 1997) were alsoobserved (13.8%). Approximately 6.5% of the sequencetypes belonged to the class Actinobacteria (Stackebrandtet al., 1997), and a smaller number of sequence types(2.3%) clustered within the Planctomyces subdivision(Stackebrandt et al., 1984). Seven cloned sequenceswere represented more than once in the library, four ofwhich clustered within the alpha-subclass of the Proteo-bacteria and three within the beta-subclass.
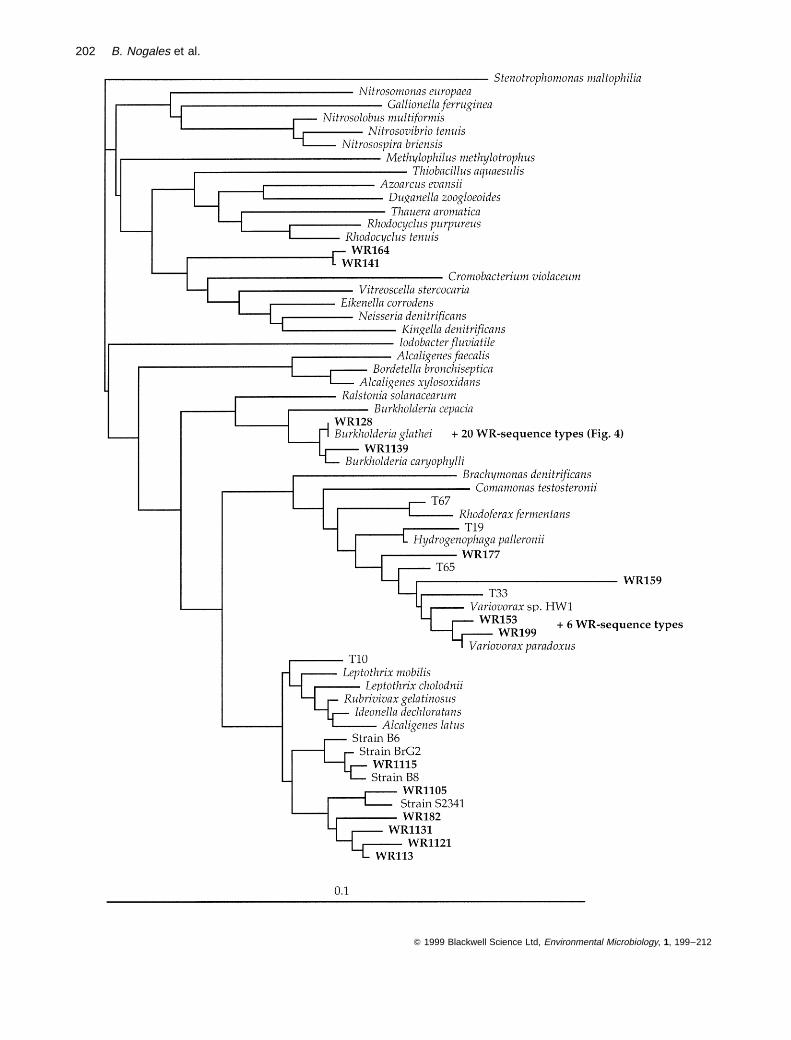
Predominance of cloned sequence types similar tosequences of species of the genera Burkholderiaand Variovorax within the beta-subclass of theProteobacteria
Most of the cloned 16S rRNA sequence types assigned tothe beta-subclass of the Proteobacteria were related tosequences of species classified within only two genera:Burkholderia (22 sequence types) and Variovorax (10sequence types). The collection of sequence types similarto those species of Burkholderia was the most abundantcluster in the clone library retrieved from Wittenberg soil(Figs 3 and 4). As shown in Fig. 4, the cloned sequencetypes were highly similar (97–99% similarity) to the 16SrRNA gene sequences of B. glathei, B. caryophylli, thephenantrene-degrading Burkholderia sp. strains N3P2,N2P5 and N2P6 isolated from soils (Mueller et al.,1997), and somewhat less similar to the sequence of thewell-described PCB degrader, Burkholderia sp. LB400(Bopp, 1986).
Variovorax-like cloned sequence types showed (withthe exception of clones WR159 and WR177) high similari-ties (above 98%) to 16S rRNA sequences of Variovoraxparadoxus, the poly(hexamethylene carbonate)-degrading
strain Variovorax sp. WFF52 (Suyama et al., 1998), andthe 2,4-dichlorophenoxyacetic acid-degrading bacteriumVariovorax sp. strain HW1 (Kamagata et al., 1997). Thisgroup of cloned sequence types was also related to clonedsequences obtained from DNA extracts of activatedsludge (Snaidr et al., 1997; see Fig. 3).
Other, less numerous, cloned sequence types asso-ciated with species of the beta-subclass of the Proteo-bacteria formed a cluster most closely related to the 16SrRNA sequence of Leptothrix mobilis (96.7–97.8% similar-ity; Fig. 3) and to the 16S rRNA sequence of an iron-oxidizing bacterium, strain BrG2 (Straub et al., 1996), anisolate from a Japanese paddy field soil, strain S23410(Mitsui et al., 1997), and several isolates from drinkingwater biofilms (Kalmbach et al., 1997). Additionally, twoclonedsequence types formedadistinct cluster with relativelylow similarities (93.6–93.9%) to the 16S rRNA sequences ofRhodocyclus purpureus and Duganella zoogloeoides.
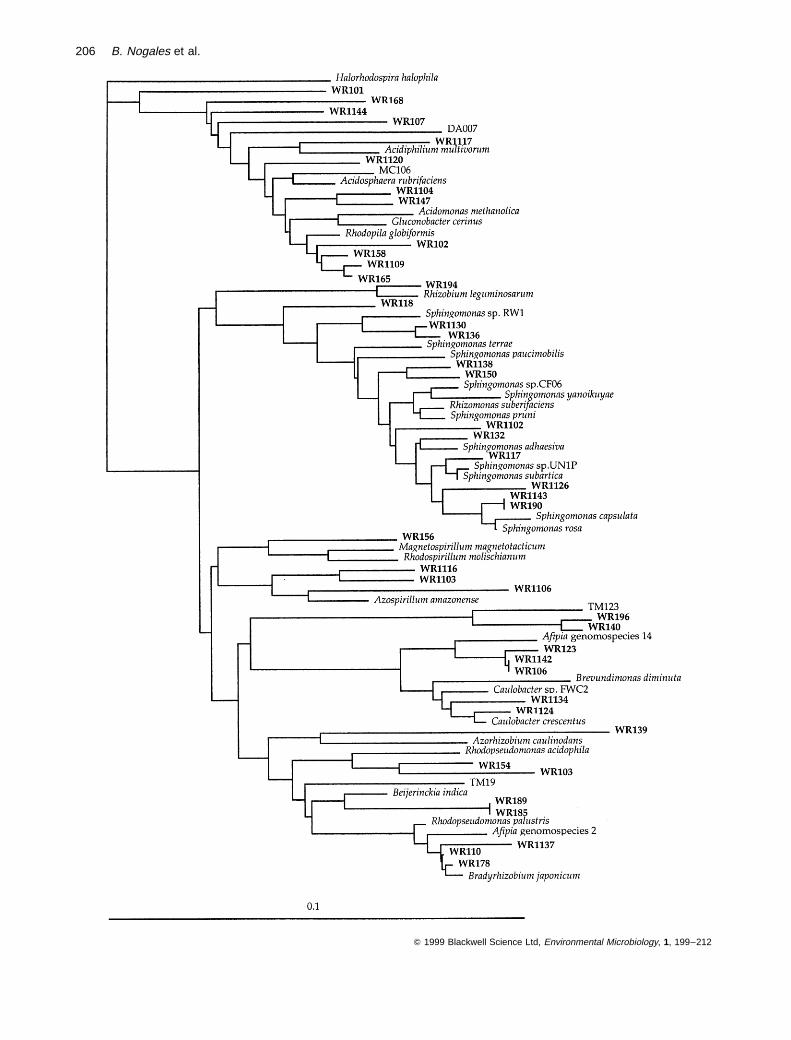
Broader diversity among the cloned sequence typesaffiliated to the alpha-subclass of the Proteobacteria
Cloned sequence types from the Wittenberg soil affiliatedwithin the alpha-subclass of the Proteobacteria showedsimilarities to the 16S rRNA sequences of species of abroad range of different genera. Most of the clonedsequence types within the alpha-subclass were relatedto the sequences of species of the genus Sphingomonas(11 sequence types) and to the Rhodopila globiformisgroup (12 sequence types), a phylogenetic group thatcontains mostly acidiphilic bacteria. Similarities amongthe cloned sequence types and the 16S rRNA sequencesof the most related described species of the genusSphingomonas ranged from 94% to 98%. As shown inFig. 5, Wittenberg cloned sequence types were most simi-lar to the sequences of S. subarctica (Nohynek et al.,1996) and the strains UN1P1 (Mueller et al., 1997),CF06 (Feng et al., 1997) and RW1 (Wittich et al., 1992),known to degrade chlorophenol, phenantrene, carbofuranand dibenzo-p-dioxin respectively. The sequence typesclustering with the Rhodopila globiformis group formed adistinct cluster moderately related to the 16S rRNAsequences of R. globiformis, Acidosphaera rubrifaciens,Acidiphilium and Gluconobacter cerinus (Fig. 5). A groupof sequence types related to Rhodopila globiformis wasalso found in 16S rRNA gene clone libraries retrievedfrom other acidic soils, such as Mount Coot-tha soil inAustralia, a German peat bog soil and the Drentse Agrassland soil in the Netherlands (Stackebrandt et al.,1993; Rheims et al., 1996a; Felske et al., 1998).
The remainder of the cloned sequence types clusteringwithin the alpha-subclass of the Proteobacteria wereobserved to be related to the 16S rRNA sequences ofspecies of different genera, such as Caulobacter (five
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Fig. 2. Relationship of total organic carbon (TOC) and PCBcontents of different soil samples from the polluted site nearWittenberg, Germany. The straight line gives the best linear fit withTOC (mg kg¹1) ¼ 0.504 ×PCB (mg kg¹1) þ 3190 (r2 ¼ 0.7687).
Diversity of metabolically active bacteria in PCB-polluted soil 201
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
202 B. Nogales et al.

cloned sequence types), Bradyrhizobium (three), Rhizo-bium (one), Rhodopseudomonas acidophila (two), Beijer-inckia indica (one) and Magnetospirillum (one) (Fig. 5),most of which are typically found in soil environments.Some of the cloned sequence types were only distantlyrelated to the sequences of any described species, as inthe case of clone WR139, most closely related to Azorhi-zobium caulinodans (86.4% similarity), and the clonedsequences WR1103, WR1106 and WR1116, which formeda cluster distantly related to Azospirillum amazonense(88.8–90.7% similarity). The closest related referencesequence to clones WR140 and WR196 was clonedsequence TM123, retrieved from a peat bog soil in Germany(Rheims et al., 1996a).
Detection of Acidobacterium capsulatum-relatedsequences in the Wittenberg soil
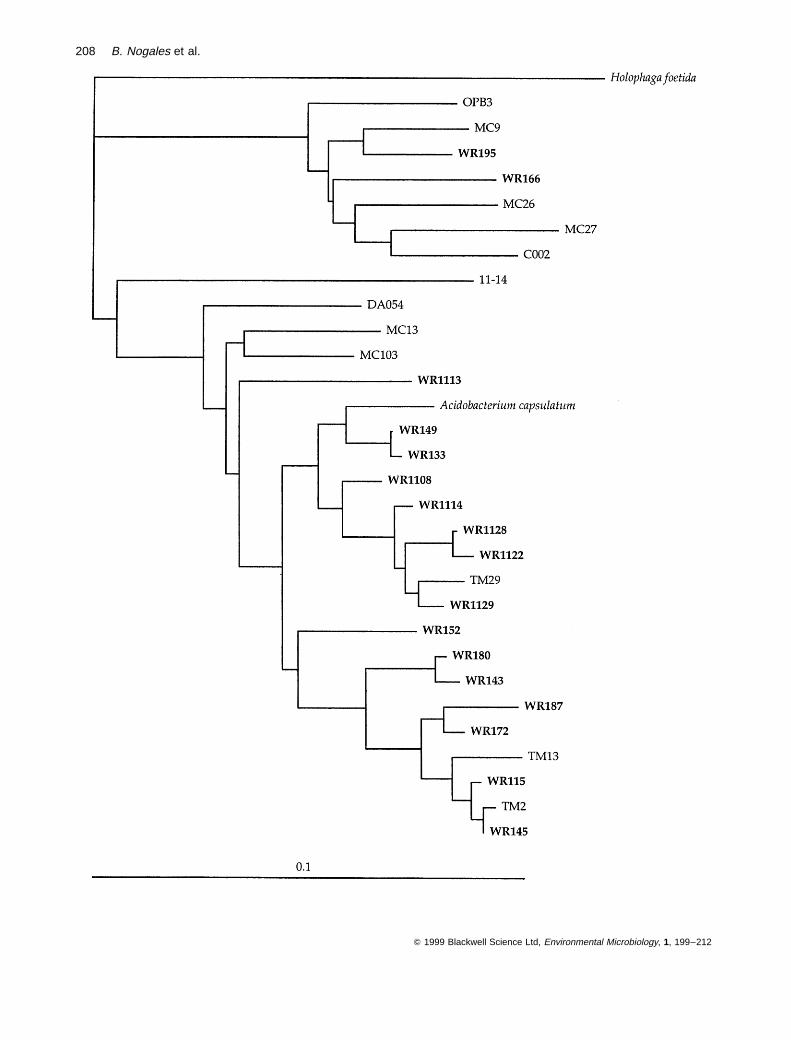
A numerous group of cloned sequence types (a total of 17,the second most abundant in the library) clustered withinthe Holophaga–Acidobacterium phylum (Ludwig et al.,1997), with the acidiphilic bacterium Acidobacterium cap-sulatum being the closest cultivated, described species(Fig. 6). Similarities of the cloned sequences to the 16SrRNA sequence of A. capsulatum ranged from 82% to96%, indicating a broad diversity of these Acidobacter-ium-related cloned sequence types. Wittenberg sequencetypes in this group were also similar to other Acidobacter-ium-related cloned 16S rRNA gene sequences retrievedfrom different non-polluted soils, i.e. TM cloned sequencesfrom a peat bog soil in Germany (Rheims et al., 1996a),MC cloned sequences from Mount Coot-tha soil in Australia(Stackebrandt et al., 1993), DA cloned sequences fromDrentse A grassland soil in The Netherlands (Felskeet al., 1998) and a cloned sequence from an acid minedrainage site in California (EMBL accession numberAF047646). The fact that representatives of these sequencetypes were also found in the Wittenberg 16S rRNA clonelibrary suggests that as yet uncultured microorganismsrelated to this species are not only widespread (Hugenholtzet al., 1998a), but also play an important role in the meta-bolic activity of the community.
Identification of other less numerous clonedsequence types
A total of nine cloned sequence types clustered within thegamma-subclass of the Proteobacteria. One of the sequence
types exhibited a high similarity to 16S rRNA sequencesof species of the genus Pseudomonas (99.5% similaritywith ‘P. pavonaceae’). A group of five cloned sequencetypes was more distantly related (95.0–96.9% similarities)to 16S rRNA sequences of strains of Nevskia ramosa, amember of the epineuston (a community living onwater–air interfaces) that produces hydrophobic surfacefilms (Sturmeyer et al., 1998). Three cloned sequencetypes (WR111, WR176 and WR1100) were more diver-gent and formed a cluster distantly related to Marinobacterand Ectothiorhodospira species sequences. The refer-ence sequence most similar to these was a clonedsequence retrieved from the roots of leguminous plants(EMBL accession number AJ006010).
Among cloned sequence types clustering within theActinobacteria, only three were related to describedorganisms (Curtobacterium luteum, Gordonia sp. strainSJS0289-JS1 and Frankia sp. strain Dryas), all membersof the subclass Actinomycetales. Other sequence typeswere most closely related to previously reported clonedsequences. Four cloned sequence types clustered nearthe species Acidimicrobium ferrooxidans and ‘CandidatusMicrothrix parvicella’ and appeared to be related to thegroup II TM cloned 16S rRNA gene sequences (Rheimset al., 1996b), to the Australian soil cloned sequenceMC58 (Stackebrandt et al., 1993) and to the grasslandcloned sequence DA079 (Felske et al., 1997). In theregion of the 16S rRNA sequenced, the cloned sequencetypes from the Wittenberg soil were observed to possessthe characteristic signature nucleotides described for thefamily Acidimicrobiaceae (Stackebrandt et al., 1997). Asingle cloned sequence type, WR1145, clustered withRubrobacter radiotolerans, the group I TM cloned sequences(Rheims et al., 1996b) and cloned sequence MC4(Stackebrandt et al., 1993). Cloned sequence WR1145possessed the signature nucleotides for Acidimicro-biaceae, except in the 408–434 nucleotide pair (E. coli16S rRNA gene sequence numbering), where a G–C pair-ing was observed rather than the typical A–T.
Three cloned sequence types from Wittenberg soil clus-tered within the 16S rRNA sequences of species of thePlanctomycetales, showing highest similarities (89–90%)to that of the Australian soil cloned sequence MC25(Liesack and Stackebrandt, 1992) and to the 16S rRNAsequences of Isosphaera pallida and other Isosphaerastrains (Ward et al., 1995), suggesting that planctomy-cetes are also present and metabolically active in thisPCB-polluted soil.
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Fig. 3. A rooted dendrogram derived from comparison of partial 16S rRNA sequence types present in Wittenberg soil affiliated within thebeta-subclass of the Proteobacteria and related sequences: clones T (Snaidr et al., 1997). Evolutionary distances and the phylogeneticrelationships were calculated by the Jukes and Cantor algorithm (1969) and the least-squares distance algorithm in the FITCH program (PHYLIP
package) respectively. The hypervariable regions in the 16S rRNA molecule (Lane, 1991) were excluded from the calculations. The sequenceof Stenotrophomonas maltophilia was used as the outgroup.
Diversity of metabolically active bacteria in PCB-polluted soil 203

Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
204 B. Nogales et al.

Finally, three of the Wittenberg cloned sequence types(WR138, WR175 and WR1135) showed very low similari-ties to any identified or described bacterium. The closestrelatives to these sequence types were two clonedsequences also retrieved from soil environments (EMBLaccession numbers AF009989 and AJ005994). However,determination of the exact phylogenetic position of thesecloned sequence types cannot be made based only onthe partial 16S rRNA sequences (Ludwig et al., 1998).
Discussion
In situ bioremediation is an important emerging technologyfor the elimination or reduction of subsurface pollutants.Decisions as to whether a site should simply be containedand monitored (natural attenuation), treated, for examplewith nutrients (biostimulation), or augmented with specia-list microorganisms exhibiting relevant metabolic prop-erties (bioaugmentation) are largely empirical. Basicknowledge about the nature of the microorganisms mosteffective in the degradation of pollutants under differentphysicochemical conditions and the nature, distribution,population densities and activities of such organisms inthe communities of sites to be remediated would undoubt-edly facilitate the development of optimal bioremediationstrategies and rational decision-making processes.
In this study, a culture-independent method, based onthe RT–PCR amplification of 16S rRNA molecules, wasused to obtain an overview of the metabolically activemembers of the bacterial community in a highly PCB-polluted soil. As growing (i.e. metabolically active) bacteriacontain more ribosomes and rRNA than resting or starvedcells (Nomura et al., 1984; Kemp et al., 1993; Bremer andDennis, 1996), the 16S rRNA RT–PCR products gener-ated from extracts of total RNA essentially reflect themetabolically active fraction of the community. A similarapproach, based on cDNA synthesis from 16S rRNA andcloning, but without the PCR amplification step after thereverse transcription, was used in early applications ofmolecular techniques to studying bacterial diversity inthe environment (Weller and Ward, 1989). Since then,the RT–PCR approach has been used to analyse sul-phate-reducing populations in a multispecies biofilm(Amann et al., 1992) and, more recently, in combinationwith electrophoretic techniques such as denaturing gradi-ent gel electrophoresis (DGGE) and thermal gradient gelelectrophoresis (TGGE), to estimate the diversity of activebacteria in marine and soil samples (Teske et al., 1996;
Felske and Akkermans, 1998). As far as we are aware,identification of the metabolically active members of asoil bacterial community through the determination andanalysis of cloned 16S rRNA sequences obtained byRT–PCR amplification from extracted RNA has not beenreported.
The analysis of the 16S rRNA clone library retrievedfrom the Wittenberg soil revealed a considerable diversitywithin the presumptive metabolically active fraction of thebacterial community, despite the acidic pH of the soiland the high concentration of PCBs. Most of the clonedsequence types obtained were related to the 16S rRNAsequences of aerobic species, and many to acidiphilicbacteria, which is consistent with the physicochemicalcharacteristics of the soil, i.e. acidic pH and sandy texture,well drained at the time of the sampling.
Five groups of sequence types, namely those related tospecies of Burkholderia, Variovorax, Sphingomonas, theRhodopila globiformis group and the Acidobacterium clus-ter, dominated the clone library. Although the compositionof microbial communities determined by the analysis oflibraries of cloned PCR-amplified sequences are not quan-titative (von Wintzingerode et al., 1997), the predominanceof these sequence types from Wittenberg soil suggeststhat the microorganisms represented by these sequencesmay play important roles in the microbial activity withinthis soil. The observation of multiple Burkholderia-,Sphingomonas- and Variovorax-related sequences alsosupports this conclusion. A striking feature of the PCB-polluted Wittenberg soil was the abundance of sequencetypes clustering within the beta-subclass of the Proteobac-teria, which are usually observed only in relatively smallnumbers in 16S rDNA clone libraries generated from soil(Stackebrandt et al., 1993; Rheims et al., 1996a; Bornemanet al., 1996; Borneman and Triplett, 1997; Zhou et al.,1997; Felske et al., 1998). It was also remarkable thatmost of the sequence types belonging to this subclasswere limited to two genera, Burkholderia and Variovorax.
An intrinsic limitation of the approach presented in thismanuscript (which is also the case for diversity studiesbased on analysis of 16S rRNA genes) is the lack of infor-mation on the physiological features of the microorgan-isms represented by the 16S rRNA sequences obtained,particularly when cloned sequences are only distantlyrelated to the reference sequences of described organ-isms. Therefore, it is not possible to draw definitive conclu-sions about the functional roles of any particularmicroorganism identified as metabolically active within
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Fig. 4. A rooted dendrogram showing the 16S rRNA relationships among the WR sequence types affiliated with the genus Burkholderia.Evolutionary distances and the phylogenetic relationships were calculated by the Jukes and Cantor algorithm (1969) and the least-squaresdistance algorithm in the FITCH program (PHYLIP package) respectively. Only ambiguous nucleotide positions were excluded from the analysis.The sequence of Ralstonia eutropha was used as the outgroup.
Diversity of metabolically active bacteria in PCB-polluted soil 205
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
206 B. Nogales et al.

the community on the basis of the 16S rRNA sequencedata alone. Despite this, it can be useful to make cautiousspeculations about some physiological characteristics ofthe microorganisms represented by the detected 16SrRNA sequences. In the case of the PCB-contaminatedsoil of Wittenberg, some of the 16S rRNA sequencetypes detected were observed to cluster among those ofspecies of well-characterized genera, such as Burkholderia,Sphingomonas, Caulobacter, Bradyrhizobium and withinthe phylogenetic range of the family Comamonadaceae(including the genus Variovorax). Some general physiolo-gical characteristics common to the organisms of thesetaxa may thus be proposed, for example they would beexpected to have an aerobic (i.e. non-fermentative),chemoorganotrophic metabolism, to be able to use arange of polysaccharides, alcohols and organic acidsand to have oligotrophic capabilities for survival under lim-ited nutrient conditions. Furthermore, as none of thecloned sequence types detected in Wittenberg soil clus-tered within the described genera of anaerobic bacteria,it may be assumed that anaerobic metabolic capabilities(including reductive dechlorination of PCBs) would notbe of high relevance in this environment (at least underthe conditions present in the site at the time of sampling).Definitive assignment of the functional roles of the micro-organisms represented by the 16S rRNA sequencesobtained will require direct analysis of metabolic activitiesof the community, for example using labelled substrates(Pelz et al., 1999).
According to the chemical analyses of the samplestaken from the Wittenberg site, the major source of carbonin the soils with high PCB contents (i.e. that analysed inthis work) is the PCBs. The observed depletion of the low-est chlorinated biphenyl congeners of the contaminatingPCB mixture in this polluted site suggests that somedegree of aerobic PCB biodegradation is occurring(although low-chlorinated congeners are more volatilethan the higher chlorinated ones, losses of PCBs by vola-tilization have been observed to be low and not to competewith biodegradation significantly; Unterman, 1996). Theisolation of bacteria from the Wittenberg site that areable to grow on biphenyl, 4-chlorobiphenyl and chloro-benzoates (W. R. Abraham, unpublished data) is consis-tent with this suggestion. Thus, some of the sequencetypes detected in the 16S rRNA library may reflect theorganisms likely to be involved, directly or indirectly, withthe utilization of PCBs as carbon and energy sources.
Consistent with this assumption is the finding that a num-ber of the sequence types detected in the PCB-pollutedWittenberg soil are related to isolates known to be ableto degrade various organic pollutants, including PCBs(Bopp, 1986; Wittich et al., 1992; Nohynek et al., 1996;Feng et al., 1997; Kamagata et al., 1997; Mueller et al.,1997; Suyama et al., 1998). However, many cloned 16SrRNA sequences were similar to those of microorganismsnot currently known to degrade PCBs. Some of these may,therefore, be new types of PCB degraders, whereasothers may play different roles in the functioning of thecommunity and use for growth other carbon sources(e.g. from plants) present in the soil in low amounts. Thismay, for example, be the case for the group of clonedsequence types related to Acidobacterium, a group repre-sented in clone libraries retrieved from many non-pollutedsoils and other environments and for which no ecologicalrole is known (Hugenholtz et al., 1998a). The overviewof the diversity and the identification of the metabolicallyactive members of the microbial community analysedshould aid the development of enrichment strategies forisolating the bacteria that can be potentially important inPCB catabolism, as well as other microorganisms thatare important for other general functional aspects withinthe community.
Experimental procedures
Site description, sampling and chemical analyses ofthe soil
Soil samples were taken from a PCB-polluted site on a typicalmoorland near Wittenberg, Germany, at different times andsampling locations. The soil contains mainly sand with someclay and has a pH of 4–5. PCBs were extracted using the fol-lowing procedure: 2 g of homogenized, dried soil was mixedwith 10 mg of PCB 153 (used as internal standard), suspendedin 5 ml of hexane, vortexed for 1 min and the hexane phaseretained. The extraction procedure was repeated threetimes. The hexane was subsequently evaporated underreduced pressure, and the residue was resuspended in500 ml of octane. Capillary gas chromatographic analysis of1 ml of the extract was performed using a Hewlett Packard5890 series II gas chromatograph, equipped with an HPUltra 2 capillary column (50 m by 0.2 mm; film thickness0.11 mm) and a FID detector. Hydrogen served as the carriergas. The temperature of the injector was set at 2508C, and thedetector temperature was 3008C. The oven programme was808C for 3 min, a ramp from 908C to 2888C at a rate of
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Fig. 5. A rooted dendrogram showing the affiliation of WR sequence types within the alpha-subclass of the Proteobacteria. Clone MC106,Mount Coot-tha, Australia (Stackebrandt et al., 1993); clones TM, peat bog, Germany (Rheims et al., 1996a); clone DA007, Drentse Agrassland, The Netherlands (Felske et al., 1998). Evolutionary distances and the phylogenetic relationships were calculated by the Jukes andCantor algorithm (1969) and the least-squares distance algorithm in the FITCH program (PHYLIP package) respectively. The hypervariableregions in the 16S rRNA molecule (Lane, 1991) were excluded from the calculations. The sequence of Halorhodospira halophila was used asthe outgroup.
Diversity of metabolically active bacteria in PCB-polluted soil 207
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
208 B. Nogales et al.

68C min¹1 and a final isothermal period of 20 min. Identifica-tion of the chromatographic peaks was made by comparisonwith commercial standards and additionally verified by GC-MS (HP 5989A quadropole mass spectrometer), using thesame conditions described above but with helium as the car-rier gas. Quantitative determination of the PCBs was madeessentially as described previously (Buthe and Denker,1995). The method was adapted to the typical distribution ofcongeners found at the site. For calibration, four different con-centrations of two reference compounds of each degree ofchlorination (from mono- to hexachlorinated congeners)were measured to obtain average peak areas for each con-centration and degree of chlorination. Different chromatogramretention time windows were defined with respect to the degreeof chlorination of the congeners. The peak areas for congenersof each degree of chlorination was determined, and their totalconcentration was calculated based on the calibration carriedout previously. PCB 153 (which has not been detected by GC-FID in the Wittenberg site) was used as an internal standard.
Homogenized, freeze-dried soil (1 mg) was combusted in aFisons EA 1108 element analyser with CHN packing foranalysis of total organic carbon (TOC) and total nitrogen(TN). The analyses were run five times.
Total RNA extraction
The sample used for total RNA extraction was taken from theupper few centimetres of the surface of a soil with an averagePCB content of 10 g kg¹1 soil, weighed and frozen at ¹708Cuntil processing. Nucleic acids were extracted from the soilusing a protocol described previously (Zhou et al., 1996),with some modifications. Soil samples (< 7.5 g) were resus-pended in 18 ml of extraction buffer (100 mM Tris-HCl,pH 8.0, 100 mM sodium EDTA, pH 8.0, 100 mM sodium phos-phate buffer, pH 8.0) containing 10 mg ml¹1 proteinase K and3 mg ml¹1 lysozyme, and incubated at 378C for 30 min on ahorizontal shaker. After this treatment, glass beads (170–180 mm in diameter) and 4.5 ml of 10% SDS (final concentra-tion) were added, and the sample was treated for 2 min in anultrasonic bath. Hexadecylmethylammonium bromide (CTAB)and NaCl were then added to final concentrations of 1% (w/v)and 1.5 M, respectively, and the samples were incubated at658C for 15 min. The samples were subsequently frozen inliquid nitrogen and thawed immediately in a water bath at658C for 10 min. This freeze–thawing step was repeatedthree times. The crude extract obtained was cleared by centri-fugation at 6000 × g for 10 min in a precooled (48C) centrifuge(Sorvall RC3C). The supernatant was poured into a separatetube, and the pellet containing soil particles and debris wasre-extracted by resuspension in 6 ml of extraction buffer and2% SDS, vortexing and incubating at 658C for 10 min. Afterclearing by centrifugation, the supernatant fluids from both
extractions were pooled and one volume of chloroform–isoamyl alcohol (24:1, v/v) was added. The mixture was sha-ken before centrifuging at 6000 × g for 10 min. The aqueousphase was recovered and extracted twice with one volumeof phenol–chloroform–isoamyl alcohol (25:24:1, v/v). Nucleicacids in the aqueous phase were precipitated subsequently bythe addition of 0.7 volumes of isopropanol, and pelleted bycentrifugation at 16 000 × g for 30 min in a Sorvall RC5Ccentrifuge. The pellet of nucleic acids was washed with 70%ethanol, dried and resuspended in 300 ml of deionizedwater. An aliquot of the sample was digested with 30 U ofRNase-free DNase I (Boehringer Mannheim) at 378C for 2 hin 10 mM sodium acetate, 0.5 mM MgSO4 (pH 5.0). After theDNase treatment, total RNA was purified using Microconmicroconcentrators 100 (Millipore), according to the manufac-turer’s instructions. Aliquots of purified and non-purified totalRNA were analysed by electrophoresis on a 1% (w/v) agarosegel. Deionized water used to prepare buffers and solutions forRNA extraction was treated with diethylpyrocarbonate(DEPC) overnight and then autoclaved at 1208C for 20 min.All the glass and plastic ware were treated with 2 N NaOHfor 30 min, washed thoroughly with DEPC-treated deionizedwater and autoclaved before use.
RT–PCR amplification of 16S rRNA
The region of 16S rRNA between nucleotide positions 27 and518 (E. coli 16S rRNA gene sequence numbering), corre-sponding to approximately one-third of the entire 16S rRNA,was targeted for RT–PCR amplification from the extractedtemplate RNA. RT–PCRs were performed with 230 ng ofthe total RNA using rTth DNA polymerase (Perkin-Elmer).Reverse transcription reactions (20 ml) contained 10 mMTris-HCl (pH 8.3), 90 mM KCl, 1 mM MnCl2, 200 mM dNTPs,5 U of rTth DNA polymerase and 0.75 mM primer DXR518(gggggggccaaCGTATTACCGCGGCTGCTGG), hybridizingat nucleotide positions 518–537 (E. coli 16S rRNA genesequence numbering). The sequences at the 58 ends of theprimers (in lower case type) were complementary tosequences in the cloning vector. Reverse transcription (RT)reactions were performed for 15 min at 708C in a GeneAmp9600 thermocycler (Perkin-Elmer). After completion of theRT reaction, 80 ml of the PCR mixture containing 10 mMTris-HCl (pH 8.3), 100 mM KCl, 0.05% Tween 20 (w/v), 5%glycerol (v/v), 0.75 mM ethylene glycol-bis(b-aminoethylether)-N,N,N8,N8-tetraacetic acid (EGTA), 1.5 mM MgCl2and 0.15 mM primer DXF27 (ccggccaaGAGTTTGATC-MTGGCTCAG), which hybridizes to the 16S rRNA corre-sponding to nucleotide positions 9–27, was added. PCRswere performed in a GeneAmp 9600 thermocycler with thefollowing conditions: an initial denaturation step at 948C for2 min followed by 30 cycles of 1 min at 948C, 1 min at 708C
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Fig. 6. A rooted dendrogram showing the WR clones clustering within the Holophaga–Acidobacterium phylum and related sequencesretrieved from other soil libraries: clones MC, Mount Coot-tha, Australia (Stackebrandt et al., 1993); clones TM, peat bog, Germany (Rheimset al., 1996a); clone DA054, Drentse A grassland, The Netherlands (Felske et al., 1998); clone 11-14, Roggenstein soil, Germany (Ludwiget al., 1997); clone C002, arid soils, Arizona (Kuske et al., 1997); clone OPB3, hot spring, Yellowstone (Hugenholtz et al., 1998b).Evolutionary distances and the phylogenetic relationships were calculated by the Jukes and Cantor algorithm (1969) and the least-squaresdistance algorithm in the FITCH program (PHYLIP package) respectively. The hypervariable regions in the 16S rRNA molecule (Lane, 1991) wereexcluded from the calculations. The sequence of Holophaga foetida was used as the outgroup.
Diversity of metabolically active bacteria in PCB-polluted soil 209

and 2 min at 728C, and a final extension step of 10 min at 728C.PCRs without a previous reverse transcription were carriedout using purified RNA as template to control for DNAcontamination.
Cloning of RT–PCR products
RT–PCRs were carried out in triplicate, and the productswere pooled before purification and cloning. RT–PCR pro-ducts were purified by electrophoresis in a 1% (w/v) low-melting-point agarose (Gibco) gel. Nucleic acid bands of theexpected size (< 530 nucleotides) were excised from thegel, and DNA was extracted as described previously(Sambrook et al., 1989). Purified RT–PCR products weretreated with 3 U of T4 DNA polymerase (New England Bio-labs) at 128C for 20 min in the presence of 100 mM dTTPand cloned as described previously (Mau, 1997). In prep-aration for cloning, the pBluescript-II KSþ cloning vector(Stratagene) was linearized with DraII restriction endo-nuclease (Boehringer Mannheim) and treated for 20 min at128C with 3 U of T4 DNA polymerase in the presence of100 mM dATP. RT–PCR products and vector were ligatedwith 400 U of T4 DNA ligase (New England Biolabs) overnightat 168C. The ligation products were dialysed against deionizedwater for 30 min and used to transform Epicurian Coli XL-1Blue electroporation-competent cells (Stratagene), using aBio-Rad electroporator at 200 V, 25 mF and a field strengthof 17 kV cm¹1. Transformant clones were selected as whitecolonies on LB agar medium (Sambrook et al., 1989) contain-ing 100 mg ml¹1 ampicillin, 20 mg ml¹1 Xgal and 24 mg ml¹1
IPTG, and subsequently streaked onto LB agar containing100 mg ml¹1 ampicillin. Lysates of transformant clones wereprepared by transferring biomass from the streaks into1.5 ml microcentrifuge tubes containing 100 ml of TE buffer(pH 8.0) and heating for 15 min at 958C. The presence of aninsert of the expected size was confirmed by PCR amplifica-tion, using primers T3 (McGraw et al., 1985) and T7 (Dunnand Studier, 1983) that anneal to the vector at positions flank-ing the insert. PCR mixtures (20 ml) contained 10 mM Tris-HCl(pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.01% (w/v) gelatin,200 mM dNTP, 0.5 mM T3 and T7 primers, 0.5 U of AmpliTaqDNA polymerase (Perkin-Elmer) and 1 ml of cell lysate.Clones containing inserts of the expected size were identifiedby electrophoresis of the PCR products in 1% (w/v) agarosegels and staining with ethidium bromide.
Sequencing of cloned RT–PCR products
The nucleotide sequences of the cloned 16S rRNA RT–PCRproducts were determined from plasmid DNA preparations.Cells of clones containing inserts of the correct size weregrown overnight in LB medium with 100 mg ml¹1 ampicillin,and the plasmid DNA was extracted using the Qiawell 8 orQiaSpin kits (Qiagen). DNA sequences were determined byTaq cycle sequencing using the PRISM Ready ReactiondRhodamine Terminator cycle sequencing kit and ABI373and 377 sequencers (PE Applied Biosystems), according tothe manufacturer’s instructions. Vector primers T3 and T7were used in the sequencing reactions. The sequence datahave been submitted to the EMBL database under accessionnumbers AJ233467 to AJ233589.
Assignment of cloned sequences to establishedphylogenetic groups
Cloned 16S rRNA sequences were compared initially withreference sequences contained in the EMBL NucleotideSequence Database (Stoesser et al., 1998) using the FASTA
program (Pearson and Lipman, 1988). Cloned sequenceswere then aligned with the most similar 16S rRNA (andrRNA gene) reference sequences, using evolutionarily con-served primary sequence and secondary structure as refer-ence (Woese et al., 1983; Gutell et al., 1985). Clonedsequences were checked for possible chimeric structures,using the CHECK_CHIMERA program (Maidak et al., 1994) andthe models for correlating primary sequence and secondarystructure. Evolutionary distances, derived from sequence-pair dissimilarities (Jukes and Cantor, 1969), were calculatedusing the DNADIST program included in the Phylogeny Inferencepackage (PHYLIP, version 3.5; Felsenstein, 1989). The least-squares distance algorithm of the FITCH program from the PHY-
LIP package was used to calculate the phylogenetic trees.
Acknowledgements
We thank Annette Kruger and Carsten Strompl, andChristian Hesse and Peter Wolff, for their excellent tech-nical assistance in the sequencing and chemical analysesrespectively. This work was supported by a grant from theGerman Federal Ministry for Science, Education andResearch (project no. 0319433C). K.N.T. thanks theFonds der Chemische Industrie for generous support.B.N. was the recipient of a postdoctoral fellowship fromthe Spanish Ministry for Education and Culture.
References
Amann, R.I., Stromley, J., Devereux, R., Key, R., andStahl, D.A. (1992) Molecular and microscopic identificationof sulfate-reducing bacteria in multispecies biofilms. ApplEnviron Microbiol 58: 614–623.
Bopp, L.H. (1986) Degradation of highly chlorinated PCBs byPseudomonas strain LB400. J Ind Microbiol 1: 23–29.
Borneman, J., and Triplett, E.W. (1997) Molecular microbialdiversity in soils from eastern Amazonia: evidence for unusualmicroorganisms and microbial population shifts associatedwith deforestation. Appl Environ Microbiol 63: 2647–2653.
Borneman, J., Skroch, P.W., O’Sullivan, K.M., Palus, J.A.,Rumjanek, N.G., Jansen, J.L., et al. (1996) Molecularmicrobial diversity of an agricultural soil in Wisconsin.Appl Environ Microbiol 62: 1935–1943.
Bouwer, E.J., Durant, N., Willson, L., Zhang, W., andCunningham, A. (1994) Degradation of xenobiotic com-pounds in situ: capabilities and limits. FEMS MicrobiolRev 15: 307–317.
Bremer, H., and Dennis, P.P. (1996) Modulation of chemicalcomposition and other parameters of the cell by growthrate. In Escherichia coli and Salmonella: Cellular andMolecular Biology. Neidhart, F.C., Curtiss III, R., Ingra-ham, J.L., Lin, E.C.C., Low, K.B., Magasanik, B., et al.(eds). Washington DC: American Society for MicrobiologyPress, pp. 1553–1569.
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
210 B. Nogales et al.

Buthe, A., and Denker, E. (1995) Qualitative and quantitativedetermination of PCB congeners by using a HT-5 GC col-umn and an efficient quadropole MS. Chemosphere 30:753–771.
Dunn, J.J., and Studier, F.W. (1983) Complete nucleotidesequence of bacteriophage T7 DNA and the locations ofT7 genetic elements. J Mol Biol 166: 477–535.
Felsenstein, J. (1989) PHYLIP – Phylogeny Inference Package(version 3.2). Cladistics 5: 164–166.
Felske, A., and Akkermans, A.D.L. (1998) Spatial homo-geneity of abundant bacterial 16S rRNA molecules ingrasslands soils. Microb Ecol 36: 31–36.
Felske, A., Rheims, H., Wolterink, A., Stackebrandt, E., andAkkermans, A.D.L. (1997) Ribosome analysis revealsprominent activity of an uncultured member of the classActinobacteria in grassland soils. Microbiology 143:2983–2989.
Felske, A., Wolterink, A., van Lis, R., and Akkermans, A.D.L.(1998) Phylogeny of the main bacterial 16S rRNAsequences in Drentse A grassland soil (The Netherlands).Appl Environ Microbiol 64: 871–879.
Feng, X., Ou, L.-T., and Ogram, A. (1997) Plasmid-mediatedmineralization of carbofuran by Sphingomonas sp. strainCF06. Appl Environ Microbiol 63: 1332–1337.
Gutell, R.R., Weiser, B., Woese, C.R., and Noller, H.F.(1985) Comparative anatomy of 16S-like ribosomal RNA.Prog Nucleic Acid Res Mol Biol 32: 155–216.
Hugenholtz, P., Goebel, B.M., and Pace, N.R. (1998a)Impact of culture-independent studies on the emergingphylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774.
Hugenholtz, P., Pitulle, C., Hershberger, K.L., and Pace, N.R.(1998b) Novel division level bacterial diversity in a Yellow-stone hot spring. J Bacteriol 180: 366–376.
Ivanov, V., and Sandell, E. (1992) Characterization of poly-chlorinated biphenyl isomers in Sovol and Trichlorobi-phenyl formulations by high-resolution gas chromatographywith electron capture detection and high-resolution gas chro-matography–mass spectrometry techniques. Environ SciTechnol 26: 2012–2017.
Jukes, T.H., and Cantor, C.R. (1969) Evolution of proteinmolecules. In Mammalian Protein Metabolism. Munro,H.M. (ed.). New York: Academic Press, pp. 21–132.
Kalmbach, S., Manz, W., and Szewzyck, U. (1997) Isolationof new bacterial species from drinking water biofilms andproof of their in situ dominance with highly specific 16SrRNA probes. Appl Environ Microbiol 63: 4164–4170.
Kamagata, Y., Fulthorpe, R.R., Tamura, K., Takami, H.,Forney, L.J., and Tiedje, J.M. (1997) Pristine environmentsharbor a new group of oligotrophic 2,4-dichlorophenoxy-acetic acid-degrading bacteria. Appl Environ Microbiol63: 2266–2272.
Kemp, P.F., Lee, S., and LaRoche, J. (1993) Estimating thegrowth rate of slowly growing marine bacteria from RNAcontent. Appl Environ Microbiol 59: 2594–2601.
Kuske, C.R., Barns, S.M., and Busch, J.D. (1997) Diverseuncultivated bacterial groups from soils of the aridsouthwestern United States that are present in manygeographic regions. Appl Environ Microbiol 63: 3614–3621.
Lane, D.J. (1991) 16S/23S rRNA sequencing. In Nucleic Acid
Techniques in Bacterial Systematics. Stackebrandt, E.,and Goodfellow, M. (eds). Chichester: John Wiley &Sons, pp. 115–175.
Liesack, W., and Stackebrandt, E. (1992) Occurrence ofnovel groups of the domain bacteria as revealed by analy-sis of genetic material isolated from an Australian terres-trial environment. J Bacteriology 174: 5072–5078.
Liu, S., and Suflita, J.M. (1993) Ecology and evolution of micro-bial populations for bioremediation. TIBTECH 11: 344–352.
Ludwig, W., Bauer, S.H., Bauer, M., Held, I., Kirchof, G.,Schulze, R., et al. (1997) Detection and in situ identificationof representatives of a widely distributed new bacterialphylum. FEMS Microbiol Lett 153: 181–190.
Ludwig, W., Strunk, O., Klugbauer, S., Klugbauer, N.,Weizenegger, M., Neumaier, J. et al. (1998) Bacterialphylogeny based on comparative sequence analysis.Electrophoresis 19: 554–568.
McGraw, N.J., Bailey, J.N., Cleaves, G.R., Dembinski, D.R.,Gocke, C.R., Joliffe, L.K., et al. (1985) Sequence and ana-lysis of the gene for bacteriophage T3 RNA polymerase.Nucleic Acids Res 13: 6753–6766.
Maidak, B.L., Larsen, N., McCaughey, M.J., Overbeek, R.,Olsen, G.J., Fogel, K., et al. (1994) The Ribosomal Data-base Project. Nucleic Acids Res 22: 3485–3487.
Mau, M.K. (1997) 16S rDNA-Sequenzanalyse und Sonden-design zur Characterisierung der Bakterienpopulation imSediment eines hochbelasteten Gewassers. PhD Thesis,Technical University Carolo-Willhelmina of Braunschweig.Gottingen: Cuvillier Verlag.
Mitsui, H., Gorlach, K., Lee, H.-J., Hattori, R., and Hattori, T.(1997) Incubation time and media requirements of cultur-able bacteria from different phylogenetic groups. J Micro-biol Methods 30: 103–110.
Mueller, J.G., Devereux, R., Santavy, D.L., Lantz, S.E.,Willis, S.G., and Pritchard, P.H. (1997) Phylogenetic andphysiological comparisons of PAH-degrading bacteriafrom geographically diverse soils. Antonie Van Leeuwen-hoek 71: 329–343.
Nohynek, L.J., Nurmiaho-Lassila, E.L., Suhonen, E.L.,Busse, H.J., Mohammadi, M., Hantula, J., et al. (1996)Description of chlorophenol-degrading Pseudomonas sp.strains KF1T, KF3 and NKF1 as a new species of thegenus Sphingomonas, Sphingomonas subarctica sp. nov.Int J Syst Bacteriol 46: 1042–1055.
Nomura, M., Gourse, R., and Baughman, G. (1984) Regula-tion of the synthesis of ribosomes and ribosomal compo-nents. Ann Rev Biochem 53: 75–117.
Pearson, W.R., and Lipman, D.J. (1988) Improved tools forbiological sequence comparison. Proc Natl Acad Sci USA85: 2444–2448.
Pelz, O., Tesar, M., Frech, G., Faude, U., Wittich, R.-M.,Moore, E.R.B., et al. (1999). Structural and functional char-acterization of a 4-chlorosalicylate-degrading bacterialconsortium. Environ Microbiol 1: (in press).
Rheims, H., Sproer, C., Rainey, F.A., and Stackebrandt, E.(1996a) A molecular approach to search for diversityamong bacteria in the environment. J Ind Microbiol 17:159–169.
Rheims, H., Rainey, F.A., and Stackebrandt, E. (1996b)Molecular biological evidence for the occurrence ofuncultured members of the actinomycete line of descent
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
Diversity of metabolically active bacteria in PCB-polluted soil 211

in different environments and geographical locations.Microbiology 142: 2863–2870.
Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989) Molecu-lar Cloning. A Laboratory Manual, 2nd edn. Cold SpringHarbor, NY: Cold Spring Harbor Laboratory Press.
Snaidr, J., Amann,R., Huber, I., Ludwig, W., and Schleifer, K.-H.(1997). Phylogenetic analysis and in situ identification ofbacteria in activated sludge. Appl Environ Microbiol 63:2884–2896.
Stackebrandt, E., Ludwig, W., Schubert, F., Klink, F.,Schlesner, H., Roggentin, F., and Hirsch, P. (1984) Mol-ecular genetic evidence for early evolutionary origin of bud-ding peptidoglycan-less eubacteria. Nature 307: 735–737.
Stackebrandt, E., Murray, R.G.E., and Truper, H.G. (1988)Proteobacteria classis nov., a name for the phylogenetictaxon that includes the ‘purple bacteria and their relatives’.Int J Syst Bacteriol 38: 321–325.
Stackebrandt, E., Liesack, W., and Goebel, B.M. (1993)Bacterial diversity in a soil sample from a subtropicalAustralian environment as determined by 16S rDNA analy-sis. FASEB J 7: 232–236.
Stackebrandt, E., Rainey, F.A., and Ward-Rainey, N. (1997)Proposal of a new hierarchic classification system, Acti-nobacteria classis nov. Int J Syst Bacteriol 47: 479–491.
Stoesser, G., Moseley, M.A., Sleep, J., McGowran, M.,Garcia-Pastor, M., and Sterk, P. (1998) The EMBL nucleo-tide sequence database. Nucleic Acids Res 26: 8–15.
Straub, K.L., Benz, M., Schink, B., and Widdel, F. (1996)Anaerobic, nitrate-dependent microbial oxidation of ferrousiron. Appl Environ Microbiol 62: 1458–1460.
Sturmeyer, H., Overmann, J., Babenzien, H.-D., andCypionka, H. (1998) Ecophysiological and phylogeneticstudies of Nevskia ramosa in pure culture. Appl EnvironMicrobiol 64: 1890–1894.
Suyama, T., Hosoya, H., and Tokiwa, Y. (1998) Bacterialisolates degrading aliphatic polycarbonates. FEMS Micro-biol Lett 161: 255–261.
Teske, A., Wawer, C., Muyzer, G., and Ramsing, N.B. (1996)Distribution of sulfate-reducing bacteria in a stratified fjord(Mariager Fjord, Denmark) as evaluated by most-probable-number counts and denaturing gradient gel electrophoresisof PCR-amplified ribosomal DNA fragments. Appl EnvironMicrobiol 62: 1405–1415.
Unterman, R. (1996) A history of PCB biodegradation. InBioremediation: Principles and Applications. Crawford,R.L., and Crawford, D.L. (eds). Cambridge: CambridgeUniversity Press, pp. 209–253.
Ward, N., Rainey, F.A., Stackebrandt, E., and Schlesner, H.(1995) Unraveling the extent of diversity within the orderPlanctomycetales. Appl Environ Microbiol 61: 2270–2275.
Weller, R., and Ward, D.M. (1989) Selective recovery of 16SrRNA sequences from natural microbial communities inthe form of cDNA. Appl Environ Microbiol 55: 1818–1822.
von Wintzingerode, F., Gobel, U.B., and Stackebrandt, E.(1997) Determination of microbial diversity in environmen-tal samples: pitfalls of PCR-based rRNA analysis. FEMSMicrobiol Rev 21: 213–229.
Wittich, R.M., Wilkes, H., Sinnwell, V., Francke, W., andFornagel, P. (1992) Metabolism of dibenzo-p-dioxin bySphingomonas sp. strain RW1. Appl Microbiol Ecol 58:1005–1010.
Woese, C.R., Gutell, R., Gupta, R., and Noller, H.F. (1983)Detailed analysis of the higher-order structure of 16S-like ribosomal ribonucleic acids. Microbiol Rev 47:621–669.
Zhou, J., Bruns, M.A., and Tiedje, J.M. (1996) DNA recoveryfrom soils of diverse composition. Appl Environ Microbiol62: 316–322.
Zhou, J., Davey, M.E., Figueras, J.B., Rivkina, E., Gilichinsky, D.,and Tiedje, J.M. (1997) Phylogenetic diversity of a bacter-ial community determined from Siberian tundra soil DNA.Microbiology 143: 3913–3919.
Q 1999 Blackwell Science Ltd, Environmental Microbiology, 1, 199–212
212 B. Nogales et al.
Copyright © 2022 FDOKUMEN




















