
The Crystal Structure of a Plant 3-Ketoacyl-CoA Thiolase Reveals the Potential for Redox Control of...
11
The Crystal Structure of a Plant 3-Ketoacyl-CoA Thiolase Reveals the Potential for Redox Control of Peroxisomal Fatty Acid b-Oxidation Ramasubramanian Sundaramoorthy 1 , Elena Micossi 1,2 Magnus S. Alphey 1 , Ve ´ ronique Germain 3 , James H. Bryce 4 Steve M. Smith 5 , Gordon A. Leonard 2 and William N. Hunter 1 * 1 Division of Biological Chemistry and Molecular Microbiology, School of Life Sciences, University of Dundee Dundee DD1 5EH, UK 2 Macromolecular Crystallography Group ESRF, BP 220, 38043 Grenoble Cedex, France 3 UMR Physiologie et Biotechnique Ve ´ge ´tables IBVM, INRA, 71 Avenue Edouard Bourleaux, 33883 Villenave d’Ornon Cedex France 4 The International Centre for Brewing and Distilling Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK 5 ARC Centre of Excellence in Plant Energy Biology. University of Western Australia Crawley, WA 6009, Australia Crystal structures of peroxisomal Arabidopsis thaliana 3-ketoacyl-CoA thiolase (AtKAT), an enzyme of fatty acid b-oxidation, are reported. The subunit, a typical thiolase, is a combination of two similar a/b domains capped with a loop domain. The comparison of AtKAT with the Saccharomyces cerevisiae homologue (ScKAT) structure reveals a different placement of subunits within the functional dimers and that a polypeptide segment forming an extended loop around the open catalytic pocket of ScKAT converts to a-helix in AtKAT, and occludes the active site. A disulfide is formed between Cys192, on this helix, and Cys138, a catalytic residue. Access to Cys138 is determined by the structure of this polypeptide segment. AtKAT represents an oxidized, previously unknown inactive form, whilst ScKAT is the reduced and active enzyme. A high level of sequence conservation is observed, including Cys192, in eukaryotic peroxisomal, but not mitochondrial or prokaryotic KAT sequences, for this labile loop/helix segment. This indicates that KAT activity in peroxisomes is influenced by a disulfide/dithiol change linking fatty acid b-oxidation with redox regulation. q 2006 Elsevier Ltd. All rights reserved. Keywords: Arabidopsis thaliana; b-oxidation; 3-ketoacyl-CoA thiolase; redox regulation; Saccharomyces cerevisiae *Corresponding author Introduction Thiolases are ubiquitous enzymes that contribute to fatty acid (FA) biosynthesis or degradation in both prokaryotes and eukaryotes. 1,2 Two thiolase classes, with distinct roles and substrate specifi- cities, have been defined. The biodegradative type I thiolases (EC 2.3.1.16), the subject of our investi- gation, use acetoacetyl-CoA or longer chain acyl- CoA derivatives as substrates, participate in FA b-oxidation and are termed 3-ketoacyl-CoA thio- lases (KAT). Type II acetoacetyl-CoA thiolases (EC 2.3.1.9) are specific for acetoacetyl-CoA as substrate 0022-2836/$ - see front matter q 2006 Elsevier Ltd. All rights reserved. Abbreviations used: amp, ampicillin; ASA, accessible surface area; CCP4, Collaborative Computational Project Number 4; CoA, coenzyme A; DPI, diffraction-component precision index; ESRF, European Synchrotron Radiation Facility; FA, fatty acid; KAT, 3-ketoacyl-CoA thiolase; LB, Luria–Bertani; MR, molecular replacement; NCS, non-crystallographic symmetry; PDB, Protein Data Bank; r.m.s.d., root-mean-square deviation; TLS, translation libration screw. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2006.03.032 J. Mol. Biol. (2006) 359, 347–357
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of The Crystal Structure of a Plant 3-Ketoacyl-CoA Thiolase Reveals the Potential for Redox Control of...

doi:10.1016/j.jmb.2006.03.032 J. Mol. Biol. (2006) 359, 347–357
The Crystal Structure of a Plant 3-Ketoacyl-CoAThiolase Reveals the Potential for Redox Controlof Peroxisomal Fatty Acid b-Oxidation
Ramasubramanian Sundaramoorthy1, Elena Micossi1,2
Magnus S. Alphey1, Veronique Germain3, James H. Bryce4
Steve M. Smith5, Gordon A. Leonard2 and William N. Hunter1*
1Division of BiologicalChemistry and MolecularMicrobiology, School of LifeSciences, University of DundeeDundee DD1 5EH, UK
2MacromolecularCrystallography GroupESRF, BP 220, 38043Grenoble Cedex, France
3UMR Physiologie etBiotechnique VegetablesIBVM, INRA, 71 AvenueEdouard Bourleaux, 33883Villenave d’Ornon CedexFrance
4The International Centre forBrewing and DistillingHeriot-Watt University,Riccarton, Edinburgh EH144AS, UK
5ARC Centre of Excellence inPlant Energy Biology.University of Western AustraliaCrawley, WA 6009, Australia
0022-2836/$ - see front matter q 2006 E
Abbreviations used: amp, ampicisurface area; CCP4, Collaborative CNumber 4; CoA, coenzyme A; DPI, dprecision index; ESRF, European SyFacility; FA, fatty acid; KAT, 3-ketoaLB, Luria–Bertani; MR, molecular rnon-crystallographic symmetry; PDr.m.s.d., root-mean-square deviationlibration screw.
E-mail address of the [email protected]
Crystal structures of peroxisomal Arabidopsis thaliana 3-ketoacyl-CoAthiolase (AtKAT), an enzyme of fatty acid b-oxidation, are reported. Thesubunit, a typical thiolase, is a combination of two similar a/b domainscapped with a loop domain. The comparison of AtKAT with theSaccharomyces cerevisiae homologue (ScKAT) structure reveals a differentplacement of subunits within the functional dimers and that a polypeptidesegment forming an extended loop around the open catalytic pocket ofScKAT converts to a-helix in AtKAT, and occludes the active site.A disulfide is formed between Cys192, on this helix, and Cys138, acatalytic residue. Access to Cys138 is determined by the structure of thispolypeptide segment. AtKAT represents an oxidized, previously unknowninactive form, whilst ScKAT is the reduced and active enzyme. A high levelof sequence conservation is observed, including Cys192, in eukaryoticperoxisomal, but not mitochondrial or prokaryotic KAT sequences, for thislabile loop/helix segment. This indicates that KAT activity in peroxisomesis influenced by a disulfide/dithiol change linking fatty acid b-oxidationwith redox regulation.
q 2006 Elsevier Ltd. All rights reserved.
Keywords: Arabidopsis thaliana; b-oxidation; 3-ketoacyl-CoA thiolase; redox
regulation; Saccharomyces cerevisiae *Corresponding authorlsevier Ltd. All rights reserve
llin; ASA, accessibleomputational Projectiffraction-component
nchrotron Radiationcyl-CoA thiolase;
eplacement; NCS,B, Protein Data Bank;; TLS, translation
ing author:
Introduction
Thiolases are ubiquitous enzymes that contributeto fatty acid (FA) biosynthesis or degradation inboth prokaryotes and eukaryotes.1,2 Two thiolaseclasses, with distinct roles and substrate specifi-cities, have been defined. The biodegradative type Ithiolases (EC 2.3.1.16), the subject of our investi-gation, use acetoacetyl-CoA or longer chain acyl-CoA derivatives as substrates, participate in FAb-oxidation and are termed 3-ketoacyl-CoA thio-lases (KAT). Type II acetoacetyl-CoA thiolases (EC2.3.1.9) are specific for acetoacetyl-CoA as substrate
d.

348 Structure of a Plant Ketoacyl-CoA Thiolase
but function in the opposite direction to type Ienzymes in several biosynthetic pathways.3–5
Fatty acids and derivatives participate in nume-rous biological events, including metabolic path-ways, endocrine, and signalling processes.6 Theyalso provide an important source of energyfollowing degradation in a sequence of enzyme-catalysed reactions termed the b-oxidation cycle.1,7,8
The name originates from oxidation that occurs atCb of a fatty acyl-coenzyme A (CoA) derivativepreceding cleavage of the Ca–Cb bond. The firststage of this highly exergonic process is FAactivation by formation of a thioester bond withCoA in a reaction catalysed by acyl-CoA synthetase.Subsequently, in reactions catalysed by acyl-CoAdehydrogenase, enoyl-CoA hydratase and 3-L-hydroxyacyl-CoA dehydrogenase, oxidation of theCa–Cb bond produces an olefin, then hydration andoxidation results in carbonyl group formation at Cb.The next stage of the cycle, the one catalysed byKAT, is cleavage of the b-keto ester in a reverseClaisen condensation resulting in a new acyl-CoAderivative truncated by two C atoms, which aresubsequently used to produce acetyl-CoA. Thesereactions proceed continuously until only acetyl-CoA is left.
Biochemical and reverse genetics methods havebeen applied to understand the physiological role ofKAT in plants. The gene encoding KAT is expressedthroughout growth and development but is stron-gest during the first days of post-germinativegrowth; that is, before the seedling becomesautotrophic.9 In Arabidopsis thaliana, it has beendemonstrated that a KAT mutant was unable tomobilize its own lipids or to germinate and growwithout an external supply of sugars, leading to theconclusion that the type I thiolases are involved inreserve lipid mobilization to generate sucrosethrough a unique type of gluconeogenesis, invol-ving enzymes located in different sub-cellularcompartments such as lipid bodies, peroxisomes,mitochondria and the cytosol.10 Within the entireneoglucogenic pathway the conversion of fatty acidto succinate takes place in peroxisomes, wherethe enzymes of FA b-oxidation and most of those ofthe glyoxylate cycles are located. It is here that thecontrol of storage fat degradation and channellingof the products towards the biosynthesis ofessential compounds such as carbohydratesoccurs.11–15
Peroxisomal KAT is produced in vivo as aprecursor carrying a cleavable N-terminal targetingsignal.16,17 The N-terminal segment is not requiredfor activity, nor does it influence the quaternarystructure, which is homodimeric in the matureconformation.18 The crystal structure of the perox-isomal KAT from the Saccharomyces cerevisiae(ScKAT) has been determined under reducingconditions and a model for the complex with thesubstrate acetoacetyl-CoA proposed based oncomparisons with biosynthetic thiolases. Thismodel, in conjunction with the available bio-chemical data allowed a plausible mechanism to
be postulated19,20 and has, in part, been sub-stantiated by mutagenesis-kinetic studies.21 Inmitochondria and bacteria, a multifunctionalenzyme provides KAT activity and structural datasuggest a role for channelling intermediates duringb-oxidation.22
Here, we report the structure of a peroxisomalKAT isoform derived from a eukaryotic organism,the plant A. thaliana (AtKAT) and comparisons ofthis structure with that of peroxisomal ScKAT. Ourresults indicate that these structures representdistinct inactive and active KAT states, respectively,and that disulfide/dithiol redox chemistry isdirectly implicated in their inter-conversion.
Results and Discussion
Structural analysis and model quality
Crystal structures of AtKAT, produced as recombi-nant protein in an Escherichia coli expression system,have been characterized in two orthorhombic forms.Form I contains a dimer in the asymmetric unit,subunit mass is approximately 47 kDa, and has beenrefined with data to 2.1 A resolution. Form II with amonomer in the asymmetric unit has been refinedusing diffraction data that extend to a resolution of2.4 A. The completeness of the data in the higherresolution shells for crystal form II drops to only 70%;nevertheless, these data were included in therefinement, since they have a beneficial impact onthe quality of the electron density maps. Therefinement statistics and stereochemical parameters(Table 1) indicate that the coordinates representacceptable medium resolution models.
The root-mean-square deviation (r.m.s.d.) oftopologically equivalent Ca atoms of the singlesubunit from crystal form II is 0.27 A and 0.18 Awith respect to subunits A and B of crystal form Iwhilst the agreement of Ca positions for the twosubunits in form I is 0.26 A. Such low valuesindicate a high degree of structural conservationand that it is only necessary to detail one structure,which by virtue of a higher diffracting resolution isthat of crystal form I.
The enzyme consists of 462 residues and con-tinuous density is observed for the main chain fromresidues 39 to 172 and from 185 to 440 in subunit A,and residues 39–176 and 184–441 in subunit B.Residues for which there was no convincingelectron density, presumably due to conformationalflexibility, were omitted from the model. The side-chain atoms of several surface residues are likewiseill defined but, when the main-chain density wasclear, these were included in the model andassigned zero occupancy. A single residue, Leu395in both subunits, is well defined by the electrondensity yet has a f/j combination in thedisallowed region of the Ramachandran plot,198/K1008 in subunit A, 138/K1028 in subunit B.This will be discussed later.

Table 1. Data collection and refinement statistics
Structure Crystal form I Crystal form II
Wavelength (A) 1.000 0.933Space group P212121 P21212Unit cell parametersa (A) 73.15 73.34b (A) 94.46 95.53c (A) 111.49 56.45
Resolution range (A) 29.2–2.1 20.0–2.4Unique reflections 44,287 16,453Redundancy 3.7 2.2Completeness 96.7 (80.6) 87.3 (69.9)hI/s(I)i 12.8 (2.2) 6.4 (1.1)Rsym (%) 7.7 (33.8) 5.9 (29.9)Wilson B (A2) 28.5 41.3Protein residues 786 396Solvent molecules 512 115R-factor (%) 20.1 21.1R-free (%) 26.3 27.2
Average B factorOverall (A2) 22.2 51.6Protein (A2) 21.9 51.7Solvent (A2) 25.9 50.5
r.m.s.d. from idealBond lengths (A) 0.012 0.012Bond angles (deg.) 1.37 1.39Cruickshank’s DPI (A) 0.17 0.24
Ramachandran plot regionsMost favoured (%) 92.4 90.7Additionally allowed (%) 7.1 8.1Generously allowed (%) 0.3 0.9Disallowed (%) 0.2 0.3
Values in parentheses pertain to the highest resolution shell(widthZ0.1 A). B is the isotropic thermal parameter. DPI,diffraction-component precision index.33
Structure of a Plant Ketoacyl-CoA Thiolase 349
Structure of the AtKAT subunitand comparisons with ScKAT
The subunit of AtKAT has a compact globularstructure of approximate dimensions 40 A!40 A!50 A, which is closely related to that of otherthiolases and condensing enzymes.20,23 The namingof the domains is altered in our description, sincethere are residues at the N terminus of AtKAT,which according to the previous nomenclaturewould be classed as belonging to the C-terminaldomain.
Approximately 60% of the residues are inelements of secondary structure, which are mappedonto the amino acid sequence of AtKAT in Figure 1and coloured corresponding to the domain in whichthey reside. For comparison the aligned sequenceand corresponding secondary structure of ScKAT isalso depicted. The AtKAT subunit is constructedfrom three domains. Residues 51–167 and 295–317form domain I, 168–294 the “loop” or lid domain II,and then residues 318–440 together with 38–50 formdomain III. Domains I and III are structurallyrelated,19 each consisting of a buried b-sheet withtwo helices on one side at the surface of the subunitand a single amphipathic helix on the other(Figure 2(a)). Domain I contains a five-strandedb-sheet in order b2-b6-b5-b3-b4 with b6 antiparallelto the others. Helices a1 and a2 lie on one side ofthe sheet at the surface of the protein with a3 buried
on the other. In domain III, a six-stranded b-sheet isformed in the order b1-b9-b13-b12-b10-b11 with b1and b13 antiparallel to the others. The a8, a9 pair isexterior with a11 buried. Domain II, the lid domain,is an extended structure covering a large surfacearea of the enzyme, and consists of helices a4, a5and a6, and a b-hairpin formed by b6–b7. Thegeneric thiolase-fold, a five-layered two-helix/sheet/two-helix/sheet/two-helix assembly, is con-structed from domains I and III aligned such thatburied at the core of the structure, and sandwichedbetween the two b-sheets, are the two amphipathichelices (a3 and a11).
A least-squares fit of six possible combinations ofthe three independent AtKATsubunits from the twocrystal forms with the two ScKAT subunits in theasymmetric unit of that structure gave a meanr.m.s.d. value of 1.38 A for 343 Ca atoms within thenarrow range of 1.34 A to 1.42 A. Selected residues,160–186, in the lid domain II of ScKAT were omittedfrom these calculations, since they clearly adopt adifferent conformation, and this will be discussedlater. The good agreement between the structuresreflects the highly conserved overall fold of the twostructures.
However, there are detailed differences betweenthe AtKAT and ScKAT subunits of note. Theseinclude an additional b-strand, b1, which extendsthe AtKAT b-sheet of domain III from five to sixstrands. Helices a2 and a5 of the ScKAT domain Iare not found in AtKAT, which results intruncations between a1–b3 and b5–a4, respec-tively. In domain II of AtKAT the structure isextended with a b-hairpin formed by b6 and b7.Most significant is the alteration in structure of asegment of polypeptide in the lid domain II. InScKAT helices a6 and a7 and the nine residuesbetween them extend out from the proteinstructure at the side of the active site cleft. InAtKAT this region of the polypeptide chain isfolded into a single helix, a4, which lies within theactive site, blocking access to catalytically impor-tant residues. As a result of this, Cys192 on a4forms a disulfide bond with the catalytic Cys138.These features will be discussed when details ofthe active site are given.
Structure of the AtKAT dimer and comparisonswith ScKAT
Previous studies of KAT enzymes indicate eitherdimeric or tetrameric structures depending onwhether type I or II enzymes are being considered.We have observed, by size-exclusion chromato-graphy, that AtKAT is predominantly a dimer insolution with some monomer present. The dimer isobserved in matrix-assisted laser desorption/ionis-ation time-of-flight mass spectrometry and persistseven in SDS-PAGE (data not shown). In crystal formI a dimer constitutes the asymmetric unit and thesubunits are related by a 2-fold axis of non-crystallographic symmetry (NCS). In form II, theasymmetric unit is a single subunit, which forms

Figure 1. The amino acid sequence and secondary structure of AtKAT. Arrows depict b-strands, cylinders a-helicesand these are coloured according to the domain in which they occur; blue for domain I, yellow domain II and red domainIII and labelled b1 to b13, a1 to a11. The aligned sequence of ScKAT with secondary structure coloured grey, is also given.Strictly conserved amino acids are shown as white letters in black boxes. This Figure was prepared using the programALINE (C. S. Bond, personal communication).
350 Structure of a Plant Ketoacyl-CoA Thiolase
the same dimer as seen in crystal form I byapplication of the crystallographic 2-fold axisparallel with c.
The dimer interface is flat with a large solvent-accessible region. Almost 40% of the atoms at theinterface are polar in character yet they participatein only nine direct inter-subunit hydrogen-bondingassociations. The functional groups are heavilyhydrated and extensive solvent-mediated networksof hydrogen bonds contribute to dimer stabiliz-ation. There are four areas of protein–proteininteractions that create the AtKAT dimer(Figure 2(b)). These involve b1 of one subunitinteracting with a3 and aligned antiparallel withb1 of the partner subunit at one end of the interface.The b1–b1 association involves few direct inter-actions but creates a solvent-filled groove allowingnumerous solvent-mediated interactions betweensubunits. At the other end of the interface region,the N-terminal segment of a2 interacts with parts ofa4 on the adjacent subunits. At the centre of theinterface the subunit–subunit association involvesloop b9–a8 interacting with residues from the turnsinto b3 and b4, and the polypeptide segmentpreceding a3.
The dimer interface regions, though not directlyinvolved in catalysis, contribute part of the activesite architecture. The loop that carries Cys138 isstabilized by a number of polar contacts and salt-
bridges from the residues of the neighbouringsubunit. For example, the side-chain of Asn135 insubunit A participates in hydrogen bonds with thecarbonyl and amide groups of Cys138 in subunit B(not shown).
Comparison of the AtKAT and ScKAT dimersreveal important differences. For both enzymes, theaccessible surface area (ASA) of a monomer treatedin isolation is approximately 9300 A2. When dimersare formed, the surface area of the interfacialregions is about 1490 A2 for ScKAT but this isreduced to only 770 A2 for AtKAT. The majordifference can be attributed to the conformation ofthe polypeptide in the lid domain comprisingresidues 160–186 of ScKAT. This segment contri-butes just over 700 A2 to the interface and makes animportant contribution to forming the ScKATdimer. In contrast, for AtKAT the correspondingpart of the polypeptide, a4, with a differentconformation does not make a significant contri-bution to the dimer interface.
With one subunit of each structure superimposedand fixed, then the rotation between the partnersubunits is estimated to be 308 with a translationaldifference of 1.0 A between the centre-of-mass ofeach subunit (Figure 3). As described earlier, thedimer interface area is heavily hydrated with fewdirect interactions between subunits. This permitsflexibility with respect to the subunit association

Figure 2. (a) Ribbon diagram of the AtKAT subunit.Elements of secondary structure are coloured accordingto domain as described for Figure 1 and several arelabelled. An asterisk (*) marks the active site, and thechain break at Pro172 is labelled. (b) The quaternarystructure viewed perpendicular to the NCS 2-fold axis ofsymmetry.
Figure 3. Structural overlay of AtKAT (red) and ScKAT(grey) dimers based on the least-squares fit of a singlesubunit A. Helices are depicted as cylinders, b-strands asarrows.
Structure of a Plant Ketoacyl-CoA Thiolase 351
and results in large structural differences whenAtKAT and ScKAT dimers are compared. Thesedifferences affect in particular the a-helices at theperiphery of the structure. The magnitude of thesedifferences is exemplified by residues at theN-terminal segment of a5, a helix in the lid domainwith a high level of sequence conservation(Figure 1). Here, equivalent Ca positions areseparated by nearly 18 A when comparing AtKATand ScKAT (Figure 3).
The active site
The active site is a deep, polar cavity formed byresidues from all three domains. It is positionedbetween the internal side of the domain II b-sheetand the N-terminal region of the buried amphi-pathic helices a3 and a11 (Figure 2(a)). Domain IIresidues form the periphery of the active site anda4, a component of domain II, binds in and occludesthe active site.
Within the thiolase superfamily, there are threestrictly conserved residues important for catalysis.
In ScKAT these are Cys125, His375 and Cys403.20
The corresponding residues in AtKAT are Cys138,in loop b4–a3 on domain I, His393 on the segmentlinking a10 and a11 of domain III, and Cys425 at theC-terminal end of b12. On the basis of comparisonswith the biosynthetic thiolases, for which a numberof enzyme ligand complexes have been deter-mined,3–5 a model for interactions formed byScKAT with substrates was proposed and thecontributions of these residues to the KAT mecha-nism suggested.20
Once substrate is bound, a Cys125/138 thiolatecould initiate catalysis by nucleophilic attack on thecarbonyl Cb, break the Ca–Cb bond and form acovalent acyl-enzyme intermediate and a Ca
carbanion. Cys403/425 may assist completion ofthe cleavage reaction by donating a proton to C2 forthe release of acetyl-CoA, the first product. In thisfirst stage of catalysis, Cys125/138 is likelyactivated by interaction with His375/393. In thesecond part of the mechanism CoA binds and reactswith the acyl-enzyme intermediate. Here, His375/393 activates CoA by abstraction of a proton fromthe thiol group to generate the nucleophile thatattacks the carbonyl Cb.
In AtKAT a well-ordered hydrophilic pocket,conserved in ScKAT, is formed by the turn betweenb12 and b13 (not shown) in which a well-definedelectron density peak was observed. On the basis ofthe chemical environment and the peak height thishas been assigned as a water molecule, not an ion,in all three subunits characterized by the two crystalforms. The thermal parameters of the watermolecules are 19.6 A and 21.6 A in crystal form I,and 44.7 A2 in form II, compared to the overallaverage thermal parameters for solvents of 25.9 Aand 50.5 A2 in the two structures. The water
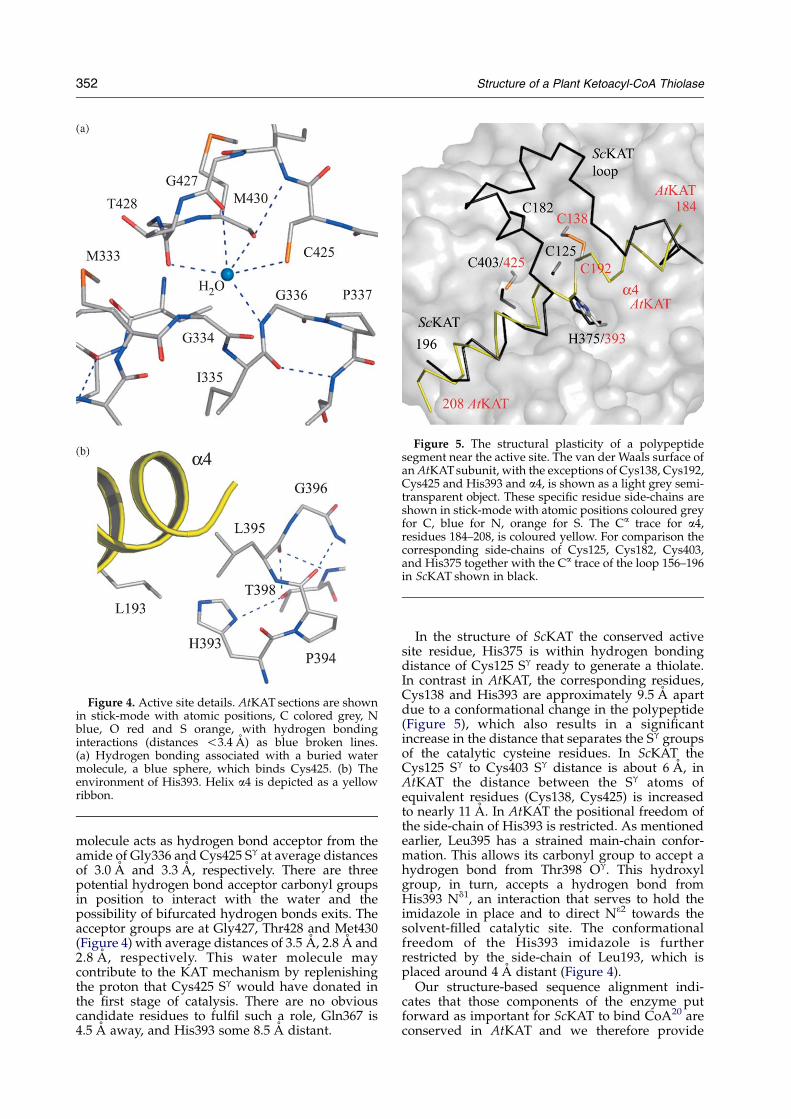
Figure 5. The structural plasticity of a polypeptidesegment near the active site. The van der Waals surface ofan AtKAT subunit, with the exceptions of Cys138, Cys192,Cys425 and His393 and a4, is shown as a light grey semi-transparent object. These specific residue side-chains areshown in stick-mode with atomic positions coloured greyfor C, blue for N, orange for S. The Ca trace for a4,residues 184–208, is coloured yellow. For comparison thecorresponding side-chains of Cys125, Cys182, Cys403,and His375 together with the Ca trace of the loop 156–196in ScKAT shown in black.
Figure 4. Active site details. AtKAT sections are shownin stick-mode with atomic positions, C colored grey, Nblue, O red and S orange, with hydrogen bondinginteractions (distances !3.4 A) as blue broken lines.(a) Hydrogen bonding associated with a buried watermolecule, a blue sphere, which binds Cys425. (b) Theenvironment of His393. Helix a4 is depicted as a yellowribbon.
352 Structure of a Plant Ketoacyl-CoA Thiolase
molecule acts as hydrogen bond acceptor from theamide of Gly336 and Cys425 Sg at average distancesof 3.0 A and 3.3 A, respectively. There are threepotential hydrogen bond acceptor carbonyl groupsin position to interact with the water and thepossibility of bifurcated hydrogen bonds exits. Theacceptor groups are at Gly427, Thr428 and Met430(Figure 4) with average distances of 3.5 A, 2.8 A and2.8 A, respectively. This water molecule maycontribute to the KAT mechanism by replenishingthe proton that Cys425 Sg would have donated inthe first stage of catalysis. There are no obviouscandidate residues to fulfil such a role, Gln367 is4.5 A away, and His393 some 8.5 A distant.
In the structure of ScKAT the conserved activesite residue, His375 is within hydrogen bondingdistance of Cys125 Sg ready to generate a thiolate.In contrast in AtKAT, the corresponding residues,Cys138 and His393 are approximately 9.5 A apartdue to a conformational change in the polypeptide(Figure 5), which also results in a significantincrease in the distance that separates the Sg groupsof the catalytic cysteine residues. In ScKAT theCys125 Sg to Cys403 Sg distance is about 6 A, inAtKAT the distance between the Sg atoms ofequivalent residues (Cys138, Cys425) is increasedto nearly 11 A. In AtKAT the positional freedom ofthe side-chain of His393 is restricted. As mentionedearlier, Leu395 has a strained main-chain confor-mation. This allows its carbonyl group to accept ahydrogen bond from Thr398 Og. This hydroxylgroup, in turn, accepts a hydrogen bond fromHis393 Nd1, an interaction that serves to hold theimidazole in place and to direct N32 towards thesolvent-filled catalytic site. The conformationalfreedom of the His393 imidazole is furtherrestricted by the side-chain of Leu193, which isplaced around 4 A distant (Figure 4).
Our structure-based sequence alignment indi-cates that those components of the enzyme putforward as important for ScKAT to bind CoA20 areconserved in AtKAT and we therefore provide

Figure 6. The proline-flip. Atoms of AtKAT are depictedas for Figure 4. The superimposed ScKAT structure has Catom positions colored cyan, N blue, and O red.
Structure of a Plant Ketoacyl-CoA Thiolase 353
support for that model. For example, four basicresidues in ScKAT, Lys48, Lys51, Arg246 andArg258 were suggested to interact with three CoAphosphate groups. At the corresponding positionsin AtKAT these residues are Lys64, Arg67, Arg265and Lys277 (Figure 1). In addition, Lys66 and Lys71of AtKAT may contribute to electrostatic attractionof the polyanionic ligand (not shown). In ScKAT thebinding of the adenine component of CoA mayinvolve hydrogen bonds with the Glu243 side-chainas well as with the Gly263 and Pro245 main-chainfunctional groups. In AtKAT, Asp262, Gly263 andIle264 offer the same possible hydrogen bondpairings. Other residues in ScKAT that mightcontribute to CoA binding using hydrogen bondsand van der Waals interactions include Phe50,Met155, Tyr159, Ser274, Val276, and Phe346. Ourstructural alignment identifies Lys66, Met168,Phe186, Ser292, Val294, and Phe364, respectively,as the equivalent residues in AtKAT (not shown).
However, the structural overlay of AtKAT andScKAT also indicates significant differences in threeadjacent sections of polypeptide that line one side ofthe active site. These parts of the enzyme structures,though highly conserved in terms of amino acidsequence, have been influenced by the presence ofthe extended a4 in AtKAT, or the linking polypep-tide between a5/a6 in ScKAT. Using AtKAT as thereference structure, differences in the b-turnbetween b12–b13 shifts the Ca position of theconserved Pro330 (Pro312 in ScKAT) by approxi-mately 5.5 A (not shown). In the polypeptidelinking b9 with a8, structural differences meanthat another conserved residue, Thr428 (Thr406 inScKAT), shifts Ca position by about 3.5 A (notshown). In the third section affected, changes in thestructure of the polypeptide linking b4 with a3 alterthe position of a catalytic cysteine. Here, the Ca
atom of Cys138 AtKAT (Cys123 ScKAT) changesposition by nearly 6 A. It has been noted that themain chain of ScKAT Gln124, the residue adjacent tothis catalytic cysteine, adopts a strained confor-mation20 with the f/j combination of 498/K1358and 518/K1338 for subunits A and B, respectively(PDB entry 1AFW). The corresponding Gln137 inAtKAT has altered position and is not in a strainedconformation, with f/j values of K738/K458 andK708/K468 in subunits A and B, respectively. Themovement of Cys138 in AtKAT appears correlatedtherefore with this release of conformational strain.
Proline, by virtue of its unique chemical struc-ture, in particular lacking the amide hydrogenutilized by amino acids in main-chain hydrogenbonding, is a poor helix-forming residue. In ScKAT,Pro184 is positioned at the start of a helix that leadsaway from the active site. However, in AtKAT thecorresponding residue (Pro195) is observed withinthe long helix a4. The overlay of AtKAT and ScKATshows that this conserved proline, with Ca atomsabout 2 A apart, is a key branch point in thepolypeptide chain (Figure 5). The f/j values forthis residue change from K578/1448 and K498/1388for Pro185 ScKAT to K628/K318 and K538/K438
for Pro195 AtKAT in subunits A and B, respectively.The structural consequence of this, what we terma proline-flip, is that the residues immediatelyN-terminal to proline run in opposite directions(Figures 5 and 6). In ScKAT, the chain is directedaway from and around the entrance to the activesite. In AtKAT, the chain continues in an a-helicalconformation (a4) and lies over and occludes theactive site. Cys192 on this helix participates indisulfide bond formation with the catalytic Cys138and this covalent link helps to hold a4 in place. Thisdisulfide linkage is absent from ScKAT, althoughthe cysteine pair (Cys125 and Cys182) is conserved(Figure 1). When the structures are superimposed,AtKAT Cys192 Sg and the equivalent residueScKAT Cys182 Sg are approximately 10.5 A apart(Figure 5).
The structures of AtKAT and ScKAT active sitesare thus very different. We propose that the ScKATstructure, with an open active site, represents areduced and active form of the enzyme, whilst theAtKAT structure represents a previously unrecog-nised oxidized and inactive form. At the N-terminalsection of AtKAT a4, a number of residues aremissing from the model, most likely due to theirmobility. Such a property would permit the poly-peptide chains of KATs to adjust their conformationsto either form the helix that blocks the active site orthe looped out structure that both allows access toand helps to form the catalytic centre.
Based on the observation that the cysteine pairsCys125/182 and Cys138/192 are conserved in bothScKAT and AtKAT, respectively, we sought toinvestigate if similar oxidized and reduced struc-tures might be possible in other KAT enzymes. Theequivalent residue of peroxisomal AtKAT Cys192 isnot conserved in any prokaryotic thiolase, irrespec-tive of type, or eukaryotic biosynthetic thiolases.Nor is this residue conserved in the multi or

Figure 7. The aligned sequencesof 14 peroxisomal KAT segmentscorresponding to AtKAT residuesAla187 to Gln205. The sequenceswere retrieved from the ExpertProtein Analysis System server(http://ca.expasy.org/) and inorder are derived from Arabidopsisthaliana, Kurokawa amakuri, Crocussativus, Magnifera indica, Helianthusannuus, Oryza sativa, Glycine max,Mus musculus, Rattus norvegicus,Homo sapiens, Saccharomyces cerevi-siae, Candida tropicalis, Yarrowialipolytica and Aspergillus fumigatus.Amino acid residues that arestrictly conserved in at least 12 ofthe sequences are shown as whiteletters in black boxes.
354 Structure of a Plant Ketoacyl-CoA Thiolase
mono-functional enzymes that encode mitochon-drial KATs. However, in 26 degradative peroxiso-mal KAT sequences that could be identified thecysteine pairing is strictly conserved. The aminoacid sequences corresponding to a segment ofAtKAT a4 are shown for 13 typical examples inFigure 7. In addition to the conserved cysteine, wenote conservation of the key residues that wouldallow for helix formation, and the proline-flip in theeukaryotic peroxisomal enzymes. This suggests thatour observations resulting from the analysis ofAtKAT are widely applicable and identifies apossible role for reversible disulfide/dithiolexchange to influence an important biologicalfunction, namely FA b-oxidation in peroxisomes.
Concluding remarks
The crystal structure of peroxisomal ScKAT wasdetermined under reducing conditions.19,20 In ourstudy, the AtKAT structure was determined in theabsence of any reducing agents. Despite deter-mined efforts we have been unable to obtaincrystals of AtKAT under reducing conditions or incomplex with a CoA derivative.
We propose that the AtKAT structure, with theactive site blocked by a4 and with a catalyticcysteine participating in a disulfide linkage with aresidue on this helix, represents an inactive form ofthe enzyme whilst the structure of ScKAT, deter-mined under reducing conditions, with an openactive site represents the active form of the enzyme.This conclusion is consistent with the observationthat optimum KAT activity is registered underreducing conditions10 and suggests likewise thatFA b-oxidation can only proceed under suchconditions. Since the peroxisomal KAT amino acidsequences in the region of a4 are so highlyconserved, this may represent a general mechanismto regulate FA b-oxidation by disulfide/dithiolredox chemistry. It will be interesting, in future
work, to investigate what physiological redoxpartners might be involved in this system toregulate KAT activity. Small molecule metabolitessuch as glutathione, or proteins such as theubiquitous thiol reductase thioredoxin, might begood candidates to investigate.
Materials and Methods
Cloning and expression of AtKAT
A gene fragment coding for a truncated AtKAT, lackingthe N-terminal peroxisomal targeting sequence of 34amino acid residues, was obtained by PCR amplificationfrom the A. thaliana ecotype Columbia [col-O] thiolasecDNA clone 91A18T7 (accession: AB008854). Two oligo-nucleotide primers were designed (forward, 5 0 GCT CATATG GCT GGG GAC AGT GCT GCA TAT C 3 0 andreverse, 5 0 TGG GGA TCC CTA GCG AGC GTC CTTGGA C 3 0) to introduce unique restriction sites for NdeIand BamHI, respectively (shown in bold). The PCRproduct was blunt-end ligated into pUC18 (SureClone;GE Healthcare) and sequenced to confirm the integrity ofthe product. The insert was isolated by digestion withNdeI-BamHI and ligated into the pET-15b vector(Novagen) creating a plasmid, which ultimately producesprotein carrying an N-terminal hexahistidine tag andthrombin cleavage site linker. This plasmid was heat-shock transformed into E. coli BL21(DE3) and selected onLuria-Bertani agar plates containing 100 mg mlK1 ampi-cillin (LB/amp). In a typical preparation, bacteria werecultured in LB/amp medium and grown at 37 8C to anabsorbance of 0.6 at 600 nm. The temperature wasreduced to 25 8C and gene expression induced with1 mM isopropyl-b-D-thiogalactopyranoside. Cell growthcontinued for 8 h before cells was harvested by centrifu-gation (3500g, 20 min, 4 8C).
Purification of recombinant His-tagged thiolaseby affinity chromatography
Cells were resuspended in 20 ml of buffer A (50 mMTris–HCl (pH 7.7), 200 mM NaCl), 5 mM benzamidine,

† DeLano, WL (2002) The PyMOL Molecular GraphicsSystem, DeLano Scientific, San Carlos, CA, USA. http://pymol.sourceforge.net
Structure of a Plant Ketoacyl-CoA Thiolase 355
5 mM lysozyme, 100 units of DNase, left on ice for 15 minand then lysed in a One-shot cell disruptor (Constant CellDisruptions Systems) at 33,000 psi. The insoluble debriswas separated by centrifugation (53,000g, 20 min, 4 8C) andthe supernatant, after passage through a 0.2 mm filter, wasapplied to a 5 ml Ni2C-resin column (Hi-trap; GEHealthcare) pre-equilibrated with buffer A. The columnwas washed with buffer A and the enzyme then eluted at aconcentration of 180 mM imidazole. Fractions wereanalysed by SDS-PAGE; those containing the enzymewere pooled, dialyzed against buffer A and the proteinthen concentrated to 20 mg mlK1. The protein concen-tration was determined spectrophotometrically using anextinction coefficient of 28,000 MK1 cmK1 (280 nm). Thehigh purity of the sample was confirmed with SDS-PAGEand matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry and the yield of enzymeestablished as approximately 15 mg lK1 of bacterial culture.
At an early stage of our study, AtKAT was subjected toadditional purification protocols as we sought to optimisepurity and crystallization properties. The histidine-tagwas removed by cleavage with thrombin, followed byanion-exchange and gel-filtration chromatography toinvestigate if they might assist. Mass spectrometry wasused to check the purity of the samples and for completeseparation of cleaved products where necessary. Ulti-mately, we observed that AtKAT carrying additional 20amino acid residues at the N terminus was perfectlyadequate for crystal growth and that no benefit accruedby further purifications.
The use of size-exclusion chromatography has servedto confirm that AtKAT is predominantly a dimer insolution. A calibration curve (data not shown) for aSuperdex 200 HiLoad 16/60 column (GE Healthcare) wasestablished using seven proteins extending from a massof 13.7–440 kDa (ribonuclease A, chymotrypsinogen,ovalbumin, fructose bisphosphate aldolase, catalase andferritin; all from GE Healthcare). AtKAT eluted as a peakcorresponding to a mass of approximately 100 kDa andan additional, much smaller peak of approximate mass45 kDa. These peaks correspond to dimer and monomerof about 94 kDa and 47 kDa, respectively, and are in goodagreement with the theoretical mass (see below).
Crystallization, data collection and processing
Crystallization trials were carried out, on AtKATcarrying the histidine-tag and on samples in which thetag had been removed by thrombin cleavage, usingcommercial screens (Hampton Research, Jena Bios-ciences). We noted no difference in the crystallizationbehaviour of samples with or without the histidine-tagand it was most efficient to work with the former.A successful condition was observed and optimised toprovide single, well-ordered crystals of dimensions0.3 mm!0.2 mm!0.1 mm. Crystals grew in hangingdrops consisting of 2 ml of protein solution and 2 ml ofreservoir containing 0.1 M Tris–HCl (pH 8.5), 300 mMMgCl2, 25% (w/v) polyethylene glycol 4000. Two crystalforms of similar morphology, orthorhombic blocks,appear under the same conditions.
Prior to X-ray exposure, crystals were cryo-protectedusing 20%(v/v) glycerol and cooled in a stream ofgaseous nitrogen at 100 K. Diffraction data weremeasured at the European Synchrotron Radiation Facility(ESRF, Grenoble, France) on charge coupled devicedetectors using station BM14 for crystal form I, andstation EH14.2 for crystal form II. The DENZO/SCALE-PACK programs24 were used to process the data (Table 1).
Both crystal forms are orthorhombic and systematicabsences indicated that form I displays space groupP212121, and form II P21212.
Structure determination and refinement
AtKAT is a homodimer and each subunit comprises 427amino acid residues of molecular mass 47 kDa. Matthewscoefficients of 2.1 A3 DaK1 and estimated solvent contentof about 40% (v/v) suggested a dimer in the asymmetricunit for crystal form I and a monomer in the asymmetricunit for crystal form II. A polyalanine model of thedimeric ScKAT,20 which on the basis of an alignmentshares 43% sequence identity with AtKAT, was used formolecular replacement (MR). A random selection ofreflections (5%) was flagged for the calculation of R-freeand used to inform decision making during structuresolution and refinement.
MR calculations, using the ScKAT model targeting formI failed to yield a solution for both subunits irrespective ofwhether a monomer or dimer search model was used.A self-rotation function gave no indication of the presenceof NCS. However, a native Patterson was calculated usingdata in the resolution range 10–3 A and displayed astrong feature, of height one half of the origin peak,placed at 0.5 along the c axis. The NCS axis, relating thetwo subunits in the asymmetric unit, is thus orientedparallel with a crystallographic 21 axis resulting inpseudo-translational symmetry. This, in combinationwith what we know now, is an MR model with significantdifferences from the true structure had served to preventstructure solution at this stage.
Crystal form II, with a monomer in the asymmetric unitwas obtained and MR with the program AMoRe25,26
identified a clear solution. After rigid body refinementand density modification (program DM),26 an interpret-able electron density map was obtained. It was quicklyapparent that the search model was in places significantlydifferent from AtKAT. The model, electron and differencedensity maps were inspected using the graphics programO,27 and refinement was carried out in REFMAC.28 Strictgeometric restraints were imposed and it proved useful toalso include TLS (translation, libration and screw) grouprefinement.29 Refinement was completed by inclusion ofordered solvent positions.
The form II structure, at an intermediate stage ofrefinement, proved to be an excellent MR model todetermine the structure of form I, which diffracted tosignificantly higher resolution. A promising MR solutionfor a dimer in the asymmetric unit was identified usingMOLREP30 and then refined applying strict geometricand, in the initial calculations, NCS restraints. When thecomplete protein structure was modelled, solventmolecules were added to complete the analysis. Theassignment of secondary structure was made using acombination of automated methods27,31 and by visualinspection. Stereochemistry was also assessed usingPROCHECK31 and statistics are presented in Table 1.ASA calculations were carried out with the programAREAIMOL of the CCP4 suite.26 The DYNDOM pro-gram32 was used to determine the difference betweensubunits when comparing the distinct AtKAT and ScKATdimers. Molecular images were prepared with PyMol†.

356 Structure of a Plant Ketoacyl-CoA Thiolase
Protein Data Bank accession codes
Coordinates and structure factors are available forimmediate release upon acceptance (2C7Y, 2C7Z).
Acknowledgements
We thank C. Bond, E. Tetaud, and D. Hall foradvice and contributions. This research was sup-ported by The Wellcome Trust, the Biotechnologyand Biological Sciences Research Council (UK)and ESRF.
References
1. Kunau, W.-H., Dommes, Y. & Schulz, H. (1995). b-Oxidation of fatty acids in mitochondria, peroxisomesand bacteria: a century of continued progress. Prog.Lipid Res. 34, 267–342.
2. Heath, R. J. & Rock, C. O. (2002). The Claisencondensation in biology. Nature Prod. Rep. 19, 581–596.
3. Modis, Y. & Wierenga, R. K. (2000). Crystallographicanalysis of the reaction pathway of Zoogloea ramigerabiosynthetic thiolase. J. Mol. Biol. 297, 1171–1182.
4. Kursula, P., Ojala, J., Lambeir, A.-M. & Wierenga, R. K.(2002). The catalytic cycle of biosynthetic thiolase: aconformational journey of an acetyl group throughfour binding modes and two oxyanion holes.Biochemistry, 41, 15543–15556.
5. Kursula, P., Sikkila, H., Fukao, T., Kondo, N. &Wierenga, R. K. (2005). High resolution crystalstructures of human cytosolic thiolase (CT): acomparison of the active sites of human CT, bacterialthiolase, and bacterial KAS I. J. Mol. Biol. 347, 189–201.
6. Reddy, J. K. & Hashimoto, T. (2001). Peroxisomalb-oxidation and peroxisome proliferator-activatedreceptor alpha: an adaptive metabolic system. Annu.Rev. Nutr. 21, 193–230.
7. Hiltunen, J. K. & Qin, Y. (2000). b-Oxidation–strategies for the metabolism of a wide variety ofacyl-CoA esters. Biochim. Biophys. Acta, 1482, 117–128.
8. Kim, J. J. & Battaile, K. P. (2002). Burning fat: thestructural basis of fatty acid b-oxidation. Curr. Opin.Struct. Biol. 12, 721–728.
9. Kato, A., Hayashi, M., Takeuchi, Y. & Nishimura, M.(1996). cDNA cloning and expression of a gene for3-ketoacyl-CoA thiolase in pumpkin cotyledons. PlantMol. Biol. 31, 843–852.
10. Germain, V., Rylott, E. L., Larson, T. R., Sherson, S. M.,Bechtold, N., Carde, J.-P. et al. (2001). Requirement for3-ketoacyl-CoA thiolase-2 in peroxisome develop-ment, fatty acid b-oxidation and breakdown oftriacylglycerol in lipid bodies of Arabidopsis seedlings.Plant J. 28, 1–12.
11. Beevers, H. (1979). Microbodies in higher plants.Annu. Rev. Plant Physiol. 30, 159–193.
12. Kindl, H. (1993). Fatty-acid degradation in plantperoxisomes–function and biosynthesis of theenzymes involved. Biochimie, 75, 225–230.
13. Hayashi, M., Toriyama, K., Kondo, M. & Nishimura, M.(1998). 2,4-Dichlorophenoxybutyric acid-resistantmutants of Arabidopsis have defects in glyoxysomalfatty acid b-oxidation. Plant Cell, 10, 183–195.
14. Oeljeklaus, S., Fischer, K. & Gerhardt, B. (2002).Glyoxysomal acetoacetyl-CoA thiolase and3-oxoacyl-CoA thiolase from sunflower cotyledons.Planta, 214, 597–607.
15. Pinfield-Wells, H., Rylott, E. L., Gilday, A. D., Graham,S., Job, K., Larson, T. R. & Graham, I. A. (2005).Sucrose rescues seedling establishment but notgermination of Arabidopsis mutants disrupted inperoxisomal fatty acid catabolism. Plant J. 43, 861–872.
16. Preisig-Muller, R. & Kindl, H. (1993). Thiolase mRNAtranslated in vitro yields a peptide with a putativeN-terminal presequence. Plant Mol. Biol. 22, 59–66.
17. Olesen, C., Thomsen, K. K., Svendsen, I. & Brandt, A.(1997). The glyoxysomal 3-ketoacyl-CoA thiolaseprecursor from Brassica napus has enzymatic activitywhen synthesized in Escherichia coli. FEBS Letters, 412,138–140.
18. Glover, J. R., Andrews, D. W. & Rachubinski, R.(1994). Saccharomyces cerevisiae peroxisomal thiolase isimported as a dimer. Proc. Natl Acad. Sci. USA, 91,10541–10545.
19. Mathieu, M., Zeelen, J. Ph., Pauptit, R. A., Erdmann, R.,Kunau, W.-H. & Wierenga, R. K. (1994). The 2.8 Acrystal structure of peroxisomal 3-ketoacyl-CoAthiolase of Saccharomyces cerevisiae: a five-layeredstructure constructed from two core domains ofidentical topology. Structure, 2, 797–808.
20. Mathieu, M., Modis, J., Zeelen, J. Ph., Engel, C. K.,Abagyan, R. A., Ahlberg, A. et al. (1997). The 1.8 Acrystal structure of the dimeric peroxisomal3-ketoacyl-CoA thiolase of Saccharomyces cerevisiae:implications for substrate binding and reactionmechanism. J. Mol. Biol. 273, 714–728.
21. Zeng, J. & Li, D. (2004). Expression and purification ofHis-tagged rat mitochondrial 3-ketoacyl-CoA thiolasewild-type and His352 mutant proteins. Protein Expr.Purif. 35, 320–326.
22. Ishikawa, M., Tsuchiya, D., Oyama, T., Tsunaka, Y. &Morikawa, K. (2004). Structural basis for channellingmechanism of a fatty acid b-oxidation multienzymecomplex. EMBO J. 23, 2745–2754.
23. Huang, W., Jia, J., Edwards, P., Dehesh, K., Schneider, G.& Lindqvist, Y. (1998). Crystal structure of b-ketoacyl-acyl carrier protein synthase II from E. coli reveals themolecular architecture of condensing enzymes. EMBOJ. 17, 1181–1191.
24. Otwinowski, Z. & Minor, W. (1997). Processing ofX-ray diffraction data collected in oscillation mode.Methods Enzymol. 276, 307–326.
25. Navaza, J. (1994). AMoRe: an automated package formolecular replacement. Acta Crystallog. sect. A, 50,157–163.
26. Collaborative Computational Project, Number 4.(1994). The CCP4 suite: programs for proteincrystallography. Acta Crystallog. sect. D, 50, 760–763.
27. Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building proteinmodels in electron-density maps and the location oferrors in these models. Acta Crystallog. sect. A, 47,110–119.
28. Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997).Refinement of macromolecular structures by themaximum-likelihood method. Acta Crystallog. sect.D, 53, 240–255.
29. Winn, M. D., Isupov, M. N. & Murshudov, G. N.(2001). Use of TLS parameters to model anisotropicdisplacements in macromolecular refinement. ActaCrystallog. sect. D, 57, 122–133.

Structure of a Plant Ketoacyl-CoA Thiolase 357
30. Vagin, A. & Teplyakov, A. (2000). An approach tomulti-copy search in molecular structures by themaximum-likelihood method. Acta Crystallog. sect. D,53, 240–255.
31. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallog. 26, 283–291.
32. Hayward, S. & Berendsen, H. J. (1998). Systematicanalysis of domain motions in proteins from confor-mational change: new results on citrate synthase andT4 lysozyme. Proteins: Struct. Funct. Genet. 30,144–154.
33. Cruikshank, D. W. J. (1999). Remarks about proteinstructure precision. Acta Crystallog. sect. D, 55,583–601.
Edited by R. Huber
(Received 16 January 2006; received in revised form 7 March 2006; accepted 15 March 2006)Available online 29 March 2006




















