
Propionyl-CoA and adenosylcobalamin metabolism in Caenorhabditis elegans: evidence for a role of...
10
Molecular Genetics and Metabolism 89 (2006) 64–73 www.elsevier.com/locate/ymgme 1096-7192/$ - see front matter 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.ymgme.2006.06.001 Propionyl-CoA and adenosylcobalamin metabolism in Caenorhabditis elegans: Evidence for a role of methylmalonyl-CoA epimerase in intermediary metabolism Randy J. Chandler a , Vijay Aswani a , Matthew S. Tsai a , Marni Falk b,c , Natasha Wehrli d , Sally Stabler e , Robert Allen e , Margaret Sedensky f , Haig H. Kazazian d , Charles P. Venditti a,¤ a Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USA b Department of Genetics, CASE School of Medicine, Cleveland, OH 44106, USA c Department of Pediatrics, CASE School of Medicine, Cleveland, OH 44106, USA d Department of Genetics, University of Pennsylvania School of Medicine, Philadelphia, PA 19114, USA e Department of Medicine, University of Colorado School of Medicine, Denver, CO 80206, USA f Department of Anesthesiology, University Hospitals of Cleveland, Cleveland, OH 44106, USA Received 25 April 2006; received in revised form 2 June 2006; accepted 2 June 2006 Available online 14 July 2006 Abstract We have utilized Caenorhabditis elegans to study human methylmalonic acidemia. Using bioinformatics, a full complement of mam- malian homologues for the conversion of propionyl-CoA to succinyl-CoA in the genome of C. elegans, including propionyl-CoA carbox- ylase subunits A and B (pcca-1, pccb-1), methylmalonic acidemia cobalamin A complementation group (mmaa-1), co(I)balamin adenosyltransferase (mmab-1), MMACHC (cblc-1), methylmalonyl-CoA epimerase (mce-1) and methylmalonyl-CoA mutase (mmcm-1) were identiWed. To verify predictions that the entire intracellular adenosylcobalamin metabolic pathway existed and was functional, the kinetic properties of the C. elegans mmcm-1 were examined. RNA interference against mmcm-1, mmab-1, mmaa-1 in the presence of pro- pionic acid revealed a chemical phenotype of increased methylmalonic acid; deletion mutants of mmcm-1, mmab-1 and mce-1 displayed reduced 1-[ 14 C]-propionate incorporation into macromolecules. The mutants produced increased amounts of methylmalonic acid in the culture medium, proving that a functional block in the pathway caused metabolite accumulation. Lentiviral delivery of the C. elegans mmcm-1 into Wbroblasts derived from a patient with mut o class methylmalonic acidemia could partially restore propionate Xux. The C. elegans mce-1 deletion mutant demonstrates for the Wrst time that a lesion at the epimerase step of methylmalonyl-CoA metabolism can functionally impair Xux through the methylmalonyl-CoA mutase pathway and suggests that malfunction of MCEE may cause meth- ylmalonic acidemia in humans. The C. elegans system we describe represents the Wrst lower metazoan model organism of mammalian pro- pionate spectrum disorders and demonstrates that mass spectrometry can be employed to study a small molecule chemical phenotype in C. elegans RNAi and deletion mutants. 2006 Elsevier Inc. All rights reserved. Keywords: Propionate metabolism; Methylmalonic acidemia; Methylmalonyl-CoA epimerase; Methylmalonyl-CoA mutase; Cobalamin; C. elegans; Methylmalonyl-CoA racemase; Mass spectrometry; RNA interference; Bioinformatics Introduction The metabolism of propionyl-CoA to succinyl-CoA via the formation and isomerization of methylmalonyl-CoA is a critical metabolic pathway in humans (Fig. 1). Defective conversion of L-methylmalonyl-CoA to succinyl-CoA in the mitochondrial matrix underlies the etiology of the hereditary methylmalonic acidemias (MIM 251000 and MIM 251100), a heterogeneous group of inborn errors of metabolism [1]. Patients deWcient in methylmalonyl-CoA mutase (MUT) or adenosylcobalamin, the enzymatic * Corresponding author. Fax: +1 3014022170. E-mail address: [email protected] (C.P. Venditti).
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Propionyl-CoA and adenosylcobalamin metabolism in Caenorhabditis elegans: evidence for a role of...

Molecular Genetics and Metabolism 89 (2006) 64–73www.elsevier.com/locate/ymgme
1096-7192/$ - see front matter ! 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.ymgme.2006.06.001
Propionyl-CoA and adenosylcobalamin metabolism in Caenorhabditis elegans: Evidence for a role of methylmalonyl-CoA
epimerase in intermediary metabolism
Randy J. Chandler a, Vijay Aswani a, Matthew S. Tsai a, Marni Falk b,c, Natasha Wehrli d, Sally Stabler e, Robert Allen e, Margaret Sedensky f, Haig H. Kazazian d, Charles P. Venditti a,¤
a Genetic Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD 20892, USAb Department of Genetics, CASE School of Medicine, Cleveland, OH 44106, USA
c Department of Pediatrics, CASE School of Medicine, Cleveland, OH 44106, USAd Department of Genetics, University of Pennsylvania School of Medicine, Philadelphia, PA 19114, USA
e Department of Medicine, University of Colorado School of Medicine, Denver, CO 80206, USAf Department of Anesthesiology, University Hospitals of Cleveland, Cleveland, OH 44106, USA
Received 25 April 2006; received in revised form 2 June 2006; accepted 2 June 2006Available online 14 July 2006
Abstract
We have utilized Caenorhabditis elegans to study human methylmalonic acidemia. Using bioinformatics, a full complement of mam-malian homologues for the conversion of propionyl-CoA to succinyl-CoA in the genome of C. elegans, including propionyl-CoA carbox-ylase subunits A and B (pcca-1, pccb-1), methylmalonic acidemia cobalamin A complementation group (mmaa-1), co(I)balaminadenosyltransferase (mmab-1), MMACHC (cblc-1), methylmalonyl-CoA epimerase (mce-1) and methylmalonyl-CoA mutase (mmcm-1)were identiWed. To verify predictions that the entire intracellular adenosylcobalamin metabolic pathway existed and was functional, thekinetic properties of the C. elegans mmcm-1 were examined. RNA interference against mmcm-1, mmab-1, mmaa-1 in the presence of pro-pionic acid revealed a chemical phenotype of increased methylmalonic acid; deletion mutants of mmcm-1, mmab-1 and mce-1 displayedreduced 1-[14C]-propionate incorporation into macromolecules. The mutants produced increased amounts of methylmalonic acid in theculture medium, proving that a functional block in the pathway caused metabolite accumulation. Lentiviral delivery of the C. elegansmmcm-1 into Wbroblasts derived from a patient with muto class methylmalonic acidemia could partially restore propionate Xux. TheC. elegans mce-1 deletion mutant demonstrates for the Wrst time that a lesion at the epimerase step of methylmalonyl-CoA metabolismcan functionally impair Xux through the methylmalonyl-CoA mutase pathway and suggests that malfunction of MCEE may cause meth-ylmalonic acidemia in humans. The C. elegans system we describe represents the Wrst lower metazoan model organism of mammalian pro-pionate spectrum disorders and demonstrates that mass spectrometry can be employed to study a small molecule chemical phenotype inC. elegans RNAi and deletion mutants.! 2006 Elsevier Inc. All rights reserved.
Keywords: Propionate metabolism; Methylmalonic acidemia; Methylmalonyl-CoA epimerase; Methylmalonyl-CoA mutase; Cobalamin; C. elegans;Methylmalonyl-CoA racemase; Mass spectrometry; RNA interference; Bioinformatics
Introduction
The metabolism of propionyl-CoA to succinyl-CoA viathe formation and isomerization of methylmalonyl-CoA is
a critical metabolic pathway in humans (Fig. 1). Defectiveconversion of L-methylmalonyl-CoA to succinyl-CoA inthe mitochondrial matrix underlies the etiology of thehereditary methylmalonic acidemias (MIM 251000 andMIM 251100), a heterogeneous group of inborn errors ofmetabolism [1]. Patients deWcient in methylmalonyl-CoAmutase (MUT) or adenosylcobalamin, the enzymatic
* Corresponding author. Fax: +1 3014022170.E-mail address: [email protected] (C.P. Venditti).

R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73 65
cofactor, accumulate methylmalonic acid in their tissuesand body Xuids, and display secondary metabolic perturba-tions such as hyperglycinemia, hyperammonemia and inter-mittent hypoglycemia [1]. Despite meticulous dietarycontrol, aVected individuals exhibit extreme metabolicinstability and suVer from severe complications, such asdevelopmental delay, renal disease, pancreatitis and meta-bolic infarction of the basal ganglia [2–5]. The pathophysi-ology of these processes and disease complications remainpoorly deWned.
A common feature of cell lines derived from MUT,MMAA, MMAB and cblD variant two deWcient patients isdiminished 1-[14C] propionate incorporation into macro-molecules, an indirect assay of propionate Xux through thepathway depicted in Fig. 1 [11,12].
The function of the methylmalonyl-CoA epimerase orracemase (MCEE) in vivo is uncertain. One older reportproposed that patients who harbor mutations at this locusare protected from developing methylmalonic acidemiabecause a free methylmalonic acid shunt bypasses the epi-merase reaction [13]. The striking conservation of the genethroughout the phyla suggests that the gene product mayplay a critical role in intermediary metabolism [14].
The methylmalonyl-CoA epimerase (mce-1) of C. ele-gans has been studied [15]. A deletion mutant has no phe-notype and exhibits relative resistance to oxidative stress.While the gene product has been demonstrated to possessmethylmalonyl-CoA epimerase activity in vitro, the bio-chemical phenotype of the mutant has not been evaluated.SpeciWcally, whether it displays a functional impairment of1-[14C]-propionate incorporation into macromolecules,suggesting an active role in propionyl- and methylmalonyl-CoA metabolism, remains unknown. Because there are nodeWnitive reports of methylmalonic acidemia patients withMCEE gene mutations, and no other animal models ofMCEE deWciency exist, the role of the MCEE gene in meth-ylmalonyl-CoA metabolism remains mysterious.
Methylmalonic acidemia has been diYcult to study inmodel organisms. Earlier eVorts have used nutritional [16]and cobalamin-analog treatments [17] to induce methylma-lonic aciduria in rats. Both of these treatments are artiWcialand produce an incomplete block, especially in the case ofnutritional deWciency, causing combined methionine syn-thase and methylmalonyl-CoA mutase functional impair-ment. Establishing tractable model organism systems tostudy methylmalonic acidemia will be required to better
Fig. 1. Major pathway of the conversion of propionyl-CoA into succinyl-CoA in human cells. The biotin-dependent enzyme, propionyl-CoA carboxylase(PCCA, PCCB) converts propionyl-CoA into D-methylmalonyl-CoA which is then racemized into L-methylmalonyl-CoA and isomerized into succinyl-CoA, a Kreb cycle intermediate. The L-methylmalonyl-CoA mutase (MUT) reaction requires adenosylcobalamin. The class and genes associated withmethylmalonic acidemia are mut (MUT), cblA (MMAA), cblB (MMAB), cblD variant 2 (unknown gene(s)), cblC (MMAHC), cblF (unknown gene(s)).Question marks indicate unknown genes or poorly deWned cellular processes [6–10,19].

66 R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73
understand the symptoms and complications associatedwith this group of disorders, develop new therapies and fur-ther deWne genes that may play a role in methylmalonyl-CoA and cobalamin metabolism. To date, knock-outmurine models of methylmalonyl-CoA mutase have dis-played a severe neonatal phenotype with uniform mortalityby 24–36 h of life [18,19]. Organisms more amenable togenetic manipulations such as yeast and Drosophila do notpossess cobalamin-dependent metabolic enzymes or utilizealternative pathways for propionyl-CoA metabolism, suchas the methylcitrate cycle [20].
In the present report, we employed a combination ofinformatic, genomic, biochemical, and metabolic analysesto identify and characterize C. elegans genes predicted toparticipate in methylmalonyl-CoA metabolism. In additionto providing direct biochemical evidence for methylmalo-nyl-CoA mutase activity and adenosylcobalamin syntheticcapacity, we show that C. elegans can be used to deWne thefunction of gene products previously suspected to partici-pate in methylmalonyl-CoA metabolism in man, particu-larly methylmalonyl-CoA epimerase. We propose that C.elegans may be used in genetic and genomic eVorts toidentify genes that inXuence cobalamin and methylmalo-nyl-CoA metabolism and demonstrate the utility of massspectrometry to study chemical phenotypes in C. elegans.
Materials and methods
Strains
C. elegans Bristol N2, RB1434 mmcm-1(ok1637), RB1347 mmab-1(ok1493), VC974 mmab-1(ok1484) and RB512 mce-1(ok243) nematodeswere obtained from the Caenorhabditis Genetics Center, University ofMinnesota, Minneapolis, MN, USA. Deletion mutants RB1434, RB1347
and RB512 were generated by the Oklahoma Medical Research Founda-tion (OMRF) Knockout Group, Oklahoma City, OK, USA. Deletionmutant VC974 was isolated by the Reverse Genetics Core facility at theUniversity of British Columbia, Vancouver, Canada. Nematodes weregrown at 20 °C on nematode growth media (NGM) plates using E. colistrain OP50 as a food source [21].
Bioinformatics and sequence analysis
The BLASTP searches were performed against WormBase(www.wormbase.org) using protein sequences from the NCBI(www.ncbi.nih.gov) known to be involved in the conversion of propionyl-CoA to succinyl-CoA in humans. Reciprocal comparisons yielded identi-cal results. The sequence accession numbers are displayed in Table 1.
Radioactive incorporation study
10–20 mg of C. elegans were incubated with 2 !Ci of [1–14C] sodiumpropionate (PerkinElmer, Boston, MA) in 500 !l of PBS with 50 !g/ml ofkanamycin for 24 h. Worms were examined for viability and re-suspendedin 500 !l of 5% trichloroacetic acid (TCA) solution, heated for 10 min at85 °C and then placed on ice for 20 min. Insoluble material was concen-trated by centrifugation at 16,060£ g for 10 min. The pellet was rinsedwith 500 !l of ice-cold 5% TCA, re-suspended in 500 !L of 1 N NaOH andheated at 85 °C for 15 min. Protein concentration was determined usingthe Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Incorporation ofradioactive isotopes was measured using Scintillation System LS6500(Beckman Instruments, Fullerton, CA).
Isolation of cDNA and C. elegans homologues
RNA was isolated from a mixed worm population using Rneasy MiniKit (Qiagen Inc., Valencia, CA). Superscript First-Strand Synthesis Systemfor RT-PCR (Invitrogen, Carlsbad, CA) was used to generate single strandcDNA libraries. The cDNAs for mmcm-1, mmaa-1, mmab-1, pcca-1, pccb-1and mce-1 were isolated by PCR using primers designed against sequencesobtained from WormBase. The forward and reverse primers were designedwith restriction enzyme sites for directional cloning; the forward primeralso contained a Kozak consensus sequence for expression. PCR products
Table 1Putative C. elegans homologues of genes involved in propionyl-CoA metabolism
The percent similarity and e-values of the p-BLAST scores between human proteins involved in propionate metabolism and putative C. elegans orthologsas well as the conserved domains between human and C. elegans proteins is shown.
Human protein C. elegans Protein Gene
Protein % Similarity/e-value
Domains present in human protein Domain(s) % Similarity/e-value
Propionyl-CoA carboxylase alpha subunit (PCCA) NP000273.1
WP:CE04451 F27D9.5 pcca-1
94.6/5.2e-215 Carbamoyl-phosphate synthase Lchain, ATP binding domain, pfam02786
89.8/2.00e-26
Biotin caboxylase C-terminal domain pfam02785
100/1.00e-35
Carbamoyl-phosphate synthase, N-terminal domain pfam00289
100/2.00e-27
Biotin-requiring enzyme pfam00364 100/5.00e-12Propionyl-CoA carboxylase beta
subunit (PCCB) NP000523.1WP:CE07269 F52E4.1 pccb-1
95.3/1.4e-212 Carboxyl transferase domain pfam01039 100/0.00e-00
Methylmalonyl-CoA Epimerase (MCE) NP115990.2
WP:CE09082 D2030.5 mce-1
79.6/2.3e-45 Glyoxalase pfam00903 100/7.00e-17
Methylmalonyl-CoA Mutase (MUT) NP000246.1
WP:CE30404 ZK1058.1 mmcm-1
97.8/7.29e-280 Methylmalonyl-CoA mutase B12 binding domain cd02071
100/5.00e-43
Methylmalonyl-CoA mutase pfam01642 100/0.00e-00Methylmalonic aciduria Type A
(MMAA) NP758454.1WP:CE31822 T02G5.13 mmaa-1
82.5/1.2e-89 ArgK protein pfam03308 100/3.00e-105
Methylmalonic aciduria Type B (MMAB-1) NP443077.1
WP:CE01159 C26E6.11 mmab-1
53.7/5.7e-35 Cobalamin adenosyltransferase pfam01923 97.6/4.00e-50
MMACHC [35] WP:CE34461 ZK546.17 cblc-1
52/5e-31 None proven

R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73 67
were cloned into pCR 2.1 TOPO vector (Invitrogen, Carlsbad, CA) usingTA cloning. Clones were screened by PCR and positive clones weresequenced.
Isolation of break point sequences in C. elegans mutants
DNA was isolated from C. elegans using DNeasy Tissue Kit (QiagenInc., Valencia, CA). The genomic DNA from each mutant underwent PCRusing the inner left and inner right primers designed by the C. elegansKnockout Project at OMRF for mutant screening. PCR products werecloned into pCR 2.1 TOPO vector (Invitrogen, Carlsbad, CA) using TAcloning. Five clones from each mutant strain were then sequenced andcompared to the genomic DNA sequence to determine the site of the dele-tion. The sequences have been deposited in WormBase under the respec-tive gene entries.
RNA interference
mmcm-1, mmaa-1, mmab-1, pcca-1, pccb-1 and mce-1 were cloned intothe RNAi vector pPD129.36 and transformed into E. coli strain OP50 [22].Feeding RNAi was performed as described [23]. The strains were grownovernight from individual colonies in LB with 100 !g/ml of ampicillin at37 °C. One milliliter of overnight culture was then placed in 30 ml of LBwith 100 !g/ml of ampicillin and grown to OD of 0.4 absorbance units at595 nm. The culture was induced with 0.4 mM IPTG for an additional 4 h.Bacteria were concentrated by centrifugation and plated on 100 mm NGMagar plate with 12.5 !g/ml of tetracycline, 100 !g/ml of ampicillin and1 mM IPTG. Strains were grown overnight at room temperature. The fol-lowing day two 100 mM plates per strain were seeded with C. elegans eggsprepared using standard methods. Nematodes were grown at 20 °C untilplates were cleared.
Loading studies
Nematodes were washed from plates (two plates per strain) after feed-ing RNAi with 6 ml phosphate buVered saline (PBS) and puriWed via asucrose gradient (30% sucrose in ultra-pure water). The animals wereplaced in 1.5 ml centrifuge tubes and suspended with 5 mM of sodium pro-pionate (Sigma) in 500 !l PBS with 50 !g/ml kanamycin incubated at 20 °Cfor 48 h. The worms were centrifuged and the supernatant was removedfor GC/MS analysis. Metabolite levels were normalized to protein concen-tration of the pellet or wet weight.
Gas chromatography/mass spectrometry (GC/MS)
Methylmalonic acid concentrations were determined using capillarygas chromatography–mass spectroscopy with stable isotopic internal cali-bration as described [24]. In some experiments, organic acids were puriWedafter perchloric acid precipitation of macromolecules and anion exchangecolumn chromatography [25].
Mitochondrial preparations
Isolation of intact mitochondria from C. elegans was performed as pre-viously described [26], with the modiWcations detailed below, to obtainwell-functioning mitochondria. A total of 1 to 3 g of freshly washed adultworms were washed twice with 10 ml MSM-E [150 ml MSM (40.08 g man-nitol, 23.96 g sucrose, 1.047 g MOPS, 3 ml 0.1 M EDTA, pH 7.4, per liter)]and re-suspended by centrifugation (300£ g, 10 min, 4 °C). A glass Potter/Elvehjem tissue grinder with a TeXon (DuPont, Wilmington, DE) pestle(Wheaton, Millville, NJ) was then used for initial rupture: 3 slow strokesat 400 rpm. Proteinase subtilisin A (Sigma, MO) was added to the homog-enate (5 mg per gram of worms) and stirred for 10 min on ice. Immediatelyafterward, the slurry was homogenized in the same apparatus as before,with 10 slow strokes at 400 rpm. An equal volume of MSM-EB (50 mlMSM-E + 0.2 g defatted bovine serum albumin) was added and thehomogenate was centrifuged (300£ g, 10 min, 4 °C). The supernatant was
pipetted into a fresh centrifuge tube, while the pellet was re-extracted inthe same manner as described above. Centrifugation (7000£ g, 10 min,4 °C) of the supernatants from both the original and re-extracted homoge-nizations yielded mitochondrial pellets, which were re-suspended in 10 mlMSM-E, centrifuged (7000£g, 10 min, 4 °C), and then re-suspended in10 ml MSM, and centrifuged as before. The Wnal mitochondrial pellet wasre-suspended in approximately 100 !l of MSM (per »2 g of fresh-washedadult worm pellet initially used). Total protein concentration was deter-mined by the Lowry assay with bovine serum albumin (0–100 !g) used asstandard [27]. Two microliters of protease inhibitor cocktail (P8340,Sigma, MO) were added per 100 !l mitochondrial preparation. Oxidativephosphorylation assays on the puriWed mitochondrial preparations veri-Wed functional activity. Isolated mitochondria were frozen at ¡80 °C priorto enzymatic analyses.
Expression of C. elegans mmcm-1 in yeast
A sequence veriWed, full-length C. elegans mmcm-1 cDNA was clonedinto the pYES-DEST52 Gateway vector (Invitrogen, Carlsbad, CA) andtransformed into strain INVSC1. The vector carries a URA selectionmarker and uses the GAL4 promoter to drive expression of the insert.Expression was induced after growth in galactose containing media andconWrmed by comparison to a LacZ expression control and enzymaticassay for the methylmalonyl-CoA mutase gene product. Lysates wereprepared by homogenization with acid-washed glass beads and subse-quent centrifugation and were used for enzyme kinetic assays asdescribed [23].
Enzymatic assays and kinetics
Methylmalonyl-CoA mutase activity was assayed using a radioactivesuccinate thiokinase-linked assay as described [28]. The assay mixture con-tained: 95 !l of D/L-[methyl-14C]methylmalonyl-CoA (150 !Ci per mmol),1 mM in 3 mM HCl], 45 !l of Tris-HCl buVer, 0.5 M, pH 9, 45 !l MgCl2,0.1 M, 45 !l GDP, 20 mM, 50 !l adenosylcobalamin (variable concentra-tion), 20 !l of succinate-CoA ligase (succinyl thiokinase) prepared at5 units/ml, yeast cell lysate or mitochondrial preparation extract (variablevolume) and water to bring the volume to 450 !l. The tubes were incubatedin a 30 °C dry block for 30 min (in dark). The reactions were stopped byacidiWcation and extracted with ethylacetate. An aliquot was then countedby liquid scintillography to determine activity. The wild-type murineenzyme, expressed in an identical fashion in yeast, served as a control forthe kinetic studies. Assays were performed in triplicate for all concentra-tion points and the kinetics repeated twice. Kinetic constants were deter-mined by nonlinear regression modeling of the kinetic data and regressionstatistics were calculated as previously described [29].
Lentiviral correction
The C. elegans mmcm-1 cDNA was cloned into pLenti6/V5-DEST(Invitrogen, Carlsbad, CA) using Gateway LR Clonase enzyme mix (Invit-rogen, Carlsbad, CA). A replication-incompetent, HIV-based lentiviruswas generated by using the ViraPower Lentiviral Expression System(Invitrogen, Carlsbad, CA). The viral construct has the mmcm-1 genedriven by the CMV promoter; the backbone also has a blasticidin cassettedriven by the E7 promoter. A control virus expressing EGFP was also pre-pared and used in parallel for correction experiments. Human muto Wbro-blast cell lines were transduced with virus containing either the C. elegansmmcm-1 or EGFP. The transduced cells were selected and expanded inDMEM with 5% fetal bovine serum containing 2.5 !g/ml blasticidin priorto 1-[14C]-propionate propionate incorporation assays.
Results
Protein Blast searches of human proteins involved inpropionate metabolism against WormBase identiWed
68 R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73
unique worm homologues for each gene product (Table 1).The homologues have been named in accordance with C.elegans nomenclature (http://biosci.umn.edu/CGC/Nomen-clature/nomenguid.htm). The high degree of similarity sug-gests that these proteins are functionally related. MUT,PCCB, PCCA, MMAA, MCEE, MMAB and MMAHChave similarities of 97.8, 95.3, 94.6, 82.5, 79.6 53.7% and 52%with the worm homologues (Table 1). The functionaldomains of the predicted protein products were also highlyconserved between species. This would be expected forMUT, PCCA and PCCB because of the high degree of sim-ilarity seen at the amino acid level. MMAA, MCEE andMMAB, which had a lower degree of similarity, also hadconservation of all known domains between species. TheC. elegans mmab-1 gene product only has a similarity of53.7%, but the adenosylcobalamin transferase domain ofthis protein displays 97.6% similarity with the same domainin the human enzyme. Such protein domain conservationmay be more indicative of proteins with similar functionthan a simple residue alignment. The high degree of similar-ity and domain conservation between the human enzymesinvolved in propionate metabolism and C. elegans proteinswould seem to be unlikely, unless enzymatic function wasconserved. As a secondary conWrmation that cobalaminmetabolic pathways might be intact in C. elegans, weperformed similar searches to identify genes that mightparticipate in methylcobalamin metabolism. Single homo-logues of methionine synthase (5-methyltetrahydrofolate–homocysteine methyltransferase) and methionine synthasereductase (MSR) appear to be present with a similar degreeof protein and domain conservation; WP: CE01609 is 80%similar to human methionine synthase (Blast e-value D 0.0)and WP: CE00868 is 50% similar to methionine synthase
reductase (Blast e-value D 1.5e-75). However, homologuesof transcobalamin I, transcobalamin II, intrinsic factor, andthe E. coli periplasmic binding protein btuF were notdetected. Queries with cubilin and megalin produced a non-speciWc alignment to a number of C. elegans genes in theEGF-like domains of these genes.
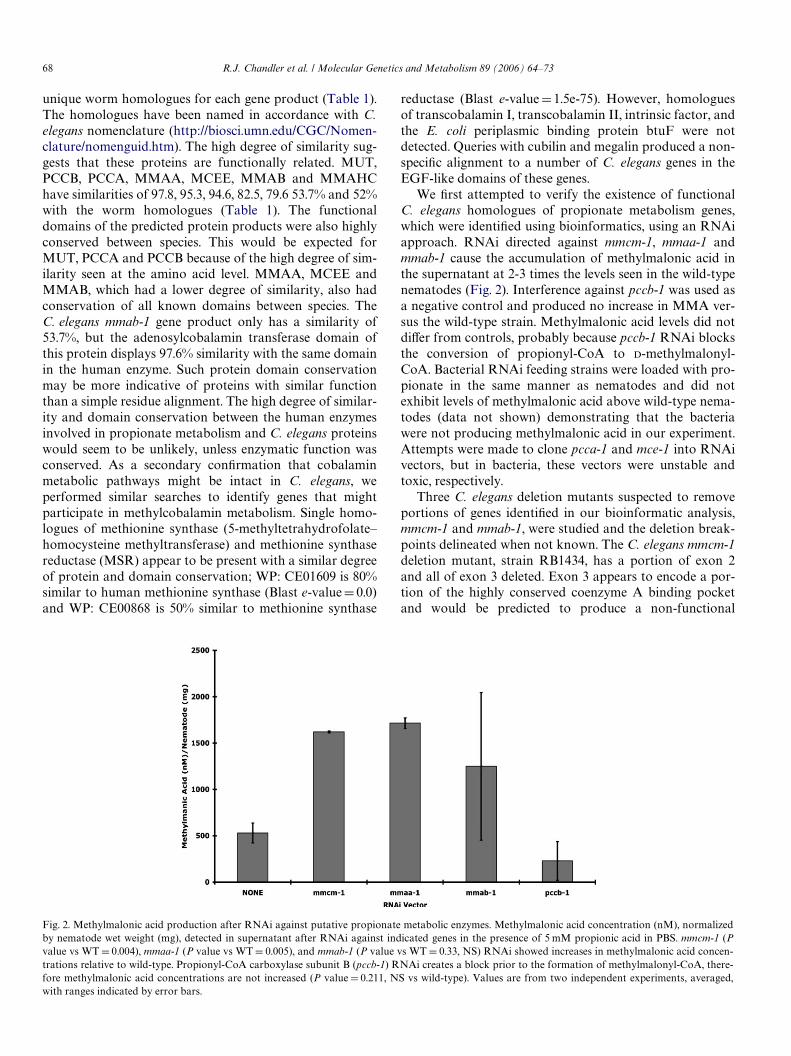
We Wrst attempted to verify the existence of functionalC. elegans homologues of propionate metabolism genes,which were identiWed using bioinformatics, using an RNAiapproach. RNAi directed against mmcm-1, mmaa-1 andmmab-1 cause the accumulation of methylmalonic acid inthe supernatant at 2-3 times the levels seen in the wild-typenematodes (Fig. 2). Interference against pccb-1 was used asa negative control and produced no increase in MMA ver-sus the wild-type strain. Methylmalonic acid levels did notdiVer from controls, probably because pccb-1 RNAi blocksthe conversion of propionyl-CoA to D-methylmalonyl-CoA. Bacterial RNAi feeding strains were loaded with pro-pionate in the same manner as nematodes and did notexhibit levels of methylmalonic acid above wild-type nema-todes (data not shown) demonstrating that the bacteriawere not producing methylmalonic acid in our experiment.Attempts were made to clone pcca-1 and mce-1 into RNAivectors, but in bacteria, these vectors were unstable andtoxic, respectively.
Three C. elegans deletion mutants suspected to removeportions of genes identiWed in our bioinformatic analysis,mmcm-1 and mmab-1, were studied and the deletion break-points delineated when not known. The C. elegans mmcm-1deletion mutant, strain RB1434, has a portion of exon 2and all of exon 3 deleted. Exon 3 appears to encode a por-tion of the highly conserved coenzyme A binding pocketand would be predicted to produce a non-functional
Fig. 2. Methylmalonic acid production after RNAi against putative propionate metabolic enzymes. Methylmalonic acid concentration (nM), normalizedby nematode wet weight (mg), detected in supernatant after RNAi against indicated genes in the presence of 5 mM propionic acid in PBS. mmcm-1 (Pvalue vs WT D 0.004), mmaa-1 (P value vs WT D 0.005), and mmab-1 (P value vs WT D 0.33, NS) RNAi showed increases in methylmalonic acid concen-trations relative to wild-type. Propionyl-CoA carboxylase subunit B (pccb-1) RNAi creates a block prior to the formation of methylmalonyl-CoA, there-fore methylmalonic acid concentrations are not increased (P value D 0.211, NS vs wild-type). Values are from two independent experiments, averaged,with ranges indicated by error bars.

R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73 69
protein. RT-PCR performed on mmcm-1 deletion mutantsdid not detect mRNA (data not shown). Based on the RT-PCR results, which showed no mmcm-1 RNA transcript,and the neonatal lethal phenotype observed in mice with asimilar deletion, the C. elegans mmcm-1 mutant would bepredicted to have no enzymatic activity. Strain RB 1347harbors a deletion of the Wrst exon of the putative mmab-1gene. The second exon of mmab-1 begins with a start codonand expression studies to characterize the RNA made bythis mutant were not pursued. The deletion of mmab-1 har-bored in strain VC974 has been described by the generatinglaboratory and appears to remove the C-terminal exon thathas high homology with the adenosyltransferase domain.Strain RB512 has previously been shown to harbor a dele-tion of the mce-1 gene [15]. Propionate incorporation andmetabolic analysis of the mce-1 mutant have not been pre-viously described.
In order to verify the phenotypes observed in our RNAitreated animals, we conducted 1-[14C]-propionate propionateincorporation studies on the C. elegans deletion mutants.1-[14C]-propionate incorporation studies, routinely used tostudy human mutant cell lines [11,12], were adapted to detectblocks in propionate metabolism of C. elegans as describedabove. Radioactive incorporation studies with the N2 strainshowed that C. elegans incorporates 1-[14C]-propionate frompropionate into protein in amounts similar to those seen inhuman cells [11§4 nmols/mg/18h] [30] (Fig. 3). Further-more, a robust stimulation of incorporation was observedwhen hydroxycobalamin (OH-Cbl) was added to themedium in excess. C. elegans mmcm-1, mmab-1 and mce-1deletion mutants all exhibited a diminished ability to incor-porate 1-[14C]-propionate into protein as compared towild-type nematodes (Fig. 3). The N-terminal deletionmutant mmab-1(ok1493) displayed a less severe block than
the C-terminal mutant mmab-1(ok1484), possibly because theC-terminal deletion removes a domain critical to mmab-1function as the sequence analysis would suggest. The mce-1,mmcm-1, and mmab-1(ok1484) mutant animals exhibiteda> 80% decrease in the ability to incorporate propionate rel-ative to wild-type animal. A similar decrease in 1-[14C]-propi-onate Xux can been seen in human muto Wbroblasts whencompared to wild-type controls.
To assess the biochemical response to precursor loading,we incubated the N2, mmcm-1, mmab-1 (ok1484, ok1493)and mce-1 mutant strains with propionic acid and mea-sured methylmalonic acid in the supernatant (Fig. 4). Pre-liminary studies suggested methylmalonic acid was easilydetected in the supernatant, which ranged in concentrationfrom 0.5–10!M and was present at higher concentrationsthan in whole cell worm extracts. While both the wild-typeand mmcm-1 animals produce an increased amount ofmethylmalonic acid after loading, the mmcm-1 mutantaccumulated 17 times more methylmalonic acid than thewild-type strain (Fig. 4). The mmab-1(ok1484, ok1493) andmce-1 mutant strains show approximately a two-foldincrease in the accumulation of methylmalonic acid in com-parison to wild-type animals. Kanamycin treatment pro-vided a bactericidal eVect on the residual feeding strain toassure bacteria had no eVect on metabolite measurements;growth assays of the bacteria that were incubated for 24 hin kanamycin showed uniform and complete sensitivity andfailed to incorporate C14 propionate or produce metabo-lites. Furthermore, metabolic measurements of the superna-tant from the wash steps of puriWcation, before and afterpropionic acid loading indicating that the bacteria were notproducing MMA (data not shown).
Attempts to demonstrate methylmalonyl-CoA mutaseenzyme activity in crude worm extracts were unsuccessful.
Fig. 3. 1-[14C]-propionate incorporation in C. elegans deletion mutants. 1-[14C]-propionate incorporation of methylmalonyl-CoA mutase mmcm-1(ok1637), methylmalonyl-CoA epimerase mce-1(ok243), and mmab-1 mutants mmab-1(ok1493) and mmab-1(ok1484) expressed as a percentage of themean propionate incorporation of wild-type (WT D 6.8 nanomoles propionate/mg protein/24 h). Values are averages from 6 (N2, mcmm-1), 5 (mce-1) and5 (mmab-1) independent experiments with triplicate or quadruplicate measurements of each sample. Error bars represent mean § SD. P-values vs wild-type: mmcm-1(ok1637) D 2.9 E¡22; mce-1(ok243) D 2.6 E¡19; mmab-1(ok1493) D 0.008; mmab-1(ok1484) D 1.4 E¡08; N2 plus OHcbl D 2.03 E¡06.
70 R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73
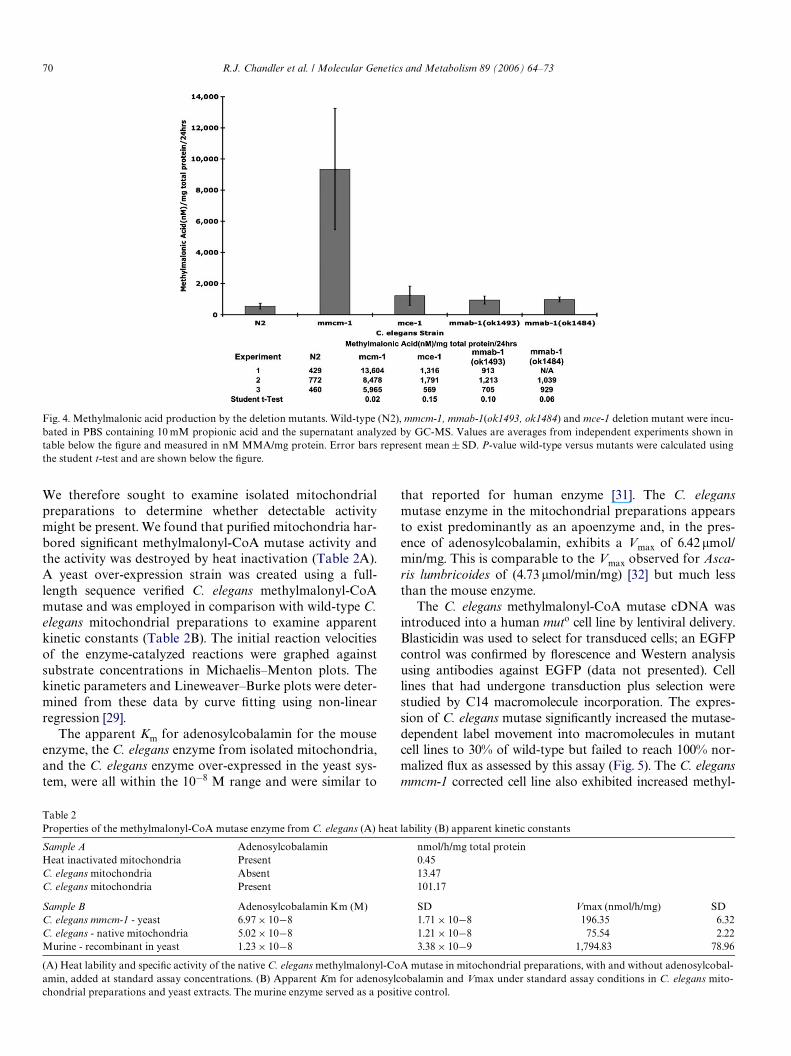
We therefore sought to examine isolated mitochondrialpreparations to determine whether detectable activitymight be present. We found that puriWed mitochondria har-bored signiWcant methylmalonyl-CoA mutase activity andthe activity was destroyed by heat inactivation (Table 2A).A yeast over-expression strain was created using a full-length sequence veriWed C. elegans methylmalonyl-CoAmutase and was employed in comparison with wild-type C.elegans mitochondrial preparations to examine apparentkinetic constants (Table 2B). The initial reaction velocitiesof the enzyme-catalyzed reactions were graphed againstsubstrate concentrations in Michaelis–Menton plots. Thekinetic parameters and Lineweaver–Burke plots were deter-mined from these data by curve Wtting using non-linearregression [29].
The apparent Km for adenosylcobalamin for the mouseenzyme, the C. elegans enzyme from isolated mitochondria,and the C. elegans enzyme over-expressed in the yeast sys-tem, were all within the 10¡8 M range and were similar to
that reported for human enzyme [31]. The C. elegansmutase enzyme in the mitochondrial preparations appearsto exist predominantly as an apoenzyme and, in the pres-ence of adenosylcobalamin, exhibits a Vmax of 6.42 !mol/min/mg. This is comparable to the Vmax observed for Asca-ris lumbricoides of (4.73 !mol/min/mg) [32] but much lessthan the mouse enzyme.
The C. elegans methylmalonyl-CoA mutase cDNA wasintroduced into a human muto cell line by lentiviral delivery.Blasticidin was used to select for transduced cells; an EGFPcontrol was conWrmed by Xorescence and Western analysisusing antibodies against EGFP (data not presented). Celllines that had undergone transduction plus selection werestudied by C14 macromolecule incorporation. The expres-sion of C. elegans mutase signiWcantly increased the mutase-dependent label movement into macromolecules in mutantcell lines to 30% of wild-type but failed to reach 100% nor-malized Xux as assessed by this assay (Fig. 5). The C. elegansmmcm-1 corrected cell line also exhibited increased methyl-
Fig. 4. Methylmalonic acid production by the deletion mutants. Wild-type (N2), mmcm-1, mmab-1(ok1493, ok1484) and mce-1 deletion mutant were incu-bated in PBS containing 10 mM propionic acid and the supernatant analyzed by GC-MS. Values are averages from independent experiments shown intable below the Wgure and measured in nM MMA/mg protein. Error bars represent mean § SD. P-value wild-type versus mutants were calculated usingthe student t-test and are shown below the Wgure.
Table 2Properties of the methylmalonyl-CoA mutase enzyme from C. elegans (A) heat lability (B) apparent kinetic constants
(A) Heat lability and speciWc activity of the native C. elegans methylmalonyl-CoA mutase in mitochondrial preparations, with and without adenosylcobal-amin, added at standard assay concentrations. (B) Apparent Km for adenosylcobalamin and Vmax under standard assay conditions in C. elegans mito-chondrial preparations and yeast extracts. The murine enzyme served as a positive control.
Sample A Adenosylcobalamin nmol/h/mg total proteinHeat inactivated mitochondria Present 0.45C. elegans mitochondria Absent 13.47C. elegans mitochondria Present 101.17
Sample B Adenosylcobalamin Km (M) SD Vmax (nmol/h/mg) SDC. elegans mmcm-1 - yeast 6.97 £ 10¡8 1.71 £ 10¡8 196.35 6.32C. elegans - native mitochondria 5.02 £ 10¡8 1.21 £ 10¡8 75.54 2.22Murine - recombinant in yeast 1.23 £ 10¡8 3.38 £ 10¡9 1,794.83 78.96

R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73 71
malonyl-CoA mutase dependent label movement after sup-plementation with hydroxycobalamin, a precursor of theactive co-factor, adenosylcobalamin.
Discussion
In these studies, we have used genetic, genomic and bio-chemical methods to establish the existence of an adeno-sylcobalamin-dependent methylmalonyl-CoA mutase in C.elegans. The conservation and extreme similarity between thevertebrate and predicted worm enzymes suggests that theentire propionyl-CoA to succinyl-CoA pathway depicted inFig. 1 is functionally intact in C. elegans. Our Wndings dem-onstrate that native C. elegans mitochondria do harbor anactive mutase that is predominantly apoenzyme in constitu-tion. The observed kinetic constants of the nematode methyl-malonyl-CoA mutase show high aYnity adenosylcobalaminbinding, as seen in vertebrate mutases and a Vmax thatapproximates what has been seen in other organisms. ThesigniWcant fraction of apoenzyme versus holoenzymeobserved in our mitochondrial preparations suggests a mod-ulation of or dependence upon cobalamin in the environmentor the food source. The accentuation of activity in wholeworm incorporation experiments after incubation in thepresence of hydroxycobalamin further demonstrates the exis-tence of adenosylcobalamin synthetic capacity in vivo andsuggests that propionate metabolism can be greatly increasedwhen the environment is cobalamin-replete. The mechanismsfor extra-cellular cobalamin uptake and transport areunclear from a bioinformatics analysis as the extracellularproteins known to participate in these processes do notappear to have C. elegans homologues. However, the synthe-sis of methylcobalamin seems likely, given the presence ofgene products that are highly similar to human vitaminB12—dependent methionine synthase and methionine syn-thase reductase. Future studies, perhaps similar to the ones
described herein, will be required to examine the functional-ity and nature of these enzymatic pathways.
A number of other lines of experimental evidence indi-cate the presence of an adenosylcobalamin metabolic path-way in C. elegans when grown under laboratory conditions.The mmcm-1 and mmab-1 deletion mutants have dimin-ished 1-[14C]-propionate incorporation compared to wild-type animals, indicating that downstream movement of thelabel from 1-[14C]-propionate into protein is impaired, pre-sumably at the step of conversion of methylmalonyl-CoAto succinyl-CoA. These metabolic blocks are consistentwith a functional defect in adenosylcobalamin synthesis inthe case of the mmab-1 mutants, and impairment in apoen-zyme synthesis in the case of the mmcm-1 deletion mutant.The deletion in the mmab-1(ok1493) mutant removes theWrst exon, which may contain an inverted repeat. The possi-bility of an internal translation product from the deletedallele with residual activity is feasible given the fact that thesecond exon begins with a start codon. The other mutantsall display severe blocks and appear to have similar activityin this assay, consistent with the proposed roles for the geneproducts in methylmalonyl-CoA metabolism.
The mmcm-1 mutant exhibits increased methylma-lonic acid production, which is accentuated in the face ofprecursor loading. The mutant consistently producedgreater than tenfold more methylmalonic acid than theN2 strain (P value D 0.02), but exhibits variabilitybetween experiments, which explains the larger standarddeviation observed in this strain versus N2. The mce-1and mmab-1 deletion mutant strains appear to havedecreased viability in propionate-containing medium butconsistently produce more methylmalonic acid than theN2 strain. Preliminary observations indicate diminishedsurvival by these strains under propionate loading, whichlikely explains the milder increases in metabolite produc-tion by these mutants, especially since the mutants show
Fig. 5. Lentiviral correction of a mutant human cell line with C. elegans methylmalonyl-CoA mutase. A full-length C. elegans mmcm-1 cDNA carried inpLENTI6 was used to transduce a human muto Wbroblast cell line. 1-[14C]-propionate incorporation was measured in triplicate and expressed as a percent-age of the mean propionate incorporation of wild-type (5.2 nanomoles 1-[14C]-propionate/mg protein/18 h). Error bars represent mean § SD. P-values:wild-type vs muto D 0.001, muto vs muto plus mmcm-1 D 0.0078; muto plus OHcbl vs muto plus mmcm-1 plus OH-cbl D 0.0001.

72 R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73
display a severe impairment in the C14 incorporationassays. The results show that the C. elegans system mightbe used to examine inducers and regulators of the meth-ylmalonyl-CoA mutase reaction by virtue of metaboliteproduction.
A surprising Wnding was that the mce-1 mutant had asevere block in C14 propionate incorporation. Althoughmutations in MUT, MMAA and MMAB have been shownto cause methylmalonic acidemia in humans, MCEE deW-
ciency has not yet been proven to cause methylmalonic aci-demia [33]. Montgomery et al. [13] administered a numberof methylmalonic acid isotopomers to vitamin B12 deWcientrats and analyzed methylmalonic acid output by mass spec-trometry. They noted a rearrangement of label in the urineafter peritoneal injection of methylmalonic acid into thecobalamin-deWcient rats, which was not observed in controlanimals. To explain the label distribution observed, theyposited the existence of a “free” methylmalonic acid shunt,which was able to bypass the epimerase reaction and pro-posed that this mechanism protected patients with MCEElesions from developing methylmalonic acidemia. Theresults we have observed are not entirely consistent with theexistence of a bypass in C. elegans because the mce-1 dele-tion mutant exhibits a block as severe as the C. elegansMCM mutant and produces increased methylmalonic acid.If a free-shunt was functionally operational over the physi-ological range of methylmalonyl-CoA concentrations,selective pressure on the MCEE gene might be lessened orremoved and conservation between species would not bepredicted. Mutations of MCEE may result in either a mildor an embryonic lethal phenotype in humans, which couldgo undetected. If a free MMA metabolic shunt exists in C.elegans, it appears to be incapable of handling the normalXux through this pathway as assessed in the assays we haveperformed. Isotopomer studies using the mutant strainsmay be useful to further examine this issue. The resultsobserved with the C. elegans mce-1 deletion mutant suggestthat humans with functional methylmalonyl-CoA epimer-ase lesions will have methylmalonic acidemia and cell linesfrom these patients will exhibit a block in 1-[14C]-propio-nate incorporation. The identiWcation and careful clinicalcharacterization of patients harboring suspected MCEEmutations will be required to resolve this issue.
We also demonstrated that the C. elegans mutase canpartially correct Wbroblasts derived from muto humanpatients. The level of correction was signiWcantly increasedover the mutant background and GFP in all instances. Thecorrected human cell line displayed similar intermediatecorrection and could be stimulated by hydroxycobalaminaddition to the media. The reason for a partial correction ofthe human cell line is uncertain, but may relate to the spe-cialized mitochondrial importation sequences found at theN-terminal end of the C. elegans gene. Other mitochondrialenzymes have been shown to have a limited cross-speciescorrection for this reason, even when the species barrier isrelatively close, such as between human and mouse [34]. IfC. elegans mitochondrial localized genes are to be used for
cross-species correction of mutant cell lines, for example, toidentify unknown cobalamin disease genes or disease genemodiWers, future studies will be required to determine opti-mum expression parameters.
We have documented that this model organism can beused to study the pathophysiology of human methylmalonicacidemia by demonstrating that the mce-1 mutant has afunctional deWcit in 1-[14C]-propionate incorporation andpredict, based on these results, that the human homolog willcause methylmalonic acidemia in humans when mutated.The metabolic lesions generate a chemical phenotype thatmight be useful if RNAi or targeted deletion mutant analy-sis could be coupled to metabolic proWling. Such an analysiscould identify mutants that produced increased amounts ofmethylmalonic acid and/or reduced 1-[14C]-propionateincorporation, and might identify new genes involved inintracellular cobalamin and propionyl-CoA metabolism,some of which may have human homologues that aremutated in patients with propionic acidemia, methylmalonicacidemia and/or homocysteinemia. The experiments pre-sented here also demonstrate that sensitive mass spectrome-try measurements can be performed on small numbers ofanimals to determine chemical phenotypes and suggest thatmetabolic proWling will be useful to study C. elegans RNAiand deletion mutants in other pathways.
Acknowledgments
This research was supported, in part, by the IntramuralResearch Program of the National Human GenomeResearch Institute, National Institutes of Health. N.W. andM.T. were supported, in part, by the MMA Research Foun-dation. The authors are grateful for the help and adviceoVered by Meera Sundaram and Robyn BarWeld, Depart-ment of Genetics, University of Pennsylvania School ofMedicine in the early stages of this project; to the OMRFfor the isolation of the mmcm-1 and mmab-1 deletionmutants and to Theresa Stiernagle, Caenorhabditis Genet-ics Center of the University of Minnesota for the provisionof C. elegans strains. M.F. and M.S were supported, in part,by NIH Grant #GM58881.
References
[1] W.A. Fenton, R.A. Gravel, D.S. Rosenblatt, Disorders of propionateand methylmalonate metabolism, in: C.R. Scriver, A.L. Beaudet, W.S.Sly, D. Valle (Eds.), The Metabolic and Molecular Bases for Inhert-ited Disease, McGraw-Hill, New York, 2001, pp. 2165–2192.
[2] E.R. Baumgarter, C. Viardot, Long-term follow-up of 77 patients with iso-lated methylmalonic acidaemia, J. Inherit. Metab. Dis. 18 (1995) 138–142.
[3] H.O. de Baulny, J.F. Benoist, O. Rigal, G. Touati, D. Rabier, J.M. Sau-dubray, Methylmalonic and propionic acidaemias: management andoutcome, J. Inherit. Metab. Dis. 28 (2005) 415–423.
[4] S.M. Matsui, M.J. Mahoney, L.E. Rosenberg, The natural history of theinherited methylmalonic acidemias, N. Engl. J. Med. 308 (1983) 857–861.
[5] S.B. van der Meer, F. Poggi, M. Spada, J.P. Bonnefont, H. Ogier, P.Hubert, E. Depondt, D. Rapoport, D. Rabier, C. Charpentier, et al., Clini-cal outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia, J. Pediatr. 125 (1994) 903–908.

R.J. Chandler et al. / Molecular Genetics and Metabolism 89 (2006) 64–73 73
[6] C.M. Dobson, T. Wai, D. Leclerc, A. Wilson, X. Wu, C. Dore, T.Hudson, D.S. Rosenblatt, R.A. Gravel, IdentiWcation of the generesponsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokary-otic gene arrangements, Proc. Natl. Acad. Sci. USA 99 (2002)15554–15559.
[7] C.M. Dobson, T. Wai, D. Leclerc, H. Kadir, M. Narang, J.P. Lerner-Ellis, T.J. Hudson, D.S. Rosenblatt, R.A. Gravel, IdentiWcation of thegene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria, Hum. Mol. Genet. 11 (2002)3361–3369.
[8] T. Suormala, M.R. Baumgartner, D. Coelho, P. Zavadakova, V. Koz-ich, H.G. Koch, M. Berghauser, J.E. Wraith, A. Burlina, A. Sewell, J.Herwig, B. Fowler, The cblD defect causes either isolated or com-bined deWciency of methylcobalamin and adenosylcobalamin synthe-sis, J. Biol. Chem. 279 (2004) 42742–42749.
[9] N. Korotkova, M.E. Lidstrom, MeaB is a component of the methyl-malonyl-CoA mutase complex required for protection of the enzymefrom inactivation, J. Biol. Chem. 279 (2004) 13652–13658.
[10] N.A. Leal, S.D. Park, P.E. Kima, T.A. Bobik, IdentiWcation of thehuman and bovine ATP:Cob(I)alamin adenosyltransferase cDNAsbased on complementation of a bacterial mutant, J. Biol. Chem. 278(2003) 9227–9234.
[11] H.F. Willard, L.M. Ambani, A.C. Hart, M.J. Mahoney, L.E. Rosen-berg, Rapid prenatal and postnatal detection of inborn errors of pro-pionate, methylmalonate, and cobalamin metabolism: a sensitiveassay using cultured cells, Hum. Genet. 34 (1976) 277–283.
[12] G.D. Morrow, B. Revsin, C. Mathews, H. Giles, A simple, rapidmethod for prenatal detection of defects in propionate metabolism,Clin. Genet. 10 (1976) 218–221.
[13] J.A. Montgomery, O.A. Mamer, C.R. Scriver, Metabolism ofmethylmalonic acid in rats. Is methylmalonyl-coenzyme a race-mase deWciency symptomatic in man? J. Clin. Invest. 72 (1983)1937–1947.
[14] T.A. Bobik, M.E. Rasche, IdentiWcation of the human methylmalonyl-CoA racemase gene based on the analysis of prokaryotic genearrangements. Implications for decoding the human genome, J. Biol.Chem. 276 (2001) 37194–37198.
[15] J. Kuhnl, T. Bobik, J.B. Procter, C. Burmeister, J. Hoppner, I. Wilde,K. Luersen, A.E. Torda, R.D. Walter, E. Liebau, Functional analysisof the methylmalonyl-CoA epimerase from Caenorhabditis elegans,Febs. J. 272 (2005) 1465–1477.
[16] E.P. Brass, A.G. Tahiliani, R.H. Allen, S.P. Stabler, Coenzyme A metab-olism in vitamin B-12-deWcient rats, J. Nutr. 120 (1990) 290–297.
[17] S.P. Stabler, E.P. Brass, P.D. Marcell, R.H. Allen, Inhibition of cobala-min-dependent enzymes by cobalamin analogues in rats, J. Clin.Invest. 87 (1991) 1422–1430.
[18] H. Peters, M. Nefedov, J. Sarsero, J. Pitt, K.J. Fowler, S. Gazeas, S.G.Kahler, P.A. Ioannou, A knock-out mouse model for methylmalonicaciduria resulting in neonatal lethality, J. Biol. Chem. 278 (2003)52909–52913.
[19] R.J. Chandler, C.P. Venditti, Genetic and genomic systems to studymethylmalonic acidemia, Mol. Genet. Metab. 86 (2005) 34–43.
[20] J.T. Pronk, A. van der Linden-Beuman, C. Verduyn, W.A. ScheVers,J.P. van Dijken, Propionate metabolism in Saccharomyces cerevisiae:implications for the metabolon hypothesis, Microbiology 140 (Pt 4)(1994) 717–722.
[21] S. Brenner, The genetics of Caenorhabditis elegans, Genetics 77(1974) 71–94.
[22] L. Timmons, D.L. Court, A. Fire, Ingestion of bacterially expresseddsRNAs can produce speciWc and potent genetic interference in Cae-norhabditis elegans, Gene 263 (2001) 103–112.
[23] E. Andrews, R. Jansen, A.M. Crane, S. Cholin, D. McDonnell, F.D.Ledley, Expression of recombinant human methylmalonyl-CoAmutase: in primary mut Wbroblasts and Saccharomyces cerevisiae,Biochem. Med. Metab. B. 50 (1993) 135–144.
[24] R.H. Allen, S.P. Stabler, D.G. Savage, J. Lindenbaum, Elevation of 2-methylcitric acid I and II levels in serum, urine, and cerebrospinal Xuidof patients with cobalamin deWciency, Metabolism 42 (1993) 978–988.
[25] R.L. Foll, A. Pleyers, G.J. Lewandovski, C. Wermter, V. Hegemann,R.J. Paul, Anaerobiosis in the nematode Caenorhabditis elegans,Comp. Biochem. Phys. B. 124 (1999) 269–280.
[26] E.B. Kayser, P.G. Morgan, C.L. Hoppel, M.M. Sedensky, Mitochon-drial expression and function of GAS-1 in Caenorhabditis elegans, J.Biol. Chem. 276 (2001) 20551–20558.
[27] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Protein measure-ment with the Folin phenol reagent, J. Biol. Chem. 193 (1951) 265–275.
[28]J.F. Kolhouse, S.P. Stabler, R.H. Allen, L-methylmalonyl-CoA mutasefrom human placenta, Methods Enzymol. 166 (1988) 407–414.
[29] E. Billo, Excel for Chemists: A Comphrensive Guide, John Wiley andSons, 2001.
[30] L.C. Worgan, K. Niles, J.C. Tirone, A. Hofmann, A. Verner, A. Sam-mak, T. Kucic, P. Lepage, D.S. Rosenblatt, Spectrum of mutations inmut methylmalonic acidemia and identiWcation of a common his-panic mutation and haplotype, Hum. Mutat. 27 (2006) 31–43.
[31] J. Janata, N. Kogekar, W.A. Fenton, Expression and kinetic charac-terization of methylmalonyl-CoA mutase from patients with the mut-phenotype: evidence for naturally occurring interallelic complementa-tion, Hum. Mol. Genet. 6 (1997) 1457–1464.
[32] Y.S. Han, J.M. Bratt, H.P. Hogenkamp, PuriWcation and characteriza-tion of methylmalonyl-CoA mutase from Ascaris lumbricoides,Comp. Biochem. Phys. B. 78 (1984) 41–45.
[33] E.S. Kang, P.J. Snodgrass, P.S. Gerald, Methylmalonyl coenzyme Aracemase defect: another cause of methylmalonic aciduria, Pediatr.Res. 6 (1972) 875–879.
[34] X. Ye, K.P. Zimmer, R. Brown, C. Pabin, M.L. Batshaw, J.M. Wilson,M.B. Robinson, DiVerences in the human and mouse amino-terminalleader peptides of ornithine transcarbamylase aVect mitochondrial importand eYcacy of adenoviral vectors, Hum. Gene Ther. 12 (2001) 1035–1046.
[35] J.P. Lerner-Ellis, J.C. Tirone, P.D. Pawelek, C. Dore, J.L. Atkinson, D.Watkins, C.F. Morel, T.M. Fujiwara, E. Moras, A.R. Hosack, G.V.Dunbar, H. Antonicka, V. Forgetta, C.M. Dobson, D. Leclerc, R.A.Gravel, E.A. Shoubridge, J.W. Coulton, P. Lepage, J.M. Rommens, K.Morgan, D.S. Rosenblatt, IdentiWcation of the gene responsible formethylmalonic aciduria and homocystinuria, cblC type, Nat. Genet.38 (2006) 93–100.




















