
The calculation of the static first and second susceptibilities of crystalline urea: A comparison of...
10
The calculation of the static first and second susceptibilities of crystalline urea: A comparison of Hartree–Fock and density functional theory results obtained with the periodic coupled perturbed Hartree–Fock/Kohn–Sham scheme Mauro Ferrero, 1,a Bartolomeo Civalleri, 1 Michel Rérat, 2 Roberto Orlando, 3 and Roberto Dovesi 1 1 Dipartimento di Chimica IFM, Università di Torino and NIS-Nanostructured Interfaces and Surfaces, Centre of Excellence, Via P. Giuria 7, 10125 Torino, Italy 2 Equipe de Chimie Physique, IPREM UMR5254, Université de Pau, 64000 Pau, France 3 Dipartimento di Scienze e Tecnologie Avanzate, Università del Piemonte Orientale, Via Bellini 25/G, 15100 Alessandria, Italy Received 4 September 2009; accepted 5 November 2009; published online 4 December 2009 The static polarizability and first hyperpolarizability tensors of crystalline urea and the corresponding first- 1 and second- 2 susceptibilities are calculated and compared to the same quantities obtained for the molecule by using the same code a development version of CRYSTAL, basis set, and level of theory. In order to separate geometrical and solid state effects, two geometries are considered for the molecule in its planar conformation: i as cut out from the bulk structure and ii fully optimized. First, the effect of basis sets on computed properties is explored at the B3LYP level by employing basis sets of increasing complexity, from 6-31Gd,p to 6-311G2df,2pd Pople’s family and from DZP to QZVPPP Thakkar/Ahlrichs/Dunning’s family on and for both the molecule and the bulk. Then, five different levels of theory, namely, SVWN local density approximation, PBE generalized gradient approximation, PBE0 and B3LYP hybrid, and Hartree–Fock are compared in combination with a TZPP basis set. Present results show that hybrid methods, in particular, B3LYP, are remarkably successful in predicting correctly both the first and second susceptibilities of urea bulk when combined at least with a triple-zeta quality basis set containing a double set of polarization functions. It is also shown that diffuse functions that are needed for molecular calculations are less crucial for the crystalline structure, as expected. Indeed, B3LYP/TZPP computed 1 and 2 tensor components aa 1 = 1.107, cc 1 = 1.459, and 2 =-0.93 a.u. are in very good agreement with experimental values. At variance with respect to previous periodic ab initio calculations, but in agreement with recent supermolecular results, the negative sign of 2 is confirmed. Overall, static linear and nonlinear optical properties such as dielectric constants, refractive, and birefringence indices and second-harmonic generation coefficient of crystalline urea are very well reproduced by present calculations. © 2009 American Institute of Physics. doi:10.1063/1.3267861 I. INTRODUCTION The coupled perturbed Hartree–Fock CPHF method has recently been implemented in a development version of the CRYSTAL program 1 to compute the polarizability and di- electric tensor of solid state systems. 2,3 The implementation is based on the CPHF equations proposed by Hurst and Dupuis 4 and adapted to the periodic boundary condition con- text with reference to a local basis set consisting of Gaussian-type atomic orbitals. 5,6 Recently, we have also implemented the CPHF calculation of the first and second hyperpolarizabilities 7 showing that the method is equally ac- curate and suitable for the treatment of zero-, one-, two-, and three-dimensional systems. Finally, a generalization from CPHF to coupled perturbed Kohn–Sham CPKS for the po- larizability tensor has been presented 8 that can be extended to the first hyperpolarizability tensor through the 2n +1 approach. This generalization includes local- local density approximation LDA, semilocal- generalized gradient ap- proximation GGA, and global hybrid-type HF/density functional theory DFT functionals. In particular, hydrid methods that include exact HF exchange appear very prom- ising because they allow to partly recover the correct asymptotic limit of the exchange-correlation potential and to partly compensate the spurious self-interaction error. Here, we investigate the accuracy of our CPHF/KS scheme in the calculation of the static hyper- polarizability of crystalline urea by exploring the effect of the basis set and of the adopted exchange-correlation functional i.e., LDA, GGA, and hybrid. Urea has been considered as a case study of the wide family of molecular crystals with application in nonlinear optics. Indeed, molecular organic crystals are among the most promising materials for linear and nonlinear optics. The theoretical ab initio investigation of their optical a Electronic mail: [email protected]. THE JOURNAL OF CHEMICAL PHYSICS 131, 214704 2009 0021-9606/2009/13121/214704/10/$25.00 © 2009 American Institute of Physics 131, 214704-1 Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of The calculation of the static first and second susceptibilities of crystalline urea: A comparison of...

The calculation of the static first and second susceptibilities of crystallineurea: A comparison of Hartree–Fock and density functional theoryresults obtained with the periodic coupled perturbedHartree–Fock/Kohn–Sham scheme
Mauro Ferrero,1,a� Bartolomeo Civalleri,1 Michel Rérat,2 Roberto Orlando,3 andRoberto Dovesi11Dipartimento di Chimica IFM, Università di Torino and NIS-Nanostructured Interfaces and Surfaces,Centre of Excellence, Via P. Giuria 7, 10125 Torino, Italy2Equipe de Chimie Physique, IPREM UMR5254, Université de Pau, 64000 Pau, France3Dipartimento di Scienze e Tecnologie Avanzate, Università del Piemonte Orientale,Via Bellini 25/G, 15100 Alessandria, Italy
�Received 4 September 2009; accepted 5 November 2009; published online 4 December 2009�
The static polarizability � and first hyperpolarizability � tensors of crystalline urea and thecorresponding first-���1�� and second-���2�� susceptibilities are calculated and compared to the samequantities obtained for the molecule by using the same code �a development version of CRYSTAL�,basis set, and level of theory. In order to separate geometrical and solid state effects, two geometriesare considered for the molecule in its planar conformation: �i� as cut out from the bulk structure and�ii� fully optimized. First, the effect of basis sets on computed properties is explored at the B3LYPlevel by employing basis sets of increasing complexity, from 6-31G�d,p� to 6-311G�2df,2pd��Pople’s family� and from DZP to QZVPPP �Thakkar/Ahlrichs/Dunning’s family� on � and � forboth the molecule and the bulk. Then, five different levels of theory, namely, SVWN �local densityapproximation�, PBE �generalized gradient approximation�, PBE0 and B3LYP �hybrid�, andHartree–Fock are compared in combination with a TZPP basis set. Present results show that hybridmethods, in particular, B3LYP, are remarkably successful in predicting correctly both the first andsecond susceptibilities of urea bulk when combined at least with a triple-zeta quality basis setcontaining a double set of polarization functions. It is also shown that diffuse functions that areneeded for molecular calculations are less crucial for the crystalline structure, as expected.Indeed, B3LYP/TZPP computed ��1� and ��2� tensor components ��aa
�1�=1.107, �cc�1�=1.459, and
��2�=−0.93 a.u.� are in very good agreement with experimental values. At variance with respect toprevious periodic ab initio calculations, but in agreement with recent supermolecular results, thenegative sign of ��2� is confirmed. Overall, static linear and nonlinear optical properties such asdielectric constants, refractive, and birefringence indices and second-harmonic generationcoefficient of crystalline urea are very well reproduced by present calculations. © 2009 AmericanInstitute of Physics. �doi:10.1063/1.3267861�
I. INTRODUCTION
The coupled perturbed Hartree–Fock �CPHF� methodhas recently been implemented in a development version ofthe CRYSTAL program1 to compute the polarizability and di-electric tensor of solid state systems.2,3 The implementationis based on the CPHF equations proposed by Hurst andDupuis4 and adapted to the periodic boundary condition con-text with reference to a local basis set consisting ofGaussian-type atomic orbitals.5,6 Recently, we have alsoimplemented the CPHF calculation of the first and secondhyperpolarizabilities7 showing that the method is equally ac-curate and suitable for the treatment of zero-, one-, two-, andthree-dimensional systems. Finally, a generalization fromCPHF to coupled perturbed Kohn–Sham �CPKS� for the po-larizability tensor has been presented8 that can be extended
to the first hyperpolarizability � tensor through the 2n+1approach. This generalization includes local- �local densityapproximation �LDA��, semilocal- �generalized gradient ap-proximation �GGA��, and global hybrid-type HF/densityfunctional theory �DFT� functionals. In particular, hydridmethods that include exact HF exchange appear very prom-ising because they allow to partly recover the correctasymptotic limit of the exchange-correlation potential and topartly compensate the spurious self-interaction error.
Here, we investigate the accuracy of our CPHF/KSscheme in the calculation of the static �hyper-� polarizabilityof crystalline urea by exploring the effect of the basis set andof the adopted exchange-correlation functional �i.e., LDA,GGA, and hybrid�. Urea has been considered as a case studyof the wide family of molecular crystals with application innonlinear optics. Indeed, molecular organic crystals areamong the most promising materials for linear and nonlinearoptics. The theoretical ab initio investigation of their opticala�Electronic mail: [email protected].
THE JOURNAL OF CHEMICAL PHYSICS 131, 214704 �2009�
0021-9606/2009/131�21�/214704/10/$25.00 © 2009 American Institute of Physics131, 214704-1
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

properties has been mostly based on molecular approaches.9
Basically, they rely on molecular calculations in which theeffect of the surrounding molecules in the crystal structure ismimicked with different strategies. A recent account on theproposed schemes was published by Champagne andBishop9 and we refer to that paper for a detailed discussion.In summary, most of those approaches are based on the cal-culation of the molecular �hyper-� polarizability and then ei-ther an additivity assumption is made to estimate the corre-sponding first and second susceptibilities of the crystallinestructure or a multiplicative factor is used to include crystalenvironment effects. Also a supermolecule approach can beadopted but it can be rather expensive and an appropriatecluster must be carefully selected. All of those approacheswork properly for weakly interacting molecules but they areinadequate to deal with hydrogen bonded molecular crystalswhere long-range dipolar interactions and polarization ef-fects take place and dominate the crystal packing. In thisrespect, crystalline urea is considered as a benchmark mo-lecular crystal. Indeed, many experimental and theoreticalworks10–18 have been done to measure and predict both first���1�� and second susceptibilities ���2��. In particular, we re-fer here to the recent work by Champagne and co-workers.18
They reported on the attempt of using a supermolecule ap-proach on a very large cluster by combining semiempiricalcalculations �i.e., TDHF/AM1� with DFT and coupled-cluster methods through a multiplicative scheme. The idea isto include electron correlation effects by means of a multi-plicative factor as obtained from high-level ab initio resultson the isolated molecule. However, the predicted suscepti-bilities, in particular, the linear response, were still underes-timated with respect to experimental data especially the lin-ear response. The authors then concluded that their approachcould be limited by its inherent inability to account for long-range cooperative effects of the crystalline structure. Thepresent full ab initio periodic approach that allows one toinclude all environmental effects due to the crystal packing istherefore the most appropriate for a correct prediction of lin-ear and nonlinear properties in hydrogen bonded molecularcrystals. To our knowledge, so far, only two other periodiccalculations were reported for crystalline urea by Lin et al.15
and Levine and Allan11 at the LDA level of theory within aplane wave/pseudopotential theoretical framework. How-ever, results were much larger than the experimental databecause of the well known deficiency of the LDA approxi-mation in reproducing correctly the band gap of solids.
In this work we show that �i� the present implementationof the CPHF and CPKS scheme for periodic systems can besuccessfully applied to molecular crystals; �ii� hybrid HF/DFT functionals, such as B3LYP, give the best performancein predicting the first- and second-order susceptibilities ofurea; �iii� as for molecular calculations, the adoptedGaussian-type basis set must be at least of triple-zeta qualityand must include diffuse s and p functions and two sets ofpolarization functions. A detailed comparison of periodic cal-culations with the results for urea molecule as both cut outfrom the crystal and relaxed in its planar conformation �C2v�is also presented.
II. COMPUTATIONAL DETAILS
Calculations were carried out with the periodic ab initioCRYSTAL06 program.1 Crystalline orbitals are represented aslinear combinations of Bloch functions �BFs� and are evalu-ated over a regular three-dimensional mesh in reciprocalspace. Each BF is built from atom-centered atomic orbitals,which are contractions �linear combinations with constantcoefficients� of Gaussian-type functions �GTFs�, each GTFbeing the product of a Gaussian times a real solid sphericalharmonic.
A. Hamiltonians and basis sets
Along with the HF method, four different DFT methodswere considered: the simplest density functional method,LDA in its SVWN parametrization,19,20 the PBE GGAfunctional,21 and two hybrid methods, B3LYP �Refs. 22–24��probably the most widely used hybrid functional in molecu-lar calculations� in its VWN5 formulation, and PBE0 �Ref.25� �also known as PBE1PBE or PBEh�. The basis set de-pendence of computed results was investigated at the B3LYPlevel of theory by using 12 molecular all-electron basis sets.The first set of six basis sets was taken from Pople’s familyof basis sets,26 namely, 6-31G�d,p�, 6-311G�d,p�,6-311G�2d,p�, 6-311G�2d,2p�, 6-311G�2df,p�, and6-311G�2df,2pd�. The second one ranges from DZP throughTZP to QZVP basis sets as proposed by Thakkar et al.27
�DZP and QZVP� and by Ahlrichs and co-workers28 �TZP�.The latter basis sets were further enriched by adding one ortwo more sets of d and f polarization functions from cc-pVXZ �X=T ,Q� Dunning’s sets to define TZPP, QZVPP, andQZVPPP basis. The number of basis functions in the unit cellranges then from 152 �6-31G�d,p�� to 568 �QZVPPP�. Thecomparison among HF and DFT methods was carried out byadopting the TZPP basis set.
B. Computational parameters
The level of accuracy in evaluating the Coulomb andexchange series is controlled by five thresholds, for whichvalues of 10−7, 10−7, 10−7, 10−7, and 10−18 were used for theCoulomb and the exchange series.1 For the QZVPPP basissets, tolerances were increased to 10−7, 10−7, 10−7, 10−12, and10−36 and a Lowdin purification scheme was adopted with ascreening on the eigenvalues of the overlap matrix of 10−4.The DFT exchange-correlation contribution is evaluated bynumerical integration over the cell volume.29 Radial and an-gular points of the atomic grid are generated through Gauss–Legendre and Lebedev quadrature schemes. A grid pruningwas adopted, as discussed in Ref. 29. In the present study a�75 974�p grid has been used that contains 75 radial pointsand a variable number of angular points, with a maximum of974 on the Lebedev surface in the most accurate integrationregion. The condition for the SCF convergence was set to10−7 on the root-mean-square �rms� variation of the densitymatrix elements between two subsequent cycles. The shrink-ing factor of the reciprocal space net was set to 4, corre-sponding to 18 reciprocal space points of the irreducible
214704-2 Ferrero et al. J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

Brillouin zone at which the Hamiltonian matrix was diago-nalized. The total energies obtained with this mesh are fullyconverged.
C. Geometry optimization
In the solid state, urea forms a noncentrosymmetric te-
tragonal crystal that belongs to the P4̄21m space group withtwo molecules in the unit cell, as shown in Fig. 1. Startingfrom the experimental crystal structure30 at 12 K �i.e.,a=5.565 Å and c=4.684 Å�, a relaxation of the atomic co-ordinates by means of analytical energy gradients31–33 wascarried out at fixed lattice constants for each method andbasis set. Lattice parameters were fixed because of the wellknown deficiency of DFT methods in dealing with weaklybound systems. In this respect, the use of extended basis sets,such as TZP or larger, leads to a large overestimation of thecell size.34
The geometry optimization of the atomic positions wasperformed by means of a quasi-Newton algorithm in whichthe quadratic step �BFGS Hessian updating scheme� is com-bined with a linear one �parabolic fit� as proposed bySchlegel.35 Convergence was tested on the rms and the ab-solute value of the largest component of the gradients and theestimated displacements. The threshold for the maximumforce, the rms force, the maximum atomic displacement, andthe rms atomic displacement on all atoms have been set to0.000 45, 0.000 30, 0.001 80, and 0.001 20 a.u., respectively.The optimization was considered complete when the fourconditions are simultaneously satisfied. The crystal symme-try was maintained during the whole optimization process.
D. Dielectric properties
Concerning the CPHF/CPKS calculation, the conver-gence was checked on the stability of the diagonal elementsof the polarizability: The process stops when these elementsdiffer by less than 10−4 in two subsequent cycles.
The computed unit cell polarizability tensor components�i.e., �ij� were transformed in the macroscopic first-ordersusceptibility tensor by using the following relation:
�ij�1� =
4�
V�ij , �1�
where V is the unit cell volume. Similarly, the second-ordersusceptibility is derived from the corresponding unit cell firsthyperpolarizability �ijk as
�ijk�2� =
2�
V�ijk. �2�
The dielectric tensor is then obtained as �ij =�ij�1�+�ij �where
�ij =1 if i= j and 0 for i� j� while the refractive indices wereevaluated according to the usual expression
nii = �ii1/2. �3�
Crystalline urea lacks the center of inversion and shows non-linear optical effects. Furthermore, it is a uniaxial crystal forwhich the first-susceptibility tensor shows two unique com-ponents �aa
�1� and �cc�1�, where a and c refer to the crystallo-
graphic axes. It is then birefringent with different refractiveindices along the two main crystallographic axes. Two bire-fringence parameters are generally used:36 the so-called lin-ear birefringence
�n = na − nc �4�
and
��1� =nc − na
na, �5�
where na corresponds to the refractive index along the a-axis�ordinary� and nc to the one along the c-axis �extraordinary�.
According to the crystal symmetry, the second-susceptibility tensor of solid urea has only one nonzero com-ponent �abc
�2� . From �abc�2� , the nonlinear coefficient d14 can be
directly obtained as d14=1 /2�abc�2� and related to second-
harmonic generation measurements.37
As in Ref. 18, also for the molecule, we adopt a coordi-nate system based on the main crystallographic directions ofcrystalline urea �a=b ,c�. For the crystal, in the present no-tation �aa�bb�=�xx�yy�, �cc=�zz, and �abc=�xyz. It must betaken into account that the crystallographic directions havedifferent orientations with respect to the Cartesian frame ofthe molecule that lies in the y-z plane with the CO bondoriented along the z-axis. In particular, while the c-axis iscorrectly oriented along the z-axis, a and b are oriented at45° with respect to x and y, respectively. Therefore, for thefree molecule, �aa is derived from �xx and �yy, and �abc from�xxz and �yyz, through a rotation of 45°.
III. RESULTS AND DISCUSSION
A. Molecular polarizabilities
Before discussing bulk dielectric properties, let us firstfocus on urea molecule. In the crystalline structure, themolecule adopts a planar conformation with C2v symmetry�Fig. 1�; in contrast, in the gas phase the most stable structurehas a C2 symmetry with the amino group assuming a pyra-midal conformation with an anticonfiguration. To make aconsistent comparison between the polarizability of the mol-
FIG. 1. Unit cell and crystal packing in solid urea as view along the c-axis.
214704-3 Dielectric properties of crystalline urea J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
ecule alone and in the solid, we refer here to the planarconformation. We also take into account relaxation effects byconsidering the polarizability of the fully optimized molecu-lar geometry �i.e., with the C2v symmetry constraint� and thegeometry as in the bulk structure.
1. Effect of the basis set on molecular polarizabilities
Table I gathers the B3LYP molecular polarizability com-puted with the different basis sets employed in the presentwork comparing the polarizability of the molecule alone andin the crystal. It is well known that polarizabilities are verysensitive to the quality of the basis set. Specific basis setshave been devised to properly predict electric properties ofmolecules as proposed by Sadlej and co-workers �see, forinstance, Ref. 38 and reference therein�. Usually, they areaugmented with rather diffuse s and p functions but alsodiffuse polarization functions �i.e., d and even f functions�are needed. In periodic system, the role of these basis func-tions is less crucial. Because of the crystal packing, the mol-ecule can take advantage from the atomic functions centeredon neighboring molecules to improve the description of thewave function. This can be seen by comparing results for thefree molecule obtained with basis sets containing very dif-fuse functions such as the aug-cc-pVTZ and the Sadlej-pVTZ basis sets that can be considered as a reference. TheB3LYP computed values of �aa ��cc� are 34.483 �41.744�a.u. and 34.766 �42.162� a.u. for the two basis sets, respec-tively. Results of Table I confirm the marked basis set depen-dence of the polarizability. In particular, the effect is largerfor �aa than �cc �i.e., the out-of-plane polarizability �xx ismore sensitive to the basis set quality�. In the molecule, therole of the diffuse functions is clearly highlighted when theTZPP basis set is enriched with the same augmented basisfunctions as in the aug-cc-pVTZ basis. The computed datapass from 28.718 and 37.129 a.u. to 33.755 and 40.778 a.u.,very close to the reference basis sets with a substantial im-provement of �aa. However, when the molecular polarizabil-ities of the molecule in the crystal are considered, the depen-
dence on the basis set is less critical. Indeed, TZPP, QZVPP,and QZVPPP results are fairly close to each other. Thisshows that in the solid the molecular polarizability is lesssensitive to the basis set when a good quality set of atomicfunctions is adopted.
Overall, computed values improve when: �i� passingfrom Pople’s to more flexible Ahlrichs/Thakkar basis sets;�ii� removing the sp-constraint �e.g., 6-311G�2df,2p� versusTZPP�; �iii� including low exponents Gaussian functionsboth of s , p-type, as from DZP through TZP to QZVP �e.g.,for carbon, the lowest exponent is less than 0.10 a.u.�, andd , f polarization functions, as from TZP to TZPP or in theseries QZVP, QZVPP, and QZVPPP �e.g., for carbon, d and flowest exponents are 0.23 and 0.49 a.u., respectively�. It isworth noting that at least two sets of polarization functions�i.e., 2d+ f functions� are necessary to obtain reasonable re-sults.
From Table I, we can also observe the following.
• Polarizabilities of the free molecule in the relaxed ge-ometry and as in the bulk differ slightly. A larger differ-ence is observed for �cc in the unrelaxed structure be-cause the effect of the crystalline environment is toelongate the CO bond that is involved in four hydrogenbonds: two along the infinite chains and two with theneighboring chains oriented in the opposite direction�see Fig. 1�. Strong dipole-dipole interactions are in-volved in the bonding along the chains thus contribut-ing to give longer CO bonds.
• The effect of the intermolecular interactions is evidentwhen considering the ratio between the polarizability ofthe molecule in the crystal and the free molecule �seeTable I� that ranges from 1.54 and 1.43 for �aa and from1.57 to 1.46 for �cc. As expected, a more significantincrease is observed along the c-axis, where the mol-ecules are oriented head to tail to form infinite chains�see Fig. 1� as a consequence of dipole-dipole interac-tions, hydrogen bonding, and charge transfer effects. In-
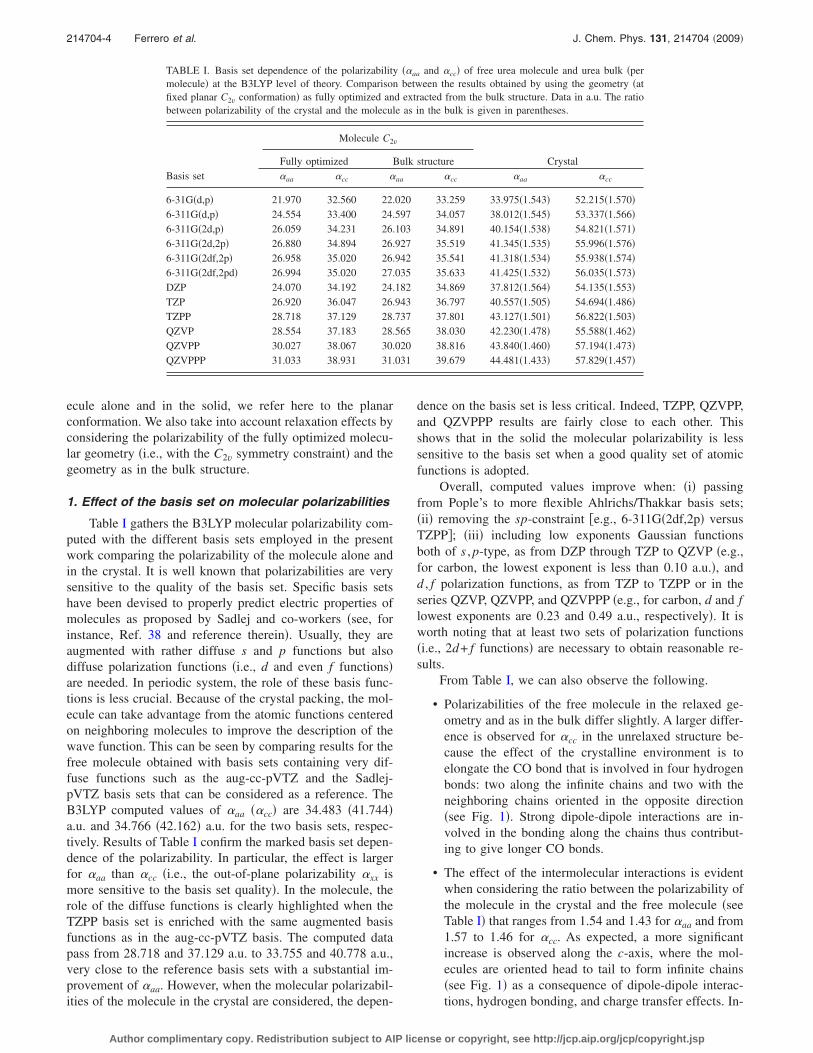
TABLE I. Basis set dependence of the polarizability ��aa and �cc� of free urea molecule and urea bulk �permolecule� at the B3LYP level of theory. Comparison between the results obtained by using the geometry �atfixed planar C2v conformation� as fully optimized and extracted from the bulk structure. Data in a.u. The ratiobetween polarizability of the crystal and the molecule as in the bulk is given in parentheses.
Basis set
Molecule C2v
CrystalFully optimized Bulk structure�aa �cc �aa �cc �aa �cc
6-31G�d,p� 21.970 32.560 22.020 33.259 33.975�1.543� 52.215�1.570�6-311G�d,p� 24.554 33.400 24.597 34.057 38.012�1.545� 53.337�1.566�6-311G�2d,p� 26.059 34.231 26.103 34.891 40.154�1.538� 54.821�1.571�6-311G�2d,2p� 26.880 34.894 26.927 35.519 41.345�1.535� 55.996�1.576�6-311G�2df,2p� 26.958 35.020 26.942 35.541 41.318�1.534� 55.938�1.574�6-311G�2df,2pd� 26.994 35.020 27.035 35.633 41.425�1.532� 56.035�1.573�DZP 24.070 34.192 24.182 34.869 37.812�1.564� 54.135�1.553�TZP 26.920 36.047 26.943 36.797 40.557�1.505� 54.694�1.486�TZPP 28.718 37.129 28.737 37.801 43.127�1.501� 56.822�1.503�QZVP 28.554 37.183 28.565 38.030 42.230�1.478� 55.588�1.462�QZVPP 30.027 38.067 30.020 38.816 43.840�1.460� 57.194�1.473�QZVPPP 31.033 38.931 31.031 39.679 44.481�1.433� 57.829�1.457�
214704-4 Ferrero et al. J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
deed, as reported by Whitten et al.,17 the dipole momentof the molecule increases from 3.83 D in the gas phaseto 6.56 D in the solid state, with the latter being esti-mated from synchrotron x-ray structure factors.39 Also,the comparison of the Born charges �i.e., charges de-rived from the first derivative of the cell polarizabilitywith respect to normal modes in the Cartesianframe40,41� of the molecule alone and in the crystal al-lows us to rationalize the enhancement of the molecularpolarizabilities. For instance, for the carbonyl group, theBorn charge on the oxygen atom increases from −0.85eto −1.50e, while carbon passes from 1.42e to 1.93e. Asimilar increment is also observed for nitrogen and hy-drogen. This shows that a large redistribution of thecharge density of the molecule takes place in the crys-talline structure. The enhancement reduces when en-riching the basis set: Extended basis sets provide a bet-ter description of the polarizability of the isolatedmolecule with respect to the crystal where it has alreadyconverged.
• Although �cc and �aa for the free molecule and the onein the crystalline structure are very different from eachother because of the crystalline environment, their ratio�i.e., �cc /�cc� is rather similar in the two systems, beingaround 1.3–1.5; it tends to reduce when enriching thebasis set. This is an evidence that larger basis sets allowa better description of the out-of-plane polarizability�i.e., �aa�. For instance, passing from DZP to QZVPPPthere is a remarkable increase in �aa while for �cc thiseffect is less evident. In this respect, results show thatbasis set convergence is more rapid for �cc than �aa forboth the molecule alone and in the crystal.
Overall, results indicate that in the trade-off betweencost and accuracy, the TZPP basis set is rather satisfactorybeing in semiquantitative agreement with the data computedwith very large basis sets. Therefore, it has been used in Sec.III A 2 as a reference to compare HF and DFT results.
2. Effect of the level of theory
The molecular polarizabilities of urea both isolated andin the crystal calculated with different DFT methods and atHF level are given in Table II. It is evident that electron
correlation effects at the DFT level are quite relevant. For thefree molecule they are of the order of 12%–22% with thelower value corresponding to the hybrid functionals, while itincreases for GGA and LDA functionals �i.e., PBE andSVWN�. An increment of the polarizabilities of 15%–20%agrees with previously reported MP2 and CCSD resultsobtained with larger basis sets by Reis et al.12 and Olejniczaket al.,18 respectively.
As already discussed, structural changes in the moleculedue to the crystal packing lead to a slight increment of thepolarizability. On the contrary, full inclusion of crystallineenvironment and electron correlation effects remarkably in-creases the polarizabilities of the molecule in the unit cell. Inthis case, electron correlation is very important giving a con-tribution of more than 20%–25% for hybrid methods, 35%–40% for GGA, and 40%–45% for LDA. The effect is morerelevant for �cc than �aa because electron correlation makeselectrostatic and charge transfer effects stronger along thec-axis. As expected, those effects are more marked at theLDA level because of the known tendency to delocalize thecharge density. This reduces when passing to GGA and hy-brid functionals, as clearly seen from Table II.
Also given in Table II is the enhancement of the polar-izabilities when the molecules are packed to form the crystal.Again, electron correlation plays a significant role. The en-hancement is larger for LDA and GGA while decreases whenadding some exact HF exchange. Although predicted polar-izabilities of urea molecule in the crystal are slightly differ-ent for PBE0 and B3LYP, nevertheless the enhancement fac-tors are the same. Interestingly, as already pointed out in Sec.III A 1, the �cc /�aa ratio is roughly independently of theadopted level of theory being �1.3 for both the isolated mol-ecule and the crystal.
B. Bulk first-order susceptibilities
1. Basis set dependence
From the data of Table I for urea crystal the static first-order susceptibility and related dielectric properties can becomputed by using Eqs. �1� and �3�. Table III reports theoptical dielectric properties of urea crystal determined at theB3LYP level as a function of the basis set size in comparisonwith experimental values for the optical dielectric tensorcomponents extrapolated to the static limit through a Sell-
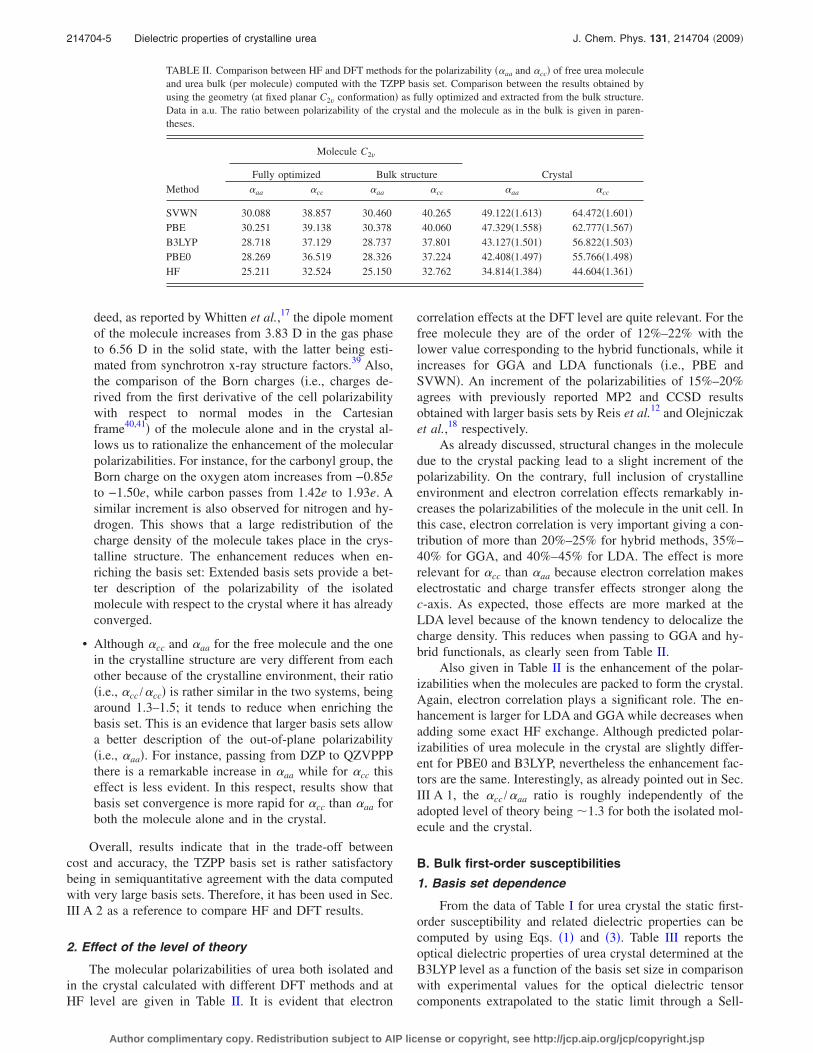
TABLE II. Comparison between HF and DFT methods for the polarizability ��aa and �cc� of free urea moleculeand urea bulk �per molecule� computed with the TZPP basis set. Comparison between the results obtained byusing the geometry �at fixed planar C2v conformation� as fully optimized and extracted from the bulk structure.Data in a.u. The ratio between polarizability of the crystal and the molecule as in the bulk is given in paren-theses.
Method
Molecule C2v
CrystalFully optimized Bulk structure�aa �cc �aa �cc �aa �cc
SVWN 30.088 38.857 30.460 40.265 49.122�1.613� 64.472�1.601�PBE 30.251 39.138 30.378 40.060 47.329�1.558� 62.777�1.567�B3LYP 28.718 37.129 28.737 37.801 43.127�1.501� 56.822�1.503�PBE0 28.269 36.519 28.326 37.224 42.408�1.497� 55.766�1.498�HF 25.211 32.524 25.150 32.762 34.814�1.384� 44.604�1.361�
214704-5 Dielectric properties of crystalline urea J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
meier equation.37 From experimental tensor components therefractive indices can be obtained as well as birefringenceparameters to be compared to computed results.
The birefringence parameters can be analyzed as indica-tors of the anisotropy of the electric response of the crystal tothe perturbation. Small basis sets lead to an unbalanced de-scription of the susceptibility favoring the cc componentwith respect to the aa one. The inclusion of more diffusefunctions and two sets of polarization functions, as in theTZPP basis set, provides a significant improvement of ��1�
and �n thus confirming that the basis set must be sufficientlyflexible to allow the proper description of the wave functionboth in the plane and out of the plane of the molecule. In thisrespect, results obtained with the largest basis set, i.e.,B3LYP/QZVPPP, are in excellent agreement with experi-mental values.
Figure 2�a� shows the deviation of computed data fromexperiment as a function of the basis set whereas in Fig. 2�b�,the dielectric tensor components are plotted as a function ofthe number of atomic orbitals in the unit cell. The two figuresshow that �i� the deviation from experiment remarkably re-duces when enriching the basis set and �ii� a near conver-gence to the complete basis set limit is reached. Hence, theQZVPPP basis set provides predictions that can be consid-ered as benchmark values for the B3LYP functional. As ex-pected from the previous discussion on the molecular polar-izabilities, Pople’s basis sets reach limit values that are farfrom the experimental data, while TZ and QZ basis sets sys-tematically improve approaching quite closely the experi-mental values, in particular, when more and more polariza-tion functions are included.
Two other aspects deserve further discussion.
• It must be pointed out that the refractive indices arecalculated from the unit cell volume �see Eq. �1�� fixedat its experimental value at 12 K. However, observedrefractive indices are usually measured at room tem-perature. The role of thermal expansion effects was thenestimated at the B3LYP level by computing the unit cellpolarizability with lattice parameters fixed at 123 and
295 K as determined from x-ray diffractionexperiments.39 The corresponding refractive indices arenaa=1.449, ncc=1.566 and naa=1.436, ncc=1.552, re-spectively. As expected, the computed values decreasewhen the volume increases. In comparison with resultsfor the 12 K unit cell, the decrease in n is larger be-tween 123 and 295 K. This is due to the substantialincrease in the cell volume in that range of temperature.
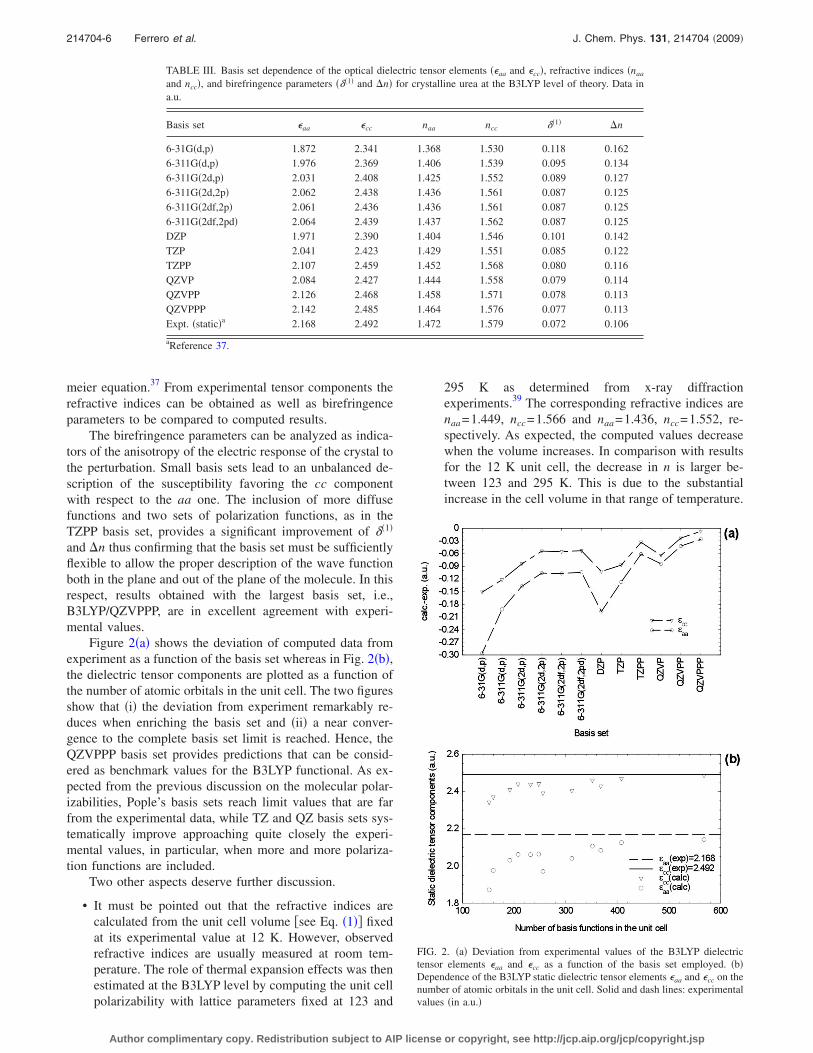
TABLE III. Basis set dependence of the optical dielectric tensor elements ��aa and �cc�, refractive indices �naa
and ncc�, and birefringence parameters ���1� and �n� for crystalline urea at the B3LYP level of theory. Data ina.u.
Basis set �aa �cc naa ncc ��1� �n
6-31G�d,p� 1.872 2.341 1.368 1.530 0.118 0.1626-311G�d,p� 1.976 2.369 1.406 1.539 0.095 0.1346-311G�2d,p� 2.031 2.408 1.425 1.552 0.089 0.1276-311G�2d,2p� 2.062 2.438 1.436 1.561 0.087 0.1256-311G�2df,2p� 2.061 2.436 1.436 1.561 0.087 0.1256-311G�2df,2pd� 2.064 2.439 1.437 1.562 0.087 0.125DZP 1.971 2.390 1.404 1.546 0.101 0.142TZP 2.041 2.423 1.429 1.551 0.085 0.122TZPP 2.107 2.459 1.452 1.568 0.080 0.116QZVP 2.084 2.427 1.444 1.558 0.079 0.114QZVPP 2.126 2.468 1.458 1.571 0.078 0.113QZVPPP 2.142 2.485 1.464 1.576 0.077 0.113Expt. �static�a 2.168 2.492 1.472 1.579 0.072 0.106
aReference 37.
FIG. 2. �a� Deviation from experimental values of the B3LYP dielectrictensor elements �aa and �cc as a function of the basis set employed. �b�Dependence of the B3LYP static dielectric tensor elements �aa and �cc on thenumber of atomic orbitals in the unit cell. Solid and dash lines: experimentalvalues �in a.u.�
214704-6 Ferrero et al. J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
Interestingly, the largest variation is observed for naa inagreement with experimental evidence that shows alarger thermal expansion for the a-axis.42 Overall, theeffect of the choice of the reference unit cell volume issmall with a percentage variation not larger than 1%.Therefore, in the present work, all reported results referto the unit cell volume at 12 K.
• From computed data, also the �=0 limit can be ob-tained by including the contribution from nuclear vibra-tional motion.41 Using the vibrational frequencies at the point29 computed at the B3LYP/TZPP level, the staticdielectric tensor components at the zero wavelengthlimit are �aa
0 =3.771 and �cc0 =2.972 a.u. It is worthy to
note that �i� the vibrational contribution is quite large,�ii� the calculation of the �=0 vibrational effect isbased on a double harmonic approximation, and �iii� itis appropriate to ignore vibration in comparing with ex-trapolated �to �=0� measurement because vibrations donot contribute in the high � limit where measurementsare made.
2. Effect of the level of theory
In Table IV the dielectric properties of crystalline ureacomputed with LDA, GGA, and hybrid methods along withHF are reported. Results are also compared to other predictedtheoretical data and experimental values. As expected fromTable II, computed results show the same trend as discussedfor the molecular polarizabilities, with the SVWN �LDA�dielectric tensor components and refractive indices beinglarger than PBE �GGA�, hybrid functionals, and HF, in order.
LDA and GGA data are also overestimated with respect toexperimental values37 while hybrid methods are in rathergood agreement, in particular, with the B3LYP functional.On the contrary, HF gives definitely underestimated refrac-tive indices highlighting then the crucial role played by elec-tron correlation for a correct prediction of the dielectric prop-erties of urea.
In the comparison between HF and DFT, an inverse cor-relation is clearly observed between the predicted dielectrictensor components and the computed band gap of crystallineurea. Reported band gap decreases in the series: HFhybridsGGALDA, while on the opposite, �aa and �cc
increase. This is not unexpected because polarizability islargely determined by the response of the electronic structureto the perturbation of the electric field and also depends onthe correct asymptotic behavior of the exchange-correlationpotential �vxc�. Also, the band gap is directly involved in theexpression of the polarizability �see Ref. 8 for details�. En-ergy level differences appear at the denominator thus lower-ing the polarizability when increasing the energy gap. HFshows the right decay but predicts a too large band gap.Unlike LDA and GGA functionals, hybrid methods reducethe self-interaction error and partly recover the correctasymptotic behavior of vxc. This, combined with a better pre-dicted band gap, leads to a good agreement with experiment.In comparing B3LYP and PBE0, it seems that a loweramount of HF exact exchange as in B3LYP �i.e., 20%� givesbetter results than for PBE0, in which it amounts to 25%.However, the reduction in exact exchange would lead to aworsen long-range decay of vxc. In this respect, newly devel-oped range-separated hybrid methods43 �i.e., long-range cor-
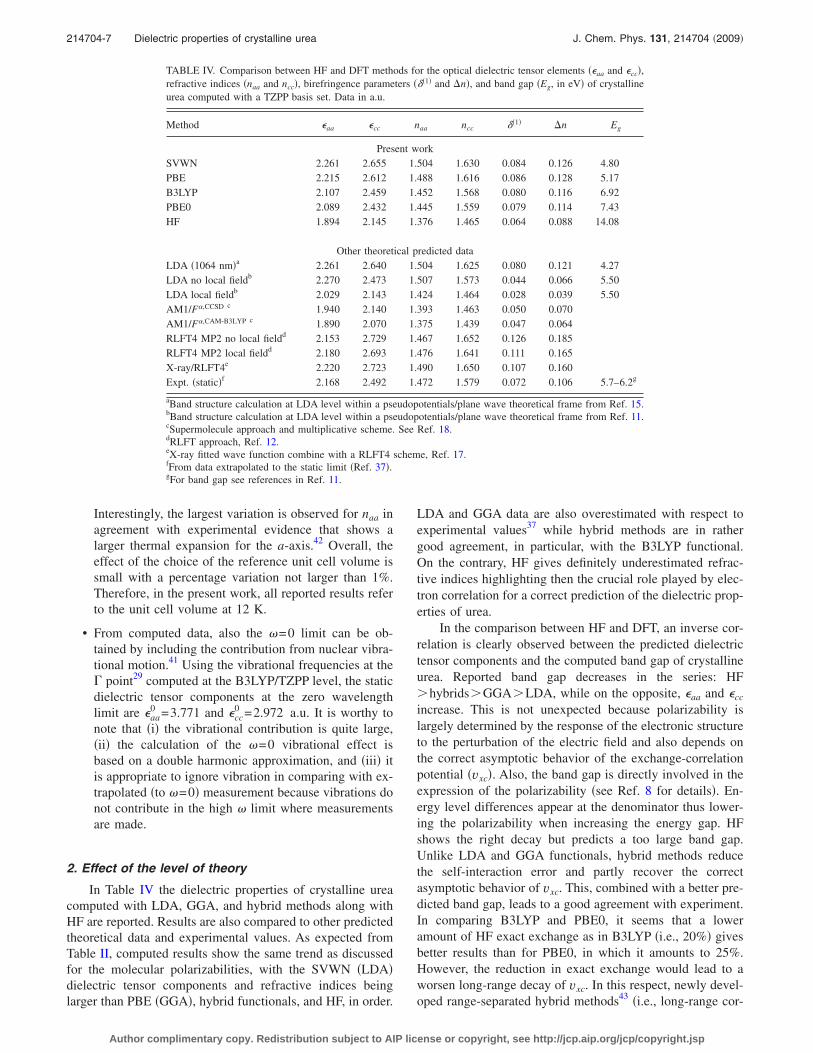
TABLE IV. Comparison between HF and DFT methods for the optical dielectric tensor elements ��aa and �cc�,refractive indices �naa and ncc�, birefringence parameters ���1� and �n�, and band gap �Eg, in eV� of crystallineurea computed with a TZPP basis set. Data in a.u.
Method �aa �cc naa ncc ��1� �n Eg
Present workSVWN 2.261 2.655 1.504 1.630 0.084 0.126 4.80PBE 2.215 2.612 1.488 1.616 0.086 0.128 5.17B3LYP 2.107 2.459 1.452 1.568 0.080 0.116 6.92PBE0 2.089 2.432 1.445 1.559 0.079 0.114 7.43HF 1.894 2.145 1.376 1.465 0.064 0.088 14.08
Other theoretical predicted dataLDA �1064 nm�a 2.261 2.640 1.504 1.625 0.080 0.121 4.27LDA no local fieldb 2.270 2.473 1.507 1.573 0.044 0.066 5.50LDA local fieldb 2.029 2.143 1.424 1.464 0.028 0.039 5.50AM1/F�,CCSD c 1.940 2.140 1.393 1.463 0.050 0.070AM1/F�,CAM-B3LYP c 1.890 2.070 1.375 1.439 0.047 0.064RLFT4 MP2 no local fieldd 2.153 2.729 1.467 1.652 0.126 0.185RLFT4 MP2 local fieldd 2.180 2.693 1.476 1.641 0.111 0.165X-ray/RLFT4e 2.220 2.723 1.490 1.650 0.107 0.160Expt. �static�f 2.168 2.492 1.472 1.579 0.072 0.106 5.7–6.2g
aBand structure calculation at LDA level within a pseudopotentials/plane wave theoretical frame from Ref. 15.bBand structure calculation at LDA level within a pseudopotentials/plane wave theoretical frame from Ref. 11.cSupermolecule approach and multiplicative scheme. See Ref. 18.dRLFT approach, Ref. 12.eX-ray fitted wave function combine with a RLFT4 scheme, Ref. 17.fFrom data extrapolated to the static limit �Ref. 37�.gFor band gap see references in Ref. 11.
214704-7 Dielectric properties of crystalline urea J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp
rected hybrids� have been shown to be more accurate thanglobal hybrid functionals,44 as B3LYP and PBE0, and de-serve to be considered in future works. On passing, we alsopoint out that the band gap is substantially independent ofthe basis set size. Also, it is worth noting that as expectedfrom discussion on basis set dependence, TZPP results areslightly underestimated and then larger basis sets should in-crease the already overestimated LDA and GGA results.
Electron correlation is important to give the right bal-ance between ordinary and extraordinary refractive indicesas expresses by ��1� and �n. At HF level, both are underes-timated while these are increased for the DFT methods. Inparticular, hybrid methods are in very good agreement withexperiment while LDA and GGA provide slightly overesti-mated results. This confirms that LDA and GGA methodsgive unbalance electron correlation effects that favor an in-crease in the crystal polarizability along the c-axis.
From Table IV, a comparison with other theoretical pub-lished data can be done. Present LDA results favorably com-pare with previously reported data, in particular, with the onefrom Ref. 15 obtained within a pseudopotential/plane wavecomputational approach, although not computed at the staticlimit. Moreover, it can be seen that the supermolecule ap-proach as adopted by Champagne and co-workers18 is notsatisfactory being largely underestimated, even if correctionswere included to take into account electron correlation ef-fects. Data calculated within the frame of the rigorous local-field theory �RLFT� both by Munn and co-workers12 at theMP2 level and by Whitten et al.17 from a x-ray fitted wavefunction are in good agreement with experiment. Eventhough, both overestimate the crystal polarizability along thec-axis showing a better agreement for �aa than �cc. It must bepointed out that the comparison with previous works couldbe partly biased by the different choices of the referencecrystalline structure adopted for the calculation. Neverthe-
less, the comparison shows that data computed at the B3LYPlevel of theory with the present fully periodic approach are inexcellent agreement with experiment.
C. Molecular hyperpolarizability and bulk second-ordersusceptibility
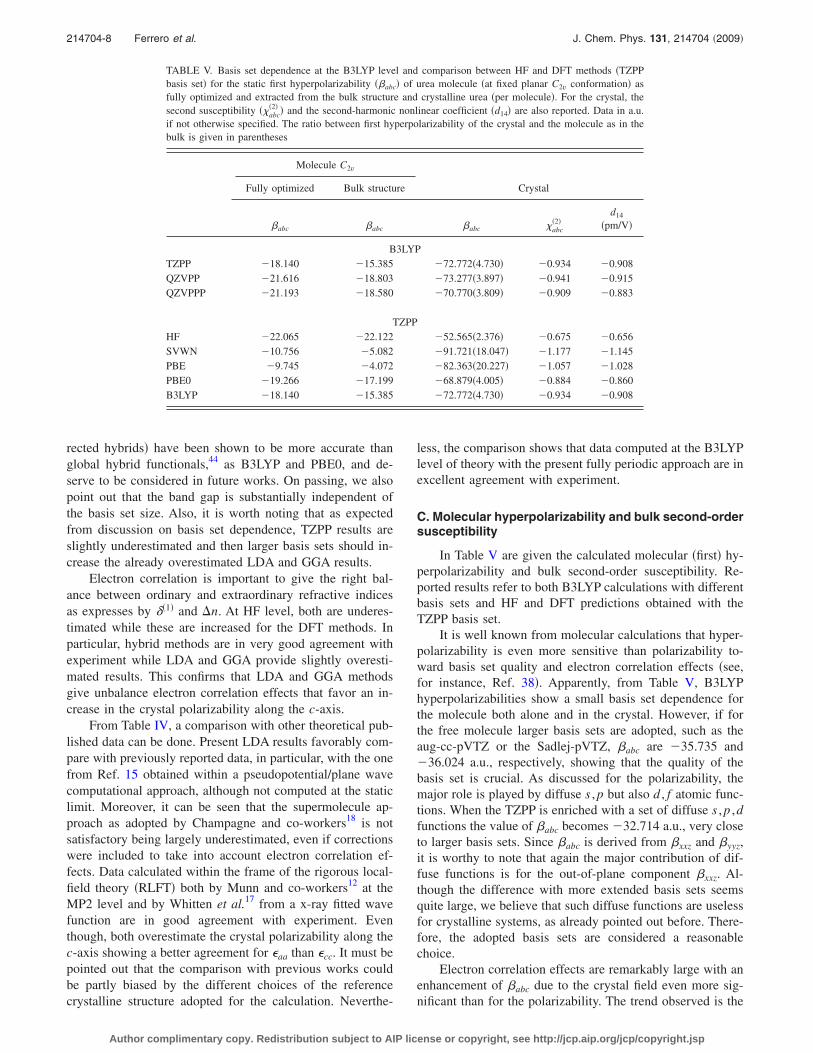
In Table V are given the calculated molecular �first� hy-perpolarizability and bulk second-order susceptibility. Re-ported results refer to both B3LYP calculations with differentbasis sets and HF and DFT predictions obtained with theTZPP basis set.
It is well known from molecular calculations that hyper-polarizability is even more sensitive than polarizability to-ward basis set quality and electron correlation effects �see,for instance, Ref. 38�. Apparently, from Table V, B3LYPhyperpolarizabilities show a small basis set dependence forthe molecule both alone and in the crystal. However, if forthe free molecule larger basis sets are adopted, such as theaug-cc-pVTZ or the Sadlej-pVTZ, �abc are �35.735 and�36.024 a.u., respectively, showing that the quality of thebasis set is crucial. As discussed for the polarizability, themajor role is played by diffuse s , p but also d , f atomic func-tions. When the TZPP is enriched with a set of diffuse s , p ,dfunctions the value of �abc becomes �32.714 a.u., very closeto larger basis sets. Since �abc is derived from �xxz and �yyz,it is worthy to note that again the major contribution of dif-fuse functions is for the out-of-plane component �xxz. Al-though the difference with more extended basis sets seemsquite large, we believe that such diffuse functions are uselessfor crystalline systems, as already pointed out before. There-fore, the adopted basis sets are considered a reasonablechoice.
Electron correlation effects are remarkably large with anenhancement of �abc due to the crystal field even more sig-nificant than for the polarizability. The trend observed is the
TABLE V. Basis set dependence at the B3LYP level and comparison between HF and DFT methods �TZPPbasis set� for the static first hyperpolarizability ��abc� of urea molecule �at fixed planar C2v conformation� asfully optimized and extracted from the bulk structure and crystalline urea �per molecule�. For the crystal, thesecond susceptibility ��abc
�2� � and the second-harmonic nonlinear coefficient �d14� are also reported. Data in a.u.if not otherwise specified. The ratio between first hyperpolarizability of the crystal and the molecule as in thebulk is given in parentheses
Molecule C2v
CrystalFully optimized Bulk structure
�abc �abc �abc �abc�2�
d14
�pm/V�
B3LYPTZPP �18.140 �15.385 �72.772�4.730� �0.934 �0.908QZVPP �21.616 �18.803 �73.277�3.897� �0.941 �0.915QZVPPP �21.193 �18.580 �70.770�3.809� �0.909 �0.883
TZPPHF �22.065 �22.122 �52.565�2.376� �0.675 �0.656SVWN �10.756 �5.082 �91.721�18.047� �1.177 �1.145PBE �9.745 �4.072 �82.363�20.227� �1.057 �1.028PBE0 �19.266 �17.199 �68.879�4.005� �0.884 �0.860B3LYP �18.140 �15.385 �72.772�4.730� �0.934 �0.908
214704-8 Ferrero et al. J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

same, with LDA, GGA, and hybrid methods being largerthan HF, in order. The inclusion of HF exact exchange inhybrid functional leads to results in between GGA and HF. Inthis case, the same arguments on the role of the band gapused for �aa�cc� also hold for �abc. For the molecule, SVWNand PBE provide very low values for �abc with respect to HFand hybrids. As already pointed out by Benková et al.45 incomparison with high-level CCSD�T� calculations, this isdue to an overall underestimation of the �xxz component atDFT level, but also �yyz results to be problematic with LDAand GGA functionals. Moreover, with those methods, thecharge density is more polarized along the molecular z-axis�i.e., along the C=O bond� than at HF level and with hy-brids.
No experimental value for the second-order susceptibil-ity is available at the static limit. Nevertheless, measure-ments of d14 at 597 and 1064 nm of 1.3�0.3 and1.2�0.1 pm /V, respectively, indicate that the dispersion tothe static limit is not significant given the substantial errorbars and the large band gap of urea.11 Therefore, this sug-gests that at the static limit a reasonable value for the second-order susceptibility of urea could be 1.0�0.2 pm /V.
Hybrid methods give slightly underestimated resultswhile GGA and LDA are close to the observed estimation.Indeed, d14 ranges from �0.86 to �1.15 pm/V for all of theDFT methods in substantial agreement with experiment andwell within the experimental uncertainty, even if the esti-mated experimental error bar is rather large being �20%.This indicates that for qualitative purposes also LDA andGGA functionals can be used to predict linear and nonlinearoptical properties of crystalline urea.
Furthermore, we point out that the sign of �abc�2� cannot be
determined from experimental measurements. Hence, ac-cording to present crystallographic directions, results showthat independent of the level of theory �abc
�2� has a negativesign. This finding is in contrast with previous periodic abinitio calculations15 but in agreement with respect to recentsupermolecular results.18
Finally, it is worthy to note that the vibrational contribu-tion to �abc
�2� might very well have an even larger effect than itdoes on �aa�cc�
�1� where it is already quite large. Although it is
not yet possible, to our knowledge, to compute it for crystal-line systems, in principle, it could be estimated as the differ-ence between measured second-harmonic generation andPockels effect.
IV. CONCLUSIONS
The present work shows that the macroscopic opticalsusceptibilities of crystalline urea can be reliably predictedthrough a fully periodic CPHF/KS approach developed forHF and DFT methods and atom-centered basis functions asimplemented in the CRYSTAL code. In particular, hybrid HF/DFT functionals, such as B3LYP, give good performance inpredicting the first- and second-order susceptibilities of urea.
We have carefully investigated the appropriate descrip-tion of the wave function employing 12 basis sets of increas-ing size and quality at the B3LYP level of theory. The basisset must be of TZ quality and include Gaussian functions
with low exponents �i.e., less than 0.1 a.u.�, compatible withthe periodic nature of the crystal, and also first- and second-order polarization functions �i.e., d and f�. In particular, atleast two sets of polarization functions �i.e., 2d , f on heavyatoms and 2p ,d on hydrogen� are needed, in agreement withevidence for molecules as demonstrated in the constructionof Sadlej’s basis sets especially devised to reproduce mo-lecular �hyper-� polarizability. As expected, dependence onthe basis set quality is more delicate for hyperpolarizability��� than polarizability ���. However, because of the periodicnature of the crystalline wave function, basis set require-ments are less critical than for molecules.
Electron correlation effects are very important for a cor-rect description of the response of crystalline urea to electricfields through its role on intermolecular interactions. ForDFT methods, the inclusion of exact exchange is shown tobe substantial to reduce the effect of the self-interaction erroron the electronic structure and partly recover the correctasymptotic limit of the exchange-correlation potential.
Overall, the present fully periodic CPHF/KS scheme of-fers, at variance with other approaches, two important advan-tages. It allows �i� the consistent inclusion of crystal environ-ment effects and �ii� the use of hybrid DFT methods. Hence,it offers an accurate computational tool to predict the linearand nonlinear optical properties of molecular crystals and, inperspective, it can be fruitfully applied for the developmentof new and improved molecular materials. Work is inprogress to extend present approach to third-order suscepti-bility, ��3�, and to include frequency dependence.
ACKNOWLEDGMENTS
Authors are grateful to Professor B. Kirtman for readingthe manuscript and for useful discussions and suggestions.R.D. acknowledges financial support from the MIUR ProjectCOFIN2007 No 200755ZKR_004�.
1 R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson,F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, Ph. D’Arco,and M. Llunell, CRYSTAL06 User’s Manual, Università di Torino, Torino,2006.
2 M. Ferrero, M. Rérat, R. Orlando, and R. Dovesi, in Computation inModern Science and Engineering, edited by T. E. Simos and G. Maroulis�American Institute of Physics, Melville, New York, 2007�, Vol. 2B.
3 M. Ferrero, M. Rérat, R. Orlando, and R. Dovesi, J. Comput. Chem. 29,1450 �2008�.
4 G. J. B. Hurst and M. Dupuis, J. Chem. Phys. 89, 385 �1988�.5 B. Kirtman, F. L. Gu, and D. M. Bishop, J. Chem. Phys. 113, 1294�2000�.
6 K. N. Kudin and G. Scuseria, J. Chem. Phys. 113, 7779 �2000�.7 M. Ferrero, M. Rérat, B. Kirtman, and R. Dovesi, J. Chem. Phys. 129,244110 �2008�.
8 M. Ferrero, M. Rérat, R. Orlando, R. Dovesi, and I. Bush, J. Phys.: Conf.Ser. 117, 012016 �2008�.
9 B. Champagne and D. M. Bishop, Adv. Chem. Phys. 126, 41 �2003�.10 J. Perez and M. Dupuis, J. Chem. Phys. 95, 6525 �1991�.11 Z. H. Levine and D. C. Allan, Phys. Rev. B 48, 7783 �1993�.12 H. Reis, M. G. Papadopoulos, and R. W. Munn, J. Chem. Phys. 109,
6828 �1998�.13 H. Reis, M. G. Papadopoulos, C. Hättig, J. G. Ángyán, and R. W. Munn,
J. Chem. Phys. 112, 6161 �2000�.14 K. Wu, J. G. Snijders, and C. Lin, J. Phys. Chem. B 106, 8954 �2002�.15 Z. Lin, Z. Wang, C. Chen, and M.-H. Lee, J. Chem. Phys. 118, 2349
�2003�.16 B. Skwara, R. W. Gora, and W. Bartkowiak, Chem. Phys. Lett. 406, 29
214704-9 Dielectric properties of crystalline urea J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp

�2005�.17 A. E. Whitten, D. Jayatilaka, and M. A. Spackman, J. Chem. Phys. 125,
174505 �2006�.18 M. Olejniczak, M. Pecul, B. Champagne, and E. Botek, J. Chem. Phys.
128, 244713 �2008�.19 P. A. M. Dirac, Proc. Cambridge Philos. Soc. 26, 376 �1930�.20 S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys. 58, 1200 �1980�.21 J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865
�1996�.22 A. D. Becke, J. Chem. Phys. 98, 5648 �1993�.23 C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B 37, 785 �1988�.24 B. Miehlich, A. Savin, H. Stoll, and H. Preuss, Chem. Phys. Lett. 157,
200 �1989�.25 C. Adamo and V. Barone, J. Chem. Phys. 110, 6158 �1999�.26 W. J. Hehre, L. Radom, P. R. Schleyer, and J. A. Pople, Ab Initio Mo-
lecular Orbital Theory �Wiley, New York, 1986�.27 A. J. Thakkar, T. Koga, M. Saito, and R. E. Hoffmeyer, Int. J. Quantum
Chem. 27, 343 �1993�.28 A. Schäfer, H. Horn, and R. Ahlrichs, J. Chem. Phys. 97, 2571 �1992�.29 F. Pascale, C. M. Zicovich-Wilson, F. López Gejo, B. Civalleri, R. Or-
lando, and R. Dovesi, J. Comput. Chem. 25, 888 �2004�.30 S. Swaminathan, B. N. Craven, and R. K. McMullan, Acta Crystallogr.,
Sect. B: Struct. Sci. 40, 300 �1984�.31 K. Doll, V. R. Saunders, and N. M. Harrison, Int. J. Quantum Chem. 82,
1 �2001�.
32 K. Doll, Comput. Phys. Commun. 137, 74 �2001�.33 K. Doll, R. Dovesi, and R. Orlando, Theor. Chem. Acc. 112, 394 �2004�.34 B. Civalleri, K. Doll, and C. M. Zicovich-Wilson, J. Phys. Chem. B 111,
26 �2007�.35 H. B. Schlegel, J. Comput. Chem. 3, 214 �1982�.36 M. J. Rosker, K. Cheng, and C. L. Tang, IEEE J. Quantum Electron. 21,
1600 �1985�.37 J.-M. Halbout, S. Blit, W. Donaldson, and C. L. Tang, IEEE J. Quantum
Electron. 15, 1176 �1979�.38 T. Pluta and A. J. Sadlej, J. Chem. Phys. 114, 136 �2001�.39 H. Birkedal, D. Madsen, R. H. Mathiesen, K. Knudsen, H.-P. Weber, P.
Pattison, and D. Schwarzenbach, Acta Crystallogr., Sect. A: Found. Crys-tallogr. 60, 371 �2004�.
40 M. Born and K. Huang, Dynamical Theory of Crystal Lattices �OxfordUniversity Press, Oxford, 1954�.
41 C. M. Zicovich-Wilson, F. J. Torres, F. Pascale, L. Valenzano, R. Or-lando, and R. Dovesi, J. Comput. Chem. 29, 2268 �2008�.
42 R. Hammond, K. Pencheva, K. J. Roberts, P. Mougin, and D. Wilkinson,J. Appl. Crystallogr. 38, 1038 �2005�.
43 B. G. Janesko, T. M. Henderson, and G. E. Scuseria, Phys. Chem. Chem.Phys. 11, 443 �2009�.
44 B. Kirtman, S. Bonness, A. Ramirez-Solis, B. Champagne, H. Matsu-moto, and H. Sekino, J. Chem. Phys. 128, 114108 �2008�.
45 Z. Benková, I. Cernusák, and P. Zaharadník, Int. J. Quantum Chem. 107,2133 �2007�.
214704-10 Ferrero et al. J. Chem. Phys. 131, 214704 �2009�
Author complimentary copy. Redistribution subject to AIP license or copyright, see http://jcp.aip.org/jcp/copyright.jsp




















