
TEMA 2. Enllaç peptídic
21
TEMA 2. Enllaç peptídic És una reacció de condensació: unió de 2 molècules perdent una molècula d’aigua. Important que els grups R no van sempre en el mateix sentit. Els carbonis α són quirals i els aa són L, per això hi ha les R alternades a dalt i abaix. L’enllaç peptídic té un caràcter parcial de doble enllaç, en veritat està en forma ressonant, els parell d’electrons es poden moure. Tot i això un enllaç doble impedeix el gir dels carbonis, per tant poden haver 2 configuracions: cis i trans Configuracions: cal trencar la molècula per formar la nova. Conformació: no cal trencar la molècula La constat de reacció d’hidròlisi no catalizada és de 10 -12 M -1 s -1 , això vol dir que és un enllaç molt estable que dura molt.
Transcript of TEMA 2. Enllaç peptídic

TEMA 2. Enllaç peptídic
És una reacció de condensació: unió de 2 molècules perdent una molècula d’aigua. Important
que els grups R no van sempre en el mateix sentit. Els carbonis α són quirals i els aa són L, per
això hi ha les R alternades a dalt i abaix.
L’enllaç peptídic té un caràcter parcial de doble enllaç, en veritat està en forma ressonant, els
parell d’electrons es poden moure. Tot i això un enllaç doble impedeix el gir dels carbonis, per
tant poden haver 2 configuracions: cis i trans
Configuracions: cal trencar la molècula per formar la nova.
Conformació: no cal trencar la molècula
La constat de reacció d’hidròlisi no catalizada és de 10-12 M-1s-1 , això vol dir que és un enllaç
molt estable que dura molt.

Els enllaços dobles en teoria són plans, els carbonis no són tetaèdrics, cada enllaç peptídic
defineix un pla. Són successions de plans, on el que pivota és la unió entre plans, límit que és
marcat per C-α, que sí que es tetraèdric i que sí que pot girar.
En els angles que poden lligar lliurement el anomenem psi (C- α amb C carbonílic) i fi (C-
α amb N). Són els angles dièdres. A més poden haver els anglès dièdres de les cadenes laterals
xi.

Les esferes verdes són cadenes laterals. En la configuració trans no hi ha cap xoc estèric, cosa
que si succeeix amb la cis, no hi caben, xoquen. Per això la relació entre configuració trans /cis
és de 1000.
L’excepció és la Prolina, hi ha poca diferència entre cis i trans i aquí la proporció és de 4.
Aquesta característica de la prolina permet que la proteïna pugui canviar de direcció.

Molts tipus de pèptids.
Llista molt llarga de noms molt coneguts.

Toxina de l’amanita faloides (bolet tòxic).
El pka del COOH i NH3 varia depenent de si està en forma d’aa lliure, de cadena peptídica o de
grup residual. EL grup carboxilat quan està en aa lliure perd el protó a pH=2, per tant és més
estable quan està a la cadena R.

Quin és el factor que modula que uns aa estiguin a dins de la proteïna o a la superfície? És la
constant dielèctrica. A l’aigua aquesta ct és de 80 i en canvi a l’interior de les proteïnes és de 6
aprox. L’entorn aquós afavoreix la forma ionitzada (l’aigua apantalla les càrrgues i s’hi troben
còmodes) i en canvi a dins de les proteïnes lo afavorit són les formes no ionitzades.
A l’interior de les proteïnes podem trobar formes electròfiles i nucleòfiles de dirents aa.
L’entorn proteic modula la reactivitat dels aa.
Anàlisi de l’efecte de diferents mutacions (canvis puntuals) sobre el pka aparent de l’histidina
64. La mutació Lys 213 (bàsica, amb càrrega +, que esta a una distància de 17.6 Angstroms de
la His, distància gran) per Thr (no caregat), el pka de la His pateix un increment, per tant ara és
més estable pq el protó aguanta més.
Tb observem 2 canvis
més destacables, fem el
contrari, treiem
càrregues negatives, de
l’Asp i del Glu, i ara
desestabilitzem la His i el
pka disminueix.
Si fem les 2 coses alhora,
treiem Asp i Glu i ho
canviem els 2 per Lys, el
pka decreix encara més.

Que passa quan fem una addició salina en un medi on hi ha proteïnes? Veiem que la solubilitat
augmenta fins un punt de màxima solubilitat i a partir d’allà decreix de cop.
Sense força iònica tenim les proteïnes despullades, tenen aigua de solvatació al voltant i poc
apantallament que fa que algunes proteïnes es puguin unir entre elles i formar agregats, però
son parcialment soluble. Quan afegim sal apantallem molt la proteïna i ara la probabilitat que
les 2 proteïnes s’acostin es molt menor. Però quan augmenta molt la sal les proteïnes perden
la capa de solvatació perque aquesta aigua ha estat segrestada per la sal i les zones
hidrofòbiques de les proteïnes tendeixen a unir-se i a precipitar.
Hi ha moltes excepcions de l’afirmació: un gen 1 prot1conformació1funció. Però serveix
bastant bé.

Les seq de proteïnes tb ens informen sobre l’evolució. Veiem els aa comuns i quins es
conserven, i podem fer arbres evolutius.
A la diagonal hi ha 0 pq es la seq comparada amb ella mateixa.

Comparació del citocrom C. Al gràfic de la dreta veiem les taxes d’evolució de 4 proteïnes que
no tenen res a veure entre elles. Veiem que la taxa de variació és molt diferent entre la
Histona H4, el citocrom C, l’hemoglobina i els fibrinopeptids. La histona H4 es poc diferent
respecte l’inici dels temps, l’ancestre comú i tot lo contrari passa amb els fibrinopeptids. Això
és pq un canvi estructural en la histona H4 seria letal ja que deixarien de funcionar la mitosi i la
meiosi, és una proteïna que accepta poques mutacions.
El citocrom C tb es molt important, en els seu cas x la respiració. És una proteïna monomèrica,
actua tot sol, no depen d’altres proteïnes i per tant accepta més mutacions i lo mateix passa
amb l’hemoglobina, que tb fa una funcio imporantissima ella sola.
Els fibrinopeptids són com parts de proteïnes i son importants però no tant vital.
La gràfica ens diu quant temps triga una proteïna a canviar cert nombre d’aa, la mesura són les
u.e.p. El temps és més gran per la histona H4 i mes petit pels fibrinopèptids.

La conformació es conserva molt més que la seqüencia. Hi ha moltes seq a la natura que dónen
lloc a la mateixa conformació proteica.
Mètodes per seqüenciar proteïnes. Normalment s’obtenen les proteïnes tal com surten del
ribosoma.
Seqüenciació d’Edman. Anar “arrencant” per mètodes químics tots els aa des de l’N terminal.
La reacció està en NH2 (és a dir, medi alcalí, forma nucleòfila), després hi ha una ciclació de la
molècula. Obtenim una molècula que conté l’aminoàcid i per altra banda la resta de la
proteïna. Llavors s’analitza per espectrofotometria la molècula ciclada, i depèn de com sigui es
pic es dedueix que es un aminoàcid o un altre. Amb aixo podriem seqüenciar tota la proteïna,
pero res té eficiencia del 100%. Si cada cicle te una eficiencia del 99%, i hi ha 35 cicles sen’s van
acumulant
subproductes que
també tenen N
terminal i es
queden també
enganxats a la
resina.

Fem 2 aliquotes i digerim la proteïna amb diferents enzims i en cada digestió obtindrem
diferents trossos. Tindrem un cromatograma de cada experiment. No sabem quin pic
correspon a cada tros.
Refent el trencaclosques podem deduir l’ordre i la seqüencia peptídica.

El mètode que més s’utilitza és el d’espectrometria de masses. Es basava en sublimar
components moleculars i s’analitzava a través d’una càmara de buit. S’han desenvolupat
mètodes per a poder-ho aplicar a proteïnes.
S’agafa la molècula, s’ionitza (posar-la a un medi on hi hagi càrregues), fem passar la molècula
per un tub de buit amb un camp magnètic. El seu desplaçament dependrà de la seva massa, i
finalment les molècules arriben a un detector que pot determinar el temps que ha trigat la
molècula desde que ha sortit.
Quan apliquem aquesta tècnia a macromolècules hi ha 2 tècniques: ESI i MALDI.
ESI. Tenim una mostra de proteïna en dissolució, però enlloc de sublimar-les es fa un spray, a
partir d’una càmara al buit amb una diferència de voltatge les molècules es desvien en un
sentit o altre depenent de la seva càrrega i de nou son detectades per un detector que fa de
“cronòmetre”. Primer les molècules (totes son la mateixa) surten en forma de gotes grans i es
van assecant. Després s’analitza el quocient m/z i podem calcular la seva massa molecular.

Anàlisi de MS per una proteïna. Els diferents pics representen diferents carregues de la
molecula. Multiplicant el m/z de cada pic per la càrrega (z) obtenim la m, i ha de sortir lo
mateix per cada pic.

S’associa una càrrega a cada pic iterativament fins que tots els pics compleixin l’equació. És un
mètode molt precís, fins a valors decimals.
I ara falta la part de seqüenciar. Són 2 experiments de MS. Tenim una proteïna digerida
(diferens trossets) i els fem passar per una cambra de separació (només fem passar un únic
fragment, varies fragments iguals, tots iguals). El que surt de la primera cambra va a una
cambra de colisió, que conté Argó, hi la molècula es trenca per l’enllaç peptídic i tindrem molts
trossos d’aquell fragment de la proteïna digerida de diferents mides.
Analitzem la mida dels fragments que surten de la cambra de colisió, i segons la diferència de
les seves masses deduïm quin aminoàcid hi ha entre mig.


Empremta dactilar peptídica. Primer es fa electroforesi de 2 dimensions, es fa una digestió
amb un enzim que talla per llocs específics. Tenim 2 divisions diferents d’un mateix genoma,
dues linies diferents. Veuriem que putse a un lloc s’expressa més una proteïna que a l’altre (al
gel). S’agafa el pic que ens interessa, retallar la zona en la q hi ha la senyal en el gel
electroforètic, solubilitzem , la tractem amb un gen digestiu i obtenim fragments de la
proteïna, llavors obtenim l’emprempta de la proteïna que tindrà uns pics únics a partir
d’espectrometria molecular.

Lo que hem fet abans era experiment humit, pq estava fet en el lab. Ho podem fer en sec, amb
base de dades. Com que la seqüencia és única, l’emprempta peptidica obtinguda al lab ha de
coincidir amb alguna proteïna que estigui enregistrada al banc de dades (sempre q la proteïna
en qüestió estigui al banc).

Síntesi de pètids. Eina important en farmacèutica. Per mètodes quimics, encara no s’ha pogut
fer amb enzims. Inconvenient que in vitro no es treballa en condicions tant suaus i específiques
com ho fan els enzims, al lab hem de forçar les condicions, poden haver reaccions paralel·les
no desitjades.
Els grups Boc són grups protectors, que eviten que es produeixi reacció en una zona de la
molècula q no volem q reaccioni.
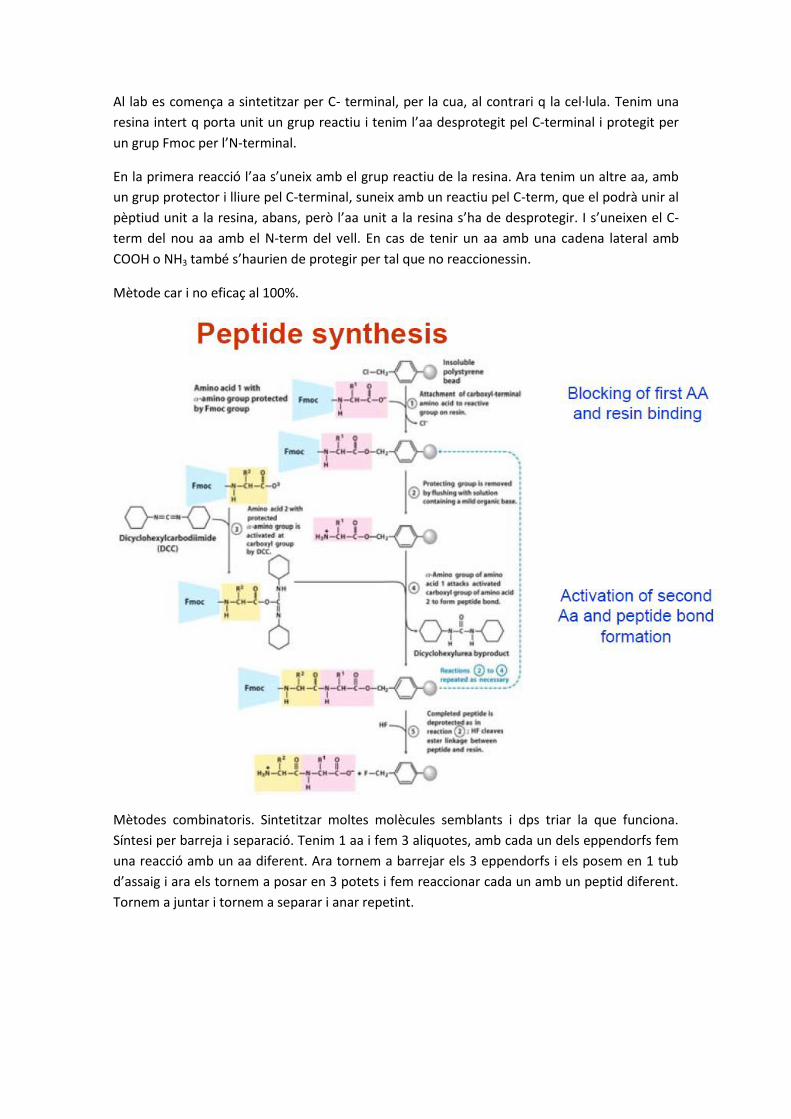
Al lab es comença a sintetitzar per C- terminal, per la cua, al contrari q la cel·lula. Tenim una
resina intert q porta unit un grup reactiu i tenim l’aa desprotegit pel C-terminal i protegit per
un grup Fmoc per l’N-terminal.
En la primera reacció l’aa s’uneix amb el grup reactiu de la resina. Ara tenim un altre aa, amb
un grup protector i lliure pel C-terminal, suneix amb un reactiu pel C-term, que el podrà unir al
pèptiud unit a la resina, abans, però l’aa unit a la resina s’ha de desprotegir. I s’uneixen el C-
term del nou aa amb el N-term del vell. En cas de tenir un aa amb una cadena lateral amb
COOH o NH3 també s’haurien de protegir per tal que no reaccionessin.
Mètode car i no eficaç al 100%.
Mètodes combinatoris. Sintetitzar moltes molècules semblants i dps triar la que funciona.
Síntesi per barreja i separació. Tenim 1 aa i fem 3 aliquotes, amb cada un dels eppendorfs fem
una reacció amb un aa diferent. Ara tornem a barrejar els 3 eppendorfs i els posem en 1 tub
d’assaig i ara els tornem a posar en 3 potets i fem reaccionar cada un amb un peptid diferent.
Tornem a juntar i tornem a separar i anar repetint.


Com trobem quin funciona entre 25.000 milions de pèptids? Ens ho explicaran a proteòmica.
Tenim mètodes, molècules, que ens serveixen x fer possible una reacció o no. Fem química
combinatòria amb un microxip. Suposem q tenim una part del microxip en el q hem ajuntat
una part d’un aa, q està protegit. En el xip dissenyem que volem una seq diferent en cada aa
del microxip. Irradiem amb llum i desbloquegem els aa q desitgem, i ara podem fer la reacció
amb un altre aa allà on volem. Així podem dissenyar pèptids dels quals sabem la seqüència.
S’iluminarà una llum verda amb el punt que funcioni, i ja sabrem la seq q té pq l’haviem
dissenyat prèviament.
La síntesis de pèotids quan es fa de manera dissenyada s’ha de fer amb molta seguertat.













![08 Tema 2 morfologia[1]](https://static.fdokumen.com/doc/165x107/6319d89ab41f9c8c6e09ee54/08-tema-2-morfologia1.jpg)






