
Tautomerism of N-unsubstituted pyrazolones (hydroxypyrazoles): MNDO and MNDO + CI study of...
18
Journal of Molecular Structure (Theochem), 258 (1992) 217-234 Elsevier Science Publishers B.V., Amsterdam 217 Tautomerism of N-unsubstituted pyrazolones (hydroxypyrazoles): MNDO and MNDO + CI study of C-substituted tautomers Venelin Enchev and Georgi D. Neykov Institute of Organic Chemistry, Bulgarian Academy of Sciences, 1113 Sofia (Bulgaria) (Received 21 May 1991; in final form 26 October 1991) Abstract The heats of formation, dipole moments, polarizabilities and ionization potentials of 96 compounds, eight theoretically possible tautomeric forms of N-unsubstituted pyrazolones (hydroxypyrazoles) and 11 of their C-substituted derivatives, are calculated by means of the MNDO method, with and without configuration interaction (CI). The MNDO+ CI results for the relative stabilities are in agreement with the available experimental data. They predict that in all cases the most stable in the vapour phase are hydroxypyrazole forms, excepting 3- and 4-phenyl substituted compounds, among which the most stable are the 2-pyrazoline-5- one and 1-pyrazoline-3-one forms, respectively. The influence of the substituents on the quantities characterizing the electronic structure is discussed. INTRODUCTION The pyrazolone derivatives find wide application in medicine, dye pro- duction and colour photography [l]. Recently, they have been used as complexation agents [2]. The N-unsubstituted pyrazolones exhibit keto- enol tautomerism and they can theoretically exist in eight tautomeric forms (Fig. 1) The tautomeric equilibrium is influenced both by external factors and by the substituents in the third and fourth positions in the five-membered ring. They can shift the equilibrium in such a direction that only one of the tautomers will be predominant. Experimental investigations have been performed on the tautomerism in 3-methyl[4-11],3-phenyl[9-141, 4-methyl [S] and 3,cdimethyl [8] substituted forms. The results have been summarized in reviews [3,15,16]. Theoretical calculations have been per- Correspondence to: V. Enchev, Institute of Organic Chemistry, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria. 0166-1260/92/$05.06 0 1992 Elsevier Science Publishers B.V. All rights reserved.
Transcript of Tautomerism of N-unsubstituted pyrazolones (hydroxypyrazoles): MNDO and MNDO + CI study of...

Journal of Molecular Structure (Theochem), 258 (1992) 217-234 Elsevier Science Publishers B.V., Amsterdam
217
Tautomerism of N-unsubstituted pyrazolones (hydroxypyrazoles): MNDO and MNDO + CI study of C-substituted tautomers
Venelin Enchev and Georgi D. Neykov
Institute of Organic Chemistry, Bulgarian Academy of Sciences, 1113 Sofia (Bulgaria)
(Received 21 May 1991; in final form 26 October 1991)
Abstract
The heats of formation, dipole moments, polarizabilities and ionization potentials of 96 compounds, eight theoretically possible tautomeric forms of N-unsubstituted pyrazolones (hydroxypyrazoles) and 11 of their C-substituted derivatives, are calculated by means of the MNDO method, with and without configuration interaction (CI). The MNDO+ CI results for the relative stabilities are in agreement with the available experimental data. They predict that in all cases the most stable in the vapour phase are hydroxypyrazole forms, excepting 3- and 4-phenyl substituted compounds, among which the most stable are the 2-pyrazoline-5- one and 1-pyrazoline-3-one forms, respectively.
The influence of the substituents on the quantities characterizing the electronic structure is discussed.
INTRODUCTION
The pyrazolone derivatives find wide application in medicine, dye pro- duction and colour photography [l]. Recently, they have been used as complexation agents [2]. The N-unsubstituted pyrazolones exhibit keto- enol tautomerism and they can theoretically exist in eight tautomeric forms (Fig. 1) The tautomeric equilibrium is influenced both by external factors and by the substituents in the third and fourth positions in the five-membered ring. They can shift the equilibrium in such a direction that only one of the tautomers will be predominant. Experimental investigations have been performed on the tautomerism in 3-methyl[4-11],3-phenyl[9-141, 4-methyl [S] and 3,cdimethyl [8] substituted forms. The results have been summarized in reviews [3,15,16]. Theoretical calculations have been per-
Correspondence to: V. Enchev, Institute of Organic Chemistry, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria.
0166-1260/92/$05.06 0 1992 Elsevier Science Publishers B.V. All rights reserved.

218
A
yt td/m
15
10
5
0 -
H
6.47
I 7.20
A B C D E F G H
Fig. 1. MNDO+CI calculated relative stabilities of the tautomers of the hydroxypyrazoles (pyrazolones) 1.
formed for tautomers of pyrazolone, and for some of its derivatives [17-221, using semiempirical (HMO, o-HMO, PPP, CNDO) methods. We have recently pointed out [23] that the MNDO+ CI method, compared to the other comprehensive semiempirical quantum chemical methods, reflects more adequately the experimental results for unsubstituted pyrazolones.
The present investigation is a theoretical study of the influence of dif- ferent substituents in the third and fourth positions in the five-membered ring on the relative stability and structure of the tautomers of pyrazolone (hydroxypyrazole) derivatives, using the MNDO method with full geometry optimization. Knowledge of the geometry, electronic structure and rela- tive stability of the different tautomeric forms may serve as a basis for understanding their biological activity and their possibilities for complex formation.
The IUPAC nomenclature denominations of the basic tautomeric forms of the compound 1 (Fig. 1) are: 5-hydroxypyrazole, A; 3-hydroxypyrazole, B; 1,3-pyrazoline-3-01, C; 2,5-pyrazoline-3-01, D; 1,4-pyrazoline-3-01, E; 2-pyr- azoline-Bone, F; 4-pyrazoline-3-one, G; 1-pyrazoline-8one, H. The investi- gations include 3-, 4- and 3,4substituted derivatives of the above tautomers, which are shown in Figs. 2-12.

219
AHf Hc&md
I.5
lo
5
0
H\o %
H
C”a 4.51
A I3 c 0 E F G
CH> HH 0. 2s
N-N W¶
i l3.93
H
Fig. 2. MNDO+CI calculated relative stabilities of the tautomers of the 3-methyl substituted hydroxypyrazoles (pyrazolones) 2.
COMPUTATIONAL METHOD
All calculations were performed by the MNDO method [24], with full geometry optimization of the compounds investigated. The optimization was first done by the Davidon-Fletcher-Powell procedure [25] and then by the Powell [26] procedure. After geometry optimization at the SCF level, additional single-point configuration interaction calculations were carried out (selected single and double excited configurations taken into account) according to the method described by Dewar and Liotard [27]. The starting geometries for the five-membered rings of all investigated tautomers A-H for the compounds 2-12 were based on results obtained for the tautomers of 1 given in our previous paper [23]. Standard values were used for bond lengths, and for angles of the substituents in the third and fourth positions in the five-membered ring. All calculations were performed using the IBM version of the AMPAC programme [28].
RESULTS AND DISCUSSION
The theoretical prediction of the tautomeric stability of a molecule [29,30] involves a determination of differences between the total SCF

5 - &33 I
7.66
O-
A B C
..O! I u 9.82
5
I
D E
1:
4.63
F G H
Fig. 3. MNDO+CI calculated relative stabilities of the tautomers of the 3-phenyl substituted hydroxypyrazoles (pyrazolones) 3.
(AESCF) + electron correlation (AE”“) energies of the tautomers, including any potential differences in their zero-point vibrational energies (AEvib) to obtain the relative internal energies at 0 K
AEW = AEsCF + AEc0” + AEvib
It should be noted that MO parameters for MNDO are optimized to reproduce the experimental heat of formation (i.e. the standard enthalpy of formation, the enthalpy change in forming a mole of compound at 25OC from its elements in their standard states) in addition to observed geome- tries (mostly at 25OC). In this sense, E SCF (defined as H,), force constants, normal vibration frequencies, etc. are all related to the values at 25’ C (not 0 K). However, the semiempirical estimations of BEti would in some cases be small [29]: for example, our calculations for AZPE of the tautomers of compound 1 are of the order of 0.09-0.94 kcal mol-‘.
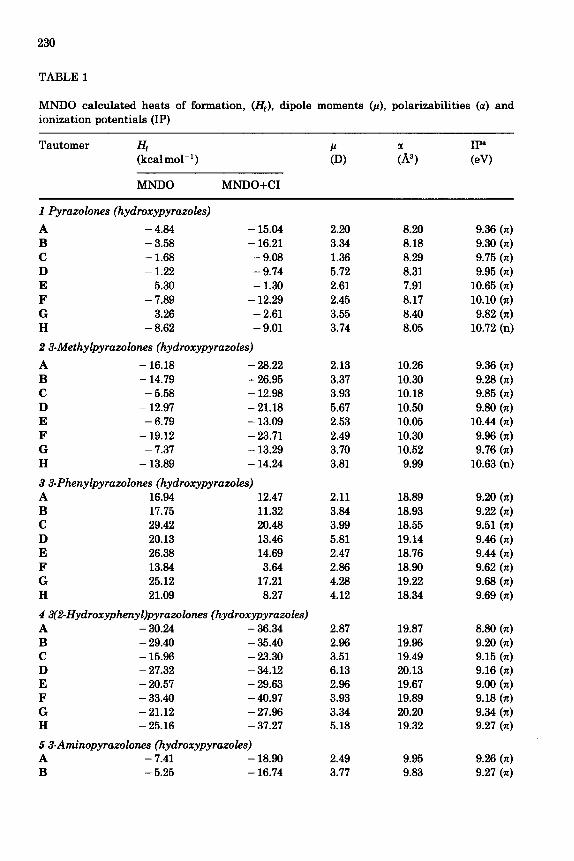
The calculated values for the heats of formation of the investigated Nunsubstituted pyrazolones 1-12 are given in Table 1.

221
AH
HCdh f
15
lo
5
0
0.67
Qy$&!!? H’ 04
T T I 5:n
4.63
I I
A B C 0 E F G H
Ok I
6.65
Fig. 4. MNDO+CI calculated relative stabilities of the tautomers of the 3(2-hydroxyphenyl) substituted hydroxypyrazoles (pyrazolones) 4.
Influence of the configuration interaction
The data concerning the heats of formation (Hf) of the tautomers A-H of all investigated compounds 1-12, calculated with (MNDO+CI) or without (MNDO) taking into account configuration interactions are given in Table 1. As can be seen from Table 1, the influence of the configuration interaction is expressed most strongly in tautomers A and B of compounds 1,2,5,7,9 and 11. In these cases, AH, (= Hymo - HfvImo+‘I) is in the range W-12.6 kcal mall’. The changes in Hf for tautomer H of compounds 3,4 and 8 is also in the same range. The influence of CI is expressed most weakly in tautomer H of all the compounds investigated, except for those of compounds 3, 8 and 12. In the 3- and 4-nitro substituted tautomers, the influence of CI is not clearly shown for certain tautomers.
Taking into account the CI in the unsubstituted tautomers of compound 1 changes the ordering of the relative stabilities of the different tautomers (see Table 1) and reconciles known theoretical and experimental results [16]. In the 3-substituted tautomers, not taking account of CI suggests tautomer F is the most stable; taking account of CI suggests tautomer A;

222
AHf ncal/md
15
lo
5
0
T 6.28
1.6:
D E
l3.b
HH ,$).
O* -6- r” /% H/ --N ad
T 3.60
F G
13.68
H
Fig. 5. MNDO+CI calculated relative stabilities of the tautomers of the 3-amino substituted hydroxypyrazoles (pyrazolones) 5.
this is in agreement with the experimental results for the 3-methyl sub- stituted tautomers [4-111. Owing to the fact that, when taking CI into account in phenyl substituted tautomers, the greatest decrease in the heat of formation is observed for H and F, tautomer F again proves to be the most stable. In this case, the theoretical predictions differ from the experi- mental results [12,14]: according to the latter tautomer F should not be present in the solution, owing to the absence of the characteristic band in the IR spectrum of 3-phenyl pyrazolone. MNDO and MNDO+CI predict tautomers G, E and C as the least stable relatively (Table 1).
In the 4-substituted tautomers, taking account of the CI does not con- siderably influence the ordering of the relative stabilities, except for the 4-phenyl substituted tautomers. In compounds 7, 9 and 10, both with and without taking account of CI, the most stable are the hydroxypyrazole forms A and B (see Table 1). In compound 8, without taking CI into account, the most stable forms are again A and B; on taking CI into account, tautomer H proves to be the most stable.
MNDO predicts tautomers G, D and E to be relatively the least stable for all investigated 4-substituted tautomers. Taking account of CI preserves the above mentioned result, except for compounds 7 and 9 (Table 1).
For the 3,4-substituted tautomers, taking account of CI has little influ-

223
AH1 d/m01
15
1 T
A 6 c D
X3.06
T 2.65
I
2
HH
0
-cxj, s “<
T 5.65
E F C H
Fig. 6. MNDO+CI calculated relative stabilities of the tautomers of the 3-nitro substituted hydroxypyrazoles (pyrazolones) 6.
ence on the ordering of the most stable tautomers (see the results for compounds 11 and 12 in Table 1).
Relative stabilities: influence of the substituents
The results for the heat of formation and relative stability discussed in this section were obtained by the MNDO+CI method, because in our previous paper [23] it has already been proved that this method reflects the known experimental data more adequately than the MNDO method [3,16]. The relative stability of the tautomers of the unsubstituted pyrazolone (hydroxypyrazole) (1) is shown in Fig. 1. In contrast to the investigations of Deschamps et al. [18] (according to which tautomer F is the most stable), MNDO+CI calculations predict that the most stable tautomer is B. This result is in agreement with the conclusion of Dorn [16] about hydroxy- pyrazole forms B and A being available in non-polar solvents. The calculated difference in the relative stabilities of the two hydroxypyrazole forms B and A is only 1.17 kcalmol-’ (Fig. 1). Small quantities of tautomer F are noticed together with B in weakly polar solvents [3]. The MNDO+CI calculations predict the difference in the relative stabilities of B and F to be 3.92 kcal mol-’ (Fig. 1). The 1-pyrazolin-3-one (H) is also known [31]. The CNDO/S calculations performed [23] using the MNDO optimized geometry

224
C
e:, T 1249
0
C’%
0,2 ?c H /
H’ N--N
8 T lcil?
E F G H
Fig. 7. MNDO+CI calculated relative stabilities of the tautomers of the Cmethyl substituted hydroxypyrazoles (pyrazolones) 7.
reproduce its absorption very well (A,, = 436 nm) in the visible region. The tautomers C and D, which must be more stable than H according to the MNDO+CI calculations (their relative stability is close to the relative stability of H), have not been synthesized yet. It ha8 been experimentally demonstrated that tautomer G (which is less stable than C and D) exists in protic solvents [3].
3Substituted tautomers (compounds 2-6)
The 5-hydroxy form is the most stable among the 3substituted tautomers (Figs. 2,5 and 6). This result is in agreement with the experimental data for non-polar solvents [8]. The tautomers of 3 and 4 are exceptions, in which the following ordering of the stabilities of the tautomeric forms has been theoretically predicted: F > H > B > A; F > H > A > B, respectively (Figs. 3 and 4). According to our calculations, the benzene (Zhydroxy- phenyl) ring is almost perpendicular to the five-membered ring. The intro- duction of a hydroxyl group in the ortho position in the benzene ring reduces the difference in the relative stabilities between these tautomers (cf. Fig. 3 and Fig. 4).

225
A HI nCal/mol
15 -
6.33
A B C f) E F G F
Fig. 8. MNDO+CI calculated relative stabilities of the tautomers of the 4-phenyl substituted hydroxypyrazoles (pyrazolones) 8.
&Substituted tautomers (compounds 7-10)
The tautomeric forms A and B (Bhydroxy and 3-hydroxy forms, respect- ively) were found to be the most stable among the 4-substituted tautomers (see Figs. 7, 9 and 10). Here again, the 4-phenyl substituted tautomers are exceptions (Fig. 8), in which the most stable is tautomer H, followed by A and B. The methyl group and the electron-withdrawing (EW) group (the nitro group) favour tautomer A (Shydroxy form); the electron-donating substituent (the amino group) favours tautomer B (the 3-hydroxy form). The difference in the relative stabilities of the tautomeric forms A and B of compounds 7 and 9, in which the substituents are the methyl and amino groups, respectively, is about 2 kcalmol-‘. The presence of an EW sub- stituent increases that difference. The benzene ring in compound 8 is situated almost perpendicularly to the pyrazolone ring (Fig. 8) and its influence on it should not differ from that of the methyl group. Neverthe- less, calculations predict tautomer H to be the most stable one.
3,4_Disubstituted tautomers (compounds 11 and 12)
The joint influence of the substituents in the third and fourth positions in the five-membered ring can be seen in Figs. 11 and 12. The influence of

226
“5 flc3.l/mol
15
10
5
3
1 2.9
c
MI
Fig. 9. ~ND~~CI calculated relative stabilities of the tautomers of the 4-amino substitute hydroxypyrazoles (pyrazolones) 9.
the nitro group in the fourth position (12) is so strong that it competes successfully with the influence of the Z-hydroxyphenyl group in the third position (cf. Figs, 4,10 and 12) and preserves the ordering A > B which is characteristic of the dnitrosubstituted tautomers. However, comparing compounds 10 and 12, the difference between the relative stabilities of these two tautomers can be seen to decrease by a factor of about 8 (see Figs. 10 and 12). Experimental data show [32] that tautomer A is predomi- nant for compound 12 in ethanol.
In 3,4-dimethyl substituted tautomers of 11, the ordering A > B > F is observed; the difference between the relative stabilities of tautomers A and B is very small (about 1 kcal mol-I). This result is in agreement with the known experimental data, according to which 11 exists as tautomers A and F [8]. The combined influence of methyl groups in the third and fourth positions leads to a decrease in the difference between the relative stabilities of A and B (see Hf for A and B in Figs. 2, ‘7 and 11).

227
AHI tK!al/d
3.5
10
5
0
A B C 0 E F G I-I
19.79
Fig. 10. MNDO+CI calculated relative stabilities of the tautomers of the 4-nitro substituted hydroxypyrazoles (pyrazolones) 10.
Dipole moments, polarizabilities and ionization potentials
The MNDO calculated values for the dipole moments p, polarizabilities 01 and ionization potentials TP for tautomers A-H of compounds l-12 are listed in Table 1.
As can be seen from Table 1, the dipole moments for the unsubstituted tautomers of 1 are in the range 1.36-3.74D, except for tautomer lD, whose calculated value of 5.73D is considerably higher. The experimental data {given in ref. 5) for the dipole moments of the unsubstitut~ tautomers lB, IF and 1G are 2.65, 2.83 and 5.03D respectively. The dipole moments of 1B and 1G calculated by us are higher by about 0.7D compared to the experi- mental data; for tautomer lF, the agreement with experiment is consider- ably better.
As can be expected, the methyl and phenyl. substituted tautomers of the studied compound f3,7,8 and 9) have dipole moments which donot differ much from those of the respective unsubstituted tautomers (see Table 1). An exception is observed for the C tautomers. For C tautomers, the dipole moments of the methyl and phenyl substituted tautomers (ZC, 3C, 7C, 8C

20
15
10
5
0
A B C 0
T /
b 1.4
E
cH’H 0. & 3 201
Y --N’
F G H
97
Fig. 11. MNDO+CI calculated relative stabilities of the tautomers of the 3,4dimethyl sub- stituted hydroxypyrazoles (pyrazolones) 11.
and 1OC) are considerably higher than the dipole moment of the unsub- stituted tautomer (1C). The difference Ap = ]A~,,~(X) - ~,,,~(x)] (X = A-H) for tautomer C when substituents are methyl and phenyl is of the order of 2.5D. Values of A,u of this order remain when the dipole moments are calculated with the CNDO/S method [33].
The polar substituents (amino, nitro and 2-hydroxyphenyl) depend to a greater degree on the distribution of electron density in the molecules, leading to an increase of the dipole moments compared to the unsubstituted tautomers. The exception is 5D, for which this difference Ap between the dipole moments of the unsubstituted and the substituted tautomers is considerably greater.
The polarizability of the molecules characterizes the ability of their

dHt -hd
15
10
5
T 4.42
D E F G H
Fig. 12. MNDO+CI calculated relative stabilities of the tautomers of the 3(2-hydroxyphenyl),4- nitro substituted hydroxypyrazoles (pyrazolonee) 12.
electron shells to become deformed under the influence of an electric field. The pola~zabilities, together with the dipole moments, determine the electric and optical properties of the substances.
The average value of the polarizability c1 is defined as
01 = f(%, + “yu + a,)
where clii are the components of the polarizability and i, j indicate respect- ive coordinates X, Y and Z. Dewar et al. [34] have shown that the MNDO results for a are systematically smaller than the experimental values. This is typical for the other semiempirical and ab initio methods with a minimal basis set.
The calculated polar&ability values of the unsubstituted tautomers (IA-1H) are similar and are in the range of 7.9--3.4A3. For the substitute tautomers, the functional groups 2-hydroxyphenyl, phenyl and nitro leads to a considerable increase of the polarizability compared to the methyl and amino groups.
For most of the tautomers with the same substitutents, the ordering is a3 > ot,, where 01~ and tr, are the polarizabilities of the tautomers with the substituents in positions 3 and 4, respectively. Tautomers of type C are exceptions for all compounds, with a3 < a,; this is also true for tautomer E when the substitutent is an amino group.
230
TABLE 1
MNDO calculated heats of formation, (IS,), dipole moments (p), polarizabilities (a) and ionization potentials (IP)
Tautomer & p u (kcalmol-‘) (D) (-w
MNDO MNDO+CI
1 Pyrazolones (hydroxypyrazoles)
A - 4.84 - 15.04 B - 3.58 - 16.21 C - 1.68 - 9.08 D - 1.22 - 9.74 E 5.30 - 1.30 F - 7.89 - 12.29 G 3.26 - 2.61 H - 8.62 - 9.01
2 EMethylpyrazolones (hydroxypyrazoles)
A - 16.18 - 28.22 B - 14.79 - 26.95 C - 5.58 - 12.98 D - 12.97 - 21.18 E - 6.79 - 13.09 F - 19.12 - 23.71 G - 7.37 - 13.29 I-I - 13.89 - 14.24
3 3-Phenylpyrazolones (hydroxypyrazoles) A 16.94 12.47 B 17.75 11.32 C 29.42 20.48 D 20.13 13.46 E 26.38 14.69 F 13.84 3.64 G 25.12 17.21 H 21.09 8.27
4 3(2-Hydroxyphenyl)pyrazolones (hydroxypyrazoles) A - 30.24 - 36.34 B - 29.40 - 35.40 C - 15.96 - 23.30 D - 27.32 - 34.12 E - 20.57 - 29.63 F - 33.40 - 40.97 G - 21.12 - 27.96 H - 25.16 - 37.27
5 EAminopyrazolones (hydroxypyrazoles) A - 7.41 - 18.90 B - 5.25 - 16.74
2.26 8.20 9.36 (n) 3.34 8.18 9.30 (n) 1.36 8.29 9.75 (a) 5.72 8.31 9.95 (n) 2.61 7.91 10.65 (R) 2.45 8.17 10.10 (K) 3.55 8.40 9.82 (n) 3.74 8.05 10.72 (n)
2.13 10.26 9.36 (n) 3.37 10.30 9.28 (x) 3.93 10.18 9.85 (n) 5.67 10.50 9.80 (K) 2.53 10.05 10.44 (a) 2.49 10.30 9.96 (n) 3.70 10.52 9.76 (x) 3.81 9.99 10.63 (n)
2.11 18.89 9.20 (x) 3.84 18.93 9.22 (n) 3.99 18.55 9.51 (a) 5.81 19.14 9.46 (n) 2.47 18.76 9.44 (n) 2.86 18.90 9.62 (n) 4.28 19.22 9.68 (x) 4.12 18.34 9.69 (x)
2.87 19.87 8.80 (a) 2.96 19.96 9.20 (n) 3.51 19.49 9.15 (7r) 6.13 20.13 9.16 (n) 2.96 19.67 9.00 (a) 3.93 19.89 9.18 (7c) 3.34 20.20 9.34 (n) 5.18 19.32 9.27 (n)
2.49 9.95 9.26 (n) 3.77 9.83 9.27 (n)

231
TABLE 1 (continued)
Tautomer H, P Ip” (kcalmol-‘) 0) (eW
MNDO MNDO+CI
C 3.61 - 3.75 D - 5.36 - 12.62 E 1.98 - 4.28 F - 10.66 - 15.10 G 0.03 - 5.73 H - 5.02 - 5.22
6 3-Nitropyrazolones (hydroxypyrazoks) A 10.80 6.09 B 13.83 11.39 C 19.18 17.22 D 16.95 14.76 E 22.99 19.15 F 10.24 8.14 G 23.69 23.01 I-I 11.98 11.74
7 kMethylpyrazolones (hydroxypyrazoles) A - 15.84 - 26.94 B - 14.34 - 25.21 C - 10.63 - 18.02 D - 6.08 - 14.45 E - 5.65 - 11.46 F - 12.61 - 16.77 G - 7.65 - 13.59 I-I - 12.73 - 13.11
8 kPhenylpyrazolones (hydroxypyrazoks) A 16.85 11.38 B 18.32 12.10 C 21.94 14.36 D 28.29 20.11 E 27.59 17.66 F 22.32 12.68 G 25.64 16.02 H 22.30 9.83
9 4Aminopyrazolones (hydroxypyrazoles) A - 6.88 - 17.07 B - 6.80 - 19.03 C - 1.06 - 8.30 D 3.23 - 1.91 E 1.49 - 4.44 F - 2.17 -6.10 G 2.46 - 3.55 H - 2.40 - 2.78
2.59 9.68 9.97 (n) 5.73 10.30 9.29 (R) 3.15 9.80 9.62 (n) 3.41 10.02 9.51 (x) 4.40 10.26 9.56 (n) 2.66 9.51 10.41 (n)
6.40 11.28 10.21 (?t) 2.33 11.51 10.21 (n) 5.25 11.35 10.72 (a) 6.59 11.84 10.89 (x) 5.60 11.00 11.71 (K) 2.71 11.36 10.97 (Ir) 1.38 11.53 10.70 (n) 3.95 11.07 11.55 (n)
2.07 10.23 9.28 (n) 3.25 10.22 9.28 (R) 3.84 10.36 9.72 (n) 5.71 10.14 9.92 (n) 2.67 10.03 10.42 (x) 2.53 10.04 10.08 (a) 3.51 10.51 9.70 (x) 3.80 9.91 10.64 (n)
2.24 18.78 9.17 (n) 3.34 18.77 9.22 (R) 4.22 18.87 9.50 (n) 6.17 18.34 9.79 (R) 2.98 18.54 9.53 (n) 2.88 18.29 9.64 (x) 3.48 19.11 9.41 (7c) 4.09 18.09 9.69 (z)
1.16 9.13 9.21 (n) 1.50 9.73 9.34 (7r) 3.15 9.91 9.70 (7r) 5.38 9.71 9.99 (x) 2.98 9.9i 9.58 (s) 1.34 9.58 10.19 (z) 2.49 10.06 9.65 (R) 4.47 9.43 10.67 (n)

232
TABLE 1 (continued)
Tautomer HI (kcal mol- ‘)
IP
(ev)
MNDO MNDO + CI
10 kNitropyrazolones (hydroxypyrazoles) A 5.94 4.34 4.26 B 8.70 7.55 4.44 c 14.72 11.91 3.86 D 23.00 21.14 3.73 E 23.47 22.28 2.54 F 14.43 14.40 3.13 G 20.61 19.13 6.48 H 15.27 15.06 3.87
11 3,kDimethylpyrazolones (hydroxypyrazoles) A - 26.12 - 36.95 1.98 B - 24.38 - 35.89 3.31 C - 15.74 - 22.84 3.83 D - 16.95 - 24.98 5.71 E - 16.76 - 22.54 2.61 F - 22.97 - 27.38 2.56 G - 16.99 - 22.80 3.67 H - 15.70 - 15.98 3.86
18 kNitro,3@hydroxyphenyl)pyrazolones (hydroxypyrazoles) A - 16.74 - 21.87 4.29 B - 15.30 - 21.47 4.23 c 0.21 - 8.17 4.15 D - 3.60 - 10.27 4.79 E - 1.43 - 7.43 2.52 F - 11.73 - 17.45 4.73 G - 5.86 - 10.45 7.77 H - 1.15 - 6.08 4.94
11.16 10.42 (n) 11.21 10.35 (x) 11.48 10.87 (x) 11.13 10.78 (n) 10.93 11.64 (x) 11.15 10.95 (x) 11.51 10.72 (x) 10.97 11.55 (n)
12.26 9.27 (R) 12.31 9.25 (R) 12.23 9.69 (n) 12.30 9.77 (n) 12.09 10.23 (n) 12.15 9.92 (n) 12.59 9.64 (n) 11.85 10.58 (n)
22.81 9.02 (n) 22.98 9.57 (n) 22.58 9.37 (n) 23.03 9.43 (R) 22.70 9.34 (x) 22.71 9.43 (77) 23.01 9.70 (n) 21.94 9.46 (n)
“The type of orbital from which the electron separates is shown in parentheses (see text).
As can be seen from Table 1, the ionization potential of the majority of the substituted tautomers is lower than the ionization potential of the respective unsubstituted tautomers. The nitro substituted tautomers A-H of compounds 6 and 10 are exceptions: their ionization potentials are systematically higher than those of the respective unsubstituted tautomers. This result can probably be ascribed to the considerable negative induction effect of the nitro group, which leads to an increase in the energy of the HOMO. The reverse effect is exerted by the phenyl and 2-hydroxyphenyl substitutents. The tautomers of compounds 3, 4 and 8 containing those substituents show ionization potentials about 0.5 eV lower than the ioniz- ation potentials of the respective unsubstituted tautomers. For tautomers

233
4F and 4H, this difference is considerably higher (0.92eV and 1.45 eV, respectively).
For the tautomers of the compound 12 (containing the 2-hydroxyphenyl and the nitro group in the five-membered ring in positions 3 and 4, respect- ively), the 2-hydroxyphenyl group exerts a dominant influence and the ionization potentials of these tautomers are nearer to those of the respective tautomers of compound 4 (see Table 1).
The type of the orbital from which the electron separates is another characteristic of the ionization potential. For non-planar systems, which most of the studied compounds are, the orbitals may be conventionally separated into n, K and 0.
As can be seen from Table 1, for all tautomers A-G of compounds 1-12, the higher occupied orbital is of type x. For most tautomers H the HOMO is of n type. An exception is observed for the compounds containing a phenyl ring as a substituent. In these cases, the higher occupied MO is of type ‘II.
SUPPLEMENTARY MATERIAL AVAILABLE
A complete set of optimized geometries of all tautomers of compounds 1-12 studied are available on request from the authors.
REFERENCES
1 2
3 4 5 6 7 8 9
10 11 12 13 14 15
16 17 18
H. Hennig, J. Tauchnitz, J. Korner and W. Schindler, J. Signal AM, 4 (1976) 125. B. Kuncheria and P. Indrasenan, Indian J. Chem., 27A (1988) 1005, and references cited therein. A.A. Solovskii, N.F. Rakova, N.E. Chebotareva and V.F. Mironov, Zh. Neorg. Khim., 34 (1989) 2966. The tautomerism of heterocycles, Adv. Heterocycl. Chem. Suppl. 1, (1976) pp. 346352. A.R. Katritzky and F.W. Maine, Tetrahedron, 20 (1964) 299, 315. A.R. Katritzky, F.W. Maine and S. Golding, Tetrahedron, 21 (1965) 1693. W.H. De Camp and J.M. Stewart, Acta Crystallogr., Sect. B, 27 (1971) 1227. J. Elguero, R. Jacguier and G. Tarrago, Bull. Sot. Chim. Fr., (1967) 3772. J. Elguero, R. Jacguier and G. Tarrago, Bull. Sot. Chim. Fr., (1967) 3780. J.M. Desmarchehelier and R.B. Johns, Org. Mass Spectrom., 2 (1969) 37. R. Jacguier, C. Petrus, E. Petrus and J. Verducci, Bull. Sot. Chim. Fr., (1970) 247. N.A. Evans, D.J. Whelan and R.B. Johns, Tetrahedron, 21 (1965) 3351. V.E. Martynov and I.B. Belov, Zh. Obshch. Khim., 33 (1963) 1092. C. Sabate-Alduy and J. Lematre, Bull. Sot. Chim. Fr., (1969) 4159. F.G. Baddar, M.F. El-Newaihy and M.R. Salem, J. Chem. Sot. C, (1969) 836. G. Kornis, Pyrazoles, pyrazolines, pyrazolones, in M. Grayson and D. Eckroth (Eds.), Kirk-Othmer Encyclopedia of Chemical Technology, 3rd edn., Wiley, New York, 1982, pp. 436453. H. Dorn, J. Prakt. Chem., 315 (1973) 382. F. Ritschl and P. Brosche, Z. Chem., 10 (1970) 475. J. Deschamps, J. Arriau and P. Parmentier, Tetrahedron, 27 (1971) 5779.

234
19 20 21
22
23 24 25
26
27 28
29 30 31 32 33 34
J. Arriau, J. Deschamps and P. Parmentier, Tetrahedron, 27 (1971) 5795. J. Arriau, M. Chailet and J. Deschamps, Tetrahedron, 27 (1971) 5807. J. Arriau, J. Deschamps, J. Teysseyre, A. Maquestiau and Y. van Haverbeke, Tetrahed- ron, 30 (1974) 1225. J. Deschamps, H. Sauvaitre, J. Arriau, A. Maquestiau, Y. van Haverbeke and R. Jac- querye, Tetrahedron Lett., (1971) 2929. V. Enchev and G.D. Neykov, Struct. Chem., 3 (1992). M.J.S. Dewar and W. Thiel, J. Am. Chem. Sot., 99 (1977) 4899, 4907. W.C. Davidon, Comput. J., 10 (1969) 406. R. Fletcher and M.J.D. Powell, Comput. J., 6 (1963) 163. M.J.D. Powell, Numerical methods for non-linear algebraic equations, AERE Harwell, 1970; VA09 N. Raevskii, implemented at the Institute of Organo-Element Compour Is, Acad. Sci. USSR, 1988. M.J.S. Dewar and D.A. Liotard, J. Mol. Struct. (Theochem), 206 (1990) 123. J.J.P. Stewart, AMPAC, QCPE program 506, Bloomington, IN, 1984. J. Kaneti, IBM version Sofia, BE 1040, 19861990. J.S. Kwiatkowski, T.J. Zielinski and R. Rein, Adv. Quantum. Chem., 18 (1986) 85. J.S. Kwiatkowski, R.J. Bartlett and W.B. Person, J. Am. Chem. Sot., 110 (1988) 2353. W. Nagata and S. Kamata, J. Chem. Sot. C, (1970) 540. Yu. Zaitzev, personal communication, 1989. G.D. Neykov and V. Enchev, Indian J. Chem., submitted for publication. M.J.S. Dewar, Y. Yamaguchi and S. Suck, Chem. Phys. Lett., 59 (1978) 541. M.J.S. Dewar and J.J.P. Stewart, Chem. Phys. Lett., 111 (1984) 416.




















