
Tobacco Industry Interference Targeting Educational Institutions
Upload
univ-paris7Category
view
5download
0
Pharmacology & Therapeutics xxx (2014) xxx–xxx
JPT-06726; No of Pages 10
Contents lists available at ScienceDirect
Pharmacology & Therapeutics
j ourna l homepage: www.e lsev ie r .com/ locate /pharmthera
Associate Editor: B. Teicher
Targeting the TGFβ pathway for cancer therapy
Cindy Neuzillet a, Annemilaï Tijeras-Raballand b, Romain Cohen b, Jérôme Cros c, Sandrine Faivre a,Eric Raymond d, Armand de Gramont d,⁎a INSERM U728 & U773 and Department of Medical Oncology, Beaujon University Hospital (AP-HP – PRES Paris 7 Diderot), 100 boulevard du Général Leclerc, 92110 Clichy, Franceb AAREC Filia Research, Translational Department, 1 place Paul Verlaine, 92100 Boulogne-Billancourt, Francec Department of Pathology, Beaujon University Hospital (AP-HP – PRES Paris 7 Diderot), 100 boulevard du Général Leclerc, 92110 Clichy, Franced New Drug Evaluation Laboratory, Centre of Experimental Therapeutics and Medical Oncology, Department of Oncology, Centre Hospitalier Universitaire Vaudois (CHUV) Lausanne, Switzerland
Abbreviations:bFGF, basic fibroblast growth factor; bi-growth factor; ECM, extracellular matrix; EMT, epitheliaSMAD, inhibitory SMAD; IFN, interferon; IL, interleukin; LONK, natural killer; NOX, NADPH oxidase; NSCLC, non-smderived growth factor; PR, partial response; PSC, pancreaTNFα, tumor necrosis factor-α; Th, T-helper; T-reg, T-regu⁎ Corresponding author at: New Drugs Evaluation Lab
Lausanne, Switzerland.E-mail address: [email protected] (A. de G
http://dx.doi.org/10.1016/j.pharmthera.2014.11.0010163-7258/© 2014 The Authors. Published by Elsevier Inc
Please cite this article as: Neuzillet, C., et adx.doi.org/10.1016/j.pharmthera.2014.11.00
a b s t r a c t
a r t i c l e i n f oKeywords:
AngiogenesisFibrosisMetastasisMicroenvironmentTGFβ inhibitorsTumor-stroma interactionsThe TGFβ signaling pathway has pleiotropic functions regulating cell growth, differentiation, apoptosis, motilityand invasion, extracellular matrix production, angiogenesis, and immune response. TGFβ signaling deregulationis frequent in tumors and has crucial roles in tumor initiation, development and metastasis. TGFβ signaling inhi-bition is an emerging strategy for cancer therapy. The role of the TGFβ pathway as a tumor-promoter or suppres-sor at the cancer cell level is still a matter of debate, due to its differential effects at the early and late stages ofcarcinogenesis. In contrast, at themicroenvironment level, the TGFβ pathway contributes to generate a favorablemicroenvironment for tumor growth and metastasis throughout all the steps of carcinogenesis. Then, targetingthe TGFβ pathway in cancer may be considered primarily as a microenvironment-targeted strategy. In thisreview, we focus on the TGFβ pathway as a target for cancer therapy. In the first part, we provide a comprehen-sive overview of the roles played by this pathway and its deregulation in cancer, at the cancer cell and microen-vironment levels. We go on to describe the preclinical and clinical results of pharmacological strategies to targetthe TGFβ pathway,with a highlight on the effects on tumormicroenvironment.We then explore the perspectivesto optimize TGFβ inhibition therapy in different tumor settings.
© 2014 The Authors. Published by Elsevier Inc. This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Contents
1. Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 02. Roles of TGFβ in cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 03. TGFβ pathway inhibitors: preclinical and clinical results . . . . . . . . . . . . . . . . . . . . . . . . . . . . 04. Summary and perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Conflicts of interest . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 01. Introduction
The transforming growth factor-β (TGFβ) superfamily consists ofmore than 30 different members including the TGFβs (comprising
shRNAi, bi-functional short hairpin Rl-to-mesenchymal transition; HCC, hH, loss of heterozygosity;MCP-1, cheall cell lung cancer; PBMC, periphertic stellate cell; OS, overall survival;latory cell; VEGF, vascular endothelialoratory, Centre of Experimental Ther
ramont).
. This is an open access article under
l., Targeting the TGFβ pathw1
three highly homologous isoforms, TGFβ1, TGFβ2, and TGFβ3),activins, NODAL, bone morphogenetic proteins (BMP), growth anddifferentiation factors (GDF) and anti-Müllerian hormone (AMH)(Wakefield & Hill, 2013). They share common properties regarding
NAi; CAF, cancer-associatedfibroblasts; CR, complete response; CTGF, connective tissueepatocellular carcinoma; HGF, hepatocyte growth factor; HSC, hepatic stellate cell; Imoattractant protein-1;MMP,matrixmetalloproteinase;MSI,microsatellite instabilityal blood mononuclear cell; PDAC, pancreatic ductal adenocarcinoma; PDGF, plateletROS, reactive oxygen specie; SD, stable disease; TGFβ, transforming growth factor-βgrowth factor.apeutics, Department of Oncology, Centre Hospitalier Universitaire Vaudois (CHUV)
the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
ay for cancer therapy, Pharmacology & Therapeutics (2014), http:/
t-h-e-irs-t-r-
-;-;
,
/

Fig. 1. Overview of the TGFβ family pathways. All of the ligands of the TGFβ family are initially synthesized and secreted as latent precursors that need to be proteolytically processed byextra-cellular pro-protein convertases to produce biologically active dimeric ligands. The signal transduction mechanism for all of them is similar. It requires ligand binding to one of thefive distinct members of constitutively activatedmembranous type II serine/threonine kinase receptors, and subsequent recruitment and transphosphorylation of one of the seven type Iserine/threonine kinase receptors, also known as activin receptor-like kinases (ALK1-7) (Wakefield & Hill, 2013). For some ligands, additional co-receptors are required for optimal ligandbinding and activation of the type I-type II receptor heterodimer. After phosphorylation, the type I receptor kinase domain becomes activated and can then activate the canonical (SMAD-dependent) pathway through phosphorylation of the receptor-regulated SMAD proteins (R-SMAD). It is classically admitted that TGFβs, activins, and NODAL signal through SMAD2 andSMAD3, and BMPs and GDFs through SMAD1, SMAD5, and SMAD8, but TGFβs can also induce phosphorylation of SMAD1 and SMAD5 (Wakefield & Hill, 2013). The R-SMADs formheteromeric complexes with SMAD4 in early endosomes, through a clathrin-dependent internalization pathway that requires accessory proteins such as SARA (Bierie & Moses, 2006;Ikushima &Miyazono, 2010). These complexes then translocate and accumulate into the nucleus and bind to site-specific recognition sequenceswithin the promoter regions of hundredsof target genes to directly regulate their transcription, both positively and negatively. Various other DNA binding co-factors such as p300, CBP, and FOXH1 can interact with SMAD com-plexes to amplify SMAD-dependent gene transcription. The TGFβ superfamily pathway activities are subject to numerous levels of regulation: interaction of the ligands with extracellularantagonists that prevent their binding to receptors; modulation of R-SMAD stability through phosphorylation byMAPKs, glycogen synthase kinase 3β (GSK3β) and cyclin-dependent ki-nases (CDKs); Smurf (SMAD-ubiquitination-regulatory factor)-dependent degradation; or expression of inhibitory SMADproteins (I-SMAD), SMAD6 and SMAD7 (Wakefield &Hill, 2013).In the non-canonical pathway, TGFβ signaling activates SMAD-independent pathways such as PI3K/AKT, MAPK pathways (ERK, JNK, and p38 MAPK), c-Src, NF-κB, FAK, Abl, or smallGTPases such as RhoA, Rac1 and Cdc42. Moreover, transversal signaling, especially at the SMAD level, allows TGFβ pathway activation to integrate signals from integrins, Wnt/β-catenin,Notch, Hedgehog, TNF-α, or EGF-dependent pathways (Watabe & Miyazono, 2009; Sakaki-Yumoto et al., 2013).
2 C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
ucture, synthesis, signal transduction mechanisms and regulation[Fig. 1] (Bierie & Moses, 2006; Watabe & Miyazono, 2009; Ikushima& Miyazono, 2010; Sakaki-Yumoto et al., 2013; Wakefield & Hill,2013).
The TGFβ superfamily signaling pathways have pleiotropic functionsregulating cell growth, differentiation, apoptosis, motility and invasion,extracellular matrix (ECM) production, angiogenesis, and immune re-sponse. They play essential roles in early embryonic development andin regulating tissue homeostasis in adults (Wakefield & Hill, 2013).Deregulated signaling by many of these members has crucial roles inboth the development of tumors and metastasis. Their influence ex-tends beyond the cancer cells themselves, to the tumor microenviron-ment and the whole organism. Furthermore, these signaling pathwaysare emerging therapeutic targets. Among the TGFβ-superfamily mem-bers, the TGFβs themselves have been the most studied and inhibitorsof this pathway are the most advanced in their clinical development.
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
The role of the TGFβ pathway as a tumor-promoter or suppressor atthe cancer cell level is still a matter of debate, due to its differential ef-fects at the early and late stages of carcinogenesis. In contrast, at themicroenvironment level, the TGFβ pathway contributes to generate afavorablemicroenvironment for tumor growth andmetastasis through-out all the steps of carcinogenesis. Then, targeting the TGFβ pathway forcancer therapy may be considered primarily as a microenvironment-targeted strategy.
In this review, we focus on the TGFβ pathway as a target for cancertherapy. In the first part, we provide a comprehensive overview of theroles played by this pathway and its deregulation in cancer, at the can-cer cell and microenvironment levels. We go on to describe the preclin-ical and clinical results of pharmacological strategies to target the TGFβpathway, with a highlight on the effects on tumor microenvironment.We then explore the perspectives to optimize TGFβ inhibition therapyin different tumor settings.
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

3C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
2. Roles of TGFβ in cancer
2.1. At the cancer cell level: dualtumor-promoter and suppressor role of TGFβ signaling
Hijacking crucial biological functions by deregulating the TGFβsignaling pathway by cancer cells has recently emerged as a leadingarea of preclinical and clinical cancer research. TGFβ expression hasbeen studied in a large panel of cancer types, including prostate, breast,lung, colorectal, pancreatic, liver, skin cancers, and gliomas (Padua &Massague, 2009). In early-stage tumors, levels of TGFβ are positively as-sociated with a favorable prognosis, while in advanced tumors, they arepositively associated with tumor aggressiveness and poor prognosis(Padua &Massague, 2009). Through these observations, it has emergedthat TGFβ pathway has both anti- and pro-tumoral activities (Inman,2011; Principe et al., 2014). In early-stage tumors, the TGFβ pathwaypromotes cell cycle arrest and apoptosis (Jakowlew, 2006; Tian et al.,2011; Drabsch & ten Dijke, 2012). In contrast, at advanced stages, bypromoting cancer cell motility, invasion, epithelial-to-mesenchymaltransition (EMT), and cell stemness, the TGFβ pathway promotestumor progression and metastasis (Jakowlew, 2006; Tian et al., 2011;Drabsch & ten Dijke, 2012). This functional switch is known as thefirst “TGFβ paradox” (Wendt et al., 2012).
The current paradigm for the role of TGFβ in carcinogenesis at thecancer cell level is that accumulation of genetic alterations in the TGFβpathway drives the pathway’s evolution from tumor-suppressive totumor-promoting activities (Jakowlew, 2006; Neuzillet et al., 2014). In-deed, tumor progression requires shutting down the tumor-suppressiveeffects of the TGFβ signaling, which can be reached by either of two gen-eralmechanisms. The first one consists in “decapitation” of the pathwayby inactivation, either through mutation or through allelic loss of het-erozygosity (LOH), of its core components: the receptors (TGFBR2,TGFBR1) or the SMAD transcription factors (SMAD2, SMAD4/DPC4)(Padua&Massague, 2009). For example, TGFBR2-inactivatingmutationsare frequently found in cancers associatedwithmicrosatellite instability(MSI), which arises from defects in themismatch repair system, and areassociated with high CpG island methylator phenotype (Markowitzet al., 1995; Ogino et al., 2007; Shima et al., 2011). The TGFBR2 gene con-tains a 10-base pair poly-adenine repeat,which is exposed to replicationerrors leading to gene inactivation specifically in MSI+ cancers, includ-ing gastric, colorectal, biliary and lung adenocarcinomas. Alternatively,SMAD4/DPC4 is found inactivated in about 50% of pancreatic ductal ad-enocarcinoma (PDAC), either by 18q LOH or mutation, and in colonand oesophagus cancers, (Padua &Massague, 2009). These genetic inac-tivations of core components of the TGFβ pathway result in eliminationof most or all of TGFβ responses including tumor-suppressive activities,and cooperate with other genetic alterations to promote tumor initia-tion and malignant progression. Mice models harboring oncogenic mu-tation (APC for colon cancer, KRAS for PDAC) combined with SMAD4 orTGFBR2 inactivation display accelerated cancer development withmore aggressive behaviour compared with mice without combinedTGFβ signaling alteration (Datto & Wang, 2000; Morris et al., 2010). Incontrast, breast, prostate cancers, melanomas, and gliomas, frequentlyretain functional TGFβ signaling but selectively “amputate” the tumorsuppressor arm downstream these core components, for examplethrough P15INK4B deletion or C/EBPβ inhibition (Padua & Massague,2009). These tumors can benefit from the remaining TGFβ activitiespromoting tumor progression and metastasis, such as invasion andEMT. Similarly, in HCC, anti-proliferative effects of TGFβ are bypassedvia mitogenic signals or impaired sensitivity to anti-growth signals(Neuzillet et al., 2014). Coulouarn et al. (Coulouarn et al., 2008) de-scribed the switch from a tumor-suppressive “early TGFβ signature”,with low endogenous levels of TGFβ and SMAD7 and strong transcrip-tional SMAD3 activity, and which was correlated with better outcomein HCC patients, to a “late TGFβ signature”, with high amounts of TGFβand SMAD7 and reduced SMAD3 signaling, associated with invasive
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
phenotype and increased tumor recurrence. The early TGFβ signature isalso characterized by expression of the DNA damage gene familyGadd45, which is involved in cell cycle arrest and apoptosis (Coulouarnet al., 2008; Dooley & ten Dijke, 2012). Given this strong induction ofanti-tumorigenic genes, tumor promoting activity of TGFβ requires a cel-lular context with imbalanced sensitivity towards pro- and anti-growthsignals (Neuzillet et al., 2014). For example, p16INK4 gene alterations arepresent in up to 90% of HCCs and favour insensitivity to anti-growth sig-nals by relieving cyclin D/CDK4,6 complex inhibition and lowering p53activation (Neuzillet et al., 2013). Once the tumor-suppressive effects ofthe TGFβ signaling shut down, it can exert its pro-tumoral and pro-metastatic activities in late steps of carcinogenesis.
To summarize, the TGFβ pathway has dual anti- and pro-tumoralroles at the cancer cell level, depending on tumor stage and genetic al-teration background, with mechanistic differences between cancermodels. This complexity, combined with intratumor genetic heteroge-neity, makes the resulting effects of TGFβ inhibition on cancer cell com-partment difficultly predictable. Moreover, the fact that TGFβ inducespro-tumoral effects although its signaling is shut down in cancer cellsrepresents a second “paradox” that leads to shift attention to themicro-environment surrounding cancer cells.
2.2. At the microenvironment level: a key thread of pro-tumoral activities
The TGFβ pathway exerts most of its pro-tumoral effects bymediat-ing tumor-stroma interactions and remodeling tumor microenviron-ment (Neuzillet et al., 2014). The stroma is a complex structurecomposed of ECM proteins (mainly, type I collagen) and various celltypes including mesenchymal cells (cancer-associated fibroblasts[CAF]), endothelial cells and pericytes, nerve cells, immune cells, andbone marrow-derived stem cells. These cell types express TGFβ recep-tors, and the TGFβ pathway can thus impacts microenvironment fibro-sis, angiogenesis, and immune cell infiltration (Neuzillet et al., 2014).The TGFβ pathway activation contributes both to the creation, from anon-tumoral environment, and to the maintenance of a favorabletumoral microenvironment.
2.2.1. Effect on tumor initiationSMAD4/DPC4 germline inactivating mutation is genetically responsi-
ble for familial juvenile polyposis, an autosomal dominant inherited con-dition characterized by the development of multiple hamartomatoustumors in the gastrointestinal tract and predisposition to cancer. Animalmodels with a loss of SMAD function have provided insight into the roleof SMADs in a variety of physiologic systems and tumors (Takaku et al.,1998; Datto & Wang, 2000). Using genetically engineered mice modelsof familial juvenile polyposis inwhich the SMAD4 gene is either specifical-ly deleted in T-cells or broadly within epithelia including the intestinalmucosa, Kim et al. (Kim et al., 2006) showed that selective disruption ofSMAD4 within the T-lineage in mice is sufficient to induce the formationof hamartomatous lesions and cancer within the gastrointestinal mucosa,contrary to epithelial-specific deletion of the SMAD4 gene. Then, SMAD4-dependent signaling in T-lymphocytes is required to maintain immunehomeostasis and cancer suppression within the gastrointestinal mucosa,and alterations of the TGFβ signaling in themicroenvironment contributeto create a favorable context for cancer development.
About 15% of human cancers emerge on underlying chronic inflam-matory diseases, e.g. colorectal cancer on colitis/Crohn's disease, gastriccancer on chronic gastritis, HCC on liver chronic hepatitis/cirrhosis,PDAC on chronic pancreatitis (Coussens & Werb, 2002). As describedin the physiological response in injured organs, TGFβ1 is produced inresponse to pro-inflammatory cytokines (e.g., tumor necrosis factor[TNFα], interleukin [IL]-1) in tumors, particularly by stromal cells, asan attempt to control the inflammatory reaction (Lopez-Novoa &Nieto, 2009). The activation of the TGFβ pathway results in increasedproduction and reduced degradation of ECM components, in particulartype I collagen, as well as mesenchymal cell proliferation and
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

4 C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
differentiation into myofibroblasts with migration and contractilecapabilities (Lopez-Novoa & Nieto, 2009; Pohlers et al., 2009; Van DeWater et al., 2013). TGFβ stimulates reactive oxygen species (ROS)production by various mechanisms (including activation of NADPHoxidases [NOX] family members) that, in turn, engage downstreamsignaling pathways (e.g., SMADs, EGFR, Src and MAPK family) resultingin expression of a subset of pro-fibrotic genes (e.g., PAI-1, connectivetissue growth factor [CTGF], TGFβ1, angiotensiogen) (Samarakoonet al., 2013). TGFβ overproduction, as a driver of the fibrotic process ofchronic phases of inflammatory diseases, precedes tumor formation andprepares a favorable microenvironment for cancer cells (Jakowlew,2006; Lopez-Novoa & Nieto, 2009).
2.2.2. Effect on tumor maintenance and progressionSome established tumors are characterized by a dense fibrotic
stroma (e.g., PDAC). CAFs and stellate cells are responsible for excessECM production and are engaged through signaling pathways in an“unholy alliance” with cancer cells (Vonlaufen et al., 2008; Coulouarn& Clement, 2014). TGFβ1 is a key mediator of this dialogue betweenCAFs or stellate cells and cancer cells. Cancer cells release mitogenicand fibrogenic stimulants (including TGFβ, PDGF, and sonic hedgehog),which activate surrounding CAFs and stellate cells. Activated CAFs andstellate cells in turn secrete various factors (including EGF, IGF-1,PDGF, FGF,MMP, and type I collagen) that promote tumor growth, inva-sion, metastasis, and resistance to chemotherapy (Duner et al., 2010).They also contribute to create a hypoxic microenvironment exerting aselection pressure toward a more invasive cancer cell phenotype(Duner et al., 2010). Overall, the stroma may act either as a barrier ora facilitator to metastatic dissemination depending on collagen I struc-ture (Levental et al., 2009).
The TGFβ pathway also plays a pro-tumoral role by promotingangiogenesis (tenDijke & Arthur, 2007; Neuzillet et al., 2014). It cooper-ates in an autocrine/paracrine fashionwith other signaling cascades, in-cluding vascular endothelial growth factor (VEGF), basic fibroblastgrowth factor (bFGF), platelet-derived growth factor (PDGF), CTGF,angiopoietin, andNotch, to regulate angiogenesis through direct or indi-rect effects on quiescence, migration, and proliferation of endothelialcells (Sakurai & Kudo, 2011). TGFβ has both pro-angiogenic and anti-angiogenic properties, depending on its expression level. Low levels ofTGFβ contribute to angiogenesis indirectly by upregulating expressionand activity of angiogenic factors (VEGF, bFGF, CTGF) and proteases,while high levels of TGFβ stimulate basement membrane reformation,recruit smooth muscle cells, increase differentiation, and inhibitendothelial cell growth (Sanchez-Elsner et al., 2001; Sakurai &Kudo, 2011). TGFβ plays a crucial role in angiogenesis regulation inhypervascularized tumors such as HCCs or gliomas (Ito et al., 1995;Roy et al., 2014).
Many lines of preclinical evidence suggest that TGFβ plays a cen-tral role in immune regulation (Wojtowicz-Praga, 2003; Teicher,2007; Yang, 2010). The immune system is responsible for the earlydetection and destruction of cancer cells. These latters can escapeimmune surveillance either by becoming immunologically invisible(i.e., cancer cell “hiding”) or by secreting cytokines that “blind” theimmune system to the presence of abnormal antigens at the cancercell surface. TGFβ1 is the most potent immunosuppressor and playsa crucial role in this process, along with IL-10. Tumor-associatedTGFβ1 downregulates the host immune response via several mecha-nisms: it (1) drives the T-helper (Th)1/Th2 balance toward the Th2immune phenotype (i.e., humoral immunity, without cytotoxic ac-tivity against cancer cells) via IL-10 as an intermediate; (2) directlyinhibits anti-tumoral Th1-type responses (i.e., cell-mediatedimmunity) and M1-type macrophages; (3) suppresses cytotoxicCD8+ T-lymphocytes, natural killer (NK) lymphocytes and dendriticcells functions; (4) generates CD4 + CD25+ T-regulatory cells (T-reg)that suppress activity of other lymphocyte populations; (5) promotesM2-type macrophages with pro-tumoral activities (Wojtowicz-Praga,
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
2003; Teicher, 2007; Truty & Urrutia, 2007; Yang, 2010; Yang et al.,2010; Achyut & Yang, 2011). This feature is of particular interest in tu-mors expressing immunogenic antigens such as melanoma (Perrotet al., 2013). TGFβ was shown to increase the expression of monocytechemoattractant protein-1 (MCP-1) and IL-10 in melanoma cells, en-hancing tumor infiltration and immunosuppression (Diaz-Valdeset al., 2011).
Solid tumors display a non-random metastatic tropism, suggest-ing that cancer cells need to be adapted to distant site to engraftand proliferate (Bhowmick, 2012). The microenvironment of boththe primary and the metastatic tumor sites can determine the abilityfor a disseminated tumor to progress. For example, TGFβ signalingcontributes to colon cancer metastasis into the liver and lungs.Calon et al. (Calon et al., 2012) demonstrated that constitutivelyTGFβ-pathway inactive KM12L4a colorectal cancer cells (homozygoteTGFBRII mutation) were unable to develop metastasis in an orthotopicxenograft mouse model, while KM12L4a engineered to secrete TGFβdeveloped lung and/or liver metastasis without any autocrine activa-tion of the TGFβ pathway in tumor cells. In addition, subcutaneous inoc-ulation of patients’ purified colorectal cancer stem cells expressingTGFβ in immunodeficient mice generated tumors that ellicited a strongTGFβ pathway activation in stromal cells at both primary and metasta-tic sites.
Finally, the role of stromal neural cells as a promoter of tumorgrowth and metastasis is receiving growing attention (Demir et al.,2012). There is emerging data regarding the role of TGFβ signalingin the interactions between nerve cells and cancer cells (Aigner &Bogdahn, 2008; Haas et al., 2009). For example, TGFβ has been shownto induce nerve growth factor expression in pancreatic stellate cells byactivation of the ALK-5 pathway (Haas et al., 2009).
To summarize, at the microenvironment level, the TGFβ pathwaymediates a broad spectrum of tumor-stroma interactions in both earlyand late tumor stages, creating a favorable microenvironment fortumor initiation, cancer cell growth and metastasis, and establishing akey thread of pro-tumoral activities throughout the steps of carcinogen-esis. This establishes interpendencies between the cancer cells that dis-play altered or no response to TGFβ, and the TGFβ-stimulated andresponsive stromal cells. Thus, TGFβ targeted therapymay affect cancercells by indirect, microenvironment-mediated,mechanisms throughoutall the steps of carcinogenesis.
3. TGFβ pathway inhibitors: preclinical and clinical results
3.1. Classification of agents targeting the TGFβ pathway for cancer therapy
Many TGFβ pathway inhibitors have been investigated in thepreclinical setting, some of which are now in clinical development.Schematically, TGFβ pathway inhibition can be realized at three levels:(i) the ligand level: antisense oligonucleotides delivered directly intra-venously or engineered into immune cells to prevent TGFβ synthesis(for example, trabedersen [AP12009], an antisense oligonucleotidetargeting TGFβ2; and Lucanix® [belagenpumatucel-L], a TGFβ2 anti-sense gene-modified allogeneic cancer cell vaccine); (ii) the ligand-receptor level: ligand-traps (TGFβ-neutralizing monoclonal antibodiesand soluble receptors) and anti-TGFβ-receptor monoclonal antibodiesto prevent ligand-receptor interaction (for example, fresolimumab, apan-TGFβ antibody; disitertide [P144], a peptidic TGFβ1 inhibitor spe-cifically designed to block the interaction with its receptor; and IMC-TR1 [LY3022859], amonoclonal antibody against TGFβRII) ; and (iii) in-tracellular level: TGFβ receptor kinase inhibitors to prevent signaltransduction (for example, galunisertib [LY2157299], a small moleculeinhibitor of TGFβRI, which is to date the most advanced TGFβ signalinginhibitor under clinical development) (Smith et al., 2012; Katz et al.,2013). Molecules under clinical development in oncology are summa-rized in Table 1.
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://
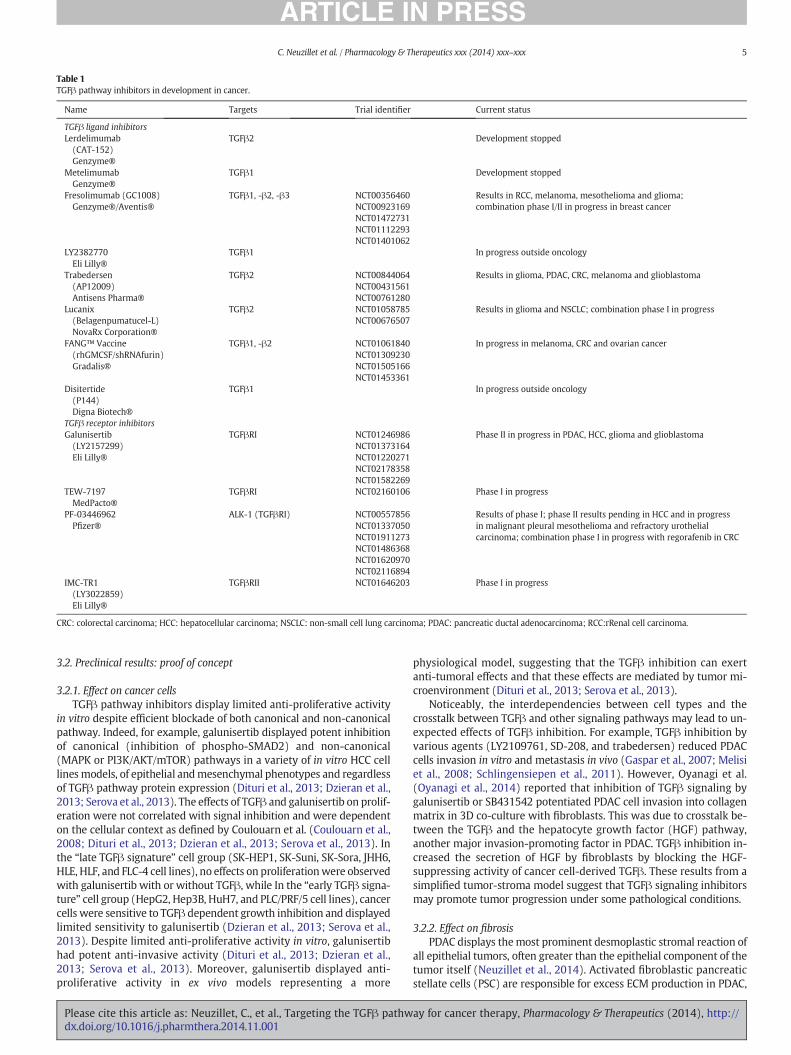
Table 1TGFβ pathway inhibitors in development in cancer.
Name Targets Trial identifier Current status
TGFβ ligand inhibitorsLerdelimumab(CAT-152)Genzyme®
TGFβ2 Development stopped
MetelimumabGenzyme®
TGFβ1 Development stopped
Fresolimumab (GC1008)Genzyme®/Aventis®
TGFβ1, -β2, -β3 NCT00356460NCT00923169NCT01472731NCT01112293NCT01401062
Results in RCC, melanoma, mesothelioma and glioma;combination phase I/II in progress in breast cancer
LY2382770Eli Lilly®
TGFβ1 In progress outside oncology
Trabedersen(AP12009)Antisens Pharma®
TGFβ2 NCT00844064NCT00431561NCT00761280
Results in glioma, PDAC, CRC, melanoma and glioblastoma
Lucanix(Belagenpumatucel-L)NovaRx Corporation®
TGFβ2 NCT01058785NCT00676507
Results in glioma and NSCLC; combination phase I in progress
FANG™ Vaccine(rhGMCSF/shRNAfurin)Gradalis®
TGFβ1, -β2 NCT01061840NCT01309230NCT01505166NCT01453361
In progress in melanoma, CRC and ovarian cancer
Disitertide(P144)Digna Biotech®
TGFβ1 In progress outside oncology
TGFβ receptor inhibitorsGalunisertib(LY2157299)Eli Lilly®
TGFβRI NCT01246986NCT01373164NCT01220271NCT02178358NCT01582269
Phase II in progress in PDAC, HCC, glioma and glioblastoma
TEW-7197MedPacto®
TGFβRI NCT02160106 Phase I in progress
PF-03446962Pfizer®
ALK-1 (TGFβRI) NCT00557856NCT01337050NCT01911273NCT01486368NCT01620970NCT02116894
Results of phase I; phase II results pending in HCC and in progressin malignant pleural mesothelioma and refractory urothelialcarcinoma; combination phase I in progress with regorafenib in CRC
IMC-TR1(LY3022859)Eli Lilly®
TGFβRII NCT01646203 Phase I in progress
CRC: colorectal carcinoma; HCC: hepatocellular carcinoma; NSCLC: non-small cell lung carcinoma; PDAC: pancreatic ductal adenocarcinoma; RCC:rRenal cell carcinoma.
5C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
3.2. Preclinical results: proof of concept
3.2.1. Effect on cancer cellsTGFβ pathway inhibitors display limited anti-proliferative activity
in vitro despite efficient blockade of both canonical and non-canonicalpathway. Indeed, for example, galunisertib displayed potent inhibitionof canonical (inhibition of phospho-SMAD2) and non-canonical(MAPK or PI3K/AKT/mTOR) pathways in a variety of in vitro HCC celllinesmodels, of epithelial andmesenchymal phenotypes and regardlessof TGFβ pathway protein expression (Dituri et al., 2013; Dzieran et al.,2013; Serova et al., 2013). The effects of TGFβ and galunisertib on prolif-eration were not correlated with signal inhibition and were dependenton the cellular context as defined by Coulouarn et al. (Coulouarn et al.,2008; Dituri et al., 2013; Dzieran et al., 2013; Serova et al., 2013). Inthe “late TGFβ signature” cell group (SK-HEP1, SK-Suni, SK-Sora, JHH6,HLE, HLF, and FLC-4 cell lines), no effects on proliferationwere observedwith galunisertib with or without TGFβ, while In the “early TGFβ signa-ture” cell group (HepG2, Hep3B, HuH7, and PLC/PRF/5 cell lines), cancercells were sensitive to TGFβ dependent growth inhibition and displayedlimited sensitivity to galunisertib (Dzieran et al., 2013; Serova et al.,2013). Despite limited anti-proliferative activity in vitro, galunisertibhad potent anti-invasive activity (Dituri et al., 2013; Dzieran et al.,2013; Serova et al., 2013). Moreover, galunisertib displayed anti-proliferative activity in ex vivo models representing a more
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
physiological model, suggesting that the TGFβ inhibition can exertanti-tumoral effects and that these effects are mediated by tumor mi-croenvironment (Dituri et al., 2013; Serova et al., 2013).
Noticeably, the interdependencies between cell types and thecrosstalk between TGFβ and other signaling pathways may lead to un-expected effects of TGFβ inhibition. For example, TGFβ inhibition byvarious agents (LY2109761, SD-208, and trabedersen) reduced PDACcells invasion in vitro and metastasis in vivo (Gaspar et al., 2007; Melisiet al., 2008; Schlingensiepen et al., 2011). However, Oyanagi et al.(Oyanagi et al., 2014) reported that inhibition of TGFβ signaling bygalunisertib or SB431542 potentiated PDAC cell invasion into collagenmatrix in 3D co-culture with fibroblasts. This was due to crosstalk be-tween the TGFβ and the hepatocyte growth factor (HGF) pathway,another major invasion-promoting factor in PDAC. TGFβ inhibition in-creased the secretion of HGF by fibroblasts by blocking the HGF-suppressing activity of cancer cell-derived TGFβ. These results from asimplified tumor-stroma model suggest that TGFβ signaling inhibitorsmay promote tumor progression under some pathological conditions.
3.2.2. Effect on fibrosisPDAC displays themost prominent desmoplastic stromal reaction of
all epithelial tumors, often greater than the epithelial component of thetumor itself (Neuzillet et al., 2014). Activated fibroblastic pancreaticstellate cells (PSC) are responsible for excess ECM production in PDAC,
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

6 C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
and TGFβ1 mediates the dialogue between PSCs and PDAC cancer cells(Apte et al., 2004). Interestingly, a study in the Panc-1 orthotopicPDAC model, showed that TGFβRI inhibition by SD-208 treatmentsignificantly decreased tumor growth and was associated with reducedfibrosis in the tumor microenvironment (Medicherla et al., 2007).
The Gianelli group (Mazzocca et al., 2010) reported similar observa-tion in a HCC xenograft mouse model. They showed that HCC cell linesproducing high levels of CTGF generated high stromogenic tumors,which was reversed by CTGF knockdown. Upon TGFβ1 stimulation,low-CTGF HCC cells formed tumors with a high stromal content andCTGF expression, which was inhibited by treatment with LY2109761.Blocking TGFβ signaling with LY2109761 or galunisertib inhibitedCTGF synthesis and release by HCC cells and reduced tumor stromalcontent by inhibiting fibroblastic cell proliferation (Mazzocca et al.,2010).
3.2.3. Effect on angiogenesisHCCs are typically hypervascularized tumors with predominant ar-
terial perfusion, and TGFβ plasma levels are positively correlated withtumor vascularity (Ito et al., 1995). TGFβ stimulate HCC cancer cells toproduce VEGF. Mazzocca et al. (Mazzocca et al., 2009) demonstratedthat the TGFβRI inhibitor LY2109761 decreased microvessel density(CD31 immunostaining) in a HCC xenograft model. Mechanistically,LY2109761 blocked VEGF gene expression in HCC cells and paracrinecrosstalk with endothelial cells, and consequently the formation of tu-moral blood vessels. This anti-angiogenic effect required functionalSMAD2/3 signaling in cancer cells. Interestingly, the anti-angiogenicand anti-tumoral effects of LY2109761 were more potent than thoseof bevacizumab, a specific anti-VEGF monoclonal antibody. Similarly,reduced vessel density after treatment with TGFβ pathway inhibitorwas reported in colorectal cancer with SD-208 (Akbari et al., 2014),and in glioblastoma models using LY2109761 (Zhang et al., 2011a,b).The angiogenic effect of TGFβ1 in this latter model may be mediatedby JNK pathway and macrophage infiltration (Yang et al., 2013).
3.2.4. Effect on immune infiltrationSome tumors express immunogenic antigens but secrete a variety of
immunosuppressive cytokines, including TGFβ or IL-10, to outgrow andevade host immune surveillance. The typical example of this immunetolerance induction ismelanoma. This gave the rational for the develop-ment of immunotherapy, aiming to reactivate the immune systemagainst these tumor antigens. TGFβ pathway inhibitors may contributeto reverse this microenvironment-induced immune suppression, byinhibiting T-reg and restoring natural killer and T cell-mediated cyto-toxicity (Teicher, 2007). A large variety of TGFβ pathway inhibitors(small molecules SB-431542, SD-108, SX-007, and SM-16; anti-TGFβantibodies; adenovirus expressing TGFβ1 or TGFβ2 shRNA; nanoscaleliposomal polymeric gels releasing TGFβ inhibitor; P144 and P17 syn-thetic small peptides inhibiting TGFβ1 and TGFβ2) have been tested inpreclinicalmodels asmonotherapy or in combinationwith other immu-notherapy (agonistic anti-TNFα receptor or anti-CD40 antibodies,adenovirus expressing interferon (IFN)β, IL-2, anti-tumor vaccine) andwere shown to restore immune response and increase the efficacyof combined immunotherapy (Uhl et al., 2004; Tran et al., 2007;Gil-Guerrero et al., 2008; Kim et al., 2008; Llopiz et al., 2009; Tanakaet al., 2010; Wilson et al., 2011; Garrison et al., 2012; Park et al., 2012;Oh et al., 2013; Xu et al., 2014). For example, in a model of advancedmelanoma, TGFβ down-regulation by means of a siRNA boosted thevaccine efficacy and inhibited tumor growth by52% comparedwith vac-cine treatment alone, as a result of increased level of tumor infiltratingCD8+ T cells and decreased level of T-reg (Xu et al., 2014). Similarly,combination delivery of TGFβ inhibitor and IL-2 by nanoscale liposomalpolymeric gels significantly delayed tumour growth, increased survivalof melanoma tumor-bearingmice, and increased the activity of NK cellsand of intratumoral-activated CD8+ T-cell infiltration (Park et al.,2012).
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
3.2.5. Effect on metastasisATGFβ-activatedmicroenvironment facilitates colon cancer cellme-
tastasis into the liver and lungs. Using an experimental model for livermetastasis by splenic injection of colorectal CT26 cancer cells constitu-tively expressing firefly luciferase in Balb/c mice, Zhang et al. (Zhanget al., 2009) showed that LY2109761 significantly reduced liver metas-tases as monitored by bioluminescence imaging when compared withvehicle control. The mean survival of LY2109761-treated mice was sig-nificantly prolonged (35.2 versus 24.5 days in control mice, p b 0.001).The overall survival at 30 days was 85.71% in LY2109761-treated miceand 0% in controlmice. Similarly, Calon et al. (Calon et al., 2012) demon-strated that galunisertib reduced phospho-SMAD2 stromal positivityand inhibited the formation of subcutaneous tumors by primary colo-rectal cancer stem cells. The authors suggest that a TGFβ-stimulatedstromal cell response, involving IL-11, potentiates colon cancer engraft-ment and growth at liver and lungmetastatic sites through activation ofthe STAT3 pathway in cancer cells. This illustrates how targeting TGFβsignaling can impact cancer cells by indirect mechanisms due to the in-terdependencies with stromal cells, through the remodeling of primarytumor and metastasis microenvironment.
The effects of TGFβ signaling at the microenvironment level aresummarized in Fig. 2.
3.3. Clinical results
Despite limited information on the microenvironmental effects ofTGFβ inhibitors in the published early phase clinical trials, some infor-mation can be highlighted.
A series of phase I and II trials targeting TGFβ2 or TGFβ1/2 expres-sion used a vaccine-based strategy (Belagenpumatucel-L and FANG vac-cines), and thus aimed to restore immune response in the tumormicroenvironment. Belagenpumatucel-L, a TGFβ2 antisense gene-modified non-viral based allogenic tumor cell vaccine, has been ex-plored in a phase II trial in NSCLC at different stages (Nemunaitis et al.,2006). Belagenpumatucel-L had an acceptable safety profile and thesurvival compared favorably with historical data, with a two-year over-all survival (OS) rate of 47% in stage IIIB and stage IV patients who re-ceived the higher doses of the vaccine. Authors showed that immuneactivation may have contributed to a favorable clinical outcome.Patients who achieved partial responses (PR) or stable diseases (SD)were more likely to display elevated IFN-γ, IL-4, and IL-6 cytokine pro-duction by peripheral blood mononuclear cells (PBMC), as well as anti-HLA seroconversion to vaccine haplotypes. Based on previous positiveclinical data with GM-CSF-secreting allogeneic tumor cells, which activ-ity may be impaired by TGFβ expression, the authors proposed to com-bine the two approaches by creating a TAG vector co-expressing GM-CSF and TGFβ2 antisense transgenes in an allogenic tumor cell vaccinestrategy (Olivares et al., 2011). Twenty-three patients with advancedsolid tumors received at least one vaccine injection in a phase I trial(Olivares et al., 2011). The majority of patients experienced SD as theirbest response and one patient had a prolonged complete response(CR); the one-year OS rate was 35%. Similarly, patients with prolongedSD or CR displayed a positive response to autologous TAG as shown bythe increased expression of IFN-γ in ELISPOT assay. As TGFβ1 andTGFβ2 isoforms have redundant functions, a vector combining GM-CSF gene expressionwith the expression of a bi-functional short hairpinRNAi (bi-shRNAi) targeting the furin convertase, which is involved inboth TGFβ1 and TGFβ2 maturation, was developed. The resultingFANG vaccine was evaluated in a phase I study with similar benefitsthan in the previous trials, i.e. FANGvaccine displayed a good safety pro-file and was able to induce an immune response that was associatedwith prolonged disease control (Senzer et al., 2012).
Fresolimumab, a human anti-TGFβ1/2/3 monoclonal antibody hasbeen tested in a phase I study involving mostly patients with advancedmelanoma (n = 28/29) (Morris et al., 2014). Fresolimumab was welltolerated; six patients had prolonged SD and one patient achieved PR.
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

Fig. 2. Summary of the effects of TGFβ at the microenvironment level. bFGF: basic fibroblast growth factor; CTGF: connective tissue growth factor; EMT epithelial-to-mesenchymal tran-sition; IL-11: interleukin-11; VEGF: vascular endothelial growth factor. Fibrosis: TGFβmediates the dialogue between cancer cells and cancer-associated fibroblastic cells (fibroblasts andstellate cells) by stimulating cancer cells to produce CTGF that has pro-fibrotic effects. Angiogenesis: TGFβ has pro-angiogenic activity by cooperatingwith and upregulating the expressionof angiogenic factors (VEGF, bFGF, CTGF) by cancer cells, with direct or indirect effects on quiescence, migration, and proliferation of endothelial cells. Metastasis: TGFβ promotes metas-tasis by enhancing EMTand cancer cellmotility and invasion. It also creates a favorablemicroenvironment for cancer cell engraftment and growth at liver and lungmetastatic sites throughmodulation of cytokines such as IL-11. Immunity: TGFβ induces a pro-tumoral state of immune tolerance by deregulating the immune balance towards immunosuppression and inflam-mation, with a predominant Th2 profile.
7C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
Various exploratory biomarkers related to the TGFβ pathway (plasmalevels of TGFβ and VEGF, tumor levels of TGFβ and TGFβ receptors,and PBMC phospho-SMAD) were analyzed, but none correlated withtumor characteristics or clinical outcome; this may be due to the smallsample size.
TβM1, a human anti-TGFβ1monoclonal antibody, has been tested in18patients in a phase I study (Cohn et al., 2014). It showed a good safetyprofile. Despite no valuable information on tumor response, a numberof potential biomarkers were investigated. At specific TβM1 doses,VEGF and bFGF plasma levels tended to decrease. Gene expression pro-filing showed that TGFβ1 pathway was activated in cancer patients’whole blood compared to normal patients. However, the short dosingduration did not translate into significant antitumor effects in thesmall number of patients investigated in this study.
PF-03446962, a fully humanmonoclonal antibody against ALK1, hasbeen tested in a phase I study, and showed some activities that sug-gested potential development in patients that failed anti VEGF/VEGFRtherapy (Goff et al., 2011). In a following phase II study as a singleagent in platimum pre-treated patients with urothelial tumors, PF-03446962 was considered inactive and the trial was stop at the first in-terim analysis for futility (Necchi et al., 2014).
In a phase IIb trial two doses of trabedersen (10 μM and 80 μM), asynthetic TGFβ2 antisens oligodeoxynucleotide, were compared to
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
standard chemotherapy (temozolomide or procarbazine/lomustine/vincristine) in 145 patients with recurrent or refractory glioblastomamultiforme or anaplastic astrocytoma (Bogdahn et al., 2011). It did notshow superiority of trabedersen in controlling tumor growth at sixmonths, which was the primary endpoint. However, delayed responseswere observed even after treatment discontinuation, and 10 μMtrabedersen tended to be superior to chemotherapy in term of two-year survival rate (p = 0.10). Median OS for 10 μM trabedersen was39.1 months compared with 35.2 months for 80 μM trabedersen and21.7 months for chemotherapy (not significant).
Preliminary results of the first phase II study of galunisertib assecond-line treatment in HCC after sorafenib (NCT01246986) werepresented at the ILCA 2013 and ASCO GI 2014 meetings (Faivre et al.,2013); 106 patients were randomized to receive galunisertib at 160 or300 mg/day. Galunisertib safety profile was suitable for patients withChild-Pugh A/B7 HCC. Median time to progression was 12 weeks.galunisertib treatment was associated with AFP responses in 24% of pa-tients, reduction in TGFβ1 and soluble E-cadherin levels, and time totumor progression was increased in patients with AFP and TGFβ1 levelsreduction from baseline. Interestingly, for patients with AFP con-centrations above 200 UI/mL, the difference in time to tumor progres-sion (TTP) and overall survival (OS) between responders and non-responders remained highly significant, suggesting that the effects of
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

8 C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
galunisertib might be more pronounced in poor prognostic patientswith elevated AFP at baseline. Further analysis is expected to launchphase III clinical trials.
Altogether, these results highlight that TGFβ inhibition may achieveprolonged disease control and that response to TGFβ inhibitors may bedelayed, as it has long been observed with immunomodulatory agents.Identification of reliable predictive biomarkers to identify whichpatients are the most likely to beneficiate from these treatments is acritical issue.
4. Summary and perspectives
TGFβ signaling inhibition is an emerging strategy for cancer therapy.As most cancer cells display altered or non-functional TGFβ signaling,TGFβ inhibitors have limited effects on these cells and exert their anti-tumoral activity mainly by affecting TGFβ responsive cells (fibroblastic,endothelial, and immune cells) in the tumor microenvironment. TGFβsignaling influences tumor microenvironment by promoting fibrosis,angiogenesis, andmetastasis, and suppressing immune-related host re-sponse. TGFβ inhibition may be considered primarily to normalizetumor microenvironment homeostasis by down-regulating stromalstimulation resulting from excess TGFβ production by tumor andtumor-related tissues, with an indirect impact on cancer cells and limit-ed expected side effects on normal tissues. This mechanism of actionillustrates how the interdepencies between cancer and stromal cellscan provide new therapeutic targets.
There are many clinical challenges to developing TGFβ inhibitors,notably timing of treatment and predictive biomarkers for patient selec-tion, in order to define in what kind of tumor microenvironment TGFβinhibition may be more beneficial. TGFβ inhibition may be of particularinterest in the early setting as a preventive strategy in tumors arising onchronic inflammation and fibrosis, in which TGFβ overproduction pre-cedes tumor formation and create a favorable microenvironment forcancer cells. This may be useful both in the primary prevention settingand as adjuvant treatment after complete resection of tumor. Of note,there may be a potential hazard of stimulating synchronous occulttumors through the inhibition of TGFβ-induced tumor suppression(particularly, with inhibitors of TGFβ receptors), depending on the pres-ence of preneoplastic condition (e.g., chronic inflammation) and geneticalteration background. However, clinical results of TGFβ inhibition inthe phase II study in HCC patients are reassuring, without evidence ofmalignant transformation from underlying cirrhotic livers (Jakowlew,2006; Faivre et al., 2013). Alternatively, TGFβ inhibition (i.e., by inhibi-tors of TGFβ ligands) may be considered to down-regulate excessTGFβ production that arises as a consequence of tumor development.Thus, TGFβ inhibitors may be preferentially used in the advancedsetting.
As TGFβ inhibitors are mainly targeting the tumor microenviron-ment, with little or dual effect on cancer cell proliferation, they shouldbe used in combination with cytotoxic agents to kill these latter cells.In addition, radiotherapy and chemotherapy can induce TGFβ activity,possibly promoting metastatic progression, and high levels of TGFβare associated with resistance to anticancer treatments (Biswas et al.,2007; Drabsch & ten Dijke, 2012). Then, combined TGFβ inhibitionmay enhance tumor sensitivity to chemotherapy and radiotherapy(Drabsch & ten Dijke, 2012). There is also a rationale for combinationwith other therapies targeting tumor microenvironment. For example,TGFβ cooperates with hypoxia to induce EMT and VEGF signalingthrough HIF-1α induction (Copple, 2010; Drabsch & ten Dijke, 2012;Mimeault & Batra, 2013). This provides a rationale for the use of TGFβpathway inhibitors in combination with or after failure of anti-angiogenic agents (tyrosine kinase inhibitors such as sunitinib or soraf-enib, or antibodies such as bevacizumab), or hypoxia-inducing proce-dures such as arterial embolization. We also described thatcombination with immunotherapies may be an option through the
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
restoration of the immune response by TGFβ inhibitors (Yang, 2010;Drabsch & ten Dijke, 2012).
Identification of reliable predictive biomarkers of response to TGFβinhibitors is also a critical issue. AFP, TGFβ1, and soluble E-cadherinlevels have been suggested to predict response in HCC (Faivre et al.,2013). More so than cancer cell characteristics, predictive power of bio-markers in terms of TGFβ inhibitor efficacymay be related to the tumormicroenvironment or a patient’s overall blood biomarker profile(Neuzillet et al., 2014). For example, patients with high intra-tumoraland/or circulating levels of TGFβmay be more likely to respond to spe-cific TGFβ inhibitors.
In conclusion, microenvironment remodeling by TGFβ, in space andtime, generates a favorable microenvironment for tumor growth andmetastasis. TGFβ inhibitors have entered clinical development in cancerpatients with encouragingfirst clinical results. The future of TGFβ inhib-itors in cancer therapy as tumor microenvironment targeting agents ispromising and opensnew challenges in terms of biomarkers and patientselection.
Conflicts of interest
Sandrine Faivre is a consultant for Merck, Pfizer, Novartis, Bayer, andLilly; and Eric Raymond is a consultant for Pfizer, Novartis, Bayer, andLilly. Other authors have no conflict of interest.
Acknowledgments
We acknowledge the Foundation Nelia & Amadeo Barleta (FNAB)and the Association pour l’Aide à la Recherche & l’Enseignement enCancérologie (AAREC).
References
Achyut, B.R., & Yang, L. (2011). Transforming growth factor-beta in the gastrointestinaland hepatic tumor microenvironment. Gastroenterology 141, 1167–1178.
Aigner, L., & Bogdahn, U. (2008). TGF-beta in neural stem cells and in tumors of thecentral nervous system. Cell Tissue Res 331, 225–241.
Akbari, A., Amanpour, S., Muhammadnejad, S., Ghahremani, M.H., Ghaffari, S.H., Dehpour,A.R., et al. (2014). Evaluation of antitumor activity of a TGF-beta receptor I inhibitor(SD-208) on human colon adenocarcinoma. Daru 22, 47.
Apte, M.V., Park, S., Phillips, P.A., Santucci, N., Goldstein, D., Kumar, R.K., et al. (2004).Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas29, 179–187.
Bhowmick, N.A. (2012). Metastatic ability: adapting to a tissue site unseen. Cancer Cell 22,563–564.
Bierie, B., & Moses, H.L. (2006). Tumourmicroenvironment: TGFbeta: themolecular Jekylland Hyde of cancer. Nat Rev Cancer 6, 506–520.
Biswas, S., Guix, M., Rinehart, C., Dugger, T.C., Chytil, A., Moses, H.L., et al. (2007). Inhibi-tion of TGF-beta with neutralizing antibodies prevents radiation-induced accelera-tion of metastatic cancer progression. J Clin Invest 117, 1305–1313.
Bogdahn, U., Hau, P., Stockhammer, G., Venkataramana, N.K., Mahapatra, A.K., Suri, A.,et al. (2011). Targeted therapy for high-grade glioma with the TGF-beta2 inhibitortrabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol13, 132–142.
Calon, A., Espinet, E., Palomo-Ponce, S., Tauriello, D.V., Iglesias, M., Cespedes, M.V., et al.(2012). Dependency of colorectal cancer on a TGF-beta-driven program in stromalcells for metastasis initiation. Cancer Cell 22, 571–584.
Cohn, A., Lahn, M.M., Williams, K.E., Cleverly, A.L., Pitou, C., Kadam, S.K., et al. (2014). Aphase I dose-escalation study to a predefined dose of a transforming growth factor-beta1 monoclonal antibody (TbetaM1) in patients with metastatic cancer. Int JOncol 45, 2221–2231.
Copple, B.L. (2010). Hypoxia stimulates hepatocyte epithelial to mesenchymal transitionby hypoxia-inducible factor and transforming growth factor-beta-dependent mecha-nisms. Liver Int 30, 669–682.
Coulouarn, C., & Clement, B. (2014). Stellate cells and the development of liver cancer:therapeutic potential of targeting the stroma. J Hepatol 60, 1306–1309.
Coulouarn, C., Factor, V.M., & Thorgeirsson, S.S. (2008). Transforming growth factor-betagene expression signature in mouse hepatocytes predicts clinical outcome inhuman cancer. Hepatology 47, 2059–2067.
Coussens, L.M., & Werb, Z. (2002). Inflammation and cancer. Nature 420, 860–867.Datto, M., & Wang, X.F. (2000). The Smads: transcriptional regulation and mouse models.
Cytokine Growth Factor Rev 11, 37–48.Demir, I.E., Friess, H., & Ceyhan, G.O. (2012). Nerve-cancer interactions in the stromal
biology of pancreatic cancer. Front Physiol 3, 97.Diaz-Valdes, N., Basagoiti, M., Dotor, J., Aranda, F., Monreal, I., Riezu-Boj, J.I., et al. (2011).
Induction of monocyte chemoattractant protein-1 and interleukin-10 by TGFbeta1 in
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

9C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
melanoma enhances tumor infiltration and immunosuppression. Cancer Res 71,812–821.
Dituri, F., Mazzocca, A., Peidro, F.J., Papappicco, P., Fabregat, I., De Santis, F., et al. (2013).Differential inhibition of the TGF-beta signaling pathway in HCC cells using theSmall molecule inhibitor LY2157299 and the D10 monoclonal antibody againstTGF-beta receptor type II. PLoS One 8, e67109.
Dooley, S., & ten Dijke, P. (2012). TGF-beta in progression of liver disease. Cell Tissue Res347, 245–256.
Drabsch, Y., & ten Dijke, P. (2012). TGF-beta signalling and its role in cancer progressionand metastasis. Cancer Metastasis Rev 31, 553–568.
Duner, S., Lopatko Lindman, J., Ansari, D., Gundewar, C., & Andersson, R. (2010). Pancreat-ic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology 10,673–681.
Dzieran, J., Fabian, J., Feng, T., Coulouarn, C., Ilkavets, I., Kyselova, A., et al. (2013). Compar-ative analysis of TGF-beta/Smad signaling dependent cytostasis in human hepatocel-lular carcinoma cell lines. PLoS One 8, e72252.
Faivre, S.J., Santoro, A., Kelley, R.K., Merle, P., Gane, E., Douillard, J.Y., et al. (2013). Ran-domized dose comparison phase II study of the oral transforming growth factor-beta (TGF-ß) receptor I kinase inhibitor LY2157299 monohydrate (LY) in patientswith advanced hepatocellular carcinoma (HCC). J Clin Oncol 31.
Garrison, K., Hahn, T., Lee, W.C., Ling, L.E., Weinberg, A.D., & Akporiaye, E.T. (2012). Thesmall molecule TGF-beta signaling inhibitor SM16 synergizes with agonistic OX40antibody to suppress established mammary tumors and reduce spontaneous metas-tasis. Cancer Immunol Immunother 61, 511–521.
Gaspar, N.J., Li, L., Kapoun, A.M., Medicherla, S., Reddy, M., Li, G., et al. (2007). Inhibition oftransforming growth factor beta signaling reduces pancreatic adenocarcinomagrowth and invasiveness. Mol Pharmacol 72, 152–161.
Gil-Guerrero, L., Dotor, J., Huibregtse, I.L., Casares, N., Lopez-Vazquez, A.B., Rudilla, F., et al.(2008). In vitro and in vivo down-regulation of regulatory T cell activity with a pep-tide inhibitor of TGF-beta1. J Immunol 181, 126–135.
Goff, L.W., Cohen, R.B., Berlin, J., Noberasco, C., Borghaei, H., Gallo-Stampino, C., et al.(2011). Phase I study of PF-03446962, a fully human mAb against ALK 1, a TGFβreceptor involved in tumor angiogenesis. J Clin Oncol 29 (Suppl.; abstr 3009).
Haas, S.L., Fitzner, B., Jaster, R., Wiercinska, E., Gaitantzi, H., Jesnowski, R., et al.(2009). Transforming growth factor-beta induces nerve growth factor expres-sion in pancreatic stellate cells by activation of the ALK-5 pathway. GrowthFactors 27, 289–299.
Ikushima, H., & Miyazono, K. (2010). TGFbeta signalling: a complex web in cancerprogression. Nat Rev Cancer 10, 415–424.
Inman, G.J. (2011). Switching TGFbeta from a tumor suppressor to a tumor promoter. CurrOpin Genet Dev 21, 93–99.
Ito, N., Kawata, S., Tamura, S., Shirai, Y., Kiso, S., Tsushima, H., et al. (1995). Positive corre-lation of plasma transforming growth factor-beta 1 levels with tumor vascularity inhepatocellular carcinoma. Cancer Lett 89, 45–48.
Jakowlew, S.B. (2006). Transforming growth factor-beta in cancer and metastasis. CancerMetastasis Rev 25, 435–457.
Katz, L.H., Li, Y., Chen, J.S., Munoz, N.M., Majumdar, A., Chen, J., et al. (2013). TargetingTGF-beta signaling in cancer. Expert Opin Ther Targets 17, 743–760.
Kim, S., Buchlis, G., Fridlender, Z.G., Sun, J., Kapoor, V., Cheng, G., et al. (2008). Systemicblockade of transforming growth factor-beta signaling augments the efficacy ofimmunogene therapy. Cancer Res 68, 10247–10256.
Kim, B.G., Li, C., Qiao, W., Mamura, M., Kasprzak, B., Anver, M., et al. (2006). Smad4signalling in T cells is required for suppression of gastrointestinal cancer. Nature441, 1015–1019.
Levental, K.R., Yu, H., Kass, L., Lakins, J.N., Egeblad, M., Erler, J.T., et al. (2009). Matrixcrosslinking forces tumor progression by enhancing integrin signaling. Cell 139,891–906.
Llopiz, D., Dotor, J., Casares, N., Bezunartea, J., Diaz-Valdes, N., Ruiz, M., et al. (2009).Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitu-mor immunotherapy. Int J Cancer 125, 2614–2623.
Lopez-Novoa, J.M., & Nieto, M.A. (2009). Inflammation and EMT: an alliance towardsorgan fibrosis and cancer progression. EMBO Mol Med 1, 303–314.
Markowitz, S., Wang, J., Myeroff, L., Parsons, R., Sun, L., Lutterbaugh, J., et al. (1995). Inac-tivation of the type II TGF-beta receptor in colon cancer cells with microsatelliteinstability. Science 268, 1336–1338.
Mazzocca, A., Fransvea, E., Dituri, F., Lupo, L., Antonaci, S., & Giannelli, G. (2010). Down-regulation of connective tissue growth factor by inhibition of transforming growthfactor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocel-lular carcinoma. Hepatology 51, 523–534.
Mazzocca, A., Fransvea, E., Lavezzari, G., Antonaci, S., & Giannelli, G. (2009). Inhibition oftransforming growth factor beta receptor I kinase blocks hepatocellular carcinomagrowth through neo-angiogenesis regulation. Hepatology 50, 1140–1151.
Medicherla, S., Li, L., Ma, J.Y., Kapoun, A.M., Gaspar, N.J., Liu, Y.W., et al. (2007). Antitumoractivity of TGF-beta inhibitor is dependent on the microenvironment. Anticancer Res27, 4149–4157.
Melisi, D., Ishiyama, S., Sclabas, G.M., Fleming, J.B., Xia, Q., Tortora, G., et al. (2008).LY2109761, a novel transforming growth factor beta receptor type I and type IIdual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis.Mol Cancer Ther 7, 829–840.
Mimeault, M., & Batra, S.K. (2013). Hypoxia-inducing factors as master regulators ofstemness properties and altered metabolism of cancer- and metastasis-initiatingcells. J Cell Mol Med 17, 30–54.
Morris, J.C., Tan, A.R., Olencki, T.E., Shapiro, G.I., Dezube, B.J., Reiss, M., et al. (2014). Phase Istudy of GC1008 (fresolimumab): a human anti-transforming growth factor-beta(TGFbeta) monoclonal antibody in patients with advanced malignant melanoma orrenal cell carcinoma. PLoS One 9, e90353.
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
Morris, J.P., 4th, Wang, S.C., & Hebrok, M. (2010). KRAS, Hedgehog, Wnt and the twisteddevelopmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 10,683–695.
Necchi, A., Giannatempo, P., Mariani, L., Fare, E., Raggi, D., Pennati, M., et al. (2014). PF-03446962, a fully-human monoclonal antibody against transforming growth-factorbeta (TGFbeta) receptor ALK1, in pre-treated patients with urothelial cancer: anopen label, single-group, phase 2 trial. Invest New Drugs 32, 555–560.
Nemunaitis, J., Dillman, R.O., Schwarzenberger, P.O., Senzer, N., Cunningham, C., Cutler, J.,et al. (2006). Phase II study of belagenpumatucel-L, a transforming growth factorbeta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lungcancer. J Clin Oncol 24, 4721–4730.
Neuzillet, C., de Gramont, A., Tijeras-Raballand, A., de Mestier, L., Cros, J., Faivre, S., et al.(2014). Perspectives of TGF-beta inhibition in pancreatic and hepatocellular carcino-mas. Oncotarget 5, 78–94.
Neuzillet, C., Hammel, P., Tijeras-Raballand, A., Couvelard, A., & Raymond, E. (2013).Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer MetastasisRev 32, 147–162.
Ogino, S., Kawasaki, T., Ogawa, A., Kirkner, G.J., Loda, M., & Fuchs, C.S. (2007). TGFBR2mu-tation is correlated with CpG island methylator phenotype in microsatelliteinstability-high colorectal cancer. Hum Pathol 38, 614–620.
Oh, S., Kim, E., Kang, D., Kim, M., Kim, J.H., & Song, J.J. (2013). Transforming growth factor-beta gene silencing using adenovirus expressing TGF-beta1 or TGF-beta2 shRNA.Cancer Gene Ther 20, 94–100.
Olivares, J., Kumar, P., Yu, Y., Maples, P.B., Senzer, N., Bedell, C., et al. (2011). Phase I trial ofTGF-beta 2 antisense GM-CSF gene-modified autologous tumor cell (TAG) vaccine.Clin Cancer Res 17, 183–192.
Oyanagi, J., Kojima, N., Sato, H., Higashi, S., Kikuchi, K., Sakai, K., et al. (2014). Inhibition oftransforming growth factor-beta signaling potentiates tumor cell invasion intocollagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp CellRes 326(2), 267–279.
Padua, D., & Massague, J. (2009). Roles of TGFbeta in metastasis. Cell Res 19, 89–102.Park, J., Wrzesinski, S.H., Stern, E., Look, M., Criscione, J., Ragheb, R., et al. (2012). Combi-
nation delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gelsenhances tumour immunotherapy. Nat Mater 11, 895–905.
Perrot, C.Y., Javelaud, D., & Mauviel, A. (2013). Insights into the transforming growthfactor-beta signaling pathway in cutaneous melanoma. Ann Dermatol 25,135–144.
Pohlers, D., Brenmoehl, J., Loffler, I., Muller, C.K., Leipner, C., Schultze-Mosgau, S., et al.(2009). TGF-beta and fibrosis in different organs – molecular pathway imprints.Biochim Biophys Acta 1792, 746–756.
Principe, D.R., Doll, J.A., Bauer, J., Jung, B., Munshi, H.G., Bartholin, L., et al. (2014). TGF-beta: duality of function between tumor prevention and carcinogenesis. J NatlCancer Inst 106, djt369.
Roy, L.O., Poirier, M.B., & Fortin, D. (2014). Transforming growth factor-beta and itsimplication in the malignancy of gliomas. Target Oncol.
Sakaki-Yumoto, M., Katsuno, Y., & Derynck, R. (2013). TGF-beta family signaling in stemcells. Biochim Biophys Acta 1830, 2280–2296.
Sakurai, T., & Kudo, M. (2011). Signaling pathways governing tumor angiogenesis.Oncology 81(Suppl. 1), 24–29.
Samarakoon, R., Overstreet, J.M., & Higgins, P.J. (2013). TGF-beta signaling in tissue fibro-sis: redox controls, target genes and therapeutic opportunities. Cell Signal 25,264–268.
Sanchez-Elsner, T., Botella, L.M., Velasco, B., Corbi, A., Attisano, L., & Bernabeu, C. (2001).Synergistic cooperation between hypoxia and transforming growth factor-betapathways on human vascular endothelial growth factor gene expression. J BiolChem 276, 38527–38535.
Schlingensiepen, K.H., Jaschinski, F., Lang, S.A., Moser, C., Geissler, E.K., Schlitt, H.J., et al.(2011). Transforming growth factor-beta 2 gene silencing with trabedersen (AP12009) in pancreatic cancer. Cancer Sci 102, 1193–1200.
Senzer, N., Barve, M., Kuhn, J., Melnyk, A., Beitsch, P., Lazar, M., et al. (2012). Phase I trial of“bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advancedcancer. Mol Ther 20, 679–686.
Serova, M., Tijeras-Raballand, A., Dos Santos, C., Muller, N., Benhadji, K.A., Paradis, V., et al.(2013). Effects of TGF-beta signaling inhibition with LY2157299 in hepatocarcinomamodels and in ex vivowhole tumor tissue samples from patient specimen. Cancer Res73(Suppl.), 1.
Shima, K., Morikawa, T., Yamauchi, M., Kuchiba, A., Imamura, Y., Liao, X., et al. (2011).TGFBR2 and BAX mononucleotide tract mutations, microsatellite instability, andprognosis in 1072 colorectal cancers. PLoS One 6, e25062.
Smith, A.L., Robin, T.P., & Ford, H.L. (2012). Molecular pathways: targeting the TGF-betapathway for cancer therapy. Clin Cancer Res 18, 4514–4521.
Takaku, K., Oshima, M., Miyoshi, H., Matsui, M., Seldin, M.F., & Taketo, M.M. (1998).Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apcgenes. Cell 92, 645–656.
Tanaka, H., Shinto, O., Yashiro, M., Yamazoe, S., Iwauchi, T., Muguruma, K., et al. (2010).Transforming growth factor beta signaling inhibitor, SB-431542, induces maturationof dendritic cells and enhances anti-tumor activity. Oncol Rep 24, 1637–1643.
Teicher, B.A. (2007). Transforming growth factor-beta and the immune response tomalignant disease. Clin Cancer Res 13, 6247–6251.
ten Dijke, P., & Arthur, H.M. (2007). Extracellular control of TGFbeta signalling in vasculardevelopment and disease. Nat Rev Mol Cell Biol 8, 857–869.
Tian, M., Neil, J.R., & Schiemann, W.P. (2011). Transforming growth factor-beta and thehallmarks of cancer. Cell Signal 23, 951–962.
Tran, T.T., Uhl, M., Ma, J.Y., Janssen, L., Sriram, V., Aulwurm, S., et al. (2007). Inhibiting TGF-beta signaling restores immune surveillance in the SMA-560 glioma model. NeuroOncol 9, 259–270.
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://

10 C. Neuzillet et al. / Pharmacology & Therapeutics xxx (2014) xxx–xxx
Truty, M.J., & Urrutia, R. (2007). Basics of TGF-beta and pancreatic cancer. Pancreatology 7,423–435.
Uhl, M., Aulwurm, S., Wischhusen, J., Weiler, M., Ma, J.Y., Almirez, R., et al. (2004). SD-208,a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growthand invasiveness and enhances immunogenicity of murine and human glioma cellsin vitro and in vivo. Cancer Res 64, 7954–7961.
Van De Water, L., Varney, S., & Tomasek, J.J. (2013). Mechanoregulation of themyofibroblast in wound contraction, scarring, and fibrosis: opportunities for newtherapeutic intervention. Adv Wound Care (New Rochelle) 2, 122–141.
Vonlaufen, A., Phillips, P.A., Xu, Z., Goldstein, D., Pirola, R.C., Wilson, J.S., et al. (2008). Pan-creatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res 68,7707–7710.
Wakefield, L.M., & Hill, C.S. (2013). Beyond TGFbeta: roles of other TGFbeta superfamilymembers in cancer. Nat Rev Cancer 13, 328–341.
Watabe, T., &Miyazono, K. (2009). Roles of TGF-beta family signaling in stem cell renewaland differentiation. Cell Res 19, 103–115.
Wendt, M.K., Tian, M., & Schiemann, W.P. (2012). Deconstructing the mechanisms andconsequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res347, 85–101.
Wilson, E.B., El-Jawhari, J.J., Neilson, A.L., Hall, G.D., Melcher, A.A., Meade, J.L., et al. (2011).Human tumour immune evasion via TGF-beta blocks NK cell activation but not sur-vival allowing therapeutic restoration of anti-tumour activity. PLoS One 6, e22842.
Please cite this article as: Neuzillet, C., et al., Targeting the TGFβ pathwdx.doi.org/10.1016/j.pharmthera.2014.11.001
Wojtowicz-Praga, S. (2003). Reversal of tumor-induced immunosuppression by TGF-betainhibitors. Invest New Drugs 21, 21–32.
Xu, Z., Wang, Y., Zhang, L., & Huang, L. (2014). Nanoparticle-delivered transforminggrowth factor-beta siRNA enhances vaccination against advanced melanoma bymodifying tumor microenvironment. ACS Nano 8, 3636–3645.
Yang, L. (2010). TGFbeta and cancer metastasis: an inflammation link. Cancer MetastasisRev 29, 263–271.
Yang, X.J., Chen, G.L., Yu, S.C., Xu, C., Xin, Y.H., Li, T.T., et al. (2013). TGF-beta1 enhancestumor-induced angiogenesis via JNK pathway and macrophage infiltration in animproved zebrafish embryo/xenograft glioma model. Int Immunopharmacol 15,191–198.
Yang, L., Pang, Y., & Moses, H.L. (2010). TGF-beta and immune cells: an important regula-tory axis in the tumor microenvironment and progression. Trends Immunol 31,220–227.
Zhang, B., Halder, S.K., Zhang, S., & Datta, P.K. (2009). Targeting transforming growthfactor-beta signaling in liver metastasis of colon cancer. Cancer Lett 277, 114–120.
Zhang, M., Herion, T.W., Timke, C., Han, N., Hauser, K., Weber, K.J., et al. (2011a). Trimodalglioblastoma treatment consisting of concurrent radiotherapy, temozolomide, andthe novel TGF-beta receptor I kinase inhibitor LY2109761. Neoplasia 13, 537–549.
Zhang, M., Kleber, S., Rohrich, M., Timke, C., Han, N., Tuettenberg, J., et al. (2011b). Block-ade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhancesradiation response and prolongs survival in glioblastoma. Cancer Res 71, 7155–7167.
ay for cancer therapy, Pharmacology & Therapeutics (2014), http://
Copyright © 2022 FDOKUMEN




















