
T Cell Responses to Human Endogenous Retroviruses in HIV1 Infection
Upload
independentCategory
view
4download
0
T-Cell Receptor Gene Therapy in Human Melanoma-BearingImmune-Deficient Mice: Human but not Mouse T Cells
Recapitulate Outcome of Clinical Studies
Trudy Straetemans,1 Miriam Coccoris,1 Cor Berrevoets,1 Elike Treffers-Westerlaken,1 Csilla E.V. Scholten,1
Debby Schipper,2 Timo L.M. ten Hagen,2 and Reno Debets1
Abstract
Adoptive cell therapy using T-cell receptor (TCR)–engineered T cells is a clinically feasible and promisingapproach to target tumors, but is currently faced with compromised antitumor efficacies in patients. Here, weextensively validated immune-deficient mice to facilitate further development of the therapeutic potential ofTCR-engineered T cells. Treatment of human melanoma-bearing SCID or NSG mice with high doses of human Tcells transduced with an hgp100/HLA-A2-specific TCR did not result in antitumor responses irrespective ofchemotherapeutic preconditioning. Imaging of human green fluorescent protein–labeled T cells demonstratedsignificant T-cell accumulation in intratumoral vasculature directly upon T-cell transfer, which was followed byloss of T cells within 72 hr. Peripheral persistence of human T cells was highly compromised and appeared relatedto T-cell differentiation. On the contrary, adoptive transfer (AT) of relatively low numbers of hgp100/HLA-A2TCR-transduced mouse T cells resulted in rapid clearance of large established human melanomas. Unexpectedlyand in contrast to reported studies with chimeric antibody receptor–engineered T cells, antitumor activity andhomeostatic expansion of T cells were independent of TCR transgene as evidenced in two SCID strains and usingtwo different human melanoma cell lines. Interestingly, the xeno-reactive melanoma response of mouse T cellsappeared to be dictated by CD4 + tumor-infiltrating lymphocytes and did not require in vitro T-cell activation,retroviral gene transfer, or subcutaneous interleukin-2 support. Taken together, AT of human but not mouse T cellsin human melanoma-bearing immune-deficient mice is in close accordance with clinical studies.
Introduction
Adoptive cell therapy using T-cell receptor (TCR) gene-modified T cells represents an attractive immunotherapy
for cancer. Generally, the endogenous repertoire of tumor-specific T cells is limited, and the infusion of T cells geneti-cally engineered with a tumor-specific TCR may providepatients with antitumor immunity. In this rapidly expandingfield, early reports have already demonstrated the successfulintroduction of tumor-specific TCRab genes into T cells,enabling them to specifically bind to and lyse antigen-positive tumor cells in vitro (Clay et al., 1999; Willemsen et al.,2000; Schaft et al., 2003). In addition, immune-competentmouse models demonstrated the antitumor activity of TCR-engineered T cells in vivo (Kessels et al., 2001; Morris et al.,2005; de Witte et al., 2008a,b). More recently, clinical trials
have provided evidence of the feasibility and therapeuticpotential of TCR-engineered T cells in patients (Morganet al., 2006; Johnson et al., 2009; Parkhurst et al., 2011; Robbinset al., 2011). In fact, TCR-engineered T cells directed againstthe human leukocyte antigen (HLA)-A2-restricted antigensMART-1, gp100, carcinoembryonic antigen (CEA), or NY-ESO-1 mediated clinical responses in patients with meta-static melanoma, colorectal, and synovial tumors. Althoughthese clinical responses were variable and based on a rela-tively small number of patients, they generally laggedbehind those observed with natural ex vivo expanded tumor-infiltrating T cells ( > 50%) (Dudley et al., 2005). Furthermore,the clinical use of high-avidity TCR-engineered T cells di-rected against antigens that are overexpressed in tumors, butalso present in normal tissues, can result in on-target toxi-cities with severe inflammation of skin, eyes, and ears (for
1Laboratory of Experimental Tumor Immunology, Department of Medical Oncology, Erasmus Medical Center–Daniel den Hoed CancerCenter, 3075 EA Rotterdam, The Netherlands.
2Laboratory of Experimental and Surgical Oncology, Department of Surgery, Erasmus Medical Center, 3015 GE Rotterdam, The Netherlands.
HUMAN GENE THERAPY 23:187–201 (February 2012)ª Mary Ann Liebert, Inc.DOI: 10.1089/hum.2010.126
187

MART-1, gp100) or colon (CEA) ( Johnson et al., 2009; Par-khurst et al., 2011).
Clinical results with TCR-engineered T cells are promis-ing, but improvements are necessary to further developclinical TCR gene therapy into an effective and safe treat-ment for cancer patients. Immune-deficient mice trans-planted with a human tumor represent a widely recognizedand successfully used model with human or mouse T cellsgene-engineered with either chimeric antibody receptors(CARs) or TCRs. These preclinical models potentially pro-vide a valuable basis to test novel strategies and translatereceptor gene therapy to the patient. With respect tohuman hematological malignancies, such as lymphoma,CAR-engineered human T cells (CAR T cells) have been re-ported to effectively mediate antitumor responses in micewith various degrees of immune deficiency, i.e., severecombined immune-deficient (SCID), nonobese diabetic SCID(NOD.SCID), SCID Beige, and NOD.SCID/il2rg–/– (NSG)mice (Brentjens et al., 2003, 2007; Kowolik et al., 2006; Savoldoet al., 2007; Cheadle et al., 2008; Milone et al., 2009; Zhao et al.,2010). With respect to human solid tumors, high numbers ofhuman CAR T cells injected intratumorally (i.t.) delayed tu-mor growth (Pinthus et al., 2003) or prolonged tumor-freesurvival (Gade et al., 2005) in immune-deficient mice. Moreinterestingly, intravenously (i.v.) injected human CAR T cellsinhibited growth of human solid tumors when mice werepreconditioned with either cyclophosphamide or irradiation(Pinthus et al., 2004; Teng et al., 2004; Westwood et al., 2005;Zhao et al., 2009; Chekmasova et al., 2010; Craddock et al.,2010). Preconditioning appeared not to be required for tumorregression in NSG mice that were treated with an i.v. injec-tion of T cells engineered with a CAR containing the CD137costimulatory domain (Carpenito et al., 2009; Zhao et al.,2010; Song et al., 2011). In addition to human CAR T cells, invarious studies mouse CAR T cells have been used suc-cessfully to target human solid tumors in nude or SCID micewithout additional treatment (Hwu et al., 1995; Darcy et al.,2000; Haynes et al., 2001, 2002a,b). Despite numerous reportson the antitumor effects of CAR T cells, there exists only alimited number of studies on the antitumor effects of TCR-engineered T cells (TCR T cells) in immune-deficient mice.Two studies by Xue and colleagues reported that i.v. injec-tion of human TCR T cells inhibited human lymphomaengraftment in NOD.SCID mice (Xue et al., 2005, 2010).Moreover, a single study by Bobisse and colleagues de-scribed that i.t., but not i.v., injection of human TCR T cellsdelayed growth of human melanoma in SCID mice (Bobisseet al., 2009). To our knowledge, there are no reports of mouseTCR T cells targeting a human tumor in immune-deficientmice. See Tables 1 and 2 for an up-to-date overview of re-ported studies on adoptive cell therapy with human andmouse T cells, respectively, genetically directed towardhuman tumors in immune-deficient mice.
Here, we evaluated immune-deficient mouse models totest the anti-human melanoma efficacy of adoptively trans-ferred human as well as mouse T cells engineered with ahuman gp100/HLA-A2-specific TCR. High numbers ofhuman TCR T cells were not efficacious against humanmelanoma cells in SCID or NSG mice irrespective of pre-treatment with cyclophosphamide with or without busulfan.In contrast, mouse TCR T cells very efficiently cleared es-tablished human melanoma, but the antitumor responses
and homeostatic expansion of mouse T cells were indepen-dent of the TCR transgene.
Materials and Methods
T cells, packaging cells, and melanoma cells
Peripheral blood mononuclear cells from healthy humandonors were isolated by centrifugation through Ficoll-Isopaque (density = 1.077 g/cm3; Amersham Pharmacia Bio-tech, Uppsala, Sweden). Transduced primary human T cellswere cultured in RPMI 1640 medium supplemented with25 mM HEPES, 200 mM L-glutamine, 10% human serum,antibiotics, and 360 IU/ml recombinant human interleukin-2(IL-2) (Proleukin; Chiron, Amsterdam, The Netherlands) andstimulated every 2 weeks with a mixture of irradiated allo-geneic feeder cells as described elsewhere (Van de Griendet al., 1984). Mouse splenocytes were cultured in completemouse medium (CMM) consisting of RPMI supplementedwith 25 mM HEPES, 200 nM L-glutamine, 10% fetal bovineserum (FBS; Greiner Bio-one Alphen a/d Rijn, The Nether-lands), 1% minimum essential medium (MEM) nonessentialamino acids, 1 mM sodium pyruvate, 50 lM b-mercaptoethanol,antibiotics, and 360 IU/ml IL-2. The human embryonic kidneycell line 293T and Phoenix-Ampho (Ph-A) were both used topackage retroviruses carrying RNA encoding TCRab, andgrown in Dulbecco’s modified Eagle’s medium with glutamine,10% FBS, 1% MEM nonessential amino acids, and antibiotics.The same medium and supplements were used to culturethe gp100/HLA-A2pos human melanoma cell lines, FM3 andBLMgp100 (BLM transfected with hgp100 cDNA).
Mice
Inbred C57BL/6 (B6), BALB/c, and BALB/cJHan�Hsd-Prkdcscid (BALB/c SCID) mice were purchased from HarlanLaboratories (Leicester, UK); B6.CB17-Prkdcscid/SzJ (B6SCID) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice werepurchased from The Jackson Laboratory (Bar Harbor,Maine). Mice were housed according to the guidelines of theErasmus Medical Center. Mice from 8 to 12 weeks of agewere used in all our experiments following approval by theExperimental Animal Committee of the Erasmus MedicalCenter (DEC consult) and carried out in accordance withinstitutional and national guidelines.
TCR transgenes and transduction of T lymphocytes
The human gp100280–288/HLA-A2-specific (gp100/A2)TCRa and b genes were introduced separately into the ret-roviral vector pBullet via NcoI and XhoI (pB-TCRa and pB-TCRb constructs) (Schaft et al., 2003). To allow detection oftransduced T cells in vivo, green fluorescent protein (GFP)DNA preceded by an internal ribosomal entry site (IRES)was inserted into the XhoI-digested pB-TCRa and pB-TCRb,resulting in pB-TCRa:GFP and pB-TCRb:GFP constructs.Human T lymphocytes of healthy donors were activatedwith anti-CD3 monoclonal antibody (mAb) and transducedwith retroviruses harboring TCRa:GFP and TCRb:GFP orGFP only (Mock). The transduction procedure was per-formed as described by Lamers and colleagues (Lamers et al.,2006b), with the only exception that TCR-encoding retro-viruses were produced by a coculture of 293T and Ph-Apackaging cells. The murinized gp100/A2 TCR (hu:mo
188 STRAETEMANS ET AL.
Ta
bl
e1.
Tr
ea
tm
en
to
fH
um
an
Tu
mo
rs
wit
hR
ec
ep
to
r-E
ng
in
ee
re
dH
um
an
TC
el
ls
in
Im
mu
ne
-D
efi
cie
nt
Mic
ea
T-c
ell
tran
sfer
Tu
mor
cell
sH
isto
log
ical
orig
inM
iceb
Rec
epto
rcR
oute
dD
osag
eT
ime
poi
nts
eA
dd
itio
nal
trea
tmen
tfA
nti
tum
orac
tiv
ity
Ref
eren
ce
Ly
mp
ho
ma
cell
sR
aji
cell
sB
LS
CID
Bei
ge
CD
19C
AR
:fi.
v.
10–2
0·
106
day
6an
d7
yes
yes
Bre
ntj
ens
etal
.,20
03N
AL
M-6
cell
sP
re-B
cell
AL
LS
CID
Bei
ge
CD
19C
AR
:fi.
v.
10–2
0·
106
day
4n
oy
esg
Dau
di
cell
sB
LN
OD
.SC
IDC
D19
CA
R:(
28)f
i.p
.5
·10
6d
ay3
yes
yes
hK
ow
oli
ket
al.,
2006
Raj
ice
lls
BL
SC
IDB
eig
eC
D19
CA
R:(
28)f
i.v
.10
·10
6d
ay6
no
yes
Bre
ntj
ens
etal
.,20
07N
AL
M-6
cell
sP
re-B
cell
AL
LS
CID
Bei
ge
CD
19C
AR
:(28
)fi.
v.
10·
106
day
2,3,
4n
oy
esh
NA
LM
-6ce
lls
Pre
-Bce
llA
LL
SC
IDB
eig
eC
D19
CA
R:(
28)f
i.v
.10
·10
6d
ay2,
8,15
,22
no
yes
Au
tolo
go
us
tum
or
cell
sE
BV
+L
CL
SC
IDC
D30
CA
R:f
ii.
v.
10·
106
bet
wee
nd
ay10
and
15y
esy
esS
avo
ldo
etal
.,20
07
L42
8ce
lls
EB
V-C
D30
+H
DS
CID
CD
30C
AR
:fi
i.p
.10
·10
6d
ay7
yes
yes
Raj
ice
lls
BL
SC
IDB
eig
eC
D19
CA
R:f
i.v
.10
·10
6d
ay6
yes
yes
Ch
ead
leet
al.,
2008
Pre
-BA
LL
cell
sA
LL
NO
D.S
CID
-b�=�
2o
rN
SG
CD
19C
AR
:(28
)(B
B)f
i.v
.1–
20·
106
bet
wee
nd
ay9
and
21y
esy
esM
ilo
ne
etal
.,20
09
BV
173
cell
sC
ML
NO
D.S
CID
WT
1-T
CR
i.v
.20
·10
6d
ay1
no
yes
Xu
eet
al.,
2005
CD
34+
leu
kem
iap
rog
enit
or
cell
sC
ML
NO
D.S
CID
WT
1-S
S-T
CR
i.v
.20
·10
6d
ay1
no
yes
Xu
eet
al.,
2010
So
lid
tum
ors
CW
R22
cell
sP
CS
CID
Bei
ge
Erb
B2
CA
R:2
8ci.
t.10
·10
6d
ay31
,32
,33
,34
,35
yes
yes
Pin
thu
set
al.,
2003
WIS
H-P
C14
cell
sP
CS
CID
Bei
ge
Erb
B2
CA
R:2
8ci.
t.50
·10
6d
ay14
,15
,16
yes
yes
CO
LO
205
cell
sC
CN
OD
.SC
IDE
rbB
2C
AR
:28f
i.v
.10
·10
6d
ay0,
1y
esy
esj
Ten
get
al.,
2004
MD
A-M
D-4
35ce
lls
BC
NO
D.S
CID
Erb
B2
CA
R:2
8fi.
v.
10·
106
day
0,1
yes
yes
j
WIS
H-P
C14
cell
sP
CS
CID
Bei
ge
Erb
B2
CA
R:2
8ci.
v.
50·
106
day
10y
esy
esk
Pin
thu
set
al.,
2004
Lu
CaP
-35
cell
sP
CS
CID
Bei
ge
Erb
B2
CA
R:2
8ci.
v.
50·
106
day
10y
esy
esk
OV
CA
R-3
cell
sO
CN
OD
.SC
IDL
eyC
AR
:28f
i.v
.10
·10
6d
ay0,
1,2,
5o
rd
ay7,
8,9,
12y
esy
esW
estw
oo
det
al.,
2005
LN
CaP
cell
sP
CS
CID
Bei
ge
PS
MA
Pz1
CA
R:f
i.t.
20·
106
bet
wee
nd
ay10
and
21n
oy
esG
ade
etal
.,20
05
LN
CaP
/C
4-2
cell
sP
CS
CID
Bei
ge
PS
MA
Pz1
CA
R:f
i.t.
20·
106
bet
wee
nd
ay15
and
30n
oy
es
M10
8ce
lls
ME
SN
SG
SS
1C
AR
:(28
)(B
B)f
i.t.
15·
106
day
46,
53n
oy
esC
arp
enit
oet
al.,
2009
M10
8ce
lls
ME
SN
SG
SS
1C
AR
:(28
)(B
B)f
i.t.
,i.
p.,
i.v
.10
·10
6d
ay43
,49
no
yes
BT
-474
cell
sB
CS
CID
Bei
ge
Erb
B2
CA
R:(
CD
8)28
(BB
)fi.
v.
1.5–
2·
106
day
10y
esy
esZ
hao
etal
.,20
09O
V-C
AR
3(M
UC
-CD
)O
VS
CID
bei
ge
MU
C-C
D4H
11C
AR
:(28
)fi.
p.,
i.v
.30
·10
6d
ay2
no
yes
Ch
ekm
aso
va
etal
.,20
10
(con
tin
ued
)
189

Ta
bl
e1.
(Co
nt
in
ue
d)
T-c
ell
tran
sfer
Tu
mor
cell
sH
isto
log
ical
orig
inM
iceb
Rec
epto
rcR
oute
dD
osag
eT
ime
poi
nts
eA
dd
itio
nal
trea
tmen
tfA
nti
tum
orac
tiv
ity
Ref
eren
ce
SK
-N-A
SN
BN
SG
GD
2C
AR
:28.
OX
40.f
i.v
.10
·10
6d
ay3,
10,
17y
esy
esl
Cra
dd
ock
etal
.,20
10M
108
ME
SN
SG
SS
1R
NA
CA
R:B
Bfm
i.t.
10–1
5·
106
day
66,
70,
74,
78n
oy
esZ
hao
etal
.,20
10S
KO
V3
OV
NS
GM
Ov
19C
AR
:(B
B)f
i.t.
,i.
p.,
i.v
.8
·10
6d
ay0,
5n
oy
esn
So
ng
etal
.,20
11S
K-2
3ce
lls
ME
LS
CID
Mel
anA
TC
RW
inn
assa
y5
·10
6d
ay0
yes
yes
Bo
bis
seet
al.,
2009
SK
-23
cell
sM
EL
SC
IDM
elan
AT
CR
i.t.
10·
106
day
5,10
,14
yes
yes
SK
-23
cell
sM
EL
SC
IDM
elan
AT
CR
i.v
.10
·10
6d
ay5,
10,
14y
esn
o
AL
L,a
cute
lym
ph
ob
last
icle
uk
emia
;BB
,CD
137
(4-1
BB
)co
stim
ula
tory
mo
lecu
le;B
C,b
reas
tca
rcin
om
a;B
D-T
CR
,bid
irec
tio
nal
TC
R;B
L,B
urk
itt
lym
ph
om
a;C
C,c
olo
nca
rcin
om
a;C
ML
,ch
ron
icm
yel
oid
leu
kem
ia;
EB
V+
LC
L,E
pst
ein
Bar
rv
iru
s–p
osi
tiv
ela
rge-
cell
lym
ph
om
a;E
BV
-CD
30+
HD
,E
BV
-CD
30+
Ho
dg
kin
lym
ph
om
a;E
rbB
2,al
sok
no
wn
asH
ER
2/N
eu,
hu
man
epid
erm
alg
row
thfa
cto
rre
cep
tor
2;G
C,
gas
tric
carc
ino
ma;
Ley
,L
ewis
Yan
tig
en;
ME
L,
mel
ano
ma;
Mel
anA
,m
elan
ocy
tean
tig
en;
ME
S,
mes
oth
elio
ma;
MO
v19
,m
on
ocl
on
alan
tib
od
ycl
on
eag
ain
sth
um
anfo
late
rece
pto
ra;
OC
,o
var
ian
carc
ino
ma;
PC
,p
rost
ate
carc
ino
ma;
PS
MA
,p
rost
ate-
spec
ific
mem
bra
ne
anti
gen
;S
S1,
anti
-mes
oth
elin
Fv
;W
T1,
Wil
ms’
tum
or
anti
gen
.aT
his
tab
lep
rov
ides
ad
etai
led
ov
erv
iew
of
stu
die
sp
erfo
rmed
wit
hei
ther
chim
eric
anti
bo
dy
-bas
edre
cep
tor
(CA
R)
or
T-c
ell
rece
pto
r(T
CR
)-en
gin
eere
dh
um
anT
cell
sin
hu
man
tum
or-
bea
rin
gim
mu
ne-
defi
cien
tm
ice.
Th
est
ud
ies
are
com
par
edw
ith
resp
ect
totu
mo
rce
lls,
mo
use
stra
ins,
anti
gen
-sp
ecifi
cre
cep
tors
,T
-cel
ltr
ansf
ers,
add
itio
nal
trea
tmen
ts,
and
anti
tum
or
resp
on
ses.
bM
ouse
stra
ins:
SC
ID,
sev
ere
com
bin
edim
mu
ne-
defi
cien
tm
ice
con
tain
ing
the
Prk
dc
mu
tati
on
;S
CID
Bei
ge,
mic
eco
nta
inin
gth
eP
rkd
can
dB
eig
em
uta
tio
n;
NO
D.S
CID
,m
ice
con
tain
ing
the
Prk
dc
mu
tati
on
on
an
on
ob
ese
dia
bet
icb
ack
gro
un
d;
NO
D.S
CID
-b–/
–,
NO
D.S
CID
mic
eco
nta
inin
ga
b2m
mu
tati
on
;N
SG
,N
OD
.SC
IDm
ice
con
tain
ing
anil
2rg
mu
tati
on
.c R
ecep
tor
form
at:
CA
Rre
cep
tors
are
chim
eric
rece
pto
rsth
atco
nsi
sto
fv
ario
us
‘‘bu
ild
ing
blo
cks’
’su
chas
(do
mai
ns
of)
Fc(
e)R
Ic(i
nsh
ort
c)o
rC
D3f
(f),
wh
ich
are
inso
me
case
sco
mb
ined
wit
hco
stim
ula
tory
mo
lecu
les
such
asC
D28
(28)
or
4-1B
B(1
BB
).S
S-T
CR
isa
cyst
ein
e-m
od
ified
TC
R.
dR
oute
ofT
-cel
lad
min
istr
atio
n:
i.p
.,in
trap
erit
on
eal;
i.t.
,in
trat
um
ora
l;i.
v.,
intr
aven
ou
s.eT
ime
po
int(
s)o
fT
-cel
ltr
ansf
erar
eaf
ter
tum
or
tran
spla
nt.
f Inca
seo
fan
add
itio
nal
trea
tmen
t,d
etai
lsar
eli
sted
her
ep
ertu
mo
rm
od
elan
dre
fere
nce
:Raj
ice
lls,
T-c
ell
exp
ansi
on
on
arti
fici
alan
tig
en-p
rese
nti
ng
cell
s(A
AP
C)
plu
sIL
-15
(Bre
ntj
ens
etal
.,20
03);
Dau
di
cell
s,2.
5G
yto
tal
bo
dy
irra
dia
tio
n(T
BI)
(Ko
wo
lik
etal
.,20
06);
auto
log
ou
stu
mo
rce
lls
and
L42
8ce
lls,
230
cGy
TB
Ian
dIL
-2su
pp
ort
(Sav
old
oet
al.,
2007
);R
aji
cell
s,cy
clo
ph
osp
ham
ide
(Ch
ead
leet
al.,
2008
);p
re-B
AL
Lce
lls,
T-c
ell
exp
ansi
on
on
CD
28b
ead
-bas
edA
AP
C(M
ilo
ne
etal
.,20
09);
CW
R22
and
WIS
H-P
C14
cell
s,IL
-2su
pp
ort
(Pin
thu
set
al.,
2003
);C
OL
O20
5an
dM
DA
-MD
-435
cell
s,2.
5G
yT
BI
(Ten
get
al.,
2004
);W
ISH
-PC
14ce
lls
and
Lu
Cap
-35
cell
s,2
Gy
TB
Ian
dIL
-2su
pp
ort
(Pin
thu
set
al.,
2004
);O
VC
AR
-3ce
lls,
2.5
Gy
TB
I(W
estw
oo
det
al.,
2005
);B
T-4
74ce
lls,
cycl
op
ho
sph
amid
ean
dIL
-2su
pp
ort
(Zh
aoet
al.,
2009
);S
K-2
3ce
lls,
IL-2
sup
po
rt(B
ob
isse
etal
.,20
09).
gC
D80
+,
bu
tn
ot
CD
80–,
NA
LM
-6tu
mo
rsw
ere
reje
cted
.hC
D19
CA
R:2
8f,
bu
tn
ot
CD
19C
AR
:f,
cau
sed
anan
titu
mo
rre
spo
nse
.i In
this
stu
dy
,E
BV
-sp
ecifi
ccy
toto
xic
Tce
lls
wer
eu
sed
ash
ost
Tce
lls
for
rece
pto
rg
ene
tran
sfer
,w
her
eas
ino
ther
stu
die
sp
oly
clo
nal
per
iph
eral
Tce
lls
wer
eu
sed
.j W
hen
mic
ew
ere
no
tir
rad
iate
d,
rece
pto
r-en
gin
eere
dT
cell
sd
idn
ot
cau
sean
anti
tum
or
resp
on
se.
kW
hen
mic
ew
ere
irra
dia
ted
or
trea
ted
wit
hcy
clo
ph
osp
ham
ide,
rece
pto
r-en
gin
eere
dT
cell
sd
idn
ot
cau
sean
anti
tum
or
resp
on
se.
l Wh
enm
ice
rece
ived
CA
RT
cell
sal
soen
gin
eere
dto
exp
ress
the
chem
ok
ine
rece
pto
rC
CR
2b,
anan
titu
mo
rre
spo
nse
was
ob
serv
ed.
mIn
this
stu
dy
,T
cell
sw
ere
elec
tro
po
rate
dw
ith
CA
R-e
nco
din
gm
RN
A.
nM
Ov
19C
AR
:1-B
Bf,
bu
tn
ot
CA
R:f
,ca
use
dan
anti
tum
or
resp
on
se.
190

Ta
bl
e2.
Tr
ea
tm
en
to
fH
um
an
Tu
mo
rs
wit
hR
ec
ep
to
r-E
ng
in
ee
re
dM
ou
se
TC
el
ls
in
Im
mu
ne
-D
efi
cie
nt
Mic
ea
T-c
ell
tran
sfer
Tu
mor
cell
sH
isto
log
ical
orig
inM
iceb
Rec
epto
r(s)
cR
oute
dD
osag
eT
ime
poi
nts
eA
dd
itio
nal
trea
tmen
tA
nti
tum
orac
tiv
ity
Ref
eren
ce
So
lid
tum
ors
IGR
OV
-1ce
lls
OC
Nu
de
MO
v18
CA
R:c
fi.
p.
10–3
0·
106
day
3n
oy
esH
wu
etal
.,19
95C
OL
O20
5ce
lls
CC
SC
IDC
EA
CA
R:c
i.v
.5
·10
6d
ay0,
1,2,
or
3gn
oy
esD
arcy
etal
.,20
00C
OL
O20
5ce
lls
CC
SC
IDC
EA
CA
R:c
or
fi.
v.
5·
106
day
0an
d1
no
yes
Hay
nes
etal
.,20
01C
OL
O20
5ce
lls
CC
SC
IDC
EA
CA
R:(
CD
28)c
i.v
.5
·10
6d
ay0
and
1n
oy
esH
ayn
eset
al.,
2002
a,b
CO
LO
205
cell
sC
CS
CID
Erb
B2
CA
R:(
CD
28)f
i.v
.0.
1–10
·10
6d
ay0,
1,an
d/
or
3n
oy
esH
ayn
eset
al.,
2002
a,b
CC
,co
lon
carc
ino
ma;
CE
A,c
arci
no
emb
ryo
nic
anti
gen
;E
rbB
2,al
sok
no
wn
asH
ER
2/N
eu,
hu
man
epid
erm
alg
row
thfa
cto
rre
cep
tor
2;M
Ov
18,m
Ab
clo
ne
agai
nst
hu
man
fola
tere
cep
tora;
OC
,ov
aria
nca
rcin
om
a.aT
his
tab
lep
rov
ides
ad
etai
led
ov
erv
iew
of
stu
die
sp
erfo
rmed
wit
hch
imer
ican
tib
od
y-b
ased
rece
pto
r(C
AR
)-ex
pre
ssin
gm
ou
seT
cell
sin
hu
man
tum
or-
bea
rin
gim
mu
ne-
defi
cien
tm
ice.
Th
ere
are
no
inv
ivo
stu
die
sre
po
rted
sofa
rw
ith
TC
R-e
ng
inee
red
mo
use
Tce
lls
dir
ecte
dto
war
dh
um
antu
mo
rsin
imm
un
e-d
efici
ent
mic
e.T
he
stu
die
sar
eco
mp
ared
wit
hre
spec
tto
tum
or
cell
s,m
ou
sest
rain
s,an
tig
en-
spec
ific
rece
pto
rs,
T-c
ell
tran
sfer
s,ad
dit
ion
altr
eatm
ents
,an
dan
titu
mo
rre
spo
nse
s.bM
ouse
stra
ins:
Nu
de
mic
e,at
hy
mic
mic
ed
ue
toa
mu
tati
on
inth
efo
xn
1g
ene;
SC
ID,
sev
ere
com
bin
edim
mu
ne-
defi
cien
tm
ice
con
tain
ing
the
Prk
dc
mu
tati
on
.c R
ecep
tor
form
at:
CA
Rre
cep
tors
are
chim
eric
rece
pto
rsth
atco
nsi
sto
fv
ario
us
‘‘bu
ild
ing
blo
cks’
’su
chas
(do
mai
ns
of)
Fc(
e)R
Ic(i
nsh
ort
c)o
rC
D3f
(f),
wh
ich
are
inso
me
case
sco
mb
ined
wit
hco
stim
ula
tory
mo
lecu
les
such
asC
D28
(28)
.dR
oute
ofT
-cel
lad
min
istr
atio
n:
i.p
.,in
trap
erit
on
eal;
i.t.
,in
trat
um
ora
l;i.
v.,
intr
aven
ou
s.eT
ime
po
int(
s)o
fT
-cel
ltr
ansf
eraf
ter
tum
or
tran
spla
nt.
f Tu
mo
r-in
filt
rati
ng
lym
ph
ocy
tes
(TIL
)d
eriv
edfr
om
am
ou
sead
eno
carc
ino
ma
(MC
38)
tum
or
wer
eu
sed
ash
ost
cell
sfo
rre
cep
tor
gen
etr
ansf
er,
wh
erea
sin
oth
erst
ud
ies
po
lycl
on
alp
erip
her
alT
cell
sw
ere
use
d.
gT
-cel
ltr
ansf
ers
are
giv
eno
ne
tofo
ur
tim
esfr
om
day
0u
nti
ld
ay3.
191

gp100/A2 TCR) was derived as described previously byPouw and colleagues (Pouw et al., 2007) and used to transducemurine T cells. Similar to the receptors used to transducehuman T cells, IRES-GFP was subcloned into the XhoI-digested hu:mo gp100/A2 TCRa and b genes. Total mousesplenocytes were isolated and activated with concanava-lin A in CMM and transduced with the retroviral vectorpBullet containing hu:mo gp100/A2 TCRab transgenes or thepSTICH:eGFP vector as control (Mock) as described previ-ously (Pouw et al., 2007).
Flow cytometry
Gene-transduced human T cells were monitored withgp100/HLA-A2 phycoerythrin (PE)-labeled tetramers(termed gp100/A2 pMHC) followed by a cell sort for gp100/A2 pMHC binding cells using a FACS-Vantage instrument(Becton Dickinson Biosciences, San Jose, CA). Surface ex-pression of TCR transgenes by human and mouse T cells wasmeasured using a PE-labeled anti-TCRVb14 mAb [Ardennomenclature (Arden et al., 1995)] (clone CAS1.1.3; BeckmanCoulter, Marseille, France). For adoptive T-cell transferstudies, we have used 3–4-week-old > 90% TCRVb14-positive,and > 80% gp100/A2-binding human T cells. TCR-transducedmouse T cells generally comprised about 30% TCRVb14-positive T cells at the day of infusion. Peripheral blood, organs,and tumors were analyzed for the presence of TCR-engineeredT cells by flow cytometry using the anti-TCRVb14 mAbin combination with GFP, PerCP-labeled anti-CD3e mAb(clone 145-2C11), and antigen-presenting cell (APC)-labeledanti-CD8a mAb (clone 53-6.7) (both from Becton DickinsonBiosciences). We analyzed the samples on a Cytomics FC-500flow cytometer using CXP software (Beckman Coulter) or aFACSCalibur using CellQuest software (Becton DickinsonBiosciences).
AT models
B6 SCID, BALB/c SCID, or NSG mice were subcutane-ously (s.c.) transplanted in the flank with either FM3 tumortissue (3 · 3 · 3 mm) or 1 · 106 BLMgp100 cells. Mice bearingestablished tumors (5 · 5 · 5 mm) or mice having a tumorplaced in a dorsal skinfold window chamber (see below)received two doses of TCR-transduced human (6–40 · 106) ormouse (1 · 106) T cells administered i.v. at days 0 and 7. Insome experiments, mice were preconditioned with cyclo-phosphamide (200 mg/kg; Sigma-Aldrich, St. Louis, MO)administered intraperitoneally (i.p.) at day - 1, preceded ornot by busulfan (16.5 lg/kg; Duchefa Farma, Haarlem, TheNetherlands) administered i.p. at day - 2 (protocol kindlyprovided by Dr. Naomi Taylor, Montpellier, France). Humanand mouse T-cell infusions were followed by 5 daily s.c.injections of recombinant human IL-2 (1 · 105 IU) starting atthe day of T-cell transfer or as indicated otherwise. Tumorsizes were measured with a caliper along the perpendicularaxes of tumors, and volumes were calculated with the for-mula 0.4 · (A2 · B) where B represents the largest diameterand A the diameter perpendicular to B (Seynhaeve et al.,2007). In separate experiments using mouse GFP T cells, weassessed the individual and combined effects of T-cell acti-vation, retroviral gene transfer, and s.c. IL-2 support ontumor growth. Blood from tail veins was collected weeklyafter the first T-cell infusion and analyzed for T cells by flow
cytometry. At the indicated time points, tumors and lym-phoid organs were isolated. Spleens and inguinal lymphnodes were isolated and processed to obtain single-cell sus-pensions by mechanic disruption over a cell strainer in PBS.Tumors were isolated, cut into small pieces, and digested inPBS containing collagenase (1 mg/ml) for 45 min at 37�Cwhile vortexing every 5 min. Then 1 ml of 0.1 mM EDTA wasadded, and the tumor was disrupted over a cell strainer.Erythrocytes in blood and organs were removed by incu-bation in erylysis buffer (154 mM NH4Cl) for 15 min on ice.Absolute cell counts were determined using Flow-CountFluorospheres (Beckman Coulter).
T-cell trafficking to tumor site using dorsalskinfold chamber
We prepared dorsal skinfold chambers according toSeynhaeve and colleagues (Seynhaeve et al., 2007). In short,mice were anesthetized with a mixture of saline, ketamine,and xylazine, injected s.c. Hair was removed from the skinon the back of the animal, and the skin was dissected in acircular area leaving the fascia and opposing skin intact.Next, the skinfold of the mouse was sandwiched betweentwo frames and fixed with two light metal bolts and sutures.A small piece of tumor tissue was transplanted in the fascia,and both sides were closed with a microscopic cover glass.During surgery, the body temperature of animals was keptconstant at 37�C, after which animals were housed individ-ually at an ambient temperature of 32�C and a humidity of70%. Twenty-one days after implantation, mice received GFPT cells. At the given time points, mice were anesthetized with3% isoflurane and fixed to a heated stage of a Leica DM-RXAmicroscope. To visualize the presence of T cells, we used aLeica I3 filter set (excitation: 450–490 nm; emission: LP 515)and photographed the GFP T cells using a Sony 3CCD DXC950 camera.
Immune histochemistry
Tumors were isolated, snap-frozen in liquid nitrogen, andstored at - 80�C until use. Tissue sections were cut at 5 lmand stained for T cells with the following primary antibodies:rat anti-mouse CD3e (clone GRJ01; R&D Systems, Abingdon,UK), rat anti-mouse CD4 (clone GK1.5), rat anti-mouse CD8a(clone 53-6.7) (both Biolegend, San Diego, CA), or rat IgGisotype control (clone 141945; R&D Systems). Goat anti-ratIgG Alexa Fluor 488 (Molecular Probes, Invitrogen, Breda,The Netherlands) was used as a secondary antibody. Air-dried cryosections were fixed with acetone for 10 min andblocked with PBS/1% bovine serum albumin for 30 minprior to immune staining. Next, sections were incubated withthe primary antibody for 1 hr, and then washed and incu-bated with the secondary antibody for 1 hr, both at roomtemperature. Finally, we stained the nuclei with DAPI (4’,6-diamidino-2-phenylindole) and embedded the sections inmounting medium containing polyvinyl alcohol (MoWiol-488; Fluka, Zwijndrecht, The Netherlands). Sections wereexamined microscopically and photographed as describedabove. Recorded photographs were analyzed using ImageTool v3 (developed by Don Wilcox, University of TexasHealth Science Center, San Antonio, TX) as described pre-viously (Seynhaeve et al., 2007). The RGB images were con-verted to gray scale, and specific fluorescence intensities
192 STRAETEMANS ET AL.
(range: 0–255) were measured with the threshold set at 20.Percentages of pixels with fluorescence intensity abovethreshold were calculated and expressed as bars.
Statistical analysis
Tumor volumes, times to tumor regression, and absolutecell counts were evaluated for statistical significance with theMann-Whitney U test using SPSS v15 software, whereasimmune histochemistry was evaluated for statistical signifi-
cance with the Student’s t test using GraphPad Prism v4software. Differences with p £ 0.05 were considered signifi-cant.
Results
Human TCR-engineered T cells do not control humanmelanoma growth in immune-deficient mice
We assessed the in vivo performance of human peripheralblood–derived T cells engineered with gp100/A2 TCR,which we had previously validated in vitro (Schaft et al.,2003). gp100/A2 TCR T cells used for AT functionally ex-pressed TCR transgenes assessed by pMHC binding andtarget cell-specific cytotoxicity (Supplementary Fig. S1A andB; Supplementary Data are available at www.liebertonline.-com/hum). Intravenous transfer of a high dose of gp100/A2TCR T cells (40 · 106 cells) into B6 SCID mice bearing anti-gen-positive human melanoma (FM3 cells) did not delaytumor growth (Fig. 1A). Preconditioning with cyclophos-phamide or a combination of cyclophosphamide and bu-sulfan significantly depleted endogenous NK cells present inB6 SCID mice (Supplementary Fig. S2), but did not stimulatethe anti-FM3 melanoma T-cell response (Fig. 1B). The lack ofantimelanoma response of human T cells, whether or not inpreconditioned mice, was supported by our inability to de-tect human T cells in peripheral blood by flow cytometry(Supplementary Fig. S3A).
To completely exclude ‘‘leakage’’ of NK cells in conven-tional SCID mice, and their potential adverse effect on T-cellengraftment, we have tested NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice, commonly known as NOD SCID GAMMA (NSG)(Shultz et al., 2005). The NSG mouse strain combines featuresof the NOD mice, the mutation SCID, and IL-2R common-cchain deficiency. Consequently, NSG mice are severely im-mune-compromised, featuring the absence of mature T or Bcells, lack of functional NK cells, and deficiency in cytokinesignaling. T-cell engraftment was improved in NSG mice asevidenced by low but significantly higher numbers of TCR Tcells in the peripheral blood at days 3 and 10 after T-cell
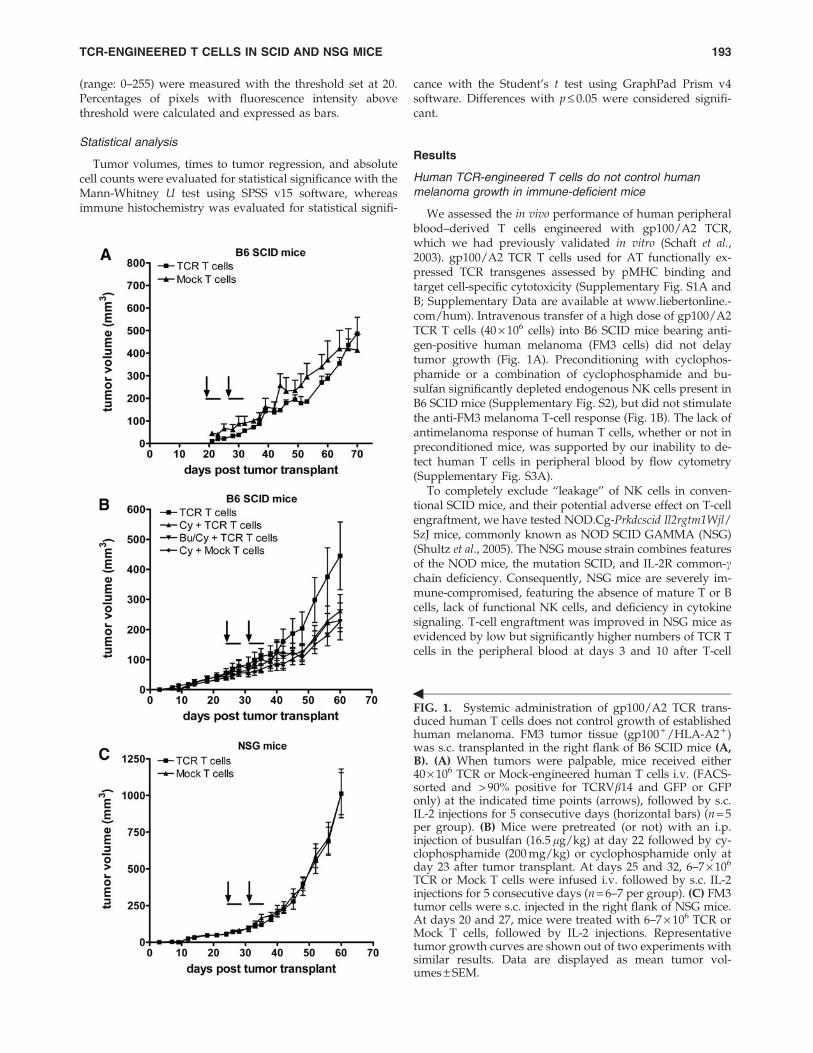
FIG. 1. Systemic administration of gp100/A2 TCR trans-duced human T cells does not control growth of establishedhuman melanoma. FM3 tumor tissue (gp100 + /HLA-A2 + )was s.c. transplanted in the right flank of B6 SCID mice (A,B). (A) When tumors were palpable, mice received either40 · 106 TCR or Mock-engineered human T cells i.v. (FACS-sorted and > 90% positive for TCRVb14 and GFP or GFPonly) at the indicated time points (arrows), followed by s.c.IL-2 injections for 5 consecutive days (horizontal bars) (n = 5per group). (B) Mice were pretreated (or not) with an i.p.injection of busulfan (16.5 lg/kg) at day 22 followed by cy-clophosphamide (200 mg/kg) or cyclophosphamide only atday 23 after tumor transplant. At days 25 and 32, 6–7 · 106
TCR or Mock T cells were infused i.v. followed by s.c. IL-2injections for 5 consecutive days (n = 6–7 per group). (C) FM3tumor cells were s.c. injected in the right flank of NSG mice.At days 20 and 27, mice were treated with 6–7 · 106 TCR orMock T cells, followed by IL-2 injections. Representativetumor growth curves are shown out of two experiments withsimilar results. Data are displayed as mean tumor vol-umes – SEM.
=
TCR-ENGINEERED T CELLS IN SCID AND NSG MICE 193

transfer (Supplementary Fig. S3B). However, adoptivelytransferred T cells did not persist beyond a time period of2 weeks past T-cell transfer and did not result in an antitu-mor response in melanoma-bearing NSG mice (Fig. 1C).
Human TCR-engineered T cells accumulatein tumor vasculature, but are lost shortly after AT
We used T-cell imaging to investigate whether the ob-served lack of peripheral persistence and antitumor responseof human TCR T cells was related to their inability to migrateto the tumor site. Mice bearing an FM3 human melanoma ina dorsal skinfold window chamber received 50 · 106 gp100/A2 TCR:GFP T cells. Directly after injection of T cells, TCR Tcells, but not mock T cells, accumulated in the tumor vas-
culature (Fig. 2A). Strikingly, the accumulated TCR T cellscompletely disappeared from the tumor site within 72 hrafter adoptive T cell (Fig. 2B). Notably, human T cells pre-dominantly showed a phenotype that corresponded to ef-fector memory and late effector T cells at the time of AT(Supplementary Fig. S4).
Murine T cells cause complete, yet TCRtransgene-independent, regression of humanmelanoma in immune-deficient mice
In the next series of experiments, we switched to gene-engineered murine T cells, which in the setting of CARsproved effective against solid tumors in SCID mice (Darcyet al., 2000; Haynes et al., 2001, 2002b,a). FM3 tumor-bearing
FIG. 2. Gp100/A2 TCR-transduced humanT cells initially accumulate in the tumorvasculature, but rapidly disappear. Micebearing a human melanoma tumor in adorsal skinfold window chamber received50 · 106 TCR/GFP- or GFP-engineeredhuman T cells i.v. at day 21 after tumortransplantation. Immediately after T-celltransfer, mice were anesthetized and fixed toa heated stage of a Leica DM-RXA micro-scope. T cells in the tumor vasculature werevisualized with a GFP filter set (excitationwavelength: 450–490 nm; emission filter: LP515). (A) TCR T cells do (a, b) and Mock (i.e.,GFP only) T cells do not (c, d) accumulate inthe tumor vasculature directly after T-cellinfusion (fluorescent fields: a and c; brightfields: b and d; magnifications: 10 · ). (B) Thepresence of T cells in the tumor vasculaturewas monitored at the following time pointsafter T-cell injection in the dorsal skinfoldwindow chamber (n = 3): 0 hr, 6 hr, 24 hr, and72 hr. Representative results of one TCR T-cell–treated mouse are shown (all fluorescentfields; magnification: 20 · ).
194 STRAETEMANS ET AL.
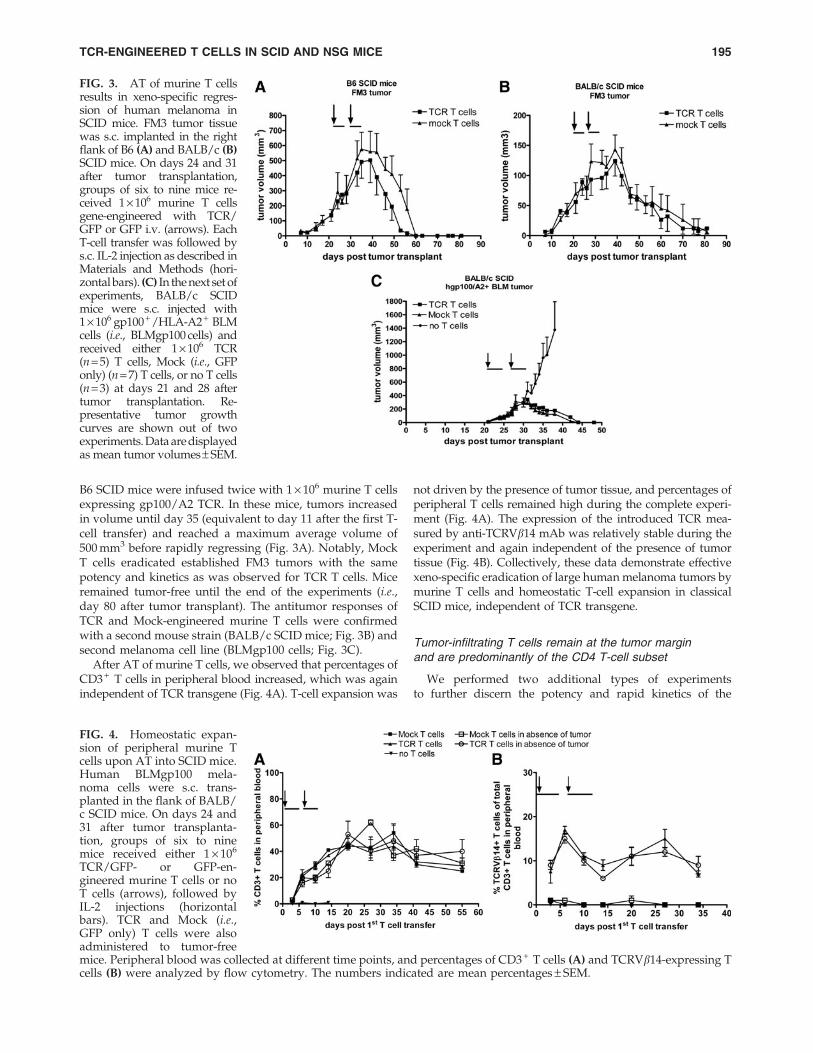
B6 SCID mice were infused twice with 1 · 106 murine T cellsexpressing gp100/A2 TCR. In these mice, tumors increasedin volume until day 35 (equivalent to day 11 after the first T-cell transfer) and reached a maximum average volume of500 mm3 before rapidly regressing (Fig. 3A). Notably, MockT cells eradicated established FM3 tumors with the samepotency and kinetics as was observed for TCR T cells. Miceremained tumor-free until the end of the experiments (i.e.,day 80 after tumor transplant). The antitumor responses ofTCR and Mock-engineered murine T cells were confirmedwith a second mouse strain (BALB/c SCID mice; Fig. 3B) andsecond melanoma cell line (BLMgp100 cells; Fig. 3C).
After AT of murine T cells, we observed that percentages ofCD3 + T cells in peripheral blood increased, which was againindependent of TCR transgene (Fig. 4A). T-cell expansion was
not driven by the presence of tumor tissue, and percentages ofperipheral T cells remained high during the complete experi-ment (Fig. 4A). The expression of the introduced TCR mea-sured by anti-TCRVb14 mAb was relatively stable during theexperiment and again independent of the presence of tumortissue (Fig. 4B). Collectively, these data demonstrate effectivexeno-specific eradication of large human melanoma tumors bymurine T cells and homeostatic T-cell expansion in classicalSCID mice, independent of TCR transgene.
Tumor-infiltrating T cells remain at the tumor marginand are predominantly of the CD4 T-cell subset
We performed two additional types of experimentsto further discern the potency and rapid kinetics of the
FIG. 3. AT of murine T cellsresults in xeno-specific regres-sion of human melanoma inSCID mice. FM3 tumor tissuewas s.c. implanted in the rightflank of B6 (A) and BALB/c (B)SCID mice. On days 24 and 31after tumor transplantation,groups of six to nine mice re-ceived 1 · 106 murine T cellsgene-engineered with TCR/GFP or GFP i.v. (arrows). EachT-cell transfer was followed bys.c. IL-2 injection as described inMaterials and Methods (hori-zontal bars). (C) In the next set ofexperiments, BALB/c SCIDmice were s.c. injected with1 · 106 gp100+/HLA-A2+ BLMcells (i.e., BLMgp100 cells) andreceived either 1 · 106 TCR(n = 5) T cells, Mock (i.e., GFPonly) (n = 7) T cells, or no T cells(n = 3) at days 21 and 28 aftertumor transplantation. Re-presentative tumor growthcurves are shown out of twoexperiments. Data are displayedas mean tumor volumes– SEM.
FIG. 4. Homeostatic expan-sion of peripheral murine Tcells upon AT into SCID mice.Human BLMgp100 mela-noma cells were s.c. trans-planted in the flank of BALB/c SCID mice. On days 24 and31 after tumor transplanta-tion, groups of six to ninemice received either 1 · 106
TCR/GFP- or GFP-en-gineered murine T cells or noT cells (arrows), followed byIL-2 injections (horizontalbars). TCR and Mock (i.e.,GFP only) T cells were alsoadministered to tumor-freemice. Peripheral blood was collected at different time points, and percentages of CD3 + T cells (A) and TCRVb14-expressing Tcells (B) were analyzed by flow cytometry. The numbers indicated are mean percentages – SEM.
TCR-ENGINEERED T CELLS IN SCID AND NSG MICE 195
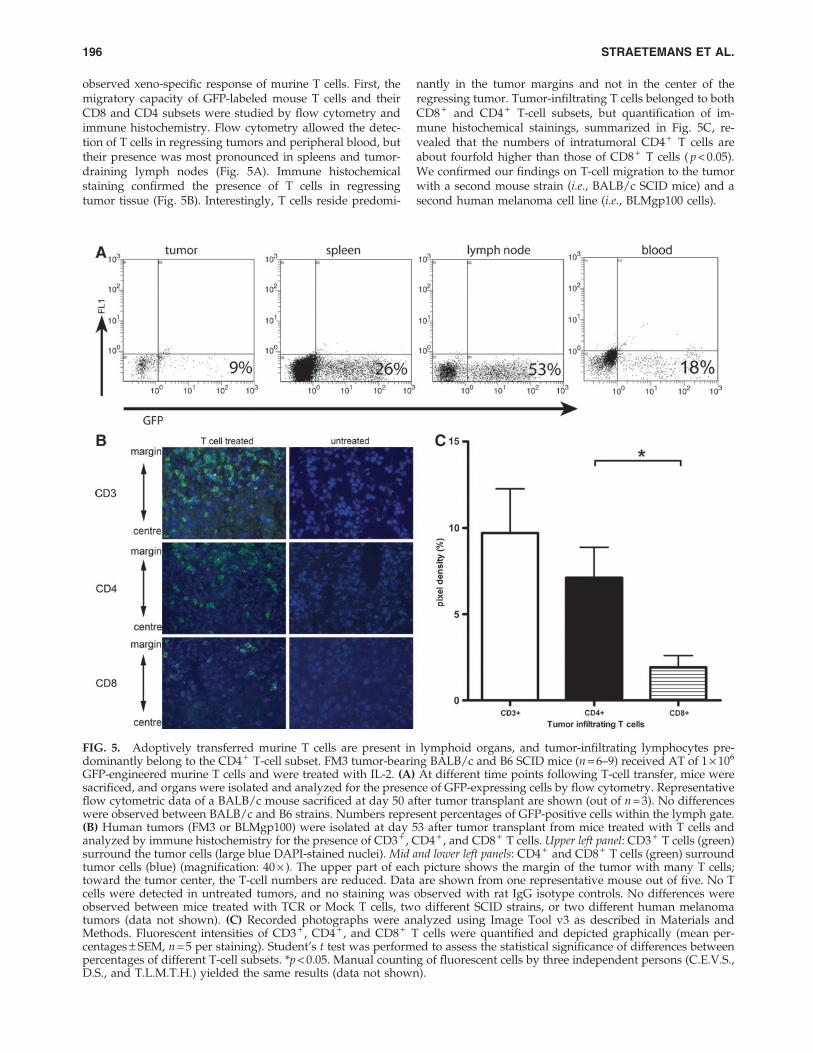
observed xeno-specific response of murine T cells. First, themigratory capacity of GFP-labeled mouse T cells and theirCD8 and CD4 subsets were studied by flow cytometry andimmune histochemistry. Flow cytometry allowed the detec-tion of T cells in regressing tumors and peripheral blood, buttheir presence was most pronounced in spleens and tumor-draining lymph nodes (Fig. 5A). Immune histochemicalstaining confirmed the presence of T cells in regressingtumor tissue (Fig. 5B). Interestingly, T cells reside predomi-
nantly in the tumor margins and not in the center of theregressing tumor. Tumor-infiltrating T cells belonged to bothCD8 + and CD4 + T-cell subsets, but quantification of im-mune histochemical stainings, summarized in Fig. 5C, re-vealed that the numbers of intratumoral CD4 + T cells areabout fourfold higher than those of CD8 + T cells ( p < 0.05).We confirmed our findings on T-cell migration to the tumorwith a second mouse strain (i.e., BALB/c SCID mice) and asecond human melanoma cell line (i.e., BLMgp100 cells).
FIG. 5. Adoptively transferred murine T cells are present in lymphoid organs, and tumor-infiltrating lymphocytes pre-dominantly belong to the CD4 + T-cell subset. FM3 tumor-bearing BALB/c and B6 SCID mice (n = 6–9) received AT of 1 · 106
GFP-engineered murine T cells and were treated with IL-2. (A) At different time points following T-cell transfer, mice weresacrificed, and organs were isolated and analyzed for the presence of GFP-expressing cells by flow cytometry. Representativeflow cytometric data of a BALB/c mouse sacrificed at day 50 after tumor transplant are shown (out of n = 3). No differenceswere observed between BALB/c and B6 strains. Numbers represent percentages of GFP-positive cells within the lymph gate.(B) Human tumors (FM3 or BLMgp100) were isolated at day 53 after tumor transplant from mice treated with T cells andanalyzed by immune histochemistry for the presence of CD3 + , CD4 + , and CD8 + T cells. Upper left panel: CD3 + T cells (green)surround the tumor cells (large blue DAPI-stained nuclei). Mid and lower left panels: CD4 + and CD8 + T cells (green) surroundtumor cells (blue) (magnification: 40 · ). The upper part of each picture shows the margin of the tumor with many T cells;toward the tumor center, the T-cell numbers are reduced. Data are shown from one representative mouse out of five. No Tcells were detected in untreated tumors, and no staining was observed with rat IgG isotype controls. No differences wereobserved between mice treated with TCR or Mock T cells, two different SCID strains, or two different human melanomatumors (data not shown). (C) Recorded photographs were analyzed using Image Tool v3 as described in Materials andMethods. Fluorescent intensities of CD3 + , CD4 + , and CD8 + T cells were quantified and depicted graphically (mean per-centages – SEM, n = 5 per staining). Student’s t test was performed to assess the statistical significance of differences betweenpercentages of different T-cell subsets. *p < 0.05. Manual counting of fluorescent cells by three independent persons (C.E.V.S.,D.S., and T.L.M.T.H.) yielded the same results (data not shown).
196 STRAETEMANS ET AL.
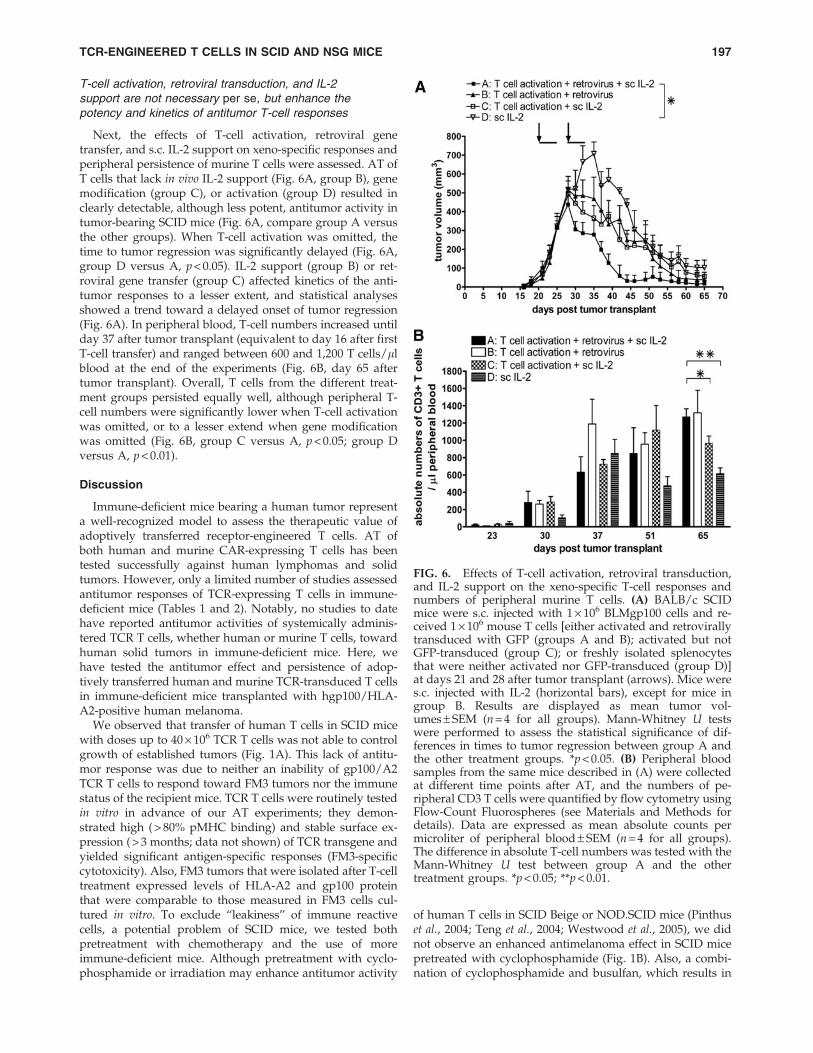
T-cell activation, retroviral transduction, and IL-2support are not necessary per se, but enhance thepotency and kinetics of antitumor T-cell responses
Next, the effects of T-cell activation, retroviral genetransfer, and s.c. IL-2 support on xeno-specific responses andperipheral persistence of murine T cells were assessed. AT ofT cells that lack in vivo IL-2 support (Fig. 6A, group B), genemodification (group C), or activation (group D) resulted inclearly detectable, although less potent, antitumor activity intumor-bearing SCID mice (Fig. 6A, compare group A versusthe other groups). When T-cell activation was omitted, thetime to tumor regression was significantly delayed (Fig. 6A,group D versus A, p < 0.05). IL-2 support (group B) or ret-roviral gene transfer (group C) affected kinetics of the anti-tumor responses to a lesser extent, and statistical analysesshowed a trend toward a delayed onset of tumor regression(Fig. 6A). In peripheral blood, T-cell numbers increased untilday 37 after tumor transplant (equivalent to day 16 after firstT-cell transfer) and ranged between 600 and 1,200 T cells/llblood at the end of the experiments (Fig. 6B, day 65 aftertumor transplant). Overall, T cells from the different treat-ment groups persisted equally well, although peripheral T-cell numbers were significantly lower when T-cell activationwas omitted, or to a lesser extend when gene modificationwas omitted (Fig. 6B, group C versus A, p < 0.05; group Dversus A, p < 0.01).
Discussion
Immune-deficient mice bearing a human tumor representa well-recognized model to assess the therapeutic value ofadoptively transferred receptor-engineered T cells. AT ofboth human and murine CAR-expressing T cells has beentested successfully against human lymphomas and solidtumors. However, only a limited number of studies assessedantitumor responses of TCR-expressing T cells in immune-deficient mice (Tables 1 and 2). Notably, no studies to datehave reported antitumor activities of systemically adminis-tered TCR T cells, whether human or murine T cells, towardhuman solid tumors in immune-deficient mice. Here, wehave tested the antitumor effect and persistence of adop-tively transferred human and murine TCR-transduced T cellsin immune-deficient mice transplanted with hgp100/HLA-A2-positive human melanoma.
We observed that transfer of human T cells in SCID micewith doses up to 40 · 106 TCR T cells was not able to controlgrowth of established tumors (Fig. 1A). This lack of antitu-mor response was due to neither an inability of gp100/A2TCR T cells to respond toward FM3 tumors nor the immunestatus of the recipient mice. TCR T cells were routinely testedin vitro in advance of our AT experiments; they demon-strated high ( > 80% pMHC binding) and stable surface ex-pression ( > 3 months; data not shown) of TCR transgene andyielded significant antigen-specific responses (FM3-specificcytotoxicity). Also, FM3 tumors that were isolated after T-celltreatment expressed levels of HLA-A2 and gp100 proteinthat were comparable to those measured in FM3 cells cul-tured in vitro. To exclude ‘‘leakiness’’ of immune reactivecells, a potential problem of SCID mice, we tested bothpretreatment with chemotherapy and the use of moreimmune-deficient mice. Although pretreatment with cyclo-phosphamide or irradiation may enhance antitumor activity
of human T cells in SCID Beige or NOD.SCID mice (Pinthuset al., 2004; Teng et al., 2004; Westwood et al., 2005), we didnot observe an enhanced antimelanoma effect in SCID micepretreated with cyclophosphamide (Fig. 1B). Also, a combi-nation of cyclophosphamide and busulfan, which results in
FIG. 6. Effects of T-cell activation, retroviral transduction,and IL-2 support on the xeno-specific T-cell responses andnumbers of peripheral murine T cells. (A) BALB/c SCIDmice were s.c. injected with 1 · 106 BLMgp100 cells and re-ceived 1 · 106 mouse T cells [either activated and retrovirallytransduced with GFP (groups A and B); activated but notGFP-transduced (group C); or freshly isolated splenocytesthat were neither activated nor GFP-transduced (group D)]at days 21 and 28 after tumor transplant (arrows). Mice weres.c. injected with IL-2 (horizontal bars), except for mice ingroup B. Results are displayed as mean tumor vol-umes – SEM (n = 4 for all groups). Mann-Whitney U testswere performed to assess the statistical significance of dif-ferences in times to tumor regression between group A andthe other treatment groups. *p < 0.05. (B) Peripheral bloodsamples from the same mice described in (A) were collectedat different time points after AT, and the numbers of pe-ripheral CD3 T cells were quantified by flow cytometry usingFlow-Count Fluorospheres (see Materials and Methods fordetails). Data are expressed as mean absolute counts permicroliter of peripheral blood – SEM (n = 4 for all groups).The difference in absolute T-cell numbers was tested with theMann-Whitney U test between group A and the othertreatment groups. *p < 0.05; **p < 0.01.
TCR-ENGINEERED T CELLS IN SCID AND NSG MICE 197

transient yet complete depletion of T cells, B cells, NK cells,and dendritic cells in immune-competent mice (personalcommunication, Dr. Naomi Taylor, Montpellier, France;Straetemans et al., manuscript submitted), did not enhancethe antimelanoma activity of TCR-engineered human T cellsin SCID mice (Fig. 1B). In more immune-deficient NSG mice(Shultz et al., 2005), we again observed no antimelanomaresponse following treatment with TCR T cells (Fig. 1C).Notably, immediately after T-cell transfer, TCR, but notMock, T cells accumulated in FM3 tumor vasculature, butthese T cells were rapidly (within 72 hr) lost (Fig. 2). T-cellpersistence in peripheral blood was negligible and not en-hanced by preconditioning and, at best, only marginallyenhanced in NSG versus SCID mice (Supplementary Fig. S3).Our data confirm a study by Bobisse and colleagues, inwhich TCR-engineered T cells showed antimelanoma activ-ity and peripheral persistence following i.t. but not i.v. ad-ministration in immune-deficient mice (Bobisse et al., 2009).
Another parameter that is potentially related to T-cellpersistence is T-cell exhaustion. Flow cytometric analysis ofmarkers of T-cell memory, activation, and costimulationshowed that the majority of T cells at the day of AT have alate T-cell differentiation phenotype (Supplementary Fig. S4).In fact, using two sets of definitions of T-cell subsets, oneincluding the adhesion molecule CD62L (according to Ka-neko et al., 2009; Wang et al., 2011) and another includingboth costimulatory markers CD27 and CD28 (according toAppay et al., 2008; Kalos et al., 2011), we observed that thesum of effector memory and end-stage T cells minimallyrepresented about 90% of T cells. This finding may argue thatunder-representation of less differentiated cells (i.e., naiveand central memory T cells) relates to an inability of humanT cells to persist and act against the tumor. In fact, clinicalstudies demonstrate that T-cell differentiation is associatedwith T-cell persistence (Huang et al., 2005), which in turn isassociated with antitumor T-cell efficacy (Robbins et al.,2004). In our current study, anti-CD3 mAb stimulation andexpansion with IL-2 may have created a bias toward TCR Tcells with a late differentiation phenotype. Notably, in aclinical phase I trial to treat metastasized renal cell carcinomawith anti-CD3 mAb-activated and IL-2-cultured CAR T cells(Lamers et al., 2006a), we observed that transduced T cells ofpatients, who all showed progressive disease, consistentlyhave a late differentiation CD8 T-cell phenotype at the time ofAT (Lamers, unpublished observations). Collectively, clinicaltrials with either CAR or TCR gene-transduced T cells usingsimilar T-cell activation and culture regimens together within vivo IL-2 support demonstrate that peripheral T-cell per-sistence, although variable, is generally limited (Kershaw et al.,2006; Lamers et al., 2006a; Morgan et al., 2006; Johnson et al.,2009; Parkhurst et al., 2011). The present model, in which NSGmice with human melanoma were treated with TCR-engineered T cells, closely recapitulates the cell-processing andpatient-treatment methods used in clinical receptor genetherapy studies and may represent a valid model to teststrategies to improve T-cell persistence and antitumor efficacy.
Two recent clinical trials provide evidence that alternativemethods for T-cell stimulation and cytokine support maybenefit clinical receptor gene therapy (Butler et al., 2011;Kalos et al., 2011; Porter et al., 2011). In one study, Butler andcolleagues adoptively transferred MART1/A2-specific T cellsthat were stimulated with CD80- and CD83-expressing arti-
ficial APCs and cultured in the presence of both IL-2 and IL-15 into patients with metastatic melanoma without pre-conditioning or in vivo IL-2 support (Butler et al., 2011). Thisstudy resulted in three mixed responses and long-lastingdisease stabilizations and one complete response out ofseven patients. In another study, Kalos and colleaguesadoptively transferred T cells that were stimulated with anti-CD3 and CD28 mAb-coated beads and gene-engineered witha CD19-specific CAR, which contained the costimulatorydomain CD137 and the accessory domain CD3f, into patientswith chronic lymphocytic leukemia who were precondi-tioned, but did not receive IL-2 support (Kalos et al., 2011).This study resulted in complete remissions in two out ofthree patients. Strikingly, in both studies post infusion, Tcells persisted long-term in the circulation, were enriched forT cells with a central memory T-cell phenotype, and retainedfunctional responses toward target antigens. Although pa-tient numbers were low, these studies clearly demonstratethe potential value of T-cell costimulation and exposure toIL-15 with respect to the persistence of adoptively trans-ferred T cells. In extension to these clinical studies, there isalso ample preclinical evidence that supports the use of naiveor central memory T-cell subsets to enhance T-cell persis-tence and antitumor efficacy of transferred T cells (Bergeret al., 2008; Hinrichs et al., 2009; Wang et al., 2011). Alongthese lines, we are currently testing T-cell stimulations withanti-CD3 and CD28 mAbs, T-cell culture with IL-15 and IL-21, and gene engineering with modified TCR formats, suchas TCRs coupled to CD3e, CD3f, and costimulatory domains,for enhanced in vivo persistence and performance (Schaftet al., 2006; Pouw et al., 2010a,b).
In the next series of AT experiments, we used mouse Tcells, in analogy to earlier studies with CAR-engineeredmouse T cells (Table 2). Our studies show that syngeneicmouse T cells can very potently and rapidly eradicate largeestablished human melanomas in SCID mice (Fig. 3). In ad-dition, mouse T cells expanded and persisted in vivo for > 6weeks after the first T-cell transfer (Fig. 4). Unexpectedly andin contrast to studies with CAR T cells, the antitumor activityand expansion of T cells were independent of the receptortransgene and represented xeno-specific T-cell responses. Infact, T-cell expansions were of a homeostatic nature and in-dependent of the cognate tumor antigen. These findingswere confirmed with two SCID strains, B6 SCID and BALB/cSCID, and two human melanoma cell lines, FM3 andBLMgp100. Notably, MHC class I expression on FM3 andBLMgp100 tumor cells is high ( > 90% of cells in vitro anddirectly after in vivo isolation; data not shown) and potentiallydrives the antimelanoma activity of mouse T cells. Smyth andcolleagues reported that cytotoxicity of mouse T cells derivedfrom human tumor-immunized mice can partially be blockedwith an anti-human MHC class I antibody in vitro (Smythet al., 1997). Along these lines, MHC class Ilow tumor cells maybe considered to lower the extent of xeno-specific responses,but recognition by TCR-engineered T cells is also likely to becompromised. Alternatively, mouse T cells with a restricted ormonoclonal TCR repertoire can be used as recipient cells forTCR gene transfer. This may prevent xeno-specific activity oftransferred T cells, but the translational value of such a modelshould be considered with caution.
To further investigate and better understand the potencyand kinetics of the xeno-specific effect of murine T cells, we
198 STRAETEMANS ET AL.

studied T-cell localization and demonstrated that mouse Tcells migrate to lymph node and spleen and persist for > 6weeks in peripheral blood (Fig. 5A). In regressing humantumors, murine T cells infiltrated the tumor site and formeda ring at the tumor margin, but were not present in thecenter of the tumor (Fig. 5B). This finding was in line withobservations reported by Hanson and colleagues (Hansonet al., 2000), who showed that in regressing tumors T cellsencircled the tumor site, but in progressively growing tu-mors T cells were scattered in patches of necrotic tumortissue. CD4 + but not CD8 + T cells constitute the predom-inant T-cell subset that infiltrated the regressing humantumors (Fig. 5C). Remarkably, this was found in bothBALB/c and B6 SCID mice despite the fact that B6 T cellspredominantly consist of CD8 + T cells (peripheral blood Tcells of BALB/c and B6 mice comprise on average 30% and80% CD8 + T cells, respectively). Although we cannot ruleout an early involvement of CD8 + T cells, our immunehistochemistry data suggest that rejection of human tumorsin SCID mice is mediated by CD4 + T cells, confirming re-ports on the CD4 dependence of T cell-mediated xenograftrejections (Pierson et al., 1989; Friedman et al., 1999). Inanother series of experiments, we observed that T-cellstimulation and, to a lesser extent, retroviral transductionand s.c. IL-2 support contributed to the potency and ki-netics of xeno-specific responses of mouse T cells (Fig. 6A).T cells that were not stimulated prior to T-cell transferpersisted well in vivo, but generally the lack of T-cell stim-ulation adversely affected T-cell numbers in peripheralblood (Fig. 6B). Although T-cell stimulation is not necessaryfor the ultimate rejection of tumors and peripheral T-cellpersistence per se, it may be of importance in case a sub-optimal TCR is used.
In conclusion, we have demonstrated that, despite reportswith CAR transgenes, immune-deficient mice have limitedvalue to assess the therapeutic efficacy of mouse T cellsengineered with TCR transgenes. With respect to TCR-engineered human T cells, stimulated with anti-CD3 mAband cultured and supported in vivo with IL-2, T-cell persis-tence is highly compromised and antitumor responses arelacking in immune-deficient mice. These observations are inclose accordance with clinical studies, are related to T-celldifferentiation, and confirm the validity of NSG mousemodels to test strategies to enhance T-cell persistence andantitumor efficacy of receptor-engineered human T cells.
Acknowledgments
The authors would like to thank Sabine van Steenbergen,Dr. Cor Lamers, and Dr. Zsolt Sebestyen for flow cytometricstainings, analyses, and interpretations. Dr. Naomi Taylor(Institut de Genetique Moleculaire de Montpellier, Mon-tpellier, France) is thanked for providing and discussing thebusulfan and cyclophosphamide preconditioning protocols.This work was financed in part by Erasmus Medical CenterTranslational Research (TR-2004) and by the European Un-ion Framework Program 6 ‘‘Adoptive Engineered T CellTargeting to Activate Cancer Killing’’ (EUFP6 ATTACK)project (number FP6-018914).
Author Disclosure Statement
There are no competing financial interests.
References
Appay, V., van Lier, R.A., Sallusto, F., and Roederer, M. (2008).Phenotype and function of human T lymphocyte subsets:consensus and issues. Cytometry A 73, 975–983.
Arden, B., Clark, S.P., Kabelitz, D., and Mak, T.W. (1995). Hu-man T-cell receptor variable gene segment families. Im-munogenetics 42, 455–500.
Berger, C., Jensen, M.C., Lansdorp, P.M., et al. (2008). Adoptivetransfer of effector CD8 + T cells derived from central memorycells establishes persistent T cell memory in primates. J. Clin.Invest. 118, 294–305.
Bobisse, S., Rondina, M., Merlo, A., et al. (2009). ReprogrammingT lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res.69, 9385–9394.
Brentjens, R.J., Latouche, J.B., Santos, E., et al. (2003). Eradicationof systemic B-cell tumors by genetically targeted human Tlymphocytes co-stimulated by CD80 and interleukin-15. Nat.Med. 9, 279–286.
Brentjens, R.J., Santos, E., Nikhamin, Y., et al. (2007). Geneti-cally targeted T cells eradicate systemic acute lym-phoblastic leukemia xenografts. Clin. Cancer Res. 13,5426–5435.
Butler, M.O., Friedlander, P., Milstein, M.I., et al. (2011). Estab-lishment of antitumor memory in humans using in vitro-educated CD8 + T cells. Sci. Transl. Med. 3, 80ra34.
Carpenito, C., Milone, M.C., Hassan, R., et al. (2009). Control oflarge, established tumor xenografts with genetically retargetedhuman T cells containing CD28 and CD137 domains. Proc.Natl. Acad. Sci. U.S.A. 106, 3360–3365.
Cheadle, E.J., Gilham, D.E., and Hawkins, R.E. (2008). Thecombination of cyclophosphamide and human T cells geneti-cally engineered to target CD19 can eradicate establishedB-cell lymphoma. Br. J. Haematol. 142, 65–68.
Chekmasova, A.A., Rao, T.D., Nikhamin, Y., et al. (2010). Suc-cessful eradication of established peritoneal ovarian tumors inSCID-Beige mice following adoptive transfer of T cells ge-netically targeted to the MUC16 antigen. Clin. Cancer Res. 16,3594–3606.
Clay, T.M., Custer, M.C., Sachs, J., et al. (1999). Efficient transferof a tumor antigen-reactive TCR to human peripheral bloodlymphocytes confers anti-tumor reactivity. J. Immunol. 163,507–513.
Craddock, J.A., Lu, A., Bear, A., et al. (2010). Enhanced tumortrafficking of GD2 chimeric antigen receptor T cells by ex-pression of the chemokine receptor CCR2b. J. Immunother. 33,780–788.
Darcy, P.K., Haynes, N.M., Snook, M.B., et al. (2000). Redirectedperforin-dependent lysis of colon carcinoma by ex vivo ge-netically engineered CTL. J. Immunol. 164, 3705–3712.
de Witte, M.A., Bendle, G.M., van den Boom, M.D., et al. (2008a).TCR gene therapy of spontaneous prostate carcinoma requiresin vivo T cell activation. J. Immunol. 181, 2563–2571.
de Witte, M.A., Jorritsma, A., Kaiser, A., et al. (2008b). Require-ments for effective antitumor responses of TCR transduced Tcells. J. Immunol. 181, 5128–5136.
Dudley, M.E., Wunderlich, J.R., Yang, J.C., et al. (2005).Adoptive cell transfer therapy following non-myeloablativebut lymphodepleting chemotherapy for the treatment of pa-tients with refractory metastatic melanoma. J. Clin. Oncol. 23,2346–2357.
Friedman, T., Shimizu, A., Smith, R.N., et al. (1999). HumanCD4 + T cells mediate rejection of porcine xenografts. J. Im-munol. 162, 5256–5262.
TCR-ENGINEERED T CELLS IN SCID AND NSG MICE 199

Gade, T.P., Hassen, W., Santos, E., et al. (2005). Targeted elimi-nation of prostate cancer by genetically directed human Tlymphocytes. Cancer Res. 65, 9080–9088.
Hanson, H.L., Donermeyer, D.L., Ikeda, H., et al. (2000). Eradi-cation of established tumors by CD8 + T cell adoptive im-munotherapy. Immunity 13, 265–276.
Haynes, N.M., Snook, M.B., Trapani, J.A., et al. (2001). Re-directing mouse CTL against colon carcinoma: superior sig-naling efficacy of single-chain variable domain chimerascontaining TCR-f vs FceRI-c. J. Immunol. 166, 182–187.
Haynes, N.M., Trapani, J.A., Teng, M.W., et al. (2002a). Rejectionof syngeneic colon carcinoma by CTLs expressing single-chainantibody receptors codelivering CD28 costimulation. J. Im-munol. 169, 5780–5786.
Haynes, N.M., Trapani, J.A., Teng, M.W., et al. (2002b). Single-chain antigen recognition receptors that costimulate potentrejection of established experimental tumors. Blood 100, 3155–3163.
Hinrichs, C.S., Borman, Z.A., Cassard, L., et al. (2009). Adoptivelytransferred effector cells derived from naive rather than centralmemory CD8 + T cells mediate superior antitumor immunity.Proc. Natl. Acad. Sci. U.S.A. 106, 17469–17474.
Huang, J., Khong, H.T., Dudley, M.E., et al. (2005). Survival,persistence, and progressive differentiation of adoptivelytransferred tumor-reactive T cells associated with tumor re-gression. J. Immunother. 28, 258–267.
Hwu, P., Yang, J.C., Cowherd, R., et al. (1995). In vivo antitumoractivity of T cells redirected with chimeric antibody/T-cellreceptor genes. Cancer Res. 55, 3369–3373.
Johnson, L.A., Morgan, R.A., Dudley, M.E., et al. (2009). Genetherapy with human and mouse T-cell receptors mediatescancer regression and targets normal tissues expressing cog-nate antigen. Blood 114, 535–546.
Kalos, M., Levine, B.L., Porter, D.L., et al. (2011). T cells withchimeric antigen receptors have potent antitumor effects andcan establish memory in patients with advanced leukemia. Sci.Transl. Med. 3, 95ra73.
Kaneko, S., Mastaglio, S., Bondanza, A., et al. (2009). IL-7 and IL-15 allow the generation of suicide gene-modified alloreactiveself-renewing central memory human T lymphocytes. Blood113, 1006–1015.
Kershaw, M.H., Westwood, J.A., Parker, L.L., et al. (2006).A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 12, 6106–6115.
Kessels, H.W., Wolkers, M.C., van den Boom, M.D., et al. (2001).Immunotherapy through TCR gene transfer. Nat. Immunol. 2,957–961.
Kowolik, C.M., Topp, M.S., Gonzalez, S., et al. (2006). CD28costimulation provided through a CD19-specific chimeric an-tigen receptor enhances in vivo persistence and antitumorefficacy of adoptively transferred T cells. Cancer Res. 66,10995–11004.
Lamers, C.H., Sleijfer, S., Vulto, A.G., et al. (2006a). Treatment ofmetastatic renal cell carcinoma with autologous T-lympho-cytes genetically retargeted against carbonic anhydrase IX:first clinical experience. J. Clin. Oncol. 24, e20–e22.
Lamers, C.H., Willemsen, R.A., van Elzakker, P., et al. (2006b).Phoenix-ampho outperforms PG13 as retroviral packagingcells to transduce human T cells with tumor-specific receptors:implications for clinical immunogene therapy of cancer. Can-cer Gene Ther. 13, 503–509.
Milone, M.C., Fish, J.D., Carpenito, C., et al. (2009). Chimericreceptors containing CD137 signal transduction domains me-
diate enhanced survival of T cells and increased antileukemicefficacy in vivo. Mol. Ther. 17, 1453–1464.
Morgan, R.A., Dudley, M.E., Wunderlich, J.R., et al. (2006).Cancer regression in patients after transfer of genetically en-gineered lymphocytes. Science 314, 126–129.
Morris, E.C., Tsallios, A., Bendle, G.M., et al. (2005). A criticalrole of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc. Natl. Acad.Sci. U.S.A. 102, 7934–7939.
Parkhurst, M.R., Yang, J.C., Langan, R.C., et al. (2011). T cellstargeting carcinoembryonic antigen can mediate regression ofmetastatic colorectal cancer but induce severe transient colitis.Mol. Ther. 19, 620–626.
Pierson, R.N., 3rd, Winn, H.J., Russell, P.S., and Auchincloss, H.,Jr. (1989). Xenogeneic skin graft rejection is especially depen-dent on CD4 + T cells. J. Exp. Med. 170, 991–996.
Pinthus, J.H., Waks, T., Kaufman-Francis, K., et al. (2003).Immuno-gene therapy of established prostate tumors usingchimeric receptor-redirected human lymphocytes. Cancer Res.63, 2470–2476.
Pinthus, J.H., Waks, T., Malina, V., et al. (2004). Adoptive im-munotherapy of prostate cancer bone lesions using redirectedeffector lymphocytes. J. Clin. Invest. 114, 1774–1781.
Porter, D.L., Levine, B.L., Kalos, M., et al. (2011). Chimeric an-tigen receptor-modified T cells in chronic lymphoid leukemia.N. Engl. J. Med. 365, 725–733.
Pouw, N., Treffers-Westerlaken, E., Kraan, J., et al. (2010a).Combination of IL-21 and IL-15 enhances tumour-specificcytotoxicity and cytokine production of TCR-transducedprimary T cells. Cancer Immunol. Immunother. 59, 921–931.
Pouw, N., Treffers-Westerlaken, E., Mondino, A., et al. (2010b).TCR gene-engineered T cell: limited T cell activation andcombined use of IL-15 and IL-21 ensure minimal differentia-tion and maximal antigen-specificity. Mol. Immunol. 47, 1411–1420.
Pouw, N.M., Westerlaken, E.J., Willemsen, R.A., and Debets, R.(2007). Gene transfer of human TCR in primary murine T cellsis improved by pseudo-typing with amphotropic and eco-tropic envelopes. J. Gene Med. 9, 561–570.
Robbins, P.F., Dudley, M.E., Wunderlich, J., et al. (2004). Cuttingedge: persistence of transferred lymphocyte clonotypes cor-relates with cancer regression in patients receiving cell transfertherapy. J. Immunol. 173, 7125–7130.
Robbins, P.F., Morgan, R.A., Feldman, S.A., et al. (2011). Tumorregression in patients with metastatic synovial cell sarcomaand melanoma using genetically engineered lymphocytes re-active with NY-ESO-1. J. Clin. Oncol. 29, 917–924.
Savoldo, B., Rooney, C.M., Di Stasi, A., et al. (2007). Epstein Barrvirus–specific cytotoxic T lymphocytes expressing the anti-CD30f artificial chimeric T-cell receptor for immunotherapy ofHodgkin disease. Blood 110, 2620–2630.
Schaft, N., Willemsen, R.A., de Vries, J., et al. (2003). Peptide finespecificity of anti-glycoprotein 100 CTL is preserved followingtransfer of engineered TCRab genes into primary human Tlymphocytes. J. Immunol. 170, 2186–2194.
Schaft, N., Lankiewicz, B., Drexhage, J., et al. (2006). T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFN-cproduction. Int. Immunol. 18, 591–601.
Seynhaeve, A.L., Hoving, S., Schipper, D., et al. (2007). Tumornecrosis factor a mediates homogeneous distribution of lipo-somes in murine melanoma that contributes to a better tumorresponse. Cancer Res. 67, 9455–9462.
200 STRAETEMANS ET AL.

Shultz, L.D., Lyons, B.L., Burzenski, L.M., et al. (2005). Humanlymphoid and myeloid cell development in NOD/LtSz-scidIL2Rcnull mice engrafted with mobilized human hemopoieticstem cells. J. Immunol. 174, 6477–6489.
Smyth, M.J., Kershaw, M.H., and Trapani, J.A. (1997). Xenos-pecific cytotoxic T lymphocytes: potent lysis in vitro and invivo. Transplantation 63, 1171–1178.
Song, D., Ye, Q., Carpenito, C., et al. (2011). In vivo persistence,tumor localization, and antitumor activity of CAR-engineeredT cells is enhanced by costimulatory signaling through CD137(4-1BB). Cancer Res. 71, 4617–4627.
Teng, M.W., Kershaw, M.H., Moeller, M., et al. (2004). Im-munotherapy of cancer using systemically delivered gene-modified human T lymphocytes. Human Gene Ther. 15,699–708.
Van de Griend, R.J., Van Krimpen, B.A., Bol, S.J., et al.(1984). Rapid expansion of human cytotoxic T cell clones:growth promotion by a heat-labile serum component andby various types of feeder cells. J. Immunol. Methods 66,285–298.
Wang, X., Berger, C., Wong, C.W., et al. (2011). Engraftment ofhuman central memory-derived effector CD8 + T cells in im-munodeficient mice. Blood 117, 1888–1898.
Westwood, J.A., Smyth, M.J., Teng, M.W., et al. (2005). Adoptivetransfer of T cells modified with a humanized chimericreceptor gene inhibits growth of Lewis-Y-expressing tumors inmice. Proc. Natl. Acad. Sci. U.S.A. 102, 19051–19056.
Willemsen, R.A., Weijtens, M.E., Ronteltap, C., et al. (2000).Grafting primary human T lymphocytes with cancer-specificchimeric single chain and two chain TCR. Gene Ther. 7, 1369–1377.
Xue, S.A., Gao, L., Hart, D., et al. (2005). Elimination of humanleukemia cells in NOD/SCID mice by WT1-TCR gene-trans-duced human T cells. Blood 106, 3062–3067.
Xue, S.A., Gao, L., Thomas, S., et al. (2010). Development of aWilms’ tumor antigen-specific T-cell receptor for clinical trials:engineered patient’s T cells can eliminate autologous leukemiablasts in NOD/SCID mice. Haematologica 95, 126–134.
Zhao, Y., Wang, Q.J., Yang, S., et al. (2009). A herceptin-basedchimeric antigen receptor with modified signaling domainsleads to enhanced survival of transduced T lymphocytes andantitumor activity. J. Immunol. 183, 5563–5574.
Zhao, Y., Moon, E., Carpenito, C., et al. (2010). Multiple injec-tions of electroporated autologous T cells expressing a chi-meric antigen receptor mediate regression of humandisseminated tumor. Cancer Res. 70, 9053–9061.
Address correspondence to:Dr. Reno Debets
Laboratory of Experimental Tumor ImmunologyDepartment of Medical Oncology
Erasmus Medical Center–Daniel den Hoed Cancer CenterGroene Hilledijk 3013075 EA Rotterdam
The Netherlands
E-mail: [email protected]
Received for publication June 25, 2010;accepted after revision September 29, 2011.
Published online: September 29, 2011.
TCR-ENGINEERED T CELLS IN SCID AND NSG MICE 201
Copyright © 2022 FDOKUMEN




















