
SYNTHETIC STUDIES OF (+)-CHETOMIN AND (–) - Mountain ...
416
DISSERTATION EPIDITHIODIOXOPIPERAZINES: SYNTHETIC STUDIES OF (+)-CHETOMIN AND (–)-SPORIDESMIN A Submitted by Timothy R. Welch Department of Chemistry In partial fulfillment of the requirements For the Degree of Doctor of Philosophy Colorado State University Fort Collins, Colorado Fall 2012 Doctoral Committee: Advisor: Robert M. Williams Tomislav Rovis John L. Wood Chris Ackerson Douglas H. Thamm
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of SYNTHETIC STUDIES OF (+)-CHETOMIN AND (–) - Mountain ...

DISSERTATION
EPIDITHIODIOXOPIPERAZINES: SYNTHETIC STUDIES OF
(+)-CHETOMIN AND (–)-SPORIDESMIN A
Submitted by
Timothy R. Welch
Department of Chemistry
In partial fulfillment of the requirements
For the Degree of Doctor of Philosophy
Colorado State University
Fort Collins, Colorado
Fall 2012
Doctoral Committee: Advisor: Robert M. Williams Tomislav Rovis John L. Wood Chris Ackerson Douglas H. Thamm

Copyright by Timothy Ryan Welch 2012
All Rights Reserved

ii
ABSTRACT
EPIDITHIODIOXOPIPERAZINES: SYNTHETIC STUDIES OF
(+)-CHETOMIN AND (–)-SPORIDESMIN A
This dissertation documents efforts toward the asymmetric total syntheses of the
natural products (+)-chetomin and (–)-sporidesmin A. Synthetic methods have been
developed to efficiently construct the dioxopiperazine core of both molecules.
Additionally, a simple epidithiodioxopiperazine has been synthesized to demonstrate a
general method for the addition of a sulfur bridge to a dioxopiperazine ring. The work
described herein, while not totally successful, provides a basis for future completion of
the asymmetric total syntheses of these two epidithiodioxopiperazines and other related
fungal metabolites.

iii
ACKNOWLEDGEMENTS
I am truly thankful for all of the supportive individuals who have motivated,
inspired, and encouraged me throughout my education. This work and much of my
personal and professional development would not have been possible without you. I
would first like to thank my advisor, Professor Robert M. Williams, for accepting me into
the Williams group and providing a dynamic research environment. It has been an honor
to work for you, and I am grateful for all of the advice and insight you have provided me
over the years. Thanks to all members of the Williams group, past and present, for all of
the ideas, encouragement, and shenanigans. Karen Kahler and Elizabeth McCoy, thank
you for making my life at CSU a little easier. Special mention must go to all of my
climbing partners; thanks for catching my numerous falls, throwing entertaining
wobblers, helping me clear my head, and inspiring me to try harder.
Without question, I would not be here were it not for two professors at KU and
several teachers from high school. Professor Brian Blagg, thank you for introducing me
to the field of organic synthesis and for your continued support. Professor Givens, it was
your skillful course instruction that first inspired me to pursue a career in academia. In
high school, I was blessed to have Kristin Seaton (Gunn), John Wachholz, and Linda
Nelson as teachers and advisors; thank you all for your encouragement and for always
challenging me to do better.
Lastly, I extend my utmost gratitude to my family. My parents taught me the
value of hard work and persistence and have always encouraged me to strive for
excellence. It was their encouragement and support that nurtured my intellectual
curiosity. Mom, Dad, Eric, and Emily, thank you for all of your love and support.

iv
TABLE OF CONTENTS
ABSTRACT ........................................................................................................................ ii
ACKNOWLEDGEMENTS ............................................................................................... iii
TABLE OF CONTENTS ................................................................................................... iv
LIST OF FIGURES .......................................................................................................... vii
LIST OF SCHEMES ....................................................................................................... xiv
Chapter 1: Epidithiodioxopiperazines
1.1: Introduction ................................................................................................1
1.2: Epidithiodioxopiperazines Derived from Phenylalanine or Tyrosine ...3
1.3: Tryptophan-Derived Epidithiodioxopiperazines ..................................13
1.4: Biosynthetic Investigations of Epidithiodioxopiperazines ...................23
1.5: Concluding Remarks ...............................................................................28
Chapter 2: Total Syntheses of Epidithiodioxopiperazines
2.1: Introduction ..............................................................................................29
2.2: Early Epidithiodioxopiperazine Syntheses (1973-1981) .......................30
2.3: Recent Epidithiodioxopiperazine Syntheses (2009-2012) .....................35
2.4: Other Relevant Syntheses: Formation of the C3-N1’ Bond .................45
2.5: Concluding Remarks ...............................................................................49

v
Chapter 3: Studies Toward the Total Synthesis of Chetomin
3.1: Introduction ..............................................................................................51
3.2: Goals and Early Studies
3.2.1: Goals of the Project ..........................................................................51
3.2.2: Iminium Ion Approach .....................................................................52
3.2.3: Pinacol-type Rearrangement ............................................................53
3.3: Evolution of Coupling Strategy
3.3.1: Witkop’s Pyrroloindole ....................................................................55
3.3.2: Indole-Aniline Coupling ..................................................................57
3.3.3: Coupling to exo-3-bromopyrroloindoline ........................................59
3.3.4: Attempted Coupling to Tetracyclic Bromide ...................................60
3.4: Epidithiodioxopiperazine Formation .....................................................62
3.5: Attempted Core Construction
3.5.1: Problematic Peptide Couplings ........................................................63
3.6: Synthesis of Heptacyclic Core
3.6.1: Synthesis of Dioxopiperazines .........................................................65
3.6.2: Initial N-Methyl Amino Acid Incorporation ....................................66
3.6.3: Bypassing the Serine Side Chain with Sarcosine ............................68
3.6.4: Completion of the Carbon Skeleton of Chetomin ...........................69
3.6.5: Attempted Epidithiodioxopiperazine Formation .............................69
3.7: Proposed Future Studies
3.7.1: Proposed Synthesis of Tetraol .........................................................70
3.7.2: Bypassing the Tetraol ......................................................................72

vi
3.7.3: Late-Stage Alkylation of Sarcosine Analogue .................................72
3.8: Concluding Remarks ...............................................................................73
Chapter 4: Synthetic Approach to Sporidesmin A
4.1: Introduction ..............................................................................................75
4.2: Retrosynthetic Analysis ...........................................................................76
4.3: Synthetic Approach to Sporidesmin A
4.3.1: Preparation of Dinitrostyrene Derivative .........................................78
4.3.2: Indole Synthesis and Sharpless Asymmetric Aminohydroxylation 80
4.3.3: Dioxopiperazine Formation .............................................................81
4.3.4: Oxidative Cyclization Attempts .......................................................83
4.3.5: Toward a Formal Synthesis of Sporidesmin A ................................86
4.4: Future Direction .......................................................................................88
4.5: Concluding Remarks ...............................................................................89
References .........................................................................................................................91
Chapter 5: Experimental Procedures ......................................................................104
Appendix I: Publications ............................................................................................346
Appendix II: Research Proposal ..................................................................................382
List of Abbreviations .....................................................................................................395

vii
LIST OF FIGURES
CHAPTER 1
Figure 1.1 Epidithiodioxopiperazines derived from tyrosine and/or phenylalanine .....2
Figure 1.2 Tryptophan-derived epidithiodioxopiperazines ...........................................2
Figure 1.3 Gliotoxins .....................................................................................................3
Figure 1.4 Redox cycling of epidithiodioxopiperazines ...............................................4
Figure 1.5 Mixed disulfide formation ...........................................................................5
Figure 1.6 Simple biologically active epidithiodioxopiperazines .................................6
Figure 1.7 Hyalodendrins and related compounds ........................................................7
Figure 1.8 Silvatins .......................................................................................................7
Figure 1.9 Aranotins ......................................................................................................8
Figure 1.10 Emethallicins ................................................................................................9
Figure 1.11 Emestrins and related metabolites .............................................................11
Figure 1.12 Epicorazines ...............................................................................................11
Figure 1.13 Scabrosin esters ..........................................................................................12
Figure 1.14 Sirodesmins ................................................................................................13
Figure 1.15 Sporidesmins ..............................................................................................15
Figure 1.16 Metabolites of the fungus Chaetomium cochliodes ...................................16
Figure 1.17 Chetoseminudins ........................................................................................16
Figure 1.18 HIF-1 hypoxia response pathway ..............................................................17
Figure 1.19 Chaetocins and related metabolites ............................................................19
Figure 1.20 Fungal metabolites related to the chaetocins .............................................20
Figure 1.21 Verticillin A and related metabolites .........................................................20
Figure 1.22 Verticillin-type epipolythiodioxopiperazines ............................................21
Figure 1.23 Leptosins ....................................................................................................23

viii
Figure 1.24 Putative epidithiodioxopiperazine gene clusters for sirodesmin PL (A) and
gliotoxin (B) ...............................................................................................26
CHAPTER 3
Figure 3.1 Chetomin and related fungal metabolites ..................................................52
CHAPTER 4
Figure 4.1 Sporidesmin A and related fungal metabolites ..........................................76
CHAPTER 5
Figure 5.1a 1H NMR spectrum of compound 3.5 .......................................................106
Figure 5.2a 1H NMR spectrum of compound 3.6 .......................................................108
Figure 5.2b 13C NMR spectrum of compound 3.6 ......................................................108
Figure 5.3a 1H NMR spectrum of compound 3.15 .....................................................110
Figure 5.4a 1H NMR spectrum of compound 3.12 .....................................................112
Figure 5.5a 1H NMR spectrum of compound 3.16 .....................................................114
Figure 5.6a 1H NMR spectrum of compound 3.17 .....................................................116
Figure 5.6b 13C NMR spectrum of compound 3.17 ....................................................116
Figure 5.7a 1H NMR spectrum of compound 3.21 .....................................................118
Figure 5.8a 1H NMR spectrum of compound 3.31 .....................................................120
Figure 5.9a 1H NMR spectrum of compound 3.32 .....................................................122
Figure 5.10a 1H NMR spectrum of compound 3.35 .....................................................124
Figure 5.11a 1H NMR spectrum of compound 3.40 .....................................................126
Figure 5.12a 1H NMR spectrum of compound 3.41 .....................................................128
Figure 5.13a 1H NMR spectrum of compound 3.42 .....................................................130
Figure 5.14a 1H NMR spectrum of (bromoethynyl)trimethysilane ..............................131
Figure 5.15a 1H NMR spectrum of compound 3.43 .....................................................133
Figure 5.16a 1H NMR spectrum of compound 3.44 .....................................................135
Figure 5.17a 1H NMR spectrum of compound 3.47 .....................................................137

ix
Figure 5.18a 1H NMR spectrum of compound 3.48 .....................................................139
Figure 5.19a 1H NMR spectrum of compound 3.56 .....................................................141
Figure 5.20a 1H NMR spectrum of compound 3.57a ...................................................143
Figure 5.21a 1H NMR spectrum of compound 3.58a ...................................................145
Figure 5.22a 1H NMR spectrum of compound 3.57c ....................................................147
Figure 5.22b 13C NMR spectrum of compound 3.57c ..................................................147
Figure 5.23a 1H NMR spectrum of compound 3.57d ...................................................149
Figure 5.23b 13C NMR spectrum of compound 3.57d ..................................................149
Figure 5.24a 1H NMR spectrum of compound 3.58d ...................................................151
Figure 5.24b 13C NMR spectrum of compound 3.58d ..................................................151
Figure 5.25a 1H NMR spectrum of compound 3.57e ....................................................153
Figure 5.25b 13C NMR spectrum of compound 3.57e ..................................................153
Figure 5.26a 1H NMR spectrum of compound 3.58e ....................................................155
Figure 5.27a 1H NMR spectrum of compound 5.1 .......................................................157
Figure 5.28a 1H NMR spectrum of compound 5.2 .......................................................159
Figure 5.28b 13C NMR spectrum of compound 5.2 ......................................................159
Figure 5.29a 1H NMR spectrum of compound 5.3 .......................................................161
Figure 5.29b 13C NMR spectrum of compound 5.3 ......................................................161
Figure 5.30a 1H NMR spectrum of compound 3.56 .....................................................163
Figure 5.31a 1H NMR spectrum of compound 3.55e ....................................................165
Figure 5.32a 1H NMR spectrum of compound 3.60 .....................................................167
Figure 5.33a 1H NMR spectrum of compound 3.61 .....................................................169
Figure 5.34a 1H NMR spectrum of compound 3.63 .....................................................172
Figure 5.34b 13C NMR spectrum of compound 3.63 ....................................................172
Figure 5.35a 1H NMR spectrum of compound 3.59 .....................................................174
Figure 5.36a 1H NMR spectrum of compound 3.34 .....................................................176
Figure 5.37a 1H NMR spectrum of compound 3.64 .....................................................178
Figure 5.38a 1H NMR spectrum of compound 3.65 .....................................................180
Figure 5.38b 13C NMR spectrum of compound 3.65 ....................................................180
Figure 5.39a 1H NMR spectrum of compound 3.66 .....................................................182
Figure 5.39b 13C NMR spectrum of compound 3.66 ....................................................182

x
Figure 5.40a 1H NMR spectrum of compound 3.67 .....................................................184
Figure 5.40b 13C NMR spectrum of compound 3.67 ....................................................184
Figure 5.41a 1H NMR spectrum of compound 3.68 .....................................................186
Figure 5.42a 1H NMR spectrum of compound 3.69 .....................................................188
Figure 5.43a 1H NMR spectrum of compound 5.4 .......................................................190
Figure 5.44a 1H NMR spectrum of compound 3.71 .....................................................192
Figure 5.45a 1H NMR spectrum of compound 5.5 .......................................................194
Figure 5.46a 1H NMR spectrum of compound 3.74 .....................................................196
Figure 5.46b 13C NMR spectrum of compound 3.74 ....................................................196
Figure 5.47a 1H NMR spectrum of compound 3.75 .....................................................198
Figure 5.47b 13C NMR spectrum of compound 3.75 ....................................................198
Figure 5.48a 1H NMR spectrum of compound 5.6 .......................................................200
Figure 5.48b 13C NMR spectrum of compound 5.6 ......................................................200
Figure 5.49a 1H NMR spectrum of compound 3.76 .....................................................203
Figure 5.49b 13C NMR spectrum of compound 3.76 ....................................................203
Figure 5.50a 1H NMR spectrum of compound 3.77 .....................................................205
Figure 5.51a 1H NMR spectrum of compound 5.7 .......................................................207
Figure 5.51b 13C NMR spectrum of compound 5.7 ......................................................207
Figure 5.52a 1H NMR spectrum of compound 3.80 .....................................................209
Figure 5.52b 13C NMR spectrum of compound 3.80 ....................................................209
Figure 5.53a 1H NMR spectrum of compound 3.81 .....................................................211
Figure 5.54a 1H NMR spectrum of compound 3.82 .....................................................214
Figure 5.55a 1H NMR spectrum of compound 5.9 .......................................................216
Figure 5.56a 1H NMR spectrum of compound 3.88 .....................................................218
Figure 5.56b 13C NMR spectrum of compound 3.88 ....................................................218
Figure 5.57a 1H NMR spectrum of compound 5.10 .....................................................220
Figure 5.58a 1H NMR spectrum of compound 3.90A ..................................................223
Figure 5.58b 13C NMR spectrum of compound 3.90A .................................................223
Figure 5.59a 1H NMR spectrum of compound 3.90B ...................................................224
Figure 5.59b 13C NMR spectrum of compound 3.90B ..................................................224
Figure 5.60a 1H NMR spectrum of compound 5.11 .....................................................226

xi
Figure 5.60b 13C NMR spectrum of compound 5.11 ....................................................226
Figure 5.61a 1H NMR spectrum of compound 4.15 .....................................................228
Figure 5.61b 13C NMR spectrum of compound 4.15 ....................................................228
Figure 5.62a 1H NMR spectrum of compound 4.17 .....................................................230
Figure 5.62b 13C NMR spectrum of compound 4.17 ....................................................230
Figure 5.63a 1H NMR spectrum of compound 4.18 .....................................................232
Figure 5.63b 13C NMR spectrum of compound 4.18 ....................................................232
Figure 5.64a 1H NMR spectrum of compound 4.19 .....................................................233
Figure 5.65a 1H NMR spectrum of compound 4.7 .......................................................235
Figure 5.65b 13C NMR spectrum of compound 4.7 ......................................................235
Figure 5.66a 1H NMR spectrum of compound 4.6 .......................................................237
Figure 5.66b 13C NMR spectrum of compound 4.6 ......................................................237
Figure 5.67a 1H NMR spectrum of compound 5.12 .....................................................239
Figure 5.67b 13C NMR spectrum of compound 5.12 ....................................................239
Figure 5.68a 1H NMR spectrum of compound 4.20 .....................................................241
Figure 5.68b 13C NMR spectrum of compound 4.20 ....................................................241
Figure 5.69a 1H NMR spectrum of compound 4.5 .......................................................243
Figure 5.69b 13C NMR spectrum of compound 4.5 ......................................................243
Figure 5.70a 1H NMR spectrum of compound 4.4 .......................................................245
Figure 5.70b 13C NMR spectrum of compound 4.4 ......................................................245
Figure 5.71a 1H NMR spectrum of compound 4.23 .....................................................247
Figure 5.71b 13C NMR spectrum of compound 4.23 ....................................................247
Figure 5.72a 1H NMR spectrum of compound 4.24 .....................................................249
Figure 5.73a 1H NMR spectrum of compound 4.25 .....................................................251
Figure 5.74a 1H NMR spectrum of compound 4.26 .....................................................253
Figure 5.74b 13C NMR spectrum of compound 4.26 ....................................................253
Figure 5.75a 1H NMR spectrum of compound 4.27 .....................................................255
Figure 5.76a 1H NMR spectrum of compound 4.28 .....................................................257
Figure 5.77a 1H NMR spectrum of compound 4.29 .....................................................259
Figure 5.77b 13C NMR spectrum of compound 4.29 ....................................................259
Figure 5.78a 1H NMR spectrum of compound 4.30 .....................................................261

xii
Figure 5.79a 1H NMR spectrum of compound 4.31 .....................................................263
Figure 5.80a 1H NMR spectrum of compound 4.32 .....................................................265
Figure 5.80b 13C NMR spectrum of compound 4.32 ....................................................265
Figure 5.81a 1H NMR spectrum of compound 4.3 .......................................................267
Figure 5.81b 13C NMR spectrum of compound 4.3 ......................................................267
Figure 5.82a 1H NMR spectrum of compound 4.40 .....................................................269
Figure 5.83a 1H NMR spectrum of compound 4.47 .....................................................271
Figure 5.84a 1H NMR spectrum of compound 4.50 .....................................................274
Figure 5.85a 1H NMR spectrum of compound 4.45 .....................................................276
Figure 5.86a 1H NMR spectrum of compound 4.46 .....................................................278
Figure 5.87a 1H NMR spectrum of compound 5.14 .....................................................281
Figure 5.88a 1H NMR spectrum of compound 5.16 .....................................................283
Figure 5.89a 1H NMR spectrum of compound 5.17 .....................................................285
Figure 5.90a 1H NMR spectrum of compound 5.18 .....................................................287
Figure 5.91a 1H NMR spectrum of compound 5.19 .....................................................289
Figure 5.92a 1H NMR spectrum of compound 5.20 .....................................................291
Figure 5.93a 1H NMR spectrum of compound 5.22 .....................................................294
Figure 5.94a 1H NMR spectrum of compound 5.23 .....................................................296
Figure 5.95a 1H NMR spectrum of compound 5.24 .....................................................298
Figure 5.96a 1H NMR spectrum of compound 5.25 .....................................................300
Figure 5.97a 1H NMR spectrum of compound 5.26 .....................................................302
Figure 5.98a 1H NMR spectrum of compound 5.27 .....................................................304
Figure 5.99a 1H NMR spectrum of compound 5.28 .....................................................306
Figure 5.100a 1H NMR spectrum of compound 5.29 .....................................................308
Figure 5.101a 1H NMR spectrum of compound 5.30 .....................................................310
Figure 5.102a 1H NMR spectrum of compound 5.31 .....................................................312
Figure 5.103a 1H NMR spectrum of compound 5.32 .....................................................314
Figure 5.104a 1H NMR spectrum of compound 5.33 .....................................................316
Figure 5.105a 1H NMR spectrum of compound 5.34 .....................................................318
Figure 5.106a 1H NMR spectrum of compound 5.35 .....................................................320
Figure 5.106b 13C NMR spectrum of compound 5.35 ....................................................320

xiii
Figure 5.107a 1H NMR spectrum of compound 5.36 .....................................................322
Figure 5.108a 1H NMR spectrum of compound 5.37 .....................................................324
Figure 5.108b 13C NMR spectrum of compound 5.37 ....................................................324
Figure 5.109a 1H NMR spectrum of compound 5.38 .....................................................326
Figure 5.109b 13C NMR spectrum of compound 5.38 ....................................................326
Figure 5.110a 1H NMR spectrum of compound 5.39 .....................................................328
Figure 5.110b 13C NMR spectrum of compound 5.39 ....................................................328
Figure 5.111a 1H NMR spectrum of compound 5.40 .....................................................330
Figure 5.111b 13C NMR spectrum of compound 5.40 ....................................................330
Figure 5.112a 1H NMR spectrum of compound 5.41 .....................................................332
Figure 5.112b 13C NMR spectrum of compound 5.41 ....................................................332
Figure 5.113a 1H NMR spectrum of compound 5.42 .....................................................334
Figure 5.113b 13C NMR spectrum of compound 5.42 ....................................................334
Figure 5.114a 1H NMR spectrum of compound 5.43 .....................................................336
Figure 5.114b 13C NMR spectrum of compound 5.43 ....................................................336
Figure 5.115a 1H NMR spectrum of compound 5.44 .....................................................338
Figure 5.115b 13C NMR spectrum of compound 5.44 ....................................................338
Figure 5.116a 1H NMR spectrum of compound 5.45a ...................................................340
Figure 5.116b 13C NMR spectrum of compound 5.45a ..................................................340
Figure 5.117a 1H NMR spectrum of compound 5.45b ...................................................341
Figure 5.117b 13C NMR spectrum of compound 5.45b ..................................................341
Figure 5.118a 1H NMR spectrum of compound 5.46 .....................................................343
Figure 5.118b 13C NMR spectrum of compound 5.46 ....................................................343
Figure 5.119a 1H NMR spectrum of compound 1.12 .....................................................345
Figure 5.119b 13C NMR spectrum of compound 1.12 ....................................................345

xiv
LIST OF SCHEMES
CHAPTER 1
Scheme 1.1 Proposed biosynthesis of gliotoxin ............................................................24
Scheme 1.2 Proposed oxepine ring formation ...............................................................24
Scheme 1.3 Proposed biosynthetic pathway of sirodesmin PL .....................................27
CHAPTER 2
Scheme 2.1 Total synthesis of (±)-sporidesmin A ........................................................31
Scheme 2.2 Synthesis of (±)-sporidesmin B .................................................................31
Scheme 2.3 Synthesis of (±)-dehydrogliotoxin .............................................................32
Scheme 2.4 Synthesis of (+)-gliotoxin ..........................................................................33
Scheme 2.5 Synthesis of (±)-hyalodendrin ....................................................................34
Scheme 2.6 Total synthesis of (±)-gliovictin and epi-gliovictin ...................................34
Scheme 2.7 Rastetter’s total synthesis of (±)-hyalodendrin ..........................................35
Scheme 2.8 Biomimetic total synthesis of (+)-11,11’-dideoxyverticillin A .................36
Scheme 2.9 Sodeoka’s total synthesis of (+)-chaetocin ................................................37
Scheme 2.10 Movassaghi’s total synthesis of (+)-chaetocin ...........................................38
Scheme 2.11 Total syntheses of chaetocin C and 12,12’-dideoxychetracin A ................39
Scheme 2.12 Synthesis of (+)-gliocladine C ...................................................................41
Scheme 2.13 Total synthesis of (+)-gliocladin B ............................................................42
Scheme 2.14 Synthesis of (+)-12-deoxybionectin A and (+)-gliocladin B .....................42
Scheme 2.15 Total synthesis of epicoccin G ...................................................................43
Scheme 2.16 Key pyrrolidine synthesis ..........................................................................44
Scheme 2.17 Completion of the total synthesis of (–)-acetylaranotin (1.35) ..................45
Scheme 2.18 Synthesis of (±)-psychotrimine via novel C3-N1’ bond formation ...........46

xv
Scheme 2.19 Total synthesis of kapakahine B ................................................................47
Scheme 2.20 Rainier’s synthetic C3-N1’ bond forming method ....................................47
Scheme 2.21 Total synthesis of kapakahine F .................................................................48
Scheme 2.22 Concise total synthesis of (+)-pestalazine B ..............................................49
CHAPTER 3
Scheme 3.1 Proposed model study for C3-N1’ bond formation ...................................52
Scheme 3.2 Attempted iminium ion formation .............................................................53
Scheme 3.3 Retrosynthesis of key intermediate 3.8 through a pinacol-type
rearrangement ............................................................................................54
Scheme 3.4 Synthesis of 2-alkynyl aniline derivative 3.17 ...........................................54
Scheme 3.5 Synthesis of 2,3-dihydrotryptophan alkyne derivative ..............................55
Scheme 3.6 Witkop’s pyrroloindole inspiration ............................................................56
Scheme 3.7 Retrosynthetic plan using pyrroloindoline .................................................56
Scheme 3.8 Attempted synthesis of 3-bromopyrroloindole ..........................................57
Scheme 3.9 Formation of key C-3 quaternary center ....................................................57
Scheme 3.10 Attempted formation of functionalized C3-N bond ...................................58
Scheme 3.11 Key alkyne synthesis ..................................................................................58
Scheme 3.12 Larock indole synthesis of tryptophan dimer .............................................59
Scheme 3.13 Coupling of 3-bromopyrroloindoline .........................................................59
Scheme 3.14 Retrosynthetic analysis of (+)-chetomin ....................................................60
Scheme 3.15 Coupling with more advanced bromopyrroloindoline ...............................61
Scheme 3.16 Optimized peptide coupling and dioxopiperazine formation .....................61
Scheme 3.17 Microwave assisted dioxopiperazine synthesis .........................................62
Scheme 3.18 Attempted coupling with indole .................................................................62
Scheme 3.19 Formation of the disulfide bridge ..............................................................63
Scheme 3.20 Coupling to 3-bromopyrroloindoline and attempted dioxopiperazine
formation ....................................................................................................64
Scheme 3.21 Attempted synthesis of alternate dioxopiperazine precursor .....................65
Scheme 3.22 Synthesis of dioxopiperazine 3.77 .............................................................66

xvi
Scheme 3.23 Synthesis of (N-Me)3 dioxopiperazine .......................................................67
Scheme 3.24 Attempted protection and deprotection of 3.82 .........................................68
Scheme 3.25 Attempted peptide coupling with N-Me,Boc-Ser(OH) ..............................68
Scheme 3.26 Synthesis of sarcosine-derived dioxopiperazine ........................................69
Scheme 3.27 Synthesis of (N-Me)3 dioxopiperazine .......................................................69
Scheme 3.28 Attempted epidithiodioxopiperazine formations .......................................70
Scheme 3.29 Key tetraols and iminium intermediates ....................................................71
Scheme 3.30 Possible oxidations ....................................................................................72
Scheme 3.31 Alternative sulfenylation ............................................................................72
Scheme 3.32 Proposed conversion of 3.91 to chetomin ..................................................73
CHAPTER 4
Scheme 4.1 Retrosynthetic analysis of (–)-sporidesmin A ............................................77
Scheme 4.2 Nitration of vanillin ....................................................................................78
Scheme 4.3 Dimethoxyindole synthesis ........................................................................79
Scheme 4.4 Synthesis of 5-chloro-dinitrostyrene derivative .........................................79
Scheme 4.5 Undesired acetal formation ........................................................................80
Scheme 4.6 Asymmetric aminohydroxylation ..............................................................80
Scheme 4.7 Preparation of (DHQD)2AQN ....................................................................81
Scheme 4.8 Formation of the dioxopiperazine and an undesired elimination ...............81
Scheme 4.9 Preparation of dipeptide .............................................................................82
Scheme 4.10 Cyclization to the dioxopiperazine ............................................................82
Scheme 4.11 Improved synthesis of dioxopiperazine 4.3 ...............................................83
Scheme 4.12 First attempts at oxidative cyclization .......................................................84
Scheme 4.13 Model cyclization study .............................................................................84
Scheme 4.14 More oxidative cyclization attempts ..........................................................85
Scheme 4.15 Progression of cyclization ..........................................................................86
Scheme 4.16 Attempted cyclization ................................................................................86
Scheme 4.17 Kishi’s final steps in the sporidesmin A synthesis .....................................87
Scheme 4.18 Preparation of N-Me,Fmoc-Ala-OH ..........................................................87

xvii
Scheme 4.19 Incorporation of N-Me-Ala ........................................................................88
Scheme 4.20 Full panel of oxidation conditions to screen ..............................................89
Scheme 4.21 Proposed epidithiodioxopiperazine formation ...........................................89

1
CHAPTER 1
Epidithiodioxopiperazines
1.1: Introduction
Nearly twenty distinct families of epidithiodioxopiperazine fungal metabolites
have been characterized since the seminal discovery of gliotoxin in 1936. This unique
class of natural products is characterized by a sulfur-bridged dioxopiperazine (1.1), a
feature generally requisite for the potent biological activity prevalent among the class.1-5
All natural epidithiodioxopiperazines discovered to date contain at least one
aromatic amino acid. Representative molecules from each family to be discussed are
shown in Figure 1.1 (tyrosine- and/or phenylalanine-derived) and Figure 1.2
(tryptophan-derived). In this chapter, we present an overview of the structures of
naturally occurring epidithiodioxopiperazines, relevant physiological properties, and
some of the more interesting of the proposed fungal biogeneses.
NN
O
O
R4
R2
R1
R3
SS
1.1
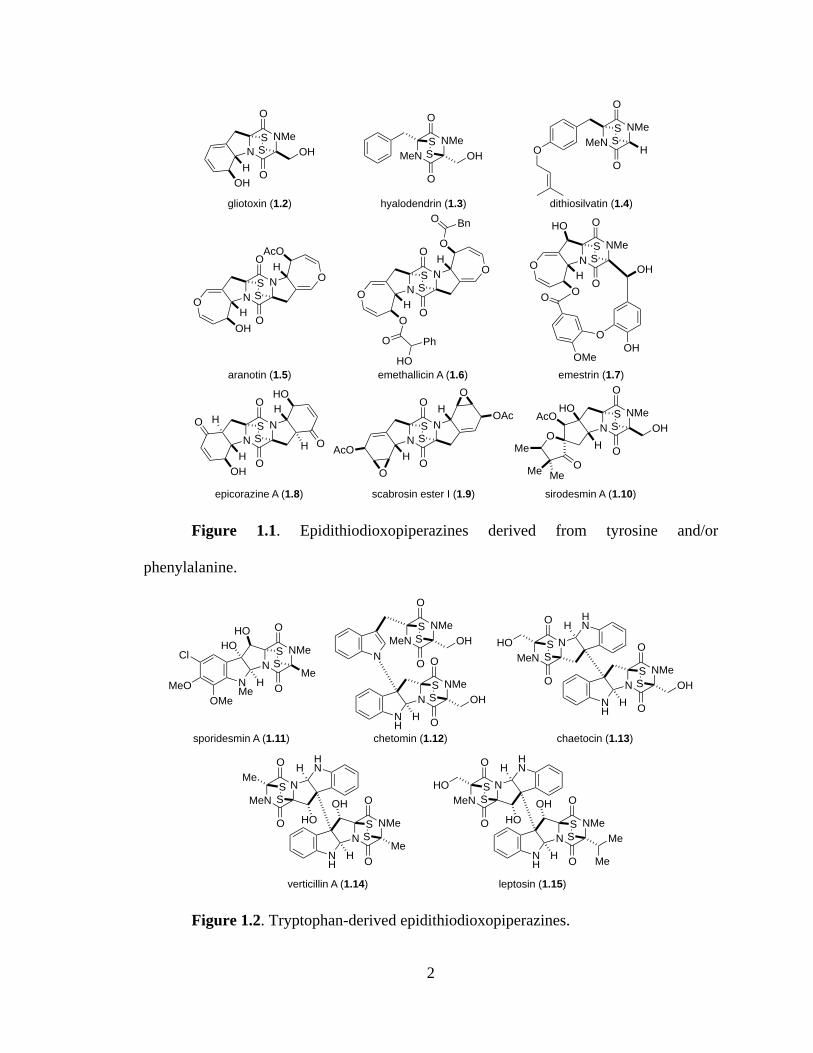
2
Figure 1.1. Epidithiodioxopiperazines derived from tyrosine and/or
phenylalanine.
Figure 1.2. Tryptophan-derived epidithiodioxopiperazines.
gliotoxin (1.2) hyalodendrin (1.3) dithiosilvatin (1.4)
NNMe
O
O
OH
OHH
SS MeN
NMe
O
O
OHSS
MeNNMe
O
OH
SS
O
NN
O
O
SS
O
O
OHH
HAcO
aranotin (1.5)
NN
O
O
SS
O
O
OH
HO
emethallicin A (1.6)
O Bn
O PhHO
NNMe
O
O
SSO
OH
emestrin (1.7)
HO
OH
OOH
O
OMe
NN
O
O
SS
epicorazine A (1.8)
HO
O
O
OH
H
H
H
H NN
O
O
SS
scabrosin ester I (1.9)
H
HO
OAc
O
AcO
sirodesmin A (1.10)
NNMe
O
O
OHSS
HO
OAcO
Me
Me MeO
H
sporidesmin A (1.11)
NNMe
O
OMe
SS
NMe
HOCl
MeOOMe
H
HO
chetomin (1.12)
NNMe
O
O
SS
NH
HOH
MeNNMe
O
O
SS
NOH
NNMe
O
O
SS
NH
HOH
NMeN
O
O
SS
HNH
HO
chaetocin (1.13)
NNMe
O
OMe
SS
NH
H
NMeN
O
OMe
SS
HNH
verticillin A (1.14)
OHHO
NNMe
O
O
SS
NH
H
NMeN
O
O
SS
HNH
leptosin (1.15)
OHHO
Me
Me
HO

3
1.2: Epidithiodioxopiperazines Derived from Phenylalanine or Tyrosine
In 1936, a novel substance with substantial antifungal and antiviral activity was
isolated from the wood fungus Gliocladium fimbriatum by Weindling and Emerson.6 A
putative structure (1.16, Figure 1.3) was proposed for the metabolite based on
degradation studies. This structure, however, could not account for some experimental
observations, leading Johnson and Woodward to propose a revised structure for gliotoxin
(1.2) in 1958.7 Absolute stereochemistry was later determined by x-ray analysis.8
Gliotoxin and related metabolites (Figure 1.3) have since been isolated from a variety of
fungi—including several Penicillium and Aspergillus species, Gliocladium,
Thermoascus, and Candida—and have been the focus of numerous synthetic and
biosynthetic studies that have formed the basis for much of the research discussed
herein.2,3
Figure 1.3. Gliotoxins.
The initial interest in the chemotherapeutic potential of gliotoxin as an antifungal
or antiviral agent waned as in vivo studies revealed gliotoxin to be generally cytotoxic.9
Moreover, gliotoxin has been implicated as a virulence factor of Aspergillus fumigatus,
NNMeO
OH
O
SS
OH1.16 dehydrogliotoxin (1.18)
N NMe
O
O OH
SS
RO H
gliotoxin (1.2, R=H)gliotoxin acetate (1.17, R=Ac)
N NMe
O
O OH
SS
HO
bisdethiodi(methylthio)gliotoxin (1.19)
N NMe
O
O OHOH H
MeS
SMe
bisdethiodi(methylthio)dehydrogliotoxin (1.20)
N NMe
O
O OHOH
MeS
SMe
N NMe
O
O OH
S3
HO H
gliotoxin E (1.21)
N NMe
O
O OH
S4
HO H
gliotoxin G (1.22)

4
the main source of invasive aspergillosis and leading cause of death in
immunocompromised patients.10 However, interest in the molecule was renewed when it
was discovered that gliotoxin displayed selective toxicity to cells of the hematopoietic
system.11,12 Specifically, gliotoxin exhibits antiproliferative activity against T and B cells
and inhibits phagocytic activity with considerable selectivity toward immune system
cells, leading to promising studies that have demonstrated that gliotoxin prevents graft-
versus-host disease after bone marrow transplantation.13
Generally, the toxicity of gliotoxin and other epidithiodioxopiperazines can be
attributed to two mechanisms: generation of reactive oxygen species (Figure 1.4) and
mixed disulfide formation (Figure 1.5). In the presence of a suitable reducing agent such
as glutathione or dithiothreitol, epidithiodioxopiperazines are reduced to the
corresponding dithiols (1.23). Autooxidation back to the disulfide (1.1) occurs with the
production of superoxide ions and hydrogen peroxide, known to cause oxidative damage
to cells.14,15 Gliotoxin has specifically been shown to induce single- and double-stranded
DNA damage in the presence of glutathione, presumably by hydroxyl radicals generated
in this redox process.16
Figure 1.4. Redox cycling of epidithiodioxopiperazines.
Evidence suggests that gliotoxin and other epidithiodioxopiperazines are also
capable of forming mixed disulfides with free thiol groups in cells.17,18 Following
NN
O
O
R4
R2
R1
R3
SS
1.1
NN
O
O
R4
R2
1.23
R1HS
R3
SH
2O22O2
cellular reductant
electron transfer
H2O2

5
incubation with radiolabelled gliotoxin, cells were shown to contain protein-bound [35S]-
gliotoxin (1.24).3 This result could be reversed if cells were treated with both gliotoxin
and excess reducing agent (dithiothreitol), suggesting a covalent interaction. Moreover,
the antiviral activity of gliotoxin is lost in the presence of excess dithiothreitol. This
evidence supports a mechanism of toxicity resulting from mixed disulfide formation
between gliotoxin and protein.19
Figure 1.5. Mixed disulfide formation.
Toxicity of epidithiodioxopiperazines would seem to rely on an intact disulfide
ring or the reduced dithiol. Indeed, dethiogliotoxin lacks the antibacterial activity of
gliotoxin. Reduction and methylation of the disulfide bridge
(bisdethiodi(methylthio)gliotoxin, 1.19) also results in a loss of antiviral activity.4,20 Not
surprisingly, the simple disulfide 1.25 and dithiol 1.26 exhibit potent biological activity,
highlighting the importance of the fragment to the observed toxicity of
epidithiodioxopiperazines.21 The great structural diversity in epidithiodioxopiperazines
may have evolved only to mask the core disulfide moiety to prevent degradation by target
organisms, rather than to impart any sort of selectivity or increased toxicity.2
N NMe
O
O OHOH H
S
SH
S
HS
N NMe
O
O OH
SS
OH H+
[35S]-gliotoxin
1.24

6
Figure 1.6. Simple biologically active epidithiodioxopiperazines.
Like gliotoxin, the hyalodendrins (Figure 1.7) are derived naturally from
phenylalanine and serine. Hyalodendrin (1.3) was originally isolated by Strunz in 1974
from Hyalodendron sp.22 The same fungus was later shown to produce the
bis(methylthio) derivative (1.29)23 and epitetrasulfide 1.28.24 Epitrithiohyalodendrin
(1.27) has only been observed as a product of the unidentified fungus NRRL 3888, along
with 1.3 and 1.29.25 Not surprisingly, hyalodendrin and the epitri- and epitetrasulfide
derivatives exhibit antibacterial activity, while the bisdethiodi(methylthio) analogue is
inactive against fungi and bacteria and relatively non-toxic to mice.22-25 Interestingly, it
was observed that hyalodendrin could be converted to epitetrasulfide 1.28 in the presence
of HCl with heating in methanol and the culture medium. Racemic tetrasulfide was
isolated when HCl was omitted from the same conditions.
Enantiomers of the hyalodendrins (except for epitrisulfide 1.27) have been
isolated from both terrestrial and marine sources. Gliovictin (1.32) was first isolated from
Helminthosporium victoriae in 1974,26 the same year that researchers at Eli Lilly reported
the isolation of the same structure (named A26771E) along with the disulfide (A26771A,
1.30) and epitetrasulfide (A26771C, 1.31) from Penicillium turbatum.27 Fenical has also
isolated gliovictin from the marine deuteromycete Asteromyces cruciatus.28 Predictably,
1.30 and 1.31 both showed antiviral and antibacterial activity, while gliovictin–lacking
sulfur atoms capable of redox cycling or mixed disulfide formation–was inactive.27
MeNNMe
O
O
H
H
SS
1.25
MeNNMe
O
O1.26
HHS
HSH

7
Figure 1.7. Hyalodendrins and related compounds.
Kawahara and coworkers reported the isolation of dithiosilvatin (1.4) and
silvathione (1.33) in 1987 from Apergillus silvaticus (Figure 1.8).29
Dioxopiperazinethiones such as 1.33 are rare and are possible intermediates in the
formation of trioxopiperazines from epidithiodioxopiperazines. The authors reported the
conversion of 1.4 to the bisdethiodi(methylthio) derivative (1.34) by reductive
methylation (NaBH4, MeI), a compound previously isolated by Hanson and O’Leary
from Gliocladium deliquescens.30
Figure 1.8. Silvatins.
The next subgroup of epidithiodioxopiperazines to be discussed is characterized
by the presence of at least one seven-membered dihydrooxepine ring, and includes the
MeNNMe
O
OssMeN
NMeO
O
sssss
MeNNMe
MeN
O
OS
NMeO
O
MeS
SMe
S
MeNNMe
O
OS MeN
NMeO
O
MeS
SMeMeN
NMeO
O
S
gliovictin (A26771E) (1.32)
A26771A (1.30)
hyalodendrin (1.3) bisdethiodi(methylthio)-hyalodendrin (1.29)
A26771C (1.31)
hyalodendrin-S4 (1.28)hyalodendrin-S3 (1.27)
OH
OH OH OH
OHOH
ssss
OH
MeNNMe
O
O
O
H
SSMeN
NMe
O
O
O
S
MeNNMe
O
O
OMeS
bisdethiodi(methylthio)silvatin (1.34)
silvathione (1.33)dithiosilvatin (1.4)
SMe

8
aranotins (Figure 1.9), emethallicins (Figure 1.10), and emestrins (Figure 1.11).
Aranotin (1.5) and acetylaranotin (1.35) have been isolated from Arachniotus aureus and
Aspergillus terreus and exhibit antiviral activity that is apparently selective for RNA
viruses such as the polio, Coxsackie (A21), rhino-, and parainfluenza viruses through
inhibition of viral RNA synthesis.31-37 Structurally, the aranotins are related to gliotoxin,
with the similarities most evident in apoaranotin (1.37). Apoaranotin can be considered a
chimera of gliotoxin and aranotin, containing the cyclohexadienol of gliotoxin and the
dihydrooxepine ring of aranotin. Asteroxepin (1.39) was the first monooxepine derivative
to be isolated and is further unique in that it contains one unsubstituted amide. This
dioxopiperazine may provide evidence for the sequence of steps in the biosynthesis of the
more complex aranotins.
Figure 1.9. Aranotins.
Closely related to the aranotins are the emethallicins (Figure 1.10). Both families
share the same absolute stereochemistry, and emethallicin A (1.6) differs apoaranotin
(1.37) only in the ester moieties. This observation was experimentally confirmed by
hydrolysis of 1.6 followed by acetylation to afford compound 1.37, identical to naturally
NN
O
O
O
OOH
SS N
NO
O
O
OOAc
SS
NN O
O
O
SS
NN
O
O
O
O
MeS
SMe
NN O
O
O
MeS
SMeN
NHO
O
OOAcH
MeS
SMe
asteroxepin (1.39)bisdethiodi(methythio)acetylapoaranotin (1.38)
bisdethiodi(methylthio)acetylaranotin (1.36)
apoaranotin (1.37)
acetylaranotin(1.35)aranotin (1.5)
AcOH
AcOH
HH
HAcO
HOAc
HOH
AcOH
AcOH
HOAc
Ph

9
occurring apoaranotin.38 Emethallicins B, D, and F (1.40, 1.42, and 1.44) share this same
monooxepine core and differ apoaranotin only in ester substitution and sulfur content of
the epipolythiodioxopiperazine ring. Emethallicin C (1.41) is symmetrical and the only
emethallicin to contain two dihydrooxepine rings, more closely resembling aranotin and
acetylaranotin.
Figure 1.10. Emethallicins.
Emethallicin A (1.6) was first isolated from Emericella heterothallica in 1989 by
Kawai and coworkers, who later reported the isolation of emethallicins B-F (1.40-1.44)
from the same fungus.38-41 Interestingly, Kawai was unable to convert synthetic
emethallicin D monoacetate (1.42, R=Ac) to the naturally occurring metabolite (1.42,
R=H). Basic hydrolytic conditions only succeeded at forming the disulfide and
N
N
O
O
OOO
O OH
H
HO
S
N
NS
O
OO
OO O
HH
SSN
N
O
OOO
O
OOH
HS
sSN
N
O
O
OOO
O O
HO
s
N
N
O
O
OOO
OO
OHH
HO
s
s
N
N
O
OOO
OO O
HH
RO
s
s
OH
s ssss
emethallicin F (1.44)emethallicin E (1.43)
emethallicin D (1.42)R= H, Ac
emethallicin C (1.41)
emethallicin B (1.40)emethallicin A (1.6)
H
H

10
tetrasulfide monoacetates of 1.6 and 1.40, respectively.39 This disproportionation of the
trisulfide is similar to a result obtained by Waring and coworkers, who similarly
converted trisulfide gliotoxin E (1.21) to the disulfide, gliotoxin (1.2), and the
tetrasulfide, gliotoxin G (1.22).42
All of the emethallicins exhibit fairly strong inhibitory activities upon histamine
release from mast cells, with IC50 values ranging from 1.0 x 10-6 to 2.0 x 10-8 M.
Generally, activity is stronger for the original emethallicins than for the acetate
derivatives. Micromolar inhibition of 5-lipoxygenase has also been reported.43
The final class of known epipolythiodioxopiperazine metabolites known to
contain at least one dihydrooxepine ring is the macrocyclic emestrins (Figure 1.11).
Emestrin (1.7) was isolated in 1985 from the fungus Emericella striata, and later from E.
quadrilineata, E. foveolata, E. acristata, and E. parvathecia.44-46 Trisulfide emestrin B
(1.45), piperazinethione aurantioemestrin (1.47), and trioxopiperazine dethiosecoemestrin
(1.48) were later isolated from E. striata.46-49 It has been postulated that the latter two
compounds are derived biosynthetically from emestrin. Emestrin displays potent
antifungal and antibacterial activity, but is also very toxic to mammals.
Recently, Kanda and coworkers reported the isolation of MPC1001 (1.46) and its
analogues (not shown) from Cladorrhinum sp. KY4922, contributing eight new members
to the emestrin family of natural products.50 MPC1001 contains a methoxy group rather
than the free phenol found in emestrin, but is otherwise structurally and stereochemically
identical. MPC1001 and its epipolysulfide analogues all showed antiproliferative activity
in the DU145 human prostate cancer cell line.50

11
Figure 1.11. Emestrins and related metabolites.
Epicorazines (Figure 1.12) have been isolated from several organisms, including
Epicoccum nigrum (epicorazine A and B, 1.8 and 1.49),51-53 E. purpurascens (epicorazine
B),54 and Stereum hirsutum (epicorazine C, 1.50).55 The only difference between 1.8 and
1.49 is the absolute stereochemistry at C6. This cis configuration is shared between 1.49
and 1.50.
Figure 1.12. Epicorazines.
Epicorazine A, B, and C display only marginal activity against Gram-positive
bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) and
vancomycin-resistant Enterococcus faecalis (VRE), and are inactive against Gram-
N
NMe
O
O
O
O
OO
MeO
HO
S S
OH
H
OH
NNMe
O
O
O
OO
O
MeO
HO
N
CHO
N
NMe
O
O
O
O
OO
MeO
HO
s
OH
H
OH
s s
S
H NMe
O
O
O
OO
O
MeO
HO
CHO
O
H
emestrin (1.7)
aurantioemestrin (1.47) desthiosecoemestrin (1.48)
emestrin B (1.45)
N
NMe
O
O
O
O
OO
MeO
MeO
S SH
OH
MPC1001 (1.46)
OH
NNO
OH H
H
S S
H
NNO
OH H
H
S S
H
epicorazine A (1.8) epicorazine B (1.49)
OO
HO HO
O
OH OH
O
NNO
OH H
H
S S
H
epicorazine C (1.50)
O
HO OH
O
OH
NNO
OS S
3822-A (1.51)
O
HO OH
O

12
negative organisms. All three exhibit antiproliferative effects against L929 mouse
fibroblast cells and K562 human leukemia cells, as well as cytotoxicity toward the HeLa
human cervical carcinoma cell line.55
The scabrosin esters were originally isolated from the lichen Xanthoparmelia
scabrosa in 1978, but it was not until 1999 that the correct structures were determined
(Figure 1.13).56,57 The isolation of these compounds was of particular interest, as this
report marked the first epidithiodioxopiperazine to be isolated from lichenized fungi.
Submicromolar activity, similar to that of gliotoxin (1.2), was observed for the scabrosins
against the murine P815 mastocytomia cell line, as well as low nanomolar activity in
MCF7 human breast carcinoma cell line. Scabrosin esters have also been shown to induce
apoptosis concomitantly with a large increase in mitochondrial membrane potential and
significant decrease in total cellular ATP. Mitochondrial ATP synthase is the proposed
cellular target of the scabrosins.58
Figure 1.13. Scabrosin esters.
The sirodesmins (Figure 1.14) were first discovered in 1977 as metabolites of
Sirodesmium diversum59 and later from the unrelated fungus Phoma lingam.60
Sirodesmins A-H (1.10, 1.56-1.60) are characterized by a spirofused tetrahydrofuran
cyclopentylpyrrolidine skeleton. Sirodesmins A-C are epimers of G and H at the
spirocenter. Notably, sirodesmin H was the first example of a naturally occurring
monosulfide.
NN
O
O
SS
scabrosin ester 1 (R1=CH3, R2=CH3, 1.9)scabrosin ester 2 (R1=CH3, R2=C3H7, 1.52)scabrosin ester 3 (R1=C3H7, R2=C3H7, 1.53)scabrosin ester 4 (R1=CH3, R2=C5H11, 1.54)scabrosin ester 5 (R1=C3H7, R2=C5H11, 1.55)H
HO
O
O
O
OR1
OR2

13
Figure 1.14. Sirodesmins.
Sirodesmins are potent antiviral agents, particularly against the rhinovirus.59
Sirodesmin G (originally named sirodesmin PL, 1.58) has specifically been shown to
exhibit activity against Gram-positive bacteria,61 although it has also been implicated as
the causative agent of blackleg disease in canola crops (along with phomalirazine, 1.61).
Metabolites 1.58 and 1.61 have both been isolated from the ascomycetous fungus
Leptosphaeria maculans, the organism known to be responsible for blackleg disease.62
1.3: Tryptophan–Derived Epidithiodioxopiperazines
The remaining epidithiodioxopiperazine alkaloids to be discussed are all derived
from tryptophan (Figure 1.2). Of this subset, the sporidesmins (Figure 1.15) are the most
densely functionalized and the only members to contain a substituted aromatic ring.
NNMe
O
H
OHAcO
Me
Me
MeO
O
OS
OH
NNMe
O
H
OHAcO
Me
Me
MeO
O
OS
OH
NNH
O
OOH
H
MeMeMe
S
O
OS
OH
s
SN
NMe
O
H
OHHO
Me
Me
MeO
O
OS
OH
S
NNMe
O
H
OHAcO
Me
Me
MeO
O
O
S
OH
S
s sNNMe
O
H
OHAcO
Me
Me
MeO
O
O
OH
sirodesmin H (1.60)
desacetyl-sirodesmin PL (1.59)sirodesmin G (sirodesmin PL, 1.58)
sirodesmin C (1.57)sirodesmin A (1.10)
phomalirazine (1.61)
sss
sN
NMe
O
H
OHAcO
Me
Me
MeO
O
O
OHsirodesmin B (1.56)

14
Sporidesmin was discovered by researchers investigating the source of the disease facial
eczema that plagued sheep in New Zealand and Australia. The disease caused extensive
liver damage in infected sheep and ultimately resulted in death. Thornton and Percival
eventually established that ingestion of pasture grasses on which the fungus Pithomyces
chartarum (previously known as Sporidesmium bakeri) was growing was the cause of the
serious disease.63,64 Sporidesmin (1.11) was isolated and implicated as the main toxic
agent produced by P. chartarum.65 The structure and absolute configuration were
subsequently determined by crystallographic means.8,66,67
As an interesting aside, veterinarians discovered that zinc sulfate doses gave
sheep protection from the effects of sporidesmin.68 Transition metals such as zinc are
now known to inhibit generation of the superoxide anion radical, with
epidithiodioxopiperazines shown to form a 2:1 complex with zinc ion.69,70
Extensive amounts of research have focused on the sporidesmins, producing the
complete characterization of all nine derivatives (1.11, 1.62-1.69). All contain a densely
functionalized, tryptophan–derived pyrroloindoline core coupled to an alanine residue.
Sporidesmin C (1.63)71 is the most unusual, containing a novel trisulfide [4.3.3] ring
system.
A great deal of chemistry applicable to most of the epipolythiodioxopiperazines
was discovered through investigations of the sporidesmins. For example, the trisulfide
sporidesmin E (1.65) is readily converted to the disulfide (1.11) upon treatment with
triphenyl phosphine. Alternatively, di- and trisulfides can be converted to tetrasulfides,
achieved using hydrogen polysulfide or dihydrogen disulfide. This was demonstrated by
the conversion of sporidesmins A (1.11) and E (1.65) into sporidesmin G (1.67).

15
Figure 1.15. Sporidesmins.
In 1944, Waksman and Bugie reported the isolation of a new antibiotic metabolite
of the fungus Chaetomium cochliodes that they named chetomin.72 It was not until 30
years later that Walter and coworkers determined the structure of chetomin (1.12),
revealing a nearly dimeric core likely formed from two molecules each of tryptophan and
serine.73 The two fragments are joined by a bond between the β-pyrrolidinoindoline
carbon and the indole nitrogen, a common feature of all five chaetocins (Figure 1.16).74
Chetomin is the only molecule in the family to contain a disulfide bridge within both
dioxopiperazine rings. Dethio-tetra(methylthio)chetomin (1.70) and chaetocochin C
(1.73) differ only in the oxidation state of the sulfurs, while chaetocochins A (1.71) and B
(1.72) are macrocyclic analogues of 1.70 and 1.73, each containing a novel 14-membered
ring.
NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
OH
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
OH
S
NNMe
N
O
OMeMeO
MeO
ClHO
H
MeS
SMe
HO
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
s
OH
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
MeSHO
NNMe
N
O
OMeMeO
MeO
ClHO
H
sOH
Me NNMe
N
O
OMeMeO
MeO
Cl
H
SS NMe
NMe
N
O
OHMeO
MeO
HO
H
SS
MeCl
HO
ss
s ss
sporidesmin G (1.67)
sporidesmin E (1.65)
sporidesmin J (1.69)sporidesmin H (1.68)
sporidesmin F (1.66)sporidesmin D (1.64)
sporidesmin C (1.63)sporidesmin B (1.62)sporidesmin A (1.11)

16
Figure 1.16. Metabolites of the fungus Chaetomium cochliodes.
Chetomin was recently isolated from Chaetomium seminudum by Fujimoto and
coworkers in 2004, along with three new metabolites named the chetoseminudins (Figure
1.17).75 Chetoseminudin A (1.74) is merely the pentasulfide homolog of chetomin. From
a biosynthetic viewpoint, the more interesting discoveries are chetoseminudins B-D
(1.75-1.77), monomeric bisdethiodi(methylthio) structures that potentially provide insight
as to the biosynthetic sequence that produces chetomin, the chaetocochins, and other
related epipolythiodioxopiperazines derived from tryptophan and serine.
Figure 1.17. Chetoseminudins.
Chetomin (1.12) has an unprecedented mechanism of action as a cancer
chemotherapeutic agent. Solid tumors must adapt to oxygen deprivation through
NH
N
NNMeS
S
O
O
OH
NMeMeN
O
HO
O
SS
chetomin (1.12)
NH
NNMe
O
O
dethio-tetra(methylthio)chetomin (1.70)
SMe
MeS
NH
NNMe
O
O
chaetocochin C (1.73)
SS
NN
NMe
O
O
SS
chaetocochin B (1.72)
N
N
NNMe
O
OSMe
MeS
O
chaetocochin A (1.71)
H
NNMeMeN
O
MeSO
SMe
H
NMeMeN
O
MeSO
SMe
N
O
NMeMeN
O
MeSO
SMe
NNMeMeN
O
MeSO
SMe
OH
HO
OHOH
HO
OH
NH
N
NNMeS
S
O
O
OH
NMeMeN
O
HO
O
SS
chetoseminudin A (1.74)
H
S NH
HNNMe
O
O
MeS
SMeOH NH
HNNH
O
O
MeS
SMeOR
chetoseminudin B (1.75) chetoseminudin C (R=H, 1.76)chetoseminudin D (R=Ac, 1.77)

17
induction of the heterodimeric transcription factor hypoxia-inducible factor 1 (HIF-1) in
order to survive. Overexpression of HIF-1 is associated with radioresistance in tumors,
increased risk of metastasis, and a poor prognosis for patients.76-78 In normal cells, the !-
subunit of HIF-1 (HIF-1!) is hydroxylated and degraded by vHL proteosome (Figure
1.18). As oxygen levels decrease and become the rate-limiting reagent in the
hydroxylation reaction, HIF-1! accumulates and binds to transcriptional coactivators
p300 and CREB binding protein (CBP). Consequent to this binding is the transcription of
proteins requisite to the survival of hypoxic cancer cells, facilitating tumor growth and
progression.79
Figure 1.18. HIF-1 hypoxia response pathway.
Chetomin has been shown to inhibit the interaction between HIF-1 and p300 both
in vitro and in cells, despite extensive surface interactions between the two proteins.
Specifically, Kung and coworkers have shown that chetomin disrupts the tertiary
structure of p300, inhibiting the transcriptional activity of HIF-1.76 No other small
molecule has been identified to mediate an antitumor response through this mechanism of
action. More recently, Hilton and coworkers proposed that chetomin and other
HIF-1!FIH-1
O2
low [O2]
HIF-1! HIF-1!
HIF-1! HIF-1!
HIF-1! HIF-1!
HIF-1!
HO
OH
OH
vHLProteosome
HIF-1!p300/CBP
transcriptiontumorgrowth
chetomin
p300/CBP
chetomin
tumordeath

18
epidithiodioxopiperazines bind zinc at the CH1 domain of p300, ultimately resulting in
ejection of a stable zinc–epidithiodioxopiperazine complex. Loss of zinc from the CH1
domain causes the previously observed disruption of the p300 tertiary structure.1,80
Chaetocin A (1.13, Figure 1.19) is a dimeric epidithiodioxopiperazine also
derived from two molecules each of tryptophan and serine. It was isolated from
Chaetomium minutum in 1970.81,82 Fifteen years passed before the penta- and hexasulfide
homologs chaetocin B and C (1.78 and 1.79) were isolated from Chaetomium spp., along
with the novel tetrasulfide chetracin A (1.80).83 In 2012, several related metabolites were
isolated from Oidiodendron truncatum, including the tetra-, penta- and hexasulfide
homologs melinacidin IV, chetracins B and C (1.82, 1.83, and 1.84), and the
dethiotetra(methylthio) derivative chetracin D (1.81).84
The three chaetocin metabolites (and likely the chetracins) can be interconverted
through either desulfurization of 1.78 and 1.79 with triphenyl phosphine to generate
chaetocin (1.13), or by sulfurization of chaetocin with phosphorus pentasulfide in carbon
disulfide to afford a mixture of chaetocins B and C.83
Recently, chaetocin A was identified as the first known inhibitor of lysine-specific
histone methyltransferases.85 Histone methylation is an important process in controlling
gene expression patterns, especially during cellular differentiation and embryonic
development. The activity of histone methyltransferases is disregulated in some tumors,
making chaetocin an attractive tool for the study of the molecular mechanism of histone
methylation.85 Additionally, melinacidin IV and chetracin B display nanomolar (3 – 54
nM) activity against five human cancer cell lines (HCT-8, Bel-7402, BGC-823, A549,
and A2780).84

19
Figure 1.19. Chaetocins and related metabolites.
Several metabolites related to the chaetocins have recently been reported,
possessing a C3-C3’ linkage to indole rather than the additional monomer found in the
chaetocins (Figure 1.20). T988 A, B, and C (1.86-1.88) were originally isolated from
Tilachlidium sp., although recently it was shown that Oidiodendron truncatum also
produces the same metabolites, in addition to oidioperazine A (1.89) and the chetracins
(1.83-1.85).75,86 Chetoseminudin C (1.76) was also isolated from O. truncatum,
suggesting that it may be a common intermediate to all of the tryptophan- and serine-
derived epipolythiodioxopiperazines discussed thus far. T988 A and B are cytotoxic to
P388 leukemia cells.86
NNMe
O
O
SS
NH
HOH
NMeN
O
O
SS
HNH
HO
chaetocin A (1.13)
NNMe
O
O
SS
NH
HOH
NMeN
O
O
SS
HNH
HO
chaetocin B (1.78)
S
NNMe
O
ONH
HOH
NMeN
O
O
SS
HNH
HO
chaetocin C (1.79)
S
SSS
NNMe
O
ONH
HOH
NMeN
O
O HNH
HO
chetracin A (1.80)
ss s
s
ss s
s
NNMe
O
ONH
HOH
NMeN
O
O HNH
HO
12,12'-dideoxychetracin A (1.81)
ss s
s
ss s
sOH
HO
melinacidin IV (x=y=2, 1.82)chetracin B (x=2, y=3, 1.83)chetracin C (x=3, y=3, 1.84)
NNMe
O
ONH
HOH
NMeN
O
O HNH
HO Sx
OHHO
Sy
chetracin D (1.85)
NNMe
O
ONH
HOH
NMeN
O
O HNH
HO
OHHO
MeS
SMe
MeS
SMe

20
Figure 1.20. Fungal metabolites related to the chaetocins.
Verticillin A (1.14, Figure 1.21) is quite similar to chaetocin A (1.13), derived
from two molecules of alanine rather than serine. Only verticillins A, D and E (1.14, 1.92
and 1.93) are symmetrical, while the two tryptophan residues of the remaining verticillins
are coupled to different amino acids on the two halves (either alanine, serine, or tyrosine).
Verticillin B (1.90), for instance, contains alanine and serine residues on the northern and
southern halves of the molecule, respectively. Verticillins A-C were isolated from
Verticillium sp., while the remaining compounds in Figure 1.21 are produced by
Gliocladium sp.87-89
Figure 1.21. Verticillin A and related metabolites.
Gliocladines A-E (Figure 1.22, 1.97-1.101) were isolated in 2005 from
Gliocladium roseum, along with verticillin A, Sch52900, and Sch52901 (1.14, 1.95, and
1.96).90 Gliocaldines A and B are simply the penta- and hexasulfide homologs of
verticillin A, thus it is not surprising that the same organism produces all three
compounds. The structures of gliocladines C and D (1.99 and 1.100) should also look
NNMe
O
ONH
HOH
HN
T988 A (1.86)
SSS
OH
NNMe
O
ONH
HOH
HN
T988 B (1.87)
OH
NNMe
O
ONH
HOH
HN
T988 C (1.88)
SS
OHMeS
SMeN
NMe
O
ONCHO
HOH
HN
oidioperazine A (1.89)
OHMeS
SMe
NNMe
O
OMe
SS
NH
H
NMeN
O
OMe
SS
HNH
verticillin A (1.14)
OHHO
NNMe
O
OR2
SS
NH
H
NMeN
O
OR1
SS
HNH verticillin B (R1=CH3, R2=CH2OH, 1.90)
verticillin C (R1=CH3, R2=CH2OH, S5 homolog 1.91)verticillin D (R1=CH(OH)CH3, R2=CH(OH)CH3, 1.92)verticillin E (R1=COCH3, R2=COCH3, 1.93)verticillin F (R1=COCH3, R2=CH(OH)CH3, 1.94)Sch52900 (R1=CH(OH)CH3, R2=CH3, 1.95)Sch52901 (R1=CH2CH3, R2=CH3, 1.96)
OHHO

21
familiar, as they are the alanine derivatives of T988 A and C (Figure 1.20). Bionectra
byssicola F120 has also been shown to produce bionectins A and B (1.102, 1.103), the
glycine and tyrosine derivatives of T988C.91
Figure 1.22. Verticillin-type epipolythiodioxopiperazines.
The dimeric subset of epipolythiodioxopiperazines increased greatly in number
with the discovery of the leptosins (Figure 1.23) from a strain of Leptosphaeria sp.
attached to the marine alga Sargassum tortile.92-94 Leptosins A-K (1.104-1.118) all
contain at least one valine residue, a feature unique among all
epipolythiodioxopiperazines to this family. Leptosins A-C, G, H, and K (1.104-1.113)
share the 12,12’-dihydroxylated, octacyclic core of the verticillins, gliocladines, and
chetracins. The C3-C3’ linkage to indole is once again produced in leptosins D-F (1.114-
1.116), the valine derivatives of the T988s, gliocladines C-E, and the bionectins. In 1994,
two remarkable additions to this family were discovered, the epimers leptosin I and J
(1.117 and 1.118).94 These two compounds are characterized by a C12-C11’ ether
linkage, introducing an additional ring to the structure that prohibits the possibility of
topoisomers. Leptosins are generally toxic to P388 leukemia cells.
NNMe
O
OMe
SS
NH
H
NMeN
O
OMe
Sx
HNH
gliocladine A (x=3, 1.97)gliocladine B (x=4, 1.98)verticillin A (x=2, 1.14)
OHHO
NNMe
O
OMe
NH
H
HN
gliocladine C (x=2, 1.99)gliocladine D (x=3, 1.100)gliocladine E (x=4, 1.101)
Sx
OH
NNMe
O
OR
NH
H
HN
bionectin A (R=H, 1.102)bionectin B (R=CH(OH)CH3, 1.103)
SS
OH

22
Figure 1.23. Leptosins.
In summary, over 100 epipolythiodioxopiperazine alkaloids have been isolated
and characterized to date. While this was intended to be a comprehensive review of all
known members of this class of natural products, some were undoubtedly and
inadvertently omitted. One cannot help but marvel at the remarkable biological activity of
the class as a whole, although it is unlikely that any epipolythiodioxopiperazine will ever
achieve therapeutic utility in the clinic due to the general cytotoxicity inherent in the
disulfide through mechanisms discussed above. Certainly others have and will continue
to argue otherwise, but it is our opinion that the true contribution to medicine will be
realized by employing epipolythiodioxopiperazines as tools for the study of novel
biological pathways. To date, the natural products chetomin (1.12), gliotoxin (1.2), and
chaetocin A (1.13) have already contributed to our understanding of the respective
biological targets of the molecules.
NNMe
O
O
Sy
NH
H
NMeN
O
O
Sx
HNH
leptosin A (x=4, y=2, 1.104)leptosin B (x=3, y=2, 1.105)leptosin C (x=2, y=2, 1.106)leptosin G (x=4, y=3, 1.107)leptosin G1 (x=3, y=3, 1.108)leptosin G2 (x=2, y=3, 1.109)leptosin H (x=2, y=4, 1.110)
OHHO
HO
Me
Me
NNMe
O
O
Sx
NH
H
NMeN
O
O
S
HNH
leptosin K (x=2, 1.111)leptosin K1 (x=3, 1.112)leptosin K2 (x=4, 1.113)
OHHO
Me
Me
Me
Me
S
N
NMeO
O
Sx
NH H
N NMe
O
OHN
H
leptosin I (R1=CH2OH, R2=H, 1.117)leptosin J (R1=H, R2=CH2OH, 1.118)
OOH
Me
Me
R2R1
NNMe
O
ONH
H
HN
leptosin D (x=2, 1.114)leptosin E (x=3, 1.115)leptosin F (x=4, 1.116)
Sx
OH
Me
Me

23
Many of the families (particularly those containing a C3-C3’ indoline linkage)
share a great deal of structural similarity beyond the epipolythiodioxopiperazine moiety
that defines the class. It is likely that hundreds of additional variants exist in nature and
have yet to be discovered. While numerous biosynthetic studies have been conducted, we
still have only a limited understanding of the ease with which nature is able to assemble
epipolythiodioxopiperazines, molecules that have for over fifty years taunted synthetic
chemists. In the last section of this chapter, we present the notable biosynthetic
discoveries reported to date.
1.4: Biosynthetic Investigations of Epidithiodioxopiperazines
Researchers have investigated the biosynthesis of gliotoxin (1.2) for nearly forty-
five years, but still very little experimental evidence exists to support the numerous
proposals. In 1958 and 1960, Suhadolnik reported the only undisputed incorporation
study, demonstrating that Trichoderma viride incorporates isotopically labeled
phenylalanine (1.119) and serine (1.120) into gliotoxin (Scheme 1.1).95,96 Walsh recently
implicated the nonribosomal peptide synthetase GliP in the catalysis of the peptide
coupling and dioxopiperazine cyclization reactions.97 Some debate has occurred as to
whether cyclo-Phe-Ser (1.121) is a biosynthetic intermediate to gliotoxin. Although
doubly labeled 1.121 is incorporated into gliotoxin by T. viride, the fungus Penicillium
terlikowskii poorly incorporates the same compound.98-100 Walsh observed that release of
the dioxopiperazine from the enzyme is slow, allowing for some speculation that further
transformations may occur to the enzyme bound compound prior to release.

24
Scheme 1.1. Proposed biosynthesis of gliotoxin.
Arene oxide 1.122 was proposed as a biosynthetic intermediate to both the
gliotoxins and aranotins by Neuss in 1968. Cyclization at the ortho position provides the
tricyclic core of gliotoxin (1.123), whereas a ring enlarging tautomerization of the arene
oxide could give the oxepine ring (1.129) characteristic of the aranotins (Scheme
1.2).33,34
Scheme 1.2. Proposed oxepine ring formation.
gliotoxin (1.2)
NNMe
O
O
OH
OHH
SS
1.123
NNH
O
O
OH
OHH
1.124
NNH
O
O
OH
OHH OH
HOGliC GliG
GSH
1.125
NNH
O
O
OH
OHH
G S
S G
NNH
O
O
OH
OHH S
NH
HN
O
CO2HO
HO2C
NH2
S
HNN
H
O
HO2CO
CO2H
NH2
NNH
O
O
OH
OHH SH
HS
1.128
NNH
O
O
OH
OHH
SS
1.126
1.127
GliT
O2 H2O2
CO2H
NH2OHHO2C
NH2
phenylalanine (1.119) serine (1.120)
+HN
NH
O
O
OHGliP GliC/GliF
O2, NADPH
HNNH
O
O
OH
1.122
O
1.121
GliJ/GliI
GliN
HNNH
O
O
OH
1.122
OHN
NH
O
O
OH
1.129
OHN
NH
O
O
OH
1.130
O
O
[O]

25
The incorporation of sulfur into epidithiodioxopiperazines was until recently
poorly understood. Sulfur transfer readily occurs among various potential donors,
including methionine, cysteine, and sodium sulfate, complicating [35S] feeding
studies.2,101 In 2011, two groups independently provided evidence supporting a GliC–
mediated bishydroxylation of a dioxopiperazine (i.e. 1.123 to 1.124).102,103 Elimination of
water to generate diiminium 1.125 could be followed by nucleophilic attack of the
cysteine thiolate residues of two glutathione (GSH) molecules, catalyzed by the
specialized glutathione S-transferase GliG.102-104 Sequential GliJ peptidase and GliI
thioesterase activity is proposed to reveal the free dithiol.102 Scharf and coworkers have
unequivocally shown that GliT catalyzes the oxidation to the disulfide (i.e. 1.128),
leaving N-methylation to complete the biosynthesis of gliotoxin.105 The order of tailoring
events is up for debate, but recent advances in genomics and proteomics tools to study
and manipulate secondary metabolite production will certainly produce much more
accurate insight as to the formation of gliotoxin in nature.
Recall that the toxicity of epidithiodioxopiperazines generally arises from redox
cycling or mixed disulfide formation (Figures 1.4 and 1.5). How is it then that producing
fungi, such as Aspergillus fumigatus, are immune to toxicity inherent in the structure of
gliotoxin? Several studies published in the last two years have provided convincing
evidence that GliT is responsible for the self-resistance of A. fumigatus to gliotoxin.105,106
Deletion of the gliT gene renders A. fumigatus mutants highly sensitive to gliotoxin,
toxicity that can be reversed by addition of glutathione. Moreover, concentrations of
reduced gliotoxin increase with the concomitant depletion of intracellular glutathione

26
levels. It is likely that alterations of the cellular redox status and mixed disulfide
formation contributes to the growth inhibition observed in the absence of gliT.
Perhaps the cytotoxicity of gliotoxin is an indirect consequence of the true role of
gliotoxin and other epidithiodioxopiperazines produced by fungi. The same redox cycling
that produces deleterious consequences in naïve organisms may, in Aspergillus
fumigatus, serve as a buffer against cellular oxidative stress.106 Gliotoxin and other
epidithiodioxopiperazines may have evolved as simple antioxidants, with the host
protected from unwanted redox cycling and mixed disulfide formation by GliT.
Sirodesmin PL (1.58) is the only other epidithiodioxopiperazine to have been
extensively studied from a biosynthetic perspective. Putative biosynthetic gene clusters
for sirodesmin and gliotoxin identified from Leptosphaeria maculans and Aspergillus
fumigatus, respectively, are given in Figure 1.24.107-109 Based on recent advances to our
understanding of gliotoxin biosynthesis and the identification of homologs in the
sirodesmin gene cluster, we are able to predict many of the experimentally unverified
steps in the biosynthesis of sirodesmin PL (Scheme 1.3).
Figure 1.24. Putative epidithiodioxopiperazine gene clusters for sirodemsin PL
(A) and gliotoxin (B).109
Feeding studies have shown that Leptosphaeria maculans incorporate labeled
tyrosine and serine into both sirodesmin PL (1.58) and presumed intermediate
phomamide (1.132).59,110-112 We propose the incorporation of sulfur to proceed

27
analogously to the gliotoxin biosynthesis, through dihydroxylation (1.133), imine
formation, and glutathione addition (1.134). Unmasking of the dithiol (1.135) and
oxidation to the disulfide (1.136) could be followed by oxidation and Claisen
rearrangement to give 1.137, which after cyclization and oxidation provides known L.
maculans metabolite phomalirazine (1.61).59,111 Oxidative spirorearrangement, N-
methylation, and ketone reduction would lead to another known metabolite, desacetyl-
sirodemin PL (1.59). Acetylation by SirH would complete the biosynthesis of sirodesmin
PL (1.58).2,65,107,109
Scheme 1.3. Proposed biosynthetic pathway of sirodesmin PL.
CO2H
NH2OHHO2C
NH2
tyrosine (1.131) serine (1.120)
+HN
NH
O
O
OH
SirD/SirP SirC
phomamide (1.132)
HO O
MeMe
HNNH
O
O
OH
1.133
O
MeMe HO
OHHN
NH
O
O
OH
1.134 (G=glutathione)
O
MeMe GS
SG
SirJ/SirISirGGSH HN
NH
O
O
OH
1.135
O
MeMe HS
SH
SirTHN
NH
O
O
OH
1.136
O
MeMe
SS HN
NH
O
O
OH
1.137
SSO
Me
MeMe
O[O]
ClaisenRearrangement
[O]
NNH
O
O
OH
phomalirazine (1.61)
SS
[O], N-Me (SirN), reduction
OMe
MeMe
OHO
H
NNMe
OH
HOHO
Me
Me
MeO
O
O
SOHS
desacetyl-sirodesmin PL (1.59)
SirH NNMe
OH
OHAcO
Me
Me
MeO
O
O
SOHS
sirodesmin G (sirodesmin PL, 1.58)

28
1.5: Concluding Remarks
Epidithiodioxopiperazine alkaloids possess an astonishing array of molecular
architecture that has for decades challenged and inspired synthetic chemists. Biosynthetic
studies presented in this chapter provide a glimpse at the efficiency and elegance with
which Nature is able to assemble these compounds. Synthetic chemists strive to mimic
and in turn better understand the mechanisms by which microorganisms are able to
produce such complexity, hoping to channel some of Nature’s efficiency into novel
synthetic pathways. In the next chapter, we present many of the synthetic advances made
toward several of the epidithiodioxopiperazines presented above.

29
CHAPTER 2
Total Syntheses of Epidithiodioxopiperazines
2.1: Introduction
Over 100 naturally occurring epidithiodioxopiperazines have been isolated.
However, relatively few have succumbed to total synthesis despite decades of effort,
highlighting the challenging synthetic nature of the class of molecules. Initial interest in
the biological activity of epidithiodioxopiperazines sparked the synthetic interest of
several groups, with six different naturally occurring members of the class yielding to
synthetic chemists between 1973 and 1981. Three decades passed before renewed interest
in epidithiodioxopiperazines arose, sparked by isolation reports of new metabolites,
exciting results from the biological community detailing novel mechanisms of action in
cells, and advances in genomics that invigorated interest in elucidating the biosynthesis
of these secondary metabolites. Between 2009 and 2012 alone, syntheses of eight
additional epidithiodioxopiperazine alkaloids have been reported. A casual glance
through Chapter 1 is enough to impress upon the reader the great degree of structural
similarity shared among and between families of epidithiodioxopiperazines. We
anticipate that in the coming decade total syntheses of entire families of this class of
fungal metabolites will be completed concomitantly with great advances in synthetic
methods and biosynthetic understanding. In this chapter, we present a comprehensive
review of total syntheses of epidithiodioxopiperazines.

30
2.2: Early Epidithiodioxopiperazine Syntheses (1973-1981)
The total synthesis of sporidesmin A was completed by Kishi and coworkers in
1973. In a series of communications Kishi described a novel strategy for the synthesis of
epidithiodioxopiperazines using a dithioacetal moiety as a protecting group for the
disulfide bridge.113-115 Thus protected, the dithioacetal is stable to acidic, basic, and
reducing conditions, allowing for the introduction of thiol groups at an early stage in a
total synthesis. Synthesis of the sporidesmins began with the treatment of
dioxopiperazine 2.1 with the dithiane derivative of p-anisaldehyde in the presence of acid
to afford dithioacetal-protected dioxopiperazine 2.2 (Scheme 2.1).113 Condensation with
acid chloride 2.3 and subsequent methoxymethyl deprotection gave compound 2.4.
Treatment of ketone 2.4 with DIBAL-H at -78 °C resulted in stereoselective reduction to
the alcohol, which was then converted into acetate 2.5 in 80% yield. Cyclization to the
diacetate (2.6) proceeded upon addition of iodosobenzene diacetate, and hydrolysis of the
acetates gave the corresponding diol. Treatment of the diol with m-chloroperbenzoic acid
(mCPBA) afforded an intermediate sulfoxide, which decomposed to the disulfide upon
exposure to strong Lewis acid, revealing (±)-sporidesmin A (1.11).
Intermediate 2.5 was also used by Kishi in a total synthesis of (±)-sporidesmin B
(Scheme 2.2).116 Reduction of the acetate gave the methylene (2.7), which underwent an
oxidative cyclization similar to that reported in the sporidesmin A synthesis to benzoate
2.8. The disulfide was revealed as described above, completing the synthesis of (±)-
sporidesmin B (1.62).

31
Scheme 2.1. Total synthesis of (±)-sporidesmin A.
Scheme 2.2. Synthesis of (±)-sporidesmin B.
Fukuyama and Kishi reported the first synthesis of dehydrogliotoxin (1.18,
Scheme 2.3) in 1973.114 Benzoic acid derivative 2.10 was coupled to dioxopiperazine 2.9
and treated with diazomethane to give methyl ester 2.11. Radical bromination with NBS
and benzoyl peroxide was followed by treatment with potassium thioacetate to give
dithioacetate 2.12. Cleavage of the thioacetates and addition of p-methoxybenzaldehyde
NNMe
O
O
MeO
AcS
p-MeOC6H4CH2S2H2,H+
NMe
COClCl
MeOOMe
+
50%
1. n-BuLi2. HCl, THF3. NaOH
NMe
Cl
MeOOMe
O
HNNMe
O
OMe
SS Ar
NNMe
O
OMe
SS ArMeO
80%
1. DIBAL-H2. Ac2O
NMe
Cl
MeOOMe
HNNMe
O
OMe
SS Ar
AcO
30%
iodosobenzenediacetate
NMe
Cl
MeOOMe
N
25%
1. NaOH, MeOH2. mCPBA
H
AcOAcO
3. BF3!Et2O
(±)-sporidesmin A (1.11)
2.1 2.2 2.3
2.4 2.5
2.6
NMe
O
OMe
SS Ar
NMe
Cl
MeOOMe
NH
HOHO
NMe
O
OMe
SS
NMe
Cl
MeOOMe
HNNMe
O
OMe
SS Ar
AcO
90%
NaCNBH3
2.5
AcOHNMe
Cl
MeOOMe
HNNMe
O
OMe
SS Ar
2.7
NMe
Cl
MeOOMe
NH
NMe
O
OMe
PhOCO
20%
(PhCO2)2, DME
4,4'-thiobis-(6-t-butyl-3-methylphenol)
SS Ar
26%
1. KOH, MeOH, THF2. mCPBA
3. BF3!Et2O NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
Me
(±)-sporidesmin B (1.62)2.8

32
and boron trifluoride etherate gave a 1:1 diasteromeric mixture of thioacetals 2.13 in
good yield. This masked disulfide is stable to a variety of conditions that the disulfide
would not otherwise survive, allowing Kishi to introduce sulfur at an early stage in the
synthesis.
Scheme 2.3. Synthesis of (±)-dehydrogliotoxin.
A three-step conversion to the primary chloride (2.14) was followed by addition
of phenyllithium to affect cyclization on the desired bridgehead carbon in 79% yield
(2.15). Addition of phenyllithium with benzyl chloromethyl ether efficiently installed the
benzyl protected serine side chain (2.16). Cleavage of the benzyl and methyl ethers and
conversion to the epidithiodioxopiperazine gave (±)-dehydrogliotoxin (1.18).
The biosynthesis of gliotoxin (1.2) is believed to proceed through the
intramolecular nucleophilic ring-opening of a phenylalanine-derived arene oxide (1.122)
and has been the subject of considerable speculation and interest. Kishi and coworkers
drew inspiration from this biogenetic hypothesis in devising a brilliant total synthesis of
gliotoxin. The total synthesis of (±)-gliotoxin was completed in 1976 utilizing the same
HNNMe
O
O
I
OMe
O OH 1. CuI, K2CO3 PhNO2, 170 °C2. CH2N2
35%
NNMe
O
O
MeO2C
OMe
+
NBS, (PhCO2)2, CCl4;
AcSK, CH2Cl2 NNMe
O
O
MeO2C
OMe
AcS
SAc
2. p-MeO-PhCHO, BF3!OEt2, CH2Cl2
72%
NNMe
O
O
MeO2C
OMe
S SAr
1. HCl, MeOH, 50 °C
61%N
NMe
O
OOMe
S SAr1. MeOH, NaBH4
2. MsCl, Et3NCl
65%
3. LiCl, DMF THF, -78 °C79%
PhLi
2.9 2.10 2.11 2.12
2.13 2.14
NNMe
O
O
S SAr
THF, -78 °C;BOMCl
71%
PhLi
2.15OMe
NNMe
O
O
S SAr
48%
1. BCl3, CH2Cl2, 0 °C2. mCPBA, CH2Cl2
2.16OBn
OBn
(±)-dehydrogliotoxin (1.18)
NNMe
O
O
OHSS
OH
3. BF3!Et2O

33
disulfide protecting strategy as employed above for the sporidesmins, and was re-
engineered in 1981 by the same route starting from optically pure dithioacetal 2.17
obtained from resolution (Scheme 2.4).117,118 Coupling of 2.17 with t-butoxy arene oxide
2.18 in the presence of triton B afforded 2.19 and 2.20 in a 2 :1 ratio. Acetylation,
deprotection, mixed anhydride formation, and reduction gave alcohol 2.21 in 77% yield
from 2.19. Primary alcohol 2.21 was converted to the chloride following mesylation, and
the secondary ester deprotected to reveal alcohol 2.22. The key stereoselective
cyclization-alkylation reaction was achieved upon addition of phenyllithium to 2.22 and
benzoxymethyl chloride, affording cycloadduct 2.23 in modest yield (53%). The primary
alcohol was revealed upon removal of the benzyl group, and the thioacetal oxidatively
removed to afford either (±)- or (+)-gliotoxin (1.2).
Scheme 2.4. Synthesis of (+)-gliotoxin.
Strunz and Kakushima followed the precedent set by Kishi and completed a total
synthesis of hyalodendrin (1.3, Scheme 2.5).119 Known thioacetal 2.24 was alkylated
HNN
O Triton B
O MeSS Ar
O
O
OtBu+N
N
O
O MeSS Ar
O
OtBu
HON
N
O
O MeSS Ar
O
OtBu
HO+
1. Ac2O2. TFA
3. ClCO2Et4. NaBH4
77%
98%
NN
O
O MeSS Ar
OH
AcO1. MsCl2. LiCl3. NaOMe
90%N
N
O
O MeSS Ar
Cl
HOPhLi, BOMCl
53%N
N
O
O MeSS ArHO H
OBn
1. BCl32. mCPBA
35%
NN
O
O MeSSHO H
OH
3. HClO4
2.17 2.182.19 2.20
2.19:2.20 = 2:1
2.21 2.22 2.23
(+)-gliotoxin (1.2)

34
sequentially with chloromethyl methyl ether and benzyl bromide, then deprotected to
reveal hyalodendrin (1.3).
Scheme 2.5. Synthesis of (±)-hyalodendrin.
In 1979 and 1980, Williams and Rastetter reported the total synthesis of gliovictin
(1.32, Scheme 2.6).120,121 Sarcosine anhydride (2.25) was converted in three steps to silyl
enol ether 2.27. Alkylation with benzyl bromide and concomitant sulfenylation and
deprotection gave methyl sulfide 2.29. Additional sulfenylation gave a 3:1 mixture of
diastereomers (2.30), favoring the undesired anti isomer. Reduction of the mixture of
aldehydes completed the total synthesis of gliovictin (1.32) and epi-gliovictin (2.31).
Scheme 2.6. Total synthesis of (±)-gliovictin and epi-gliovictin.
A synthesis of hyalodendrin (1.3, Scheme 2.7) was achieved from silyl enol ether
2.28, used previously in the gliovictin synthesis.120 Addition of monoclinic sulfur to the
MeNNMe
O
O
S SAr
2.24
1. n-BuLi; MOMCl2. n-BuLi; BnBr3. mCPBA4. HClO45. BCl3, -70 °C
5% (±)-hyalodendrin (1.3)
MeNNMe
O
O
OHSS
MeNNMe
O
O
EtOCHO, NaOMeMeN
NMe
O
O
1. KOtBu
O
H
H
MeNNMe
O
O
LDA, -78 °C;2. TBDPSCl
H
OTBDPS BnBr
MeNNMe
O
O
LDA, -78 °C;
H
OTBDPS MeSSMe, -78 °C MeNNMe
O
O
1. Et3N
O
H 2. MeSCl, -100 °C
MeS
H
MeNNMe
O
O
LiAl(t-BuO)3HMeS
SMeCHO
95%80% (3 steps)
87%
MeNNMe
O
OMeS
SMeOH
MeNNMe
O
OMeS
SMeOH
(±)-epi-gliovictin (2.31)40%
(±)-gliovictin (1.32)16%
2.25 2.26 2.27
2.28 2.29
2.30, anti:syn = 3:1
+

35
enolate of 2.28 followed by reductive workup provided thiol 2.32. Conversion to the
enolic methyl disulfide, deprotection of the silyl group, and sulfenylation with
triphenylmethyl chlorodisulfide gave a mixture of diastereomers, unfortunately favoring
the undesired anti isomer (2.34). Reduction and oxidation of 2.34 gave hyalodendrin (1.3)
in 29% yield.
Scheme 2.7. Rastetter’s total synthesis of (±)-hyalodendrin.
2.3: Recent Epidithiodioxopiperazine Syntheses (2009-2012)
Several total syntheses of epidithiodioxopiperazines have been reported in the last
three years. Movassaghi and coworkers were the first to publish in this modern synthetic
revival, reporting the synthesis of (+)-11,11’-dideoxyverticillin A (2.43, Scheme 2.8).122
The authors showed that the amine resulting from cleavage of the N-Boc carbamate of
2.35 was readily cyclized to dioxopiperazine 2.36 in morpholine. Exposure of 2.36 to
bromine produced 3-bromopyrroloindoline 2.37, and the amides were subsequently
methylated upon treatment with iodomethane. Addition of tris(triphenylphosphine)cobalt
chloride gave the desired dimeric intermediate 2.38. After extensive investigation
searching for appropriate stereoselective hydroxylation conditions, the dimer was
eventually oxidized with bis(pyridine)silver(I) permanganate to a fragile octacyclic
MeNNMe
O
O 1. LDA, -78 °C; !-sulfur2. HCl
H
OTBDPS 3. NaBH4
1. MeSCl, Et3N, -78 °C2. HCl, H2O
Ph3CSSCl, Et3N
>99%
2.28 2.32
2.33
MeNNMe
O
O
H
OTBDPS
HS
MeNNMe
O
O
O
H
S
H
MeS
MeNNMe
O
OS
SCHO
2.34, anti:syn = 2:1
MeS
SCPh3
-78 °C1. NaBH4
2. KI3
(±)-hyalodendrin (1.3)
MeNNMe
O
O
OHSS
51%
29%
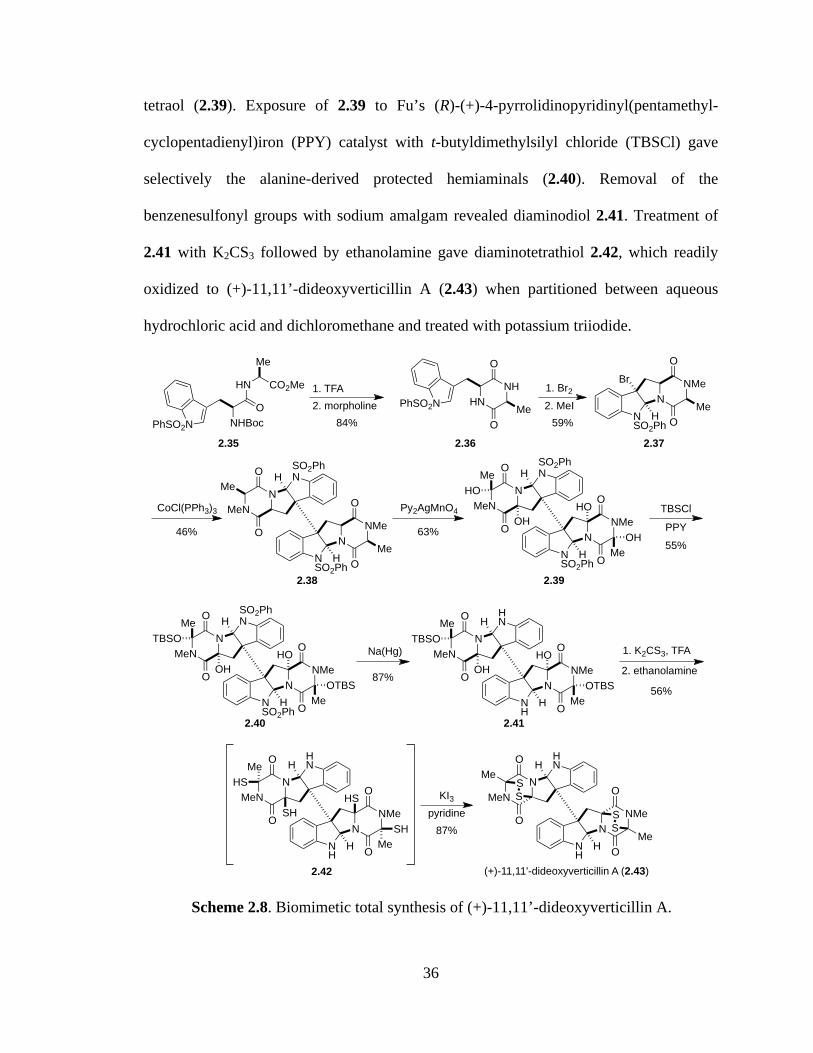
36
tetraol (2.39). Exposure of 2.39 to Fu’s (R)-(+)-4-pyrrolidinopyridinyl(pentamethyl-
cyclopentadienyl)iron (PPY) catalyst with t-butyldimethylsilyl chloride (TBSCl) gave
selectively the alanine-derived protected hemiaminals (2.40). Removal of the
benzenesulfonyl groups with sodium amalgam revealed diaminodiol 2.41. Treatment of
2.41 with K2CS3 followed by ethanolamine gave diaminotetrathiol 2.42, which readily
oxidized to (+)-11,11’-dideoxyverticillin A (2.43) when partitioned between aqueous
hydrochloric acid and dichloromethane and treated with potassium triiodide.
Scheme 2.8. Biomimetic total synthesis of (+)-11,11’-dideoxyverticillin A.
NHBocO
HN
Me
CO2Me
PhSO2N
1. TFA
84%PhSO2N
59%
HNNH
O
OMe
1. Br2
NSO2Ph
NNMeBr
H
O
OMe2. MeI
46%
CoCl(PPh3)3
NSO2Ph
NNMe
H
O
OMe
SO2PhN
NMeN
H
O
OMe
63%
Py2AgMnO4
NSO2Ph
NNMe
H
O
O Me
SO2PhN
NMeN
H
O
OMeHO
OHHO
OH55%
TBSCl
NSO2Ph
NNMe
H
O
O Me
SO2PhN
NMeN
H
O
OMeTBSO
OHHO
OTBS
PPY
87%
Na(Hg)
NH
NNMe
H
O
O Me
HN
NMeN
H
O
OMeTBSO
OHHO
OTBS 56%
1. K2CS3, TFA
2. ethanolamine
NH
NNMe
H
O
O Me
HN
NMeN
H
O
OMeHS
SHHS
SH 87%
KI3pyridine
NH
NNMe
H
O
O
HN
NMeN
H
O
OMe
Me
SS
SS
(+)-11,11'-dideoxyverticillin A (2.43)
2.35 2.36 2.37
2.38 2.39
2.40 2.41
2.42
2. morpholine

37
Chaetocin (1.13) is quite similar in structure to 11,11’-dideoxyverticillin A,
derived from two molecules of serine rather than alanine. Substituted dioxopiperazine
2.46 was prepared in five steps from N-Me,Cbz-D-Ser (2.44) and D-Trp-OMe
(2.45).123,124 Bromocyclization gave tetracycle 2.47, converted to the tribromide (2.48)
under radical conditions and hemiaminal 2.49 following addition of water. Movassaghi’s
reductive dimerization conditions using a Co(I) complex afforded the desired dimer
(2.50) in modest yield. Addition of 2.50 to condensed hydrogen sulfide and BF3⋅OEt2
formed the tetrathiol, oxidized to the bis(disulfide) upon addition of iodine to complete
the synthesis of (+)-chaetocin (1.13).
Scheme 2.9. Sodeoka’s total synthesis of (+)-chaetocin.
Sodeoka originally planned to form the tetraol from the product resulting from
dimerization of 2.47. It was noted that this dimer was not stable to the radical conditions
employed above, so the synthetic plan was altered to that shown in Scheme 2.9 to
NNMe
O
O
SS
NH
HOH
NMeN
O
O
SS
HNH
HO
(+)-chaetocin (1.13)
NNMe
O
ONBoc
HOTBS
NMeN
O
O BocNH
TBSO
2.50
OH
HOOH
HO
NNMe
O
ONBoc
HOTBS
OH
HOBr
NNMe
O
ONBoc
HOTBS
Br
BrBr
NNMe
O
ONBoc
HOTBS
H
HBr
HNNMe
O
ONBoc
OTBS
NMeCbzHO2C
HONH
NH2
CO2Me
+ NBS
88%
5 steps
69%
NBS, V-70 H2O
47%
CoCl(PPh3)3
55%
H2S, BF3!OEt2;I2
44%
2.442.45 2.46
2.47 2.48 2.49

38
circumvent this problem. Recall that Movassaghi had similar difficulties in forming
related tetraol 2.39 and eventually settled on the mild oxidation discussed above.
Mere months after Sodeoka reported the first total synthesis of chaetocin,
Movassaghi published the synthesis of (+)-chaetocin (Scheme 2.10), the hexasulfide (+)-
chaetocin C (1.79), and the octasulfide (+)-12,12’-dideoxychetracin A (1.81, Scheme
2.11), all from the natural amino acids L-serine and L-tryptophan.125
Scheme 2.10. Movassaghi’s total synthesis of (+)-chaetocin.
Bromocyclization of dioxopiperazine 2.51 gave endo-tetracyclic bromide 2.52. N-
methylation, protecting group exchange, and key reductive radical dimerization afforded
the dimeric octacycle (2.53). Selective tetrahydroxylation was achieved using
Py2AgMnO4. Tetraol 2.54 is analogous to 2.50 used in Sodeoka’s synthesis, but
NNMe
O
O
SS
NH
H
NMeN
O
O
SS
HNH
(+)-chaetocin (1.13)
NNMe
O
ONH
HOAc
NMeN
O
O HNH
AcOO
O O
Me Me
SSCPh3
SS
Ph3C
O
MeMe
NNMe
O
ON HOAc
NMeN
O
O SO2PhNH
AcOO
O O
Me Me
S
S
O
MeMe
O
Me
Me
O
Me
MeN
NMe
O
ONOAc
NMeN
O
O SO2PhNH
AcOHO
OH
HOOH
NNMe
O
ON H
NMeN
O
O SO2PhNHH
H
HH
PhO2SH
PhO2S
NNH
O
ONSO2Ph
Br
HHN
NH
O
ONSO2Ph
OAc
AcO
OTBDPS OTBDPS
Br2
59%
1. MeI, LHMDS2. HF!py; AcCl3. CoCl(PPh3)3
36%
Py2AgMnO4
55%
1. H2S, TFA
53%2. i-PrCOCl
1. h" (350 nm), L-ascorbic acid, 1,4-dimethoxy- naphthalene
46%
2. N2H4, 0 °C; TrSSCl, Et3N
1. BF3!OEt2, 2,6- di-t-Bu-4-Me-py., Et3SiH
75%2. Otera's cat.
OH
OH
2.51 2.522.53
2.54 2.55
2.56
PhO2S

39
interestingly the acetate allows for differentiation of the hemiaminals, resulting in
regioselective substitution of hydrogen sulfide. Protection of resulting thiohemiaminal as
the dithioisobutyrate (2.55) served both to prevent opening of the hemiaminal under polar
protic conditions and to activate the hemiaminal to mild ionization in future steps. Mild
deprotection of the sulfonyl group was followed by chemoselective hydrazinolysis and
addition of triphenylmethanesulfenyl chloride to give disulfane 2.56. Ionization of the
isobutyrates and cyclization to the epidithiodioxopiperazines with concomitant loss of a
triphenylmethyl cation was followed by removal of the acetates using Otera’s catalyst to
afford (+)-chaetocin (1.13).
Chaetocin C (1.79) and 12,12’-dideoxychetracin A (1.81), the epitri- and
epitetrathiodioxopiperazine analogues of chaetocin, were similarly synthesized from a
common intermediate (Scheme 2.11).125
Scheme 2.11. Total syntheses of chaetocin C and 12,12’-dideoxychetracin A.
NNMe
O
ONH
HOAc
NMeN
O
O HNH
AcOO
O O
Me Me
SSn
CPh3
SSn
Ph3C
O
MeMe
NNMe
O
ONH
HOAc
NMeN
O
O HNH
AcOO
O O
Me Me
S
S
O
MeMe
O
Me
Me
O
Me
Me
(n=2): N2H4, 0 °C;TrSSCl, Et3N (86%)
or
2.57
(n=3): N2H4, 0 °C;TrSSSCl, Et3N (74%)
2.58 (n=2)2.59 (n=3)
NNMe
O
O
Sm
N H
NMeN
O
O
Sm
NHO
R
OR
2.60 (m=3, R=CF3)2.61 (m=4, R=H)
(m=3): TFAA, di-t-Bu-4-Me-py.; BF3!OEt2 (91%)
or(m=4): HCO2Ac; BF3!OEt2 (60%)
(m=3): Otera's cat., 90 °C (95%)
or(m=4): HCl, MeOH
(52%)
OAc
AcO
NNMe
O
O
Sm
NH
H
NMeN
O
O
Sm
HNH
(+)-chaetocin C (1.79), m=3(+)-12,12'-dideoxychetracin A (1.81), m=4
OH
HO

40
Hydrazinolysis of diaminodithioisobutyrate 2.57 was followed by treatment with
the corresponding sulfur source to give 2.58 or 2.59. It was necessary to protect the
indoline nitrogens as the trifluoroacetates before addition of Lewis acid to form the
polysulfide bridges (2.60 and 2.61), presumably to prevent decomposition pathways
encountered over longer reactions times necessary to the formation of the larger
polysulfide bridges. Global deprotection gave either (+)-chaetocin C (1.79) or (+)-12,12’-
dideoxychetracin A (1.81).
In 2011, Overman and coworkers reported the total synthesis of (+)-gliocladine C
(1.99, Scheme 2.12).126 Known 2-indolinone 2.62 (prepared from isatin and indole) was
reduced and Boc-protected to afford compound 2.63. Conversion to the oxindole ester
(2.64) proceeded efficiently upon treatment with 2,2,2-trichloro-1,1-dimethylethyl
chloroformate, triethyl amine, and 10 mol % of Fu’s (S)-(–)-4-pyrrolidinopyrindinyl-
(pentamethylcyclopentadienyl)iron catalyst. The oxindole was elaborated to indoline 2.65
over four steps in 54% yield. Aldol condensation with the lithium enolate of the
piperazinedione provided exclusively the Z isomer of 2.66, which readily cyclized to
hexacycle 2.67 upon treatment with BF3·OEt2. Grignard addition, silyl protection of the
resultant alcohol, asymmetric dihydroxylation, and acetylation gave advanced
intermediate 2.68. Addition of the epimeric mixture of silyl ethers to condensed hydrogen
sulfide and BF3·OEt2 gave the dithiol, oxidized to the disulfide upon exposure to oxygen.
Removal of the acetate revealed (+)-gliocladine C (1.99).

41
Scheme 2.12. Synthesis of (+)-gliocladine C.
Boyer and Movassaghi recently completed the total synthesis of two compounds
related to the gliocladines. The most direct route to the first, (+)-gliocladin B (2.73), is
detailed in Scheme 2.13.127 Bromocyclization proceeded as above, providing tetracycle
2.70 in good yield with excellent stereoselectivity (97:3 endo:exo). A Friedel–Crafts type
coupling of N-TIPS-5-bromoindole with 2.70 formed the desired 3-3’ bond. Several
indole derivatives were screened, and the 5-bromo derivative was found to give the best
yield and selectivity. The bromine and silyl protecting group were removed and the
resulting compound oxidized to diol 2.72. Exposure of this diol to sodium thiomethoxide
and trifluoracetic acid gave the bismethylthio derivative. (+)-Gliocladin B (2.73) was
revealed upon removal of the benzenesulfonyl group.
NNMe
O
OMe
NH
H
HN
(+)-gliocladine C (1.99)
S
OH
NH
O
OHHN
NBoc
O
HBocN
NBoc
O
BocN O
NBoc
OMe
BocN O
H
1. TFA, Et3SiH
2. Boc2O, DMAP; MeOH
68%
88%, 98:2 er
O
OCl ClCl
Cl
Et3N, Fu's cat., 40 °C
1. NaBH42. HC(OMe)3, PPTS, 65 °C3. LiBH4-MeOH, r.t. to 40 °C4. Dess-Martin
54%
O CCl3
MeMe
2.62 2.63 2.64
2.65
LDA, -78 °C;
75%
NNMe
O
OMeO
2.65, -78 °C; AcOH, -78 °C to r.t.
NBoc
OMe
BocN
N
NMe
OMe
O
O
80%-78 °C to -40 °C
BF3·OEt2
2.66
NNMe
O
OO
NBoc
H
BocN
62%
3. AD-mix-!, H2NSO2Me, K2OsO4·2H2O, (DHQ)2PHAL, >14:1 dr)4. Ac2O, DMAP
1. MeMgCl, -78 °C (9:1 dr)2. TBSOTf, DMAP, Et3N (3:2 dr)
NNMe
O
ONBoc
H
BocN AcOAcO
OTBSMe 47%
2. La(OTf)3, 40 °C
1. H2S, BF3·OEt2; O2
S
2.67
2.68

42
Scheme 2.13. Total synthesis of (+)-gliocladin B.
Although it has not to date been isolated from a natural source, it is worth noting
that the authors were able to convert diol 2.72 into the disulfide (+)-12-deoxybionectin A
(2.76) using conditions analogous to those discussed above in the chaetocin syntheses
(Scheme 2.14).127 Disulfide 2.76 is also easily converted to (+)-gliocladin B (2.73) by
reduction of the disulfide with sodium borohydride in the presence of iodomethane.
Scheme 2.14. Synthesis of (+)-12-deoxybionectin A and (+)-gliocladin B.
Epiccocin G (2.84) lacks a disulfide bridge and was thus not discussed in Chapter
1. Nonetheless, the structure is remarkably similar to that of epicorazine A (1.8), and a
discussion of Nicolaou’s recent total synthesis is certainly relevant in this context.128
NNMe
O
ONSO2Ph
Br
HHN
NMe
O
ONSO2Ph
Br2
75%
2.69 2.70
NNMe
O
ONSO2Ph
H
AgBF4, DTBMP, 0 °C
83%
2.71
TIPSN
BrTIPSN
Br
NNMe
O
ONSO2Ph
H
2.72
HN
OH
HO1. H2, Pd/C; Et3N·3HF2. n-Bu4NMnO4
41%
1. MeSNa, TFA-MeNO2
2. h! (350 nm), 1,4-di- methoxynaphthalene, ascorbic acid, sodium ascorbate
68%
NNMe
O
ONH
H
(+)-gliocladin B (2.73)
HN
SMe
MeS
NNMe
O
ONSO2Ph
H
2.72
HN
OH
HO
NNMe
O
ONH
H
2.74
HN
O
S
O Me
MeO
Me
Me
1. H2S, TFA-CH2Cl2, 0 °C; i-PrCOCl, pyr.2. h! (350 nm), 1,4-di- methoxynaphthalene, ascorbic acid, sodium ascorbate
47%
N2H4, 0 °C;Ph3CSCl, Et3N, 0 °C
81%
NNMe
O
ONH
H
2.75
HN
OH
SSCPh3
Hf(OTf)4
NNMe
O
ONH
H
HN
SS
NaBH4, MeI,pyr.
80%80% NNMe
O
ONH
H
(+)-gliocladin B (2.73)
HN
SMe
MeS
(+)-12-deoxybionectin A (2.76)

43
Hydroxy enone 2.77 (prepared in two steps from N-Boc-tyrosine) was converted in four
steps to hydroxy methyl ester 2.78 (Scheme 2.15). Separate deprotections to either the
amine or carboxylic acid gave two derivatives that were coupled to form amide 2.79.
Deprotection and cyclization to the dioxopiperazine was followed by conversion to the
bistrifluoroacetate (2.80), which eliminated to bisdiene 2.81 upon exposure to Pd(PPh3)4
catalyst. Treatment with S8 and NaHMDS gave a mixture of oligosulfenylated
compounds (2.82) that were readily converted to the bisdimethylthio compound (2.83)
upon reduction with sodium borohydride and addition of iodomethane. Bisendoperoxide
2.85 was generated on reaction with singlet oxygen, and addition of DBU induced a
Kornblum–DeLaMare rearrangement to give a bishydroxy enone. Reduction to the
ketone completed the total synthesis of epicoccin G (2.84).
Scheme 2.15. Total synthesis of epicoccin G.
NBoc
CO2Me
OH
HO
1. Ac2O2. Zn, AcOH3. DBU4. NaBH4, CeCl3
NBoc
CO2Me
H
HHO
1. TFA (a) or LiOH (b)2. BOP-Cl, a + b
NBoc
H
HHO NMeO2C
H
H OH
O1. TFA; Et3N2. (CF3CO)2O
N
H
HF3COCO N
O
OH
H
OCOCF3
N
H
H N
O
OH
H
Pd(PPh3)4
N
H
H N
O
OH
H
S8, NaHMDS;2.81, NaHMDS
SSnS
SSnS
2.82, n=0-6
N
H
H N
O
OH
H
NaBH4; MeI
SMe
MeS
1. O2, h!, TPP; DBU2. H2, Pd(OH)2
N
H
H N
O
OH
HSMe
MeSOH
O
OH
O
N
H
H N
O
OH
HSMe
MeS
OO
OO
H
HB:
:B
via:
2.77 2.78
2.79 2.80
2.81 2.83
epicoccin G (2.84) 2.85
47%84%
53%90%
58% (3 steps)
45%

44
The last epidithiodioxopiperazine total synthesis to be discussed was reported
recently by the Reisman group, who completed an elegant synthesis of (–)-acetylaranotin
(1.35).129 Pyrrolidine 2.88 was synthesized with >98% ee by (1,3)-dipolar cycloaddition
of cinnamaldimine 2.87 to t-butyl acrylate (2.86) and subsequent cleavage of the t-butyl
ester (Scheme 2.16). Protection of the amine and ozonolytic cleavage of the alkene
provided lactone 2.89 in good yield. Ethynylmagnesium bromide was added to the
hydroxylactone to form the hydroxy acid, which upon addition of triphenylphosphine and
DIAD underwent Mitsunobu lactonization to 2.90. Reduction to the diol (2.91) was
followed by bis-TBS protection of the alcohols and selective deprotection of the primary
alcohol. The aldehyde obtained following oxidation with Dess–Martin periodinane was
efficiently converted to chlorohydrin 2.92.
Scheme 2.16. Key pyrrolidine synthesis.
Formation of the dihydrooxepine (2.93) was realized upon treatment of alkyne
2.92 with catalytic [Rh(cod)-Cl]2 and tris(4-fluorophenyl)phosphine (Scheme 2.17).129
Elimination of HCl and hydrolysis of the ester gave acid 2.94, while deprotection of the
Teoc group and HCl elimination of the same compound gave amine 2.95. BOP-Cl–
mediated coupling of 2.94 and 2.95 delivered amide 2.96, which readily cyclized to
dioxopiperazine 2.97 after TBAF·(t-BuOH)4–induced desilylation. Synthesis of (–)-
N CO2Et
t-BuO2C
+1. CuI, Brucin-OL, DBU2. TFA, Et3SiH
39%NH
HO2C
CO2EtPh·TFA
2.88 (>98% ee)
NTeoc
CO2EtO
H
H
HO
O1. Teoc-OSu, Et3N2. O3, -78 °C; DMS
77%
NTeoc
CO2EtO
H
HO
-78 °C to 0 °C;PPh3, DIAD, 0 °C
76%
MgBr
NaBH4
86% NTeoc
CO2EtHHO
HO
NTeoc
CO2EtHTBSO
HO Cl1. TBSOTf, 2,6-lutidine, 0 °C2. AcOH3. DMP, pyr.4. pyrrolidine·TFA, NCS; NaBH4
58%
2.86
2.87 2.89
2.90 2.91 2.92

45
acetylaranotin (1.35) was completed following formation of tetrasulfide 2.98,
bisacetylation, mild reduction to the dithiol, and oxidation to the natural disulfide. This
publication marked the first total synthesis of a dihydrooxepine-containing
epidithiodioxopiperazine natural product.
Scheme 2.17. Completion of the total synthesis of (–)-acetylaranotin (1.35).
2.4: Other Relevant Syntheses: Formation of the C3-N1’ Bond
Recall that chetomin (1.12) contains a C3-N1’ linkage between two indole–
derived fragments. This unique structural feature was synthetically unprecedented when
we began our synthetic efforts toward a total synthesis of chetomin. In the last few years,
two different methods for the formation of this bond were reported in the literature. We
NN
O
O
O
OOH
(!)-acetylaranotin(1.35)
HO H H
NTeoc
CO2EtHTBSO
HO Cl
2.92
[Rh(cod)Cl]2,(4-FC6H4)3P,
85 °C
88% NTeoc
CO2EtH
ClO
TBSO
1. LiCl, Li2CO3, 100 °C2. Me3SnOH, 80 °C
1. TBAF, 0 °C2. LiCl, Li2CO3, 100 °C
48%
55%
2.94
2.95
BOP-Cl
NTeoc
CO2HH
O
TBSO
NH
CO2EtH
O
TBSO
87%
2.93
N H
O
OTBSN CO2EtH
O
TBSOO
Teoc
TBAF·(t-BuOH)4, 70 °C, N2 atm
76%
NN
O
O
O
OOH
HO H H
S8, NaHMDS;2.97, NaHMDS;
40%
S4
NaHMDS
1. AcCl, DMAP2. Et3N; O2
HS SH NN
O
O
O
OOAc
AcO H HS S
2.96 2.97
2.9832%

46
feel it prudent to discuss in the remainder of this chapter methods for the formation of the
challenging C3-N1’ bond as it relates to the synthesis of chetomin.
In 2008, Newhouse and Baran reported the total synthesis of (±)-psychotrimine
(Scheme 2.18).130,131 The key C3-N1’ bond forming reaction was completed by treating
tryptamine derivative 2.99 with N-iodosuccinimide and 2-iodoaniline to afford compound
2.100. Larock indole synthesis with alkyne 2.101 completed the carbon framework for
the top tryptamine fragment (2.102). A total synthesis of psychotrimine (2.103) was
completed in two steps from 2.102.
Scheme 2.18. Synthesis of (±)-psychotrimine via novel C3-N1’ bond formation.
Baran used the same method in the synthesis of kapakahine B (2.108, Scheme
2.19).130,132 In this instance, dipeptide 2.104 was cyclized and coupled to 2-iodoaniline to
give 2.105. Indole formation by a Larock annulation with TES-alkyne 2.106 gave indole
2.107, converted in several steps to the natural product kapakahine B (2.108).
NH
NHCO2Me
I
NH2
NIS
67%NH
NCO2Me
NH
H
I
2.992.100
Br
NH
NCO2Me
N
H
2.102
Br
Br
NHCO2Me
TMS
NHCO2Me
Pd(II)
85%
NH
NMe
N
HN
NHMe
MeHN
2.101
psychotrimine (2.103)
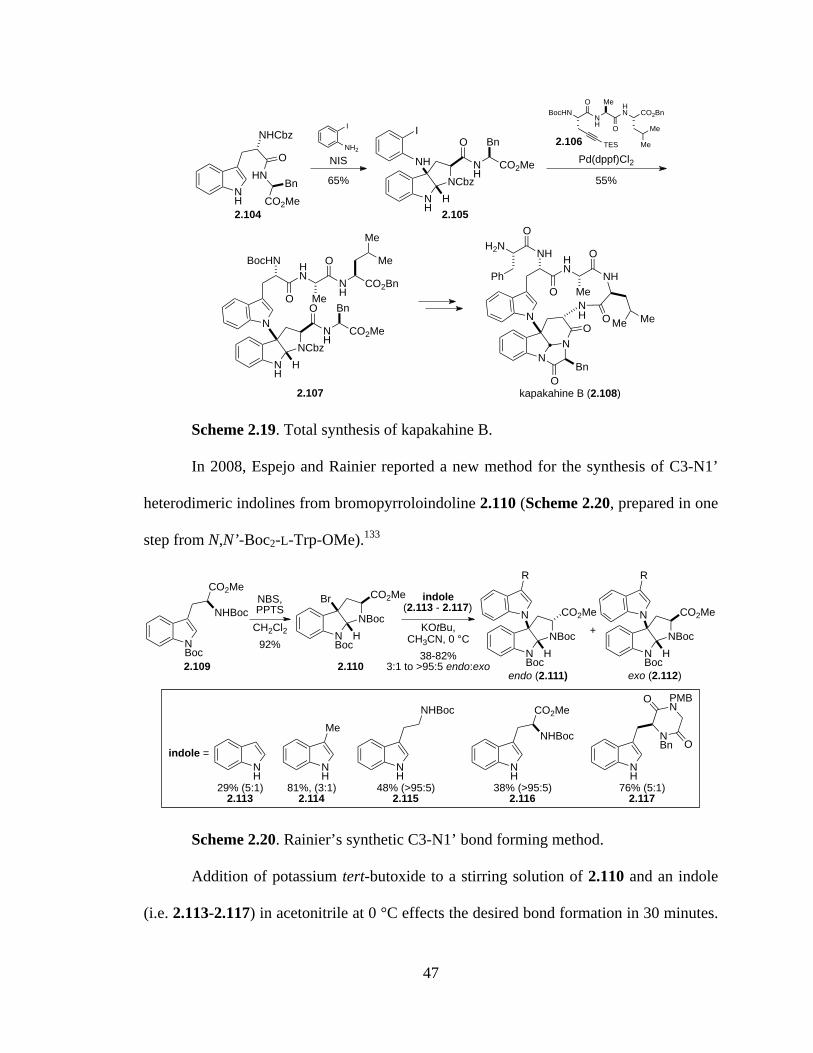
47
Scheme 2.19. Total synthesis of kapakahine B.
In 2008, Espejo and Rainier reported a new method for the synthesis of C3-N1’
heterodimeric indolines from bromopyrroloindoline 2.110 (Scheme 2.20, prepared in one
step from N,N’-Boc2-L-Trp-OMe).133
Scheme 2.20. Rainier’s synthetic C3-N1’ bond forming method.
Addition of potassium tert-butoxide to a stirring solution of 2.110 and an indole
(i.e. 2.113-2.117) in acetonitrile at 0 °C effects the desired bond formation in 30 minutes.
NH
NHCbz
HNO
CO2MeBn
I
NH2
NIS
65%NH
NCbz
NH
H
I
NH
O
CO2Me
Bn
BocHN NH
HN CO2Bn
TES
O Me
O
Me
Me
Pd(dppf)Cl255%
NH
NCbz
N
H
NH
O
CO2Me
Bn
BocHN HN
O Me
O
NH
CO2Bn
Me
Me
N
N
NH HN
O Me
O
NH
N
OBn
O
NOH
Me Me
OH2N
Ph
2.104 2.105
2.106
2.107 kapakahine B (2.108)
NBoc
NBoc
Br
H
CO2Me indole(2.113 - 2.117)
KOtBu, CH3CN, 0 °C
NBoc
NBoc
N
H
CO2Me
R
NBoc
NBoc
N
H
CO2Me
R
NBoc
CO2Me
NHBocNBS, PPTS
CH2Cl292%
38-82%3:1 to >95:5 endo:exo
endo (2.111) exo (2.112)2.109 2.110
NH
NH
Me
NH
NHBoc
NH
CO2Me
NHBoc
NH
NBn
PMBN
O
O
indole =
29% (5:1)2.113
81%, (3:1)2.114
48% (>95:5)2.115
38% (>95:5)2.116
76% (5:1)2.117
+

48
This method is far more general and cost effective than that reported by Baran, as a
variety of indole derivatives can be coupled directly without the added Larock annulation
step.
The utility of this method was demonstrated through the total synthesis of
kapakahine F (2.121, Scheme 2.21).134 Pyrroloindoline dimer 2.118 was synthesized as
described above and transformed to phenylalanine derivative 2.119 over several steps.
This substrate was originally intended to serve as a model for the subsequent trimethyl
aluminum induced rearrangement, but unfortunately the product resulting from the
coupling of 2.110 to a tryptophan derivative decomposed under the rearrangement
conditions. Thus, indole 2.120 was ultimately used to complete the synthesis of
kapakahine F (2.121).
Scheme 2.21. Total synthesis of kapakahine F.
The final total synthesis to be discussed in this chapter featuring a C3-N1’ bond
forming step is that of (+)-pestalazine B (2.126, Scheme 2.22), a natural product with
similar carbon framework to chetomin (1.12).135 Pestalazine B is unique in this discussion
in that, like chetomin, it is enantiomeric at the pyrroloindoline core compared to the
NBoc
NBoc
Br
H
CO2Me
KOtBu, 0 °CNBoc
NBoc
N
H
CO2Me
82%5:1 endo:exo
indole
NH
N
N
H
O
NH
CO2Et
Bn
AlMe3
0 °C to 30 °C
80% (1.5:1)N
N
N
OBn
NH2
N
N
NH2 HN
O Me
O
NH
N
OBn
O
NOH
MeMe
kapakahine F (2.121)
2.110 2.118 2.119
2.120
SO2Ph

49
above examples. Accordingly, D-tryptophan was selected as the starting material,
converted to bromopyrroloindoline 2.122 using known methods. Rainier’s coupling
methodology was employed to join 2.122 with dioxopiperazine 2.123 to form key
intermediate 2.124. Removal of the Boc groups and peptide coupling produced
tetrapeptide 2.125. Treatment of 2.125 with diethyl amine resulted in concomitant Fmoc
deprotection and cyclization to the naturally occurring dioxopiperazine, completing the
total synthesis of pestalazine B (2.126).
Scheme 2.22. Concise total synthesis of (+)-pestalazine B.
2.5: Concluding Remarks
Epidithiodioxopiperazine alkaloids possess an astonishing array of molecular
architecture and, with that, corresponding synthetic challenges to construct such
substances. The biosynthesis of many of the natural metabolites touched on in Chapter 1
has certainly inspired many of the chemists cited in this chapter, as several have sought to
exploit insights from Nature’s strategic bond constructions in a synthetic laboratory
NBoc
NBoc
N
H
CO2Me
NH
HNO
O
Ph
NBoc
NBoc
Br
H
CO2MeKOtBu, 12 °C
30%
2.122
NH
NH
HN
O
O
Ph
2.123
2.124
+
1. TMSI, 0 °C2. N-Fmoc-D-Leu, HATU, Et3N
NH
N
N
H
CO2Me
NH
HNO
O
Ph
2.125
O
NHFmoc
MeMeNH
N
N
H
NH
HNO
O
Ph
NH
O
OMe
Me
(+)-pestalazine B (2.126)
Et2NH
48% (3 steps)

50
context. Our own synthetic efforts toward chetomin and sporidesmin A as described in
the following chapters were undertaken before all of the recent reports were published,
although insight and inspiration were drawn from each as new developments in the
modern synthetic renaissance of epidithiodioxopiperazines emerged.

51
CHAPTER 3
Studies Toward the Total Synthesis of Chetomin
3:1: Introduction
Our interest in chetomin stems both from its novel mechanism of action as a
potential cancer chemotherapeutic agent and the synthetic challenge posed by the unique
architecture of the molecule. Chetomin (1.12) and related metabolites isolated from
Chaetomium sp. (Figure 3.1) are the only known epidithiodioxopiperazine alkaloids to
contain a nearly dimeric structure joined by a C3-N1’ linkage.73,74 The densely
functionalized structure contains five tetrasubstituted carbons and six stereocenters. The
synthetic challenge imposed by the epidithiodioxopiperazine rings is also not to be
overlooked, as the disulfide bridge is sensitive to oxidative, reductive, basic, and strongly
acidic conditions.
3.2: Goals and Early Studies
3.2.1: Goals for the Project
The primary goal of this project was to develop a scalable, divergent synthesis of
(+)-chetomin (1.12). We hoped to bring in as much functionality as possible when
forming the C3-N1’ bond to exploit the pseudo-dimeric architecture. Introduction of the
disulfide bridges would be reserved for a late stage in the synthesis to circumvent the
troubling sensitivity of the functional group. However, no precedent existed in the
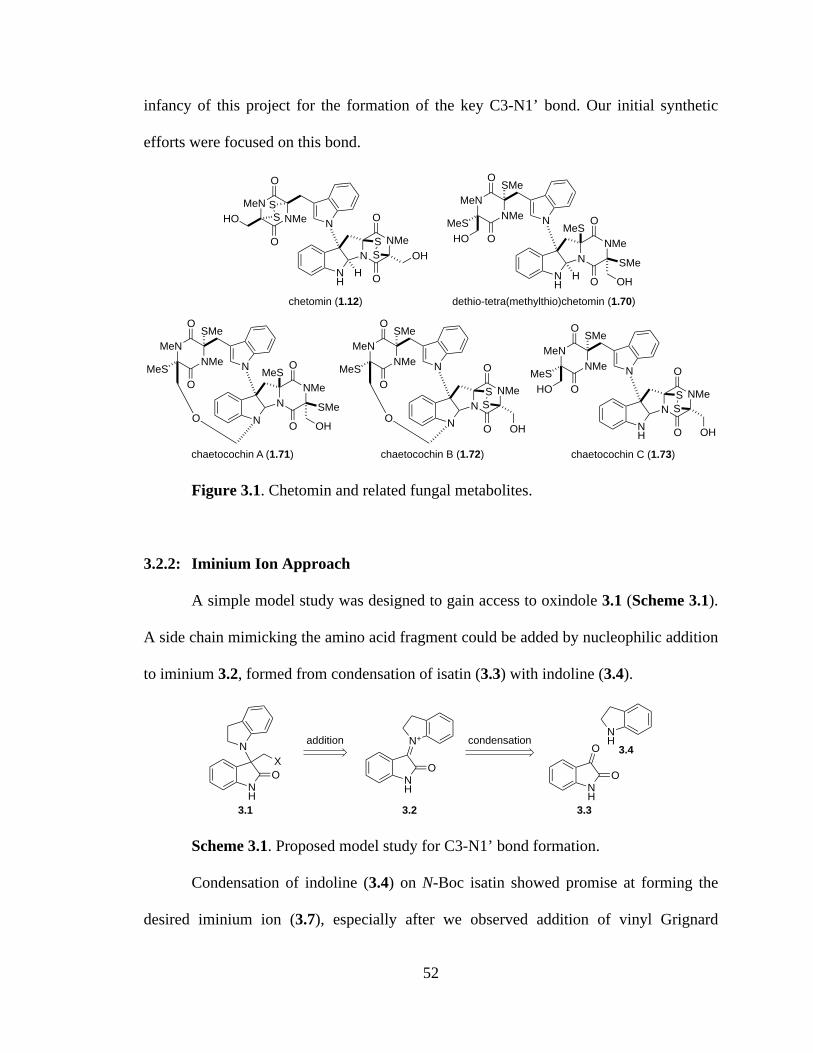
52
infancy of this project for the formation of the key C3-N1’ bond. Our initial synthetic
efforts were focused on this bond.
Figure 3.1. Chetomin and related fungal metabolites.
3.2.2: Iminium Ion Approach
A simple model study was designed to gain access to oxindole 3.1 (Scheme 3.1).
A side chain mimicking the amino acid fragment could be added by nucleophilic addition
to iminium 3.2, formed from condensation of isatin (3.3) with indoline (3.4).
Scheme 3.1. Proposed model study for C3-N1’ bond formation.
Condensation of indoline (3.4) on N-Boc isatin showed promise at forming the
desired iminium ion (3.7), especially after we observed addition of vinyl Grignard
NH
N
NNMeS
S
O
O
OH
NMeMeN
O
HO
O
SS
chetomin (1.12)
NH
NNMe
O
O
dethio-tetra(methylthio)chetomin (1.70)
SMe
MeS
NH
NNMe
O
O
chaetocochin C (1.73)
SS
NN
NMe
O
O
SS
chaetocochin B (1.72)
N
N
NNMe
O
OSMe
MeS
O
chaetocochin A (1.71)
H
NNMeMeN
O
MeSO
SMe
H
NMeMeN
O
MeSO
SMe
N
O
NMeMeN
O
MeSO
SMe
NNMeMeN
O
MeSO
SMe
OH
HO
OHOH
HO
OH
NH
N+
NH
O
ONH
ONH
N
O
addition condensation
X
3.1 3.2 3.3
3.4

53
reagent to the condensation product (Scheme 3.2). However, further structural analysis
by NMR revealed that water was not eliminated in the condensation, and hemiaminal 3.6
was instead formed through direct addition of indoline to the C3 carbonyl of isatin. Vinyl
Grignard reagent was adding to the lactam carbonyl carbon. Numerous conditions were
attempted to effect elimination of water, but all failed to produce the desired iminium ion
(3.7).
Scheme 3.2. Attempted iminium ion formation.
Looking back, we did achieve our goal of forming a tetrasubstituted carbon on an
indole surrogate. However, no reasonable plan existed for the incorporation of the amino
acid residue, let alone with any stereocontrol.
3.2.3: Pinacol-type Rearrangement
Inspired by past Williams group chemistry and aryl pinacol-type rearrangements
recently reported by Movassaghi, we next envisioned forming the C3 tetrasubstituted
center through a pinacol-type rearrangement of tryptophan dimer 3.9 to oxindole 3.8
(Scheme 3.3).136-138 The amino acid fragment of indole 3.9 could be installed through
alkylation of 3.10, which was expected to arise from copper-mediated indole cyclization
NH
Boc2O, DMAP
NBoc
THF, 0 °-r.t.
O
O
O
O
NH
4Å m.s.toluene, reflux
K2CO3
NBoc
O
NHO
NBoc
O
N+
3.3 3.5
3.4
3.6 3.7
70%>99%
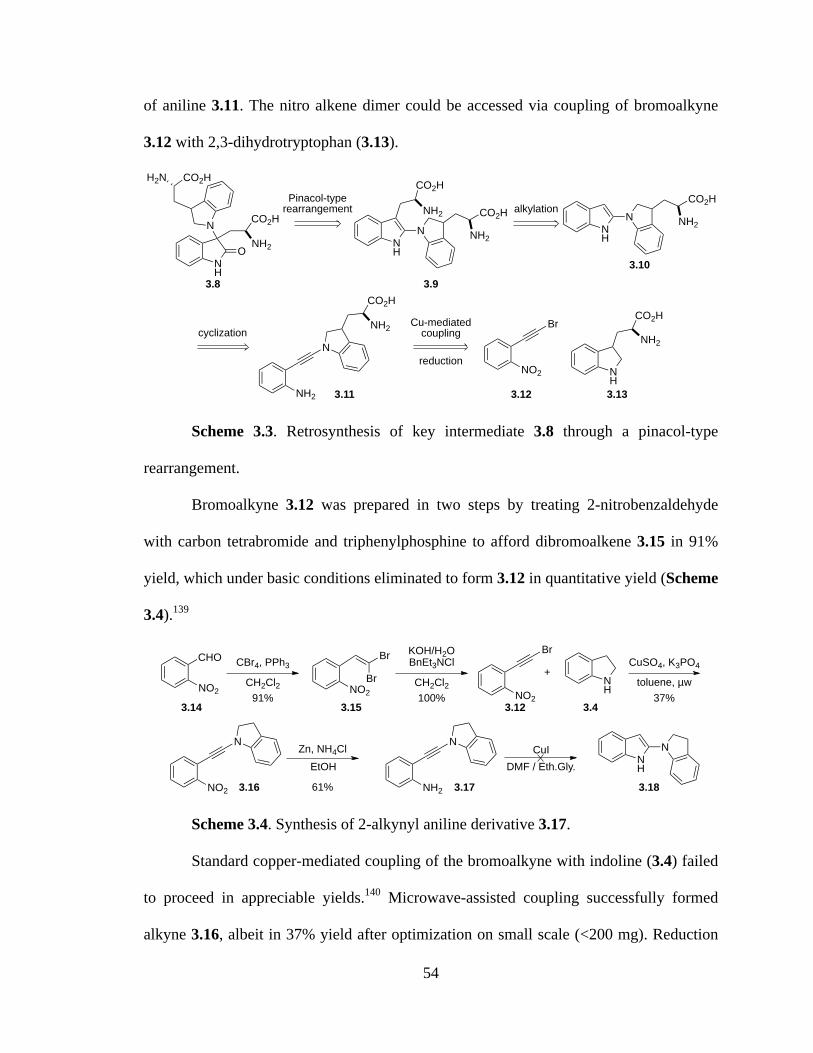
54
of aniline 3.11. The nitro alkene dimer could be accessed via coupling of bromoalkyne
3.12 with 2,3-dihydrotryptophan (3.13).
Scheme 3.3. Retrosynthesis of key intermediate 3.8 through a pinacol-type
rearrangement.
Bromoalkyne 3.12 was prepared in two steps by treating 2-nitrobenzaldehyde
with carbon tetrabromide and triphenylphosphine to afford dibromoalkene 3.15 in 91%
yield, which under basic conditions eliminated to form 3.12 in quantitative yield (Scheme
3.4).139
Scheme 3.4. Synthesis of 2-alkynyl aniline derivative 3.17.
Standard copper-mediated coupling of the bromoalkyne with indoline (3.4) failed
to proceed in appreciable yields.140 Microwave-assisted coupling successfully formed
alkyne 3.16, albeit in 37% yield after optimization on small scale (<200 mg). Reduction
Cu-mediatedcouplingcyclization
alkylationPinacol-type
rearrangement
NH
O
NNH2
CO2H
CO2HH2N
NH
N NH2
CO2HNH2
CO2H
NH
N NH2
CO2H
NH2
N
NH2
CO2H
NO2
Br
NH
NH2
CO2H
reduction
3.8 3.9
3.10
3.11 3.12 3.13
NO2
BrCuSO4, K3PO4
NH
+
NO2
N
toluene, µw
CHO
NO2CH2Cl2
CBr4, PPh3Br
BrNO2
CH2Cl2
KOH/H2OBnEt3NCl
Zn, NH4Cl
NH2
N
EtOH DMF / Eth.Gly. NH
NCuI
3.15 3.12
3.16 3.17 3.18
3.1491% 100% 37%
61%
3.4

55
of the nitro group using zinc and ammonium chloride gave aniline 3.17 in 61% yield,
although copper iodide mediated indole formation failed to produce the desired indole
(3.18).
Protected 2,3-dihydrotryptophan was synthesized in parallel to this model study to
be used in place of indoline (3.4) above. N-Cbz tryptophan was treated with iodomethane
to form the methyl ester (3.20) in quantitative yield (Scheme 3.5). Reduction of 3.20
using BH3·Me2S in THF and TFA gave 2,3-dihydrotryptophan derivative 3.21 in 79%
yield. Coupling using the previously optimized conditions afforded alkyne 3.22 in 45%
yield, although reduction to aniline 3.23 gave only trace amounts of the desired product.
Scheme 3.5. Synthesis of 2,3-dihydrotryptophan alkyne derivative.
3.3: Evolution of Coupling Strategy
3.3.1: Witkop’s Pyrroloindole
At this point, we took a step back and looked to nature for inspiration. It is
unlikely that fungi synthesize chetomin by such contrived routes. If chetomin is derived
naturally from two molecules each of tryptophan and serine, why not start with
tryptophan and serine–optically pure, readily available compounds? Moreover, it is likely
NH
NHCbz
CO2H
MeI, KHCO3
DMFNH
NHCbz
CO2Me
BH3·Me2STHF/TFA
NH
NHCbz
CO2Me
CuSO4, K3PO4
toluene,µw
NO2
NNHCbz
CO2Me
Zn, NH4ClEtOH
NH2
NNHCbz
CO2Me
3.12,
3.20 3.21
3.22 3.23
100% 79%
45% trace
3.19

56
that the pyrroloindoline core is formed in nature prior to coupling with the second half of
the molecule, if not concomitantly. Witkop’s pyrroloindole (3.24) was identified as a
synthetically useful intermediate in the synthesis of chetomin that could be formed
readily from tryptophan.141 Halogenation of the indole double bond would provide a
handle that could be substituted with another indole, ideally tryptophan, all with the
potential for some stereocontrol (Scheme 3.6).
Scheme 3.6. Witkop’s pyrroloindole inspiration.
Retrosynthetically, we could form advanced intermediate 3.26 from the coupling
of bromopyrroloindoline 3.28 and a suitably protected tryptophan derivative (3.27,
Scheme 3.7). Both of these coupling partners could be synthesized in a few steps from
tryptophan.
Scheme 3.7. Retrosynthetic plan using pyrroloindoline.
Tryptophan methyl ester (3.30) was protected as the trifluoroacetate amide (3.31),
then cyclized to pyrroloindole 3.32 using t-BuOCl in 63 % overall yield (Scheme 3.8).
Treatment of 3.32 with sodium hydride and NBS did not provide the desired
bromopyrroloindole.142
NH
NAc
CO2Me
NNAc
CO2MeX
Witkop's pyrroloindole (3.24) 3.25
NR'
NR'
CO2RBr
H
NH
R'HN
RO2C
NR'
N
NR'
CO2R
NR'
NHR'
CO2R
3.29
halocyclizationcoupling
RO2C
R'HN
3.26 3.28
3.27

57
Scheme 3.8. Attempted synthesis of 3-bromopyrroloindole.
3.3.2: Indole–Aniline Coupling
This stepwise procedure to 3.33 was put on hold after a report from the Baran
group was published, showing an analogous sequence that showed promise at reducing
our synthesis of 3.26 to one step. Baran showed that treatment of N-Boc-Trp-OMe (3.34)
and 2-iodoaniline with N-iodosuccinimide in acetonitrile afforded the desired C-N bond
and C-3 quaternary center in one pot (Scheme 3.9).130-132
Scheme 3.9. Formation of key C-3 quaternary center.
We repeated this chemistry using indoline in place of 2-iodoaniline, but only
starting material was recovered (Scheme 3.10). Ideally, this reaction would be done with
tryptophan or 2,3-dihydrotryptophan, eliminating the need for a Larock indole synthesis
with the iodoaniline. To our disappointment, the reaction only worked as originally
reported with 2-iodoaniline, despite numerous attempts at the reaction with indoline (3.4),
aniline (3.37), or 7-iodoindoline143 (3.36).
NH
NH2
CO2Me
EtCO2CF3
MeOH, Et3NNH
NH
CO2Me
O CF3
t-BuOClCH2Cl2, Et3N
NH
NCOCF3
CO2MeNaH, NBS
NNCOCF3
CO2MeBr
100%
3.31
63%
3.32 3.33
3.30
NH
NHBoc
CO2Meo-Iodoaniline,
NISCH3CN
-42°C to -30°CNH
NBoc
CO2MeNH
I
H78%
3.353.34

58
Scheme 3.10. Attempted formation of functionalized C3-N bond.
A suitable TMS-acetylene derivative was synthesized from L-serine methyl ester
(3.39) and TMS-acetylene (Scheme 3.11). Serine methyl ester (3.39) was N-Boc
protected (3.40) and the alcohol converted to the iodide (3.41) upon treatment with
triphenylphosphine, imidazole, and iodine. TMS-acetylene bromide (prepared in one
step, 72% yield) was coupled to serine-derived halide 3.41 to afford the desired alkyne
(3.42) in 40% yield.132
Scheme 3.11. Key alkyne synthesis.
Alkyne 3.42 was subjected to Larock annulation with iodoaniline 3.12 to form
indole 3.43. Saponification to the diacid (3.44) proceeded in good yield. While this route
afforded access to advanced intermediate 3.44 in decent yields, it suffered from high step
count due to the necessary Larock annulation and provided access to a diastereomer of
chetomin. We again looked back to the convergent, stepwise approach that we had
NH
NH
NHBoc
CO2Me
CH3CN-42°C to -30°C
NH
NBoc
CO2MeN
NH
I
NH2
NISR1
R2
R3
+
or
or H3.34
3.4
3.36
3.373.38
MeO2C NH2
OH Boc2OCH3CN, Et3N MeO2C NHBoc
OH PPh3, imidazoleI2, CH2Cl2
MeO2C NHBoc
I
CuCN, Zn MeO2C NHBoc
Br TMS
TMS
78% 3.40 55%
3.41 40% 3.42
3.39

59
previously envisioned (Scheme 3.7) and attempted the desired sequence in a stepwise
manner.
Scheme 3.12. Larock indole synthesis of tryptophan dimer.
3.3.3: Coupling to exo-3-bromopyrroloindoline
N-Cbz-Trp-OMe (3.20) was synthesized in >99% yield from tryptophan and the
indole nitrogen protected as the Boc carbamate (3.46). Per a report published by Espejo
and Rainier in 2008, addition of NBS and PPTS to 3.46 afforded exo-3-
bromopyrroloindoline 3.47 in 89% yield (Scheme 3.13).133,144 Deprotonation by KOtBu
in the presence of indole formed the C3-N bond in 28% yield and resulted in
epimerization of the methyl ester to the thermodynamically more favorable endo position
(3.48).133
Scheme 3.13. Coupling of 3-bromopyrroloindoline.
NH
NBoc
CO2MeN
NHBoc
CO2Me
LiOHTHF, H2O
NH
NBoc
CO2HN
NHBoc
CO2H
H H70%
3.43
92%
3.44
NH
NBoc
CO2MeNH
I
H
3.35
DMF
Pd(OAc)2, NaOAc,LiCl
3.42
NH
NH2
CO2H
CbzClMeOH, NaHCO3
NH
NHCbz
CO2H
MeI, KHCO3
DMFNH
NHCbz
CO2Me
Boc2O, NaOHBu4NHSO4
CH2Cl2
NBoc
NHCbz
CO2Me
NBoc
NCbz
CO2MeBrNBS, PPTS
CH2Cl2
indole,KOtBuCH3CN
NBoc
NCbz
CO2MeN
HH
100%
3.19
100%
3.2098%
89%
3.46 3.47
28%
3.48
3.45

60
This result evolved into our current strategy for the formation of the C3-N1’ bond.
The stereochemistry of 3.48 matches the enantiomer of what would be needed for a
synthesis of chetomin (i.e. 3.51, Scheme 3.14). We could either form the
bromopyrroloindoline from L-tryptophan and to it couple D-tryptophan and D-serine to
synthesize the enantiomer of chetomin, or we could synthesize the D-tryptophan-derived
pyrroloindoline (3.52) and use the natural amino acid isomers for the remaining three
couplings in a synthesis of (+)-chetomin (1.12). Initially, however, only the cheaper L-
isomers were used for all four fragments as conditions were worked out for the remaining
steps.
Scheme 3.14. Retrosynthetic analysis of (+)-chetomin.
3.3.4: Attempted Coupling to Tetracyclic Bromide
The next goal was to test a more divergent approach. Ideally, the dioxopiperazine
ring could be formed prior to coupling with indole (Scheme 3.15).
NH
NNH
O
O
OTBSH
N
NMe
MeN
O
O
OTBS
tetra-thiolation
C3-N1' coupling(L-Trp)
HOOH
HO
OH
oxidation
NNMe
O
O
SS
NH
HOH
MeNNMe
O
O
SS
NOH
(+)-chetomin (1.12)
NH
NNMe
O
O
OTBSH
N
NMe
MeN
O
O
OTBS
L-Ser (2 eq), cyclization,
N-methylation
NH
NHCO2Me
H
N
CO2Me
NHBoc
NBoc
NBocCO2Me
Br
H NH
NH2
CO2H
D-tryptophan (3.53)
bromo-cyclization
3.52
3.49 3.50
3.51

61
Scheme 3.15. Coupling with more advanced bromopyrroloindoline.
Unfortunately, synthesis of 3.55 was more challenging than expected. Peptide
coupling of tryptophan derivative (3.56) with N-Me-L-Ser-OMe initially suffered from
poor yield. Eventually, the coupling was optimized using EDCI as the coupling reagent to
afford dipeptide 3.57a in good, reproducible yield (Scheme 3.16). Cyclization to the
dioxopiperazine (3.58a) failed under methods traditionally used in the Williams group
(i.e. refluxing toluene with catalytic 2-hydroxy pyridine following Boc deprotection).
However, microwave heating of neat N-Boc dipeptide 3.57a at 180 °C effected
deprotection and cyclization to the dioxopiperazine (3.58a) in two minutes. The
recovered product did not require any purification before proceeding to the next step.
Scheme 3.16. Optimized peptide coupling and dioxopiperazine formation.
Gratifyingly, this procedure was generally applicable to a variety of related
dipeptides. Dioxopiperazines 3.58a-e were all synthesized with consistently good yield
on scales up to 500 mg (Scheme 3.17).
Several of the dioxopiperazines were cyclized to the corresponding
bromopyrroloindolines (3.55, Scheme 3.18). Despite numerous attempts, coupling of
indole failed using conditions analogous to those first used in Scheme 3.13.
NN
NR2
OR3
O
O
Br
HR1N
NNR2
OR3
O
O
N
HR1
3.54 3.55
NHN
NMeOH
O
OSO2PhNNHBoc
NMe
O
SO2Ph
CO2Me
OH
NNHBoc
SO2Ph
CO2H N-Me-L-Ser-OMe,EDCI, iPr2NEt
CH2Cl2
3.56 3.57a 3.58a
µw (180 °C)neat
86% 94%

62
Scheme 3.17. Microwave assisted dioxopiperazine synthesis.
Scheme 3.18. Attempted coupling with indole.
3.4: Epidithiodioxopiperazine Formation
While unsuccessfully attempting to couple indole or tryptophan to 3.55, we also
began to explore methods for the introduction of sulfur to the same dioxopiperazine.
Epidithiodioxopiperazine 3.63, essentially a monomer of chetomin, was synthesized by a
general method recently described by the Sodeoka group in Japan (Scheme 3.19).123,124
Benzenesulfonamide protection of L-tryptophan (3.59) gave compound 3.56, to which L-
Ser-OMe was coupled to afford dipeptide 3.57d. The primary alcohol was protected as
the silyl ether (3.57e) and suffered microwave-induced deprotection and cyclization to
dioxopiperazine 3.58e. Treatment with bromine affected the bromocyclization to endo-
pyrroloindoline 3.55e. Addition of iodomethane successfully methylated the secondary
amide, but also resulted in the loss of the silyl ether to primary alcohol 3.60. After
reprotecting the alcohol, diol 3.62 was formed following radical bromination and
NNHBoc
N
O
R1
CO2Me
OR3
µw (180 °C)neat
NHN
NR2
OR3
O
OR1
R2
75-98%
3.57a (R1=SO2Ph, R2=Me, R3=H)3.57b (R1=H, R2=Me, R3=H)3.57c (R1=H, R2=H, R3=H)3.57d (R1=SO2Ph, R2=H, R3=H)3.57e (R1=SO2Ph, R2=H, R3=TBS)
3.58a-e
NHN
NR2
OR3
O
OR1 NN
NR2
OR3
O
O
Br
HR1 N
NNR2
OR3
O
O
N
HR1
Br2CH3CN
indole,KOtBuCH3CN
3.58 3.55 3.5438-63%

63
substitution of 3.61. Addition of the diol to condensed hydrogen sulfide and BF3·OEt2
and oxidation of the resultant disulfide with iodine gave epidithiodioxopiperazine 3.63.
Scheme 3.19. Formation of the disulfide bridge.
3.5: Attempted Core Construction
3.5.1: Problematic Peptide Couplings
With a method for the formation of an epidithiodioxopiperazine in hand, we went
back to the method described in Scheme 3.13 for construction of the key C3-N1’ bond.
N-Boc-tryptophan methyl ester (3.34) was prepared and further protected as the
benzenesulfonamide (3.64, Scheme 3.20). Cyclization to the bromopyrroloindoline
(3.65) proceeded readily upon treatment with NBS and PPTS, and coupling of 3.65 with
N-Boc-Trp-OMe (3.34) gave 3.66 in poor yield (34%). Saponification to diacid 3.67 was
followed by peptide coupling with serine methyl ester to give peptide 3.68. TBS
NH
CO2H
NHBoc 1) LHMDS2) PhSO2Cl
NSO2Ph
CO2H
NHBocL-Ser-OMe,
EDCIiPr2NEt, CH2Cl2 N
HPhO2SN NHBoc
O
CO2Me
OH
TBSCl, imidDMF N
HPhO2SN NHBoc
O
CO2Me
OTBSµw, 180°C
NSO2Ph
HNNH
O
O
OTBS
NSO2Ph
NNH
O
O
OTBS
Br
Br2
MeCN
NSO2Ph
NNMe
O
O
OH
BrK2CO3, MeIacetone
NSO2Ph
NNMe
O
O
OTBS
BrTBSCl
1. NBS, V-70
NSO2Ph
NNMe
O
O
OTBS
Br
OH
OH
2. H2O, MeCN1. BF3!OEt2, H2S2. I2
NSO2Ph
NNMe
O
O
OH
BrSS
3.59 3.56 3.57d
3.57e 3.58e
3.55e 3.60 3.61
3.62 3.63
79% 66%
90%98%
38%
98%47%
30% 42%

64
protection of the primary alcohols proceeded in good yield, although cyclization to the
dioxopiperazines (3.70) using the previously described microwave conditions failed, as
did the reaction with traditional thermal conditions.
Scheme 3.20. Coupling to 3-bromopyrroloindoline and attempted
dioxopiperazine formation.
Deprotection of the Boc groups of intermediate 3.66 and peptide couplings on the
resultant amines gave tripeptide 3.71 (Scheme 3.21). Intriguingly, formation of the
tetrapeptide of 3.66 was never achieved, despite numerous attempts with varying
coupling conditions and protecting groups on the amino acid functionalities.
NH
CO2H
NHBocDMF, KHCO3
NH
CO2Me
NHBocBu4NHSO4,
NaOH
MeI PhSO2Cl
NSO2Ph
CO2Me
NHBoc
CH2Cl2
NBS, PPTS
NSO2Ph
NBocCO2Me
BrNH
CO2Me
NHBoc
KOtBu
EDCI, iPr2NEt,L-Ser-OMe
NSO2Ph
NBoc
NCO2Me
MeO2C
BocHN
NSO2Ph
NBoc
NBocHN O
NH CO2Me
OH
O
NH
MeO2C
HO
THF/H2O
NSO2Ph
NBoc
NCO2H
HO2C
BocHN
LiOH
TBSCl, imidDMF
NSO2Ph
NBoc
NBocHN O
NH CO2Me
OTBS
O
NH
MeO2C
TBSO
µw, 180°C
NSO2Ph
N
NNH
NH
HN
O
OTBSO
O
O
TBSO
3.59 3.34 3.64
3.65
3.66
3.67 3.68
3.69 3.70
3.34
95%>99%
>99% 34% 97%
33% 87%CH2Cl2

65
Scheme 3.21. Attempted synthesis of alternate dioxopiperazine precursor.
3.6: Synthesis of Heptacyclic Core
3.6.1: Synthesis of Dioxopiperazines
We reasoned that the failed cyclization attempts in Scheme 3.20 and peptide
coupling problems in Scheme 3.21 could be due to steric congestion caused by the
indoline protecting group. Thus, N,N’-Boc2-D-Trp-OMe was prepared from D-tryptophan
(3.73, Scheme 3.22). Bromocyclization proceeded stereoselectively to exo-
bromopyrroloindoline 3.74. Coupling with N-Boc-L-Trp-OMe gave endo-intermediate
3.75 in decent yield, while deprotection of the carbamate gave the key intermediate for
peptide coupling. In the absence of an aniline protecting group, peptide coupling
proceeded in 44% yield to tetrapeptide 3.76. With the Fmoc amines, deprotection and
cyclization to dioxopiperazine 3.77 proceeded in one pot upon treatment with morpholine
in THF with gentle heating. At this point, all of the amides needed to be methylated prior
to the introduction of sulfur, which first required protection of the aniline nitrogen.
Selective Boc protection at this position was never achieved, nor was protection of
tetrapeptide 3.76 to 3.78.
NSO2Ph
NBoc
NCO2Me
MeO2C
BocHN
NSO2Ph
NH
NCO2Me
MeO2C
NH
2. N-Cbz-L-Ser-OTBS EDCI, iPr2NEt
O
TBSOCbzHN
3.66 3.71
1. TFA
73%

66
Scheme 3.22. Synthesis of dioxopiperazine 3.77.
3.6.2: Initial N-Methyl Amino Acid Incorporation
It was clear that N-methyl tryptophan and serine derivatives needed to be prepared
to avoid late stage N-methylation. Months of laborious chemistry went in to preparing
these seemingly simple derivatives, and ultimately bromopyrroloindoline 3.74 was
coupled to N-Me,Boc-L-Trp-OMe (Scheme 3.23). Deprotection of the two Boc groups
gave 3.80, a key intermediate used extensively in the remainder of this chapter. Peptide
coupling with N-Me,Boc-Ser(OBn) gave tetrapeptide 3.81, which was cyclized to
NBoc
NBocCO2Me
Br
KOtBu, MeCNN-Boc-L-Trp-OMe
NH
NH2
CO2H
3. NBS
1. SOCl2, MeOH2. Boc2O
H
NBoc
NBocCO2Me
H
N
CO2Me
NHBoc
58%
2. N-Fmoc-Ser(OTBS), T3P, iPr2NEt
NH
NCO2Me
H
N
CO2Me
HNO
NHFmoc
OTBS
O
NHFmoc
OTBS
1. TFA
NH
NNH
O
O
OTBSH
N
NH
HN
O
O
OTBS
morpholineTHF
44% 85%
conditionsBoc2O, Et3N, MeOHBoc2O, EtOHBoc2O, Et3N, DMFBoc2O, Et3N, THFBoc2O, DMAP, CH3CN
NBoc
NNH
O
O
OTBSH
N
NH
HN
O
O
OTBS
3.79
NRNRNRNRInseparable
Boc2O, DMAP, CH3CN(inseparable mixture)
NBoc
NCO2Me
H
N
CO2Me
HNO
NHFmoc
OTBS
O
NHFmoc
OTBS
3.73 3.74
3.75
3.76 3.77
3.78
45%

67
dioxopiperazine 3.82 in excellent yield after Boc deprotection and addition of
morpholine.
Scheme 3.23. Synthesis of (N-Me)3 dioxopiperazine.
Initially we attempted to again protect the aniline nitrogen before subjecting the
material to radical bromination conditions. All attempts to Boc or TBS protect 3.82 failed
(Scheme 3.24). We decided to move forward to try the bromination anyway on diol 3.84,
but the benzyl ether resisted all deprotection attempts.
A serine derivative lacking the benzyl group was added to diamine 3.80, but this
coupling failed to give the desired tetrapeptide (Scheme 3.25). The benzyl group seemed
to be a poor choice of protecting group for this intermediate, necessitating the synthesis
of N-Me,Fmoc-Ser(OTBS).
NBoc
NBocCO2Me
Br
2. TFA
1. N-Me,Boc-L-Trp-OMe KOtBu, MeCN
H
NH
NHCO2Me
H
N
CO2Me
NHMe
40%
N-Me,Boc-Ser(OBn),T3P, iPr2NEt
42%
3.80
NH
NCO2Me
H
N
CO2Me
MeNO
NMeBoc
OBn
O
NMeBoc
OBn
3.81
NH
NNMe
O
O
OBnH
N
NMe
MeN
O
O
OBn
2. morpholine, THF1. TFA
98%
3.82
3.74
CH2Cl2
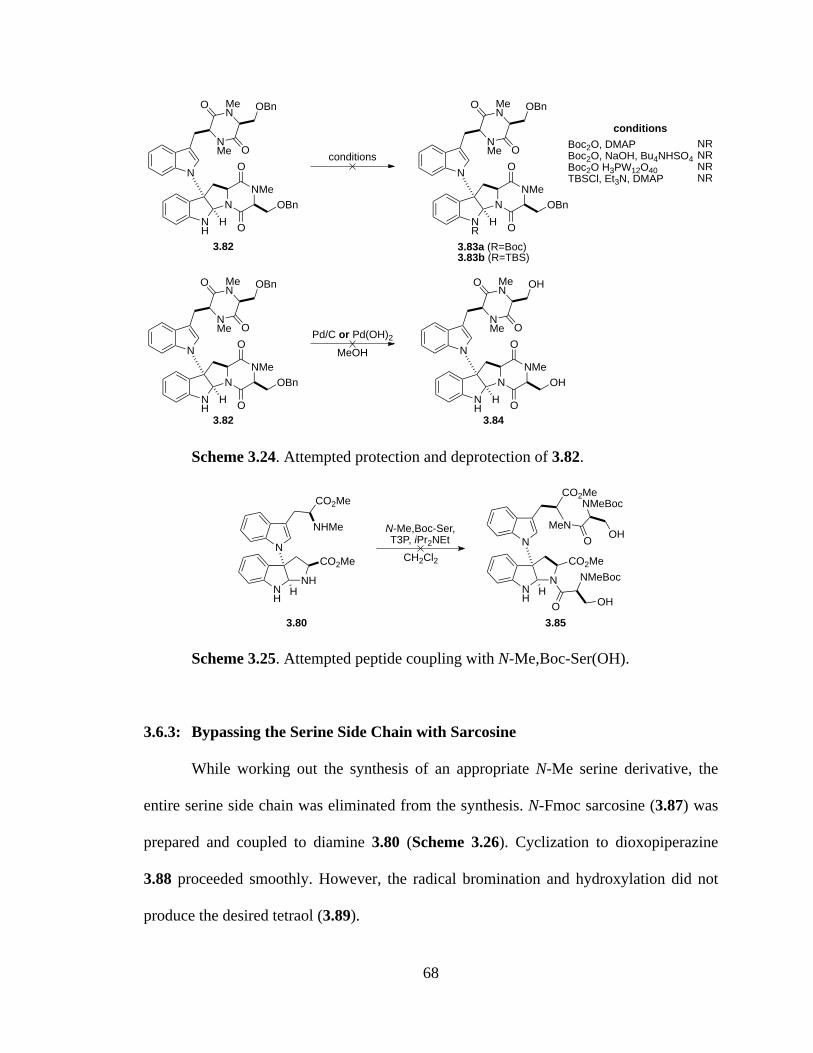
68
Scheme 3.24. Attempted protection and deprotection of 3.82.
Scheme 3.25. Attempted peptide coupling with N-Me,Boc-Ser(OH).
3.6.3: Bypassing the Serine Side Chain with Sarcosine
While working out the synthesis of an appropriate N-Me serine derivative, the
entire serine side chain was eliminated from the synthesis. N-Fmoc sarcosine (3.87) was
prepared and coupled to diamine 3.80 (Scheme 3.26). Cyclization to dioxopiperazine
3.88 proceeded smoothly. However, the radical bromination and hydroxylation did not
produce the desired tetraol (3.89).
NH
NNMe
O
O
OBnH
N
NMe
MeN
O
O
OBn
conditions
NR
NNMe
O
O
OBnH
N
NMe
MeN
O
O
OBn
3.83a (R=Boc)3.83b (R=TBS)
NH
NNMe
O
O
OBnH
N
NMe
MeN
O
O
OBn
Pd/C or Pd(OH)2
NH
NNMe
O
O
OHH
N
NMe
MeN
O
O
OH
MeOH
conditionsBoc2O, DMAPBoc2O, NaOH, Bu4NHSO4Boc2O H3PW12O40TBSCl, Et3N, DMAP
NRNRNRNR
3.82
3.82 3.84
NH
NHCO2Me
H
N
CO2Me
NHMe
NH
NCO2Me
H
N
CO2Me
MeNO
NMeBoc
OH
O
NMeBoc
OH
N-Me,Boc-Ser,T3P, iPr2NEt
3.80 3.85
CH2Cl2
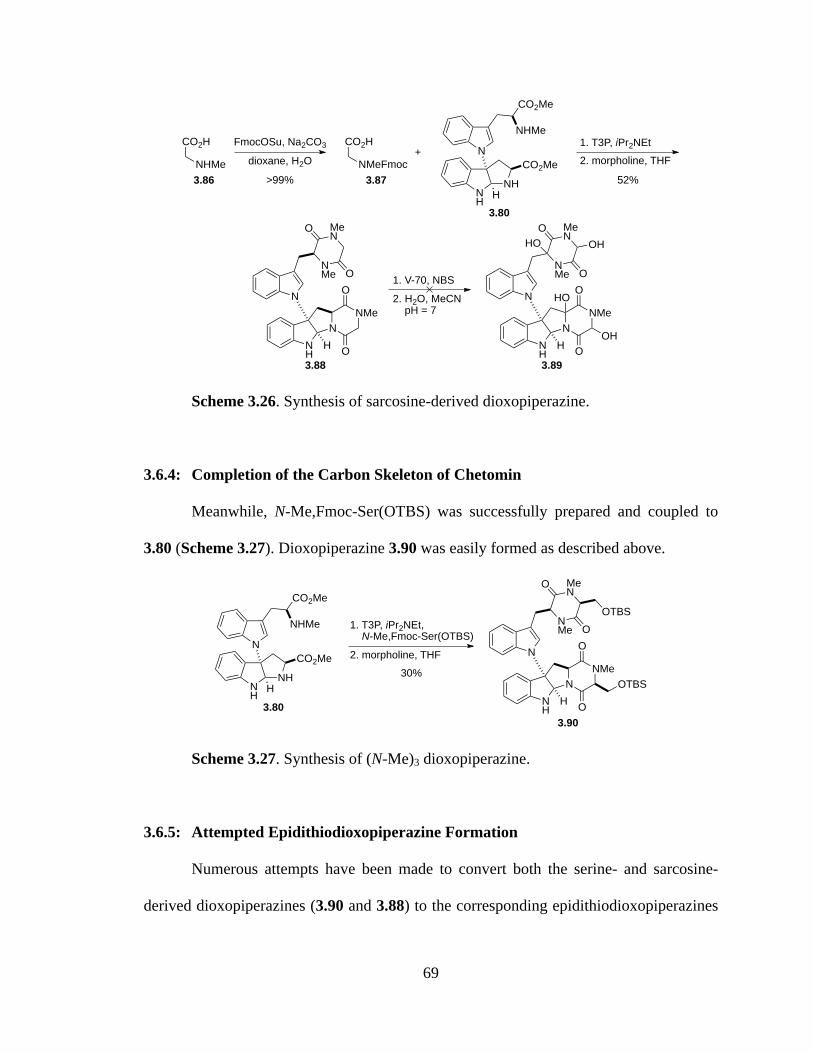
69
Scheme 3.26. Synthesis of sarcosine-derived dioxopiperazine.
3.6.4: Completion of the Carbon Skeleton of Chetomin
Meanwhile, N-Me,Fmoc-Ser(OTBS) was successfully prepared and coupled to
3.80 (Scheme 3.27). Dioxopiperazine 3.90 was easily formed as described above.
Scheme 3.27. Synthesis of (N-Me)3 dioxopiperazine.
3.6.5: Attempted Epidithiodioxopiperazine Formation
Numerous attempts have been made to convert both the serine- and sarcosine-
derived dioxopiperazines (3.90 and 3.88) to the corresponding epidithiodioxopiperazines
CO2H
NHMe
FmocOSu, Na2CO3 CO2H
NMeFmocdioxane, H2O>99%
+1. T3P, iPr2NEt2. morpholine, THF
NH
NHCO2Me
H
N
CO2Me
NHMe
52%
NH
NNMe
O
OH
N
NMe
MeN
O
O 1. V-70, NBS2. H2O, MeCN pH = 7
NH
NNMe
O
OH
N
NMe
MeN
O
O
HO OH
OH
HO
3.86 3.87
3.80
3.88 3.89
1. T3P, iPr2NEt, N-Me,Fmoc-Ser(OTBS)2. morpholine, THF
NH
NHCO2Me
H
N
CO2Me
NHMe
30%
NH
NNMe
O
OH
N
NMe
MeN
O
O
OTBS
OTBS
3.903.80

70
(1.12 and 3.91, Scheme 3.28). Much to our disappointment, both have refused to yield to
our continued efforts.
Scheme 3.28. Attempted epidithiodioxopiperazine formations.
3.7: Proposed Future Studies
3.7.1: Proposed Synthesis of Tetraol
We have successfully synthesized the carbon framework of chetomin. The final
key to a total synthesis is the oxidation of this core (3.90 or 3.88) to tetraols 3.92 or 3.89
(Scheme 3.29). Numerous examples (see Chapter 2) suggest that this compound should
readily form intermediate iminium ions such as 3.93 and 3.94. Addition of sulfur should
occur at the less hindered faces of the dioxopiperazines.122-125,127
NH
NNMe
O
OH
N
NMe
MeN
O
O
OTBS
OTBS
NNMe
O
O
SS
NH
HOH
MeNNMe
O
O
SS
NOH
1. NBS, V-70; H2O2. H2S; I2
NH
NNMe
O
OH
N
NMe
MeN
O
O
NNMe
O
O
SS
NH
H
MeNNMe
O
O
SS
N
3.90 1.12
3.88 3.91
1. NBS, V-70; H2O2. H2S; I2

71
Scheme 3.29. Key tetraols and iminium intermediates.
Synthesis of the tetraols should be possible either by the method previously
attempted (radical bromination and substitution) or by mild oxidation with
bis(pyridine)silver(I) permanganate (Scheme 3.30). In the synthesis of gliocladin B, n-
Bu4NMnO4 was also used as an oxidant for this reaction.127 Repeated trials will be
necessary to produce optimal conditions, requiring sufficient time, material, and
persistence. Three potential problems may complicate this route: (1) The free indoline
nitrogen may require protection if it proves to be reactive under the proposed radical or
oxidation conditions. This has previously been a difficult protection, and while a Boc or
silyl group would be ideal, a trifluoroacetate group would also be appropriate (although it
would introduce an additional deprotection step). (2) The desired hemiaminal may very
well be unstable and require additional functionalization after it is formed before sulfur
can be introduced. (3) The serine-derived bis(dioxopiperazine) (3.90) is susceptible to
elimination of the primary alcohol.
NH
NNMe
O
OH
N
NMe
MeN
O
O
OTBS
NH
NNMe
O
OH
N
NMe
MeN
O
O
3.92
3.89
HOOH
OTBS
OH
HO
HO OH
OH
HO
NH
NNMe
O
OH
N
NMe
MeN
O
O
OTBS
OTBS
NH
NNMe
O
OH
N
NMe
MeN
O
O
3.94
3.93

72
Scheme 3.30. Possible oxidations.
3.7.2: Bypassing the Tetraol
It would also be worth attempting the addition of sulfur directly to
dioxopiperazines 3.90 and 3.88. Both Nicolaou and Reisman took this approach in their
respective epidithiodioxopiperazine syntheses, using S8 and NaHMDS (Scheme
3.31).128,129
Scheme 3.31. Alternative sulfenylation.
3.7.3: Late-Stage Alkylation of Sarcosine Analogue
Lastly, if access to sarcosine-derived epidithiodioxopiperazine 3.91 proves to be
the most efficient route, conversion to chetomin can occur using inspiration from Kishi’s
early syntheses of sporidesmin and gliotoxin (Scheme 3.32).113-118 Reduction of the
NH
NNMe
O
OH
N
NMe
MeN
O
O
R
R
3.90 (R=CH2OTBS)3.88 (R=H)
NH
NNMe
O
OH
N
NMe
MeN
O
O
R
R
3.92 (R=CH2OTBS)3.89 (R=H)
HOOH
OH
HO
NBS, V-70; H2Oor
Py2AgMnO4
NNMe
O
OR
SS
NH
H
MeNNMe
O
OR
SS
N
1.12 (R=CH2OTBS)3.91 (R=H)
NH
NNMe
O
OH
N
NMe
MeN
O
O
R
R
3.90 (R=CH2OTBS)3.88 (R=H)
1. S8, NaHMDS2. deprotection3. I2

73
disulfide bridges and protection as the thioacetals would give 3.95. Alklation with
BOMCl would install the protected serine side chain (3.96), which could be deprotected
and the disulfide revealed to give chetomin (1.12).
Scheme 3.32. Proposed conversion of 3.91 to chetomin.
3.8: Concluding Remarks
It will be difficult to leave this project behind with the total synthesis of chetomin
so near to completion. However, short of a completed synthesis, we did achieve the
remaining goals for this project in the synthesis of dioxopiperazines 3.88 and 3.90. The
key C3-N1’ bond formation works for a variety of substrates and is a convergent
approach to the core structure. A variety of peptide couplings were optimized for this
system, producing di- and tetrapeptides in respectable yields. Cyclization of the
dioxopiperazine rings is now performed consistently and in high yields. Moreover, the
entire sequence is scalable and allows quick access to gram quantities of material. All
NNMe
O
O
SS
NH
H
MeNNMe
O
O
SS
N
3.91
NNMe
O
ONH
H
MeNNMe
O
O
SS
N
3.95
Ar
SS
Ar1. NaBH4
2. p-MeO-PhCHO
NNMe
O
ONH
H
MeNNMe
O
O
SS
N
3.96
Ar
SS
Ar
OBn
OBn
PhLi, BOMCl
NNMe
O
O
SS
NH
HOH
MeNNMe
O
O
SS
NOH
1.12
1. BCl32. mCPBA3. BF3·OEt2

74
that remains is synthesis of the disulfide bridges to complete a total synthesis of (+)-
chetomin.

75
CHAPTER 4
Synthetic Approach to Sporidesmin A
4:1: Introduction
In 2010, we had the opportunity to write a book chapter on biomimetic syntheses
of tryptophan-derived dioxopiperazines, in which Kishi’s elegant synthesis of (±)-
sporidesmin A (1.11) was featured (Figure 4.1).145 We were drawn to the densely
functionalized structure of sporidesmin A, including the polysubstituted aromatic ring,
pyrroloindoline core, hydroxyl groups at C3 and C12, and the epidiothiodioxopiperazine
ring. We hoped that chemistry developed for the synthesis of chetomin (see Chapter 3)
could be applied to an improved, stereoselective synthesis of sporidesmin A.
As detailed in Chapter 1, sporidesmin A was isolated from Pithomyces chartarum
in 1959 as part of a forty-year nationwide search for the cause of facial eczema, a disease
that was plaguing sheep herds of New Zealand.63-65 Facial eczema causes extensive liver
damage in sheep and has been responsible for massive economic losses in New Zealand
since the late 1890s. A dental nurse allegedly discovered that the addition of zinc sulfate
to drinking water could prevent the disease, a measure still endorsed by veterinarians.146
Epidithiodioxopiperazines are known to form a 2:1 complex with zinc ions, inhibiting the
generation of the superoxide anion radical and likely providing the observed protective
effects of zinc against sporidesmin.69,70 The toxicity of sporidesmin toward sheep was
responsible for many of the early advances made in the study of

76
epidithiodioxopiperazines, including the isolation and characterization of sporidesmin
and related metabolites (Figure 4.1).
Figure 4.1. Sporidesmin A and related fungal metabolites.
No reasonable argument can be made advocating that a new synthesis of
sporidesmin A will have any direct impact on mankind, medicine, or even sheep herds in
New Zealand. Our interest is purely academic. Sporidesmin A is a synthetically
challenging molecule, and pursuit of a total synthesis will allow us both to explore the
scope of similar reactions employed toward the total synthesis of chetomin and to expand
our general understanding of the reactivity of epidithiodioxopiperazine alkaloids.
4.2: Retrosynthetic Analysis
The primary goal of this project was to develop a scalable total synthesis of (–)-
sporidesmin A (1.11). Compared to chetomin (1.12), sporidesmin A presents several
unique synthetic challenges, the most significant of which is a highly substituted aromatic
NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
OH
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
SS
OH
S
NNMe
N
O
OMeMeO
MeO
ClHO
H
MeS
SMe
HO
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
s
OH
Me NNMe
N
O
OMeMeO
MeO
ClHO
H
MeSHO
NNMe
N
O
OMeMeO
MeO
ClHO
H
sOH
Me NNMe
N
O
OMeMeO
MeO
Cl
H
SS NMe
NMe
N
O
OHMeO
MeO
HO
H
SS
MeCl
HO
ss
s ss
sporidesmin G (1.67)
sporidesmin E (1.65)
sporidesmin J (1.69)sporidesmin H (1.68)
sporidesmin F (1.66)sporidesmin D (1.64)
sporidesmin C (1.63)sporidesmin B (1.62)sporidesmin A (1.11)

77
ring. We hoped to develop an efficient synthesis of the requisite substituted L-tryptophan
derivative and to apply the previously used bromocyclization reaction (Scheme 3.13) to
form the pyrroloindoline core.
As with chetomin, we planned to introduce the disulfide bridge of (–)-sporidesmin
A (1.11) at a late stage in the synthesis from hemiaminal 4.1 (Scheme 4.1). The
hemiaminal could be formed by oxidation of tetracycle 4.2 after forming the
pyrroloindoline core through oxidative cyclization and N-methylation of dioxopiperazine
4.3. This intermediate is similar to many of the dioxopiperazines prepared in the previous
chapter and was envisioned to arise via coupling of 4.4 with alanine, followed by
cyclization.
Scheme 4.1. Retrosynthetic analysis of (–)-sporidesmin A.
One key to an asymmetric synthesis is the preparation of β-hydroxytryptophan
derivative 4.4. We hoped to introduce both stereocenters and the amino acid portion of
this intermediate through Sharpless asymmetric aminohydroxylation of unsaturated ester
4.5. This ester could be formed by Wittig olefination of the aldehyde product of a
CHO
OMeHO
OMeMeO
Cl
NO2
NO2
OMeMeO
Cl
NH
OMeMeO
Cl
NMe
CO2Me
OMeMeO
Cl
NMe
CO2MeHONHCbz
NMe
Cl
MeOOMe
HNNH
O
OMe
TBSO
NMe
Cl
MeOOMe
NH
TBSOTBSO
NMe
O
OMe
(!)-sporidesmin A (1.11)
NMe
Cl
MeOOMe
NH
HOHO
NMe
O
OMe
SS
NMe
Cl
MeOOMe
NH
TBSOTBSO
NMe
O
O
Me
HO
OH
thiolation oxidation
oxidativecyclization
peptidecoupling
amino-hydroxylation
alkylation,Wittig
indoleformation substitution
4.1 4.2
4.3 4.4 4.5
4.6 4.7 vanillin (4.8)

78
Vilsmeier–Haack reaction of indole 4.6. Modified dinitrostyrene Batcho–Leimgruber
conditions were planned for the formation of the indole from styrene 4.7, prepared in
several precedented steps from vanillin (4.8).
4.3: Synthetic Approach to Sporidesmin A
4.3.1: Preparation of Dinitrostyrene Derivative
Nitration of O-acetyl vanillin (4.9) afforded 4.10 when fuming nitric acid (>90%)
was used as the nitronium source (Scheme 4.2). Fuming nitric acid was originally
prepared fresh by distillation from a slurry of potassium nitrate and concentrated sulfuric
acid, but this procedure seemed quite dangerous on scales producing more than 50 mL of
nitric acid, prompting us to obtain the reagent from a commercial source.
Scheme 4.2. Nitration of vanillin.
Following nitration, 4.10 was heated to reflux in aqueous KOH to afford phenol
4.11, then O-methylation with dimethyl sulfate gave 4.12 in 99% yield (Scheme 4.3).
The Henry reaction was used to prepare dinitrostyrene derivative 4.13 through addition of
nitromethane to the aldehyde and subsequent elimination of water. Indole 4.14 was then
prepared in low yield using modified Batcho–Leimgruber conditions.147
Originally, we had envisioned preparing chloroindole 4.6 from indole 4.14.
However, it seemed more prudent to chlorinate vanillin, as we would be less concerned
with yield and could minimize the amount of chlorine gas used. Accordingly, 5-Cl-
CHO
OMeHO
Ac2O, Et3NDMAPTHF
CHO
OMeAcO
conditionsCHO
OMeAcO NO2
conditionsHNO3 (70%)H2SO4, HNO3HNO3 (>90%)
NRNR47%
4.8 4.9 4.1090%
yield

79
vanillin (4.15) was prepared by bubbling chlorine gas through a solution of vanillin in
acetic acid (Scheme 4.4).148 Nitration, deacetylation, and methylation proceeded in good
yield to provide aldehyde 4.18. The Henry reaction gave dinitrostyrene 4.7 in low
yield.149
Scheme 4.3. Dimethoxyindole synthesis.
Scheme 4.4. Synthesis of 5-chloro-dinitrostyrene derivative.
We determined that the small amount of ethyl acetal 4.19 formed during the
methylation reaction was detrimental to the yield of the Henry reaction (Scheme 4.5). To
circumvent this problem, the crude mixture of 4.18 and 4.19 was treated with TFA in
chloroform to cleave the acetal, affording pure 4.18. Use of clean material greatly
increased the ease of recrystallization and yield of the product of the Henry reaction (4.7).
CHO
OMeAcO NO2
KOHCHO
OMeHO NO2
Me2SO4, NaOHCHO
OMeMeO NO2
MeNO2, NaOH
OMeMeO NO2
NO2Fe, SiO2, AcOHPhCH3, reflux
OMeMeO N
H
H2O, reflux H2O, EtOH
H2O, MeOH
4.10 4.11 4.12
4.13 4.14
47% (2 steps) 99%
21% 7%
CHO
OMeHO
1. Cl2, AcOH2. Ac2O, Et3N DMAP, THF
CHO
OMeAcO
HNO3 (>90%)Cl
CHO
OMeHO
Cl
NO2OMe
MeO
Cl
NO2
NO2CHO
OMeMeO
Cl
NO2
CHO
OMeAcO
Cl
NO2
KOHH2O, reflux
Me2SO4, NaOHH2O, EtOH
MeNO2, NaOHH2O, MeOH
<10%
4.8 4.15 4.16
4.17 4.18 4.7
64%76% (2 steps)
>99%

80
Scheme 4.5. Undesired acetal formation.
4.3.2: Indole Synthesis and Sharpless Asymmetric Aminohydroxylation
Synthesis of indole 4.6 proceeded in good yield using modified Batcho–
Leimgruber conditions (Scheme 4.6). Following N-methylation, an aryl aldehyde was
installed at C3 via the Vilsmeier–Haack reaction.150 This aldehyde (4.20) was
immediately converted to the unsaturated ester (4.5) upon Wittig reaction with the
requisite phosphonium ylide. Sharpless asymmetric aminohydroxylation gave β-
hydroxytryptophan derivative 4.4, albeit in modest yield.151
Scheme 4.6. Asymmetric aminohydroxylation.
While it is commercially available, we opted to prepare the ligand for the
Sharpless reaction as described in the literature (Scheme 4.7).152 Acylation of
CHO
OMeHO
Cl
NO2
OMeMeO
Cl
NO2
NO2
CHO
OMeMeO
Cl
NO2
Me2SO4, NaOHH2O, EtOH
MeNO2, NaOHH2O, MeOH
+
OMeMeO
Cl
NO2
OEt
OEt
TFACHCl3
CHO
OMeMeO
Cl
NO2
64%
4.17 4.18 4.19
4.18 4.7
>99%
Fe, SiO2, AcOHPhCH3, reflux
OMeMeO
1. MeI
Ph3P=CHCO2Me
Cl
NH
OMeMeO
Cl
NMe
CO2Me K2OsO2(OH)4,(DHQD)2AQN (4.24),
CbzNClNa
OMeMeO
Cl
NMe
CO2MeHONHCbz
n-PrOH, H2O
91%
35%
OMeMeO
Cl
NMe
CHO
OMeMeO
Cl
NO2
NO2
2. POCl3, DMF; NaOH, H2O
PhCH3, reflux
4.7 4.6 4.20
4.5 4.4
93%
80%

81
difluorobenzene gave dione 4.23, which was alkylated to form the dihydroquinidinyl
anthraquinone ligand (4.24) in good yield.
Scheme 4.7. Preparation of (DHQD)2AQN.
4.3.3: Dioxopiperazine Formation
β-hydroxytryptophan derivative 4.4 was protected as the silyl ether (4.25) and the
Cbz group cleaved to reveal the free amine (4.26, Scheme 4.8). EDCI-mediated coupling
of N-Boc-Ala-OH provided dipeptide 4.27 in 59% yield.
Scheme 4.8. Formation of the dioxopiperazine and an undesired elimination.
Attempts to form the desired dioxopiperazine directly from 4.27 using the
microwave conditions described in the previous chapter resulted in decomposition.
O
O
O 1. AlCl3, reflux, 48 h
F
F
+2. polyphosphoric acid, 140°C
O
O F
F
DHQD, n-BuLiTHF
O
O DHQD
DHQD(DHQD)2AQN (4.24)4.21 4.22 4.23
14%78%
NMe
Cl
MeOOMe
HNNH
O
OMe
H2, Pd/C
EDCI, iPr2NEt,N-Boc-Ala-OH
NMe
Cl
MeOOMe
HNNHBoc
CO2Me
OMe
TBSO
2-OH-pyridine
OMeMeO
Cl
NMe
CO2MeHONHCbz
OMeMeO
Cl
NMe
CO2MeTBSONHCbzTBSCl, imid
OMeMeO
Cl
NMe
CO2MeTBSONH2
NMe
Cl
MeOOMe
HNNH2
CO2Me
OMe
TBSOTMSOTf,
2,6-lutadineCH2Cl2
PhCH3, reflux
4.4 4.25 4.26
4.27 4.28
4.29
DMF>99%
MeOH94%
CH2Cl259% 59%
61%

82
Following more traditional methods, we first removed the Boc group, then heated the
crude product (4.28) in toluene in the presence of 2-hydroxypyridine. The
dioxopiperazine was formed, but unfortunately the conditions also caused elimination of
the silyl ether to afford 4.29.
An alternative cyclization precursor was synthesized from 4.25 by saponification
of the methyl ester and coupling of L-Ala-OMe (Scheme 4.9). The low yielding coupling
step forced a quick screen of coupling reagents, through which T3P was identified as the
most efficient of those tested, providing dipeptide 4.31 in a mere 34% yield.
Scheme 4.9. Preparation of dipeptide.
The unoptimized reaction allowed us access to enough material to attempt the
cyclization reaction. Dipeptide 4.31 was converted to the free amine and heated gently in
toluene with catalytic 2-hydroxypyridine (Scheme 4.10). A mixture of the elimination
product (4.29) and the desired dioxopiperazine (4.3) was obtained in poor yield.
Scheme 4.10. Cyclization to the dioxopiperazine.
OMeMeO
Cl
NMe
CO2MeTBSONHCbz
NMe
Cl
MeOOMe
NHCbz
TBSO O
NH
CO2Me
Me
OMeMeO
Cl
NMe
CO2HTBSONHCbzLiOH
H2O, THF
L-Ala-OMe,iPr2NEt,
conditions
conditionsEDCIHATUT3P
6%29%34%
4.25 4.30 4.31
CH2Cl287%
OP O P
OPO
OO
!T3P
NMe
Cl
MeOOMe
NHCbz
TBSO O
NH
CO2Me
Me
NMe
Cl
MeOOMe
HNNH
O
OMe2. 2-OH-pyridine
PhCH3, 50 °C NMe
Cl
MeOOMe
HNNH
O
OMe
TBSO
1. H2, Pd/C +
22%4.31 4.29 4.3

83
We returned to amine 4.26 and to it coupled N-Fmoc-L-Ala (Scheme 4.11).
Again, our standard EDCI-mediated coupling produced dipeptide 4.32 in very poor yield.
Use of T3P nearly doubled the yield and greatly simplified purification of 4.32. Gentle
heating of the dipeptide in THF and morpholine resulted in concomitant Fmoc
deprotection and cyclization to the desired dioxopiperazine (4.3).
Scheme 4.11. Improved synthesis of dioxopiperazine 4.3.
4.3.4: Oxidative Cyclization Attempts
Elated to finally have access to dioxopiperazine 4.3, we pressed forward and
attempted the oxidative cyclization to the pyrroloindoline. Treatment of 4.3 with NBS or
bromine resulted in products containing two bromine atoms. We turned to the conditions
reported in Kishi’s synthesis, but only isolated starting material from the reaction mixture
(Scheme 4.12).113 Alcohol 4.34 was expected to result from aqueous conditions, but only
a small amount of 4.35, formed by oxidative fission of the indole double bond and
elimination of TBSOH, was recovered with starting material.153
N-Fmoc-L-Ala,iPr2NEt, x
NMe
Cl
MeOOMe
HNNHFmoc
CO2Me
OMe
TBSO
OMeMeO
Cl
NMe
CO2MeTBSONH2
CH2Cl2x=EDCI, 30%x=T3P, 59%
NMe
Cl
MeOOMe
HNNH
O
OMe
TBSO
morpholineTHF, 35 °C
4.26 4.32 4.3
78%

84
Scheme 4.12. First attempts at oxidative cyclization.
Dioxopiperazine 4.3 was precious material at this point in the synthesis, and we
certainly did not have large enough quantities to attempt a variety of conditions.
Therefore, simple dioxopiperazine 4.36 was prepared and subjected numerous times to
variations of the above conditions (Scheme 4.13). Unfortunately, model compound 4.36
also failed to react. Dioxopiperazine 4.36 is nearly insoluble in most solvents, so we were
never able to determine if this was the true cause of the failure. The unique electronics of
dioxopiperazine 4.3 and the poor solubility of any readily accessible surrogate forced us
to study the cyclization using advanced material.
Scheme 4.13. Model cyclization study.
We next attempted the oxidative cyclization using DMDO in acetone and
dichloromethane (Scheme 4.14).154,155 No product (4.34 or 4.39) was detected from the
NMe
Cl
MeOOMe
HN
TBSONH
O
OMe
NMe
Cl
MeOOMe
NH
AcOTBSO
NH
O
OMe
PhI(OAc)2
Me2S, MeCN
NMe
Cl
MeOOMe
HN
TBSONH
O
OMe
NMe
Cl
MeOOMe
NH
HOTBSO
NH
O
OMe
PhI(OAc)2
H2O, MeCNNHAc
Cl
MeOOMe
O
NH
HNO Me
O
4.3
4.3
4.33
4.344.35
NH
HNNH
O
O NH
NH
AcO NH
O
O
PhI(OAc)2
Me2S, MeCN
NH
HNNH
O
O NH
NH
HO NH
O
O
PhI(OAc)2
H2O, MeCN
OTBS OTBS
OTBS OTBS
4.36 4.37
4.36 4.38
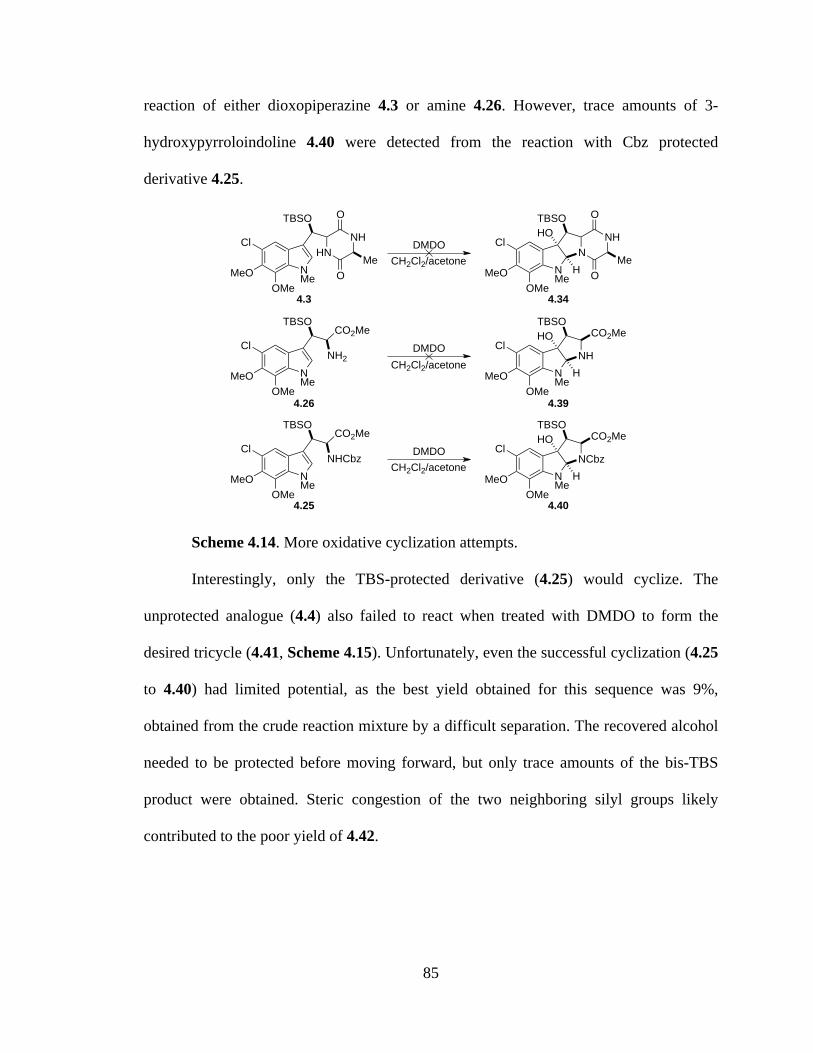
85
reaction of either dioxopiperazine 4.3 or amine 4.26. However, trace amounts of 3-
hydroxypyrroloindoline 4.40 were detected from the reaction with Cbz protected
derivative 4.25.
Scheme 4.14. More oxidative cyclization attempts.
Interestingly, only the TBS-protected derivative (4.25) would cyclize. The
unprotected analogue (4.4) also failed to react when treated with DMDO to form the
desired tricycle (4.41, Scheme 4.15). Unfortunately, even the successful cyclization (4.25
to 4.40) had limited potential, as the best yield obtained for this sequence was 9%,
obtained from the crude reaction mixture by a difficult separation. The recovered alcohol
needed to be protected before moving forward, but only trace amounts of the bis-TBS
product were obtained. Steric congestion of the two neighboring silyl groups likely
contributed to the poor yield of 4.42.
NMe
Cl
MeOOMe
HN
TBSONH
O
OMe
NMe
Cl
MeOOMe
NH
HOTBSO
NH
O
OMe
DMDOCH2Cl2/acetone
NMe
Cl
MeOOMe
TBSO
NMe
Cl
MeOOMe
NHH
HOTBSO
CO2MeDMDO
CH2Cl2/acetone
CO2Me
NH2
NMe
Cl
MeOOMe
TBSO
NMe
Cl
MeOOMe
NCbzH
HOTBSO
CO2MeDMDO
CH2Cl2/acetone
CO2Me
NHCbz
4.3 4.34
4.26 4.39
4.25 4.40

86
Scheme 4.15. Progression of cyclization.
We also attempted the cyclization using other oxidants, including Davis’s
oxaziridine and benzoyl peroxide (Scheme 4.16).116,156 Only starting material was
recovered, with no trace of the desired products (4.34 and 4.43).
Scheme 4.16. Attempted cyclizations.
4.3.5: Toward a Formal Synthesis of Sporidesmin A
These repeated attempts at forming the requisite 3-hydroxypyrroloindoline were
frustrating, especially considering that Kishi had accomplished the same reaction using a
NMe
Cl
MeOOMe
NCbzH
HOHO
CO2Me
DMDOCH2Cl2/acetone
OMeMeO
Cl
NMe
CO2MeHONHCbz
TBSCl, imid
OMeMeO
Cl
NMe
CO2MeTBSONHCbz
>99%
NMe
Cl
MeOOMe
NCbzH
HOTBSO
CO2Me
DMDOCH2Cl2/acetone 9%
TBSCl, imidNMe
Cl
MeOOMe
NCbzH
TBSOTBSO
CO2Me
4.4 4.25
4.41 4.40 4.42
trace
NMe
Cl
MeOOMe
HN
TBSONH
O
OMe
NMe
Cl
MeOOMe
NH
HOTBSO
NH
O
OMe
Davis's oxaziridineCH2Cl2
NMe
Cl
MeOOMe
HN
TBSONH
O
OMe
NMe
Cl
MeOOMe
NH
PhOCOTBSO
NH
O
OMe
(PhCO2)2
DME
4.3 4.34
4.3 4.43

87
similar substrate. That said, it should be noted that in the original sporidesmin synthesis,
the authors only recovered diacetate 2.6 in 30% yield and completed the synthesis using
authentic diacetate prepared from the degradation of sporidesmin A (Scheme 4.17).113
One can only conclude that they also were unable to access appreciable quantities of the
pyrroloindoline. Moreover, in Kishi’s later synthesis of sporidesmin B, benzoyl peroxide
was used as the oxidant instead of iodosobenzene diacetate, yielding only 20% of the
desired oxidative cyclization product.116 We recognized that intermediate 2.5 reported in
the Kishi sporidesmin A synthesis could be intercepted from alcohol 4.4.
Scheme 4.17. Kishi’s final steps in the sporidesmin A synthesis.
A suitable N-Me alanine derivative needed to be prepared for the formal synthesis
of sporidesmin A. N-Fmoc-Ala-OH (4.44) was converted to the oxazolidinone (4.45),
then reduced to afford N-Me,Fmoc-Ala-OH (4.46, Scheme 4.18).157
Scheme 4.18. Preparation of N-Me,Fmoc-Ala-OH.
Alcohol 4.4 was acetylated by standard procedures to give 4.47. Amine 4.48 was
revealed following deprotection of the Cbz group, then coupled to N-Me alanine
NMe
Cl
MeOOMe
HNNMe
O
OMe
SS Ar
AcO
30% NMe
Cl
MeOOMe
N
25%
1. NaOH, MeOH2. mCPBA
H
AcOAcO
3. BF3!Et2O
(±)-sporidesmin A (1.11)
2.5 2.6
NMe
O
OMe
SS Ar
NMe
Cl
MeOOMe
NH
HOHO
NMe
O
OMe
SS
PhI(OAc)2
NHFmoc
Me CO2H
(HCHO)n, p-TsOH
PhCH3, reflux
CF3CO2H, Et3SiHOFmocN
MeO CHCl3
NMeFmoc
Me CO2H4.44 4.45 4.4655% (2 steps)

88
derivative 4.46 to afford dipeptide 4.49. This substrate, however, did not cyclize to the
dioxopiperazine when heated in morpholine and THF. Only 4.50, the product resulting
from Fmoc-deprotection and elimination of acetic acid, was recovered.
Scheme 4.19. Incorporation of N-Me-Ala.
4.4: Future Direction
Given more time, I would like to prepare bis(N,N’-Cbz) tryptophan derivatives
4.51 and 4.53 from indole 4.6 (Scheme 4.20). The bromocyclizations that were carried
out for the synthesis of chetomin were all performed on tryptophan derivatives bearing an
electron-withdrawing group on the indole nitrogen. Cyclization to the pyrroloindoline
should be more favorable with these substrates than for the N-Me derivatives. Experience
thus far with the oxidative cyclization reaction shows that a complex mixture of products
will likely be recovered from each reaction, so sufficient starting material will be required
to obtain adequate characterization of the reaction products.
OMeMeO
Cl
NMe
CO2MeHONHCbz
N-Fmoc,Me-Ala-OH,iPr2NEt, T3P morpholine
NMe
Cl
MeOOMe
HN
AcO
NMeFmocCO2Me
OMe THF N
Me
Cl
MeOOMe
HNNHMe
CO2Me
OMe
OMeMeO
Cl
NMe
CO2MeAcONHCbzAc2O H2, Pd/C
OMeMeO
Cl
NMe
CO2MeAcONH2
4.4 4.47 4.48
4.49 4.50
56%70%
MeOH
CH2Cl218%

89
Scheme 4.20. Full panel of oxidation conditions to screen.
The proposed installation of the epidithiodioxopiperazine ring may also be
problematic, especially considering the attempts made in the chetomin synthesis. Still, we
propose forming (–)-sporidesmin A (1.11) through oxidation of the disulfide prepared by
reaction of hemiaminal 4.1 with hydrogen sulfide (Scheme 4.21). The hemiaminal could
be prepared from dioxopiperazine 4.2 or a related derivative.
Scheme 4.21. Proposed epidithiodioxopiperazine formation.
4.5: Concluding Remarks
In summary, we have developed an asymmetric approach toward the synthesis of
(–)-sporidesmin A. Advanced dioxopiperazine 4.3 can be prepared in decent overall yield
and is theoretically three to four steps away from the target natural product. Moreover,
OMeMeO
Cl
NCbz
CO2MeTBSONHCbz
NCbz
Cl
MeOOMe
NCbzH
ROTBSO
CO2Me
NCbz
Cl
MeOOMe
HN
TBSONH
O
OMe
NCbz
Cl
MeOOMe
NH
ROTBSO
NH
O
OMe
conditions
conditions
conditions
a. PhI(OAc)2, Me2Sb. PhI(OAc)2, H2Oc. DMDOd. Davis's oxaziridinee. (PhCO2)2f. N-PSP; mCPBAg. Br2; AgCN, H2Oh. O2, methylene blue, sunlamp; Me2S
AcHHHCOPhHHH
R
4.51 4.52
4.53 4.54
NMe
Cl
MeOOMe
NH
TBSOTBSO
NMe
O
OMe
(!)-sporidesmin A (1.11)
NMe
Cl
MeOOMe
NH
HOHO
NMe
O
OMe
SS
NMe
Cl
MeOOMe
NH
TBSOTBSO
NMe
O
O
Me
HO
OH
thiolation oxidation
4.1 4.2

90
many of the synthetic advances made over the course of this synthesis were applied in
our approach to chetomin. Particularly, use of N-Fmoc amino acids as precursors to
dioxopiperazine rings eliminated a deptrotection step from each synthesis while
improving the cyclization yields. While it is difficult to leave this project behind
unfinished, I have no doubt that capable hands will pick it up in the future to achieve the
total synthesis of (–)-sporidesmin A.

91
REFERENCES
1. Cook, K. M.; Hilton, S. T.; Mecinovic, J.; Motherwell, W. B.; Figg, W. D.; Schofield, C. J. Epidithiodiketopiperazines Block the Interaction between Hypoxia-Inducible Factor-1alpha (Hif-1alpha) and P300 by a Zinc Ejection Mechanism. J. Biol. Chem. 2009, 284, 26831-26838.
2. Gardiner, D. M.; Waring, P.; Howlett, B. J. The Epipolythiodioxopiperazine (Etp) Class of Fungal Toxins: Distribution, Mode of Action, Functions and Biosynthesis. Microbiology 2005, 151, 1021-1032.
3. Waring, P.; Beaver, J. Gliotoxin and Related Epipolythiodioxopiperazines. Gen. Pharmacol. 1996, 27, 1311-1316.
4. Waring, P.; Eichner, R. D.; Mullbacher, A. The Chemistry and Biology of the Immunomodulating Agent Gliotoxin and Related Epipolythiodioxopiperazines. Med. Res. Rev. 1988, 8, 499-524.
5. Iwasa, E.; Hamashima, Y.; Sodeoka, M. Epipolythiodiketopiperazine Alkaloids: Total Syntheses and Biological Activities. Isr. J. Chem. 2011, 51, 420-433.
6. Weindling, R.; Emerson, O. H. The Isolation of a Toxic Substance from the Culture Filtrate of Trichoderma. Phytopathology 1936, 26, 1068-1070.
7. Bell, M. R.; Johnson, J. R.; Wildi, B. S.; Woodward, R. B. The Structure of Gliotoxin. J. Am. Chem. Soc. 1958, 80, 1001-1001.
8. Beecham, A. F.; Fridrichsons, J.; Mathieson, A. M. Structure and Absolute Configuration of Gliotoxin and Absolute Configuration of Sporidesmin. Tetrahedron Lett. 1966, 3131.
9. Jordan, T. W.; Cordiner, S. J. Fungal Epipolythiodioxopiperazine Toxins Have Therapeutic Potential and Roles in Disease. Trends Pharmacol. Sci. 1987, 8, 144-149.
10. Kwon-Chung, K. J.; Sugui, J. A. What Do We Know About the Role of Gliotoxin in the Pathobiology of Aspergillus Fumigatus? Med. Mycol. 2009, 47 Suppl 1, S97-103.
11. Mullbacher, A.; Eichner, R. D. Immunosuppression Invitro by a Metabolite of a Human Pathogenic Fungus. Proc. Natl. Acad. Sci., Biol. 1984, 81, 3835-3837.
12. Mullbacher, A.; Waring, P.; Eichner, R. D. Identification of an Agent in Cultures of Aspergillus-Fumigatus Displaying Anti-Phagocytic and Immunomodulating Activity Invitro. J. Gen. Microbiol. 1985, 131, 1251-1258.

92
13. Mullbacher, A.; Moreland, A. F.; Waring, P.; Sjaarda, A.; Eichner, R. D. Prevention of Graft-Versus-Host Disease by Treatment of Bone-Marrow with Gliotoxin in Fully Allogeneic Chimeras and Their Cyto-Toxic T-Cell Repertoire. Transplantation 1988, 46, 120-125.
14. Munday, R. Studies on the Mechanism of Toxicity of the Mycotoxin, Sporidesmin I. Generation of Superoxide Radical by Sporidesmin. Chem.-Biol. Interact. 1982, 41, 361-374.
15. Chai, C. L.; Waring, P. Redox Sensitive Epidithiodioxopiperazines in Biological Mechanisms of Toxicity. Redox Rep. 2000, 5, 257-264.
16. Eichner, R. D.; Waring, P.; Geue, A. M.; Braithwaite, A. W.; Mullbacher, A. Gliotoxin Causes Oxidative Damage to Plasmid and Cellular DNA. J. Biol. Chem. 1988, 263, 3772-3777.
17. Cordiner, S. J.; Jordan, T. W. Inhibition by Sporidesmin of Hepatocyte Bile Acid Transport. Biochem. J. 1983, 212, 197-204.
18. Mason, J. W.; Kidd, J. G. Effects of Gliotoxin and Other Sulfur-Containing Compounds on Tumor Cells in Vitro; with Observations on the Mechanism of Action of Gliotoxin. J. Immunol. 1951, 66, 99-106.
19. Hurne, A. M.; Chai, C. L.; Waring, P. Inactivation of Rabbit Muscle Creatine Kinase by Reversible Formation of an Internal Disulfide Bond Induced by the Fungal Toxin Gliotoxin. J. Biol. Chem. 2000, 275, 25202-25206.
20. Trown, P. W.; Bilello, J. A. Mechanism of Action of Gliotoxin: Elimination of Activity by Sulfhydryl Compounds. Antimicrob. Agents Chemother. 1972, 2, 261-266.
21. Mullbacher, A.; Waring, P.; Tiwari-Palni, U.; Eichner, R. D. Structural Relationship of Epipolythiodioxopiperazines and Their Immunomodulating Activity. Mol. Immunol. 1986, 23, 231-235.
22. Stillwell, M. A.; Magasi, L. P.; Strunz, G. M. Production, Isolation, and Antimicrobial Activity of Hyalodendrin, a New Antibiotic Produced by a Species of Hyalodendron. Can. J. Microbiol. 1974, 20, 759-764.
23. Strunz, G. M.; Heissner, C. J.; Kakushima, M.; Stillwell, M. A. Metabolites of Hyalodendron Sp - Bisdethiodi(Methylthio)Hyalodendrin. Can. J. Chem. 1974, 52, 325-326.
24. Strunz, G. M.; Kakushima, M.; Stillwell, M. A. Epitetrathiodioxopiperazine with 3s,6s Configuration from Hyalodendron Sp. Can. J. Chem. 1975, 53, 295-297.
25. DeVault, R. L.; Rosenbrook, W., Jr. A Novel Class of Diketopiperazines. J. Antibiot. 1973, 26, 532-534.

93
26. Dorn, F.; Arigoni, D. Gliovictin, New Metabolite of Helminthosporium-Victoriae. Experientia 1974, 30, 134-135.
27. Michel, K. H.; Chaney, M. O.; Jones, N. D.; Hoehn, M. M.; Nagarajan, R. Epipolythiopiperazinedione Antibiotics from Penicillium Turbatum. J. Antibiot. 1974, 27, 57-64.
28. Shin, J. H.; Fenical, W. Isolation of Gliovictin from the Marine Deuteromycete Asteromyces-Cruciatus. Phytochemistry 1987, 26, 3347.
29. Kawahara, N.; Nozawa, K.; Nakajima, S.; Kawai, K. Studies on Fungal Products 13. Isolation and Structures of Dithiosilvatin and Silvathione, Novel Dioxopiperazine Derivatives from Aspergillus-Silvaticus. J. Chem. Soc., Perkin Trans. 1 1987, 2099-2101.
30. Hanson, J. R.; Oleary, M. A. New Piperazinedione Metabolites of Gliocladium-Deliquescens. J. Chem. Soc., Perkin Trans. 1 1981, 218-220.
31. Nagarajan, R.; Huckstep, L. L.; Lively, D. H.; Delong, D. C.; Marsh, M. M.; Neuss, N. Aranotin and Related Metabolites from Arachniotus Aureus I. Determination of Structure. J. Am. Chem. Soc. 1968, 90, 2980.
32. Nagarajan, R.; Neuss, N.; Marsh, M. M. Aranotin and Related Metabolites 3. Configuration and Conformation of Acetylaranotin. J. Am. Chem. Soc. 1968, 90, 6518.
33. Neuss, N.; Boeck, L. D.; Brannon, D. R.; Cline, J. C.; DeLong, D. C.; Gorman, M.; Huckstep, L. L.; Lively, D. H.; Mabe, J.; Marsh, M. M.; Molloy, B. B.; Nagarajan, R.; Nelson, J. D.; Stark, W. M. Aranotin and Related Metabolites from Arachniotus Aureus (Eidam) Schroeter Iv. Fermentation, Isolation, Structure Elucidation, Biosynthesis, and Antiviral Properties. Antimicrob. Agents Chemother. 1968, 8, 213-219.
34. Neuss, N.; Nagarajan, R.; Molloy, B. B.; Huckstep, L. L. Aranotin and Related Metabolites 2. Isolation Characterization and Structures of 2 New Metabolites. Tetrahedron Lett. 1968, 4467.
35. Trown, P. W.; Lindh, H. F.; Milstrey, K. P.; Gallo, V. M.; Mayberry, B. R.; Lindsay, H. L.; Miller, P. A. Ll-S88-Alpha, an Antiviral Substance Produced by Aspergillus Terreus. Antimicrob. Agents Chemother. 1968, 8, 225-228.
36. Cosulich, D. B.; Nelson, N. R.; Van den Hende, J. H. Crystal and Molecular Structure of L-S88alpha, an Antiviral Epidithiapiperazinedione Derivative from Aspergillus Terreus. J. Am. Chem. Soc. 1968, 90, 6519.
37. Murdock, K. C. Antiviral Agents. Chemical Modifications of a Disulfide Antibiotic, Acetylaranotin. J. Med. Chem. 1974, 17, 827-835.

94
38. Kawahara, N.; Nakajima, S.; Yamazaki, M.; Kawai, K. Structure of a Novel Epidithiodioxopiperazine, Emethallicin a, a Potent Inhibitor of Histamine Release from Emericella Heterothallica. Chem. Pharm. Bull. 1989, 37, 2592-2595.
39. Kawahara, N.; Nozawa, K.; Yamazaki, M.; Nakajima, S.; Kawai, K. Structures of Novel Epipolythiodioxopiperazines, Emethallicins B, C, and D, Potent Inhibitors of Histamine Release, from Emericella Heterothallica. Chem. Pharm. Bull. 1990, 38, 73-78.
40. Kawahara, N.; Nozawa, K.; Nakajima, S.; Kawai, K.; Yamazaki, M. Novel Epitetrathiodioxopiperazines, Emethallicin-B and Emethallicin-C, as Potent Inhibitors of Compound 48/80-Induced Histamine-Release, from Emericella-Heterothallica. J. Chem. Soc., Chem. Commun. 1989, 951-952.
41. Kawahara, N.; Nozawa, K.; Yamazaki, M.; Nakajima, S.; Kawai, K. Studies on Fungal Products 32. Novel Epidithiodioxopiperazines, Emethallicin-E and Emethallicin-F, from Emericella-Heterothallica. Heterocycles 1990, 30, 507-515.
42. Waring, P.; Eichner, R. D.; Tiwaripalni, U.; Mullbacher, A. Gliotoxin-E - a New Biologically-Active Epipolythiodioxopiperazine Isolated from Penicillium-Terlikowskii. Aust. J. Chem. 1987, 40, 991-997.
43. Ueno, Y.; Umemori, K.; Niimi, E.; Tanuma, S.; Nagata, S.; Sugamata, M.; Ihara, T.; Sekijima, M.; Kawai, K.; Ueno, I.; et al. Induction of Apoptosis by T-2 Toxin and Other Natural Toxins in Hl-60 Human Promyelotic Leukemia Cells. Nat. Toxins 1995, 3, 129-137.
44. Seya, H.; Nozawa, K.; Nakajima, S.; Kawai, K. I.; Udagawa, S. I. Studies on Fungal Products 8. Isolation and Structure of Emestrin, a Novel Antifungal Macrocyclic Epidithiodioxopiperazine from Emericella-Striata - X-Ray Molecular-Structure of Emestrin. J. Chem. Soc., Perkin Trans. 1 1986, 109-116.
45. Seya, H.; Nakajima, S.; Kawai, K.; Udagawa, S. Structure and Absolute-Configuration of Emestrin, a New Macrocyclic Epidithiodioxopiperazine from Emericella-Striata. J. Chem. Soc., Chem. Commun. 1985, 657-658.
46. Kawai, K.; Nozawa, K.; Seya, H.; Kawahara, N.; Udagawa, S.; Nakajima, S. Studies on Fungal Products 12. Structure of Aurantioemestrin from Emericella-Striata. Heterocycles 1987, 26, 475-479.
47. Kawahara, N.; Nozawa, K.; Nakajima, S.; Kawai, K. Aurantioemestrin from Emericella-Striata and Silvathione from Aspergillus-Silvaticus, Possible Key Intermediates from Epidithiodioxopiperazines to Trioxopiperazines. J. Chem. Soc., Chem. Commun. 1986, 1495-1496.
48. Seya, H.; Nozawa, K.; Udagawa, S.; Nakajima, S.; Kawai, K. Studies on Fungal Products 9. Dethiosecoemestrin, a New Metabolite Related to Emestrin, from Emericella-Striata. Chem. Pharm. Bull. 1986, 34, 2411-2416.

95
49. Nozawa, K.; Udagawa, S. I.; Nakajima, S.; Kawai, K. I. Studies on Fungal Products 14. Emestrin-B, a New Epitrithiodioxopiperazine, from Emericella-Striata. Chem. Pharm. Bull. 1987, 35, 3460-3463.
50. Onodera, H.; Hasegawa, A.; Tsumagari, N.; Nakai, R.; Ogawa, T.; Kanda, Y. Mpc1001 and Its Analogues: New Antitumor Agents from the Fungus Cladorrhinum Species. Org. Lett. 2004, 6, 4101-4104.
51. Deffieux, G.; Gadret, M.; Leger, J. M.; Carpy, A. Crystal Structure of Original Fungal Metabolite of 3,6-Epidithio-2,5-Dioxopiperazine Group - Epicorazine-A. Acta Crystallogr. B. 1977, 33, 1474-1478.
52. Deffieux, G.; Baute, M. A.; Baute, R.; Filleau, M. J. New Antibiotics from Fungus Epicoccum-Nigrum 2. Epicorazine-a - Structure Elucidation and Absolute Configuration. J. Antibiot. 1978, 31, 1102-1105.
53. Deffieux, G.; Filleau, M. J.; Baute, R. New Antibiotics from Fungus Epicoccum-Nigrum 3. Epicorazine-B - Structure Elucidation and Absolute Configuration. J. Antibiot. 1978, 31, 1106-1109.
54. Brown, A. E.; Finlay, R.; Ward, J. S. Antifungal Compounds Produced by Epicoccum-Purpurascens against Soil-Borne Plant Pathogenic Fungi. Soil Biol. Biochem. 1987, 19, 657-664.
55. Kleinwachter, P.; Dahse, H. M.; Luhmann, U.; Schlegel, B.; Dornberger, K. Epicorazine C, an Antimicrobial Metabolite from Stereum Hirsutum Hki 0195. J. Antibiot. 2001, 54, 521-525.
56. Begg, W. R.; Elix, J. A.; Jones, A. J. Nonacyclic Amides from Lichens of Genus Xanthoparmelia. Tetrahedron Lett. 1978, 1047-1050.
57. Ernst-Russell, M. A.; Chai, C. L. L.; Hurne, A. M.; Waring, P.; Hockless, D. C. R.; Elix, J. A. Structure Revision and Cytotoxic Activity of the Scabrosin Esters, Epidithiopiperazinediones from the Lichen Xanthoparmelia Scabrosa. Aust. J. Chem. 1999, 52, 279-283.
58. Moerman, K. L.; Chai, C. L. L.; Waring, P. Evidence That the Lichen-Derived Scabrosin Esters Target Mitochondrial Atp Synthase in P388d1 Cells. Toxicol. Appl. Pharmacol. 2003, 190, 232-240.
59. Curtis, P. J.; Greatbanks, D.; Hesp, B.; Cameron, A. F.; Freer, A. A. Sirodesmins a, B, C, and G, Antiviral Epipolythiopiperazine-2,5-Diones of Fungal Origin - X-Ray Analysis of Sirodesmin-a Diacetate. J. Chem. Soc., Perkin Trans. 1 1977, 180-189.
60. Ferezou, J. P.; Riche, C.; Quesneauthierry, A.; Pascardbilly, C.; Barbier, M.; Bousquet, J. F.; Boudart, G. Structures of 2 Toxins Isolated from Fungus Cultures

96
Phoma-Lingam Tode - Sirodesmin Pl and Deacetylsirodesmin Pl. Nouv. J. Chim. 1977, 1, 327-334.
61. Boudart, G. Antibacterial Activity of Sirodesmin Pl Phytotoxin: Application to the Selection of Phytotoxin-Deficient Mutants. Appl. Environ. Microbiol. 1989, 55, 1555-1559.
62. Elliott, C. E.; Gardiner, D. M.; Thomas, G.; Cozijnsen, A.; Van De Wouw, A.; Howlett, B. J. Production of the Toxin Sirodesmin Pl by Leptosphaeria Maculans During Infection of Brassica Napus. Mol. Plant Pathol. 2007, 8, 791-802.
63. Thornton, R. H.; Sinclair, D. P. Sporidesmium Bakeri and Facial Eczema of Sheep in the Field. Nature 1959, 184, 1327-1328.
64. Thornton, R. H.; Percival, J. C. A Hepatotoxin from Sporidesmium Bakeri Capable of Producing Facial Eczema Diseases in Sheep. Nature 1959, 183, 63.
65. Synge, R. L. M.; White, E. P. Sporidesmin - a Substance from Sporidesmium-Bakeri Causing Lesions Characteristic of Facial Eczema. Chem. Ind. 1959, 1546-1547.
66. Fridrichsons, J.; Mathieson, A. M. Structure of Methylene Dibromide Adduct of Sporidesmin at - 150 Degrees C. Acta Crystallogr. 1965, 18, 1043.
67. Fridrichsons, J.; Mathieson, A. M. The Structure of Sporidesmin - Causative Agent of Facial Eczema in Sheep. Tetrahedron Lett. 1962, 1265-1268.
68. Smith, B. L.; Embling, P. P.; Towers, N. R.; Wright, D. E.; Payne, E. The Protective Effect of Zinc Sulphate in Experimental Sporidesmin Poisoning of Sheep. New Zealand veterinary journal 1977, 25, 124-127.
69. Waring, P.; Egan, M.; Braithwaite, A.; Mullbacher, A.; Sjaarda, A. Apoptosis Induced in Macrophages and T Blasts by the Mycotoxin Sporidesmin and Protection by Zn2+ Salts. Int. J. Immunopharmacol. 1990, 12, 445-457.
70. Munday, R. Studies on the Mechanism of Toxicity of the Mycotoxin Sporidesmin 3. Inhibition by Metals of the Generation of Superoxide Radical by Sporidesmin. J. Appl. Toxicol. 1984, 4, 182-186.
71. Hodges, R.; Shannon, J. S. Isolation and Structure of Sporidesmin C. Aust. J. Chem. 1966, 19, 1059.
72. Waksman, S. A.; Bugie, E. Chaetomin, a New Antibiotic Substance Produced by Chaetomium Cochliodes I. Formation and Properties. J. Bacteriol. 1944, 48, 527-530.
73. Mcinnes, A. G.; Taylor, A.; Walter, J. A. Structure of Chetomin. J. Am. Chem. Soc. 1976, 98, 6741-6741.

97
74. Li, G. Y.; Li, B. G.; Yang, T.; Yan, J. F.; Liu, G. Y.; Zhang, G. L. Chaetocochins a-C, Epipolythiodioxopiperazines from Chaetomium Cochliodes. J. Nat. Prod. 2006, 69, 1374-1376.
75. Fujimoto, H.; Sumino, M.; Okuyama, E.; Ishibashi, M. Immunomodulatory Constituents from an Ascomycete, Chaetomium Seminudum. J. Nat. Prod. 2004, 67, 98-102.
76. Kung, A. L.; Zabludoff, S. D.; France, D. S.; Freedman, S. J.; Tanner, E. A.; Vieira, A.; Cornell-Kennon, S.; Lee, J.; Wang, B.; Wang, J.; Memmert, K.; Naegeli, H. U.; Petersen, F.; Eck, M. J.; Bair, K. W.; Wood, A. W.; Livingston, D. M. Small Molecule Blockade of Transcriptional Coactivation of the Hypoxia-Inducible Factor Pathway. Cancer Cell 2004, 6, 33-43.
77. Staab, A.; Loeffler, J.; Said, H. M.; Diehlmann, D.; Katzer, A.; Beyer, M.; Fleischer, M.; Schwab, F.; Baier, K.; Einsele, H.; Flentje, M.; Vordermark, D. Effects of Hif-1 Inhibition by Chetomin on Hypoxia-Related Transcription and Radiosensitivity in Ht 1080 Human Fibrosarcoma Cells. BMC Cancer 2007, 7, 213.
78. Mohammed, K. A.; Jadulco, R. C.; Bugni, T. S.; Harper, M. K.; Sturdy, M.; Ireland, C. M. Strongylophorines: Natural Product Inhibitors of Hypoxia-Inducible Factor-1 Transcriptional Pathway. J. Med. Chem. 2008, 51, 1402-1405.
79. Melillo, G. Hypoxia-Inducible Factor 1 Inhibitors. Methods Enzymol. 2007, 435, 385-402.
80. Block, K. M.; Wang, H.; Szabo, L. Z.; Polaske, N. W.; Henchey, L. K.; Dubey, R.; Kushal, S.; Laszlo, C. F.; Makhoul, J.; Song, Z.; Meuillet, E. J.; Olenyuk, B. Z. Direct Inhibition of Hypoxia-Inducible Transcription Factor Complex with Designed Dimeric Epidithiodiketopiperazine. J. Am. Chem. Soc. 2009, 131, 18078-18088.
81. Hauser, D.; Weber, H. P.; Sigg, H. P. Isolation and Configuration of Chaetocin. Helv. Chim. Acta 1970, 53, 1061-1073.
82. Weber, H. P. Molecular Structure and Absolute Configuration of Chaetocin. Acta Crystallogr. B. 1972, B 28, 2945.
83. Saito, T.; Suzuki, Y.; Koyama, K.; Natori, S.; Iitaka, Y.; Kinoshita, T. Chetracin-a and Chaetocin-B and Chaetocin-C, 3 New Epipolythiodioxopiperazines from Chaetomium Spp. Chem. Pharm. Bull. 1988, 36, 1942-1956.
84. Li, L.; Li, D.; Luan, Y.; Gu, Q.; Zhu, T. Cytotoxic Metabolites from the Antarctic Psychrophilic Fungus Oidiodendron Truncatum. J. Nat. Prod. 2012, 75, 920-927.

98
85. Greiner, D.; Bonaldi, T.; Eskeland, R.; Roemer, E.; Imhof, A. Identification of a Specific Inhibitor of the Histone Methyltransferase Su(Var)3-9. Nat. Chem. Biol. 2005, 1, 143-145.
86. Feng, Y.; Blunt, J. W.; Cole, A. L.; Munro, M. H. Novel Cytotoxic Thiodiketopiperazine Derivatives from a Tilachlidium Sp. J. Nat. Prod. 2004, 67, 2090-2092.
87. Minato, H.; Matsumoto, M.; Katayama, T. Studies on the Metabolites of Verticillium Sp. Structures of Verticillins a, B, and C. J. Chem. Soc., Perkin Trans. 1 1973, 17, 1819-1825.
88. Joshi, B. K.; Gloer, J. B.; Wicklow, D. T. New Verticillin and Glisoprenin Analogues from Gliocladium Catenulatum, a Mycoparasite of Aspergillus Flavus Sclerotia. J. Nat. Prod. 1999, 62, 730-733.
89. Chu, M.; Truumees, I.; Rothofsky, M. L.; Patel, M. G.; Gentile, F.; Das, P. R.; Puar, M. S.; Lin, S. L. Inhibition of C-Fos Proto-Oncogene Induction by Sch 52900 and Sch 52901, Novel Diketopiperazine Produced by Gliocladium Sp. J. Antibiot. 1995, 48, 1440-1445.
90. Dong, J. Y.; He, H. P.; Shen, Y. M.; Zhang, K. Q. Nematicidal Epipolysulfanyldioxopiperazines from Gliocladium Roseum. J. Nat. Prod. 2005, 68, 1510-1513.
91. Zheng, C. J.; Kim, C. J.; Bae, K. S.; Kim, Y. H.; Kim, W. G. Bionectins a-C, Epidithiodioxopiperazines with Anti-Mrsa Activity, from Bionectra Byssicola F120. J. Nat. Prod. 2006, 69, 1816-1819.
92. Takahashi, C.; Minoura, K.; Yamada, T.; Numata, A.; Kushida, K.; Shingu, T.; Hagishita, S.; Nakai, H.; Sato, T.; Harada, H. Potent Cytotoxic Metabolites from a Leptosphaeria Species - Structure Determination and Conformational-Analysis. Tetrahedron 1995, 51, 3483-3498.
93. Takahashi, C.; Numata, A.; Ito, Y.; Matsumura, E.; Araki, H.; Iwaki, H.; Kushida, K. Leptosins, Antitumor Metabolites of a Fungus Isolated from a Marine Alga. J. Chem. Soc., Perkin Trans. 1 1994, 1859-1864.
94. Takahashi, C.; Numata, A.; Matsumura, E.; Minoura, K.; Eto, H.; Shingu, T.; Ito, T.; Hasegawa, T. Leptosins I and J, Cytotoxic Substances Produced by a Leptosphaeria Sp. Physico-Chemical Properties and Structures. J. Antibiot. 1994, 47, 1242-1249.
95. Winstead, J. A.; Suhadolnik, R. J. Biosynthesis of Gliotoxin 2. Further Studies on the Incorporation of Carbon-14 and Tritium-Labeled Precursors. J. Am. Chem. Soc. 1960, 82, 1644-1647.

99
96. Suhadolnik, R. J.; Chenoweth, R. G. Biosynthesis of Gliotoxin 1. Incorporation of Phenylalanine-1-C-14 and Phenylalanine-2-C-14. J. Am. Chem. Soc. 1958, 80, 4391-4392.
97. Balibar, C. J.; Walsh, C. T. Glip, a Multimodular Nonribosomal Peptide Synthetase in Aspergillus Fumigatus, Makes the Diketopiperazine Scaffold of Gliotoxin. Biochemistry 2006, 45, 15029-15038.
98. Bulock, J. D.; Leigh, C. Biosynthesis of Gliotoxin. J. Chem. Soc., Chem. Commun. 1975, 628-629.
99. Kirby, G. W.; Patrick, G. L.; Robins, D. J. Cyclo-(L-Phenylalanyl-L-Seryl) as an Intermediate in Biosynthesis of Gliotoxin. J. Chem. Soc., Perkin Trans. 1 1978, 1336-1338.
100. Macdonald, J. C.; Slater, G. P. Biosynthesis of Gliotoxin and Mycelianamide. Can. J. Biochem. 1975, 53, 475-478.
101. Kirby, G. W.; Rao, G. V.; Robins, D. J. New Co-Metabolites of Gliotoxin in Gliocladium-Virens. J. Chem. Soc., Perkin Trans. 1 1988, 301-304.
102. Davis, C.; Carberry, S.; Schrettl, M.; Singh, I.; Stephens, J. C.; Barry, S. M.; Kavanagh, K.; Challis, G. L.; Brougham, D.; Doyle, S. The Role of Glutathione S-Transferase Glig in Gliotoxin Biosynthesis in Aspergillus Fumigatus. Chem. Biol. 2011, 18, 542-552.
103. Scharf, D. H.; Remme, N.; Habel, A.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Hortschansky, P.; Brakhage, A. A.; Hertweck, C. A Dedicated Glutathione S-Transferase Mediates Carbon-Sulfur Bond Formation in Gliotoxin Biosynthesis. J. Am. Chem. Soc. 2011, 133, 12322-12325.
104. Scharf, D. H.; Heinekamp, T.; Remme, N.; Hortschansky, P.; Brakhage, A. A.; Hertweck, C. Biosynthesis and Function of Gliotoxin in Aspergillus Fumigatus. Appl. Microbiol. Biotechnol. 2012, 93, 467-472.
105. Scharf, D. H.; Remme, N.; Heinekamp, T.; Hortschansky, P.; Brakhage, A. A.; Hertweck, C. Transannular Disulfide Formation in Gliotoxin Biosynthesis and Its Role in Self-Resistance of the Human Pathogen Aspergillus Fumigatus. J. Am. Chem. Soc. 2010, 132, 10136-10141.
106. Schrettl, M.; Carberry, S.; Kavanagh, K.; Haas, H.; Jones, G. W.; O'Brien, J.; Nolan, A.; Stephens, J.; Fenelon, O.; Doyle, S. Self-Protection against Gliotoxin--a Component of the Gliotoxin Biosynthetic Cluster, Glit, Completely Protects Aspergillus Fumigatus against Exogenous Gliotoxin. PLoS Pathog. 2010, 6, e1000952.

100
107. Gardiner, D. M.; Cozijnsen, A. J.; Wilson, L. M.; Pedras, M. S.; Howlett, B. J. The Sirodesmin Biosynthetic Gene Cluster of the Plant Pathogenic Fungus Leptosphaeria Maculans. Mol. Microbiol. 2004, 53, 1307-1318.
108. Gardiner, D. M.; Howlett, B. J. Bioinformatic and Expression Analysis of the Putative Gliotoxin Biosynthetic Gene Cluster of Aspergillus Fumigatus. FEMS Microbiol. Lett. 2005, 248, 241-248.
109. Fox, E. M.; Howlett, B. J. Biosynthetic Gene Clusters for Epipolythiodioxopiperazines in Filamentous Fungi. Mycol. Res. 2008, 112, 162-169.
110. Ferezou, J. P.; Quesneauthierry, A.; Barbier, M.; Kollmann, A.; Bousquet, J. F. Structure and Synthesis of Phomamide, a New Piperazine-2,5-Dione Related to the Sirodesmins, Isolated from the Culture-Medium of Phoma-Lingam Tode. J. Chem. Soc., Perkin Trans. 1 1980, 113-115.
111. Bulock, J. D.; Clough, L. E. Sirodesmin Biosynthesis. Aust. J. Chem. 1992, 45, 39-45.
112. Kremer, A.; Li, S. M. A Tyrosine O-Prenyltransferase Catalyses the First Pathway-Specific Step in the Biosynthesis of Sirodesmin Pl. Microbiology 2010, 156, 278-286.
113. Kishi, Y.; Nakatsuka, S.; Fukuyama, T.; Havel, M. Total Synthesis of Sporidesmin-A. J. Am. Chem. Soc. 1973, 95, 6493-6495.
114. Kishi, Y.; Fukuyama, T.; Nakatsuka, S. Total Synthesis of Dehydrogliotoxin. J. Am. Chem. Soc. 1973, 95, 6492-6493.
115. Kishi, Y.; Fukuyama, T.; Nakatsuka, S. New Method for Synthesis of Epidithiodiketopiperazines. J. Am. Chem. Soc. 1973, 95, 6490-6492.
116. Nakatsuka, S.; Fukuyama, T.; Kishi, Y. Total Synthesis of D,L-Sporidesmin B. Tetrahedron Lett. 1974, 1549-1552.
117. Fukuyama, T.; Kishi, Y. Total Synthesis of Gliotoxin. J. Am. Chem. Soc. 1976, 98, 6723-6724.
118. Fukuyama, T.; Nakatsuka, S.; Kishi, Y. Total Synthesis of Gliotoxin, Dehydrogliotoxin and Hyalodendrin. Tetrahedron 1981, 37, 2045-2078.
119. Strunz, G. M.; Kakushima, M. Total Synthesis of (+/-) Hyalodendrin. Experientia 1974, 30, 719-720.
120. Williams, R. M.; Rastetter, W. H. Syntheses of the Fungal Metabolites (+/-)-Gliovictin and (+/-)-Hyalodendrin. J. Org. Chem. 1980, 45, 2625-2631.

101
121. Williams, R. M.; Rastetter, W. H. Efficient Synthesis of D,L-Gliovictin - Construction of the Hydroxymethyl Moiety Via a 3-Formyl-2,5-Piperazinedione. Tetrahedron Lett. 1979, 1187-1190.
122. Kim, J.; Ashenhurst, J. A.; Movassaghi, M. Total Synthesis of (+)-11,11'-Dideoxyverticillin A. Science 2009, 324, 238-241.
123. Iwasa, E.; Hamashima, Y.; Fujishiro, S.; Higuchi, E.; Ito, A.; Yoshida, M.; Sodeoka, M. Total Synthesis of (+)-Chaetocin and Its Analogues: Their Histone Methyltransferase G9a Inhibitory Activity. J. Am. Chem. Soc. 2010, 132, 4078-4079.
124. Iwasa, E.; Hamashima, Y.; Fujishiro, S.; Hashizume, D.; Sodeoka, M. Total Syntheses of Chaetocin and Ent-Chaetocin. Tetrahedron 2011, 67, 6587-6599.
125. Kim, J.; Movassaghi, M. General Approach to Epipolythiodiketopiperazine Alkaloids: Total Synthesis of (+)-Chaetocins a and C and (+)-12,12'-Dideoxychetracin A. J. Am. Chem. Soc. 2010, 132, 14376-14378.
126. DeLorbe, J. E.; Jabri, S. Y.; Mennen, S. M.; Overman, L. E.; Zhang, F. L. Enantioselective Total Synthesis of (+)-Gliocladine C: Convergent Construction of Cyclotryptamine-Fused Polyoxopiperazines and a General Approach for Preparing Epidithiodioxopiperazines from Trioxopiperazine Precursors. J. Am. Chem. Soc. 2011, 133, 6549-6552.
127. Movassaghi, M.; Boyer, N. Concise Total Synthesis of (+)-Gliocladins B and C. Chem. Sci. 2012, 3, 1798-1803.
128. Nicolaou, K. C.; Totokotsopoulos, S.; Giguere, D.; Sun, Y. P.; Sarlah, D. Total Synthesis of Epicoccin G. J. Am. Chem. Soc. 2011, 133, 8150-8153.
129. Codelli, J. A.; Puchlopek, A. L. A.; Reisman, S. E. Enantioselective Total Synthesis of (-)-Acetylaranotin, a Dihydrooxepine Epidithiodiketopiperazine. J. Am. Chem. Soc. 2012, 134, 1930-1933.
130. Newhouse, T.; Lewis, C. A.; Eastman, K. J.; Baran, P. S. Scalable Total Syntheses of N-Linked Tryptamine Dimers by Direct Indole-Aniline Coupling: Psychotrimine and Kapakahines B and F. J. Am. Chem. Soc. 2010, 132, 7119-7137.
131. Newhouse, T.; Baran, P. S. Total Synthesis of (+/-)-Psychotrimine. J. Am. Chem. Soc. 2008, 130, 10886-10887.
132. Newhouse, T.; Lewis, C. A.; Baran, P. S. Enantiospecific Total Syntheses of Kapakahines B and F. J. Am. Chem. Soc. 2009, 131, 6360-6361.
133. Espejo, V. R.; Rainier, J. D. An Expeditious Synthesis of C(3)-N(1') Heterodimeric Indolines. J. Am. Chem. Soc. 2008, 130, 12894-12895.

102
134. Espejo, V. R.; Rainier, J. D. Total Synthesis of Kapakahine E and F. Org. Lett. 2010, 12, 2154-2157.
135. Perez-Balado, C.; de Lera, A. R. Concise Total Synthesis and Structural Revision of (+)-Pestalazine B. Org. Biomol. Chem. 2010, 8, 5179-5186.
136. Greshock, T. J.; Williams, R. M. Improved Biomimetic Total Synthesis of D,L-Stephacidin A. Org. Lett. 2007, 9, 4255-4258.
137. Movassaghi, M.; Schmidt, M. A.; Ashenhurst, J. A. Stereoselective Oxidative Rearrangement of 2-Aryl Tryptamine Derivatives. Org. Lett. 2008, 10, 4009-4012.
138. Poriel, C.; Lachia, M.; Wilson, C.; Davies, J. R.; Moody, C. J. Oxidative Rearrangement of Indoles: A New Approach to the Efhg-Tetracyclic Core of Diazonamide A. J. Org. Chem. 2007, 72, 2978-2987.
139. Yao, P. Y.; Zhang, Y.; Hsung, R. P.; Zhao, K. A Sequential Metal-Catalyzed C-N Bond Formation in the Synthesis of 2-Amido-Indoles. Org. Lett. 2008, 10, 4275-4278.
140. Istrate, F. M.; Buzas, A. K.; Jurberg, I. D.; Odabachian, Y.; Gagosz, F. Synthesis of Functionalized Oxazolones by a Sequence of Cu(Ii)- and Au(I)-Catalyzed Transformations. Org. Lett. 2008, 10, 925-928.
141. Ohno, M.; Spande, T. F.; Witkop, B. Cyclization of Tryptophan and Tryptamine Derivatives to 2,3-Dihydropyrrolo[2,3-B]Indoles. J. Am. Chem. Soc. 1970, 92, 343-348.
142. Fuchs, J. R.; Funk, R. L. Total Synthesis of (+/-)-Perophoramidine. J. Am. Chem. Soc. 2004, 126, 5068-5069.
143. Yamada, Y.; Arima, S.; Okada, C.; Akiba, A.; Kai, T.; Harigaya, Y. Preparation of 7-Halo-Indoles by Thallation of N-Formylindoline and Their Attempted Use for Synthesis of the Right-Hand Segment of Chloropeptin. Chem. Pharm. Bull. 2006, 54, 788-794.
144. Lopez, C. S.; Perez-Balado, C.; Rodriguez-Grana, P.; de Lera, A. R. Mechanistic Insights into the Stereocontrolled Synthesis of Hexahydropyrrolo[2,3-B]Indoles by Electrophilic Activation of Tryptophan Derivatives. Org. Lett. 2008, 10, 77-80.
145. Welch, T. R.; Williams, R. M. Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids. In Biomimetic Organic Synthesis, Poupon, E.; Nay, B., Eds. Wiley-VCH: 2011, 117-148.
146. Di Menna, M. E.; Smith, B. L.; Miles, C. O. A History of Facial Eczema (Pithomycotoxicosis) Research. New Zeal. J. Agr. Res. 2009, 52, 345-376.

103
147. Rege, P. D.; Tian, Y.; Corey, E. J. Studies of New Indole Alkaloid Coupling Methods for the Synthesis of Haplophytine. Org. Lett. 2006, 8, 3117-3120.
148. Ojo, B.; Findsen, L. A.; Igarashi, N.; Kong, B.; Chowdhury, B. K. Synthesis and Bronchodilatory Activity of Four New Derivatives of Deoxyvasicine. Drug Des. Discovery 1996, 14, 1-14.
149. Blunt, J. W.; Erasmuson, A. F.; Ferrier, R. J.; Munro, M. H. G. Syntheses of Haptens Related to the Benzenoid and Indole Portions of Sporidesmin-a - C-13 Nmr-Spectra of Indole-Derivatives. Aust. J. Chem. 1979, 32, 1045-1054.
150. Ishikawa, H.; Elliott, G. I.; Velcicky, J.; Choi, Y.; Boger, D. L. Synthesis of (-)- and Ent-(+)-Vindoline and Related Alkaloids. J. Am. Chem. Soc. 2006, 128, 10596-10612.
151. Koketsu, K.; Oguri, H.; Watanabe, K.; Oikawa, H. Identification and Stereochemical Assignment of the Beta-Hydroxytryptophan Intermediate in the Echinomycin Biosynthetic Pathway. Org. Lett. 2006, 8, 4719-4722.
152. Becker, H.; Sharpless, K. B. A New Ligand Class for the Asymmetric Dihydroxylation of Olefins. Angew. Chem., Int. Ed. 1996, 35, 448-451.
153. Ritchie, R.; Saxton, J. E. Studies on Indolic Mold Metabolites - Total Synthesis of L-Prolyl-2-Methyltryptophan Anhydride and Deoxybrevianamide-E. Tetrahedron 1981, 37, 4295-4303.
154. Zhao, L.; May, J. P.; Huang, J.; Perrin, D. M. Stereoselective Synthesis of Brevianamide E. Org. Lett. 2012, 14, 90-93.
155. Schkeryantz, J. M.; Woo, J. C. G.; Siliphaivanh, P.; Depew, K. M.; Danishefsky, S. J. Total Synthesis of Gypsetin, Deoxybrevianamide E, Brevianamide E, and Tryprostatin B: Novel Constructions of 2,3-Disubstituted Indoles. J. Am. Chem. Soc. 1999, 121, 11964-11975.
156. Greshock, T. J.; Grubbs, A. W.; Williams, R. M. Concise, Biomimetic Total Synthesis of D,L-Marcfortine C. Tetrahedron 2007, 63, 6124-6130.
157. Luo, Y.; Evindar, G.; Fishlock, D.; Lajoie, G. A. Synthesis of N-Protected N-Methyl Serine and Threonine. Tetrahedron Lett. 2001, 42, 3807-3809.

104
CHAPTER 5
Experimental Procedures
Unless otherwise noted, all reagents were obtained from commercial suppliers
and were used without further purification. All air or moisture sensitive reactions were
performed under a positive pressured of argon in flame-dried glassware. Tetrahydrofuran
(THF), toluene, diethyl ether (Et2O), dichloromethane, benzene (PhH), acetonitrile
(MeCN), triethylamine (Et3N), pyridine, diisopropyl amine, methanol (MeOH),
dimethylsulfoxide (DMSO), and N,N-dimethylformamide (DMF) were obtained from a
dry solvent system (Ar degassed solvents delivered through activated alumina columns,
positive pressure of argon). Column chromatography was performed on Merck silica gel
Kieselgel 60 (230-400 mesh). Melting points were determined in open-end capillary
tubes and are uncorrected. 1HNMR and 13CNMR spectra were recorded on Varian 300, or
400 MHz spectrometers. Chemical shifts are reported in ppm relative to CHCl3 at δ 7.27
(1HNMR) and δ 77.23 (13CNMR). Mass spectra were obtained on Fisons VG Autospec.
IR spectra were obtained from thin films on a NaCl plate using a Bruker Tensor 27 FT-IR
spectrometer. Optical rotations were collected at 589 nm on a Rudolph Research
automatic polarimeter Autopol III.

105
Tert-butyl 2,3-dioxoindoline-1-carboxylate (3.5). To a solution of isatin (4.0 g, 27.2
mmol) in THF (100 mL) at 0°C was added Boc2O (7.1 g, 32.6 mmol) and DMAP (195
mg, 1.6 mmol). The resulting mixture was stirred overnight at r.t. under Ar, then
concentrated under reduced pressure. The product was recrystallized in EtOAc/hexanes
to afford 3.5 (4.7 g, 70% yield).
1H-NMR (300 MHz; DMSO-d6): δ 7.92 (d, J = 8.2 Hz, 1H), 7.76-7.65 (m, 2H), 7.29 (t, J
= 7.5 Hz, 1H), 1.56 (s, 9H).
REF: TRW-I-175, TRW-I-179.
NBoc
O
O
3.5
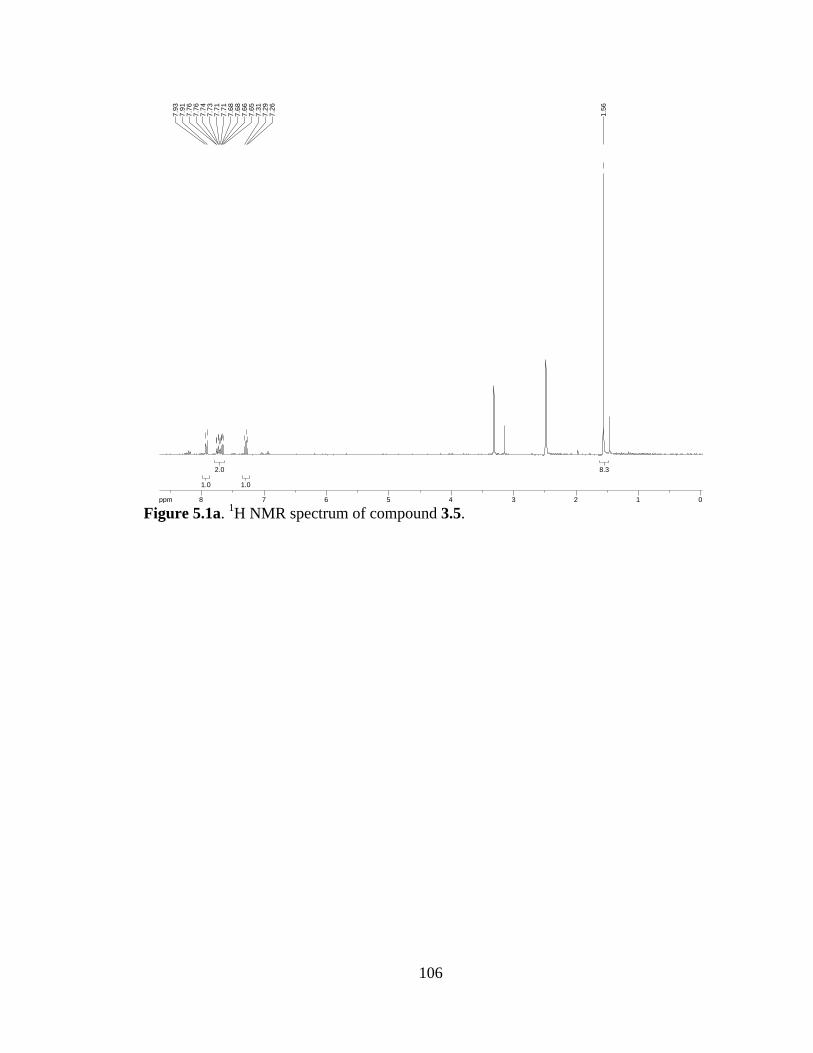
106
Figure 5.1a. 1H NMR spectrum of compound 3.5.
8.38.3
1.01.0
2.02.0
1.01.0
ppm0 01122334455667788-19.97
1.56
7.26
7.29
7.31
7.65
7.66
7.68
7.68
7.71
7.71
7.73
7.74
7.76
7.76
7.91
7.93

107
Boc-Hemiaminal (3.6). To a flame-dried RBF was added 3.5 (500 mg, 2.02 mmol) and
toluene (20 mL). Indoline (134 µL, 1.2 mmol), K2CO3 (372 mg, 2.7 mmol), and 4 Å M.S.
(5 g) were subsequently added, and the resulting suspension heated to reflux for 12 h
under Ar. The reaction mixture was filtered through celite and the fitrate concentrated
under reduced pressure to afford 419 mg of 3.6 as a tan solid (100% yield) that was taken
on without further purification.
1H-NMR (400 MHz; CDCl3): δ 10.59 (s, 1H), 8.55 (d, J = 8.7 Hz, 1H), 8.26 (d, J = 8.1
Hz, 1H), 7.74 (dd, J = 8.1, 1.4 Hz, 1H), 7.57 (dd, J = 8.6, 7.2 Hz, 1H), 7.24-7.20 (m, 1H),
7.13-7.11 (m, 1H), 7.03-6.99 (m, 1H), 3.97 (t, J = 8.4 Hz, 2H), 3.15 (t, J = 8.3 Hz, 2H),
1.54 (s, 9H); 13C-NMR (101 MHz; CDCl3): δ 193.7, 163.4, 152.9, 144.1, 141.7, 137.0,
133.9, 132.0, 129.2, 128.0, 125.5, 121.7, 119.3, 117.7, 81.4, 48.1, 28.49, 28.42.
REF: TRW-I-180, TRW-I-185, TRW-I-201.
NBoc
O
NHO
3.6
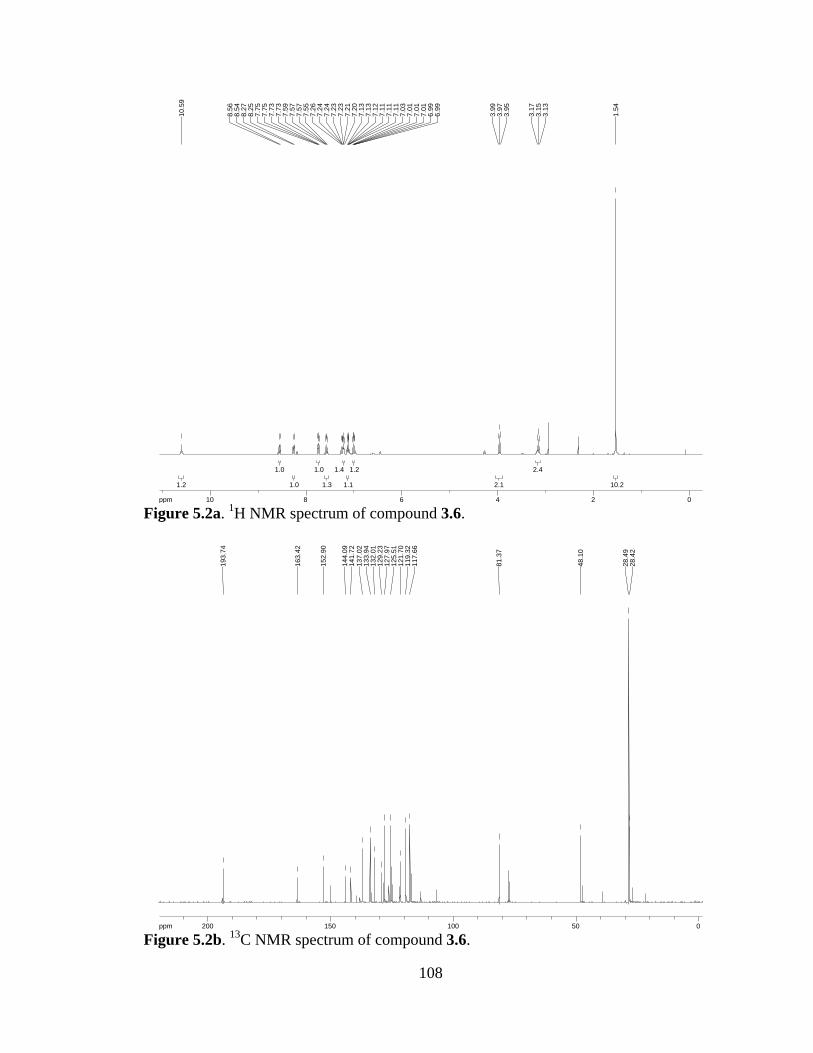
108
Figure 5.2a. 1H NMR spectrum of compound 3.6.
Figure 5.2b. 13C NMR spectrum of compound 3.6.
10.210.2
2.42.4
2.12.1
1.21.2
1.11.1
1.41.4
1.31.3
1.01.0
1.01.0
1.01.0
1.21.2
ppm0 0224466881010-17.45
1.54
3.13
3.15
3.17
3.95
3.97
3.99
6.99
6.99
7.01
7.01
7.01
7.03
7.11
7.11
7.11
7.12
7.13
7.13
7.20
7.21
7.23
7.23
7.24
7.24
7.26
7.55
7.57
7.57
7.59
7.73
7.73
7.75
7.75
8.25
8.27
8.54
8.56
10.5
9
ppm0 05050100100150150200200-298.35
28.4
228
.49
48.1
0
81.3
7
117.
6611
9.32
121.
7012
5.51
127.
9712
9.23
132.
0113
3.94
137.
0214
1.72
144.
09
152.
90
163.
42
193.
74

109
1-(2,2-dibromovinyl)-2-nitrobenzene (3.15). 2-nitrobenzaldehyde (3.14, 2.0 g, 13.2
mmol) and CBr4 (6.57 g, 19.8 mmol) were dissolved in CH2Cl2 (100 mL). The resulting
solution was cooled to 0 °C and a solution of PPh3 (10.38 g, 39.6 mmol) in CH2Cl2 (80
mL) slowly added. The reaction was stirred an additional 30 minutes at 0 °C, then
concentrated under reduced pressure. The residue was taken up in chloroform (30 mL),
filtered, and the solid washed with chloroform (2 x 30 mL). The filtrate was concentrated,
then purified by SiO2 chromatography (8:1 hexanes:EtOAc) to afford 3.15 (3.69 g, 91%
yield).
1H-NMR (300 MHz; CDCl3): δ 8.12 (dd, J = 8.2, 1.3 Hz, 1H), 7.78 (s, 1H), 7.71-7.65
(m, 1H), 7.60-7.51 (m, 2H).
REF: TRW-I-286, TRW-I-337, TRW-I-363.
Br
BrNO2
3.15

110
Figure 5.3a. 1H NMR spectrum of compound 3.15.
2.02.0
1.11.1
1.01.0
1.01.0
ppm0 011223344556677889-19.97
7.51
7.52
7.54
7.54
7.55
7.57
7.57
7.58
7.58
7.58
7.60
7.65
7.66
7.68
7.68
7.68
7.70
7.71
7.78
8.10
8.11
8.13
8.14

111
1-(bromoethynyl)-2-nitrobenzene (3.12). To a solution of dibromoalkene 3.15 (3.60 g,
11.7 mmol) and BnEt3NCl (1.33 g, 5.85 mmol) in CH2Cl2 (46 mL) at 0 °C was added a
solution of KOH (30 g) in 23 mL of H2O. The resulting solution was stirred for 1 h at 0
°C, then extracted with CH2Cl2. The organic extracts were dried over Na2SO4, filtered,
concentrated under reduced pressure, and purified by SiO2 chromatography (10-20%
EtOAc in hexanes) to afford 3.12 in quantitative yield.
1H-NMR (300 MHz; CDCl3): δ 8.06 (dd, J = 8.2, 1.4 Hz, 1H), 7.66 (dd, J = 7.7, 1.7 Hz,
1H), 7.58 (td, J = 7.5, 1.4 Hz, 1H), 7.49 (td, J = 7.8, 1.6 Hz, 1H).
REF: TRW-I-291, TRW-I-338, TRW-I-364.
NO2
Br
3.12

112
Figure 5.4a. 1H NMR spectrum of compound 3.12.
1.01.0
1.11.1
1.11.1
1.01.0
ppm1 122334455667788-19.97
7.46
7.47
7.49
7.49
7.51
7.52
7.56
7.56
7.58
7.59
7.61
7.61
7.65
7.65
7.67
7.68
8.05
8.05
8.07
8.08

113
1-((2-nitrophenyl)ethynyl)indoline (3.16). Bromoalkyne 3.12 (185 mg, 0.82 mmol) was
dissolved in toluene (2.75 mL). Indoline (110 µL, 0.98 mmol), K3PO4 (418 mg, 1.97
mmol), CuSO4⋅5H2O (40 mg, 0.16 mmol), and 1,10-phenanthroline (60 mg, 0.33 mmol)
were added successively. The resulting suspension was heated to 120 °C for 4 h in a
microwave. Upon completion of the reaction, the solution was cooled and diluted with
ether, then filtered through celite. The filtrate was concentrated under reduced pressure
and purified by SiO2 chromatography (10-20% EtOAc in hexanes) to afford 3.16 (80 mg,
37% yield).
1H-NMR (300 MHz; CDCl3): δ 8.38 (d, J = 7.9 Hz, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.68
(d, J = 9.1 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H), 7.30-7.26 (m, 2H), 7.19 (td, J = 10.2, 6.6 Hz,
2H), 4.72 (t, J = 8.3 Hz, 2H), 3.32 (t, J = 8.3 Hz, 2H).
REF: TRW-I-328, TRW-I-334, TRW-I-342, TRW-I-343, TRW-I-353, TRW-I-360,
TRW-I-362, TRW-I-385.
NO2
N
3.16
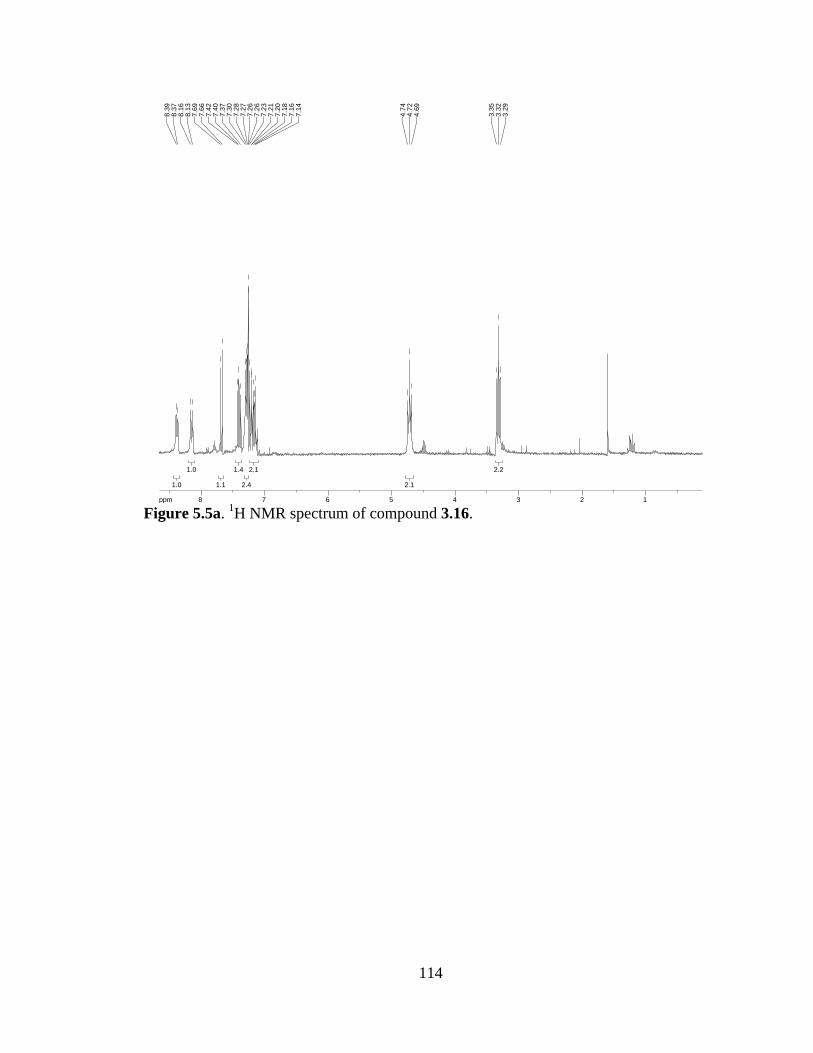
114
Figure 5.5a. 1H NMR spectrum of compound 3.16.
2.22.2
2.12.1
2.12.1
2.42.4
1.41.4
1.11.1
1.01.0
1.01.0
ppm1 122334455667788-19.973.
293.
323.
35
4.69
4.72
4.74
7.14
7.16
7.18
7.20
7.21
7.23
7.26
7.26
7.27
7.28
7.30
7.37
7.40
7.42
7.66
7.69
8.13
8.16
8.37
8.39

115
2-(indolin-1-ylethynyl)aniline (3.17). Nitroalkyne 3.16 was dissolved in a 4 to 1
acetone/water mixture (4.3 mL). Zinc dust (141 mg, 2.15 mmol) and NH4Cl (228 mg,
4.30 mmol) were added successively, and the resulting suspension stirred briskly for 10
minutes at r.t. The mixture was concentrated under reduced pressure, diluted with EtOAc,
washed with water, washed with brine, dried over Na2SO4 and concentrated. Purification
by SiO2 chromatography (3:1 hexanes:EtOAc, 1:1 hexanes:EtOAc, then 100% EtOAc)
afforded pure aniline derivative 3.17 (61 mg, 61% yield).
1H-NMR (300 MHz; CDCl3): δ 8.33 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 15.6 Hz, 2H), 7.20-
7.04 (m, 4H), 6.72 (dd, J = 7.4, 2.3 Hz, 2H), 4.39-4.14 (m, 2H), 3.96 (td, J = 10.4, 6.1
Hz, 1H), 3.54 (td, J = 10.4, 7.0 Hz, 1H), 3.21-2.99 (m, 2H); 13C-NMR (75 MHz; CDCl3):
δ 131.5, 130.1, 129.1, 127.9, 124.98, 124.95, 122.2, 118.8, 117.6, 117.3, 112.5, 71.6,
47.5, 28.4.
REF: TRW-I-331, TRW-I-336, TRW-I-352, TRW-I-357, TRW-I-361, TRW-I-366.
NH2
N
3.17
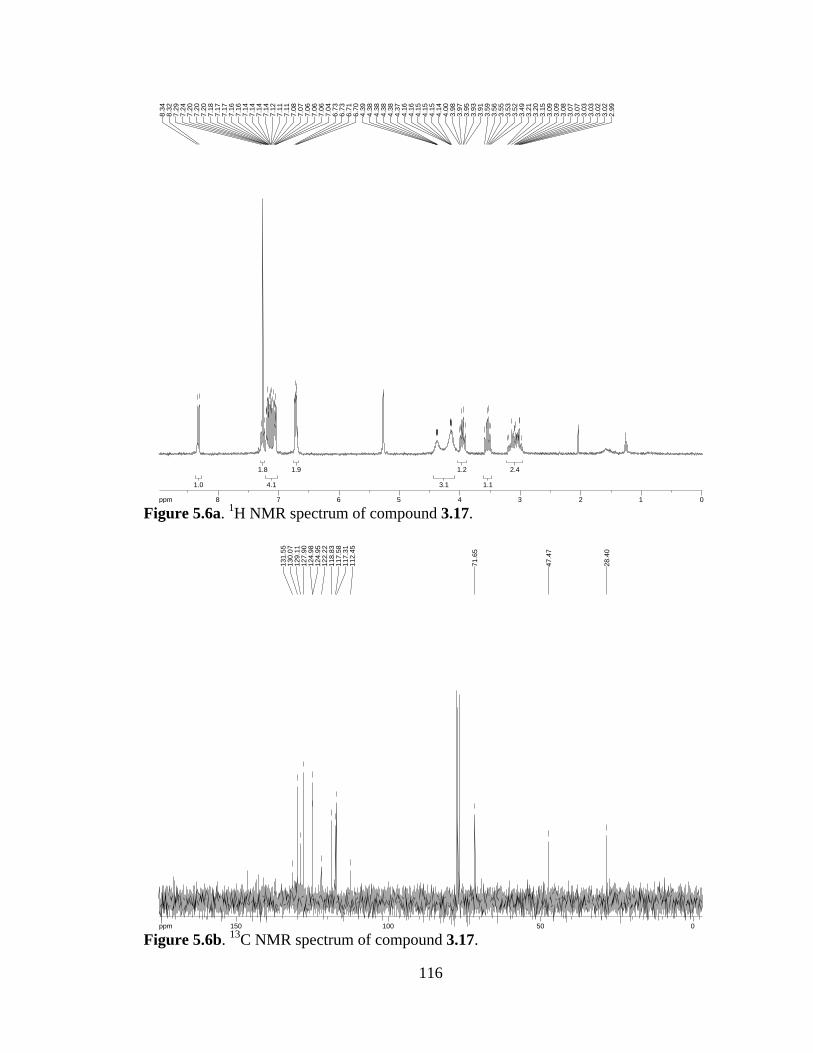
116
Figure 5.6a. 1H NMR spectrum of compound 3.17.
Figure 5.6b. 13C NMR spectrum of compound 3.17.
2.42.4
1.11.1
1.21.2
3.13.1
1.91.9
4.14.1
1.81.8
1.01.0
ppm0 01122334455667788-19.97
2.99
3.02
3.02
3.03
3.03
3.07
3.07
3.08
3.09
3.09
3.15
3.20
3.21
3.49
3.52
3.53
3.55
3.56
3.59
3.91
3.93
3.95
3.97
3.98
4.00
4.14
4.15
4.15
4.15
4.16
4.16
4.37
4.38
4.38
4.38
4.38
4.39
6.70
6.71
6.73
6.73
7.04
7.06
7.06
7.06
7.07
7.08
7.11
7.11
7.12
7.14
7.14
7.14
7.14
7.16
7.16
7.17
7.17
7.18
7.20
7.20
7.20
7.24
7.29
8.32
8.34
ppm0 05050100100150150-303.86
28.4
0
47.4
7
71.6
5
112.
4511
7.31
117.
5811
8.83
122.
2212
4.95
124.
9812
7.90
129.
1113
0.07
131.
55

117
Methyl 3-(benzyloxycarbonylamino)-4-(indolin-3-yl)butanoate (3.21). N-Cbz-L-Trp-
OMe (3.52 g, 10.0 mmol) was dissolved in THF/TFA (16.5 mL/16.5 mL) and cooled to 0
°C. BH3⋅Me2S was added dropwise and the resulting solution stirred 15 min. at 0 °C.
Water (33 mL) was added and the mixture stirred 15 min. at r.t. The solvent was removed
in vacuo and TFA azeotroped off with toluene (3x). The residue was taken up in EtOAc,
washed with 1M NaOH (3x), water (1x), and brine (1x) successively. The organic layer
was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by
SiO2 chromatography (2:1 – 1:1 hexanes:EtOAc) to afford indoline 3.21 (2.81 g, 79%
yield).
1H-NMR (300 MHz; CDCl3): δ 7.36 (s, 5H), 7.03 (t, J = 7.1 Hz, 2H), 6.74-6.71 (m, 1H),
6.64 (dd, J = 9.5, 1.8 Hz, 1H), 5.37 (dd, J = 26.1, 8.8 Hz, 1H), 5.12 (s, 2H), 4.53-4.44 (m,
1H), 3.74-3.68 (m, 4H), 3.36-3.22 (m, 2H), 2.11-2.06 (m, 2H).
REF: TRW-I-369.
NH
CO2Me
NHCbz
3.21

118
Figure 5.7a. 1H NMR spectrum of compound 3.21.
2.42.4
2.42.4
4.34.3
1.11.1
2.12.1
1.01.0
1.11.1
1.01.0
1.81.8
5.05.0
ppm1 1223344556677-19.97
2.06
2.08
2.08
2.08
2.10
2.11
3.22
3.25
3.25
3.31
3.33
3.34
3.36
3.36
3.36
3.68
3.71
3.74
4.44
4.45
4.46
4.46
4.46
4.48
4.48
4.49
4.51
4.52
4.53
5.12
5.31
5.34
5.40
5.43
6.63
6.63
6.66
6.66
6.71
6.73
6.74
7.01
7.03
7.06
7.36

119
(S)-methyl 3-(1H-indol-3-yl)-2-(2,2,2-trifluoroacetamido)propanoate (3.31). To a
solution of L-Trp-OMe·HCl (1.0 g, 3.92 mmol) in MeOH was added trifluoromethyl
propionate (0.94 mL, 7.84 mmol) and Et3N (1.1 mL, 7.84 mmol). After stirring 1 h at r.t.,
the reaction mixture was concentrated and the resulting residue dissolved in CH2Cl2. The
organic phase was washed with ammonium hydroxide (5%, 1x) and brine (1x), then dried
over Na2SO4 and concentrated to afford 1.23 g of the title compound, carried forward as
crude (>99% yield).
1H-NMR (300 MHz; CDCl3): δ 8.25-8.23 (bs, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.37 (d, J =
8.1 Hz, 1H), 7.21 (td, J = 7.5, 1.1 Hz, 1H), 7.13 (td, J = 7.4, 1.0 Hz, 1H), 6.98 (d, J = 2.4
Hz, 1H), 6.87-6.85 (bs, 1H), 4.94 (dt, J = 7.8, 5.0 Hz, 1H), 3.73 (s, 3H), 3.42 (d, J = 5.1
Hz, 2H).
REF: TRW-I-423, TRW-I-425.
NH
NH
CO2Me
O CF3
3.31

120
Figure 5.8a. 1H NMR spectrum of compound 3.31.
2.12.1
3.03.0
1.01.0
0.80.8
1.01.0
1.01.0
1.11.1
1.11.1
1.11.1
1.01.0
ppm1 122334455667788-19.973.
413.
43
3.73
4.91
4.92
4.93
4.94
4.95
4.96
6.85
6.85
6.87
6.87
6.87
6.97
6.98
7.10
7.11
7.13
7.13
7.15
7.16
7.18
7.19
7.21
7.21
7.23
7.24
7.36
7.39
7.48
7.50
8.23
8.23
8.24
8.24
8.24
8.24
8.24
8.25

121
(S)-methyl 1-(2,2,2-trifluoroacetyl)-1,2,3,8-tetrahydropyrrolo[2,3-b]indole-2-
carboxylate (3.32). To a solution of 3.31 (1.07 g, 3.4 mmol) in CH2Cl2 (34 mL) and Et3N
(1.9 mL, 13.6 mmol) at 0 °C was added t-BuOCl (0.41 mL, 3.4 mmol) dropwise. The
resulting solution was allowed to warm to r.t. O/N with stirring. Water was added and the
product extracted in to Et2O. The combined organic layers were washed with brine, dried
over Na2SO4, and concentrated. Recrystallization from EtOAc/hexanes provided the pure
title compound (670 mg, 63% yield).
1H-NMR (300 MHz; CDCl3): δ 9.17-9.16 (bs, 1H), 7.39 (td, J = 6.7, 2.6 Hz, 2H), 7.17
(ddd, J = 7.0, 4.3, 2.2 Hz, 2H), 5.55 (dt, J = 9.7, 1.8 Hz, 1H), 3.82 (s, 3H), 3.74 (dd, J =
14.8, 9.7 Hz, 1H), 3.36 (dd, J = 14.8, 2.3 Hz, 1H).
REF: TRW-I-424, TRW-I-427.
NH
NCOCF3
CO2Me
3.32

122
Figure 5.9a. 1H NMR spectrum of compound 3.32.
1.11.1
1.31.3
3.33.3
1.11.1
1.91.9
2.22.2
1.01.0
ppm2 233445566778899-19.97
3.33
3.34
3.38
3.39
3.70
3.73
3.75
3.78
3.82
5.53
5.53
5.54
5.56
5.57
5.57
7.15
7.15
7.16
7.17
7.18
7.18
7.19
7.36
7.37
7.38
7.39
7.40
7.41
9.16
9.16
9.17

123
(2S,3aR,8aS)-1-tert-butyl 2-methyl 3a-((2-iodophenyl)amino)-3,3a,8,8a-
tetrahydropyrrolo[2,3-b]indole-1,2(2H)-dicarboxylate (3.35). N-Boc-Trp-OMe (100
mg, 0.31 mmol) was dissolved in acetonitrile (6 mL), then 2-iodoaniline (81 mg, 0.37
mmol) added and the resulting solution cooled to -45 °C. Freshly recrystallized NIS (113
mg, 0.50 mmol) was added dropwise over 1 h as a solution in acetonitrile (1.5 mL). The
reaction was allowed to warm to -35 °C as it stirred an additional hour. The entire
reaction mixture was poured into a separatory funnel containing sat’d Na2S2O4 and
EtOAc. The product was extracted in EtOAc (3x) and the combined organic layers
washed with brine, dried over Na2SO4, and concentrated. Crude 3.35 was purified by
SiO2 chromatography (5-10% EtOAc in hexanes) to afford pure 3.35 in 78% yield (130
mg).
1H-NMR (300 MHz; CDCl3): δ 7.64 (t, J = 7.8 Hz, 1H), 7.12 (dt, J = 13.7, 7.3 Hz, 2H),
6.99 (t, J = 7.7 Hz, 1H), 6.75-6.65 (m, 2H), 6.40 (t, J = 7.6 Hz, 1H), 6.30 (dd, J = 16.2,
8.2 Hz, 1H), 5.89 (d, J = 34.7 Hz, 1H), 4.63 (d, J = 25.6 Hz, 1H), 4.39 (ddd, J = 25.8, 8.3,
4.3 Hz, 1H), 3.82 (s, 3H), 2.72 (qd, J = 15.3, 6.6 Hz, 2H), 1.46 (d, J = 30.4 Hz, 9H).
REF: TRW-I-435, TRW-I-441.
NH
NBoc
CO2MeNH
I
H
3.35
124
Figure 5.10a. 1H NMR spectrum of compound 3.35.
9.09.0
2.12.1
2.82.8
1.21.2
1.21.2
1.01.0
0.90.9
1.21.2
1.91.9
1.01.0
2.02.0
1.01.0
ppm0 011223344556677-19.97
1.42
1.52
2.63
2.66
2.68
2.71
2.74
2.76
2.79
2.80
3.82
4.33
4.34
4.35
4.37
4.41
4.43
4.44
4.45
4.59
4.68
5.82
5.94
6.26
6.28
6.31
6.34
6.38
6.40
6.43
6.65
6.67
6.70
6.73
6.75
6.96
6.99
7.01
7.07
7.10
7.12
7.14
7.17
7.61
7.64
7.67

125
L-serine-OMe (3.39). To a solution of L-serine (50.0 g, 0.48 mol) in MeOH (450 mL) at
0 °C was added SOCl2 (35 mL, 0.48 mol). The reaction was allowed to warm to r.t. as it
stirred O/N. The resulting solution was concentrated, taken up in ether, filtered, and the
solid washed with ether. The crude solid material was recrystallized in MeOH/ether to
afford pure crystalline methyl ester 3.39 as the HCl salt.
REF: TRW-I-462, TRW-II-118.
N-Boc-Ser-OMe (3.40). Boc anhydride (12.2 g, 56 mmol) was dissolved in acetonitrile
(190 mL), then Et3N (24 mL, 168 mmol) and L-Ser-OMe·HCl (8.74 g, 56 mmol) added.
The reaction was allowed to stir for 6 h at r.t., then CH2Cl2 (950 mL) and 1M HCl (665
mL) added. The organic layer was separated and washed with sat’d NaHCO3, then dried
over Na2SO4 and concentrated to afford the title material, used as crude (10.01 g, 82%
yield).
1H-NMR (300 MHz; CDCl3): δ 5.43 (m, J = 1.6, 0.8 Hz, 1H), 4.40-4.38 (m, 1H), 3.97-
3.91 (m, 2H), 3.79 (s, 3H), 2.20-2.19 (m, 1H), 1.46 (s, 9H).
REF: TRW-I-299, TRW-I-439.
MeO2C NH2
OH
3.39
MeO2C NHBoc
OH
3.40

126
Figure 5.11a. 1H NMR spectrum of compound 3.40.
9.49.4
0.90.9
3.03.0
2.12.1
0.90.9
0.90.9
ppm0 011223344556677-19.97
1.46
2.19
2.19
2.19
2.20
2.20
3.79
3.91
3.91
3.92
3.93
3.94
3.94
3.95
3.96
3.97
4.38
4.38
4.39
4.39
4.39
4.40
4.40
4.40
4.40
5.42
5.42
5.42
5.42
5.43
5.43
5.43
5.43
5.44
5.44
5.44
5.44
5.45
5.45

127
Methyl 2-(tert-butoxycarbonylamino)-3-iodopropanoate (3.41). To a solution of Ph3P
(2.13 g, 8.13 mmol) and imidazole (553 mg, 8.13 mmol) in CH2Cl2 (30 mL) at 0 °C was
added I2 (2.07 g, 8.13 mmol) in 3 portions. The reaction mixture was warmed to r.t. for
10 minutes, then cooled to 0 °C before adding dropwise a solution of Boc-Ser-OMe (1.42
g, 6.5 mmol) in 10 mL CH2Cl2. The resulting solution was stirred 1 h at 0 °C, then
allowed to warm to r.t. as it stirred an additional 1.5 h. The crude reaction mixture was
filtered through a plug of silica with 1:1 EtOAC/hexanes. The filtrate was concentrated
under reduced pressure and purified by SiO2 chromatography (5-20% EtOAc in hexanes)
to afford iodide 3.41 (1.17 g, 55% yield).
1H-NMR (300 MHz; CDCl3): δ 5.36-5.33 (m, 1H), 4.55-4.50 (m, 1H), 3.80 (s, 3H), 3.56
(dq, J = 9.8, 5.0 Hz, 2H), 1.46 (s, 9H).
REF: TRW-I-196, TRW-I-205, TRW-I-243, TRW-I-440
MeO2C NHBoc
I
3.41

128
Figure 5.12a. 1H NMR spectrum of compound 3.41.
10.210.2
2.02.0
3.03.0
0.90.9
0.90.9
ppm1 1223344556677-19.97
1.46
3.52
3.54
3.56
3.57
3.58
3.61
3.62
3.80
4.50
4.50
4.50
4.51
4.52
4.53
4.54
4.55
5.33
5.33
5.34
5.34
5.34
5.34
5.34
5.34
5.35
5.35
5.36
5.36
5.36
5.36

129
(S)-methyl 2-((tert-butoxycarbonyl)amino)-5-(trimethylsilyl)pent-4-ynoate (3.42). A
flask (Flask A) containing CuCN (123 mg, 1.37 mmol) and LiCl (115 mg, 2.74 mmol)
was heated to 150 °C under vacuum for 2 h, then cooled to r.t. DMF (4 mL) was added
and the suspension sonicated until full dissolution was observed. This solution was then
cooled to -20 °C. A second flask (Flask B) containing Zn dust (358 mg, 5.47 mmol) was
heated under vaccum, cooled to r.t., filled with Ar, then DMF (1.75 mL) and
dibromoethane (26 µL, 0.30 mmol) added. The suspension was heated to 80 °C for 30
min, then cooled to r.t. before addition of TMSCl (19 µL, 0.15 mmol). Following an
additional 30 min of stirring, N-Boc-Ser(I)-OMe (3.41, 500 mg, 1.52 mmol) was added
slowly as a solution in a minimal amount of DMF. After complete addition, this
suspension was transferred to Flask A. The combined reaction mixture was allowed to
stir for 15 min at -20 °C, then TMS-acetylene bromide (269 mg, 1.52 mmol) added
dropwise. The reaction mixture was allowed to warm to r.t. O/N. Water was added (15
mL) and the product extracted in EtOAc. The combined organic layers were washed with
brine, dried over Na2SO4, and concentrated. SiO2 chromatography (3:1 hexanes/EtOAc)
afforded the pure title compound (180 mg, 40% yield, stains with vanillin).
1H-NMR (300 MHz; CDCl3): δ 5.28 (d, J = 8.4 Hz, 1H), 4.43-4.37 (m, 1H), 3.70 (s, 3H),
2.75-2.59 (m, 2H), 1.39 (s, 9H), 0.07 (s, 8H).
REF: TRW-I-444, TRW-I-452, TRW-I-469.
MeO2C NHBoc
TMS
3.42

130
Figure 5.13a. 1H NMR spectrum of compound 3.42.
8.58.5
9.09.0
2.02.0
2.82.8
0.80.8
0.80.8
ppm-1 -10011223344556-19.97
0.07
1.39
2.59
2.61
2.65
2.67
2.68
2.69
2.73
2.75
3.70
4.37
4.39
4.40
4.40
4.42
4.43
5.27
5.30

131
(Bromoethynyl)trimethylsilane. To a solution of TMS-acetylene (5 mL, 35.5 mmol) in
acetone (120 mL) was added AgNO3 (604 mg, 3.55 mmol) and NBS (6.94 g, 39 mmol).
After stirring 3 h, the reaction was quenched by addition of ice water. The product was
extracted in pentane and the combined organic layers washed with brine, dried over
Na2SO4, and concentrated (bath at 0 °C) to afford pure (bromoethynyl)trimethylsilane
(4.51 g, 72% yield).
1H-NMR (300 MHz; CDCl3): δ 0.18 (s, 9H).
REF: TRW-I-442, TRW-I-457.
Figure 5.14a. 1H NMR spectrum of (bromoethynyl)trimethysilane.
Br TMS
9.09.0
ppm0 011223344-19.97
0.18

132
(2S,3aR,8aS)-1-tert-butyl 2-methyl 3a-(3-((S)-2-((tert-butoxycarbonyl)amino)-3-
methoxy-3-oxopropyl)-1H-indol-1-yl)-3,3a,8,8a-tetrahydropyrrolo[2,3-b]indole-
1,2(2H)-dicarboxylate (3.43). A flask containing 3.35 (300 mg, 0.56 mmol), 3.42 (370
mg, 1.24 mmol), NaOAc (321 mg, 3.92 mmol), LiCl (24 mg, 0.56 mmol), and Pd(OAc)2
(25 mg, 0.11 mmol) was vacated and backfilled with Ar. DMF (3 mL) was added and the
suspension sonicated for 15 minutes while bubbling Ar through reaction. Three freeze-
pump-thaw cycles were performed to complete the degassing procedure. The reaction
mixture was heated to 100 °C for 24 h, then diluted with toluene and concentrated. The
crude residue was taken up in EtOAc, filtered through celite, and washed with 3M HCl.
The organic fraction was further washed with brine, dried over Na2SO4, and concentrated
to afford the title compound (3.43, 250 mg, 70% yield).
1H-NMR (300 MHz; CDCl3): δ 7.62 (d, J = 8.1 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.35-
7.25 (m, 3H), 7.14 (d, J = 7.4 Hz, 1H), 6.92 (d, J = 7.5 Hz, 1H), 6.77 (d, J = 7.7 Hz, 1H),
6.64 (s, 1H), 6.05-5.91 (m, 1H), 5.02 (d, J = 7.9 Hz, 1H), 4.55-4.53 (m, 1H), 4.28-4.20
(m, 1H), 3.80 (d, J = 3.1 Hz, 3H), 3.58-3.47 (m, 3H), 3.36 (dd, J = 13.1, 9.3 Hz, 1H),
3.15-3.14 (m, 1H), 2.98 (dd, J = 13.2, 7.8 Hz, 1H), 1.42 (complex, J = 28.4 Hz, 18H).
REF: TRW-I-453, TRW-I-458.
NH
NBoc
CO2MeN
NHBoc
CO2Me
H
3.43

133
Figure 5.15a. 1H NMR spectrum of compound 3.43.
17.717.7
0.90.9
1.31.3
0.90.9
2.52.5
2.92.9
0.80.8
0.70.7
0.70.7
0.90.9
0.70.7
1.01.0
1.01.0
1.01.0
2.82.8
1.01.0
1.01.0
ppm0 0112233445566778-19.97
1.33
1.42
1.52
2.95
2.97
2.99
3.02
3.14
3.14
3.15
3.32
3.35
3.37
3.40
3.47
3.49
3.58
3.80
3.81
4.20
4.22
4.23
4.24
4.25
4.28
4.28
4.53
4.54
4.55
4.55
5.01
5.03
5.91
6.04
6.05
6.64
6.75
6.78
6.91
6.94
7.13
7.15
7.25
7.26
7.26
7.28
7.30
7.33
7.35
7.51
7.53
7.61
7.63

134
(2S,3aR,8aS)-1-(tert-butoxycarbonyl)-3a-(3-((S)-2-((tert-butoxycarbonyl)amino)-2-
carboxyethyl)-1H-indol-1-yl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole-2-
carboxylic acid (3.44). To a solution of 3.43 (100 mg, 0.136 mmol) in THF and MeOH
(2:1, 0.40 mL:0.20 mL) was added LiOH (20 mg, 0.80 mmol). After stirring under Ar
O/N, 10% KHSO4 was added until the solution was acidic, then the product extracted in
EtOAc. The combined organic extracts were washed with brine, dried over Na2SO4, and
concentrated. Crude 3.44 recovered as such was carried forward to the next reaction (90.4
mg, 93% yield).
1H-NMR (300 MHz; CDCl3): δ 7.55-7.53 (m, 1H), 7.17 (d, J = 8.4 Hz, 2H), 7.06-7.00
(m, 1H), 6.90-6.86 (m, 1H), 6.79-6.62 (m, 3H), 6.48-6.32 (m, 1H), 6.24-6.13 (m, 1H),
4.63-4.34 (m, 2H), 3.56-3.47 (m, 1H), 3.20-3.18 (m, 1H), 3.04-2.82 (m, 2H), 1.50-1.38
(m, 18H).
REF: TRW-I-456, TRW-I-460.
NH
NBoc
CO2HN
NHBoc
CO2H
H
3.44
135
Figure 5.16a. 1H NMR spectrum of compound 3.44.
19.019.0
1.81.8
0.60.6
1.31.3
2.02.0
0.70.7
0.60.6
3.23.2
1.21.2
1.51.5
2.12.1
1.01.0
ppm0 011223344556677-19.97
1.38
1.40
1.50
2.82
2.82
2.84
2.87
2.87
3.02
3.04
3.04
3.18
3.18
3.18
3.19
3.19
3.19
3.20
3.20
3.20
3.20
3.47
3.47
3.47
3.48
3.48
3.48
3.49
3.50
3.51
3.51
3.53
3.53
3.53
3.53
3.55
3.56
4.34
4.36
4.36
4.36
4.57
4.60
4.63
6.13
6.14
6.24
6.32
6.32
6.45
6.45
6.45
6.46
6.46
6.47
6.47
6.47
6.47
6.48
6.48
6.62
6.65
6.69
6.69
6.70
6.72
6.74
6.76
6.79
6.86
6.87
6.87
6.88
6.88
6.90
7.00
7.04
7.04
7.05
7.06
7.15
7.18
7.53
7.55
7.55
7.55

136
(2S,3aR,8aR)-1-benzyl 8-tert-butyl 2-methyl 3a-bromo-3,3a-dihydropyrrolo[2,3-
b]indole-1,2,8(2H,8aH)-tricarboxylate (3.47). To a solution of N-Cbz,N’-Boc-Trp-OMe
(1.0 g, 2.2 mmol) in CH2Cl2 (100 mL) was added PPTS (552 mg, 2.2 mmol) and NBS
(392 mg, 2.2 mmol). The resulting solution was stirred O/N at r.t, then diluted with
CH2Cl2, washed with brine, dried over Na2SO4, and concentrated. Crude 3.47 was
purified by SiO2 chromatography (25% EtOAc in hexanes) to afford pure material in
88% yield (1.03 g).
1H-NMR (300 MHz; CDCl3): δ 7.37-7.28 (m, 8H), 7.12 (td, J = 7.5, 0.7 Hz, 1H), 6.43 (s,
1H), 5.19-5.16 (m, 2H), 3.97 (dd, J = 10.3, 6.5 Hz, 1H), 3.69-3.41 (m, 3H), 3.26 (dd, J =
12.7, 6.5 Hz, 1H), 2.84 (dd, J = 12.7, 10.3 Hz, 1H), 1.52 (s, 9H).
REF: TRW-I-477.
NBoc
NCbz
CO2MeBr
H
3.47

137
Figure 5.17a. 1H NMR spectrum of compound 3.47.
14.314.3
1.21.2
1.41.4
3.23.2
1.21.2
1.81.8
1.01.0
1.31.3
7.77.7
ppm1 1223344556677-19.97
1.52
2.81
2.84
2.85
2.88
3.22
3.25
3.27
3.29
3.41
3.66
3.66
3.67
3.67
3.68
3.69
3.69
3.94
3.97
3.98
4.00
5.16
5.17
5.17
5.18
5.18
5.18
5.19
5.19
6.43
7.10
7.10
7.12
7.12
7.15
7.15
7.26
7.27
7.28
7.28
7.28
7.28
7.29
7.30
7.31
7.32
7.32
7.32
7.33
7.34
7.35
7.37

138
(2R,3aR,8aR)-1-benzyl 8-tert-butyl 2-methyl 3a-(1H-indol-1-yl)-3,3a-
dihydropyrrolo[2,3-b]indole-1,2,8(2H,8aH)-tricarboxylate (3.48).
Bromopyrroloindoline 3.47 (500 mg, 0.94 mmol) was dissolved in acetonitrile (20 mL),
and to it added indole (74 mg, 0.63 mmol). The suspension was cooled to 0 °C, then
KOtBu (141 mg, 1.26 mmol) added slowly. Once the reaction was deemed complete by
TLC, the mixture was quenched by addition of sat’d NaHCO3 and the product extracted
in CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4,
concentrated, and purified by SiO2 chromatography (3:1 hexanes:EtOAc) to afford pure
3.48 (100 mg, 28% yield).
1H-NMR (300 MHz; CDCl3): δ 7.79-7.75 (bs, 1H), 7.65-7.62 (m, 1H), 7.44-7.27 (m,
8H), 7.19-7.15 (m, 3H), 6.96 (d, J = 3.0 Hz, 1H), 6.44 (d, J = 3.4 Hz, 1H), 5.30-5.20 (m,
2H), 4.97 (d, J = 8.9 Hz, 1H), 3.57 (dd, J = 13.0, 9.3 Hz, 1H), 3.22 (s, 3H), 3.07 (d, J =
13.1 Hz, 1H), 1.47 (bs, 9H).
REF: TRW-I-482, TRW-I-484.
NBoc
NCbz
CO2MeN
H3.48

139
Figure 5.18a. 1H NMR spectrum of compound 3.48.
8.48.4
0.90.9
2.62.6
0.90.9
0.80.8
1.71.7
0.70.7
1.01.0
2.72.7
8.18.1
1.01.0
1.01.0
ppm1 1223344556677-19.97
1.47
1.47
3.05
3.09
3.22
3.54
3.57
3.58
3.61
4.96
4.99
5.20
5.24
5.27
5.27
5.27
5.28
5.28
5.29
5.29
5.29
5.29
5.29
5.30
5.30
5.30
6.43
6.45
6.95
6.96
7.15
7.17
7.17
7.19
7.27
7.29
7.36
7.41
7.44
7.62
7.63
7.65
7.75
7.76
7.76
7.76
7.76
7.76
7.77
7.77
7.77
7.78
7.78
7.78
7.78
7.79

140
N-Boc-L-tryptophan-N-SO2Ph (3.56). N-Boc-Trp (14.8 g, 48.7 mmol) was dissolved in
THF (100 mL) and the resulting solution cooled to -78 °C. LHMDS (146 mL, 146.1
mmol) was added slowly over 5 minutes. The reaction was stirred at -78 °C for 1 h, then
PhSO2Cl (7.5 mL, 58.4 mmol) added in one portion. After stirring an additional 2 h at -78
°C, the reaction was quenched by addition of 1:1 AcOH:EtOAc then allowed to warm to
r.t. The suspension was diluted with 1M HCl and the product extracted in EtOAc. The
combined organic layers were dried and concentrated to afford crude 3.56, purified by
SiO2 chromatography (5% AcOH, 45% hexanes, 50% CH2Cl2) to afford pure 3.56 (17.12
g, 79% yield).
1H-NMR (300 MHz; DMSO-d6): δ 12.64-12.63 (m, 1H), 7.90-7.87 (m, 2H), 7.61 (t, J =
7.8 Hz, 2H), 7.56-7.50 (m, 2H), 7.25-7.20 (m, 2H), 7.16-7.13 (m, 2H), 4.20-4.17 (m, 1H),
3.11 (dd, J = 14.8, 4.2 Hz, 1H), 2.95 (dd, J = 14.8, 10.0 Hz, 1H), 1.31 (s, 9H).
REF: TRW-II-176, TRW-II-251.
NNHBoc
SO2Ph
CO2H
3.56

141
Figure 5.19a. 1H NMR spectrum of compound 3.56.
9.49.4
1.01.0
1.01.0
0.90.9
2.42.4
2.42.4
2.32.3
2.22.2
2.32.3
0.90.9
ppm0 02244668810101212-19.97
1.31
2.91
2.94
2.96
2.99
3.08
3.10
3.13
3.15
4.17
4.19
4.19
4.19
4.20
7.13
7.13
7.16
7.20
7.22
7.23
7.25
7.25
7.27
7.27
7.32
7.50
7.53
7.56
7.56
7.59
7.61
7.64
7.87
7.88
7.89
7.90
12.6
312
.63
12.6
312
.64
12.6
4

142
Dipeptide (3.57a). To a solution of N-Me-L-serine-OMe (2.0 g, 15 mmol) in CH2Cl2 (75
mL) was added iPr2NEt (2.61 mL, 15 mmol). After stirring 15 minutes, the suspension
was added to a stirring solution of 3.56 (6.66 g, 15 mmol) and EDCI (2.88 g, 15 mmol) in
CH2Cl2. The reaction was stirred O/N at r.t., then concentrated and purified through a
plug of SiO2 (100% EtOAc) to afford 3.57a (86% yield).
1H-NMR (300 MHz; DMSO-d6): δ 8.40 (d, J = 7.4 Hz, 1H), 7.87 (t, J = 8.8 Hz, 2H),
7.67 (dt, J = 21.1, 6.5 Hz, 2H), 7.54 (t, J = 7.7 Hz, 2H), 7.29 (complex, 2H), 6.95 (d, J =
8.9 Hz, 1H), 5.14 (dt, J = 11.2, 5.6 Hz, 1H), 4.47 (d, J = 5.7 Hz, 1H), 4.38-4.34 (m, 1H),
3.77-3.70 (m, 1H), 3.60 (s, 3H), 3.07-3.01 (m, 1H), 2.88-2.80 (m, 1H), 2.48 (s, 3H), 1.25
(s, 9H), 1.06 (s, 1H).
REF: TRW-II-129.
NNHBoc
NMe
O
SO2Ph
CO2Me
OH
3.57a

143
Figure 5.20a. 1H NMR spectrum of compound 3.57a.
1.11.1
6.76.7
1.81.8
0.80.8
0.90.9
3.03.0
0.90.9
1.41.4
0.60.6
1.01.0
0.70.7
2.32.3
1.71.7
2.42.4
2.42.4
0.70.7
ppm0 01122334455667788-19.97
1.06
1.25
2.48
2.80
2.83
2.83
2.84
2.84
2.88
2.88
3.01
3.01
3.02
3.02
3.02
3.03
3.06
3.06
3.07
3.07
3.60
3.70
3.71
3.72
3.73
3.74
3.75
3.77
3.77
4.34
4.35
4.36
4.37
4.37
4.38
4.38
4.46
4.48
5.10
5.12
5.14
5.16
5.18
6.93
6.96
7.24
7.26
7.28
7.30
7.33
7.51
7.54
7.56
7.62
7.63
7.65
7.68
7.70
7.73
7.84
7.87
7.89
8.39
8.41

144
Dioxopiperazine (3.58a). Dipeptide 3.57a (500 mg) was transferred to a microwave
tube, then heated neat in the microwave at maximum power to a safe temperature of 180
°C (microwave set to ‘open-vessel’). After cooling, the residue was suspended in CH2Cl2,
transferred to a vial, and concentrated to afford dioxopiperazine 3.58a (94% yield).
1H-NMR (300 MHz; DMSO-d6): δ 8.15 (d, J = 2.4 Hz, 1H), 8.03 (d, J = 2.0 Hz, 1H),
7.89 (dd, J = 24.0, 7.8 Hz, 3H), 7.64 (t, J = 3.6 Hz, 2H), 7.56 (d, J = 4.0 Hz, 1H), 7.33-
7.27 (m, 2H), 5.14 (t, J = 5.7 Hz, 1H), 4.47 (d, J = 5.6 Hz, 1H), 4.05-4.00 (m, 1H), 3.74
(d, J = 2.1 Hz, 1H), 3.65-3.60 (m, 1H), 3.42 (t, J = 4.2 Hz, 1H), 3.24 (dd, J = 59.1, 5.0
Hz, 3H).
REF: TRW-II-132.
NHN
NMeOH
O
OSO2Ph3.58a

145
Figure 5.21a. 1H NMR spectrum of compound 3.58a.
2.32.3
1.31.3
0.90.9
0.80.8
0.70.7
0.60.6
1.01.0
2.12.1
1.31.3
2.12.1
3.03.0
0.80.8
0.80.8
ppm1 122334455667788-19.97
3.14
3.15
3.33
3.35
3.41
3.42
3.44
3.60
3.60
3.64
3.65
3.65
3.73
3.74
4.00
4.00
4.00
4.01
4.01
4.01
4.01
4.02
4.03
4.04
4.04
4.04
4.05
4.05
4.46
4.48
5.13
5.14
5.16
7.27
7.28
7.29
7.30
7.33
7.33
7.53
7.55
7.56
7.57
7.58
7.59
7.63
7.64
7.66
7.84
7.86
7.92
7.94
8.02
8.03
8.14
8.15

146
(S)-methyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-(1H-indol-3-yl)propan-amido)-3-
hydroxypropanoate (3.57c). To a solution of L-Ser-OMe·HCl (2.56 g, 16.4 mmol) in
CH2Cl2 (80 mL) was added iPr2NEt (2.85 mL, 16.4 mmol). After stirring 15 minutes, N-
Boc-L-Trp (5.0 g, 16.4 mmol) and EDCI (3.15 g, 16.4 mmol) were added. The reaction
was stirred O/N at r.t., then concentrated and purified through a plug of SiO2 (100%
EtOAc) to afford 3.57c (5.71 g, 86% yield).
1H-NMR (400 MHz; CDCl3): δ 8.69 (bs, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.23 (d, J = 8.0
Hz, 1H), 7.06 (t, J = 7.4 Hz, 1H), 7.01-6.97 (m, 2H), 5.34 (d, J = 7.4 Hz, 1H), 4.42 (bs,
2H), 3.70 (bs, 2H), 3.54 (s, 3H), 3.16 (bs, 2H), 1.30 (s, 9H); 13C-NMR (101 MHz;
CDCl3): δ 172.4, 170.6, 155.8, 136.2, 127.5, 123.5, 122.0, 119.4, 118.5, 111.4, 109.8,
80.4, 62.5, 54.8, 52.6, 28.23, 28.14, 28.08.
REF: TRW-II-043.
NH
NHBocNH
O
CO2Me
OH
3.57c
147
Figure 5.22a. 1H NMR spectrum of compound 3.57c.
Figure 5.22b. 13C NMR spectrum of compound 3.57c.
8.98.9
2.42.4
3.03.0
2.02.0
2.02.0
1.01.0
2.52.5
1.11.1
1.11.1
1.01.0
1.01.0
ppm1 122334455667788-16.04
1.30
3.16
3.54
3.70
3.70
3.70
4.42
5.33
5.35
6.97
6.97
6.99
7.01
7.05
7.06
7.08
7.22
7.24
7.51
7.53
8.69
ppm0 05050100100150150-253.79
28.0
828
.14
28.2
3
52.5
654
.85
62.4
6
80.3
7
109.
8411
1.37
118.
5311
9.38
121.
9612
3.55
127.
51
136.
25
155.
77
170.
5717
2.39

148
Dipeptide (3.57d). To a solution of L-Ser-OMe·HCl (6.0 g, 38.5 mmol) in CH2Cl2 (200
mL) was added iPr2NEt (6.7 mL, 38.5 mmol). After stirring 15 minutes, the suspension
was added to a stirring solution of 3.56 (17.12 g, 38.5 mmol) and EDCI (7.39 g, 38.5
mmol) in CH2Cl2. The reaction was stirred O/N at r.t., then concentrated and purified
through a plug of SiO2 (100% EtOAc) to afford 3.57d (13.78 g, 66% yield).
1H-NMR (300 MHz; CDCl3): δ 7.96 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 7.1 Hz, 2H), 7.52
(t, J = 7.0 Hz, 2H), 7.44 (dd, J = 13.3, 5.5 Hz, 3H), 7.31 (t, J = 7.1 Hz, 1H), 6.96 (d, J =
6.7 Hz, 1H), 5.24-5.21 (m, 1H), 4.59-4.56 (m, 1H), 4.44 (q, J = 7.2 Hz, 1H), 3.89 (dt, J =
1.3, 0.7 Hz, 2H), 3.72 (s, 3H), 3.18 (d, J = 6.0 Hz, 2H), 1.40 (s, 9H); 13C-NMR (101
MHz; CDCl3): δ 171.9, 170.7, 155.9, 138.0, 135.0, 133.7, 130.8, 129.2, 126.6, 124.72,
124.70, 123.2, 119.6, 117.9, 113.5, 80.3, 62.4, 54.7, 52.6, 28.16, 28.00.
REF: TRW-II-107, TRW-II-178, TRW-II-253.
NSO2Ph
NHBocNH
O
CO2Me
OH
3.57d
149
Figure 5.23a. 1H NMR spectrum of compound 3.57d.
Figure 5.23b. 13C NMR spectrum of compound 3.57d.
8.98.9
2.02.0
3.33.3
2.22.2
1.31.3
1.21.2
0.90.9
0.90.9
1.31.3
3.03.0
2.42.4
2.02.0
1.01.0
ppm1 122334455667788-19.97
1.40
3.17
3.19
3.72
3.89
3.89
3.89
3.89
3.89
4.40
4.43
4.45
4.47
4.56
4.57
4.57
4.58
4.59
5.21
5.24
5.24
5.24
5.24
6.95
6.97
7.29
7.31
7.34
7.40
7.43
7.45
7.46
7.50
7.52
7.54
7.84
7.87
7.94
7.97
ppm0 05050100100150150200-253.79
28.0
028
.16
52.6
054
.73
62.4
2
80.2
9
113.
4511
7.94
119.
5612
3.22
124.
7012
4.72
126.
6412
9.18
130.
8513
3.71
134.
9713
7.97
155.
87
170.
7317
1.94

150
(3S,6S)-3-(hydroxymethyl)-6-((1-(phenylsulfonyl)-1H-indol-3-yl)methyl)piperazine-
2,5-dione (3.58d). The general cyclization procedure given for compound 3.58a was
repeated.
1H-NMR (300 MHz; DMSO-d6): δ 8.14-7.86 (m, 4H), 7.64-7.56 (m, 4H), 7.30-7.28 (m,
J = 5.5, 0.8 Hz, 2H), 5.13-5.13 (m, 1H), 4.38-4.03 (m, 1H), 3.74-3.60 (m, 2H), 3.43-3.33
(m, 2H), 3.15-2.85 (m, 2H); 13C-NMR (101 MHz; DMSO): δ 167.5, 167.3, 137.6, 137.2,
134.8, 131.4, 130.2, 126.9, 125.9, 123.6, 120.4, 118.5, 113.40, 113.31, 63.0, 57.3, 54.7,
50.9.
REF: TRW-II-063, TRW-II-186.
NSO2Ph
HNNH
OH
O
O
3.58d

151
Figure 5.24a. 1H NMR spectrum of compound 3.58d.
Figure 5.24b. 13C NMR spectrum of compound 3.58d.
2.02.0
2.32.3
2.02.0
1.11.1
0.70.7
1.91.9
4.14.1
3.93.9
ppm0 0112233445566778899-19.97
2.85
2.85
2.85
3.14
3.15
3.15
3.15
3.15
3.33
3.33
3.34
3.34
3.42
3.43
3.60
3.60
3.74
3.74
3.74
4.03
4.36
4.37
4.38
5.13
5.13
5.13
5.13
5.13
7.28
7.28
7.28
7.30
7.30
7.30
7.56
7.57
7.63
7.64
7.64
7.86
7.87
7.91
7.91
7.92
8.14
8.14
8.14
ppm0 05050100100150150-253.79
50.8
854
.72
57.3
2
63.0
0
113.
3111
3.40
118.
4712
0.45
123.
6312
5.94
126.
9313
0.17
131.
3613
4.81
137.
2213
7.60
167.
2716
7.50

152
Dipeptide (3.57e). Alcohol 3.57d (2.9 g, 5.3 mmol) was dissolved in DMF (13 mL), and
to it added imidazole (864 mg, 12.7 mmol) and TBSCl (2.08 g, 13.8 mmol). The reaction
stirred O/N at r.t., then was concentrated, dissolved in EtOAc, washed with H2O (3x),
washed with brine, dried, and concentrated to afford silylether 3.57e (3.14 g, 90% yield).
1H-NMR (300 MHz; DMSO-d6): δ 8.14 (d, J = 7.8 Hz, 1H), 7.85 (t, J = 7.9 Hz, 3H),
7.65 (t, J = 7.2 Hz, 2H), 7.58-7.50 (m, 3H), 7.33-7.20 (m, 2H), 7.00 (d, J = 8.8 Hz, 1H),
4.46-4.40 (m, 1H), 4.34-4.30 (m, 1H), 3.86 (dd, J = 10.3, 4.7 Hz, 1H), 3.74 (dd, J = 10.3,
4.9 Hz, 1H), 3.60 (s, 3H), 3.05-2.99 (m, 1H), 2.89-2.80 (m, 1H), 1.25 (s, 9H), 0.82 (s,
9H), 0.00 (d, J = 3.2 Hz, 6H); 13C-NMR (101 MHz; CDCl3): δ 170.6, 170.2, 138.2,
135.1, 133.7, 130.8, 129.2, 126.7, 124.9, 124.6, 123.3, 119.6, 117.6, 113.6, 63.3, 54.2,
52.4, 28.20, 28.10, 25.70, 25.61, 18.1, -5.63, -5.77.
REF: TRW-II-196, TRW-II-222, TRW-II-254.
NSO2Ph
NHBocNH
O
CO2Me
OTBS
3.57e

153
Figure 5.25a. 1H NMR spectrum of compound 3.57e.
Figure 5.25b. 13C NMR spectrum of compound 3.57e.
6.36.3
8.98.9
8.58.5
1.21.2
1.21.2
3.43.4
1.21.2
1.31.3
1.01.0
1.21.2
1.01.0
2.42.4
3.23.2
2.32.3
3.33.3
1.01.0
ppm0 011223344556677889-19.97
-0.0
00.
01
0.82
1.25
2.80
2.84
2.84
2.86
2.88
2.89
2.99
2.99
3.00
3.01
3.04
3.04
3.05
3.05
3.05
3.60
3.71
3.73
3.75
3.76
3.84
3.86
3.87
3.89
4.30
4.30
4.31
4.31
4.32
4.33
4.33
4.33
4.34
4.34
4.34
4.34
4.40
4.42
4.42
4.44
4.46
4.46
6.99
7.02
7.20
7.21
7.23
7.25
7.26
7.27
7.28
7.30
7.30
7.32
7.33
7.50
7.53
7.55
7.58
7.58
7.62
7.65
7.67
7.83
7.85
7.88
8.13
8.16
ppm0 05050100100150150-253.79
-5.7
7-5
.63
18.0
525
.61
25.7
028
.10
28.2
0
52.4
254
.19
63.3
0
113.
6011
7.62
119.
5712
3.28
124.
5912
4.87
126.
7012
9.19
130.
7513
3.69
135.
1213
8.16
170.
2017
0.63

154
Dioxopiperazine (3.58e). Dipeptide 3.57e (500 mg) was transferred to a microwave tube,
then heated neat in the microwave at maximum power to a safe temperature of 180 °C
(microwave set to ‘open-vessel’). After cooling, the residue was suspended in CH2Cl2,
transferred to a vial, and concentrated to afford dioxopiperazine 3.57e (2.47 g over 6
batches, 98%).
1H-NMR (300 MHz; DMSO-d6): δ 7.92-7.83 (m, 4H), 7.64 (t, J = 3.7 Hz, 2H), 7.58-7.52
(m, 4H), 7.33-7.28 (m, 1H), 7.25-7.20 (m, 1H), 4.15-4.05 (m, 1H), 3.84-3.77 (m, 1H),
3.61-3.43 (m, 2H), 3.16-2.94 (m, 2H), 0.79 (s, 9H), -0.02 (s, 3H), -0.06 (s, 3H).
REF: TRW-II-226, TRW-II-255.
NSO2Ph
HNNH
OTBS
O
O
3.58e

155
Figure 5.26a. 1H NMR spectrum of compound 3.58e.
3.53.5
3.03.0
9.69.6
2.32.3
2.22.2
0.90.9
1.01.0
1.11.1
1.21.2
3.93.9
1.91.9
4.24.2
ppm0 01122334455667788-19.97
-0.0
6-0
.02
0.79
2.94
3.00
3.00
3.01
3.01
3.02
3.03
3.03
3.03
3.08
3.10
3.11
3.12
3.13
3.13
3.14
3.16
3.43
3.45
3.47
3.53
3.54
3.54
3.56
3.57
3.57
3.61
4.05
4.05
4.05
4.05
4.06
4.06
4.06
4.07
4.07
4.08
4.08
4.15
4.15
7.20
7.22
7.23
7.23
7.24
7.25
7.25
7.25
7.28
7.28
7.30
7.33
7.52
7.53
7.53
7.55
7.56
7.57
7.58
7.63
7.64
7.66
7.83
7.84
7.85
7.85
7.87
7.90
7.90
7.92

156
Dipeptide (5.1). Alcohol 3.57c (2.0 g, 4.9 mmol) was dissolved in DMF (12 mL), and to
it added imidazole (802 mg, 11.8 mmol) and TBSCl (1.92 g, 12.7 mmol). The reaction
stirred O/N at r.t., then was concentrated, dissolved in EtOAc, washed with H2O (3x),
washed with brine, dried, and concentrated to afford silylether 5.1 (2.38 g, 93% yield).
1H-NMR (300 MHz; CDCl3): δ 8.18 (bs, J = 0.4 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.34
(d, J = 8.0 Hz, 1H), 7.20-7.18 (m, 1H), 7.16-7.11 (m, 2H), 6.55-6.52 (m, 1H), 5.19-5.16
(m, 1H), 4.54 (broad, J = 7.0, 4.1 Hz, 2H), 3.95 (dd, J = 10.1, 2.7 Hz, 1H), 3.66 (s, 3H),
3.62-3.58 (m, 1H), 3.31-3.30 (m, 1H), 3.21 (dd, J = 14.6, 6.9 Hz, 1H), 1.42 (s, 9H), 0.77
(s, 9H), -0.07 (s, 3H), -0.10 (s, 3H).
REF: TRW-III-196.
NH
NHBocNH
O
CO2Me
OTBS
5.1

157
Figure 5.27a. 1H NMR spectrum of compound 5.1.
3.13.1
3.03.0
9.49.4
9.49.4
1.21.2
1.01.0
0.80.8
3.33.3
1.11.1
1.91.9
0.80.8
1.01.0
2.22.2
0.90.9
1.21.2
1.11.1
1.01.0
ppm-1 -1001122334455667788-19.97
-0.1
0-0
.07
0.77
1.42
3.17
3.19
3.22
3.24
3.30
3.30
3.31
3.31
3.31
3.31
3.58
3.58
3.59
3.59
3.59
3.59
3.60
3.60
3.61
3.61
3.61
3.61
3.62
3.62
3.66
3.93
3.93
3.96
3.97
4.52
4.53
4.54
4.56
5.16
5.16
5.16
5.16
5.17
5.18
5.18
5.18
5.18
5.18
5.19
6.52
6.52
6.54
6.54
6.54
6.55
7.11
7.13
7.13
7.15
7.16
7.18
7.18
7.20
7.33
7.36
7.64
7.67
8.18
8.18

158
(3S,6S)-3-((1H-indol-3-yl)methyl)-6-(((tert-
butyldimethylsilyl)oxy)methyl)piperazine-2,5-dione (5.2). The general cyclization
procedure given for compound 3.58a was repeated.
1H-NMR (300 MHz; DMSO-d6): δ 10.88 (m, 1H), 7.95 (s, 1H), 7.60-7.48 (m, 1H), 7.29
(t, J = 7.8 Hz, 1H), 7.05-6.93 (m, 2H), 4.09-3.97 (m, 1H), 3.82-3.70 (m, 1H), 3.52-3.43
(m, 1H), 3.27-3.14 (m, 2H), 2.79 (d, J = 47.6 Hz, 1H), 0.81 (s, 9H), -0.02--0.05 (m, 6H);
13C-NMR (101 MHz; DMSO): δ 167.6, 165.5, 136.5, 127.9, 124.4, 121.3, 118.96,
118.77, 111.7, 109.5, 65.4, 57.5, 55.9, 31.5, 26.33, 26.24, 26.11, 26.05, 25.99, 18.7, -5.0.
REF: TRW-II-187.
NH
HNNH
OTBS
O
O
5.2
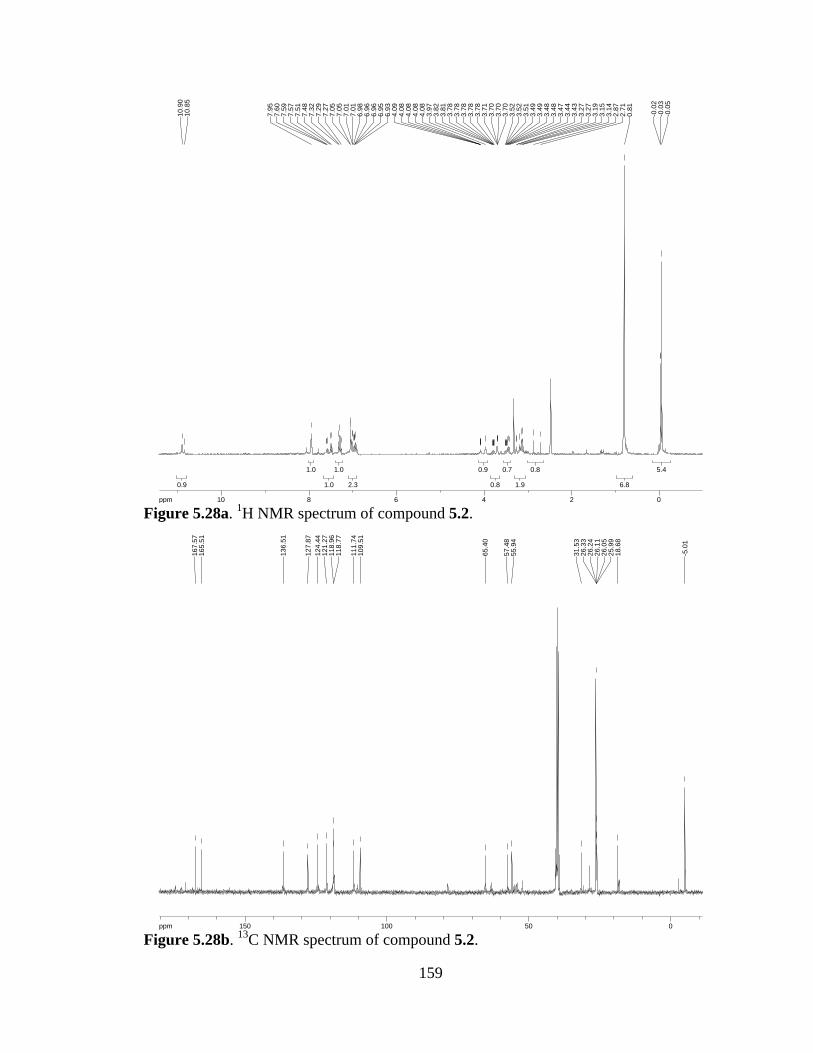
159
Figure 5.28a. 1H NMR spectrum of compound 5.2.
Figure 5.28b. 13C NMR spectrum of compound 5.2.
5.45.4
6.86.8
0.80.8
1.91.9
0.70.7
0.80.8
0.90.9
2.32.3
1.01.0
1.01.0
1.01.0
0.90.9
ppm0 0224466881010-19.97
-0.0
5-0
.03
-0.0
2
0.81
2.71
2.87
3.14
3.15
3.19
3.27
3.27
3.43
3.44
3.47
3.48
3.48
3.49
3.49
3.51
3.52
3.52
3.70
3.70
3.70
3.71
3.78
3.78
3.78
3.78
3.78
3.81
3.82
3.97
4.08
4.08
4.08
4.08
4.09
6.93
6.95
6.96
6.96
6.98
7.01
7.01
7.05
7.05
7.27
7.29
7.32
7.48
7.51
7.57
7.59
7.60
7.95
10.8
510
.90
ppm0 05050100100150150-253.79
-5.0
1
18.6
825
.99
26.0
526
.11
26.2
426
.33
31.5
3
55.9
457
.48
65.4
0
109.
5111
1.74
118.
7711
8.96
121.
2712
4.44
127.
87
136.
51
165.
5116
7.57

160
tert-butyl 3-(((2S,5S)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3,6-dioxopiperazin-2-
yl)methyl)-1H-indole-1-carboxylate. The general cyclization procedure given for
compound 3.58a was repeated.
1H-NMR (300 MHz; CDCl3): δ 8.01 (d, J = 8.2 Hz, 1H), 7.47 (dd, J = 7.7, 0.4 Hz, 1H),
7.35 (s, 1H), 7.23-7.10 (m, 2H), 4.16-4.12 (m, 1H), 3.87-3.84 (m, 3H), 3.63 (dd, J = 10.1,
3.1 Hz, 1H), 3.31 (dd, J = 10.1, 5.8 Hz, 2H), 3.05 (dd, J = 14.4, 8.8 Hz, 1H), 1.54 (s, 9H),
0.75 (s, 9H), -0.09 (s, 6H); 13C-NMR (101 MHz; CDCl3): δ 173.3, 171.6, 135.4, 130.61,
130.58, 130.50, 128.6, 124.8, 121.0, 120.2, 89.7, 70.5, 62.7, 60.5, 54.7, 54.5, 36.5, 33.7,
31.4, 24.1, 0.06, -0.00.
REF: TRW-III-187.
NBoc
HNNH
OTBS
O
O
5.3
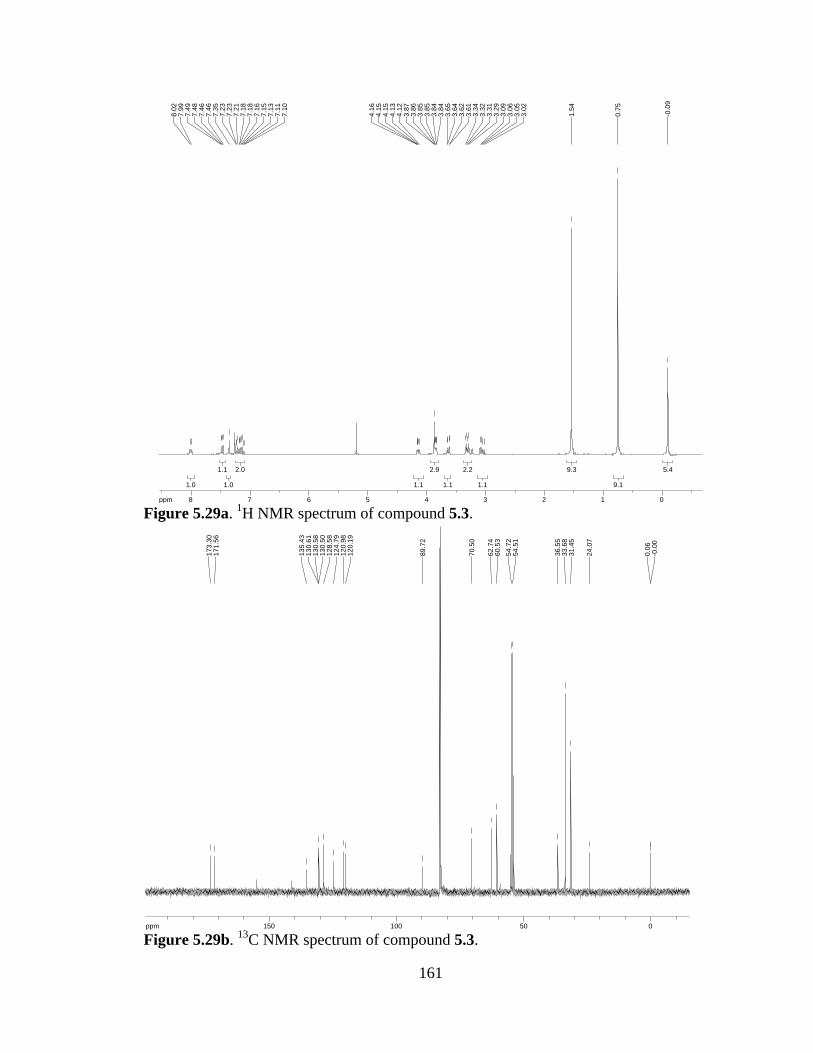
161
Figure 5.29a. 1H NMR spectrum of compound 5.3.
Figure 5.29b. 13C NMR spectrum of compound 5.3.
5.45.4
9.19.1
9.39.3
1.11.1
2.22.2
1.11.1
2.92.9
1.11.1
2.02.0
1.01.0
1.11.1
1.01.0
ppm0 01122334455667788-14.99
-0.0
9
0.75
1.54
3.02
3.05
3.06
3.09
3.29
3.31
3.32
3.34
3.61
3.62
3.64
3.65
3.84
3.84
3.85
3.85
3.86
3.87
4.12
4.13
4.15
4.15
4.16
7.10
7.11
7.13
7.15
7.16
7.18
7.18
7.21
7.23
7.23
7.35
7.46
7.46
7.48
7.49
7.99
8.02
ppm0 05050100100150150-253.79
-0.0
00.
06
24.0
7
31.4
533
.68
36.5
5
54.5
154
.72
60.5
362
.74
70.5
0
89.7
2
120.
1912
0.98
124.
7912
8.58
130.
5013
0.58
130.
6113
5.43
171.
5617
3.30

162
N-Boc-L-tryptophan-N’-SO2Ph (3.56). Compound 3.59 (14.8 g, 48.7 mmol) was
dissolved in THF (100 mL) and the resulting solution cooled to -78 °C. LHMDS (146
mL, 146.1 mmol) was added slowly over 5 minutes. The reaction was stirred at -78 °C
for 1 h, then PhSO2Cl (7.5 mL, 58.4 mmol) added in one portion. After stirring an
additional 2 h at -78 °C, the reaction was quenched by addition of 1:1 AcOH:EtOAc then
allowed to warm to r.t. The suspension was diluted with 1M HCl and the product
extracted in EtOAc. The combined organic layers were dried and concentrated to afford
crude 3.56, purified by SiO2 chromatography (5% AcOH, 45% hexanes, 50% CH2Cl2) to
afford pure 3.56 (17.12 g, 79% yield).
1H-NMR (300 MHz; DMSO-d6): δ 12.64-12.63 (m, 1H), 7.90-7.87 (m, 2H), 7.61 (t, J =
7.8 Hz, 2H), 7.56-7.50 (m, 2H), 7.25-7.20 (m, 2H), 7.16-7.13 (m, 2H), 4.20-4.17 (m, 1H),
3.11 (dd, J = 14.8, 4.2 Hz, 1H), 2.95 (dd, J = 14.8, 10.0 Hz, 1H), 1.31 (s, 9H).
REF: TRW-II-176, TRW-II-251.
NSO2Ph
CO2H
NHBoc
3.56

163
Figure 5.30a. 1H NMR spectrum of compound 3.56.
9.49.4
1.01.0
1.01.0
0.90.9
2.42.4
2.42.4
2.32.3
2.22.2
2.32.3
0.90.9
ppm0 02244668810101212-19.97
1.31
2.91
2.94
2.96
2.99
3.08
3.10
3.13
3.15
4.17
4.19
4.19
4.19
4.20
7.13
7.13
7.16
7.20
7.22
7.23
7.25
7.25
7.27
7.27
7.32
7.50
7.53
7.56
7.56
7.59
7.61
7.64
7.87
7.88
7.89
7.90
12.6
312
.63
12.6
312
.64
12.6
4

164
Bromopyrroloindoline (3.55e). A solution of dioxopiperazine 3.58e (2.47 g, 4.68 mmol)
in MeCN (50 mL) was cooled to 0 °C and to it added bromine (0.962 mL, 18.7 mmol)
dropwise. After stirring 5 minutes, 1:1 Na2S2O3:NaHCO3 was added and the product
extracted in EtOAc. The combined organic layers were washed with brine, dried over
Na2SO4 and concentrated. Purification by SiO2 chromatgraphy (10/40/50 iPrOH/hexanes/
CH2Cl2) afforded pure pyrroloindoline 3.55e in 38% yield (1.09 g).
1H-NMR (300 MHz; DMSO-d6): δ 8.06 (s, 1H), 7.87-7.84 (m, 2H), 7.66-7.53 (m, 4H),
7.42 (d, J = 2.3 Hz, 2H), 7.27-7.22 (m, 1H), 6.46 (s, 1H), 4.05 (d, J = 1.0 Hz, 1H), 3.77-
3.75 (m, 2H), 3.73-3.68 (m, 2H), 3.60-3.56 (m, 2H), 1.01 (d, J = 6.1 Hz, 9H), -0.02 (s,
6H).
REF: TRW-II-228, TRW-II-256.
NSO2Ph
NNH
O
O
OTBS
Br
3.55e

165
Figure 5.31a. 1H NMR spectrum of compound 3.55e.
1.81.8
1.81.8
1.81.8
1.21.2
0.80.8
0.90.9
1.81.8
4.34.3
2.22.2
1.01.0
ppm0 01122334455667788-19.97
-0.0
2
1.00
1.02
3.56
3.59
3.59
3.60
3.60
3.68
3.68
3.70
3.70
3.71
3.71
3.73
3.75
3.76
3.77
4.05
4.05
6.46
7.22
7.23
7.23
7.24
7.24
7.26
7.27
7.42
7.43
7.53
7.55
7.58
7.60
7.62
7.64
7.66
7.84
7.85
7.87
7.87
8.06

166
N-Me-bromopyrroloindoline (3.60). Dioxopiperazine 3.55e (2.09 g, 3.4 mmol) was
dissolved in acetone (50 mL). Potassium carbonate (11.73 g, 85 mmol) and MeI (27.5
mL, 442 mmol) were added, the flask covered with foil, and the reaction allowed to stir at
r.t. under Ar for 5 d. The crude reaction mixture was concentrated, partitioned between
EtOAc and H2O (200 mL each), and the product extracted in EtOAc. The combined
organic layers were dried and concentrated. SiO2 chromatography (10/40/50
iPrOH/hexanes/CH2Cl2) gave the pure title compound (1.69 g, 98% yield).
1H-NMR (300 MHz; CDCl3): δ 7.89-7.71 (m, J = 1.6 Hz, 2H), 7.69 (d, J = 8.2 Hz, 1H),
7.58-7.52 (m, 1H), 7.44 (ddd, J = 8.4, 6.8, 1.4 Hz, 2H), 7.38-7.32 (m, 2H), 7.21 (d, J =
7.0 Hz, 1H), 6.54 (d, J = 28.9 Hz, 1H), 4.08-3.78 (m, 4H), 3.17 (ddd, J = 28.8, 12.6, 5.2
Hz, 1H), 2.98 (s, 3H), 2.89-2.76 (m, 1H).
REF: TRW-II-235, TRW-II-258.
NSO2Ph
NNMe
O
O
OH
Br
3.60

167
Figure 5.32a. 1H NMR spectrum of compound 3.60.
1.31.3
3.23.2
1.31.3
4.64.6
1.01.0
0.90.9
1.91.9
2.32.3
1.01.0
0.90.9
2.02.0
ppm0 01122334455667788-19.97
2.76
2.77
2.78
2.81
2.82
2.83
2.87
2.89
2.98
3.09
3.11
3.14
3.15
3.19
3.21
3.23
3.25
3.78
3.79
3.82
3.82
3.83
3.90
3.91
3.92
3.92
3.93
3.94
3.95
3.96
3.96
3.97
3.97
3.99
3.99
4.00
4.00
4.01
4.02
4.03
4.04
4.05
4.07
4.08
6.48
6.58
7.22
7.26
7.32
7.33
7.33
7.34
7.34
7.35
7.35
7.36
7.36
7.36
7.38
7.38
7.41
7.42
7.43
7.44
7.44
7.46
7.46
7.52
7.53
7.53
7.55
7.55
7.56
7.58
7.68
7.71
7.85
7.85
7.86
7.86
7.87
7.87
7.89
7.89

168
OTBS-N-Me-bromopyrroloindoline (3.61). The same general procedure for TBS
protection was followed as for 3.57e. Instead of carrying the crude product forward as
before, it was necessary for the subsequent radical bromination to purify the product by
SiO2 chromatography (20-60% EtOAc in hexanes) to afford 3.61 (47% yield).
1H-NMR (300 MHz; CDCl3): δ 7.89 (dd, J = 8.1, 1.0 Hz, 2H), 7.70 (dd, J = 7.9, 0.6 Hz,
1H), 7.56-7.53 (m, 1H), 7.48-7.43 (m, 2H), 7.35-7.32 (m, 2H), 7.21-7.16 (m, 1H), 6.63
(s, 1H), 4.28 (dd, J = 10.2, 1.3 Hz, 1H), 4.06 (s, 1H), 3.94 (dd, J = 10.1, 2.7 Hz, 1H), 3.78
(dd, J = 12.4, 4.5 Hz, 1H), 3.09 (dd, J = 12.0, 4.5 Hz, 1H), 2.91 (s, 3H), 2.73 (t, J = 12.2
Hz, 1H), 0.89 (s, 9H), 0.10 (s, 6H).
REF: TRW-II-236, TRW-II-242.
NSO2Ph
NNMe
O
O
OTBS
Br
3.61

169
Figure 5.33a. 1H NMR spectrum of compound 3.61.
6.86.8
10.410.4
1.11.1
3.33.3
1.21.2
1.11.1
1.31.3
1.21.2
1.11.1
1.01.0
1.21.2
2.32.3
2.42.4
1.21.2
0.90.9
2.02.0
ppm0 01122334455667788-19.97
0.10
0.89
2.69
2.73
2.78
2.91
3.06
3.08
3.10
3.12
3.75
3.77
3.79
3.81
3.92
3.93
3.95
3.96
4.06
4.26
4.27
4.30
4.30
6.63
7.16
7.18
7.18
7.18
7.19
7.21
7.21
7.32
7.32
7.32
7.34
7.34
7.35
7.35
7.35
7.43
7.45
7.48
7.48
7.53
7.54
7.54
7.55
7.56
7.56
7.69
7.69
7.72
7.72
7.87
7.88
7.90
7.90

170
Diol (3.62). To a solution of Br-dioxopiperazine 3.61 (710 mg, 1.14 mmol) in CCl4 (40
mL) was added NBS (406 mg, 2.28 mmol) and V-70 (89 mg, 0.29 mmol). The resulting
suspension was stirred 8 h. at r.t., then filtered through a cotton plug, the solid washed
with CCl4, and the combined filtrate concentrated. The resulting residue was dissolved in
1:1 MeCN:pH 7 phosphate buffer (58 mL) and stirred 3 h. at r.t. The product was
extracted in EtOAc (3x), washed with brine, dried, and concentrated to afford the diol in
97% yield (720 mg), carried forward to the next step as the crude material.
REF: TRW-II-244, 245, TRW-II-368, 370.
NSO2Ph
NNMe
O
O
OTBS
Br
OH
OH
3.62

171
Epidithiodioxopiperazine (3.63). Hydrogen sulfide (ca. 8 mL) gas was condensed in a
sealed tube capped with a rubber septum at -78 °C. Diol 3.62 (125 mg, 0.19 mmol) and
BF3·OEt2 (0.119 mL, 0.95 mmol) were added as a solution in CH2Cl2 (7.6 mL). The
rubber septum was removed and the sealed tube capped with a teflon cap and placed
behind a blast shield. The reaction mixture stirred at r.t. for 90 min., recooled to -78 °C,
uncapped and sealed with a rubber septum, then allowed to warm to r.t. with 3 NaOH and
1 bleach trap in place as the gas vented. The resulting solution was diluted with EtOAc,
washed with sat’d NH4Cl, and the product extracted in EtOAc. Iodine (100 mg, 2 eq) was
added to the EtOAc extracts and the mixture allowed to stir 1 min. 10% Na2S2O3 was
added and the product extracted in EtOAc, dried, and concentrated. Purification by SiO2
chromatography (1:1 hexanes:EtOAc) afforded pure epidithiodioxopiperazine 3.63 in
42% yield (44.9 mg).
1H-NMR (300 MHz; CDCl3): δ 7.78 (dd, J = 8.4, 1.1 Hz, 2H), 7.64 (d, J = 8.2 Hz, 1H),
7.52-7.49 (m, 1H), 7.41-7.36 (m, 4H), 7.28-7.26 (m, 1H), 6.49 (s, 1H), 4.33 (d, J = 12.8
Hz, 1H), 4.18 (d, J = 12.7 Hz, 1H), 3.85 (d, J = 15.3 Hz, 1H), 3.21 (d, J = 15.3 Hz, 1H),
3.13 (s, 3H); 13C-NMR (101 MHz; CDCl3): δ 176.9, 163.7, 134.83, 134.82, 134.3, 133.8,
132.8, 131.23, 131.22, 131.0, 123.9, 114.6, 72.4, 66.2, 37.6, 31.3, 26.9, 23.7, 20.0;
HRMS (ESI-APCI): [M+H]+ 567.9670 calcd for C21H19BrN3O5S3, found: 567.9641. REF:
TRW-II-246, 247, TRW-II-372, 373.
NSO2Ph
NNMe
O
O
OH
BrSS
3.63
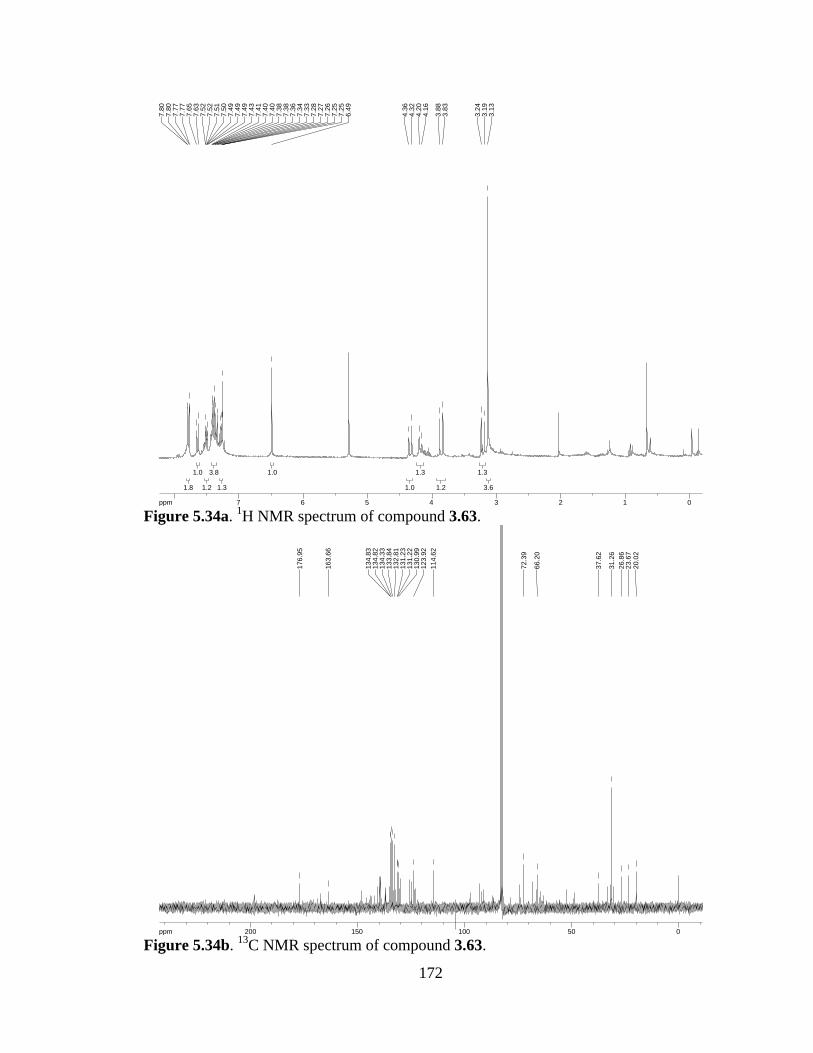
172
Figure 5.34a. 1H NMR spectrum of compound 3.63.
Figure 5.34b. 13C NMR spectrum of compound 3.63.
3.63.6
1.31.3
1.21.2
1.31.3
1.01.0
1.01.0
1.31.3
3.83.8
1.21.2
1.01.0
1.81.8
ppm0 0112233445566778-19.973.
133.
193.
24
3.83
3.88
4.16
4.20
4.32
4.36
6.49
7.25
7.25
7.26
7.27
7.28
7.33
7.34
7.36
7.38
7.38
7.40
7.40
7.41
7.43
7.49
7.49
7.49
7.50
7.51
7.52
7.52
7.63
7.65
7.77
7.77
7.80
7.80
ppm0 05050100100150150200200-253.79
20.0
223
.67
26.8
631
.26
37.6
2
66.2
0
72.3
9
114.
6212
3.92
130.
9913
1.22
131.
2313
2.81
133.
8413
4.33
134.
8213
4.83
163.
66
176.
95

173
N-Boc-L-tryptophan (3.59). To a solution of L-tryptophan (50 g, 245 mmol) in 1:1
THF:H2O (700 mL) was added NaOH (10.8 g, 270 mmol) and Boc2O (58.9 g, 270
mmol). The resulting solution was stirred O/N at r.t. The volume was reduced and the
product extracted in CH2Cl2. The aqueous layer was acidified with 1M HCl to pH 4, and
the product extracted in CH2Cl2. The combined organic layers were dried over Na2SO4
and concentrated to afford 3.59 (70.23 g, 94% yield).
1H-NMR (300 MHz; CDCl3): δ 7.50 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.09-
6.98 (m, 2H), 6.96 (s, 1H), 4.45 (t, J = 5.4 Hz, 1H), 3.28-3.17 (m, 2H), 1.31 (s, 9H).
REF: TRW-II-252.
NH
CO2H
NHBoc
3.59

174
Figure 5.35a. 1H NMR spectrum of compound 3.59.
9.19.1
2.02.0
0.80.8
1.11.1
1.91.9
1.31.3
1.01.0
ppm0 011223344556677-19.97
1.31
3.17
3.19
3.22
3.23
3.26
3.26
3.27
3.27
3.28
3.28
4.43
4.45
4.47
6.96
6.98
7.01
7.01
7.03
7.04
7.06
7.06
7.08
7.09
7.25
7.28
7.49
7.51

175
N-Boc-L-tryptophan-OMe (3.34). To a solution of N-Boc-L-tryptophan (30 g, 98.7
mmol) in DMF (200 mL) at 0 °C was added MeI (9.2 mL, 148 mmol) and KHCO3 (19.7
g, 197 mmol). After stirring O/N at r.t., the mixture was diluted with ether, washed with
H2O (2x), 1M KHSO4 (1x), and brine (1x). The combined organic layers were dried and
concentrated to afford the title compound (29.86 g, 95% yield).
1H-NMR (300 MHz; CDCl3): δ 8.10-8.08 (m, 1H), 7.55 (d, J = 7.9 Hz, 1H), 7.36 (d, J =
8.0 Hz, 1H), 7.22-7.09 (m, 2H), 7.01 (d, J = 2.4 Hz, 1H), 5.09-5.06 (m, 1H), 4.68-4.62
(m, 1H), 3.68 (s, 3H), 3.29 (d, J = 5.2 Hz, 2H), 1.43 (s, 9H).
REF: TRW-II-273, TRW-II-285, TRW-II-349.
NH
CO2Me
NHBoc
3.34

176
Figure 5.36a. 1H NMR spectrum of compound 3.34.
9.49.4
1.81.8
2.92.9
0.90.9
0.80.8
0.90.9
1.91.9
1.11.1
1.11.1
1.01.0
ppm0 01122334455667788-19.97
1.43
3.28
3.30
3.68
4.62
4.62
4.62
4.62
4.62
4.63
4.64
4.64
4.64
4.65
4.65
4.65
4.66
4.66
4.66
4.66
4.67
4.68
4.68
4.68
5.06
5.06
5.06
5.08
5.09
7.01
7.01
7.09
7.10
7.12
7.14
7.15
7.17
7.17
7.19
7.19
7.20
7.22
7.22
7.34
7.37
7.54
7.57
8.08
8.08
8.08
8.08
8.08
8.09
8.09
8.09
8.10

177
N-Boc-L-tryptophan-OMe-N’-SO2Ph (3.64). N-Boc-L-tryptophan-OMe (10.0 g, 31.4
mmol), NaOH (3.77 g, 94.2 mmol), and Bu4NHSO4 (544 mg, 1.6 mmol) were suspended
in CH2Cl2 (150 mL), then PhSO2Cl (4.8 mL, 37.7 mmol) added dropwise. After stirring 6
h. at r.t., aqueous NH4Cl (300 mL) and EtOAc (300 mL) were added and the product
extracted in EtOAc (3x). The combined organic extracts were dried and concentrated to
afford the title compound (14.4 g, >99% yield).
1H-NMR (300 MHz; CDCl3): δ 7.97 (d, J = 8.2 Hz, 1H), 7.84 (d, J = 7.5 Hz, 2H), 7.56-
7.50 (m, 1H), 7.47-7.44 (m, 2H), 7.42 (d, J = 7.2 Hz, 1H), 7.36 (s, 1H), 7.31-7.20 (m,
2H), 5.05 (dt, J = 7.9, 0.6 Hz, 1H), 4.66-4.59 (m, 1H), 3.61 (s, 3H), 3.27-3.11 (m, 2H),
1.44 (s, 9H).
REF: TRW-II-263, TRW-II-275, TRW-II-350.
NSO2Ph
CO2Me
NHBoc
3.64

178
Figure 5.37a. 1H NMR spectrum of compound 3.64.
8.88.8
1.91.9
2.72.7
0.90.9
0.90.9
2.12.1
1.01.0
0.70.7
2.12.1
1.11.1
1.81.8
1.01.0
ppm1 122334455667788-19.97
1.44
3.11
3.11
3.11
3.12
3.12
3.12
3.13
3.13
3.13
3.14
3.15
3.15
3.15
3.15
3.15
3.16
3.17
3.17
3.18
3.18
3.19
3.19
3.20
3.20
3.20
3.22
3.25
3.25
3.25
3.25
3.26
3.27
3.27
3.27
3.27
3.27
3.61
4.59
4.59
4.59
4.59
4.60
4.60
4.60
4.61
4.62
4.63
4.63
4.63
4.63
4.64
4.65
4.65
4.65
4.66
5.03
5.04
5.04
5.06
5.06
5.06
7.20
7.20
7.23
7.25
7.25
7.26
7.26
7.28
7.29
7.31
7.31
7.36
7.41
7.43
7.44
7.44
7.44
7.44
7.46
7.47
7.47
7.50
7.50
7.51
7.52
7.53
7.54
7.55
7.56
7.83
7.85
7.95
7.98

179
Bromopyrroloindoline (3.65). To a solution of N-Boc-L-tryptophan-OMe-N’-SO2Ph (42
g, 91.5 mmol) in CH2Cl2 (1000 mL) was added NBS (16.29 g, 91.5 mmol) and PPTS
(22.97 g, 91.5 mmol). The reaction was stirred O/N at r.t., then washed with 10%
NaHCO3 and 10% Na2S2O4 (1:1), and the organic extract concentrated to afford 3.65 in
quantitative yield.
1H-NMR (300 MHz; CDCl3): δ 7.83-7.79 (bs, 2H), 7.57 (d, J = 8.0 Hz, 1H), 7.46 (ddd, J
= 8.4, 6.4, 1.6 Hz, 1H), 7.34 (td, J = 7.7, 1.4 Hz, 3H), 7.27-7.25 (m, 1H), 7.17 (td, J = 7.5,
1.0 Hz, 1H), 6.33-6.31 (bs, 1H), 3.85-3.80 (m, 1H), 3.73 (s, 3H), 3.06 (dd, J = 12.6, 5.9
Hz, 1H), 2.81-2.73 (m, 1H), 1.54-1.53 (bs, 9H); 13C-NMR (101 MHz; CDCl3): δ 171.9,
138.0, 133.8, 133.4, 130.9, 129.2, 128.6, 128.3, 127.3, 126.6, 124.9, 124.3, 123.9, 123.3,
113.6, 86.7, 80.0, 59.4, 52.4, 28.26, 28.17, 27.8, 21.0, 14.2.
REF: TRW-II-264, TRW-II-277, TRW-II-351.
NSO2Ph
NBocCO2Me
Br
3.65

180
Figure 5.38a. 1H NMR spectrum of compound 3.65.
Figure 5.38b. 13C NMR spectrum of compound 3.65.
9.39.3
1.11.1
1.11.1
3.03.0
1.01.0
0.90.9
1.11.1
1.91.9
3.13.1
1.31.3
1.01.0
1.91.9
ppm0 0112233445566778-19.97
1.53
1.53
1.53
1.53
1.54
1.54
2.73
2.73
2.77
2.77
2.80
2.81
3.03
3.05
3.08
3.10
3.73
3.80
3.80
3.82
3.82
3.83
3.85
6.31
6.31
6.31
6.31
6.31
6.31
6.32
6.32
6.32
6.32
6.32
6.33
6.33
7.14
7.15
7.17
7.17
7.19
7.20
7.25
7.25
7.25
7.26
7.26
7.27
7.32
7.32
7.34
7.35
7.37
7.37
7.43
7.44
7.46
7.46
7.47
7.48
7.49
7.56
7.59
7.79
7.79
7.79
7.80
7.80
7.80
7.80
7.80
7.81
7.81
7.81
7.82
7.82
7.82
7.82
7.83
ppm0 05050100100150150200200-253.79
28.1
728
.26
52.2
852
.43
59.4
4
76.7
677
.08
77.4
0
113.
6412
3.28
123.
8912
4.90
126.
6112
8.28
128.
5912
9.21
130.
8713
3.39
133.
76
171.
91

181
Indole (3.66). Bromopyrroloindoline 3.65 (16.41 g, 30.6 mmol) was dissolved in
CH3CN, and to the resulting solution added N-Boc-L-tryptophan-OMe (6.49 g, 20.4
mmol). The solution was cooled to 0 °C, then KOtBu (4.57 g, 40.8 mmol) added. After
stirring 1 h at 0 °C, the reaction was quenched with NaHCO3, extracted in CH2Cl2,
washed with brine, dried, and concentrated. Purification by SiO2 chromatography (40%
EtOAc in hexanes) afforded 3.66 in 34% yield (5.36 g).
1H-NMR (300 MHz; CDCl3): δ 7.85-7.79 (m, 1H), 7.85-7.79 (m, 1H), 7.60-7.43 (m,
2H), 7.60-7.43 (m, 2H), 7.34 (t, J = 7.7 Hz, 3H), 7.34 (t, J = 7.7 Hz, 3H), 7.22-7.01 (m,
7H), 7.22-7.01 (m, 7H), 6.85 (s, 1H), 6.85 (s, 1H), 6.31 (bs, 1H), 6.31 (s, 1H), 5.06 (bs,
1H), 5.06 (s, 1H), 4.64-4.61 (m, 1H), 4.64-4.61 (m, 1H), 3.73 (s, 3H), 3.73 (s, 3H), 3.68
(s, 3H), 3.68 (s, 3H), 3.28-3.21 (m, 2H), 3.28-3.21 (m, 2H), 3.07 (dd, J = 12.6, 5.9 Hz,
1H), 3.07 (dd, J = 12.6, 5.9 Hz, 1H), 2.77 (t, J = 11.6 Hz, 1H), 2.77 (t, J = 11.6 Hz, 1H),
1.58-1.55 (bs, 9H), 1.58-1.55 (m, 9H), 1.43 (s, 9H), 1.43 (s, 9H); 13C-NMR (101 MHz;
CDCl3): δ 172.4, 161.2, 154.9, 143.2, 133.7, 132.9, 132.2, 131.61, 131.58, 131.52, 130.5,
128.9, 128.5, 128.1, 127.0, 126.46, 126.35, 125.2, 124.1, 122.4, 121.82, 121.76, 120.24,
120.17, 119.57, 119.43, 114.8, 82.2, 59.44, 59.36, 54.4, 53.3, 52.1, 51.6, 37.9, 37.7,
28.18, 28.14, 28.08, 28.01, 27.99, 27.97, 27.94, 27.93, 27.91, 27.79; HRMS (ESI-
APCI+): [M+Na]+ 797.2832 calcd for C40H46N4NaO10S, found: 797.2829.
REF: TRW-II-266, TRW-II-278, TRW-II-352.
NSO2Ph
NBoc
NCO2Me
MeO2C
BocHN
3.66

182
Figure 5.39a. 1H NMR spectrum of compound 3.66.
Figure 5.39b. 13C NMR spectrum of compound 3.66.
9.59.5
8.88.8
1.01.0
1.21.2
2.42.4
2.72.7
3.43.4
1.01.0
0.90.9
0.90.9
0.70.7
6.96.9
2.62.6
1.81.8
0.90.9
ppm0 01122334455667788-19.97
1.43
1.55
1.55
1.55
1.55
1.56
1.56
1.58
1.58
2.73
2.77
2.81
3.04
3.06
3.08
3.10
3.21
3.21
3.21
3.23
3.23
3.26
3.28
3.28
3.68
3.73
4.61
4.63
4.63
4.64
5.06
6.31
6.85
7.01
7.01
7.06
7.06
7.10
7.10
7.10
7.10
7.14
7.15
7.17
7.19
7.21
7.21
7.22
7.32
7.34
7.37
7.43
7.44
7.46
7.46
7.46
7.49
7.49
7.51
7.54
7.54
7.54
7.56
7.56
7.59
7.59
7.60
7.79
7.80
7.80
7.82
7.83
7.85
7.85
ppm50 50100100150150-253.79
27.7
927
.91
27.9
327
.94
27.9
727
.99
28.0
128
.08
28.1
428
.18
37.7
137
.92
51.5
952
.15
53.3
454
.35
59.3
659
.44
82.2
2
114.
7611
9.43
119.
5712
0.17
120.
2412
1.76
121.
8212
2.43
124.
1012
5.16
126.
3512
6.46
126.
9912
8.09
128.
4912
8.93
130.
5013
1.52
131.
5813
1.61
132.
2013
2.91
133.
6514
3.17
154.
8816
1.16
172.
42

183
Diacid (3.67). To a solution of 3.66 (5.35 g, 6.9 mmol) in 2:1 THF:H2O (24 mL) was
added LiOH (828 mg, 34.5 mmol). The reaction stirred O/N before acidifying with 10%
KHSO4. The product was extracted in EtOAc, and the combined organic extracts washed
with brine, dried, and concentrated. SiO2 chromatography (1:1 hexanes:EtOAc) gave the
pure diacid (5.01 g, 97% yield).
1H-NMR (300 MHz; CDCl3): δ 9.51 (bs, 2H), 7.70-7.67 (m, 1H), 7.55 (dt, J = 7.0, 0.8
Hz, 1H), 7.42-7.38 (m, 1H), 7.19-7.13 (m, 8H), 6.95-6.88 (m, 2H), 6.70 (d, J = 5.8 Hz,
1H), 6.06-5.93 (m, 1H), 4.99-4.92 (m, 1H), 4.54-4.40 (m, 1H), 3.43-3.34 (m, 1H), 3.19-
3.05 (m, 1H), 2.81-2.72 (m, 2H), 1.56 (s, 8H), 1.42 (d, J = 11.7 Hz, 9H); 13C-NMR (101
MHz; CDCl3): δ 178.03, 177.95, 159.41, 159.30, 146.7, 141.6, 137.56, 137.44, 136.8,
136.3, 135.3, 134.47, 134.33, 132.8, 132.4, 131.7, 130.8, 130.4, 129.5, 128.35, 128.18,
126.2, 123.97, 123.91, 123.64, 123.63, 123.58, 123.54, 123.52, 123.50, 123.47, 113.45,
113.42, 98.4, 86.33, 86.25, 83.81, 83.68, 64.5, 63.3, 32.0, 31.8.
REF: TRW-II-268, TRW-II-446.
NSO2Ph
NBoc
NCO2H
HO2C
BocHN
3.67

184
Figure 5.40a. 1H NMR spectrum of compound 3.67.
Figure 5.40b. 13C NMR spectrum of compound 3.67.
9.09.0
8.38.3
1.71.7
1.01.0
0.90.9
0.90.9
1.41.4
0.90.9
1.11.1
2.02.0
8.28.2
1.31.3
1.21.2
1.31.3
2.12.1
ppm0 011223344556677889910-19.97
1.40
1.44
1.56
2.72
2.73
2.76
2.76
2.77
2.77
2.77
2.78
2.78
2.80
2.80
2.81
2.81
2.81
2.81
3.05
3.05
3.05
3.06
3.06
3.06
3.08
3.14
3.14
3.14
3.15
3.15
3.16
3.18
3.19
3.34
3.35
3.35
3.35
3.37
3.37
3.38
3.38
3.38
3.38
3.38
3.39
3.39
3.39
3.41
3.42
3.42
3.42
3.43
4.40
4.41
4.41
4.42
4.42
4.51
4.51
4.51
4.52
4.52
4.52
4.53
4.53
4.53
4.53
4.54
4.92
4.92
4.92
4.93
4.93
4.93
4.93
4.94
4.94
4.96
4.96
4.96
4.96
4.96
4.97
4.99
4.99
4.99
4.99
5.93
5.93
5.93
6.02
6.02
6.03
6.03
6.03
6.04
6.04
6.05
6.05
6.05
6.06
6.69
6.71
6.88
6.88
6.90
6.92
6.92
6.93
6.93
6.93
6.93
6.93
6.94
6.94
6.95
6.95
7.13
7.14
7.14
7.16
7.16
7.16
7.17
7.17
7.17
7.17
7.18
7.18
7.18
7.19
7.19
7.19
7.37
7.38
7.40
7.42
7.42
7.42
7.54
7.54
7.54
7.56
7.56
7.57
7.67
7.70
7.70
7.70
9.51
ppm0 05050100100150150-253.79
31.8
432
.05
63.2
564
.49
83.6
883
.81
86.2
586
.33
98.4
211
3.42
113.
4512
3.47
123.
5012
3.52
123.
5412
3.58
123.
6312
3.64
123.
9112
3.97
126.
1612
8.18
128.
3512
9.46
130.
3613
0.81
131.
6713
2.40
132.
8113
4.33
134.
4713
5.28
136.
2613
6.81
137.
4413
7.56
141.
6214
6.74
159.
3015
9.41
177.
9517
8.03

185
Tetrapeptide (3.68). To a solution of L-serine-OMe·HCl (2.09 g, 13.4 mmol) in CH2Cl2
(70 mL) was added iPr2NEt (2.3 mL, 13.4 mmol). After stirring 15 minutes, the
suspension was added to a stirring solution of 3.67 (5.01 g, 6.69 mmol) and EDCI (2.57
g, 13.4 mmol) in CH2Cl2. The reaction was stirred O/N at r.t., then concentrated and
purified through a plug of SiO2 (100% EtOAc) to afford 3.68 (2.11 g, 33% yield).
1H-NMR (300 MHz; DMSO-d6): δ 8.10-8.08 (broad, 1H), 7.73-7.58 (m, 5H), 7.44-7.31
(m, 3H), 7.10-7.07 (m, 3H), 6.83-6.74 (m, 1H), 6.44-6.42 (m, 1H), 5.04-4.86 (m, 3H),
4.34-4.22 (m, 3H), 3.61-3.43 (m, 9H), 3.02-2.83 (m, 1H), 2.76-2.51 (m, 2H), 1.34-1.27
(m, 18H).
REF: TRW-II-270, TRW-II-447.
NSO2Ph
NBoc
NBocHN O
NH CO2Me
OH
O
NH
MeO2C
HO
3.68

186
Figure 5.41a. 1H NMR spectrum of compound 3.68.
13.713.7
2.02.0
1.11.1
8.88.8
2.92.9
2.62.6
0.70.7
1.11.1
2.62.6
2.82.8
4.84.8
1.01.0
ppm1 122334455667788-19.97
1.27
1.29
1.33
1.34
2.51
2.51
2.51
2.52
2.52
2.52
2.65
2.65
2.65
2.66
2.72
2.72
2.73
2.75
2.75
2.76
2.76
2.76
2.76
2.76
2.83
2.83
2.83
2.84
2.84
2.84
2.85
2.96
2.96
2.97
2.97
2.97
2.98
2.98
3.01
3.01
3.02
3.43
3.44
3.44
3.44
3.45
3.45
3.45
3.45
3.45
3.46
3.46
3.57
3.61
4.22
4.22
4.22
4.23
4.23
4.23
4.23
4.32
4.32
4.33
4.34
4.34
4.86
4.86
4.86
4.87
4.87
4.87
4.87
4.88
4.88
4.89
5.02
5.02
5.02
5.03
5.04
5.04
5.04
5.04
5.04
6.42
6.43
6.43
6.43
6.43
6.44
6.44
6.44
6.44
6.74
6.77
6.77
6.77
6.80
6.80
6.80
6.83
6.83
7.07
7.07
7.07
7.08
7.08
7.08
7.08
7.09
7.09
7.09
7.10
7.10
7.31
7.31
7.31
7.32
7.32
7.32
7.33
7.33
7.39
7.41
7.43
7.43
7.43
7.44
7.44
7.44
7.58
7.59
7.59
7.61
7.61
7.61
7.61
7.70
7.71
7.72
7.72
7.72
7.73
7.73
8.08
8.08
8.10
8.10

187
TBS2-Tetrapeptide (3.69). To a solution of diol 3.68 (2.11 g, 2.22 mmol) in DMF (12
mL) was added imidazole (728 mg, 10.7 mmol) and TBSCl (1.74 g, 11.5 mmol). The
reaction mixture stirred O/N at r.t., then was concentrated. The crude residue was taken
up in EtOAc and washed with water (3x), brine (1x), dried over Na2SO4, and
concentrated to afford the title compound (2.27 g, 87% yield).
1H-NMR (300 MHz; DMSO-d6): δ 8.02-7.99 (m, 1H), 7.72-7.42 (m, 6H), 7.37-7.21 (m,
2H), 7.13-6.86 (m, 4H), 6.48-6.39 (m, 1H), 4.92-4.89 (m, 1H), 4.43-4.39 (m, 1H), 4.21-
4.06 (m, 2H), 3.82-3.69 (m, 3H), 3.63-3.58 (m, 6H), 3.00-2.89 (m, 1H), 2.86-2.80 (m,
1H), 2.66-2.52 (m, 3H), 1.31 (dd, J = 22.1, 8.9 Hz, 18H), 0.81 (s, 18H), -0.01--0.03 (m,
12H).
REF: TRW-II-272.
NSO2Ph
NBoc
NBocHN O
NH CO2Me
OTBS
O
NH
MeO2C
TBSO
3.69

188
Figure 5.42a. 1H NMR spectrum of compound 3.69.
12.612.6
18.018.0
14.214.2
3.23.2
0.70.7
0.60.6
6.16.1
3.13.1
1.61.6
1.01.0
0.90.9
1.11.1
3.93.9
2.12.1
6.26.2
1.11.1
ppm0 01122334455667788-19.97
-0.0
3-0
.02
-0.0
10.
811.
261.
291.
341.
362.
522.
522.
522.
542.
552.
552.
552.
652.
652.
652.
662.
802.
802.
812.
812.
822.
832.
832.
832.
832.
842.
842.
842.
862.
892.
892.
902.
942.
942.
952.
952.
952.
962.
962.
992.
992.
993.
003.
583.
623.
633.
693.
693.
723.
743.
743.
803.
824.
064.
064.
074.
074.
204.
204.
204.
214.
214.
394.
394.
404.
404.
414.
424.
424.
424.
434.
434.
894.
894.
914.
914.
914.
914.
914.
926.
396.
396.
396.
406.
406.
406.
406.
416.
416.
466.
476.
476.
476.
476.
486.
866.
896.
896.
896.
896.
896.
966.
966.
967.
027.
037.
037.
037.
037.
047.
047.
047.
127.
127.
127.
137.
217.
227.
227.
227.
227.
227.
367.
367.
377.
377.
427.
437.
457.
557.
557.
567.
567.
567.
567.
607.
607.
677.
677.
697.
727.
727.
727.
997.
998.
018.
028.
028.
028.
02

189
Diamine (5.4). To a solution of 3.66 (2 g, 2.58 mmol) in CH2Cl2 (50 mL) at 0 °C was
added TFA (4.8 mL). The resulting solution was warmed to r.t. and stirred for 2 h under
Ar. The reaction was quenched with sat’d NaHCO3, and the product extracted in CH2Cl2.
The combined organic layers were dried and concentrated to afford 5.4 in quantitative
yield (1.48 g).
1H-NMR (300 MHz; DMSO-d6): δ 7.69 (d, J = 7.4 Hz, 2H), 7.63-7.58 (m, 2H), 7.50-
7.36 (m, 6H), 7.20-7.15 (m, 1H), 7.09-7.04 (m, 1H), 6.99-6.98 (m, 1H), 6.82-6.75 (m,
2H), 5.89 (d, J = 4.1 Hz, 1H), 4.51 (t, J = 4.2 Hz, 1H), 4.33-4.29 (m, 1H), 3.78 (s, 1H),
3.52 (d, J = 6.2 Hz, 3H), 3.39-3.32 (m, 3H), 2.95-2.78 (m, 2H), 2.71-2.65 (m, 2H),
HRMS (ESI-APCI-): [M-H]- 574.1886 calcd for C30H30N4O6S, found: 574.1867.
REF: TRW-II-282, TRW-II-289, TRW-II-292, TRW-II-300, TRW-II-335, TRW-II-353.
NSO2Ph
NH
NCO2Me
MeO2C
H2N
5.4

190
Figure 5.43a. 1H NMR spectrum of compound 5.4.
1.71.7
2.02.0
2.42.4
3.23.2
1.01.0
1.11.1
1.01.0
0.90.9
2.32.3
1.31.3
1.31.3
1.21.2
6.36.3
2.22.2
2.02.0
ppm0 01122334455667788-19.97
2.65
2.65
2.69
2.69
2.69
2.70
2.71
2.78
2.81
2.81
2.81
2.83
2.88
2.90
2.95
2.95
3.32
3.33
3.33
3.37
3.37
3.37
3.39
3.39
3.51
3.53
3.78
4.29
4.30
4.30
4.31
4.32
4.32
4.33
4.33
4.50
4.51
4.52
5.88
5.90
6.75
6.77
6.80
6.80
6.82
6.82
6.98
6.98
6.99
7.04
7.05
7.06
7.06
7.09
7.15
7.17
7.17
7.19
7.20
7.36
7.41
7.41
7.43
7.44
7.44
7.46
7.47
7.50
7.58
7.60
7.60
7.61
7.61
7.63
7.68
7.71

191
Tripeptide (3.71). N-Cbz-L-serine(OTBS) (2.52 g, 7.14 mmol), iPr2NEt (1.3 mL, 7.14
mmol), and EDCI (1.37 g, 7.14 mmol) were added to a solution of 5.4 (2.05 g, 3.57
mmol) in CH2Cl2 (20 mL). The reaction stirred O/N, then was concentrated and purified
through an SiO2 plug to afford pure 3.71 (2.37 g, 73% yield).
1H-NMR (300 MHz; DMSO-d6): δ 8.48-8.37 (m, 1H), 7.70-7.58 (m, 5H), 7.48-7.42 (m,
3H), 7.37-7.29 (m, 8H), 7.09-6.99 (m, 3H), 6.79-6.71 (m, 2H), 5.90-5.87 (m, 1H), 5.03-
4.96 (m, 2H), 4.49-4.40 (m, 2H), 4.31-4.29 (m, 1H), 4.23-4.19 (m, 1H), 3.78 (s, 1H),
3.64-3.61 (m, 1H), 3.53 (d, J = 4.4 Hz, 3H), 3.41-3.34 (m, 1H), 3.19 (d, J = 9.1 Hz, 3H),
3.01-2.93 (m, 1H), 2.68-2.64 (m, 1H), 0.78 (d, J = 10.3 Hz, 9H), -0.02 (s, 6H); HRMS
(ESI-APCI): [M+H]+ 910.3517 calcd for C47H56N5O10SSi, found: 910.3543.
REF: TRW-II-354, TRW-II-341, TRW-II-359.
NSO2Ph
NH
NCO2Me
MeO2C
NH
O
TBSOCbzHN
3.71

192
Figure 5.44a. 1H NMR spectrum of compound 3.71.
6.86.8
9.09.0
1.21.2
1.41.4
2.82.8
0.70.7
3.13.1
1.41.4
1.41.4
1.11.1
1.11.1
2.32.3
2.22.2
0.90.9
2.22.2
3.13.1
8.48.4
3.23.2
5.05.0
1.31.3
ppm0 01122334455667788-19.97
-0.0
20.
760.
792.
642.
642.
662.
672.
672.
672.
682.
682.
682.
932.
932.
942.
942.
952.
952.
962.
962.
982.
982.
993.
003.
003.
003.
013.
173.
203.
523.
533.
613.
613.
623.
633.
633.
643.
643.
643.
643.
784.
194.
214.
214.
214.
214.
224.
234.
234.
294.
304.
314.
314.
314.
404.
404.
484.
494.
494.
494.
964.
994.
995.
035.
875.
885.
885.
895.
895.
906.
716.
726.
726.
726.
726.
736.
736.
786.
786.
796.
796.
996.
997.
067.
087.
097.
297.
317.
377.
377.
427.
447.
447.
447.
487.
507.
507.
587.
617.
617.
637.
637.
677.
677.
677.
697.
707.
70

193
(R)-tert-butyl 3-(2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxopropyl)-1H-indole-
1-carboxylate (5.5). Powdered NaOH (19.0 g, 475 mmol) was added to a stirring
suspension of D-Trp-OMe·HCl (24.14 g, 95 mmol) and Bu4NHSO4 (3.23 g, 9.5 mmol) in
CH2Cl2 (950 mL). After stirring 2.5 h, Boc2O (62.1 g, 285 mmol) was added, and the
resulting suspension stirred O/N at r.t. The mixture was filtered through celite,
concentrated, and purified by SiO2 chromatography (9:1 hexanes:EtOAc) to afford N,N’-
Boc2-D-Trp-OMe (18.53 g, 47% yield).
1H-NMR (300 MHz; CDCl3): δ 8.11 (d, J = 7.4 Hz, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.39
(s, 1H), 7.27 (td, J = 16.1, 8.4 Hz, 2H), 5.11 (d, J = 7.2 Hz, 1H), 4.65 (q, J = 6.4 Hz, 1H),
3.69 (s, 3H), 3.22 (qd, J = 13.5, 5.5 Hz, 2H), 1.66 (s, 9H), 1.43 (s, 9H).
REF: TRW-III-171, TRW-III-239.
NBoc
NHBoc
CO2H
5.5

194
Figure 5.45a. 1H NMR spectrum of compound 5.5.
9.99.9
10.510.5
2.22.2
3.33.3
0.90.9
0.90.9
2.62.6
1.11.1
1.21.2
1.01.0
ppm1 1223344556677889-14.99
1.43
1.66
3.14
3.15
3.19
3.21
3.23
3.25
3.28
3.29
3.69
4.62
4.63
4.66
4.68
5.10
5.12
7.20
7.23
7.25
7.28
7.31
7.33
7.39
7.47
7.50
8.09
8.12

195
Bromopyrroloindoline (3.74). To a solution of N,N’-Boc2-D-Trp-OMe (18.53 g, 44.3
mmol) in CH2Cl2 (1000 mL) was added NBS (7.89 g, 44.3 mmol) and PPTS (11.12 g,
44.3 mmol). The resulting solution was allowed to stir at r.t. O/N. The reaction mixture
was washed with brine, the organic layer dried (Na2SO4) and concentrated. Purification
by SiO2 chromatography (30% EtOAc in hexanes) gave the desired product in 96% yield
(21.20 g).
[α]D = +150.0 (CH2Cl2); 1H-NMR (400 MHz; CDCl3): δ 7.53-7.52 (bs, 1H), 7.34-7.24
(m, 2H), 7.09 (t, J = 7.6 Hz, 1H), 6.37 (s, 1H), 3.86 (dd, J = 10.3, 6.3 Hz, 1H), 3.71 (s,
3H), 3.18 (dd, J = 12.6, 6.3 Hz, 1H), 2.79 (dd, J = 12.6, 10.3 Hz, 1H), 1.56 (s, 9H), 1.37
(bs, 9H); 13C-NMR (101 MHz; CDCl3): δ 171.46, 171.41, 171.0, 152.1, 141.5, 130.6,
124.37, 124.35, 123.2, 83.8, 82.2, 59.4, 52.3, 36.4, 28.6, 28.2, 24.1, 21.0, 14.2; IR (NaCl,
film) 2980, 1719, 1395, 1334, 1163, 753 cm-1; HRMS (ESI-APCI): [M+Na]+ 519.1107
calcd for C22H29BrN2NaO6, found: 519.1102.
REF: TRW-III-174, TRW-III-246.
NBoc
NBocCO2Me
Br
H
3.74
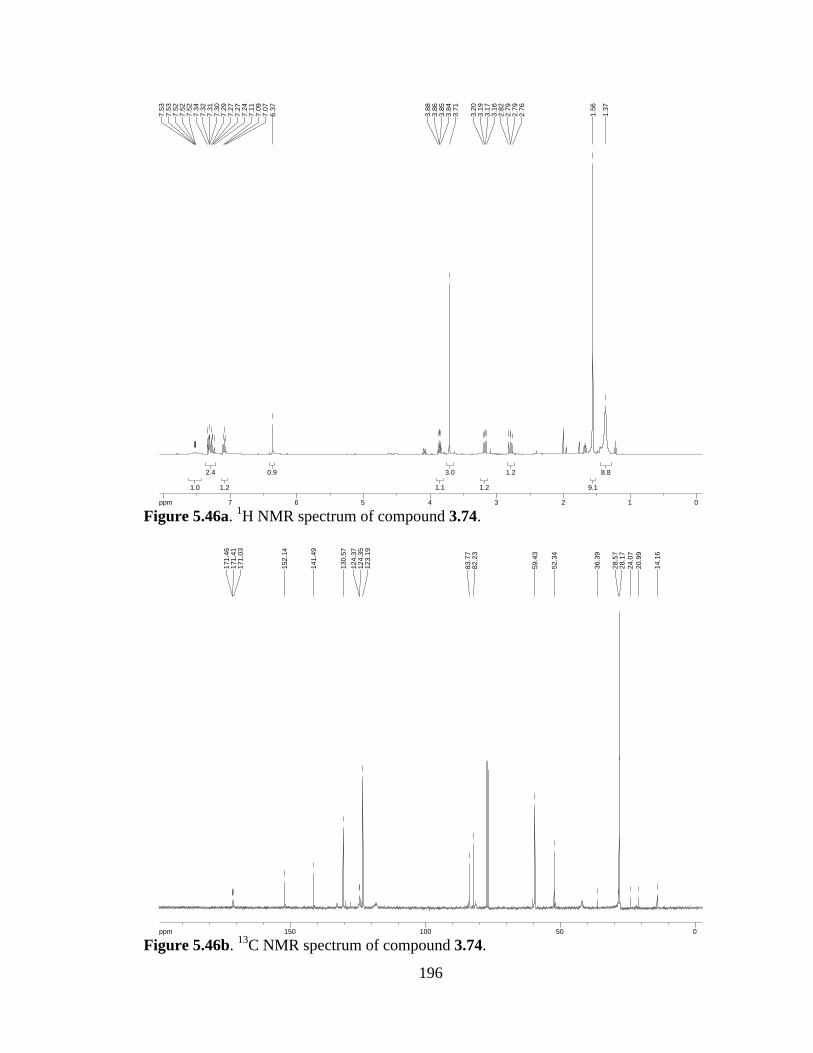
196
Figure 5.46a. 1H NMR spectrum of compound 3.74.
Figure 5.46b. 13C NMR spectrum of compound 3.74.
8.88.8
9.19.1
1.21.2
1.21.2
3.03.0
1.11.1
0.90.9
1.21.2
2.42.4
1.01.0
ppm0 0112233445566778-16.04
1.37
1.56
2.76
2.79
2.79
2.82
3.16
3.17
3.19
3.20
3.71
3.84
3.85
3.86
3.88
6.37
7.07
7.09
7.11
7.24
7.27
7.27
7.29
7.30
7.31
7.32
7.34
7.52
7.52
7.52
7.53
7.53
ppm0 05050100100150150-253.79
14.1
6
20.9
924
.07
28.1
728
.57
36.3
9
52.3
4
59.4
3
82.2
383
.77
123.
1912
4.35
124.
3713
0.57
141.
49
152.
14
171.
0317
1.41
171.
46

197
(2S,3aS,8aS)-1,8-di-tert-butyl 2-methyl 3a-(3-((S)-2-((tert-butoxycarbonyl)amino)-3-
methoxy-3-oxopropyl)-1H-indol-1-yl)-3,3a-dihydropyrrolo[2,3-b]indole-
1,2,8(2H,8aH)-tricarboxylate (3.75). KOtBu (3.02 g, 27 mmol) was added to a solution
of bromopyrroloindoline 3.74 (10.0 g, 20.2 mmol) and N-Boc-Trp-OMe (4.29 g, 13.5
mmol) in CH3CN (1000 mL) at 0 °C at stirred 1h under Ar. NaHCO3 was added and the
product extracted in CH2Cl2, washed with brine, dried (Na2SO4), and concentrated.
Purification by SiO2 chromatography (40% EtOAc in hexanes) afforded pure dipeptide
(5.47 g, 55% yield).
[α]D = -7.2 (CH2Cl2); 1H-NMR (400 MHz; CDCl3): δ 7.70-7.64 (bs, 1H), 7.51 (t, J = 7.5
Hz, 1H), 7.37-7.32 (m, 1H), 7.26 (s, 1H), 7.20-7.05 (m, J = 7.8 Hz, 4H), 6.74 (s, 1H),
6.67 (s, 1H), 4.97 (t, J = 6.4 Hz, 1H), 4.88 (d, J = 7.2 Hz, 1H), 4.53 (s, 1H), 3.58 (s, 2H),
3.53-3.46 (m, 2H), 3.20 (s, 3H), 3.13 (s, 3H), 1.51-1.48 (m, 18H), 1.39 (d, J = 7.7 Hz,
9H); 13C-NMR (101 MHz; CDCl3): δ 172.7, 171.0, 155.0, 151.9, 143.6, 134.7, 131.00,
130.89, 124.80, 124.78, 122.9, 122.31, 122.30, 122.0, 120.0, 119.55, 119.37, 111.5,
109.7, 82.1, 79.7, 72.57, 72.54, 60.3, 54.2, 52.1, 28.29, 28.17; IR (NaCl, film) 2979,
1716, 1457, 1207, 739 cm-1; HRMS (ESI-APCI): [M+Na]+ 757.3425 calcd for
C39H50N4NaO10, found: 757.3427.
REF: TRW-III-183, TRW-III-194.
NBoc
NBocCO2Me
H
N
CO2Me
NHBoc
3.75
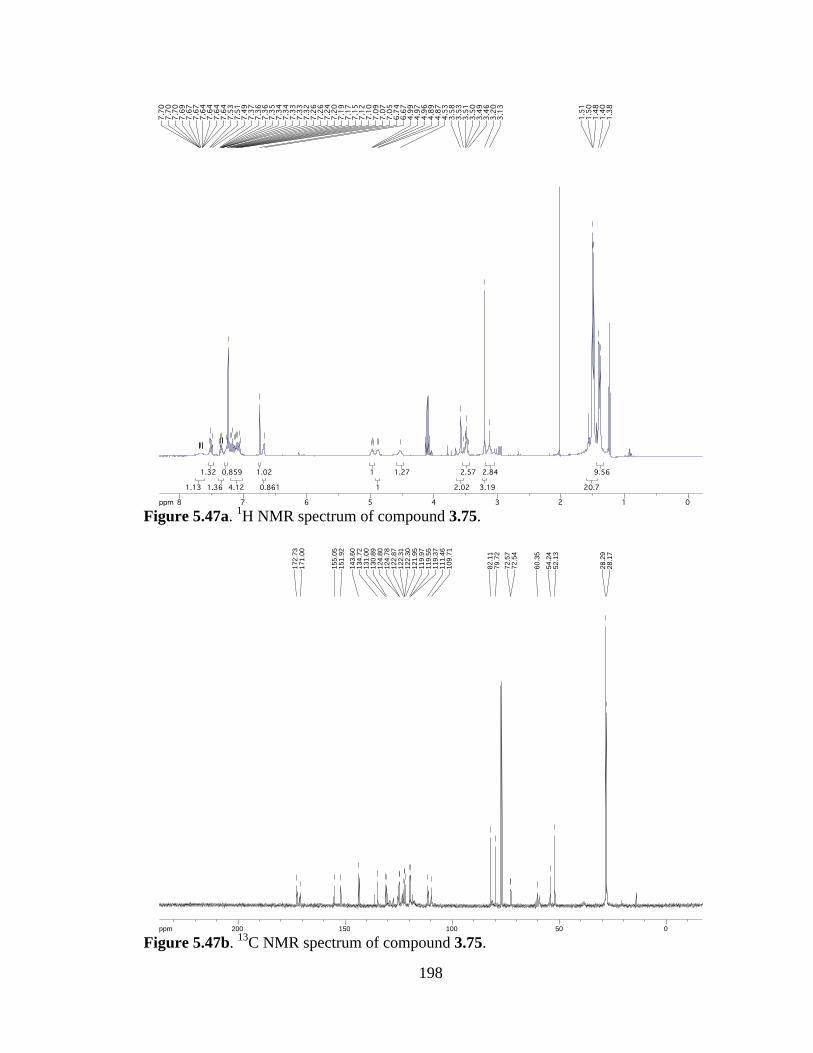
198
Figure 5.47a. 1H NMR spectrum of compound 3.75.
Figure 5.47b. 13C NMR spectrum of compound 3.75.
9.569.56
20.720.7
2.842.84
3.193.19
2.572.57
2.022.02
1.271.27
11
11
0.8610.861
1.021.02
4.124.12
0.8590.859
1.361.36
1.321.32
1.131.13
ppm0 01122334455667788-16.04
1.38
1.40
1.48
1.50
1.51
3.13
3.20
3.46
3.49
3.50
3.51
3.53
3.58
4.53
4.87
4.89
4.96
4.97
4.99
6.67
6.74
7.05
7.07
7.09
7.10
7.12
7.15
7.17
7.19
7.20
7.24
7.26
7.26
7.32
7.33
7.33
7.34
7.34
7.35
7.36
7.36
7.37
7.49
7.51
7.53
7.64
7.64
7.64
7.64
7.67
7.67
7.69
7.70
7.70
7.70
ppm0 05050100100150150200200-253.79
28.1
728
.29
52.1
354
.24
60.3
5
72.5
472
.57
79.7
282
.11
109.
7111
1.46
119.
3711
9.55
119.
9712
1.95
122.
3012
2.31
122.
8712
4.78
124.
8013
0.89
131.
0013
4.72
143.
6015
1.92
155.
05
171.
0017
2.73

199
(2S,3aS,8aS)-methyl 3a-(3-((S)-2-amino-3-methoxy-3-oxopropyl)-1H-indol-1-yl)-
1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole-2-carboxylate (5.6). TFA (15 mL) was
slowly added to a solution of the Boc-protected compound 3.75 (5.47 g, 7.5 mmol) in
CH2Cl2 (150 mL) at 0 °C. The reaction mixture was allowed to warm to r.t. as it stirred
O/N. NaHCO3 was added and the product extracted into CH2Cl2. The combined organic
extracts were dried (Na2SO4) and concentrated to afford the deprotected product (3.02 g,
93% yield), used without further purification.
[α]D = +80.0 (CH2Cl2); 1H-NMR (400 MHz; CDCl3): δ 7.52 (dt, J = 6.6, 3.0 Hz, 1H),
7.40-7.37 (m, 1H), 7.11-6.99 (m, 5H), 6.70 (t, J = 7.4 Hz, 1H), 6.62 (dd, J = 7.9, 3.4 Hz,
1H), 5.44 (s, 1H), 4.14 (dd, J = 7.8, 2.6 Hz, 1H), 3.72 (t, J = 6.0 Hz, 1H), 3.60 (d, J =
17.0 Hz, 3H), 3.40 (ddd, J = 13.0, 7.9, 2.3 Hz, 1H), 3.29 (s, 3H), 3.15 (dt, J = 14.4, 4.3
Hz, 1H), 2.95-2.86 (m, 2H), 1.99-1.90 (bs, 3H); 13C-NMR (101 MHz; CDCl3): δ 175.6,
173.8, 149.7, 135.2, 130.5, 129.80, 129.76, 127.5, 125.4, 125.0, 121.8, 119.5, 119.2,
112.1, 110.9, 109.7, 81.8, 75.7, 60.3, 55.0, 52.1, 40.6, 30.5, 28.4; IR (NaCl, film) 3373,
2950, 1735, 1459, 1207, 744 cm-1; HRMS (ESI-APCI): [M+H]+ 435.2032 calcd for
C24H27N4O4, found: 435.2036.
REF: TRW-III-184, TRW-III-195.
NH
NHCO2Me
H
N
CO2Me
NH2
5.6

200
Figure 5.48a. 1H NMR spectrum of compound 5.6.
Figure 5.48b. 13C NMR spectrum of compound 5.6.
3.23.2
2.02.0
1.41.4
2.72.7
1.11.1
3.03.0
1.61.6
1.21.2
1.01.0
1.11.1
1.21.2
5.45.4
1.11.1
1.31.3
ppm0 0112233445566778-16.04
1.90
1.91
1.91
1.92
1.92
1.93
1.93
1.93
1.99
1.99
2.86
2.87
2.88
2.89
2.90
2.90
2.91
2.93
2.95
3.12
3.13
3.14
3.15
3.16
3.17
3.29
3.37
3.37
3.39
3.39
3.40
3.40
3.42
3.42
3.58
3.62
3.71
3.72
3.74
4.13
4.14
4.15
4.16
5.44
6.60
6.61
6.62
6.63
6.68
6.70
6.72
6.99
7.00
7.03
7.04
7.06
7.06
7.07
7.08
7.09
7.11
7.37
7.38
7.39
7.39
7.40
7.51
7.51
7.52
7.53
7.54
ppm0 05050100100150150-253.79
28.4
430
.54
40.6
4
52.0
655
.00
60.2
9
75.7
2
81.8
3
109.
7311
0.85
112.
0811
9.19
119.
5412
1.83
124.
9612
5.43
127.
5412
9.76
129.
8013
0.48
135.
25
149.
69
173.
7917
5.58

201
(2S,3aS,8aR)-methyl 3a-(3-((S)-2-((S)-2-((((9H-fluoren-9-yl)methoxy)-
carbonyl)amino)-3-((tert-butyldimethylsilyl)oxy)propanamido)-3-methoxy-3-
oxopropyl)-1H-indol-1-yl)-1-((S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)-amino)-3-
((tert-butyldimethylsilyl)oxy)propanoyl)-1,2,3,3a,8,8a-hexahydro-pyrrolo[2,3-
b]indole-2-carboxylate (3.76). Diamine 5.6 (1.03 g, 2.4 mmol), N-Fmoc-L-Ser(OTBS)-
OH (2.12 g, 4.8 mmol), T3P (50% by weight in DMF, 3.51 g, 5.5 mmol), iPr2NEt (2.1
mL, 12 mmol), and CH2Cl2 (24 mL) were combined and stirred under Ar O/N at 35 °C.
Water was added, the product extracted in EtOAc, the combined extracts washed with
brine, dried (Na2SO4), concentrated, and purified by SiO2 chromatography (10-25%
EtOAc in hexanes) to afford the tetrapeptide (2.15 g, 70% yield).
[α]D = +61.9 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.77 (d, J = 7.4 Hz, 4H), 7.60 (d,
J = 8.0 Hz, 5H), 7.41 (t, J = 7.3 Hz, 5H), 7.35-7.30 (m, 4H), 7.26-7.15 (m, 4H), 6.82-6.67
(m, 2H), 5.68 (d, J = 7.7 Hz, 1H), 5.42 (s, 1H), 4.90 (s, 1H), 4.40-4.33 (m, 4H), 4.24-4.10
(m, 3H), 4.05-3.88 (m, 2H), 3.74 (s, 1H), 3.63 (d, J = 13.3 Hz, 3H), 3.20 (s, 4H), 2.95 (s,
1H), 0.94-0.82 (m, 18H), 0.08 (dd, J = 35.4, 2.8 Hz, 12H); 13C-NMR (101 MHz; CDCl3):
δ 177.5, 177.3, 175.7, 175.35, 175.20, 160.8, 156.0, 150.0, 149.48, 149.34, 149.30, 147.1,
146.9, 140.3, 137.11, 137.00, 135.96, 135.82, 133.25, 133.17, 132.6, 131.5, 130.75,
130.70, 130.64, 130.60, 130.57, 130.4, 130.1, 127.8, 125.65, 125.58, 125.48, 125.1,
124.7, 117.28, 117.22, 116.21, 116.13, 114.93, 114.83, 84.8, 78.3, 73.0, 72.78, 72.75,
NH
NCO2Me
H
N
CO2Me
HNO
NHFmoc
OTBS
O
NHFmoc
OTBS
3.76

202
72.62, 70.8, 68.7, 65.3, 59.06, 58.90, 58.4, 57.7, 56.0, 52.75, 52.72, 52.69, 35.3, 33.9,
33.4, 31.8, 31.52, 31.43, 24.1, 23.75, 23.71, 0.23, 0.11, 0.08, 0.03, -0.01; IR (NaCl, film)
2953, 1724, 1673, 1450, 1253, 1107, 740 cm-1; HRMS (ESI-APCI): [M+H]+ 1281.5764
calcd for C72H85N6O12Si2, found: 1281.5759.
REF: TRW-III-185, TRW-III-196, TRW-III-202.
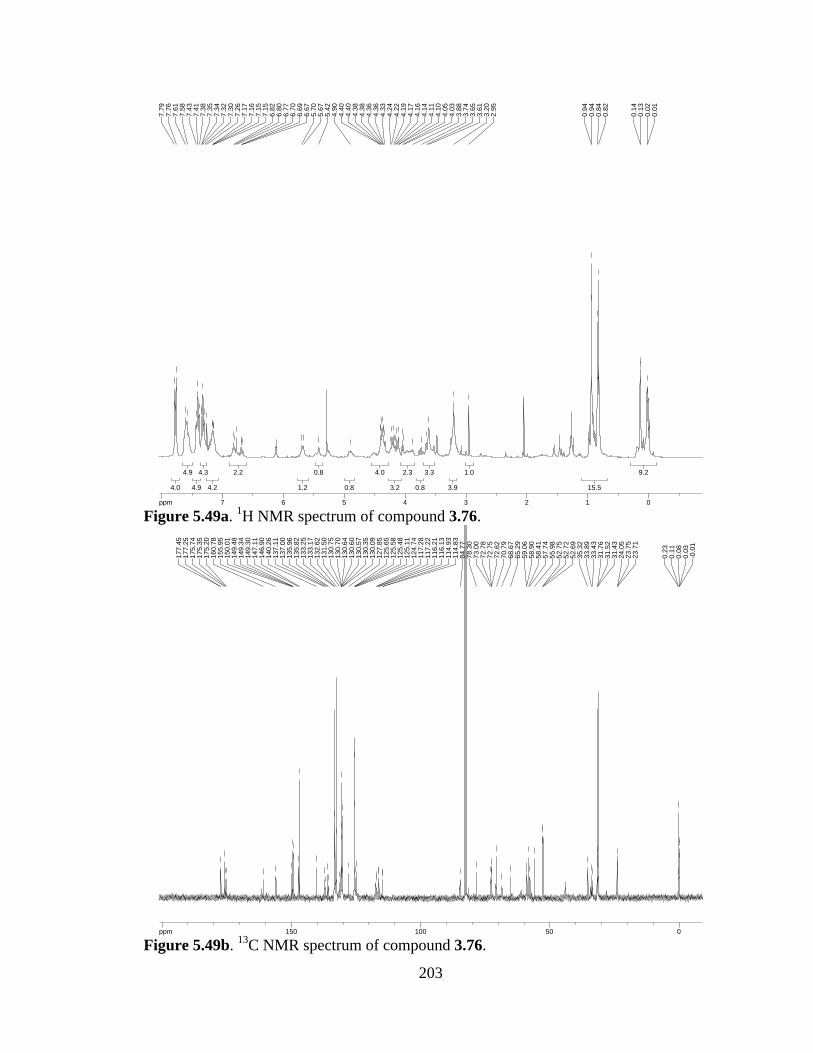
203
Figure 5.49a. 1H NMR spectrum of compound 3.76.
Figure 5.49b. 13C NMR spectrum of compound 3.76.
9.29.2
15.515.5
1.01.0
3.93.9
3.33.3
0.80.8
2.32.3
3.23.2
4.04.0
0.80.8
0.80.8
1.21.2
2.22.2
4.24.2
4.34.3
4.94.9
4.94.9
4.04.0
ppm0 0112233445566778-14.99
0.01
0.02
0.13
0.14
0.82
0.84
0.94
0.94
2.95
3.20
3.61
3.65
3.74
3.88
4.03
4.05
4.10
4.11
4.14
4.16
4.17
4.19
4.22
4.24
4.33
4.36
4.36
4.38
4.38
4.40
4.40
4.90
5.42
5.67
5.70
6.67
6.69
6.70
6.77
6.80
6.82
7.15
7.15
7.16
7.17
7.26
7.30
7.32
7.34
7.35
7.38
7.41
7.43
7.58
7.61
7.76
7.79
ppm0 05050100100150150200-253.79
-0.0
10.
030.
080.
110.
23
23.7
123
.75
24.0
531
.43
31.5
231
.76
33.4
333
.89
35.3
252
.69
52.7
252
.75
55.9
857
.74
58.4
158
.90
59.0
665
.29
68.6
770
.79
72.6
272
.75
72.7
873
.00
78.3
084
.77
114.
8311
4.93
116.
1311
6.21
117.
2211
7.28
124.
7412
5.11
125.
4812
5.58
125.
6512
7.85
130.
0913
0.35
130.
5713
0.60
130.
6413
0.70
130.
7513
1.50
132.
6213
3.17
133.
2513
5.82
135.
9613
7.00
137.
1114
0.26
146.
9014
7.11
149.
3014
9.34
149.
4815
0.01
155.
9516
0.78
175.
2017
5.35
175.
7417
7.25
177.
45

204
Dioxopiperazine2 (3.77). Tetrapeptide 3.76 (2.18 g, 1.7 mmol) and morpholine (17 mL)
were dissolved in THF (70 mL) and heated to 35 °C. After stirring O/N, the reaction
mixture was concentrated and purified by SiO2 chromatography (5% MeOH in CH2Cl2)
to afford pure dioxopiperazine 3.77 (1.11 g, 85% yield).
1H-NMR (400 MHz; CDCl3): δ 7.68 (d, J = 8.0 Hz, 1H), 7.56 (s, 1H), 7.29 (d, J = 7.4
Hz, 1H), 7.16 (t, J = 7.4 Hz, 1H), 7.09 (d, J = 7.8 Hz, 1H), 7.00 (d, J = 7.7 Hz, 1H), 6.81-
6.79 (m, 2H), 6.63 (d, J = 17.9 Hz, 1H), 6.42 (s, 1H), 6.13 (d, J = 3.5 Hz, 1H), 5.53 (d, J
= 3.5 Hz, 1H), 4.83 (dd, J = 11.1, 6.1 Hz, 1H), 4.37-4.35 (m, 1H), 4.25 (q, J = 5.4 Hz,
2H), 4.11 (t, J = 3.8 Hz, 1H), 4.03 (dd, J = 10.0, 3.5 Hz, 1H), 3.85 (dd, J = 19.1, 8.2 Hz,
2H), 3.72 (d, J = 14.7 Hz, 2H), 3.59 (dd, J = 9.9, 8.0 Hz, 1H), 3.54 (s, 1H), 3.34-3.29 (m,
1H), 3.19 (dd, J = 14.6, 9.8 Hz, 1H), 2.83 (dd, J = 14.7, 11.4 Hz, 1H), 0.96 (s, 18H), 0.16
(s, 12H); HRMS (ESI-APCI): [M+Na]+ 795.3698 calcd for C40H56N6NaO6Si2, found:
795.3696.
REF: TRW-III-191, TRW-III-198.
NH
NNH
O
O
OTBSH
N
NH
HN
O
O
OTBS
3.77

205
Figure 5.50a. 1H NMR spectrum of compound 3.77.
10.710.7
15.715.7
1.31.3
1.11.1
0.90.9
0.80.8
1.31.3
2.42.4
1.81.8
0.90.9
1.01.0
1.81.8
1.21.2
1.01.0
0.90.9
1.21.2
0.90.9
1.41.4
2.72.7
1.01.0
1.01.0
1.11.1
1.31.3
1.01.0
1.41.4
ppm0 0112233445566778-16.04
0.16
0.96
2.80
2.83
2.84
2.87
3.16
3.18
3.19
3.22
3.29
3.30
3.34
3.54
3.57
3.59
3.59
3.61
3.71
3.74
3.82
3.83
3.86
3.88
4.02
4.03
4.04
4.05
4.11
4.11
4.12
4.23
4.24
4.26
4.27
4.35
4.35
4.35
4.37
4.81
4.83
4.84
4.86
5.52
5.53
6.13
6.14
6.42
6.60
6.65
6.79
6.80
6.81
6.99
7.01
7.08
7.10
7.15
7.16
7.18
7.28
7.30
7.33
7.56
7.67
7.69

206
(2S,3aS,8aS)-1,8-di-tert-butyl 2-methyl 3a-(3-((S)-2-((tert-butoxycarbonyl)
(methyl)amino)-3-methoxy-3-oxopropyl)-1H-indol-1-yl)-3,3a-dihydro-pyrrolo[2,3-
b]indole-1,2,8(2H,8aH)-tricarboxylate (5.7). KOtBu (3.04 g, 27.2 mmol) was added to
a solution of bromopyrroloindoline 3.74 (10.08 g, 20.3 mmol) and N-Me,Boc-Trp-OMe
(4.50 g, 13.6 mmol) in CH3CN (1000 mL) at 0 °C and stirred 1h under Ar. NaHCO3 was
added and the product extracted in CH2Cl2, washed with brine, dried (Na2SO4), and
concentrated. Purification by SiO2 chromatography (10-20% EtOAc in hexanes) afforded
pure dipeptide (5.41 g, 53% yield).
[α]D = -3.1 (CH2Cl2); 1H-NMR (400 MHz; CDCl3): δ 7.68-7.67 (bs, 1H), 7.56 (d, J = 7.5
Hz, 1H), 7.35-7.32 (m, 2H), 7.23 (s, 1H), 7.16-7.08 (m, 3H), 6.84 (d, J = 15.2 Hz, 1H),
6.74 (d, J = 3.3 Hz, 1H), 4.87-4.86 (bs, 1H), 4.73-4.56 (broad, 2H), 3.69 (broad, J = 7.1
Hz, 3H), 3.49-3.43 (m, 1H), 3.29 (dd, J = 15.0, 4.9 Hz, 1H), 3.20 (s, 3H), 3.02-2.91 (m,
2H), 2.67-2.59 (m, 3H), 1.57-1.11 (m, 27H); 13C-NMR (101 MHz; CDCl3): δ 171.7,
170.9, 151.9, 134.7, 130.97, 130.82, 129.9, 125.0, 123.3, 122.2, 119.88, 119.86, 119.2,
111.6, 110.6, 82.0, 79.9, 72.59, 72.57, 59.9, 59.32, 59.30, 52.12, 52.09, 52.05, 52.00,
51.97, 34.6, 31.5, 28.19, 28.17, 25.2, 22.6, 20.7, 14.1; IR (NaCl, film) 2978, 1703, 1482,
1394, 1157, 738 cm-1; HRMS (ESI-APCI): [M+Na]+ 771.3581 calcd for C40H52N4NaO10,
found: 771.3600.
REF: TRW-III-211, TRW-III-258.
NBoc
NBocCO2Me
H
N
CO2Me
NMeBoc
5.7
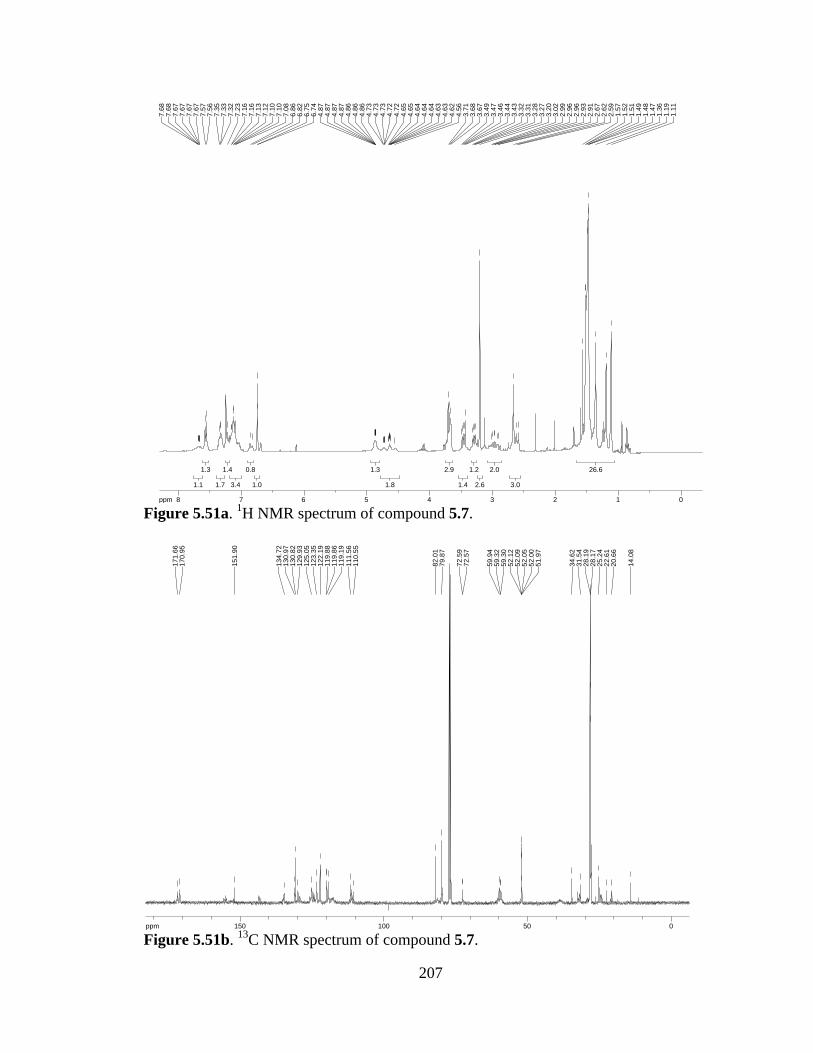
207
Figure 5.51a. 1H NMR spectrum of compound 5.7.
Figure 5.51b. 13C NMR spectrum of compound 5.7.
26.626.6
3.03.0
2.02.0
2.62.6
1.21.2
1.41.4
2.92.9
1.81.8
1.31.3
1.01.0
0.80.8
3.43.4
1.41.4
1.71.7
1.31.3
1.11.1
ppm0 01122334455667788-16.04
1.11
1.19
1.36
1.47
1.48
1.49
1.51
1.52
1.57
2.59
2.62
2.67
2.91
2.93
2.96
2.96
2.99
3.02
3.20
3.27
3.28
3.31
3.32
3.43
3.44
3.46
3.47
3.49
3.67
3.68
3.71
4.56
4.62
4.63
4.63
4.64
4.64
4.64
4.65
4.65
4.72
4.72
4.73
4.73
4.73
4.86
4.86
4.86
4.87
4.87
4.87
4.87
6.74
6.75
6.82
6.86
7.08
7.10
7.10
7.12
7.13
7.16
7.16
7.23
7.32
7.33
7.35
7.56
7.57
7.67
7.67
7.67
7.67
7.68
7.68
ppm0 05050100100150150-253.79
14.0
8
20.6
622
.61
25.2
428
.17
28.1
931
.54
34.6
2
51.9
752
.00
52.0
552
.09
52.1
259
.30
59.3
259
.94
72.5
772
.59
79.8
782
.01
110.
5511
1.56
119.
1911
9.86
119.
8812
2.19
123.
3512
5.05
129.
9313
0.82
130.
9713
4.72
151.
90
170.
9517
1.66

208
(2S,3aS,8aS)-methyl 3a-(3-((S)-3-methoxy-2-(methylamino)-3-oxopropyl)-1H-indol-
1-yl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole-2-carboxylate (3.80). TFA (11 mL)
was slowly added to a solution of Boc-protected material 5.7 (4.11 g, 5.49 mmol) in
CH2Cl2 (110 mL) at 0 °C. The reaction mixture was allowed to warm to r.t. as it stirred
O/N. NaHCO3 was added and the product extracted into CH2Cl2. The combined organic
extracts were dried (Na2SO4) and concentrated to afford the deprotected product (2.35 g,
96% yield), used without further purification.
[α]D = +73.3 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.61-7.58 (m, 1H), 7.46-7.43 (m,
1H), 7.18-7.05 (m, 6H), 6.77 (t, J = 7.4 Hz, 1H), 6.68 (dd, J = 7.9, 0.9 Hz, 1H), 5.51 (s,
1H), 4.55 (bs, 1H), 4.21 (d, J = 7.6 Hz, 1H), 3.61 (d, J = 9.5 Hz, 3H), 3.53-3.46 (m, 2H),
3.36 (d, J = 1.1 Hz, 3H), 3.10-2.93 (m, 4H), 2.35 (s, 3H); 13C-NMR (101 MHz; CDCl3): δ
174.7, 173.8, 149.69, 149.66, 135.26, 135.23, 130.4, 129.7, 127.58, 127.54, 125.3,
125.00, 124.89, 121.8, 119.5, 119.30, 119.13, 112.1, 110.74, 110.71, 109.55, 109.49,
82.0, 75.72, 75.69, 63.71, 63.69, 60.2, 52.1, 51.7, 40.76, 40.70, 34.68, 34.62, 28.91,
28.81; IR (NaCl, film) 2950, 1734, 1459, 1206, 742 cm-1; HRMS (ESI-APCI): [M+H]+
449.2189 calcd for C25H29N4O4, found: 449.2179.
REF: TRW-III-216, TRW-III-267.
NH
NHCO2Me
H
N
CO2Me
NHMe
3.80

209
Figure 5.52a. 1H NMR spectrum of compound 3.80.
Figure 5.52b. 13C NMR spectrum of compound 3.80.
3.63.6
4.04.0
3.03.0
2.42.4
3.13.1
1.01.0
0.90.9
1.01.0
1.01.0
1.11.1
6.06.0
1.11.1
1.31.3
ppm0 0112233445566778-14.99
2.35
2.93
2.96
2.97
2.98
3.06
3.08
3.09
3.10
3.35
3.36
3.46
3.46
3.48
3.50
3.52
3.53
3.60
3.63
4.20
4.22
4.54
4.55
4.55
4.55
5.51
6.67
6.67
6.70
6.70
6.74
6.77
6.79
7.05
7.05
7.11
7.12
7.13
7.15
7.16
7.16
7.18
7.43
7.44
7.46
7.58
7.59
7.59
7.59
7.60
7.61
ppm0 05050100100150150200-253.79
28.8
128
.91
34.6
234
.68
40.7
040
.76
51.7
052
.05
60.2
363
.69
63.7
1
75.6
975
.72
81.9
9
109.
4910
9.55
110.
7111
0.74
112.
1011
9.13
119.
3011
9.51
121.
8012
4.89
125.
0012
5.28
127.
5412
7.58
129.
6613
0.42
135.
26
149.
6614
9.69
173.
7917
4.72

210
(2S,3aS,8aR)-methyl 3a-(3-((S)-2-((S)-3-(benzyloxy)-2-((tert-
butoxycarbonyl)(methyl)amino)-N-methylpropanamido)-3-methoxy-3-oxopropyl)-
1H-indol-1-yl)-1-((S)-3-(benzyloxy)-2-((tert-
butoxycarbonyl)(methyl)amino)propanoyl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-
b]indole-2-carboxylate (3.81). The diamine (3.80, 430 mg, 0.96 mmol), N-Me,Boc-L-
Ser(OBn)-OH (591 mg, 1.91 mmol), T3P (50% by weight in DMF, 1.41 g, 2.21 mmol),
iPr2NEt (0.835 mL, 4.8 mmol), and CH2Cl2 (10 mL) were combined and stirred under Ar
O/N at 35 °C. Water was added, the product extracted in EtOAc, the combined extracts
washed with brine, dried (Na2SO4), concentrated, and purified by SiO2 chromatography
(60% EtOAc in hexanes) to afford the tetrapeptide (420 mg, 42% yield).
1H-NMR (300 MHz; CDCl3): δ 7.58-7.55 (m, 1H), 7.41-7.15 (m, 16H), 6.99 (s, 1H),
6.76-6.69 (m, 1H), 6.26-6.02 (m, 1H), 5.42-5.12 (m, 3H), 4.96-4.86 (m, 1H), 4.61-4.41
(m, 5H), 3.71-3.54 (m, 7H), 3.40-3.35 (m, 2H), 3.18 (d, J = 16.2 Hz, 5H), 2.93-2.67 (m,
8H), 1.42 (t, J = 8.2 Hz, 18H); HRMS (ESI-APCI): [M+H]+ 1031.5130 calcd for
C57H71N6O12, found: 1031.5126.
REF: TRW-III-231, TRW-III-237, TRW-III-253.
NH
NCO2Me
H
N
CO2Me
MeNO
NMeBoc
OBn
O
NMeBoc
OBn
3.81

211
Figure 5.53a. 1H NMR spectrum of compound 3.81.
18.018.0
7.67.6
4.74.7
2.12.1
6.96.9
4.74.7
1.01.0
3.33.3
1.01.0
1.21.2
1.01.0
16.316.3
1.41.4
ppm0 01122334455667788-14.99
1.40
1.41
1.45
2.67
2.82
2.85
2.93
3.15
3.21
3.35
3.36
3.36
3.40
3.54
3.56
3.57
3.57
3.57
3.64
3.66
3.71
4.41
4.42
4.56
4.56
4.61
4.86
4.86
4.86
4.87
4.87
4.95
4.96
5.12
5.12
5.12
5.13
5.18
5.20
5.25
5.35
5.40
5.42
5.42
6.02
6.03
6.04
6.24
6.25
6.25
6.26
6.69
6.70
6.71
6.72
6.75
6.75
6.76
7.15
7.26
7.29
7.30
7.31
7.31
7.41
7.55
7.55
7.58

212
(2S,3aS,8aR)-methyl 1-((S)-3-(benzyloxy)-2-(methylamino)propanoyl)-3a-(3-((S)-2-
((S)-3-(benzyloxy)-N-methyl-2-(methylamino)propanamido)-3-methoxy-3-
oxopropyl)-1H-indol-1-yl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole-2-
carboxylate (5.8). TFA (0.2 mL) was slowly added to a solution of Boc-protected
material 3.81 (103 mg, 0.1 mmol) in CH2Cl2 (2 mL) at 0 °C. The reaction mixture was
allowed to warm to r.t. as it stirred O/N. NaHCO3 was added and the product extracted
into CH2Cl2. The combined organic extracts were dried (Na2SO4) and concentrated to
afford the deprotected product (81 mg, 98% yield), used without further purification.
REF: TRW-III-233, TRW-III-243, TRW-III-254.
NH
NCO2Me
H
N
CO2Me
MeNO
NHMe
OBn
O
NHMe
OBn
5.8

213
Dioxopiperazine2 (3.82). Tetrapeptide 5.8 (81 mg, 0.098 mmol) and morpholine (1 mL)
were dissolved in THF (4 mL) and heated to 35 °C. After stirring O/N, the reaction
mixture was concentrated and purified by SiO2 chromatography (5% MeOH in CH2Cl2)
to afford pure dioxopiperazine (75 mg, >99% yield).
1H-NMR (300 MHz; CDCl3): δ 7.53-7.05 (m, 23H), 6.91 (t, J = 7.1 Hz, 2H), 6.76-6.71
(m, 2H), 6.46-6.34 (m, 2H), 5.72 (d, J = 4.1 Hz, 1H), 4.99-4.92 (m, 1H), 4.63-4.52 (m,
3H), 4.29-4.23 (m, 2H), 3.96 (dd, J = 9.7, 4.1 Hz, 2H), 3.69-3.54 (m, 5H), 3.26-3.21 (m,
2H), 3.08 (s, 3H), 2.87 (d, J = 3.9 Hz, 2H), 2.71 (dd, J = 12.2, 2.8 Hz, 1H), 2.21 (s, 3H);
HRMS (ESI-APCI): [M+Na]+ 789.3377 calcd for C45H46N6NaO6, found: 789.3379.
REF: TRW-III-235, TRW-III-245, TRW-III-257.
NH
NNMe
O
O
OBnH
N
NMe
MeN
O
O
OBn
3.82
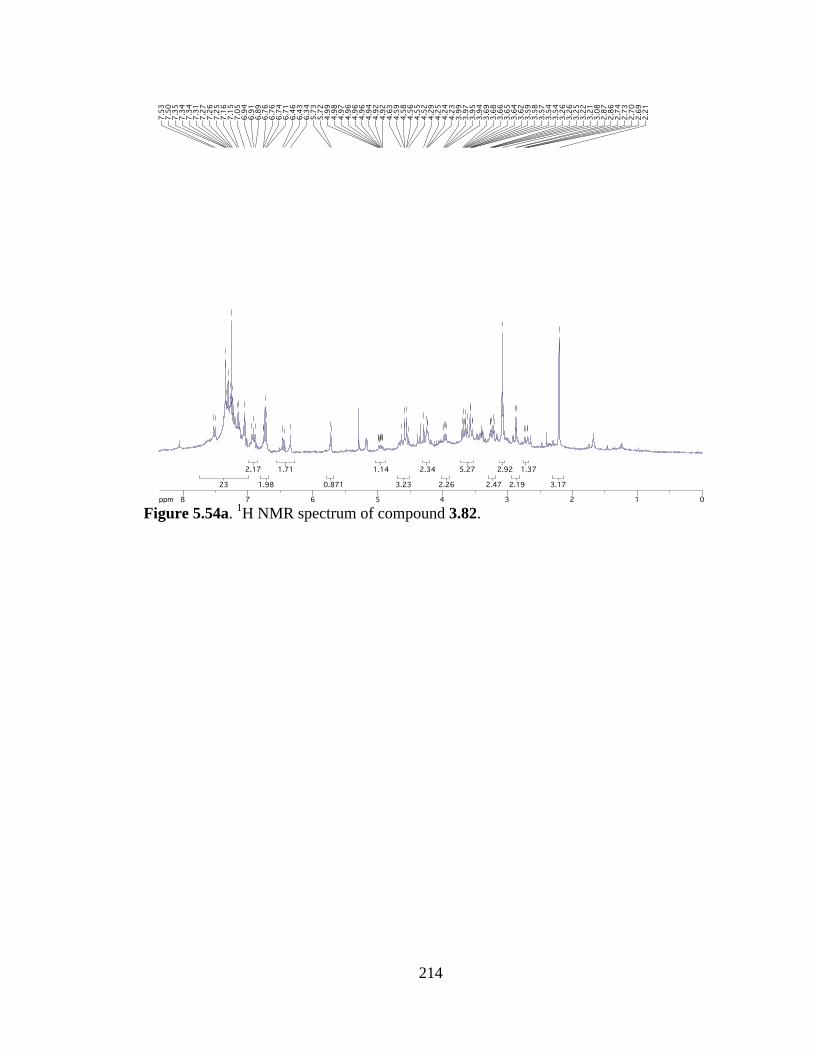
214
Figure 5.54a. 1H NMR spectrum of compound 3.82.
3.173.17
1.371.37
2.192.19
2.922.92
2.472.47
5.275.27
2.262.26
2.342.34
3.233.23
1.141.14
0.8710.871
1.711.71
1.981.98
2.172.17
2323
ppm0 01122334455667788-14.99
2.21
2.69
2.70
2.73
2.74
2.86
2.87
3.08
3.21
3.22
3.25
3.26
3.26
3.54
3.54
3.57
3.58
3.59
3.62
3.64
3.65
3.66
3.68
3.69
3.94
3.95
3.97
3.99
4.23
4.24
4.25
4.29
4.52
4.55
4.56
4.58
4.59
4.63
4.92
4.92
4.94
4.96
4.96
4.96
4.97
4.98
4.99
5.72
5.73
6.34
6.43
6.46
6.71
6.74
6.76
6.76
6.89
6.91
6.94
7.05
7.15
7.16
7.25
7.26
7.27
7.31
7.34
7.34
7.35
7.50
7.53

215
(2S,3aS,8aR)-methyl 3a-(3-((S)-2-(2-((((9H-fluoren-9-
yl)methoxy)carbonyl)(methyl)amino)-N-methylacetamido)-3-methoxy-3-oxopropyl)-
1H-indol-1-yl)-1-(2-((((9H-fluoren-9-yl)methoxy)carbonyl)(methyl)amino)acetyl)-
1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole-2-carboxylate (5.9). The diamine (3.80,
896 mg, 2.0 mmol), N-Fmoc-sarcosine-OH (1.24 g, 4.0 mmol), T3P (50% by weight in
DMF, 2.93 g, 4.6 mmol), iPr2NEt (1.7 mL, 10.0 mmol), and CH2Cl2 (20 mL) were
combined and stirred under Ar O/N at 35 °C. Water was added, the product extracted in
EtOAc, the combined extracts washed with brine, dried (Na2SO4), concentrated, and
purified by SiO2 chromatography (50-100% EtOAc in hexanes) to afford tetrapeptide 5.9
(1.17 g, 57% yield).
[α]D = +92.1 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.78-7.74 (m, 4H), 7.63-7.59 (m,
6H), 7.35 (dd, J = 19.2, 7.0 Hz, 8H), 7.16-7.04 (m, 5H), 6.78-6.65 (m, 2H), 6.10-6.00 (m,
1H), 4.54-4.20 (m, 7H), 3.99-3.86 (m, 2H), 3.72-3.61 (m, 5H), 3.37-3.33 (m, 3H), 3.23-
3.08 (m, 6H), 2.99-2.90 (m, 4H), 2.80-2.56 (m, 4H); IR (NaCl, film) 2951, 1703, 1451,
1229, 740 cm-1; HRMS (ESI-APCI): [M+H]+ 1035.4293 calcd for C61H59N6O10, found:
1035.4285.
REF: TRW-III-292.
NH
NCO2Me
H
N
CO2Me
MeNO
NMeFmoc
O
NMeFmoc
5.9

216
Figure 5.55a. 1H NMR spectrum of compound 5.9.
4.34.3
4.24.2
6.36.3
2.62.6
4.94.9
1.81.8
7.17.1
0.70.7
1.71.7
5.35.3
8.48.4
6.06.0
4.04.0
ppm1 12233445566778-14.99
2.56
2.59
2.66
2.66
2.70
2.80
2.90
2.95
2.97
2.99
3.08
3.09
3.10
3.23
3.33
3.34
3.34
3.34
3.34
3.35
3.36
3.36
3.37
3.37
3.37
3.61
3.62
3.72
3.86
3.86
3.87
3.87
3.87
3.88
3.92
3.99
4.20
4.20
4.20
4.21
4.22
4.22
4.29
4.30
4.39
4.51
4.52
4.52
4.52
4.54
6.00
6.00
6.07
6.10
6.65
6.65
6.65
6.66
6.68
6.68
6.68
6.69
6.70
6.77
6.78
7.00
7.04
7.12
7.14
7.16
7.31
7.33
7.38
7.40
7.59
7.59
7.61
7.63
7.63
7.74
7.76
7.78

217
(5aR,10bS,11aS)-10b-(3-(((S)-1,4-dimethyl-3,6-dioxopiperazin-2-yl)methyl)-1H-
indol-1-yl)-2-methyl-2,3,5a,6,11,11a-hexahydro-1H-pyrazino[1',2':1,5]pyrrolo[2,3-
b]indole-1,4(10bH)-dione (3.88). Tetrapeptide 5.9 (1.0 g, 0.97 mmol) and morpholine
(10 mL) were dissolved in THF (40 mL) and heated to 35 °C. After stirring O/N, the
reaction mixture was concentrated and purified by SiO2 chromatography (2-6% MeOH in
CH2Cl2) to afford pure dioxopiperazine 3.88 (470 mg, 92% yield).
[α]D = +145.5 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.49 (t, J = 7.3 Hz, 1H), 7.26-
7.15 (m, 2H), 7.09-6.91 (m, 3H), 6.79-6.73 (m, 2H), 6.01 (dd, J = 51.3, 3.1 Hz, 1H), 5.45
(dd, J = 53.9, 3.8 Hz, 1H), 4.57-4.49 (m, 1H), 4.18 (t, J = 3.2 Hz, 1H), 3.84-3.46 (m, 4H),
3.23-3.12 (m, 2H), 3.06-2.98 (m, 6H), 2.68 (ddd, J = 15.3, 11.5, 4.2 Hz, 1H), 2.33 (d, J =
32.5 Hz, 3H), 2.06-1.94 (m, 1H); 13C-NMR (101 MHz; CDCl3): δ 166.3, 166.0, 164.6,
164.2, 146.7, 135.4, 130.1, 129.1, 128.1, 127.8, 124.7, 124.4, 122.8, 122.5, 120.2, 119.7,
112.4, 110.2, 108.3, 82.6, 82.0, 74.1, 62.7, 57.2, 53.5, 40.1, 39.7, 33.5, 32.9, 32.0; IR
(NaCl, film) 2931, 1664, 1459, 1324, 1259 cm-1; HRMS (ESI-APCI): [M+H]+ 527.2407
calcd for C29H31N6O4, found: 527.2402.
REF: TRW-III-293.
NH
NNMe
O
OH
N
NMe
MeN
O
O
3.88
218
Figure 5.56a. 1H NMR spectrum of compound 3.88.
Figure 5.56b. 13C NMR spectrum of compound 3.88.
1.11.1
3.43.4
1.41.4
6.16.1
1.91.9
4.34.3
1.21.2
0.90.9
1.01.0
1.01.0
2.42.4
3.33.3
2.02.0
1.01.0
ppm1 1223344556677-14.99
1.94
2.00
2.06
2.27
2.38
2.63
2.64
2.67
2.68
2.69
2.72
2.73
2.98
3.02
3.05
3.06
3.12
3.13
3.15
3.16
3.18
3.20
3.21
3.23
3.46
3.48
3.51
3.52
3.53
3.57
3.57
3.61
3.62
3.63
3.66
3.68
3.71
3.76
3.78
3.84
4.17
4.18
4.19
4.49
4.51
4.53
4.54
4.57
5.36
5.37
5.54
5.55
5.92
5.93
6.09
6.11
6.73
6.75
6.76
6.76
6.77
6.79
6.89
6.91
6.93
6.96
6.99
7.01
7.05
7.07
7.09
7.15
7.17
7.20
7.22
7.22
7.25
7.26
7.26
7.47
7.49
7.52
ppm0 05050100100150150-250.03
32.0
032
.86
33.5
439
.74
40.0
7
53.5
457
.21
62.6
5
74.1
0
82.0
082
.64
108.
3411
0.19
112.
4011
9.68
120.
2212
2.55
122.
8312
4.39
124.
7112
7.84
128.
1112
9.12
130.
1413
5.36
146.
73
164.
2016
4.64
166.
0516
6.33

219
Tetrapeptide (5.10). The diamine (3.80, 327 mg, 0.73 mmol), N-Me,Fmoc-L-
Ser(OTBS)-OH (663 g, 1.46 mmol), T3P (50% by weight in DMF, 1.07 g, 1.68 mmol),
iPr2NEt (0.635 mL, 5.0 mmol), and CH2Cl2 (7.3 mL) were combined and stirred under Ar
O/N at 35 °C. Water was added, the product extracted in EtOAc, the combined extracts
washed with brine, dried (Na2SO4), concentrated, and purified by SiO2 chromatography
(35% EtOAc in hexanes) to afford tetrapeptide 5.10 (351 mg, 36% yield).
[α]D = -1.5 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.78-7.71 (m, 4H), 7.61-7.53 (m,
5H), 7.41-7.15 (m, 14H), 6.93-6.66 (m, 3H), 5.13-5.07 (m, 2H), 4.41 (d, J = 6.8 Hz, 2H),
4.16-4.01 (m, 5H), 3.93-3.89 (m, 1H), 3.75-3.66 (m, 5H), 3.40-3.35 (m, 2H), 3.25 (d, J =
10.7 Hz, 2H), 3.09-2.94 (m, 5H), 2.82-2.81 (m, 4H), 2.72-2.66 (m, 2H), 2.34 (dd, J = 6.3,
3.2 Hz, 1H), 2.10-1.95 (m, 2H), 0.87-0.81 (m, 18H), 0.09--0.02 (m, 12H); IR (NaCl,
film) 2953, 1696, 1651, 1451, 1317, 1154, 740 cm-1; HRMS (ESI-APCI): [M+H]+
1323.6234 calcd for C75H91N6O12Si2, found:1323.6253.
REF: TRW-III-299, TRW-III-314.
NH
NCO2Me
H
N
CO2Me
MeNO
NHMeFmoc
O
NMeFmoc
5.10
OTBS
OTBS

220
Figure 5.57a. 1H NMR spectrum of compound 5.10.
12.012.0
18.818.8
1.71.7
1.21.2
2.02.0
4.24.2
5.45.4
1.91.9
1.81.8
5.05.0
1.01.0
5.15.1
1.91.9
2.32.3
3.03.0
14.914.9
5.25.2
4.44.4
ppm0 0112233445566778-14.99
-0.0
2-0
.02
0.02
0.08
0.09
0.81
0.82
0.85
0.86
0.87
1.95
2.05
2.09
2.10
2.33
2.34
2.35
2.36
2.66
2.71
2.72
2.81
2.82
2.94
2.98
2.99
3.06
3.09
3.23
3.27
3.35
3.35
3.36
3.36
3.40
3.66
3.67
3.69
3.69
3.70
3.71
3.74
3.75
3.89
3.90
3.90
3.93
4.01
4.06
4.09
4.11
4.12
4.13
4.16
4.40
4.42
5.07
5.08
5.08
5.10
5.11
5.11
5.13
6.66
6.68
6.71
6.71
6.74
6.74
6.77
6.78
6.81
6.83
6.86
6.87
6.91
6.93
7.15
7.15
7.16
7.17
7.26
7.26
7.28
7.29
7.30
7.31
7.31
7.36
7.38
7.40
7.41
7.53
7.54
7.57
7.59
7.60
7.61
7.61
7.71
7.71
7.73
7.76
7.78

221
(3S,5aR,10bS,11aS)-3-(((tert-butyldimethylsilyl)oxy)methyl)-10b-(3-(((2S,5S)-5-
(((tert-butyldimethylsilyl)oxy)methyl)-1,4-dimethyl-3,6-dioxopiperazin-2-yl)methyl)-
1H-indol-1-yl)-2-methyl-2,3,5a,6,11,11a-hexahydro-1H-
pyrazino[1',2':1,5]pyrrolo[2,3-b]indole-1,4(10bH)-dione (3.90). Tetrapeptide 5.10 (300
mg, 0.227 mmol) and morpholine (2.3 mL) were dissolved in THF (9 mL) and heated to
35 °C. After stirring O/N, the reaction mixture was concentrated and purified by SiO2
chromatography (4% MeOH in CH2Cl2) to afford pure dioxopiperazine 3.90 (153 mg,
83% yield).
3.90A: [α]D = +56.6 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.51 (d, J = 7.9 Hz, 1H),
7.18 (t, J = 7.6 Hz, 1H), 7.05 (d, J = 11.2 Hz, 2H), 6.90 (t, J = 6.1 Hz, 2H), 6.69 (t, J =
7.5 Hz, 2H), 6.50 (d, J = 8.4 Hz, 1H), 5.70 (s, 1H), 4.80 (dd, J = 11.9, 5.9 Hz, 1H), 4.33-
4.09 (m, 4H), 3.76 (d, J = 10.4 Hz, 1H), 3.66 (dt, J = 9.9, 4.9 Hz, 2H), 3.50-3.43 (m, 2H),
3.37 (t, J = 4.8 Hz, 1H), 3.19 (dd, J = 14.8, 4.6 Hz, 1H), 3.10 (s, 3H), 3.02 (s, 3H), 2.31
(s, 3H), 0.82 (s, 9H), 0.74 (s, 9H), 0.04 (d, J = 4.6 Hz, 6H), -0.11 (d, J = 4.3 Hz, 6H); 13C-
NMR (101 MHz; CDCl3): δ 167.9, 166.5, 165.73, 165.62, 146.6, 135.6, 130.2, 129.5,
124.4, 123.1, 122.5, 120.2, 112.3, 110.0, 108.6, 82.9, 62.6, 62.2, 62.0, 60.5, 59.9, 56.9,
40.7, 32.5, 30.4, 30.0, 27.2, 25.64, 25.52, 18.1, 17.9, -5.60, -5.66, -5.69; IR (NaCl, film)
2930, 2857, 1662, 1461, 1256, 1116 cm-1; HRMS (ESI-APCI): [M+H]+ 815.4348 calcd
for C43H63N6O6Si2, found:815.4352.
NH
NNMe
O
OH
N
NMe
MeN
O
O
OTBS
OTBS
3.90

222
3.90B: [α]D = +80.0 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.60 (d, J = 7.9 Hz, 1H),
7.30 (s, 1H), 7.15 (td, J = 7.7, 1.0 Hz, 1H), 7.07 (t, J = 7.4 Hz, 1H), 6.99-6.94 (m, 1H),
6.88 (d, J = 7.4 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 6.69-6.63 (m, 2H), 5.91 (d, J = 2.6 Hz,
1H), 5.59 (d, J = 2.6 Hz, 1H), 4.57-4.52 (m, 1H), 4.26-4.19 (m, 2H), 4.03 (dd, J = 10.8,
2.7 Hz, 1H), 3.87-3.77 (m, 2H), 3.68-3.61 (m, 2H), 3.40 (dd, J = 9.4, 5.6 Hz, 2H), 3.26-
3.21 (m, 1H), 2.99 (d, J = 9.6 Hz, 6H), 2.71 (s, 3H), 2.60-2.55 (m, 1H), 0.84 (s, 9H), 0.74
(s, 9H), -0.01--0.08 (m, 12H); 13C-NMR (101 MHz; CDCl3): δ 166.0, 164.56, 164.55,
164.35, 146.7, 135.6, 130.1, 129.5, 123.1, 122.6, 120.4, 119.5, 112.1, 111.2, 109.7, 82.7,
65.1, 64.2, 63.7, 63.3, 60.6, 56.8, 41.5, 33.8, 33.1, 31.1, 30.7, 25.9, 25.5, 18.4, 18.0, -
5.38, -5.40, -5.71, -5.74; IR (NaCl, film) 2930, 2857, 1654, 1462, 1255, 1112 cm-1;
HRMS (ESI-APCI): [M+H]+ 815.4348 calcd for C43H63N6O6Si2, found:815.4349.
REF: TRW-III-301, TRW-III-315.
223
Figure 5.58a. 1H NMR spectrum of compound 3.90A.
Figure 5.58b. 13C NMR spectrum of compound 3.90A.
7.57.5
5.85.8
10.110.1
10.510.5
2.92.9
3.43.4
2.92.9
1.31.3
1.21.2
1.91.9
2.02.0
1.21.2
4.44.4
1.01.0
0.90.9
0.90.9
2.12.1
2.22.2
1.91.9
1.41.4
1.21.2
ppm0 01122334455667788-14.99
-0.1
2-0
.10
0.04
0.05
0.74
0.82
2.31
3.02
3.10
3.15
3.17
3.20
3.22
3.36
3.37
3.39
3.43
3.43
3.46
3.47
3.48
3.50
3.53
3.62
3.64
3.66
3.68
3.69
3.74
3.78
4.09
4.10
4.13
4.13
4.19
4.19
4.23
4.29
4.29
4.32
4.33
4.77
4.79
4.81
4.83
5.70
6.48
6.51
6.67
6.69
6.72
6.88
6.90
6.92
7.00
7.03
7.07
7.16
7.18
7.21
7.49
7.52
ppm0 05050100100150150-250.03
-5.6
9-5
.66
-5.6
0
17.9
218
.12
25.5
225
.64
27.2
230
.04
30.4
232
.51
40.6
8
56.9
459
.88
60.5
561
.98
62.2
262
.59
82.8
6
108.
6311
0.04
112.
2812
0.19
122.
4612
3.14
124.
4112
9.53
130.
1613
5.56
146.
56
165.
6216
5.73
166.
5016
7.87
224
Figure 5.59a. 1H NMR spectrum of compound 3.90B.
Figure 5.59b. 13C NMR spectrum of compound 3.90B.
10.710.7
8.48.4
9.29.2
1.11.1
2.72.7
5.85.8
1.31.3
2.12.1
2.12.1
2.42.4
1.01.0
1.71.7
0.80.8
0.80.8
0.80.8
1.81.8
1.11.1
1.01.0
1.01.0
1.01.0
1.31.3
0.90.9
1.01.0
ppm0 011223344556677-14.99
-0.0
8-0
.05
-0.0
1-0
.01
0.74
0.84
2.55
2.56
2.60
2.71
2.97
3.00
3.21
3.22
3.24
3.25
3.26
3.38
3.40
3.41
3.43
3.61
3.62
3.64
3.67
3.68
3.77
3.78
3.81
3.82
3.85
3.85
3.86
3.87
4.01
4.02
4.04
4.05
4.19
4.20
4.21
4.22
4.23
4.25
4.26
4.52
4.53
4.53
4.56
4.57
5.58
5.59
5.90
5.91
6.63
6.66
6.69
6.69
6.75
6.78
6.87
6.89
6.94
6.94
6.97
6.99
6.99
7.04
7.07
7.09
7.12
7.12
7.14
7.15
7.17
7.17
7.30
7.59
7.62
ppm0 05050100100150150-250.03
-5.7
4-5
.71
-5.4
0-5
.38
18.0
218
.35
25.5
525
.89
30.7
331
.13
33.0
533
.76
41.4
7
56.8
260
.59
63.3
463
.66
64.1
765
.06
82.6
5
109.
6811
1.21
112.
1511
9.52
120.
3812
2.57
123.
1512
9.46
130.
1213
5.60
146.
66
164.
3516
4.55
164.
5616
5.96

225
5-chlorovanillin (5.11). Vanillin (25 g, 164 mmol) was dissolved in AcOH (150 mL),
then chlorine gas bubbled through the solution. After a significant amount of white solid
had precipitated, the solution was filtered and the solid washed with hexanes. To the
AcOH filtrate was added an additional batch of vanillin (25 g), and chlorine gas again
bubbled through the solution. The resulting precipitate was filtered, washed with hexanes,
combined with the first yield, and dried to afford 5-chlorovanillin (39.30 g, 64% yield).
1H-NMR (400 MHz; acetone-d6): δ 9.83 (s, 1H), 7.58 (d, J = 1.7 Hz, 1H), 7.43 (d, J =
1.7 Hz, 1H), 3.98 (s, 3H); 13C-NMR (101 MHz; acetone): δ 189.5, 148.7, 129.2, 125.6,
120.0, 108.7, 56.0.
REF: TRW-II-413, TRW-II-463, TRW-II-479, TRW-III-076.
CHO
OMeHO
5.11
Cl
226
Figure 5.60a. 1H NMR spectrum of compound 5.11.
Figure 5.60b. 13C NMR spectrum of compound 5.11.
3.283.28
1.161.16
0.8280.828
1.011.01
ppm0 0224466881010-16.043.98
7.43
7.43
7.57
7.58
9.83
ppm0 05050100100150150200200-253.79
28.32
28.52
29.29
29.48
55.96
108.68
119.97
125.57
129.23
148.62
148.81
189.47
205.40

227
5-chlorovanillin-OAc (4.15). To a solution of 5-chlorovanillin (58.5 g, 313 mmol) in
THF (1000 mL) was added Ac2O (35.5 mL, 376 mmol), Et3N (65.5 mL, 470 mmol), and
DMAP (200 mg). The resulting solution was stirred O/N, then concentrated. The product
was extracted in CH2Cl2 from 1M HCl, the combined organic extracts washed with brine,
dried over Na2SO4, and concentrated to give 4.15 (71.45 g, >99% yield).
1H-NMR (400 MHz; acetone-d6): δ 9.98 (s, 1H), 7.67 (d, J = 1.7 Hz, 1H), 7.59 (d, J =
1.7 Hz, 1H), 3.97 (s, 3H), 2.36 (s, 3H); 13C-NMR (101 MHz; acetone): δ 190.0, 166.8,
153.6, 141.3, 135.4, 128.5, 123.2, 110.4, 56.2, 19.2; HRMS (ESI-APCI): [M-H]-
229.0268 calcd for C10H9ClO4, found: 229.9854.
REF: TRW-II-415, TRW-II-464, TRW-II-481, TRW-III-078.
CHO
OMeAcO
Cl
4.15
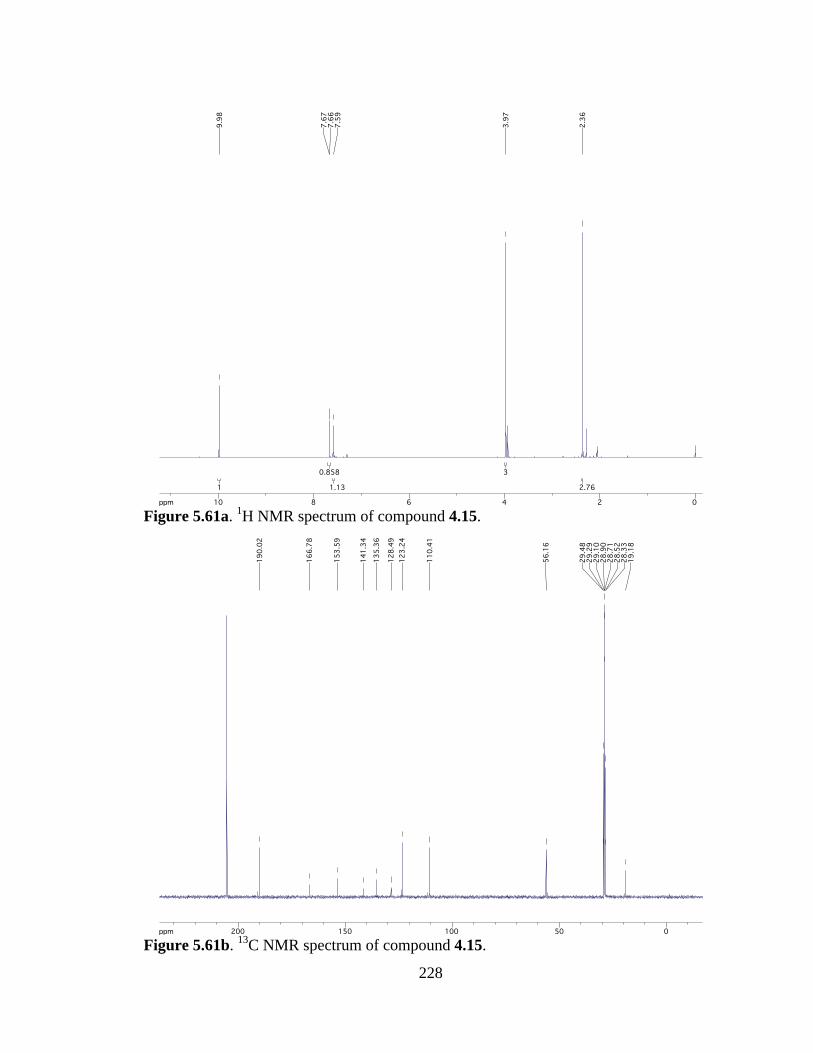
228
Figure 5.61a. 1H NMR spectrum of compound 4.15.
Figure 5.61b. 13C NMR spectrum of compound 4.15.
2.762.76
33
1.131.13
0.8580.858
11
ppm0 0224466881010-16.04
2.36
3.97
7.59
7.66
7.67
9.98
ppm0 05050100100150150200200-253.79
19.18
28.33
28.52
28.71
28.90
29.10
29.29
29.48
56.16
110.41
123.24
128.49
135.36
141.34
153.59
166.78
190.02

229
5-chloro-2-nitrovanillin (4.17). Nitric acid (fuming, 125) was chilled in an ethylene
glycol/dry ice bath, then 4.15 (28 g) added in small batches such that the internal
temperature did not rise above 5 °C. Following the addition, the reaction was stirred for 2
additional hours at -15 °C. The reaction mixture was carefully poured into ice water, and
the resulting bright orange precipitate filtered and rinsed with cold water until the
washings were pH neutral. This solid (combined from 2x28 g batches) was dissolved in
2M KOH (500 mL) and the solution heated to a boil for 10 min. After cooling to r.t., the
solution was poured into ice-cold concentrated HCl, and the resulting precipitate filtered,
washed with cold water, and dried to afford 4.17 (43.21 g, 76% yield).
1H-NMR (400 MHz; acetone-d6): δ 9.82 (s, 1H), 7.94 (s, 1H), 3.95 (s, 3H); 13C-NMR
(101 MHz; acetone): δ 186.0, 153.0, 140.9, 129.6, 123.2, 119.6, 98.5, 62.4; HRMS (-
APCI): [M-H]- 229.9856 calcd for C8H6ClNO5, found: 229.9860.
REF: TRW-II-417,419; TRW-II-424,425; TRW-II-484, TRW-III-080.
Nitric acid (fuming). Potassium nitrate (500 g) was added to concentrated sulfuric acid
(500 mL) in a flask equipped with a distillation column, and the resulting suspension
heated in an oil bath at 170 °C. Fuming nitric acid (75 mL) was collected over a 4 hour
period. This can be purchased commercially from VWR.
CHO
OMeHO
Cl
NO2
4.17
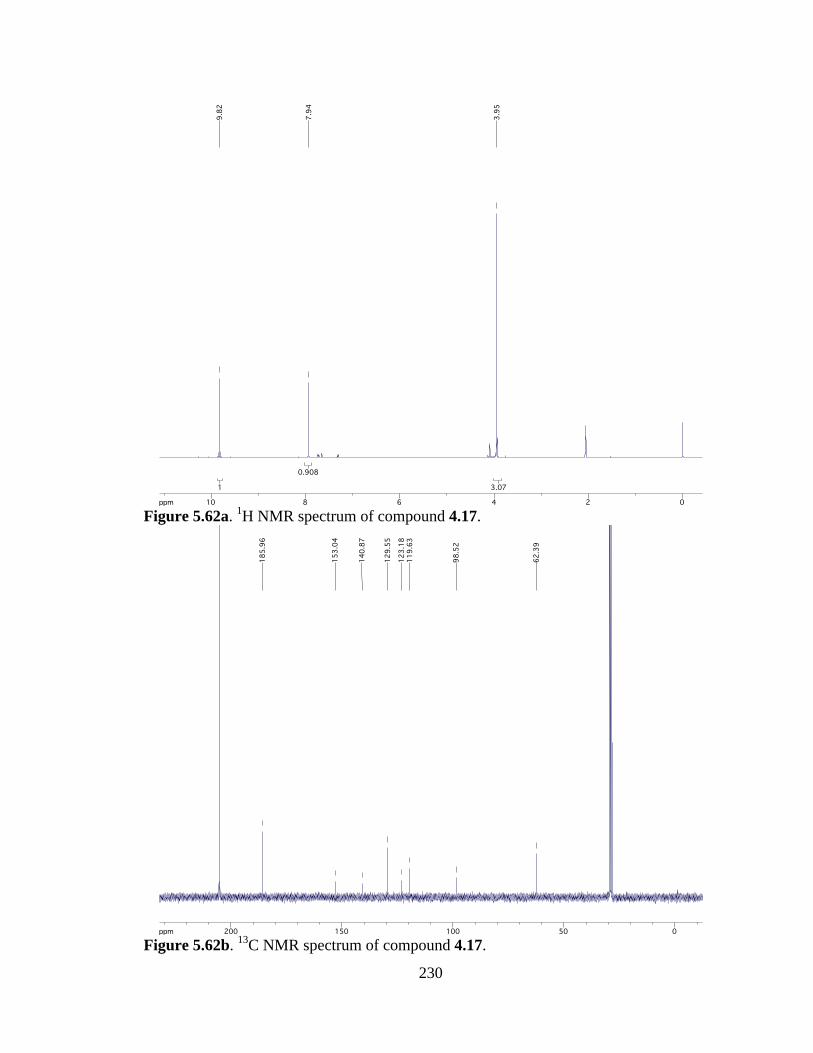
230
Figure 5.62a. 1H NMR spectrum of compound 4.17.
Figure 5.62b. 13C NMR spectrum of compound 4.17.
3.073.07
0.9080.908
11
ppm0 0224466881010-16.043.95
7.94
9.82
ppm0 05050100100150150200200-253.79
62.39
98.52
119.63
123.18
129.55
140.87
153.04
185.96

231
5-chloro-3,4-dimethoxy-2-nitrobenzaldehyde (4.18). Phenol 4.17 (15.0 g, 64.7 mmol)
was dissolved in absolute EtOH (26.3 mL), then Me2SO4 (26.3 mL, 278 mmol) added.
The resulting solution was immersed in an ice/brine bath before adding a solution of
NaOH (11.1 g, 278 mmol) in water (13.2 mL) dropwise. The reaction was heated to 50
°C for 3 hours, then cooled to r.t. and diluted with water. The product was extracted into
ether, and the organic extracts dried and concentrated to afford the title compound (15.9
g, >99% yield).
1H-NMR (400 MHz; CDCl3): δ 9.80 (d, J = 17.5 Hz, 1H), 7.73 (s, 1H), 4.05 (s, 3H), 4.01
(s, 3H); 13C-NMR (101 MHz; CDCl3): δ 184.7, 155.3, 151.4, 131.6, 126.7, 123.1, 120.3,
62.7, 61.4; HRMS (-APCI): [M-H]- 245.0091 calcd for C10H9ClN2O6, found: 245.0080.
4.19: 1H-NMR (300 MHz; CDCl3): δ 7.45 (s, 1H), 5.58 (s, 1H), 3.97 (s, 3H), 3.93 (s,
3H), 3.65-3.50 (m, 4H), 1.22 (t, J = 7.1 Hz, 6H).
REF: TRW-II-421, TRW-II-429, TRW-II-485, TRW-III-081.
CHO
OMeMeO
Cl
NO2
4.18

232
Figure 5.63a. 1H NMR spectrum of compound 4.18.
Figure 5.63b. 13C NMR spectrum of compound 4.18.
66
0.9710.971
1.111.11
ppm0 0224466881010-16.044.01
4.05
7.73
9.78
9.82
ppm0 05050100100150150200-253.79
61.42
61.53
62.71
120.35
123.05
126.73
131.57
151.41
155.26
184.72

233
Figure 5.64a. 1H NMR spectrum of compound 4.19.
6.16.1
4.14.1
3.33.3
3.33.3
0.90.9
1.01.0
ppm0 0224466881010-19.97
1.20
1.22
1.25
3.50
3.51
3.53
3.53
3.56
3.57
3.58
3.60
3.62
3.63
3.64
3.65
3.93
3.97
5.58
7.45
OMeMeO
Cl
NO2
OEt
OEt
4.19

234
Dinitrostyrene (4.7). Aldehyde 4.18 (13.64 g, 55.4 mmol) was suspended in EtOH (180
mL), and to it added nitromethane (4.4 mL, 77.6 mmol). The mixture was cooled to -15
°C, then a solution of KOH (13.3 g, 238 mmol) in water (18 mL) and EtOH (180 mL)
added dropwise over 2 hours. Upon complete addition of the KOH solution, the reaction
was acidified with concentrated HCl (34 mL), diluted with H2O (200 mL), and the
product extracted into chloroform. The organic extracts were dried and concentrated. To
the resulting oil was added 55 mL acetic anhydride and sodium acetate (5.5 g). The
solution was heated to reflux for 30 min., then poured into water and stirred. Water was
decanted from the brown paste then EtOH added and heated to dissolve the crude
product. Pure nitrostyrene crystals were recovered (10.3 g, 64% yield).
1H-NMR (400 MHz; DMSO-d6): δ 8.33 (d, J = 13.3 Hz, 1H), 8.15 (s, 1H), 7.63 (d, J =
13.4 Hz, 1H), 3.98 (s, 3H), 3.97 (s, 3H); 13C-NMR (101 MHz; DMSO): δ 152.3, 146.3,
145.0, 142.2, 131.5, 129.8, 124.9, 119.3, 63.1, 61.9; IR (NaCl, film) 1639, 1519 cm-1;
HRMS (-APCI): [M-H]- 288.0149 calcd for C10H9ClN2O6, found: 288.0141.
REF: TRW-II-426, TRW-II-431, TRW-II-489, TRW-III-003, TRW-III-011, TRW-III-
084.
OMeMeO
Cl
NO2
NO2
4.7
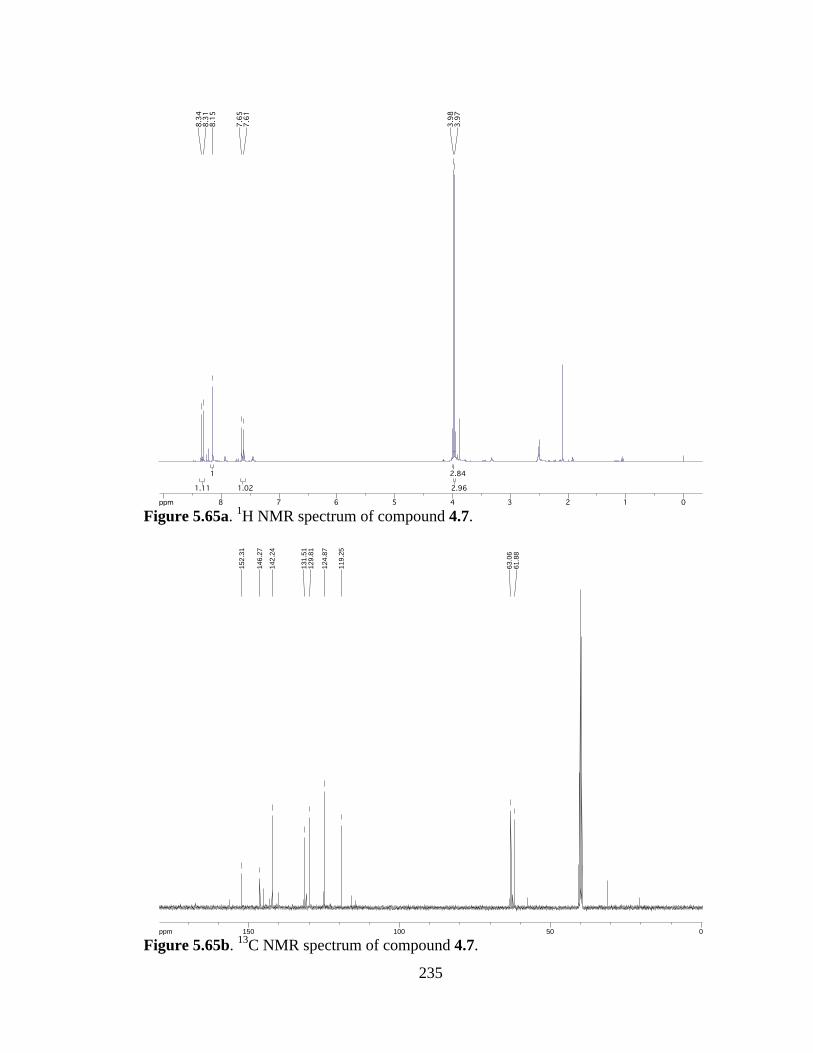
235
Figure 5.65a. 1H NMR spectrum of compound 4.7.
Figure 5.65b. 13C NMR spectrum of compound 4.7.
2.962.96
2.842.84
1.021.02
11
1.111.11ppm0 011223344556677889-16.04
3.97
3.98
7.61
7.65
8.15
8.31
8.34
ppm0 05050100100150150-253.79
61.8
863
.06
119.
25
124.
87
129.
8113
1.51
142.
2414
6.27
152.
31

236
5-chloro-6,7-dimethoxy-1H-indole (4.6). A suspension of 4.7 (380 mg, 1.3 mmol), SiO2
(3.8 g, 1000 wt%), Fe powder (1.37 g, 24.4 mmol), AcOH (7.6 mL), and PhCH3 (13 mL)
was heated to reflux for 3 h, then cooled to r.t. and diluted with CH2Cl2. The mixture was
filtered and the solid washed with CH2Cl2. The filtrate was washed with NaHSO3 (5%
aq), NaHCO3 (sat’d aq), and brine, then dried and concentrated. The resulting oil was
dissolved in CH2Cl2 (13 mL) and SiO2 added until the suspension was just fluid. After
stirring under Ar for 30 min, the suspension was filtered and the solid washed with
MeOH. The filtrate was concentrated and the product deemed sufficiently pure as crude
(250 mg, 91% yield).
1H-NMR (400 MHz; CDCl3): δ 8.40 (bs, 1H), 7.27 (s, 1H), 7.03 (t, J = 2.8 Hz, 1H), 6.32
(t, J = 2.6 Hz, 1H), 3.94 (s, 3H), 3.82 (s, 3H).; 13C-NMR (101 MHz; CDCl3): δ 191.9,
142.7, 139.1, 130.6, 128.8, 121.1, 115.8, 102.5, 61.4, 61.0; IR (NaCl, film) 3357, 2939,
2831, 1655 cm-1; HRMS (ESI-APCI+): [M+H]+ 212.0478 calcd for C10H11ClNO2, found:
212.0472.
REF: TRW-II-430, TRW-II-434, TRW-II-490, TRW-III-086.
OMeMeO
Cl
NH
4.6
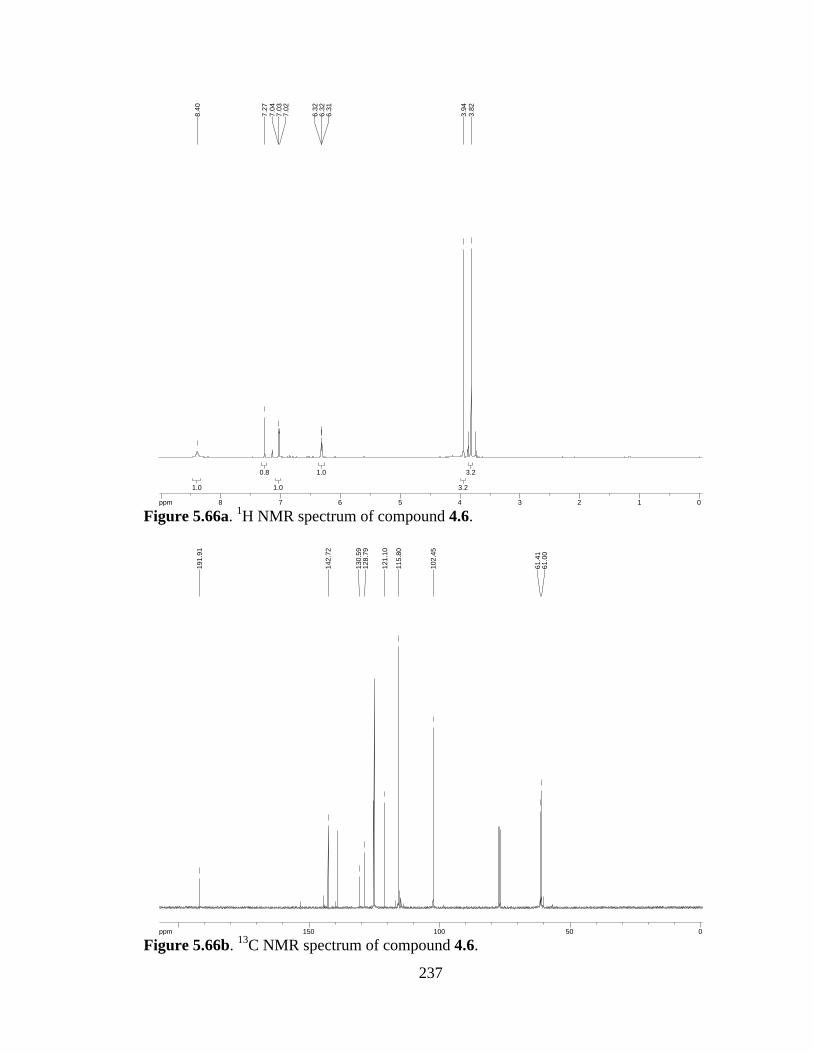
237
Figure 5.66a. 1H NMR spectrum of compound 4.6.
Figure 5.66b. 13C NMR spectrum of compound 4.6.
3.23.2
3.23.2
1.01.0
1.01.0
0.80.8
1.01.0
ppm0 011223344556677889-16.04
3.82
3.94
6.31
6.32
6.32
7.02
7.03
7.04
7.27
8.40
ppm0 05050100100150150200-253.79
61.0
061
.41
102.
45
115.
80
121.
10
128.
7913
0.59
142.
72
191.
91

238
5-chloro-6,7-dimethoxy-1-methyl-1H-indole (5.12). To a solution of indole 4.6 (126
mg, 0.59 mmol) in DMF (1.5 mL) at 0 °C was slowly added NaH (47 mg). After stirring
30 minutes, iodomethane (0.044 mL, 0.71 mmol) was added dropwise, and the reaction
stirred an additional 30 minutes at 0 °C. The reaction was warmed to r.t. and stirred 5 h
before adding water. The product was extracted in EtOAc, washed with water (2x) and
brine, then dried and concentrated to afford the title compound, deemed sufficiently pure
as crude (133 mg, >99% yield).
1H-NMR (300 MHz; CDCl3): δ 7.35 (s, 1H), 6.93 (d, J = 3.1 Hz, 1H), 6.34 (d, J = 3.1
Hz, 1H), 4.02 (s, 3H), 3.98 (s, 3H), 3.93 (s, 3H); 13C-NMR (101 MHz; CDCl3): δ 143.7,
140.6, 131.0, 126.9, 120.6, 116.0, 115.7, 100.6, 61.8, 61.2, 35.4; IR (NaCl, film) 2928,
1048 cm-1; HRMS (ESI-APCI+): [M+H]+ 226.0635 calcd for C11H13ClNO2, found:
226.0618.
REF: TRW-II-433, TRW-II-437, TRW-III-087.
OMeMeO
Cl
NMe
5.12

239
Figure 5.67a. 1H NMR spectrum of compound 5.12.
Figure 5.67b. 13C NMR spectrum of compound 5.12.
3.23.2
3.33.3
3.33.3
1.01.0
1.01.0
1.01.0
ppm1 12233445566778-19.97
3.93
3.98
4.02
6.33
6.34
6.92
6.93
7.35
ppm0 05050100100150150-253.79
35.4
3
61.2
261
.83
100.
64
115.
7411
6.05
120.
62
126.
8913
1.03
140.
6214
3.68

240
5-chloro-6,7-dimethoxy-1-methyl-1H-indole-3-carbaldehyde (4.20). DMF (6.5 mL)
was cooled to 0 °C, then POCl3 (0.586 mL, 6.42 mmol) added dropwise. Indole 5.12
(1.32 g, 5.84 mmol) was added dropwise as a solution in DMF. After warming the
mixture to 35 °C and stirring an additional 1.5 h, ice water was added followed by 2M
NaOH (15 mL). The resulting solution was boiled for 10 min, then cooled and diluted
with EtOAc. The organic layer was washed with water and brine, then dried and
concentrated to give the aldehyde (1.38 g, 93% yield), used as crude in the next reaction.
1H-NMR (400 MHz; CDCl3): δ 9.76 (s, 1H), 7.96 (s, 1H), 7.44 (s, 1H), 3.95 (s, 3H), 3.94
(s, 3H), 3.82 (s, 3H); 13C-NMR (101 MHz; CDCl3): δ 183.9, 145.4, 140.92, 140.89,
129.6, 124.5, 123.1, 117.26, 117.20, 61.9, 61.1, 36.7; IR (NaCl, film) 2930, 2853, 1656,
1044 cm-1; HRMS (ESI-APCI+): [M+H]+ 254.0584 calcd for C12H13ClNO3, found:
254.0581.
REF: TRW-II-436, TRW-II-439, TRW-III-090.
OMeMeO
Cl
NMe
CHO
4.20
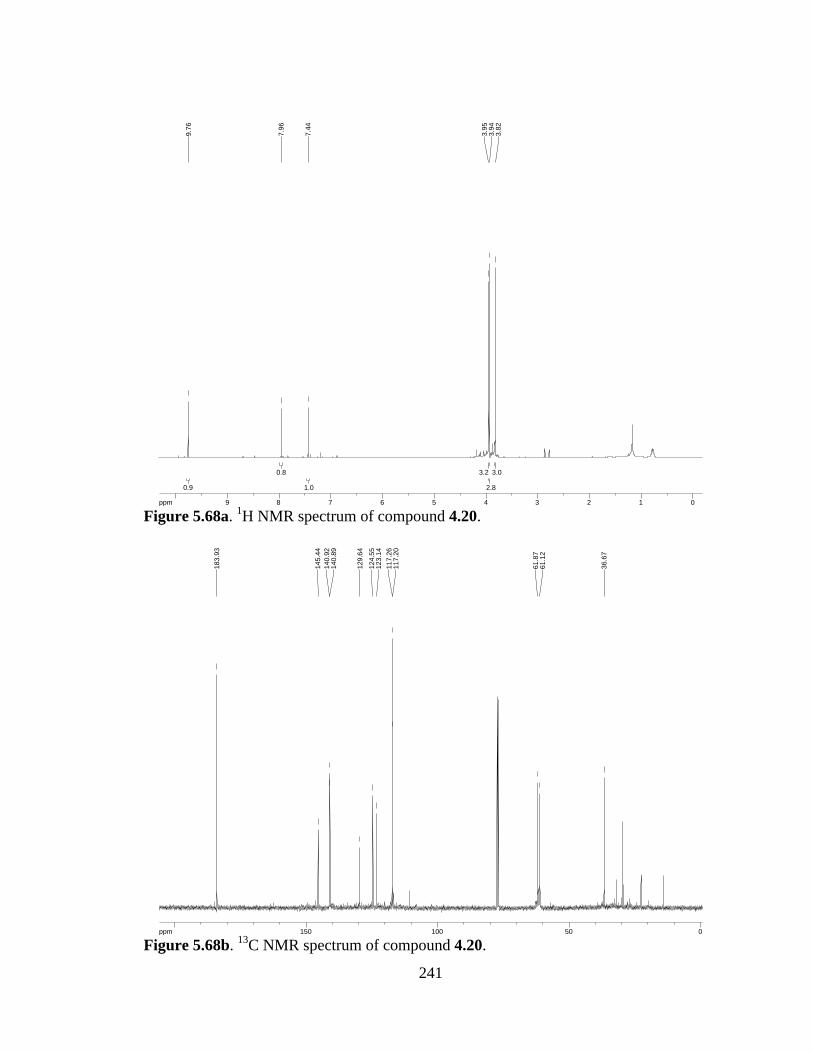
241
Figure 5.68a. 1H NMR spectrum of compound 4.20.
Figure 5.68b. 13C NMR spectrum of compound 4.20.
3.03.0
2.82.8
3.23.2
1.01.0
0.80.8
0.90.9
ppm0 011223344556677889910-16.043.
823.
943.
95
7.44
7.96
9.76
ppm0 05050100100150150200-253.79
36.6
7
61.1
261
.87
117.
2011
7.26
123.
1412
4.55
129.
64
140.
8914
0.92
145.
44
183.
93

242
(E)-methyl 3-(5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-yl)acrylate (4.5). To a
solution of aldehyde 4.20 (133 mg, 0.52 mmol) in toluene (1.8 mL) was added
methyl(triphenyl-phosphoranylidene)acetate (347 mg, 1.04 mmol). The resulting solution
was heated to reflux O/N, then concentrated and purified by SiO2 chromatography (9:1 –
4:1 – 2:1 hexanes:EtOAc) to afford olefin 4.5 (128 mg, 80% yield).
1H-NMR (400 MHz; CDCl3): δ 7.67 (d, J = 16.0 Hz, 1H), 7.51 (s, 1H), 7.11 (s, 1H), 6.20
(d, J = 16.0 Hz, 1H), 3.93 (s, 3H), 3.89 (s, 3H), 3.83 (s, 3H), 3.71 (s, 3H); 13C-NMR (101
MHz; CDCl3): δ 168.4, 144.7, 141.0, 137.2, 134.8, 129.9, 124.3, 122.8, 115.7, 112.5,
111.4, 61.8, 61.2, 51.4, 36.1; IR (NaCl, film) 2940, 1736, 1626, 1043 cm-1; HRMS (ESI-
APCI+): [M+H]+ 310.0846 calcd for C15H17ClNO4, found: 310.0840.
REF: TRW-II-441, TRW-II-443, TRW-III-091.
OMeMeO
Cl
NMe
CO2Me
4.5
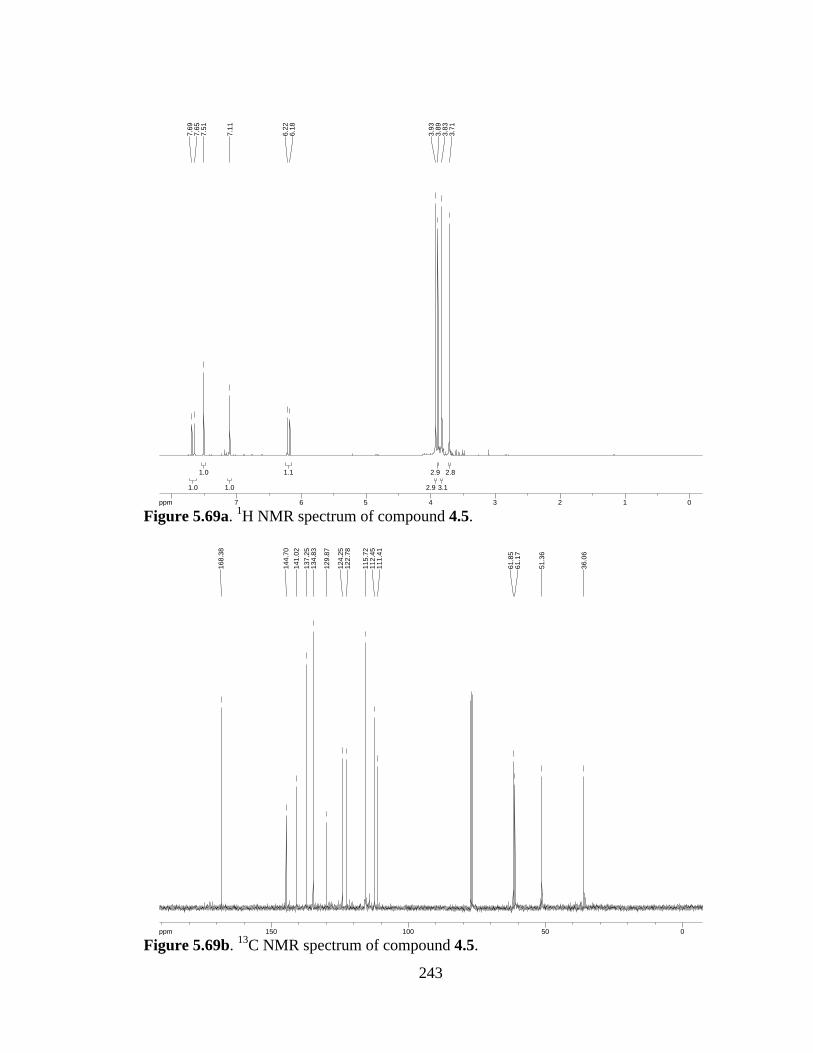
243
Figure 5.69a. 1H NMR spectrum of compound 4.5.
Figure 5.69b. 13C NMR spectrum of compound 4.5.
2.82.8
3.13.1
2.92.9
2.92.9
1.11.1
1.01.0
1.01.0
1.01.0
ppm0 0112233445566778-16.04
3.71
3.83
3.89
3.93
6.18
6.22
7.11
7.51
7.65
7.69
ppm0 05050100100150150-253.79
36.0
6
51.3
6
61.1
761
.85
111.
4111
2.45
115.
72
122.
7812
4.25
129.
87
134.
8313
7.25
141.
0214
4.70
168.
38

244
(2S,3R)-methyl 2-(((benzyloxy)carbonyl)amino)-3-(5-chloro-6,7-dimethoxy-1-
methyl-1H-indol-3-yl)-3-hydroxypropanoate (4.4). To a solution of benzyl carbamate
(178 mg, 1.18 mmol) in 0.4 M NaOH (2.5 mL) and n-propanol (2 mL) in a flask covered
with foil was added fresh t-butyl hypochlorite (0.135 mL, 1.20 mmol). After stirring 5
min, (DHQD)2AQN (14 mg, 0.016 mmol) in n-propanol (0.5 mL), olefin 4.5 (100 mg,
0.32 mmol) in n-propanol (1 mL), and K2OsO2(OH)4 (5 mg, 0.013 mmol) in 0.4 M
NaOH (0.5 mL) were added successively, and the resulting solution stirred O/N. Sodium
bisulfite (0.2 g) was added and the product extracted into EtOAc. The organic layer was
washed with water, then brine, dried over Na2SO4, and concentrated. SiO2
chromatography (10:1 – 1:1 hexanes:EtOAc) gave pure tryptophan derivative 4.4 (54 mg,
35% yield).
[α]D = -20.9 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.40 (s, 1H), 7.32 (s, 5H), 6.92
(s, 1H), 5.77 (d, J = 9.2 Hz, 1H), 5.46-5.45 (bs, 1H), 5.06 (s, 2H), 4.69-4.65 (m, 1H), 3.98
(s, 3H), 3.89 (s, 3H), 3.88 (d, J = 9.3 Hz, 3H), 3.78 (s, 3H), 2.69 (bs, 1H); 13C-NMR (101
MHz; CDCl3): δ 166.1, 144.6, 140.9, 133.75, 133.73, 128.66, 128.60, 128.46, 128.43,
128.28, 128.18, 128.07, 126.3, 122.6, 114.2, 108.6, 72.9, 67.3, 66.9, 61.9, 61.2, 52.3,
36.2; IR (NaCl, film) 3346, 2952, 1723, 1465, 1218, 1040 cm-1; HRMS (ESI-APCI+):
[M+Na]+ 499.1248 calcd for C23H25ClN2NaO7, found: 499.1249.
REF: TRW-II-459, TRW-II-461, TRW-III-093.
OMeMeO
Cl
NMe
CO2MeHONHCbz
4.4
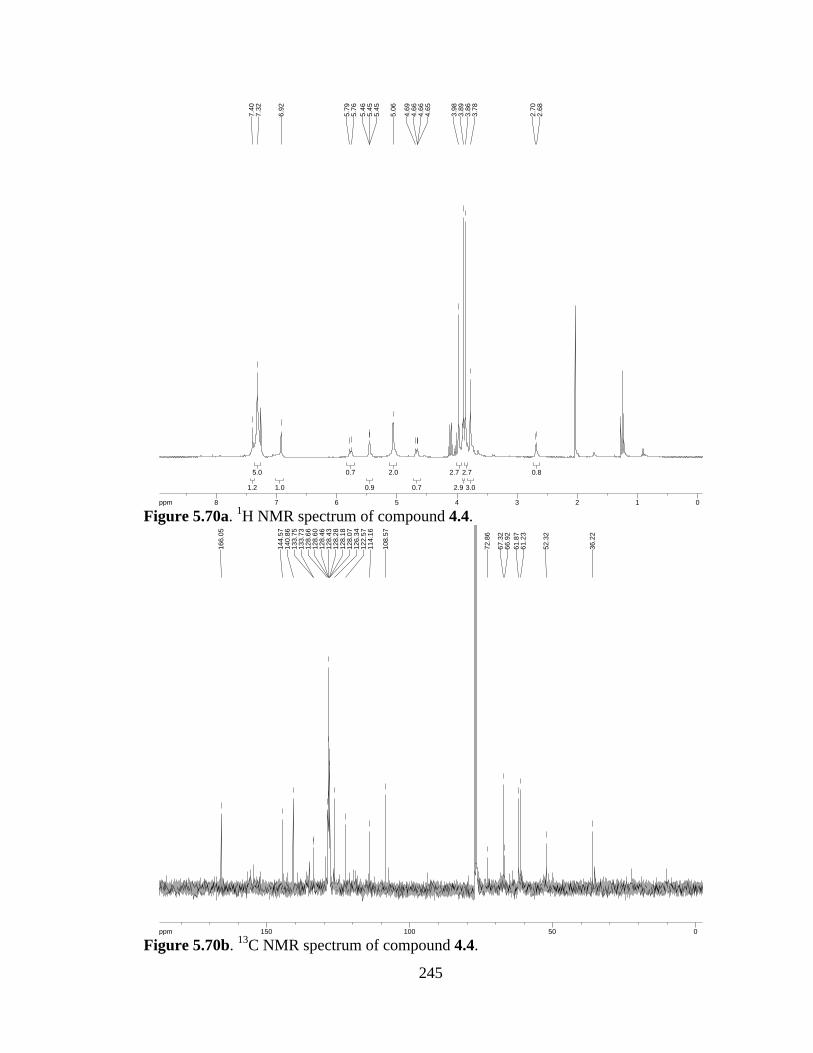
245
Figure 5.70a. 1H NMR spectrum of compound 4.4.
Figure 5.70b. 13C NMR spectrum of compound 4.4.
0.80.8
3.03.0
2.72.7
2.92.9
2.72.7
0.70.7
2.02.0
0.90.9
0.70.7
1.01.0
5.05.0
1.21.2
ppm0 01122334455667788-19.97
2.68
2.70
3.78
3.86
3.89
3.98
4.65
4.66
4.66
4.69
5.06
5.45
5.45
5.46
5.76
5.79
6.92
7.32
7.40
ppm0 05050100100150150-253.79
36.2
2
52.3
2
61.2
361
.87
66.9
267
.32
72.8
6
108.
57
114.
1612
2.57
126.
3412
8.07
128.
1812
8.28
128.
4312
8.46
128.
6012
8.66
133.
7313
3.75
140.
8614
4.57
166.
05

246
1,4-difluoroanthracene-9,10-dione (4.23). To a solution of phthalic anhydride (10 g,
67.6 mmol) in 1,4-difluorobenzene (68 mL) was added AlCl3 (36 g, 270 mmol). The
resulting solution was stirred at reflux for 48h before removing the solvent by distillation.
HCl (1M, 400 mL) was added and the product extracted into CH2Cl2 (3x). The combined
organic extracts were filtered through a cotton plug and concentrated. The crude residue
was taken up in chloroform (40 mL) and to it added hexanes (100 mL). The solution was
cooled to -20 °C then filtered to collect the resultant precipitate. After drying the
precipitate under vacuum, polyphosphoric acid (50 mL) was added and the mixture
heated to 140 °C for 2h. The mixture was poured over approximately 400 g ice and
allowed to warm to r.t. while stirring. Solid K2CO3 was added to neutralize the solution,
then the product was extracted into CH2Cl2. The combined organic extracts were dried
(Na2SO4) and concentrated. The residue was dissolved in CH2Cl2, filtered through basic
alumina using CH2Cl2 as the eluent, and concentrated to afford the title compound (920
mg).
1H-NMR (400 MHz; CDCl3): δ 8.21-8.16 (m, 2H), 7.76-7.72 (m, 2H), 7.43-7.39 (m,
2H); 13C-NMR (101 MHz; CDCl3): δ 134.3, 133.2, 127.0, 124.7, 124.48, 124.34.
REF: TRW-II-451, TRW-III-079.
O
O F
F4.23
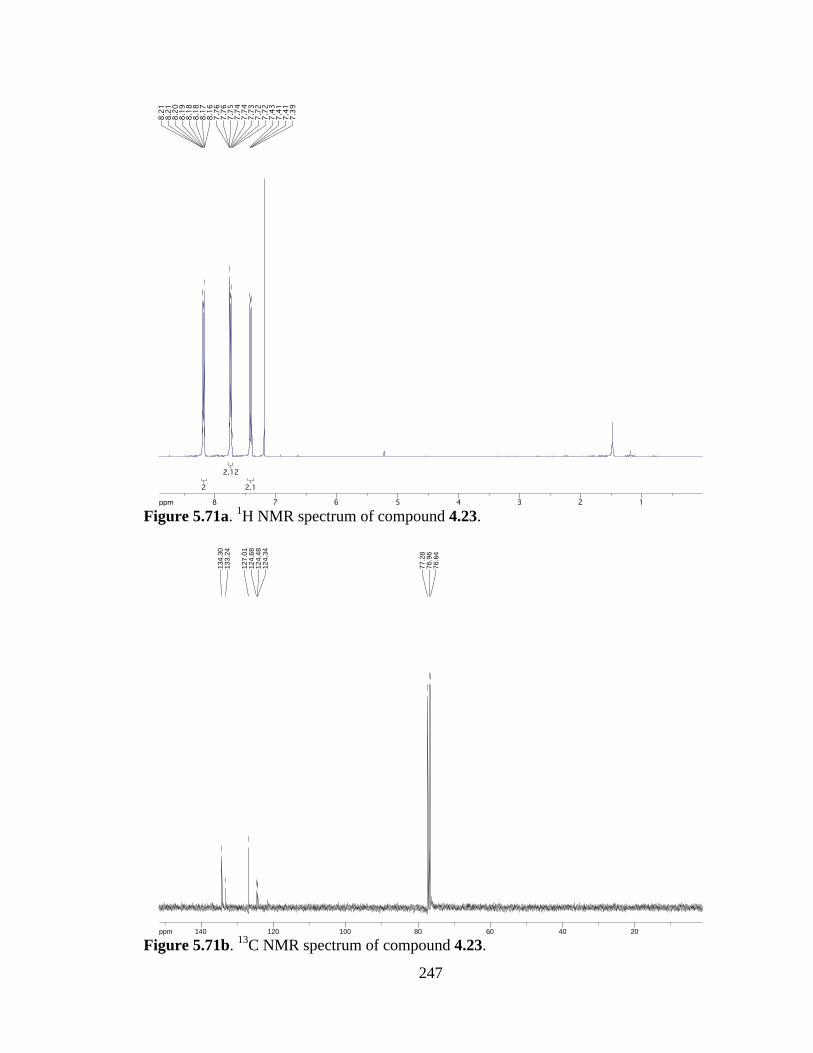
247
Figure 5.71a. 1H NMR spectrum of compound 4.23.
Figure 5.71b. 13C NMR spectrum of compound 4.23.
2.12.1
2.122.12
22
ppm1 122334455667788-16.04
7.39
7.41
7.41
7.43
7.72
7.72
7.73
7.74
7.74
7.75
7.76
7.76
8.16
8.17
8.18
8.18
8.19
8.20
8.21
8.21
ppm20 20404060608080100100120120140140-253.79
76.6
476
.96
77.2
8
124.
3412
4.48
124.
6812
7.01
133.
2413
4.30

248
(DHQD)2AQN (4.24). To a solution of DHQD (3.1 g, 9.5 mmol) in THF (19 mL) at -50
°C was added nBuLi (1.6M in hexanes, 6.0 mL, 9.5 mmol) dropwise over 10 min. After
stirring 15 min at -50 °C, the reaction mixture was warmed to 0 °C and the difluoro
compound added (920 mg, 3.8 mmol). The reaction was allowed to warm to r.t. as it
stirred O/N under Ar, then warmed to 40 °C and stirred an additional 2h. EtOAc was
added and the reaction quenched by addition of sat’d NaHCO3. The product was
extracted in EtOAc, dried (Na2SO4), concentrated, and purified by SiO2 chromatography
(5% MeOH, 0.5% NH4OH, in CHCl3) to afford (DHQD)2AQN (2.55 g, 78% yield).
1H-NMR (300 MHz; CDCl3): δ 8.63 (d, J = 4.5 Hz, 2H), 8.22 (dd, J = 5.8, 3.3 Hz, 2H),
8.01 (d, J = 9.2 Hz, 2H), 7.79 (dd, J = 5.8, 3.3 Hz, 2H), 7.49 (d, J = 4.5 Hz, 3H), 7.37
(dd, J = 9.2, 2.5 Hz, 3H), 6.80 (bs, 2H), 5.28 (s, 2H), 3.95 (s, 6H), 3.28-3.26 (bs, 2H),
3.00-2.53 (broad, 10H), 1.83 (bs, 2H), 1.61-1.45 (broad, 12H), 0.84 (t, J = 7.3 Hz, 6H).
REF: TRW-II-457, TRW-III-002, TRW-III-082.
O
O DHQD
DHQD(DHQD)2AQN (4.24)

249
Figure 5.72a. 1H NMR spectrum of compound 4.24.
6.36.3
12.312.3
2.12.1
10.410.4
2.02.0
6.06.0
2.12.1
1.81.8
2.72.7
3.33.3
1.91.9
1.81.8
1.81.8
1.61.6
ppm0 0112233445566778899-19.97
0.82
0.84
0.86
1.45
1.46
1.46
1.46
1.47
1.54
1.55
1.55
1.56
1.56
1.58
1.59
1.59
1.61
1.83
1.83
2.53
2.53
2.76
2.77
2.88
2.88
2.88
2.88
2.89
2.91
2.91
2.91
2.91
2.92
3.00
3.26
3.26
3.27
3.27
3.27
3.27
3.27
3.28
3.28
3.95
5.28
6.80
6.80
7.35
7.36
7.38
7.39
7.48
7.50
7.78
7.79
7.80
7.81
8.00
8.03
8.21
8.22
8.23
8.24
8.62
8.64

250
(2S,3R)-methyl 2-(((benzyloxy)carbonyl)amino)-3-((tert-butyldimethylsilyl) oxy)-3-
(5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-yl)propanoate (4.25). Alcohol 4.4 (296
mg, 0.62 mmol) was dissolved in DMF (1.6 mL), and to it added imidazole (102 mg, 1.5
mmol) and TBSCl (242 mg, 1.6 mmol). After stirring O/N, the solution was diluted with
EtOAc, washed with water (3x), brine (1x), dried over Na2SO4, and concentrated to give
the title compound (366 mg, >99% yield).
1H-NMR (300 MHz; CDCl3): δ 7.36 (s, 1H), 7.32 (s, 5H), 6.83 (s, 1H), 5.49 (dd, J =
12.6, 2.3 Hz, 1H), 5.07 (s, 2H), 4.98 (s, 1H), 4.47 (dd, J = 9.6, 2.1 Hz, 1H), 4.00 (s, 3H),
3.91 (s, 3H), 3.89 (s, 3H), 3.79 (s, 3H), 0.86 (s, 9H), 0.09 (s, 6H); HRMS (ESI-APCI+):
[M+Na]+ 613.2113 calcd for C29H39ClN2NaO7Si, found: 613.2109.
REF: TRW-II-460, TRW-II-465, TRW-III-096.
OMeMeO
Cl
NMe
CO2MeTBSONHCbz
4.25
251
Figure 5.73a. 1H NMR spectrum of compound 4.25.
5.85.8
9.49.4
2.92.9
3.33.3
2.72.7
3.43.4
0.70.7
1.11.1
1.91.9
1.31.3
0.90.9
4.74.7
1.01.0
ppm0 011223344556677-19.97
0.09
0.86
3.79
3.89
3.91
4.00
4.45
4.45
4.48
4.49
4.98
5.07
5.46
5.47
5.50
5.51
6.83
7.32
7.36

252
(2S,3R)-methyl 2-amino-3-((tert-butyldimethylsilyl)oxy)-3-(5-chloro-6,7-dimethoxy-
1-methyl-1H-indol-3-yl)propanoate (4.26). To a suspension of 4.25 (366 mg, 0.61
mmol) and Pd/C (37 mg) in MeOH (3 mL) was added hydrogen gas. After stirring O/N,
the reaction was flushed with argon and filtered through celite to afford the free amine
(262 mg, 94% yield).
[α]D = +6.4 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.32 (s, 1H), 6.90 (s, 1H), 5.40 (d,
J = 1.8 Hz, 1H), 4.85-4.63 (m, 1H), 4.00 (s, 3H), 3.94 (s, 3H), 3.90 (s, 3H), 3.75 (s, 3H),
2.24-2.14 (bs, 2H), 0.88 (s, 9H), -0.01 (s, 3H), -0.15 (s, 3H); 13C-NMR (101 MHz;
CDCl3): δ 173.3, 143.8, 140.7, 129.3, 129.0, 123.9, 120.6, 114.8, 114.3, 70.4, 61.8, 61.1,
60.8, 52.0, 35.5, 25.66, 25.61, 25.52, 18.0, -4.6, -5.6; IR (NaCl, film) 3332, 2953, 2858,
1750, 1464, 1038 cm-1; HRMS (ESI-APCI+): [M+Na]+ 479.1745 calcd for
C21H33ClN2NaO5Si, found: 479.1746.
REF: TRW-II-462, TRW-II-468, TRW-III-098.
OMeMeO
Cl
NMe
CO2MeTBSONH2
4.26
253
Figure 5.74a. 1H NMR spectrum of compound 4.26.
Figure 5.74b. 13C NMR spectrum of compound 4.26.
2.72.7
2.92.9
9.09.0
2.02.0
2.92.9
2.82.8
2.92.9
3.43.4
1.31.3
0.90.9
0.80.8
1.01.0
ppm0 011223344556677-14.99
-0.1
5-0
.01
0.88
2.14
2.14
2.15
2.15
2.16
2.16
2.17
2.17
2.17
2.18
2.19
2.19
2.19
2.20
2.20
2.21
2.21
2.23
2.23
2.24
3.75
3.90
3.94
4.00
4.63
4.64
4.66
4.84
4.85
5.39
5.40
6.90
6.90
7.32
ppm0 05050100100150150-253.79
-5.6
2-4
.57
18.0
2
25.5
225
.61
25.6
6
35.5
0
51.9
8
60.7
661
.12
61.7
5
70.3
9
114.
2811
4.82
120.
5812
3.95
128.
9612
9.32
140.
6814
3.84
173.
34

254
(2S,3R)-methyl 2-((S)-2-((tert-butoxycarbonyl)amino)propanamido)-3-((tert-butyl-
dimethylsilyl)oxy)-3-(5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-yl)propanoate
(4.27). To a solution of amine 4.26 (262 mg, 0.57 mmol) in CH2Cl2 (3 mL) was added N-
Boc-ala-OH (108 mg, 0.57 mmol), iPr2NEt (0.1 mL, 0.57 mmol), and EDCI (110 mg,
0.57 mmol). The reaction was stirred at r.t. O/N, the concentrated and purified by SiO2
chromatography (10% - 25% - 50% EtOAc in hexanes) to afford 4.27 (210 mg, 59%
yield).
1H-NMR (300 MHz; CDCl3): δ 7.36 (s, 1H), 6.86 (s, 1H), 6.73-6.70 (bs, 1H), 5.53 (d, J
= 2.0 Hz, 1H), 4.97-4.96 (bs, 1H), 4.67 (dd, J = 9.1, 1.7 Hz, 1H), 4.19-4.15 (bs, 1H), 3.99
(s, 3H), 3.92 (s, 3H), 3.90 (s, 3H), 3.78 (s, 3H), 1.45 (s, 9H), 1.24 (dd, J = 7.1, 0.8 Hz,
3H), 0.89 (s, 9H), 0.01 (s, 3H), -0.15 (s, 3H).
REF: TRW-II-466, TRW-II-471.
NMe
Cl
MeOOMe
HNNHBoc
CO2Me
OMe
TBSO
4.27

255
Figure 5.75a. 1H NMR spectrum of compound 4.27.
2.72.7
2.62.6
9.19.1
3.03.0
8.98.9
2.72.7
2.92.9
2.92.9
2.92.9
1.21.2
0.90.9
0.90.9
0.90.9
0.70.7
0.90.9
1.01.0
ppm0 0112233445566778-19.97
-0.1
50.
01
0.89
1.23
1.23
1.25
1.26
1.45
3.78
3.90
3.92
3.99
4.15
4.15
4.17
4.17
4.18
4.18
4.18
4.18
4.18
4.19
4.65
4.65
4.68
4.68
4.96
4.96
4.96
4.97
4.97
4.97
5.52
5.53
6.70
6.70
6.70
6.71
6.72
6.72
6.72
6.72
6.73
6.86
7.36

256
(2S,3R)-methyl 2-((S)-2-aminopropanamido)-3-((tert-butyldimethylsilyl)oxy)-3-(5-
chloro-6,7-dimethoxy-1-methyl-1H-indol-3-yl)propanoate (4.28). Compound 4.27
(105 mg, 0.167 mmol) was dissolved in CH2Cl2 (1.7 mL), then 2,6-lutadine (0.039 mL,
0.334 mmol) added. The solution was cooled to 0 °C, then TMSOTf (0.045 mL, 0.251
mmol) added. After warming to r.t., the reaction was monitored by TLC and once
complete, quenched with sat’d NH4Cl. The product was extracted into CH2Cl2, the
organic extracts dried and concentrated, and the product purified by SiO2
chromatography to afford the title compound (52 mg, 59% yield).
1H-NMR (300 MHz; CDCl3): δ 7.36 (s, 1H), 6.92 (s, 1H), 5.53 (s, 1H), 4.79-4.77 (bs,
2H), 4.59 (d, J = 8.8 Hz, 1H), 3.97 (s, 3H), 3.90 (s, 3H), 3.88 (s, 3H), 3.82 (d, J = 7.4 Hz,
1H), 3.74 (s, 3H), 1.29 (d, J = 8.4 Hz, 3H), 0.87 (s, 9H), -0.01 (s, 3H), -0.16 (s, 3H).
REF: TRW-II-476; for TFA version, see TRW-II-474.
NMe
Cl
MeOOMe
HNNH2
CO2Me
OMe
TBSO
4.28

257
Figure 5.76a. 1H NMR spectrum of compound 4.28.
3.03.0
2.72.7
9.19.1
2.72.7
2.92.9
1.21.2
2.72.7
2.72.7
2.72.7
1.01.0
2.02.0
1.01.0
1.01.0
1.41.4
ppm0 0112233445566778-19.97
-0.1
6-0
.01
0.87
1.27
1.30
3.74
3.80
3.83
3.88
3.90
3.97
4.57
4.60
4.77
4.77
4.78
4.78
4.78
4.79
4.79
5.53
6.92
7.36

258
(S,Z)-3-((5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-yl)methylene)-6-
methylpiperazine-2,5-dione (4.29). To a solution of amine 4.28 (69 mg, 0.167 mmol) in
toluene (5 mL) was added a small amount of 2-OH-pyridine. The solution was heated to
reflux O/N, then cooled to r.t. The solvent was decanted off and the remaining solid
washed with toluene and dried to afford pure 4.29 (37 mg, 61% yield).
1H-NMR (300 MHz; DMSO-d6): δ 9.43 (s, 1H), 8.32 (s, 1H), 7.87 (s, 1H), 7.43 (s, 1H),
6.81 (s, 1H), 4.11 (q, J = 6.9 Hz, 1H), 3.96 (s, 3H), 3.93 (s, 3H), 3.81 (s, 3H), 1.32 (d, J =
6.8 Hz, 3H); 13C-NMR (101 MHz; DMSO): δ 167.9, 160.77, 160.73, 144.2, 141.0, 133.1,
128.4, 126.3, 123.8, 120.9, 114.2, 107.2, 106.2, 62.4, 61.4, 50.7, 36.0, 19.9; HRMS (ESI-
APCI+): [M+H]+ 364.1064 calcd for C17H19ClN3O4, found: 364.1075.
REF: TRW-II-475, TRW-II-477.
NMe
Cl
MeOOMe
HNNH
O
OMe
4.29

259
Figure 5.77a. 1H NMR spectrum of compound 4.29.
Figure 5.77b. 13C NMR spectrum of compound 4.29.
3.33.3
3.13.1
3.13.1
3.03.0
1.11.1
1.11.1
1.11.1
1.11.1
1.01.0
0.90.9
ppm0 0112233445566778899-19.97
1.31
1.33
3.81
3.93
3.96
4.08
4.10
4.12
4.15
6.81
7.43
7.87
8.32
9.43
ppm0 05050100100150150-253.79
19.8
6
35.9
7
50.7
3
61.4
262
.41
106.
1610
7.21
114.
17
120.
8712
3.79
126.
2612
8.40
133.
09
140.
9914
4.19
160.
7316
0.77
167.
85

260
(2S,3R)-2-(((benzyloxy)carbonyl)amino)-3-((tert-butyldimethylsilyl)oxy)-3-(5-chloro-
6,7-dimethoxy-1-methyl-1H-indol-3-yl)propanoic acid (4.30). To a solution of methyl
ester 4.25 (682 mg, 1.2 mmol) in THF/H2O (2:1, 2.6:1.3 mL) was added LiOH (144 mg,
6.0 mmol) and the resulting suspension stirred O/N. The reaction mixture was acidified
with 10% KHSO4, then the product extracted in EtOAc. The combined organic extracts
were washed with brine, dried over Na2SO4, and concentrated to afford 4.30 (545 mg,
82% yield).
1H-NMR (300 MHz; CDCl3): δ 7.37-7.28 (m, 6H), 6.87 (bs, 1H), 5.62-5.58 (broad, 1H),
5.10-5.00 (m, 2H), 4.70 (s, 1H), 4.51-4.49 (bs, 1H), 3.99-3.89 (broad, 9H), 0.91-0.86 (m,
9H), 0.10-0.01 (m, 6H).
REF: TRW-III-023.
OMeMeO
Cl
NMe
CO2HTBSONHCbz
4.30

261
Figure 5.78a. 1H NMR spectrum of compound 4.30.
6.06.0
9.09.0
9.49.4
1.01.0
0.60.6
2.12.1
1.41.4
1.01.0
6.56.5
ppm0 011223344556677-19.97
0.01
0.10
0.10
0.86
0.87
0.88
0.91
0.91
3.89
3.99
4.49
4.50
4.50
4.50
4.50
4.50
4.50
4.51
4.70
5.00
5.01
5.01
5.01
5.02
5.02
5.02
5.03
5.10
5.58
5.58
5.59
5.59
5.61
5.61
5.61
5.61
5.62
5.62
5.62
5.62
6.87
6.87
6.87
7.28
7.34
7.34
7.36
7.37

262
(S)-methyl 2-((2S,3R)-2-(((benzyloxy)carbonyl)amino)-3-((tert-
butyldimethylsilyl)oxy)-3-(5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-
yl)propanamido)propanoate (4.31). To a solution of acid 4.30 (100 mg, 0.173 mmol),
L-Ala-OMe·HCl (24 mg, 0.173 mmol), and iPr2NEt (0.075 mL, 0.433 mmol) in CH2Cl2
(2 mL) was added T3P (50 wt % in DMF, 127 mg of solution). The resulting solution
was stirred O/N under Ar at r.t., then quenched by addition of water. The product was
extracted in EtOAc and the combined organic layers dried over Na2SO4 and concentrated
to afford the crude material. Purification by SiO2 chromatography (10-40% EtOAc in
hexanes) gave 4.31 (39 mg, 34% yield).
1H-NMR (300 MHz; CDCl3): δ 7.58 (d, J = 7.4 Hz, 1H), 7.35 (d, J = 3.1 Hz, 5H), 7.04
(s, 1H), 5.56 (dd, J = 29.0, 5.4 Hz, 2H), 5.13-5.10 (broad, 3H), 4.76-4.74 (bs, 1H), 4.57-
4.50 (m, 1H), 3.98 (s, 3H), 3.89 (d, J = 3.7 Hz, 6H), 3.78 (s, 3H), 1.42 (d, J = 7.2 Hz,
3H), 0.94 (s, 9H), 0.06 (d, J = 48.3 Hz, 6H).
REF: TRW-III-026.
NMe
Cl
MeOOMe
NHCbz
TBSO O
NH
CO2Me
Me
4.31
263
Figure 5.79a. 1H NMR spectrum of compound 4.31.
6.36.3
9.19.1
2.82.8
3.03.0
6.16.1
3.23.2
1.31.3
1.01.0
2.82.8
1.91.9
0.80.8
7.57.5
0.90.9
ppm0 011223344556677-19.97
-0.0
20.
14
0.94
1.40
1.43
3.78
3.88
3.89
3.98
4.50
4.51
4.53
4.55
4.57
4.74
4.74
4.75
4.75
4.75
4.75
4.76
4.76
5.10
5.13
5.50
5.51
5.59
5.62
7.04
7.28
7.34
7.35
7.57
7.60

264
(2S,3R)-methyl 2-((S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)propan-
amido)-3-((tert-butyldimethylsilyl)oxy)-3-(5-chloro-6,7-dimethoxy-1-methyl-1H-
indol-3-yl)propanoate (4.32). To a solution of amine 4.26 (100 mg, 0.22 mmol) in
CH2Cl2 (2.2 mL) was added N-Fmoc-Ala (68 mg, 0.22 mmol), iPr2NEt (0.1 mL, 0.55
mmol), and T3P (160 mg 50 wt% solution in DMF, 0.25 mmol). The resulting solution
was allowed to stir O/N at r.t. under Ar, then quenched with water and the product
extracted in EtOAc. The combined organic layers were dried over Na2SO4, filtered, and
concentrated. Purification by SiO2 chromatography (1:1 hexanes:EtOAc) afforded 4.32 in
59% yield (97 mg).
[α]D = -3.9 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.76 (d, J = 7.5 Hz, 2H), 7.58 (d, J
= 7.4 Hz, 2H), 7.40 (d, J = 7.5 Hz, 2H), 7.36 (s, 1H), 7.31 (d, J = 7.4 Hz, 2H), 6.85 (s,
1H), 6.65 (s, 1H), 5.54 (d, J = 1.4 Hz, 1H), 5.41-5.38 (m, 1H), 4.68 (dd, J = 9.0, 1.7 Hz,
1H), 4.33-4.27 (m, 2H), 3.97 (s, 3H), 3.89 (s, 3H), 3.86 (s, 3H), 3.79 (s, 3H), 1.36 (d, J =
6.8 Hz, 3H), 0.89 (s, 9H), 0.02 (s, 3H), -0.13 (s, 3H); 13C-NMR (101 MHz; CDCl3): δ
171.4, 165.8, 156.4, 144.4, 143.7, 143.3, 141.27, 141.15, 140.9, 135.1, 128.3, 127.78,
127.70, 127.5, 127.10, 127.06, 126.5, 124.87, 124.82, 122.4, 119.98, 119.92, 117.9,
113.9, 108.1, 67.0, 61.8, 61.2, 52.3, 51.5, 47.1, 36.0, 25.61, 25.48, 25.3, 17.94, 17.91, -
3.6; IR (NaCl, film) 3302, 2953, 2857, 1714, 1462, 1254, 1040 cm-1; HRMS (ESI-
APCI+): [M+Na]+ 772.2797 calcd for C39H48ClN3NaO8Si, found: 772.2799.
REF: TRW-III-056, TRW-III-064, TRW-III-113.
NMe
Cl
MeOOMe
HNNHFmoc
CO2Me
OMe
TBSO
4.32
265
Figure 5.80a. 1H NMR spectrum of compound 4.32.
Figure 5.80b. 13C NMR spectrum of compound 4.32.
2.82.8
2.82.8
9.39.3
2.52.5
2.62.6
2.82.8
2.92.9
3.13.1
2.12.1
0.90.9
0.70.7
1.01.0
0.70.7
0.90.9
1.91.9
1.21.2
2.02.0
1.91.9
2.02.0
ppm0 01122334455667788-19.97
-0.1
30.
02
0.89
1.34
1.37
3.79
3.86
3.89
3.97
4.27
4.28
4.31
4.33
4.33
4.33
4.66
4.67
4.69
4.70
5.38
5.38
5.38
5.41
5.41
5.54
5.54
6.63
6.66
6.85
7.30
7.32
7.36
7.39
7.42
7.57
7.60
7.75
7.77
ppm0 05050100100150150-253.79
-3.6
1
17.9
117
.94
25.2
625
.48
25.6
1
35.9
8
47.1
151
.47
52.2
8
61.2
161
.83
67.0
4
108.
0511
3.90
117.
8711
9.92
119.
9812
2.45
124.
8212
4.87
126.
5012
7.06
127.
1012
7.48
127.
7012
7.78
128.
2613
5.08
140.
8714
1.15
141.
2714
3.32
143.
6814
4.44
156.
39
165.
75
171.
44

266
(6S)-3-((R)-((tert-butyldimethylsilyl)oxy)(5-chloro-6,7-dimethoxy-1-methyl-1H-
indol-3-yl)methyl)-6-methylpiperazine-2,5-dione (4.3). Dipeptide 4.32 (350 mg, 0.47
mmol) was dissolved in THF (20 mL), then morpholine (4.7 mL) added. The resulting
solution was stirred O/N at r.t., then concentrated. The crude residue was purified by SiO2
chromatography (10% methanol in CH2Cl2) to afford 181 mg of the title dioxopiperazine
(78% yield).
[α]D = -5.8 (CH2Cl2); 1H-NMR (300 MHz; CDCl3): δ 7.31 (s, 1H), 6.98 (s, 1H), 5.67 (s,
2H), 4.13 (q, J = 6.7 Hz, 1H), 4.05 (d, J = 1.5 Hz, 1H), 4.02 (s, 3H), 3.97 (s, 3H), 3.91 (s,
3H), 3.76 (s, 1H), 1.61 (d, J = 7.0 Hz, 3H), 0.88 (s, 9H), 0.03 (s, 3H), -0.03 (s, 3H); 13C-
NMR (101 MHz; CDCl3): δ 166.5, 160.4, 140.9, 136.1, 129.7, 127.4, 125.7, 122.7, 114.6,
108.6, 61.9, 61.2, 51.6, 51.3, 36.02, 36.00, 25.8, 25.6, 25.3, 20.4, 17.9, -1.6, -3.6; IR
(NaCl, film) 3225, 2932, 1681, 1449, 1259, 1042 cm-1; HRMS (ESI-APCI+): [M+Na]+
518.1854 calcd for C23H34ClN3NaO5Si, found: 518.1852.
REF: TRW-III-057, TRW-III-065.
NMe
Cl
MeOOMe
HNNH
O
OMe
TBSO
4.3
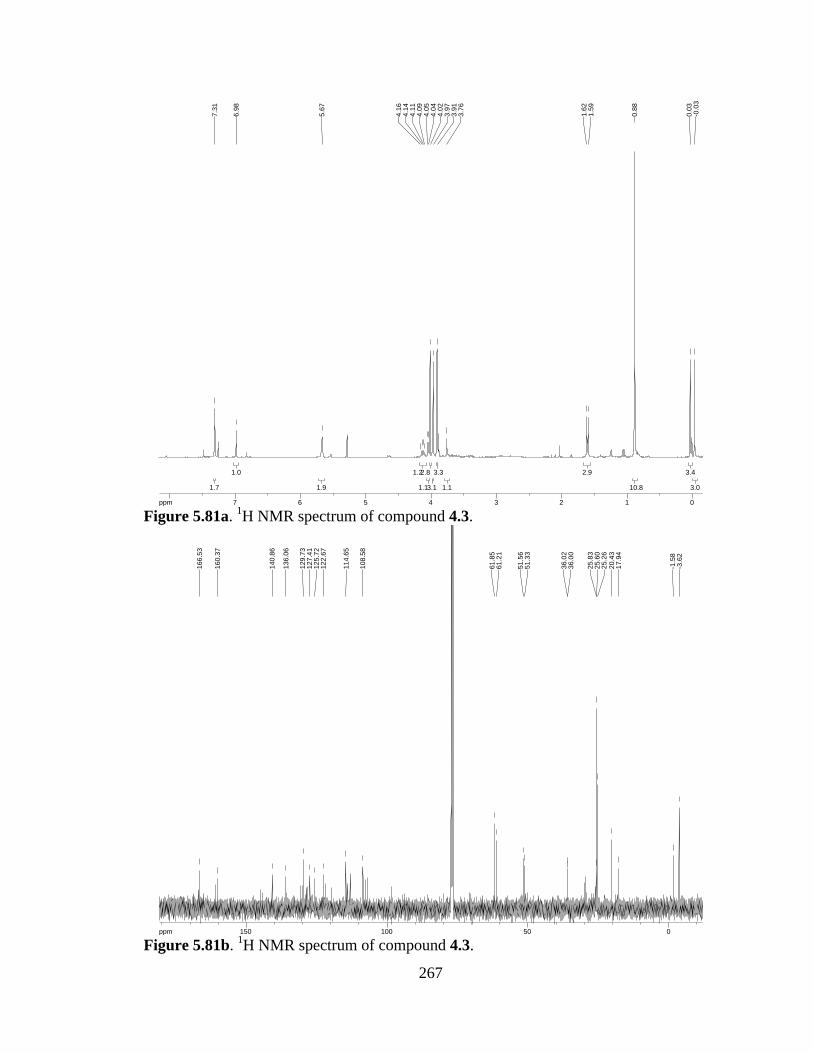
267
Figure 5.81a. 1H NMR spectrum of compound 4.3.
Figure 5.81b. 1H NMR spectrum of compound 4.3.
3.03.0
3.43.4
10.810.8
2.92.9
1.11.1
3.33.3
3.13.1
2.82.8
1.11.1
1.21.2
1.91.9
1.01.0
1.71.7
ppm0 0112233445566778-19.97
-0.0
30.
03
0.88
1.59
1.62
3.76
3.91
3.97
4.02
4.04
4.05
4.09
4.11
4.14
4.16
5.67
6.98
7.31
ppm0 05050100100150150-253.79
-3.6
2-1
.58
17.9
420
.43
25.2
625
.60
25.8
3
36.0
036
.02
51.3
351
.56
61.2
161
.85
108.
58
114.
65
122.
6712
5.72
127.
4112
9.73
136.
06
140.
86
160.
37
166.
53

268
(2S,3S,3aS,8aR)-1-benzyl 2-methyl 3-((tert-butyldimethylsilyl)oxy)-5-chloro-3a-
hydroxy-6,7-dimethoxy-8-methyl-3,3a,8,8a-tetrahydropyrrolo[2,3-b]indole-1,2(2H)-
dicarboxylate (4.40). Indole 4.25 (70 mg, 0.118 mmol) was dissolved in CH2Cl2 (6 mL)
and the resulting solution cooled to -78 °C. Freshly prepared DMDO (0.056 M in
acetone, 3 mL, 0.165 mmol) was added and the reaction mixture allowed to warm to r.t.
O/N. Sodium thiosulfate (sat’d aq) was added and the product extracted in CH2Cl2. The
combined organic extracts were washed with brine, dried over Na2SO4, and concentrated.
Purification by pTLC (40% EtOAc in hexanes) afforded pure 4.25 (6.2 mg, 9% yield).
1H-NMR (400 MHz; CDCl3): δ 7.46-7.40 (m, 5H), 7.05 (t, J = 9.1 Hz, 1H), 5.59 (s, 1H),
5.42-5.37 (m, 2H), 5.11-5.08 (m, 1H), 4.67-4.61 (m, 1H), 3.97-3.87 (m, 6H), 3.55 (s,
3H), 3.39 (s, 3H), 0.96 (s, 9H), 0.04 (d, J = 36.9 Hz, 6H); HRMS (ESI-APCI+): [M+H]+
607.2242 calcd for C29H40ClN2O8Si, found: 607.2240.
REF: TRW-III-107, TRW-III-110.
NMe
Cl
MeOOMe
NCbzH
HOTBSO
CO2Me
4.40
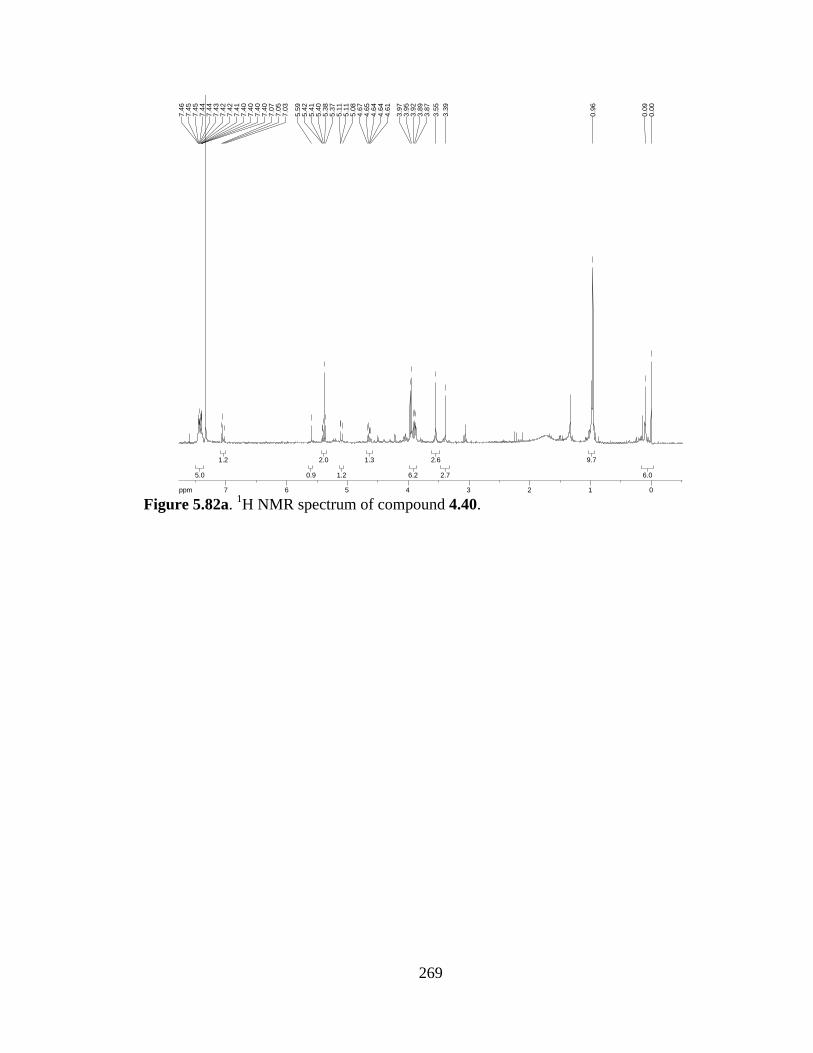
269
Figure 5.82a. 1H NMR spectrum of compound 4.40.
6.06.0
9.79.7
2.72.7
2.62.6
6.26.2
1.31.3
1.21.2
2.02.0
0.90.9
1.21.2
5.05.0
ppm0 011223344556677-16.04
0.00
0.09
0.96
3.39
3.55
3.87
3.89
3.92
3.95
3.97
4.61
4.64
4.64
4.65
4.67
5.08
5.11
5.11
5.37
5.38
5.40
5.41
5.42
5.59
7.03
7.05
7.07
7.40
7.40
7.40
7.40
7.41
7.42
7.42
7.43
7.44
7.44
7.45
7.45
7.46

270
(2S,3R)-methyl 3-acetoxy-2-(((benzyloxy)carbonyl)amino)-3-(5-chloro-6,7-
dimethoxy-1-methyl-1H-indol-3-yl)propanoate (4.47). To a solution of alcohol 4.4
(250 mg, 0.52 mmol) in THF (2.5 mL) was added Ac2O (0.059 mL, 0.62 mmol), Et3N
(0.109 mL, 0.78 mmol), and DMAP (small amt). The resulting solution was stirred at r.t.
O/N, then concentrated and 1M HCl added. The product was extracted in CH2Cl2,
washed with brine, dried over Na2SO4, and concentrated. The crude material was purified
by SiO2 chromatography (1:1 hexanes:EtOAc) to afford 4.47 (150 mg, 56% yield).
1H-NMR (300 MHz; CDCl3): δ 7.38-7.35 (broad, 6H), 6.92 (d, J = 38.1 Hz, 1H), 5.61-
5.54 (m, 1H), 5.18 (s, 1H), 5.09-5.06 (m, 2H), 4.82 (dd, J = 9.7, 3.7 Hz, 1H), 4.00-3.98
(m, 3H), 3.92-3.87 (m, 6H), 3.75 (d, J = 7.0 Hz, 3H), 2.17 (s, 3H).
REF: TRW-III-137.
OMeMeO
Cl
NMe
CO2MeAcONHCbz
4.47

271
Figure 5.83a. 1H NMR spectrum of compound 4.47.
2.92.9
3.13.1
7.17.1
3.23.2
0.50.5
1.81.8
0.90.9
1.01.0
0.80.8
7.07.0
ppm1 1223344556677-14.99
2.17
3.74
3.76
3.81
3.87
3.89
3.90
3.90
3.91
3.92
3.98
3.99
4.00
4.80
4.81
4.83
4.84
5.06
5.08
5.09
5.18
5.54
5.58
5.58
5.58
5.60
5.61
6.84
6.97
7.35
7.38

272
(2S,3R)-methyl 3-acetoxy-2-amino-3-(5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-
yl)propanoate (4.48). Compound 4.47 (150 mg, 0.29 mmol) was dissolved in MeOH
(1.5 mL) and to it added Pd/C (15 mg). The suspension was treated with H2 (atm) O/N,
then filtered through celite and concentrated to afford crude 4.48, used as crude in the
next step.
REF: TRW-III-141.
(3R)-methyl 2-((S)-2-((((9H-fluoren-9-
yl)methoxy)carbonyl)(methyl)amino)propanamido)-3-acetoxy-3-(5-chloro-6,7-
dimethoxy-1-methyl-1H-indol-3-yl)propanoate (4.49). Crude 4.48 (78 mg, 0.203
mmol) was dissolved in CH2Cl2 (2 mL) and to it added N-Me,Fmoc-Ala (66 mg, 0.203
mmol), iPr2NEt (0.088 mL, 0.233 mmol), and T3P (150 mg 50 wt% solution in DMF,
0.233 mmol). The resulting solution was allowed to stir O/N at r.t. under Ar, then
quenched with water and the product extracted in EtOAc. The combined organic layers
were dried over Na2SO4, filtered, and concentrated. Purification by SiO2 chromatography
afforded 4.49 in 18% yield (25 mg). A clean NMR could not be obtained for this sample.
REF: TRW-III-145.
OMeMeO
Cl
NMe
CO2MeAcONH2
4.48
NMe
Cl
MeOOMe
HN
AcO
NMeFmocCO2Me
OMe
4.49

273
(S,Z)-methyl 3-(5-chloro-6,7-dimethoxy-1-methyl-1H-indol-3-yl)-2-(2-
(methylamino)propanamido)acrylate (4.50). Dipeptide 4.49 (25 mg, 0.037 mmol) was
dissolved in THF (1.5 mL), then morpholine (0.37 mL) added. The resulting solution was
stirred O/N at r.t., then concentrated. The crude residue was purified by SiO2
chromatography (10% methanol in CH2Cl2) to afford the title product as a 1:1 mixture of
diasteromers (trace yield).
1H-NMR (400 MHz; CDCl3): δ 7.44 (d, J = 17.9 Hz, 2H), 6.86 (d, J = 4.3 Hz, 2H), 5.07
(dt, J = 15.5, 4.8 Hz, 2H), 4.84-4.77 (m, 2H), 3.98 (s, 3H), 3.96 (s, 3H), 3.90 (s, 3H), 3.89
(s, 3H), 3.77 (s, 3H), 3.71 (s, 2H), 3.66 (s, 3H), 3.32 (s, 3H), 3.27 (s, 3H), 2.40 (s, 3H),
2.29 (s, 3H), 1.18 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 7.0 Hz, 3H); HRMS (ESI-APCI+):
[M+H]+ 410.1483 calcd for C19H25ClN3O5, found: 410.1471.
NMe
Cl
MeOOMe
HNNHMe
CO2Me
OMe
4.50

274
Figure 5.84a. 1H NMR spectrum of compound 4.50.
33
3.133.13
2.892.89
2.842.84
3.033.03
2.642.64
2.792.79
2.392.39
2.812.81
2.952.95
3.283.28
2.832.83
2.652.65
1.791.79
1.871.87
1.941.94
2.052.05
ppm0 011223344556677-16.04
0.93
0.95
1.17
1.19
2.29
2.40
3.27
3.32
3.66
3.71
3.77
3.89
3.90
3.96
3.98
4.77
4.78
4.80
4.80
4.82
4.84
5.04
5.05
5.07
5.08
5.09
5.10
6.85
6.86
7.42
7.46

275
(S)-(9H-fluoren-9-yl)methyl 4-methyl-5-oxooxazolidine-3-carboxylate (4.45). To a
solution of N-Fmoc-Ala (5.0 g, 16.1 mmol) in PhMe (50 mL) was added
paraformaldehyde (3.15 g, 105 mmol) and p-TsOH (247 mg, 1.3 mmol). The resulting
suspension was heated to reflux for 1 h with a Dean-Stark trap in place. Upon cooling,
EtOAc was added and the organic phase washed with sat’d NaHCO3, dried over Na2SO4,
and concentrated to afford 4.45, carried forward as crude.
1H-NMR (300 MHz; CDCl3): δ 7.78 (d, J = 7.3 Hz, 2H), 7.54 (d, J = 7.4 Hz, 2H), 7.38
(dt, J = 26.6, 7.4 Hz, 4H), 5.38-5.14 (broad, 3H), 4.61-4.60 (broad, 2H), 4.25 (t, J = 5.6
Hz, 1H), 1.51-1.14 (broad, 3H).
REF: TRW-III-125.
OFmocN
MeO
4.45

276
Figure 5.85a. 1H NMR spectrum of compound 4.45.
3.63.6
1.31.3
2.32.3
2.62.6
4.34.3
2.12.1
2.02.0
ppm1 12233445566778-14.99
1.14
1.14
1.14
1.15
1.15
1.16
1.16
1.16
1.17
1.17
1.49
1.50
1.50
1.50
1.50
1.51
1.51
1.51
4.23
4.25
4.26
4.60
4.60
4.60
4.61
4.61
4.61
5.14
5.14
5.15
5.32
5.33
5.34
5.34
5.34
5.35
5.35
5.35
5.36
5.36
5.37
5.37
5.37
5.37
5.38
7.31
7.33
7.36
7.40
7.42
7.44
7.53
7.55
7.77
7.79

277
N-Me,Fmoc-L-Ala-OH (4.46). Compound 4.45 (crude, directly from previous reaction)
was dissolved in CHCl3 (4.1 mL) and to the resulting solution added TFA (4.1 mL) and
Et3SiH (1.0 mL). After stirring 10 h, additional Et3SiH (3.0 mL) was added. The reaction
stirred another 24 h, then was concentrated and TFA removed by repeated azeotropic
distillations with toluene. The crude product was recrystallized from ether/EtOAc to
afford pure 4.46 (2.87 g, 55% yield over 2 steps).
1H-NMR (300 MHz; CDCl3): δ 7.77 (d, J = 7.2 Hz, 2H), 7.61-7.53 (m, 2H), 7.43-7.29
(m, 4H), 4.76 (dq, J = 84.6, 7.2 Hz, 1H), 4.44 (qd, J = 11.3, 9.0 Hz, 2H), 4.28 (t, J = 6.9
Hz, 1H), 2.92 (s, 3H), 1.42 (dd, J = 28.0, 7.2 Hz, 3H).
REF: TRW-III-126.
NMeFmoc
Me CO2H4.46
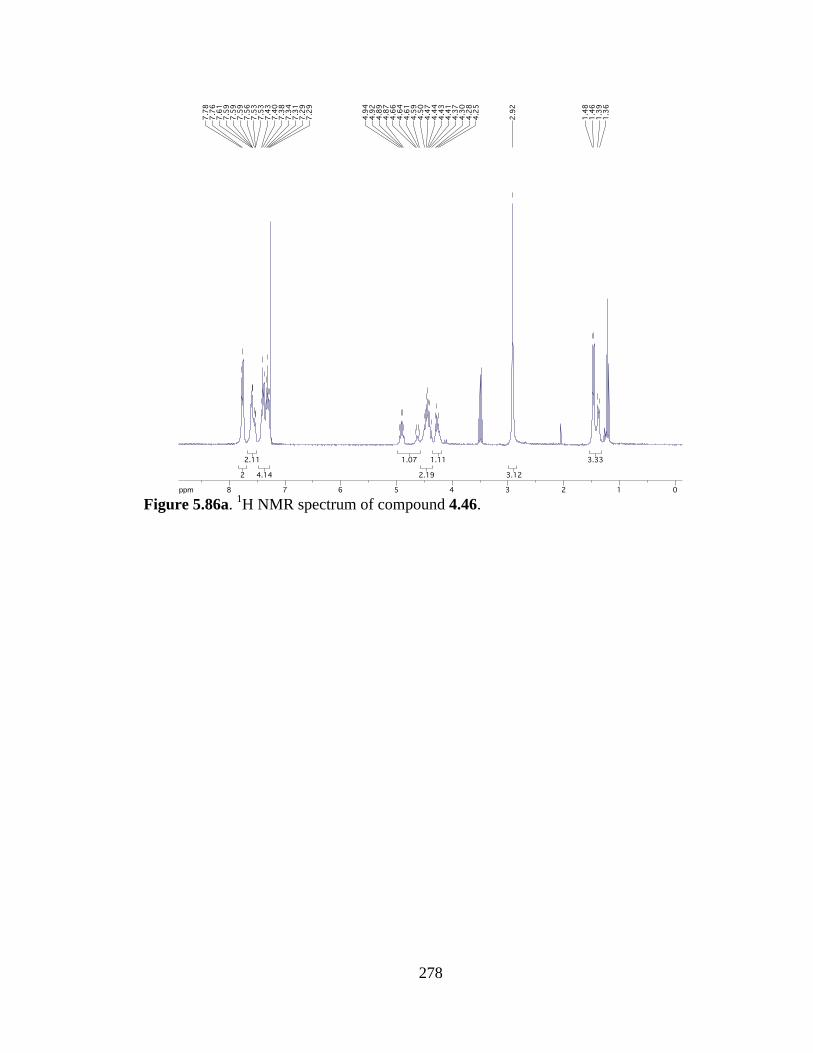
278
Figure 5.86a. 1H NMR spectrum of compound 4.46.
3.333.33
3.123.12
1.111.11
2.192.19
1.071.07
4.144.14
2.112.11
22
ppm0 01122334455667788-14.99
1.36
1.39
1.46
1.48
2.92
4.25
4.28
4.30
4.37
4.41
4.43
4.44
4.47
4.50
4.59
4.61
4.64
4.66
4.87
4.89
4.92
4.94
7.29
7.29
7.31
7.34
7.38
7.40
7.43
7.53
7.53
7.56
7.59
7.59
7.59
7.61
7.76
7.78

279
N-Bn-L-Ser-OH (5.13). To a solution of serine (14.7 g, 140 mmol) in 2M NaOH (70
mL) was added freshly distilled benzaldehyde (14.0 mL, 138 mmol). After stirring for 1 h
at r.t., the reaction mixture was cooled to 0 °C and NaBH4 (1.5 g, 40 mmol) added
portionwise such that the internal temperature did not exceed 10 °C. The resulting
solution was stirred 30 min at 5 °C, 1 h at r.t., then recooled to 0 °C. Additional NaBH4
(1.5 g, 40 mmol) was added portionwise as before. The reaction was allowed to warm to
r.t. as it stirred O/N. The crude reaction mixture was washed with ether (3x), then the
aqueous phase acidified to pH 5 with HCl. The resulting precipitate was filtered and
washed with water, then dried O/N to afford the crude product (6.55 g, 24% yield).
REF: TRW-III-288.
CO2H
NHBnHO
5.13

280
N-Me,Bn-L-Ser-OH (5.14). N-Bn-Ser (5.13, 2.49 g, 12.8 mmol) was dissolved in formic
acid (1.6 mL, 38.4 mmol) and formaldehyde (37% in H2O, 1.1 mL, 15.4 mmol) and
heated to 90 °C for 2 h. The reaction mixture was concentrated, then acetone added and a
solid triturated out of the solution. The suspension was concentrated, acetone added, and
the trituration process repeated 3x. Following the final concentration, acetone was again
added and the suspension allowed to stir 1 h at r.t, then 3 h at -20 °C. The solid was
filtered, washed with cold acetone, and dried to afford 5.14 (1.47 g, 55% yield).
1H-NMR (300 MHz; CD3OD): δ 7.58-7.45 (m, 5H), 4.52 (d, J = 12.6 Hz, 1H), 4.37 (d, J
= 12.7 Hz, 1H), 4.12 (qd, J = 11.8, 5.6 Hz, 2H), 3.73 (t, J = 3.5 Hz, 1H), 2.89 (s, 3H).
REF: TRW-III-289.
CO2H
NMeBnHO
5.14
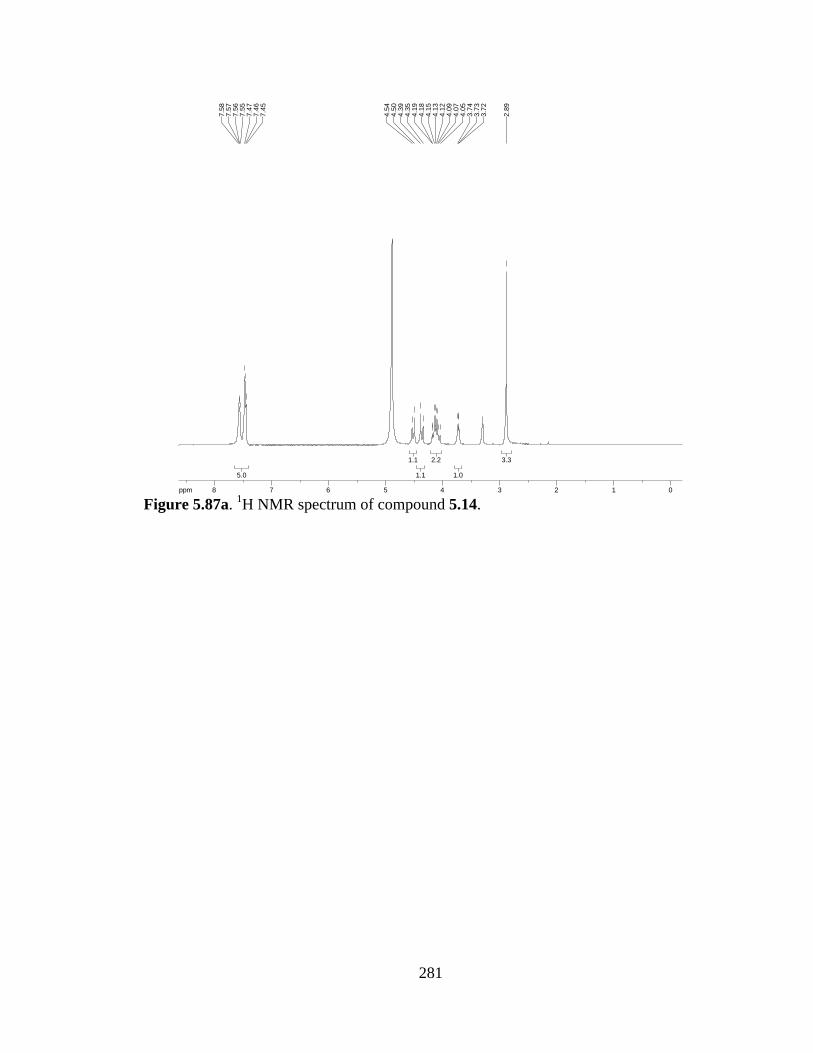
281
Figure 5.87a. 1H NMR spectrum of compound 5.14.
3.33.3
1.01.0
2.22.2
1.11.1
1.11.1
5.05.0
ppm0 01122334455667788-14.99
2.89
3.72
3.73
3.74
4.05
4.07
4.09
4.12
4.13
4.15
4.18
4.19
4.35
4.39
4.50
4.54
7.45
7.46
7.47
7.55
7.56
7.57
7.58

282
N-Me-L-Ser-OH (5.15). 5.14 (1.47 g, 7.0 mmol) was dissolved in MeOH (70 mL) and to
it added Pd(OH)2 (700 mg). The resulting suspension was treated with H2 (55 psi) for 40
h, then filtered through celite and concentrated to afford the title compound (660 mg,
79% yield), carried forward as crude.
REF: TRW-III-291.
N-Me-L-Ser(OTBS)-OH (5.16). To a solution of 5.15 (660 mg, 5.55 mmol) in DMF (12
mL) was added TBSCl (923 mg, 6.11 mmol) and imidazole (755 mg, 11.1 mmol). The
resulting solution was allowed to stir at r.t. O/N, then concentrated. To the crude residue
was added H2O and hexanes (1:1) and the resulting suspension stirred 4 h. The solid was
filtered, rinsed with hexanes, and dried to give 5.16 (685 mg, 53% yield).
1H-NMR (300 MHz; CD3OD): δ 4.09 (d, J = 4.1 Hz, 2H), 3.50 (t, J = 4.1 Hz, 1H), 2.72
(s, 3H), 0.92 (s, 9H), 0.12 (s, 6H).
REF: TRW-III-296.
CO2H
NHMeHO
5.15
CO2H
NHMeTBSO
5.16

283
Figure 5.88a. 1H NMR spectrum of compound 5.16.
5.35.3
9.09.0
3.03.0
1.01.0
2.02.0
ppm0 011223344-14.99
0.12
0.92
2.72
3.49
3.50
3.52
4.08
4.10

284
N-Me,Fmoc-L-Ser(OTBS)-OH (5.17). To a suspension of 5.16 (375 mg, 1.61 mmol) in
Na2CO3 (aq, 10%, 3.2 mL) and dioxane (1.3 mL) at 0 °C was added dropwise FmocOSu
(570 mg, 1.69 mmol) as a solution in dioxane (2.1 mL). The resulting solution was
allowed to warm to r.t. as it stirred O/N. After concentration, the crude mixture was
diluted with water, acidified to pH 3-4 with 10% KHSO4, and the product extracted in
EtOAc. The combined organic layer was washed with brine, dried over Na2SO4, and
concentrated to afford 5.17 (663 mg, 90% yield).
1H-NMR (300 MHz; CDCl3): δ 7.77 (d, J = 7.6 Hz, 2H), 7.61 (d, J = 7.2 Hz, 2H), 7.36
(dt, J = 27.2, 7.4 Hz, 4H), 4.82 (t, J = 5.9 Hz, 1H), 4.49-4.40 (m, 2H), 4.30-4.22 (m, 1H),
4.14-4.05 (m, 2H), 3.88 (t, J = 5.1 Hz, 1H), 3.04 (d, J = 24.1 Hz, 3H), 0.86 (s, 9H), 0.08-
0.03 (m, 6H); HRMS (ESI-APCI+): [M+H]+ 456.2206 calcd for C25H34NO5Si, found
456.2200.
REF: TRW-III-298.
CO2H
NMeFmocTBSO
5.17
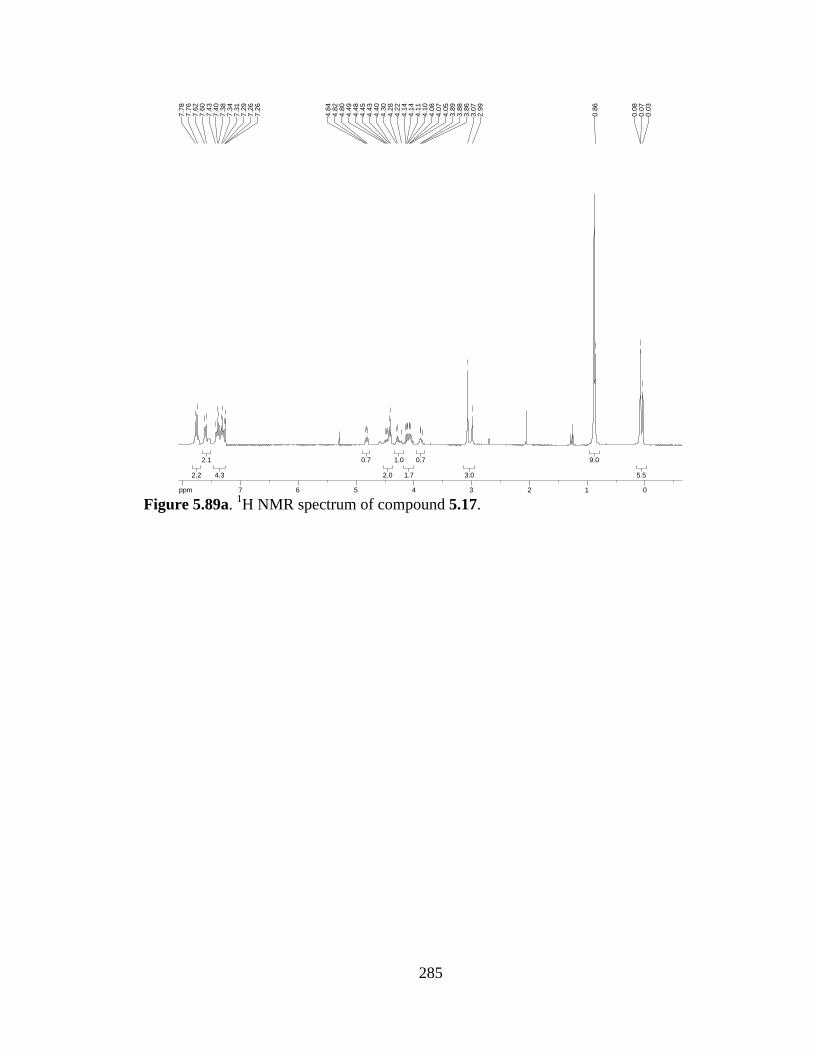
285
Figure 5.89a. 1H NMR spectrum of compound 5.17.
5.55.5
9.09.0
3.03.0
0.70.7
1.71.7
1.01.0
2.02.0
0.70.7
4.34.3
2.12.1
2.22.2
ppm0 0112233445566778-14.99
0.03
0.07
0.08
0.86
2.99
3.07
3.86
3.88
3.89
4.05
4.07
4.08
4.10
4.11
4.14
4.14
4.22
4.28
4.30
4.40
4.43
4.45
4.48
4.49
4.80
4.82
4.84
7.26
7.26
7.29
7.31
7.34
7.38
7.40
7.43
7.60
7.62
7.76
7.78

286
N-Me,Bn-L-Trp-OMe (5.18). To a solution of L-Trp-OMe·HCl (500 mg, 2 mmol) in
MeOH (20 mL) and Et3N (1.67 mL) was added PhCHO (212 µL, 2.1 mmol). The
resulting mixture was stirred at r.t. for 1 h, then NaBH3CN (132 mg, 2.1 mmol) added.
The reaction mixture was stirred overnight at r.t., at which time paraformaldehyde (180
mg, 2 mmol) was added. After full dissolution (ca. 5 h), PhCHO (132 mg, 2.1 mmol) was
added and the reaction stirred overnight. The crude reaction mixture was concentrated
and the residue dissolved in EtOAc, filtered through celite, and concentrated. Purification
by SiO2 chromatography (5-30% EtOAc in hexanes) afforded diprotected amine 5.18
(300 mg, 47% yield).
1H-NMR (300 MHz; CDCl3): δ 8.11 (s, 1H), 7.55 (d, J = 7.9 Hz, 1H), 7.35 (t, J = 4.3 Hz,
6H), 7.23 (t, J = 6.9 Hz, 1H), 7.14 (t, J = 6.9 Hz, 1H), 7.02 (d, J = 2.2 Hz, 1H), 3.92 (d, J
= 13.5 Hz, 1H), 3.82 (dd, J = 9.1, 5.8 Hz, 1H), 3.72-3.68 (m, 4H), 3.42 (dd, J = 14.4, 9.1
Hz, 1H), 3.17 (ddd, J = 14.3, 5.8, 0.6 Hz, 1H), 2.44 (s, 3H).
REF: TRW-I-278, TRW-I-284.
NH
CO2Me
NMeBn
5.18
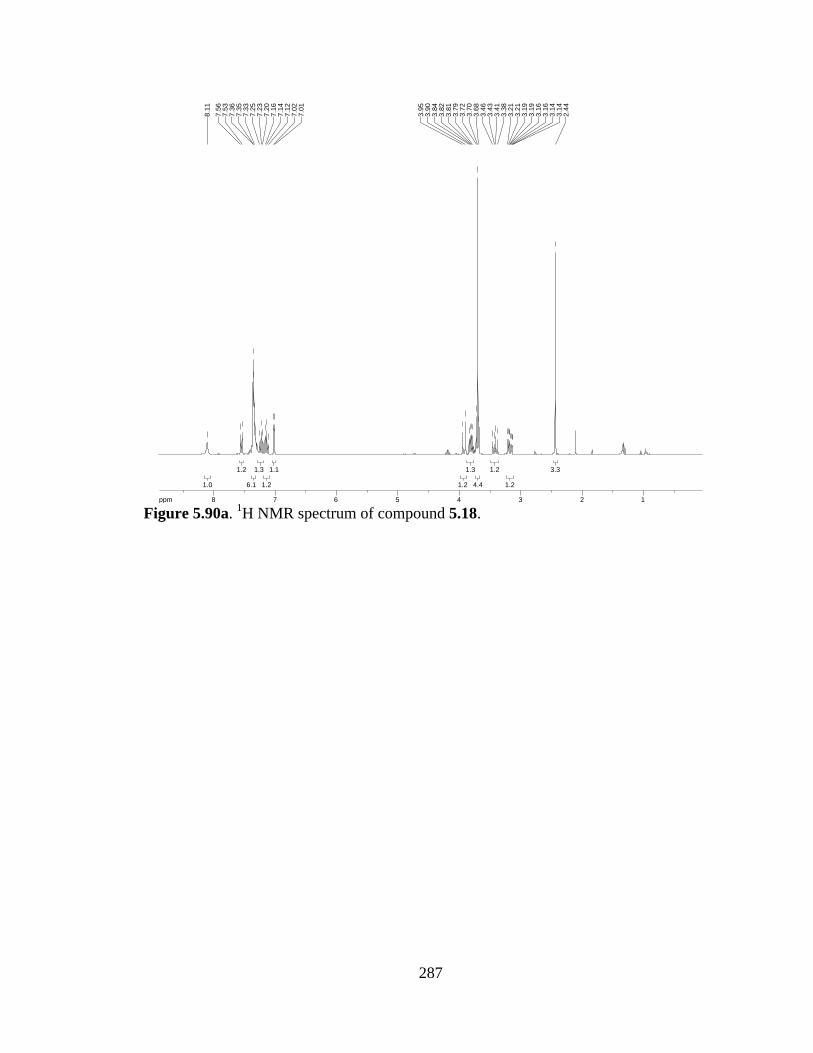
287
Figure 5.90a. 1H NMR spectrum of compound 5.18.
3.33.3
1.21.2
1.21.2
4.44.4
1.31.3
1.21.2
1.11.1
1.21.2
1.31.3
6.16.1
1.21.2
1.01.0
ppm1 122334455667788-19.97
2.44
3.14
3.14
3.16
3.16
3.19
3.19
3.21
3.21
3.38
3.41
3.43
3.46
3.68
3.70
3.72
3.79
3.81
3.82
3.84
3.90
3.95
7.01
7.02
7.12
7.14
7.16
7.20
7.23
7.25
7.33
7.35
7.36
7.53
7.56
8.11

288
N-Me-L-Trp-OMe (5.19). To a degassed solution of 5.18 (300 mg, 0.93 mmol) in
MeOH (9 mL) was added Pd(OH)2/C (150 mg). The resulting suspension was treated
with H2 (55 psi) for 18 h, then filtered through celite and concentrated to afford
methylamine 5.19 (215 mg, 99.5% yield), deemed sufficiently pure to carry forward
without further purification.
1H-NMR (300 MHz; CDCl3): δ 8.26 (s, 1H), 7.61 (d, J = 7.7 Hz, 1H), 7.36-7.33 (m, 1H),
7.15 (tdd, J = 14.4, 6.5, 1.2 Hz, 2H), 7.04 (d, J = 2.3 Hz, 1H), 3.66 (s, 3H), 3.57 (t, J =
6.6 Hz, 1H), 3.16 (qd, J = 13.0, 6.6 Hz, 2H), 2.38 (s, 3H), 2.05-1.99 (bs, 1H).
REF: TRW-I-282.
NH
CO2Me
NHMe
5.19

289
Figure 5.91a. 1H NMR spectrum of compound 5.19.
1.41.4
3.423.42
2.492.49
1.141.14
2.982.98
11
2.42.4
1.281.28
1.061.06
0.950.95
ppm1 1223344556677889-14.99
1.99
2.00
2.00
2.01
2.01
2.02
2.02
2.02
2.03
2.03
2.04
2.04
2.05
2.05
2.38
3.09
3.11
3.13
3.16
3.17
3.19
3.22
3.24
3.55
3.57
3.59
3.66
7.04
7.04
7.09
7.10
7.12
7.12
7.14
7.15
7.16
7.17
7.19
7.19
7.21
7.22
7.33
7.33
7.36
7.60
7.62
8.26

290
N-Me,Boc-L-Trp-OMe (5.20). To a solution of 5.19 (6.40 g, 27.6 mmol) in
water/dioxane (1:1, 110 mL) was added NaOH (1.10 g, 27.6 mmol) and Boc anhydride
(7.22 g, 33.1 mmol). The resulting solution was allowed to stir O/N at r.t., then acidified
with HCl to pH 6-7. The product was extracted in CHCl3 and the organic phase dried
over Na2SO4 and concentrated. Crude material was purified by SiO2 chromatography
(25% EtOAc in hexanes) to afford 5.20 (4.50 g, 49% yield).
1H-NMR (300 MHz; CDCl3): δ 8.07-8.06 (m, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.36 (d, J =
7.5 Hz, 1H), 7.16 (dt, J = 21.1, 7.3 Hz, 2H), 7.06-7.00 (broad, 1H), 5.07-4.73 (m, 1H),
3.76 (s, 3H), 3.48-3.40 (m, 1H), 3.27-3.11 (m, 1H), 2.75 (d, J = 14.1 Hz, 3H), 1.29 (d, J =
61.0 Hz, 9H).
REF: TRW-III-209.
NH
CO2Me
NMeBoc
5.20
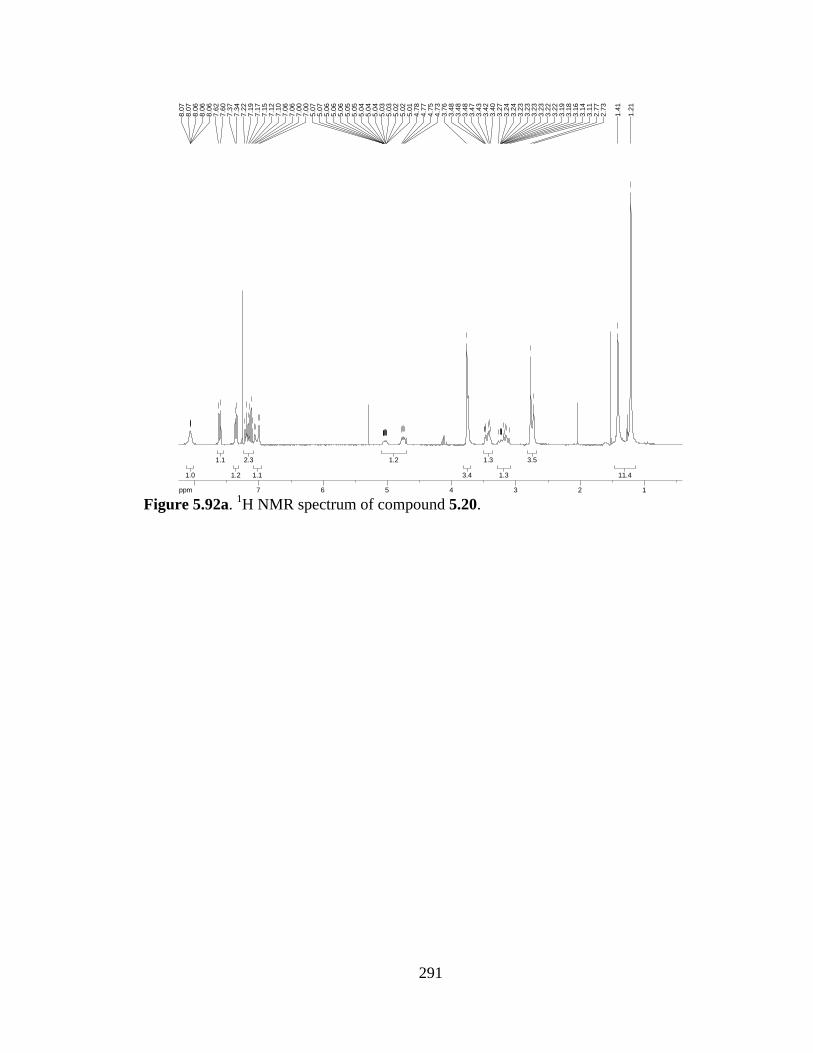
291
Figure 5.92a. 1H NMR spectrum of compound 5.20.
11.411.4
3.53.5
1.31.3
1.31.3
3.43.4
1.21.2
1.11.1
2.32.3
1.21.2
1.11.1
1.01.0
ppm1 12233445566778-14.99
1.21
1.41
2.73
2.77
3.11
3.14
3.16
3.18
3.19
3.22
3.22
3.23
3.23
3.23
3.23
3.24
3.24
3.27
3.40
3.42
3.43
3.47
3.48
3.48
3.48
3.76
4.73
4.75
4.77
4.78
5.01
5.02
5.02
5.03
5.03
5.04
5.04
5.04
5.05
5.05
5.06
5.06
5.06
5.07
5.07
7.00
7.00
7.06
7.06
7.10
7.12
7.15
7.17
7.19
7.22
7.34
7.37
7.60
7.62
8.06
8.06
8.06
8.07
8.07

292
L-Ser-OMe (5.21). To a solution of L-serine (50.0 g, 0.48 mol) in MeOH (450 mL) at 0
°C was added SOCl2 (35 mL, 0.48 mol). The reaction was allowed to warm to r.t. as it
stirred O/N. The resulting solution was concentrated, taken up in ether, filtered, and the
solid washed with ether. The crude solid material was recrystallized in MeOH/ether to
afford pure crystalline methyl ester 5.21 as the HCl salt.
REF: TRW-II-118.
CO2MeH2N
OH
5.21

293
N-Bn,Me-L-Ser-OMe (5.22). To a solution of L-Ser-OMe·HCl (10.0 g, 64.1 mmol) in
MeOH (500 mL) and Et3N (50.0 mL, 385 mmol) was added PhCHO (6.8 mL, 67.3
mmol). The resulting mixture was stirred at r.t. for 1 h, then NaBH3CN (4.24 g, 67.3
mmol) added. The reaction mixture was stirred overnight at r.t., at which time
paraformaldehyde (5.78 g, 64.1 mmol) was added. After full dissolution (ca. 5 h),
NaBH3CN (4.24 g, 67.3 mmol) was added and the reaction stirred overnight. The crude
reaction mixture was concentrated and the residue dissolved in EtOAc, filtered through
celite, and concentrated. Purification by SiO2 chromatography (2:1 hexanes:EtOAc)
afforded diprotected amine 5.22 (7.41 g, 52% yield).
1H-NMR (300 MHz; CDCl3): δ 7.37-7.29 (m, 5H), 4.69 (d, J = 5.9 Hz, 1H), 3.79-3.71
(m, 5H), 3.54 (dd, J = 8.8, 6.6 Hz, 1H), 2.78 (dd, J = 8.6, 3.0 Hz, 1H), 2.33 (s, 3H).
REF: TRW-II-019, TRW-II-120, TRW-III-208.
CO2MeBnMeN
OH
5.22

294
Figure 5.93a. 1H NMR spectrum of compound 5.22.
3.03.0
0.80.8
1.01.0
5.65.6
0.70.7
6.46.4
ppm0 0112233445566778-14.99
2.33
2.76
2.77
2.79
2.80
3.52
3.54
3.55
3.57
3.71
3.72
3.73
3.73
3.74
3.75
3.76
3.79
3.84
3.88
4.68
4.70
7.29
7.31
7.33
7.33
7.36
7.37

295
N-Me-L-Ser-OMe (5.23). To a degassed solution of 5.22 (3.7 g, 16.6 mmol) in MeOH
(140 mL) was added Pd(OH)2 (1.85 g). The resulting suspension was treated with H2 (55
psi) for 18 h, then filtered through celite and concentrated to afford methylamine 5.23
(99.5% yield), deemed sufficiently pure to carry forward without further purification.
1H-NMR (300 MHz; CDCl3): δ 3.82-3.77 (m, 4H), 3.63 (dd, J = 10.9, 6.1 Hz, 1H), 3.28
(t, J = 5.2 Hz, 1H), 2.43 (s, 3H).
REF: TRW-II-020, TRW-II-123.
CO2MeMeHN
OH
5.23

296
Figure 5.94a. 1H NMR spectrum of compound 5.23.
3.33.3
1.01.0
1.11.1
4.14.1
ppm0 011223344556677-14.99
2.43
3.26
3.28
3.30
3.60
3.62
3.64
3.66
3.77
3.79
3.81
3.82

297
3-(1H-indol-3-yl)-2-(methylamino)propan-1-ol (5.24). To a suspension of LAH (813
mg, 21.4 mmol) in THF (20 mL) at 0°C was added methyl ester 5.19 (1.70 g, 5.35 mmol)
dropwise. The resulting mixture was heated to reflux and stirred O/N. After cooling to
0°C, 15% NaOH (8 mL) was added and the reaction stirred an additional 1 h at r.t. The
suspension was filtered through celite and the filtrate dried over Na2SO4 before
concentrating to afford 1.09 g (100% yield) of methylamine alcohol 5.24.
1H-NMR (300 MHz; CDCl3): δ 8.76 (s, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.34 (d, J = 8.0
Hz, 1H), 7.23-7.10 (m, 2H), 6.92 (s, 1H), 3.71 (dd, J = 11.5, 2.1 Hz, 1H), 3.47 (dd, J =
10.9, 4.3 Hz, 1H), 2.98-2.90 (m, 3H), 2.41 (s, 3H).
REF: TRW-I-294, TRW-I-310, TRW-I-323, TRW-I-327.
NH
NHMe
5.24
OH
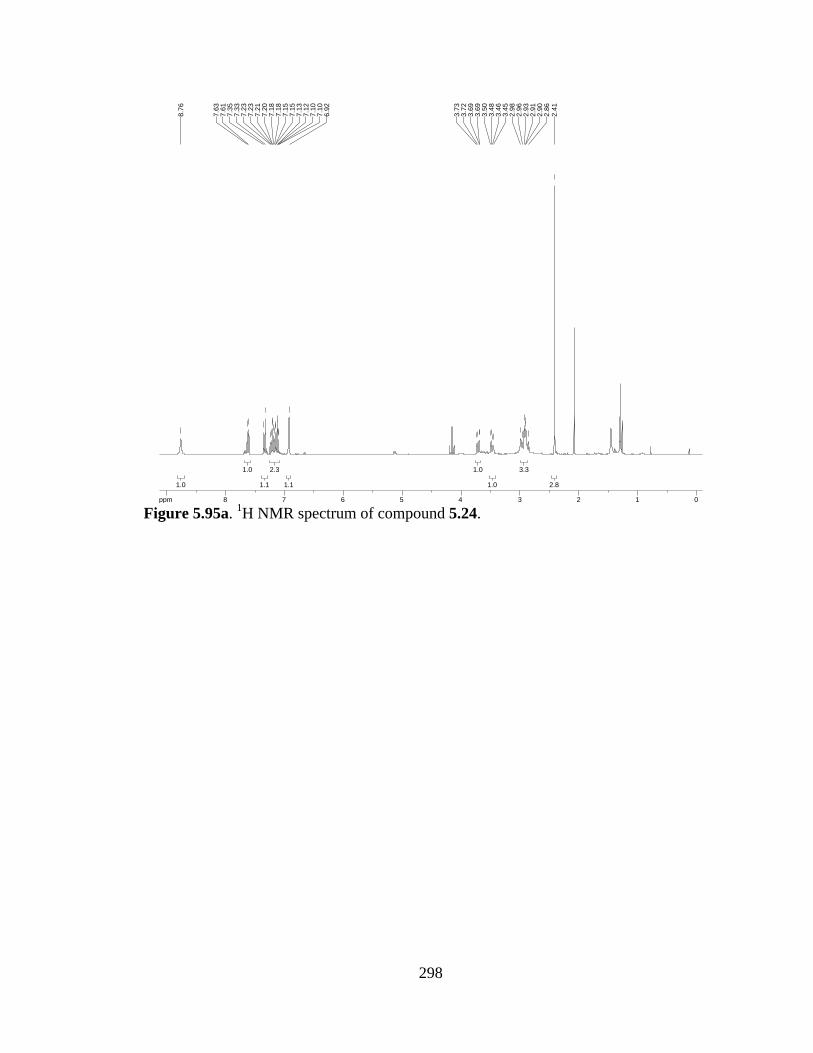
298
Figure 5.95a. 1H NMR spectrum of compound 5.24.
2.82.8
3.33.3
1.01.0
1.01.0
1.11.1
2.32.3
1.11.1
1.01.0
1.01.0
ppm0 011223344556677889-19.97
2.41
2.86
2.90
2.91
2.93
2.96
2.98
3.45
3.46
3.48
3.50
3.69
3.69
3.72
3.73
6.92
7.10
7.10
7.12
7.13
7.15
7.15
7.18
7.18
7.20
7.21
7.23
7.23
7.33
7.35
7.61
7.63
8.76

299
N-Cbz-L-Ser-OH (5.25). To a solution of L-Ser (10.0 g, 95 mmol) in saturated aqueous
NaHCO3 (380 mL) was added CbzCl (15 mL, 100 mmol). The resulting solution was
stirred O/N at r.t., then acidified to ph 4 with 1M HCl. The product was extracted in
EtOAc (3x), and the combined organic layers dried and concentrated to afford N-Cbz-L-
Ser-OH (21.78 g, 96% yield).
1H-NMR (300 MHz; CD3OD): δ 7.38-7.28 (m, 5H), 5.10 (s, 2H), 4.27 (t, J = 4.3 Hz,
1H), 3.91-3.80 (m, 2H).
REF: TRW-II-337, TRW-II-398.
CO2HCbzHN
OH
5.25
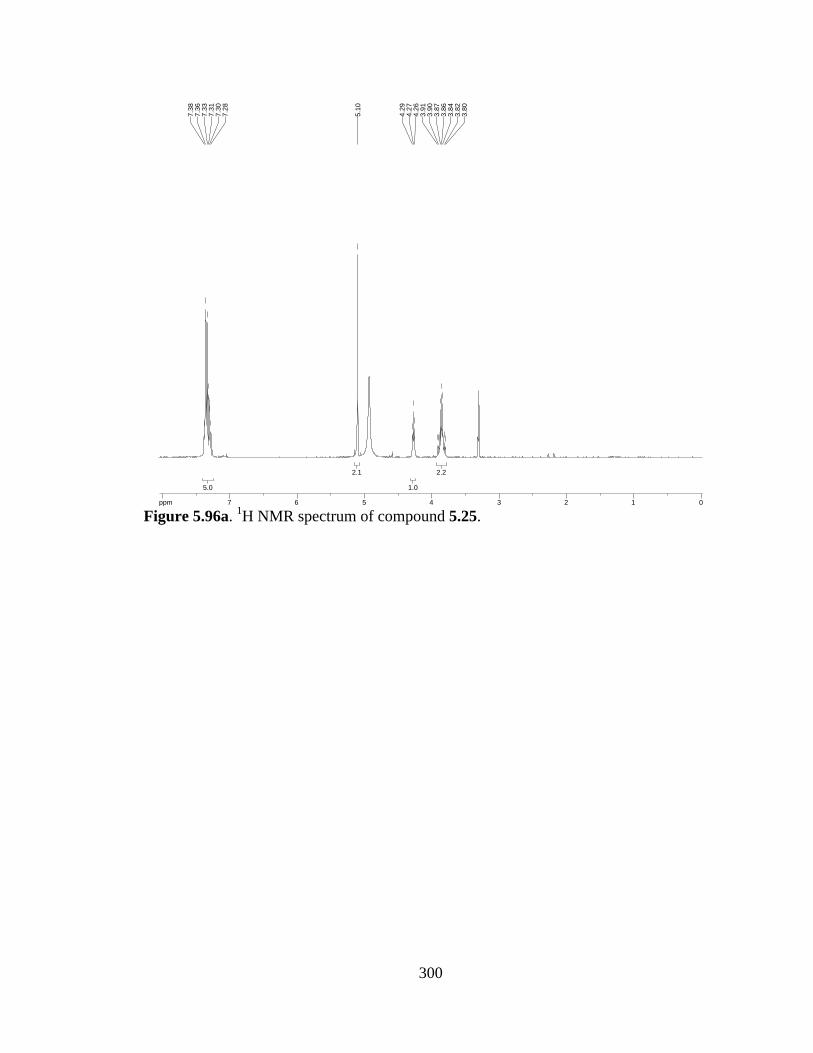
300
Figure 5.96a. 1H NMR spectrum of compound 5.25.
2.22.2
1.01.0
2.12.1
5.05.0
ppm0 0112233445566778-19.97
3.80
3.82
3.84
3.86
3.87
3.90
3.91
4.26
4.27
4.29
5.10
7.28
7.30
7.31
7.33
7.36
7.38

301
N-Cbz-L-Ser(OTBS)-OH (5.26). N-Cbz-L-Ser-OH (10.0 g, 41.8 mmol), TBSCl (6.95 g,
46.0 mmol), and imidazole (5.68 g, 83.6 mmol) were dissolved in DMF (100 mL). The
resulting solution was stirred at r.t. under Ar for 48 h before concentrating. The residue
was suspended in hexanes and the product extracted into 5% aqueous NaHCO3. KHSO4
(1M) was added to acidify the aqueous layer to pH 3, from which the product was
extracted into EtOAc. The combined organic extracts were washed with brine, dried over
Na2SO4, and concentrated to afford N-Cbz-L-Ser(OTBS)-OH (10.04 g, 68% yield).
1H-NMR (300 MHz; CDCl3): δ 7.33-7.30 (m, 5H), 5.08 (d, J = 3.4 Hz, 2H), 4.33-4.31
(m, 1H), 4.05 (dd, J = 10.2, 2.8 Hz, 1H), 3.82 (dd, J = 10.2, 2.8 Hz, 1H), 0.82 (d, J = 2.8
Hz, 9H), -0.02 (s, 6H).
REF: TRW-II-198, TRW-II-338, TRW-II-399.
CO2HCbzHN
OTBS
5.26

302
Figure 5.97a. 1H NMR spectrum of compound 5.26.
6.06.0
8.98.9
1.31.3
1.11.1
1.11.1
2.42.4
5.15.1
ppm0 011223344556677-19.97
-0.0
2
0.81
0.82
3.80
3.81
3.83
3.84
4.03
4.04
4.06
4.07
4.31
4.32
4.32
4.33
5.07
5.08
7.26
7.30
7.31
7.32
7.33

303
L-Trp-OMe·HCl (5.27). To a solution of L-tryptophan (25 g, 123 mmol) in MeOH (250
mL) at 0 °C was added SOCl2 (8.9 mL, 123 mmol) dropwise. The reaction was allowed
to warm to r.t. as it stirred O/N. Upon completion, the reaction was concentrated, then
MeOH (2x) and ether (3x) separately added and the mixture concentrated to thoroughly
dry the crude product. Pure L-tryptophan-OMe·HCl crystals were recovered following
recrystallization in MeOH/ether (25.08 g, 80% yield).
1H-NMR (300 MHz; CD3OD): δ 10.62 (bs, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.39 (d, J =
8.1 Hz, 1H), 7.20 (d, J = 2.3 Hz, 1H), 7.17-7.03 (m, 2H), 4.33 (dd, J = 7.4, 5.5 Hz, 1H),
3.79 (s, 3H), 3.42 (dq, J = 24.1, 7.9 Hz, 2H).
REF: TRW-II-391.
NH
CO2Me
NH2
5.27

304
Figure 5.98a. 1H NMR spectrum of compound 5.27.
2.02.0
3.03.0
1.01.0
2.12.1
0.90.9
1.01.0
1.01.0
0.80.8
ppm0 0224466881010-19.97
3.30
3.34
3.36
3.39
3.42
3.44
3.47
3.49
3.79
4.30
4.32
4.33
4.35
4.91
7.03
7.04
7.06
7.06
7.08
7.09
7.11
7.12
7.14
7.14
7.16
7.17
7.20
7.20
7.38
7.40
7.52
7.55
10.6
210
.62
10.6
210
.63
10.6
3

305
N-Cbz-L-Trp-OMe (5.28). To a suspension of L-tryptophan-OMe·HCl (15 g, 58.8
mmol) in dioxane (75 mL) at 0 °C was added 1M NaOH (60 mL). CbzCl (9.7 mL, 64.7
mmol) was added, followed by a second portion of 1M NaOH (60 mL). The reaction
mixture was allowed to stir for 20 minutes at 0 °C, then diluted with EtOAc. The organic
layer was separated, washed with 1M HCl and brine, then dried and concentrated to
afford the title compound.
1H-NMR (300 MHz; CDCl3): δ 8.22 (bs, 1H), 7.53 (d, J = 7.9 Hz, 1H), 7.34 (bs, 6H),
7.26-7.17 (m, 2H), 7.13-7.08 (m, 1H), 6.93 (s, 1H), 5.39 (d, J = 8.1 Hz, 1H), 5.11 (d, J =
4.4 Hz, 2H), 4.77-4.70 (m, 1H), 3.68 (s, 3H), 3.32 (d, J = 5.3 Hz, 2H).
REF: TRW-II-448.
NH
CO2Me
NHCbz
5.28
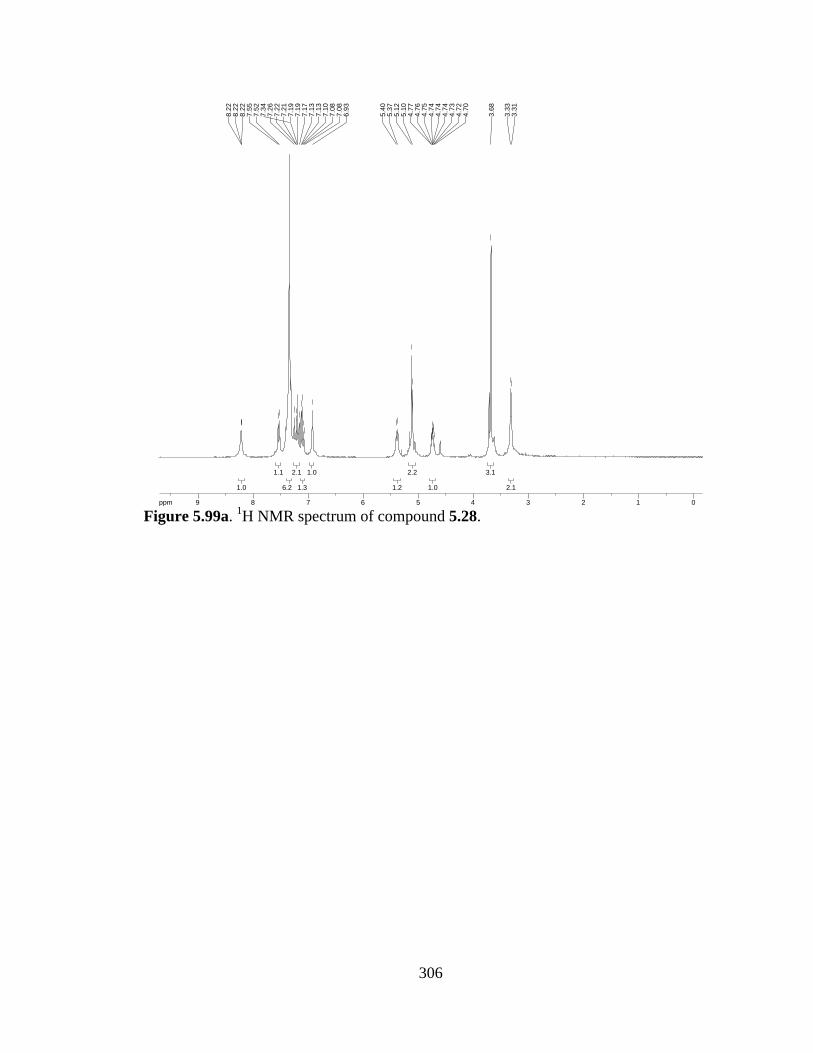
306
Figure 5.99a. 1H NMR spectrum of compound 5.28.
2.12.1
3.13.1
1.01.0
2.22.2
1.21.2
1.01.0
1.31.3
2.12.1
6.26.2
1.11.1
1.01.0
ppm0 0112233445566778899-19.973.
313.
33
3.68
4.70
4.72
4.73
4.74
4.74
4.74
4.75
4.76
4.77
5.10
5.12
5.37
5.40
6.93
7.08
7.08
7.10
7.13
7.13
7.17
7.19
7.19
7.21
7.22
7.26
7.34
7.52
7.55
8.22
8.22
8.22

307
N’-SO2Ph-N-Cbz-L-Trp-OMe (5.29). Phenylsulfonyl chloride (4.4 mL, 34.1 mmol) was
added dropwise to a suspension of N-Cbz-L-Trp-OMe (10 g, 28.4 mmol), NaOH (3.4 g,
85.2 mmol), and Bu4NHSO4 (483 mg, 1.42 mmol) in CH2Cl2 (140 mL). The reaction was
allowed to stir O/N at r.t., then to it added concentrated NH4Cl and EtOAc. The organic
layer was removed and the product further extracted into EtOAc (2x), then the combined
organic extracts dried and concentrated to afford the title compound (13.31 g, 95% yield).
1H-NMR (300 MHz; CDCl3): δ 8.05 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.3 Hz, 1H), 7.81
(d, J = 7.8 Hz, 2H), 7.64 (d, J = 8.0 Hz, 1H), 7.37 (sextet, J = 7.7 Hz, 9H), 7.20 (d, J =
7.4 Hz, 1H), 5.31 (d, J = 7.9 Hz, 1H), 5.16-5.06 (m, 2H), 4.70 (q, J = 6.6 Hz, 1H), 3.62
(s, 3H), 3.22 (t, J = 5.6 Hz, 2H).
REF: TRW-II-452.
NSO2Ph
CO2Me
NHCbz
5.29
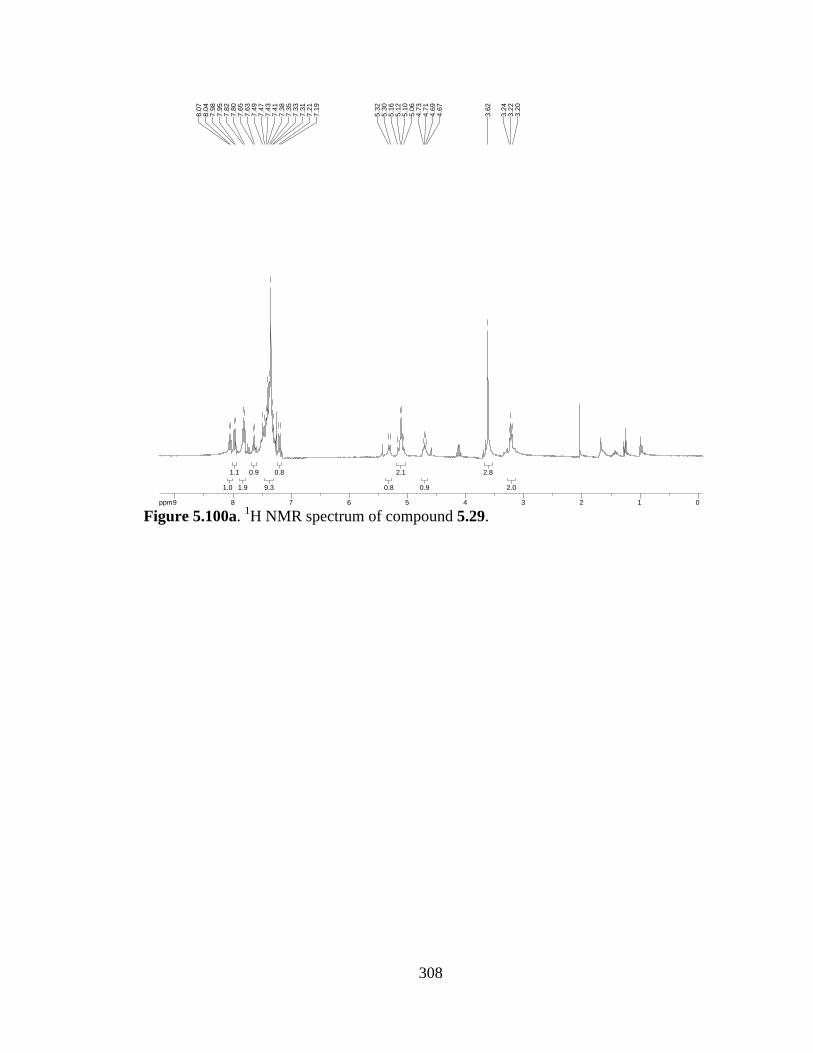
308
Figure 5.100a. 1H NMR spectrum of compound 5.29.
2.02.0
2.82.8
0.90.9
2.12.1
0.80.8
0.80.8
9.39.3
0.90.9
1.91.9
1.11.1
1.01.0
ppm0 0112233445566778899-19.97
3.20
3.22
3.24
3.62
4.67
4.69
4.71
4.73
5.06
5.10
5.12
5.16
5.30
5.32
7.19
7.21
7.31
7.33
7.35
7.38
7.41
7.43
7.47
7.49
7.63
7.65
7.80
7.82
7.95
7.98
8.04
8.07

309
Bromopyrroloindoline (5.30). N’-SO2Ph-N-Cbz-L-Trp-OMe (13.31 g, 27 mmol) was
dissolved in CH2Cl2 (400 mL), and to the resulting solution added NBS (4.81 g, 27
mmol) and PPTS (6.78 g, 27 mmol). The reaction was allowed to stir O/N at r.t. NaHCO3
(10% aq.) and Na2S2O4 (10% aq.) were added in a 1:1 mixture, the organic layer
removed, dried, and purified (SiO2 chromatography, 1:1 H:E) to afford the title
compound (11.46 g, 74% yield).
1H-NMR (300 MHz; CDCl3): δ 7.63 (d, J = 8.1 Hz, 1H), 7.50-7.45 (m, 3H), 7.36-7.34
(m, 8H), 7.26-7.25 (m, 2H), 7.19 (d, J = 7.4 Hz, 1H), 6.36-6.29 (bs, 1H), 5.42-5.22
(broad, 2H), 3.90-3.84 (m, 1H), 3.69-3.52 (broad, 3H), 3.09 (dd, J = 12.8, 6.0 Hz, 1H),
2.82-2.74 (m, 1H).
REF: TRW-II-453.
NSO2Ph
NCbzCO2Me
Br
H
5.30
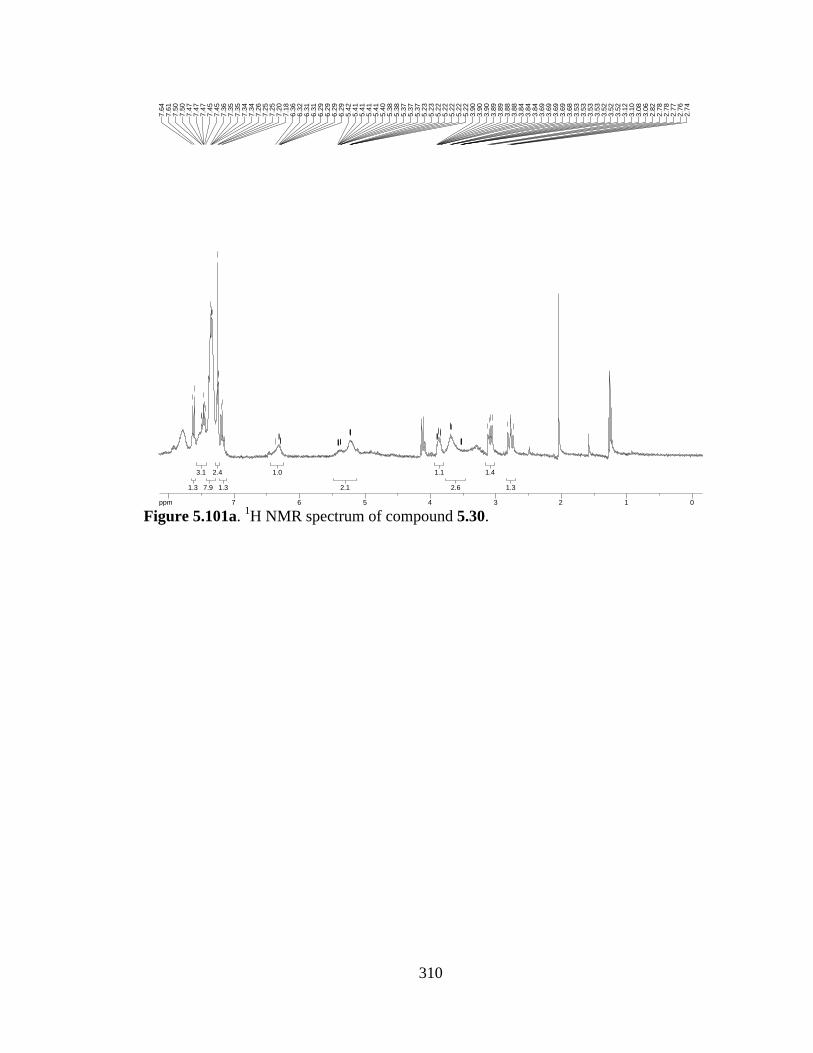
310
Figure 5.101a. 1H NMR spectrum of compound 5.30.
1.31.3
1.41.4
2.62.6
1.11.1
2.12.1
1.01.0
1.31.3
2.42.4
7.97.9
3.13.1
1.31.3
ppm0 0112233445566778-19.97
2.74
2.76
2.77
2.78
2.78
2.82
3.06
3.08
3.10
3.12
3.52
3.52
3.52
3.53
3.53
3.53
3.53
3.68
3.69
3.69
3.69
3.69
3.84
3.84
3.84
3.88
3.88
3.89
3.89
3.90
3.90
3.90
5.22
5.22
5.22
5.22
5.22
5.23
5.23
5.37
5.37
5.37
5.38
5.38
5.40
5.41
5.41
5.41
5.41
5.42
6.29
6.29
6.29
6.29
6.31
6.31
6.32
6.36
7.18
7.20
7.25
7.25
7.26
7.34
7.34
7.35
7.35
7.36
7.45
7.45
7.47
7.47
7.47
7.50
7.50
7.61
7.64

311
Methyl 3-bromo-2-(tert-butoxycarbonylamino)propanoate (5.31). To a solution of N-
Boc-Ser-OMe (2.0 g, 9.1 mmol) in THF (50 mL) at 0 °C was added CBr4 (4.54 g, 13.7
mmol) and PPh3 (3.59 g, 13.7 mmol). The reaction was stirred at 0 °C for 10 minutes,
then warmed to r.t. and stirred 4 h under Ar. The crude mixture was filtered through a bed
of celite, diluted with ether, and washed with water and brine. The organic phase was
dried over Na2SO4, filtered, and concentrated. The crude residue was purified by SiO2
chromatography (3:1 hexanes:EtOAC) to afford bromide 5.31 (1.64 g, 64% yield).
1H-NMR (300 MHz; CDCl3): δ 5.42-5.39 (m, 1H), 4.78-4.73 (m, 1H), 3.81 (s, 3H), 3.71
(dd, J = 10.4, 3.4 Hz, 1H), 1.46 (s, 9H).
REF: TRW-I-191.
CO2MeBocHN
Br
5.31

312
Figure 5.102a. 1H NMR spectrum of compound 5.31.
8.88.8
1.01.0
3.03.0
0.80.8
0.70.7
ppm0 011223344556677-19.97
1.46
3.69
3.70
3.72
3.73
3.81
4.73
4.74
4.74
4.75
4.76
4.76
4.76
4.77
4.78
4.78
5.39
5.39
5.39
5.40
5.40
5.40
5.41
5.41
5.41
5.41
5.42
5.42

313
Methyl 2-(benzyloxycarbonylamino)-3-hydroxypropanoate (5.32). To a solution of
Ser-OMe·HCl (5.0 g, 32.1 mmol) in MeOH:NaHCO3 (1:1, 120 mL) was added CbzCl
(5.75 g, 33.7 mmol). The reaction mixture was stirred at r.t. for 48 h, then 1 M HCl
added. The crude mixture was concentrated and the resulting residue dissolved in EtOAc.
The organic phase was washed with 1 M NaOH, dried over Na2SO4, filtered, and
concentrated under reduced pressure to afford Cbz-amine 5.32 (8.12 g, 100% yield) as a
clear oil, carried forward without additional purification.
1H-NMR (300 MHz; CDCl3): δ 7.37-7.34 (m, 5H), 5.72-5.67 (m, 1H), 5.13 (s, 2H), 4.47-
4.43 (m, 1H), 4.01-3.93 (m, 2H), 3.79 (s, 3H).
REF: TRW-I-256.
CO2MeCbzHN
OH
5.32

314
Figure 5.103a. 1H NMR spectrum of compound 5.32.
3.03.0
2.12.1
0.90.9
1.91.9
0.80.8
4.74.7
ppm0 011223344556677-19.97
3.79
3.93
3.93
3.93
3.93
3.93
3.94
3.94
3.94
3.95
3.95
3.95
3.96
3.96
3.96
3.97
3.97
3.97
3.97
3.97
3.97
3.99
3.99
4.01
4.43
4.44
4.44
4.44
4.44
4.45
4.45
4.46
4.46
4.47
4.47
5.13
5.67
5.67
5.68
5.68
5.68
5.68
5.68
5.69
5.69
5.69
5.69
5.69
5.70
5.70
5.70
5.70
5.70
5.71
5.71
5.71
5.72
7.34
7.34
7.35
7.36
7.37
7.37

315
3-benzyl 4-methyl 2,2-dimethyloxazolidine-3,4-dicarboxylate (5.33). To a solution of
Cbz-Ser-OMe (7.56 g, 30.0 mmol) in dry acetone (100 mL) was added 2,2-
dimethoxypropane (40 mL), BF3·OEt2 (3.8 mL, 30.0 mmol), and Na2SO4 (10 g). The
reaction mixture stirred overnight at r.t. under Ar, and then Et3N (12 mL) was added. The
resulting suspension was filtered and the filtrate concentrated under reduced pressure.
The product was extracted with ether (3 x 75 mL), washed with NaHCO3, washed with
brine, dried over Na2SO4, and concentrated. The crude oil was purified by SiO2
chromatography (5-50% EtOAc in hexanes) to afford 5.33 (7.20 g, 82% yield) as a
mixture of rotamers. Crude oil was likely pure enough to carry forward without
purification.
1H-NMR (300 MHz; CDCl3): δ 7.36-7.30 (m, 5H), 5.20-5.02 (m, 2H), 4.52 (ddd, J =
23.2, 6.6, 2.7 Hz, 1H), 4.19-4.07 (m, 2H), 3.69 (d, J = 40.4 Hz, 3H), 1.68 (d, J = 20.1 Hz,
3H), 1.58 (d, J = 9.5 Hz, 3H).
REF: TRW-I-257.
O
CbzN
CO2Me
MeMe
5.33
316
Figure 5.104a. 1H NMR spectrum of compound 5.33.
3.13.1
3.33.3
3.33.3
2.42.4
1.11.1
2.22.2
5.05.0
ppm1 1223344556677-19.97
1.49
1.56
1.59
1.64
1.71
3.64
3.77
4.07
4.08
4.10
4.11
4.12
4.13
4.14
4.14
4.16
4.16
4.17
4.19
4.46
4.47
4.48
4.49
4.54
4.55
4.56
4.57
5.02
5.06
5.15
5.18
5.19
5.20
7.30
7.31
7.32
7.32
7.34
7.35
7.36

317
Benzyl 4-(hydroxymethyl)-2,2-dimethyloxazolidine-3-carboxylate (5.34). To a
solution of ester 5.33 (1.0 g, 3.4 mmol) in THF (12 mL) at -10 °C was added NaBH4 (517
mg, 13.6 mmol). The resulting mixture was stirred for 30 minutes at -10 °C, then MeOH
(5 mL) added dropwise. The reaction was allowed to warm to r.t. while it stirred
overnight under Ar. Water was added and the resulting suspension stirred for 30 minutes,
concentrated under reduced pressure, diluted with brine, and the product extracted with
EtOAc (3 x 15 mL). The combined organic layers were washed with brine, dried over
Na2SO4, and concentrated to afford alcohol 5.34 (900 mg, 100% yield), deemed
sufficiently pure to carry forward without additional purification.
1H-NMR (300 MHz; CDCl3): δ 7.36 (s, 5H), 5.17-5.12 (m, 2H), 4.15-4.13 (m, 1H), 4.04-
3.98 (m, 2H), 3.83-3.59 (m, 3H), 3.39-3.36 (bs, 1H), 1.65-1.47 (m, 6H).
REF: TRW-I-260, TRW-I-263, TRW-I-267.
O
CbzN
MeMe
5.34OH

318
Figure 5.105a. 1H NMR spectrum of compound 5.34.
7.17.1
0.60.6
3.03.0
2.12.1
0.90.9
2.12.1
5.05.0
ppm0 011223344556677-19.97
1.47
1.54
1.62
1.65
3.36
3.36
3.36
3.37
3.37
3.37
3.37
3.38
3.38
3.39
3.39
3.59
3.59
3.59
3.70
3.70
3.70
3.71
3.79
3.79
3.80
3.82
3.82
3.83
3.98
3.98
3.99
4.00
4.01
4.03
4.03
4.03
4.04
4.13
4.14
4.14
4.14
4.14
4.14
4.15
4.15
4.15
5.12
5.12
5.13
5.13
5.17
7.36

319
Benzyl 4-(iodomethyl)-2,2-dimethyloxazolidine-3-carboxylate (5.35). To a solution of
Ph3P (550 mg, 2.1 mmol) and imidazole (143 mg, 2.1 mmol) in CH2Cl2 (10 mL) at 0 °C
was added I2 (533 mg, 2.1 mmol) in 3 portions. The reaction mixture was warmed to r.t.
for 10 minutes, then cooled to 0 °C before adding dropwise a solution of alcohol 5.34
(450 mg, 1.7 mmol) in 5 mL CH2Cl2. The resulting solution was stirred 1 h at 0 °C, then
allowed to warm to r.t. as it stirred an additional 1.5 h. The crude reaction mixture was
filtered through a plug of silica with 1:1 EtOAC/Hexanes. The filtrate was concentrated
under reduced pressure and purified by SiO2 chromatography (5-20% EtOAc in hexanes)
to afford iodide 5.35 (30 mg, 8% yield).
1H-NMR (300 MHz; CDCl3): δ 7.36 (s, 5H), 5.15 (s, 2H), 4.25-4.19 (m, 1H), 4.08-4.02
(m, 2H), 3.54-3.35 (m, 1H), 3.21-3.10 (m, 1H), 1.62 (d, J = 20.1 Hz, 3H), 1.47 (d, J =
19.3 Hz, 3H); 13C-NMR (75 MHz; CDCl3): δ 128.9, 128.0, 67.69, 67.58, 67.0, 59.5, 58.9,
28.1, 27.2, 24.7, 23.2, 6.8, 6.5.
REF: TRW-I-261, TRW-I-266.
O
CbzN
MeMe
5.35I

320
Figure 5.106a. 1H NMR spectrum of compound 5.35.
Figure 5.106b. 13C NMR spectrum of compound 5.35.
3.33.3
3.13.1
1.31.3
1.11.1
2.22.2
1.01.0
2.12.1
5.05.0
ppm0 011223344556677-19.97
1.43
1.49
1.58
1.64
3.10
3.13
3.14
3.16
3.17
3.18
3.19
3.21
3.21
3.21
3.35
3.38
3.38
3.39
3.51
3.51
3.53
3.54
3.54
3.54
4.02
4.04
4.05
4.08
4.19
4.20
4.22
4.23
4.23
4.24
4.25
5.15
7.36
ppm0 05050100100150150200-303.86
6.46
6.77
23.2
424
.67
27.2
128
.08
58.8
959
.46
67.0
367
.58
67.6
9
127.
9712
8.87

321
Benzyl 4-(bromomethyl)-2,2-dimethyloxazolidine-3-carboxylate (5.36). To a solution
of alcohol 5.34 (450 mg, 1.7 mmol) in THF (9 mL) at 0 °C was added CBr4 (862 mg, 2.6
mmol) and PPh3 (681 mg, 2.6 mmol). The reaction was stirred at 0 °C for 10 minutes,
then warmed to r.t. and stirred 4 h under Ar. The crude mixture was concentrated, diluted
with ether, filtered through a bed of celite, and concentrated. The crude residue was
purified by SiO2 chromatography (5-20% EtOAc in hexanes) to afford bromide 5.36 (460
mg, 82% yield).
1H-NMR (300 MHz; CDCl3): δ 7.35 (s, 5H), 5.15 (s, 2H), 4.20-4.15 (m, 1H), 4.12-4.07
(m, 1H), 3.99 (ddd, J = 7.0, 5.1, 2.3 Hz, 1H), 3.68-3.49 (m, 1H), 3.30 (dt, J = 15.3, 10.0
Hz, 1H), 1.60 (d, J = 20.2 Hz, 3H), 1.48 (d, J = 19.8 Hz, 3H).
REF: TRW-I-262, TRW-I-269.
O
CbzN
MeMe
5.36Br

322
Figure 5.107a. 1H NMR spectrum of compound 5.36.
3.23.2
3.33.3
1.11.1
1.11.1
1.11.1
1.11.1
1.11.1
2.02.0
5.05.0
ppm1 1223344556677-19.97
1.44
1.51
1.56
1.63
3.25
3.28
3.30
3.31
3.33
3.36
3.49
3.50
3.50
3.52
3.53
3.53
3.54
3.65
3.65
3.68
3.68
3.97
3.98
3.98
3.99
4.00
4.01
4.02
4.07
4.08
4.10
4.10
4.10
4.11
4.12
4.15
4.16
4.18
4.19
4.21
5.15
7.35

323
3,5,6-trichloroindole (5.37). NCS (1.75 g, 13.2 mmol) was added in one portion to a
solution of 5,6-dichloroindole (2.45 g, 13.2 mmol) in DMF (65 mL) at rt. The reaction
was stirred for 3 h at rt, and brine (50 mL) was added. The mixture was extracted with
EtOAc (3 x 50 mL). The combined organic layers were washed with water (100 mL),
brine (100 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue
was purified by flash chromatography eluting with hexanes/EtOAc (9:1) to give 1.95 g
(67%) of 5.37 as a brown solid.
1H NMR (300 MHz, CDCl3) δ 8.11 (bs, 1 H), 7.71 (s, 1 H), 7.49 (s, 1 H), 7.20 (d, J = 2.6
Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 133.7, 127.5, 125.3, 125.1, 122.9, 119.7, 113.2,
106.5; IR (neat) 3442, 1448,1266, 1011 cm-1; HRMS (ES/ApCl) calcd for C8H3NCl3 (M-
H) 217.9337, found 217.9347.
NH
Cl
Cl
Cl
5.37
324
Figure 5.108a. 1H NMR spectrum of compound 5.37.
Figure 5.108b. 13C NMR spectrum of compound 5.37.

325
5,6-dichloro-2-(2-methylbut-3-en-2-yl)-1H-indole (5.38). Solid 5.37 (1.95 g, 8.84
mmol) was added in one portion to a solution of freshly prepared prenyl-9-BBN (26.52
mmol) and Et3N (3.12 g, 30.94 mmol, 4.30 mL) in THF (53 mL) at rt. The reaction was
stirred for 3 h, and then quenched with sat. NaHCO3 (50 mL). The organic layer was
separated and the aqueous layer was extracted with Et2O (3 x 50 mL). The combined
organic layers were washed with H2O (100 mL), brine (100 mL), and dried (Na2SO4),
and concentrated under reduced pressure. The residue was purified by flash
chromatography eluting with hexanes/EtOAc (99:1) to give 1.80 g (80%) of 5.38 as a
yellow oil.
1H NMR (300 MHz, CDCl3) δ 7.87 (bs, 1 H), 7.59 (s, 1 H), 7.38 (s, 1 H), 6.23 (m, 1 H),
6.01 (dd, J = 17.3, 10.6 Hz, 1 H), 5.15 (d, J = 10.6 Hz, 1 H), 5.09 (d, J = 17.3 Hz, 1 H),
1.46 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 148.2, 145.6, 134.9, 128.5, 125.0, 123.7,
121.1, 113.0, 112.0, 97.6, 38.5, 27.5; IR (neat) 3231, 2927, 1453, 1102 cm-1; HRMS
(FAB) calcd for C13H13NCl2 (M+) 253.0425, found 253.0428.
NH
Cl
Cl MeMe
5.38

326
Figure 5.109a. 1H NMR spectrum of compound 5.38.
Figure 5.109b. 13C NMR spectrum of compound 5.38.

327
1-(5,6-dichloro-2-(2-methylbut-3-en-2-yl)-1H-indol-3-yl)-N,Ndimethylmethanamine
(5.39). A solution of Me2NH (0.78 mL, 40% solution in H2O, 6.00 mmol) and a solution
of CH2O (0.47 mL, 37% solution in H2O, 6.00 mmol) were sequentially added to AcOH
(2 mL) and the reaction stirred for 1 h. Neat 5.38 (1.45 g, 5.71 mmol) was added, and the
reaction was stirred for 12 h at rt. 2N NaOH was added until pH ≈ 12. The reaction was
extracted with Et2O (3 x 20 mL). The combined organic layers were washed with H2O
(50 mL), brine (50 mL), and dried (Na2SO4), and concentrated under reduced pressure to
give 1.66 g (90%) of 5.39 as a yellow oil which was used without further purification.
1H NMR (400 MHz, CDCl3) δ 7.86 (bs, 1 H), 7.76 (s, 1 H), 7.32 (s, 1 H), 6.08 (dd, J =
17.6, 10.0 Hz, 1 H), 5.14 (d, J = 10.0 Hz, 1 H), 5.12 (d, J = 17.6 Hz, 1 H), 3.49 (s, 2 H),
2.16 (s, 6 H), 1.52 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 145.7, 143.4, 132.9, 130.4,
124.9, 123.3, 120.6, 112.7, 111.8, 109.0, 54.1, 45.5, 39.5, 27.2; IR (neat) 3320, 2928,
1463, 1016 cm-1; HRMS (+TOF) calcd for C16H21N2Cl2 (M+H) 311.1082, found
311.1020.
NH
Cl
Cl MeMe
NMe2
5.39
328
Figure 5.110a. 1H NMR spectrum of compound 5.39.
Figure 5.110b. 13C NMR spectrum of compound 5.39.

329
Ethyl 2-amino-3-(5,6-dichloro-2-(2-methylbut-3-en-2-yl)-1H-indol-3-yl)–propanoate
(5.40). PBu3 (346 mg, 1.72 mmol, 0.43 mL) was added to a solution of 5.39 (1.0 g, 3.43
mmol) and N-(Diphenylmethylene)glycine ethyl ester (832 mg, 3.12 mmol) in CH3CN
(30 mL) at rt. The reaction was heated to reflux and was stirred under Ar for 12 h. The
reaction was cooled to rt and concentrated under reduced pressure. The residue was
purified by flash chromatography eluting with hexanes/EtOAc (4:1) to give 1.19 g of a
yellow oil which was dissolved in CH2Cl2 (25 mL). 1N HCl was added and the reaction
was vigorously stirred for 12 h at rt. The organic layer was separated and the aqueous
layer was extracted with CH2Cl2 (2 x 25 mL). The combined organic layers were
concentrated under reduced pressure. The residue was purified by flash chromatography
eluting with MeOH/CH2Cl2 (1:99-5:95) to give 848 mg (67%) of 5.40 as a yellow oil.
1H NMR (400 MHz, CDCl3) δ 7.89 (bs, 1 H), 7.59 (s, 1 H), 7.34 (s, 1 H), 6.08 (dd, J =
17.2, 10.8 Hz, 1 H), 5.16 (d, J = 10.8 Hz, 1 H), 5.15 (d, J = 17.2 Hz, 1 H), 4.09 (comp, 2
H), 3.73 (m, 1 H), 3.20 (dd, J = 14.8, 5.6 Hz, 1 H), 2.96 (dd, J = 14.8, 8.8 Hz, 1 H), 1.53
(s, 6 H), 1.16 (t, J = 7.2 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ 170.6, 145.6, 142.7,
133.1, 129.9, 125.3, 123.6, 119.9, 112.9, 112.0, 107.3, 61.2, 56.0, 39.5, 31.0, 27.8, 14.3;
IR (neat) 3368, 2973, 2927, 1729, 1463, 1199 cm-1; HRMS (TOF-) calcd for
C18H21N2O2Cl2 (M-H) 367.0986, found 367.0975.
NH
Cl
Cl Me Me
NH2
CO2Et
5.40
330
Figure 5.111a. 1H NMR spectrum of compound 5.40.
Figure 5.111b. 13C NMR spectrum of compound 5.40.

331
2-(tert-butoxycarbonylamino)-3-(5,6-dichloro-2-(2-methylbut-3-en-2-yl)-1Hindol-3-
yl)propanoic acid (5.41). 0.5M NaOH (4.5 mL, 2.25 mmol) and Boc2O (586 mg, 2.69
mmol) were sequentially added to a solution of 5.40 (848 mg, 2.24 mmol) in dioxane (5
mL). The reaction was stirred at rt for 3 h, and then concentrated under reduced pressure
to remove dioxane. 10% KHSO4 was added until pH ≈ 2, and the solution was extracted
with EtOAc (3 x 25 mL). The combined organic layers were dried (Na2SO4) and
concentrated under reduced pressure. The residue was dissolved in 2:1 THF/H2O (18
mL), and LiOH (280 mg, 11.25 mmol) was added in one portion. The reaction stirred at rt
for 12 h. 10% KHSO4 was added until pH ≈ 2, and the solution was extracted with
EtOAc (3 x 25 mL). The combined organic layers were dried (Na2SO4) and concentrated
under reduced pressure to afford 902 mg (91%) of a yellow solid which was used without
further purification.
1H NMR (300 MHz, CDCl3) δ 11.0 (bs, 1 H), 8.19 (m, 1 H), 6.63 (m, 1 H), 7.31 (m, 1 H),
6.80 (bs, 1 H), 6.08 (m, 1 H), 5.18 (comp, 2 H), 4.56 (bs, 1 H), 3.37 (bs, 1 H), 3.14 (m, 1
H), 1.52 (s, 6 H), 1.28-1.02 (comp, 9 H); 13C NMR (75 MHz, CDCl3) δ 177.3, 176.3,
170.4, 156.6, 155.3, 145.4, 142.9, 133.0, 129.9, 129.7, 128.4, 125.0, 124.9, 123.5, 123.3,
119.7, 119.5, 112.9, 112.8, 106.6, 106.0, 81.4, 80.3, 55.4, 54.7, 39.2, 29.8, 28.6, 28.2,
27.8, 27.5; IR (neat) 3335, 2976, 2932, 1697, 1461 cm-1; HRMS (TOF-) calcd for
C21H25N2O4Cl2 (M-H) 439.1197, found 439.1185.
NH
Cl
Cl Me Me
NHBoc
CO2H
5.41
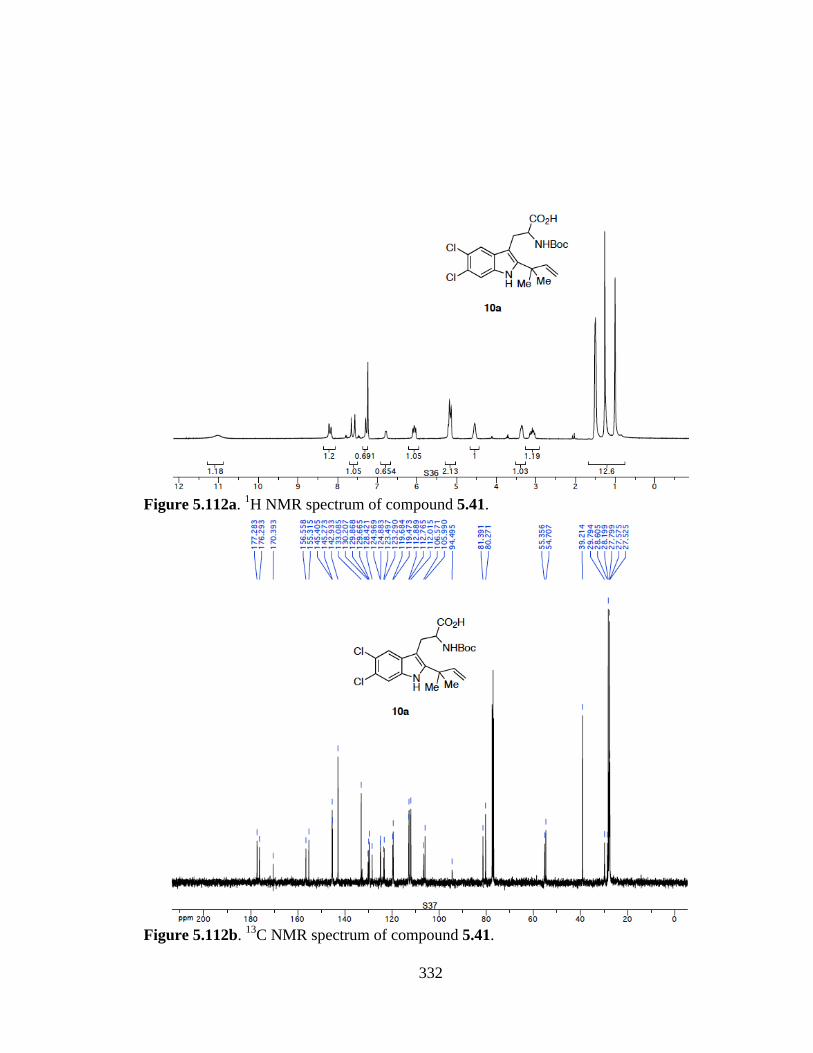
332
Figure 5.112a. 1H NMR spectrum of compound 5.41.
Figure 5.112b. 13C NMR spectrum of compound 5.41.

333
(2S,3R)-ethyl-1-(2-(tert-butoxycarbonylamino)-3-(5,6-dichloro-2-(2-methylbut-3-en-
2-yl)-1H-indol-3-yl)propanoyl)-3-hydroxypyrrolidine-2-carboxylate (5.42). To a
solution of 5.41 (280 mg, 0.63 mmol) in CH3CN (6 mL) was sequentially added HATU
(361 mg, 0.95 mmol), iPr2NEt (327 mg, 2.54 mmol), and cis-3-hydroxy-L-proline ethyl
ester HCl (217 mg, 1.11 mmol). The reaction was stirred at rt for 3 h, and 2N HCl (10
mL) was added. The mixture was extracted with CH2Cl2 (3 x 25 mL), and the combined
organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue
was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to give 252 mg
(68%) of 5.42 as a colorless oil.
1H NMR (300 MHz, CDCl3) δ 8.73- 8.47 (m, 1 H), 7.58-7.32 (comp, 2 H), 6.11 (m, 1 H),
5.50 (m, 1 H), 5.17 (comp, 2 H), 4.63-3.04 (comp, 9 H), 1.57-1.14 (comp, 18 H); 13C
NMR (75 MHz, CDCl3) δ 172.1, 169.3, 169.1, 169.0, 155.0, 154.8, 154.3, 145.7, 144.8,
144.7, 143.7, 143.3, 142.9, 133.1, 132.7, 129.9, 129.5, 129.2, 124.9, 124.7, 123.1, 119.6,
112.9, 112.6, 112.0, 105.9, 105.6, 79.6, 79.3, 71.7, 70.4, 70.1, 63.8, 63.3, 62.0, 61.4, 60.6,
53.0, 52.4, 45.2, 44.4, 44.3, 39.2, 33.5, 32.6, 30.8, 30.5, 30.2, 28.3, 28.0, 27.7, 21.1, 18.4,
14.2, 14.0; IR (neat) 3359, 2978, 2932, 1694, 1633, 1500, 1455 cm-1; HRMS (TOF-)
calcd for C28H36N3O6Cl2 (M-H) 580.1987, found 580.1976.
NHCl
NHBoc
NO
Me Me
Cl
EtO2C OH
5.42
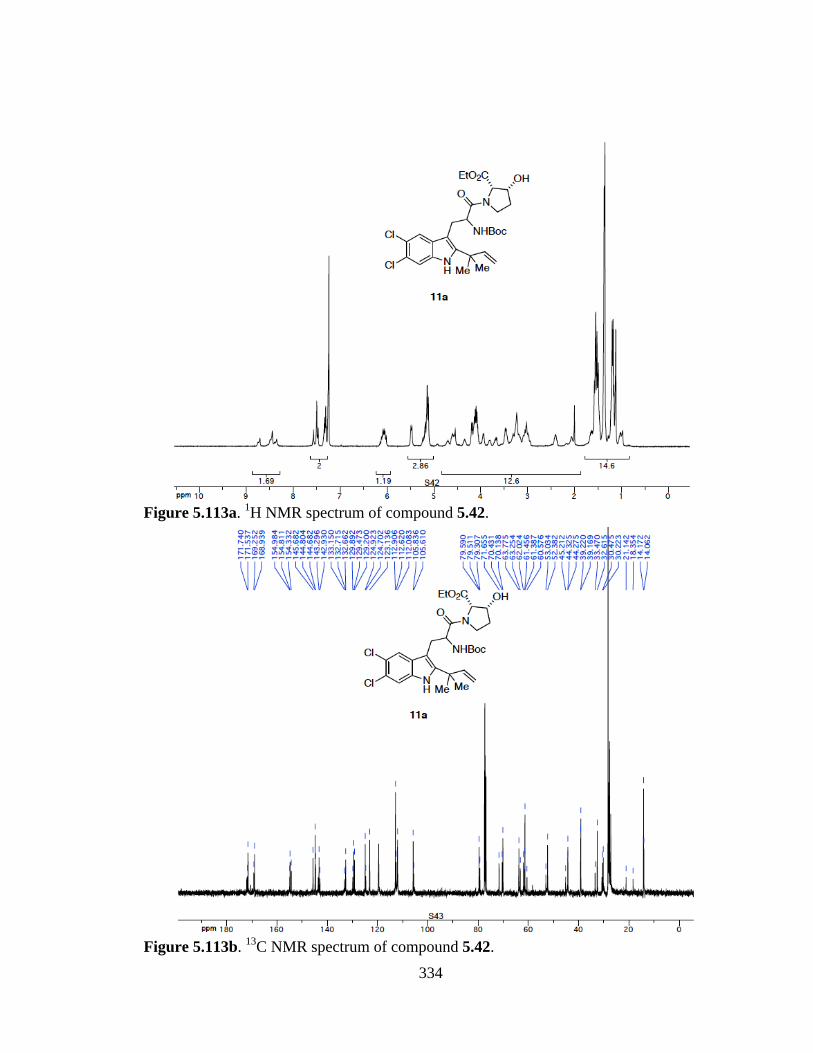
334
Figure 5.113a. 1H NMR spectrum of compound 5.42.
Figure 5.113b. 13C NMR spectrum of compound 5.42.

335
(8R,8aS)-3-((5,6-dichloro-2-(2-methylbut-3-en-2-yl)-1H-indol-3-yl)methyl)-8-
hydroxyhexahydropyrrolo[1,2-a]pyrazine-1,4-dione (5.43). TFA (0.86 mL, 11.16
mmol) was added to a solution of 5.42 (252 mg, 0.43 mmol) in CH2Cl2 (9 mL). The
reaction was stirred for 3 h, and sat. NaHCO3 (30 mL) was added. The mixture was
extracted with EtOAc (3 x 20 mL), and the combined organic layers were dried (Na2SO4)
and concentrated under reduced pressure. The residue was dissolved in toluene (9 mL)
and 2-hydroxypyridine (9 mg, 0.09 mmol) was added. The reaction was refluxed under
Ar for 12 h, cooled to rt, and concentrated under reduced pressure. The residue was
dissolved in CH2Cl2 (30 mL) and washed with 1N HCl (30 mL). The organic layer was
dried (Na2SO4) and concentrated under reduced pressure to afford 156 mg (85%) of a
beige solid which was used without further purification.
1H NMR (300 MHz, CDCl3) δ 8.14 (s, 1 H), 7.54 (s, 1 H), 7.43 (s, 1 H), 6.10 (dd, J =
17.4, 10.6 Hz, 1 H), 5.72 (bs, 1 H), 5.22 (d, J = 10.6 Hz, 1 H), 5.17 (d, J = 17.4 Hz, 1 H),
4.71-2.10 (comp, 9 H), 1.55 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 167.7, 165.5, 145.0,
144.1, 133.2, 128.8, 126.1, 124.3, 119.0, 113.6, 112.6, 104.5, 71.0, 64.7, 54.5, 44.2, 39.1,
30.3, 27.8, 26.3; IR (neat) 3352, 2973, 1647, 1459 cm-1; HRMS (TOF+) calcd for
C21H24N3O3Cl2 (M+) 436.1189, found 436.1167.
NHCl
NH
NHO
O
Me Me
Cl
OHH
5.43
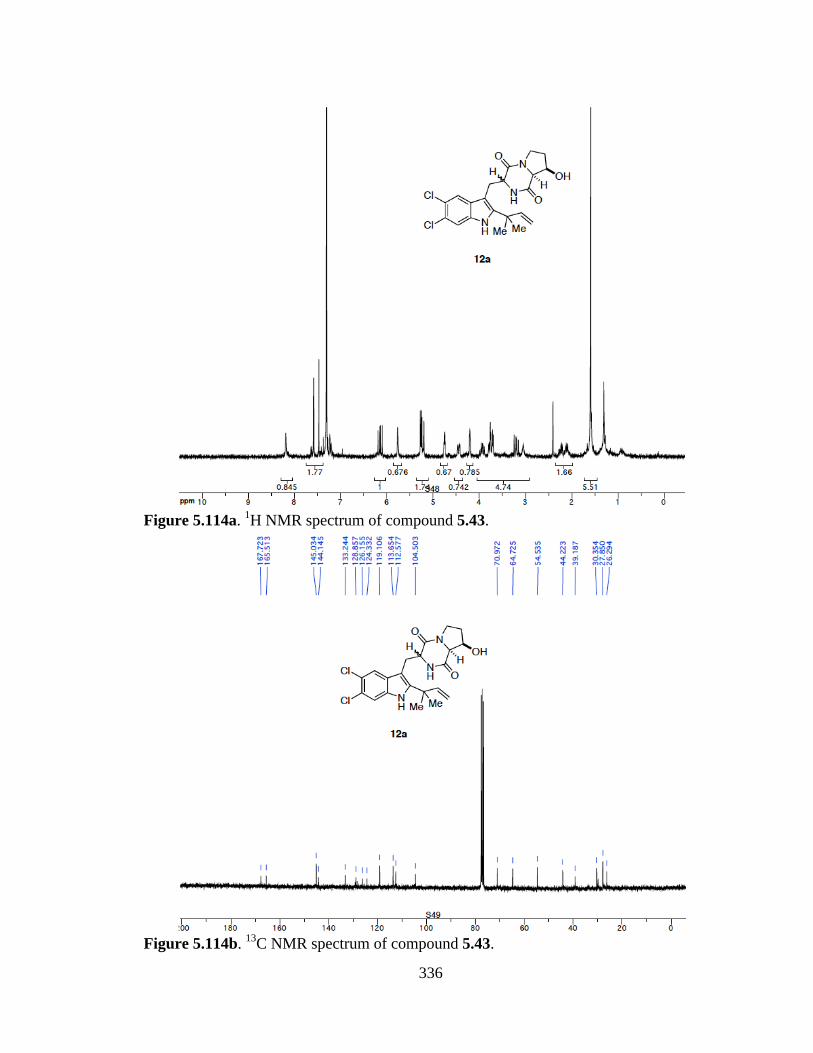
336
Figure 5.114a. 1H NMR spectrum of compound 5.43.
Figure 5.114b. 13C NMR spectrum of compound 5.43.

337
3-((5,6-dichloro-2-(2-methylbut-3-en-2-yl)-1H-indol-3-yl)methyl)-2,3,6,7-
tetrahydropyrrolo[1,2-a]pyrazine-1,4-dione (5.44). DEAD (419 mg soln., 40% in
toluene, 0.44 mL, 0.96 mmol) was added to a solution of 5.43 (140 mg, 0.32 mmol) in
CH2Cl2 (10 mL). The reaction was stirred for 5 min, and PBu3 (194 mg, 0.96 mmol) was
added. The reaction was stirred for 3 h, and then concentrated under reduced pressure.
The residue was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to
give 86 mg (64%) of 5.44 as a pale yellow oil.
1H NMR (300 MHz, CDCl3) δ 8.72 (s, 1 H), 7.52 (s, 1 H), 7.33 (s, 1 H), 6.05 (comp, 3
H), 5.09 (d, J = 17.3 Hz, 1 H), 5.09 (d, J = 10.6 Hz, 1 H), 4.42 (m, 1 H), 4.01 (comp, 2
H), 3.57 (dd, J = 14.6, 4.0 Hz, 1 H), 3.15 (dd, J = 14.6, 10.6 Hz, 1 H), 2.73 (comp, 2 H),
1.46 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 162.4, 156.8, 145.4, 144.3, 133.4, 132.8,
128.7, 125.5, 123.7, 119.4, 119.2, 112.6, 112.4, 104.3, 60.5, 57.2, 45.7, 39.2, 30.7, 27.8;
IR (neat) 3323, 2971, 1681, 1644, 1446 cm-1; HRMS (TOF+) calcd for C21H22N3O2Cl2
(M+H) 418.1084, found 418.1089.
NHCl
NH
NHO
O
Me Me
Cl
5.44
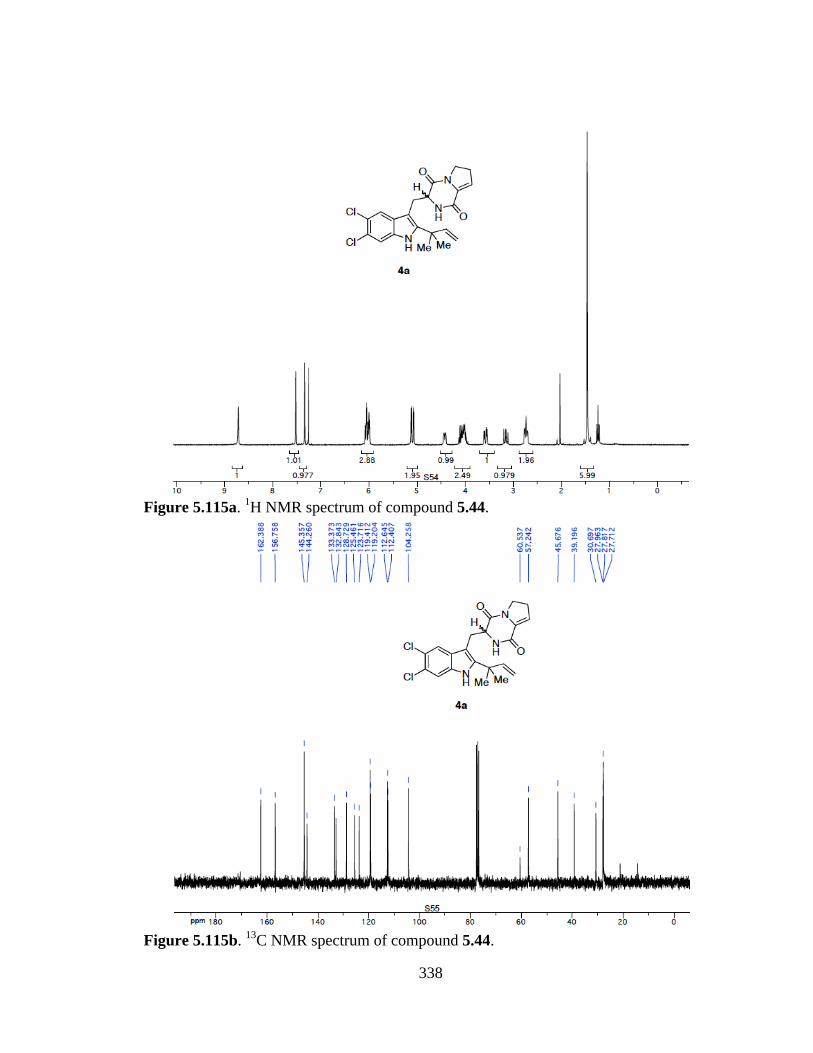
338
Figure 5.115a. 1H NMR spectrum of compound 5.44.
Figure 5.115b. 13C NMR spectrum of compound 5.44.

339
Preparation of Cycloadducts 5.45a and 5.45b. To a solution of 5.44 (86 mg, 0.21
mmol) in MeOH (16 mL) at 0 °C was added 20% aqueous KOH (4 mL). The reaction
was warmed to room temperature (rt) and was stirred for 12 h. The reaction was
quenched with sat. NH4Cl (30 mL) and extracted with CH2Cl2 (3x30 mL). The combined
organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue
was triturated with CHCl3 (30 mL), and the suspension was filtered to provide 53 mg
(60%) of 5.45a as a white amorphous solid. Concentration of the filtrate gave 25 mg
(29%) of 5.45b as a white amorphous solid.
Data for major isomer 5.45a: 1H NMR (300 MHz, CD3OD) δ 7.55 (s, 1 H), 7.40 (s, 1 H),
3.57 (d, J ) 15.5 Hz, 1 H), 3.47 (m, 1 H), 2.74 (d, J ) 15.5 Hz, 1 H), 2.60 (m, 1 H), 2.20-
1.96 (comp, 6 H), 1.35 (s, 3 H), 1.11 (s, 3 H); 13C NMR (75 MHz, CD3OD) δ 175.7,
171.3, 144.3, 137.2, 128.1, 125.4, 123.3, 119.8, 113.1, 104.8, 68.3, 61.6, 50.8, 45.2, 36.2,
31.7, 30.1, 28.6, 25.4, 24.9, 22.3; IR (neat) 1663, 1428 cm-1; HRMS (TOF+) calcd for
C21H22N3O2Cl2 (M + H) 418.1084, found 418.1084.
Data for minor isomer 5.45b: 1H NMR (300 MHz, CDCl3) δ 9.78 (s, 1 H), 7.49 (s, 1 H),
7.33 (s, 1 H), 3.71 (d, J ) 17.8 Hz, 1 H), 3.47 (comp, 2 H), 3.25 (s, 1 H), 2.78 (d, J ) 17.8
Hz, 1 H), 2.67 (m, 1 H), 2.18- 1.78 (comp, 6 H), 1.24 (s, 3H), 1.16 (s, 3 H); 13C NMR (75
MHz, CD3OD /CDCl3 (1:9)) δ 173.7, 169.6, 142.6, 135.7, 127.1, 125.0, 122.9, 119.1,
112.4, 102.9 67.2, 61.5, 45.8, 44.3, 34.8, 32.6, 29.8, 29.1, 28.4, 24.5, 23.2; IR (neat)
1676, 1453 cm-1; HRMS (TOF+) calcd for C21H22N3O2Cl2 (M + H) 418.1084, found
418.1079.
OH
N N
HNMe Me Cl
H Cl
O
OH
N N
HNMe Me Cl
H Cl
O5.45a 5.45b
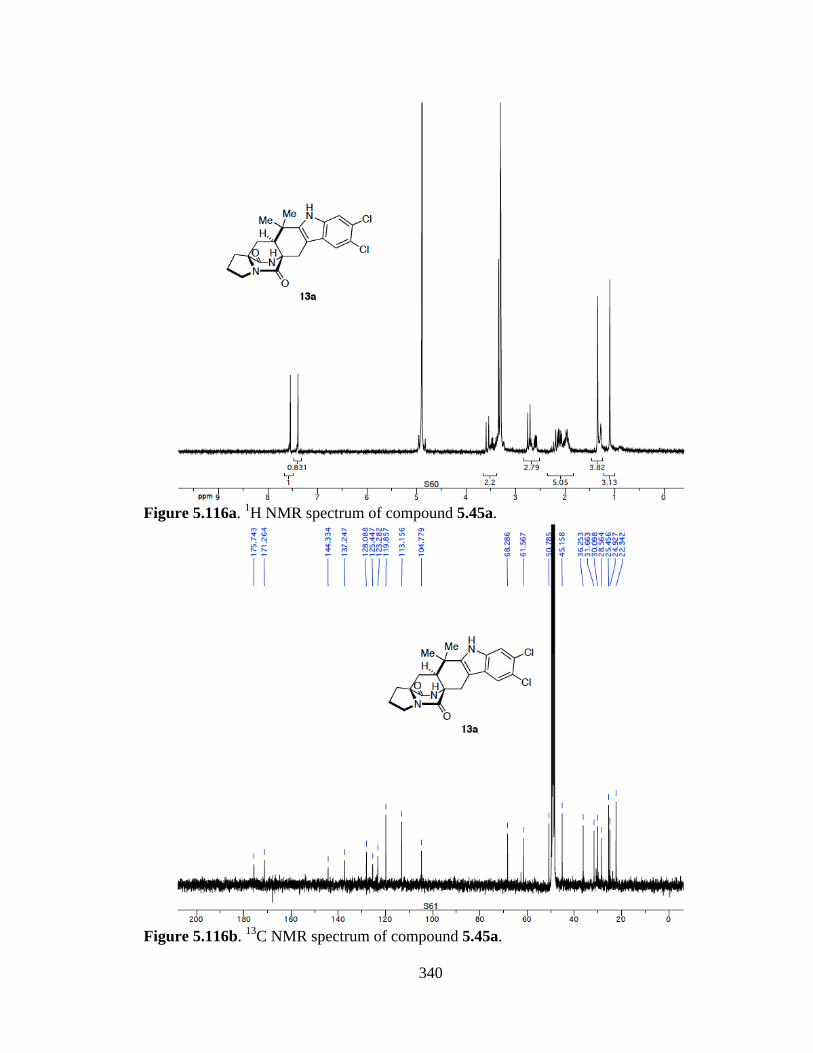
340
Figure 5.116a. 1H NMR spectrum of compound 5.45a.
Figure 5.116b. 13C NMR spectrum of compound 5.45a.
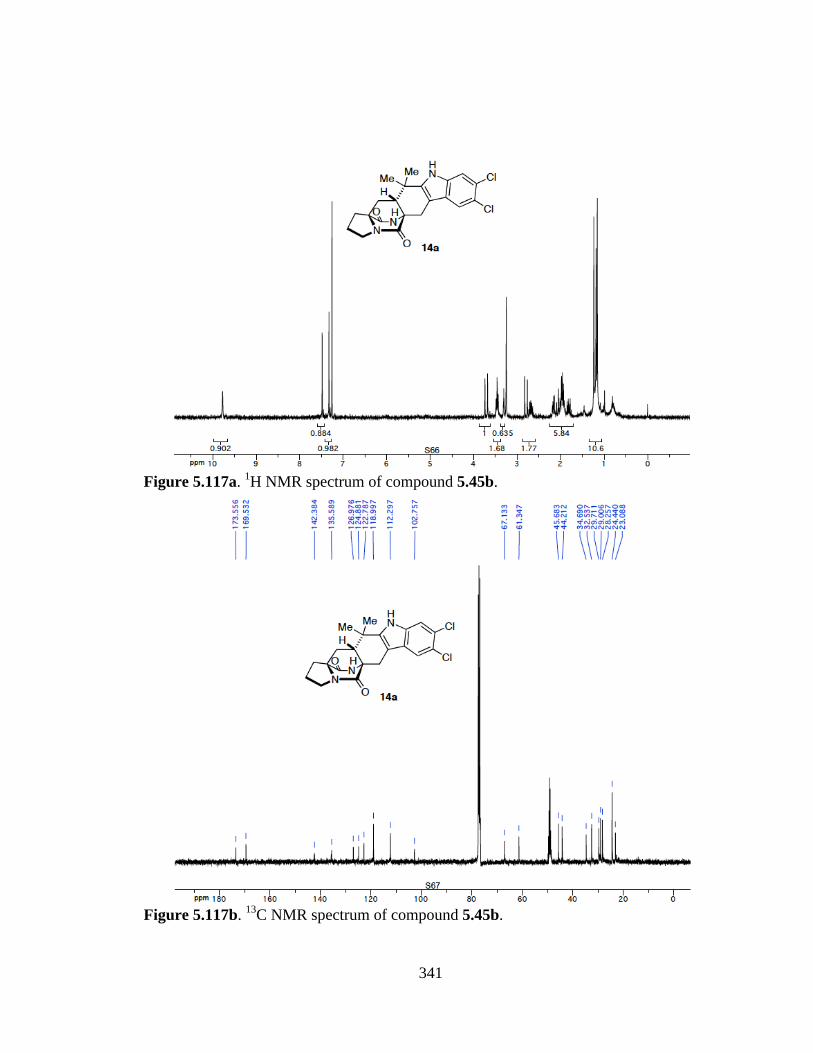
341
Figure 5.117a. 1H NMR spectrum of compound 5.45b.
Figure 5.117b. 13C NMR spectrum of compound 5.45b.

342
Synthesis of Malbrancheamide (5.46). DIBAL-H (0.70 mL, 1 M in toluene, 0.70 mmol)
was added to a suspension of 5.45a (15 mg, 0.036 mmol) in toluene (7 mL) at rt. The
reaction was stirred at rt for 12 h, whereupon finely powdered Na2SO4-10H2O was added
until bubbling ceased. The mixture was filtered with a medium porosity fritted funnel
washing with EtOAc (50 mL) and MeOH (50 mL), and the filtrate was concentrated
under reduced pressure. The residue was purified by flash chromatography eluting with
MeOH/ CH2Cl2 (2:98) to give 12 mg (80%) of 5.46 as a white amorphous solid.
1H NMR (400 MHz, CD3OD) δ 7.48 (s, 1 H), 7.40 (s, 1 H), 3.43 (d, J ) 10.3 Hz, 1 H),
3.06 (m, 1 H), 2.85 (comp, 2 H), 2.54 (m, 1 H), 2.27 (dd, J ) 10.2, 1.5 Hz, 1 H), 2.20-1.18
(comp, 6 H), 1.43 (s, 3 H), 1.34 (s, 3 H); 13C NMR (100 MHz, CD3OD) δ 176.6, 145.1,
137.3, 128.2, 125.3, 123.2, 119.6, 113.1, 104.7, 66.1, 59.4, 57.4, 55.4, 48.5, 35.5, 32.4,
30.6, 30.0, 28.1, 24.2, 23.5; IR (neat) 3226, 1658, 1460 cm-1; HRMS (TOF+) calcd for
C21H24N3OCl2 (M + H) 404.1291, found 404.1290.
OH
N N
HNMe Me Cl
H Cl
5.46
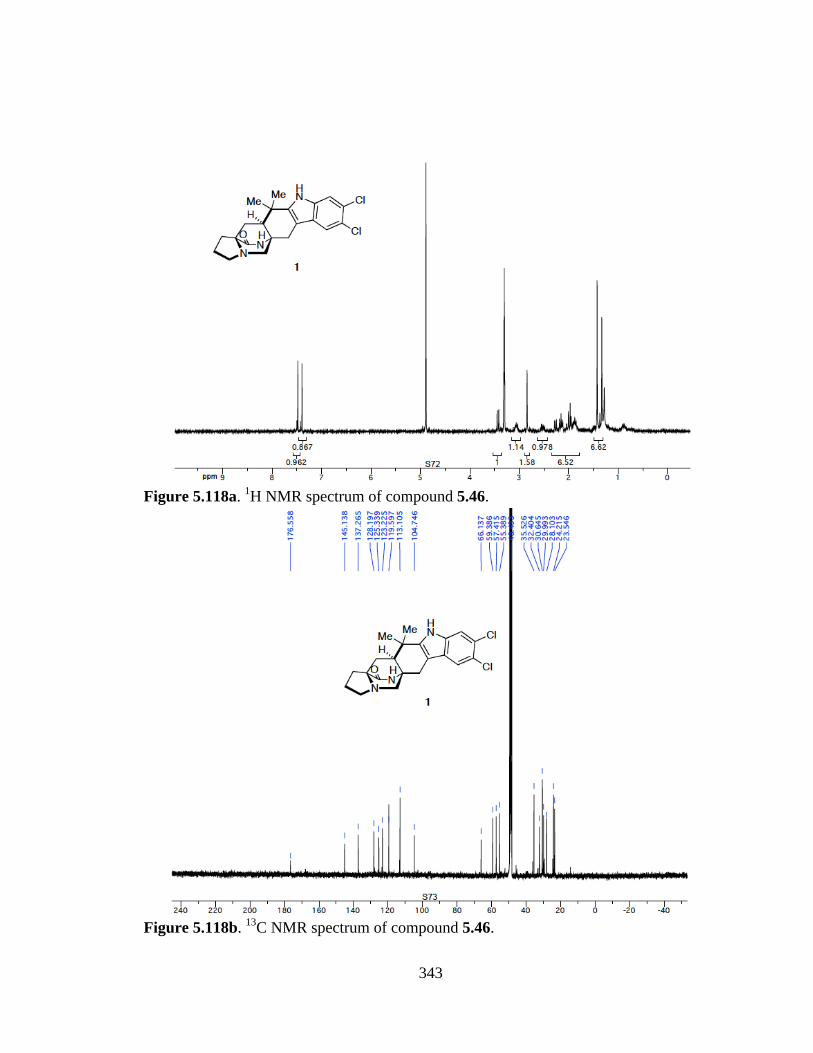
343
Figure 5.118a. 1H NMR spectrum of compound 5.46.
Figure 5.118b. 13C NMR spectrum of compound 5.46.

344
Chetomin (1.12). Authentic sample purchased from Santa Cruz Biotechnology.
1H-NMR (400 MHz; CDCl3): δ 7.66 (dd, J = 6.1, 3.0 Hz, 1H), 7.35-7.33 (m, 1H), 7.31-
7.27 (m, 2H), 7.23-7.22 (m, 2H), 7.18 (s, 1H), 6.95 (t, J = 7.4 Hz, 1H), 6.80 (d, J = 8.0
Hz, 1H), 6.21 (s, 1H), 4.43 (s, 1H), 4.39 (d, J = 4.7 Hz, 1H), 4.35 (s, 1H), 4.31 (d, J = 3.5
Hz, 1H), 4.28 (s, 1H), 3.87 (d, J = 15.6 Hz, 1H), 3.71 (d, J = 15.4 Hz, 1H), 3.20 (s, 3H),
3.16 (s, 3H), 3.10 (s, 1H), 2.96 (s, 3H); 13C-NMR (101 MHz; CDCl3): δ 166.8, 165.51,
165.48, 163.2, 148.3, 134.0, 131.4, 130.4, 127.2, 126.5, 125.0, 122.8, 120.60, 120.40,
119.2, 111.4, 111.2, 107.7, 80.0, 76.5, 76.1, 74.7, 73.7, 73.5, 61.3, 60.7, 42.6, 28.2, 27.44,
27.34, 27.1.
FOR NMR FILES, SEE Tim/NMR/robot/trw-III-chetomin.
See also (for characterization data): Fujimoto, H.; Sumino, M.; Okuyama, E.; Ishibashi,
M. Immunomodulatory Constituents from an Ascomycete, Chaetomium Seminudum. J.
Nat. Prod. 2004, 67, 98-102.
chetomin (1.12)
NNMe
O
O
SS
NH
HOH
MeNNMe
O
O
SS
NOH
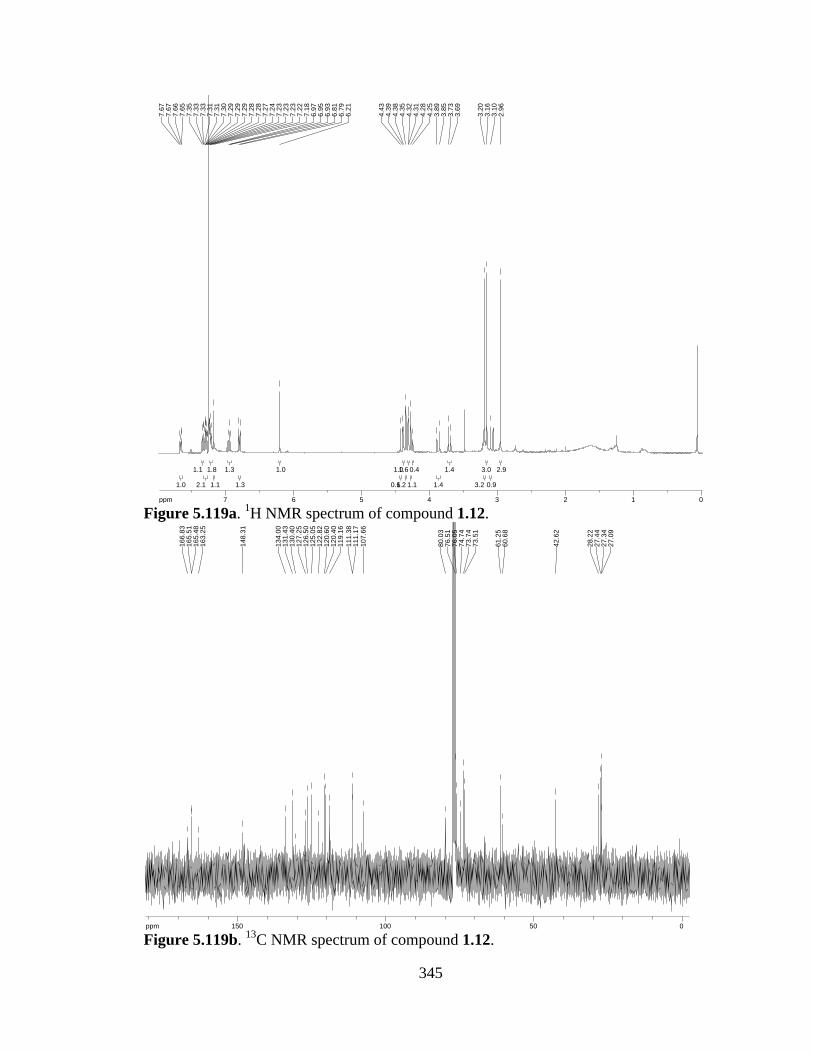
345
Figure 5.119a. 1H NMR spectrum of compound 1.12.
Figure 5.119b. 13C NMR spectrum of compound 1.12.
2.92.9
0.90.9
3.03.0
3.23.2
1.41.4
1.41.4
0.40.4
1.11.1
1.61.6
1.21.2
1.01.0
0.60.6
1.01.0
1.31.3
1.31.3
1.11.1
1.81.8
2.12.1
1.11.1
1.01.0
ppm0 011223344556677-16.042.
963.
103.
163.
20
3.69
3.73
3.85
3.89
4.25
4.28
4.31
4.32
4.35
4.38
4.39
4.43
6.21
6.79
6.81
6.93
6.95
6.97
7.18
7.22
7.23
7.23
7.23
7.24
7.27
7.28
7.28
7.29
7.29
7.29
7.30
7.31
7.31
7.33
7.33
7.35
7.65
7.66
7.67
7.67
ppm0 05050100100150150-253.79
27.0
927
.34
27.4
428
.22
42.6
2
60.6
861
.25
73.5
173
.74
74.7
476
.05
76.5
180
.03
107.
6611
1.17
111.
3811
9.16
120.
4012
0.60
122.
8212
5.05
126.
5012
7.25
130.
4013
1.43
134.
00
148.
31
163.
2516
5.48
165.
5116
6.83

117
4Biomimetic Synthesis of Alkaloids Derivedfrom Tryptophan: Dioxopiperazine AlkaloidsTimothy R. Welch and Robert M. Williams
4.1Introduction
Countless secondary metabolic indole alkaloids produced in both marine and ter-restrial fungi are derived from tryptophan. Our crude attempts to synthesize someof the vast array of structurally diverse natural alkaloids only serves to showcase theefficiency and elegance with which Nature is able assemble the same molecules.Still, we strive to mimic and in turn better understand the mechanisms inside thecell that are able to produce molecular architecture of such synthetic complexity.
Moreover, it has often been found advantageous to exploit Nature’s evolutionarycreative design of alkaloids in search of new compounds of therapeutic potential.The rich subclass of biologically active, tryptophan-derived dioxopiperazines foundin nature has inspired medicinal chemists to use the dioxopiperazine core indrug design efforts to mimic the interactions of natural peptides while reducingsusceptibility to metabolic amide bond cleavage. Furthermore, a dioxopiperazineshould pay a small entropic penalty upon binding to a target in comparison to ananalogous peptide, as a direct consequence of the reduced conformational mobilityinherent in the dioxopiperazine ring. In this chapter, we present a brief review of aselect group of (partly) biomimetic syntheses of tryptophan-derived dioxopiperazinealkaloids (Figure 4.1). In most of these syntheses, a single step or key transformationhas been deemed to constitute the ‘‘biomimetic’’ aspect of that particular work. Asthe actual biosynthetic pathways to most, if not all, of the alkaloid natural productscovered here are either unknown or known only in part we have attempted, whereappropriate, to point out the particular biomimetic step or transformation.
4.2Prenylated Indole Alkaloids
Birch, Wright, and Russell first isolated brevianamide A from Penicillium brevi-compactum in 1969 [1–3]. Several years later, Birch and coworkers determinedthat brevianamide A was biosynthetically derived from tryptophan, proline, and
Biomimetic Organic Synthesis, First Edition. Edited by Erwan Poupon and Bastien Nay.! 2011 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2011 by Wiley-VCH Verlag GmbH & Co. KGaA.
346
APPENDIX I Publications

118 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NH
NH
O N
OH H
brevianamide F
NN
O
OMe
Me
HN
O
spirotryprostatin B
N
Me MeNH
NOOH
H
NMe Me
HOH
okaramine N
NN
NH
HN
O
O
Me
Me
Me
Me
HO
OH
H
H
gypsetin
O
H
NN
HNMe Me Cl
H Cl
malbrancheamide (+)-versicolamide B
O
NHO
NN
OMe
Me
MeMe
O
H
H
NHN N
OO
Me
NH
O
ent-alantrypinone
NN
O
O MeSSHO H
OH(+)-gliotoxin
Section 4.2.1
Section 4.2.2
Section 4.2.3 Section 4.3 Section 4.3.1
Figure 4.1 Representative molecules discussed in this chapter.
mevalonic acid through feeding experiments (Scheme 4.1) [4]. Furthermore, Birchshowed that radiolabeled brevianamide F was incorporated into 5. It was postu-lated at the time and later supported with experimental evidence by Williams andcoworkers that deoxybrevianamide E was also a biosynthetic precursor [5].
NH
CO2HH3HN
H
14CH2
NH2
CO2H
14CH2
O
OHMe
O
[2-14C]-D,L-tryptophan(1)
[5-3H]-L-proline(2)
[2-14C]-D,L-mevalonicacid lactone (3)
NH
14CH2NH
O N
OH H
3H
brevianamide F (4)
O
H
NNH
O
(+)-brevianamide A (5)
NH
OMe(14C)
Me
NH
NH
O N
OH H
MeMe
3H3H
deoxybrevianamide E (6)
Scheme 4.1 Proposed biosynthesis of the brevianamides.
Since these early studies, Williams has developed a proposal for the biosyn-thesis of the brevianamides, with that of brevianamide E shown in Scheme 4.2.Derived from tryptophan and proline, 6 was proposed to undergo oxidation tohydroxyindolenine 7. Irreversible nucleophilic ring closure was supposed to leadto brevianamide E (8), supported by incorporation of [8-3H2]6 into 8 in significantradiochemical yield [5].
347

4.2 Prenylated Indole Alkaloids 119
NH
NH
O N
OH H
MeMe
3H3H
deoxybrevianamide E (6)
[ox]
N
NH
O N
OH H
MeMe
HOnucleophilic
additionN
N
NH O
O
Me
Me
H
H
brevianamide E (8)
HO
7
Scheme 4.2 Proposed biosynthesis of brevianamide E.
4.2.1Dioxopiperazines Derived from Tryptophan and Proline
Brevianamide F (12) was first isolated in 1972 and is one of the simplesttryptophan-derived dioxopiperazine natural products. It is readily synthesizedthrough amino acid coupling of N-Boc-tryptophan (9) to proline ethyl ester (10),Boc-deprotection, and ring closure, in modest overall yield (Scheme 4.3) (R.M.Williams, unpublished results).
NH
CO2HH
NH
EtO2CHATU, iPr2NEt
MeCN+
NH
NHBoc
O N 1. TFA, CH2Cl2
2. 2-OH-pyridinePhCH3
EtO2C
NH
NH
O N
OH H
brevianamide F (12)
NHBoc
9 10
11
62%
1:1 = cis:trans
56%
Scheme 4.3 Biomimetic synthesis of brevianamide F.
Brevianamide F lacks only the reverse prenyl group found in deoxybrevianamideE, which has been synthesized by Kametani and coworkers en route tobrevianamide E (Scheme 4.4) [6]. N-Benzyloxycarbonyl-l-proline (13) wassubjected to Schotten–Baumann conditions with dimethyl aminomalonateto give amide 14. Debenzyloxycarbonylation of 14 followed by heating withcatalytic 2-hydroxypyridine effected cyclization to dioxopiperazine 15 in 93%yield. Condensation with indole 16 gave a separable mixture of diastereomers,individually hydrolyzed to the corresponding free acids (17). Heating of the desireddiastereomer in dioxane gave deoxybrevianamide E (18) and its epimer (19) in 29and 55% yield, respectively. Irradiation of methanolic 18, containing Rose Bengalin the presence of oxygen, followed by addition of dimethyl sulfide, resulted in thebiomimetic hydroxylation of deoxybrevianamide E, furnishing brevianamide E (8)and 20 as a separable mixture of diastereomers.
Nineteen years later, a more efficient synthesis of brevianamide E was com-pleted by Danishefsky and coworkers [7]. The synthesis commenced with C3chlorination of the known phthaloylated tryptophan derivative 21, followed byaddition of fresh prenyl-9-borabicyclo[3.3.1]nonane (prenyl-9-BBN) to the resultant3-chloroindolenine (Scheme 4.5). Hydrazinolysis in ethanol provided amino ester
348

120 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
CO2BzNHO2C
H2NCO2Me
CO2Me 1. Pd/C, H2
2. 70 °C, 2-OH-pyridine (cat)
HNN
O
O
CO2MeH
H
HN
NO
CO2MeCO2Me
H
H
CO2Bz
NH
NMe2
MeMe
+
1. NaH, "HNHNMe
Me
N
O
OH
HO2C
2. NaOH HNHNMe
Me
N
O
OH
HNHNMe
Me
N
O
OH
+
H Hdioxane, "
deoxybrevianamide E (18)
HNHNMe
Me
N
O
OH
HRose Bengal
deoxybrevianamide E (18)
hn, O2; Me2SN
N
NH O
O
Me
Me
HOH
H
brevianamide E (8)
13 14 15 16
17 19
NN
NH O
O
Me
Me
HOH
H
bis(epi)brevianamide E (20)
+
69%
93%
22%
84%
18:19 = 1:1.9
63%8:20 = 2:1
Scheme 4.4 Kametani’s total synthesis of deoxybrevianamide E and brevianamide E.
NH2HN
H
MeMe
1. Boc-proline,BOPCl
52%
2. TFA; NH3, MeOHHNHN
H
MeMe
N
O
OH
CH2Cl2, acetone
76%
NN
NH O
O
Me
Me
HOH
H
NN
NH O
O
Me
Me
HOH
H
brevianamide E (8)
bis(epi)brevianamide E (20)
deoxybrevianamide E (18)
NPhth
CO2Me
CO2Me
CO2Me
NH
H1. tBuOCl, Et3N
THF, #78 °C
NPhthHN
H
MeMe
95%
N2H4, EtOH, rt
65%
21 22
23
Me
Me
9-BBN2.
DMDO
Scheme 4.5 Danishefsky’s total synthesis of brevianamide E.
23 in 65% yield, which was coupled to N-Boc-l-proline, deprotected, and cyclizedto afford deoxybrevianamide E (18) in 52% yield. Compound 18 was elaboratedto brevianamide E (8) and bis(epi)brevianamide E (20) in a ratio of !1 : 5 upontreatment with dimethyldioxirane (DMDO) in a biomimetic oxidative cyclizationsequence reminiscent of the original Kametani work discussed above.
The structural similarities between deoxybrevianamide E and the then newlyisolated natural products tryprostatin A and B did not escape notice of Danishefsky
349

4.2 Prenylated Indole Alkaloids 121
and coworkers. While prenylation failed in attempts to use a reverse prenylboranenucleophile directly as in the method used to synthesize 22 above, a solutionwas found in treating chloroindolenine 24 with tri(n-butyl)prenylstannane andBCl3 to afford the desired prenyl functionality at C2 in excellent yield (Scheme 4.6).Phthalimide deprotection, peptide coupling, Boc-deprotection, and cyclization wereachieved to afford tryprostatin B (30) in 43% overall yield [7].
NPhth
CO2Me
CO2Me
HN
H tBuOCl,CCl4
NPhth
CO2MeCO2Me CO2Me
N
HCl
n-Bu3Sn
Me
Me+ BCl3
MeMe
BCl2
NPhthHN
H
MeMe
N2H4 H2O
82%
NH2HN
H
MeMe
N-Boc-L-Pro-FHNHN
H
MeMe
ONBoc
1. TMSI, MeCN
67%94%
HNHN
H
MeMe
N
O
O
H
tryprostatin B (30)
CH2Cl2
21 24
25 26
27 28
29
Et3N
MeOH, CH2Cl2
CH2Cl2, NaHCO3 2. NH3, MeOH
83%
•
Scheme 4.6 Biomimetic total synthesis of tryprostatin B.
Danishefsky and coworkers also completed the total synthesis of the spirooxin-dole spirotryprostatin B [8]. l-Tryptophan methyl ester was converted into theoxindole derivative 32, followed by addition of prenyl aldehyde under basicconditions to afford an inseparable four-component mixture of spirooxindoles(33–36, Scheme 4.7). Peptide coupling and subsequent treatment of the mix-ture with lithium bis(trimethylsilyl)amide (LHMDS) followed by selenylationpresumably gave phenyl selenide mixture 38. Oxidative elimination produceda mixture from which 39 was separated and elaborated to spirotryprostatin B (40)via Boc-deprotection and base-induced cyclization.
Danishefsky took a markedly different approach in the synthesis of spirotrypro-statin A [9]. A potentially biomimetic Pictet–Spengler reaction of tryptophanderivative 42 with thioaldehyde 41 as a masked isoprene equivalent gave thedesired cis-tetrahydrocarboline (43) with marginal selectivity (Scheme 4.8).N-Bromosuccinimide (NBS)-mediated oxidative rearrangement proceeded viaintermediate 44 to the oxindole was followed by deprotection of the carbamate togive amine 45. The modest yield of the sequence (57%) reflects the susceptibility ofthe oxindole to electrophilic aromatic bromination under the spiro-rearrangementconditions. Peptide coupling and Troc-deprotection resulted in cyclization to thedioxopiperazine, after which oxidation and sulfoxide elimination revealed theprenyl group to afford selectively spirotryprostatin A (48).
The synthesis of notoamide J was completed by Williams and coworkers, startingwith the Boc-protection of 7-hydroxyindole (Scheme 4.9) [10]. Chlorination at C3was followed by reverse prenylation of the resultant 3-chloroindolenine 51 to afford
350

122 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NH
CO2Me CO2Me
CO2Me
CO2MeCO2Me
CO2Me
CO2MeCO2Me CO2Me
NH2•HCl NH2•HCl
H
DMSO, 12N HCl
NH
H
O
Me
Me
CHO
Et3N, py.
NHHN
O
Me
Me
NHHN
O
Me
Me
H H
NHHN
O
Me
Me
H
NHHN
O
Me
Me
H
NBoc
CO2HH
BOPCl
73% (2 steps)2:3:3:3
NN
O
OMe
Me
HN
O
NHN
O
Me
H
OMe
NBoc
LHMDS NHN
O
Me
PhSe
OMe
NBoc
DMDO
NHN
O
Me
OMe
NBoc
1. TFA, CH2Cl2
2. Et3N
7% (5 steps)
31 32
33 34 35 36
37 38
39 spirotryprostatin B (40)
AcOH, PhOH
+ + +
PhSeCl
Scheme 4.7 Danishefsky’s synthesis of spirotryprostatin B.
52. The corresponding gramine was prepared by treating 52 with formaldehyde anddimethylamine, and subsequent Somei–Kametani coupling and imine hydrolysisgave tryptophan derivative 54 in good yield. Protection of the free amine as theBoc-carbamate and ester hydrolysis gave 55, which was coupled to proline ethylester in the presence of O-(7-azabenzotriazol-1-yl)-N,N,N",N"-tetramethyluroniumhexafluorophosphate (HATU) to afford amide 56. Cyclization to dioxopiperazine57 was followed by a biomimetic oxidation sequence accompanied by pinacol-typerearrangement to the oxindoles notoamide J (58) and 3-epi-notoamide J (59) in a2 : 1 separable mixture.
4.2.2Dioxopiperazine Derived from Tryptophan and Amino Acids other than Proline
Corey and coworkers designed a succinct synthesis of okaramine N, featuringa Pd-promoted dihydroindoloazocine formation [11]. Readily available trypto-phan derivative 60 was reduced to indoline 61 and subsequently prenylatedvia copper(I)-catalyzed alkylation with butyne 62 (Scheme 4.10). Treatment with
351

4.2 Prenylated Indole Alkaloids 123
PhS
H
Me
Me
O N H
CO
2Me
CO
2Me
CO
2Me
NH
2
MeO
H
1. T
FA
, 4Å
CH
2Cl 2
2. (
Boc
) 2O
MeC
N, E
t 3N
+N H
NB
oc
MeO
Me
Me
SP
h
H
74%
57%
(2
step
s)
NH
HNO
SP
h
Me
Me
CO
2Me
MeO
H
H
Tro
cN
ClO
CH
+1.
CH
2Cl 2
, Et 3
N
2. Z
no , TH
F, M
eOH
3.N
aIO
4, M
eOH
H2O
4. P
hCH
3,"
57%
(4
step
s)
NH
N
O
Me
MeO
H H
N
O OH
RhC
l 3. 3
H2O
EtO
H,"
45%
NH
N
O Me
Me
MeO
H H
N
O OH
spiro
tryp
rost
atin
A (
48)
N H
NB
oc
MeO
Me
Me
SP
h
HB
r
HO
H
NB
S, T
HF
AcO
H, H
2O
TF
A, C
H2C
l 2
2:1,
cis:
trans
41
4243
44
45
46
47
Sche
me
4.8
Dan
ishe
fsky
’ssy
nthe
sis
ofsp
irotr
ypro
stat
inA
.
352

124 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
N HH
O
Boc
2O, D
MA
P
MeC
NN H
Boc
O
NC
S
DM
FN H
Boc
O
Cl
Et 3
N, T
HF
N HB
ocO
Me M
e
HN
Me 2
, CH
2O
AcO
HN H
Me M
e
NM
e 2NC
O2E
t Ph Ph
1.
PB
u 3, M
eCN
,2.
1 M
HC
l, T
HF
N H
Me
Me
NH
2
CO
2Et
Boc
OB
ocO
1. B
oc2O
, 1M
NaO
Hdi
oxan
e
2. L
iOH
, TH
F/H
2O
N H
Me
Me
NH
Boc
CO
2H
Boc
O
Me
Me
9-B
BN
4950
5152
5354
55
N H
Me
Me
NH
Boc
ON
Boc
O
1. T
FA
, CH
2Cl 2
2. 2
-OH
-pyr
idin
eP
hCH
3N H
EtO
2C
EtO
2C
56
1. L
iOH
, TH
F/H
2O
2.
N H
Me
Me
N H
ON
OH
H
HO
57
N H
N H
ON
O
H
O
MeMe
HO
SNO
nB
u OO
H
noto
amid
e J
(58)
N H
N H
ON
O
H
O
MeMe
HO
H
59
+
70%
83%
48%
84%
76%
83%
59%
53%
cis:
trans
= 1
:1
CH
2Cl 2
47%
58:5
9 =
2:1
HA
TU
,iP
r 2N
Et
MeC
N
Sche
me
4.9
Will
iam
s’bi
omim
etic
synt
hesi
sof
noto
amid
eJ.
353

4.2 Prenylated Indole Alkaloids 125
CO2Me
CO2Me
CO2Me
NHBocNH
NaBH3CN,AcOH
60% NHBocNH
1. CuCl, iPr2NEt
87%
2. DDQ; H2,quinoline, Pd/C
MeOAcMe
NHBocN81%Me
Me
1. SOCl2
2. LiOH; Fmoc-Cl
CO2H
NHFmocNMe
Me64
60 61
63
62
Scheme 4.10 Synthesis of the N-reverse-prenylated tryptophan derivative.
2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) effected the dehydrogenation ofthe indoline, and the resulting tryptophan derivative 63 was deprotected, saponified,and reprotected as the N-Fmoc derivative (64).
The synthesis of okaramine N was completed through reductive amination of3-methyl-buten-2-al onto l-tryptophan methyl ester, followed by coupling of theresultant product (65) with acid 64 to form the desired tetracycle (Scheme 4.11).Treatment of 66 with Pd(OAc)2 provided the eight-membered ring (67) in modestyield (38%). The free amine obtained upon Fmoc cleavage underwent cyclization toform dioxopiperazine 68 in 95% yield. A potentially biomimetic oxidative cyclizationwas effected by treating 68 with N-methyltriazolinedione (MTAD), which reactedselectively with the N-unsubstituted indole subunit, and after photooxidation and
CO2Me
NH2NH
CO2Me
HNNH Me
Me
MeMe
CHODCM;
NaBH4, MeOH 70%
BOPCl,64
NH
NMeO2C
MeMe
O
FmocHN
N
MeMe
38%
Pd(OAc)2, O2
N O
FmocHN
N
MeMe
MeO2C
Me MeNH
95%
Et2NH
N
N
MeMe
Me MeNH
HNO
OH
H
70%
MTAD; O2,sunlamp,
methylene blue;
N
Me MeNH
NOO
HHMe2S; "
NMe Me
HOH
okaramine N (69)
31 65
66
67 68
Scheme 4.11 Completion of the total synthesis of okaramine N.
354

126 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
CO2Me
NH2NH
NaOH, Boc2O,
(n-Bu)4NHSO4
91%CO2Me
NHBocBocN
N-PSP,PPTS
93% BocN NBoc
CO2Me
SePh
BocN NBoc
CO2Me
SePh
H
+
71 (major) exo 72 (minor) endo
60%
MeOTf,2,6-di(t-
butyl)pyridine
Sn(n-Bu)3 BocN NBoc
CO2R
Me Me
18:1 (a :b)
83%
TMSI
NH NH
CO2Me
Me Me
NaOHTHF/MeOH/H2O
73 (a) R=Me
74 (a) R=H98%
1. 74, BOPCl,Et3N NH N
Me Me
2. TMSI34%
N NH
O
O H
H
MeMeamauromine (76)
31 70
75
Scheme 4.12 Danishefsky’s total synthesis of amauromine.
then reduction formed a hydroxylated octacycle that was directly converted intookaramine N (69) via thermolysis in 70% yield.
Amauromine was synthesized as shown in Scheme 4.12, starting with thebis(Boc) protection of l-tryptophan [12]. Conversion into the selenide by re-action with N-phenylselenophthalimide (N-PSP) in the presence of pyridiniump-toluenesulfonate (PPTS) afforded a mixture of 71 and 72 (9 : 1). Photolysis ofthe mixture in the presence of prenyltri(n-butyl)tin produced a mixture of reverseprenylated pyrrolodinoindolines, the desired major diastereomer of which wasseparated by crystallization from hexanes. The Boc groups were globally cleavedusing iodotrimethylsilane (TMSI) to afford the ester (75). Coupling of 75 withacid 74 gave the amide, which readily cyclized to the desired dioxopiperazine (76,amauromine) upon treatment with TMSI.
Danishefsky and coworkers completed a total synthesis of gypsetin in the samefashion as their effort on brevianamide E [7]. The reverse prenylated amine, 23,was synthesized as shown above (Scheme 4.5). Boc protection and cleavage of themethyl ester afforded acid 77, which was coupled to amine 23, deprotected, andcyclized to dioxopiperazine 78 (Scheme 4.13). Treatment with DMDO effected thebiomimetic oxidative conversion into the natural product gypsetin (79).
4.2.3Bicyclo[2.2.2]diazaoctanes
In 1970, Sammes proposed a hetero-Diels–Alder cycloaddition to be the biosyn-thetic origin of the bicyclo[2.2.2]diazaoctane core found in brevianamides A and B(Scheme 4.14) [13].
Support for this proposal was observed upon treating dihydroxypyrazine 82 withdimethyl acetylenedicarboxylate (83) or with norbornadiene (84) to give cycloadducts85 or 86, respectively (Scheme 4.15) [13].
355
4.2 Prenylated Indole Alkaloids 127
NPhth
CO2Me
NH
H1. t BuOCl, Et3N
THF, #78 °C
2. Me
Me 9-BBNNPhth
CO2Me
HN
H
MeMe
95%
N2H4, EtOH, rt
65% NH2
CO2Me
HN
H
MeMe
1. (Boc)2O, Et3N THF2. LiOH, MeOH, THF
94%
NHBoc
CO2H
HN
H
MeMe
1. BOPCl, CH2Cl2
2. TFA, CH2Cl23. NH3MeOH
87%
HNHN
H
MeMe
NH
O
O
NHMe
Me
HCH2Cl2,acetone
NN
NH
HN
O
O
Me
Me
Me
Me
HO
OH
H
H
gypsetin (79)40%
(+ 18% all syn-isomer + 20% double anti -isomer)
DMDO
2122
23
77
78
Scheme 4.13 Synthesis of gypsetin.
NN
O
OH
MeMe
O
NNH
O
MeMe
80 81
Scheme 4.14 Proposed hetero-Diels–Alder formation of bicyclo[2.2.2]diazaoctanes.
N
N OH
MeHO
PhH2CNH
HN O
O
MeO2C
CO2Me
Me
CH2Ph
NH
HN
O
OCH2Ph
Me
MeO2C CO2Me
DMF, r.t.
82
85
86
84
83
Scheme 4.15 Sammes’ model study of proposed cycloaddition.
Williams and coworkers expanded on the pioneering work of Sammes withthe following biosynthetic proposal for the brevianamides [5]. DeoxybrevianamideE (18) was thought to undergo oxidation to hydroxyindolenine 7, which couldundergo a pinacol-type rearrangement to indoxyl 87 (Scheme 4.16). Subsequenttwo-electron oxidation and enolization to azadiene 88, followed by an intramolecularhetero-Diels–Alder reaction was, following from the original proposal of Sammes,envisioned to give the natural products (+)-brevianamide A and (+)-brevianamide
356

128 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NH
NH
O N
OH H
MeMe
deoxybrevianamide E (18)
[ox]
N
NH
O N
OH H
MeMe
HOrearrangement N
H
O N
OH H
MeMe NH
O
2e# ox
NH+
O N
OMeMe NH
OFe3+
enolization
NNH+O
O#
MeMe
H
HN O
N
H+N
Me
H
HNO
#OMe
O
O
H
NNH
O(+)-brevianamide A (89)
NH
OMe
Me
O
H
NNH
O(+)-brevianamide B (90)
N
O
Me MeH
7 87
8888a 88b
Scheme 4.16 Biosynthetic proposal for the brevianamides.
B. The pseudo-enantiomorphic relationship between the two natural productswas envisaged to arise from the equilibrium between conformers 88a and 88b,which undergo cycloaddition to give 89 and 90, respectively. Ab initio studiesof the two transition states demonstrated that 88a is the more stable of thetwo, which is consistent with the observed product ratios of 89 and 90. Thetheoretical insights published by Domingo et al. lend support to the proposalof a biosynthetic intramolecular Diels–Alder reaction of intermediate 88 [14].Unfortunately, experimental support for this pathway has been elusive to secureand thus it remains a speculative biogenetic construction.
The total synthesis of d,l-brevianamide B demonstrated the first congru-ent application of a biomimetic Diels–Alder reaction used to form the bicy-clo[2.2.2]diazaoctane core common to the series of prenylated dioxopiperazines tobe discussed in this section [15]. 9-Epi-deoxybrevianamide E (19) was convertedinto the lactim ether (91) and oxidized to give the Diels–Alder precursor 92(Scheme 4.17). Treatment with aqueous methanolic KOH induced tautomeriza-tion to azadiene 93, which underwent a potentially biomimetic intramolecularDiels–Alder cycloaddition to give a mixture of diastereomers (94 and 95, 2 : 1).Oxidation, pinacol-type rearrangement, and lactim ether deprotection of the minordiastereomer (95) afforded brevianamide B (90) in 65% overall yield from 96. Thisstudy was one of the first to experimentally support the biogenetic origin of thecore bicyclo[2.2.2] ring system as arising via a dioxopiperazine that undergoes a nettwo-electron oxidation to an azadiene moiety.
357

4.2 Prenylated Indole Alkaloids 129
NH
MeMe
NHN
O
OH
NH
MeMe
NN
O
OMeH
NH
MeMe
NN
O
OMe
NH
MeMe
NN
O
OMe
MeOH
NN
MeMe
O 95
HN
MeO
H
NN
MeMe
O94
HN
MeOH
NN
MeMe
O
96
N
HO O
H
NNH
O(±)-brevianamide B (90)
N
O
MeMeH
Me3OBF4KOH
MeOH
H2O
9119 92 93
60%94:95 = 2:1
+
1. mCPBA
2. HCl, 99%
1. NaOMe
2. HCl, 65%
69%
DDQ
40%
Scheme 4.17 Application of the proposed biomimetic Diels–Alder reaction.
Williams and coworkers also applied the intramolecular Diels–Alder reaction tothe racemic synthesis of VM55599 [16]. The reverse-prenylated tryptophan deriva-tive 97 was coupled to !-methyl-!-hydroxyproline ethyl ester 98 to afford dipeptide99 (Scheme 4.18). N-Boc deprotection and cyclization afforded dioxopiperazine 100,and elimination with thionyl chloride gave enamide 101. Formation of the lactimether (102) and treatment with aqueous KOH gave azadiene 103, which sponta-neously suffered intramolecular Diels–Alder reaction to give a separable mixture ofall four possible racemic diastereomers (104–107, 2.6 : 3.7 : 1.0 : 1.6, respectively).Cleavage of the lactim ether and diisobutylaluminum hydride (DIBAL-H) reductiongave VM55599 in 73% yield from 105.
The synthesis of VM55599 allowed for assignment of the absolute stereochem-istry of the molecule, which places the methyl group at the !-position of theproline residue syn- to the bridging isoprene moiety [16]. In stark contrast, theanalogous methyl group of paraherquamide A is anti to the bridging isopreneunit. Scheme 4.19 outlines a possible unified biosynthesis for VM55599 and para-herquamide A, arising from dimethylallyl pyrophosphate (DMAPP), l-isoleucine,and l-tryptophan. If a Diels–Alder cycloaddition is to be invoked, approach of theisoprene moiety must occur from the same face as the methyl group on the prolinering for synthesis of VM55599, and from the opposite face to the methyl group togive paraherquamide A. The diastereofacial selectivity of the Diels–Alder reactiongave a preponderance of the syn-relative stereochemistry alpha to the gem-dimethylgroup in both molecules. As VM55599 is a very minor metabolite of Penicilliumsp. IMI332995, it is plausible that cyclization of 112#114 is preferred and furthermetabolization gives paraherquamide A, whereas the minor cycloaddition via 113produces VM55599 as a dead-end shunt metabolite. As shown in Scheme 4.18
358

130 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NH
CO2HBocHN
MeMe
BOPCl
TFA$HN
EtO2CMeHO
98
83%NH
BocHN
Me Me
ON
CO2Et
MeOH
1. TFA
2. 2-OH-pyr.", 95%
97 99
NH
MeMe
NHN
O
O
100
MeOH
SO2Cl, py.
75%
NH Me
Me
NHN
O
O
101
Me
Me3OBF4
72%
NH
MeMe
NN
O
OMe
102
Me
KOH
NH
MeMe
NN
O
OMe
103
Me
MeO
NN
HN MeMe
O
HH
Me
MeO
NN
HN MeMe
O
HH
Me
MeO
NN
HN MeMe
O
MeH
H
MeO
NN
HN MeMe
O
MeH
H
O
H
NN
HN Me
Me
HMe
H
VM55599 (108)
1. HCl2. PhCH3, "
3. DIBAL-H73%104
106
105
107
104:105:106:107 = 2.6 : 3.7 : 1.0 : 1.6
Scheme 4.18 Williams’ biomimetic total synthesis of VM55599.
HN
HO2C
H2N
Me
Me
PPO
CO2H
NH2
Me
Me
DMAPP (109)
L-Ile (110) L-Trp (111)
OH
NN
HNMe Me
HMe
O
H
NN
HN MeMe
HMe
H
X
HO
NN
HN MeMe
MeH
X
VM55599 (108)minor
O
H
NN
HNMe
Me
HH
Me
X
syn-selective [4+2]
OMe
NHO
NN
MeH
HMeMe
O
O
MeMe
[ox]SAM
DMAPP
114major
paraherquamide A (115)
112 113
Scheme 4.19 Proposed biosynthesis of paraherquamide A and VM55599.
359

4.2 Prenylated Indole Alkaloids 131
above, the intrinsic diastereofacial bias of the Diels–Alder reaction is modest atbest, giving a slight excess (1.47 : 1) of cycloaddition from the same face as themethyl group, and favoring the syn-relative stereochemistry to the extent of 2.4 : 1.Such observations suggest that the biosynthesis may rely on protein organizationof the precyclization conformers to stereoselectively produce the syn-isomers.
A report from Liebscher and coworkers showed promise in terms of improvingthe diastereoselectivity of the Diels–Alder cycloaddition, using neutral conditionsto prepare the azadiene in contrast to the basic conditions employed by Williams[17]. Compound 118 was prepared by a Horner–Wadsworth–Emmons reactionof aldehyde 116 with phosphonate 117 (Scheme 4.20). Treatment of 118 withneat acetyl chloride for 20 days gave the Diels–Alder product 119 as a singlediastereomer in 48% yield.
NMOM
CHO
MeMe
HNN
O
OP
(MeO)2
O
H117
KOtBuN
NH
NO
O
MeMe
118
H
116
AcCl
20 d O
H
NN
O
HN MeMe
H
119one diastereomer
MOM48%
Scheme 4.20 Liebscher’s Diels–Alder work.
The precedent set by Liebscher’s work was applied to an asymmetric totalsynthesis of VM55599 [18]. The loss of stereochemistry observed at the prolinemethyl group in 101 above led Williams to employ a dehydrotryptophan derivativeas the Diels–Alder azadiene precursor, allowing for the preparation of VM55599 inan enantioselective fashion. Williams has demonstrated that the !-methylprolineresidue of paraherquamide A and VM55599 are biosynthetically derived from l-Ile.In a biomimetic construct, l-Ile was converted into optically pure !-methylproline120 using Hoffman–Loffler–Freytag conditions in 45% yield (Scheme 4.21).
MeMe
CO2HH2NNBoc
Me
CO2Et
45%L-Ile (110) 120
S
S SS LiOH
96%NBoc
Me
CO2H
121
HNN
O
O
Me
122
1. EDCI, HOBtEt3N
ClH3N CO2Me
2. TFA; Et3N3. PhCH3, "
88%
NaH,MeSCH2Cl
62%
NN
O
O
Me
123
SMe N
CHO
MeMe
NaHMDS87%
116
NMe Me
124
NN
MeO
O
MeSHO
H
NH
MeMe
125
N
NH
OO
Me
1. MeI, NaHCO3, 92%
2. 50% aq.
MOM
MOMHCO2H, 70%
Scheme 4.21 Key dioxopiperazine synthesis.
360

132 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NH
MeMe
125
N
NH
OO
Me
AcCl
14 days
NH
MeMe
126
N
NAcO
O
MeH
NH Me
Me
127
N
NAcO
O
Me
H+
O
H
NN
O
HN Me
Me
HMe
H
O
H
NN
O
HN MeMe
HMe
H
S
SO
H
NN
O
HNMe
Me
HH
Me
O
H
NN
O
HNMe
Me
HH
Me
S
S129 (35%)
131 (15%)
128 (10%)
130 (0%)
O
H
NN
HN Me
Me
HMe
HS
(–)VM55599 (108)
DIBAL-H
68%
Scheme 4.22 Completion of the asymmetric total synthesis of ($)-VM55599.
Cleavage of the ethyl ester, peptide coupling to glycine methyl ester hydrochloride,Boc deprotection, and cyclization gave dioxopiperazine 122 in good overall yield.Protection of the secondary amide and subsequent condensation with aldehyde 116gave an epimeric mixture of dioxopiperazines, which gave selectively (Z)-isomer125 following deprotection and dehydration.
Following Liebscher’s protocol, 125 was treated with acetyl chloride for 14 days,yielding a mixture of three diastereomers (Scheme 4.22). The reaction is thoughtto proceed by initial acylation to the O-acyl lactim 126, tautomerization to azadiene127, which suffers intramolecular Diels–Alder reaction from three of the four pos-sible diastereomeric transition states, followed by loss of acetate to give compounds128–131. The major diastereomer 129 was treated with excess DIBAL-H to effectreduction to ($)-VM55599 (108). Interestingly, cycloadduct 130 was not observedfrom the cycloaddition reaction. Penicillium sp. produces paraherquamide A inlarge excess over VM55599 (>600 : 1), so it is surprising that this provocative bio-genetic precursor to paraherquamide A is not observed in laboratory cycloadditionreactions. Despite the structural differences between the laboratory and biologicalDiels–Alder precursors, the intrinsic facial selectivity of the cyclization does notappear to mimic the bias toward paraherquamide stereochemistry one wouldexpect given the observed product ratios of the fungal metabolites.
As many of the bicyclo[2.2.2]diazaoctane natural products differ only at thesubstitution of the indole ring, a convergent approach utilizing a Fischer indolesynthesis was undertaken in an alternative racemic synthesis of brevianamideB [19]. This work had the objective of validating other possible biosynthetic pathways
361

4.2 Prenylated Indole Alkaloids 133
O
Me Me
S S
EtO2C H
nBuLi, CuI
76%132
O
Me Me
134
EtO2CS S
LiOH
99%
O
Me Me
135
HO2CS S
NH
CONH2
H
BOPCl, iPrNEt
77%
136
O
Me Me
137
S SN
O
O NH2H
NCS, AgNO3
88%
138:139 = 45:43
O
Me Me
138
N
O
O NH2H
O
AlCl3
EtOAc
48%
NNH
O
O O
Me Me
H
+
139
O
H
NN
Me Me
H
O
O
140
1. PhNHNH2
2. ZnCl2, 58%O
H
NN
Me Me
H
O141
HN
1. mCPBA
2. NaOH, 16%O
H
NNH
O(±)-brevianamide B (90)
N
O
Me MeH
133
Scheme 4.23 Concise synthesis of brevianamide B.
to reach the oxidation state of the azadiene. In the present instance, the "-ketoamidespecies (138) was posed to serve as a surrogate for the possible biosynthetic oxidativedeamination of the tryptophan moiety and coupling to a proline amide species(Scheme 4.23). Conjugate addition of carboxylate 133 to ketone 132 gave ester 134in 76% yield. Saponification, peptide coupling with l-proline amide, and dithianedeprotection gave a mixture of the uncyclized amide 138 and dioxopiperazine139. Aluminum trichloride was added to the mixture to give the Diels–Aldercycloadduct (140) in exclusively the anti-configuration, whereas mixtures of boththe syn- and anti-cycloadducts were observed in previous syntheses of VM55599and brevianamide B discussed above. A Fischer indole synthesis was completed bytreatment of 140 with phenyl hydrazine followed by ZnCl2, affording 141 in goodyield. Oxidation and pinacol-type rearrangement of this known intermediate gavebrevianamide B. While an appealing convergent approach towards the synthesisof other related bicyclo[2.2.2]diazaoctanes, the utility of this strategy is limited tothose natural products containing the anti-stereochemistry observed in formationof compound 140, the remainder of which must be able to withstand the harshconditions of the Fischer indole synthesis.
The same biomimetic Diels–Alder disconnection was exploited in the total syn-thesis of stephacidin A [20]. Starting with reverse prenylated tryptophan derivative142 (prepared in eleven steps from 6-hydroxyproline), dipeptide 144 was preparedthrough bis(2-oxo-3-oxazolindinyl)phosphinic chloride (BOPCl) mediated coupling
362

134 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
step
haci
din
A (
150)
OH
NN
O
H NM
eM
eO
Me
Me
N HO
Me M
e
N H
N
HHO
O
OH
Me
Me
145
N HO
Me M
e
N H
NHO
O
Me
Me
146
N HO
Me M
e
N
NHO
OM
e
Me
Me
147
MeOH
NN
O
H NM
eM
eO
Me
Me
N HO
Me M
e
NH
Fm
oc
NHO M
eM
e
144
EtO
2CO
H
N HO
Me M
e
NH
Fm
oc
CO
2H
Me
Me
142
N H·H
Cl
EtO
2CHO
143
BO
PC
l,iP
rNE
t 254
%
N HO 95%
PB
u 3, D
EA
D
91%
Me 3
OB
F4
Cs 2
CO
3
81%
KO
H, M
eOH
86%
148:
149
= 1:
2.4
148
MeOH
NN
O
H NM
eM
eO
Me
Me
149
+H
HC
l
96%
Sche
me
4.24
Synt
hesi
sof
step
haci
din
A.
363

4.2 Prenylated Indole Alkaloids 135
with cis-3-hydroxyproline ethyl ester (143, Scheme 4.24). Fmoc deprotection resultedin the cyclization to dioxopiperazine 145, which upon treatment with tributyl phos-phine and diethyl azodicarboxylate (DEAD) underwent Mitsunobu dehydration togive enamide 146. Formation of the lactim ether (147) was followed by intramolec-ular Diels–Alder reaction to give a mixture of epimers enriched with the syn-isomer(149, 2.4 : 1). Deprotection of 149 gave stephacidin A (150) in excellent yield.
Williams found that the bicyclo[2.2.2]diazaoctane core could be accessed directlyfrom compound 145 by treating it with excess PBu3 and DEAD, effecting the dehy-dration, tautomerization, and Diels–Alder reaction in one pot to afford stephacidinA and its epimer (Scheme 4.25) [21].
NHO
MeMe
NH
N
HHO
O
OH
Me Me
145
PBu3
DEAD
40 °C64%
stephacidin A (150)
O
H
NN
O
HN MeMeO
Me
Me
H
+ epi
150:epi = 2.4:1
Scheme 4.25 Improved biomimetic synthesis of stephacidin A.
Myers and coworkers, in the course of their total synthesis of avrainvillamide,discovered that synthetic ($)-152 spontaneously dimerized to stephacidin B underseveral mild conditions, including addition of triethylamine or exposure to silica gel[22, 23]. Baran and coworkers later determined that stephacidin A could be readilyconverted into avrainvillamide through reduction to indoline 151 followed bySomei oxidation (Scheme 4.26). In accord with Myers’ observations, dimerizationto stephacidin B occurred readily upon exposure to silica gel, triethylamine,or on evaporation from dimethyl sulfoxide (DMSO) [24–26]. Myers has furtherdemonstrated that the observed biological activity of stephacidin B may be due tothe formation of 152 from 153 in vivo [22].
Stephacidin A is of particular biogenetic interest, as both enantiomers havebeen isolated in nature: (+)-stephacidin A from Aspergillus ochraceus [27] andfrom a marine-derived Aspergillus sp. [28] and ($)-stephacidin A from terrestrialAspergillus versicolor [29–31]. Operating under the assumption that the biosynthe-sis of stephacidin A proceeds through a common, achiral intermediate, Williamsand coworkers have proposed notoamide S (154) as the point of divergence in thetwo biosyntheses [32]. Oxidation of 154 could give achiral azadiene 155, whichis postulated to undergo intramolecular Diels–Alder reaction to give either (+)-or ($)-stephacidin A depending on the enantiofacial selectivity of the reaction(Scheme 4.27).
Tryptophan derivative 142 used in the above synthesis of stephacidin A was alsoemployed in the biomimetic total synthesis of marcfortine C (164) [33]. Pipecolicacid derivative 156 was coupled to acid 142 to form amide 157, which underwentcyclization to the dioxopiperazine following Fmoc-deprotection to give 158 as
364

136 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
stephacidin A (150)
O
H
NN
O
HN MeMeO
MeMe
H
N O
MeMe
Me Me
NNO
O
H
N
O MeMe
MeMe O
OH
H
HNO
N O
H
O
H
NN
O
HN MeMeO
MeMe
H
O
H
NN
O
N MeMeO
MeMe
H
O
avrainvillamide (152)
stephacidin B (153)
NaBH3CN
AcOH
SeO2, H2O2
SiO2
151
Scheme 4.26 Biomimetic conversion of stephacidin A into stephacidin B.
(#)-stephacidin A, (#)-150
O
H
NN
O
HN MeMeO
Me
Me
H
(+)-stephacidin A, (+)-150
O
H
NN
O
HNMe
Me O
Me
Me
H
NH
NH
NO
OH
H
Me MeHO
MeMe
NH
N
NO
OH
Me MeHO
MeMe
notoamide S (154)
[ox]
Aspergillusversicolor
Aspergillussp.
IMDA
[ox]
155(achiral)
Scheme 4.27 Proposed biosynthesis of (+)- and ($)-stephacidin A through notoamide S.
an inconsequential mixture of diastereomers (Scheme 4.28). The biomimeticDiels–Alder reaction preferred the syn-diastereomer 160 in 2.4-fold excess, asexpected. Excess DIBAL-H selectively reduced the tertiary amide of 160, and aminesalt formation followed by a biomimetic oxidative rearrangement gave marcfortineC (164).
Malbrancheamide and malbrancheamide B were synthesized via the biomimeticDiels–Alder reaction of enamides 165 and 166 to afford both syn-cycloadducts 167
365

4.2 Prenylated Indole Alkaloids 137
N H
CO
2H
NH
Fm
oc
OM
eM
e
Me
Me
N HC
O2E
t
OH
N H
NH
Fm
oc
ON
EtO
2C
OM
eM
e
Me
Me
OH
N HO
Me
Me
Me
Me
N H
NO
OHH
OH
Bu 3
P, D
EA
D
40°C
60%
159:
160
= 1:
2.4
+OH
NN
O
H NM
eM
eO
Me M
e
OH
NN
O
H NM
eM
eO
Me M
e
HH
OH
NN
H NM
eM
eO
Me M
e
H
NS
OnB
u OO
OH
NN
H NM
eM
eO
Me M
e
H
H OT
s
+m
arcf
ortin
e C
(16
4)
OH NNH
NH
Me
Me
O
OM
e
Me
142
156
157
158
159
160
BO
P,i
Pr 2
NE
t77
%94
%O N H
DIB
AL-
H
89%
PP
TS
77%
161
162
163
Sche
me
4.28
Tota
lsy
nthe
sis
ofm
arcf
ortin
eC
.
366

138 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NHCl
NH
NHO
O
Me Me
X
165: X = Cl166: X = H
O
H
NN
HNMe Me Cl
H Cl
malbrancheamide B (172)
O
H
NN
HNMe Me Cl
H
O
H
NN
HNMe Me Cl
H Cl
O
H
NN
HNMe Me Cl
H
O
O
DIBAL-H
80%
DIBAL-H
74%
X = Cl
89%167:168 = 2:1
167
+ epi (168)
169
+ epi (170)
X = H
86%169:170 = 1.8:1
KOH
MeOH
malbrancheamide (171)
Scheme 4.29 Completion of the total syntheses of the malbrancheamides.
and 169, as well as the anti-epimers 168 and 170 (Scheme 4.29) [34]. Treatmentof the syn-cycloadducts with excess DIBAL-H gave either malbrancheamide ormalbrancheamide B.
As the malbrancheamides were the first of this family of prenylated indolealkaloids to possess a halogenated indole ring, Williams and coworkers probed thebiosynthesis to establish the timing of the chlorination event [35]. Malbrancheamideis proposed to arise from tryptophan, proline, and dimethylallyl diphosphate,leading to deoxybrevianamide E (18, Scheme 4.30). Oxidation of 18 could giveintermediate 174, which is expected to suffer intramolecular Diels–Alder reaction
NH
CO2H
NH2
Me
Me
OPP
HN
HO2C
NH
NH
NO
OH
H
Me Me NH
N
NX
OH
Me Me
[ox]
O
H
NN
HNMe Me
H
X
IMDA
174, X = O175, X = H2
176, X = O
redO
H
NN
HNMe
Me
Hhalogenase
O
H
NN
HNMe Me
H
Cl
malbrancheamide B (172)
halogenaseO
H
NN
HNMe Me
H
Cl
malbrancheamide (171)
Cl
111
173
109 18
177
Scheme 4.30 Proposed biosynthesis of the malbrancheamides.
367

4.2 Prenylated Indole Alkaloids 139
ON H
OM
e Me
HN
NO
O
Me
Me
H
OH
H
178
ON H
OM
e Me
HN
NO
O
Me
Me
H
180
ON H
O
Me M
e
HN
NO
O
Me
Me
H
OH
H
179
ON H
O
Me M
e
HN
NO
O
Me
Me
H
181
+
CH
2Cl 2
80%
178:
179
= 3:
1
DE
AD
, PB
u 3
CH
2Cl 2
71%
DE
AD
, PB
u 3
CH
2Cl 2
78%
163S
N
nBu
O
OO
N HO
Me M
e
N H
N
HHO
O
OH
Me
Me
145
Sche
me
4.31
Synt
hesi
sof
key
oxin
dole
s.
368

140 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
(IMDA) to cycloadduct 176. Reduction of the tertiary amide would provide premal-brancheamide (177). Alternatively, 18 could suffer reduction of the tertiary amideto 175, providing 177 directly upon cycloaddition. Premalbrancheamide is pro-posed to undergo subsequent halogenation events to give both malbrancheamideB and malbrancheamide. In feeding studies, labeled dioxopiperazine 176 andpremalbrancheamide (177) were added to separate cultures of Malbranchea au-rantiaca, but interestingly only premalbrancheamide 177 was incorporated intomalbrancheamide B. This suggests that reduction of the tertiary amide mustprecede Diels–Alder construction of the bicyclo[2.2.2]diazaoctane core through anintermediate analogous to monooxopiperazine 175.
Williams has reported that–like stephacidin A and notoamide B, which areproduced in Nature as distinct enantiomers–the minor metabolite versicolamideB is likewise produced as distinct enantiomers in different strains of Aspergillus sp.The asymmetric syntheses of both (+)- and ($)-versicolamide B (182) have recentlybeen accomplished by deploying compound 145, previously used in the synthesisof stephacidin A (Scheme 4.31). Oxaziridine oxidation of 145 and pinacol-typerearrangement consequent to oxidation of the indole gave a 3 : 1 separable mixtureof 178 and 179 [36]. As previously reported, treatment with tributyl phosphine andDEAD induced Mitsunobu dehydration to give enamides 180 and 181.
Treatment of 180 and 181 individually with potassium hydroxide in methanolinduced the intramolecular Diels–Alder reaction to afford either (+)- or($)-versicolamide B (182), along with the minor anti-diastereomers (+)-183 or($)-183 (Scheme 4.32). As previous biomimetic Diels–Alder reactions containingindole-based azadienes display syn-selectivity (typically !2.5 : 1 syn : anti), theexclusive selectivity for the anti-products in the versicolamide B syntheses is ofparticular interest. The authors suggest that the anti-preference stems from astable transition state leading to the anti-cycloadduct when an oxindolic azadieneis employed, whereas the syn- and anti-transition states arising from an indolicazadiene are of roughly equal stability [14, 37].
O NH
OMe
Me
HN
NO
O
MeMe H
180
O NH
O
Me
Me
HN
NO
O
MeMe H
181
KOH
MeOH75%
182:183 = 1.4:1
KOH
MeOH72%
182:183 = 1.4:1
(+)-versicolamideB, (+)-182
O
NHO
NN
OMe
Me
MeMe
O
O
HNO
NN
OMe
Me
MeMe
O
(+)-183
H H+
H H
(#)-versicolamide B, (#)-182 (#)-183
O
HNO
NNO
MeMe
MeMe
O
O
NHO
NN O
MeMe
MeMe
O
H H+
H H
Scheme 4.32 Completion of the asymmetric syntheses of the versicolamides.
369

4.3 Non-prenylated Indole Alkaloids 141
While the above syntheses have differed in the approach to the azadiene, allshare a common biomimetic intramolecular Diels–Alder reaction to give thebicyclo[2.2.2]diazaoctane core featured in the molecules presented in this section.
4.3Non-prenylated Indole Alkaloids
Hart and coworkers have expressed interest in the biosynthesis of the fumiquina-zoline family of alkaloid natural products, and reported a biomimetic synthesisof ent-alantrypinone as the initial effort at a synthetic program to access morecomplex, related substances [38]. l-Tryptophan methyl ester was coupled to isatoicanhydride to afford amide 184 in good yield (Scheme 4.33). Acylation of 184 withacyl chloride 186 (prepared in two steps from S-methyl-l-cysteine, 185) underSchotten–Baumann conditions furnished diamide 187, which underwent cyclode-hydration to iminobenzoxazine 188. Treatment with excess Li[Me3AlSPh] effectedthe rearrangement to quinazolinone 189, and Fmoc deprotection was accompaniedby amide formation to give 190. Oxidation of 190 provided the sulfoxide, which wasconverted into enamide 191 upon heating in benzene with triphenylphosphine.Trifluoroacetic acid induced the conversion into bicycle 192, presumably throughintramolecular electrophilic attack of an intermediate N-acyliminium ion ontoindole. Conversion into the oxindole proceeded via oxidative rearrangement of 192with NBS to give the polybrominated indolinone, which was hydrogenolyzedover platinum on carbon to give ent-alantrypinone (193), along with ent-17-epi-alantrypinone (194).
Three dimeric tryptophan-derived dioxopiperazines have succumbed tobiomimetic total syntheses, all completed by Movassaghi and coworkers, namely,(+)-WIN 64821, ($)-ditryptophenaline, and (+)-11,11"-dideoxyverticillin A [39, 40].Syntheses of the former two began with cleavage of the Boc carbamate to effect thecyclization to dioxopiperazine 196 (Scheme 4.34) [40]. Treatment with brominethen gave a separable mixture of two diastereomers, endo-(+)-197 and exo-($)-198.endo-Bromide (+)-197 was carried on to (+)-WIN 64821 (201) by, first, treatmentwith tris(triphenylphosphine)cobalt chloride to afford the dimerized product 199,which was globally deprotected upon exposure to samarium diiodide. (+)-WIN64821 was thus obtained in 75% yield. Similarly, ($)-ditryptophenaline (202) wassynthesized following methylation of exo-($)-198, dimerization, and deprotectionto give the product in 79% yield.
4.3.1Epidithiodioxopiperazines
(+)-11,11"-Dideoxyverticillin A (211) differs from dimers 201 and 202 in that it islikely derived from l-tryptophan and l-alanine, rather than from l-phenylalanine.Additionally, the dioxopiperazines are bridged by a disulfide, adding a difficultchallenge to the synthetic construction of the molecule. Similar to their previous
370

142 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
isat
oic
anhy
drid
e
97%
CO
2Me
NH
2N
H"
96%
NH
N H
MeO
2CO
NH
2
NH
N H
MeO
2CO
HN
NH
Fm
ocSM
e
HO
ON
H2
SM
e1.
Fm
ocC
l
89%
2. S
OC
l 2C
l
ON
HF
moc
SM
e
DC
M
H2O
, NaH
CO
3
80%
Ph 3
P, I
2, D
CM
76%
Li[M
e 3A
lSP
h]
NH
N
MeO
2CN
O
Fm
ocH
NS
Me
NH
N
MeO
2C
N
O
Fm
ocH
NS
Me
94%
pipe
ridin
e
NH
NN
O
H NS
Me
O
79%
1.m
CP
BA
2. P
h 3P
,"
NH
NN
O
H NO
89%
TF
A
NH
NN
OO
Me
NH
74%
193:
194
= 1:
1.5
1. N
BS
, TH
F,
TF
A, H
2O
NH
NN
OO
Me
2. H
2, P
t/C
NH
NN
OO
Me
HN
O
NH
O+
ent-1
7-ep
iala
ntry
pino
ne (
194)
ent-a
lant
rypi
none
(19
3)
O
3118
4
185
186
187
188
189
190
191
192
iPr 2
NE
t
Sche
me
4.33
Synt
hesi
sof
ent-a
lant
rypi
none
.
371

4.3 Non-prenylated Indole Alkaloids 143
NH
BocO
HN
PhS
O2N
TF
A;
mor
phol
ine
80%
PhS
O2N
86%
HN
NH
O O
Br 2
N SO
2Ph
SO
2PhN
NH
Br
H
O O
48%
CoC
l(PP
h 3) 3
OM
eO
N SO
2PhN
NH
H
O O
SO
2Ph
NN
HN
H
OO
75%
Sm
I 2, N
MP
N H
NN
H
H
O O
H NN
HN
H
OO (+)-
WIN
648
21(2
01)
NN
NH
Br
H
O O
197
48%
2. C
oCl(P
Ph 3
) 3
NN
NM
e
H
O O
SO
2Ph SO
2Ph
NN
MeN
H
OO
79%
Sm
I 2, N
MP
N H
NN
Me
H
O O
H NN
MeN
H
OO
(#)-
ditr
ypto
phen
alin
e (2
02)
198
1. M
eI
195
196
197
198
199
200
Sche
me
4.34
Con
cise
tota
lsy
nthe
ses
of(+
)-W
IN64
821
and
($)-
ditr
ypto
phen
alin
e.
372

144 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
NHBocO
HN
Me
CO2Me
PhSO2N
TFA;morpholine
84%PhSO2N
59%
HNNH
O
OMe
1. Br2
NSO2Ph
NNMeBr
H
O
OMe2. MeI
46%
CoCl(PPh3)3
NSO2Ph
NNMe
H
O
OMe
SO2PhN
NMeN
H
O
OMe
63%
Py2AgMnO4
NSO2Ph
NNMe
H
O
OMe
SO2PhN
NMeN
H
O
OMe
HO
OHHO
OH55%
TBSCl
NSO2Ph
NNMe
H
O
OMe
SO2PhN
NMeN
H
O
OMe
TBSO
OHHO
OTBS
PPY
87%
Na(Hg)
NH
NNMe
H
O
OMe
HN
NMeN
H
O
OMe
TBSO
OHHO
OTBS 56%
1. K2CS3, TFA
2. ethanolamine
NH
NNMe
H
O
OMe
HN
NMeN
H
O
OMe
HS
SHHS
SH 87%
KI3
pyridine
NH
NNMe
H
O
O
HN
NMeN
H
O
OMe
Me
SS
SS
(+)-11,11%-dideoxyverticillin A (211)
203 204 205
206 207
208 209
210
Scheme 4.35 Biomimetic total synthesis of (+)-11,11"-dideoxyverticillin A.
work, Movassaghi and coworkers showed that cleavage of the N-Boc carbamate wasaccompanied by cyclization to dioxopiperazine 204 (Scheme 4.35) [39]. Exposure of204 to bromine produced the 3-bromopyrroloindoline, and the amides were sub-sequently methylated upon treatment with iodomethane. Reductive dimerizationwith the cobalt(I) complex as before gave the desired dimeric intermediate 206. Thedimer was oxidized with bis(pyridine)-silver(I) permanganate to octacycle 207, andexposure to Fu’s (R)-(+)-4-pyrrolidinopyridinyl(pentamethylcyclopentadienyl)iron(PPY) catalyst with t-butyldimethylsilyl chloride (TBSCl) gave selectively thealanine-derived protected hemiaminals of 208. Removal of the benzenesulfonylgroups with sodium amalgam revealed diaminodiol 209. Treatment of 209 withK2CS3 followed by ethanolamine gave diaminotetrathiol 210, which readily oxi-dized to (+)-11,11"-dideoxyverticillin A (211) when partitioned between aqueoushydrochloric acid and dichloromethane and treated with potassium triiodide.
373

4.3 Non-prenylated Indole Alkaloids 145
The total synthesis of sporidesmin A was completed by Kishi and coworkersin 1973. In a series of communications, Kishi described a novel strategy for thesynthesis of epidithiodioxopiperazines using a dithioacetal moiety as a protectinggroup for the disulfide bridge [41–43]. Thus protected, the dithioacetal is stableto acidic, basic, and reducing conditions, allowing for the introduction of thiolgroups at an early stage in a total synthesis. Synthesis of the sporidesmins be-gan with the treatment of dioxopiperazine 212 with the dithiane derivative ofp-anisaldehyde in the presence of acid to afford dithioacetal-protected dioxopiper-azine 213 (Scheme 4.36) [43]. Condensation with acid chloride 214 and subsequentmethoxymethyl deprotection gave compound 215. Treatment of ketone 215 withDIBAL-H at $78 %C resulted in stereoselective reduction to the alcohol, which wasthen converted into acetate 216 in 80% yield. Cyclization to the diacetate (217)proceeded upon addition of iodosobenzene diacetate, and hydrolysis of the acetatesgave the corresponding diol. Treatment of the diol with m-chloroperbenzoic acid(mCPBA) afforded an intermediate sulfoxide, which decomposed to the disulfideupon exposure to strong Lewis acid, revealing (±)-sporidesmin A (218).
NNMe
O
O
MeO
AcS
p-MeOC6H4CH2S2H2,H+
NMe
COClCl
MeOOMe
+
50%
1. BuLi2. HCl, THF
3. NaOH
NMe
Cl
MeOOMe
O
HNNMe
O
OMe
SS Ar
NNMe
O
OMe
SS ArMeO
80%
1. DIBAL-H2. Ac2O
NMe
Cl
MeOOMe
HNNMe
O
OMe
SS Ar
AcO
30%
iodosobenzenediacetate
NMe
Cl
MeOOMe
N
25%
1. NaOH, MeOH2. mCPBA
H
AcOAcO
3. BF3$Et2O
(±)-sporidesmin A (218)
212 213 214
215 216
217
NMe
O
OMe
SS Ar
NMe
Cl
MeOOMe
NH
HOHO
NMe
O
OMe
SS
Scheme 4.36 Total synthesis of (±)-sporidesmin A.
The biosynthesis of gliotoxin is believed to proceed through the intramolecularnucleophilic ring-opening of a phenylalanine-derived arene oxide and has beenthe subject of considerable speculation and interest. Kishi and coworkers drewinspiration from these biogenetic hypotheses in devising a brilliant total synthesisof gliotoxin. The total synthesis of (±)-gliotoxin was completed in 1976 utilizingthe same disulfide protecting strategy as deployed above for the sporidesmins,and was re-engineered in 1981 by the same route starting from optically pure
374

146 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
HNN
O Triton B
O Me
SS Ar
O
O
OtBu+N
N
O
O Me
SS Ar
OOtBu
HO NN
O
O Me
SS Ar
OOtBu
HO+
1. Ac2O2. TFA
3. ClCO2Et4. NaBH2
77%
98%
NN
O
O Me
SS Ar
OH
AcO
1. MsCl2. LiCl
3. NaOMe90%
NN
O
O Me
SS Ar
Cl
HO
PhLi,PhCH2OCH2Cl
53%
NN
O
O Me
SS ArHO H
OCH2OPh
1. BCl32. mCPBA
35%
NN
O
O Me
SSHO H
OH
3. HClO4
219 220221 222
221:222 = 2:1
223 224
225 (+)-gliotoxin (226)
Scheme 4.37 Kishi’s total synthesis of (±)- and (+)-gliotoxin.
dithioacetal 219 obtained from resolution (Scheme 4.37) [44, 45]. Coupling of 219with t-butoxy arene oxide 220 in the presence of triton B afforded 221 and 222 in a2 : 1 ratio. Acylation, deprotection, mixed anhydride formation, and reduction gavealcohol 223 in 77% yield. Alcohol 223 was converted into the chloride followingmesylation, and then deprotected to reveal alcohol 224. The key stereoselectivecyclization–alkylation reaction was achieved upon addition of phenyllithium to 224and phenoxymethyl chloride, affording cycloadduct 225 in modest yield (53%). Theprimary alcohol was revealed upon removal of the benzyl ether, and the thioacetaloxidatively removed to afford either (±)- or (+)-gliotoxin (226).
4.4Conclusion
Dioxopiperazine alkaloids cover an astonishing array of molecular architectureand, with that, corresponding synthetic challenges to construct such substances.The biosynthesis of many of the natural substances touched on in this chapter has,in some instances, been studied going back several decades, and many workershave sought to exploit insights from Nature’s strategic bond constructions in asynthetic laboratory context. Advances in whole genome sequencing have broughtnew and invigorated interest in elucidating the biosynthesis of structurally intrigu-ing and biomedically relevant secondary metabolites. The insights to be gainedfrom educated guess work on what specific compounds might lie along a pos-sible biosynthetic pathway, traditionally accomplished by isolation and structural
375

References 147
elucidation to map metabolite co-occurrence in conjunction with isotopically la-beled precursor incorporation experiments, is in the process of giving way to amuch higher resolution picture of secondary metabolism in microorganisms andplants. With the advent of powerful new genomics and proteomics tools to studyand manipulate secondary metabolite production, advances in our understandingof Nature’s creative synthetic palette will surely explode in the coming years.The fruits of these insights will undoubtedly be extensively exploited by syntheticchemists working at the forefront of complex molecule synthesis. In addition, manynew natural products–the biosynthetic intermediates themselves, often isolated intrace amounts if at all–will provide and constitute worthy new synthetic targetsand substrates for various important applications.
Acknowledgment
R.M.W. is grateful to the National Institutes of Health (GM068011; CA70375;CA85419) for financial support.
References
1. Birch, A.J. and Russell, R.A. (1972)Tetrahedron, 28, 2999–3008.
2. Birch, A.J. and Wright, J.J. (1969)J. Chem. Soc., Chem. Commun.,644–645.
3. Birch, A.J. and Wright, J.J. (1970) Tetra-hedron, 26, 2329–2344.
4. Baldas, J., Birch, A.J., and Russell, R.A.(1974) J. Chem. Soc., Perkin Trans. 1,50–52.
5. Sanz-Cervera, J.F., Glinka, T., andWilliams, R.M. (1993) Tetrahedron, 49,8471–8482.
6. Kametani, T., Kanaya, N., and Ihara,M. (1980) J. Am. Chem. Soc., 102,3972–3975.
7. Schkeryantz, J.M., Woo, J.C.G.,Siliphaivanh, P., Depew, K.M., andDanishefsky, S.J. (1999) J. Am. Chem.Soc., 121, 11964–11975.
8. von Nussbaum, F. and Danishefsky,S.J. (2000) Angew. Chem., Int. Ed., 39,2175–2178.
9. Edmondson, S., Danishefsky, S.J.,Sepp-Lorenzino, L., and Rosen, N.(1999) J. Am. Chem. Soc., 121,2147–2155.
10. Finefield, J.M. and Williams, R.M.(2010) J. Org. Chem., 75, 2785–2789.
11. Baran, P.S., Guerrero, C.A., and Corey,E.J. (2003) J. Am. Chem. Soc., 125,5628–5629.
12. Depew, K.M., Marsden, S.P., Zatorska,D., Zatorski, A., Bornmann, W.G., andDanishefsky, S.J. (1999) J. Am. Chem.Soc., 121, 11953–11963.
13. Porter, A.E.A. and Sammes, P.G. (1970)J. Chem. Soc., Chem. Commun., 1103.
14. Domingo, L.R., Sanz-Cervera, J.F.,Williams, R.M., Picher, M.T., andMarco, J.A. (1997) J. Org. Chem., 62,1662–1667.
15. Williams, R.M., Sanz-Cervera, J.F.,Sancenon, F., Marco, J.A., and Halligan,K. (1998) J. Am. Chem. Soc., 120,1090–1091.
16. Stocking, E.M., Sanz-Cervera, J.F., andWilliams, R.M. (2000) J. Am. Chem. Soc.,122, 1675–1683.
17. Jin, S.D., Wessig, P., and Liebscher, J.(2001) J. Org. Chem., 66, 3984–3997.
18. Sanz-Cervera, J.F. and Williams,R.M. (2002) J. Am. Chem. Soc., 124,2556–2559.
19. Adams, L.A., Valente, M.W.N., andWilliams, R.M. (2006) Tetrahedron, 62,5195–5200.
376

148 4 Biomimetic Synthesis of Alkaloids Derived from Tryptophan: Dioxopiperazine Alkaloids
20. Greshock, T.J., Grubbs, A.W.,Tsukamoto, S., and Williams, R.M.(2007) Angew. Chem. Int. Ed., 46,2262–2265.
21. Greshock, T.J. and Williams, R.M.(2007) Org. Lett., 9, 4255–4258.
22. Herzon, S.B. and Myers, A.G. (2005)J. Am. Chem. Soc., 127, 5342–5344.
23. Myers, A.G. and Herzon, S.B. (2003)J. Am. Chem. Soc., 125, 12080–12081.
24. Baran, P.S., Guerrero, C.A.,Ambhaikar, N.B., and Hafensteiner,B.D. (2005) Angew. Chem. Int. Ed., 44,606–609.
25. Baran, P.S., Guerrero, C.A.,Hafensteiner, B.D., and Ambhaikar,N.B. (2005) Angew. Chem. Int. Ed., 44,3892–3895.
26. Baran, P.S., Hafensteiner, B.D.,Ambhaikar, N.B., Guerrero, C.A., andGallagher, J.D. (2006) J. Am. Chem. Soc.,128, 8678–8693.
27. Qian-Cutrone, J., Huang, S., Shu, Y.Z.,Vyas, D., Fairchild, C., Menendez, A.,Krampitz, K., Dalterio, R., Klohr, S.E.,and Gao, Q. (2002) J. Am. Chem. Soc.,124, 14556–14557.
28. Kato, H., Yoshida, T., Tokue, T., Nojiri,Y., Hirota, H., Ohta, T., Williams, R.M.,and Tsukamoto, S. (2007) Angew. Chem.Int. Ed., 46, 2254–2256.
29. Deyrup, S.T., Swenson, D.C., Gloer, J.B.,and Wicklow, D.T. (2006) J. Nat. Prod.,69, 608–611.
30. Mudur, S.V., Gloer, J.B., andWicklow, D.T. (2006) J. Antibiot., 59,500–506.
31. Shim, S.H., Swenson, D.C., Gloer, J.B.,Dowd, P.F., and Wicklow, D.T. (2006)Org. Lett., 8, 1225–1228.
32. McAfoos, T.J., Li, S., Tsukamoto, S.,Sherman, D.H., and Williams, R.M.(2010) Heterocycles, 82, 461–472.
33. Greshock, T.J., Grubbs, A.W., andWilliams, R.M. (2007) Tetrahedron, 63,6124–6130.
34. Miller, K.A., Welch, T.R., Greshock,T.J., Ding, Y.S., Sherman, D.H., andWilliams, R.M. (2008) J. Org. Chem., 73,3116–3119.
35. Ding, Y.S., Greshock, T.J., Miller, K.A.,Sherman, D.H., and Williams, R.M.(2008) Org. Lett., 10, 4863–4866.
36. Miller, K.A., Tsukamoto, S., andWilliams, R.M. (2009) Nat. Chem., 1,63–68.
37. Domingo, L.R., Zaragoza, R.J., andWilliams, R.M. (2003) J. Org. Chem., 68,2895–2902.
38. Hart, D.J. and Magomedov, N.A. (2001)J. Am. Chem. Soc., 123, 5892–5899.
39. Kim, J., Ashenhurst, J.A., andMovassaghi, M. (2009) Science, 324,238–241.
40. Movassaghi, M., Schmidt, M.A., andAshenhurst, J.A. (2008) Angew. Chem.Int. Ed., 47, 1485–1487.
41. Kishi, Y., Fukuyama, T., and Nakatsuk,S. (1973) J. Am. Chem. Soc., 95,6492–6493.
42. Kishi, Y., Fukuyama, T., and Nakatsuk,S. (1973) J. Am. Chem. Soc., 95,6490–6492.
43. Kishi, Y., Nakatsuk, S., Fukuyama, T.,and Havel, M. (1973) J. Am. Chem. Soc.,95, 6493–6495.
44. Fukuyama, T. and Kishi, Y. (1976) J.Am. Chem. Soc., 98, 6723–6724.
45. Fukuyama, T., Nakatsuka, S., and Kishi,Y. (1981) Tetrahedron, 37, 2045–2078.
377

Biomimetic Total Synthesis of Malbrancheamide andMalbrancheamide B
Kenneth A. Miller,† Timothy R. Welch,† Thomas J. Greshock,† Yousong Ding,‡David H. Sherman,‡ and Robert M. Williams*,†,§
Department of Chemistry, Colorado State UniVersity, Fort Collins, Colorado 80523-1872, UniVersity ofColorado Cancer Center, Aurora, Colorado, 80045, and Life Sciences Institute, and Departments of
Medicinal Chemistry, Chemistry, Microbiology & Immunology, The UniVersity of Michigan,210 Washtenaw AVenue, Ann Arbor, Michigan 48109-2216
ReceiVed January 17, 2008
The biomimetic total syntheses of both malbrancheamide and malbrancheamide B are reported. Thesynthesis of the two monochloro species enabled the structure of malbrancheamide B to be unambiguouslyassigned. The syntheses each feature an intramolecular Diels-Alder reaction of a 5-hydroxypyrazin-2(1H)-one to construct the bicyclo[2.2.2]diazaoctane core, which has also been proposed as the biosyntheticroute to these compounds.
Introduction
Our research group has exhibited a long-standing interest inthe synthesis and biosynthetic study of a number of uniqueprenylated indole alkaloids containing a characteristic bicyclo-[2.2.2]diazaoctane core.1 This class of natural products includessuch highly biologically active fungal metabolites as theparaherquamides,2 brevianamides,3 notamides,4 and stephaci-dins,5 among others, which we have shown all arise biogeneti-cally from tryptophan, mevalonate-derived isoprene units, and
proline or derivatives of proline.1 Sammes originally proposedthat the bicyclo[2.2.2]diazaoctane core common to all of thesenatural products arises in Nature via an intramolecular hetero-Diels-Alder reaction of a 5-hydroxypyrazin-2(1H)-one,6 andwork from this laboratory has extensively supported such aproposal.1 In fact, we have applied such a [4+2] hetero-Diels-Alder cycloaddition strategy to the total synthesis of several ofthese prenylated indole alkaloids, including stephacidin A,7brevianamide B,8 marcfortine C,9 notoamide B,7b and VM55599.10
Malbrancheamide (1)11 and malbrancheamide B (2) wererecently isolated from Malbranchea aurantiaca RRC1813, a† Colorado State University.
‡ The University of Michigan.§ University of Colorado Cancer Center.(1) (a) Williams, R. M.; Cox, R. J. Acc. Chem. Res. 2003, 36, 127. (b)
Williams, R. M. Chem. Pharm. Bull. 2002, 50, 711. (c) Williams, R. M.;Stocking, E. M.; Sanz-Cevera, J. F. Top. Curr. Chem. 2000, 209, 98.
(2) (a) Yamazaki, M.; Okuyama, E. Tetrahedron Lett. 1981, 22, 135.(b) Ondeyka, J. G.; Goegelman, R. T.; Schaeffer, J. M.; Kelemen, L.; Zitano,L. J. Antibiot. 1990, 43, 1375. (c) Liesch, J. M.; Wichmann, C. F. J. Antibiot.1990, 43, 1380. (d) Banks, R. M.; Blanchflower, S. E.; Everett, J. R.; Manfer,B. R.; Reading, C. J. J. Antibiot. 1997, 50, 840.
(3) (a) Birch, A. J.; Wright, J. J. J. Chem. Soc. Chem. Commun. 1969,644. (b) Birch, A. J.; Wright, J. J. Tetrahedron 1970, 26, 2329. (c) Birch,A. J.; Russell, R. A. Tetrahedron 1972, 28, 2999.
(4) Kato, H.; Yoshida, T.; Tokue, T.; Nojiri, Y.; Hirota, H.; Ohta, T.;Williams, R. M.; Tsukamoto, S. Angew. Chem., Int. Ed. 2007, 46, 2254.
(5) (a) Qian-Cutrone, J.; Huang, S.; Shu, Y.-Z.; Vyas, D.; Fairchild, C.;Menendez, A.; Krampitz, K.; Dalterio, R.; Klohr, S. E.; Gao, Q. J. Am.Chem. Soc. 2002, 124, 14556. (b) Qian-Cutrone, J.; Krampitz, K.; Shu,Y.-Z.; Chang, L. P. U.S. Patent 6,291,461, 2001.
(6) (a) Porter, A. E. A.; Sammes, P. G. J. Chem. Soc. Chem. Commun.1970, 1103. (b) Baldas, J.; Birch, A. J.; Russell, R. A. J. Chem. Soc., PerkinTrans I 1974, 50.
(7) (a) Greshock, T. J.; Williams, R. W. Org. Lett. 2007, 9, 4255. (b)Greshock, T. J.; Grubbs, A. W; Tsukamoto, S.; Williams, R. W. Angew.Chem., Int. Ed. 2007, 46, 2262.
(8) (a) Williams, R. M.; Sanz-Cervera, J. F.; Sancenon, F.; Marco, J.A.; Halligan, K. J. Am. Chem. Soc. 1998, 120, 1090. (b) Williams, R. M.;Sanz-Cervera, J. F.; Sancenon, F.; Marco, J. A.; Halligan, K. Bioorg. Med.Chem. 1998, 6, 1233. (c) Sanz-Cervera, J. F.; Williams, R. M.; Marco, J.A.; Lopez-Sanchez, J. M.; Gonzalez, F.; Martınez, M. E.; Sancenon, F.Tetrahedron 2000, 56, 6345. (d) Adams, L. A.; Valente, M. W. N.; Williams,R. M. Tetrahedron 2006, 62, 5195.
(9) Greshock, T. J.; Grubbs, A. W.; Williams, R. M. Tetrahedron 2007,63, 6124.
(10) (a) Stocking, E. M.; Sanz-Cervera, J. F.; Williams, R. M. J. Am.Chem. Soc. 2000, 122, 1675. (b) Sanz-Cervera, J. F.; Williams, R. M. J.Am. Chem. Soc. 2002, 124, 2556.
3116 J. Org. Chem. 2008, 73, 3116-311910.1021/jo800116y CCC: $40.75 © 2008 American Chemical Society
Published on Web 03/18/2008
378

fungus collected on bat detritus collected in a cave in Mexicoby Mata and co-workers. These new substances are the firstalkaloids in this class of prenylated indole alkaloids to containa halogenated indole ring (Figure 1). The lack of a tertiary amidein the bicyclo[2.2.2]diazaoctane core also serves to characterizethe malbrancheamides. In addition to these notable structuralfeatures, malbrancheamide has been shown to be a calmodulin(CaM) antagonist that inhibits the activity of CaM-dependentphosphodiesterase (PDE1) in a concentration dependent man-ner.11 The chemotherapeutic potential of PDE1 inhibitorsincludes applications in the treatment of neurodegenerativediseases, cancers, and vascular diseases, due to the effect onintracellular cAMP and cGMP concentrations.12 New pharma-cological properties of malbrancheamide may be discoveredthrough the study of malbrancheamide, malbrancheamide B, andother analogs, as specific PDE1 inhibitors are scarce and theexact function of the enzyme has not been fully characterized.
Although compelling spectroscopic evidence indicated thatthe structure of 1 was as shown,11 the precise structure ofmalbrancheamide B (2) was less certain. Isolation and structuralcharacterization of 2 indicated the presence of a single chlorineon the indole ring, and further, preliminary biosynthetic experi-ments indicated that malbrancheamide B (2) is a putativebiosynthetic precursor to 1,13 which is thought to arise bysequential halogenation events. However, the question as towhether malbrancheamide B (2) was constituted as the 5-chloroor the 6-chloro derivative was unclear from the preliminarycharacterization data due to the scarce supply of the naturalmaterial. With these issues at the forefront, we undertook thesynthesis of both natural substances to determine the exactidentity of malbrancheamide B. To this end, we envisioned thatmalbrancheamide (1) and malbrancheamide B (2) would arisefrom the aforementioned hetero-IMDA of the 5-hydroxypyrazin-2(1H)-one 3, which we could access by enolization andtautomerization of the enamide 4 (Scheme 1). Using chemistrypreviously established in our laboratory,7a we planned onassembling the enamide 4 from a reverse prenylated tryptophan5, which could be obtained in a few steps from the correspond-ing chlorinated indole 6.
Results and Discussion
Installation of the reverse prenyl group at the indole 2-positionwas carried out using a two-step protocol developed byDanishefsky and co-workers.14 Chlorination at the 3-position
of indoles 6a-c15 using NCS in DMF gave 7a-c (Scheme 2),which were treated with prenyl-9-BBN in the presence of Et3Nto afford the reverse prenylated indoles 8a-c.14 The corre-sponding gramines 9a-c were prepared by treating 8a-c withformaldehyde and dimethylamine, and subsequent Somei-Kametani coupling16 and imine hydrolysis gave the tryptophanderivatives 5a-c in good yields.
The free amine moieties in 5a-c were protected as thecorresponding BOC-carbamates followed by ester hydrolysisunder standard conditions to yield acids 10a-c (Scheme 3).Coupling of cis-3-hydroxyproline ethyl ester with the tryptophanderivatives 10a-c in the presence of HATU delivered theamides 11a-c as inseparable mixtures of diastereomers. Treat-ment of 11a-c with TFA led to carbamate deprotection andthe resulting amino esters were immediately cyclized to thecorresponding diketopiperazines 12a-c after refluxing with2-hydroxypyridine. Dehydration of 12a-c under Mitsunobuconditions gave the enamides 4a-c, which would serve as therespective IMDA substrates.17
Treatment of the enamides 4a-c with aqueous KOH inMeOH gave intermediate hydroxy-azadienes by enolization andtautomerizaion, and subsequent IMDA gave mixtures of 13a-cand 14a-c favoring the desired syn-isomers 13a-c in ratios of(2-1.6):1 (Scheme 4). The observed preference for the IMDAto provide the syn-isomers 13a-c as the major products mirrors
(11) (a) Martinez-Luis, S.; Rodriguez, R.; Acevedo, L.; Gonzalez, M.C.; Lira-Rocha, A.; Mata, R. Tetrahedron 2006, 62, 1817. (b) Figueroa,M.; del Carmen Gonzalez, M.; Mata, R. Unpublished results.
(12) (a) Zhu, H. J.; Wang, J. S.; Guo, Q. L.; Jiang, Y.; Liu, G. Q. Biol.Pharm. Bull. 2005, 28, 1974. (b) Leisner, T. M.; Liu, M. J.; Jaffer, Z. M.;Chernoff, J.; Parise, L. V. J. Cell Biol. 2005, 170, 465.
(13) Ding, Y.; Sherman, D. H.; Williams, R. M. Unpublished results.(14) Schkeryantz, J. M.; Woo, J. C. G.; Siliphaivanh, P.; Depew, K. M.;
Danishefsky, S. J. J. Am. Chem. Soc. 1999, 121, 11964.
(15) For the preparation of 5,6-dichloroindole see: Bromidge, S. M.; etal. J. Med. Chem. 1998, 41, 1598.
(16) (a) Somei, M.; Karasawa, Y.; Kaneko, C. Heterocycles 1981, 16,941. (b) Kametani, T.; Kanaya, N.; Ihara, M. J. Chem. Soc., Perkin Trans.1 1981, 959.
(17) Curiously, attempts to effect the one-step dehydration/IMDA reactionsequence from 12a-c directly to 13a-c + 14a-c failed under the sameconditions used successfully for stephacidin A (ref 7a) and marcfortine C(ref 9).
FIGURE 1. Malbrancheamide and malbrancheamide B.
SCHEME 1. Retrosynthetic Plan
SCHEME 2. Reverse Prenylated Tryptophan Derivatives
Synthesis of Malbrancheamide and Malbrancheamide B
J. Org. Chem, Vol. 73, No. 8, 2008 3117
379

the preference we have noted for this cycloaddition in thepast.7-9 Considering that previous optimization efforts toimprove the syn:anti ratio in related IMDAs were not productivewhen a variety of solvents and temperatures were studied, weelected to simply separate the major isomers 13a-c and continuethe total syntheses without further optimization.7-9
Completion of the syntheses required selective reduction ofthe tertiary amide in the presence of the secondary amide, andto that end, 13a-c were treated with excess DIBAL-H,18 whichcleanly provided malbrancheamide (1) from 12a (80%) andmalbrancheamide B (2) from 12c (74%) (Scheme 5). Synthetic1 was identical in all respects (1H, 13C, HRMS) to the naturalproduct.11 Comparison of the 1H NMR spectrum of 15, the5-chloro derivative, to that of natural malbrancheamide Brevealed significant differences in the aromatic region revealingthat the correct structure contained a 6-chloroindole ring.
Gratifyingly, synthetic 2 was identical in all respects (1H,HRMS) to natural malbrancheamide B.
It is striking that the initial halogenation of the indole ringduring the biosynthesis of malbrancheamide B, occurs at theless-activated 6-position as opposed to the more electron-rich5-position. Studies to clone and express the putative halogenasefrom M. aurantiaca are currently under investigation in theselaboratories.
In summary, the first total synthesis of malbrancheamide (1)and malbrancheamide B (2) have been completed in twelve stepsin 5.3% and 8.2% overall yield, respectively. In addition, thestructure of malbrancheamide B (2) was confirmed though thesynthesis of both the 5-chloro and the 6-chloro regioisomers.Experiments to establish the biosynthetic relationship between1 and 2 and their putative progenitors are in progress and willbe reported in due course.
Experimental Section
Representative Procedure for the Hetero-Diels-Alder Reac-tion of Enamides 4. Preparation of Cycloadducts 13a and 14a.To a solution of 4a (86 mg, 0.21 mmol) in MeOH (16 mL) at 0 °Cwas added 20% aqueous KOH (4 mL). The reaction was warmedto room temperature (rt) and was stirred for 12 h. The reactionwas quenched with sat. NH4Cl (30 mL) and extracted with CH2-Cl2 (3 ! 30 mL). The combined organic layers were dried (Na2-SO4) and concentrated under reduced pressure. The residue wastriturated with CHCl3 (30 mL), and the suspension was filtered toprovide 53 mg (60%) of 13a as a white amorphous solid.Concentration of the filtrate gave 25 mg (29%) of 14a as a whiteamorphous solid. Data for major isomer 13a: 1H NMR (300 MHz,CD3OD) ! 7.55 (s, 1 H), 7.40 (s, 1 H), 3.57 (d, J ) 15.5 Hz, 1 H),3.47 (m, 1 H), 2.74 (d, J ) 15.5 Hz, 1 H), 2.60 (m, 1 H), 2.20-1.96 (comp, 6 H), 1.35 (s, 3 H), 1.11 (s, 3 H); 13C NMR (75 MHz,CD3OD) ! 175.7, 171.3, 144.3, 137.2, 128.1, 125.4, 123.3, 119.8,113.1, 104.8, 68.3, 61.6, 50.8, 45.2, 36.2, 31.7, 30.1, 28.6, 25.4,24.9, 22.3; IR (neat) 1663, 1428 cm-1; HRMS (TOF+) calcd forC21H22N3O2Cl2 (M + H) 418.1084, found 418.1084. Data for minorisomer 14a: 1H NMR (300 MHz, CDCl3) ! 9.78 (s, 1 H), 7.49 (s,1 H), 7.33 (s, 1 H), 3.71 (d, J ) 17.8 Hz, 1 H), 3.47 (comp, 2 H),3.25 (s, 1 H), 2.78 (d, J ) 17.8 Hz, 1 H), 2.67 (m, 1 H), 2.18-1.78 (comp, 6 H), 1.24 (s, 3H), 1.16 (s, 3 H); 13C NMR (75 MHz,CD3OD /CDCl3 (1:9)) ! 173.7, 169.6, 142.6, 135.7, 127.1, 125.0,122.9, 119.1, 112.4, 102.9 67.2, 61.5, 45.8, 44.3, 34.8, 32.6, 29.8,29.1, 28.4, 24.5, 23.2; IR (neat) 1676, 1453 cm-1; HRMS (TOF+)calcd for C21H22N3O2Cl2 (M + H) 418.1084, found 418.1079.
Cycloadducts 13b and 14b. Prepared from 4b in to give 43%of 13b as a white amorphous solid and 27% of 14b as a whiteamorphous solid according to the representative procedure describedabove for 13a and 14a. Data for major isomer 13b: 1H NMR (300MHz, CD3OD) ! 7.54 (s, 1 H), 7.44 (s, 1 H), 7.22 (d, J ) 8.6 Hz,1 H), 7.04 (d, J ) 8.6 Hz, 1 H), 3.68 (d, J ) 15.4 Hz, 1 H), 3.55-3.40 (comp, 2 H), 2.77 (d, J ) 15.4 Hz, 1 H), 2.76 (m, 1 H), 2.61(m, 1 H), 2.25-1.92 (comp, 5 H), 1.37 (s, 3 H), 1.12 (s, 3 H); 13CNMR (100 MHz, CD3OD) ! 175.8, 171.4, 143.4, 136.8, 129.2,125.2, 122.1, 118.2, 112.8, 104.5, 68.3, 61.6, 50.8, 45.2, 36.2, 31.7,30.1, 28.7, 25.5, 25.0, 22.4; IR (neat) 1678, 1441 cm-1; HRMS(18) Fukuyama, T.; Liu, G. J. Am. Chem. Soc. 1996, 118, 7426-7427.
SCHEME 3. Enamide Diels-Alder Precursors
SCHEME 4. Hetero-IMDA Reactions
SCHEME 5. Amide Reductions
Miller et al.
3118 J. Org. Chem., Vol. 73, No. 8, 2008
380

(TOF+) calcd for C21H23N3O2Cl (M + H) 384.1473, found384.1460. Data for minor isomer 14b: 1H NMR (300 MHz, CD3-OD) ! 7.91 (s, 1 H), 7.41 (d, J ) 2.0 Hz, 1 H), 7.23 (d, J ) 8.6Hz, 1 H), 7.01 (dd, J ) 8.6, 2.0 Hz, 1H), 3.71 (d, J ) 17.6 Hz,1H), 3.53 (comp, 2 H), 2.90 (d, J ) 17.6 Hz, 1 H), 2.70 (m, 1 H),2.23-1.89 (comp, 6 H), 1.34 (s, 3 H), 1.27 (s, 3 H); 13C NMR(100 MHz, CD3OD) ! 175.4, 171.7, 143.4, 136.8, 129.7, 125.3,122.1, 118.1, 112.8, 103.8, 68.6, 62.7, 47.3, 45.2, 35.9, 33.3, 30.8,29.9, 28.7, 25.4, 24.8, 23.8; IR (neat) 1675, 1461 cm-1; HRMS(TOF+) calcd for C21H23N3O2Cl (M + H) 384.1473, found384.1468.
Cycloadducts 13c and 14c. Prepared from 4c in to give 55%of 13c as a white amorphous solid and 31% of 14c as a whiteamorphous solid according to the representative procedure describedabove for 13a and 14a; data for major isomer 13c: 1H NMR (400MHz, DMSO-d6) ! 8.75 (s, 1 H), 7.40-6.98 (comp, 3 H), 3.42 (d,J ) 15.3 Hz, 1 H), 3.30 (comp, 2 H), 2.68 (d, J ) 15.3 Hz, 1 H),2.50 (comp, 1 H), 2.10-1.70 (comp, 6 H), 1.27 (s, 3 H), 0.99 (s,3 H); 13C NMR (100 MHz, DMSO-d6) ! 173.0, 168.4, 142.0, 136.8,125.3, 125.2, 118.9, 118.5, 110.4, 103.8, 66.0, 59.6, 48.9, 43.6,34.6, 30.0, 28.7, 27.9, 24.0, 23.7, 21.6; IR (neat) 1671, 1409 cm-1;HRMS (TOF+) calcd for C21H23N3O2Cl (M+H) 384.1473, found384.1470. Data for minor isomer 14c: 1H NMR (400 MHz, CD3-OD/CDCl3 (1:9)) ! 9.58 (s, 1 H), 7.33 (d, J ) 8.4 Hz, 1 H), 7.23(d, J ) 1.8 Hz, 1 H), 6.97 (dd, J ) 8.4, 1.8 Hz, 1 H), 3.75 (d, J )17.8 Hz, 1 H), 3.47 (comp, 2 H), 3.30 (bs, 1 H), 2.84 (d, J ) 17.8Hz, 1 H), 2.70 (m, 1 H), 2.23-1.77 (comp, 6 H), 1.25 (s, 3 H),1.18 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) ! 172.43, 169.0,142.0, 136.8, 125.8, 125.3, 119.0, 118.5, 110.4, 103.1, 79.2, 66.4,60.5, 45.4, 43.7, 34.2, 31.6, 28.6, 27.7, 24.0, 22.5; IR (neat) 1672,1410 cm-1; HRMS (TOF+) calcd for C21H23N3O2Cl (M + H)384.1473, found 384.1468.
Representative Procedure for the Selective Reduction ofTertiary Amides with Excess DIBAL-H. Synthesis of Mal-brancheamide (1). DIBAL-H (0.70 mL, 1 M in toluene, 0.70mmol) was added to a suspension of 13a (15 mg, 0.036 mmol) intoluene (7 mL) at rt. The reaction was stirred at rt for 12 h,whereupon finely powdered Na2SO4‚10H2O was added untilbubbling ceased. The mixture was filtered with a medium porosityfritted funnel washing with EtOAc (50 mL) and MeOH (50 mL),and the filtrate was concentrated under reduced pressure. Theresidue was purified by flash chromatography eluting with MeOH/CH2Cl2 (2:98) to give 12 mg (80%) of 1 as a white amorphoussolid: 1H NMR (400 MHz, CD3OD) ! 7.48 (s, 1 H), 7.40 (s, 1 H),3.43 (d, J ) 10.3 Hz, 1 H), 3.06 (m, 1 H), 2.85 (comp, 2 H), 2.54(m, 1 H), 2.27 (dd, J ) 10.2, 1.5 Hz, 1 H), 2.20-1.18 (comp, 6H), 1.43 (s, 3 H), 1.34 (s, 3 H); 13C NMR (100 MHz, CD3OD) !176.6, 145.1, 137.3, 128.2, 125.3, 123.2, 119.6, 113.1, 104.7, 66.1,59.4, 57.4, 55.4, 48.5, 35.5, 32.4, 30.6, 30.0, 28.1, 24.2, 23.5; IR
(neat) 3226, 1658, 1460 cm-1; HRMS (TOF+) calcd for C21H24N3-OCl2 (M + H) 404.1291, found 404.1290.
Isomalbrancheamide B (15). Prepared from 13b in 68% yieldas a white amorphous solid according to the representativeprocedure described above for 1: 1H NMR (400 MHz, DMSO-d6)! 8.39 (s, 1 H), 7.33 (s, 1 H), 7.26 (d, J ) 8.5 Hz, 1 H), 7.01 (d,J ) 8.5 Hz, 1 H), 3.33 (d, J ) 7.2 Hz, 1 H), 3.26 (d, J ) 9.9 Hz,1 H), 2.76 (s, 2 H), 2.42 (m, 1 H), 2.15-1.73 (comp, 7 H), 1.33 (s,3 H), 1.27 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) ! 173.1. 143.4,134.9, 127.7, 122.8, 120.4, 116.7, 112.1, 103.4, 64.1, 58.6, 55.3,53.9, 47.0, 34.0, 31.1, 30.0, 28.7, 26.6, 23.7, 22.5; IR (neat) 3311,1637, 1458 cm-1; HRMS (TOF+) calcd for C21H25N3OCl (M +H) 370.1680, found 370.1675.
Malbrancheamide B (2). Prepared from 13c in 74% yield as awhite amorphous solid according to the representative proceduredescribed above for 1: 1H NMR (400 MHz, DMSO-d6) ! 8.41 (s,1 H), 7.32 (d, J ) 8.4 Hz, 1 H), 7.27 (d, J ) 1.7 Hz, 1 H), 6.95(dd, J ) 8.4, 1.7 Hz, 1 H), 3.36 (s, 1 H), 3.27 (d, J ) 10.0 Hz, 1H), 2.95 (m, 1 H), 2.76 (s, 2 H), 2.43 (m, 1 H), 2.13 (d, J ) 9.9Hz, 1 H), 2.10-1.70 (comp, 6 H), 1.32 (s, 3 H), 1.26 (s, 3 H); 13CNMR (100 MHz, DMSO-d6) ! 173.1, 142.6, 136.8, 125.3, 125.2,118.7, 118.5, 110.3, 103.7, 64.1, 58.5, 55.3, 53.9, 47.0, 34.0, 31.1,30.0, 28.7, 26.6, 23.7, 22.5; IR (neat) 3297, 1652, 1457 cm-1;HRMS (TOF+) calcd for C21H25N3OCl (M + H) 370.1680, found370.1670.
Acknowledgment. Financial support from the NationalInstitutes of Health (CA70375 to R.M.W) and the Hans W.Vahlteich Professorship (to D.H.S.) is gratefully acknowledged.We are grateful to Prof. Rachel Mata and Dr. Marıa del CarmenGonzalez of the Departamento de Farmacia, Facultad deQuımica, Universidad Nacional Autonoma de Mexico forproviding 1H NMR spectra of natural malbrancheamide andmalbrancheamide B as well as an authentic specimen ofmalbrancheamide. We also acknowledge Prof. Mata for sharinga pre-print of their manuscript describing the structural elucida-tion of malbrancheamide B (ref 11b). We thank Dr. AnthonyE. Glenn of the USDA for providing M. aurantiaca RRC1813strain (originally obtained from Dr. Marıa del Carmen Gonza-lez). Mass spectra were obtained on instruments supported bythe NIH Shared Instrumentation Grant GM49631.
Supporting Information Available: Spectroscopic data andexperimental details for the preparation of all new compounds aswell as copies of 1H NMR and 13C NMR spectra. This material isavailable free of charge via the Internet at http://pubs.acs.org.
JO800116Y
Synthesis of Malbrancheamide and Malbrancheamide B
J. Org. Chem, Vol. 73, No. 8, 2008 3119
381

Principal Investigator: Welch, Timothy R. 6.4
Postdoctoral Fellowship Application 2012
Plans for Work Under Fellowship
B. Research Plan: Targeting STAT5 in Prostate Cancer Cells with DNA Binding Polyamides
I. Specific Aims Prostate cancer was responsible for approximately 33720 deaths in 2011.1 While patients with metastatic prostate cancer typically respond to surgical or medicinal androgen deprivation initially, all will eventually progress to an androgen independent state termed castration-resistant prostate cancer (CRPC). The progression of prostate cancer cells to the androgen independent phenotype occurs concomitantly with an increase in the constitutive activation of the transcription factors androgen receptor (AR) and signal transducer and activator of transcription 5 (STAT5).2 AR is established as a relevant target for the treatment of CRPC, and several therapies targeting the AR signaling pathway are available or in development.3,4 While the exact role of STAT5 in CRPC is not as well defined, it appears to be equally important to prostate cancer cell growth and survival, as evidenced by the following: (1) STAT5 is overactive in 95% of CRPCs,5 (2) STAT5 activity is associated with highly aggressive (high Gleason grade) prostate cancers,6,7 and (3) inhibition of STAT5 by siRNA results in massive apoptotic cell death in human prostate cancer cell lines.8 STAT5 represents a promising target for the treatment of CRPC, a disease for which no effective therapy currently exists. To date, no small molecule inhibitors of STAT5 transcriptional activity have been reported. Our long term goal is to develop small molecule inhibitors of STAT5 function for the treatment of malignancies displaying aberrant STAT5 activity. The overall objective of this proposal is to determine the therapeutic potential of DNA binding pyrrole-imidazole polyamides that target STAT5 for the treatment of CRPC. We hypothesize that polyamides can be designed to downregulate the expression of a subset of genes required for CRPC cell survival. Polyamides containing N-methylpyrrole (Py) and N-methylimidazole (Im) comprise the only known class of programmable small molecules capable of binding specific DNA sequences.9,10 Previous studies from the Dervan group have shown that polyamides are cell permeable,11-13 access nuclear chromatin,14-16 inhibit specific protein–DNA interfaces,17 and modulate gene expression in cell culture.18-23 Polyamides represent a promising approach toward the pursuit of small molecule inhibitors of STAT5 transcription for the treatment of CRPC. The specific aims proposed below will determine the suitability of this approach while contributing to the overall understanding of STAT5 signaling in prostate cancer.
AIM 1. Synthesize a series of Py/Im polyamides designed to target the STAT5 response element and evaluate their DNA binding interactions in vitro. Polyamides designed to target the consensus STAT5 binding sequence 5’-TTCNNNGAA-3’† will be synthesized by solid phase methodology. DNA binding interactions of the polyamides will be evaluated in vitro using melting temperature analysis and electrophoretic mobility shift assay (EMSA).
AIM 2. Examine nuclear localization and characterize gene regulatory potency of polyamides.
Laser-scanning confocal microscopy will be used to examine cellular uptake and localization of high-affinity polyamides in several cancer cell lines. The expression of STAT5 regulated oncogenes BCL-2,24-27 BCL-XL,28,29 and CYCLIN-D12,8,30 in prostate cancer cells will be examined following polyamide treatment using quantitative real-time RT-PCR (qRT-PCR). The global effects of polyamides that are shown to modify gene expression will then be studied
† Abbreviations for degenerate nucleotides: N = any base; W = A or T.
382
APPENDIX II Research Proposal

Principal Investigator: Welch, Timothy R. 6.5
Postdoctoral Fellowship Application 2012
with chromatin immunoprecipitation coupled with sequencing (ChIP-seq) and mRNA sequencing (RNA-seq).
AIM 3. Investigate polyamide induced apoptosis in prostate cancer cell lines and in mice
bearing xenograft tumors. General cell death will be analyzed following polyamide treatment in both normal and prostate cancer cell lines. Polyamide effects on C4-2 tumor growth will be studied in a murine model at the Caltech Animal Facility.
The aims in this proposal represent a new approach toward the development of inhibitors of STAT5 regulated gene expression. The proposed experiments will provide thorough and systematic understanding of the activity, mechanism, and specificity of polyamides in prostate cancer. If successful, the proposed research could produce the first known specific inhibitors of STAT5 regulated gene expression for the treatment of CRPC. II. Background and Significance A limited set of transcription factors has been implicated in a large number of diverse oncogenic signaling pathways.31 Selective inhibition of a single transcription factor would thus influence the expression of numerous upstream oncogenes, many of which are currently individually targeted by discrete inhibitors. Signal transducer and activator of transcription (STAT) proteins are overactive in a variety of cancers, promote cancer cell survival and proliferation, and are susceptible to inhibition, making them ideal targets for cancer therapy.32-34 STATs are latent cytoplasmic proteins that, once activated by tyrosine kinase phosphorylation, dimerize, traffic to the nucleus, and bind specific response elements of target gene promoters (Figure 1).32,35-37 In normal cells, STAT activation has a role in cell survival, angiogenesis, immune function, and in controlling cell growth.38,39 However, disregulation of this activity can contribute to tumor cell growth and proliferation.32,40 Persistent activity of STAT5, a member of the STAT family of proteins, is associated with prostate cancer,5,6,8,41,42 lung, head, and neck cancers,43 and several types of leukemia44-47 and lymphomas.35,48 The link to prostate cancer is particularly strong, with 95% of castration-resistant prostate cancers (CRPC) displaying constitutive STAT5 activity.5 Active STAT5 in prostate cancer predicts early disease recurrence and is associated with high Gleason grade prostate cancers characterized by an aggressive phenotype and poor patient outcome.6,7 Furthermore, active STAT5 has been shown to increase the motility of prostate cancer cells, enhancing the intrinsic ability of the disease to metastasize.49 STAT5 is a generic term for two highly homologous protein isoforms, 94-kDa STAT5a and 92-kDa STAT5b.50 Both isoforms share a conserved DNA binding domain that permits binding of STAT5 to consensus motifs (5’-TTCNNNGAA-3’) found in target gene promoters, including the oncogenes encoding anti-apoptotic (BCL-XL, BCL-2) and pro-proliferative (Cyclin-D1) proteins.2,8,24-30,51 STAT5 binding to this consensus motif
Figure 1. Canonical STAT5 signaling pathway.
383

Principal Investigator: Welch, Timothy R. 6.6
Postdoctoral Fellowship Application 2012
found at non-canonical recognition sites has additionally been shown to activate several novel and potentially oncogenic genes, including TNFRSF13b, MKP-1, and C3ar1.52 Numerous studies have shown that inhibition of STAT5 results in massive apoptotic cell death in human prostate cancer cell lines, regardless of the inhibitory method employed (adenoviral expression of dominant-negative mutants of STAT5, siRNA, or antisense oligonucleotides).2,8,41,53 STAT5 inhibition by antisense oligonucleotides has specifically been shown to suppress the expression of Cyclin-D1 and BCL-XL in LNCaP, C4-2, and DU145 prostate cancer cell lines at both the mRNA and protein levels, resulting in increased apoptotic death and decreased cell growth.2,8 In mouse xenograft models, the progression of CRPC was delayed significantly by STAT5 inhibition with antisense oligonucleotides.2 These findings imply prostate cancers depend on persistent STAT5 activation for cell growth and survival. Chemotherapeutic small molecules targeting STAT5 signaling could thus have a substantial impact on the treatment of CRPC, a disease for which no effective pharmacological therapy currently exists. STAT5 inhibitors published to date indiscriminately downregulate STAT5 regulated transcription and lack practical therapeutic potential. Herein, we describe a new, promising approach for the selective inhibition of STAT5 mediated gene expression in prostate cancer, using DNA binding polyamides to disrupt the STAT5–DNA interface. Polyamides containing N-methylpyrrole (Py) and N-methylimidazole (Im) comprise a class of programmable small molecules that can be designed to bind specific DNA sequences with affinities
Figure 2. Molecular recognition of the minor groove of DNA by a hairpin polyamide. (A) Model of an eight ring hairpin polyamide (ImImPyPy-(R)H2N-!-PyPyPyPy-"-IPA) bound to a 5’-TGGAAA-3’ sequence. Putative hydrogen bonds are represented by dashed lines. (B) X-ray crystal structure of a cyclic polyamide (blue) in complex with dsDNA (0.95Å resolution). (C) Ball-and-stick model of the polyamide in 2A. Closed and open circles represent N-methylimidazole and N-methylpyrrole monomers, respectively. Diamonds, "-alanine; triangle with positive charge, triamine linker; curved line substituted with positive charge, #-amino-substituted !-turn. The isophthalic acid derived tail is represented by a hexagon.
!
NN
O
N H
NN
NO H
N
O NH
N NO
H
5' 3'
T
G
A
A
A
G
A
T
T
T
C
C
N
ONH
N
N OH
N
ONH
N
N O
H
NH
O
NH3
H
H
NH
NH
IPA + +
W G G W W W
W C C W W W
5' -
3' -
- 3'
- 5'
A B
C
!-turntargetsW"W
Py/PytargetsW"W
Im/Pytargets
G"C
#-alatargetsW"W
O
-O
O
384

Principal Investigator: Welch, Timothy R. 6.7
Postdoctoral Fellowship Application 2012
comparable to native DNA binding proteins.9 Sequence specificity of Py/Im polyamides is determined by side-by-side pairing of the heterocyclic amino acids to form distinct hydrogen bonds to nucleotides in the minor groove of DNA (Figure 2).54 Im/Py pairs distinguish G!C from C!G, whereas Py/Py pairs degenerately bind A!T and T!A.10 Hairpin polyamides designed as such have been shown to bind DNA with high affinities,55 inhibit interactions with DNA binding proteins,17 bind chromatin,14-16 and traffic to the nucleus in a variety of cell types.11-13 Recently, analyses have shown that 8-ring cyclic polyamides allosterically perturb the DNA helix through a 4Å widening of the minor groove and consequent major groove compression.56 This significant change provides a mechanistic basis for disruption of protein–DNA interfaces by hairpin polyamides. To evaluate the specific inhibition of STAT5 mediated gene expression by cell permeable, sequence specific DNA binding polyamides, we propose the synthesis, biochemical evaluation, and biological study of a small library of Py/Im polyamides designed to specifically disrupt the interactions of STAT5 with regulatory regions of pro-proliferative and anti-apoptotic genes. III. Preliminary Studies This proposal builds upon known chemical and biological techniques developed in the Dervan group. The study highlighted here shows precedence for the use of polyamides in the modulation of endogenous gene expression (Figure 3). In the study, hypoxia inducible factor (HIF-1#) binding to the sequence 5’-WWWCGW-3’ found in the vascular endothelial growth factor (VEGF) hypoxia response element (HRE) was perturbed by a hairpin polyamide.21,22 The polyamide reduced occupancy of HIF-1# at the VEGF HRE and suppressed VEGF mRNA expression. Only a subset of genes induced by deferoxamine (DFO)—a small molecule used to mimic hypoxia in cells—was inhibited by polyamide, whereas HIF-1# siRNA and the DNA-binding natural product echinomycin inhibited most of the DFO induced transcripts. This demonstrated the relative selectivity of Py/Im polyamides compared to other methods used to perturb HIF-1# activity. A second investigation key to the design of this proposal was detailed in a series of publications from the Dervan group in which the nuclear localization of a library of over 120 polyamide-fluorophore conjugates was described.11-13 Cellular uptake profiles of the polyamides were assayed in 13 cell lines using confocal microscopy. The reported nuclear uptake trends suggest the following structural features to be conducive to uptake: an eight-ring hairpin or cyclic core, a positive charge on the amino substituent of the !-aminobutyric acid (GABA) turn, and either a conjugated fluorescein fluorophore (FITC) or isophthalic acid (IPA) moiety on the hairpin tail. Polyamides targeting 5’-WGWWGW-3’ and 5’-WGGWWW-3’ similar to those proposed below were shown to have strong nuclear localization in PC-3 and LNCaP prostate cancer cell lines.11,12 The transition from cell culture work to murine models is an important next step in Py/Im polyamide research that is focused on biomedical applications of the technology. Recent studies undertaken in the Dervan laboratory have provided basic pharmacokinetic analyses of polyamides in C57/BL6 wild type mice.57 Micromolar blood levels of Py/Im polyamides were achieved for multiple hours following a single injection of the compound subcutaneously or intraperitoneally. An example is shown in Figure 4a (a representative HPLC trace) and 4B (a representative pharmacokinetic profile for a hairpin Py/Im polyamide). Decline in hairpin polyamide concentration was observed over the course of multiple hours,
Figure 3. Polyamide mediated inhibition of HIF-1 DNA binding to the HRE suppresses VEGF expression. (The square symbol represents chlorothiophene.)
! !" HIF-1
IPA + +
G C A T A C G T G G5' -
3' -
- 3'
- 5'C G T A T G C A C C
HRE
VEGFtranscription
XX
385

Principal Investigator: Welch, Timothy R. 6.8
Postdoctoral Fellowship Application 2012
with a peak concentration noted between 1-3h depending on route of administration. Compound plasma levels were below detection level 24h after the injection. The polyamide was well tolerated by the animals. Multiple administrations (i.p. or s.c. at 120 nmol per injection per animal) did not result in any significant reduction in body weight. Weight loss is a commonly used criterion for health deterioration in mice (Figure 4C, D).57 The Dervan group has recently gained broad experience with xenograft experimentation in immunocompromised mice, encompassing the engraftment of human cancer cell lines A549, LNCaP, U251, and T47D. FITC-labeled polyamides injected subcutaneously on the flank opposite the tumor were shown by confocal microscopy to access the tumor derived cell nuclei. Moreover, gene expression changes have been observed in engrafted A549 tumors following polyamide treatment.58 The studies outlined above demonstrate the validity of targeting the transcription factor–DNA interface as a means of modulating gene expression. Furthermore, evaluation of nuclear uptake trends allows for the rational design of new polyamides that are structurally optimized for nuclear localization. The Dervan group has also recently developed a gram-scale synthesis of hairpin polyamides and shown that polyamides are bioavailable in mice, eliminating two major obstacles in the evaluation of polyamides in animal models.59,60 Tumor engraftment protocols, established tolerable dosing conditions, and general animal care and handling expertise found in the Dervan group will be invaluable during in vivo testing of the proposed STAT5-targeting polyamides. We are confident the methods established by these studies can be extended to the proposed research and will increase the probability of success. IV. Research Design and Methods An estimated timeline for completion of the specific aims is given in Figure 5. The proficiency of the Dervan group with these techniques coupled with the outstanding equipment, facilities, and expertise found at Caltech will provide strong support for the success of this research project. We are confident that the proposed objectives will be thoroughly pursued during the planned three year period.
A B
C D
Figure 4. Pharmacokinetic data for hairpin polyamide 1 in C57/BL6 wild type mice. (A) HPLC analysis of a blood sample collected retro-orbitally 1.5 h post intraperitoneal (i.p.) injection of 1. Plasma was isolated by centrifugation and pre-cleared from protein by methanol precipitation. (4 = internal reference standard). (B) Plasma levels of 1 as a percent of reference standard 4 following i.p. or subcutaneous (s.c.) injection. (C, D) Weight of four individual mice in response to polyamide dosing regimen. 120 nmol doses were administered either i.p. (C) or s.c. (D) at intervals denoted by arrows in the plot. The solid red line indicates the 15% weight loss mark considered an endpoint criterion for the mice.
386

Principal Investigator: Welch, Timothy R. 6.9
Postdoctoral Fellowship Application 2012
AIM 1. Synthesize a series of Py/Im polyamides designed to target the STAT5 response element
and evaluate their DNA binding interactions in vitro. Three oncogenes shown in the literature to be functionally activated by STAT5 and to contain a STAT5 binding site characterized at the sequence level were selected as targets for polyamide inhibition (Table 1). The three degenerate base pairs found in the middle of the consensus STAT5 binding sequence (5’-TTCNNNGAA-3’) represent the great degree of diversity of the binding sequences on target genes, allowing for the design of polyamides with greater DNA site sequence specificity than the native transcription factor. Three polyamides (Figure 6) are proposed to bind the sequences 5’-WGWWGW-3’ (1, found in BCL-XL), 5’-WWCGGW-3’ (2, found in BCL-XL), and 5’-WGGWWW-3’ (3, found in BCL-2 and CYCLIN-D1).27,30 Polyamide 3 is also expected to bind non-canonical STAT5 binding sites found in MKP-1, TNFRSF13b, and C3ar1.52 Mismatch controls will be prepared for each compound by substituting a Py for an Im (structures not shown). Polyamides 1-3 and the three mismatch controls will be synthesized using solid-phase methodology established in the Dervan laboratory.18,61,62 Preparative reverse-phase high performance liquid chromatography (RP-HPLC) will be used to purify crude reaction products. Chemical purity of polyamides will be determined by analytical HPLC and the chemical composition of each confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF).
The ability of synthesized polyamides 1-3 to bind DNA will be determined using melting temperature (Tm) analysis. The degree to which both match and mismatch polyamides stabilize 14 base pair DNA duplexes containing the appropriate match sequence will be measured. Previous results have shown a correlation between the increase in Tm of DNA duplexes and DNA binding affinity.63-65 Polyamides
Gene Function STAT5 Binding Sequence BCL-XL anti-apoptotic 5’-GCATTTCGGAGAAGACG-3’ BCL-2 anti-apoptotic 5’-CAAGTTCCAGGAAAGCG-3’
CYCLIN-D1 pro-proliferative 5’-GGCGTTCTTGGAAATGC-3’
Table 1. Promoter sequences for selected genes demonstrated to be STAT5 dependent. The consensus sequence is specified in bold.
Figure 6. Proposed library of polyamide inhibitors of STAT5. Structural abbreviations are defined in Figure 2.
IPA + +
W G G W W W
W C C W W WIPA + + IPA + +
W W C G G W
W W G C C W
W G W W G W
W C W W C W3'
3'
5'
5'
1 2
3'
3'
5'
5'
3'
3'
5'
5'
3
Found in promoters of: BCL-XL Found in promoters of: BCL-XL Found in promoters of: BCL-2CYCLIN-D1
Figure 5. Predicted timetable for the proposed research.
Synthesis (2 months)
Characterization of DNA binding (6 months)
Confocal microscopy (2 months)
qRT-PCR (6 months)
ChIP-seq and RNA-seq (13 months)
Apoptosis assay
(6 months)
Tumor xenograft studies
(18 months)
Year 1 Year 2 Year 3
387

Principal Investigator: Welch, Timothy R. 6.10
Postdoctoral Fellowship Application 2012
identified to bind DNA with high affinity will be subjected to electrophoretic mobility shift assay (EMSA) to evaluate their ability to disrupt STAT5 binding to their cognate sequences. Published protocols from the Dervan laboratory will be followed using commercially available STAT5 and 32P-labelled oligonucleotides purchased from Abcam and Integrated DNA Technologies, respectively.19-21,63
AIM 2. Examine nuclear localization and characterize gene regulatory potency of polyamides. Fluorescent (FITC) derivatives of polyamides 1-3 will be evaluated in cell culture to examine their uptake and localization properties. Cells treated with varying concentrations of fluorescent polyamides will be examined using confocal microscopy as previously described.11-13 Several prostate cancer cell lines will be studied to establish general cellular uptake trends, including LNCaP, C4-2, DU145, and PC-3 (Table 2). All have been previously used in the Dervan laboratory or are commercially available and will be cultured and propagated according to recommended American Type Culture Collection (ATCC) procedures. Polyamides will then be screened for the ability to suppress gene expression in cells by quantitative real-time RT-PCR (qRT-PCR).22 Specifically, we will examine the mRNA expression profiles of STAT5 regulated genes BCL-XL, BCL-2, and CYCLIN-D1 in the presence of polyamides following induction of STAT5 activity by interleukin 3 (IL-3) in four different prostate cancer cell lines. A reduction in IL-3 induced expression is expected if a polyamide disrupts STAT5 activation of the gene under investigation. Mismatch polyamides will serve as a negative control, while STAT5 siRNA known to inhibit expression of all three target genes will be used as a positive control for comparison.8 Since constitutive STAT5 activation is most prevalent in cells expressing an androgen independent phenotype, cell lines for this experiment were selected based on androgen dependence (Table 2). Polyamide binding is expected to impart a greater change on gene expression in androgen independent cell lines C4-2 and DU145 than on androgen sensitive LNCaP cells. PC-3 cells do not express STAT5 and are included as a negative control for STAT5 regulated gene expression. The global effects of polyamide treatment on prostate cancer cells will be investigated through chromatin-immunoprecipitation coupled with sequencing (ChIP-seq) and cellular mRNA sequencing (RNA-seq). To probe the mechanism of polyamide induced gene suppression in prostate cancer cells, ChIP-seq will be conducted to assess STAT5 occupancy in nuclear chromatin. In the absence of polyamide or in the presence of the mismatched control, amplification of DNA isolated from anti-STAT5 antibody (purchased from Santa Cruz) pull-down is expected, demonstrating normal STAT5–DNA binding. A decrease in pull-down efficiency from polyamide treated cells would support an inhibitory mechanism in which displacement of STAT5 is attributable to polyamide binding to gene promoters. Isolated DNA will be submitted to high throughput sequencing using the Illumina Genome Analyzer at Caltech. Sequencing results mapped to the human genome will be compared between the untreated, mismatch control, and match polyamide experiments to identify specific genomic sites susceptible to polyamide-mediated STAT5 displacement. A consensus polyamide binding site will also be characterized, providing valuable insight as to the sequence specificity of the polyamide. The global effect of polyamides on gene expression can be evaluated using RNA-seq. STAT5 activity will be induced in cells with IL-3, then either (1) left untreated, or treated with (2) mismatch polyamide, (3) polyamide, or (4) STAT5 siRNA. Total mRNA will be isolated from each experiment, sequenced, and scanned for a 2-fold or greater alteration in expression. Our hypothesis that polyamides can be designed to selectively inhibit
Cell Line
Androgen Dependence
STAT5 (+/-)
Commercial source
LNCaP sensitive + ATCC C4-2 independent + UroCor Labs
DU145 independent + ATCC PC-3 independent - ATCC
Table 2. Proposed prostate cancer cell lines.
388

Principal Investigator: Welch, Timothy R. 6.11
Postdoctoral Fellowship Application 2012
a subset of STAT5 regulated genes will be supported if fewer IL-3 induced transcripts are affected by polyamide than by siRNA. A sequence specific mechanism of polyamide mediated perturbation is also supported if the mismatch control suppresses fewer induced gene transcripts than the matched polyamide. AIM 3. Investigate polyamide induced apoptosis in prostate cancer cell lines and in mice bearing
xenograft tumors. The effect on cell apoptosis of the proposed polyamides will be evaluated in each of the prostate cancer cell lines mentioned above. Cells will be incubated with varying concentrations of polyamide, then the effect on cell cycle population analyzed by flow cytometry, caspase-3 activity, and cleaved poly ADP ribose polymerase (PARP) expression.2,53 Polyamide induced apoptosis would be characterized by dose-dependent increases in the fraction of cells in the sub-G0/G1 phases of the cell cycle, caspase-3 activity, and in the expression of cleaved PARP. STAT5-negative PC-3 cells will serve as a necessary negative control. The selectivity of polyamide induction of apoptosis in cancerous versus normal cell lines will be determined by comparing these results to those found in an analogous experiment on HUVEC and NHDF cell lines. Polyamide effects on the C4-2 cell line will be further studied in an in vivo murine model. All experiments are to be conducted at the Caltech Animal Facility following the experimental procedures outlined in the IACUC protocol of the Dervan laboratory (#1638-11, date of approval: 9/21/11). Male immunocompromised mice will be injected subcutaneously (s.c.) with up to 107 C4-2 cells in PBS. Following a 1-2 week tumor engraftment period, Py/Im polyamides will be injected intraperitoneally (i.p.) at doses not exceeding 40 mg/kg over the course of up to 12 weeks. The animals will be injected with polyamide up to three times a week with a resting period of at least onea day between injections. A vehicle-treated reference control group will be maintained throughout the experiment. Recent investigations conducted in the Dervan laboratory showed that micromolar concentrations of polyamide in murine blood could be achieved following i.p. injection of polyamides with varying sequences.57,58 Tumor volume will be monitored at least once a week using caliper measurements. Retro-orbital blood samples will be collected weekly and levels of tumor-specific secreted factor PSA measured by enzyme-linked immunosorbent assay (ELISA). After four weeks of polyamide treatment, the mice will be sacrificed by asphyxiation (CO2 chamber, 60%). Tumors will be excised and sectioned for histological blood vessel quantification. RNA-seq experiments will further be conducted to determine global polyamide effects on the tumor transcriptome in comparison to the vehicle treated group. Potential Problems with this Proposal Three possible problems with this proposal are presented and addressed. (i) The proposed polyamides are not sufficiently specific to be considered therapeutic candidates for the
selective inhibition of STAT5 in the context of the human genome. The human genome likely contains over a million potential binding sites for a compound that targets six base pair sequences. However, many of these sites are inaccessible in the context of nuclear chromatin. Boyle and coworkers have shown using DNase I hypersensitivity maps that only 2.1% of the genome is accessible in CD4+ T-cells.66 Moreover, polyamide binding at non-promoter sequences of accessible DNA does not affect transcription, as RNA polymerase dislodges polyamides from DNA during transcription.67 Despite the prevalence of six base pair sequences targeted by 8-ring polyamides in the genome, the HIF-1 studied mentioned above shows that polyamides have a relatively small effect on global gene expression.
389

Principal Investigator: Welch, Timothy R. 6.12
Postdoctoral Fellowship Application 2012
(ii) The STAT family of proteins share a similar DNA binding domain, so polyamides designed to target a six base pair STAT5 sequence will invariably target other members of the family of transcription factors. STAT5 and STAT3 site sequences share the most
homology (5’-TTCNNNGAA-3’ and 5’-TTCCNGGAA-3’, respectively) among the STAT family.68 Polyamides 1 and 2 target STAT consensus sequences with at least two mismatched sites and are expected to be selective for STAT5 over STAT3. Polyamide 3 matches the STAT3 consensus when N=W and will likely inhibit some binding of both STAT3 and STAT5 to the cognate sequence. Comparison of the gene regulatory activity of polyamides 1-3 will allow us to analyze selectivity of the proposed polyamides. Binding specificity could be improved by expanding the Py/Im core to target seven or eight base pair sequences (Figure 7). However, disruption of STAT3 activated gene expression would not necessarily be an undesired consequence of polyamide binding, as constitutive STAT3 activity has also been implicated in the malignant phenotype of numerous cancers.32 For this reason, polyamide 3 was deliberately included in this proposal.
(iii) Inhibition of STAT5 target gene expression is not selective for cancer cells. CRPC tumors are dependent upon persistent STAT5 activity for malignant cell growth and survival. It has been suggested that even partial inhibition of STAT5 mediated transcription would be fatal to tumor cells as a consequence of this increased STAT5 dependence.32 Normal cells could plausibly survive with low levels of active STAT5 or utilize other survival pathways. Indeed, even STAT5 null mice remain viable, despite impaired mammary gland development and abnormalities in hematopoiesis.36,69 Conversely, disruption of STAT5 signaling in tumor cells causes massive apoptosis. Aim 3 will provide a direct comparison of polyamide treatment on normal versus cancerous cells. Closing Remarks The primary aim of this proposal is the development of a small library of polyamides designed to inhibit the binding of STAT5 to its cognate promoter site sequences. We hypothesize that the disruption of STAT5 binding events should suppress gene expression necessary for tumor cell growth and survival, resulting in apoptosis of CRPC cells. Inhibition of STAT5 transcriptional activity at the protein–DNA interface represents a new approach toward the treatment of CRPC. Accordingly, extensive experimentation has been proposed to fully elucidate the binding affinities and specificities of polyamides 1-3 for the cognate DNA sequences, to determine if the polyamides are able to traffic to the nucleus, and to quantify the effect of polyamide treatment on gene expression in prostate cancer cells. The ability of polyamides to reduce the viability of cancer cells will then be examined in cell lines and in mice bearing xenograft tumors. Results of these studies will reveal the therapeutic potential of polyamides in the treatment of CRPC and contribute to the understanding of STAT5 signaling in prostate cancer cells.
Figure 7. Polyamide modifications: extended hairpins targeting 7 bp sequences of CYCLIN-D1 promoter.
IPA + +
IPA + +
G W W C W W G G W W
C W W G W W C C W W
4
3'
3'
5'
5'
W C W W G G W W W W
W G W W C C W W W W
5
3'
3'
5'
5'
390

Principal Investigator: Welch, Timothy R. 6.13
Postdoctoral Fellowship Application 2012
Literature Cited 1. Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: the impact of eliminating
socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011, 61, 212-236.
2. Thomas, C.; Zoubeidi, A.; Kuruma, H.; Fazli, L.; Lamoureux, F.; Beraldi, E.; Monia, B. P.; MacLeod, A. R.; Thuroff, J. W.; Gleave, M. E. Transcription factor Stat5 knockdown enhances androgen receptor degradation and delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther 2011, 10, 347-359.
3. Antonarakis, E. S.; Carducci, M. A.; Eisenberger, M. A. Novel targeted therapeutics for metastatic castration-resistant prostate cancer. Cancer Lett 2010, 291, 1-13.
4. Ryan, C. J.; Tindall, D. J. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol 2011, 29, 3651-3658.
5. Tan, S. H.; Dagvadorj, A.; Shen, F.; Gu, L.; Liao, Z.; Abdulghani, J.; Zhang, Y.; Gelmann, E. P.; Zellweger, T.; Culig, Z.; Visakorpi, T.; Bubendorf, L.; Kirken, R. A.; Karras, J.; Nevalainen, M. T. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res 2008, 68, 236-248.
6. Li, H.; Ahonen, T. J.; Alanen, K.; Xie, J.; LeBaron, M. J.; Pretlow, T. G.; Ealley, E. L.; Zhang, Y.; Nurmi, M.; Singh, B.; Martikainen, P. M.; Nevalainen, M. T. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res 2004, 64, 4774-4782.
7. Li, H.; Zhang, Y.; Glass, A.; Zellweger, T.; Gehan, E.; Bubendorf, L.; Gelmann, E. P.; Nevalainen, M. T. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res 2005, 11, 5863-5868.
8. Dagvadorj, A.; Kirken, R. A.; Leiby, B.; Karras, J.; Nevalainen, M. T. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin Cancer Res 2008, 14, 1317-1324.
9. Dervan, P. B.; Edelson, B. S. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr Opin Struct Biol 2003, 13, 284-299.
10. Trauger, J. W.; Baird, E. E.; Dervan, P. B. Recognition of DNA by designed ligands at subnanomolar concentrations. Nature 1996, 382, 559-561.
11. Best, T. P.; Edelson, B. S.; Nickols, N. G.; Dervan, P. B. Nuclear localization of pyrrole-imidazole polyamide-fluorescein conjugates in cell culture. Proc Natl Acad Sci U S A 2003, 100, 12063-12068.
12. Edelson, B. S.; Best, T. P.; Olenyuk, B.; Nickols, N. G.; Doss, R. M.; Foister, S.; Heckel, A.; Dervan, P. B. Influence of structural variation on nuclear localization of DNA-binding polyamide-fluorophore conjugates. Nucleic Acids Res 2004, 32, 2802-2818.
13. Nickols, N. G.; Jacobs, C. S.; Farkas, M. E.; Dervan, P. B. Improved nuclear localization of DNA-binding polyamides. Nucleic Acids Res 2007, 35, 363-370.
14. Dudouet, B.; Burnett, R.; Dickinson, L. A.; Wood, M. R.; Melander, C.; Belitsky, J. M.; Edelson, B.; Wurtz, N.; Briehn, C.; Dervan, P. B.; Gottesfeld, J. M. Accessibility of nuclear chromatin by DNA binding polyamides. Chem Biol 2003, 10, 859-867.
15. Suto, R. K.; Edayathumangalam, R. S.; White, C. L.; Melander, C.; Gottesfeld, J. M.; Dervan, P. B.; Luger, K. Crystal structures of nucleosome core particles in complex with minor groove DNA-binding ligands. J Mol Biol 2003, 326, 371-380.
16. Edayathumangalam, R. S.; Weyermann, P.; Gottesfeld, J. M.; Dervan, P. B.; Luger, K. Molecular recognition of the nucleosomal "supergroove". Proc Natl Acad Sci U S A 2004, 101, 6864-6869.
17. Gottesfeld, J. M.; Neely, L.; Trauger, J. W.; Baird, E. E.; Dervan, P. B. Regulation of gene expression by small molecules. Nature 1997, 387, 202-205.
391

Principal Investigator: Welch, Timothy R. 6.14
Postdoctoral Fellowship Application 2012
18. Chenoweth, D. M.; Harki, D. A.; Phillips, J. W.; Dose, C.; Dervan, P. B. Cyclic pyrrole-imidazole polyamides targeted to the androgen response element. J Am Chem Soc 2009, 131, 7182-7188.
19. Muzikar, K. A.; Nickols, N. G.; Dervan, P. B. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc Natl Acad Sci U S A 2009, 106, 16598-16603.
20. Nickols, N. G.; Dervan, P. B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci U S A 2007, 104, 10418-10423.
21. Nickols, N. G.; Jacobs, C. S.; Farkas, M. E.; Dervan, P. B. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem Biol 2007, 2, 561-571.
22. Olenyuk, B. Z.; Zhang, G. J.; Klco, J. M.; Nickols, N. G.; Kaelin, W. G., Jr.; Dervan, P. B. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc Natl Acad Sci U S A 2004, 101, 16768-16773.
23. Dickinson, L. A.; Burnett, R.; Melander, C.; Edelson, B. S.; Arora, P. S.; Dervan, P. B.; Gottesfeld, J. M. Arresting cancer proliferation by small-molecule gene regulation. Chem Biol 2004, 11, 1583-1594.
24. Buitenhuis, M.; Baltus, B.; Lammers, J. W.; Coffer, P. J.; Koenderman, L. Signal transducer and activator of transcription 5a (STAT5a) is required for eosinophil differentiation of human cord blood-derived CD34+ cells. Blood 2003, 101, 134-142.
25. Wierenga, A. T.; Vellenga, E.; Schuringa, J. J. Maximal STAT5-induced proliferation and self-renewal at intermediate STAT5 activity levels. Mol Cell Biol 2008, 28, 6668-6680.
26. Weber-Nordt, R. M.; Egen, C.; Wehinger, J.; Ludwig, W.; Gouilleux-Gruart, V.; Mertelsmann, R.; Finke, J. Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood 1996, 88, 809-816.
27. Li, G.; Miskimen, K. L.; Wang, Z.; Xie, X. Y.; Brenzovich, J.; Ryan, J. J.; Tse, W.; Moriggl, R.; Bunting, K. D. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood 2010, 115, 1416-1424.
28. Gesbert, F.; Griffin, J. D. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood 2000, 96, 2269-2276.
29. Horita, M.; Andreu, E. J.; Benito, A.; Arbona, C.; Sanz, C.; Benet, I.; Prosper, F.; Fernandez-Luna, J. L. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J Exp Med 2000, 191, 977-984.
30. Ehret, G. B.; Reichenbach, P.; Schindler, U.; Horvath, C. M.; Fritz, S.; Nabholz, M.; Bucher, P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem 2001, 276, 6675-6688.
31. Darnell, J. E. Transcription factors as targets for cancer therapy. Nature Reviews Cancer 2002, 2, 740-749.
32. Yu, H.; Jove, R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 2004, 4, 97-105.
33. Frank, D. A. STAT signaling in cancer: insights into pathogenesis and treatment strategies. Cancer Treat Res 2003, 115, 267-291.
34. Turkson, J.; Jove, R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613-6626.
35. Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta 2011, 1815, 104-114.
36. Levy, D. E.; Darnell, J. E., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002, 3, 651-662.
37. Darnell, J. E., Jr. STATs and gene regulation. Science 1997, 277, 1630-1635.
392

Principal Investigator: Welch, Timothy R. 6.15
Postdoctoral Fellowship Application 2012
38. Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607-2611.
39. Nevalainen, M. T.; Ahonen, T. J.; Yamashita, H.; Chandrashekar, V.; Bartke, A.; Grimley, P. M.; Robinson, G. W.; Hennighausen, L.; Rui, H. Epithelial defect in prostates of Stat5a-null mice. Lab Invest 2000, 80, 993-1006.
40. Libermann, T. A.; Zerbini, L. F. Targeting transcription factors for cancer gene therapy. Curr Gene Ther 2006, 6, 17-33.
41. Ahonen, T. J.; Xie, J.; LeBaron, M. J.; Zhu, J.; Nurmi, M.; Alanen, K.; Rui, H.; Nevalainen, M. T. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem 2003, 278, 27287-27292.
42. Dagvadorj, A.; Collins, S.; Jomain, J. B.; Abdulghani, J.; Karras, J.; Zellweger, T.; Li, H.; Nurmi, M.; Alanen, K.; Mirtti, T.; Visakorpi, T.; Bubendorf, L.; Goffin, V.; Nevalainen, M. T. Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology 2007, 148, 3089-3101.
43. Xi, S.; Zhang, Q.; Dyer, K. F.; Lerner, E. C.; Smithgall, T. E.; Gooding, W. E.; Kamens, J.; Grandis, J. R. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem 2003, 278, 31574-31583.
44. Lin, T. S.; Mahajan, S.; Frank, D. A. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene 2000, 19, 2496-2504.
45. Schwaller, J.; Parganas, E.; Wang, D.; Cain, D.; Aster, J. C.; Williams, I. R.; Lee, C. K.; Gerthner, R.; Kitamura, T.; Frantsve, J.; Anastasiadou, E.; Loh, M. L.; Levy, D. E.; Ihle, J. N.; Gilliland, D. G. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol Cell 2000, 6, 693-704.
46. Huang, M.; Dorsey, J. F.; Epling-Burnette, P. K.; Nimmanapalli, R.; Landowski, T. H.; Mora, L. B.; Niu, G.; Sinibaldi, D.; Bai, F.; Kraker, A.; Yu, H.; Moscinski, L.; Wei, S.; Djeu, J.; Dalton, W. S.; Bhalla, K.; Loughran, T. P.; Wu, J.; Jove, R. Inhibition of Bcr-Abl kinase activity by PD180970 blocks constitutive activation of Stat5 and growth of CML cells. Oncogene 2002, 21, 8804-8816.
47. Levis, M.; Allebach, J.; Tse, K. F.; Zheng, R.; Baldwin, B. R.; Smith, B. D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885-3891.
48. Kelly, J. A.; Spolski, R.; Kovanen, P. E.; Suzuki, T.; Bollenbacher, J.; Pise-Masison, C. A.; Radonovich, M. F.; Lee, S.; Jenkins, N. A.; Copeland, N. G.; Morse, H. C., 3rd; Leonard, W. J. Stat5 synergizes with T cell receptor/antigen stimulation in the development of lymphoblastic lymphoma. J Exp Med 2003, 198, 79-89.
49. Gu, L.; Vogiatzi, P.; Puhr, M.; Dagvadorj, A.; Lutz, J.; Ryder, A.; Addya, S.; Fortina, P.; Cooper, C.; Leiby, B.; Dasgupta, A.; Hyslop, T.; Bubendorf, L.; Alanen, K.; Mirtti, T.; Nevalainen, M. T. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer 2010, 17, 481-493.
50. Liu, X.; Robinson, G. W.; Gouilleux, F.; Groner, B.; Hennighausen, L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A 1995, 92, 8831-8835.
51. Horvath, C. M.; Wen, Z.; Darnell, J. E., Jr. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev 1995, 9, 984-994.
52. Basham, B.; Sathe, M.; Grein, J.; McClanahan, T.; D'Andrea, A.; Lees, E.; Rascle, A. In vivo identification of novel STAT5 target genes. Nucleic Acids Res 2008, 36, 3802-3818.
53. Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M. P.; Addya, S.; Fortina, P.; Dasgupta, A.; Hyslop, T.; Bubendorf, L.; Nevalainen, M. T. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential
393

Principal Investigator: Welch, Timothy R. 6.16
Postdoctoral Fellowship Application 2012
role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol 2010, 176, 1959-1972.
54. Chenoweth, D. M.; Dervan, P. B. Structural basis for cyclic Py-Im polyamide allosteric inhibition of nuclear receptor binding. J Am Chem Soc 2010, 132, 14521-14529.
55. Hsu, C. F.; Phillips, J. W.; Trauger, J. W.; Farkas, M. E.; Belitsky, J. M.; Heckel, A.; Olenyuk, B. Z.; Puckett, J. W.; Wang, C. C.; Dervan, P. B. Completion of a Programmable DNA-Binding Small Molecule Library. Tetrahedron 2007, 63, 6146-6151.
56. Chenoweth, D. M.; Dervan, P. B. Allosteric modulation of DNA by small molecules. Proc Natl Acad Sci U S A 2009, 106, 13175-13179.
57. Raskatov, J. A.; Hargrove, A. E.; So, A. Y.; Dervan, P. B. Pharmacokinetics of Py-Im Polyamides Depend on Architecture: Cyclic versus Linear. J Am Chem Soc 2012, Submitted.
58. Raskatov, J. A.; Nickols, N. G.; Dervan, P. B. Unpublished results, 2012. 59. Chenoweth, D. M.; Harki, D. A.; Dervan, P. B. Solution-phase synthesis of pyrrole-imidazole
polyamides. J Am Chem Soc 2009, 131, 7175-7181. 60. Harki, D. A.; Satyamurthy, N.; Stout, D. B.; Phelps, M. E.; Dervan, P. B. In vivo imaging of
pyrrole-imidazole polyamides with positron emission tomography. Proc Natl Acad Sci U S A 2008, 105, 13039-13044.
61. Baird, E. E.; Dervan, P. B. Solid phase synthesis of polyamides containing imidazole and pyrrole amino acids. J Am Chem Soc 1996, 118, 6141-6146.
62. Belitsky, J. M.; Nguyen, D. H.; Wurtz, N. R.; Dervan, P. B. Solid-phase synthesis of DNA binding polyamides on oxime resin. Bioorg Med Chem 2002, 10, 2767-2774.
63. Dose, C.; Farkas, M. E.; Chenoweth, D. M.; Dervan, P. B. Next generation hairpin polyamides with (R)-3,4-diaminobutyric acid turn unit. J Am Chem Soc 2008, 130, 6859-6866.
64. Pilch, D. S.; Poklar, N.; Baird, E. E.; Dervan, P. B.; Breslauer, K. J. The thermodynamics of polyamide-DNA recognition: hairpin polyamide binding in the minor groove of duplex DNA. Biochemistry 1999, 38, 2143-2151.
65. Muzikar, K. A.; Meier, J. L.; Gubler, D. A.; Raskatov, J. A.; Dervan, P. B. Expanding the Repertoire of Natural Product-Inspired Ring Pairs for Molecular Recognition of DNA. Org Lett 2011, ASAP.
66. Boyle, A. P.; Davis, S.; Shulha, H. P.; Meltzer, P.; Margulies, E. H.; Weng, Z.; Furey, T. S.; Crawford, G. E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311-322.
67. Gottesfeld, J. M.; Belitsky, J. M.; Melander, C.; Dervan, P. B.; Luger, K. Blocking transcription through a nucleosome with synthetic DNA ligands. J Mol Biol 2002, 321, 249-263.
68. Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V. B.; Wong, E.; Orlov, Y. L.; Zhang, W.; Jiang, J.; Loh, Y. H.; Yeo, H. C.; Yeo, Z. X.; Narang, V.; Govindarajan, K. R.; Leong, B.; Shahab, A.; Ruan, Y.; Bourque, G.; Sung, W. K.; Clarke, N. D.; Wei, C. L.; Ng, H. H. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106-1117.
69. Teglund, S.; McKay, C.; Schuetz, E.; van Deursen, J. M.; Stravopodis, D.; Wang, D.; Brown, M.; Bodner, S.; Grosveld, G.; Ihle, J. N. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 1998, 93, 841-850.
394

395
LIST OF ABBREVIATIONS
Ac2O Acetic anhydride
AcCl Acetyl Chloride
AcOH Acetic acid
AIBN Azobisisobutyronitrile
AQN Anthraquinone
ATP Adenosine-5’-triphosphate
9-BBN 9-Borabicyclo[3.3.1]nonane
Bn Benzyl
BnBr Benzyl bromide
Boc tert-Butoxycarbonyl
Boc2O Di-tert-butyldicarbonate
BOMCl Benzyloxymethyl chloride
BOP (Benzotriazol-1-yloxy)tris(dimethylamino) phophonium
hexafluorophosphate
CbzCl Benzyl chloroformate
cod 1,5-Cyclooctadiene
CREB cAMP response element-binding
d Day
Davis’s oxaziridine 2-(Phenylsulfonyl)-3-phenyloxaziridine
DEAD Diethyl azodicarboxylate
Dess-Martin or DMP Triacetoxy o-iodoxybenzoic acid
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DHQD Dihydroquinidine
DIBAL-H Diisobutylaluminium hydride
DMAP 4-(Dimethylamino)-pyridine

396
DMDO Dimethyldioxirane
DME Dimethoxyethane
DMF Dimethylformamide
DMS Dimethylsulfide
DMSO Dimethyl sulfoxide
dppf 1,1’-Bis(diphenylphosphino)ferrocene
DTBMP 2,6-di-tert-butyl-4-methylpyridine
EDCI N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide
ee Enantiomeric excess
e.r. Enantiomeric ratio
Et3N Triethylamine
EtOAc Ethyl Acetate
Et2O Diethyl ether
EtOH Ethanol
Fmoc Fluorenylmethyloxycarbonyl
FmocOSu 9-Fluorenylmethyl N-succinimidyl carbonate, N-(9-
fluorenylmethoxycarbonyloxy)succinimide
GSH Glutathione
h Hour
HATU O-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium
hexafluorophosphate
HIF-1 Hypoxia-inducible factor 1
hν Irradiation with light
iBu Isobutyl
iPr Isopropyl
iPr2NEt N,N-Diisopropylethylamine
Imid Imidazole
KOtBu Potassium tert-butoxide
LDA Lithium N,N-diisopropylamide
LHMDS Lithium bis(trimethylsilyl)amide
mCPBA m-Chloroperbenzoic acid

397
Me Methyl
MeCN Acetonitrile
MeI Methyl iodide
MeOH Methanol
min Minute
MOMCl Methyl chloromethyl ether
MRSA Methicillin-resistant Staphylococcus aureus
MsCl Methanesulfonyl chloride
NADPH Nicotinamide adenine dinucleotide phosphate
NaHMDS Sodium bis(trimethylsilyl)amide
NBS N-Bromosuccinimide
NCS N-Chlorosuccinimide
NIS N-Iodosuccinimide
nBuLi n-Butyllithium
N-PSP Diphenyl diselenide
O/N Overnight
Pd(OAc)2 Palladium(II) acetate
Pd/C Palladium on carbon
Ph Phenyl
PhI(OAc)2 Iodobenzene diacetate
PhMe Toluene
PPTS Pyridinium p-toluenesulfonate
PPY (R)-(+)-4-pyrrolidinopyridinyl(pentamethylcyclo-
pentadienyl)iron
Py. or Pyr. Pyridine
No Rxn No reaction was observed
RT or r.t. Room temperature
Sat’d Saturated aqueous solution
T3P Propane phosphonic acid anhydride
TBAF Tetrabutyl ammonium fluoride
TBDMSCl or TBSCl tert-Butyldimethylsilyl chloride

398
TBDPS tert-Butyldiphenylsilyl
TBSOTf tert-Butyldimethylsilyl trifluoromethanesulfonate
Teoc 2-(trimethylsilyl)ethoxycarbonyl
TES Triethylsilyl
Tf Trifluoromethanesulfonate
TFA Trifluoroacetic acid
TFAA Trifluoroacetic anhydride
Tf2O Trifluoromethanesulfonyl anhydride
THF Tetrahydrofuran
TIPS Triisopropylsilyl
TLC Thin layer chromatography
TMS Trimethylsilyl
TMSOTf Trimethylsilyl trifluoromethylsulfonate
Tr Trityl (triphenylmethyl)
Triton B Benzyltrimethylammonium hydroxide
pTsOH or pTSA p-Toluenesulfonic acid
µw Microwave irradiation
V-70 2,2’-Azobis(4-methoxy-2,4-dimethyl valeronitrile)
VRE Vancomycin-resistant Enterococcus




















