
Synthesis and characterizations of N , N ′-bis(diphenylphosphino)-2-(aminomethyl)aniline...
7
Synthesis and characterizations of N,N-bis(diphenylphosphino)ethylaniline derivatives and X-ray crystal structure of palladium (II), platinum (II) complexes Feyyaz Durap a, * , Nermin Biricik a , Bahattin Gu ¨mgu ¨m a , Saim O ¨ zkar b , Wee Han Ang c , Zhaofu Fei c , Rosario Scopelliti c a Department of Chemistry, University of Dicle, 21280 Diyarbakir, Turkey b Department of Chemistry, METU, 06531 Ankara, Turkey c Institut de Sciences et Inge ´ nierie Chimiques, Ecole Polytechnique Fe ´de ´rale de Lausanne, CH-1015 Lausanne, Switzerland Received 27 July 2007; accepted 5 September 2007 Available online 5 November 2007 Abstract N,N-Bis(diphenylphosphino)ethylaniline compounds, [Ph 2 P] 2 N-C 6 H 4 -C 2 H 5 , with ethyl groups at the ortho- and para-positions have been synthesized. Oxidation of the aminophosphines with hydrogen peroxide, elemental sulfur and selenium gave the corresponding oxi- des, sulfides and selenides [Ph 2 P(E)] 2 N-C 6 H 4 -C 2 H 5 (E = O, S, Se). Complexes [MCl 2 {(Ph 2 P) 2 N-C 6 H 4 -(C 2 H 5 )}] (M = Pd, Pt) and [Cu{(Ph 2 P) 2 N-C 6 H 4 -C 2 H 5 } 2 ]PF 6 were obtained by the reaction of N,N-bis(diphenylphosphino)ethylaniline with [MCl 2 (COD)] (M = Pd, Pt) and [Cu(MeCN) 4 ]PF 6 . The new compounds were characterized by NMR, IR spectroscopy and microanalysis. In addition, representative solid-state structures of the palladium and platinum complexes were determined using single crystal X-ray diffraction analyses. Ó 2007 Elsevier Ltd. All rights reserved. Keywords: Aminophosphine; Synthesis; Oxidation; Complexation; X-ray structures 1. Introduction The chemistry of aminophosphines containing direct P–N bonds is one of the most challenging areas in main group chemistry [1]. Among the numerous aminophos- phines, bis(diphenylphosphino)alkylaniline derivatives are particularly interesting due to their relatively high stability and their ability to chelate transition metals such as palla- dium, platinum and copper [2,3]. Many aminophosphine ligands and their complexes have been investigated as co-catalysts in a number of catalytic processes [4,5]. Some aminophosphines and derivatives have also found applica- tion as anticancer drugs [6], herbicides and antimicrobial agents, as well as neuroactive agents [7,8]. The synthesis of bis(diphenylphosphino)alkylaniline is usually achieved in high yield via the aminolysis of aniline [9,10], which allows for the incorporation of additional functional groups such as O and N donors [11,12], or p-donors such as allyl groups [13]. The presence of the bulky groups attached to the phosphorus center renders the aminophos- phine more stable against hydrolysis. The substituents at the aniline backbone can also play an important role in determining the outcome of the products [14]. In a recent study [15], we found that electron-donating groups often lead to the exclusive formation of aminophosphines [P(III)–N–P(III)], whereas electron-withdrawing groups, such as nitrile and trifluoromethyl at the ortho-position can lead to formation of iminobiphosphine [P(III)– P(V)@N] species. 0277-5387/$ - see front matter Ó 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.poly.2007.09.011 * Corresponding author. Tel.: +90 4122488550; fax: +90 4122488300. E-mail address: [email protected] (F. Durap). www.elsevier.com/locate/poly Available online at www.sciencedirect.com Polyhedron 27 (2008) 196–202
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Synthesis and characterizations of N , N ′-bis(diphenylphosphino)-2-(aminomethyl)aniline...

Available online at www.sciencedirect.com
www.elsevier.com/locate/poly
Polyhedron 27 (2008) 196–202
Synthesis and characterizationsof N,N-bis(diphenylphosphino)ethylaniline derivatives
and X-ray crystal structure of palladium (II),platinum (II) complexes
Feyyaz Durap a,*, Nermin Biricik a, Bahattin Gumgum a, Saim Ozkar b,Wee Han Ang c, Zhaofu Fei c, Rosario Scopelliti c
a Department of Chemistry, University of Dicle, 21280 Diyarbakir, Turkeyb Department of Chemistry, METU, 06531 Ankara, Turkey
c Institut de Sciences et Ingenierie Chimiques, Ecole Polytechnique Federale de Lausanne, CH-1015 Lausanne, Switzerland
Received 27 July 2007; accepted 5 September 2007Available online 5 November 2007
Abstract
N,N-Bis(diphenylphosphino)ethylaniline compounds, [Ph2P]2N-C6H4-C2H5, with ethyl groups at the ortho- and para-positions havebeen synthesized. Oxidation of the aminophosphines with hydrogen peroxide, elemental sulfur and selenium gave the corresponding oxi-des, sulfides and selenides [Ph2P(E)]2N-C6H4-C2H5 (E = O, S, Se). Complexes [MCl2{(Ph2P)2N-C6H4-(C2H5)}] (M = Pd, Pt) and[Cu{(Ph2P)2N-C6H4-C2H5}2]PF6 were obtained by the reaction of N,N-bis(diphenylphosphino)ethylaniline with [MCl2(COD)](M = Pd, Pt) and [Cu(MeCN)4]PF6. The new compounds were characterized by NMR, IR spectroscopy and microanalysis. In addition,representative solid-state structures of the palladium and platinum complexes were determined using single crystal X-ray diffractionanalyses.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Aminophosphine; Synthesis; Oxidation; Complexation; X-ray structures
1. Introduction
The chemistry of aminophosphines containing directP–N bonds is one of the most challenging areas in maingroup chemistry [1]. Among the numerous aminophos-phines, bis(diphenylphosphino)alkylaniline derivatives areparticularly interesting due to their relatively high stabilityand their ability to chelate transition metals such as palla-dium, platinum and copper [2,3]. Many aminophosphineligands and their complexes have been investigated asco-catalysts in a number of catalytic processes [4,5]. Someaminophosphines and derivatives have also found applica-tion as anticancer drugs [6], herbicides and antimicrobial
0277-5387/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.poly.2007.09.011
* Corresponding author. Tel.: +90 4122488550; fax: +90 4122488300.E-mail address: [email protected] (F. Durap).
agents, as well as neuroactive agents [7,8]. The synthesisof bis(diphenylphosphino)alkylaniline is usually achievedin high yield via the aminolysis of aniline [9,10], whichallows for the incorporation of additional functionalgroups such as O and N donors [11,12], or p-donors suchas allyl groups [13]. The presence of the bulky groupsattached to the phosphorus center renders the aminophos-phine more stable against hydrolysis. The substituents atthe aniline backbone can also play an important role indetermining the outcome of the products [14]. In a recentstudy [15], we found that electron-donating groups oftenlead to the exclusive formation of aminophosphines[P(III)–N–P(III)], whereas electron-withdrawing groups,such as nitrile and trifluoromethyl at the ortho-positioncan lead to formation of iminobiphosphine [P(III)–P(V)@N] species.

F. Durap et al. / Polyhedron 27 (2008) 196–202 197
Herein, we describe the synthesis of new N,N-bis(diph-enylphosphino)ethylaniline with an ethyl group at theortho- and para-positions. In order to study their reactivi-ties, we prepared their corresponding complexes withselected transition metals (Cu, Pd, Pt), and also the amino-phosphine oxides, sulfides and selenides. The compoundswere fully characterized using multi-nuclear NMR spectro-scopic methods and the solid-state structures of two of theproducts were established by single crystal X-ray diffrac-tion analyses.
2. Experimental
All reactions were performed under argon unless other-wise stated. Ph2PCl and the ethyl anilines were purchasedfrom Fluka and used directly. The starting materials[MCl2(COD)] (M = Pd, Pt, COD = 1,5-cyclooctadiene)[16,17] and [Cu(MeCN)4]PF6 [18] were prepared accordingto the literature procedures. Solvents were dried using theappropriate reagents and distilled prior to use. Infraredspectra were recorded as KBr pellets in the range 4000–400 cm�1 on a Mattson 1000 ATI UNICAM FT-IR spec-trometer. 1H NMR (400.1 MHz) spectra were recorded ona Bruker AC 400 spectrometer, and 31P–{1H} NMR spec-tra at 162 MHz with d referenced to external 85% H3PO4.Microanalysis was carried out on a Fisons EA 1108CHNS-O instrument.
2.1. General procedure for the synthesis of
N,N-bis(diphenylphosphino)ethylanilines
2.1.1. [(Ph2P)2N-C6H4-C2H5] (1, 2)
N,N-Bis(diphenylphosphino)ethylanilines were preparedby the aminolysis reaction of H2N-C6H4-C2H5 (1: 2-C2H5;2: 4-C2H5) with two equivalents of Ph2PCl according to theprocedure given in the literature [19].
2.2. General procedure for the synthesis of the chalcogenides
2.2.1. N,N-Bis(diphenyloxophosphino)ethylanilines
[(Ph2P(O))2N-C6H4-C2H5] (1a, 2a)
A THF solution (10 mL) of N,N-bis(diphenylphosphi-no)ethylaniline (0.20 g, 0.420 mmol) and aqueous H2O2
(30% w/w, 0.083 mL) was stirred for 2 h at room tempera-ture. The solution was evaporated to dryness underreduced pressure and the white solids 1a and 2a wereobtained.
N,N-Bis(diphenyloxophosphino)-2-ethylaniline (1a):Yield: 0.065 g, 25%, mp 180–181 �C. 1H NMR (CDCl3) d(ppm): 6.85–7.89 (m, 24H, Ar H), 2.72–2.78 (q, 2H, CH2,3JHH = 7.4 Hz), 0.90–0.94 (t, 3H, CH3, 3JHH = 7.3 Hz).31P–{1H} NMR (CDCl3) d (ppm): 23.6 (s). Selected IR, m(cm�1): 901 (P–N–P), 1219 (PO). Anal. Calc. forC32H29NP2O2: C, 73.70; H, 5.56; N, 2.68. Found: C,73.56; H, 5.47; N, 2.46%.
N,N-Bis(diphenyloxophosphino)-4-ethylaniline (2a):Yield: 0.18 g, 70%, mp 215–217 �C. 1H NMR (CDCl3) d
(ppm): 6.74–7.88 (m, 24H, Ar H), 2.32–2.38 (q, 2H, CH2,3JHH = 7.8 Hz), 0.96–1.00 (t, 3H, CH3, 3JHH = 7.6 Hz).31P–{1H} NMR (CDCl3) d (ppm): 24.3 (s). Selected IR, m(cm�1): 909 (P–N–P), 1213 (PO). Anal. Calc. forC32H29NP2O2: C, 73.70; H, 5.56; N, 2.68. Found: C,73.84; H, 5.36; N, 2.59%.
2.2.2. N,N-Bis(diphenylthiophosphino)ethylanilines
[(Ph2P(S))2N-C6H4-C2H5] (1b, 2b)
To solid N,N-bis(diphenylphosphino)ethylaniline(0.20 g, 0.420 mmol) and S8 (0.027 g, 0.840 mmol) wasadded THF (20 mL) and this was refluxed for 6 h. The vol-ume was concentrated in vacuum to ca. 1–2 mL and addi-tion of n-hexane (20 mL) gave 1b and 2b as white solidswhich were collected by suction filtration.
N,N-Bis(diphenylthiophosphino)-2-ethylaniline (1b):Yield: 0.117 g, 52%, mp 172–174 �C. 1H NMR (CDCl3) d(ppm): 6.85–8.13 (m, 24H, Ar H), 2.43–2.48 (q, 2H, CH2,3JHH = 7.4 Hz), 0.85–0.89 (t, 3H, CH3, 3JHH = 7.5 Hz).31P–{1H} NMR (CDCl3) d (ppm): 68.3 (s). Selected IR, m(cm�1): 862 (P–N–P), 650 (P–S). Anal. Calc. forC32H29NP2S2: C, 69.44; H, 5.24; N, 2.53; S, 11.57. Found:C, 69.63; H, 5.18; N, 2.73; S, 11.62%.
N,N-Bis(diphenylthiophosphino)-4-ethylaniline (2b):Yield: 0.132 g, 58%, mp 196–197 �C. 1H NMR (CDCl3) d(ppm): 6.68–8.09 (m, 24H, Ar H), 2.32–2.34 (q, 2H, CH2,3JHH = 7.8 Hz), 0.97–1.00 (t, 3H, CH3, 3JHH = 7.6 Hz).31P–{1H} NMR (CDCl3) d (ppm): 68.8 (s). Selected IR, m(cm�1): 882 (P–N–P), 658 (P–S). Anal. Calc. forC32H29NP2S2: C, 69.44; H, 5.24; N, 2.53; S, 11.57. Found:C, 69.31; H, 5.30; N, 2.24; S, 11.41%.
2.2.3. N,N-Bis(diphenylselenophosphino)ethylanilines
[(Ph2P(Se))2N-C6H4-C2H5] (1c, 2c)
To solid N,N-bis(diphenylphosphino)ethylaniline(0.20 g, 0.420 mmol) and grey Se (0.064 g, 0.840 mmol)was added THF (20 mL) and this was refluxed for 6 h.The volume was concentrated in vacuum to ca. 1–2 mLand addition of n-hexane (20 mL) gave 1c and 2c whichwere collected by suction filtration.
N,N-Bis(diphenylselenophosphino)-2-ethylaniline (1c):Yield: 0.155 g, 59%, mp 174–175 �C. 1H NMR (CDCl3) d(ppm): 6.87–8.15 (m, 24H, Ar H), 2.38–2.44 (q, 2H, CH2,3JHH = 7.4 Hz), 0.88–0.91 (t, 3H, CH3, 3JHH = 7.4 Hz).31P–{1H} NMR (CDCl3) d (ppm): 67.5, J(PSe): 788 Hz.Selected IR, m (cm�1): 855 (P–N–P), 579 (P–Se). Anal. Calc.for C32H29NP2Se2: C, 59.35; H, 4.48; N, 2.16. Found: C,59.34; H, 4.22; N, 2.03%.
N,N-Bis(diphenylselenophosphino)-4-ethylaniline (2c):Yield: 0.176 g, 67%, mp 182–184 �C. 1H NMR (CDCl3) d(ppm): 6.68–8.19 (m, 24H, Ar H), 2.32–2.35 (q, 2H, CH2,3JHH = 7.5 Hz), 0.96–1.00 (t, 3H, CH3, 3JHH = 7.5 Hz).31P–{1H} NMR (CDCl3) d (ppm): 69.6, J(PSe): 782 Hz.Selected IR, m (cm�1): 876 (P–N–P), 565 (P–Se). Anal. Calc.for C32H29NP2Se2: C, 59.35; H, 4.48; N, 2.16. Found: C,59.48; H, 4.53; N, 2.07%.

198 F. Durap et al. / Polyhedron 27 (2008) 196–202
2.3. General procedure for the synthesis of the complexes
2.3.1. Dichloro{N,N-bis(diphenylphosphino)ethylaniline}-
palladium (II) (1d, 2d)
A solution of [PdCl2(COD)] (0.049 g, 0.172 mmol)and N,N-bis(diphenylphosphino) ethylaniline (0.086 g,0.175 mmol) in CH2Cl2 (10 mL) was stirred for 1.5 h. Thevolume was concentrated in vacuum to ca. 1–2 mL andaddition of diethyl ether (20 mL) gave 1d and 2d as yellowsolids which were collected by suction filtration and driedin vacuum.
Dichloro{N,N-bis(diphenylphosphino)-2-ethylaniline}-palladium (II) (1d): Yield: 0.094 g, 85%, mp >300 �C. 1HNMR (CDCl3) d (ppm): 6.57–7.94 (m, 24H, Ar H), 1.38–1.41 (q, 2H, CH2, 3JHH = 7.4 Hz), 0.91–0.95 (t, 3H, CH3,3JHH = 7.5 Hz). 31P–{1H} NMR (CDCl3) d (ppm): 37.2(s). Selected IR, m (cm�1): 900 (P–N–P). Anal. Calc. forC32H29NP2PdCl2: C, 60.86; H, 4.59; N, 2.22. Found: C,60.77; H, 4.39; N, 2.05%.
Dichloro{N,N-bis(diphenylphosphino)-4-ethylaniline}-palladium (II) (2d): Yield: 0.10 g, 97%, mp >300 �C. 1HNMR (CDCl3) d (ppm): 6.43–7.92 (m, 24H, Ar H); 2.52–2.54 (q, 2H, CH2, 3JHH = 7.5 Hz), 1.15–1.18 (t, 3H, CH3,3JHH = 7.5 Hz). 31P–{1H} NMR (CDCl3) d (ppm): 34.8(s). Selected IR, m (cm�1): 913 (P–N–P). Anal. Calc. forC32H29NP2PdCl2: C, 60.86; H, 4.59; N, 2.22. Found: C,60.66; H, 4.38; N, 2.33%. Slow diffusion of diethyl etherinto a CH2Cl2 solution of 2d over 48 h gave crystals suit-able for X-ray diffraction.
2.3.2. Dichloro{N,N-bis(diphenylphosphino)ethylaniline}-
platinum (II) (1e, 2e)
A solution of [PtCl2(COD)] (0.034 g, 0.0860 mmol) andN,N-bis(diphenylphosphino)ethylaniline (0.0427 g, 0.0875mmol) in CH2Cl2 (10 mL) was stirred for 1.5 h. Thevolume was concentrated in vacuum to ca. 1–2 mL andaddition of diethyl ether (20 mL) gave 1e and 2e as whitesolids which were collected by suction filtration and driedin vacuum.
Dichloro{N,N-bis(diphenylphosphino)-2-ethylaniline}-platinum (II) (1e): Yield: 0.056 g, 86%, mp 339–341 �C.1H NMR (CDCl3) d (ppm): 6.48–7.93 (m, 24H, Ar H),1.21–1.25 (q, 2H, CH2, 3JHH = 7.1 Hz), 0.23–0.27 (t, 3H,CH3, 3JHH = 7.2 Hz). 31P–{1H} NMR (CDCl3) d (ppm):22.3, J(PtP): 3348.6 Hz. Selected IR, m (cm�1): 900 (P–N–P). Anal. Calc. for C32H29NP2PtCl2: C, 50.86; H, 3.84; N,1.85. Found: C, 50.84; H, 3.63; N, 1.67%. Slow diffusionof diethyl ether into a CHCl3 solution of 1e over 48 h gavecrystals suitable for X-ray crystallography.
Dichloro{N,N-bis(diphenylphosphino)-4-ethylaniline}-platinum (II) (2e): Yield: 0.057 g, 88%, mp 339–340 �C.1H NMR (CDCl3) d (ppm): 6.36–7.90 (m, 24H, Ar H),2.51–2.56 (q, 2H, CH2, 3JHH = 7.5 Hz), 1.14–1.17 (t, 3H,CH3, 3JHH = 7.5 Hz). 31P–{1H} NMR (CDCl3) d (ppm):20.6, J(PtP): 3324 Hz. Selected IR, m (cm�1): 906 (P–N–P).Anal. Calc. for C32H29NP2PtCl2: C, 50.86; H, 3.84; N,1.85. Found: C, 50.71; H, 3.78; N, 1.76%.
2.3.3. Bis[N,N-bis(diphenylphosphino)ethylaniline]copper
(I) hexafluorophosphates (1f, 2f)To a solution of [Cu(MeCN)4]PF6 (0.071 g, 0.191 mmol)
in CH2Cl2 (20 mL) was added N,N-bis(diphenylphosphi-no)ethylaniline (0.185 g, 0.380 mmol) and the resultingsolution was stirred for 2 h. The volume was concentratedin vacuum to ca. 1–2 mL and addition of diethyl ether(20 mL) gave 1f and 2f as white solids which were collectedby suction filtration and dried in vacuum.
Bis[N,N-bis(diphenylphosphino)-2-ethylaniline]copper (I)
hexafluorophosphate (1f ): Yield: 0.183 g, 83%, mp 204–205 �C. 1H NMR (CDCl3) d (ppm): 6.87–8.15 (m, 24H,Ar H), 1.21–1.27 (q, 2H, CH2, 3JHH = 7.6 Hz), 0.02–0.05(t, 3H, CH3, 3JHH = 7.5 Hz). 31P–{1H} NMR (CDCl3) d(ppm): 87.7 (s), d(PF6): �144.3(m); J(PF6): 1418 Hz. SelectedIR, m (cm�1): 843 (P–N–P). Anal. Calc. for C64H58N2-P5F6Cu: C, 64.73; H, 4.89; N, 2.36. Found: C, 64.69; H,5.02; N, 2.21%.
Bis[N,N-bis(diphenylphosphino)-4-ethylaniline]copper (I)
hexafluorophosphate (2f ): Yield: 0.20 g, 91%, mp 339–340 �C. 1H NMR (CDCl3) d (ppm): 6.68–8.19 (m, 24H,Ar H), 2.45–2.51 (q, 2H, CH2, 3JHH = 7.6 Hz), 1.08–1.12(t, 3H, CH3, 3JHH = 7.6 Hz). 31P–{1H} NMR (CDCl3) d(ppm): 88.2 (s), d(PF6): �144.3(m); J(PF6): 1424 Hz. SelectedIR, m (cm�1): 848 (P–N–P). Anal. Calc. for C64H58N2P5-F6Cu: C, 64.73; H, 4.89; N, 2.36. Found: C, 64.79; H,4.95; N, 2.41%.
2.3.4. Structure determination in the solid state
Data collection for 1e and 2d was performed on anOxford-Kuma Xcalibur diffractor with a Sapphire CCDarea detector at 140(2) K and data reduction was per-formed using CrysAlis RED [20]. The structures were solvedby direct methods using SIR-92 [21], and refined by full-matrix least-squares refinement (against F2) using SHELXTL
[22,23], with all non-hydrogen atoms refined anisotropi-cally. The hydrogen atoms were placed in their geometri-cally generated positions and treated as riding on theirparent atoms with Uiso(H) = 1.2Ueq(C), except for the ter-minal H-atoms on C8 where Uiso(H) = 1.5Ueq(C). Empiri-cal absorption corrections were applied using DELABS [24].Relevant crystallographic data are compiled in Table 1and graphical representations of the structures were madewith DIAMOND [25].
3. Results and discussion
N,N-Bis(diphenylphosphino)ethylanilines, (Ph2P)2N-C6H4-C2H5 (1: 2-C2H5; 2: 4-C2H5) were prepared from H2-N-C6H4-C2H5 and Ph2PCl by aminolysis in CH2Cl2 inaccordance with our previous reports [19] (Scheme 1).
The reaction was monitored by 31P–{1H} NMR spec-troscopy. In contrast to reactions of diphenylphosphoruschloride (Ph2PCl) with anilines bearing electron-withdraw-ing groups [15], the reaction mixture of Ph2PCl with 2-ethy-laniline or 4-ethylaniline gave a single resonance at d(P)61.6 ppm for 1 and 68.5 ppm for 2, and no formation of

NPPh2
Ph2P
NPPh2
Ph2P
EE
C2H5 C2H5+ 2E
THF
Scheme 2. Oxidation of (Ph2P)2N-C6H4-C2H5 with aqueous H2O2
(1a, 2a), elemental sulfur (1b, 2b) and powder selenium (1c, 2c).
Table 1Crystallographic data for compounds 1e Æ CHCl3 and 2d Æ CH2Cl2
Compound 1e Æ CHCl3 2d Æ CH2Cl2
Chemicalformula
C32H29Cl3NP2Pt Æ CHCl3 C32H29Cl3NP2Pd Æ CH2Cl2
Formula weight 874.86 751.73Crystal system monoclinic monoclinicSpace group P21/c P21/na (A) 11.9722(6) 10.2901(10)b (A) 14.6887(4) 14.8786(15)c (A) 19.9617(10) 21.665(2)a (�) 90 90b (�) 106.536(5) 98.332(9)c (�) 90 90Volume (A3) 3365.2(3) 3282.0(6)Z 4 4Dcalc (g cm�3) 1.727 1.521F(000) 1712 1520l (mm�1) 4.687 1.013Temperature (K) 140(2) 140(2)Wavelength (A) 0.71073 0.71073Measured
reflections19557 19146
Uniquereflections
5737 5489
Uniquereflections[I > 2r(I)]
4218 1781
Number of data/restraints/parameters
5737/0/380 5489/192/371
Ra [I > 2r(I)] 0.0264 0.0480wR2
a (all data) 0.0547 0.1051Goodness-of-fit
(GOF)b0.891 0.677
a R =P
iFoj � jFci/PjFoj, wR2 = {
P[w(F2
o � F2c)2]/
P[w(F2
o)2]}1/2.b GOF = {
P[w(F2
o � F2c)2]/(n � p)}1/2 where n is the number of data and
p is the number of parameters refined.
NH2N
Ph2P PPh2
C2H5C2H5
+ 2Ph2PClEt3N/CH2Cl2
+ 2Et3NHCl0 oC
Scheme 1. Syntheses of N,N-bis(diphenylphosphino)ethylanilines by theaminolysis reaction of H2N-C6H4-C2H5with Ph2PCl (1: 2-C2H5; 2:4-C2H5).
F. Durap et al. / Polyhedron 27 (2008) 196–202 199
iminobiphosphine species (Ph2P–PPh2@NC6H4(C2H5))could be observed. The 31P–{1H} NMR signal of com-pound 1 is shifted upfield compared to that of 2, althoughthey both lie in the expected region of 60–70 ppm [26]. Thecompounds are air-stable solids but gradually undergodecomposition on exposure to moisture.
Oxidation of 1 and 2 with aqueous hydrogen peroxide,elemental sulfur and selenium gave the corresponding oxi-des 1a, 2a, sulfides 1b, 2b and selenides 1c, 2c (Scheme 2).The 31P NMR spectrum of 1a and 2a displays one singletat 23.6 ppm and 24.3 ppm, respectively, suggesting thetwo phosphorus atoms are chemically equivalent in bothcompounds. Similarly, singlet resonances were also foundin the 31P NMR spectra of 1b and 2b (68.3 ppm and
68.8 ppm) and 1c, 2c (67.5 ppm and 69.6 ppm). Thecomposition of the oxidized derivatives 1a, 2a, 1b, 2b, 1c
and 2c were further confirmed by using microanalysisand IR spectroscopy, and were found to be in good agree-ment with the theoretical values [12,13,15].
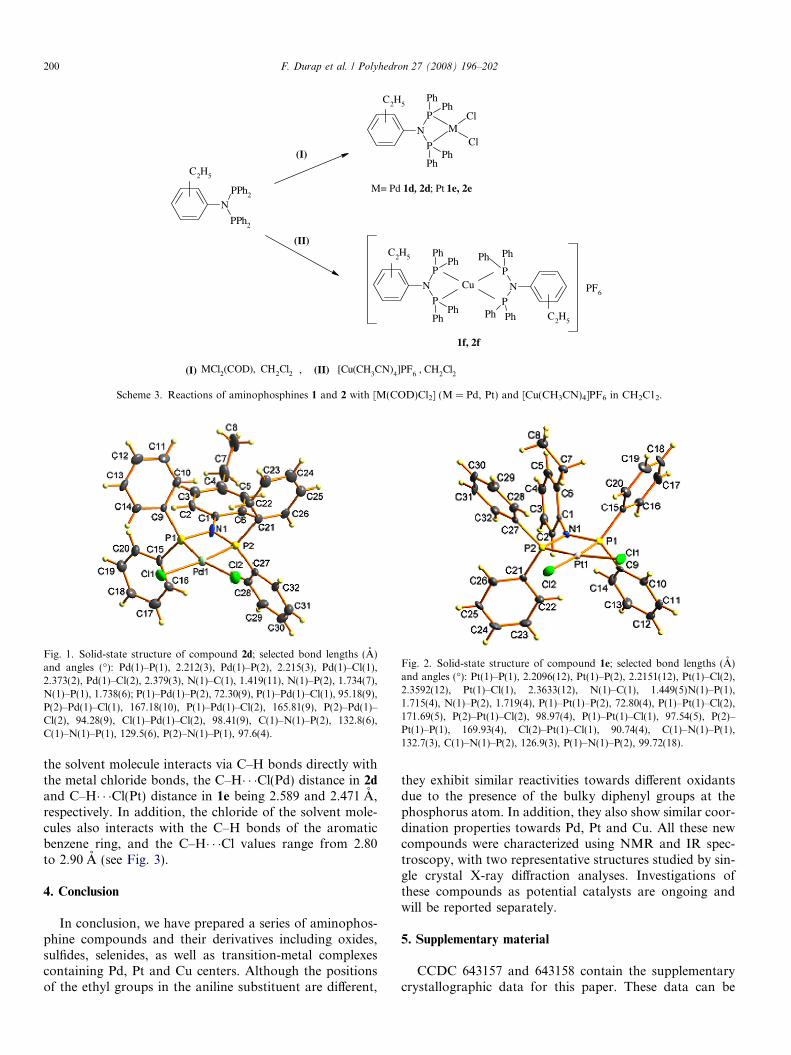
Reactions of compounds 1 and 2 with [M(COD)Cl2](M = Pd, Pt; COD = 1,5-cyclooctadiene) and [Cu(CH3-CN)4]PF6 in CH2C12 gave the corresponding complexes1d, 2d, 1e, 2e [M{(Ph2P)2N-C6H4-C2H5}Cl2] (M = Pd; Pt)and 1f, 2f [Cu{(Ph2P)2N-C6H4-C2H5}2]PF6 in good yields(Scheme 3).
The 31P NMR chemical shifts of complexes 1d and 2d
(37.2 ppm and 34.8 ppm) are within the range expectedfor structurally similar complexes where an electron-with-drawing group is attached to the aniline ring [27]. Thechemical shifts of 1e, 2e, 1f and 2f in the 31P NMR spectraare also within the expected range and they show minorsolvent dependence [13]. In the 1H NMR spectra, thesecomplexes display overlapped multiplets in the region6.70–8.10 ppm for the aromatic H-atoms, and a quartetat 2.50 ppm and a triplet at 1.15 ppm for the hydrogenatoms of the ethyl group.
Single crystals of 1e and 2d suitable for X-ray diffractionanalyses were obtained by slow diffusion of diethyl etherinto a solution of the complexes in chloroform and dichlo-romethane, respectively. They both crystallized as 1:1 sol-vent adducts and the crystallographic data of 1e Æ CHCl3and 2d Æ CH2Cl2 are listed in Table 1.
The molecular representation of complexes 1e and 2d
are shown in Figs. 1 and 2, with key bond parameters givenin the figure captions. In general, the key bond lengths andangles of complexes 1e and 2d in the solid state are verysimilar, with minimal structural differences due to the relo-cation of the ethyl groups. The P–N bond distances of1.734(7) and 1.738(6) A in complex 2d are essentially thesame, but they are longer than that of the platinum com-plex 1e [1.715(4) and 1.719(4) A], although they are withinthe expected value range on comparison to similar struc-tures [27]. Both complexes form metallacycles, i.e. P–N–P–Pd and P–N–P–Pt, respectively, that are nearly planarwith torsion angles P–N–P–Pd of 0.5(4)� in 2d and P–N–P–Pt of 5.78(17)� in 1e. Despite the difference of the twometal atoms, the P–Pd–P angle in 2d [72.30(9)�] is nottoo different from that of the P–Pt–P angle in 1e [72.80(4)�].
It is interesting to note that compounds 1e and 2d crys-tallised under similar conditions, both forming 1:1 adductswith solvent molecules, presumably driven by H-bondinginteractions with the solvent molecules. In both instances,
NPPh2
PPh2
MP Cl
ClN
P
Ph
PhPh
Ph
MCl2(COD), CH2Cl2 [Cu(CH3CN)4]PF6 , CH2Cl2
PF6N
P
P
Ph
PhPh
Ph
N
P
P
Ph
PhPh
Ph
Cu
C2H5
C2H5
C2H5
C2H5
(I)
(II)
(I) (II),
M= Pd 1d, 2d; Pt 1e, 2e
1f, 2f
Scheme 3. Reactions of aminophosphines 1 and 2 with [M(COD)Cl2] (M = Pd, Pt) and [Cu(CH3CN)4]PF6 in CH2C12.
Fig. 2. Solid-state structure of compound 1e; selected bond lengths (A)and angles (�): Pt(1)–P(1), 2.2096(12), Pt(1)–P(2), 2.2151(12), Pt(1)–Cl(2),2.3592(12), Pt(1)–Cl(1), 2.3633(12), N(1)–C(1), 1.449(5)N(1)–P(1),1.715(4), N(1)–P(2), 1.719(4), P(1)–Pt(1)–P(2), 72.80(4), P(1)–Pt(1)–Cl(2),171.69(5), P(2)–Pt(1)–Cl(2), 98.97(4), P(1)–Pt(1)–Cl(1), 97.54(5), P(2)–Pt(1)–P(1), 169.93(4), Cl(2)–Pt(1)–Cl(1), 90.74(4), C(1)–N(1)–P(1),132.7(3), C(1)–N(1)–P(2), 126.9(3), P(1)–N(1)–P(2), 99.72(18).
Fig. 1. Solid-state structure of compound 2d; selected bond lengths (A)and angles (�): Pd(1)–P(1), 2.212(3), Pd(1)–P(2), 2.215(3), Pd(1)–Cl(1),2.373(2), Pd(1)–Cl(2), 2.379(3), N(1)–C(1), 1.419(11), N(1)–P(2), 1.734(7),N(1)–P(1), 1.738(6); P(1)–Pd(1)–P(2), 72.30(9), P(1)–Pd(1)–Cl(1), 95.18(9),P(2)–Pd(1)–Cl(1), 167.18(10), P(1)–Pd(1)–Cl(2), 165.81(9), P(2)–Pd(1)–Cl(2), 94.28(9), Cl(1)–Pd(1)–Cl(2), 98.41(9), C(1)–N(1)–P(2), 132.8(6),C(1)–N(1)–P(1), 129.5(6), P(2)–N(1)–P(1), 97.6(4).
200 F. Durap et al. / Polyhedron 27 (2008) 196–202
the solvent molecule interacts via C–H bonds directly withthe metal chloride bonds, the C–H� � �Cl(Pd) distance in 2d
and C–H� � �Cl(Pt) distance in 1e being 2.589 and 2.471 A,respectively. In addition, the chloride of the solvent mole-cules also interacts with the C–H bonds of the aromaticbenzene ring, and the C–H� � �Cl values range from 2.80to 2.90 A (see Fig. 3).
4. Conclusion
In conclusion, we have prepared a series of aminophos-phine compounds and their derivatives including oxides,sulfides, selenides, as well as transition-metal complexescontaining Pd, Pt and Cu centers. Although the positionsof the ethyl groups in the aniline substituent are different,
they exhibit similar reactivities towards different oxidantsdue to the presence of the bulky diphenyl groups at thephosphorus atom. In addition, they also show similar coor-dination properties towards Pd, Pt and Cu. All these newcompounds were characterized using NMR and IR spec-troscopy, with two representative structures studied by sin-gle crystal X-ray diffraction analyses. Investigations ofthese compounds as potential catalysts are ongoing andwill be reported separately.
5. Supplementary material
CCDC 643157 and 643158 contain the supplementarycrystallographic data for this paper. These data can be

Fig. 3. Hydrogen bonding interactions in 2d (left) and 1e (right).
F. Durap et al. / Polyhedron 27 (2008) 196–202 201
obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallo-graphic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or e-mail: [email protected].
Acknowledgments
We thank The Council of Dicle University (DUAPK-05-FF-19 and DUAPK-06-FF-41 projects) for financialsupport and Ecole Polytechnique Federale de Lausannefor X-ray structure analyses.
References
[1] (a) F.R. Hartley, The Chemistry of Organophosphorus Compounds,vol. 1, Wiley, Manchester, 1990;(b) N.N. Greenwood, A. Earnshaw, Chemistry of the Elements,Pergamon Press, Oxford, 1984, p. 619.
[2] (a) M.S. Balakrishna, V. Sreenivasa Reddy, S.S. Krishnamurthy, J.F.Nixon, J.C.T.R. Burckett St. Laurent, Coord. Chem. Rev. 129 (1994) 1;(b) F. Agbossou, J.F. Carpentier, F. Hapiot, Coord. Chem. Rev. 178–180 (1998) 1615;(c) M.R.I. Zubiri, J.D. Woollins, Comment Inorg. Chem. 24 (5–6)(2003) 189;(d) N. Biricik, F. Durap, B. Gumgum, Z. Fei, R. Scopelliti, TransitionMet. Chem. 32 (2007) 877.
[3] (a) T. Appleby, J.D. Woollins, Coord. Chem. Rev. 235 (2002) 121;(b) Z. Fei, P.J. Dyson, Coord. Chem. Rev. 249 (2005) 2056.
[4] (a) I. Bachert, P. Braunstein, R. Hasselbring, New J. Chem. 20 (1996)993;(b) I. Bachert, P. Braunstein, M.K. McCart, F. Fabrizi de Biani, F.Lashi, P. Zanello, G. Kickelbick, U. Schubert, J. Organomet. Chem.573 (1999) 47;(c) I. Bachert, I. Bartussek, P. Braunstein, E. Guillon, J. Rose, G.Kickelbick, J. Organomet. Chem. 580 (1999) 257;(d) B. Gumgum, N. Biricik, F. Durap, _I. Ozdemir, N. Gurbuz, W.H.Ang, P.J. Dyson, Appl. Organomet. Chem. 21 (2007) 711.
[5] I.M.R. Zubiri, M.L. Clarke, D.F. Foster, D.J. Cole-Hamilton,A.M.Z. Slawin, J.D. Woollins, J. Chem. Soc., Dalton Trans. 969(2001).
[6] J. Reedijk, J. Chem. Soc., Chem. Commun. 801 (1996).[7] (a) D.J. Birdsall, J. Green, T.Q. Ly, J. Novosad, M. Necas, A.M.Z.
Slawin, J.D. Woollins, Z. Zak, Eur. J. Inorg. Chem. 1445 (1999);
(b) P. Bhattacharyya, T.Q. Ly, A.M.Z. Slawin, J.D. Woollins,Polyhedron 20 (2001) 1803.
[8] P. Kafarski, P. Mastalerz, Beitr. Wriskstorfforch 21 (1984).[9] (a) Z. Fei, R. Scopelliti, P.J. Dyson, Eur. J. Inorg. Chem. (2003) 3527;
(b) Z. Fei, R. Scopelliti, Paul J. Dyson, Eur. J. Inorg. Chem. (2004)530;(c) N. Biricik, Z. Fei, R. Scopelliti, P.J. Dyson, Eur. J. Inorg. Chem.(2004) 4232;(d) I. Fernandez, F. Breher, P.S. Pregosin, Z. Fei, P.J. Dyson, Inorg.Chem. 44 (2005) 7616.
[10] K.G. Gaw, M.B. Smith, A.M.Z. Slawin, New J. Chem. 24 (2000) 429.[11] K.G. Gaw, M.B. Smith, J.W. Steed, J. Organomet. Chem. 664 (2002)
294.[12] (a) Q. Zhang, S.M. Aucott, A.M.Z. Slawin, J.D. Woollins, Eur. J.
Inorg. Chem. 7 (2002) 1635;(b) M.L. Clarke, G.L. Holliday, A.M.Z. Slawin, J.D. Woollins, J.Chem. Soc., Dalton Trans. 6 (2002) 1093;(c) A.M.Z. Slawin, M. Wainwright, J.D. Woollins, J. Chem. Soc.,Dalton Trans. 4 (2002) 513;(d) N. Biricik, Z. Fei, R. Scopelliti, P.J. Dyson, Helv. Chim. Acta 86(2003) 3281;(e) Z. Fei, D. Zhao, N. Biricik, R. Scopelliti, P.J. Dyson, Inorg.Chem. 44 (2004) 2228;(f) A. Baysal, M. Aydemir, F. Durap, B. Gumgum, S. Ozkar, L.T.Yıldırım, Polyhedron 26 (2007) 3373.
[13] (a) A.M.Z. Slawin, H.L. Milton, J. Wheatley, J.D. Woollins,Polyhedron 23 (2004) 3125;(b) A.M.Z. Slawin, J. Wheatley, J.D. Woollins, Phosphorus, Sulfur,Silicon Relat. Elem. 179 (4–5) (2004) 991;(c) A.M.Z. Slawin, J. Wheatley, J.D. Woollins, Eur. J. Inorg. Chem.(2005) 713;(d) B. Gumgum, O. Akba, F. Durap, L.T. Yıldırım, D. Ulku, S.Ozkar, Polyhedron 25 (2006) 3133.
[14] Z. Fei, R. Scopelliti, P.J. Dyson, Inorg. Chem. 42 (2003) 2125.[15] Z. Fei, R. Scopelliti, P.J. Dyson, J. Chem. Soc., Dalton Trans. (2003)
2772.[16] D. Drew, J.R. Doyle, Inorg. Synth. 13 (1972) 47.[17] J.X. McDermott, J.F. White, G.M. Whitesides, J. Am. Chem. Soc. 98
(1976) 6521.[18] G.J. Kubas, Inorg. Synth. 19 (1979) 90.[19] N. Biricik, F. Durap, C. Kayan, B. Gumgum, Heteroatom Chem. 18
(6) (2007) 613.[20] Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2002.[21] A. Altomare, M.C. Burla, G. Camalli, G. Cascarano, C. Giacovazzo,
A. Gualiardi, G. Polidori, J. Appl. Crystallogr. 27 (1994) 435.[22] G.M. Sheldrick, University of Gottingen, Gottingen, Germany, 1997.[23] G.M. Sheldrick, Bruker AXS, Inc., Madison, WI 53719, USA, 1997.

202 F. Durap et al. / Polyhedron 27 (2008) 196–202
[24] DIAMOND 3.0a, Crystal Impact GbR, Bonn, Germany.[25] N. Walker, D. Stuart DELABS, Acta Crystallogr., Sect. A 39 (1983) 158.[26] Z. Fei, D. Zhao, N. Biricik, R. Scopelliti, P.J. Dyson, Inorg. Chem. 44
(2004) 2228.
[27] (a) H.L. Milton, M.V. Wheatley, A.M.Z. Slawin, J.D. Woollins,Inorg. Chem. Commun. 7 (10) (2004) 1106;(b) Z. Fei, W.H. Ang, D. Zhao, R. Scopelliti, P.J. Dyson, Inorg.Chim. Acta 359 (2006) 2635.













![N -[4-( N -Cyclohexylsulfamoyl)phenyl]acetamide](https://static.fdokumen.com/doc/165x107/632f4f4de68feab59a0210b7/n-4-n-cyclohexylsulfamoylphenylacetamide.jpg)



![Stacking patterns of thieno[3,2- b ]thiophenes functionalized by sequential palladium-catalyzed Suzuki and Heck cross-coupling reactions](https://static.fdokumen.com/doc/165x107/6344b1c438eecfb33a063498/stacking-patterns-of-thieno32-b-thiophenes-functionalized-by-sequential-palladium-catalyzed.jpg)


