
Surfactant-assisted size control of hydroxyapatite nanorods for bone tissue engineering
8
Colloids and Surfaces B: Biointerfaces 116 (2014) 666–673 Contents lists available at ScienceDirect Colloids and Surfaces B: Biointerfaces journal homepage: www.elsevier.com/locate/colsurfb Surfactant-assisted size control of hydroxyapatite nanorods for bone tissue engineering Nguyen Kim Nga a,∗ , Luu Truong Giang a , Tran Quang Huy b , Pham Hung Viet c , Claudio Migliaresi d a School of Chemical Engineering, Hanoi University of Science and Technology, 1 Dai Co Viet Road, Hanoi, Viet Nam b National Institute of Hygiene and Epidemiology, 1 Yersin Street, Hanoi, Viet Nam c Research Center for Environmental Technology and Sustainable Development, Hanoi University of Science, 334 Nguyen Trai Street, Hanoi, Viet Nam d Department of Industrial Engineering and BIOtech Research Centre, University of Trento, Via Mesiano, 77, I-38123 Trento, Italy article info Article history: Received 29 July 2013 Received in revised form 26 October 2013 Accepted 2 November 2013 Available online 12 November 2013 Keywords: Hydroxyapatite nanorods Pluronic SBF Bioactivity Bone tissue engineering abstract This study presents the physicochemical characterization of the pluronic surfactant-assisted size control of hydroxyapatite (HAp) nanorods for bone tissue engineering (BTE). Rod-shaped HAp nanoparticles were synthesized via a simple route by hydrothermal treatment and with the assistance of the tri- block co-polymer PEO 20 -PPO 70 -PEO 20 (P123). The films of poly (d, l) lactic acid (PDLLA) were prepared as a substrate to spread synthesized HAp nanorods. Powder X-ray diffraction (XRD), field electron scanning microscopy, Fourier transform infrared spectroscopy, nitrogen adsorption isotherms, and energy-dispersive X-ray spectroscopy were used to characterize the structure and composition of the HAp samples. Results showed that regular rod-shaped HAp nanoparticles (with a mean length of 120 nm and a mean width of 28 nm) were successfully produced. Moreover, synthesized HAp nanorods revealed the rapid formation of bone-like apatite with a distinctive morphology, similar to flower-like apatite; the formation was observed as early as 7 days after incubation in stimulated body fluids. This study is a posi- tive addition to the ongoing research on the preparation of HAp nanostructures toward the development of biocompatible composite scaffolds for BTE applications. © 2013 Elsevier B.V. All rights reserved. 1. Introduction Bone tissue engineering (BTE) has attracted considerable scientific attention in the fields of nanomedicine and biotechnol- ogy because it offers a more promising method for bone repair and regeneration than traditional methods (e.g., allografts and autografts) [1]. BTE uses scaffolding materials as template for cell interactions and formation of extracellular matrix, thereby providing structural support to the newly formed tissue [2]. Till date, a number of biomaterials have been investigated for possible use in BTE scaffolds, and these biomaterials can be classified into the following categories according to their composition: biodegradable polymers [3–9], bioactive inorganics [10,11], and polymer/bioactive ceramic composites [12–20]. Calcium phos- phates with Ca/P ratios of 1.5–2.0 and belonging to the group of bioactive inorganics have three main compounds namely, tetraphosphate Ca 4 P 2 O 9 , hydroxyapatite Ca 10 (PO 4 ) 6 (OH) 2 , and tricalcium phosphate Ca 3 (PO 4 ) 2 . Among them, hydroxyapatite (HAp) is thermodynamically the most stable calcium phosphate ∗ Corresponding author. Tel.: +84 4 38680 110; fax: +84 4 38680 070. E-mail address: [email protected] (N.K. Nga). in physiological environments [10] and a major component of bones and teeth. HAp has been widely studied alone or as filler in polymeric composites for BTE applications because of its chemical similarity to inorganic components of bone matrix, strong affinity for host hard tissues, and osteoconductivity [6]. A number of stud- ies have focused on investigating the formation of nanoscale HAp for additional applications, such as producing restorable scaffolds that can be replaced by endogenous hard tissues over time [21]. Several wet chemistry methods (e.g., co-precipitation, hydrothermal, and ultrasonic) assisted by organic molecules (e.g., sodium dodecylbenzene sulfonate, sodium dodecyl sulfate [22], block copolymer poly(lactide-co-glycolide)-block- monomethoxy (polyethylene glycol) and polyvinyl pyrrolidone (PVP) [23], cetyltrimethylammonium bromide (CTAB) [24], polyvinyl alcohol [25], and polyethylene glycol 400/CTAB [26], CTAB/n-pentanol/n-hexane/water [27], double-hydrophilic block copolymer poly-(vinylpyrrolidone)-b-poly(vinylpyrrolidone-alt- maleic anhydride)-b-poly-(vinylpyrrolidone) [28]) have been studied to produce rod-shaped (or needle-like) HAp nanopar- ticles. A study has suggested that better osteoconductivity can be achieved if the synthetic HAp resembles bone minerals in composition, size, and morphology [29]. In a previous study, we successfully prepared HAp nanorods via hydrothermal technique 0927-7765/$ – see front matter © 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.colsurfb.2013.11.001
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Surfactant-assisted size control of hydroxyapatite nanorods for bone tissue engineering

St
NCa
b
c
d
a
ARRAA
KHPSBB
1
soaacpduibppott(
0h
Colloids and Surfaces B: Biointerfaces 116 (2014) 666–673
Contents lists available at ScienceDirect
Colloids and Surfaces B: Biointerfaces
journa l homepage: www.e lsev ier .com/ locate /co lsur fb
urfactant-assisted size control of hydroxyapatite nanorods for boneissue engineering
guyen Kim Ngaa,∗, Luu Truong Gianga, Tran Quang Huyb, Pham Hung Vietc,laudio Migliaresid
School of Chemical Engineering, Hanoi University of Science and Technology, 1 Dai Co Viet Road, Hanoi, Viet NamNational Institute of Hygiene and Epidemiology, 1 Yersin Street, Hanoi, Viet NamResearch Center for Environmental Technology and Sustainable Development, Hanoi University of Science, 334 Nguyen Trai Street, Hanoi, Viet NamDepartment of Industrial Engineering and BIOtech Research Centre, University of Trento, Via Mesiano, 77, I-38123 Trento, Italy
r t i c l e i n f o
rticle history:eceived 29 July 2013eceived in revised form 26 October 2013ccepted 2 November 2013vailable online 12 November 2013
eywords:ydroxyapatite nanorods
a b s t r a c t
This study presents the physicochemical characterization of the pluronic surfactant-assisted size controlof hydroxyapatite (HAp) nanorods for bone tissue engineering (BTE). Rod-shaped HAp nanoparticleswere synthesized via a simple route by hydrothermal treatment and with the assistance of the tri-block co-polymer PEO20-PPO70-PEO20 (P123). The films of poly (d, l) lactic acid (PDLLA) were preparedas a substrate to spread synthesized HAp nanorods. Powder X-ray diffraction (XRD), field electronscanning microscopy, Fourier transform infrared spectroscopy, nitrogen adsorption isotherms, andenergy-dispersive X-ray spectroscopy were used to characterize the structure and composition of the
luronicBFioactivityone tissue engineering
HAp samples. Results showed that regular rod-shaped HAp nanoparticles (with a mean length of 120 nmand a mean width of 28 nm) were successfully produced. Moreover, synthesized HAp nanorods revealedthe rapid formation of bone-like apatite with a distinctive morphology, similar to flower-like apatite; theformation was observed as early as 7 days after incubation in stimulated body fluids. This study is a posi-tive addition to the ongoing research on the preparation of HAp nanostructures toward the developmentof biocompatible composite scaffolds for BTE applications.
. Introduction
Bone tissue engineering (BTE) has attracted considerablecientific attention in the fields of nanomedicine and biotechnol-gy because it offers a more promising method for bone repairnd regeneration than traditional methods (e.g., allografts andutografts) [1]. BTE uses scaffolding materials as template forell interactions and formation of extracellular matrix, therebyroviding structural support to the newly formed tissue [2]. Tillate, a number of biomaterials have been investigated for possiblese in BTE scaffolds, and these biomaterials can be classified
nto the following categories according to their composition:iodegradable polymers [3–9], bioactive inorganics [10,11], andolymer/bioactive ceramic composites [12–20]. Calcium phos-hates with Ca/P ratios of 1.5–2.0 and belonging to the groupf bioactive inorganics have three main compounds namely,
etraphosphate Ca4P2O9, hydroxyapatite Ca10(PO4)6(OH)2, andricalcium phosphate Ca3(PO4)2. Among them, hydroxyapatiteHAp) is thermodynamically the most stable calcium phosphate∗ Corresponding author. Tel.: +84 4 38680 110; fax: +84 4 38680 070.E-mail address: [email protected] (N.K. Nga).
927-7765/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.colsurfb.2013.11.001
© 2013 Elsevier B.V. All rights reserved.
in physiological environments [10] and a major component ofbones and teeth. HAp has been widely studied alone or as filler inpolymeric composites for BTE applications because of its chemicalsimilarity to inorganic components of bone matrix, strong affinityfor host hard tissues, and osteoconductivity [6]. A number of stud-ies have focused on investigating the formation of nanoscale HApfor additional applications, such as producing restorable scaffoldsthat can be replaced by endogenous hard tissues over time [21].
Several wet chemistry methods (e.g., co-precipitation,hydrothermal, and ultrasonic) assisted by organic molecules(e.g., sodium dodecylbenzene sulfonate, sodium dodecylsulfate [22], block copolymer poly(lactide-co-glycolide)-block-monomethoxy (polyethylene glycol) and polyvinyl pyrrolidone(PVP) [23], cetyltrimethylammonium bromide (CTAB) [24],polyvinyl alcohol [25], and polyethylene glycol 400/CTAB [26],CTAB/n-pentanol/n-hexane/water [27], double-hydrophilic blockcopolymer poly-(vinylpyrrolidone)-b-poly(vinylpyrrolidone-alt-maleic anhydride)-b-poly-(vinylpyrrolidone) [28]) have beenstudied to produce rod-shaped (or needle-like) HAp nanopar-
ticles. A study has suggested that better osteoconductivity canbe achieved if the synthetic HAp resembles bone minerals incomposition, size, and morphology [29]. In a previous study, wesuccessfully prepared HAp nanorods via hydrothermal technique
es B: B
ablcsops[ab[
osBbssttfitdHa
2
2
osCPM(pBD
2
Ct(bvss2wfie3bto3
rafi
N.K. Nga et al. / Colloids and Surfac
ssisted by CTAB; the HAp nanorods exhibited high cellular affinityut with wide size distributions (mean diameter of 17–58 nm and
ength of 85–439 nm) [24]. Surfactants (e.g., CTAB) are efficienthemicals in modifying the desired size and shape of nano-tructures because of their self-assembly into rod-like micellesr lamellar structures at high concentrations to improve theirroperties [24,30]. As reported, the wide size distributions ofynthetic HAp can reduce the mechanical properties of scaffolds31]. Despite recent advances in the synthesis of HAp for BTEpplications, the products still face major drawbacks related toioactivities, fracture toughness, and other mechanical properties21].
In this study, we focus on the synthesis and characterizationf HAp nanoparticles that resemble bone minerals with narrowize distributions to develop biocompatible composite scaffolds forTE applications. Rod-shaped HAp nanoparticles were synthesizedy triblock co-polymer PEO20-PPO70-PEO20 (P123) to control theirizes. P123 is a non-ionic chemical that has been used to assist theynthesis of rod-like nanostructures [32]. A possible mechanism forhe formation of rod-shaped HAp nanoparticles was also proposedo identify the effect of P123 on the formation of HAp nanorods. Thelms of poly (d, l) lactic acid (PDLLA) were prepared as substrateso spread HAp nanorods and test their bioactivity. Bioactivity wasetermined by assessing the formation of apatite on the surface ofAp/PDLLA films upon incubation in stimulated body fluids (SBF)fter 1, 3, and 7 days.
. Experimental
.1. Chemicals
All reagents were of analytical grade and used as received with-ut further purification. Calcium chloride dihydrate (CaCl2·2H2O),odium monophosphate dihydrate (Na2HPO4·2H2O), NaOH,2H5OH, CHCl3, hydrochloric acid (HCl), the pluronic co-polymerEO20-PPO70-PEO20 (P123), NaCl, NaHCO3, KCl, K2HPO4·3H2O,gCl2·6H2O, Na2SO4, and tris-hydroxymethyl aminomethane
(HOCH2)3CNH2) were obtained from Sigma–Aldrich. PDLLA wasurchased from Boehringer Ingelheim (Ingelheim, Germany).uffer solutions (pH 4, 7, and 9) were purchased from Merck.eionized water was used to prepare all solutions and reagents.
.2. Synthesis and characterization of HAp nanoparticles
For a typical experiment and in the absence of P123, 2.53 gaCl2·2H2O was dissolved in 86 mL deionized water and broughto a pH ranging from 9.5 to 11 by adding a 0.4 weight percentwt%) NaOH solution. 0.2 M Na2HPO4 was added into the solutiony drops to a Ca/P molar ratio of 1.67. The resulting mixture wasigorously stirred at 40 ◦C for 4 h to form white suspension. Theuspension was then transferred to a 200 mL teflon-lined stainlessteel autoclave, and the suspension was maintained at 180 ◦C for4 h. White precipitate was collected and washed several timesith deionized water before being dried at 60 ◦C overnight and,nally, calcinated at 500 ◦C for 1 h. In a typical reaction in the pres-nce of P123, a mixture of a specific amount of P123 (either 1, 2, org) and 2.53 g CaCl2·2H2O were dissolved in 86 mL deionized watery stirring for 30 min to produce clear gel. The following steps arehe same as those for the synthesis without P123. Finally, a seriesf samples was produced and denoted 0P123, 1P123, 2P123, andP123 based on the amount of P123 used.
Powder X-ray diffraction patterns of synthesized samples wereecorded on a Siemens D5005 diffractometer through CuK� radi-tion (� = 0.15406 nm). Powder morphology was examined byeld emission scanning microscopy (S4800, Hitachi, Japan). The
iointerfaces 116 (2014) 666–673 667
average diameter and length of nanorods were measured throughscanning electron microscopic (SEM) images. Fourier transforminfrared spectra (FTIR) were recorded on a Nicolet 6700 spec-trometer by KBr pellet technique in the range of 4000–400 cm−1
with a resolution of 4 cm−1. All measurements were conductedat room temperature. The chemical composition of HAp parti-cles was determined with a JEOL JED-2300 Analysis Station with aZAF (atomic number absorption fluorescence) quantitative analysisprogram. Nitrogen adsorption–desorption isotherms were mea-sured at −196 ◦C with a Micromeritics ASAP 2020 apparatus. Thetotal surface areas were calculated by the Brunauer–Emmett–Teller(BET) method, whereas pore size distributions were determined bythe Barrett–Joyner–Halenda (BJH) method.
2.3. Production and characterization of HAp/PDLLA films
The preparation of HAp/PDLLA films was described in our pre-vious report [24]. To prepare PDLLA films, we cast 0.3 g PDLLAdissolved in 3 mL CHCl3 on a 55 mm aluminum plate. The filmswere air-dried under a chemical hood for 3 h and vacuum-driedfor 24 h to remove all solvent. A suspension of C2H5OH with 30 mgof HAp nanorods was dispersed on the resulting PDLLA films. Afterbeing placed at 55–60 ◦C for 2 h, the composite films of HAp/PDLLAwere vacuum-dried for another 3 h and stored in a desiccator untilused. Three different samples of HAp nanorods (0P123, 1P123, and2P123) were used, which resulted in three different HAp/PDLLAfilms denoted as F 0P123, F 1P123, and F 2P123. The morpholo-gies of resulting films and the presence of HAp nanorods on thesubstrate were checked by FE-SEM imaging at low and high mag-nifications.
2.4. In vitro bioactivity tests
SBF with ion concentrations similar to those in human bloodplasma was prepared according to the method reported in otherstudies [33] and filtered by a 0.22 �m Millipore filter system toeliminate bacterial contamination. The SBF was preserved in a plas-tic bottle in a refrigerator at 4 ◦C. Prior to in vitro experiments,all films of HAp/PDLLA were cut into disks with 5 mm diameter,sterilized in 70% ethanol at 4 ◦C overnight, and dried under a lami-nar airflow hood. Two specimens for each kind of film (F 0P123,F 1P123, and F 2P123) were soaked in 10 mL of SBF in closedpolyethylene containers at 37 ◦C. The SBF solution was replacedevery 2.5 days. After 1, 3, and 7 days of soaking, the specimenswere removed from the SBF, rinsed with deionized water, and driedin warm flowing air. All operations were conducted in a laminarairflow hood to avoid microorganism contamination. The apatitemineralization of HAp/PDLLA films was assessed through FE-SEMand energy-dispersive X-ray spectroscopy (EDXS).
3. Results and discussion
3.1. Characterization of HAp nanoparticles
Four samples were synthesized with different P123 concentra-tions (one, non-assisted-P123 and the other three, assisted-P123samples). The main characteristics of representative samples aresummarized in Table 1. The XRD patterns of representative sam-ples are presented in Fig. 1. The diffraction peaks can be indexed tothe hexagonal lattice of HAp (JCPDS No. 9-432), whereas the char-acteristic peaks are at (0 0 2), (1 0 2), (2 1 0), (2 1 1), (1 1 2), (3 0 0),(2 0 2), and (3 0 1). No characteristic peak of other phases (e.g.,
tricalcium phosphate and tetracalcium phosphate) was observed,which would confirm that a pure HAp phase was produced for allsamples. The morphology of the HAp samples was then identifiedby FE-SEM images. Rod-shaped particles with a smooth surface
668 N.K. Nga et al. / Colloids and Surfaces B: Biointerfaces 116 (2014) 666–673
Table 1Characteristics of representative HAp samples.
Sample codes Mean diameter ± SD (nm) Mean length ± SD (nm) Aspect ratios Ca/P Morphology
0P123 103 ± 26 585 ± 315 5.68 1.55 Rod-like shape1P123 74 ± 25 383 ± 152 5.18 1.61 Rod-like shape2P123 28 ± 5 120 ± 32
SD: standard deviation; n = 50.
Fig. 1. XRD patterns of HAp samples at different concentrations of P123: (a) 0P123,(b) 1P123, and (c) 2P123 calcinated at 500 ◦C for 1 h.
Fig. 2. FE-SEM images of representative HAp samples synthesized at different concentrainset shows particle size distribution of 2P123.
4.29 1.66 Rod-like shape
were produced for the synthesized samples, independent of P123concentration (Fig. 2 and Fig. S1, Supporting material). Accordingto Fig. 2a and Table 1, large and irregular rod-shaped HAp particles(mean width of 103 nm and mean length of 585 nm) were obtainedfor non-assisted P123 samples (0P123 samples). Obtained assistedP123 samples with increasing P123 concentrations can result in asignificant decrease in the size of HAp nanoparticles. 1P123 sam-ple (Fig. 2b) shows considerably smaller rod-shaped particles than0P123 samples; the mean diameter is 74 nm and the mean lengthis 383 nm. Furthermore, the FE-SEM image (Fig. 2c) and particlesize distributions of 2P123 demonstrate that these samples havethe smallest and most uniform rod-shaped particles (their sizesare mainly in the range of 25–30 nm in width and 100 nm–130 nmin length) among the assisted P123 samples. However, a furtherincrease of P123 concentration produced larger and irregular rod-shaped HAp. As shown in Fig. S1, irregular HAp particles with mean
width of 82 nm and mean length of 365 nm were formed for 3P123samples. These observations confirm that the synthesis in the pres-ence of P123 does not affect the final morphology of nanorods butsignificantly affects their sizes. 2 g of P123 should be optimal for thetions of P123: (a) 0P123, (b) 1P123, and (c) 2P123 calcinated at 500 ◦C for 1 h. The
N.K. Nga et al. / Colloids and Surfaces B: Biointerfaces 116 (2014) 666–673 669
Fc(
ptrtnPPCs0Cr
popma4abd6istt
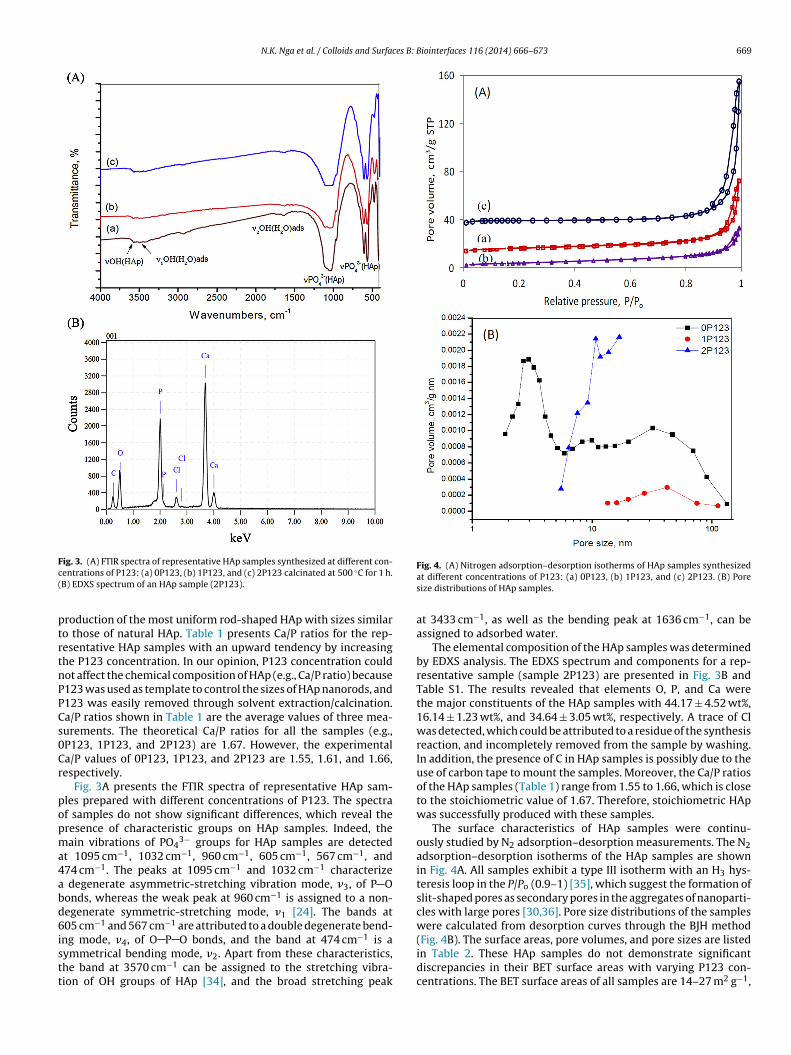
ig. 3. (A) FTIR spectra of representative HAp samples synthesized at different con-entrations of P123: (a) 0P123, (b) 1P123, and (c) 2P123 calcinated at 500 ◦C for 1 h.B) EDXS spectrum of an HAp sample (2P123).
roduction of the most uniform rod-shaped HAp with sizes similaro those of natural HAp. Table 1 presents Ca/P ratios for the rep-esentative HAp samples with an upward tendency by increasinghe P123 concentration. In our opinion, P123 concentration couldot affect the chemical composition of HAp (e.g., Ca/P ratio) because123 was used as template to control the sizes of HAp nanorods, and123 was easily removed through solvent extraction/calcination.a/P ratios shown in Table 1 are the average values of three mea-urements. The theoretical Ca/P ratios for all the samples (e.g.,P123, 1P123, and 2P123) are 1.67. However, the experimentala/P values of 0P123, 1P123, and 2P123 are 1.55, 1.61, and 1.66,espectively.
Fig. 3A presents the FTIR spectra of representative HAp sam-les prepared with different concentrations of P123. The spectraf samples do not show significant differences, which reveal theresence of characteristic groups on HAp samples. Indeed, theain vibrations of PO4
3− groups for HAp samples are detectedt 1095 cm−1, 1032 cm−1, 960 cm−1, 605 cm−1, 567 cm−1, and74 cm−1. The peaks at 1095 cm−1 and 1032 cm−1 characterizedegenerate asymmetric-stretching vibration mode, �3, of P O
onds, whereas the weak peak at 960 cm−1 is assigned to a non-egenerate symmetric-stretching mode, �1 [24]. The bands at05 cm−1 and 567 cm−1 are attributed to a double degenerate bend-
−1
ng mode, �4, of O P O bonds, and the band at 474 cm is aymmetrical bending mode, �2. Apart from these characteristics,he band at 3570 cm−1 can be assigned to the stretching vibra-ion of OH groups of HAp [34], and the broad stretching peakFig. 4. (A) Nitrogen adsorption–desorption isotherms of HAp samples synthesizedat different concentrations of P123: (a) 0P123, (b) 1P123, and (c) 2P123. (B) Poresize distributions of HAp samples.
at 3433 cm−1, as well as the bending peak at 1636 cm−1, can beassigned to adsorbed water.
The elemental composition of the HAp samples was determinedby EDXS analysis. The EDXS spectrum and components for a rep-resentative sample (sample 2P123) are presented in Fig. 3B andTable S1. The results revealed that elements O, P, and Ca werethe major constituents of the HAp samples with 44.17 ± 4.52 wt%,16.14 ± 1.23 wt%, and 34.64 ± 3.05 wt%, respectively. A trace of Clwas detected, which could be attributed to a residue of the synthesisreaction, and incompletely removed from the sample by washing.In addition, the presence of C in HAp samples is possibly due to theuse of carbon tape to mount the samples. Moreover, the Ca/P ratiosof the HAp samples (Table 1) range from 1.55 to 1.66, which is closeto the stoichiometric value of 1.67. Therefore, stoichiometric HApwas successfully produced with these samples.
The surface characteristics of HAp samples were continu-ously studied by N2 adsorption–desorption measurements. The N2adsorption–desorption isotherms of the HAp samples are shownin Fig. 4A. All samples exhibit a type III isotherm with an H3 hys-teresis loop in the P/Po (0.9–1) [35], which suggest the formation ofslit-shaped pores as secondary pores in the aggregates of nanoparti-cles with large pores [30,36]. Pore size distributions of the sampleswere calculated from desorption curves through the BJH method
(Fig. 4B). The surface areas, pore volumes, and pore sizes are listedin Table 2. These HAp samples do not demonstrate significantdiscrepancies in their BET surface areas with varying P123 con-centrations. The BET surface areas of all samples are 14–27 m2 g−1,
670 N.K. Nga et al. / Colloids and Surfaces B:
Table 2Surface characteristics of representative HAp samples.
Sample codes SBETa (m2 g−1) VBJH
b (cm3 g−1) Dpc (nm)
0P123 21 0.095 3; 10; 311P123 14 0.039 422P123 27 0.16 12
a
bswdada
3
b(a
1
Tacscastadeai
astsHdboPetctimitw
3
tbi
BET surface area.b Total pore volume determined using desorption branch of the isotherms.c Peak pore sizes from the pore size distributions.
ut the samples differ in the structures of their pores. Among threeamples studied, sample 2P123 has narrow pore size distributionith a maximum peak of 12 nm, whereas sample 0P123 shows aisordered pore size distribution with three maximum peaks ofpproximately 3, 10, and 31 nm. Sample 1P123 has a broad pore sizeistribution, which ranges from 20 nm to 80 nm and concentratedt approximately 42 nm.
.2. Possible mechanism for HAp particle formation
A possible mechanism for the formation of HAp nanorods maye proposed based on the results obtained (Fig. 5). Without P123Fig. 5A) and at high pH, the reaction between the solutions of CaCl2nd Na2HPO4 yields HAp via the following reaction:
0Ca2+ + 6PO43− + 2OH− → Ca10(PO4)6(OH)2
he resulting HAp was hydrothermally treated at 180 ◦C, calcinatedt 500 ◦C, and produced large irregularly rod-shaped HAp parti-les (with mean width of 103 nm and mean length of 585 nm) ashown in Fig. 2a and Table 1. Without hydrothermal treatment afteralcination at 500 ◦C, the aggregated spherical HAp particles withverage diameters of 350 nm were obtained instead of the rod-haped particles (Fig. S2). These results confirm that hydrothermalreatment at 180 ◦C provides energy for the HAp nuclei to grownd produce elongated HAp crystals. This crystal growth is possiblyue to the intrinsic crystal habit caused by the difference in latticenergy between the different crystal planes of HAp [37]. Withoutny control, the obtained HAp crystals are extremely long and haverregular size.
Theoretically, P123, a non-ionic surfactant can be self-ssembled into polymeric micelles with various morphologies (e.g.,pherical, cylindrical, or lamellar structures) at high concentra-ions above the critical micelle concentration [36]. The size andhape of micelles determine the morphology of the synthesizedAp particles. In aqueous solution, P123 forms nano-sized cylin-rical micelles that function as “nanoreactors”. Ca2+ can preferablyind first with the hydrophilic head groups on the inner surfacef the resulting micelles through hydrogen bonds and then, withO4
3−/OH− to form the P123-HAp complex that provides the nec-ssary topology that in turns directs mineral growth and leads tohe formation of elongated HAp nanocrystals (Fig. 5B). At low con-entrations of P123 (e.g., 1P123 samples), a number of HAp nucleihat are unbound to micelles, later grow uncontrollably to producerregularly shaped and large HAp crystals. Meanwhile, the sizes of
icelles increase as the P123 concentration increases. The increasen the size of micelles results in an increase in the sizes of HAp par-icles. The most uniform HAp particles that resemble bone mineralsere obtained with the suitable concentration of 2 g.
.3. In vitro bioactivity of synthesized HAp/PDLLA films in SBF
An essential characteristic of biomaterials is their ability to bindo living bone by forming a bone-like apatite layer on its surfaceoth in vitro and in vivo. The powder form of the HAp particles
s unfavorable when working with SBF. Thus, the bioactivity of
Biointerfaces 116 (2014) 666–673
the HAp samples was evaluated through in vitro tests by soak-ing films of HAp/PDLLA in SBF. Fig. S3 represents the SEM imagesof HAp/PDLLA composite films with low and high magnificationsbefore immersion into SBF. Generally, the surfaces of the compos-ite films are rough. The images with high magnification confirm afull cover of rod-shaped HAp particles with smooth morphology onthe surface of the substrate.
The formation of a bone-like apatite layer on the HAp/PDLLAfilms was investigated through FE-SEM measurements after 1, 3,and 7 days of soaking in SBF. Fig. S4 shows representative FE-SEMimages of F 0P123 films after soaking in SBF for 1 and 7 days andthat of F 1P123 films after soaking in SBF for 3 and 7 days. Com-pared with films examined before soaking, these composite filmsdid not show significant changes in their morphologies during theinitial stage of soaking (1–3 days). However, a few bundles of HApparticles were found because of the aggregation of such particleson the surface of the composite films after the initial (1–3 days)immersion in SBF (e.g., after 1 day for the F 0P123 film and after3 days for the F 1P123 film), as shown in Fig. S4(a) and (d). After7 days of soaking in SBF, both composite samples exhibited mor-phological changes, which are different from those before soaking.As shown in high-resolution FE-SEM images in Fig. S4(c) and (f),their morphologies were rougher with scant deposition of needle-like mineral crystals (160 nm in length and 20 nm in diameter forF 0P123; 120 nm in length and 20 nm in diameter for F 1P123). Suchan observation revealed the induction of a mineral phase on the sur-face of these films after 7 days in SBF. FE-SEM images of F 2P123films after 1, 3, and 7 days of treatment in SBF are presented inFig. 6. During incubation in SBF, the morphology of F 2P123 com-posite films drastically changed with the appearance of abundantdeposits. In particular, the intense formation of spherical mineralparticles (mean diameter of 2 �m) composed of tiny needle-likecrystals was already observed after 1 day of immersion in SBF(Fig. 6a and b). The same tendency was seen after 3 days (Fig. 6cand d), but the size of the spherical particles with needle-like crys-tallites increased with longer soaking time (their mean diameterincreased to 3 �m). Thereafter (beyond 3 days), the mineral phasewas largely spread on the surface of F 2P123 films. FE-SEM imageswith different magnifications (Fig. 6e and f) reveal that a coral-likemineral layer of numerous tiny needle-like crystals was depositedon the entire surface of F 2P123 films after 7 days in SBF. FE-SEMimages with higher resolution (Fig. 6g and h) indicate the formationof a flower-like mineral layer composed of tiny needle-like crystal-lites on the entire surface of F 2P123. This interesting morphologyof the mineral layer is typical in bone-like apatite [33]. The givenresults reveal the complete formation of the mineral layer on thesurface of F 2P123 even for short soaking periods, which indicatesits highest in vitro bioactive response among the three compositefilms studied. As reported in past studies, mineralization involvesthe nucleation and growth of bone-like apatite onto biomaterials inSBF [38], which is mainly associated with the uptake of calcium andphosphate ions from the SBF solution. The composite film (F 2P123)composed of HAp nanoparticles (2P123) with the smallest sizesand the highest BET surface area among the studied HAp samples(Tables 1 and 2) certainly resulted in its high uptake for calcium andphosphate ions, thereby exhibiting the highest bioactivity. Conse-quently, the composite films (F 0P123 and F 1P123) consisted ofthe HAp nanoparticles with large dimensions and low BET surfaceareas (both against their uptake capacity of calcium and phosphateions) show considerably lower bioactivity.
The Ca/P ratio is a key characteristic used to identify the mineralphase among biologically relevant minerals. To further analyze the
chemical composition of the released mineral layer, EDXS analysiswas conducted for the typical composite films after SBF treatment.The results indicated that the main elements (i.e., Ca, P, and O)were found in the mineral layer and C was also detected as a trace
N.K. Nga et al. / Colloids and Surfaces B: Biointerfaces 116 (2014) 666–673 671
les in
epatrFptFi3bpC(saiamrCs
Fig. 5. Possible mechanism for the formation of (A) HAp nanopartic
lement (not shown). Table S2 presents the Ca/P ratios of the com-osite films after 7 days of soaking in SBF for the F 0P123 filmnd after 1, 3, and 7 days for F 2P123 films. As shown in Table S2,he Ca/P ratio for F 0P123 after 7 days of treatment is 1.55, whicheveals the formation of a new apatite phase on the surface of the0P123 in SBF. However, the intensity of the carbon peak on EDXSatterns of F 0P123 before and after treatment in SBF increased,hereby suggesting that the apatite produced was carbonated. For2P123 films, the Ca/P ratio showed an increasing trend with
ncreasing soaking time; the Ca/P ratio was 1.36, 1.47, and 1.57 for 1,, and 7 days of SBF, respectively. As shown in the literature, a num-er of other calcium phosphate minerals (e.g., amorphous calciumhosphate Cax(PO4)y·zH2O (ACP), dicalcium phosphate dihydrateaHPO4·2H2O (DCPD), octacalcium phosphate Ca8H2(PO4)6·5H2OOCP), tricalcium phosphate �- and �-Ca3·(PO4)2 (TCP), and Mg-ubstituted tricalcium phosphate (Ca,Mg)3(PO4)2·(Mg-TCP)) canlso be produced under conditions similar to those that form apatiten vitro. These minerals can be transformed from one type tonother depending on the pH and composition of the biological
icro-environment and can be distinguished through their Ca/Patio through EDXS [37]. Therefore, based on EDXS analyses anda/P ratios (Table S2), OCP and TCP were probably released on theurfaces of F 2P123 films after 1 and 3 days of soaking, respectively.
the absence of P123 and (B) HAp nanorods in the presence of P123.
The highest value of 1.57 can be assigned to a bone-like apatite layerproduced in F 2P123 after 7 days. Considering the increase in theintensity of the carbon peak for EDXS patterns before and after 7days of immersion in SBF for F 2P123, we find that the carbonatedapatite layer was already produced on the surface of F 2P123 after7 days of immersion in SBF.
Table S3 compares the in vitro bioactivity of HAp nanorods asprepared in this study with those reported in previous studies [39].The formation of new bone-like mineral was already detected after1 day of incubation in SBF for the prepared HAp nanorods in thisstudy; it was detected only after 45 days for HAp nanorods, HApnanospheres, and HAp fibrous microparticles in past studies [39].Moreover, after a quick period of incubation in SBF (7 days), a highlyinteresting and distinctive morphology of bone-like apatite resem-bling the flower-like apatite was exclusively observed for the HApnanorods prepared in this study (Fig. 6g and h). The in vitro bioactiv-ity of the prepared HAp nanorods in this study that is more efficientthan the other HAp particles should be attributed to their closemorphology (e.g., size and shape) to natural HAp.
As shown in Table S3, the HAp nanoparticles prepared in thisstudy have a mean width of 28 nm and a mean length of 120 nm,which is extremely similar to those of bone minerals. The otherHAp particles possess morphologies that are considerably different

672 N.K. Nga et al. / Colloids and Surfaces B: Biointerfaces 116 (2014) 666–673
r soak
fbsolpam
Fig. 6. SEM images of HAp/PDLLA composite film (F 2P123) afte
rom those of the prepared HAp nanorods and the mineral part ofone. For instance, the best HAp particles in the previous study havepherical and near-spherical shape (approximately 50 nm in size)r extremely short nanorods (average width of 60 nm and average
ength of 110 nm) [39]. Based on the comparison of the results of theresent and previous study, the size and shape of HAp particles havecrucial function in their bioactivity and biocompatibility in pro-oting the bone-like apatite phase. The morphological similarity ofing in SBF for (a and b) 1 day, (c and d) 3 days and (e–h) 7 days.
synthetic HAp particles can facilitate both their in vitro and in vivobioactivity.
4. Conclusion
This study demonstrates that rod-shaped hydroxyapatite par-ticles resembling bone minerals are easily synthesized withcontrollable sizes with a suitable concentration of the pluronic

es B: B
swaIftatit
A
fn
t7m
A
f2
R
[[[
[[[[[[
[[
[[[
[
[[[[[[
[[[[
[
[
N.K. Nga et al. / Colloids and Surfac
urfactant P123. The HAp nanorods revealed a uniform rod shapeith sizes similar to those of bone minerals (mean width of 28 nm
nd mean length of 120 nm), Ca/P of 1.66, and stoichiometric HAp.n vitro bioactivity tests showed that the HAp nanorods induce rapidormation of bone-like apatite after an extremely short soakingime in SBF, which makes them excellent candidates for bone repairnd reconstruction. This study can provide an efficient protocol forhe controlled synthesis of HAp nanorods similar to bone mineralsn view of developing biocompatible composite scaffolds for boneissue engineering.
cknowledgments
This study was funded by the Vietnam National Foundationor Science and Technology Development (NAFOSTED) under grantumber 104.02-2012.42.
The authors would like to thank Dr. Anne-Lise Haenni, Insti-ut Jaques Monod Institute/CNRS – Université Paris Diderot – Paris, France, for English correction and helpful comments on theanuscript.
ppendix A. Supplementary data
Supplementary data associated with this article can beound, in the online version, at http://dx.doi.org/10.1016/j.colsurfb.013.11.001.
eferences
[1] K.E. Healy, R.E. Guldberg, J. Musculoskelet. Neuronal Interact. 7 (2007) 328.[2] D. Puppi, F. Chiellini, A.M. Piras, E. Chiellini, Prog. Polym. Sci. 35 (2010) 403.
[3] Z. Ma, Z. Mao, C. Gao, Colloids Surf. B: Biointerfaces 60 (2007) 137.[4] X. Wen, P.A. Tresco, Biomaterials 27 (2006) 3800.[5] Y.C. Kuo, C.F. Yeh, Colloids Surf. B: Biointerfaces 82 (2011) 624.[6] H.R. Pant, P. Risal, C.H. Park, L.D. Tijing, Y.J. Jeong, C.S. Kim, Colloids Surf. B:Biointerfaces 102 (2013) 152.
[
[[
iointerfaces 116 (2014) 666–673 673
[7] Y.R.V. Shin, C.N. Chen, S.W. Tsai, Y.J. Wang, O.K. Lee, Stem Cells 24 (2006)2391.
[8] S.Y. Shin, H.N. Park, K.H. Kim, M.H. Lee, Y.S. Choi, Y.J. Park, Y.M. Lee, I.C. Rhyu,S.B. Han, S.J. Lee, C.P. Chung, J. Periodontol. 76 (2005) 1778.
[9] Y.C. Kuo, C.C. Wang, Colloids Surf. B: Biointerfaces 84 (2011) 63.10] Q. Fu, E. Saiz, M.N. Rahaman, A.P. Tomsia, Mater. Eng. C 31 (2011) 1245.11] R.Z. LeGeros, Chem. Rev. 108 (2008) 4742.12] Y.M. Ha, T. Amna, M.H. Kim, H.C. Kim, M.S. Hassan, M.S. Khil, Colloids Surf. B:
Biointerfaces 102 (2013) 795.13] R. Zhang, P.X. Ma, Macromol. Biosci. 4 (2004) 100.14] H. Wang, Y. Li, Y. Zuo, J. Li, et al., Biomaterials 28 (2007) 3338.15] H.W. Kim, J. Biomed. Mater. Res. 83 (2007) 169.16] Y. Zhang, K.E. Tanner, J. Mater. Sci. Mater. Med. 19 (2008) 761.17] J.P. Chen, Y.S. Chang, Colloids Surf. B: Biointerfaces 86 (2011) 169.18] Z. Li, L. Yubao, Y. Aiping, P. Xuelin, W. Xuejiang, et al., J. Mater. Sci. Mater. Med.
16 (2005) 213.19] C. Du, F.Z. Cui, Q.L. Feng, X.D. Zhu, J. Biomed. Mater. Res. 42 (1998) 540.20] Y. Zhang, J.R. Venugopal, A. El-Turki, S. Ramakrishna, B. Su, C.T. Lim, Biomaterials
29 (2008) 4314.21] H. Zhou, J. Lee, Acta Biomater. 7 (2011) 2769.22] A. Wang, D. Liu, H. Yin, et al., Mater. Sci. Eng. C 27 (2007) 865.23] G.J. Wu, L.Z. Zhou, K.W. Wang, F. Chen, Y. Sun, Y.R. Duan, Y.J. Zhu, H.C. Gu, J.
Colloid Interface Sci. 345 (2010) 427.24] N.K. Nguyen, M. Leoni, D. Maniglio, C. Migliaresi, J. Biomater. Appl. 28 (1) (2013)
49.25] S. Mollazadeh, J. Javadpour, A. Khavandi, Ceram. Int. 33 (2007) 1579.26] Y. Liu, D. Hou, G. Wang, Mater. Chem. Phys. 86 (2004) 69.27] K. Lin, J. Chang, R. Cheng, M. Ruan, Mater. Lett. 61 (2007) 1683.28] X. Yao, H. Yao, G. Li, Y. Li, J. Mater. Sci. 45 (2010) 1930.29] S. Gay, S. Arostegui, J. Lemaitre, Mater. Sci. Eng. C 29 (2009) 172.30] N.K. Nga, P.T. Hong, T.D. Lam, T.Q. Huy, J. Colloid Interface Sci. 398 (2013)
210.31] A. Banerjee, A. Bandyopadhyay, S. Bose, Mater. Sci. Eng. C 27 (2007) 729.32] Y. Li, D. Li, Z. Xu, J. Mater. Sci. 44 (2009) 1258.33] T. Kokubo, H. Takadama, Biomaterials 27 (2006) 2907.34] C. Li, L. Zhao, J. Han, R. Wang, C. Xiong, X. Xie, J. Colloid Interface Sci. 360 (2011)
341.35] S.J. Gregg, K.S.W. Sing, Adsorption, Surface Area and Porosity, 2nd ed., Academic
Press, London, 1982.36] E.S. Lee, Y.T. Oh, Y.S. Youn, M. Nam, B. Park, J. Yun, J.H. Kim, H.T. Song, K.T. Oh,
Colloids Surf. B: Biointerfaces 82 (2011) 190.37] L.C. Palmer, C.J. Newcomb, S.R. Kaltz, E.D. Spoerke, S.I. Stupp, Chem. Rev. 108
(2008) 4754.38] F. Sun, H. Zhou, J. Lee, Acta Biomater. 7 (2011) 3813.39] M. Sadat-Shojai, M.T. Khorasani, A. Jamshidi, J. Cryst. Growth 361 (2012) 73.




















