
Substantial changes in gene expression of Wnt, MAPK and TNF pathways induced by TGF- 1 in...
10
Substantial changes in gene expression of Wnt, MAPK and TNFa pathways induced by TGF-b1 in cervical cancer cell lines Judith N.Kloth , Gert Jan Fleuren, Jan Oosting, Renee X.de Menezes 1 , Paul H.C.Eilers 1 , Gemma G.Kenter 2 and Arko Gorter Department of Pathology, 1 Department of Medical Statistics and 2 Department of Gynecology, Leiden University Medical Center, Leiden, The Netherlands To whom correspondence should be addressed. Tel: þ31 71 526 65 96; Fax: þ31 71 524 81 58; Email: [email protected] Transforming growth factor-beta 1 (TGF-b1) is a potent inhibitor of epithelial cell proliferation. During the devel- opment of cervical carcinoma however, an increase in pro- duction of TGF-b1 is accompanied by decreased sensitivity for the growth-limiting effect of TGF-b1. TGF-b1 has an anti-proliferative effect on cells of the immune system and thus can be advantageous for tumor progression. The aim of the present study was to determine the effect of TGF-b1 on mRNA expression profile of genes in pathways involved in cell growth and cell death, in cervical carcinoma cell lines with different sensitivity to TGF-b1. For this purpose, we have investigated changes in gene expression in TGF-b1 stimulated cervical cancer cell lines with high (CC10B), intermediate (SiHa) and low (HeLa) sensitivity to the anti- proliferative effect of TGF-b1, at timepoints 0, 6, 12 and 24 h. Microarray analysis, using Affymetrics focus arrays, rep- resenting 8973 genes, was used to measure gene expression. In our study novel target genes involved in tumor necrosis factor alpha (TNFa), mitogen-activated protein kinase (MAPK) and wingless type (Wnt) pathways in response to TGF-b1 were found. Substantial differences in gene expression between TGF-b1 sensitive and insensitive cell lines were observed involving genes in TNFa, MAPK, Wnt and Smad pathways. Since these pathways are implicated in cell proliferation and cell death, these pathways may play a role in determining the overall sensitivity of a cell to TGF-b1 induced cell growth inhibition. The results were subsequently validated by quantitative real-time PCR. Increased resistance to TGF-b1 induced cell growth inhibi- tion was correlated with an elevated production of TGF-b1 by the cell lines, as measured by enzyme linked immuno- sorbent assay. TGF-b1 production did not inhibit cell growth, since blocking TGF-b1 protein by anti-TGF-b had no effect on cell proliferation. TGF-b1 excretion by tumor cells more likely contributes to paracrine stimula- tion of tumor development. Introduction Transforming growth factor-beta 1 (TGF-b1) plays an essen- tial role in cellular processes such as proliferation, differenti- ation, embryonic development, angiogenesis, wound healing and inhibition of epithelial cell growth. During cancer devel- opment, however, the majority of tumor cells become either partly or completely resistant to TGF-b1 growth inhibition and TGF-b1-induced apoptotic responses (1,2). In cervical carcin- oma, extracellular levels of TGF-b1 are increased in late stage of malignancy (3,4), thereby contributing to invasion, meta- stasis and inhibition of host-tumor immune responses (5–7). Defective TGF-b1 signaling, due to inactivating mutations of TGF-b receptors and Smad signaling molecules, has been reported in cervical carcinoma (8,9). However, only a few cervical cancers showed mutations in TGF-b receptors or Smad (10,11) and this does not explain the loss of growth inhibition in the majority of cervical cancers. Hence, other mechanisms, besides functional loss of TGF-b receptor I, TGF-b receptor II and Smad, are likely to exist. In different types of cancers, a defect in the TGF-b1 pathway downstream of Smad signaling has been suggested (12) and crosstalk of TGF-b1–Smad signaling with other pathways have been demonstrated (13,14). Mitogen-activated protein kinases (MAPK), phosphatidylinositol-3 kinase (PI3K) and Rho signaling via TGF-b1, with or without crosstalk with Smad signaling, have shown to be involved in the inhibition of apoptosis in cancer cells, invasion and metastatic processes (13,15). In addition, crosstalk between TGF-b1 and wingless type (Wnt) (16,17), tumor necrosis factor a (TNFa) (18), interferon g (IFNg) (19) and epidermal growth factor (EGF) (20) have been demonstrated, which may influence tumorigen- esis. This suggests that differential regulation of TGF-b1 sig- naling, other than attenuation of TGF-b1–Smad signaling by reduced expression or functional loss of Smad pathway com- ponents, may determine the magnitude of the TGF-b1 response. Only a few studies investigated large-scale gene expression changes after stimulation with TGF-b1 in cancer cells. These studies focused primarily on epithelial to mesenchym trans- ition (EMT) processes and did not address loss of cell growth inhibition (21,22). The aim of the present study was to deter- mine the effect of TGF-b1 on mRNA expression profile of genes in pathways involved in cell growth and cell death, in cervical carcinoma cell lines with different sensitivity to TGF-b1. For this purpose we used cDNA microarrays to measure differences in gene expression between cervical can- cer cell lines with low (HeLa), intermediate (SiHa) and high (CC10B) sensitivity to TGF-b1. Materials and methods Cell lines and cultures Human cervical cancer cell lines, HeLa, SiHa (ATCC, Rockville, MD) and CC10B (23) were maintained in RPMI 1640 (Life Technologies, Grand Island, Abbreviations: AffyPLM, Affy-Probe Level Model; CDK, cyclin dependent kinase; ECM, extracellular matrix; EGF, epidermal growth factor; EMT, epithelial to mesenchym transition; ERK, extracellular response kinase; FGF, fibroblast growth factor; GO, gene ontology; IFNg, interferong; MAPK, mitogen-activated protein kinases; NFkB, nuclear factor kappa B; PI3K, phosphatidylinositol-3 kinase; QRT–PCR, quantitative real-time PCR; RMA, Robust Multi-Chip Average; SAPK, stress activated protein kinase; SSCG, statistically significant changes in gene expression; TGF-b1, transforming growth factor b1; TNFa, tumor necrosis factor a; Wnt, wingless type. Carcinogenesis vol.26 no.9 # Oxford University Press 2005; all rights reserved. 1493 Carcinogenesis vol.26 no.9 pp.1493–1502, 2005 doi:10.1093/carcin/bgi110 Advance Access publication May 5, 2005 by guest on June 4, 2013 http://carcin.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Substantial changes in gene expression of Wnt, MAPK and TNF pathways induced by TGF- 1 in...

Substantial changes in gene expression of Wnt, MAPK and TNFapathways induced by TGF-b1 in cervical cancer cell lines
Judith N.Kloth�, Gert Jan Fleuren, Jan Oosting,Renee X.de Menezes1, Paul H.C.Eilers1,Gemma G.Kenter2 and Arko Gorter
Department of Pathology, 1Department of Medical Statistics and2Department of Gynecology, Leiden University Medical Center, Leiden,The Netherlands
�To whom correspondence should be addressed. Tel: þ31 71 526 65 96;Fax: þ31 71 524 81 58;Email: [email protected]
Transforming growth factor-beta 1 (TGF-b1) is a potentinhibitor of epithelial cell proliferation. During the devel-opment of cervical carcinoma however, an increase in pro-duction of TGF-b1 is accompanied by decreased sensitivityfor the growth-limiting effect of TGF-b1. TGF-b1 has ananti-proliferative effect on cells of the immune system andthus can be advantageous for tumor progression. The aimof the present study was to determine the effect of TGF-b1on mRNA expression profile of genes in pathways involvedin cell growth and cell death, in cervical carcinoma celllines with different sensitivity to TGF-b1. For this purpose,we have investigated changes in gene expression in TGF-b1stimulated cervical cancer cell lines with high (CC10B),intermediate (SiHa) and low (HeLa) sensitivity to the anti-proliferative effect ofTGF-b1, at timepoints0, 6, 12and24h.Microarray analysis, using Affymetrics focus arrays, rep-resenting 8973 genes, was used to measure gene expression.In our study novel target genes involved in tumor necrosisfactor alpha (TNFa), mitogen-activated protein kinase(MAPK) and wingless type (Wnt) pathways in response toTGF-b1 were found. Substantial differences in geneexpression between TGF-b1 sensitive and insensitive celllines were observed involving genes in TNFa, MAPK, Wntand Smad pathways. Since these pathways are implicatedin cell proliferation and cell death, these pathways mayplay a role in determining the overall sensitivity of a cellto TGF-b1 induced cell growth inhibition. The results weresubsequently validated by quantitative real-time PCR.Increased resistance to TGF-b1 induced cell growth inhibi-tion was correlated with an elevated production of TGF-b1by the cell lines, as measured by enzyme linked immuno-sorbent assay. TGF-b1 production did not inhibit cellgrowth, since blocking TGF-b1 protein by anti-TGF-bhad no effect on cell proliferation. TGF-b1 excretion bytumor cells more likely contributes to paracrine stimula-tion of tumor development.
Introduction
Transforming growth factor-beta 1 (TGF-b1) plays an essen-tial role in cellular processes such as proliferation, differenti-ation, embryonic development, angiogenesis, wound healingand inhibition of epithelial cell growth. During cancer devel-opment, however, the majority of tumor cells become eitherpartly or completely resistant to TGF-b1 growth inhibition andTGF-b1-induced apoptotic responses (1,2). In cervical carcin-oma, extracellular levels of TGF-b1 are increased in late stageof malignancy (3,4), thereby contributing to invasion, meta-stasis and inhibition of host-tumor immune responses (5–7).Defective TGF-b1 signaling, due to inactivating mutations
of TGF-b receptors and Smad signaling molecules, has beenreported in cervical carcinoma (8,9). However, only a fewcervical cancers showed mutations in TGF-b receptorsor Smad (10,11) and this does not explain the loss of growthinhibition in the majority of cervical cancers.Hence, other mechanisms, besides functional loss of TGF-b
receptor I, TGF-b receptor II and Smad, are likely to exist. Indifferent types of cancers, a defect in the TGF-b1 pathwaydownstream of Smad signaling has been suggested (12) andcrosstalk of TGF-b1–Smad signaling with other pathwayshave been demonstrated (13,14). Mitogen-activated proteinkinases (MAPK), phosphatidylinositol-3kinase (PI3K) andRhosignaling via TGF-b1, with or without crosstalk with Smadsignaling, have shown to be involved in the inhibition ofapoptosis in cancer cells, invasion and metastatic processes(13,15). In addition, crosstalk between TGF-b1 and winglesstype (Wnt) (16,17), tumor necrosis factor a (TNFa) (18),interferon g (IFNg) (19) and epidermal growth factor (EGF)(20) have been demonstrated, which may influence tumorigen-esis. This suggests that differential regulation of TGF-b1 sig-naling, other than attenuation of TGF-b1–Smad signaling byreduced expression or functional loss of Smad pathway com-ponents,may determine themagnitude of the TGF-b1 response.Only a few studies investigated large-scale gene expression
changes after stimulation with TGF-b1 in cancer cells. Thesestudies focused primarily on epithelial to mesenchym trans-ition (EMT) processes and did not address loss of cell growthinhibition (21,22). The aim of the present study was to deter-mine the effect of TGF-b1 on mRNA expression profile ofgenes in pathways involved in cell growth and cell death,in cervical carcinoma cell lines with different sensitivity toTGF-b1. For this purpose we used cDNA microarrays tomeasure differences in gene expression between cervical can-cer cell lines with low (HeLa), intermediate (SiHa) and high(CC10B) sensitivity to TGF-b1.
Materials and methods
Cell lines and cultures
Human cervical cancer cell lines, HeLa, SiHa (ATCC, Rockville, MD) andCC10B (23) were maintained in RPMI 1640 (Life Technologies, Grand Island,
Abbreviations: AffyPLM, Affy-Probe Level Model; CDK, cyclin dependentkinase; ECM, extracellular matrix; EGF, epidermal growth factor;EMT, epithelial to mesenchym transition; ERK, extracellular response kinase;FGF, fibroblast growth factor; GO, gene ontology; IFNg, interferong; MAPK,mitogen-activated protein kinases; NFkB, nuclear factor kappa B; PI3K,phosphatidylinositol-3 kinase; QRT–PCR, quantitative real-time PCR; RMA,Robust Multi-Chip Average; SAPK, stress activated protein kinase; SSCG,statistically significant changes in gene expression; TGF-b1, transforminggrowth factor b1; TNFa, tumor necrosis factor a; Wnt, wingless type.
Carcinogenesis vol.26 no.9 # Oxford University Press 2005; all rights reserved. 1493
Carcinogenesis vol.26 no.9 pp.1493–1502, 2005doi:10.1093/carcin/bgi110Advance Access publication May 5, 2005
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

NY), supplemented with 10% fetal calf serum (FCS), streptomycin andpenicillin at 37�C in a humid atmosphere containing 5% CO2.
Growth inhibition study
Cells were plated onto 24-well plates (Greiner, Frickenhausen, Germany) at adensity of 3–5 � 103 cells/well in 0.5 ml culture medium. The following day,cells were incubated with different amounts of human recombinant TGF-b1ranging from 0.0001–1 ng/ml (Sigma, Saint Louis, MO). Each day recombin-ant TGF-b1 was added to the cells over a period of 6 days. Cells were countedevery second day in triplicate using a CASY1 cell counter (Scharfe System,Reutlingen, Germany). Growth kinetics experiments were repeated at least twotimes.
To examine growth arrest, induced by a cell’s self-produced TGF-b1,TGF-b antibody (2G7) in a concentration ranging from 0.1 to 10 mg/ml wasadded to the cell culture.
TGF-b1 enzyme linked immunosorbent assay (ELISA)
For quantification of TGF-b1 protein in cell culture supernatant, the Quanti-kine Immunoassay (R&D Systems Europe, Abingdon, UK) was performedaccording to the manufacturer’s instructions. Cells were cultured for 2, 4, 8 and16 days, starting with 50 000 cells in 4 ml of medium for all cell lines. TGF-b1in supernatants was activated by adding 1 N HCl (100 ml/500 ml sample) to thesamples for 10 min and subsequently neutralized by the addition of 1.2 NNaOH containing 0.5 M HEPES (100 ml/500 ml sample). To correct for theTGF-b1 present in the medium, the OD450 values of the HCl treated mediumwere subtracted from the OD450 value of the supernatants. All samples wereassayed as triplicates.
RNA extraction
Total RNA was isolated from cells that were grown to �60% confluence in250 ml culture flasks (Greiner, Frickenhausen, Germany) using TRIzol reagent(Gibco BRL/Life Technologies, Breda, The Netherlands). The total RNA wasphenol–chloroform-extracted, ethanol precipitated and cleaned with Rneasycleanup system columns (Qiagen GmbH, Hilden, Germany).
Oligonucleotide microarray and data analysis
Affymetrics focus arrays (Santa Clara, CA), representing 8793 humansequences from the NCBI Refseq database, were used for mRNA expressionprofiling according to manufacturer’s instructions. Briefly, 10 mg ofpurified RNA was transcribed by Superscript II reverse transcriptase (LifeTechnologies, Grand Island, NY) using T7- (dT)24 primer containing aT7 RNA polymerase promoter. After synthesis of the second complementaryDNA (cDNA) strand, this product was used in an in vitro transcription reactionto generate biotinylated complementary RNA (cRNA) using the Bioarray RNAtranscript labeling kit (Enzo, Farmingdale, NY). Focus arrays were hybridizedwith 10 mg of biotinylated RNA. After hybridization, microarrays werewashed and stained on an Affymetrics fluidics station and scannedwith an argon ion confocal laser. Data files of all microarrays were normalizedusing sequence information in Robust Multi-Chip Average (GCRMA) method(24,25). Quality assurance was performed on the arrays using Affy-ProbeLevel Model (AffyPLM) (26). Microarrays for each cell line were performedin duplicate for all conditions. Normalized data were statistically analyzedfor each cell line separately. A change in gene expression during thefour time points was analyzed by one-way ANOVA using R language forstatistical computing. Genes that showed significant changes (P � 0.01)in expression pattern were selected for further analysis by functionalgrouping on gene ontology (GO) terms and by screening the public availabledatabases (http://www.ncbi.nlm.nih.gov and http://www.ncbi.nlm.nih.gov/omim).
Real-time quantitative PCR
2 mg of total RNA was transcribed to cDNA with reverse transcriptase ofSuperscript II (Life Technologies). Amplification reactions were performedwith qPCR Core kit for Sybr Green (Eurogentec Hampshire, UK) according tomanufacturer’s protocol. Fluorescent PCR analysis was performed using theBIO-RAD iCycler (BIO-RAD, Hercules, CA). The following PCR conditionswere used: 10 min at 95�C, followed by 40 cycles of 15 s at 95�C and 1 minat the appropriate annealing temperature. Genes for normalization of expres-sion data were selected on the basis of constant high microarray gene expres-sion data during the four time points. Primers were either designed withBeacon designer (BIO-RAD), retrieved from Real Time PCR Primer andProbe Database (http://medgen.ugent.be/rtprimerdb) or ordered (Tebu-Bio,Heerhugowaard, The Netherlands). Expression of the genes of interest wasnormalized by geometric averaging of multiple internal control genes using theGenorm program (27). Out of five normalization genes the best three wereselected within this program: CPSF6, EEF1A1 and RPL13. Relative quanti-fication was performed using standard curves, followed by adjustment withthe normalization factor, which is calculated by the Genorm program. Primer
sequences are listed in Table I, apart from commercially available primers forCDKN1A and NFkB2.
Results
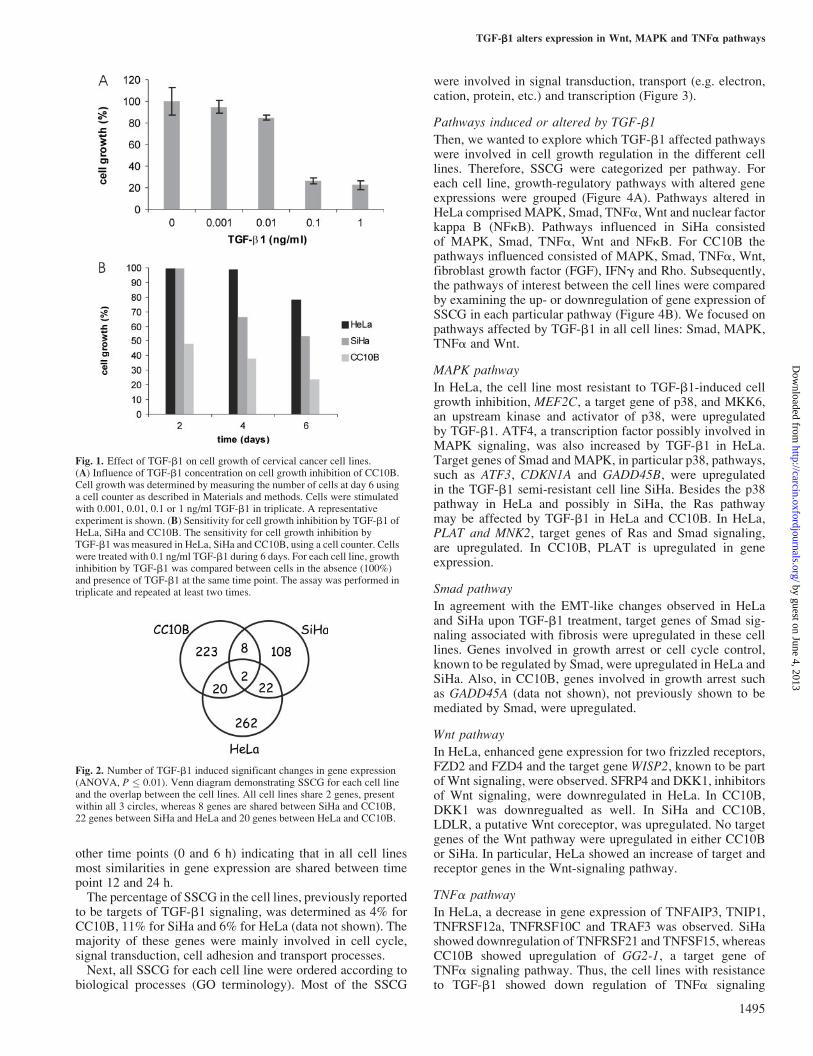
Cell growth response of CC10B, SiHa and HeLa to TGF-b1The degree of TGF-b1 sensitivity was determined in cervicalcancer cell lines, CC10B, SiHa and HeLa at day 2, 4 and6 of cell growth after exposure to TGF-b1, in a concentrationranging from 0.001 to 1 ng/ml. For all cell lines 0.1 ng/mlTGF-b1 gave plateau values in cell growth inhibition (datashown for cell line CC10B, Figure 1A). TGF-b1 induced cellgrowth inhibition was evident from day 1 onwards for CC10B,after 2 days for SiHa and after 4 days for HeLa. After 6 daysof TGF-b1 treatment, CC10B showed �75% decrease in cellgrowth, SiHa, 50% and HeLa only 25%, demonstrating thatCC10B was most, SiHa intermediate and HeLa least sensitiveto TGF-b1-induced growth inhibition (Figure 1B). Only HeLaand SiHa showed an EMT-like morphology upon TGF-b1treatment (data not shown).
Expression profile of CC10B, SiHa and HeLa after TGF-b1stimulation
To determine the pathways involved in TGF-b1-induced cellgrowth regulation, CC10B, SiHa and HeLa were stimulatedwith 0.1 ng/ml TGF-b1 for 0, 6, 12 or 24 h for profiling bymicroarray. Sampling for each cell line was performed induplicate for statistical analysis. The reliability of the dataafter normalization was confirmed by AffyPLM-analysisindicating that arrays between the four time points (0, 6, 12and 24 h) for each cell line were comparable (data not shown).For each cell line statistically significant changes (P � 0.01)in gene expression (SSCG) were selected and calculated byone-way ANOVA analysis. The number of SSCG varied percell line: for CC10B 253, for SiHa 140 and for HeLa 306(Figure 2).To investigate at which time point TGF-b1 stimulation re-
sulted in the majority of changes in gene expression, the SSCGwere clustered in a hierarchical tree for each cell line (data notshown). Most TGF-b1-induced gene expression changes occurafter 6 h. Time points 12 and 24 h cluster separately from the
Table I. Primers used in QRT–PCR
Gene Primers
CPSF6 sense (50–30) AAGATTGCCTTCATGGAATTGAGCPSF6 antisense (30–50) TCGTGATCTACTATGGTCCCTCTCTEEF1A1 sense (50–30) CTGGCAAGGTCACCAAGTCTEEF1A1 antisense (30–50) CCGTTCTTCCACCACTGATTRPL13a sense (50–30) CCTGGAGGAGAAGAGGAAAGAGARPL13a antisense (30–50) TTGAGGACCTCTGTGTATTTGTCAAATF4 sense (50–30) CCAACAACAGCAAGGAGGATATF4 antisense (30–50) GTGTCATCCAACGTGGTCAGTNFRSF21 sense (50–30) CCACAGCTCAGCCAGAACAGTNFRSF21 antisense (30–50) CGGTGGCACGGTCAACATTNFRSF12a sense (50–30) CCAAGCTCCTCCAACCACAATNFRSF12a antisense (30–50) TGGGGCCTAGTGTCAAGTCTDKK1 sense (50–30) TCCGAGGAGAAATTGAGGAADKK1 antisense (30–50) CCTTCTTGTCCTTTGGTGTGAGADD45B sense (50–30) CCAGTCCTTCTGCTGTGACAACGADD45B antisense (30–50) TCCGTGTGAGGGTTCGTGACTGFBI sense (50–30) GCCCTGAGAGACCTGCTGAACTGFBI antisense (30–50) TCGCCTTCCCGTTGATAGTGAGPAI-1 sense (50–30) CCTGGGAATGACCGACATGTPAI-1 antisense (50–30) TGCGGGCTGAGACTATGACA
J.N.Kloth et al.
1494
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
other time points (0 and 6 h) indicating that in all cell linesmost similarities in gene expression are shared between timepoint 12 and 24 h.The percentage of SSCG in the cell lines, previously reported
to be targets of TGF-b1 signaling, was determined as 4% forCC10B, 11% for SiHa and 6% for HeLa (data not shown). Themajority of these genes were mainly involved in cell cycle,signal transduction, cell adhesion and transport processes.Next, all SSCG for each cell line were ordered according to
biological processes (GO terminology). Most of the SSCG
were involved in signal transduction, transport (e.g. electron,cation, protein, etc.) and transcription (Figure 3).
Pathways induced or altered by TGF-b1
Then, we wanted to explore which TGF-b1 affected pathwayswere involved in cell growth regulation in the different celllines. Therefore, SSCG were categorized per pathway. Foreach cell line, growth-regulatory pathways with altered geneexpressions were grouped (Figure 4A). Pathways altered inHeLa comprised MAPK, Smad, TNFa, Wnt and nuclear factorkappa B (NFkB). Pathways influenced in SiHa consistedof MAPK, Smad, TNFa, Wnt and NFkB. For CC10B thepathways influenced consisted of MAPK, Smad, TNFa, Wnt,fibroblast growth factor (FGF), IFNg and Rho. Subsequently,the pathways of interest between the cell lines were comparedby examining the up- or downregulation of gene expression ofSSCG in each particular pathway (Figure 4B). We focused onpathways affected by TGF-b1 in all cell lines: Smad, MAPK,TNFa and Wnt.
MAPK pathway
In HeLa, the cell line most resistant to TGF-b1-induced cellgrowth inhibition, MEF2C, a target gene of p38, and MKK6,an upstream kinase and activator of p38, were upregulatedby TGF-b1. ATF4, a transcription factor possibly involved inMAPK signaling, was also increased by TGF-b1 in HeLa.Target genes of Smad and MAPK, in particular p38, pathways,such as ATF3, CDKN1A and GADD45B, were upregulatedin the TGF-b1 semi-resistant cell line SiHa. Besides the p38pathway in HeLa and possibly in SiHa, the Ras pathwaymay be affected by TGF-b1 in HeLa and CC10B. In HeLa,PLAT and MNK2, target genes of Ras and Smad signaling,are upregulated. In CC10B, PLAT is upregulated in geneexpression.
Smad pathway
In agreement with the EMT-like changes observed in HeLaand SiHa upon TGF-b1 treatment, target genes of Smad sig-naling associated with fibrosis were upregulated in these celllines. Genes involved in growth arrest or cell cycle control,known to be regulated by Smad, were upregulated in HeLa andSiHa. Also, in CC10B, genes involved in growth arrest suchas GADD45A (data not shown), not previously shown to bemediated by Smad, were upregulated.
Wnt pathway
In HeLa, enhanced gene expression for two frizzled receptors,FZD2 and FZD4 and the target gene WISP2, known to be partof Wnt signaling, were observed. SFRP4 and DKK1, inhibitorsof Wnt signaling, were downregulated in HeLa. In CC10B,DKK1 was downregualted as well. In SiHa and CC10B,LDLR, a putative Wnt coreceptor, was upregulated. No targetgenes of the Wnt pathway were upregulated in either CC10Bor SiHa. In particular, HeLa showed an increase of target andreceptor genes in the Wnt-signaling pathway.
TNFa pathway
In HeLa, a decrease in gene expression of TNFAIP3, TNIP1,TNFRSF12a, TNFRSF10C and TRAF3 was observed. SiHashowed downregulation of TNFRSF21 and TNFSF15, whereasCC10B showed upregulation of GG2-1, a target gene ofTNFa signaling pathway. Thus, the cell lines with resistanceto TGF-b1 showed down regulation of TNFa signaling
Fig. 1. Effect of TGF-b1 on cell growth of cervical cancer cell lines.(A) Influence of TGF-b1 concentration on cell growth inhibition of CC10B.Cell growth was determined by measuring the number of cells at day 6 usinga cell counter as described in Materials and methods. Cells were stimulatedwith 0.001, 0.01, 0.1 or 1 ng/ml TGF-b1 in triplicate. A representativeexperiment is shown. (B) Sensitivity for cell growth inhibition by TGF-b1 ofHeLa, SiHa and CC10B. The sensitivity for cell growth inhibition byTGF-b1 was measured in HeLa, SiHa and CC10B, using a cell counter. Cellswere treated with 0.1 ng/ml TGF-b1 during 6 days. For each cell line, growthinhibition by TGF-b1 was compared between cells in the absence (100%)and presence of TGF-b1 at the same time point. The assay was performed intriplicate and repeated at least two times.
Fig. 2. Number of TGF-b1 induced significant changes in gene expression(ANOVA, P � 0.01). Venn diagram demonstrating SSCG for each cell lineand the overlap between the cell lines. All cell lines share 2 genes, presentwithin all 3 circles, whereas 8 genes are shared between SiHa and CC10B,22 genes between SiHa and HeLa and 20 genes between HeLa and CC10B.
TGF-b1 alters expression in Wnt, MAPK and TNFa pathways
1495
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

components, in contrast to the cell line most sensitive to TGF-b1, which showed upregulation of a TNFa target gene.
Other pathways
Pathways that were not shared between all the cell lines wereRho, IFN, FGF and NFkB. As for the Rho family of smallGTPases, Rac2 was upregulated in HeLa, whereas CDC42EP2and ARHGEF3 were downregulated in CC10B. Componentsof the IFN signaling pathway were downregulated inCC10B, whereas FGF signaling components were upregulatedin CC10B. Mediators of the NFkB pathway were downregu-lated in HeLa, which coincides with the downregulation ofTNFa pathway genes. NFkB2 was transiently upregulated inSiHa.
Real-time quantitative PCR verification of microarray data
To validate the microarray results, representative gene pro-ducts of different pathways were subjected to quantitativereal-time PCR (QRT–PCR). The change, observed with thistechnique, confirmed results obtained by the microarrays,although variations in magnitude of the change between thetwo technologies were observed. Twelve SSCG verified in thecell lines exhibited TGF-b1-induced changes in gene expres-sion that correlated with those observed in the microarrayexperiments (Figure 5). The respective genes were: DKK1and ATF4 for HeLa and CC10B, NFkB2 for HeLa and SiHa,TNFRSF12a for HeLa, TNFRSF21, GADD45B, TGFBI, PAI-1and CDKN1A for SiHa.
TGF-b1 production and TGF-b1 growth inhibition
Finally, we investigated the association between decreasedsensitivity to TGF-b1-mediated cell growth inhibition of thecell lines and increased TGF-b1 production.
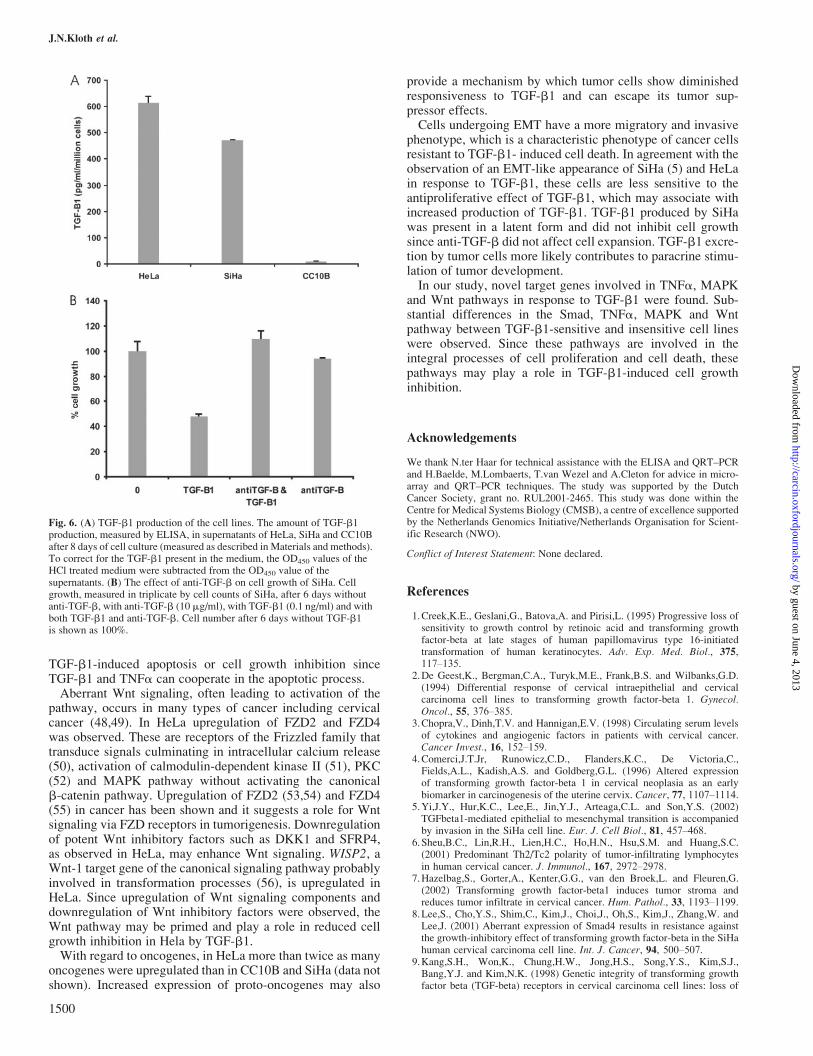
TGF-b1 production was measured in the supernatants ofCC10B, SiHa and HeLa cells after 2, 4, 8 and 16 days of cellculture. Only HeLa and SiHa excreted TGF-b1. HeLa, whichwas most resistant to TGF-b1-induced growth inhibition,showed the highest production of TGF-b1 (Figure 6A). Sub-sequently, the effect of the produced TGF-b1 on cell growthwas determined. For this purpose SiHa cells were cultured withanti-TGF-b. Anti-TGF-b did not alter cell growth kinetics,suggesting that the produced TGF-b1 is latent TGF-b1 anddoes not affect cell growth (Figure 6B).
Discussion
The mechanisms by which cancer cells lose their TGF-b1-induced growth inhibition are not fully understood. Defects inTGF-b1 signal transduction components, thereby contributingto loss of Smad signaling, does appear in some tumor types,but does not explain the loss of growth inhibition by TGF-b1in most cancers, including cervical cancer (28). TGF-b1 canactivate signaling pathways involved in growth control inde-pendently of Smads (15,29–31). The role of these non-Smadsignaling cascades remains to be better characterized. Bymeans of gene expression profiling, we have investigated dif-ferences in TGF-b1-induced response using cervical cancercell lines with different TGF-b1 sensitivity.In all cell lines many SSCG participated in signal transduc-
tion and transcription processes. Substantial parts of SSCGgenes were involved in Smad, MAPK, TNFa and Wnt path-ways. These pathways involved in cell growth and mainten-ance processes are associated with TGF-b1 signaling.Smad signaling is the main route through which TGF-b1
transduces its effects on cell cycle control. There is evident
Fig. 3. Biological processes demonstrating numbers of TGF-b1-induced SSCG. Bars indicate proportions of SSCG for each cell line; HeLa is depicted in black,SiHa in grey and CC10B in white. The bars are categorized by GO terms.
J.N.Kloth et al.
1496
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

upregulation of genes that act via Smad activation and areinvolved in cell cycle control such as CDKN1A, GADD45Band ATF3 (32,33) in SiHa and CDKN1C and TP53 in HeLa. InCC10B, GADD45A (34) and TP73L (data not shown), nega-tive regulators of cell cycle were upregulated, although thesehave not been described before as targets of Smad signaling.Besides genes involved in cell cycle regulation or apoptosisin HeLa, genes involved in cell division were upregulated,namely cyclin B2 and G2 (data not shown). Thus, in allcell lines, genes that negatively regulate cell growth wereinduced, but only in HeLa significant upregulation of positive
regulators of cell growth were observed, which may explainthe resistance to TGF-b1-induced cell growth inhibition.MAPK can be activated by TGF-b1 via TGF-b1-activating
kinase 1 (TAK1) (35,36), which is a potent activator of theStress activated protein kinases (SAPK/JNK) (37) and thep38 pathway (37–39). In HeLa, increase in gene expressionof MKK6, the main MAPK that activates p38, and MEF2C,a p38 downstream transcription factor (40), was observed.In a recent report, enhancement of a proliferative pathwayin HeLa by TGF-b1 was shown involving the activation ofMAPKs and probably not involving Smad activation (41).
Fig. 4. TGF-b1 induced alterations in gene expression. (A) Expression profiles of potential TGF-b1 target genes. Changes in gene expression after TGF-b1stimulation of HeLa (on the left), SiHa (in the middle) and CC10B (on the right), grouped by pathway. Dark colors, relative to time point 0, represent upregulationof gene expression, whereas light colors represent downregulation of SSCG. (B) TGF-b1 induced changes in gene expression of pathways in HeLa, SiHa andCC10B. Pathways, shown in light gray boxes, are associated with TGF-b1 signaling and involved in regulation of cell proliferation. The boxes below the pathwaysshow in red, genes which are upregulated in expression, in blue, genes which are downregulated and in black, genes which show evident upregulation anddownregulation during the four time points compared with time point 0 h. In HeLa, SSCG are observed in MAPK, Smad, Wnt, TNFa, NFkB and Rho pathways,in SiHa, SSCG are observed in MAPK, Smad, TNFa and NFkB pathways and in CC10B, SSCG were found in MAPK, Smad, TNFa, IFN, FGF and Rhopathways.
TGF-b1 alters expression in Wnt, MAPK and TNFa pathways
1497
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

Fig. 4. Continued.
J.N.Kloth et al.
1498
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

They reported that in SiHa, a growth inhibitory pathway invol-ving activation of Smads and a low level of MAPK activationwas observed. In agreement with these results, we found moreprominent upregulation of specific MAPK targets and signal-ing molecules in HeLa from our microarray data. However,Maliekal et al. (41) showed extracellular response kinase(ERK) and JNK activation in HeLa, whereas our microarraydata supported p38 activation and possibly ERK activation inHeLa. Our results and these observations, showing a strongTGF-b1-dependent activation of MAPK and insufficient Smadactivation by TGF-b1 in HeLa, suggest that this activationpattern may hamper the growth inhibitory effect of TGF-b1.
In HeLa we observed downregulation of TNFa receptors,signaling molecules and target proteins, of which TRAF3(42,43) and TNFRSF12a (44) have been associated, withapoptotic processes or inhibition of cell growth. In SiHa,downregulation of TNFSF15 (45) and TNFRSF21 (46) genesinvolved in cell death and suppression of cell growth wasobserved. In the TGF-b1 sensitive cell line CC10B no down-regulation of TNFa signaling proteins was observed. Incontrast, upregulation of a TNFa target gene, GG2-1 (47)was observed. In view of the TGF-b1 resistance of HeLa andSiHa, downregulation of receptors or signaling componentsinvolved in death signaling could be a way to impede
Fig. 5. Correlation of expression profile between microarray and QRT–PCR. The relative expression levels of SSCG; DKK1, ATF4, TNFRSF12a, TNFRSF21,GADD45B, TGFBI, NFkB2, PAI-1 and CDKN1A were measured with microarray and QRT–PCR at 0, 6, 12 and 24 h of TGF-b1 stimulation. Data are normalizedto internal control genes (see Materials and methods). The dotted line represents gene expression analyzed by QRT–PCR, the straight line represents geneexpression generated by microarray.
TGF-b1 alters expression in Wnt, MAPK and TNFa pathways
1499
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
TGF-b1-induced apoptosis or cell growth inhibition sinceTGF-b1 and TNFa can cooperate in the apoptotic process.Aberrant Wnt signaling, often leading to activation of the
pathway, occurs in many types of cancer including cervicalcancer (48,49). In HeLa upregulation of FZD2 and FZD4was observed. These are receptors of the Frizzled family thattransduce signals culminating in intracellular calcium release(50), activation of calmodulin-dependent kinase II (51), PKC(52) and MAPK pathway without activating the canonicalb-catenin pathway. Upregulation of FZD2 (53,54) and FZD4(55) in cancer has been shown and it suggests a role for Wntsignaling via FZD receptors in tumorigenesis. Downregulationof potent Wnt inhibitory factors such as DKK1 and SFRP4,as observed in HeLa, may enhance Wnt signaling. WISP2, aWnt-1 target gene of the canonical signaling pathway probablyinvolved in transformation processes (56), is upregulated inHeLa. Since upregulation of Wnt signaling components anddownregulation of Wnt inhibitory factors were observed, theWnt pathway may be primed and play a role in reduced cellgrowth inhibition in Hela by TGF-b1.With regard to oncogenes, in HeLa more than twice as many
oncogenes were upregulated than in CC10B and SiHa (data notshown). Increased expression of proto-oncogenes may also
provide a mechanism by which tumor cells show diminishedresponsiveness to TGF-b1 and can escape its tumor sup-pressor effects.Cells undergoing EMT have a more migratory and invasive
phenotype, which is a characteristic phenotype of cancer cellsresistant to TGF-b1- induced cell death. In agreement with theobservation of an EMT-like appearance of SiHa (5) and HeLain response to TGF-b1, these cells are less sensitive to theantiproliferative effect of TGF-b1, which may associate withincreased production of TGF-b1. TGF-b1 produced by SiHawas present in a latent form and did not inhibit cell growthsince anti-TGF-b did not affect cell expansion. TGF-b1 excre-tion by tumor cells more likely contributes to paracrine stimu-lation of tumor development.In our study, novel target genes involved in TNFa, MAPK
and Wnt pathways in response to TGF-b1 were found. Sub-stantial differences in the Smad, TNFa, MAPK and Wntpathway between TGF-b1-sensitive and insensitive cell lineswere observed. Since these pathways are involved in theintegral processes of cell proliferation and cell death, thesepathways may play a role in TGF-b1-induced cell growthinhibition.
Acknowledgements
We thank N.ter Haar for technical assistance with the ELISA and QRT–PCRand H.Baelde, M.Lombaerts, T.van Wezel and A.Cleton for advice in micro-array and QRT–PCR techniques. The study was supported by the DutchCancer Society, grant no. RUL2001-2465. This study was done within theCentre for Medical Systems Biology (CMSB), a centre of excellence supportedby the Netherlands Genomics Initiative/Netherlands Organisation for Scient-ific Research (NWO).
Conflict of Interest Statement: None declared.
References
1.Creek,K.E., Geslani,G., Batova,A. and Pirisi,L. (1995) Progressive loss ofsensitivity to growth control by retinoic acid and transforming growthfactor-beta at late stages of human papillomavirus type 16-initiatedtransformation of human keratinocytes. Adv. Exp. Med. Biol., 375,117–135.
2.De Geest,K., Bergman,C.A., Turyk,M.E., Frank,B.S. and Wilbanks,G.D.(1994) Differential response of cervical intraepithelial and cervicalcarcinoma cell lines to transforming growth factor-beta 1. Gynecol.Oncol., 55, 376–385.
3.Chopra,V., Dinh,T.V. and Hannigan,E.V. (1998) Circulating serum levelsof cytokines and angiogenic factors in patients with cervical cancer.Cancer Invest., 16, 152–159.
4.Comerci,J.T.Jr, Runowicz,C.D., Flanders,K.C., De Victoria,C.,Fields,A.L., Kadish,A.S. and Goldberg,G.L. (1996) Altered expressionof transforming growth factor-beta 1 in cervical neoplasia as an earlybiomarker in carcinogenesis of the uterine cervix. Cancer, 77, 1107–1114.
5.Yi,J.Y., Hur,K.C., Lee,E., Jin,Y.J., Arteaga,C.L. and Son,Y.S. (2002)TGFbeta1-mediated epithelial to mesenchymal transition is accompaniedby invasion in the SiHa cell line. Eur. J. Cell Biol., 81, 457–468.
6.Sheu,B.C., Lin,R.H., Lien,H.C., Ho,H.N., Hsu,S.M. and Huang,S.C.(2001) Predominant Th2/Tc2 polarity of tumor-infiltrating lymphocytesin human cervical cancer. J. Immunol., 167, 2972–2978.
7.Hazelbag,S., Gorter,A., Kenter,G.G., van den Broek,L. and Fleuren,G.(2002) Transforming growth factor-beta1 induces tumor stroma andreduces tumor infiltrate in cervical cancer. Hum. Pathol., 33, 1193–1199.
8.Lee,S., Cho,Y.S., Shim,C., Kim,J., Choi,J., Oh,S., Kim,J., Zhang,W. andLee,J. (2001) Aberrant expression of Smad4 results in resistance againstthe growth-inhibitory effect of transforming growth factor-beta in the SiHahuman cervical carcinoma cell line. Int. J. Cancer, 94, 500–507.
9.Kang,S.H., Won,K., Chung,H.W., Jong,H.S., Song,Y.S., Kim,S.J.,Bang,Y.J. and Kim,N.K. (1998) Genetic integrity of transforming growthfactor beta (TGF-beta) receptors in cervical carcinoma cell lines: loss of
Fig. 6. (A) TGF-b1 production of the cell lines. The amount of TGF-b1production, measured by ELISA, in supernatants of HeLa, SiHa and CC10Bafter 8 days of cell culture (measured as described in Materials and methods).To correct for the TGF-b1 present in the medium, the OD450 values of theHCl treated medium were subtracted from the OD450 value of thesupernatants. (B) The effect of anti-TGF-b on cell growth of SiHa. Cellgrowth, measured in triplicate by cell counts of SiHa, after 6 days withoutanti-TGF-b, with anti-TGF-b (10 mg/ml), with TGF-b1 (0.1 ng/ml) and withboth TGF-b1 and anti-TGF-b. Cell number after 6 days without TGF-b1is shown as 100%.
J.N.Kloth et al.
1500
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

growth sensitivity but conserved transcriptional response to TGF-beta.Int. J. Cancer, 77, 620–625.
10.Maliekal,T.T., Antony,M.L., Nair,A., Paulmurugan,R. andKarunagaran,D. (2003) Loss of expression, and mutations of Smad 2and Smad 4 in human cervical cancer. Oncogene, 22, 4889–4897.
11.Chen,T., de Vries,E.G., Hollema,H., Yegen,H.A., Vellucci,V.F.,Strickler,H.D., Hildesheim,A. and Reiss,M. (1999) Structural alterationsof transforming growth factor-beta receptor genes in human cervicalcarcinoma. Int. J. Cancer, 82, 43–51.
12.Dunfield,L.D., Dwyer,E.J. and Nachtigal,M.W. (2002) TGF beta-inducedSmad signaling remains intact in primary human ovarian cancer cells.Endocrinology, 143, 1174–1181.
13. Janda,E., Lehmann,K., Killisch,I., Jechlinger,M., Herzig,M.,Downward,J., Beug,H. and Grunert,S. (2002) Ras and TGF betacooperatively regulate epithelial cell plasticity and metastasis: dissectionof Ras signaling pathways. J. Cell Biol., 156, 299–313.
14.Calonge,M.J. and Massague,J. (1999) Smad4/DPC4 silencing andhyperactive Ras jointly disrupt transforming growth factor-beta antipro-liferative responses in colon cancer cells. J. Biol. Chem., 274,33637–33643.
15.Bhowmick,N.A., Ghiassi,M., Bakin,A., Aakre,M., Lundquist,C.A.,Engel,M.E., Arteaga,C.L. and Moses,H.L. (2001) Transforming growthfactor-beta1 mediates epithelial to mesenchymal transdifferentiationthrough a RhoA-dependent mechanism. Mol. Biol. Cell, 12, 27–36.
16.Nishita,M., Hashimoto,M.K., Ogata,S., Laurent,M.N., Ueno,N.,Shibuya,H. and Cho,K.W. (2000) Interaction between Wnt and TGF-beta signalling pathways during formation of Spemann’s organizer.Nature, 403, 781–785.
17.Attisano,L. and Labbe,E. (2004) TGFbeta and Wnt pathway cross-talk.Cancer Metastasis Rev., 23, 53–61.
18.Bitzer,M., von Gersdorff,G., Liang,D., Dominguez-Rosales,A., Beg,A.A.,Rojkind,M. and Bottinger,E.P. (2000) A mechanism of suppression ofTGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev., 14,187–197.
19.Ulloa,L., Doody,J. and Massague,J. (1999) Inhibition of transforminggrowth factor-beta/SMAD signalling by the interferon-gamma/STATpathway. Nature, 397, 710–713.
20.Afrakhte,M., Moren,A., Jossan,S., Itoh,S., Sampath,K., Westermark,B.,Heldin,C.H., Heldin,N.E. and ten Dijke,P. (1998) Induction of inhibitorySmad6 and Smad7 mRNA by TGF-beta family members. Biochem.Biophys. Res. Commun., 249, 505–511.
21.Xie,L., Law,B.K., Aakre,M.E., Edgerton,M., Shyr,Y., Bhowmick,N.A.and Moses,H.L. (2003) Transforming growth factor beta-regulated geneexpression in a mouse mammary gland epithelial cell line. Breast CancerRes., 5, R187–R198.
22.Zavadil,J., Bitzer,M., Liang,D., Yang,Y.C., Massimi,A., Kneitz,S., Piek,E.and Bottinger,E.P. (2001) Genetic programs of epithelial cell plasticitydirected by transforming growth factor-beta. Proc. Natl Acad. Sci. USA,98, 6686–6691.
23.Koopman,L.A., Szuhai,K., van Eendenburg,J.D., Bezrookove,V.,Kenter,G.G., Schuuring,E., Tanke,H. and Fleuren,G.J. (1999)Recurrent integration of human papillomaviruses 16, 45 and 67 neartranslocation breakpoints in new cervical cancer cell lines. Cancer Res.,59, 5615–5624.
24.Bolstad,B.M., Irizarry,R.A., Astrand,M. and Speed,T.P. (2003) Acomparison of normalization methods for high density oligonucleotidearray data based on variance and bias. Bioinformatics, 19, 185–193.
25. Irizarry,R.A., Hobbs,B., Collin,F., Beazer-Barclay,Y.D., Antonellis,K.J.,Scherf,U. and Speed,T.P. (2003) Exploration, normalization, andsummaries of high density oligonucleotide array probe level data.Biostatistics, 4, 249–264.
26.Gautier,L., Cope,L., Bolstad,B.M. and Irizarry,R.A. (2004) Affy–analysisof Affymetrix GeneChip data at the probe level. Bioinformatics, 20,307–315.
27.Vandesompele,J., De Preter,K., Pattyn,F., Poppe,B., Van Roy,N., DePaepe,A. and Speleman,F. (2002) Accurate normalization of real-timequantitative RT-PCR data by geometric averaging of multiple internalcontrol genes. Genome Biol., 3, 1–12.
28.Chu,T.Y., Lai,J.S., Shen,C.Y., Liu,H.S. and Chao,C.F. (1999) Frequentaberration of the transforming growth factor-beta receptor II gene in celllines but no apparent mutation in pre-invasive and invasive carcinomas ofthe uterine cervix. Int. J. Cancer, 80, 506–510.
29.Mulder,K.M. and Morris,S.L. (1992) Activation of p21ras by transforminggrowth factor beta in epithelial cells. J. Biol. Chem., 267, 5029–5031.
30.Sano,Y., Harada,J., Tashiro,S., Gotoh-Mandeville,R., Maekawa,T. andIshii,S. (1999) ATF-2 is a common nuclear target of Smad and TAK1
pathways in transforming growth factor-beta signaling. J. Biol. Chem.,274, 8949–8957.
31.Hocevar,B.A., Brown,T.L. and Howe,P.H. (1999) TGF-beta inducesfibronectin synthesis through a c-Jun N-terminal kinase-dependent,Smad4-independent pathway. EMBO J., 18, 1345–1356.
32.Kang,Y., Chen,C.R. and Massague,J. (2003) A self-enabling TGFbetaresponse coupled to stress signaling: Smad engages stress response factorATF3 for Id1 repression in epithelial cells. Mol. Cell, 11, 915–926.
33.Fan,F., Jin,S., Amundson,S.A., Tong,T., Fan,W., Zhao,H., Zhu,X.,Mazzacurati,L., Li,X., Petrik,K.L., Fornace,A.J.Jr, Rajasekaran,B. andZhan,Q. (2002) ATF3 induction following DNA damage is regulated bydistinct signaling pathways and over-expression of ATF3 proteinsuppresses cells growth. Oncogene, 21, 7488–7496.
34.Fornace,A.J.,Jr., Nebert,D.W., Hollander,M.C., Luethy,J.D.,Papathanasiou,M., Fargnoli,J. and Holbrook,N.J. (1989) Mammaliangenes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol. Cell. Biol., 9, 4196–4203.
35.Yamaguchi,K., Shirakabe,K., Shibuya,H., Irie,K., Oishi,I., Ueno,N.,Taniguchi,T., Nishida,E. and Matsumoto,K. (1995) Identification of amember of the MAPKKK family as a potential mediator of TGF-betasignal transduction. Science, 270, 2008–2011.
36.Shibuya,H., Yamaguchi,K., Shirakabe,K., Tonegawa,A., Gotoh,Y.,Ueno,N., Irie,K., Nishida,E. and Matsumoto,K. (1996) TAB1: an activatorof the TAK1 MAPKKK in TGF-beta signal transduction. Science, 272,1179–1182.
37.Shirakabe,K., Yamaguchi,K., Shibuya,H., Irie,K., Matsuda,S.,Moriguchi,T., Gotoh,Y., Matsumoto,K. and Nishida,E. (1997) TAK1mediates the ceramide signaling to stress-activated protein kinase/c-JunN-terminal kinase. J. Biol. Chem., 272, 8141–8144.
38.Moriguchi,T., Kuroyanagi,N., Yamaguchi,K., Gotoh,Y., Irie,K., Kano,T.,Shirakabe,K., Muro,Y., Shibuya,H., Matsumoto,K., Nishida,E. andHagiwara,M. (1996) A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem., 271,13675–13679.
39.Wang,W., Zhou,G., Hu,M.C., Yao,Z. and Tan,T.H. (1997) Activation ofthe hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activatedc-Jun N-terminal kinase (JNK) pathway by transforming growth factorbeta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF betasignal transduction. J. Biol. Chem., 272, 22771–22775.
40.Han,J., Jiang,Y., Li,Z., Kravchenko,V.V. and Ulevitch,R.J. (1997)Activation of the transcription factor MEF2C by the MAP kinase p38 ininflammation. Nature, 386, 296–299.
41.Maliekal,T.T., Anto,R.J. and Karunagaran,D. (2004) Differential activa-tion of Smads in HeLa and SiHa cells that differ in their response totransforming growth factor-beta. J. Biol. Chem., 279, 36287–36292.
42.VanArsdale,T.L., VanArsdale,S.L., Force,W.R., Walter,B.N.,Mosialos,G., Kieff,E., Reed,J.C. and Ware,C.F. (1997) Lymphotoxin-beta receptor signaling complex: role of tumor necrosis factor receptor-associated factor 3 recruitment in cell death and activation of nuclearfactor kappaB. Proc. Natl Acad. Sci. USA, 94, 2460–2465.
43.Eliopoulos,A.G., Dawson,C.W., Mosialos,G., Floettmann,J.E., Rowe,M.,Armitage,R.J., Dawson,J., Zapata,J.M., Kerr,D.J., Wakelam,M.J.,Reed,J.C., Kieff,E. and Young,L.S. (1996) CD40-induced growth inhibi-tion in epithelial cells is mimicked by Epstein–Barr virus-encodedLMP1: involvement of TRAF3 as a common mediator. Oncogene, 13,2243–2254.
44.Nakayama,M., Ishidoh,K., Kayagaki,N., Kojima,Y., Yamaguchi,N.,Nakano,H., Kominami,E., Okumura,K. and Yagita,H. (2002) Multiplepathways of TWEAK-induced cell death. J. Immunol., 168, 734–743.
45.Zhai,Y., Ni,J., Jiang,G.W., Lu,J., Xing,L., Lincoln,C., Carter,K.C.,Janat,F., Kozak,D., Xu,S., Rojas,L., Aggarwal,B.B., Ruben,S., Li,L.Y.,Gentz,R. and Yu,G.L. (1999) VEGI, a novel cytokine of the tumornecrosis factor family, is an angiogenesis inhibitor that suppresses thegrowth of colon carcinomas in vivo. FASEB J., 13, 181–189.
46.Pan,G., Bauer,J.H., Haridas,V., Wang,S., Liu,D., Yu,G., Vincenz,C.,Aggarwal,B.B., Ni,J. and Dixit,V.M. (1998) Identification and functionalcharacterization of DR6, a novel death domain-containing TNF receptor.FEBS Lett., 431, 351–356.
47.Zhang,H.G., Hyde,K., Page,G.P., Brand,J.P., Zhou,J., Yu,S., Allison,D.B.,Hsu,H.C. and Mountz,J.D. (2004) Novel tumor necrosis factor alpha-regulated genes in rheumatoid arthritis. Arthritis Rheum., 50, 420–431.
48.Shinohara,A., Yokoyama,Y., Wan,X., Takahashi,Y., Mori,Y., Takami,T.,Shimokawa,K. and Tamaya,T. (2001) Cytoplasmic/nuclear expressionwithout mutation of exon 3 of the beta-catenin gene is frequent in thedevelopment of the neoplasm of the uterine cervix. Gynecol. Oncol., 82,450–455.
TGF-b1 alters expression in Wnt, MAPK and TNFa pathways
1501
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from

49. Imura,J., Ichikawa,K., Takeda,J. and Fujimori,T. (2001) Beta-cateninexpression as a prognostic indicator in cervical adenocarcinoma. Int. J.Mol. Med., 8, 353–358.
50.Slusarski,D.C., Corces,V.G. and Moon,R.T. (1997) Interaction of Wntand a Frizzled homologue triggers G-protein-linked phosphatidylinositolsignalling. Nature, 390, 410–413.
51.Kuhl,M., Sheldahl,L.C., Malbon,C.C. and Moon,R.T. (2000) Ca(2þ)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzledhomologs and promotes ventral cell fates in Xenopus. J. Biol. Chem., 275,12701–12711.
52.Sheldahl,L.C., Park,M., Malbon,C.C. and Moon,R.T. (1999) Proteinkinase C is differentially stimulated by Wnt and Frizzled homologs in aG-protein-dependent manner. Curr. Biol., 9, 695–698.
53.Kirikoshi,H., Sekihara,H. and Katoh,M. (2001) Expression profiles of10 members of Frizzled gene family in human gastric cancer. Int. J.Oncol., 19, 767–771.
54.Tanaka,S., Akiyoshi,T., Mori,M., Wands,J.R. and Sugimachi,K. (1998) Anovel frizzled gene identified in human esophageal carcinoma mediatesAPC/beta-catenin signals. Proc. Natl Acad. Sci. USA, 95, 10164–10169.
55.Wissmann,C., Wild,P.J., Kaiser,S., Roepcke,S., Stoehr,R.,Woenckhaus,M., Kristiansen,G., Hsieh,J.C., Hofstaedter,F.,Hartmann,A., Knuechel,R., Rosenthal,A. and Pilarsky,C. (2003) WIF1, acomponent of the Wnt pathway, is down-regulated in prostate, breast, lungand bladder cancer. J. Pathol., 201, 204–212.
56.Zoubine,M.N., Banerjee,S., Saxena,N.K., Campbell,D.R. andBanerjee,S.K. (2001) WISP-2: a serum-inducible gene differentiallyexpressed in human normal breast epithelial cells and in MCF-7 breasttumor cells. Biochem. Biophys. Res. Commun., 282, 421–425.
Received January 4, 2005; revised April 13, 2005;accepted April 26, 2005
J.N.Kloth et al.
1502
by guest on June 4, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from




















