
Research Costs Investigated: A Study Into the Budgets of ...
Upload
u-picardieCategory
view
1download
0
THEO CHEM
ELSEVIER Journal of Molecular Structure (Theochem) 368 (1996) 67-80
Structures of oxyfluoroaluminates in molten cryolite-alumina mixtures investigated by DFT-based calculations1
Gerard S. Picard”‘*, FrCdCric C. Bouyera, Michel Leroyb, Yves Bertaudc, S. BouvetC
“Laboratoire d’Electrochimie et de Chimie Analytique, Groupe Rtactivitk en Milieu Ioniques Liquides, Unit6 associie au Centre National de
la Recherche Scienti&ue no. 216, Ecole Nationale Supkrieure de Chimie de Paris, I1 rue Pierre et Marie Curie, 75231 Paris CEDEX 05, France
bAluminium Pechiney AL WAL, Centr ‘Alp, BP 27, 38340 Voreppe, France
“Pechiney Centre de Recherches de Voreppe, Centr’Alp, BP 27, 38340 Voreppe, France
Received 1 November 1995; accepted 29 March 1996
Abstract
Aluminum is produced throughout the world using the HCroult-Hall process, which consists in the electrolysis of alumina dissolved in cryolite melts. This dissolution of alumina gives rise to the formation of oxyfluoroaluminates, the structures of which vary with the composition of the electrolyte mixture. Up to now there has been no real consensus as to the structural entities of these fluoroaluminate ions. The lack of knowledge of the ionic species arising from the dissolution of alumina in molten cryolite has led us to undertake structural, energetic and vibrational studies of these species using the methods of computational chemistry.
Conformational and vibrational analyses of the ionic complexes were performed with the DGauss software on Cray C98 and C916 machines located at Cray Research, Eagan, MN, USA. A systematic study was carried out on the complexes [AlOF,]‘” (1 5 n 5 5), [A120F.J4-” (4 5 x 5 9), [A1202FX]2-” (2 5 x I 7), which are those most commonly suggested for interpreted experimental measurements. The A140FIo and A&OF? complexes were also investigated because of their possible occurrence in the electrorefining process of aluminum.
Some of the studied complexes are much more stable than the others and so are the complexes most probably involved in the electrowinning process of aluminum. These complexes are AlO&, A120g- and A1202e-. Their infrared spectra have also been calculated. Concerning the two complexes A140FIo and A&O@, we find that oxygen is three-coordinated and not four- coordinated. The last atom of aluminum is stabilized by two fluoride bridges.
This computational approach has enabled us to eliminate the majority of the structures suggested in the literature for oxyfluoroaluminate ions. Currently we are examining in our laboratory the effects of cations on the anionic complexes.
Keywords: Aluminium; Cryolite; Fluoride; Modelling; Molten salts; Oxyfluorides
1. Introduction * Corresponding author. 1 Presented at the Second Electronic Computational Chemistry
Conference. This issue along with any supplimentary material can be accessed from the THEOCHEM HomePage at URL:http:// www.elsevier.nl/locate/theochem
The chemistry of molten aluminum chlorides (in connection with aluminum industry processes) has been under investigation in our laboratory since
0166-1280/96/$15.00 0 1996 Elsevier Science B.V. All rights reserved PII SO166-1280(96)04611-S

68 G.S. Picard et ai.iJournal of Molecular Structure (Theochem) 368 (1996) 67-80
1979. In fact, the first research efforts were initiated with the objective of obtaining pure aluminum chlor- ide for eventual use as a feed in the ALCOA chloride process of electrowinning of aluminum developed at that time [l]. The dehydration of hexahydrated alumi- num chloride [2] and chlorination procedures for it and for industrial alumina-especially those invol- ving phosgene-were investigated [3]. Moreover, it was the beginning of our research efforts on selective chlorination using several gaseous mixtures of various chlorinating powers’ [4-lo]. This was applied in par- ticular to the chemical treatment of bauxites [4,5].
These researches were extended both to determin- ing the stabilities of aluminum oxychloride and alu- mina [ll] and to analyzing the role of the fluoride anion in the conditional solubility of alumina in chlor- ide melts [12], with the idea of a possible mixed chlor- ide-fluoride based process for electrowinning of aluminum. In a similar manner, we studied the elec- trorefining process electrochemically and developed a new low temperature process3 [13-151.
More recently, much research work has been and is currently being performed on the chemistry involved in the Heroult-Hall process. In particular, electroche- mical impedance spectroscopic (E.I.S.) studies were undertaken for studying the discharge of oxyfluoroa- luminates [16,17] and thus to conceive and develop an electrochemical sensor for determining the amount of alumina dissolved in cryolite melts and to determine the kinetics of dissolution as a function of the nature of the alumina involved [18]. E.I.S. was also used for testing the electrochemical behavior of various carbon anodes in these melts. In addition, a molten salt poten- tiometric method was developed for characterizing industrial aluminas [19].
All these studies suffer from a lack of information about the structures of the species involved. For this reason we have undertaken computational investiga- tions in the domain of the aluminum industry in order to complement our previous studies. In fact, model- ling techniques appear to be a highly promising tool for obtaining structural, energetic and vibrational information and also electronic properties for various systems [20] and have already been used in our
2See page: http://alcyone.enscp.jussieu.fr/Pages/LECA/ GP/euchem_talk_94.html.
3 See WWW page: http://alcyone.enscp.jussieu.fr/Pages/J..ECA/ GP/GRC*95html.
laboratory to investigate the chemistry in melts related to the industrial electrorefining [21-231 and electro- winning [24-281 processes of aluminum.
2. Computational details
Our theoretical calculations were performed with the DGauss [29] software, version 2.3.1, using the Unichem interface developed by Cray Research. This DFT software involves an N3 level of computa- tion (N denotes the number of atomic orbitals) and, in addition, takes explicitly into account the exchange and correlation effects. At the same level of calcula- tion, the Hartree-Fock method would lead to more time consuming calculations (N5). For this reason, the DFT methodologies are more tractable and we consider them as a good alternative to which we turned for computational chemistry. All calculations were done on a Cray C90 (8 and 16 processors) series computer located in Eagan, MN, USA with an Internet gateway, using our Silicon Graphics Iris Indigo R4400 workstations for visualizing and analyzing the results. Geometrical optimizations were per- formed at the GGA (generalized gradient approxima- tion) level using the Becke [30] and Lee-Yang-Parr [31] correlation and exchange functionals. We took the finest grid (Tight) and we verified the consistency of the stationary point obtained by calculating the Hessian matrix. The triple zeta plus polarization (TZP) basis set was employed. All geometry optimi- zations were performed without any fixed symmetry. The symmetries determined and reported in the fol- lowing tables and figures are those obtained after a full optimization (without any restriction). Mulliken charges and dipolar moments were also computed to help us in interpreting the discharge process at carbon anodes. In addition, we employed the DMol software (from Biosym/MSI) at the same level of accuracy in order to compare the computational results on the single AlOF complex.
We employed the cluster method to treat some complexes that might be present in the industrial elec- trochemical batch. For highly charged species, the model could not correctly represent the physical sys- tem. What we want to point out is that some com- plexes autodissociate by rejecting one fluoride ion. The energy gained by this dissociation is much

G.S. Picard et al./Joumal of Molecular Structure (Theochem) 368 (19%) 67-80 69
more notable that that corresponding to any stabiliz- ing effect of counter cations. In fact, a geometry opti- mization of OAlF$ with one Na’ (i.e. NaOAl@-) showed that the Al atom is always fivefold coordi- nated (surrounded by four fluorides and one oxygen), as previously thought through an optimization on OAlF$. This demonstrates the instability of such a complex. Nevertheless, we did not use HOMO- LUMO for any interpretation.
In order to compare all the energies of all the com- plexes, we preferred to calculate the binding energy, which is the difference in total energy of the complex with the corresponding monatomic ions (A13+, F-, 02-). Within the DFT context, 02- surely has no meaning; nevertheless, we used binding energies in order to have the same reference for all complexes. In that way, relative stabilities can be compared directly.
3. Results and discussion.
Despite the great number of studies, the exact struc- ture of oxyfluoroaluminates is still controversial [32]. The more recent contributions are due to Gilbert et al. [33], who suggest Al-O-Al bridges from Raman spectroscopic measurements, and to Kvande [34]. Our approach was essentially a systematic and com- puterized study of the stabilities of the whole series of the oxyfluoroaluminates envisaged up to now for explaining various experimental results. In order to take some account of the effect of the alumina con- centration in cryolite melts, we first studied the series
of AlOF, (charge l-x) supposed to occur at low alu- mina concentration in melts, and then those contain- ing Al-O-AI bridges such as A120c” and A1202Fz-‘, which are expected to occur at high alumina concen- trations in molten cryolite.
3.1. AlOF, series
First of all we check the consistency of our quan- tum approach. For that, we compare the results of our calculations performed with DGauss (analytical) aad DMol (numerical) software on the structure (A), moment of inertia Ba (cm-‘) and vibrational frequen- cies (cm-‘) of the most simple structure (AlOF) of the series with the corresponding values available in the literature [35]. Values reported in Table 1 show that the structures (and the moment of inertia) are in good agreement with the Janaf data whatever the level of calculation and the software used. Concerning the vibrational frequencies, the results obtained with the GGA level of calculations agree better with the experimental values except for the low energy range (which is currently one of the problems of DFT).
The optimized structures of the AlOF, complexes are represented in Fig. 1 (which also gives the dipole moments in Debye). The Al-F and Al-O bond lengths increase with the number of fluoride ions, but in contrast the absolute values of the dipole moment decrease. The maximum coordination num- ber of Al is equal to five (one oxygen bond and four fluoride bonds). Any attempt at adding one extra fluor- ide failed (even when adding one counter cation to stabilize the structure).
Table 1 Comparison of the structures, moments of inertia and vibrational frequencies and dipolar moments of AlOF (obtained from DGauss/DMol calculations with the values reported in JANAF
Origill Structure Vibrational frequencies Dipolar moments
JANAF O-AI = 1.61 A; AI-F = 1.63 & Ba = 0.184214 cm-’ 386(2~75(1~1022(1) (15.195791 x lo-r9 g cm’)
DMol (LDA-JMW) G-AI = 1.62 A; At-F = 1.63 .k, Bs = 0.180526 cm-’ 195(2)-716(1~1138(1) 2.97 (15.506219 x 1O-39 g cm*)
DMol (NLDA-BLYP) G-AI = 1.63 A; Al-F = 1.68 A, Ba = 0.176137 cm-’ 184(2)-685(1>-1090(l) 2.92 (15.892621 x JO-” g cm*)
DGauss (LDA-VWN) O-At = 1.61 A, AI-F - 1.64 A, Bs = 0.182887 cm-r 222(2)-725(1hl148(1) 2.90 (15.306016 x 1O-39 g cm*)
DGauss (NLDA-BLYF’) O-Al = 1.63 & Al-F = 1.67 A, Ba = 0.178358 cm-r 217(2)-695(1)-1107(l) 2.81 (15.694686 1O-39 g cm’)

70 GS. Picard et aLlJournal of Molecular Structure (Theochem) 368 (19%) 67-80
=I .52 D 120P
0 St Aluninun 8
Oxygen a Fluorine
Fig. 1. Structures and dipolar moments of AlOF, (charge: 1 - n).
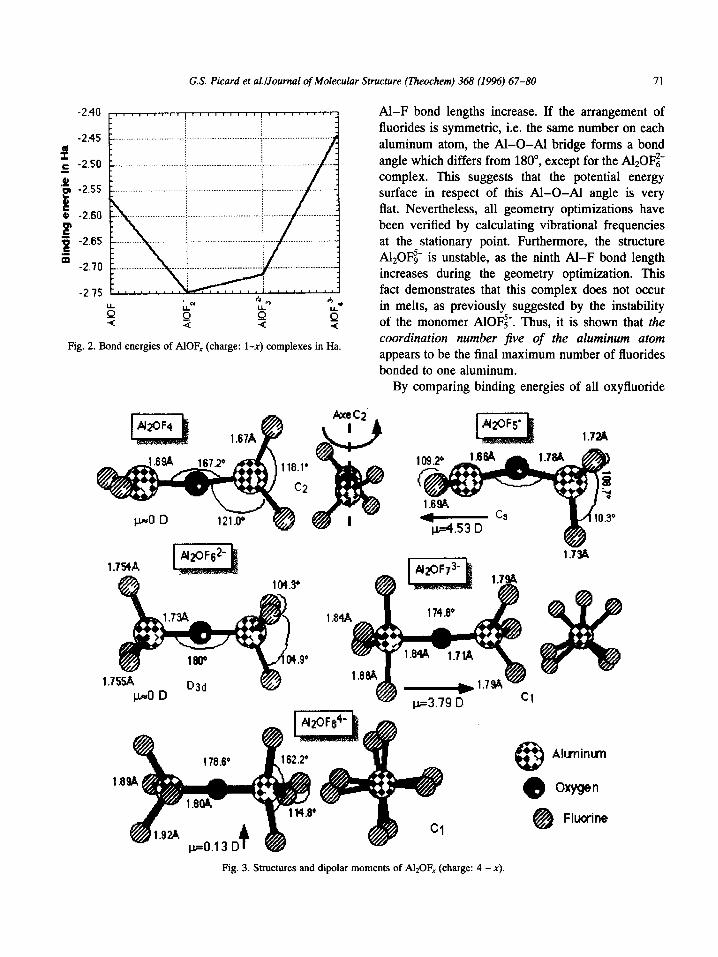
Bond energies of these complexes were determined by subtracting from the total energy of the complex under consideration the total energies of A13’ (-240.39182 Ha), O*- (-74.79007 Ha) and F- (-99.87150 Ha) so that all relative stabilities could be compared directly. The total energies and bond energies for the first series of complexes are reported in Table 2 and Fig. 2.
It appears from these studies that AlOF; is the most stable of all the anionic complexes. This is in agree- ment with the work of Boner [36] and of Qiu and Xie [37]. The AlOF:- ion could also be envisaged (possi- ble effect of the counter cation), as done by Holm [38]. However, Holm also envisaged the formation of the octahedral AlOF=f- complex, which appears from our results not to have to be considered.
Table 2
Total energies and bond energies (Ha) for the AlOF, series
Species Total energies Bond energies
AIOF -417.61795 -2.56455 AlOF; -517.67395 -2.74905 AlOF;- -617.51061 -2.71421 Aloe- -717.11149 -2.44358 AlOF$ -816.92630 -2.38690
On the basis of our results, it is clearly shown that the maximum coordination of the aluminum atom is five, with the threefold coordinated aluminum com- pound AlOF; being the most stable within the whole series. As for molten cryolite, Robert et al. [39] sug- gested that molten pure cryolite is composed mainly of AlF:- and AlFi with the minor compound AlF$-.
Except for pure fluoride and chloride aluminum complexes which are sixfold coordinated, the new (recently shown by Raman spectroscopic measure- ments) fivefold coordinated aluminum complex is generally proposed as being the most stable aluminum coordination in pure halide melts. With the presence of oxygen in baths, this divalent atom reduces the coordination of aluminum, and structures in which Al is less than five-coordinated are preferentially sta- bilized. The structure most likely to occur is that with a three-coordinated Al, AlOF;, in which the Al-O bond could be seen as a double bond.
3.2. The A&OF, series
All structures are depicted in Fig. 3 and total ener- gies and bond energies are given in Table 3. As the number of fluoride ions in the structures increases, all
-2.40
-2.45
P E -2.50
i -2.55
b Q, -2.60
i -2.65
m -2.70
-2.75
G.S. Picard et aLlJournal of Molecular Structure (Theochem) 368 (1996) 67-80 71
!,,““‘,‘!“1’)1”‘_ Al-F bond lengths increase. If the arrangement of fluorides is symmetric, i.e. the same number on each aluminum atom, the Al-O-Al bridge forms a bond angle which differs from MO”, except for the A120Fz- complex. This suggests that the potential energy
_ surface in respect of this Al-O-Al angle is very flat. Nevertheless, all geometry optimizations have been verified by calculating vibrational frequencies at the stationary point. Furthermore, the structure A120Fs- is unstable, as the ninth Al-F bond length increases during the geometry optimization. This fact demonstrates that this complex does not occur
9
’ s-4 $4 *) 5 5
A-
5
in melts, as previously suggested by the instability of the monomer AlOg-. Thus, it is shown that the
Fig. 2. Bond energies of AlOF, (charge: l-x) complexes in Ha. coordination number five of the aluminum atom appears to be the final maximum number of fluorides bonded to one aluminum.
By comparing binding energies of all oxyfluoride
1.754A
1.X5&
ClpOD D3d
1.694
1c3 ~4.53 D
174.6'
p3.79 D -
9 Oxygen
@ Fluorine
Fig. 3. Structures and dipolar moments of A&OF, (charge: 4 - x).

72 G.S. Picard et aLlJournal of Molecular Structure (Theochem) 368 (1996) 67-80
Table 3 Table 4
Total and bond energies (Ha) for the A120Fx series Total and bond energies (Ha) for the A1202Fx series
Species Total energies Bond energies
&OF4 -960.04922 -4.98949
M2OF; -1060.10808 -5.17685
A120F;- -1160.02811 -5.22538
Al2oG- -1259.70318 -5.02895
.4l2m- -1359.26197 -4.71623
Species Total energies Bond energies
A202F2 -835.4375 1 -5.33072
~2025 -935.49459 -5.51629
A1202F:- -1035.38972 -5.53992
ti202e -1135.05648 -5.33518
.4l202F;1- -1234.59245 -4.99965
complexes, A120Fz-, which is the fourfold coordi- nated aluminum complex, appears to be the most
stable complex among the whole series. These results are in agreement with those of Kvande [34] and of Sterten [40] for molten cryolite at low alumina
concentrations.
the oxygen being the center of a square and the alu- minum atoms at the top. These geometry optimiza-
tions lead to the same final structure, the one depicted in Fig. 5.
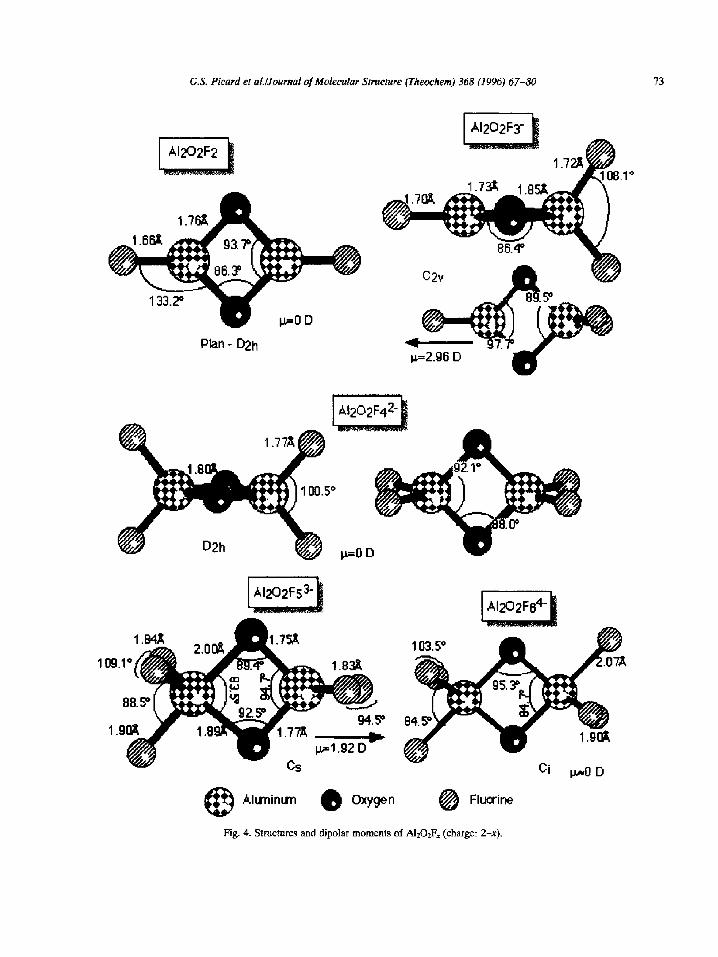
3.3. The A1202Fx series
All structures are doubly bridged with two Al-O- Al links (see Fig. 4), prior evidence for which was
given by Gilbert et al. [33] from Raman spectroscopic measurements. As the number of fluorides has increased, it appears that at most only three fluorides
are bonded to one aluminum atom. Once more, we finally find the maximum number of five fluorides bonded to one aluminum.
The linal conformation provides evidence of a coor- dination of three for the o.xygen atom, whereas we
expect a fourfold-coordinated oxygen complex. The last aluminum atom is rejected from the cluster but stabilized by two fluoride bridges, as shown by the
three-dimensional density representation of Fig. 5. There is no covalent O-Al bond (with the fourth alumi- num atom) but two strong covalent Al-F-Al bridges.
Table 4 gives total energies and also bond energies. It emerges that A1202F2- is the most stable complex within the whole series of oxyfluoride anions. Again,
these results are in agreement with experimental ones, in particular with those of Kvande and of Sterten, concerning the structures of oxyfluorides anions in
cryolite melts at high alumina concentrations.
In order to see if the activity of fluorides changes
the coordination of oxygen, we get rid of two fluoride ions, and the species AlhOFF has been optimized. As
depicted in Fig. 6, one aluminum atom is finally bonded not to the oxygen atom but linked with another aluminum atom with one fluoride bridge within the structure. This structure suggest a coordi- nation number of three for the oxygen atom. In con-
clusion, even if we expected four O-Al bonds, only the threefold-coordinated oxygen complex is stable. The fourth aluminum atom is stabilized with Al-F-
Al bridges if the activity of the fluoride anion in mixed chloride-fluoride melts allows this.
3.4. A140F8 and Al&FtO
During the electrorefining of aluminum (mixed chloride-fluoride melts), it has been shown [41] that small amounts of oxides are dissolved in the molten mixtures.
4. Conclusion
A ratio of one oxygen atom for four aluminum atoms has been determined by chemical analyses of the bath [41]. We attempted to model the structure of such a complex with the assumption of an entire oxyfluoride compound without a counter cation first. Several initial geometries were chosen, one with the oxygen atom being the center of a tetrahedron and all aluminum atoms being at the top, and the other with
Several facts and remarks emerge from our results.
In particular, three major factors make these theoreti- cal approach appealing: (i) this anionic approach allows rapid screening to eliminate some oxyfluoride complexes; (ii) AlzOFt- and Al20& appear to be the most stable entities in the melts, as shown by Kvande and by Sterten (respectively at low and at high alumina concentrations in the cryolite); and (iii) the maximum coordination offve for the aluminum atom, which eliminates some structures previously proposed
G.S. Picard et &Journal of Molecular Structure (Theo&em) 368 (1996) 67-80 73
Fig. 4. Structures and dipolar moments of A1202F, (charge: 2-x).

74 G.S. Picard et aLlJournal of Molecular Structure (Theo&em) 368 (1%) 6740
Fig. 5. Structures and Mulliken charges (in em fraction) of A&OFIo (charge: 0).
(e.g. AlO@-, Holm [38]) and the maximum coordina- tion of three for the oxygen atom in the A140F10 clus- ter. Thus, it appears that the fourfold-coordinated aluminum complexes are the most stable except for the AlOF, series monomers, for which the threefold- coordinated Al complex is stable.
4.1. Cryolite-alumina mixtures
A deeper analysis can be performed on the relative stabilities of complexes from the different series, lead- ing to some information about the corresponding equilibrium.
Fig. 6. Structures and Mulliken charges (in e- fraction) of AL,OFs (charge: 2+).

G.S. Picard et al.IJournal of Molecular Structure (Theochem) 368 (1996) 67-80 75
In particular, the equilibrium relative to the dimerization of AlOK can be written as follows
2AlOF; = A&O& -
From the binding energy values (Table 2 and Table 4) we can calculate a dimerization energy Ed = -0.04182 Ha, i.e. -109.8 kJ mol-‘. This high value for Ed means that the structure A120&- is likely to be favoured.
Concerning the equilibrium
AlOF; + AlF; = A120F;-
of A1203 solutions in molten cryolite [33], could be found only in the cases of the AlzOF, and A1202F, complexes. Bands observed at 530 cm-’ and 310 cm-’ could be related to some A1202F, com- plexes. The occurrence of Al-O-Al bridges seems now to be well established, in agreement with the reports of Gilbert [33], Rolin and Bernard [42] and Forland and Ratjke [43]. A further analysis taking into account the effect of the cation is currently in progress in our laboratory.
4.4. Further investigations the corresponding energy E (calculated from the bind- ing energies reported in Table 2 and Table 3, and from the binding energy of All?+calculated with DGauss with the same level of accuracy-of -2.4721374 Ha) is equal to -0.00419 Ha, i.e. E = -11 kJ mol-‘, which is very weak. So, because of both the composition and the temperature of cryolite-alumina mixtures, we can envisage modifications near the anode involving this type of equilibrium. To sum up, the oxygen-containing species which probably occur in cryolite-alumina melts are AlOF;, A120F~- and Al202Fi-. This is in agreement with the conclusion of Forland and Ratkje [43] but, in addition, our work clarifies the nature and structure of the com- plexes and quantifies their stabilities.
4.4.1. Model improvements
4.2. Electrorejining bath
Looking at Fig. 5, we can consider the A140Fi0 complex as being formed by combining the AlF; ion with the AlsOE cation. This last complex can also be recognized in the A&O@ cluster. This suggests the possibility of a new series of complexes (not yet reported in the literature): A130F4, (with x = 2,3 or 4).
Currently there are several major shortcomings of studies of oxyfluoride anions: (i) there is no diffuse orbital set to take into account the delocalized elec- trons, especially for charged ions; (ii) we do not take into account the effect of counter cations (as for pure cryolite species [28]), which will be the subject of a forthcoming paper. Nevertheless, this is the first attempt to model oxyfluoride species, and the current success of DFT results will stimulate research in these areas. One of the most critical but also most difficult obstacles is the lack in our computations of diffise functions. However, the results are generally good if the charges of the ions are not too large [44], e.g. for the first complexes of the series. In addition, we have established (in work to be published) that diffuse functions do not improve calculations on simple anionic entities of cryolite, especially for predicting reaction energies.
Our point of view is thus to take into account the effects of counter cations, which could provide at least a heuristic guideline on how to improve our first com- putational results. To this end, the approach will point out energy reaction shifts due to cations effects.
4.3. Spectroscopic data 4.4.2. Anode process (Hkoult-Hall)
All vibrational frequencies and infrared intensities On the basis of our results, investigations of anionic were determined (see Appendix), and could with species and, especially, calculations of dipolar advantage be used to help in interpreting the experi- moments provide evidence of orientations of mental spectra obtained for cryolite-alumina melts oxyfluoride complexes near the carbon anode in the systems. Although the effect of the cation was not aluminum electrowimiing process. In fact, it is known taken into account, a rough analysis of frequencies that a strong electric field arises in the double layer relative to the whole series of all the anionic com- plexes shows that the wavenumbers 460 cm-’ and
near the electrode. Because of this electric field, polar
185 cm-‘, complexes have their proper orientation, the one that
which appear in the experimental spectra forces the dipole moment and the double layer electric

76 G.S. Picard et aLlJournal of Molecular Structure (Theochem) 368 (1996) 67-80
field to be aligned. Thus, fluoro-oxide anions diffuse towards the electrode in a certain way. We have remarked that, for monomer oxides (AlOF, series), the fluorides face towards the electrode. We showed this fact even for the A120FX and A1202FX series. In contrast, if we look at the HOMO orbital, the one which is involved in the oxidation of complexes and thus in the discharge mechanism, this orbital is delo- calized on the oxygen atom. It is thus obvious that complexes have to change their orientation with respect to the electrode surface. This computerized approach for the reaction mechanism of the anodic reaction is currently being investigated with the help of our previous E.I.S. studies on this reaction [16,17].
Acknowledgements
The authors thank Pechiney for financial support. One author (F.B.) thanks Pechiney and ADEME for providing him with a doctoral grant. The authors are very grateful to Ch. Henriet from Cray Research France for his help in using Unichem/DGauss packages running on Cray machines in Eagan. The authors also thank J.-M. Cense for helping them to use his MolView and MolDraw software.
Appendix A: All vibrational frequencies and infrared intensities are given below for each compound. Results from DGauss 2.3.1 (TZP basis sets) at the GGA level of computation (Becke88 and Lee-Yang- Parr for the exchange and correlation functionals)
AIOF Mode Freq.
6 7 8 9
0AlI;I Mode
222.36 222.36 725.14
1147.62
Freq.
7 277.51 8 287.52 9 315.75 10 604.24 11 698.47 12 990.21
Intensl Intens, Intenskm a.“. D/A mol-’ 0.0904 2.0846 88.08 0.0904 2.0846 88.08 0.0211 0.4857 20.52 0.0953 2.1994 92.94
Intens/a.u. Intens, Ink&km D/A mol -’
0.0103 0.2378 10.05 0.0383 0.8841 37.36 0.0761 1.7546 74.14 0.0251 0.5786 24.45 0.1635 3.7713 159.36 0.1522 3.5117 148.39
OAlF:- Mode
7 8 9 10 11 12 13 14 15
OAK- Mode
7 8 9 10 11 12 13 14 15 16 17 18
AWF4 Mode
7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
‘42oF;
Mode
7 8 9 10 11 12 13 14 15
Freq.
203.69 208.89 301.88 317.70 320.97 509.78 576.24 577.86 856.30
Intens/ Intens, Intens/km a.u. D/A mol-’ 0.0008 0.0195 0.83 0.0022 0.0499 2.11 0.0131 0.3013 12.73 0.0195 0.4498 19.00 0.0191 0.4408 18.63 0.0279 0.6434 27.19 0.1791 4.1317 174.58 0.1749 4.0348 170.49 0.1949 4.4957 189.96
Freq.
85.47 161.07 164.99 224.86 260.47 271.19 298.45 312.16 343.40 412.03 489.22 747.04
Intens/ Intens, Intensh a.u. D/A mol“ 0.0003 0.0077 0.33 0.0011 0.0261 1.10 0.0012 0.0282 1.19 0.0631 1.4562 61.53 0.0730 1.6836 71.14 0.0010 0.0223 0.94 0.0035 0.0797 3.37 0.0016 0.0377 1.59 0.1516 3.4980 147.81 0.0292 0.6725 28.42 0.1965 4.5336 191.57 0.1909 4.4039 186.09
Freq. Intensl Intens, Intenskm ax. D/A mol-’
66.52 0.0019 0.0446 1.89 90.39 0.0011 0.0264 1.11
105.11 0.0001 0.0023 0.10 190.25 0.0032 0.0741 3.13 246.04 0.0220 0.5074 21.44 248.21 0.0566 1.3052 55.15 251.77 0.0205 0.4731 19.99 335.63 0.1473 3.3972 143.55 345.72 0.1451 3.3479 141.47 459.06 0.0055 0.1267 5.35 709.84 0.0837 1.9302 81.56 825.27 0.0090 0.2083 8.80 880.87 0.1729 3.9896 168.58 882.74 0.1709 3.9430 166.61
1103.21 0.5798 13.3767 565.23
Freq.
51.08 66.40 85.13
176.94 180.97 229.82 242.13 274.31 295.12
Intens/ Intens, Intens/km a.u. D/A mole’ 0.0006 0.0128 0.54 0.0005 0.0124 0.52 0.0001 0.0012 0.05 0.0037 0.0849 3.59 0.0009 0.0218 0.92 0.0101 0.2326 9.83 0.0003 0.0062 0.26 0.0868 2.0020 84.59 0.0226 0.5204 21.99

G.S. Picard et aLlJournal of Molecular Struchcre (Theochem) 368 (1996) 67-80 77
323.02 0.0611 1.4105 59.60 345.29 0.1006 2.3220 98.11 470.05 0.0463 1.0680 45.13 623.31 0.1480 3.4139 144.26 735.28 0.2018 4.6555 196.72 742.40 0.1694 3.9080 165.13 744.73 0.0172 0.3968 16.77 804.48 0.1978 4.5639 192.85
1073.41 0.4544 10.4823 442.93
28 29 30
AI*OF;f‘
Mode
608.64 0.2403 5.5428 234.21 615.75 0.0378 0.8718 36.84 951.38 0.4893 11.2886 477.00
Freq.
7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33
~202F2
48.26 83.32 92.17
106.19 111.23 133.30 136.95 170.93 243.57 249.92 260.89 275.10 292.82 294.52 316.60 321.13 342.37 345.36 346.58 390.74 435.62 463.02 466.23 498.16 503.53 572.70 844.81
Mode Freq.
7 8 9 10 11 12 13 14 15 16 17 18
~202s
Mode
138.59 173.95 220.53 237.55 363.57 377.34 614.09 641.25 708.67 779.71 902.45 957.61
7 90.32 8 162.92 9 192.70 10 212.73 11 225.94 12 300.25
16 17
18 19 20 21 22 23 24
A120F:- Mode Freq.
7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27
A120F;- Mode
78.92 91.66 96.10
173.48 182.51 200.33 225.44 226.13 288.32 293.47 323.41 347.58 347.73 445.85 583.64 664.31 671.57 673.62 680.75 702.99
1010.64
Freq.
7 15.06 8 40.32 9 80.48 10 102.74 11 127.71 12 173.02 13 182.52 14 211.02 15 252.72 16 289.57 17 301.95 18 305.84 19 310.75 20 330.47 21 344.48 22 350.40 23 419.25 24 501.99 25 522.52 26 558.76 27 604.81
Intens, Intenskm
D/A mol-’ 0.0039 0.16 0.0034 0.14 0.0163 0.69 0.0026 0.11 0.0176 0.74 0.0028 0.12 0.0126 0.53 0.0000 0.00 0.0297 1.25 0.0120 0.51 0.1120 4.73 0.1025 4.33 0.0031 0.13 0.0023 0.10 0.0222 0.94 0.0010 0.04 1.2713 53.72 0.2105 8.90 0.1435 6.07 0.0069 0.29 6.9619 294.17 1.8630 78.72 2.4750 104.58 9.8820 417.56
10.4646 442.18 0.0023 0.10
12.7736 539.75
a.u. 0.0002 0.0001 0.0007 0.0001 0.0008 0.0001 0.0005 0.0000 0.0013 0.0005 0.0049 0.0044 0.0001 0.0001 0.0010 0.0000 0.0551 0.0091 0.0062 0.0003 0.3018 0.0808 0.1073 0.4283 0.4536
Intens/ Intens, a.“. WA 0.0000 0.0004 0.0000 0.0008 o.ooo5 0.0121 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0010 0.0222 0.0023 0.0538 0.0000 0.0000 0.0000 0.0000 0.1233 2.8447 0.0554 1.2774 0.0509 1.1735 0.0000 0.0000 0.1802 4.1566 0.0000 0.0003 0.0001 0.0022 0.4033 9.3044 0.3997 9.2218 0.0000 0.0002 0.5690 13.1266
mol-’ 0.02 0.03 0.51 0.00 0.00 0.00 0.94 2.27 0.00 0.00
120.20 53.98 49.59
0.00 175.63
0.01 0.09
393.16 389.66
0.01 554.66
Intens/
El01 0.0003 0.0000 0.0004 0.0006 0.0006 0.0003 0.0010 0.0092 0.0008 0.0072 0.0113 0.0028 0.0899 0.0191 0.0196 0.0017 0.2692 0.2690 0.1867 0.2173
Intens, Intenskm D/A moI_’ 0.0014 0.06 0.0075 0.32 0.0008 0.03 0.0098 0.41 0.0144 0.61 0.0147 0.62 0.0068 0.29 0.0229 0.97 0.2130 9.00 0.0175 0.74 0.1654 6.99 0.2613 11.04 0.0653 2.76 2.0739 87.63 0.4397 18.58 0.4525 19.12 0.0392 1.66 6.2116 262.47 6.2049 262.19 4.3081 182.04 5.0127 211.81
Intens, Intenskm WA mol-’ 0.1104 4.66 0.6638 28.05 0.0000 0.00 0.0000 0.00 3.9346 166.25 0.0000 0.00 0.7289 30.80 0.0009 0.04 0.0001 0.00 6.5096 275.06
12.0437 508.90 0.0091 0.39
a.u.
0.0288 0.0000 0.0000 0.1705 0.0000 0.0316 0.0000
0.2822 0.5220 0.0004
Intens, lntenskm D/A mol-’ 0.0136 0.57 0.0690 2.91 0.3070 12.97 0.3639 15.37 0.1166 4.93 0.1723 7.28
a.u. 0.0006 0.0030 0.0133 0.0158 0.0051 0.0075
78 G.S. Picard et aLlJournal of Molecular Structure (Theochem) 368 (19%) 67-80
13
14
15
16
17
18
19
20
21
AN&- Mode
353.83 0.0871 2.0098 84.93
392.47 0.0037 0.0853 3.60
535.29 0.0819 1.8899 79.86
570.40 0.0107 0.2468 10.43
678.31 0.0645 1.4885 62.90
730.61 0.2269 5.2347 221.19
744.51 0.2921 6.7384 284.73
818.62 0.2319 5.3511 226.11
908.35 0.1682 3.8810 163.99
“W&- Mode Freq.
Freq.
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
AN&- Mode
77.31
115.16
201.82
207.62
220.35
231.22
258.37
312.64
363.93
388.68
542.21
584.90
619.38
628.76
641.43
718.66
735.78
772.94
Intens/ Intens, Intenskrn
a.“. WA mol“
O.OitO6 0.0149 0.63
0.0001 0.0016 0.07
0.0000 0.0001 0.00
0.0000 0.0011 0.05
0.0000 0.0001 0.00
0.0194 0.4467 18.88
0.0251 0.5792 24.47
0.0000 0.0001 0.00
0.0285 0.6566 27.74
0.0000 0.0000 0.00
0.0299 0.6896 29.14
0.0000 0.0001 0.00
0.0000 0.0006 0.03
0.0000 0.0005 0.02
0.3426 7.9031 333.94
0.3740 8.6278 364.56
0.6461 14.9071 629.89
0.0000 0.0003 0.01
7 25.87
8 84.97
9 92.55
10 126.69
11 129.72
12 145.42
13 159.69
14 214.97
15 221.47
16 234.33
17 250.86
18 274.38
19 303.64
20 319.47
21 326.27
22 363.99
23 383.92
24 447.45
25 460.70
26 492.04
27 536.42
28 558.58
29 655.75
30 676.36
au.
0.0003
0.0014
0.0007
0.0001
0.0004
0.0003
0.1331
0.0099
0.0000
0.0001
0.0011
0.0001
0.0127
0.0000
0.0049
0.0001
0.2627
0.0005
0.0001
0.3369
0.0001
0.4204
0.0003
0.4705
References
Freq.
7 62.71
8 97.32
9 142.87
10 164.32
11 187.16
12 209.41
13 233.39
14 268.84
15 285.06
16 296.79
17 352.12
18 357.86
19 382.88
20 435.81
21 470.11
22 529.15
23 558.59
24 564.40
25 588.62 26 749.04
27 794.70
au.
0.0002
0.0000
0.0001
0.0014
0.0026
0.0014
0.0012
0.0057
0.0009
0.0162
0.0047
0.0009
0.0013
0.3121
0.0730
0.0226
0.1976
0.2982
0.1810
0.2309
0.2137
Intens,
WA 0.0056
0.0011
0.0020
0.0313
0.0605
0.0325
0.0275
0.1325
Intens/km
mol-’
0.24
0.05
0.08
1.32
2.56
1.37
1.16
5.60
0.87
15.81
4.57
0.84
1.29
304.27
71.13
21.99
192.61
290.72
176.44
225.13
208.32
[l] ALCOA, US Patents 3554893 (1973); 3 822 195 (1975);
3 893 899 (1975).
[2] G.S. Picard, F. Sean and B. Tremillon, Attempt at low-tem-
perature dehydration of aluminum chloride hexahydrate in the
presence of molten chlorides, Bull. Sot. Chim. Fr. I (9-10)
(1981) 353.
[3] F. SCon, G.S. Picard and B. Trbmillon, Industrial aluminas and
hexahydrated aluminum chloride chlorinations by phosgene in
lithium chloride-potassium chloride eutectic melt at 470°C
Electrochim. Acta, 28(2) (1983) 209.
0.3743
0.1081
0.0198
0.0305
7.2008
1.6835
0.5204
4.5582
6.8802
4.1757 5.3278
4.9301
[4] F. Stan, Reactions d’echange de l’ion oxyde dam I’eutectique
LiCl-KC1 a 470°C. Application a la chloruration selective
d’oxydes mCtalliques en milieux chlorures fondus, These de
Doctorat d’Etat, Paris VI, 1981.
[5] F. Sbon, G.S. Picard, B. Tremillon and Y. Bertaud, Selective
chlorination of metal oxide mixtures of natural or synthetic
origin, Ahtminium Pechiney, Fr. Patent 2514028 (1981).
[6] G.S. Picard, F. Sdon and B. Trtmillon, Selective chlorination
of oxides in suspension in molten chlorides, Proc. 1st Int.
Symp. Molten Salt Chem. and Technol., Kyoto, April 1983;
Molten Salt Comm. Electrochem. Sot. Jpn., Ed., Kyoto, 1983, pp. 49-52.
[7] G.S. Picard, F. SCon and B. Tremillon, Prediction of the
Intens, Intens/km
WA mol-’
0.0064 0.27
0.0333 1.41
0.0158 0.67
0.0828 0.12
0.0098 0.42
0.0076 0.32
3.0706 129.75
0.2282 9.64
0.0800 0.00
0.0017 0.07
0.0252 1.07
0.0026 0.11
0.2933 12.40
0.0005 0.02
0.1121 4.74
0.0013 0.05
6.0605 256.08
0.0122 0.52
0.0026 0.11
7.7716 328.39
0.0027 0.12
9.6990 409.83
0.0060 0.25
10.8559 458.71

G.S. Picard et aLlJournal of Molecular Structure (Theochem) 368 (19%) 67-80 79
selective chlorination of oxides by gaseous mixtures in molten
lithium chloride-potassium chloride eutectic, in M. Blander,
D.S. Newman, M.L. Saboungi, G. Mamantov and K. Johnson
@is)., Molten Salts, Electrochemical Society Softbound Pro-
ceedings Series, Vol. 84-2, 1984, pp. 694-706. [8] B.L. TrCmillon and G.S. Picard, Effect of gaseous chlorinating
reagents on solubilization of metal oxides in chloride melts,
Xiyou Jinshu (Rare Metals), 4(2) (1985) 19.
[9] B.L. Trtmillon and G.S. Picard, Chemical solubilization of
metal oxides and sulfides in chloride melts by means of chlor-
inating agents, in G. Mamantov and R. Marassi (Eds.), Molten
Salt Chemistry-An Introduction and Selected Applications,
NATO ASI Ser., Ser. C: Math. Phys. Sci., Vol. 202, D. Reidel,
1987, pp. 305-327 (Proc. Adv. Study Inst. on Molten Salt
Chemistry, Camerino, Italy, August 3-15, 1986, under the
auspices of NATO and the University of Camerino).
[lo] G.S. Picard, Three-dimensional reactivity diagrams for reac-
tion chemistry in molten chohides, Molten Salt Forum, l-2
(1993/94) 25.
[ll] G.S. Picard, F. Sean and B. Tremillon, Oxoacidity reactions in
molten lithium chloride + potassium chloride eutectic (at
470°C): potentiometric study of the equilibria of exchange of
oxide ion between aluminum(II1) systems and carbonate and
water systems, J. Electroanal. Chem. Interfacial Electrochem.,
102(l) (1979) 65.
[12] G.S Picard, F. SCon, B. TrCmillon and Y. Bertaud, Effect of the
addition of fluoride on the conditional solubility of alumina in
lithium chloride-potassium chloride eutectic melt, Electro-
chim. Acta, 25(11) (1980) 1453.
[13] V. Villard, Etude de processus chimiques et electrochimiques
conduisant a la conception d’un nouveau procede basse temp
Brature de raffinage de l’aluminium en milieux sels fondus, Th
bse de Doctorat de I’UniversitC de Paris VI, 1989.
[14] G.S. Picard, V. Villard, Y. Bertaud, J.-M. Hitter and M. Leroy,
Procddt d’electroraffinage sequentiel de I’aluminium, Alumi-
nium Pechiney, Fr. Patent 2655 055 (1989).
[15] G.S. Picard, V.A. Villard, D.M. Ferry, J.-M. Hitter and Y.J.
Bertaud, Conceiving a new low-temperature aluminum elec-
trorefining process and testing its feasibility, in CL. Hussey,
S.N. Flengas, J.S. Wilkes and Y.Ito (Eds.), Molten Salts, The
Electrochemical Society Softbound Proceedings Series, Vol.
90-17,1990, pp. 492-514.
[16] E. Prat, Etude par voltammetrie cyclique et impedancemetrie
de reactions d’oxydation mettant en jeu les ions oxyfluoroalu-
minate en milieu cryolithique a lOOO”C-Application a I’am
tlioration du pro&de Hall-Heroult d’obtention Clectrolytique
de I’aluminium, These de Doctorat de l’universitt de Paris VI,
1987.
[17] G.S. Picard, E.C. Prat, Y.J. Bertaud and M.J. Leroy, Eviden-
cing the electrochemical mechanism at carbon/bath interface
by means of impedance measurement: an improved approach
of the aluminum reduction process, in R.D. Zabreznik (Ed.),
Light Met. (Warrendale, Pa.), (1987) 507; Proc. TMS Light
Metal Committee, 116th Annual Meeting, Denver, CO, Feb-
ruary 24-26, 1987.
[18] G.S. Picard, Y. Bertaud, E. Prat and M. Leroy, Electrochemi-
cal method and apparatus for determination of oxide ions
dissolved in a halide-melt bath, Ahrminium Pechiney, Fr.
Patent, 2605 410 (1986).
[19] G.S. Picard, F. SCon and Y. Bertaud, A molten salt potentio-
metric method for characterizing industrial ahrminas, Electro-
chim. Acta, 27(3) (1982) 401.
(201 G.S. Picard and F.C. Bouyer, Molecular modelling: an
analytical tool with a predictive character for investigating
reactivity in molten salt media, in F. Bemardi and J.-L. Rivail
(Eds.), Proc. 1st Eur. Conf. on Computational Chemistry,
held in Nancy, France, 1994, J. Am. Inst. Phys., 330 (1995)
295.
[21] F.C. Bouyer, Etude analytique, par des techniques de mod
Clisation, de l’intluence de cations alcalins ou alcalino-terreux
sur la stabilite et la reactivitt de complexes de I’aluminiu-
m(II1) avec des ions halogenures ou oxydes, These de Doctorat
de I’Universitt de Paris VI, 1995.
[22] F.C. Bouyer, G.S. Picard and J.-J. Legendre, Structural,
energetic and vibrational studies on Al(III)-CI(-l)-F(-1)
complexes based on LDF calculations, C.R. Acad. Sci., Ser.
II, 318 (1994) 455.
[23] F. Bouyer, G. Picard and J.J. Legendre, Density functional
calculations for predicting structures and vibrational fre-
quencies of aluminum chloride-fluorides, J. Mol. Struct.
(Theochem), 330 (1995) 217.
[24] F.-C. Bouyer, G.S. Picard and J.-J. Legendre, Geometrical and
spectroscopical characterizations of some complex entities of
aluminum(II1) with fluoride ions by LDF-based calculations,
Int. J. Quantum Chem., 52 (1994) 927.
[25] F.C. Bouyer, G.S. Picard, J.-J. Legendre, S. Bouvet and Y.
Bertaud, Bond energy investigations for aluminum(III)-fluor-
ide complexes relative to cryolitic melts. Part I: Potential
energy surface for the dissociation of pure isolated complexes,
in C.L. Hussey, D.S. Newman, G. Mamantov and Y. Ito (Eds.),
Proc. 9th Int. Symp. on Molten Salts, held in San Francisco,
May 22-27, 1994, The Electrochemical Society Softbound
Proceedings Series, Vol. 94-13, 1994, pp. 34-41.
[26] F.C. Bouyer and G.S. Picard, Evidence of cations’ influence
for structural rearrangements in cryolitic melts, in Proc. Fast
Elementary Processes in Chemical and Biological Systems,
Congrts de la SociCtt Francaise de Chimie, Division de Chi-
mie Physique, Universitt des Sciences et Technologies de
Lille, 26-30 juin 1995, J. Am. Inst. Phys., 364 (1996) 532.
(271 F.C. Bouyer, G.S. Picard and J.-J. Legendre, Computational
chemistry: a way to obtain spectroscopic and thermodynamic
data for exotic compounds, XIth Int. Conf.on Computers in
Chemical Research and Education, Paris, France, July 17-21,
1995, J. Chem. Inf. Comput. Sci., in press.
[28] F.C. Bouyer, G.S. Picard and J.-J. Legendre, Computational
and analytical chemistry: a methodology to study chemical
reactions between sodium, calcium and aluminum fluorides
in molten cryolite, 6th Int. Conf. on Application of Density
Functional Theory in Chemistry and Physics, E.N.S.C.P.,
Paris, August 29-September lst, 1995, hit. J. Quantum
Chem., in press.
[29] J. Andzelm and E. Wimmer, Density functional Gaussian-
type-orbital approach to molecular geometries, vibrations,
and reaction energies, J. Chem. Phys., 96 (2) (1992) 1280.

80 G.S. Picard et aLlJournal of Molecular Structure (Theochem) 368 (1996) 6740
[30] A.D. Becke, Density-functional exchange energy approxima- tion with correct asymptotic behavior, Pbys. Rev. A, 38(2) (1988), 3098; Basis-set-free density-functional quantum chemistry, Int. J. Quantum Chem.: Quantum. Chem. Symp., 23 (1989) 599.
[31] C. Lee, W. Yang and R.G. Parr, Development of the Colle- Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 37(2) (1988) 785.
[32] K. Grjotheim, C. Krohn, M. Malinovsky, K. Matiasovsky and J. Thonstad, Aluminum Electrolysis-Fundamental of the Hall-H6roult process, 2nd edn., Aluminum-Verlag, D ilsseldorf, 1982, p. 122.
[33] B. Gilbert, G. Mamantov and G.M. Begun, Raman spectra of AI,4 solutions in molten cryolite and other aluminum fluoride containing melts, Inorg. Nucl. Chem. Len., 12 (1976) 415.
[34] H. Kvande, The structure of afumina dissolved in cryolite melts, Light Met. (Warrendale, Pa.), 2 (1986) 4.51.
[35] JANAF Thermochemical Tables, J. Phys. Chem. Ref. Data, 14 (Suppl. l), American Institute of Physics, New York, 1985.
[36] J.E. Boner, Beitrag xur Kenntnis der elektrometallurgie des Aluminiums, Helv. Chim. Acta, 33(146) (1950) 1137.
[37] 2. Qiu and G. Xie, Computer simulation of the structure of the cryolite-alumina melts, Light Met. (Warrendale, Pa.), (1989)
315; G. Xie, Z. Qiu, C. Xu and N.Chen, Computerized simula- tion of the structure of cryolite-alumina melts, Chin. J. Met. Sci. Technol., 5 (1989) 426.
[38] J.L. Holm, Thermodynamic properties of molten cryolite and other fluoride mixtures, Dr. Tech. Dissertation, University of Trondheim, Norway (1971).
[39] E. Robert, T. Mateme, E. Tixhon and B. Gilbert, Effect of calcium fluoride addition to molten NaF-AlFs mixtures: a s$rdy by Raman spectroscopy, Vibr. Spectrosc., 6 (1993) 71.
[40] A. Sterten, Structural entities in NaF-AlFs melts containing alumina, Electrochim. Acta, 25 (1980) 1673,
[41] Pechiney source. [42] M. Rolin and M. Bernard, Le diagramme binaire cryolithe-
alumine. II-Schema g&&al d’ionisation des oxydes en solu- tion dans la cryolithe fondue. Le cas particulier de l’alumine, Bull. Sot. Chim. Fr., (1962) 939.
[43] T. Forland and SK. Ratkje, AIuminium-oxygen containing species in fluoride melts, Acta Chem. Stand., 27 (1973) 1883.
[44] P. Hebant and G.S. Picard, Equilibrium between molecular and ionic species in pure molten LiCl and in LiCl+MCl (M - Na, K, Rb) melts investigated by computational chem- istry, J. Mol. Struct. (Theochem), 358 (1995) 39.
Copyright © 2022 FDOKUMEN




















