
Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and...
10
ORIGINAL ARTICLE Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and migration CP Lim 1 , T-T Phan 2,3 , IJ Lim 2 and X Cao 1 1 Institute of Molecular and Cell Biology, Proteos, Singapore; 2 Department of Surgery, National University of Singapore, Singapore and 3 Department of Bioengineering, National University of Singapore, Singapore Keloids, partially considered as benign tumors, represent the most extreme example of cutaneous scarring that uniquely afflicts humans as a pathological response to wound healing. It is characterized by excessive deposition of collagen and other extracellular matrix components by dermal fibroblasts. Upon cutaneous injury, cocktails of chemokines, cytokines and growth factors are secreted temporally and spatially to direct appropriate responses from neutrophils, macrophages, keratinocytes and fibro- blasts to facilitate normal wound healing. Signal transdu- cer and activator of transcription 3 (Stat3) is an oncogene and a latent transcription factor activated by various cytokines and growth factors. We investigated the possible role of Stat3 in keloid scar pathogenesis by examining skin tissue and cultured fibroblasts from keloid-scarred patients. We observed enhanced expression and phosphor- ylation of Stat3 in keloid scar tissue, and in cultured keloid fibroblasts (KFs) in vitro. Increased activation of Janus kinase (Jak)2, but not Jak1, was detected in KFs, and suppression of Jak2 by its inhibitor repressed Stat3 Y705 phosphorylation. Inhibition of Stat3 expression and phosphorylation by short interfering RNA or Cucurbitacin I resulted in the loss of collagen production, impaired proliferation and delayed cell migration in KFs. We show, for the first time, a role of Stat3 in keloid pathogenesis. Inhibitors of Stat3 may be useful therapeutic strategies for the prospective treatment of keloid scars. Oncogene (2006) 25, 5416–5425. doi:10.1038/sj.onc.1209531; published online 17 April 2006 Keywords: Stat3; keloid; scar; fibroblasts; collagen; migration Introduction Wound healing is a regulated, yet complex, event including the breakdown of fibrin clots, degradation of extracellular matrix (ECM), promotion of angiogenesis, and the migration and proliferation of keratinocytes and fibroblasts. In the event of excessive wound healing scar formation occurs, ranging from hypertrophic scars to keloids. The term ‘keloid’ was coined in 1806 to describe the crab claw-like appearance of the scar (Yang et al., 2003). Keloids are strictly defined as scars that spread beyond the boundaries of the original wound and do not regress spontaneously (Cosman et al., 1961). In contrast, hypertrophic scars do not develop beyond the periphery of wound, with spontaneous softening and flattening from remodeling over time (Ragoowansi et al., 2001). These scars afflict humans exclusively, and the inciting skin trauma can range from small injuries such as ear piercing and abrasions to more severe trauma such as surgery or burns. Keloids are aggressive in nature and Africans and Asians appear to be more susceptible (Tuan and Nichter, 1998). Treatment and management of keloid scars have been difficult, owing to their recurrence, and although a myriad of treatments are available none are fully effective (Tuan and Nichter, 1998; Alster and Tanzi, 2003; Mustoe, 2004). Keloid scars are typified by overexuberant deposition of collagen and other ECM components such as fibronectin by keloid fibroblasts (KFs) (Diegelmann et al., 1979; Babu et al., 1989), with overproduction of collagen types I and III mRNA in keloid tissues (Naitoh et al., 2001). Keloid fibroblasts reportedly displayed an increased rate of proliferation upon wounding (Calderon et al., 1996). At the cellular level, increased levels of cytokines and growth factors, or their receptors, and sensitization of mitogenic response to these secreted factors after wounding in KFs have been suggested to play a role in keloid pathogenesis (Calderon et al., 1996). Transforming growth factor (TGF)-b1 and TGF-b2 were found to be elevated in KFs (Lee et al., 1999), and increased sensitivity to TGF-b1 in KFs increased fibronectin (Babu et al., 1992) and collagen production, and cell proliferation (Younai et al., 1994). Platelet-derived growth factor (PDGF)a receptor expression was shown to be enhanced in KFs, which corresponded to increased mitotic response upon exposure to all three PDGF isoforms (Haisa et al., 1994). Overexpression of insulin- like growth factor-I receptor detected in KFs appeared to enhance their invasiveness, but not fibroproliferation (Yoshimoto et al., 1999). Elevated interleukin (IL)-6 Received 10 October 2005; revised 25 January 2006; accepted 20 February 2006; published online 17 April 2006 Correspondence: Dr X Cao, Cell Signaling Lab, Institute of Molecular and Cell Biology, 61 Biopolis Drive, Proteos, Singapore 138673, Singapore. E-mail: [email protected] Oncogene (2006) 25, 5416–5425 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Stat3 contributes to keloid pathogenesis via promoting collagen production, cell proliferation and...

ORIGINAL ARTICLE
Stat3 contributes to keloid pathogenesis via promoting collagen production,
cell proliferation and migration
CP Lim1, T-T Phan2,3, IJ Lim2 and X Cao1
1Institute of Molecular and Cell Biology, Proteos, Singapore; 2Department of Surgery, National University of Singapore, Singaporeand 3Department of Bioengineering, National University of Singapore, Singapore
Keloids, partially considered as benign tumors, representthe most extreme example of cutaneous scarring thatuniquely afflicts humans as a pathological response towound healing. It is characterized by excessive depositionof collagen and other extracellular matrix components bydermal fibroblasts. Upon cutaneous injury, cocktails ofchemokines, cytokines and growth factors are secretedtemporally and spatially to direct appropriate responsesfrom neutrophils, macrophages, keratinocytes and fibro-blasts to facilitate normal wound healing. Signal transdu-cer and activator of transcription 3 (Stat3) is an oncogeneand a latent transcription factor activated by variouscytokines and growth factors. We investigated the possiblerole of Stat3 in keloid scar pathogenesis by examiningskin tissue and cultured fibroblasts from keloid-scarredpatients. We observed enhanced expression and phosphor-ylation of Stat3 in keloid scar tissue, and in culturedkeloid fibroblasts (KFs) in vitro. Increased activation ofJanus kinase (Jak)2, but not Jak1, was detected in KFs,and suppression of Jak2 by its inhibitor repressed Stat3Y705 phosphorylation. Inhibition of Stat3 expression andphosphorylation by short interfering RNA or CucurbitacinI resulted in the loss of collagen production, impairedproliferation and delayed cell migration in KFs. We show,for the first time, a role of Stat3 in keloid pathogenesis.Inhibitors of Stat3 may be useful therapeutic strategiesfor the prospective treatment of keloid scars.Oncogene (2006) 25, 5416–5425. doi:10.1038/sj.onc.1209531;published online 17 April 2006
Keywords: Stat3; keloid; scar; fibroblasts; collagen;migration
Introduction
Wound healing is a regulated, yet complex, eventincluding the breakdown of fibrin clots, degradation of
extracellular matrix (ECM), promotion of angiogenesis,and the migration and proliferation of keratinocytes andfibroblasts. In the event of excessive wound healing scarformation occurs, ranging from hypertrophic scars tokeloids. The term ‘keloid’ was coined in 1806 to describethe crab claw-like appearance of the scar (Yang et al.,2003). Keloids are strictly defined as scars that spreadbeyond the boundaries of the original wound and do notregress spontaneously (Cosman et al., 1961). In contrast,hypertrophic scars do not develop beyond the peripheryof wound, with spontaneous softening and flatteningfrom remodeling over time (Ragoowansi et al., 2001).These scars afflict humans exclusively, and the incitingskin trauma can range from small injuries such as earpiercing and abrasions to more severe trauma such assurgery or burns.
Keloids are aggressive in nature and Africansand Asians appear to be more susceptible (Tuan andNichter, 1998). Treatment and management of keloidscars have been difficult, owing to their recurrence, andalthough a myriad of treatments are available none arefully effective (Tuan and Nichter, 1998; Alster andTanzi, 2003; Mustoe, 2004). Keloid scars are typified byoverexuberant deposition of collagen and other ECMcomponents such as fibronectin by keloid fibroblasts(KFs) (Diegelmann et al., 1979; Babu et al., 1989), withoverproduction of collagen types I and III mRNA inkeloid tissues (Naitoh et al., 2001). Keloid fibroblastsreportedly displayed an increased rate of proliferationupon wounding (Calderon et al., 1996). At the cellularlevel, increased levels of cytokines and growth factors,or their receptors, and sensitization of mitogenicresponse to these secreted factors after wounding inKFs have been suggested to play a role in keloidpathogenesis (Calderon et al., 1996). Transforminggrowth factor (TGF)-b1 and TGF-b2 were found tobe elevated in KFs (Lee et al., 1999), and increasedsensitivity to TGF-b1 in KFs increased fibronectin(Babu et al., 1992) and collagen production, and cellproliferation (Younai et al., 1994). Platelet-derivedgrowth factor (PDGF)a receptor expression was shownto be enhanced in KFs, which corresponded to increasedmitotic response upon exposure to all three PDGFisoforms (Haisa et al., 1994). Overexpression of insulin-like growth factor-I receptor detected in KFs appearedto enhance their invasiveness, but not fibroproliferation(Yoshimoto et al., 1999). Elevated interleukin (IL)-6
Received 10 October 2005; revised 25 January 2006; accepted 20February 2006; published online 17 April 2006
Correspondence: Dr X Cao, Cell Signaling Lab, Institute of Molecularand Cell Biology, 61 Biopolis Drive, Proteos, Singapore 138673,Singapore.E-mail: [email protected]
Oncogene (2006) 25, 5416–5425& 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00
www.nature.com/onc

(Xue et al., 2000) and vascular endothelial growth factor(Wu et al., 2004) expression have also been observed inKFs.
Signal transducer and activator of transcription 3(Stat3) is an oncogene and a latent transcription factorthat is involved in diverse processes, including cellproliferation and migration, inflammation, immuneresponse and cell survival. It is activated by variousgrowth factors and cytokines, including PDGF and IL-6(Darnell, 1997), by Tyr705 phosphorylation, whichenables the dimerization via its phosphotyrosine residueand reciprocal SH2 domain of its dimer partner. Stat3can homodimerize, or heterodimerize with Stat1, andthe dimer then translocates to the nucleus, binds totarget DNA sequences and regulates the transcription oftarget genes (Darnell et al., 1994). Whereas Tyr705phosphorylation is essential for Stat3 activation, Ser727phosphorylation was shown to be required for maximalgene activation (Wen et al., 1995). Tyr705 phosphoryla-tion in cytokine receptors that lack intrinsic tyrosinekinase activity is mediated by receptor-associated Januskinases (Jaks) (Darnell, 1997). Overexpression of aconstitutively activated Stat3 caused cell transformationand tumorigenesis in mice (Bromberg et al., 1999).Activation of Stat3 has been detected in various cancersand tumor-derived cell lines (reviewed by Bowman et al.,2000). On the other hand, Stat3 knockout resulted inembryonic lethality (Takeda et al., 1997), promptingfurther conditional tissue- or cell-specific knockoutanalyses (Takeda and Akira, 2000). In particular, Stat3expression disrupted in keratinocytes using Cre recom-binase driven by keratin 5-specific promoter showedimpaired wound healing and growth factor-dependentmigration of Stat3-deficient epidermal cells in mice,although the development of epidermis and hair folliclesappeared normal and proliferation remained unaffected(Sano et al., 1999).
In this paper, we hypothesized a role for Stat3 inkeloid scar pathogenesis as Stat3 was previously shownto function in wound healing (Sano et al., 1999).Moreover, Stat3 is a key molecule activated by variouscytokines and growth factors, ligands that are known tobe secreted by various cell components in responseto tissue injury during wound healing. Our resultsshowed an enhancement of Stat3 expression and/orphosphorylation in keloid tissues in vivo, as well as incultured proliferating KFs. We detected concomitantincreased activation of Jak2, but not Jak1, in KFscompared to normal dermal fibroblasts. Inhibition ofTyr705 Stat3 phosphorylation was observed by Jak2inhibitor, AG 490, corroborating our hypothesis thatJak2 mediates Stat3 phosphorylation. Finally, wedemonstrated that the increased collagen production,cell proliferation and migration in KFs were suppressedby Stat3 short interfering RNA (siRNA) and aninhibitor of Jak2/Stat3, Cucurbitacin I (Blaskovichet al., 2003). Taken together, these data reveal animportant role of Stat3 in the pathogenesis of keloidscars. Stat3 inhibition suppresses KF activity, and mayserve as a therapeutic target for the treatment of keloidfibrosis.
Results
Enhanced Stat3 expression and phosphorylation in keloidscar tissue vs normal skin tissue in vivoA thin layer of epidermis underlies a very thin layer ofkeratin in normal skin. In contrast, the keratin andepidermal layers of keloid tissue are thicker, as shown bythe hematoxylin–eosin staining of paraffin sections(Figure 1a). The dermis is also considerably thicker,with compacted and irregular connective tissue com-pared to normal skin, which appears more uniformlystacked and regular.
To examine the expression of Stat3, cryosections fromnormal skin and skin overlying keloid scar weresubjected to immunofluorescence using a polyclonalStat3 (C-20) antibody, performed in parallel with anti-rabbit immunoglobulin (Ig)G as controls. As illustratedin Figure 1b, Stat3 could be detected in both normalskin and keloid tissue, whereas weak fluorescence wasobserved with anti-rabbit IgG antibody. Stat3 expres-sion was easily detected in the epidermal layer in bothskin types, which is predominantly populated withkeratinocytes. In the dermal layer, the connective tissueis interspersed with cellular components comprisedprimarily of fibroblasts, which are involved in thedeposition of collagen fibers and other ECM compo-nents. A small population of fibroblast cells in thedermal layer of normal skin also showed Stat3 expres-sion, whereas a much higher number of fibroblasts inkeloid samples 25, 26, 31 and 48 showed Stat3expression (Figure 1b and data not shown).
Activation of Stat3 was also examined using phos-phoStat3 (pStat3) monoclonal antibody, performed inparallel with anti-mouse IgG as controls. Weak pStat3staining was observed in normal skin, whereas activa-tion of Stat3 was significantly increased in both dermisand epidermis in keloid 26 (Figure 1c).
To further verify the data above, tissue lysates ofnormal skin from five individuals and keloid scars fromeight individuals were investigated for phosphorylationand expression of Stat3. Three samples from normalskin showed low Tyr705 Stat3 phosphorylation, whereastwo showed moderate Tyr705 Stat3 phosphorylation(Figure 1d). In contrast, all keloid tissue lysates, exceptfor one, showed moderate to very high Tyr705 Stat3phosphorylation. The degree of Tyr705 Stat3 phosphor-ylation correlated well with the level of Stat3 expression.As for Ser727 Stat3 phosphorylation, six out of eightkeloid tissue samples showed an increase in Ser727 Stat3phosphorylation compared to all five normal skinsamples.
Increased Stat3 activation in keloid fibroblasts comparedto normal skin fibroblastsThe epidermis is populated with 95% keratinocytes and5% non-keratinocyte cells comprising melanocytes,Langerhans cells and Merkel cells (Edmondson et al.,2003), whereas fibroblasts are the predominant cells inthe dermis, along with some endothelial and mast cells.As we observed increased Tyr705 Stat3 phosphorylationin the dermal cells in keloid tissue compared to normal
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5417
Oncogene

skin, we further investigated the activation of Stat3 inprimary fibroblast cells derived from normal skinand keloid tissues. Two normal fibroblast (NF) andthree KF strains were examined for Stat3 activation.Tyr705 phosphorylation of Stat3 was elevated from 2.3-to 4.3-fold, with an average of three-fold, in KFscompared to NFs (Figure 2a). Tyr phosphorylation ofStat1 and Stat5 was also examined, but was undetect-able in any of the samples. Signal transducers andactivators of transcription 1 protein expression waspresent, whereas Stat5 expression was weak, in all of thesamples.
To further examine the kinetics of Tyr705 and Ser727Stat3 phosphorylation, NFs and KFs were cultured for5 days under normal growth conditions. Five differentsets of NF and KF were examined, and two are
presented here as representatives. In NF5, very weakTyr705 Stat3 phosphorylation was observed at day 1,increased at day 2 and sustained until day 5 (Figure 2b,left panel). In contrast, strong Tyr705 Stat3 phosphor-ylation was observed in KF48 at day 1, which increasedfrom day 2 to day 4, and decreased to day 1 level at day5. The Ser727 Stat3 phosphorylation profile was similarto Tyr705 Stat3 phosphorylation in both NF5 andKF48. In addition, an elevated Stat3 expression wasobserved in KF48 compared to NF5. This kinetics inNF5 and KF48 is similarly observed in NF4 and KF8(Figure 2b, right panel), and KF8 also showed anenhanced pY705 Stat3 and pS727 Stat3 compared toNF4.
Stat3 phosphorylation was further examined inserum-free condition. We observed strong constitutive
Figure 1 Stat3 expression and activation are elevated in keloid skin tissues. (a) Hematoxylin–eosin stain of paraffin tissue sections ofnormal skin (NS1) and keloid scar (KS28). D, E and K indicate dermis, epidermis and keratin, respectively. (b) Cryosections of normalskin and keloid scar tissue were stained with either anti-rabbit immunoglobulinG (a-rabbit IgG) or polyclonal anti-Stat3 (C-20) (a-Stat3) and counterstained with 4,6-diamidino-2-phenylindole (DAPI). (c) Cryosections were probed with anti-mouse IgG (a-mouseIgG) or monoclonal anti-phosphoStat3 (pStat3) (a-pStat3) and counterstained with DAPI. Pictures were taken together with phasecontrast and all scale bars represent 50 mM. (d) Equal amounts of tissue lysates from normal skin (NS) and keloid samples (KS) weresubjected to Western blot analysis with anti-pY705 Stat3 antibody. The blot was stripped and reprobed with anti-pS727 Stat3, andsubsequently with anti-Stat3 and anti-actin.
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5418
Oncogene

Tyr705 and Ser727 Stat3 phosphorylation in KF48throughout the 5 days, compared to very weakphosphorylation in NF4 (Figure 2c). Although actinexpression was similar in all samples, Stat3 expressionwas elevated in KF48 compared to NF4. Nevertheless, adramatic increase in the Tyr705 Stat3 phosphorylationin KF48 was clearly observed after normalization toStat3 expression. Overall, phosphorylations and expres-sion of Stat3 were augmented in KFs and tissue lysatescompared to the samples from normal skin. Tyr705 andSer727 Stat3 phosphorylations in KFs also occur in aconstitutive and serum-independent manner.
Jak2, but not Jak1, activation is increased in keloidfibroblastsJanus kinase (Jak)1 and Jak2 are known Tyr kinasesthat phosphorylate Stat3 in cytokine signaling. We
examined whether Jaks contribute to the enhancedpTyr705 Stat3 in KF. Nearly confluent NFs andKFs were maintained in serum-free media and har-vested, and activation of endogenous Jak1 and Jak2 wasexamined in Western blot analysis using anti-phospho-Jak1 (pJak1) or anti-phosphoJak2 (pJak2) antibody.As shown in Figure 3a, increased pJak2 was detectedin KF48 compared to NF103, whereas pJak1 signalhardly detectable in either normal fibroblasts orKFs, although Jak1 expression was present in bothcell types. We also examined Src and epidermal growthfactor receptor (EGFR), as they are reported to beable to phosphorylate Stat3 also. However, phosphoSrc(Tyr416) and phosphoEGFR (Tyr845) signalswere undetectable in both NF and KF (data notshown).
To confirm that Jak2 mediates Stat3 Tyr phosphor-ylation, NF103 and KF48 were incubated with tyrphos-tin AG 490, a Jak2 tyrosine kinase inhibitor. pTyr705Stat3 was inhibited by AG 490, but not by dimethylsulfoxide (DMSO) control, as shown in Figure 3b. Thiscorroborates the finding that Jak2 mediates Tyr705Stat3 phosphorylation in both normal and keloid skinfibroblasts.
Figure 3 Jak2 mediates Tyr705 Stat3 phosphorylation. (a) Stat3phosphorylation by Jak2, but not Jak1. Equal amounts of total celllysates from normal fibroblast, NF103 and keloid fibroblast, KF48,were resolved by sodium dodecyl sulfate–polyacrylamide gelelectrophoresis (SDS–PAGE) and Western blot analysis wasperformed using phosphoJak1 (pJak1) or phosphoJak2 (pJak2)antibodies. The blot was stripped and reprobed with Jak1 and Jak2antibodies, respectively, and subsequently with anti-actin fornormalization. Arrows indicate the position of the protein bands.(b) NF103 and KF48 were serum-starved for 24 h and incubatedwith either dimethyl sulfoxide (DMSO) or 50mM AG 490 foranother 27 h. Equal amounts of total cell lysates were resolved inSDS–PAGE and blotted with pY705 Stat3 antibody in Westernblot analysis. The blot was stripped and reprobed with Stat3 andactin antibodies.
Figure 2 Enhanced Stat3 phosphorylation and expression inkeloid fibroblasts (KFs). (a) Western blot analyses of total celllysates from normal fibroblasts (NFs) and KFs with equal amountsof protein, and antibodies as indicated. The graph on the upperright illustrates the pY705 Stat3 level normalized to Stat3expression, whereas the graph on the lower right depicts anaverage of the normalized pY705 Stat37s.d. of the KFs and NFspresented. (b) Normal fibroblasts, NF5 and NF4, and KFs, KF48and KF8, were cultured in serum-free media for 2 days beforestimulating with 10% fetal bovine serum. Total cell lysates wereharvested at day 1–5, and phosphorylations of Stat3 were analysedby Western blot after loading equal amounts of protein. (c) NF4and KF48 fibroblasts were incubated in serum-free Dulbecco’smodified Eagles’s medium for 2 days before harvesting daily over 5days to investigate the serum-independent phosphorylation ofStat3. Equal amounts of total cell lysates were subjected to Westernblot analysis with antibodies as indicated. Arrowheads indicate theposition of the protein bands.
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5419
Oncogene

Inhibition of Stat3 inhibits collagen production and Stat3target genesSecretion of collagen by fibroblasts occurs during theprocess of tissue remodeling in wound healing, andenhanced collagen production has been implicated infibrosis (Singer and Clark, 1999). Indeed, collagenproduction was elevated in six out of eight keloid tissuesamples examined, whereas the other two showed asimilar expression to those from five normal skinsamples (Figure 4a).
To investigate whether Stat3 plays any role in keloidpathogenesis, KFs were infected with retrovirus expres-sing Stat3 siRNAs. Of the four different targets for Stat3siRNA selected, three displayed an inhibitory effect onStat3 expression, namely Stat3 siRNA 1, 2 and 4, butnot Stat3 siRNA 3, in a 293T-based amphotropicpackaging cell line (Figure 4b, left panel). Inhibition ofStat3 expression by these three Stat3 siRNAs correlatedwith the inhibition of collagen production in KF48(Figure 4b, right panel). Inhibition of Stat3 was alsoevaluated by incubating KF48 for 30 min with variousdoses of Cucurbitacin I, an inhibitor of Jak2/Stat3activation (Blaskovich et al., 2003), or with DMSO as acontrol, followed by further incubation in normalgrowth media for 48 h. As shown in Figure 4c, a dose-dependent inhibition of pTyr705 Stat3 and collagenproduction was observed by Cucurbitacin I, witheffective concentration as low as 1 mM. A dose-depen-dent decrease of Stat3 expression was also observed,although actin expression was rather similar in allsamples. In addition, inhibition of Stat3 also resulted ina decreased expression of two Stat3 target genes(Bromberg et al., 1999), cell cycle regulator cyclin D1and antiapoptotic Bcl-XL (Figure 4c). Taken together,inhibition of Stat3 resulted in diminished collagenproduction and Stat3 target genes.
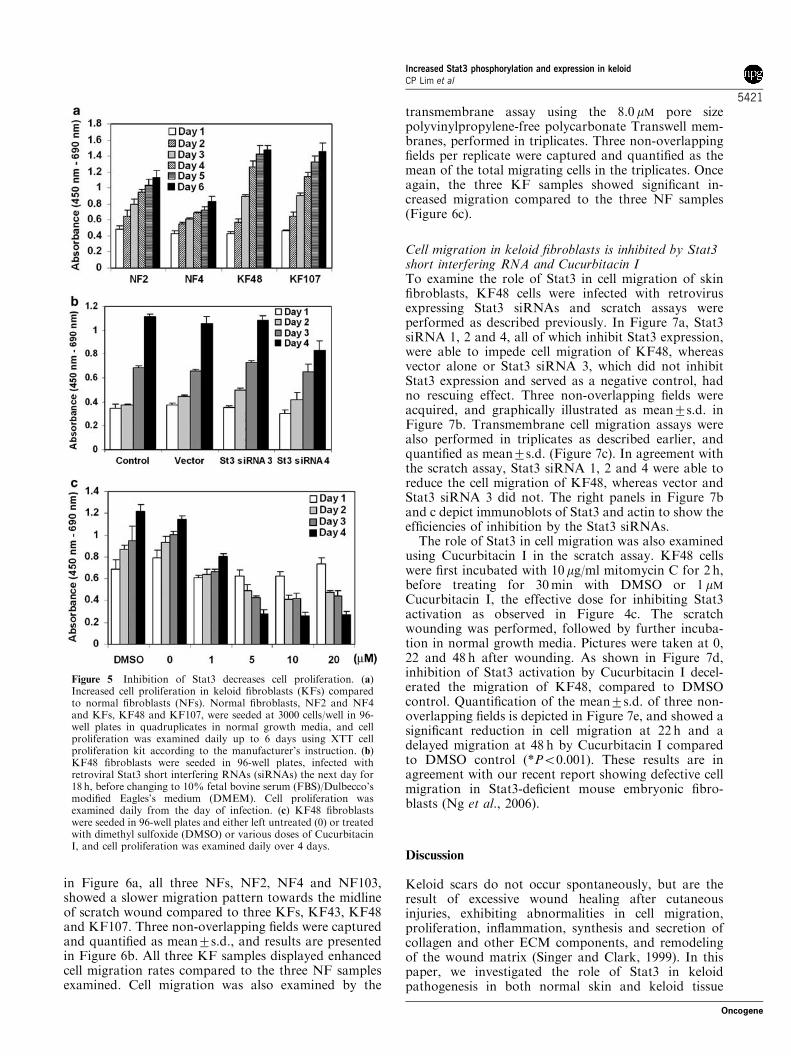
Inhibition of Stat3 decreases cell proliferationEnhanced fibroblast proliferation is a characteristic ofkeloid tissue. In agreement with this, KFs grown innormal culture conditions proliferated faster than NFs, asobserved in KF48 and KF107 compared to NF2 andNF4 (Figure 5a). To investigate whether Stat3 is involvedin the enhanced KF proliferation, KF48 was infected withretrovirus expressing Stat3 siRNAs, and cell proliferationwas examined. As shown in Figure 5b, inhibition of Stat3by Stat3 siRNA 4 decreased cell proliferation of KF48,compared to a non-infected control, vector control andStat3 siRNA 3, which do not inhibit Stat3. Cellproliferation was also investigated by treating KF48 cellswith various doses of Cucurbitacin I or with DMSOcontrol before incubating in normal growth media andexamined daily over 4 days. Cucurbitacin I concentrationas low as 1mM was able to inhibit cell proliferation ofKF48 compared to DMSO and untreated control (0mM),whereas Cucurbitacin I exposure at 5mM and aboveseemed to cause cell death (Figure 5c).
Increased cell migration in keloid fibroblastWound healing is a complex multi-step process. Itinvolves the migration and proliferation of keratinocytes
and fibroblasts to resurface areas of skin loss. Wefurther examined the migration potential of NFs andKFs using the scratch assay. Cells were first cultured tofull confluence, and treated with 10 mg/ml mitomycin Cfor 2 h before scratch-wounding with a yellow pipettetip. Pictures were taken at 0 and 14 h later. As observed
Figure 4 Inhibition of Stat3 decreases collagen production andStat3 target genes. (a) Tissue lysates from normal skin (NS) andkeloid samples (KS) were analysed for collagen production inWestern blot analysis using equal amounts of protein. (b) Fourtargets of Stat3 short interfering RNA (siRNA) were cloned intopSuper.retro.puro vector and transfected into amphotropic 293T-based retroviral packaging cell line, including vector alone. KF48cells were subsequently infected with retrovirus harvested 48 h aftertransfection. Total cell lysates from both amphotropic packagingcell line (left panel) and KF48 (right panel) were harvested andequal amounts of protein were analysed for Stat3, actin andcollagen expressions in Western blot analyses. The inhibition ofcollagen was further illustrated in the graph at the bottom rightpanel after normalization to actin by densitometry. (c) KF48 cellswere left untreated (0mM), or treated with dimethyl sulfoxide(DMSO) or various doses of Cucurbitacin I (1, 5, 10 or 20mM) for30 min, followed by further incubation in 10% fetal bovine serum/Dulbecco’s modified Eagles’s medium for 48 h before harvesting.Equal amounts of total cell lysates were resolved by sodiumdodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE)and examined for the phosphorylation and expression of Stat3,expression of cyclin D1, Bcl-XL, actin and production of collagenin Western blot analyses. Arrows/arrowheads indicate the positionof the protein bands.
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5420
Oncogene
in Figure 6a, all three NFs, NF2, NF4 and NF103,showed a slower migration pattern towards the midlineof scratch wound compared to three KFs, KF43, KF48and KF107. Three non-overlapping fields were capturedand quantified as mean7s.d., and results are presentedin Figure 6b. All three KF samples displayed enhancedcell migration rates compared to the three NF samplesexamined. Cell migration was also examined by the
transmembrane assay using the 8.0 mM pore sizepolyvinylpropylene-free polycarbonate Transwell mem-branes, performed in triplicates. Three non-overlappingfields per replicate were captured and quantified as themean of the total migrating cells in the triplicates. Onceagain, the three KF samples showed significant in-creased migration compared to the three NF samples(Figure 6c).
Cell migration in keloid fibroblasts is inhibited by Stat3short interfering RNA and Cucurbitacin ITo examine the role of Stat3 in cell migration of skinfibroblasts, KF48 cells were infected with retrovirusexpressing Stat3 siRNAs and scratch assays wereperformed as described previously. In Figure 7a, Stat3siRNA 1, 2 and 4, all of which inhibit Stat3 expression,were able to impede cell migration of KF48, whereasvector alone or Stat3 siRNA 3, which did not inhibitStat3 expression and served as a negative control, hadno rescuing effect. Three non-overlapping fields wereacquired, and graphically illustrated as mean7s.d. inFigure 7b. Transmembrane cell migration assays werealso performed in triplicates as described earlier, andquantified as mean7s.d. (Figure 7c). In agreement withthe scratch assay, Stat3 siRNA 1, 2 and 4 were able toreduce the cell migration of KF48, whereas vector andStat3 siRNA 3 did not. The right panels in Figure 7band c depict immunoblots of Stat3 and actin to show theefficiencies of inhibition by the Stat3 siRNAs.
The role of Stat3 in cell migration was also examinedusing Cucurbitacin I in the scratch assay. KF48 cellswere first incubated with 10 mg/ml mitomycin C for 2 h,before treating for 30 min with DMSO or 1 mM
Cucurbitacin I, the effective dose for inhibiting Stat3activation as observed in Figure 4c. The scratchwounding was performed, followed by further incuba-tion in normal growth media. Pictures were taken at 0,22 and 48 h after wounding. As shown in Figure 7d,inhibition of Stat3 activation by Cucurbitacin I decel-erated the migration of KF48, compared to DMSOcontrol. Quantification of the mean7s.d. of three non-overlapping fields is depicted in Figure 7e, and showed asignificant reduction in cell migration at 22 h and adelayed migration at 48 h by Cucurbitacin I comparedto DMSO control (*Po0.001). These results are inagreement with our recent report showing defective cellmigration in Stat3-deficient mouse embryonic fibro-blasts (Ng et al., 2006).
Discussion
Keloid scars do not occur spontaneously, but are theresult of excessive wound healing after cutaneousinjuries, exhibiting abnormalities in cell migration,proliferation, inflammation, synthesis and secretion ofcollagen and other ECM components, and remodelingof the wound matrix (Singer and Clark, 1999). In thispaper, we investigated the role of Stat3 in keloidpathogenesis in both normal skin and keloid tissue
Figure 5 Inhibition of Stat3 decreases cell proliferation. (a)Increased cell proliferation in keloid fibroblasts (KFs) comparedto normal fibroblasts (NFs). Normal fibroblasts, NF2 and NF4and KFs, KF48 and KF107, were seeded at 3000 cells/well in 96-well plates in quadruplicates in normal growth media, and cellproliferation was examined daily up to 6 days using XTT cellproliferation kit according to the manufacturer’s instruction. (b)KF48 fibroblasts were seeded in 96-well plates, infected withretroviral Stat3 short interfering RNAs (siRNAs) the next day for18 h, before changing to 10% fetal bovine serum (FBS)/Dulbecco’smodified Eagles’s medium (DMEM). Cell proliferation wasexamined daily from the day of infection. (c) KF48 fibroblastswere seeded in 96-well plates and either left untreated (0) or treatedwith dimethyl sulfoxide (DMSO) or various doses of CucurbitacinI, and cell proliferation was examined daily over 4 days.
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5421
Oncogene

sections, as well as in fibroblast cell cultures. Our datashowed that Stat3 expression and phosphorylation areenhanced in keloid tissue sections, tissue lysates andfibroblasts (Figures 1b–d and 2), and Stat3 Tyrphosphorylation is mediated by Jak2 (Figure 3). Inhibi-tion of Stat3 using Stat3 siRNA or by an inhibitor ofJak2/Stat3 activation, Cucurbitacin I, decreased thecollagen production, proliferation and migration ofKFs, to a level comparable to that of NFs (Figures 4–7).
Fibroblasts are responsible for the synthesis, deposi-tion and remodeling of the ECM. We report here thatoverexpression and phosphorylation of Stat3 plays anovel role in the excessive collagen deposition byfibroblasts, leading to fibrotic scars such as keloids.Inhibition of Stat3 expression by siRNA or Cucurbita-cin I showed a corresponding decline in collagenexpression (Figure 4b and c), suggesting that collagenproduction may be regulated by Stat3 at the transcrip-tional level. Preliminary scanning of the promoterregion of both collagen type I a1, COL1A1, andcollagen type III a1, COL3A1, revealed some putativeStat3 binding sites (CP Lim and X Cao, unpublished). Itwill be interesting to investigate whether Stat3 has adirect effect on the transcription of procollagen type Iand III, the two collagens that are found to be highlysynthesized in keloids (Abergel et al., 1985; Naitoh et al.,2001).
The process of cutaneous wound healing can bearbitrarily divided into three phases – inflammation,tissue formation (re-epithelialization, formation of
granulation tissue, neovascularization) and tissue remo-deling, events that are governed by a well-orchestratedtemporal and spatial secretion of various chemokines,cytokines and growth factors by different cell compo-nents (Singer and Clark, 1999; Werner and Grose,2003). Stat3 is a key molecule activated in response tomost of these ligands. Here, we observed enhancedexpression and phosphorylation of Stat3 in keloidtissues and fibroblasts, suggesting a lack of a stop signalor its downregulation, and/or an incessant stimulationby cytokines or growth factors. Indeed, some cytokinesthat activate Stat3 have been reported to be enhanced inkeloid tissue such as IL-6 (Xue et al., 2000). On the otherhand, whether there is any mutation in Stat3 in keloidtissues that predisposes to its overexpression remains tobe determined. As Stat3 is involved in all the differentphases of wound healing, we speculate that normalregulation of Stat3 is required throughout the process,and dysregulation of Stat3 signaling will tip the balancetowards impaired wound healing and fibrosis. Activa-tion of Stat3 by proinflammatory cytokines alsosuggests a role of Stat3 in the progression of scarringat the inflammation phase, as fetal tissues that lackinflammation undergo perfect scarless wound healing,and scarring ensued in the presence of an acuteinflammatory infiltrate (Yang et al., 2003).
Owing to their autonomous nature, keloids have alsobeen considered as benign tumors. Stat3, on the otherhand, possesses oncogenic potential, as overexpressionof a constitutively dimerized/active Stat3 mutant
Figure 6 Increased cell migration in keloid fibroblasts (KFs) compared to normal fibroblasts (NFs). (a) Three NFs, NF2, NF4 andNF103, and three KFs, KF43, KF48 and KF107, were grown in 60 mm dishes until confluent, and treated with 10mg/ml mitomycin Cfor 2 h, before a scratch wound was introduced using a yellow pipette tip. Cells were incubated for a further 14 h in normal growthmedia. Pictures were taken at 0 and 14 h after wounding. (b) Pictures from three non-overlapping fields from (a) were taken and cellsmigrated into the wound site were quantified. Data are presented as mean7s.d. *Po0.05 indicates KF43 vs NF4 and NF103, whereas**Po0.005 and ***Po0.001 refer to three NFs vs KF48 and KF107, respectively, by Student’s t-test. (c) Cell migration was alsoexamined using Transwell assay, performed in triplicates. KF48 cells were seeded in serum-free Dulbecco’s modified Eagles’s medium(DMEM) at the top of the insert with 10% fetal bovine serum (FBS)/DMEM as chemoattractant in the bottom chamber. Cells fromthree non-overlapping fields from each replicate that migrated from top to bottom of the insert after 48 h were counted and the mean ofthe triplicates7s.d. was presented in the graph. *Po0.001 and **Po0.005 indicate three NFs vs KF43, and KF48 and KF107,respectively, by Student’s t-test.
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5422
Oncogene

(Stat3C) caused cellular transformation and tumorformation (Bromberg et al., 1999), and Stat3 was shownto be essential for developing skin cancer in mice (Chanet al., 2004). Recently, overexpression of Stat3C in A549carcinoma cells followed by microarray analyses re-vealed that Stat3 regulated genes are common to bothwound healing and cancer, including cell invasion/migration, angiogenesis and remodeling of ECM, basedon the concept that tumors are ‘wounds that do notheal’ (Dauer et al., 2005). This supports our observationthat hyperactive Stat3 plays a role in wound healinggone awry, as seen in keloids. However, there is still adistinct difference between wound healing and skincancer, as cancer cells metastasize and scars do not.
In summary, we have shown for the first time a novelrole of Stat3 in the pathogenesis of keloid scars.
Inhibitors of Stat3 expression or phosphorylation wereable to suppress KF collagen production, cell prolifera-tion and migration to a level comparable to NFs.Besides siRNA and Cucurbitacin I, the efficacy of otherStat3 inhibitors could additionally be explored, includ-ing other pharmacological agents such as CucurbitacinQ, which is an analog of Cucurbitacin I and a selectiveStat3 inhibitor (Sun et al., 2005), STA-21 (Song et al.,2005), Stat3 decoy oligonucleotide (Leong et al., 2003),or phosphotyrosyl peptides (Turkson et al., 2001), all ofwhich either exhibited antitumor activity, increasedapoptosis and inhibited proliferation or suppressed celltransformation of cancer-derived or v-Src-transformedcell lines. These potential cancer therapeutic agents thattarget the inhibition of Stat3 may also be useful for theprospective clinical treatment of keloid scars.
Figure 7 Decreased cell migration owing to inhibition of Stat3. (a) KF48 cells were infected with retrovirus expressing Stat3 shortinterfering RNA (siRNA), and scratch-wound assay was performed as described previously in Figure 6a. Pictures were taken at 0 and15 h after wounding. (b) Data represent mean7s.d. of cells migrated into the wounding site from three non-overlapping fields from (a).*Po0.005 refers to Stat3 siRNA 1, 2 and 4 vs vector, by Student’s t-test, and NS indicates not significant. Inhibition of Stat3 by Stat3siRNAs is shown in the Western blot at the right panel. (c) KF48 cells were infected with retrovirus harboring Stat3 siRNAs andsimilar cell migration assay using Transwell inserts was performed as mentioned in Figure 6c. *Po0.001 in comparison to vector byStudent’s t-test, and n.s. indicates not significant. Inhibition of Stat3 by Stat3 siRNAs is shown in the Western blot at the right panel.(d) KF48 cells were pretreated with 10 mg/ml mitomycin C for 2 h, followed by either dimethyl sulfoxide (DMSO) or 1mM CucurbitacinI for 30 min, before cells were wounded using yellow pipette tips. Cells were further incubated in 10% fetal bovine serum (FBS)/Dulbecco’s modified Eagles’s medium (DMEM) normal growth media, and pictures were taken at 0, 22 and 48 h after wounding. (e)Data represent mean7s.d. of cells migrated into wounding sites from three non-overlapping fields from (d). *Po0.001 by Student’s t-test.
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5423
Oncogene

Materials and methods
Normal and keloid tissue/fibroblastsEthical approvals from the National University of SingaporeInstitutional Review Board and National Healthcare GroupDomain Specific Review Boards, and informed consent frompatients, were obtained before surgical excision of skin andkeloid tissue. All patients had received no prior treatment forkeloid lesions. A full history was taken and physicalexamination was performed in addition to color-slide photodocumentation of all keloid lesions. The procedure for theisolation and culture of fibroblasts from skin specimens was asdescribed previously (Lim et al., 2002). Isolated fibroblastswere grown in Dulbecco’s modified Eagles’s medium (DMEM)containing 4500 mg/ml glucose, supplemented with 10% fetalbovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillinand 100 ng/ml streptomycin. Normal skin and keloid scartissue, and normal and keloid-derived fibroblasts, used in thiswork are listed in Supplementary data.
For the isolation of tissue lysates for Western blot analysis,frozen tissue specimens were lysed in lysis buffer containing20 mM Tris-HCl, pH 7.5, 1% Triton X-100, 100 mM NaCl,0.5% Nonidet P-40 and 1 mg/ml protease inhibitor cocktail(Boehringer Mannheim, Germany). Equal amounts of proteinswere resolved by sodium dodecyl sulfate–polyacrylamide gelelectrophoresis in Western blot analysis.
Antibodies and reagentsPolyclonal anti-phosphoTyr705 Stat3, monoclonal anti-phosphoSer727 Stat3, anti-pJak1 (Tyr1022/1023), anti-pJak2(Tyr1007/1008), anti-phosphoSrc (Tyr416), anti-phospho-EGFR (Tyr845) and anti-Bcl-XL antibodies were purchasedfrom Cell Signaling Technology, whereas monoclonal pStat3(B7) (sc-8059), polyclonal Stat3 (C-20) (sc-482), monoclonalcyclin D1 (sc-20044), anti-mouse IgG (sc-2025) and anti-rabbitIgG (sc-2027) antibodies were acquired from Santa CruzBiotechnology Inc. (Santa Cruz, CA, USA). Monoclonalanti-Stat3 antibody was purchased from BD TransductionLaboratories (Lexington, KY, USA). Polyclonal anti-actinantibody and mitomycin C were purchased from Sigma (SaintLouis, MO, USA) and monoclonal anti-human collagen I, II,III was purchased from MONOSAN (Uden, Netherlands).AG 490 was obtained from Calbiochem (San Diego, CA,USA), whereas Cucurbitacin I was acquired from Galchimia(A Coruna, Spain).
Hematoxylin–eosin stainingTissue sections were fixed with 4% paraformaldehyde inphosphate-buffered saline (PBS), embedded in paraffin andsectioned. Tissue sections were stained with hematoxylin for 5–8 min, washed with running water, quickly destained with acidethanol containing 1% hydrochloric acid and 70% ethanol,washed with running water and counterstained with eosin–ethanol for 3–5 min. Tissue sections were then rehydrated bygradual immersion in 70, 80, 95 and 100% ethanol, clearedwith xylene and finally mounted in entellan. Pictures weretaken with Leica DM4000 B microscope.
ImmunofluorescenceCryosections were fixed with 4% paraformaldehyde in PBS for15–30 min at room temperature, washed thrice with 0.1%Triton X-100 in PBS for 10 min each, and blocked with FDB(Fluorescence Dilution Buffer) containing 5% normal goatserum, 2% FBS, 2% bovine serum albumin, 1 mM CaCl2 and1 mM MgCl2 in PBS. Tissue sections were washed as above andincubated for 1 h at room temperature, with primary antibody
diluted 1:200 in FDB to 1mg/ml for monoclonal pStat3 (B7),polyclonal Stat3 (C-20), anti-mouse IgG or anti-rabbit IgGantibodies. Tissue sections were washed again as described andincubated for 1 h at room temperature, with either FluoroLinkCy3-labeled goat anti-rabbit IgG (PA43004; AmershamBiosciences, Pistacaway, NJ, USA) or anti-mouse IgG-Cy3(AP124C; Chemicon International, Tencala, CA, USA)diluted at 1:400 in FDB. Sections were washed again andmounted with Vectashield mounting medium containing4,6-diamidino-2-phenylindole (DAPI) (H-1200; VectorLaboratories Inc., Burlingame, CA, USA). Pictures weretaken with Leica DM4000 B microscope.
XTT proliferation assayCells seeded at 3000 cells/well in 96-well plates and grown innormal growth conditions, or infected with retrovirus expres-sing Stat3 siRNAs, were assayed for proliferation using theXTT sodium 30-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro) benzene sulfonic acid hydrate prolifera-tion kit (Roche, Mannheim, Germany) according to themanufacturer’s instructions. The readings were taken at450 nm and referenced against 690 nm, 2 h after the additionof the XTT reagent. Each sample was performed atquadruplicates in two to three independent experiments. Dataare presented as mean7s.d.
Migration assayFor migration assay using the scratch method, cells weregrown to confluence and incubated with 10mg/ml mitomycin Cfor 2 h, before a scratch wound was introduced using a yellowpipette tip. Cells were further incubated in normal growthmedia and pictures were taken at 0, 14, 15, 22 or 48 h afterwounding. Three non-overlapping fields were captured byAxiovert 135 microscope from Zeiss (Jena, Germany) and cellsmigrated into the wound site were counted and presented asmean7s.d.
Migration assay was also performed using the Transwell8.0mM pore size polycarbonate membrane (Corning Inc.,Acton, MA, USA). 1� 104 cells/well were seeded at the topof the insert in 0.2 ml serum-free DMEM, with 0.5 ml 10%FBS/DMEM in the bottom chamber as chemoattractant.After 24 h, cells were washed once in cold PBS, beforefixing with 4% paraformaldehyde in PBS for 15 min atroom temperature. Cells were permeabilized with 0.1%Triton X-100 in PBS for 3 min at room temperature,and stained with hematoxylin for 10 min at room temperature,followed by destaining with water thrice. Cells from thetop of the inserts were removed by cotton swabs, and insertswere left to dry. The inserts were mounted onto slidesand three non-overlapping fields of cells migrated to thebottom of the inserts were captured using Leica DM4000 Bmicroscope.
Retroviral Stat3 short interfering RNA Four different targetsagainst Stat3 (Genbank Accession number NM_139276) wereselected and cloned into BglII and XhoI into pSUPER.retro.puro from OligoEngine, Seattle, WA, USA and se-quenced. The four targets are named Stat3 siRNA 1(nucleotides (nt) 461–480), Stat3 siRNA 2 (nt 1264–1283),Stat3 siRNA 3 (nt 364–383) and Stat3 siRNA 4 (nt 1662–1681). The corresponding primers with a 9-nt short hairpin inthe middle (bold) are F1 50-GATCCCCAGTCGAATGTTCTCTATCATTCAAGAGATGATAGAGAACATTCGACTTTTTTC-30 and R1 50-TCGAGAAAAAAGTCGAATGTTCTCTATCATCTCTTGAATGATAGAGAACATTCGACTGGG-30, F2 50-GATCCCCGGCGTCCAGTTCACTACTA
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5424
Oncogene

TTCAAGAGATAGTAGTGAACTGGACGCCTTTTTC-30
and R2 50-TCGAGAAAAAGGCGTCCAGTTCACTACTATCTCTTGAATAGTAGTGAACTGGACGCCGGG-30, F350-GATCCCCGATTGGGCATATGCGGCCATTCAAGAGA
TGGCCGCATATGCCCAATCTTTTTC-30 and R3 50-TCGAGAAAAAGATTGGGCATATGCGGCCATCTCTTGAA
TGGCCGCATATGCCCAATCGGG-30, and F4 50-GATCCCCGCGTCCATCCTGTGGTACATTCAAGAGATGTACCACAGGATGGACGCTTTTTC-30 and R4 50-TCGAGAAAAAGCGTCCATCCTGTGGTACATCTCTTGAATGTACCACAGGATGGACGCGGG-30. These vectors containingStat3 siRNA or empty vector were transfected into 293T-based amphotropic packaging cell line (Phoenix-Ampho) for7–9 h, and the amphotropic retroviruses were harvested 48 hlater, pelleted to remove non-adherent cells and cellular debris,and filtered through a 0.45 mM cellulose acetate membrane.The KFs were infected overnight in the presence of 4mg/mlpolybrene, and replaced with fresh 10% FBS/DMEM thefollowing day. Cells were assayed for Western blot 48 h after
infection, or XTT cell proliferation or migration assay, whichwas performed by replating the cells into 8.0 mM Transwellmembrane and fixed 24 h later as described above.
Acknowledgements
We are grateful to Dr Garry Nolan for the retroviral Phoenixpackaging cell line. We thank Dr Birgit Lane for helpfuldiscussions, Zheng Qi, Guo Ke, Li Jie and Bin Qi from theHistology Unit at the Institute of Molecular and Cell Biology,Singapore, for their kind assistance. The research work at theInstitute of Molecular and Cell Biology was supported by theAgency of Science, Technology and Research (A*STAR),Singapore. This research was also supported by A*STAR-Biomedical Research Council (BMRC) grant 02/1/21/19/106(to Lim IJ and Phan TT). Xinmin Cao is also an adjunctprofessor at the Department of Biochemistry, NationalUniversity of Singapore, Singapore.
References
Abergel RP, Pizzurro D, Meeker CA, Lask G, Matsuoka LY,Minor RR et al. (1985). J Invest Dermatol 84: 384–390.
Alster TS, Tanzi EL. (2003). Am J Clin Dermatol 4: 235–243.Babu M, Diegelmann R, Oliver N. (1989). Mol Cell Biol 9:
1642–1650.Babu M, Diegelmann R, Oliver N. (1992). J Invest Dermatol
99: 650–655.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti
SM. (2003). Cancer Res 63: 1270–1279.Bowman T, Garcia R, Turkson J, Jove R. (2000). Oncogene 19:
2474–2488.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell
RG, Albanese C et al. (1999). Cell 98: 295–303.Calderon M, Lawrence WT, Banes AJ. (1996). J Surg Res 61:
343–347.Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N,
Takeda J et al. (2004). J Clin Invest 114: 720–728.Cosman B, Crikelair GF, Gaulin JC, Lattes R. (1961). Plast
Reconstr Surg 27: 335–358.Darnell Jr JE. (1997). Science 277: 1630–1635.Darnell Jr JE, Kerr IM, Stark GR. (1994). Science 264:
1415–1421.Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R et al.
(2005). Oncogene 24: 3397–3408.Diegelmann RF, Cohen IK, McCoy BJ. (1979). J Cell Physiol
98: 341–346.Edmondson SR, Thumiger SP, Werther GA, Wraight CJ.
(2003). Endocr Rev 24: 737–764.Haisa M, Okochi H, Grotendorst GR. (1994). J Invest
Dermatol 103: 560–563.Lee TY, Chin GS, Kim WJ, Chau D, Gittes GK, Longaker
MT. (1999). Ann Plast Surg 43: 179–184.Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC
et al. (2003). Proc Natl Acad Sci USA 100: 4138–4143.Lim IJ, Phan TT, Bay BH, Qi R, Huynh H, Tan WT et al.
(2002). Am J Physiol Cell Physiol 283: C212–C222.
Mustoe TA. (2004). BMJ 328: 1329–1330.Naitoh M, Hosokawa N, Kubota H, Tanaka T, Shirane H,
Sawada M et al. (2001). Biochem Biophys Res Commun 280:1316–1322.
Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V et al.(2006). J Cell Biol 172: 245–257.
Ragoowansi R, Cornes PG, Glees JP, Powell BW, Moss AL.(2001). Br J Plast Surg 54: 504–508.
Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, MiuraH et al. (1999). EMBO J 18: 4657–4668.
Singer AJ, Clark RA. (1999). N Engl J Med 341: 738–746.Song H, Wang R, Wang S, Lin J. (2005). Proc Natl Acad Sci
USA 102: 4700–4705.Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D,
Sebti SM. (2005). Oncogene 24: 3236–3245.Takeda K, Akira S. (2000). Cytokine Growth Factor Rev 11:
199–207.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M,
Yoshida N et al. (1997). Proc Natl Acad Sci USA 94: 3801–3804.
Tuan TL, Nichter LS. (1998). Mol Med Today 4: 19–24.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E et al.
(2001). J Biol Chem 276: 45443–45455.Wen Z, Zhong Z, Darnell Jr JE. (1995). Cell 82: 241–250.Werner S, Grose R. (2003). Physiol Rev 83: 835–870.Wu Y, Zhang Q, Ann DK, Akhondzadeh A, Duong HS,
Messadi DV et al. (2004). Am J Physiol Cell Physiol 286:C905–C912.
Xue H, McCauley RL, Zhang W. (2000). J Surg Res 89:74–77.
Yang GP, Lim IJ, Phan TT, Lorenz HP, Longaker MT.(2003). Wound Repair Regen 11: 411–418.
Yoshimoto H, Ishihara H, Ohtsuru A, Akino K, Murakami R,Kuroda H et al. (1999). Am J Pathol 154: 883–889.
Younai S, Nichter LS, Wellisz T, Reinisch J, Nimni ME, TuanTL. (1994). Ann Plast Surg 33: 148–151.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Increased Stat3 phosphorylation and expression in keloidCP Lim et al
5425
Oncogene




















