
Liquiphant M/S with electronic insert FEL57 + Nivotester ...

www.BioTechniques.com681Vol. 47 | No. 2 | 2009
Research Reports
Introduction Large insert DNA libraries, such as bacterial artificial chromosome (BAC) libraries, have served a pivotal role for a number of genetic and genomic purposes, including map-based cloning, construction of physical maps, and whole-genome sequencing. BAC end sequences (BESs) were originally proposed as the primary scaffold in the sequencing of the human genome, serving as the primary means for selecting minimally overlapping clones for sequencing (1). In a BAC-by-BAC sequencing approach, BESs aided in the construction of physical maps and minimal tile paths (MTPs), anchoring of the physical and genetic maps, and in the assembly of sequences. The BAC-by-BAC strategy has been used for sequencing the genomes of several model eukaryotic organisms, including human (2), mouse (3), Arabidopsis thaliana (4) and rice (5).
In addition, BES data provide a first glimpse at the composition of unsequenced genomes (6). In this approach, BESs are usually analyzed by means of querying the BES using BLAST algorithms (7) against DNA and protein sequence databases.
Identities are assigned to the BES on the basis of sequence similarity with the reference sequences. A match to a database entry is considered significant when the ‘expect (E) value’ or the ‘BLAST score’—two parameters that reflect the degree of sequence similarity between the queried and reference sequences—surpasses a specified cutoff value.
An important quality parameter in BES data is the actual sequence length. This can significantly affect the BLAST search results and, consequently, the informativeness of the analysis. Presumably, the probability that a BES finds a significant match in a database (for a given E-value cutoff) is higher if longer BESs are queried.
BES data are also a source of molecular markers useful for mapping and breeding (8). The identification of potential markers in the BES, such as simple sequence repeats (SSRs), is usually done by using specific software. The proba-bility of detecting useful SSR markers in short BESs is lower than if longer (>500 bp) BESs are used as non-repetitive sequences f lanking the SSR are needed for designing primers.
For these reasons it is important to develop a procedure for generating longer BESs. To date, all the large-scale BAC end sequencing projects have used direct sequencing from purified recombinant plasmids. This technology has typically yielded 400–650 nucleotides of BES, in BAC libraries from species as diverse as human (9), mouse (10), A. thaliana (11), rice (12), and papaya (6).
Splinkerettes (13) enable PCR amplifi-cation of DNA sequences that lie between a single known primer and a nearby restriction site. The advantage of the splin-kerette adaptor is that it forms a hairpin loop in one of the strands, which prevents amplification in the absence of the known primer, thus minimizing the possibility of non-specific amplifications. This feature has made splinkerettes a robust tool for characterizing unknown DNA regions that are adjacent to known sequences (14,15).
Here we report on the development of a novel splinkerette-based method, suitable for high-throughput application, for gener-ating longer end sequences from large insert library clones, using as a model a carrot (Daucus carota L.) BAC library.
SplinkBES: a splinkerette-based method for generating long end sequences from large insert DNA librariesPablo F. Cavagnaro1, 2, Douglas Senalik1, 3, and Philipp W. Simon1, 3
1Department of Horticulture, University of Wisconsin, Madison, WI, USA, 2Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Mendoza, Argentina, 3USDA-Agricultural Research Service, Vegetable Crops Research Unit, University of Wisconsin, Madison, WI, USA
BioTechniques 47:681-690 ( July 2009) doi 10.2144/000113122 Keywords: BAC end sequence; splinkerette; Daucus carota; BAC library
Supplementary material for this article is available at www.BioTechniques.com/article/113122.
We report on the development of a novel splinkerette-based method for generating long end sequences from large insert library clones, using a carrot (Daucus carota L.) BAC library as a model. The procedure involves digestion of the BAC DNA with a 6-bp restriction enzyme, followed by ligation of splinkerette adaptors that serve as primer-annealing sites for PCR amplification of the BAC ends. The resulting amplicons are sequenced from both directions, and when overlapping, the pairs of sequences are assembled, originating two types of BAC end sequences (BESs): ungapped and gapped. The average sequence length for ungapped and gapped BESs was 698 and 1055 nucleotides, respectively, with an overall average length of 838 nucleotides. This is considerably higher than the average length typically obtained by direct end sequencing. Through the analysis of actual and in silico–generated BES of different lengths from carrot and five model organisms, we demonstrated that longer BESs are more informative, since they had more matches to the GenBank database and contained more simple sequence repeats (SSRs). A pilot high-throughput procedure is proposed for splinkerette-based end sequencing (SplinkBES). This method may contribute to generating more robust BES analy-sis and provide a richer source of BES-derived markers for genomics, mapping, and breeding.
www.BioTechniques.com682Vol. 47 | No. 2 | 2009
Research Reports
Materials and methods BAC library, vector, and splinkerette componentsA BamHI BAC library of carrot (16) was used as source of large insert DNA clones. The BamHI cloning site of the vector pIndigoBAC-5 (Epicenter Technol-ogies, Madison, WI, USA) is flanked 5′ and 3′ by the manufacturer’s described pIndigoBAC-5 forward and reverse sequencing primers, which we termed VL1 (for vector left primer 1) and VR1 (for vector right primer 1), respectively. Two additional nested primers, VL2 (5′-GCCAGTGA ATTGTA ATAC-G AC TC AC TAT-3 ′ ) a nd V R 2 (5′-CACACAGGA A ACAGCTAT GACCATGATT-3′), were designed -57 and +96 bases from the cloning site, respectively, and used for sequencing of the insert ends. The splinkerettes were made by duplexing the top strand 5′-CGAATCGTAACCGTTCGTAC-GAGAATTCGTACGAGAATCGCT-GTCCTCTCCAACGAGCCAAGG-3′ and the bottom strand 5′-XXXXCCTTG-GCTCGTTTTTTTTTGCAAAAA-3′, where XXXX is the overhang compatible with the cohesive ends left by the restriction enzyme used (e.g., AATT for MunI, GATC for BglII, or TCGA for SalI). The annealing reaction contained both oligonucleotides (40 µM each), 10 mM Tris-HCl (pH 7.4), 5 mM MgCl2, and was performed in a thermocycler by heating the solution to 65°C for 10 min and cooling 1°C/min to 4°C. The external and internal (nested) splinkerette primers, Splk1 (5′-CGAATCGTAACCGTTCGTAC-GAGAA-3′) and Splk2 (5′-TTCGTAC-GAGAATCGCTGTCCTCTCC-3′), used in PCR and sequencing reactions, respectively, were the same as originally described (13).
BAC plasmid DNA extractionBAC DNA was extracted using a modified alkaline lysis method from 1.5-mL overnight cultures in a 96-well plate format as described by Kim et al. (17). This extraction method yielded ~1.6–2.0 µg of digestable BAC DNA.
Digestion, ligation to the splinkerette, PCR amplification, and sequencing of BAC end ampliconsFive microliters of BAC DNAs (~0.4 µg) were digested to completion with one of the three following restriction enzymes (REs) with 6-bp recognition sequences: BglII, MunI, and SalI. Digestions were performed in a final volume of 20 µL, by incubation at 37°C for 3 h. The BAC vector used (pIndigoBAC-5)
had no restriction site for these enzymes in its sequence between the VL1 and VR1 primer annealing sites. Two microliters of the digests (~40 ng DNA) were then ligated to the splinkerette in a 10-µL ligation reaction containing 0.5 µM adaptors, 1 unit T4 DNA ligase, and 1× ligase buffer.
For the primary PCR using VL1 and Splk1 primers, an initial preheating step of 4 min at 94°C was used followed by a touchdown procedure of 50 s at 94°C, 12 cycles of 45 s at temperatures decreasing from 64 to 58°C (0.5°C decremental steps), and 72°C for 3 min; followed by 35 cycles of 50 s at 94°C, 45 s at 58°C, and 3 min at 72°C; and a final extension step at 72°C for 5 min. Reactions with
VR1 and Splk1 primers were performed similarly, except that the annealing temperature decreased from 62 to 56°C in the touchdown procedure, and was 56°C in the last 35 cycles. All reactions were performed in a final volume of 20 µL using 1.5 µL ligation product, 0.2 µM each primer, 0.2 mM each dNTP, 0.5 unit of Taq DNA polymerase, 1× PCR buffer, and 2.5 mM MgCl2. Ten-microliter aliquots of the PCR products were resolved by 1.2% agarose gel electrophoresis. The remaining PCR products were treated with exonuclease I and shrimp alkaline phosphatase (ExoSAP) (Fermentas Inc., Hanover, MD, USA) to remove remaining dNTPs and primers in the PCR mixture,
0
5
10
15
20
25
30
< 500 500- 999
1000-1499
1500-1999
2000-2499
2500-3000
>3000
Bgl IIMun ISal I
0
20
40
60
80
10 0
Bgl II
Mun I
* * * * * * *
Band size (bp)
3000
500 3000 500
Amplicon size (bp)
Fre
quen
cy (
%)
x
x
x
x
x
x
x
x
Bgl II Mun I Sal I
Bgl II Mun I
Bgl II Mun I Sal I
Suc
cess
rat
e (%
)
PCR with primers Splk1 and VL1
Treat PCR products with exonuclease I and phosphatase and direct sequence with nested primers Splk2 and VL2
Trim vector, splinkerette, and low-quality
sequences and merge overlapping regions to extend the BAC-end
sequence (BES)
BAC clone, restricted with enzyme x, is ligated to the splinkerette
Unmerged BESMerged BES
[gap]
A
B
C
D
E
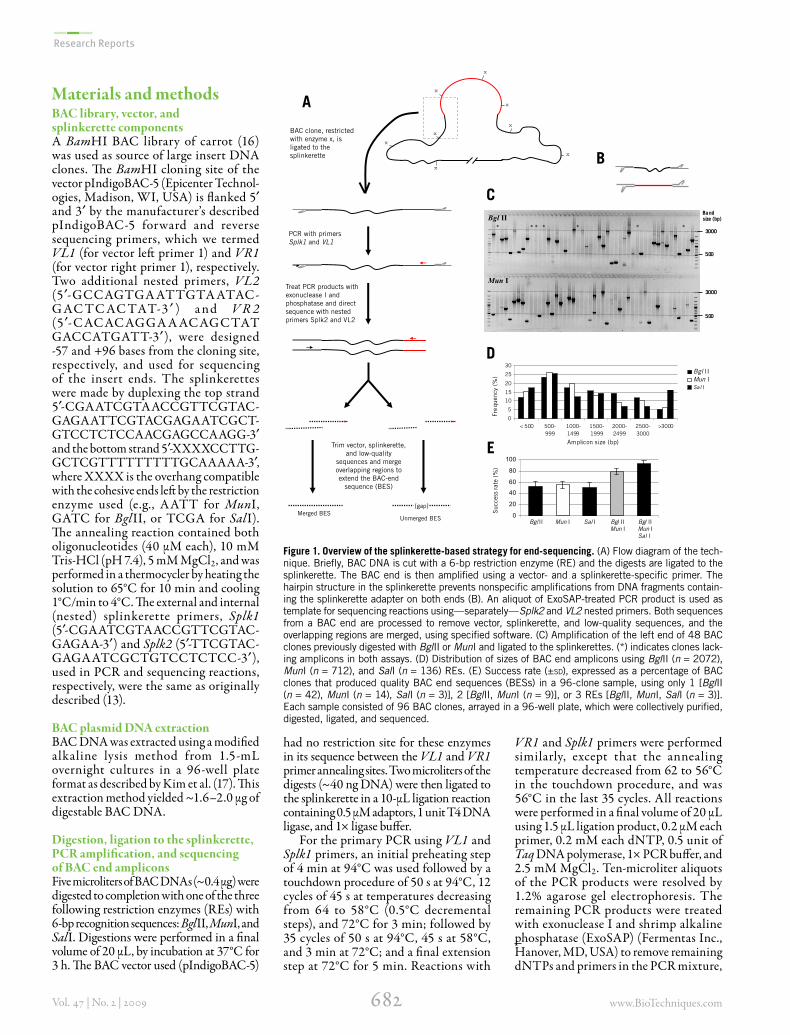
Figure 1. Overview of the splinkerette-based strategy for end-sequencing. (A) Flow diagram of the tech-nique. Briefly, BAC DNA is cut with a 6-bp restriction enzyme (RE) and the digests are ligated to the splinkerette. The BAC end is then amplified using a vector- and a splinkerette-specific primer. The hairpin structure in the splinkerette prevents nonspecific amplifications from DNA fragments contain-ing the splinkerette adapter on both ends (B). An aliquot of ExoSAP-treated PCR product is used as template for sequencing reactions using—separately—Splk2 and VL2 nested primers. Both sequences from a BAC end are processed to remove vector, splinkerette, and low-quality sequences, and the overlapping regions are merged, using specified software. (C) Amplification of the left end of 48 BAC clones previously digested with BglII or MunI and ligated to the splinkerettes. (*) indicates clones lack-ing amplicons in both assays. (D) Distribution of sizes of BAC end amplicons using BglII (n = 2072), MunI (n = 712), and SalI (n = 136) REs. (E) Success rate (±sd), expressed as a percentage of BAC clones that produced quality BAC end sequences (BESs) in a 96-clone sample, using only 1 [BglII (n = 42), MunI (n = 14), SalI (n = 3)], 2 [BglII, MunI (n = 9)], or 3 REs [BglII, MunI, SalI (n = 3)]. Each sample consisted of 96 BAC clones, arrayed in a 96-well plate, which were collectively purified, digested, ligated, and sequenced.

www.BioTechniques.com684Vol. 47 | No. 2 | 2009
Research Reports
following the manufacturer’s instructions. The samples were then diluted 5-fold in TE buffer (10 mM Tris pH 7.0, 1 mM EDTA) and 1.5 µL were used as template in 10-µL sequencing reactions containing
1 µL of BigDye Terminator V.3.1 (Applied Biosystems, Foster City, CA USA), 1.5 µL of sequencing buffer (ABI) and 1 µM of primer. The left BAC end amplicons were sequenced, in both directions, using the nested vector and splinkerette primers VL2 and Splk2, respectively. Similarly, VR2 and Splk2 primers were used to sequence the right BAC end amplicons. Cycle sequencing conditions were 60 cycles of 15 s at 95°C, 20 s at 55°C, and 2 min at 58°C. The reaction products were purified with CleanSeq magnetic beads (Agencourt, Beverly, MA, USA) and separated on an ABI 3730xl capillary sequencer (Applied Biosystems) at the Biotechnology Center at the University of Wisconsin (Madison, WI, USA). All the steps, from digestion to sequencing, were performed in 96-well plate format.
Sequence data processingSequence data were collected using using an ABI 3730xl with included acqui-sition software (Applied Biosystems). PHRED software (18,19) was used for base calling and assignment of base quality values, and vector, splinkerette, and low quality sequences were removed using the program LUCY (20). Quality sequences were defined as those having at least 100 nucleotides with PHRED-quality values of ≥20, of sequence other than vector or splinkerette. After removal of non-insert DNA, the two sequences from each BAC end amplicon were assembled when possible (i.e., the two sequences were aligned and assembled into one based on their overlapping regions), using the program CAP3 (21). When the BAC end amplicons exceeded 1.5 kb, the two sequences (from the vector and splin-kerette sides, respectively) did not overlap. Therefore, a gap remained, and they could not be assembled. Consequently, this process resulted in two types of BAC end sequences, designated as ungapped and gapped BES. When a second RE was used to generate BES from a previously-analyzed set of BAC clones (e.g., using first BglII and then MunI), the BES from both REs were assembled using CAP3.
Effect of BES lengthTo investigate whether sequence length had an effect on BES data analysis, carrot BESs >1 kb were selected (resulting in a set of 408 BESs with an average length of 1286 nucleotides) and queried against the NCBI nr/nt database using BLASTN algorithms with a cutoff E-value of ≤10-10. The same set of BES was also screened for presence of SSRs with di-, tri-, tetra-, penta- and hexanucleotide motifs spanning at least
12 nucleotides, using the program MISA (22). To simulate the effect of different sequence lengths on both BLAST searches and SSR discovery, the BESs were sequen-tially trimmed, from the non-vector side, producing shorter sequences from 1000 to 100 nucleotides, in 100-nucleotide decre-ments, and analyzed in the same way. The percentage of BESs significantly matching sequences from the NCBI database, as well as the percentage of BESs containing SSRs, were recorded and plotted against the BES length.
For comparison purposes, and to have a larger data set, we downloaded the entire genome sequence of five model species [Arabidopsis thaliana (ftp://ftp.ncbi.nih.gov/genomes/Arabidopsis_thaliana), Oryza sativa (ftp://ftp.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_4.0), Homo sapiens (ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/Assembled_chromosomes), Mus musculus (ftp://ftp.ncbi.nih.gov/genomes/M_musculus/Assembled_chromosomes), and Caenorhab-ditis elegans (ftp://ftp.sanger.ac.uk/pub/wormbase/live_release/genomes/elegans/sequences/dna)] in May 2008, and compu-tationally simulated the construction of BamHI BAC libraries, generating 10,000 in silico BESs (isBESs) of 1286 nucleotides for each species. Briefly, a random BamHI site in the genome was selected, and a search was made for another BamHI site in proximity to a point ~100 kb away from this site, searching randomly up or downstream. In silico BAC sizes were restricted to 90–110 kb. A more detailed description of the method and the perl script used to generate the isBESs is provided in Supplementary Materials 1. The isBESs from each species were then sequen-tially trimmed from the 3′ end, as described (in the previous paragraph) and subjected to the same analysis as the carrot BESs. Because the nr/nt database contains sequences from the entire genome of the selected species, to avoid self-matches we excluded from the searched database (NCBI nr/nt) those sequences from the same genus as the query, using, for example, the command “nucleotide NOT Arabidopsis[ORGN]” for BLASTN searches of Arabidopsis thaliana isBESs.
Results The basic techniqueThe basic SplinkBES (splinkerette-based end-sequencing) procedure involves digestion of the BAC DNA, ligation to the splinkerettes, PCR amplification of the BAC end, sequencing of the BAC end amplicon in both directions, followed by the assembly of each pair of sequences
Splk1/VL1 Splk1/VR1 Splk1/VL 1 Splk1/VR1
Splk2 VL 2 Splk2 VR2 Splk2 VL 2 Splk2 VR2
Bgl II Mun I
L R L R
[gap]
[gap]
BAC-end sequence analysis
A
B
C
D
E
F
G
Figure 2. Utilization of the splinkerette-based method for high-throughput BAC end-sequencing. The software used for processing of the se-quences is shown in brackets. (A) Grow BAC clones overnight in 1.5-mL cultures in 96-well plates. (B) Purify BAC DNA using Unifilters and Uniplates as described by Kim et al. (17). (C) Transfer aliquots of BAC DNA (~0.4 μg in 5 μL) to two PCR 96-well plates and digest with BglII and MunI, respectively. Then ligate the digests to the respective splinkerette adaptors. (D) PCR amplify the left (L) and right (R) BAC ends, using Splk1/VL1 and Splk1/VR1 primer pairs, respec-tively, using as template the BglII- and MunI-digested/ligated DNAs. Treat PCR products with exonuclease I + shrimp alkalyne phosphatase (ExoSAP). (E) Sequence the BAC end ampli-cons in both directions, using nested splinker-ette (Splk2) and vector primers (VL2, VR2). (F) Call bases [Phred] and trim vector, splinkerette, and low-quality sequences [LUCY], and merge overlapping regions of sequences from the same BAC end amplicon [CAP3]. (G) For each insert terminal end, merge overlapping regions of the end sequences obtained with BglII and MunI to close gaps and extend the BES [CAP3].

www.BioTechniques.com686Vol. 47 | No. 2 | 2009
Research Reports
(Figure 1A). A prerequisite is that the restriction enzyme (RE) used should not have restriction sites in the vector region flanked by the left and right vector primers (e.g., VL1 and VR1 for pIndigoBAC-5 vector). Since our goal was to obtain large amplicons and, consequently, long BESs, we used REs with 6-bp recognition sequences (instead of 5- or 4-bp cutters), considering a theoretical average digest size of 4096 bp, which can be amplified by PCR.
Most of the BAC end amplicons were in the size range of 500–3000 bp (this fraction representing 82%, 78%, and 66% of the amplicons obtained using BglII, MunI, and SalI, respectively), with the 500–999 bp category being the most frequent class for all 3 restriction enzymes used (Figure 1, C and D). The average amplicon size was 1494 bp for BglII (n = 2072), 1405 bp for MunI (n = 712) and 1726 bp for SalI (n = 136). These values are much lower than the expected average size (~4 kb) based on the theoretical cutting frequency for a 6-bp RE (assuming a random distribution of the nucleotides in the genome). This bias may result from the fact that, under regular PCR conditions using common DNA polymerases, fragments larger than 5–6 kb are generally not amplified. The observation that our overall range of amplicon sizes was 100–5000 bp supports this hypothesis.
As expected, only one band was amplified in the majority (>90%) of the clones (Figure 1C). When a second PCR product was observed in the gel for a given clone, end sequencing with the nested splinkerette (Splk2) or vector primers (VL2 or VR2) generally yielded clean sequence chromatograms, suggesting that one of the originally observed bands was a nonspecific amplicon generated by mispriming of Splk1 and/or VL1/VR1. Since these products lacked the annealing site for the nested primers, only the specific amplicons from the primary PCR served as template for the sequencing reaction.
Sequencing from opposite sides of the BAC end amplicons, using the vector and splinkerette nested primers, generated pairs of sequences for each BAC end. After sequence processing, which included trimming of vector-, splinkerette- and low quality sequences, overlapping regions were merged to the extent possible for each pair of end-sequences, resulting in either “ungapped” or “gapped” BESs (Figure 1A). From our data set, we were able to assemble sequences from BAC end amplicons up to 1500 bp long. These fragments ≤1500 bp accounted for 53%, 62%, and 55% of the total amplicons generated using BglII, MunI, and SalI,
respectively (Figure 1D). Based on this, it is expected that at least half of the BESs will be ungapped. Indeed, of the 1244 BAC end amplicons that were sequenced from both directions, we were able to assemble 60.8% (757/1244) of the sequence pairs, whereas 39.2% (487/1244) could not be assembled (gapped BESs) (Table 1).
The average sequence length, after quality processing, for ungapped and gapped BESs was 698 and 1055 nucleotides, respec-tively, with an overall average length of 838 nucleotides (Table 1). To our knowledge, this value is considerably higher than the average length obtained in large-scale BAC end sequencing projects reported to date. As a comparison, the average BES length was 477, 406–485, 576, and 611 nucle-otides for human (9), mouse (10), Arabi-dopsis (NCBI-GSS database, June 2008), and rice (12), respectively.
Success rate and throughput/automationUnder our conditions, success rate (expressed as the percentage of quality BES obtained from a sample set of 96 BAC clones), using a single 6-bp cutter, ranged from 50.3% (BglII) to 53.2% (MunI) (Figure 1E). Both the actual RE recognition sequence and the genome’s GC content can affect the size of digests and consequently the size of the BAC end amplicons, therefore affecting success rate. Probability calculations (Supplementary Material 2) predict that in a genome with 38.8% GC content, such as observed for carrot (16), using a 6-base RE recognizing 33% GC, such as BglII or MunI, 56% of the splinkerette products will be in the size range of 200–3000 nucleotides (48% and 50% observed for BglII and MunI, respec-tively). An RE recognizing 66% GC such as SalI only has 30% of products in this range (42% observed). A 5-base RE recognizing 50% GC, such as XhoII, should produce 77% of the products in this size range.
When a set of BAC clones was digested separately with each of two restriction enzymes (namely, BglII and MunI) and their amplicons sequenced, the success rate increased to 79.3 ± 6.1% (Figure 1, C and E). This value is comparable to the success rate obtained using direct BAC end-sequencing in mouse (79–83%) (10), human and Arabidopsis (80%) (11), tomato (80.9%) (23), and papaya (78.1% (6), but lower than reported in rice wild
species O. nivara (91%), O. rufipogon (97%), and O. brachyantha (91%) (24); common bean (93%) (25); and honey bee (87%) (26). The use of a second RE not only increased the success rate by ~28% but also increased the actual length of some BESs generated with the first RE, by either closing gaps in the gapped BES or extending the ungapped BES. Figure 2 illustrates these two scenarios.
Automation and throughputness are important parameters in large-scale BAC end-sequencing projects. We tested a high-throughput procedure for splinkerette-based BAC end sequencing using two RE, BglII and MunI (Figure 2). Under this scheme, sets of putative BAC end amplicons were immediately exoSAP-treated and sequenced without previous confirmation of PCR results by gel electrophoresis. Resulting successful BESs generated with both REs were assembled with CAP3. A bioinformatic pipeline was developed for processing and analysis of the sequences (data not shown).
All the steps in the SplinkBES method were performed in a 96-well plate format and could be automated relatively easily. Moreover, once the BAC DNA has been isolated, the procedure can be scaled up to a 384-well plate format for the digestion-to-sequencing steps, thus increasing the number of samples that are simultaneously analyzed.
When 3 REs were used (BglII, MunI, and SalI), BESs were obtained in 90.6 ± 4.2% of the BAC clones (Figure 1E). Despite the high success rate, the use of a third RE is not cost-effective, since most of the BAC end amplicons—and conse-quently also the BESs—are redundant to the previous two RE used, with only a small increase in success rate.
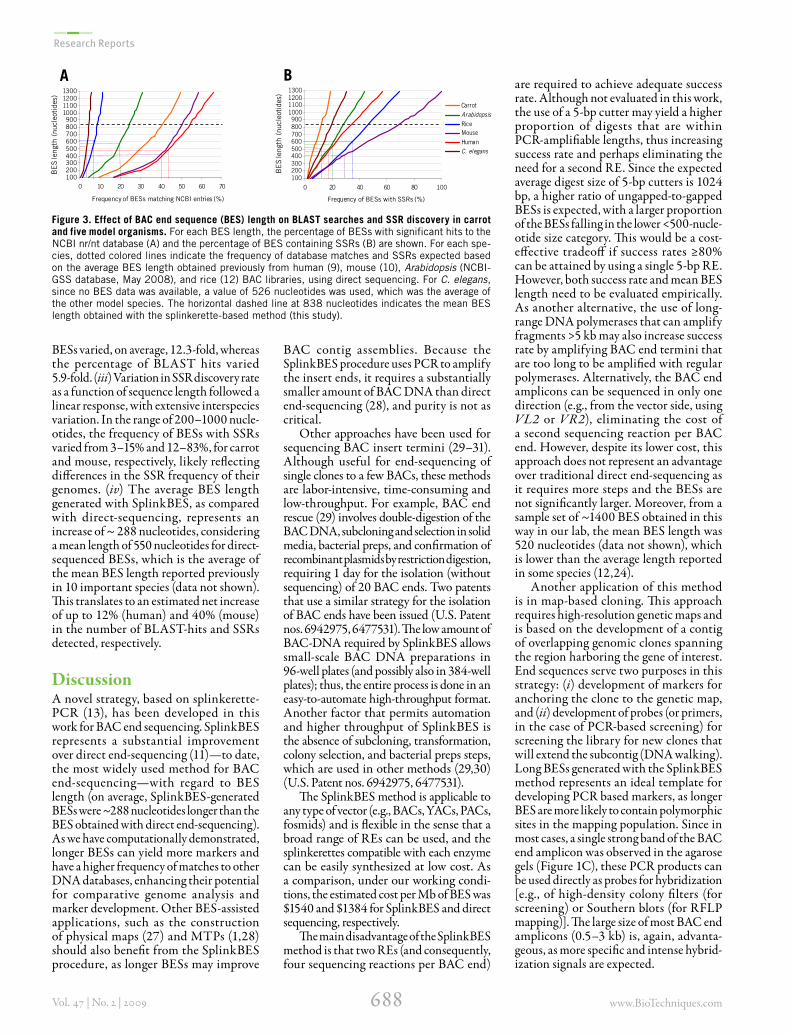
Longer end sequences are more informativeThe analysis of isBESs of different lengths clearly demonstrated the advantages of generating longer BESs (Figure 3). Our results showed that: (i) for all the species analyzed, longer BESs had more matches to the database and contained more SSRs. (ii) SSR discovery was more affected by sequence length than the BLAST search results. This is based upon our observation that in the size range of 100–1000 nucle-otides, the frequency of SSR-containing
Table 1. Type, frequency, and length of BESs generated with SplinkBES
No. of BESs % Total length Avg. length
Unmerged BESs 487 39.2 518,840 1055
Merged BESs 757 60.8 528,611 698
Total 1244 100 1,042,581 838
www.BioTechniques.com688Vol. 47 | No. 2 | 2009
Research Reports
BESs varied, on average, 12.3-fold, whereas the percentage of BLAST hits varied 5.9-fold. (iii) Variation in SSR discovery rate as a function of sequence length followed a linear response, with extensive interspecies variation. In the range of 200–1000 nucle-otides, the frequency of BESs with SSRs varied from 3–15% and 12–83%, for carrot and mouse, respectively, likely reflecting differences in the SSR frequency of their genomes. (iv) The average BES length generated with SplinkBES, as compared with direct-sequencing, represents an increase of ~ 288 nucleotides, considering a mean length of 550 nucleotides for direct-sequenced BESs, which is the average of the mean BES length reported previously in 10 important species (data not shown). This translates to an estimated net increase of up to 12% (human) and 40% (mouse) in the number of BLAST-hits and SSRs detected, respectively.
DiscussionA novel strategy, based on splinkerette-PCR (13), has been developed in this work for BAC end sequencing. SplinkBES represents a substantial improvement over direct end-sequencing (11)—to date, the most widely used method for BAC end-sequencing—with regard to BES length (on average, SplinkBES-generated BESs were ~288 nucleotides longer than the BES obtained with direct end-sequencing). As we have computationally demonstrated, longer BESs can yield more markers and have a higher frequency of matches to other DNA databases, enhancing their potential for comparative genome analysis and marker development. Other BES-assisted applications, such as the construction of physical maps (27) and MTPs (1,28) should also benefit from the SplinkBES procedure, as longer BESs may improve
BAC contig assemblies. Because the SplinkBES procedure uses PCR to amplify the insert ends, it requires a substantially smaller amount of BAC DNA than direct end-sequencing (28), and purity is not as critical.
Other approaches have been used for sequencing BAC insert termini (29–31). Although useful for end-sequencing of single clones to a few BACs, these methods are labor-intensive, time-consuming and low-throughput. For example, BAC end rescue (29) involves double-digestion of the BAC DNA, subcloning and selection in solid media, bacterial preps, and confirmation of recombinant plasmids by restriction digestion, requiring 1 day for the isolation (without sequencing) of 20 BAC ends. Two patents that use a similar strategy for the isolation of BAC ends have been issued (U.S. Patent nos. 6942975, 6477531). The low amount of BAC-DNA required by SplinkBES allows small-scale BAC DNA preparations in 96-well plates (and possibly also in 384-well plates); thus, the entire process is done in an easy-to-automate high-throughput format. Another factor that permits automation and higher throughput of SplinkBES is the absence of subcloning, transformation, colony selection, and bacterial preps steps, which are used in other methods (29,30) (U.S. Patent nos. 6942975, 6477531).
The SplinkBES method is applicable to any type of vector (e.g., BACs, YACs, PACs, fosmids) and is flexible in the sense that a broad range of REs can be used, and the splinkerettes compatible with each enzyme can be easily synthesized at low cost. As a comparison, under our working condi-tions, the estimated cost per Mb of BES was $1540 and $1384 for SplinkBES and direct sequencing, respectively.
The main disadvantage of the SplinkBES method is that two REs (and consequently, four sequencing reactions per BAC end)
are required to achieve adequate success rate. Although not evaluated in this work, the use of a 5-bp cutter may yield a higher proportion of digests that are within PCR-amplifiable lengths, thus increasing success rate and perhaps eliminating the need for a second RE. Since the expected average digest size of 5-bp cutters is 1024 bp, a higher ratio of ungapped-to-gapped BESs is expected, with a larger proportion of the BESs falling in the lower <500-nucle-otide size category. This would be a cost-effective tradeoff if success rates ≥80% can be attained by using a single 5-bp RE. However, both success rate and mean BES length need to be evaluated empirically. As another alternative, the use of long-range DNA polymerases that can amplify fragments >5 kb may also increase success rate by amplifying BAC end termini that are too long to be amplified with regular polymerases. Alternatively, the BAC end amplicons can be sequenced in only one direction (e.g., from the vector side, using VL2 or VR2), eliminating the cost of a second sequencing reaction per BAC end. However, despite its lower cost, this approach does not represent an advantage over traditional direct end-sequencing as it requires more steps and the BESs are not significantly larger. Moreover, from a sample set of ~1400 BES obtained in this way in our lab, the mean BES length was 520 nucleotides (data not shown), which is lower than the average length reported in some species (12,24).
Another application of this method is in map-based cloning. This approach requires high-resolution genetic maps and is based on the development of a contig of overlapping genomic clones spanning the region harboring the gene of interest. End sequences serve two purposes in this strategy: (i) development of markers for anchoring the clone to the genetic map, and (ii) development of probes (or primers, in the case of PCR-based screening) for screening the library for new clones that will extend the subcontig (DNA walking). Long BESs generated with the SplinkBES method represents an ideal template for developing PCR based markers, as longer BES are more likely to contain polymorphic sites in the mapping population. Since in most cases, a single strong band of the BAC end amplicon was observed in the agarose gels (Figure 1C), these PCR products can be used directly as probes for hybridization [e.g., of high-density colony filters (for screening) or Southern blots (for RFLP mapping)]. The large size of most BAC end amplicons (0.5–3 kb) is, again, advanta-geous, as more specific and intense hybrid-ization signals are expected.
100200300400500600700800900
1000110012001300
0 10 20 30 40 50 60 70
100200300400500600700800900
1000110012001300
0 20 40 60 80 100
CarrotArabidopsisRiceMouseHumanC. elegans
Frequency of BESs with SSRs (%)
Frequency of BESs matching NCBI entries (%)
BE
S le
ngth
(nu
cleo
tide
s)
BE
S le
ngth
(nu
cleo
tide
s)
A B
Figure 3. Effect of BAC end sequence (BES) length on BLAST searches and SSR discovery in carrot and five model organisms. For each BES length, the percentage of BESs with significant hits to the NCBI nr/nt database (A) and the percentage of BES containing SSRs (B) are shown. For each spe-cies, dotted colored lines indicate the frequency of database matches and SSRs expected based on the average BES length obtained previously from human (9), mouse (10), Arabidopsis (NCBI-GSS database, May 2008), and rice (12) BAC libraries, using direct sequencing. For C. elegans, since no BES data was available, a value of 526 nucleotides was used, which was the average of the other model species. The horizontal dashed line at 838 nucleotides indicates the mean BES length obtained with the splinkerette-based method (this study).
100200300400500600700800900
1000110012001300
0 10 20 30 40 50 60 70
100200300400500600700800900
1000110012001300
0 20 40 60 80 100
CarrotArabidopsisRiceMouseHumanC. elegans
Frequency of BESs with SSRs (%)
Frequency of BESs matching NCBI entries (%)
BE
S le
ngth
(nu
cleo
tide
s)
BE
S le
ngth
(nu
cleo
tide
s)

www.BioTechniques.com689Vol. 47 | No. 2 | 2009
Research Reports
With the rise of next-generation sequencing (NGS) technologies such as 454 (Roche Applied Science, Indianapolis, IN, USA), Illumina (San Diego, CA, USA) and SOLiD (Applied Biosystems), which can generate millions of short reads (35–250 nucleotides) in only a few hours or days at relatively low cost (32), whole-genome sequencing in many eukaryotes is now a realistic possibility. However, de novo sequencing and assembly of large and complex, highly-repetitive genomes, such as those commonly found in plant species (33), requires a backbone structure (i.e., an MTP) onto which the sequence reads are assembled. Because BESs are instrumental in the construction of physical maps (27) and MTPs (1,34), and facilitate the assembly of whole-genome shotgun sequence data (35), the use of longer BESs (e.g., using SplinkBES) may also contribute to achieving more accurate assemblies in these applications.
AcknowledgmentsThe authors gratefully acknowledge H.R. Kim for valuable advice on the BAC DNA extraction procedure and end sequencing. This work was supported by the Agricul-
tural Research Service, United States Department of Agriculture.
The authors declare no competing interests.
References 1. Venter, J.C., H.O. Smith, and L. Hood. 1996.
A new strategy for genome sequencing. Nature 381:364-366.
2. International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409:860-921.
3. Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420:520-562.
4. The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796-815.
5. International Rice Genome Sequencing Project. 2005. The map-based sequence of the rice genome. Nature 436:793-800.
6. Lai, C.W.J., Q. Yu, S. Hou, R.L. Skelton, M.R. Jones, K.L.T. Lewis, J. Murray, M. Eustice, et al. 2006. Analysis of papaya BAC end sequences reveals first insights into the organization of a fruit tree genome. Mol. Genet. Genomics 276:1-12.
7. Altschul, S.F., W. Gish, W. Miller, E.W. Myers, and D.J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410.
8. Frelichowski, J.E., M.B. Palmer, D. Main, J.P. Tomkins, R.G. Cantrell, D.M. Stelly, J. Yu, R.J. Kohel, and M. Ulloa. 2006. Cotton genome mapping with new microsatellites from Acala ‘Maxxa’ BAC ends. Mol. Genet. Genomics 275:479-491.
9. Zhao, S. 2000. Human BAC ends. Nucleic Acids Res. 28:129-132.
10. Zhao, S., S. Shatsman, B. Ayodeji, K. Geer, T. Getahun, M. Krol, E. Gebregeorgis, A. Shvartsbeyn, et al. 2001. Mouse BAC ends quality assessment and sequence analysis. Genome Res. 11:1736-1745.
11. Kelley, J.M., C.E. Field, M.B. Craven, D. Bocskai, U.J. Kim, S.D. Rounsley, and M.D. Adams. 1999. High throughput direct end sequencing of BAC clones. Nucleic Acids Res. 27:1539-1546.
12. Yuan, Q., J. Quackenbush, R. Sultana, M. Pertea, S.L. Salzberg, and R.C. Buell. 2001. Rice bioinformatics. Analysis of rice sequence data and leveraging the data to other species. Plant Physiol. 125:1166-1174.
13. Devon, R.S., D.J. Porteous, and A.J. Brookes. 1995. Splinkerettes – improved vector-ettes for greater efficiency in PCR walking. Nucleic Acids Res. 23:1644-1645.
14. Mikkers, H., J. Allen, P. Knipscheer, L. Romeyn, A. Hart, E. Vink, and A. Berns. 2002. High throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat. Genet. 32:153-159.
15. Lund, A.H., G. Turner, A. Trubetskoy, E. Verhoeven, E. Wientjens, D. Hulsman, R. Russell, R.A. DePinho, et al. 2002. Genome
Reach over
125,000Life Science
Professionals Life science marketing lists with more life.For more information, call (212) 520-2729
If you are looking to maximize your mar-keting efforts to professionals who work inall aspects of pharma and biotech, youneed a resource that gives you access toyour best responders — and that’s InformaLife Science mailing lists.
Cultivated from our targeted subscriberbase of life science professionals, our listsgive you the best chance of delivering yourbrand and offer to this market.
Let us locate the right decision-makers inyour market segment:
• Over 125,000 life science professionals
• $25 billion in purchasing power
• Segmentation including function, title and laboratory technique
• worldwide reach
Let Informa Life Sciences mailing lists spearhead your next campaign
Give your marketing programs a LIFT!
ILSD_AD_8.375x5.5_half_H 10/10/07 9:46 AM Page 1
Informa_Life_Science_half_h.indd 1 2/1/08 12:26:56 PM

www.BioTechniques.com690Vol. 47 | No. 2 | 2009
Research Reports
wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat. Genet. 32:160-165.
16. Cavagnaro, P.F., S.-M. Chung, M. Szklarczyk, D. Grzebelus, D. Senalik, A.E. Atkins, and P.W. Simon. 2009. Character-ization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol. Genet. Genomics. 10.1007/s00438-008-0411-9.
17. Kim, H., P. San Miguel, W. Nelson, K. Collura, M. Wissotski, J.G. Walling, J.P. Kim, S.A. Jackson, et al. 2007. Comparative physical mapping between Oryza sativa (AA genome type) and O. punctata (BB genome type). Genetics 176:379-390.
18. Ewing, B. and P. Green. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186-194.
19. Ewing, B., L. Hillier, M.C. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175-185.
20. Chou, H.H. and M.H. Holmes. 2001. DNA sequence quality trimming and vector removal. Bioinformatics 17:1093-1094.
21. Huang, X. and A. Madan. 1999. CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
22. Thiel, T., W. Michalek, R.K. Varshney, and A. Graner. 2003. Exploiting EST databases for the development and characterization of gene derived SSR markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106:411-422.
23. Budiman, M.A., L. Mao, T.C. Wood, and R.A. Wing. 2000. A deep-coverage tomato BAC library and prospects toward development of an STC framework for genome sequencing. Genome Res. 10:129-136.
24. Wing, R.A., J. Ammiraju, M. Luo, H. Kim, Y. Yu, D. Kudrna, J.L. Goicoechea, W. Wang, et al. 2005. The Oryza Map Alignment Project: the golden path to unlocking the genetic potential of wild rice species. Plant Mol. Biol. 59:53-62.
25. Schlueter, J.A., J.L. Goicoechea, K. Collura, N. Gill, J.-Y. Lin, Y. Yu, D. Kudrna, A. Zuccolo, et al. 2008. BAC-end sequence analysis and a draft physical map of the common bean (Phaseolus vulgaris L.) genome. Trop. Plant Biol. 1:40-48.
26. Tomkins, J.P., M. Luo, G.C. Fang, D. Main, J.L. Goicoechea, M. Atkins, D.A. Frisch, R.E. Page, et al. 2002. New genomic resources for the honey bee (Apis mellifera L.): development of a deep-coverage BAC library and a preliminary STC database. Genet. Mol. Res. 1:306-316
27. International Bovine BAC Mapping Consortium. 2007. A physical map of the bovine genome. Genome Biol. 8:R165.
28. Chibana, H., B.B. Magee, S. Grindle, Y. Ran, S. Scherer, and P.T. Magee. 1998. A physical map of chromosome 7 of Candida albicans. Genetics 149:1739-1752.
29. R ipoll, P.-J., D.M. O’Sullivan, K.J. Edwards, and M. Rodgers. 2000. Technique for cloning and sequencing the ends of bacterial artificial chromosome inserts. BioTechniques 29:271-276.
30. Huang, Y., D.-P. Liu, M. Wu, and C.-C. Liang. 2001. A fast and efficient method for isolation of the BAC end. Mol. Biotechnol. 19:215-217.
31. Tomkins, J.P., Y. Yu, H. Miller-Smith, D.A. Frisch, S.S. Woo, and R.A. Wing. 1999. A bacterial artificial chromosome library for sugarcane. Theor. Appl. Genet. 99:419-424.
32. Mardis, E.R. 2008. The impact of next-generation sequencing technology on genetics. Trends Genet. 24:133-141.
33. Bennett, M.D. and J.B. Smith. 1976. Nuclear DNA amounts in angiosperms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 274:227-274.
34. Muel ler, L . A., S.D. Tan ksley, J.J. Giovannoni, J. van Eck, S. Stack, D. Choi, B.D. Kim, M. Chen, et al. 2005. The tomato sequencing project, the first cornerstone of the International Solanaceae Project (SOL). Comp. Funct. Genomics 6:153-158.
35. Warren, R.L., D. Varabei, D. Platt, X. Huang, D. Messina, S.P. Yang, J.W. Kronstad, M. Krzywinski, et al. 2006. Physical map-assisted whole-genome shotgun sequence assemblies. Genome Res. 16:768-775.
Received 24 September 2008; accepted 30 January 2009.
Address correspondence to Philipp W. Simon, USDA, ARS, Vegetable Crops Research Unit, Department of Horticulture, University of Wisconsin, 1575 Linden Drive, Madison, WI, 53706, USA. e-mail: [email protected]
Best Products, Best Performance, Best Protection
www.nuaire .com
Best Products, Best Performance, Best ProtectionBest Products, Best Performance, Best Protection
Where Quality meets E� ciency
®
NuAire, Inc.® | 2100 Fernbrook Lane | Plymouth, MN 55447 Ph: 1.800.328.3352 | Fx: 763.553.0459
™
NuAire’s® CellGard ES™ NU-480 Class II, Type A2 Biological Safety
Cabinet uses the best of our existing technologies and the best of
the new DC ECM motor technology to provide lower energy costs,
longer fi lter life, and reduced noise and vibration. NuAire® uses
the largest HEPA fi lters with the most pleats per square inch; the
TouchLink™ airfl ow control system; internal exhaust damper; and
individually selected, optimally determined forward-curved fans
for each model size/width. CellGard ES™ Biological Safety Cabinets
provide long-term reliability; outstanding performance; superior
quality; and cost saving technologies.
Our dedication to quality, performance, service, and customer
satisfaction, makes NuAire® the perfect choice - a real Value
NuAire_CellGardES.indd 1 6/15/09 1:49:41 PM
Copyright © 2022 FDOKUMEN




















