
Specific inhibition as the kinetic principle of the Fischer-Tropsch synthesis
12
Topics in Catalysis 2 (1995) 223-234 223 Specific inhibition as the kinetic principle of the Fischer-Tropsch synthesis Hans Schulz, Eric van Steen a and Michael Claeys Engler-Bunte-lnstitut, UniversitdtKarlsruhe, Kaiserstrasse 12, 76128Karlsruhe, Germany aDepartment of Chemical Engineering, Universityof Cape Town, Rondebosch 7700, South Africa FT CO-hydrogenation on cobalt catalysts has been performed in a well stirred continuously operated slurry reactor with and without addition of n-octene-I at different CO partial pres- sures. Kinetic data have been determined with the help of a mechanistic model. Increasing CO partial pressure inhibits individual reaction steps selectively, in particular associative methane desorption, the steps of chain termination (as paraffins and olefins likewise),whereas in second- ary olefin conversion hydrogenation is inhibited drastically, however double bond shift is even promoted. Secondary hydrogenation is concluded to proceed on different sites than chain growth. Keywords: FT synthesis; specific inhibition; cobalt catalysts; slurry reactor; olefin addition; secondary reactions 1. Introduction The principle of catalysis is frequently pictured as opening gates for the mass flow towards new directions and, respectively, new compounds by lowering activa- tion energies and allowing for intermediate states of low energy. This implies low- ering of reaction temperature and higher conversions in equilibrium controlled reactions. A different view of creating new pathways in catalytic reactions is by installing barriers on the main route. Obstacles on pathways of catalytic conversion, provid- ing specific inhibition, can be steric or spacious constraints like in zeolite cat- alysis, specifically on the surface located additives in the catalyst composition or strongly adsorbed compounds in the reaction mixture. Specific inhibition has recently been proposed to be the dominating principle in Fischer-Tropsch synthesis [1], which rules the formation of long chain aliphatic compounds from CO plus H2 in a unique set of reactions on the catalyst surface. Of course, evaluation of this concept requires knowledge, respectively assump- tions, about surface bonded intermediates, the elemental reaction steps and a kinetic model. Experimental verification affords precise determination of product © J.C. Baltzer AG, SciencePublishers
Transcript of Specific inhibition as the kinetic principle of the Fischer-Tropsch synthesis

Topics in Catalysis 2 (1995) 223-234 223
Specific inhibition as the kinetic principle of the Fischer-Tropsch synthesis
Hans Schulz, Eric van Steen a and Michael Claeys
Engler-Bunte-lnstitut, Universitdt Karlsruhe, Kaiserstrasse 12, 76128 Karlsruhe, Germany a Department of Chemical Engineering, University of Cape Town,
Rondebosch 7700, South Africa
FT CO-hydrogenation on cobalt catalysts has been performed in a well stirred continuously operated slurry reactor with and without addition of n-octene-I at different CO partial pres- sures. Kinetic data have been determined with the help of a mechanistic model. Increasing CO partial pressure inhibits individual reaction steps selectively, in particular associative methane desorption, the steps of chain termination (as paraffins and olefins likewise), whereas in second- ary olefin conversion hydrogenation is inhibited drastically, however double bond shift is even promoted. Secondary hydrogenation is concluded to proceed on different sites than chain growth.
Keywords: FT synthesis; specific inhibition; cobalt catalysts; slurry reactor; olefin addition; secondary reactions
1. I n t r o d u c t i o n
The principle of catalysis is frequently pictured as opening gates for the mass flow towards new directions and, respectively, new compounds by lowering activa- tion energies and allowing for intermediate states of low energy. This implies low- ering of reaction temperature and higher conversions in equilibrium controlled reactions.
A different view of creating new pathways in catalytic reactions is by installing barriers on the main route. Obstacles on pathways of catalytic conversion, provid- ing specific inhibition, can be steric or spacious constraints like in zeolite cat- alysis, specifically on the surface located additives in the catalyst composi t ion or strongly adsorbed compounds in the reaction mixture.
Specific inhibition has recently been proposed to be the dominating principle in Fischer-Tropsch synthesis [1], which rules the formation of long chain aliphatic compounds from CO plus H2 in a unique set of reactions on the catalyst surface.
Of course, evaluation of this concept requires knowledge, respectively assump- tions, about surface bonded intermediates, the elemental reaction steps and a kinetic model. Experimental verification affords precise determination of product
© J.C. Baltzer AG, Science Publishers

224 H. Schulz et al. / Specific inhibition, kinetic principle of FTsynthesis
composit ion and a kinetically well defined reaction technology. Then this study intends to contribute to the shift in understanding of the Fischer-Tropsch synthesis from a kind of mystery to believe in to a scientifically proven finely structured unique combination of catalytic surface events.
2. Exper imenta l
Experiments of CO-hydrogenation have been performed in a well-mixed con- tinuously operated autoclave reactor with the catalyst particles being suspended in squalane [2]. The mass streams of hydrogen and carbon monoxide to the reactor were precisely controlled and measured. A constant pressure was obtained with the help of a pressure controlled make up stream of argon, fed to the gaseous product stream behind the reactor. A reference gas stream, containing N2 and cyclopro- pane, was also added to the hot gaseous product stream for the determination of CO conversion and yields of individual compounds from the gas chromatograms. A continuous stream of products, consisting of liquids, vapors and gases, was con- tinuously withdrawn from the reactor and the liquid fraction separated in a hot trap held at reaction temperature. Only one of the reaction parameters (CO partial pressure or reaction temperature in this investigation) was varied, while the other intensive parameters (PH2, PH20 and T or Pco) were kept constant. When a desired reaction condition attained the stationary state after about 80-100 h, gas/vapor- ampoule samples [3] were taken from the gaseous product stream and all relevant data for establishing the mass balance were recorded. In order to examine second- ary reactions of ~-olefins, a stream of n-octene-1 was added to the syngas via a saturator while maintaining the reaction conditions constant. A survey of the reac- tion conditions and some results concerning the added olefin is given in table 1.
The cobalt catalysts were prepared by precipitation with a NH3-solution from nitrate solutions (one exception: ReC13-solution) in which the Aerosil (Aerosil 200, Degussa, Hanau) was suspended at a temperature closely below the boiling tem- perature. The precipitate was washed, dried (15 h, 100°C), ground to particles below 100 gm diameter and reduced at 550°C with hydrogen in a fixed bed reactor until no further reduction water could be detected (30 h). The final compositions of the catalysts were 100Co : 19Re : 100Aerosil and 100Co : 10MgO : 3ThO2 : 100Aerosil (mass ratios). For the synthesis experiments catalyst amounts as equivalent to 2 g of cobalt were transferred to the slurry reactor, which already con- tained 270 ml squalane as solvent, respectively suspension liquid.
Gas-chromatography of the ampoule samples was performed off-line [3]. Degree of conversion and the yields of all compounds in the gaseous product stream were directly calculated from the chromatograms by referring to the peaks of the compounds, nitrogen and cyclopropane, which had been introduced with the reference stream. The capillary chromatograms of organic compounds covered
H. Schulz et aL / Specific inhibition, kinetic principle of FTsynthesis 225
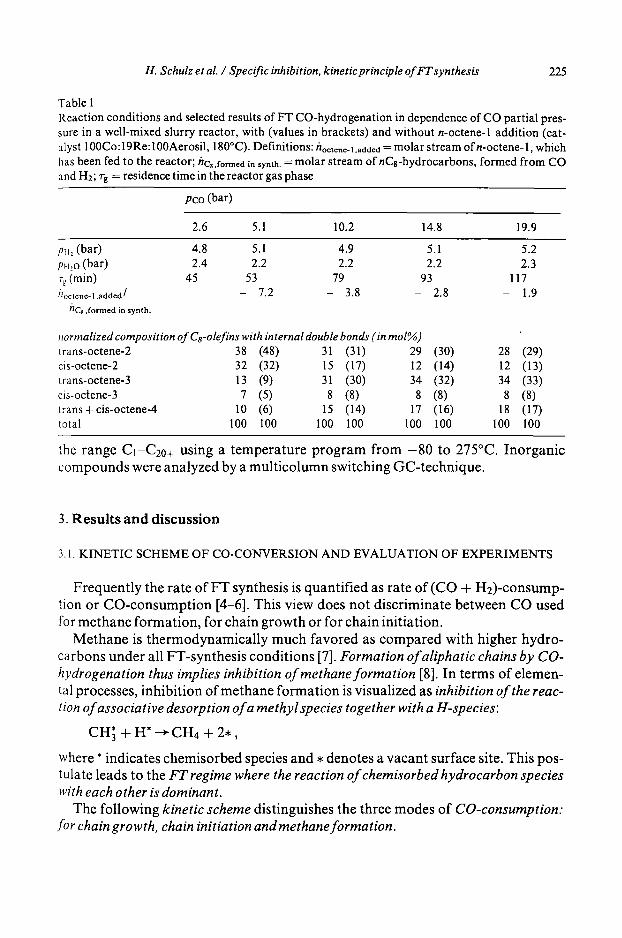
Table 1 Reaction conditions and selected results of FT CO-hydrogenation in dependence of CO partial pres- sure in a well-mixed slurry reactor, with (values in brackets) and without n-octene-1 addition (cat- alyst 100Co: 19Re: 100Aerosil, 180 ° C). Definitions: hoctenc-t.addcd = molar stream of n-octene- 1, which has been fed to the reactor; hc,,fo,~ed in symh. = molar stream of nCs-hydrocarbons, formed from CO and H2; 7-g = residence time in the reactor gas phase
Pco (bar)
2.6 5.1 10.2 14.8 19.9
pH, (bar) 4.8 5. I 4.9 5.1 5.2 pH,o (bar) 2.4 2.2 2.2 2.2 2.3 Tg (min) 45 53 79 93 117 h o c t e n e . l , a d d e d / - - 7.2 - 3.8 - 2.8 - 1.9
hC8 ,formed in synth.
normalized composition of Cs-olefins with internal double bonds (in tool%) trans-octene-2 38 (48) 31 (31) 29 (30) 28 (29) cis-octene-2 32 (32) 15 (17) 12 (14) 12 (13) trans-octene-3 13 (9) 31 (30) 34 (32) 34 (33) cis-octene-3 7 (5) 8 (8) 8 (8) 8 (8) trans+cis-octene-4 10 (6) 15 (14) 17 (16) 18 (17) total 100 I00 I00 100 100 100 100 100
the r ange C1-C20+ us ing a t e m p e r a t u r e p r o g r a m f r o m - 8 0 to 275°C. I n o r g a n i c c o m p o u n d s were a n a l y z e d by a m u l t i c o l u m n swi tch ing G C - t e c h n i q u e .
3. R e s u l t s a n d d i s c u s s i o n
3.1. KINETIC SCHEME OF CO-CONVERSION AND EVALUATION OF EXPERIMENTS
F r e q u e n t l y the ra te o f F T synthes is is quan t i f i ed as ra te o f ( C O + H2) -consu rnp- tion or C O - c o n s u m p t i o n [4-6]. This v iew does no t d i sc r imina te be tween C O used for m e t h a n e f o r m a t i o n , for cha in g r o w t h or fo r cha in in i t ia t ion.
M e t h a n e is t h e r m o d y n a m i c a l l y m u c h f a v o r e d as c o m p a r e d wi th h igher h y d r o - ca rbons u n d e r all FT- syn thes i s cond i t ions [7]. Formation ofaliphatic chains by CO- hydrogenation thus implies inhibition o f methane formation [8]. In t e r m s o f e l emen- tal processes , inh ib i t ion o f m e t h a n e f o r m a t i o n is v isual ized as inhibition o f the reac- tion o f associative desorption o f a methyl species together with a H-species:
CH~ + H* ~ CH4 + 2 . ,
where * ind ica tes c h e m i s o r b e d species and • deno tes a v a c a n t sur face site. Th is pos - tulate leads to the FT regime where the reaction o f chemisorbed hydrocarbon species with each other is dominant.
The fo l lowing kinetic scheme dis t inguishes the three m o d e s o f CO-consumption: for chain growth, chain initiation andmethane formation.

226 H. Schulz et aL / Specific inhibition, kinetic principle o f FTsynthesis
C O (1!, CO* (2) C* (3) C H * (4) CH~ (5) CH; (6_~) C H 4
(7)l+R" (8)1+CH;
R - C H ~ C H 3 - C H ~
This kinetic scheme is much simplified, not taking into account detailed aspects of the Fischer-Tropsch reaction as chain branching, chain termination as olefin, par- affin or oxygen compound, readsorption of olefins and oxygenates, double bond shift in olefins and the generally to be expected chain length dependence of reaction rates of all kinds of organic surface species. It as well implies methane formation to occur only on the same kind of sites as chain growth.
At the stationary state of conversion, as pertinent for a continuously oper- ated well-mixed tank reactor, the rates (1) to (4) are equal (overall rate of CO- consumption),
rl ~ r2 ~ r3 z r 4.
Alternative pathways for the CH~ intermediate are the desorption as methane (reac- tion (6)) or the reaction with CH~ (reaction (8)) to form the ethyl intermediate,
r5 = r6 + r8.
Thus the overall rate of CO-consumption (rl) is split into the rates of consumption for chain growth ( r 7 ) , chain start ( rs ) and methane formation ( r6 ) ,
rl = r6 + 1"7 -~- r8 •
These individual rates can be calculated from the experimental values of CO-con- sumption and product composition evaluated as fractions of carbon in CH4, in CH2-groups and in one CH3-group per molecule of all the organic product com- pounds.
Inhibition as postulated to govern the overall and individual rates of the Fischer-Tropsch conversion is assumed to depend predominantly on carbon mon- oxide partial pressure and temperature (and in the case of iron catalysts also car- bide formation and alkali surface spreading). Accordingly the results of synthesis experiments with cobalt catalysts are evaluated below as reaction rates in the assumed kinetic scheme with reaction temperature and CO partial pressure as parameters.
3.2. RATE OF CO-CONSUMPTION
Partial pressure of carbon monoxide generally reduces the rate of CO-consump- tion, as recognized in several proposed kinetic equations [4-6]. The experimental dependency, determined in this investigation, where the other important partial pressures are kept constant, is shown in fig. 1, together with the influence of reac- tion temperature, at constant values of all the main partial pressures. With the two

H. Schulz et al. / Specific inhibition, kinetic principle of FT synthesis 227
=" 0.3 o
E .~ o.2
o W O ~
O ~ O A o E
m
_•.....-...- Co-Re-Aerosil (18o'c. pH2=5.obar. PH20=2.2bar)
Co-Mg-Th-Aerosil o. , . ,oo , o. ,o . , , , , ) o ,
I I I I I I I /
0 10 20 30 40
CO Partial pressure, bar
0.3
0.2 J Co-Mg-Th-Aerosil
(pco,2.5bar. pN2=$.0bar, pH20=l .Sbsff)
i I i I i I
180 200 220
Temperature, °C
Fig. 1. Influence of CO partial pressure (left) and reaction temperature (right) on the rate of CO-con- sumption (per gram cobalt); slurry phase synthesis with two different cobalt catalysts (see section
"experimental").
cobalt catalysts applied, the reaction rate decreases by a factor of about 4, when increasing the CO partial pressure from 0.5 to ca. 10 bar. Towards higher CO par- tial pressure (up to 40 bar), the reaction rate decreases only slightly, indicating no further inhibitive effects, caused by the strongly adsorbed CO-reactant. Reaction rate decreases with temperature as anticipated.
3.3. METHANE SELECTIVITY
The pioneering observation by Sabatier and Senderens [9] of methane formation via CO-hydrogenation on the metals nickel and cobalt at a comparatively high reaction temperature preceded the "gasoline synthesis" by Fischer and Tropsch [I0], which, however, only resulted in satisfactory yields of liquid products after successful "catalyst engineering" by Fischer and Koch [11] leading to the highly active "standard" precipitated cobalt catalyst 100Co-18ThOz-100Kieselguhr, which allowed initial reaction temperatures below 180°C and suppression of ample methane formation.
Inhibition of methane formation is simply quantified as methane selectivity, defined as the fraction of carbon in methane in relation to the carbon in all of the organic product compounds. Referring to the above kinetic scheme, methane selec- tivity (ScH4) is equal to the ratio of the rates r6 and r],
r6 SCH4 = - - .
rl
Methane inhibition is additionally specified by the ratio of reaction rates of methane formation (r6) to that of CO consumption for starting chains (r8): r6/rs.
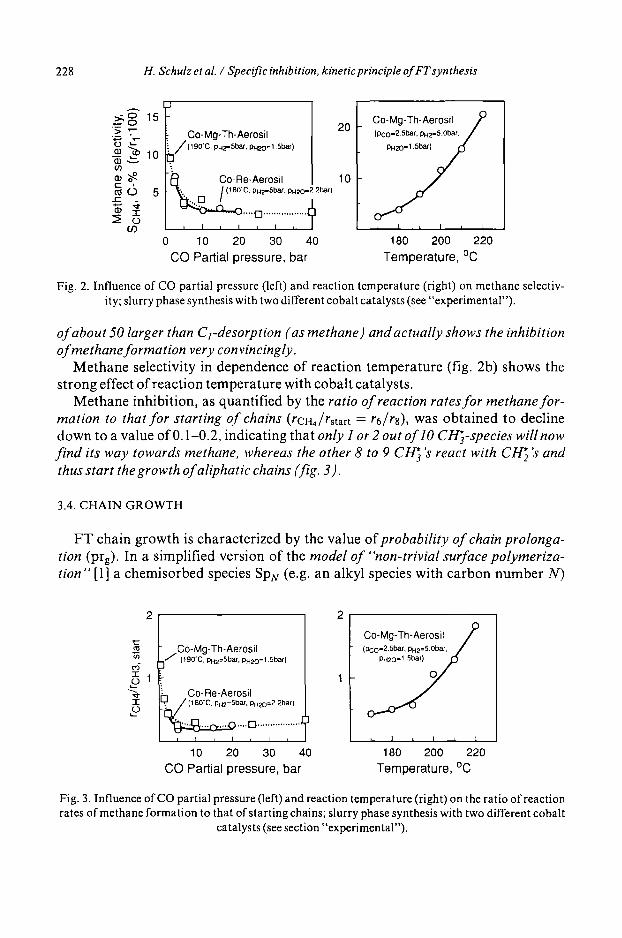
Methane selectivity decreases in the range of experiments of this work with increasing CO partial pressure from about 17 to 2 C% (fig. 2a). The only 2 C% selectivity of methane indicates the probability of chain prolongation to be by a factor
228 H. Schulz et al. / Specific inhibition, kinetic principle of FTsynthesis
o
O3
t-
O3
15 Co-Mg-Th-Aerosil
.1/{190"C. pH2=5bar, PH20=l .Sbar) 1 o
Co-Re-Aerosil 5 ~-. / (18o'c. p.2-s~,. ~.~o=22~,)
. . . . . . .
10 20 30 40 CO Partial pressure, bar
20
10
Co-Mg-Th-Aerosil ? (Pco-257. r. PH2 =5.0bar, J
Y n I i I I I
180 200 220 Temperature, °C
Fig. 2. Influence of CO partial pressure (left) and reaction temperature (right) on methane selectiv- ity; slurry phase synthesis with two different cobalt catalysts (see "experimental").
of about 50 larger than C l-desorption (as methane) and actually shows the inhibition of methane formation very convincingly•
Methane selectivity in dependence of reaction temperature (fig. 2b) shows the strong effect of reaction temperature with cobalt catalysts•
Methane inhibition, as quantified by the ratio of reaction rates for methane for- mation to that for starting of chains (rCHa/rstart = r6/rs), was obtained to decline down to a value of 0.1-0.2, indicating that only I or 2 out of lO CI-l~3-species will now find its way towards methane, whereas the other 8 to 9 CI-I~ 's react with Ct-1~2 's and thus start the growth of aliphatic chains (fig• 3).
3.4. CHAIN GROWTH
FT chain growth is characterized by the value of probability of chain prolonga- tion (prg). In a simplified version of the model of "non-trivial surface polymeriza- tion" [1] a chemisorbed species SpN (e.g. an alkyl species with carbon number N)
2 2
t~
e6
3E
jCo-Mg-Th-Aerosil (190"C. PH2=SbaL pN20=l.5baf)
] Co-Re-Aerosil ] ,.~(180"C. pH2=5ba,r, PH20=2.2bar)
~ . . . , E 3 ................. [ I I n I I I n
10 20 30 40 CO Partial pressure, bar
r I I ! i I
180 200 220 Temperature, °C
Fig. 3. Influence of CO partial pressure (left) and reaction temperature (right) on the ratio of reaction rates of methane formation to that of starting chains; slurry phase synthesis with two different cobalt
catalysts (see section "experimental").

H. Schulz et al. / Specific inhibition, kinetic principle of FTsynthesis 229
has two options to react: (1) by chain prolongation (to species SPN+I) or (2) by de- sorption (to product PN)
PN ) Sp N //~ desorption
N,~ growth
SpN+I
In the model which refers to the "gradientless" steady state of conversion, the total of product moles is normalized to 100 and the probabilities of a chain growth inter- mediate SpN to desorb or to grow further are equal to 1,
prg ÷ prd = 1.00.
Assuming chain growth probability and accordingly chain termination probabil- ity to be independent of surface species carbon number, leads to the simple Flory polymerization kinetics as describing the product composition by only one param- eter, the chain propagation probability Pgr. For evaluation of the rules of product formation in Fischer-Tropsch synthesis, the basic Flory kinetics are now extended for chain branching and product desorption in several modes as paraffins, olefins, a]kanals and alkanols and each sort of surface reaction has basically to be regarded as carbon number dependent. Additionally, reversibility of product desorption may prevail and further side reactions may occur. These deviations from the ideal model are then discriminated and quantified by extending the model adequately.
Under the condition of our slurry phase conversion experiments, composition of the gas/vapor product was representative up to carbon number Nc = 17. At higher values of Nc increasingly larger fractions of the product did accumulate in the solvent (squalane). For chain propagation probability quantification, the molar carbon-number-product-fractions in the range Nc7 to Nc,5 were used.
Fig. 4 shows probability of chain growth in dependence of CO partial pressure. The value for chain termination (prdes = p r g - 1) declines from ca. 20% at
Q .
~ . . ¢ -
12_.~ t - " O
100 100
~ .....12" ................. [
90 -Mcl-Th-Aerosil . . . , = , = ,
(180"C, pH2,=5baL P N 2 0 = 2 . 2 b a t )
70 , I , I , i , 70 10 20 30 40
CO Partial pressure, bar
90
80 ( P c o = 2 . S b ~ , p H Z = 5 0 b a L
p H 2 0 = 1 . 5 b a r )
I i I I I
180 200 220
Temperature, °C
Fig. 4. Influence of CO partial pressure (left) and reaction temperature (right) on probability of chain growth; slurry phase synthesis with two different cobalt catalysts (see section "experimental").

230 H. Schulz et al. / Specific inhibition, kinetic principle of FT synthesis
Pco = 3 bar to about 4% at about Pco = 20 bar. This proves the inhibition of chain termination by carbon monoxide. At levels of CO partial pressure higher than 20 bar no further increase of inhibition is noticed.
Appreciable deviations from simple Flory polymerization kinetics are to be dis- tinguished in experimental diagrams of chain growth probability as a function of carbon number (see fig. 5). Flory kinetics would just represent horizontal lines. As compared to this, several deviations are pertinent. A typical curve, as obtained for cobalt at "normal pressure" (ca. 1 bar, H 2 / C O = 2 reproduced from ref. [12]), is also being represented.
The curve for "normal pressure synthesis" (fig. 5, right) is characterized by a low value of prgl (high methane selectivity), a decline of prg in the range of carbon number Nc -- 2 to Nc = 4 (which is attributed to a decrease of the desorption rate constant with increasing carbon number) and a slight increase of prg in the range of Nc = 12 to Nc = 15 which is explained by increasing readsorbability of product molecules. Readsorption of product olefins has been earlier materialized by addi- tion of radioactively labelled olefins to the synthesis gas [13] for cobalt catalysts. Evidently, chain length of the olefins should have an effect on olefin readsorption. The impact of intraparticle diffusion on olefin readsorption with supported ruthe- nium catalysts has been recently reported convincingly by Madon and Iglesia [14]. However, different kinetics are to be expected with small catalyst particles in the slurry phase (as in this investigation). Secondary olefin reactions in FT-synthesis are of course strongly dependent on reaction conditions and as well on catalyst composition. Iron has earlier been observed to exhibit a low tendency for second- ary olefin conversions [13]. Accordingly, ct-olefins (the main primary FT-products) can be selectively obtained on iron catalysts [15]. They are the main constituents in the products of the hydrocoal and the synthol processes. Differently shaped curves are obtained by increase of the CO partial pressure (see fig. 5 left): In particu- lar an increase of prg is observed in the range Nc -- 3 to Nc -- 6 and higher Nc's. The first explanation is again, additional contributions to prg as resulting from rever- looF
~6~80
I R I o pco= 2.4 b~
100
80
60
40
~ 2 (fixed bed reactor), Ref[12] , , , i I , , , i I i i , , I
5 10 15 5 10 15 Carbon number, N C Carbon number, N C
Fig. 5. Chain growth probability prg as a function of carbon number for FT slurry phase experiments at varied CO partial pressure (left) and FT fixed bed experiments (right) [12] with cobalt catalysts.
H. Schulz et al. / Specific inhibition, kinetic principle of FT synthesis 231
sibility o f olefin desorption. Thus increasing CO partial pressure modulates the F T product distribution not only via inhibition o f chain termination but as well via pro- rooting olefin readsorption.
3.5. OLEFINICITY, SECONDARY HYDROGENATION AND OLEFIN ISOMERIZATION
Desorpt ion of hydrocarbons from Fischer-Tropsch sites generally produces a- olefins and paraffins in a molar ratio of 70-80 to 30-20% as pictured in the follow- ing mechanistic scheme:
- H ~ R - C H = C H 2 ca. 7 0 - 8 0 %
R - C H z - C H ~
,,,~14. R - C H E - C H 3 + 2 , ca. 2 0 - 3 0 %
This pr imary selectivity has been previously observed with iron and cobalt cat- alysts as well [15-17] and was deduced from the indicatively shaped carbon number plots of olefins among the hydrocarbons and of t~-olefins in the fraction of olefins as well. Wi th iron catalysts primary olefin selectivity increased slightly with reac- tion temperature and decreased slightly with total pressure [15]. FT synthesis with cobalt was more sensitive for secondary olefin reactions than synthesis with iron [17].
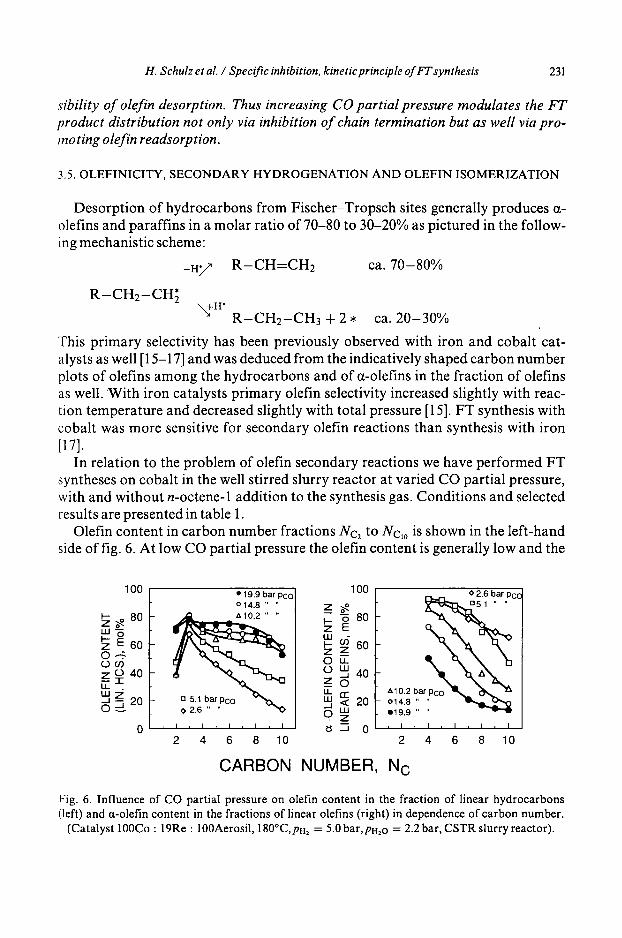
In relation to the problem of olefin secondary reactions we have performed FT syntheses on cobalt in the well stirred slurry reactor at varied CO partial pressure, with and without n-octene-1 addition to the synthesis gas. Condit ions and selected results are presented in table 1.
Olefin content in carbon number fractions Nc2 to Nc,0 is shown in the left-hand side of fig. 6. At low CO partial pressure the olefin content is generally low and the
100 100
z ~ 80 IJJ z 60 oct) ZTO 40 if- . Nz O ~..~ 20
• 19.9 bat PCO o 14.8 " "
, I I I I I
2 4 6 8 10
CARBON
z ~
O u_
_ZO
02.6 bar Pc(
A10.2 b, a r ~ ~ o14.8 o19.9 ....
I I I I I
2 4 6 8 10
NUMBER, N C
Fig. 6. Influence of CO partial pressure on olefin content in the fraction of linear hydrocarbons (left) and ct-olefin content in the fractions of linear olefins (right) in dependence of carbon number.
(Catalyst 100Co : 19Re : 100Aerosil, 180°C,pH2 = 5.0 bar, pH2o = 2.2 bar, CSTR slurry reactor).

232 H. Schulz et al. / Specific inhibition, kinetic principle of FTsynthesis
shape of the curve with a maximum at C3 proves strong secondary hydrogena- tion. At high CO partial pressure in the range Nc = 3 to Arc = 7 the olefin content represents almost a horizontal line proving primary olefinicity.
The curves in the right-hand side of fig. 6, of a-olefin content in all n-olefin iso- mers, as a function of carbon number, are low at high olefinicity.
It is concluded from these results that on cobalt highpartialpressure of CO inhi- bits secondary olefin hydrogenation, however favors secondary double bond shift (in contrast to the behavior of iron catalysts [15]).
This conclusion has been verified via experiments with and without octene-I addition to the synthesis gas (see table 1). The degree of added octene-1 conversion is shown in the left-hand side of fig. 7 (indicating an increase with increasing CO- partial pressure) and the selectivity of added octene-1 conversion is shown in the right-hand side of fig. 7. At Pco = 20 bar selectivity of secondary hydrogenation is inhibited to almost zero, whereas selectivity of secondary olefin isomerization increases to ca. 60%. At these conditions olefin incorporation into growing chains attains about 30%. The almost equal compositions of the C8 olefin internal iso- mers produced via synthesis from CO and H2 and the respective olefins obtained via double shift (see table 1) lead to the conclusion that pathways of secondary FT olefin conversion are quite comparable to reactions of the added olefin in these experiments.
These results differ from those obtained by Madon and Iglesia [14] with impreg- nated ruthenium catalysts. In their experiments strong intraparticle mass transfer was observed, governing olefin readsorption for further chain growth. Increasing paraffinicity with carbon number was attributed to multiple readsorption. Correla- tions with double bond shift were not reported.
The following kinetic scheme is suited to discuss the interrelations between sec- ondary olefin hydrogenation and isomerization:
U_ O Z o_ 03 rr LU > Z 0 0
100
80
o 60
cl
4o ffl
J ~60
___°4o
I t r t 0
0 5 10 15 20 0
x / Incorporation " ~ _ _ ..A.- - -&
5 10 15 20
PARTIAL PRESSURE OF CO, bar
Fig. 7. Influence of CO partial pressure on conversion (left) and selectivity of hydrogenation, double bond isomerization and incorporation (right) for n-octene-1 which had been added to the synthesis gas. (Catalyst 100Co: 19Re:100Aerosil, 180°C,pH~ = 5.0 bar, pH2o = 2.2 bar, CSTR slurry reactor.)

H. Schulz et al. / Specific inhibition, kinetic principle of FT synthesis 233
R--CH2--CH~CH2 ~ +H* ~, R--CH2--CH2--CH[
R--CH~CH--CH3 ~-H*'~ R--CH2--CH--CH3 +H* ~ R--CH2--CH2--CH3 +H*
R'--CH~CH--CH2--CH3 ~ ~ R--CH--CH2--CH3 +H/~
The secondstep of H-addition to form the paraffin is more demanding andmuch inhib- ited by CO. The first step of H-addition is comparatively easy and highly reversible, leading to a stepwise shift of the double bond from the chains end to the interior of the chain.
Inhibition of secondary hydrogenation by CO is not associated with a signifi- cant increase of primary olefinicity of the FT product (compare fig. 7). It must therefore be concluded that secondary olefin hydrogenation on cobalt proceeds.on different sites than FT chain growth.
Another conclusion to be drawn from these experiments is that olefin readsorp- tion on FT sites for further chain growth, generally occurs at the terminating C-atom of the chain, because no significant change in amount and distribution of the branched isomers with carbon numbers higher than the added olefin (Nc > 8) have been observed. This leads to the picture of hydrocarbon chains being very preferen- tially terminally bonded to the surface which again should be explained by strong competitive chemisorption, steric demands and respective inhibition.
4. C o n c l u d i n g r emarks
The concept of Fischer-Tropsch synthesis, being basically determined through specific inhibition of elemental reaction steps, has been substantiated through the results of this investigation. Several detailed conclusions have been drawn which are valuable for understanding the Fischer-Tropsch regime and its side reactions and might as well be useful for concepts of catalyst engineering and improvement of technical processes.
Acknowledgement
Part of this work has been supported by the Deutsche Forschungsge- meinschaft, SFB 250, which is gratefully acknowledged.
References
[1] H. Schulz, K. Beck and E. Erich, Proc. Methane Conversion Symposium, Auckland 1987, Stud. Surf. Sci. Catal., Vol. 36, eds. D. Bibby, C. Chang, R. Howe and S. Yurchak (Elsevier, Amsterdam, 1988) p. 457.
[2] H. Schulz, E. van Steen and M. Claeys, Proc. Natural Gas Conversion Symp., Sydney 1993.

234 H. Schulz et al. / Specific inhibition, kinetic principle of FTsynthesis
[3] H. Schulz and S. Nehren, Erd61 und Kohle-Erdgas-Petrochemie 39 (1986) 93. [4] I. Yates and C. Satterfield, Energy and Fuels 5 (1991) 168. [5] B. Wojciechowski, Catal. Rev. Sci. Eng. 30 (1988) 678. [6] M. Dry, in: Catalysis Science and Technology, Vol. 1, eds. J. Anderson and M. Boudard
(Springer, Berlin, 1984). [7] H. Schulz, in: Ullmanns Encycklopddie der technischen Chemie, Vol. 14, 4th Ed. (Verlag
Chemic, Weinheim, 1977) p. 329. [8] H. Schulz, Erd61 und Kohle 30 (1977) 123. [9] P. Sabatier and J.B. Senderens, J. Soc. Chem. Ind. 21 (1902) 504.
[I 0] F. Fischer and H. Tropsch, Brennstoff-Chem. 7 (1926) 97. [1 I] F. Fischer and H. Koch, Brennstoff-Chem. 13 (1932) 61. [ 12] H. Pichler, H. Schulz and H. Elstner, Brennstoff-Chem. 48 (1967) 78. [ 13] H. Schulz, B. Rao and H. Elstner, Erd61 und Kohle 22 (1970) 651. [14] R.J. Madon and E. Iglesia, J. Catal. 139 (I 993) 576. [15] H. Schulz and H. G6kcebay, in: Catalysis of Organic Reactions, ed. J. Kosak (Dekker, New
York, 1984)p. 153. [I 6] H. Schulz, E. Erich, H. Gorre and E. van Steen, Catal. Lett. 7 (1990) 157. [17] H. Schulz and H. Achtsnit, Rev. Portug. Quim. 19 (I 977) 317.




















