
Isolation of a human-like antibody fragment (scFv) that neutralizes ricin biological activity
Upload
independentCategory
view
0download
0
DNA Repair 4 (2005) 271–277
Brief report
Shiga toxin 1 and ricin inhibit the repair of H2O2-induced DNA singlestrand breaks in cultured mammalian cells
Piero Sestilia,b,∗, Roberta Alfieric, Domenica Carnicellid, Chiara Martinellib, Luigi Barbierid,Fiorenzo Stirped, Mara Bonellic, Pier Giorgio Petroninic, Maurizio Brigottid
a Istituto di Farmacologia e Farmacognosia, Universit`a degli Studi di Urbino “Carlo Bo”, Via S. Chiara 27, 61029 Urbino (PU), Italyb Istituto di Ricerca sulla Attivit`a Motoria, Universita degli Studi di Urbino “Carlo Bo”, 61029 Urbino (PU), Italy
c Dipartimento di Medicina Sperimentale, Sezione di Patologia Molecolare e Immunologia, Universit`a degli Studi di Parma, 43100 Parma, Italyd Dipartimento di Patologia Sperimentale, Universit`a degli Studi di Bologna, 40126 Bologna, Italy
Received 7 July 2004; accepted 9 September 2004Available online 18 November 2004
Abstract
, but alsof
lesionsg pletion ofc
tween direct( articular,w ed tissuesi©
K
1
tdttdt
ii
oopscat-facetic A
d that
wn in
ndliald al-ry
1d
A growing body of evidence suggests that ribosome-inactivating proteins (RIPs) remove adenine moieties not only from rRNArom DNA – an effect leading to DNA damage in cultured cells.
We herein report that two distinct RIPs of bacterial (shiga toxin 1, Stx1) and plant (ricin) origin, inhibit the repair of the DNAenerated by hydrogen peroxide in cultured human cells. This effect is unrelated either to inhibition of protein synthesis or to deellular antioxidant defenses and is likely to derive from direct interactions with cellular DNA repair machinery.Therefore, the genotoxicity of these toxins on mammalian cells seems to be a complex phenomenon resulting from the balance be
DNA damaging activity), indirect (DNA repair inhibition) effects and the eventual presence of other DNA damaging species. In pith regard to Stx1, it could be hypothesized that Stx-producing bacteria increase the risk of transformation of surrounding, inflam
n the course of human infections.2004 Elsevier B.V. All rights reserved.
eywords:Ribosome inactivating proteins; DNA damage; DNA repair; Hydrogen peroxide; Genotoxicity
. Introduction
Shiga toxin (Stx) fromShigella dysenteriaeand the struc-urally and functionally related cytotoxins (Stx1, Stx2) pro-uced by someEscherichia coliserotypes have a main role in
he pathogenesis of human diseases such as bacillary dysen-ery, enterohemorrhagic colitis and hemolytic uremic syn-rome[1]. Ricin, fromRicinus communisseeds, is highly
oxic and has been used in criminal and terrorist acts[2,3].These toxins belong to the large family of ribosome-
nactivating proteins (RIPs) with RNA-N-glycosidase activ-ty which irreversibly inactivate eukaryotic ribosomes by re-
∗ Corresponding author. Tel.: +39 0722 303414; fax: +39 0722 303401.E-mail address:[email protected] (P. Sestili).
moving a specific adenine from a highly conserved lpresent in 28S rRNA[1,2]. Both Stx1 and ricin, known atype 2 RIPs, consist of a single A chain containing thealytic site and a second B chain which binds to cell surreceptors mediating endocytosis. In Stx1, the enzymachain is non-covalently associated to five B chains.
For almost two decades it has been largely assumeRIPs acted only on 28S rRNA within ribosomes[4,5]. Lately,however, all plant RIPs and Stxs tested have been shovitro to remove adenine residues from DNA[6,7] leading tothe formation of apurinic sites.
More recently[8], ricin and Stx1 have also been fouto induce DNA lesions in human umbilical vein endothecells (HUVEC), as evaluated by alkaline-halo assay ankaline filter elution. The nature of the nuclear DNA inju
568-7864/$ – see front matter © 2004 Elsevier B.V. All rights reserved.oi:10.1016/j.dnarep.2004.09.007

272 P. Sestili et al. / DNA Repair 4 (2005) 271–277
observed in HUVEC is consistent with the enzymatic activ-ity (adenine release) of the toxins on nucleic acids in vitro,involving mainly the formation of apurinic sites.
Since nuclear toxicity of ricin and of Stx1 might implicatetight interactions with DNA, we have investigated whetherStx1 and ricin might interfere with other mechanisms regu-lating DNA homeostasis, such as DNA repair. At this purpose,we have studied the effect of sub-DNA-damaging concentra-tions of Stx1 and ricin on the ability of HUVEC and U937human promonocytic cells to repair the DNA lesions inflictedby the potentially genotoxic agent hydrogen peroxide and bythe alkylating agent methyl methane sulphonate (MMS).
2. Materials and methods
2.1. Reagents
Ricin (the two-chain RIP from the seeds ofRicinus com-munis) was purified as described by Nicolson et al.[9]. Stx1,obtained from a prototype Stx1 producerEscherichia coliC600 (H19J) gently supplied by Dr. Alison O’Brien (Depart-ment of Microbiology and Immunology, USU of the HealthSciences, Bethesda, U.S.A.), was purified as described byRyd et al.[10]. l-[4,5-3H]leucine (2.5 mCi/mmol) was ob-t K.).D fromC s ofa l Co.(
2
inga and5 no-c up-p etalb eo ret lturem A( .
2a
oa aks( pro-p erera DTAa oateds erei A,
∼pH 13, for 20 min), washed and stained with 10�g/mlethidium bromide. DNA was visualized using an OlympusBX 51 fluorescence microscope (Olympus, Tokio, Japan) andthe resulting images were taken and processed with a Hama-matsu CCD 5985 camera (Hamamatsu Italy S.p.a., Milan,Italy) coupled with a Macintosh computer using the pub-lic domain NIH Image program. The level of DNA singlestrand breakage was quantified by calculating the nuclearspreading factor (NSF) value, which represents the ratio be-tween the area of the halo and that of the nucleus, from about50 randomly selected cells/experiment/treatment condition.Data are expressed as relative NSF (rNSF), calculated by sub-tracting the NSF of control cells from that of treated cells. Insome experiments DNA damage and repair have been deter-mined with the comet microgel assay, which has been carriedout as described in[12].
2.4. Determination of cell survival and apoptosis
Cell survival was determined using a growth inhibitionassay described in[13]. After treatments HUVEC were har-vested by trypsinization, plated in duplicate at a density of5× 104/35 mm dish, and grown for 5 days at 37◦C. Cellproliferation and viability were determined by counting thecells after proper dilution in a hemocytometer; cell death wasm lue.A xami-n inedc
2
t ratioo wasmi letem here[
2p
ctsw Aebi[
3
xicmi C.IV -p le e
ained from Amersham Pharmacia Biotech (Bucks, U.isposable plastics for laboratory use were obtainedostar (Broadway, Cambridge, MA, U.S.A.). Reagentnalytical grade were purchased from Sigma ChemicaSt. Louis, MO, U.S.A.).
.2. Cell cultures and treatment conditions
HUVEC were maintained in M199 medium containntibiotics, 1.4 mM glutamine, 10% fetal bovine serum0�g/ml endothelial cell growth factor. Human promoytic U937 cells were cultured in RPMI 1640 medium slemented with antibiotics, 1.2 mM glutamine and 10% fovine serum. Cells were grown at 37◦C in an atmospherf 95% air and 5% CO2. Treatments with MMS, exposu
o RIPs and repair incubations were performed in cuedium; treatments with H2O2 were performed in Saline
0.145 M NaCl, 5 mM KCl, 10 mM NaHCO3, 5 mM glucose)
.3. Determination of DNA damage and repair by thelkaline-halo assay
The alkaline-halo assay described in[11] was used tssess either the initial level of DNA single strand breSSBs), or the residual proportion of SSBs following apriate repair times. Briefly, after treatments the cells wesuspended at 2.0× 104 cells/100�l in 1.5% low-meltinggarose in phosphate-buffered saline containing 5 mM End immediately sandwiched between an agarose-clide and a coverslip. After complete gelling the slides wmmersed in an alkaline buffer (0.1 M NaOH/1 mM EDT
onitored by counting cells permeated by 0.1% trypan bpoptosis was assessed by fluorescence microscopy eation of the nuclear morphology of Hoechst 33342-staells.
.5. Determination of protein synthesis
The rate of protein synthesis expressed as the percenf nmoles/mg of protein in treated versus control cellseasured by the incorporation of 0.4 mMl-[3H] leucine dur-
ng a 10-min incubation of the cell monolayers in compinimal essential medium, as described in detail elsew
14].
.6. Determination of catalase and glutathioneeroxidase (GPx) activity
Catalase and GPx (total activity) activities in cell extraere assayed spectrophotometrically by the methods of
15] and Lawrence and Burke[16], respectively.
. Results
Hydrogen peroxide is a potentially cyto- and genotoetabolite found in all aerobic cells[17] and it efficiently
nduces DNA SSBs in mammalian cells, including HUVEndeed, using the alkaline halo assay[11], nuclei from HU-EC treated with 150�M H2O2 for 10 min showed, as comared to nuclei from control cells (Fig. 1A), a marked radiaxpansion (halo) of DNA (Fig. 1B), a phenomenon indicativ
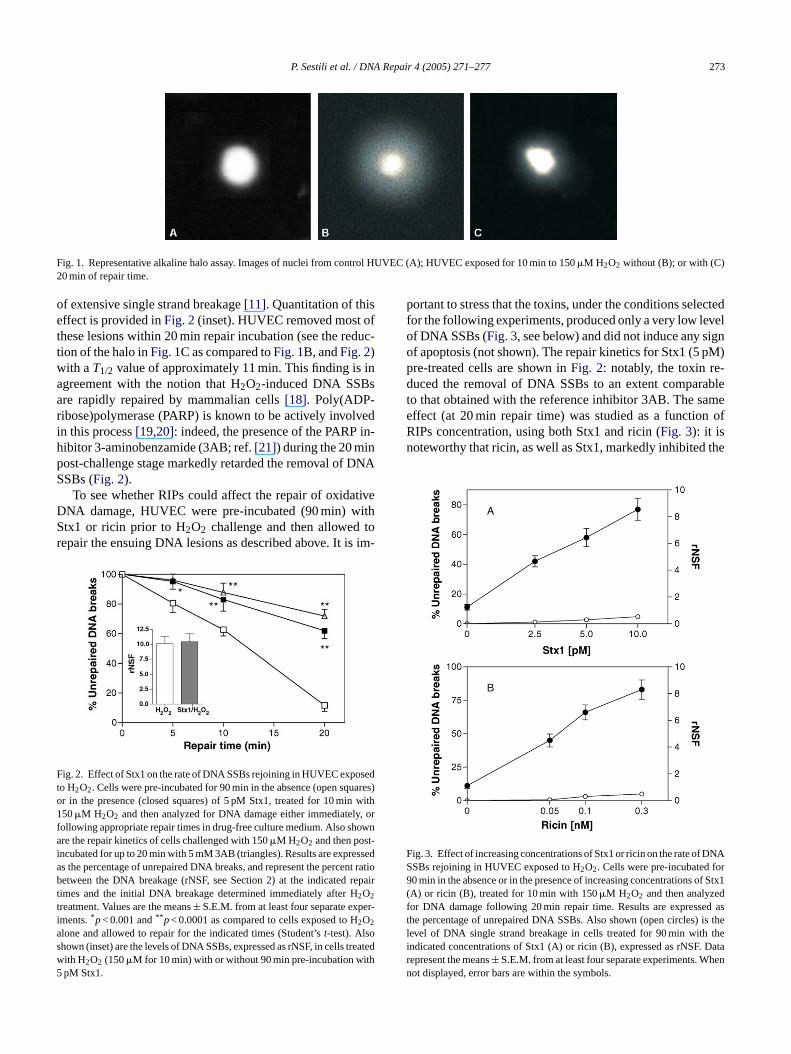
P. Sestili et al. / DNA Repair 4 (2005) 271–277 273
Fig. 1. Representative alkaline halo assay. Images of nuclei from control HUVEC (A); HUVEC exposed for 10 min to 150�M H2O2 without (B); or with (C)20 min of repair time.
of extensive single strand breakage[11]. Quantitation of thiseffect is provided inFig. 2(inset). HUVEC removed most ofthese lesions within 20 min repair incubation (see the reduc-tion of the halo inFig. 1C as compared toFig. 1B, andFig. 2)with aT1/2 value of approximately 11 min. This finding is inagreement with the notion that H2O2-induced DNA SSBsare rapidly repaired by mammalian cells[18]. Poly(ADP-ribose)polymerase (PARP) is known to be actively involvedin this process[19,20]: indeed, the presence of the PARP in-hibitor 3-aminobenzamide (3AB; ref.[21]) during the 20 minpost-challenge stage markedly retarded the removal of DNASSBs (Fig. 2).
To see whether RIPs could affect the repair of oxidativeDNA damage, HUVEC were pre-incubated (90 min) withStx1 or ricin prior to H2O2 challenge and then allowed torepair the ensuing DNA lesions as described above. It is im-
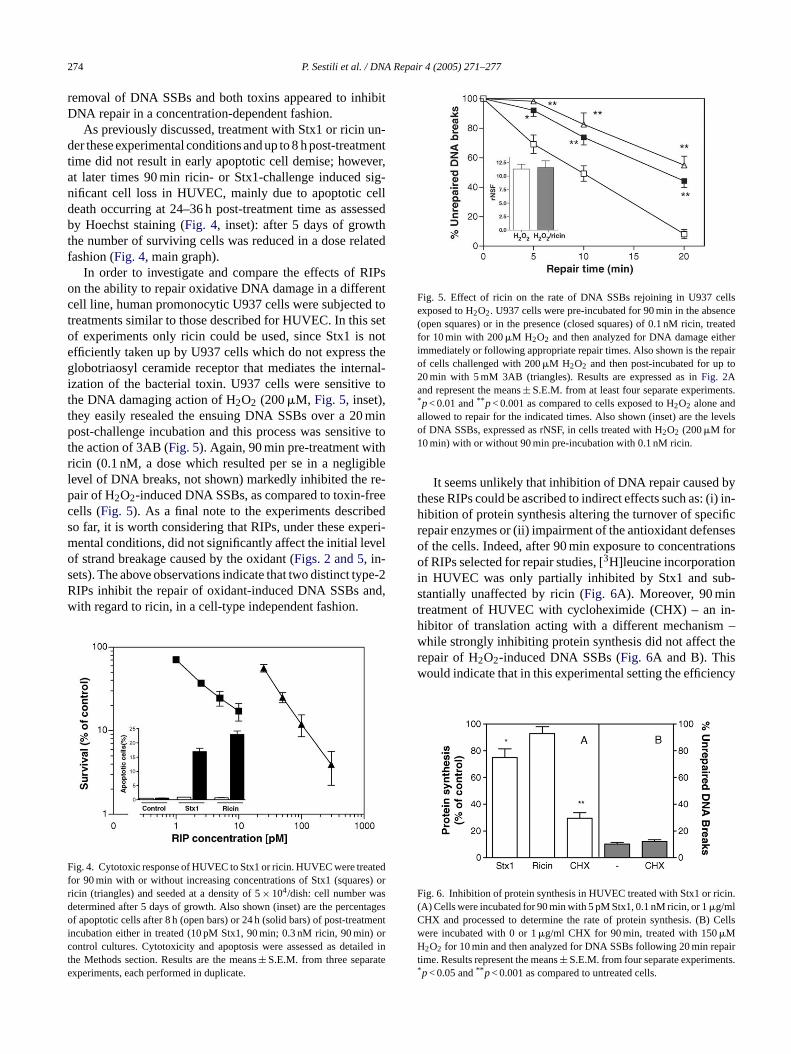
F sedt ares)o with1 y, orf owna t-i sseda nt ratiob irtt er-ias reatedw5
portant to stress that the toxins, under the conditions selectedfor the following experiments, produced only a very low levelof DNA SSBs (Fig. 3, see below) and did not induce any signof apoptosis (not shown). The repair kinetics for Stx1 (5 pM)pre-treated cells are shown inFig. 2: notably, the toxin re-duced the removal of DNA SSBs to an extent comparableto that obtained with the reference inhibitor 3AB. The sameeffect (at 20 min repair time) was studied as a function ofRIPs concentration, using both Stx1 and ricin (Fig. 3): it isnoteworthy that ricin, as well as Stx1, markedly inhibited the
F NASSBs rejoining in HUVEC exposed to H2O2. Cells were pre-incubated for90 min in the absence or in the presence of increasing concentrations of Stx1(A) or ricin (B), treated for 10 min with 150�M H2O2 and then analyzedfor DNA damage following 20 min repair time. Results are expressed asthe percentage of unrepaired DNA SSBs. Also shown (open circles) is thelevel of DNA single strand breakage in cells treated for 90 min with theindicated concentrations of Stx1 (A) or ricin (B), expressed as rNSF. Data
ig. 2. Effect of Stx1 on the rate of DNA SSBs rejoining in HUVEC expoo H2O2. Cells were pre-incubated for 90 min in the absence (open squr in the presence (closed squares) of 5 pM Stx1, treated for 10 min50�M H2O2 and then analyzed for DNA damage either immediatel
ollowing appropriate repair times in drug-free culture medium. Also shre the repair kinetics of cells challenged with 150�M H2O2 and then pos
ncubated for up to 20 min with 5 mM 3AB (triangles). Results are expres the percentage of unrepaired DNA breaks, and represent the perceetween the DNA breakage (rNSF, see Section2) at the indicated repa
imes and the initial DNA breakage determined immediately after H2O2
reatment. Values are the means± S.E.M. from at least four separate expments.*p< 0.001 and** p< 0.0001 as compared to cells exposed to H2O2
lone and allowed to repair for the indicated times (Student’st-test). Alsohown (inset) are the levels of DNA SSBs, expressed as rNSF, in cells t
ith H2O2 (150�M for 10 min) with or without 90 min pre-incubation withpM Stx1.rn
ig. 3. Effect of increasing concentrations of Stx1 or ricin on the rate of D
epresent the means± S.E.M. from at least four separate experiments. Whenot displayed, error bars are within the symbols.
274 P. Sestili et al. / DNA Repair 4 (2005) 271–277
removal of DNA SSBs and both toxins appeared to inhibitDNA repair in a concentration-dependent fashion.
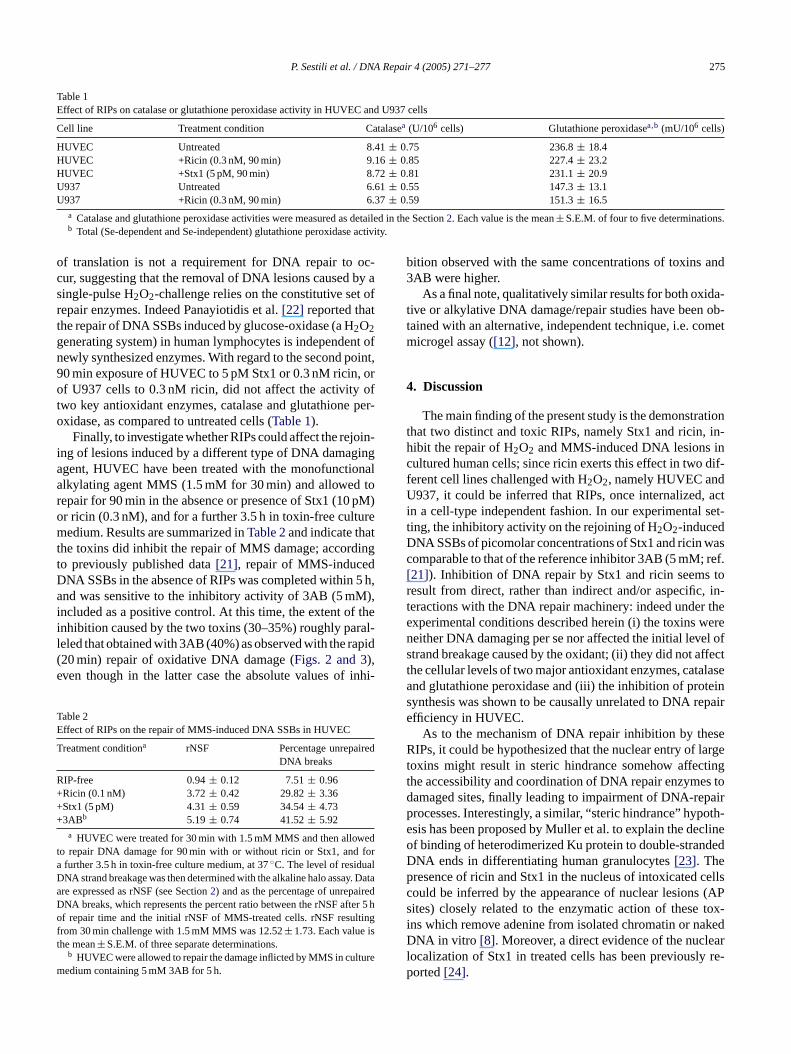
As previously discussed, treatment with Stx1 or ricin un-der these experimental conditions and up to 8 h post-treatmenttime did not result in early apoptotic cell demise; however,at later times 90 min ricin- or Stx1-challenge induced sig-nificant cell loss in HUVEC, mainly due to apoptotic celldeath occurring at 24–36 h post-treatment time as assessedby Hoechst staining (Fig. 4, inset): after 5 days of growththe number of surviving cells was reduced in a dose relatedfashion (Fig. 4, main graph).
In order to investigate and compare the effects of RIPson the ability to repair oxidative DNA damage in a differentcell line, human promonocytic U937 cells were subjected totreatments similar to those described for HUVEC. In this setof experiments only ricin could be used, since Stx1 is notefficiently taken up by U937 cells which do not express theglobotriaosyl ceramide receptor that mediates the internal-ization of the bacterial toxin. U937 cells were sensitive tothe DNA damaging action of H2O2 (200�M, Fig. 5, inset),they easily resealed the ensuing DNA SSBs over a 20 minpost-challenge incubation and this process was sensitive tothe action of 3AB (Fig. 5). Again, 90 min pre-treatment withricin (0.1 nM, a dose which resulted per se in a negligiblelevel of DNA breaks, not shown) markedly inhibited the re-p reec beds peri-m velos pe-2R nd,w
F atedf s) orr sd tageso tmenti ) orc iled int tee
Fig. 5. Effect of ricin on the rate of DNA SSBs rejoining in U937 cellsexposed to H2O2. U937 cells were pre-incubated for 90 min in the absence(open squares) or in the presence (closed squares) of 0.1 nM ricin, treatedfor 10 min with 200�M H2O2 and then analyzed for DNA damage eitherimmediately or following appropriate repair times. Also shown is the repairof cells challenged with 200�M H2O2 and then post-incubated for up to20 min with 5 mM 3AB (triangles). Results are expressed as inFig. 2Aand represent the means± S.E.M. from at least four separate experiments.*p< 0.01 and** p< 0.001 as compared to cells exposed to H2O2 alone andallowed to repair for the indicated times. Also shown (inset) are the levelsof DNA SSBs, expressed as rNSF, in cells treated with H2O2 (200�M for10 min) with or without 90 min pre-incubation with 0.1 nM ricin.
It seems unlikely that inhibition of DNA repair caused bythese RIPs could be ascribed to indirect effects such as: (i) in-hibition of protein synthesis altering the turnover of specificrepair enzymes or (ii) impairment of the antioxidant defensesof the cells. Indeed, after 90 min exposure to concentrationsof RIPs selected for repair studies, [3H]leucine incorporationin HUVEC was only partially inhibited by Stx1 and sub-stantially unaffected by ricin (Fig. 6A). Moreover, 90 mintreatment of HUVEC with cycloheximide (CHX) – an in-hibitor of translation acting with a different mechanism –while strongly inhibiting protein synthesis did not affect therepair of H2O2-induced DNA SSBs (Fig. 6A and B). Thiswould indicate that in this experimental setting the efficiency
F cin.(C CellswH pairt ts.*
air of H2O2-induced DNA SSBs, as compared to toxin-fells (Fig. 5). As a final note to the experiments descrio far, it is worth considering that RIPs, under these exental conditions, did not significantly affect the initial lef strand breakage caused by the oxidant (Figs. 2 and 5, in-ets). The above observations indicate that two distinct tyIPs inhibit the repair of oxidant-induced DNA SSBs aith regard to ricin, in a cell-type independent fashion.
ig. 4. Cytotoxic response of HUVEC to Stx1 or ricin. HUVEC were treor 90 min with or without increasing concentrations of Stx1 (squareicin (triangles) and seeded at a density of 5× 104/dish: cell number waetermined after 5 days of growth. Also shown (inset) are the percenf apoptotic cells after 8 h (open bars) or 24 h (solid bars) of post-trea
ncubation either in treated (10 pM Stx1, 90 min; 0.3 nM ricin, 90 minontrol cultures. Cytotoxicity and apoptosis were assessed as detahe Methods section. Results are the means± S.E.M. from three separaxperiments, each performed in duplicate.
ig. 6. Inhibition of protein synthesis in HUVEC treated with Stx1 or riA) Cells were incubated for 90 min with 5 pM Stx1, 0.1 nM ricin, or 1�g/mlHX and processed to determine the rate of protein synthesis. (B)ere incubated with 0 or 1�g/ml CHX for 90 min, treated with 150�M
2O2 for 10 min and then analyzed for DNA SSBs following 20 min reime. Results represent the means± S.E.M. from four separate experimenp< 0.05 and** p< 0.001 as compared to untreated cells.
P. Sestili et al. / DNA Repair 4 (2005) 271–277 275
Table 1Effect of RIPs on catalase or glutathione peroxidase activity in HUVEC and U937 cells
Cell line Treatment condition Catalasea (U/106 cells) Glutathione peroxidasea,b (mU/106 cells)
HUVEC Untreated 8.41± 0.75 236.8± 18.4HUVEC +Ricin (0.3 nM, 90 min) 9.16± 0.85 227.4± 23.2HUVEC +Stx1 (5 pM, 90 min) 8.72± 0.81 231.1± 20.9U937 Untreated 6.61± 0.55 147.3± 13.1U937 +Ricin (0.3 nM, 90 min) 6.37± 0.59 151.3± 16.5
a Catalase and glutathione peroxidase activities were measured as detailed in the Section2. Each value is the mean± S.E.M. of four to five determinations.b Total (Se-dependent and Se-independent) glutathione peroxidase activity.
of translation is not a requirement for DNA repair to oc-cur, suggesting that the removal of DNA lesions caused by asingle-pulse H2O2-challenge relies on the constitutive set ofrepair enzymes. Indeed Panayiotidis et al.[22] reported thatthe repair of DNA SSBs induced by glucose-oxidase (a H2O2generating system) in human lymphocytes is independent ofnewly synthesized enzymes. With regard to the second point,90 min exposure of HUVEC to 5 pM Stx1 or 0.3 nM ricin, orof U937 cells to 0.3 nM ricin, did not affect the activity oftwo key antioxidant enzymes, catalase and glutathione per-oxidase, as compared to untreated cells (Table 1).
Finally, to investigate whether RIPs could affect the rejoin-ing of lesions induced by a different type of DNA damagingagent, HUVEC have been treated with the monofunctionalalkylating agent MMS (1.5 mM for 30 min) and allowed torepair for 90 min in the absence or presence of Stx1 (10 pM)or ricin (0.3 nM), and for a further 3.5 h in toxin-free culturemedium. Results are summarized inTable 2and indicate thatthe toxins did inhibit the repair of MMS damage; accordingto previously published data[21], repair of MMS-inducedDNA SSBs in the absence of RIPs was completed within 5 h,and was sensitive to the inhibitory activity of 3AB (5 mM),included as a positive control. At this time, the extent of theinhibition caused by the two toxins (30–35%) roughly paral-leled that obtained with 3AB (40%) as observed with the rapid(e inhi-
TE
T ed
R+++
edt fora lD y. Dataa iredD ter 5 ho ltingf ist
turem
bition observed with the same concentrations of toxins and3AB were higher.
As a final note, qualitatively similar results for both oxida-tive or alkylative DNA damage/repair studies have been ob-tained with an alternative, independent technique, i.e. cometmicrogel assay ([12], not shown).
4. Discussion
The main finding of the present study is the demonstrationthat two distinct and toxic RIPs, namely Stx1 and ricin, in-hibit the repair of H2O2 and MMS-induced DNA lesions incultured human cells; since ricin exerts this effect in two dif-ferent cell lines challenged with H2O2, namely HUVEC andU937, it could be inferred that RIPs, once internalized, actin a cell-type independent fashion. In our experimental set-ting, the inhibitory activity on the rejoining of H2O2-inducedDNA SSBs of picomolar concentrations of Stx1 and ricin wascomparable to that of the reference inhibitor 3AB (5 mM; ref.[21]). Inhibition of DNA repair by Stx1 and ricin seems toresult from direct, rather than indirect and/or aspecific, in-teractions with the DNA repair machinery: indeed under theexperimental conditions described herein (i) the toxins wereneither DNA damaging per se nor affected the initial level ofstrand breakage caused by the oxidant; (ii) they did not affectt lasea teins paire
seR arget tingt s tod pairp oth-e clineo dedDp cellsc (APs tox-i kedD earl re-p
20 min) repair of oxidative DNA damage (Figs. 2 and 3),ven though in the latter case the absolute values of
able 2ffect of RIPs on the repair of MMS-induced DNA SSBs in HUVEC
reatment conditiona rNSF Percentage unrepairDNA breaks
IP-free 0.94± 0.12 7.51± 0.96Ricin (0.1 nM) 3.72± 0.42 29.82± 3.36Stx1 (5 pM) 4.31± 0.59 34.54± 4.733ABb 5.19± 0.74 41.52± 5.92
a HUVEC were treated for 30 min with 1.5 mM MMS and then allowo repair DNA damage for 90 min with or without ricin or Stx1, andfurther 3.5 h in toxin-free culture medium, at 37◦C. The level of residuaNA strand breakage was then determined with the alkaline halo assare expressed as rNSF (see Section2) and as the percentage of unrepaNA breaks, which represents the percent ratio between the rNSF aff repair time and the initial rNSF of MMS-treated cells. rNSF resu
rom 30 min challenge with 1.5 mM MMS was 12.52± 1.73. Each valuehe mean± S.E.M. of three separate determinations.
b HUVEC were allowed to repair the damage inflicted by MMS in culedium containing 5 mM 3AB for 5 h.
he cellular levels of two major antioxidant enzymes, catand glutathione peroxidase and (iii) the inhibition of proynthesis was shown to be causally unrelated to DNA refficiency in HUVEC.
As to the mechanism of DNA repair inhibition by theIPs, it could be hypothesized that the nuclear entry of l
oxins might result in steric hindrance somehow affeche accessibility and coordination of DNA repair enzymeamaged sites, finally leading to impairment of DNA-rerocesses. Interestingly, a similar, “steric hindrance” hypsis has been proposed by Muller et al. to explain the def binding of heterodimerized Ku protein to double-stranNA ends in differentiating human granulocytes[23]. Theresence of ricin and Stx1 in the nucleus of intoxicatedould be inferred by the appearance of nuclear lesionsites) closely related to the enzymatic action of thesens which remove adenine from isolated chromatin or naNA in vitro [8]. Moreover, a direct evidence of the nucl
ocalization of Stx1 in treated cells has been previouslyorted[24].

276 P. Sestili et al. / DNA Repair 4 (2005) 271–277
Another possibility might involve direct inhibition ofDNA repair enzymes: oxidative DNA damage results in a va-riety of lesions, including direct single/double-strand breaksand oxidative damage to bases and sugar phosphates, whichare processed and repaired by a large number of enzymesincluding some involved in base excision repair (BER)[25].Indeed, the fact that Stx1 and ricin have also been shownto inhibit the repair of MMS-induced DNA breaks wouldsuggest a negative interaction with BER machinery: pos-sible candidates of RIPs inhibitory activity could then bethe enzymes processing oxidative and/or alkylative dam-age, such as OGG1, MPG, NTH, NEIL1-3, APE1, XRCC1,Pol �, DNA ligase 1 or 3 and PARP[25,26]. Particularly,PARP and XRCC1 rapidly assemble after H2O2 treatmentin nuclear foci of damage and, as in the case of cells ex-posed to alkylating agents, the post-translational modifica-tion of target proteins that consists of the synthesis of ADP-ribose polymers catalysed by PARP does occur[27]. In-terestingly some RIPs, including ricin, have been recentlyshown to catalyze the release of adenines from poly(ADP-ribosyl)ated PARP in vitro[28]: it is worth considering thatan extensive depurination might promote precocious degra-dation of poly(ADP-ribose) polymer[28]. Since the effectsof PARP are directly dependent on the extent of poly(ADP-ribosyl)ation it is reasonable and attractive to hypothesizet ei-t DP-r dentn es ofD ivea
olei acil-l ure-m motet ducet1 -c urnmr tiona activeo ysa tesw e tod ida-tti theD mu-t rio isc Stx1,w NA[ -p sedr the
transformation is the result of a cell damage which obviouslyis non-lethal, the finding that a proportion of intoxicated cellsmight overcome RIPs challenge (Fig. 4) could be toxicologi-cally relevant. As a final note, it is worth considering that Stx1might also inhibit the repair of the DNA lesions generated byagents different from reactive oxygen species such as specificdietary-related mutagens, whose net effect in carcinogenesiscan be profoundly modified by cellular responses to DNAdamage through, in particular, the efficiency of DNA repairmechanisms[30].
In conclusion, although further studies are needed to clar-ify the mechanism responsible for the effects herein de-scribed, the present work demonstrates a novel biologicallyrelevant activity of ricin and Stx1, namely inhibition of therepair of oxidatively- and alkylatively-induced DNA lesionsand, to the best of our knowledge, this is the first timethat a bacterial toxin is found to inhibit DNA repair in cul-tured mammalian cells. As a consequence, the possibilitythat Shiga toxin-producing bacteria increase the risk of trans-formation of surrounding, inflamed tissues in the course ofhuman infections merits further investigation. In this light,case–control studies on the association between Stxs-relateddiseases and cancer should be encouraged.
A
te ofM inga
R
toxin-1
ins
cts inuary
.bo-
entmes:ys.
, F.ome-ic
S.ase,
.NA
ells,
hat RIPs might accelerate in vivo deadenylation ofher automodified PARP, or of specific acceptor poly(Aibosyl)ated proteins, and profoundly affect PARP-depenuclear processes such as the repair of different typNA lesions resulting from both alkylative and oxidatttack.
From a pathological point of view Stxs play a pivotal rn the aetiology of some important human diseases: bary dysentery, enterohemorrhagic colitis and hemolytic
ic syndrome. It has been demonstrated that Stxs prohe adhesion of leukocytes to endothelial cells and inhe production of pro-inflammatory cytokines (TNF-�, IL-�, IL-6 and IL-8) by a large variety of cells[1]. These loally produced factors promote inflammation which in tight trigger oxidative stress. Recently, Ohshima et al.[29]
eviewed the relationships between infections, inflammand carcinogenesis, where the damage induced by rexygen species, including H2O2, on inflamed tissues plan important role. H2O2 is released at the inflammation sihere, apart from its physiological role, it may contributamage of surrounding tissues by inducing lipid perox
ion, and damage to proteins, RNA and DNA[18]. Repairinghese lesions is of primary importance for therestitutio adntegrumof inflamed tissues: in particular, impairment ofNA repair machinery may lead to increased genetic
ations and carcinogenesis. Therefore, a similar scenaonceivable in human infections where the presence ofhich is simultaneously capable of directly damaging D
8], of promoting inflammation[1] and of inhibiting the reair of oxidative DNA damage, may result in an increaisk of transformation of target cells; in this regard, since
cknowledgements
The Authors are grateful to Dr. Ivana Scovassi (Instituolecular Genetics CNR, Pavia, Italy) for critically readnd commenting on the manuscript.
eferences
[1] J.C. Paton, A.W. Paton, Pathogenesis and diagnosis of Shigaproducing Escherichia coli infections, Clin. Microbiol. Rev. 1(1998) 450–479.
[2] L. Barbieri, M.G. Battelli, F. Stirpe, Ribosome-inactivating protefrom plants, Biochim. Biophys. Acta 1154 (1993) 237–282.
[3] S. Lyall, Threats and responses: Britain; arrest of terror suspeLondon turns up a deadly toxin, The New York Times, 8 (Jan2003) A10.
[4] Y. Endo, K. Tsurugi, RNAN-glycosidase activity of ricin A-chainMechanism of action of the toxic lectin ricin on eukaryotic risomes, J. Biol. Chem. 262 (1987) 8128–8130.
[5] Y. Endo, K. Tsurugi, J.M. Lambert, The site of action of six differribosome-inactivating proteins from plants on eukaryotic ribosothe RNA N-glycosidase activity of the proteins, Biochem. BiophRes. Commun. 150 (1988) 1032–1036.
[6] L. Barbieri, P. Valbonesi, E. Bonora, P. Gorini, A. BolognesiStirpe, Polynucleotide:adenosine glycosidase activity of ribosinactivating proteins: effect on DNA, RNA and poly(A), NucleAcids Res. 25 (1997) 518–522.
[7] L. Barbieri, P. Valbonesi, M. Brigotti, L. Montanaro, F. Stirpe,Sperti, Shiga-like toxin I is a polynucleotide:adenosine glycosidMol. Microbiol. 29 (1998) 661–662.
[8] M. Brigotti, R. Alfieri, P. Sestili, M. Bonelli, P.G. Petronini, AGuidarelli, L. Barbieri, F. Stirpe, S. Sperti, Damage to nuclear Dinduced by Shiga toxin 1 and ricin in human endothelial cFASEB J. 16 (2002) 365–372.

P. Sestili et al. / DNA Repair 4 (2005) 271–277 277
[9] G.L. Nicolson, J. Blaustein, M.E. Etzler, Characterization of twoplant lectins fromRicinus communisand their quantitative interactionwith a murine lymphoma, Biochemistry 13 (1974) 196–204.
[10] M. Ryd, H. Alfredsson, L. Blomberg, A. Andersson, A.A. Lind-berg, Purification of Shiga toxin by alpha-d-galactose-(1-4)-beta-d-galactose-(1-4)-beta-d-glucose-(1) receptor ligand-based chromatog-raphy, FEBS Lett. 258 (1989) 320–322.
[11] P. Sestili, O. Cantoni, Osmotically driven radial diffusion of single-stranded DNA fragments on an agarose bed as a convenient mea-sure of DNA strand scission, Free Radic. Biol. Med. 26 (1999)1019–1026.
[12] P. Sestili, A. Guidarelli, M. Dacha, O. Cantoni, Quercetin pre-vents DNA single strand breakage and cytotoxicity caused bytert-butylhydroperoxide: free radical scavenging versus iron chelatingmechanism, Free Radic. Biol. Med. 25 (1998) 196–200.
[13] O. Cantoni, P. Sestili, G. Brandi, F. Cattabeni, Thel-histidine-mediated enhancement of hydrogen peroxide-induced cytotoxicity isa general response in cultured mammalian cell lines and is alwaysassociated with the formation of DNA double strand breaks, FEBSLett. 353 (1994) 75–78.
[14] P.G. Petronini, M. Tramacere, A. Mazzini, G. Piedimonte, L. Silvotti,A.F. Borghetti, Hyperosmolarity-induced stress proteins in chick em-bryo fibroblasts, Exp. Cell. Res. 172 (1987) 450–462.
[15] H. Aebi, Catalase in vitro, Methods Enzymol. 105 (1984) 121–126.[16] R.A. Lawrence, R.F. Burk, Glutathione peroxidase activity in
selenium-deficient rat liver, Biochem. Biophys. Res. Commun. 71(1976) 952–958.
[17] J.P. Kehrer, The Haber–Weiss reaction and mechanisms of toxicity,Toxicology 149 (2000) 43–50.
[18] M.O. Bradley, L.C. Erickson, Comparison of the effects of hydro-NActa
[ P-ns,
[ oxiahate-
ribose polymerase activity, and nicotinamide adenine dinucleotideand adenosine triphosphate contents in cultured endothelial cells andfibroblasts, J. Cell. Physiol. 140 (1989) 177–185.
[21] P. Sestili, G. Spadoni, C. Balsamini, I. Scovassi, F. Cattabeni, E. Du-ranti, O. Cantoni, D. Higgins, C. Thomson, Structural requirementsfor inhibitors of poly(ADP-ribose) polymerase, J. Cancer Res. Clin.Oncol. 116 (1990) 615–622.
[22] M. Panayiotidis, O. Tsolas, D. Galaris, Glucose oxidase-producedH2O2 induces Ca2+-dependent DNA damage in human periph-eral blood lymphocytes, Free Radic. Biol. Med. 26 (1999) 548–556.
[23] C. Muller, S. Monferran, A.C. Gamp, P. Calsou, B. Salles, Inhibitionof Ku heterodimer DNA end binding activity during granulocytic dif-ferentiation of human promyelocytic cell lines, Oncogene 20 (2001)4373–4382.
[24] A. Suzuki, H. Doi, F. Matsuzawa, S. Aikawa, K. Takiguchi, H.Kawano, M. Hayashida, S. Ohno, Bcl-2 antiapoptotic protein me-diates verotoxin II-induced cell death: possible association betweenbcl-2 and tissue failure by E. coli O157:H7, Genes Dev. 14 (2000)1734–1740.
[25] G. Slupphaug, B. Kavli, H.E. Krokan, The interacting pathways forprevention and repair of oxidative DNA damage, Mutat. Res. 531(2003) 231–251.
[26] M.S. Satoh, G.G. Poirier, T. Lindahl, Dual function for poly(ADP-ribose) synthesis in response to DNA strand breakage, Biochemistry33 (1994) 7099–7106.
[27] K.W. Caldecott, XRCC1 and DNA strand break repair, DNA Repair(Amst.) 2 (2003) 955–969.
[28] L. Barbieri, M. Brigotti, P. Perocco, D. Carnicelli, M. Ciani,L. Mercatali, F. Stirpe, Ribosome-inactivating proteins depurinate
ans-8–
[ am-003)
[ pl. 3)
gen peroxide and X-ray irradiation on toxicity, mutation, and Ddamage/repair in mammalian cells (V-79), Biochim. Biophys. A654 (1981) 135–141.
19] D. D’Amours, S. Desnoyers, I. D’Silva, G.G. Poirier, Poly(ADribosyl)ation reactions in the regulation of nuclear functioBiochem. J. 342 (Pt. 2) (1999) 249–268.
20] A.F. Junod, L. Jornot, H. Petersen, Differential effects of hyperand hydrogen peroxide on DNA damage, polyadenosine diphosp
poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase and have trforming activity for 3T3 fibroblasts, FEBS Lett. 538 (2003) 17182.
29] H. Ohshima, M. Tatemichi, T. Sawa, Chemical basis of inflmation-induced carcinogenesis, Arch. Biochem. Biophys. 417 (23–11.
30] R. Goldman, P.G. Shields, Food mutagens, J. Nutr. 133 (Sup(2003) 965S–973S.
Copyright © 2022 FDOKUMEN




















