
Cervical Stenosis & Myelopathy - North American Spine Society
Upload
independentCategory
view
1download
0
J Physiol 589.18 (2011) pp 4491–4510 4491
The
Jou
rnal
of
Phys
iolo
gy
SAP97 directs NMDA receptor spine targeting and synapticplasticity
Dong Li2, Christian G. Specht1,3, Clarissa L. Waites1, Charlotte Butler-Munro2, Sergio Leal-Ortiz1,Janie W. Foote2, David Genoux4, Craig C. Garner1 and Johanna M. Montgomery2
1Department of Psychiatry and Behavioral Sciences, Nancy Pritzker Laboratory, Stanford University, Palo Alto, CA 94304-5485, USA2Centre for Brain Research and Department of Physiology, University of Auckland, New Zealand3Ecole Normale Superieure, U1024, 46 rue d’Ulm, 75005 Paris, France4Brain Mind Institute, Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland
Abstract SAP97 is a multidomain scaffold protein implicated in the forward trafficking andsynaptic localization of NMDA- and AMPA-type glutamate receptors. Alternative splicing ofSAP97 transcripts gives rise to palmitoylated αSAP97 and L27-domain containing βSAP97isoforms that differentially regulate the subsynaptic localization of GluR1 subunits of AMPAreceptors. Here, we examined whether SAP97 isoforms regulate the mechanisms underlyinglong-term potentiation (LTP) and depression (LTD) and find that both α- and β-forms of SAP97impair LTP but enhance LTD via independent isoform-specific mechanisms. Live imaging of α-and βSAP97 revealed that the altered synaptic plasticity was not due to activity-dependent changesin SAP97 localization or exchange kinetics. However, by recording from pairs of synapticallycoupled hippocampal neurons, we show that αSAP97 occludes LTP by enhancing the levels ofpostsynaptic AMPA receptors, while βSAP97 blocks LTP by reducing the synaptic localizationof NMDA receptors. Examination of the surface pools of AMPA and NMDA receptors indicatesthat αSAP97 selectively regulates the synaptic pool of AMPA receptors, whereas βSAP97 regulatesthe extrasynaptic pools of both AMPA and NMDA receptors. Knockdown of βSAP97 increasesthe synaptic localization of both AMPA and NMDA receptors, showing that endogenous βSAP97restricts glutamate receptor expression at excitatory synapses. This isoform-dependent differentialregulation of synaptic versus extrasynaptic pools of glutamate receptors will determine how manyreceptors are available for the induction and the expression of synaptic plasticity. Our data supporta model wherein SAP97 isoforms can regulate the ability of synapses to undergo plasticity bycontrolling the surface distribution of AMPA and NMDA receptors.
(Resubmitted 18 July 2011; accepted 18 July 2011; first published online 18 July 2011)Corresponding author J. M. Montgomery: Centre for Brain Research, University of Auckland, New Zealand. Email:[email protected] and C. C. Garner: Department of Psychiatry and Behavioral Sciences, Nancy PritzkerLaboratory, Stanford University, 1201 Welch Road, Palo Alto, CA 94304-5485, USA. Email: [email protected]
Abbreviations AMPAR, AMPA receptor; CaMKII, calcium–calmodulin-dependent kinase II; cLTD, chemical LTD;cLTP, chemical LTP; LTD, long-term depression; LTP, long-term potentation; NMDAR, NMDA receptor; PSD, post-synaptic density; SAP97, synapse associated protein of 97 kDa; shRNA, short hairpin RNA.
D. Li, C. G. Specht, C. C. Garner and J. M. Montgomery contributedequally to this work.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society DOI: 10.1113/jphysiol.2011.215566

4492 D. Li and others J Physiol 589.18
Introduction
Long-term potentiation (LTP) and depression (LTD) aretwo major forms of synaptic plasticity expressed in thehippocampus (Bliss & Collingridge, 1993; Malenka &Nicoll, 1999). The N-methyl-D-aspartate-type glutamatereceptor (NMDAR) plays a key role in synaptic plasticity byacting as a coincidence detector to trigger the recruitmentof α-amino-3-hydroxy-5-methyl-4-isoxazole propionicacid receptors (AMPARs) into the synaptic membrane(Malenka & Nicoll, 1999; Malinow & Malenka, 2002;Citri & Malenka, 2008). Thus NMDARs are critical forthe induction of synaptic plasticity while AMPARs areresponsible for its expression.
The recruitment of receptors to synapses depends ontheir interaction with scaffold proteins at the postsynapticdensity (PSD) (Montgomery et al. 2004; Specht & Triller,2008). PSD-95 and SAP97 are central organizers of thePSD, simultaneously binding to receptors and to otherscaffold or regulator proteins through PDZ, SH3 andGK interaction domains (Montgomery et al. 2004). BothPSD-95 and SAP97 bind the AMPAR subunit GluR1,although PSD-95 also requires the auxiliary proteinstargazin (Leonard et al. 1998; Cai et al. 2002; Fukata et al.2005). PSD-95 and SAP97 undergo alternative N-terminalsplicing that gives rise to the α variants which contain apalmitoylation sequence, or β variants with an L27 proteininteraction domain (Muller et al. 1995; Mori et al. 1998;Chetkovich et al. 2002; McLaughlin et al. 2002; Schluteret al. 2006; Waites et al. 2009). The N-terminal sequencesof SAP97 and PSD-95 control their dynamic properties(Chetkovich et al. 2002; Nakagawa et al. 2004; Waites et al.2009). For example, the synaptic exchange rates of α- andβSAP97 differ widely, consistent with their associationwith structures localized within or outside the PSD,respectively (Waites et al. 2009). On many levels, αSAP97mimics the properties of αPSD-95 (Nakagawa et al. 2004;Schluter et al. 2006; Waites et al. 2009). Nonetheless, giventhat αPSD-95 is the main expressed PSD-95 variant andβSAP97 the main form of SAP97, it appears that functionalspecialization has occurred between the two proteins(Chetkovich et al. 2002; Schluter et al. 2006; Waites et al.2009). In contrast to αPSD-95, βSAP97 has multiple rolesin the trafficking of receptor subunits, participating in theforward trafficking of AMPARs (Sans et al. 2001; Jeyifouset al. 2009), forming part of a multiprotein traffickingcomplex for Kir2 potassium channels (Leonoudakis et al.2004), and trafficking NMDARs to synapses via Golgioutposts (Jeyifous et al. 2009).
Our previous work revealed that αSAP97 and βSAP97differentially regulate the levels, dynamics and sub-synaptic localization of AMPARs (Waites et al. 2009).Given the importance of AMPARs in the expressionof synaptic plasticity, we examined how SAP97 iso-forms could regulate synaptic plasticity mechanisms. By
recording from synaptic connections between individualhippocampal neurons, we demonstrate that α- andβSAP97 independently regulate LTP at hippocampalsynapses by altering the synaptic and extrasynapticlocalization of AMPARs and NMDARs respectively. Ourdata reveal that these isoform specific SAP97-inducedchanges in glutamate receptor localization have thepotential to control the plasticity of hippocampalsynapses.
Methods
Expression constructs
A restriction fragment (NheI/XbaI) containing thesequence of rat βSAP97 (splice variant I1b, I3 and I5),fused at its N-terminus with enhanced green fluorescentprotein (EGFP; Clontech, containing F64L/S65T) viathe linker sequence SGLRSRAQASNS, was subclonedinto the XbaI/XbaI backbone of the lentiviral vectorpFUGW (Lois et al. 2002). This resulted in plasmidpFU-EG-rSAP97I3 (EMBL accession no. AM710296;http://www.ebi.ac.uk/embl), for the expression of thefusion protein EGFP-βSAP97. Furthermore, we usedexpression constructs for αSAP97-EGFP (Schluter et al.2006) and βSAP97-EGFP (Waites et al. 2009), alsobased on pFUGW and tagged at their C-terminuswith EGFP. The shRNA construct used to specificallyknock down the expression of βSAP97 was targettedto the unique L27 domain of βSAP97. Both theβSAP97 shRNA and the scrambled control constructshave been described previously (Jeyifous et al. 2009).Sequence analysis reveals that αSAP97 has no uniquedomains: αSAP97 is identical to βSAP97 with theexception of the palmitoylation sequence, and αSAP97shares this palmitoylation sequence with other MAGUKs(specifically αPSD93). Therefore it was not possibleto create an shRNA that specifically targets αSAP97.Rescue of shRNA mediated knockdown was achievedby coexpressing an shRNA-resistant βSAP97∗-EGFP andshRNA-SAP97 (S12) in the pFU-rSAP97I3-EG vector.Silent mutations in codons encoding N-terminal aminoacid residues 6, 7, 8 and 9 of βSAP97-EGFP was achievedby PCR with oligonucleotides containing the followingsequence ATGCCGGTCCGGAAGCAgGAcACaCAaAGA(bold: start codon; lower case: nucleotide changes). Notethat this sequence is unique to βSAP97 and present near itsL27 domain. The shRNA-SAP97 (S12) under the H1 poly-merase III promoters was subcloned into the BsiW1-Pac1sites upstream of the ubiquitin promoter/βSAP97∗-EGFPexpression cassette. Translocation studies (see Fig. 3) wereconducted with yellow-fluorescent protein (YFP)-taggedrat CaMKIIα (a kind gift from J. Tsui) (Tsui et al. 2005).
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4493
Hippocampal neuron culture
Primary neuron cultures were prepared according toa modified Banker culture protocol (Banker & Goslin,1998). Briefly, hippocampi from Sprague–Dawley ratembryos (E18–19) were dissociated in 0.05% trypsin(Gibco, no. 25300) and plated on poly-L-lysine coatedcoverslips (Carolina Biological Supply Co., Burlington,NC, USA) at a density of 165 cells mm−2. Coverslipswere inverted after 1 h, placed over a glial feederlayer and maintained at 37◦C/5% CO2 in Neuro-basal medium (Gibco, no. 21103) containing 2 mM
GlutaMAX (Gibco, no. 35050) and B27 (Gibco,no. 17504-044). Neuron cultures were infected withlentivirus expression constructs at DIV0–2 and usedfor experiments on DIV14–17. Alternatively, cultureswere transfected by Ca3(PO4)2 precipitation (on DIV7–9)or lipofectamine 2000 (Invitrogen) and used forexperiments on DIV12–15. Most imaging experiments(localization studies, time-lapse imaging and fluorescencerecovery after photobleaching (FRAP)) were done onlentivirus-infected neuron cultures, while the electro-physiological experiments were performed exclusivelyon transfected neurons. Control neurons were eitheruntransfected neurons plated on the same coverslips asneurons transfected with α- or βSAP97, or neurons trans-fected with GFP alone. No significant difference in AMPARor NMDAR EPSC amplitude was observed betweenuntransfected and GFP-transfected neurons (Waites et al.2009).
Hippocampal organotypic slice culture
Hippocampal slice cultures were prepared from P7 malerat pups as previously described (Montgomery et al.2001). Briefly, 400 mm hippocampal sections were grownin MEM + 40% horse serum at 37◦C on nitrocellulosemembranes. At 3 DIV slices were moved to 34◦Cand maintained for 1–2 weeks. Slices were induced toexpress αSAP97-EGFP or EGFP-βSAP97 by lentiviralinfection at 1 DIV. Paired whole cell recordings fromCA3 pyramidal neurons were then performed at 7–11DIV with the postsynaptic cell within the CA3 pair beingGFP-positive indicative of expression of αSAP97-EGFP orEGFP-βSAP97.
Neuron transfection
A volume of 60 μl containing 2 μg DNA and7.5 μl 2 M CaCl2 was added dropwise to 60 μlof 2 × HBS buffer (274 mM NaCl, 10 mM KCl,1.4 mM Na2HPO4, 15 mM glucose, 42 mM Hepes, pH7.1). After 20 min incubation in the dark, the pre-cipitate was pipetted onto a coverslip of culturedneurons in 1 ml conditioned medium containing 50 μM
DL-2-amino-5-phosphonopentanoic acid (APV) and10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX).Neurons were incubated for 30 min at 37◦C/5% CO2,rinsed three times with 2 ml pre-warmed HBSS and trans-ferred back into culture dishes.
Lentivirus production
HEK cells were grown to confluence and cotransfectedwith pFUGW plasmids (10 μg) together with the helperplasmids VSVg (5 μg) and �8.9 (7.5 μg) in 1.5 mlOpti-MEM (Gibco, no. 51985), using 60 μl lipofectamine2000 (Invitrogen, no. 11668) according to the supplier’sprotocol. Cells were transfected and maintained in neuro-basal medium containing GlutaMAX and B27 at 32◦C/5%CO2. The medium was exchanged once after 24 h and thevirus was harvested in culture medium after ∼55 h andstored at −80◦C. Generally, lentivirus titres were in therange of 108 ml−1, as estimated by the number of infectedHEK cells expressing EGFP fluorescence.
Imaging buffers
Buffers used for live imaging were based on Tyrodesolution (120 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 25 mM
Hepes pH 7.4, 30 mM glucose, sterile filtered). For controlexperiments, neurons were incubated in buffer solutioncontaining 2 mM MgCl2, 0.5 μM tetrodotoxin (TTX), 1 μM
strychnine, 50 μM APV and 10 μM CNQX, to fully blockneuronal activity. To induce neuronal activity, we usedconditions similar to those described by Ivanov et al.(2006). For the direct activation of NMDARs, we applied25 μM NMDA/10 μM glycine in the presence of 0.5 μM
TTX and 1 μM strychnine (= cLTD treatment). Enhancedsynaptic activity was achieved through the applicationof 20 μM bicuculline/200 μM glycine in the presenceof 1 μM strychnine (= cLTP). For this application,prior incubations in control buffer were done in theabsence of TTX. Similar treatments with NMDA or highconcentrations of glycine have been shown to induce LTDor LTP in cultured hippocampal neurons, respectively (Luet al. 2001).
Synaptic localization assay
Coverslips with hippocampal cultures expressingαSAP97-EGFP or EGFP-βSAP97 were transferred intoTyrode control solution containing Mg2+, TTX (notfor subsequent bicuculline/glycine treatment), strychine,APV and CNQX at 37◦C. After 5 min, the solutionwas exchanged with NMDA/glycine (cLTD) or withbicuculline/glycine containing solution (cLTP), or withthe identical control solution (see imaging buffercompositions). After 10 min the neurons were fixed for
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4494 D. Li and others J Physiol 589.18
10 min at 37◦C in 0.1 M sodium phosphate buffer pH7.4, containing 4% paraformaldehyde and 1% sucrose.To correct for the expression levels of individual neurons,we quantified the synaptic accumulation of SAP97, i.e.the mean fluorescence intensity of synaptic spines relativeto the fluorescence in dendritic shafts, rather than theabsolute levels of synaptic SAP97.
Live imaging
Time-lapse imaging was done on a scanning confocalmicroscope (custom-built by N. Ziv and S. Smith ona Zeiss Axiovert 100TV) with 488 nm and 514 nmlasers (Coherent; Sapphire 488–20CDRH and Compass215M-20) and a 40× objective (1.3 NA; Zeiss PlanNeofluar), using OpenView software (by N. Ziv) forimage acquisition. Coverslips with primary neurons weremounted in a perfusion chamber, maintained at 37◦C andperfused with Tyrode-based solutions.
Fluorescence recovery after photobleaching (FRAP)
Synaptic puncta of EGFP-tagged SAP97 were bleachedto ∼20% of initial fluorescence intensity by multiplescanning passes (15–20×) of a high intensity 488 nm laserbeam. The fluorescence recovery was imaged for up to30 min, initially every 30 s (14 frames) and then every5 min (5 frames). Intensity values of bleached puncta ateach time point were normalized to their fluorescenceintensity prior to bleaching (I t/Ipre) and to non-bleachedcontrol puncta (Inb,t/Inb,pre) in the same field of view.To calculate mean recovery traces the dynamic range ofindividual experiments was adjusted by setting the firstvalue after photobleaching to zero. Thus, FRAP data werenormalized according to the equations:
F t = I tInb , pre
IpreInb,t
and
F norm = F t − F 0
F pre − F 0
where Fnorm is the normalized fluorescence intensity, F t
the intensity at time t , F0 at time t = 0 and Fpre = 1 theintensity prior to photobleaching. The mean experimentaldata were fitted with an exponential equation with twocomponents, as described previously (Tsuriel et al. 2006):
f (t) = a(
1 − e− tτ1
)+ (1 − a)
(1 − e− t
τ2
)
where τ1 and τ2 are the time constants and a and(1 – a) are the relative fractions of fluorescence in thetwo pools. The theoretical parameters were extractedby minimizing the sum of squared residuals, using a
macro written in Excel (by N. Ziv). For time-lapseexperiments, images were acquired every 3 min for upto 33 min (12 frames). Intensity values of synaptic SAP97puncta were normalized to their fluorescence intensityprior to bleaching (I t/Ipre), and expressed as meanintensity ± SEM.
Immunocytochemistry
Immunocytochemistry to detect α-actinin (Sigma,1:2000) was performed as previously described (Cheyne& Montgomery, 2008; Waites et al. 2009). Primaryantibody binding was performed overnight at 4◦C.Secondary antibody was performed for 1 h at roomtemperature. Coverslips were washed and mounted ontoslides (Vectashield) for imaging. Pyramidal neurons wereidentified by morphology and Z-stack (0.5 μm) images ofdendrites were obtained on a Zeiss Axioskop with a CCDdigital camera using a 63× oil objective. Image analysiswas performed using ImageJ. Z-stacks were converted to8-bit, merged into maximum projections and backgroundsubtracted to remove the diffuse protein expression withindendrites. Images in which staining was dim or the back-ground was high were excluded from the analysis. Imageswere manually thresholded to select only puncta thatwere greater than 2-fold above image background. Punctawere analysed using the Analyze Particles function inImageJ so that the average puncta intensity and numberof puncta could be determined. All data were normalizedto parallel controls and presented as means ± SEM weren is the number of fields analysed. Statistical analysis wasperformed using Student’s t tests (one-tailed distribution,unequal variance).
Electrophysiology
Dual whole cell recordings were performed at DIV9–14on primary dissociated hippocampal cultures and atDIV7–11 on hippocampal organotypic slice culturestransfected with αSAP97-EGFP or βSAP97-EGFP aspreviously described (Montgomery et al. 2001; Waiteset al. 2009). Briefly, hippocampal cultures were transferredto a recording chamber on a Zeiss Axioskop 2FS andvisualized under infrared differential interference contrastmicroscopy. Cultures were perfused at room temperatureat a rate of 2 ml min−1 with artificial cerebrospinal fluid(ACSF, in mM: 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2,1 Na2HPO4, 26.2 NaHCO3, 11 glucose). Presynapticuntransfected neurons were held in current clamp andinduced to fire action potentials at 0.1 or 0.2 Hz bybrief current injection (typically 20–50 pA for 20 ms).Postsynaptic neurons (GFP-positive) were held in voltageclamp at −65 mV. Series resistance (Rs) was continuouslymonitored and recordings in which Rs varied by more
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4495
than 20% were excluded from analysis. Internal solutionconsisted of (in mM): 120 potassium gluconate (pre-synaptic cell) or 120 caesium gluconate (postsynapticcell), 40 Hepes, 5 MgCl2, 0.3 NaGTP, 2 NaATP, 5QX314 (postsynaptic cell only), pH 7.2 with KOH orCsOH. Evoked AMPAR- and NMDAR-mediated EPSCswere measured as previously described (Montgomeryet al. 2001, 2005; Montgomery & Madison, 2002;Waites et al. 2009). Isolated NMDAR EPSCs weremeasured at +40 mV in ACSF containing 10 μM 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX). AMPAR and NMDAR EPSCs inresponse to presynaptic action potentials were collectedat 0.1 or 0.2 Hz. LTP and LTD were induced 5 min afterthe initiation of the postsynaptic whole cell recordingusing either pharmacological stimulation of NMDARs asdescribed above for live imaging or by pairing presynapticaction potentials with postsynaptic depolarization(Montgomery et al. 2001, 2005; Montgomery &Madison, 2002). To measure surface AMPAR andNMDAR-mediated currents, exogenous AMPA (1 mM) orNMDA (1 mM) was applied with a picospritzer (pressure2 bar, pulse 200 ms). The micropipette was placed at astandard distance of 200 μm from the dendrites of α- orβSAP97-EGFP transfected neurons. The peak amplitudesof AMPAR- and NMDAR-mediated surface currentswere measured by whole cell patch clamp as detailedabove. Online data acquisition and offline analysis forall electrophysiology experiments was performed withpCLAMP (Clampex v9.2). Statistical significance ofchanges in AMPAR and NMDAR EPSC amplitudes wastested using Student’s t test with a level of significance setat P < 0.05.
Results
SAP97 isoforms regulate synaptic plasticityin hippocampal neurons
SAP97 is known to be critical for the trafficking of AMPA-and NMDA-type glutamate receptors to synapses (Sanset al. 2001; Jeyifous et al. 2009) and in directing the sub-synaptic localization of AMPARs at excitatory synapses(Schluter et al. 2006; Jeyifous et al. 2009; Waites et al. 2009).Our previous data show that αSAP97 drives AMPARs intothe PSD, whereas βSAP97 drives AMPARs to extrasynapticsites (Waites et al. 2009). We therefore hypothesized thatSAP97 isoforms are prime candidates to regulate changesin AMPAR expression that are known to occur with LTPand LTD. In order to determine the potential role ofSAP97 isoforms in synaptic plasticity, we employed acombination of electrophysiology and live cell imagingin the dissociated hippocampal cell culture preparation.Dissociated cultures and pharmacological induction ofplasticity were employed to enable us to visualize synaptic
protein dynamics and to compare these data to theelectrophysiogical changes seen with the same inductionprotocols.
LTP and LTD were induced in dissociated hippocampalcultures by pharmacological stimulation of NMDARs.Using paired whole-cell recordings from pyramidalneurons, we measured the amplitude of evoked excitatorypostsynaptic currents (EPSCs) at synaptic connectionsbetween individual hippocampal neurons before, duringand after the induction of chemical LTP (cLTP; 20 μM
bicuculline + 200 μM glycine, in the presence of 1 μM
strychnine to avoid activation of glycine receptors) orchemical LTD (cLTD; 25 μM NMDA + 10 μM glycine +1 μM strychnine, see Methods).
In control hippocampal neurons, application ofbicuculline and glycine induced an increase in theamplitude of the AMPAR EPSCs (Fig. 1A). Potentiationof the AMPAR-mediated currents developed graduallyover the first 5 min after the pharmacological inductionof plasticity and was sustained for the length of allpaired recordings (n = 13 pairs). Twenty minutes aftercLTP induction, the amplitude of the AMPAR EPSCs wasincreased to an average of 144.3 ± 16.4% of the baselinecurrent amplitude (P < 0.01; n = 13 pairs). This cLTPwas NMDAR dependent, as application of the NMDARantagonist APV (50 μM) during cLTP induction preventedthe increase in AMPAR EPSC amplitude (average AMPAREPSC amplitude was 94.7 ± 2.4% of baseline 20 min aftercLTP + APV, n = 5 pairs).
We next examined whether the expression of twoSAP97 isoforms, α- and βSAP97, altered this plasticity.These two N-terminal SAP97 isoforms were selectedbased on previous data showing that the palmitoylatedαSAP97 recruits GluR1 containing AMPARs into thePSD and consequently increases basal synaptic trans-mission, while the L27 containing βSAP97 redirectsGluR1 containing AMPARs into a peri-PSD compartmentwithin dendritic spines, thus reducing basal synaptictransmission (Waites et al. 2009). Indeed, as expectedthe baseline levels of evoked AMPAR EPSC amplitudeswere higher in neurons over-expressing αSAP97 andreduced in βSAP97-expressing neurons compared tocontrol neurons, as reported previously (Waites et al.2009). Average baseline AMPAR EPSC amplitude was419.9 ± 54 pA in αSAP97-expressing neurons (n = 22pairs) and 62.9 ± 16.1 pA in βSAP97-expressing neurons(n = 21 pairs). To assess the impact of α- and βSAP97on the ability of excitatory synapses to undergoLTP, we measured EPSC amplitudes between pairs ofhippocampal neurons in which the postsynaptic cellsexpressed EGFP-tagged αSAP97 or βSAP97. In contrastto untransfected neurons, we found that cLTP couldnot be induced in hippocampal neurons expressingαSAP97-EGFP or βSAP97-EGFP (Fig. 1A). In neuronsexpressing αSAP97, cLTP induction resulted in a decrease
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4496 D. Li and others J Physiol 589.18
in the amplitude of AMPAR EPSCs. Twenty minutes afterthe induction of cLTP, the AMPAR EPSC amplitudeswere 78.0 ± 13.9% of baseline control amplitudes (n = 10pairs; Fig. 1A). Similarly, in neurons expressing βSAP97,cLTP induction resulted in a decrease in the amplitudeof AMPAR EPSCs. Twenty minutes after the inductionof cLTP, AMPAR EPSC amplitudes were decreased to58.0 ± 13.7% of baseline current amplitudes (P < 0.01;n = 13 pairs). These data reveal that the overexpressionof either SAP97 isoform impairs the induction and/or theexpression of cLTP.
We next examined the ability of αSAP97 and βSAP97to modulate long-term depression (LTD). In controlneurons, the induction of cLTD produced an immediateand long-lasting depression of AMPAR-mediated EPSCs(Fig. 1B). AMPAR EPSC amplitudes were significantlydecreased to 78.4 ± 14.8% of baseline current amplitudes(measured 20 min after cLTD induction; Fig. 1B; P < 0.01;n = 7 pairs). This cLTD was blocked by the NMDARantagonist APV, demonstrating its NMDAR dependence(average AMPAR EPSC amplitude was 106.3 ± 1.2% ofbaseline currents 20 min after cLTD + APV; n = 5 pairs).
cLTD could also be induced in neurons expressingeither αSAP97 or βSAP97 (Fig. 1B). As in control neurons,the amplitude of AMPAR EPSCs decreased immediatelyand this decrease was sustained in both αSAP97 andβSAP97-expressing hippocampal neurons for the lengthof the paired recordings. AMPAR EPSC amplitudeswere 45.7 ± 11.1% of baseline current amplitudesin αSAP97-expressing neurons (n = 12 pairs; 20 minafter cLTD induction). Similarly in βSAP97-expressingneurons, cLTD induction resulted in a significant decrease
Figure 1. N-terminal SAP97 splice variants alter synapticplasticity in hippocampal neuronsA, AMPAR EPSC amplitudes measured from paired recordingsbetween control hippocampal neurons or between neuronsexpressing α- or βSAP97-EGFP in the postsynaptic cell. cLTP wasinduced 5 min after attaining each paired recording by a 5 minapplication of 20 μM bicuculline and 200 μM glycine + 1 μM
strychnine. EPSC amplitudes are normalized to baseline, pre-cLTPAMPAR EPSC amplitudes. Top panels, representative postsynaptictraces, overlaid, in control, α- or βSAP97-EGFP-expressing neuronsbefore and after cLTP. One example of a presynaptic action potentialtrace is shown for each group. B, AMPAR EPSC amplitudes measuredfrom paired recordings between hippocampal neurons as describedin A. cLTD was induced by a 5 min application of 25 μM NMDA and10 μM glycine + 1 μM strychnine and paired recordings were heldfor up to 20 min post-cLTD induction. C, surface AMPAR currentamplitudes were measured by focal application of 1 mM AMPA incontrol, α- and βSAP97-expressing neurons immediately prior to and20 min after the induction of cLTP or cLTD. Top, sample traces ofsurface AMPAR-mediated currents measured in control, α- andβSAP97-expressing neurons. Bottom, data were normalized topre-cLTP or cLTD current amplitudes and are expressed as apercentage of the baseline surface AMPAR current.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4497
in AMPAR EPSC amplitudes to 59.6 ± 8.3% (n = 8 pairs)of baseline current amplitudes. The level of depression inαSAP97- and βSAP97-expressing hippocampal neuronswas significantly greater than the LTD expressed in controlneurons (P < 0.01). These data show that neither SAP97isoform prevents the pharmacological induction of LTD.However, as reported above, we observed a dramaticdifference in the baseline levels of evoked AMPAREPSC amplitudes in neurons over-expressing αSAP97(higher) and βSAP97 (reduced) compared to controlneurons (Fig. 1A), as reported previously (Waites et al.2009). These differences support the general conclusionthat N-terminal SAP97 isoforms differentially affect thesynaptic levels of AMPARs, and also imply that the changesin establishing LTP and/or LTD could have different under-lying mechanisms.
As differences in glutamate receptor trafficking havebeen suggested to occur in dissociated versus slicehippocampal preparations (Shi et al. 1999; Waiteset al. 2009), we sought to determine whether αSAP97and βSAP97 also altered the ability of synapses toexpress plasticity in hippocampal slices. We performedpaired whole cell recordings from CA3 pyramidal cellpairs in which the postsynaptic neuron was expressingeither EGFP-tagged α- or βSAP97. On average, baselineAMPAR-mediated EPSCs in control pyramidal cell pairswere 16.0 ± 2.3 pA, compared with 27.3 ± 8.4 pA and14.0 ± 2.4 pA in pairs in which the postsynaptic neuronexpressed αSAP97 or βSAP97 respectively. The increase inAMPAR EPSC amplitude in αSAP97-expressing neuronswas significant (P < 0.01), demonstrating that αSAP97exerts similar effects in dissociated and hippocampal slicein vitro preparations. The AMPAR EPSC amplitude inβSAP97-expressing neurons was not significantly differentfrom controls. The smaller magnitude of the change inaverage AMPAR EPSC amplitude in the slice culturesmay reflect the lower expression levels of α- and βSAP97induced by lentiviral infection. However, we also noteda decrease in the probability of βSAP97-expressing CA3pyramidal cell pairs having an evident AMPAR EPSCindicative of a synaptically connected pair (27.3% inβSAP97-expressing neurons, compared with 38.5% incontrol slices and 53.8% in αSAP97-expressing slices),suggesting that AMPAR-mediated currents may bedecreased to undetectable levels or that CA3 pyramidalneuron connectivity is decreased in βSAP97-expressingneurons. We then induced LTP or LTD in each connectedCA3 pyramidal cell pair by our electrical synapticstimulation protocols described previously, resulting inrobust LTP and LTD (Supplemental Fig. S1; Montgomeryet al. 2001, 2005, Montgomery & Madison, 2002).Twenty minutes after the LTP pairing protocol, averageAMPAR EPSC amplitude was 210.2 ± 71.0% of baselineamplitude in control CA3 pyramidal cell pairs (n = 5pairs, Fig. S1A). The induction of LTP between pyramidal
cell pairs in hippocampal organotypic slices was alsoaltered by the expression of αSAP97 or βSAP97 butto a differing degree from that observed with cLTP indissociated hippocampal cultures. Twenty minutes afterthe induction of LTP, average AMPAR EPSC amplitudewas 126.4 ± 7.3% and 86.9 ± 5.6% of the baseline EPSCamplitude in pyramidal cell pairs in which the post-synaptic neuron expressed α- (n = 6 pairs) or βSAP97(n = 5 pairs), respectively. Thus αSAP97 impaired andβSAP97 prevented the induction of LTP in hippocampalslices. Induction of LTD with 1 Hz presynaptic stimulationfor 5 min induced robust LTD in control, αSAP97and βSAP97-expressing neurons (Fig. S1B). AMPAREPSC amplitudes were 47.1 ± 10.6%, 24.8 ± 6.3% and29.0 ± 4.6% of baseline current amplitudes in control(n = 5 pairs), αSAP97- (n = 6 pairs) and βSAP97 (n = 6pairs)-expressing neurons, respectively. The magnitudeof the LTD induced by 1 Hz stimulation was strongercompared to that induced by chemical stimulation indissociated hippocampal cultures (Fig. 1B). Interestingly,the relative amounts of LTD expressed in control, α- andβSAP97-expressing neurons showed the same relationshipin both forms of LTD. That is, the level of electricallyinduced LTD in αSAP97- and βSAP97-expressinghippocampal neurons was significantly greater than thatexpressed in control neurons (P < 0.05 in both cases),as was observed in response to the induction of cLTD(Fig. 1B).
SAP97 isoforms differentially alter synaptic andextrasynaptic AMPAR pools
Our above analysis of αSAP97- and βSAP97-inducedchanges in LTP and LTD were measured as changesin synaptic AMPAR-mediated currents. Since we havepreviously shown that a significant proportion ofAMPARs in βSAP97-expressing neurons are located inthe extrasynaptic membrane, we examined whether thesize of the surface pool of receptors was altered by theinduction of synaptic plasticity in neurons expressingαSAP97 orβSAP97. Any changes observed in the size of thesurface AMPAR currents could then be compared to thechanges previously observed in synaptic AMPAR currents(Fig. 1A and B), providing insights into the redistributionof synaptic and extrasynaptic AMPAR pools duringsynaptic plasticity. In these experiments, we measuredthe amplitude of the surface AMPAR-mediated currentin response to focal application of AMPA prior to and20 min after cLTP or cLTD treatment (Fig. 1C). In controlneurons, we found that the amplitude of the surfaceAMPAR-mediated current was not significantly alteredby cLTP (average current amplitude was 90.6 ± 11.7% ofbaseline current amplitude 20 min after cLTP induction;n = 6; P = 0.08; Fig. 1C), despite the LTP-induced increase
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4498 D. Li and others J Physiol 589.18
in the amplitude of the synaptic AMPAR-mediatedcurrents (Fig. 1A). These data are consistent with thehypothesis that under control conditions, the increase insynaptic AMPAR current measured during LTP resultsfrom the recruitment of extrasynaptic AMPARs tosynapses (Makino & Malinow, 2009).
We next examined how the surface pool of AMPARswas altered in neurons expressing αSAP97 or βSAP97.Consistent with βSAP97 inducing a higher level oftotal surface AMPARs localized to extrasynaptic sites(Waites et al. 2009), baseline surface current amplitudeswere significantly higher in βSAP97-expressing neurons(Fig. 1C) (average current amplitude was 198.6 ± 45.3 pAin βSAP97 neurons versus 107.7 ± 25.5 pA in αSAP97neurons; P < 0.001; n = 6), as previously reported (Waiteset al. 2009). Following the pharmacological inductionof LTP, we found that the amplitude of the surfaceAMPAR current was decreased in αSAP97-expressingneurons. Twenty minutes after the induction of LTPaverage current amplitude was 76.2 ± 13.5% of baselinecurrents (P < 0.05; n = 6; Fig. 1C), a decrease similar tothat occuring in the synaptic pool (Fig. 1A). In contrast,the surface AMPAR-mediated current amplitude didnot change with cLTP in βSAP97 neurons (95.7 ± 4.4%of baseline AMPAR current amplitude, P > 0.1, n = 6;Fig. 1C), despite the significant decrease in synapticAMPARs with cLTP (Fig. 1A), suggesting that βSAP97facilitates an increase in the size of the extrasynaptic poolof AMPARs with cLTP.
We also examined the amplitude of the surface AMPARmediated current in response to the induction of cLTD.Here we found that cLTD resulted in a decrease inthe amplitude of the surface AMPAR-mediated currentsin control, αSAP97- and βSAP97-expressing neurons(control: 80.1 ± 11.3% of baseline AMPAR currentamplitude; αSAP97: 64.6 ± 5.5% of baseline AMPARcurrent amplitude; βSAP97: 65.14 ± 13.4% of baselineAMPAR current amplitude; Fig. 1C, P < 0.01 in allcases). This decrease in the size of the total surfaceAMPAR-mediated currents is similar to the changes seen inAMPAR-mediated synaptic currents in control, αSAP97-and βSAP97-expressing neurons (Fig. 1B), suggesting thatwith LTD expression both synaptic and extrasynapticpools of AMPARs are changing in parallel and to a similardegree.
Synaptic plasticity does not alter the synapticlocalization of SAP97 isoforms
Activity-dependent changes in synaptic strength have beendirectly linked to changes in the number of receptorslocalized in the PSD. If α- and βSAP97 are involved indirecting AMPAR movement, then changes in α- andβSAP97 localization in response to synaptic activity may
be responsible for the re-distribution of AMPARs duringcLTP or cLTD. This concept is supported by severalstudies indicating that both the synaptic levels (Mauceriet al. 2004) and the protein dynamics (Nakagawa et al.2004) of SAP97 may be regulated by synaptic activity.We designed a set of experiments to assess whether ourcLTP or cLTD protocols caused a redistribution of SAP97isoforms that could explain the observed changes ofsynaptic plasticity in α- and βSAP97-expressing neurons(Fig. 2). Initially, we examined the synaptic levels ofEGFP-tagged α- and βSAP97 using time-lapse imaging.The intensity of synaptic SAP97 puncta were measuredprior to, during and after the induction of cLTP orcLTD (Fig. 2A–C). No significant changes in the levelsof α- or βSAP97 were detected either during or after thecLTP treatment (αSAP97: 99 ± 1.5% after 4 min of cLTPtreatment and 99 ± 6.5% at 20 min after the treatment;βSAP97: 97 ± 2.0% after 4 min, 101 ± 5.4% after 20 min;Fig. 2B).
In contrast, there was a significant decrease in thefluorescence intensity of EGFP-βSAP97, and to a lesserextent of αSAP97-EGFP, during the induction of cLTD(αSAP97: 88 ± 6.1% after 4 min of cLTD treatment,P = 0.2; βSAP97: 63 ± 2.3%; P < 0.001, Mann–WhitneryU test; Fig. 2A and C). These data imply that conditionsthat stimulate the reduction of synaptic AMPARs (i.e.cLTD) are perhaps causing the simultaneous removal ofα- and βSAP97 from the spine. However, we also notedthat this loss in fluorescence was reversible and was notassociated with a long-term change in synaptic levels ofαSAP97-EGFP or EGFP-βSAP97 (αSAP97: 103 ± 4.5%at 20 min after cLTD treatment; βSAP97: 101 ± 1.3%;Fig. 2C). Upon closer inspection, we noticed that the lossof fluorescence during the addition of NMDA/glycineoccurred throughout the entire cell including dendrites(Fig. 2A) and soma (not shown), putting into doubtthat an actual redistribution of the protein was takingplace as had been previously proposed (Nakagawa et al.2004). Instead, we hypothesized that the loss of EGFPfluorescence was due to the acidification of the post-synaptic neuron during depolarization (Chesler, 2003)and thus due to quenching of the fluorophore (EGFP).In the presence of 25 μM NMDA, the intracellular pH ofprimary neurons decreases to about 6.5, recovering over atime course of minutes (Irwin et al. 1994). Furthermore,the fluorescence of EGFP is strongly pH dependent, witha pK a ≈ 6 and a decrease by �pHi ≈ −0.5 leading to areduction of fluorescence intensity of 20% (Kneen et al.1998). These observations suggest that pH-dependentquenching of EGFP-based fluorophores is a confoundingfactor when using protocols that induce a prolongeddepolarisation of neurons.
To test this quenching hypothesis, we repeatedthe time-lapse experiment with neurons expressingYFP-CaMKIIα (Fig. 3), which is known to be recruited
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4499
Figure 2. SAP97 localization is unchanged by cLTPor cLTDA–C, lentivirus-infected neuronal cultures expressingαSAP97-EGFP or EGFP-βSAP97 were imaged for 5 minunder baseline conditions, during 5 min application ofbicuculline/glycine (cLTP treatment) or NMDA/glycine(cLTD) and after drug washout. A, sample images ofEGFP-βSAP97 in spines (DIV15), imaged before (3 mintime point shown; left image) and during NMDA/glycineapplication (9 min; centre image) and after washout(18 min timepoint; right image). B, quantification oftime-lapse imaging data of αSAP97- (grey trace, n = 13cells) and βSAP97-expressing neurons (black, n = 24)before, during and after cLTP induction, expressed asmeans ± SEM. No significant changes in mean spinelevels of SAP97 were detected at 3 versus 33 min timepoints; Mann–Whitney U test. C, application of NMDAand glycine was used to induce cLTD in neuronsexpressing αSAP97-EGFP (grey, n = 6) or EGFP-βSAP97(black, n = 11). The treatment caused a temporaryquenching of EGFP fluorescence, particularly inβSAP97-expressing neurons (see also Fig. 2A), but hadno long-term effect on synaptic SAP97 levels(means ± SEM, Mann-Whitney U test). D and E, cLTP orcLTD treatments were applied for 10 min in neuronsexpressing αSAP97-EGFP or EGFP-βSAP97 and the cellswere immediately fixed. After fixation, the fluorescenceintensity in spines relative to the shaft was quantified.For the cLTP application, TTX was omitted from thepre-incubation buffer (see methods). D, quantificationof the spine/shaft ratio of αSAP97-EGFP fluorescence(DIV15; mean ± SEM; n ≥ 11 cells with 11 synapticpuncta per cell; right panel). αSAP97 enrichment didnot change by either treatment (P = 0.8;Mann–Whitney U test, two-tailed). E, similarly, the cLTPand cLTD treatments produced no significant changes inthe levels of βSAP97 in synaptic spines (n ≥ 14 randomfields of view with ≥ 27 synaptic puncta per cell,analysis blind to condition; right panel).
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4500 D. Li and others J Physiol 589.18
to synapses by the activation of NMDARs (Shen & Meyer,1999). Following the addition of NMDA/glycine to theimaging buffer, the YFP fluorescence was quickly lostin all cellular compartments (Fig. 3A). Furthermore, theloss/quenching of YFP-CaMKIIα was more pronouncedthan that observed with EGFP-βSAP97, as expected fora fluorophore with a substantially higher pH sensitivityat physiological levels (Llopis et al. 1998). Moreover,after blocking excitatory neurotransmission with APVand CNQX, the fluorescence slowly recovered over aperiod of about 5 min (Fig. 3B). In a second experiment,neurons expressing YFP-CaMKIIα were stimulated withbicuculline/glycine or NMDA/glycine for 5 min and thenimmediately fixed (Fig. 3C). Here, we observed the trans-location of YFP-CaMKIIα fluorescence into spines withboth treatments, as reported previously (Shen & Meyer,1999). These data clearly show that the apparent loss offluorescence of YFP-CaMKIIα following the addition ofNMDA/glycine is largely due to quenching and not to aredistribution of the protein. This quenching effect is likely
to have caused the reduction of YFP-βSAP97 fluorescenceduring NMDA application observed in an earlierstudy (Nakagawa et al. 2004). In our experiments thequenching by NMDA was more pronounced in neuronsexpressing EGFP-βSAP97 compared to αSAP97-EGFP(Fig. 2). This difference may result from α and βSAP97occupying different subsynaptic compartments that maybe differentially affected by pH changes (Degiorgis et al.2008; Waites et al. 2009).
To evaluate the effect of cLTP and cLTD on the synapticlocalization of EGFP-taggedα- andβSAP97 independentlyof quenching, we immediately fixed SAP97-expressingneurons after incubation in Tyrode buffer containingeither bicuculline/glycine or NMDA/glycine (Fig. 2D andE). We found that neither cLTP nor cLTD inducedsignificant changes in the localization of αSAP97 orβSAP97 (αSAP97: control spine/shaft levels 2.8 ± 0.2, LTD2.9 ± 0.4, LTP 2.8 ± 0.4; βSAP97: control 2.5 ± 0.2, LTD2.4 ± 0.1, LTP 2.5 ± 0.2; n ≥ 11 fields of view), indicatingthat the induction of synaptic plasticity does not change
Figure 3. cLTP and cLTD treatments causeYFP-CaMKIIα translocation into synapticspinesA, time-lapse images of YFP-CaMKIIαtranslocation into spines. Sample images ofdendritic segments (DIV15) acquired during3 min baseline conditions (left image; 1 mintime point shown, see B), after application of25 μM NMDA/10 μM glycine (centre image;6 min), and after addition of 50 μM APV/10 μM
CNQX (right image; 11 min time point). B,quantification of data shown in A. Mean spineintensity levels were reduced during NMDAapplication (n = 6 puncta). This quenchingoccurred in all cellular compartments and wasnot related to a redistribution of theYFP-CaMKIIα. After addition of the glutamatereceptor blockers APV and CNQX, thefluorescence recovered over a period of 5 min.Note that the spine intensities after recoveryexceed the levels prior to NMDA application asa result of CaMKIIα translocation (compareright and left image in A). C, neuronstransfected with YFP-CaMKIIα were incubatedwith NMDA/glycine (cLTD) or bicuculline/glycine(cLTP) for 5 min before fixation and imageacquistion (left panel) (DIV12). The enrichmentof YFP-CaMKIIα in synapses was quantified asspine versus shaft fluorescence intensity (rightpanel) and showed that both treatmentsproduced a translocation of CaMKIIα intodendritic spines (mean ± SEM; n = 3 cells with≥17 synaptic puncta per cell; control:shaft/spine ratio 0.51 ± 0.06; cLTD:0.93 ± 0.07; cLTP: 0.78 ± 0.12).
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4501
the steady-state synaptic levels of either of the two SAP97isoforms.
Synaptic plasticity does not alter the exchange rate ofSAP97
Our previous imaging studies revealed that thesteady-state levels of synaptic scaffold proteins suchas SAP97 are composed of soluble, membrane- andcytoskeleton-associated pools (Waites et al. 2009). Withregard to changes in synaptic strength, the most importantis the size of the synaptic fraction of the scaffold proteinsas this pool largely dictates the number and distributionof binding sites for neurotransmitter receptors within thePSD (Bredt & Nicoll, 2003; Bats et al. 2007; Waites et al.2009). As such, shifts in the ratio of PSD-associated versussoluble pools of scaffold proteins could have a significantimpact on receptor docking sites and synaptic strength.We therefore explored whether the induction of synapticplasticity altered the exchange kinetics of EGFP-taggedα- or βSAP97 by performing fluorescence recovery afterphotobleaching (FRAP) experiments (Fig. 4).
Since the expression of long-term changes in synapticstrength are acquired over time, we evaluated whether theexchange kinetics of αSAP97-EGFP and EGFP-βSAP97were changed 20 min after the pharmacological inductionof synaptic plasticity (Fig. 4C and D). However, we could
not detect any changes in the exchange kinetics of α- orβSAP97 after the induction of LTP or cLTD comparedto control conditions. Therefore, the dynamics of α- andβSAP97 seem to be independent not only of the acutelevel of synaptic activity but were also unchanged bythe chemical induction of LTP or LTD in these cultures,putting into doubt to what extent synaptic plasticity isactually involved in the regulation of SAP97 dynamicsand vice versa. Taken together our data demonstrate thatconditions that alter the synaptic strength in our systemdo not change the synaptic levels or protein dynamics ofαSAP97 or βSAP97. This implies that the observed failureto induce LTP in neurons overexpressing α- or βSAP97is not linked to changes in the dynamic properties of thescaffold proteins.
SAP97 regulates the synaptic localization of NMDARs
The activation of NMDARs is critical for the inductionof many forms of synaptic plasticity. As LTP wasabsent in both αSAP97- and βSAP97-expressing neurons,we investigated whether SAP97 isoforms altered thesynaptic expression of NMDARs. Using paired wholecell recordings, we first examined whether evokedNMDAR-mediated EPSCs between individual pairs ofpyramidal neurons were altered by the overexpressionof αSAP97 or βSAP97 in the postsynaptic neuron.
Figure 4. SAP97 dynamics are unchanged during induction and expression of cLTD or cLTPA and B, the exchange of synaptic αSAP97-EGFP and EGFP-βSAP97 was measured by FRAP during and afterthe application of NMDA/glycine (cLTD) or bicuculline/glycine (cLTP) or under control conditions. Sample imagesof αSAP97-EGFP in lentivirus-infected hippocampal neurons (control condition) show the limited recovery of ableached αSAP97 punctum (A, arrowhead) compared to the faster exchange of βSAP97 (B). C and D, cLTD orcLTP were induced in lentivirus-infected neurons expressing αSAP97-EGFP or EGFP-βSAP97. Fluorescence recoveryof bleached synaptic SAP97 was recorded with a delay of 20 min after drug washout. C, fluorescence recoveryof αSAP97-EGFP after cLTD (dark grey trace, n = 22) or cLTP treatment (light grey, n = 13) did not vary from theexchange kinetics observed during blocked synaptic activity (control, black, n = 27). D, quantification of FRAPshowed that the recovery of synaptic EGFP-βSAP97 was unchanged between control conditions (black trace,n = 20) and the neurons that had undergone cLTP or cLTD treatments followed by a 20 min delay (light and darkgrey traces, n = 20 for both conditions).
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4502 D. Li and others J Physiol 589.18
Isolated synaptic NMDAR-mediated currents weremeasured between individual cultured hippocampalpyramidal neurons in response to presynaptic actionpotentials.
In control neurons, NMDAR-mediated EPSCamplitudes remained constant throughout the lengthof the paired recordings (minimum of 20 min; averageNMDAR EPSC amplitude was 30.30 ± 1.28 pA, n = 10pairs; Fig. 5A). Interestingly, we found that αSAP97 andβSAP97 had differential effects on NMDAR-mediatedEPSCs. In neurons expressing αSAP97, NMDAR EPSCamplitudes were not significantly different from controlEPSCs (29.77 ± 0.78 pA, n = 12 pairs; P > 0.1). However,NMDAR EPSC amplitude was remarkably decreasedin neurons expressing βSAP97 (9.41 ± 0.61 pA; n = 13pairs; Fig. 5A), which was significantly decreased fromboth control and αSAP97-expressing neurons (P < 0.001in both cases). This reduction in NMDAR-mediatedEPSC amplitude in βSAP97-expressing neurons wasnot a result of a decrease in synapse number as wefound no effect of either αSAP97 or βSAP97 on spinedensity in hippocampal neurons: in βSAP97-expressingneurons α-actinin density was 1.18 ± 0.13 μm−1, whichwas not significantly different from control neurons(1.01 ± 0.09 μm−1; P > 0.05) or αSAP97-expressingneurons (0.98 ± 0.16 μm−1; P > 0.05).
In order to determine whether the surface expressionof functional NMDARs, comprising both synapticand extrasynaptic pools, is altered in αSAP97- versusβSAP97-expressing neurons, we measured the surfaceNMDAR-mediated currents in hippocampal neurons byfocal application of NMDA. Interestingly, despite thefact that βSAP97 decreases synaptic NMDARs, neuronsexpressing βSAP97 were more responsive to exogenousNMDA than those expressing αSAP97. NMDA-evoked
current amplitudes were 112.47 ± 17.90 pA inβSAP97-expressing neurons, significantly higher thanboth αSAP97-expressing neurons and control neurons(αSAP97: 52.29 ± 5.38 pA, control: 60.00 ± 5.84 pA; bothsignificantly different from βSAP97-expressing neurons,P < 0.001; Fig. 5C). Therefore, while βSAP97-expressingneurons have fewer functional NMDARs at the synapsefor the detection of synaptically released glutamate,βSAP97 supports a significantly higher total surfaceexpression of functional NMDARs.
Specific knockdown of βSAP97 increases synapticexpression of AMPA and NMDA receptors
To determine whether βSAP97 negatively controls thesize of the synaptic pool of NMDARs in the endogenoussituation, we used a short hairpin RNA (shRNA) tospecifically downregulate the expression of endogenousβSAP97 in hippocampal neurons. Our previous studieshave shown that this shRNA suppresses the expressionof βSAP97 by more than 90% (Jeyifous et al. 2009).In contrast, previous studies have designed shRNA tothe common GUK domain of SAP97 or utilized Dlgh1mice lacking any functional SAP97 protein and thereforeresult in knockdown of all SAP97 isoforms (Howardet al. 2010). We found that specific knockdown ofβSAP97 reverses the inhibitory effect previously observedon synaptic NMDAR-mediated EPSCs and results ina significant increase in synaptic NMDARs (Fig. 6A).Paired recordings revealed that the amplitude of NMDAREPSCs in shRNA-expressing neurons were significantlyhigher than control neurons and in neurons expressing ascrambled shRNA (average NMDAR EPSC amplitude inshRNA-expressing neurons was 115.44 ± 47.03 pA (n = 6
Figure 5. βSAP97 is a negative regulator of synaptic NMDARsA, NMDAR EPSC amplitudes measured from paired recordings between pyramidal neurons in hippocampal cultures.NMDAR EPSCs were measured at +40 mV and paired recordings were maintained for a minimum of 20 min. Inset:representative traces of NMDAR EPSCs in control, α- and βSAP97-expressing neurons. Five postsynaptic traces areshown overlaid for each group, with 1 example of a presynaptic action potential. B, surface NMDAR-mediatedcurrents evoked by focal application of NMDA to α- and βSAP97-expressing neurons. Right: example traces ofsurface NMDAR-mediated currents shown overlaid for control, α- and βSAP97-expressing neurons.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4503
pairs), compared with 33.87 ± 8.15 pA in control neurons(n = 5 pairs) and 33.50 ± 5.95 pA in neurons expressinga scrambled shRNA (n = 5 pairs); P < 0.01). Similarly,knockdown of βSAP97 reversed the previously observeddecrease in synaptic AMPARs and also resulted in anincrease in synaptic AMPARs (average AMPAR EPSCamplitude was 554.86 ± 287.42 pA in shRNA-expressingneurons (n = 6 pairs), compared with 141.18 ± 40.10 pAin control neurons (n = 5 pairs) and 139.38 ± 21.35 pA inneurons expressing the scrambled shRNA (n = 5 pairs);P < 0.01; Fig. 6B). The original effect of βSAP97, that is adecrease in both AMPAR and NMDAR EPSC amplitudecompared with controls, could be rescued by expressionof a modified βSAP97∗ that is resistant to knockdown(see Methods; Fig. 6A and B). Together these data revealthat endogenous βSAP97 decreases the synaptic pools ofNMDARs and AMPARs.
We also measured surface (i.e. synaptic + extrasynaptic)NMDAR- and AMPAR-mediated currents in βSAP97shRNA-expressing neurons with exogenous applicationof NMDA or AMPA, respectively. In response toexogenous application of NMDA, average surfaceNMDAR current amplitude was 136.98 ± 13.49 pA(n = 21 neurons) in βSAP97 shRNA-expressing neurons.In response to exogenous application of AMPA,average surface AMPAR current amplitude was435.12 ± 59.57 pA (n = 24 neurons). Neither the surfaceNMDAR- nor AMPAR-mediated current amplitudes inβSAP97 shRNA-expressing neurons were significantlydifferent from the synaptic NMDAR or AMPAREPSC amplitudes in βSAP97 shRNA-expressing neurons(NMDAR EPSC 115.44 ± 47.03 pA Fig. 6A, AMPAR EPSC554.86 ± 287.42 pA Fig. 6B; P > 0.05). Together these dataare consistent with the redistribution of NMDA and
Figure 6. Knockdown of endogenous βSAP97 releases extrasynaptic receptors to the synaptic poolA, knockdown of endogenous βSAP97 results in a significant increase in the amplitude of NMDAR-mediated EPSCsthat was not observed in the shRNA scrambled controls. Expression of the altered form βSAP97∗ that is resistant toknockdown rescued the phenotype observed in βSAP97-expressing neurons. Example traces of NMDAR-mediatedEPSCs are shown for control (untransfected), shRNA βSAP97 and βSAP97∗ with a representative action potential.Scale bars: 50 pA/50 ms and 20 mV/20 ms. B, knockdown of endogenous βSAP97 also significantly increases theamplitude of AMPAR mediated EPSCs. Expression of the altered form βSAP97∗ that is resistant to knockdownrescued the phenotype observed in βSAP97-expressing neurons. In both A and B, average NMDAR and AMPAREPSCs were measured from paired whole cell recordings in which the postsynaptic cells were either untransfected(controls), or expressing shRNA βSAP97, scrambled shRNA or βSAP97∗ (see Methods). Example traces ofAMPAR-mediated EPSCs are shown for control (untransfected), shRNA βSAP97 and βSAP97∗ with a representativeaction potential. Scale bars: 100 pA/100 ms and 20 mV/20 ms.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society
4504 D. Li and others J Physiol 589.18
AMPA receptors from extrasynaptic to synaptic sites inthe absence of βSAP97.
SAP97 isoforms alter the distribution of NMDARsduring synaptic plasticity
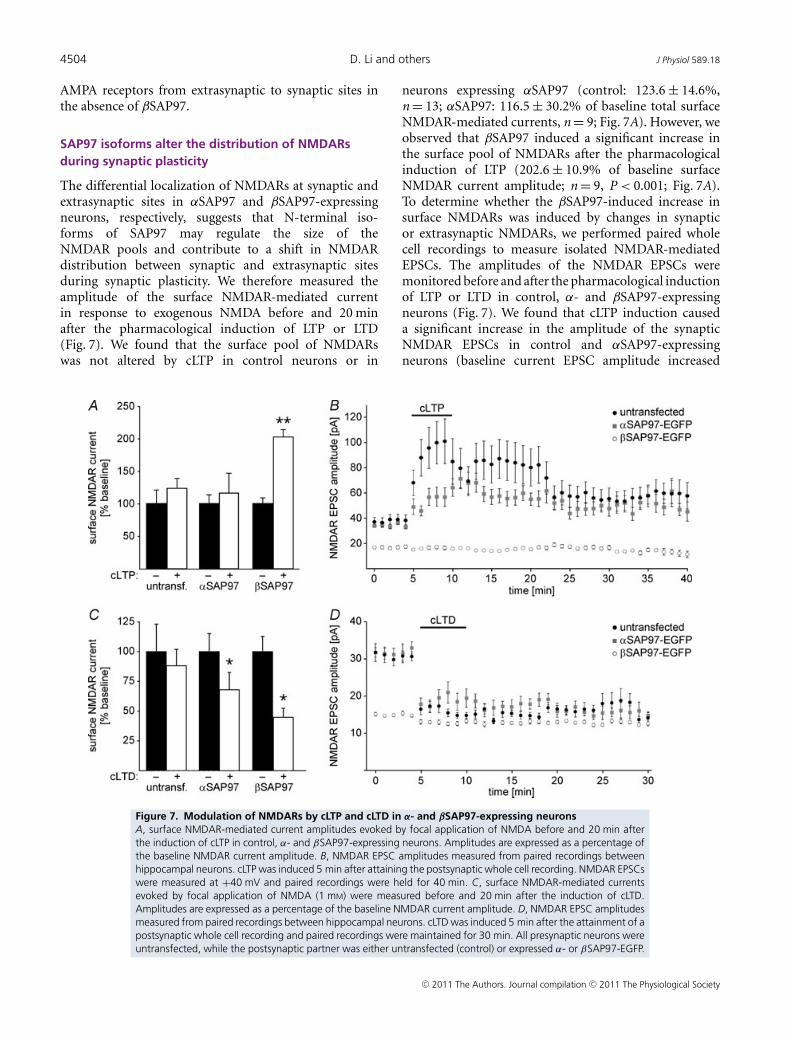
The differential localization of NMDARs at synaptic andextrasynaptic sites in αSAP97 and βSAP97-expressingneurons, respectively, suggests that N-terminal iso-forms of SAP97 may regulate the size of theNMDAR pools and contribute to a shift in NMDARdistribution between synaptic and extrasynaptic sitesduring synaptic plasticity. We therefore measured theamplitude of the surface NMDAR-mediated currentin response to exogenous NMDA before and 20 minafter the pharmacological induction of LTP or LTD(Fig. 7). We found that the surface pool of NMDARswas not altered by cLTP in control neurons or in
neurons expressing αSAP97 (control: 123.6 ± 14.6%,n = 13; αSAP97: 116.5 ± 30.2% of baseline total surfaceNMDAR-mediated currents, n = 9; Fig. 7A). However, weobserved that βSAP97 induced a significant increase inthe surface pool of NMDARs after the pharmacologicalinduction of LTP (202.6 ± 10.9% of baseline surfaceNMDAR current amplitude; n = 9, P < 0.001; Fig. 7A).To determine whether the βSAP97-induced increase insurface NMDARs was induced by changes in synapticor extrasynaptic NMDARs, we performed paired wholecell recordings to measure isolated NMDAR-mediatedEPSCs. The amplitudes of the NMDAR EPSCs weremonitored before and after the pharmacological inductionof LTP or LTD in control, α- and βSAP97-expressingneurons (Fig. 7). We found that cLTP induction causeda significant increase in the amplitude of the synapticNMDAR EPSCs in control and αSAP97-expressingneurons (baseline current EPSC amplitude increased
Figure 7. Modulation of NMDARs by cLTP and cLTD in α- and βSAP97-expressing neuronsA, surface NMDAR-mediated current amplitudes evoked by focal application of NMDA before and 20 min afterthe induction of cLTP in control, α- and βSAP97-expressing neurons. Amplitudes are expressed as a percentage ofthe baseline NMDAR current amplitude. B, NMDAR EPSC amplitudes measured from paired recordings betweenhippocampal neurons. cLTP was induced 5 min after attaining the postsynaptic whole cell recording. NMDAR EPSCswere measured at +40 mV and paired recordings were held for 40 min. C, surface NMDAR-mediated currentsevoked by focal application of NMDA (1 mM) were measured before and 20 min after the induction of cLTD.Amplitudes are expressed as a percentage of the baseline NMDAR current amplitude. D, NMDAR EPSC amplitudesmeasured from paired recordings between hippocampal neurons. cLTD was induced 5 min after the attainment of apostsynaptic whole cell recording and paired recordings were maintained for 30 min. All presynaptic neurons wereuntransfected, while the postsynaptic partner was either untransfected (control) or expressed α- or βSAP97-EGFP.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4505
from 37.85 ± 6.4 pA to 58.5 ± 25.9 pA in control neurons30 min post-cLTP, n = 6 pairs, and from 34.18 ± 8.27 pAto 55.57 ± 9.4 pA in αSAP97-expressing neurons 30 minpost-cLTP, n = 8 pairs; Fig. 7B). In contrast, no significantchange occurred in the amplitude of the NMDAR EPSCsin βSAP97-expressing neurons (EPSC amplitudes were16.7 ± 2.1 pA and 13.4 ± 1.97 pA, prior to and 30 minafter LTP, respectively, n = 10 pairs; Fig. 7B). Therefore, theLTP-induced increase in total surface NMDAR mediatedcurrents in βSAP97-expressing neurons must result froman increase in the size of the extrasynaptic pool ofNMDARs.
We also examined how the pharmacological inductionof LTD altered the amplitude of surface NMDAR-mediatedcurrents in control, α- and βSAP97-expressing neurons(Fig. 7C). Five minutes after cLTD induction, surfaceNMDAR-mediated currents were significantly decreasedin control neurons (average total NMDAR-mediatedsurface current was 55.6 ± 13.1% of baseline, P < 0.01,n = 6; not shown), but recovered to near baseline levels20 min after the pharmacological induction of LTD(Fig. 7C: 87.8 ± 13.4% of baseline current, P > 0.05,n = 6). Similarly, 5 min after cLTD induction, we foundthat surface NMDAR-mediated currents were significantlydecreased in α- and βSAP97-expressing neurons (αSAP97:56.2 ± 14.8% baseline current, P < 0.01, n = 6; βSAP97:48.8 ± 6.6% baseline current, P < 0.01, n = 9; not shown).However in α- and βSAP97-expressing neurons, thedepression of NMDAR-mediated currents did not recover,such that 20 min after cLTD induction total surfaceNMDAR-mediated currents were still significantly belowbaseline levels (αSAP97: 67.6 ± 14.6% and βSAP97:44.8 ± 7.2% of baseline currents, n = 6 and 9, respectively,P < 0.01 in both cases; Fig. 7C).
To determine whether these changes inNMDAR-mediated currents occurred at synapticreceptors, we performed paired whole cell recordingsto measure the amplitude of isolated NMDAR EPSCsin control, α- and βSAP97-expressing neurons. Wefound that NMDAR EPSC amplitudes decreasedsignificantly in control neurons, as previously describedfor electrically evoked LTD (Montgomery & Madison,2002; Montgomery et al. 2005). Similarly, in α- andβSAP97-expressing neurons, we found that NMDAREPSC amplitudes decreased significantly and thatthis decrease persisted for the length of the pairedrecording (≥35 min; Fig. 7D; control: average baselineEPSC amplitude decreased from 31.51 ± 2.59 pA to12.68 ± 1.66 pA at 25 min post-LTD, n = 7 pairs; αSAP97:from 31.42 ± 3.28 pA to 16.14 ± 3.94 pA at 25 minpost-LTD, n = 6 pairs; βSAP97: from 14.93 ± 0.81 pA to11.87 ± 0.95 pA at 25 min post-LTD, n = 7 pairs). Thus,in response to cLTD the size of the synaptic pool ofNMDARs decreases in control, α- and βSAP97-expressingneurons, albeit to varying degrees. These decreases are
likely to contribute to the observed reduction in surfaceNMDAR-mediated currents, but the differences weobserved in the changes of these two pools of NMDARsagain reflect the differential regulation of synaptic andextrasynaptic NMDARs by αSAP97 and βSAP97.
Discussion
In this study we have examined how N-terminal SAP97isoforms regulate the induction and the expression ofsynaptic plasticity, and whether this occurs throughactivity-dependent regulation of the number of synapticbinding sites, or through the differential localization ofNMDARs and AMPARs. The use of pharmacologicalprotocols to induce plasticity enabled us to directlyexamine how the same protocols influence synapsefunction via electrophysiology and protein dynamics usinglive cell imaging. Our data suggest that the capacityto induce and express synaptic plasticity is at least inpart determined by how many receptors are bound toeither the PSD-dominant palmitoylatedαSAP97, inducingincreased clustering of AMPARs within the PSD, orto extrasynaptic L27 domain-containing βSAP97 thatsequesters both AMPARs and NMDARs at extrasynapticsites.
αSAP97 occludes synaptic potentiation by increasingsynaptic AMPARs
α- and βSAP97 appear to prevent LTP via differentmechanisms. As NMDAR-mediated synaptic currentswere entirely normal in αSAP97-expressing neurons, theseneurons possess the ability to induce LTP. We believe thatthe lack or the reduction of LTP is due to an increasedsteady-state localization of AMPARs in the PSD by αSAP97(Fig. 8) (Waites et al. 2009). Therefore LTP and αSAP97overexpression appear to engage the same mechanismsto increase synaptic strength and the expression of LTPis occluded by αSAP97. As LTP was not accompaniedby a change in αSAP97 exchange kinetics, this revealsthat the number of αSAP97 AMPAR binding sites doesnot change, but that during LTP αSAP97 promotes theselective localization of AMPARs to the synaptic pool.
βSAP97 regulates plasticity by sequestering NMDARsoutside of synapses
Our observation that βSAP97 prevents LTP, decreasessynaptic NMDARs and increases surface NMDARs revealsthat βSAP97 localizes NMDARs to extrasynaptic sites,as it does with AMPARs (Waites et al. 2009). This issupported by converse experiments where in the absenceof βSAP97, synaptic AMPARs and NMDARs are increasedwhereas extrasynaptic receptors are decreased, indicating
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4506 D. Li and others J Physiol 589.18
that a property of endogenous βSAP97 is to exclude thesereceptors from the synapse (Fig. 8). These data have ledus to the conclusion that βSAP97 is a negative regulatorof synaptic pools of NMDARs as well as AMPARs,consequently preventing the induction of LTP.
A previous model proposed that βSAP97 functionsto either (a) facilitate the delivery of AMPARs to theextrasynaptic membrane from intracellular pools or(b) trigger the release of AMPARs that are retainedin an extrasynaptic pool so they can be translocatedto synapses (Rumbaugh et al. 2003; Schluter et al.2006). While this model was in relation to AMPARs,our independent measurement of synaptic and surfaceNMDARs and AMPARs has enabled us to distinguishbetween these two possibilities. The observed highextrasynaptic levels of glutamate receptors are consistentwith βSAP97 facilitating the delivery of AMPARs andNMDARs to the extrasynaptic membrane from intra-
Figure 8. Model of SAP97 isoform-dependent regulation ofsynaptic glutamate receptor localizationOur physiological data reveal that α- and β-isoforms of SAP97 playstrikingly different roles in controlling the localization of AMPA- andNMDA-type glutamate receptors at synaptic and extrasynaptic sites.A, βSAP97 creates docking sites for AMPA and NMDA receptors outof the PSD, sequestering AMPA and NMDA receptors atextrasynaptic sites where they are unable to contribute to theinduction and expression of LTP. The net result is a greater shift inreceptor localization to the extrasynaptic sites. Both receptor typesare able to freely diffuse away from the synaptic site, however, andbe internalised in response to LTD. B, αSAP97 creates docking sitesfor AMPA receptors in the PSD, shifting the localization of AMPAreceptors into the synapse and increasing synaptic strength. Noeffect of αSAP97 on NMDAR localization or function was evident inour studies.
cellular pools. However, our data indicate that thesereceptors are sequestered outside of the synapse, pre-venting the induction of LTP and the movement ofextrasynaptic AMPARs into synaptic sites.
Previous studies have produced conflicting data onthe effect of βSAP97, and the deletion or knockdownof SAP97, on AMPAR and NMDAR EPSCs (Nakagawaet al. 2004; Schluter et al. 2006); (Howard et al.2010). A major reason for these discrepancies lies inthe differing strategies employed to knockdown theexpression of SAP97. In previous studies, only the effectsof βSAP97 overexpression were examined. However, in theloss-of-function experiments the expression of all SAP97isoforms was decreased by knockdown/knockout targetedto the GUK domain or exon 4 of SAP97 (Nakagawaet al. 2004; Howard et al. 2010). Therefore the combinedsynaptic effects of all SAP97 isoforms were reflected inthese data. In the current study, we designed an shRNAto specifically target βSAP97, enabling us to exclusivelydetermine the role of this dominant isoform in regulatingthe extrasynaptic pools of AMPA and NMDA receptors.Moreover, the extracellular stimulation employed in theprevious studies may have activated both synaptic andextrasynaptic receptors, preventing the specific effect onextrasynaptic receptors being identified. Furthermore, thedata discrepancies could also be caused by I3/I4/I5 insertswhich are present in SAP97 (SAP97 with I3 and I5 wasused in the present study; not stated in other studies)and also the level and temporal expression profiles ofβSAP97 having different effects on AMPAR and NMDARcurrents (Howard et al. 2010). The fact that βSAP97 isalso important in the forward trafficking of glutamatereceptors (Sans et al. 2001; Jeyifous et al. 2009) addsyet another degree of complexity to the regulation ofreceptors by βSAP97. Our previously observed reductionin NR1 puncta intensity in βSAP97-deficient hippocampalneurons (Jeyifous et al. 2009) is likely to reflect a decrease inβSAP97-bound extrasynaptic surface receptors. However,by specifically decreasing βSAP97 expression in the pre-sent study, we further show that independent of itsrole in receptor trafficking, βSAP97 shifts the dynamicequilibrium of glutamate receptors from a synaptic to anextrasynaptic location.
Glutamate receptors have previously been proposed tobe differentially regulated in hippocampal organotypicslices in comparison to dissociated hippocampal cultures,with GluR1-containing AMPARs being inserted uponstimulation in slices but under basal conditions indissociated cultures (Shi et al. 1999). Our data supportthis conclusion in that we were able to induce LTP inαSAP97-expressing neurons in hippocampal slices butnot in dissociated cultures, suggesting that a proportionof GluR1-containing receptors remained intracellular inthe slice preparation and were inserted in response toLTP stimuli. As a result LTP was partially occluded by
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4507
the expression of αSAP97, but was fully occluded in thedissociated culture system where GluR1 readily moves tothe surface under basal conditions. Intriguingly, LTP wasalso blocked by the expression of βSAP97 in hippocampalorganotypic slices, consistent with our conclusion fromdissociated hippocampal cultures that βSAP97 preventsthe movement of receptors into the synaptic space that isrequired for the expression of LTP.
The observed differences in synaptic AMPAR- andNMDAR-mediated synaptic currents induced by theexpression of α- or β-SAP97 do not appear to resultfrom differential effects on synapse number as weobserved no effect of either αSAP97 or βSAP97 onspine density in hippocampal neurons or in surfaceGluR1 puncta (Waites et al. 2009). In addition,the significant decrease in the amplitude of theAMPAR- and NMDAR-mediated synaptic currents inβSAP97-expressing neurons, and the selective increase inthe amplitude of the AMPAR- but not NMDAR-mediatedsynaptic currents in αSAP97-expressing neurons is notconsistent with SAP97 isoforms increasing presynapticglutamate release. Effects of SAP97 on presynapticfunction have previously been observed in response tochronic overexpression of βSAP97 in vivo from E16–P8,but presynaptic changes were not observed in immatureneurons acutely expressing βSAP97 (Howard et al. 2010).Thus, our results likely reflect that N-terminal SAP97isoforms regulate receptor targeting but not presynapticfunction in early development.
α- and β-isoforms also exist for the MAGUK proteinsPSD-95 and PSD-93. As the dominant SAP97 variantexpressed at synapses is the β-isoform, and αPSD-95 themain form of PSD-95 (Chetkovich et al. 2002; Schluteret al. 2006), functional specialization may occur at thesynapse as αPSD-95 stabilizes AMPARs in the PSD,while βSAP97 stabilizes receptors in the extrasynapticpool. Similar to αSAP97, the α-isoform of PSD-95has been shown to promote the synaptic clustering ofAMPARs (Schluter et al. 2006), suggesting that there issome functional redundancy between α-isoforms withregards to LTP. The strong effects produced by βSAP97expression or knockdown on AMPARs and NMDARsdemonstrate that the L27 domain of βSAP97 bestowsa unique and functionally important set of propertiesto this isoform. Our data also imply that the L27domains in PSD-93 and PSD-95 may similarly performfunctions that modulate the ratios of synaptic versusextrasynaptic receptor complexes and thus may regulatesynaptic plasticity in unexpected ways.
Following cLTP treatment, βSAP97 induced an increasein the extrasynaptic NMDAR pool, revealing that the sizeof extrasynaptic pools of receptors can also be regulatedby plasticity. As there was no parallel increase in thesynaptic NMDARs, these extrasynaptic receptors do notappear to merge with synaptic receptor pools as occurs
during LTP in control neurons. Currently the role of thelarge extrasynaptic NMDAR pool in βSAP97-expressingneurons is unknown, but extrasynaptic signalling throughNMDARs has been shown to be detrimental, activatingcell death pathways and contributing to pathologicalconditions (Groc et al. 2009; Milnerwood et al. 2010).Therefore βSAP97 may play a role in the altered NMDARsignalling and trafficking observed in conditions suchas Alzheimer’s and Huntington’s disease, schizophreniaand addiction (Marcello et al. 2007; Groc et al. 2009;Milnerwood et al. 2010).
As LTD could still be induced in α- andβSAP97-expressing neurons, N-terminal SAP97 isoformsallow receptor movement away from synaptic sites. Incontrol, α- and βSAP97-expressing neurons, depressionof the amplitude of AMPAR- and NMDAR-mediatedcurrents was observed in the synaptic and the surfacepools, reflecting a reduction in the size of both thesynaptic and extrasynaptic receptor pools during LTD.This is consistent with the movement of AMPARs andNMDARs to membrane sites lateral to the PSD andtheir subsequent internalization at extrasynaptic end-ocytic zones to drive synapses to a depressed state(Blanpied et al. 2002; Montgomery & Madison, 2002;Ashby et al. 2004; Racz et al. 2004; Montgomery et al. 2005;Petrini et al. 2009). The internalization of NMDARs wasmost pronounced in neurons expressing βSAP97, wherewe observed a strong reduction of the surface receptorlevels after LTD. This may result from the high baselinelevels of extrasynaptic surface NMDARs in these neuronsthat facilitate NMDAR-dependent cLTD induction andreceptor movement to extrasynaptic endocytic sites.
Synaptic plasticity does not affect the dynamics ordistribution of N-terminal SAP97 isoforms
Previous studies have shown contradictory results withrespect to the activity dependence of SAP97 localizationand dynamics. Treatment of hippocampal neurons withNMDA has been suggested to recruit βSAP97 to thesynapse by CaMKII phosphorylation at Ser39 (Mauceriet al. 2004), or conversely to lead to the dissipation ofβSAP97 from synapses (Nakagawa et al. 2004). In contrast,we have shown that neither α- nor βSAP97 localizationor dynamics was altered by the chemical LTP or LTDprotocols. These differences could be due to fluoresencequenching described earlier, the βSAP97 isoform usedin these studies (βSAP97 containing the I3/I5 inserts inour experiments; not stated in the other reports), or thelevel of SAP97 overexpression in comparison to the lowlevels expressed in the current study by lentiviral infection.Another possibility is the differences in concentration andlength of treatment with NMDA utilized in the previousstudies (50 μM for 15 min or 3 μM for 20 min). It is not
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4508 D. Li and others J Physiol 589.18
known what, if any, changes in synaptic strength wereinduced by these protocols. In contrast, the protocols usedin the current study were utilized in both imaging andelectrophysiology experiments, and were shown in controlneurons to induce LTP or LTD and to cause the trans-location of CaMKII to synapses.
Ultimately, we aimed to determine whether there isa relationship between SAP97 dynamics and synapticplasticity. We found little evidence that either α or βSAP97alter their distribution or exchange kinetics in responseto changes in synaptic activity. This implies that theyexert their effects on synaptic function by creating surfaceglutamate receptor docking sites within the PSD andextrasynaptic space. At present, we find no evidence tosupport a model wherein either α- or βSAP97 performsa chaperone function for these receptors. This is notto say that receptor binding to these scaffold proteinsis not regulated. Our cLTD data clearly demonstratethat surface AMPA and NMDA receptors can uncouplefrom the cytoskeletal matrix and be internalized. Sucha model is consistent with studies showing that bothAMPA and NMDA receptor binding to scaffold proteinsis regulated by post-translational modifications suchas phosphorylation and that SAP97 isoforms use theirmultidomain structure to create tertiary complexes withboth kinase and phospatases (AKAP/calcineurin) capableof responding to changes in synaptic signalling (Colledgeet al. 2000; Tavalin et al. 2002; Montgomery et al. 2004;Dell’Acqua et al. 2006; Shepherd & Huganir, 2007). Takentogether, our data show that α- and βSAP97 do notregulate synaptic plasticity via activity-dependent changesin their synaptic localization or dynamics, but rather area platform for regulating the tethering of receptors insynaptic or extrasynaptic compartments.
Conclusions
We have performed parallel functional and imagingstudies that revealed that N-terminal α and β splicevariants of SAP97 differentially regulate the synapticlocalization of AMPARs and NMDARs, resulting in majorchanges in the ability of neurons to express synapticplasticity. Our data show that αSAP97 is importantfor localizing AMPARs in the PSD and increases thesynaptic strength, but that these receptors are readilyremoved from the synapse in response to LTD. Incontrast, βSAP97 appears to be a negative regulator ofsynaptic potentiation, favouring a shift towards synapticdepression by sequestering both AMPARs and NMDARsat extrasynaptic sites. As a result βSAP97 can blockand/or modulate both the induction and the expression ofsynaptic plasticity. Together these data reveal that synapticSAP97 isoforms can play functionally distinct roles in
regulating glutamate receptor levels at synapses and assuch influence synaptic plasticity mechanisms.
References
Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL &Henley JM (2004). Removal of AMPA receptors (AMPARs)from synapses is preceded by transient endocytosis ofextrasynaptic AMPARs. J Neurosci 24, 5172–5176.
Banker G & Goslin K (1998). Culturing Nerve Cells. MIT Press,Cambridge, MA.
Bats C, Groc L & Choquet D (2007). The interaction betweenStargazin and PSD-95 regulates AMPA receptor surfacetrafficking. Neuron 53, 719–734.
Blanpied TA, Scott DB & Ehlers MD (2002). Dynamics andregulation of clathrin coats at specialized endocytic zones ofdendrites and spines. Neuron 36, 435–449.
Bliss TV & Collingridge GL (1993). A synaptic model ofmemory: long-term potentiation in the hippocampus.Nature 361, 31–39.
Bredt DS & Nicoll RA (2003). AMPA receptor trafficking atexcitatory synapses. Neuron 40, 361–379.
Cai C, Coleman SK, Niemi K & Keinanen K (2002). Selectivebinding of synapse-associated protein 97 to GluR-Aα-amino-5-hydroxy-3-methyl-4-isoxazole propionatereceptor subunit is determined by a novel sequence motif. JBiol Chem 277, 31484–31490.
Chesler M (2003). Regulation and modulation of pH in thebrain. Physiol Rev 83, 1183–1221.
Chetkovich DM, Bunn RC, Kuo SH, Kawasaki Y, Kohwi M &Bredt DS (2002). Postsynaptic targeting of alternativepostsynaptic density-95 isoforms by distinct mechanisms. JNeurosci 22, 6415–6425.
Cheyne JE & Montgomery JM (2008). Plasticity-dependentchanges in metabotropic glutamate receptor expression atexcitatory hippocampal synapses. Mol Cell Neurosci 37,432–439.
Citri A & Malenka RC (2008). Synaptic plasticity: multipleforms, functions, and mechanisms.Neuropsychopharmacology 33, 18–41.
Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL &Scott JD (2000). Targeting of PKA to glutamate receptorsthrough a MAGUK-AKAP complex. Neuron 27, 107–119.
Degiorgis JA, Galbraith JA, Dosemeci A, Chen X & Reese TS(2008). Distribution of the scaffolding proteins PSD-95,PSD-93, and SAP97 in isolated PSDs. Brain Cell Biol 35,239–250.
Dell’Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES &Gomez LL (2006). Regulation of neuronal PKA signalingthrough AKAP targeting dynamics. Eur J Cell Biol 85,627–633.
Fukata Y, Tzingounis AV, Trinidad JC, Fukata M, BurlingameAL, Nicoll RA & Bredt DS (2005). Molecular constituents ofneuronal AMPA receptors. J Cell Biol 169, 399–404.
Groc L, Bard L & Choquet D (2009). Surface trafficking ofN-methyl-D-aspartate receptors: physiological andpathological perspectives. Neuroscience 158, 4–18.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

J Physiol 589.18 SAP97 regulates synaptic plasticity 4509
Howard MA, Elias GM, Elias LA, Swat W & Nicoll RA (2010).The role of SAP97 in synaptic glutamate receptor dynamics.Proc Natl Acad Sci U S A 107, 3805–3810.
Irwin RP, Lin SZ, Long RT & Paul SM (1994).N-methyl-D-aspartate induces a rapid, reversible, andcalcium-dependent intracellular acidosis in cultured fetal rathippocampal neurons. J Neurosci 14, 1352–1357.
Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-AriY & Medina I (2006). Opposing role of synaptic andextrasynaptic NMDA receptors in regulation of theextracellular signal-regulated kinases (ERK) activity incultured rat hippocampal neurons. J Physiol 572, 789–798.
Jeyifous O, Waites CL, Specht CG, Fujisawa S, Schubert M, LinEI, Marshall J, Aoki C, de Silva T, Montgomery JM, GarnerCC & Green WN (2009). SAP97 and CASK mediate sortingof NMDA receptors through a previously unknown secretorypathway. Nat Neurosci 12, 1011–1019.
Kneen M, Farinas J, Li Y & Verkman AS (1998). Greenfluorescent protein as a noninvasive intracellular pHindicator. Biophys J 74, 1591–1599.
Leonard AS, Davare MA, Horne MC, Garner CC & Hell JW(1998). SAP97 is associated with theα-amino-3-hydroxy-5-methylisoxazole-4-propionic acidreceptor GluR1 subunit. J Biol Chem 273, 19518–19524.
Leonoudakis D, Conti LR, Radeke CM, McGuire LM &Vandenberg CA (2004). A multiprotein trafficking complexcomposed of SAP97, CASK, Veli, and Mint1 is associatedwith inward rectifier Kir2 potassium channels. J Biol Chem279, 19051–19063.
Llopis J, McCaffery JM, Miyawaki A, Farquhar MG & Tsien RY(1998). Measurement of cytosolic, mitochondrial, and GolgipH in single living cells with green fluorescent proteins. ProcNatl Acad Sci U S A 95, 6803–6808.
Lois C, Hong EJ, Pease S, Brown EJ & Baltimore D (2002).Germline transmission and tissue-specific expression oftransgenes delivered by lentiviral vectors. Science 295,868–872.
Lu W, Man H, Ju W, Trimble WS, MacDonald JF & Wang YT(2001). Activation of synaptic NMDA receptors inducesmembrane insertion of new AMPA receptors and LTP incultured hippocampal neurons. Neuron 29, 243–254.
Makino H & Malinow R (2009). AMPA receptor incorporationinto synapses during LTP: the role of lateral movement andexocytosis. Neuron 64, 381–390.
Malenka RC & Nicoll RA (1999). Long-term potentiation – adecade of progress? Science 285, 1870–1874.
Malinow R & Malenka RC (2002). AMPA receptor traffickingand synaptic plasticity. Annu Rev Neurosci 25, 103–126.
Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A,Epis R, Borroni B, Cattabeni F, Sala C, Padovani A & Di LucaM (2007). Synapse-associated protein-97 mediatesα-secretase ADAM10 trafficking and promotes its activity. JNeurosci 27, 1682–1691.
Mauceri D, Cattabeni F, Di Luca M & Gardoni F (2004).Calcium/calmodulin-dependent protein kinase IIphosphorylation drives synapse-associated protein 97 intospines. J Biol Chem 279, 23813–23821.
McLaughlin M, Hale R, Ellston D, Gaudet S, Lue RA & Viel A(2002). The distribution and function of alternatively splicedinsertions in hDlg. J Biol Chem 277, 6406–6412.
Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM,Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK,Hayden MR, Murphy TH & Raymond LA (2010). Earlyincrease in extrasynaptic NMDA receptor signaling andexpression contributes to phenotype onset in Huntington’sdisease mice. Neuron 65, 178–190.
Montgomery JM & Madison DV (2002). State-dependentheterogeneity in synaptic depression between pyramidal cellpairs. Neuron 33, 765–777.
Montgomery JM, Pavlidis P & Madison DV (2001). Pairrecordings reveal all-silent synaptic connections and thepostsynaptic expression of long-term potentiation. Neuron29, 691–701.
Montgomery JM, Selcher JC, Hanson JE & Madison DV (2005).Dynamin-dependent NMDAR endocytosis during LTD andits dependence on synaptic state. BMC Neurosci 6, 48.
Montgomery JM, Zamorano PL & Garner CC (2004).MAGUKs in synapse assembly and function: an emergingview. Cell Mol Life Sci 61, 911–929.
Mori K, Iwao K, Miyoshi Y, Nakagawara A, Kofu K, Akiyama T,Arita N, Hayakawa T & Nakamura Y (1998). Identification ofbrain-specific splicing variants of the hDLG1 gene andaltered splicing in neuroblastoma cell lines. J Hum Genet 43,123–127.
Muller BM, Kistner U, Veh RW, Cases-Langhoff C, Becker B,Gundelfinger ED & Garner CC (1995). Molecularcharacterization and spatial distribution of SAP97, a novelpresynaptic protein homologous to SAP90 and theDrosophila discs-large tumor suppressor protein. J Neurosci15, 2354–2366.
Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T,Hayashi Y & Sheng M (2004). Quaternary structure, proteindynamics, and synaptic function of SAP97 controlled by L27domain interactions. Neuron 44, 453–467.
Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD & Choquet D(2009). Endocytic trafficking and recycling maintain a poolof mobile surface AMPA receptors required for synapticpotentiation. Neuron 63, 92–105.
Racz B, Blanpied TA, Ehlers MD & Weinberg RJ (2004). Lateralorganization of endocytic machinery in dendritic spines. NatNeurosci 7, 917–918.
Rumbaugh G, Sia GM, Garner CC & Huganir RL (2003).Synapse-associated protein-97 isoform-specific regulation ofsurface AMPA receptors and synaptic function in culturedneurons. J Neurosci 23, 4567–4576.
Sans N, Racca C, Petralia RS, Wang YX, McCallum J &Wenthold RJ (2001). Synapse-associated protein 97selectively associates with a subset of AMPA receptors earlyin their biosynthetic pathway. J Neurosci 21,7506–7516.
Schluter OM, Xu W & Malenka RC (2006). AlternativeN-terminal domains of PSD-95 and SAP97 governactivity-dependent regulation of synaptic AMPA receptorfunction. Neuron 51, 99–111.
Shen K & Meyer T (1999). Dynamic control of CaMKIItranslocation and localization in hippocampal neurons byNMDA receptor stimulation. Science 284, 162–166.
Shepherd JD & Huganir RL (2007). The cell biology of synapticplasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol23, 613–643.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society

4510 D. Li and others J Physiol 589.18
Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ,Svoboda K & Malinow R (1999). Rapid spine delivery andredistribution of AMPA receptors after synaptic NMDAreceptor activation. Science 284, 1811–1816.
Specht CG & Triller A (2008). The dynamics of synapticscaffolds. Bioessays 30, 1062–1074.
Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL &Scott JD (2002). Regulation of GluR1 by the A-kinaseanchoring protein 79 (AKAP79) signaling complex sharesproperties with long-term depression. J Neurosci 22,3044–3051.
Tsui J, Inagaki M & Schulman H (2005).Calcium/calmodulin-dependent protein kinase II (CaMKII)localization acts in concert with substrate targeting to createspatial restriction for phosphorylation. J Biol Chem 280,9210–9216.
Tsuriel S, Geva R, Zamorano P, Dresbach T, Boeckers T,Gundelfinger ED, Garner CC & Ziv NE (2006). Local sharingas a predominant determinant of synaptic matrix moleculardynamics. PLoS Biol 4, e271.
Waites CL, Specht CG, Hartel K, Leal-Ortiz S, Genoux D, Li D,Drisdel RC, Jeyifous O, Cheyne JE, Green WN, MontgomeryJM & Garner CC (2009). Synaptic SAP97 isoforms regulateAMPA receptor dynamics and access to presynapticglutamate. J Neurosci 29, 4332–4345.
Author contributions
Electrophysiology experiments were performed and analysed inthe Montgomery lab by D.L., C.B.M. and J.M.M. Supportingelectrophysiology experiments were performed by D.G. Allimaging experiments and analysis were performed in theGarner lab by C.G.S. and C.L.W. Minor immunocytochemistryexperiments were performed by J.W.F. and D.L. SAP97constructs were built by C.G.S., S.O. and C.C.G. J.M.M., C.C.G.,D.L., C.G.S. and C.L.W. conceived and designed the experiments.The manuscript was written by J.M.M., C.C.G., D.L. and C.G.S.All authors have approved the final version of the manuscript.
Acknowledgements
We would like to thank members of the Montgomery and Garnerlabs for helpful discussion. This work was supported by NIHgrant no. DA016758 to C.C.G., NIH NRSA to C.L.W., a DFGpostdoctoral fellowship to C.G.S., a UOA doctoral scholarshipto D.L., and Auckland Medical Research Foundation, LotteriesHealth Board and Maurice and Phyllis Paykel Trust to J.M.M.
C© 2011 The Authors. Journal compilation C© 2011 The Physiological Society
Copyright © 2022 FDOKUMEN




















