
Role of Cadmium in the Regulation of AR Gene Expression and Activity
13
Role of Cadmium in the Regulation of AR Gene Expression and Activity MARY BETH MARTIN, H. JAMES VOELLER, EDWARD P. GELMANN, JIANMING LU, ELLY-GERALD STOICA, ELIJAH J. HEBERT, RONALD REITER, BALJIT SINGH, MARK DANIELSEN, ELIZABETH PENTECOST, AND ADRIANA STOICA Departments of Biochemistry and Molecular Biology (M.B.M., J.L., M.D., E.P., A.S.) and Oncology (M.B.M., H.J.V., E.P.G., E.-G.S., E.J.H., R.R., B.S., A.S.), Lombardi Cancer Center, School of Nursing and Health Studies (A.S.), Georgetown University, Washington, D.C. 20007 Treatment of human prostate cancer cells, LNCaP, with cad- mium stimulated cell growth. There was a 2.4-fold increase in the population of cells in the S G 2 M phase by d 4 and a 2.7-fold increase in cell number by d 8. The metal decreased the concentration of AR protein and mRNA (80 and 60%, re- spectively) and increased the expression of prostate-specific antigen and the homeobox gene, NKX 3.1 (6-fold) that was blocked by an antiandrogen. In addition, cadmium activated the AR in mouse L cells containing an MMTV-luciferase re- porter gene (4-fold increase) and in COS-1 cells transfected with wild-type AR and an MMTV-CAT reporter gene (7-fold increase). Cadmium also activated a chimeric receptor (GAL- AR) containing the hormone-binding domain of AR. The metal bound to AR with an equilibrium dissociation constant of 1.19 10 10 M. Cadmium blocked the binding of androgen to the receptor but did not alter its affinity (dissociation con- stant 2.8 10 10 M), suggesting that the metal is an inhibitor of hormone binding. In castrated animals, a single, low, en- vironmentally relevant dose of cadmium (20 g/kg body weight) increased the wet weight of the prostate (1.97- to 3-fold) and the seminal vesicle complex (approximately 1.5- fold) and increased the expression of the androgen-regulated gene, probasin (27-fold). The in vivo effects were also blocked by an antiandrogen. (Endocrinology 143: 263–275, 2002) P ROSTATE CANCER IS the most common cancer in men and the second leading cause of cancer-related deaths in American men (1). The risk factors for prostate cancer include age, endocrine status, genetic susceptibility, occu- pation, ethnicity, race, UV radiation, diet, and environmental factors (2). The importance of the latter in prostate cancer is supported by migration studies. When men from countries with a low prostate cancer incidence (Asia) migrate to the United States, a country with a high incidence of the disease, they acquire a risk that approaches the risk of American men (3–5). Although the factors responsible for the increased risk are not known, several studies suggest that exposure to the heavy metal cadmium may play a role in the etiology of the disease (6 –13). The growth and development of the prostate gland is under the control of androgenic sex steroids (14 –16). The effects of androgens are mediated by the AR, which regulates the expression of genes involved in the growth as well as in the secretory function of the gland. Androgens may also play a central role in the development of prostate cancer. A causal relationship between androgens and prostate cancer devel- opment is supported by the androgen-sensitivity and re- sponse to hormonal therapy of many prostate cancers (15, 16). Though the precise mechanism by which androgens affect prostate carcinogenesis is unknown, several hypoth- eses have been proposed. Androgens are strong promoters of carcinogenesis, acting via AR-mediated mechanisms, and may enhance the carcinogenicity of endogenous estrogen metabolites and estrogen- and prostatitis-generated reactive oxygen species and possible weak environmental carcino- gens. All these processes seem to be modulated by a variety of environmental factors such as diet and life style habits, as well as by genetic determinants such as hereditary suscep- tibility and polymorphic genes that encode for steroid re- ceptors and enzymes involved in the metabolism and acti- vation of steroid hormones (15, 17, 18). The AR is a member of a superfamily of genes that encode for steroid/thyroid hormone receptors. Members of the re- ceptor family are ligand-dependent nuclear transcription factors, which have a similar modular structure (19). The N-terminal domain of the receptor is involved in the regu- lation of transcription and is the least conserved domain between members of the receptor family. The central region is a short, well-conserved cysteine-rich region, which corre- sponds to the DNA-binding domain. The C-terminal region of the receptor is the moderately conserved hormone-bind- ing domain, which is composed of -helices that form a ligand-binding pocket (20 –27). Recent studies from this laboratory suggest that cadmium has potent estrogen-like activity. The metal binds with high affinity to the hormone-binding domain of ER- and acti- vates the receptor (28). The interaction of cadmium with ER- involves several amino acids, some of which are also conserved in the hormone-binding domain of the AR (20, 25). In this study, we asked whether cadmium also mimics the effects of androgens by activating the AR and thereby poses a health risk. Abbreviations: ARA 70 , AR-associated protein 70; BN, nonspecific binding; BT, total binding; B S , specific binding; CAT, chloramphenicol acetyl transferase; CPA, cyproterone acetate; DHT, dihydrotestosterone; IMEM, improved MEM; K d , dissociation constant; PSA, prostate-specific antigen; RNase, ribonuclease. 0013-7227/02/$03.00/0 Endocrinology 143(1):263–275 Printed in U.S.A. Copyright © 2002 by The Endocrine Society 263
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Role of Cadmium in the Regulation of AR Gene Expression and Activity

Role of Cadmium in the Regulation of AR GeneExpression and Activity
MARY BETH MARTIN, H. JAMES VOELLER, EDWARD P. GELMANN, JIANMING LU,ELLY-GERALD STOICA, ELIJAH J. HEBERT, RONALD REITER, BALJIT SINGH, MARK DANIELSEN,ELIZABETH PENTECOST, AND ADRIANA STOICA
Departments of Biochemistry and Molecular Biology (M.B.M., J.L., M.D., E.P., A.S.) and Oncology (M.B.M., H.J.V., E.P.G.,E.-G.S., E.J.H., R.R., B.S., A.S.), Lombardi Cancer Center, School of Nursing and Health Studies (A.S.), GeorgetownUniversity, Washington, D.C. 20007
Treatment of human prostate cancer cells, LNCaP, with cad-mium stimulated cell growth. There was a 2.4-fold increase inthe population of cells in the S � G2M phase by d 4 and a2.7-fold increase in cell number by d 8. The metal decreasedthe concentration of AR protein and mRNA (80 and 60%, re-spectively) and increased the expression of prostate-specificantigen and the homeobox gene, NKX 3.1 (6-fold) that wasblocked by an antiandrogen. In addition, cadmium activatedthe AR in mouse L cells containing an MMTV-luciferase re-porter gene (4-fold increase) and in COS-1 cells transfectedwith wild-type AR and an MMTV-CAT reporter gene (7-foldincrease). Cadmium also activated a chimeric receptor (GAL-
AR) containing the hormone-binding domain of AR. The metalbound to AR with an equilibrium dissociation constant of1.19 � 10�10 M. Cadmium blocked the binding of androgen tothe receptor but did not alter its affinity (dissociation con-stant � 2.8 � 10�10 M), suggesting that the metal is an inhibitorof hormone binding. In castrated animals, a single, low, en-vironmentally relevant dose of cadmium (20 �g/kg bodyweight) increased the wet weight of the prostate (1.97- to3-fold) and the seminal vesicle complex (approximately 1.5-fold) and increased the expression of the androgen-regulatedgene, probasin (27-fold). The in vivo effects were also blockedby an antiandrogen. (Endocrinology 143: 263–275, 2002)
PROSTATE CANCER IS the most common cancer in menand the second leading cause of cancer-related deaths
in American men (1). The risk factors for prostate cancerinclude age, endocrine status, genetic susceptibility, occu-pation, ethnicity, race, UV radiation, diet, and environmentalfactors (2). The importance of the latter in prostate cancer issupported by migration studies. When men from countrieswith a low prostate cancer incidence (Asia) migrate to theUnited States, a country with a high incidence of the disease,they acquire a risk that approaches the risk of American men(3–5). Although the factors responsible for the increased riskare not known, several studies suggest that exposure to theheavy metal cadmium may play a role in the etiology of thedisease (6–13).
The growth and development of the prostate gland isunder the control of androgenic sex steroids (14–16). Theeffects of androgens are mediated by the AR, which regulatesthe expression of genes involved in the growth as well as inthe secretory function of the gland. Androgens may also playa central role in the development of prostate cancer. A causalrelationship between androgens and prostate cancer devel-opment is supported by the androgen-sensitivity and re-sponse to hormonal therapy of many prostate cancers (15,16). Though the precise mechanism by which androgensaffect prostate carcinogenesis is unknown, several hypoth-eses have been proposed. Androgens are strong promoters
of carcinogenesis, acting via AR-mediated mechanisms, andmay enhance the carcinogenicity of endogenous estrogenmetabolites and estrogen- and prostatitis-generated reactiveoxygen species and possible weak environmental carcino-gens. All these processes seem to be modulated by a varietyof environmental factors such as diet and life style habits, aswell as by genetic determinants such as hereditary suscep-tibility and polymorphic genes that encode for steroid re-ceptors and enzymes involved in the metabolism and acti-vation of steroid hormones (15, 17, 18).
The AR is a member of a superfamily of genes that encodefor steroid/thyroid hormone receptors. Members of the re-ceptor family are ligand-dependent nuclear transcriptionfactors, which have a similar modular structure (19). TheN-terminal domain of the receptor is involved in the regu-lation of transcription and is the least conserved domainbetween members of the receptor family. The central regionis a short, well-conserved cysteine-rich region, which corre-sponds to the DNA-binding domain. The C-terminal regionof the receptor is the moderately conserved hormone-bind-ing domain, which is composed of �-helices that form aligand-binding pocket (20–27).
Recent studies from this laboratory suggest that cadmiumhas potent estrogen-like activity. The metal binds with highaffinity to the hormone-binding domain of ER-� and acti-vates the receptor (28). The interaction of cadmium withER-� involves several amino acids, some of which are alsoconserved in the hormone-binding domain of the AR (20, 25).In this study, we asked whether cadmium also mimics theeffects of androgens by activating the AR and thereby posesa health risk.
Abbreviations: ARA70, AR-associated protein 70; BN, nonspecificbinding; BT, total binding; BS, specific binding; CAT, chloramphenicolacetyl transferase; CPA, cyproterone acetate; DHT, dihydrotestosterone;IMEM, improved MEM; Kd, dissociation constant; PSA, prostate-specificantigen; RNase, ribonuclease.
0013-7227/02/$03.00/0 Endocrinology 143(1):263–275Printed in U.S.A. Copyright © 2002 by The Endocrine Society
263

Materials and MethodsTissue culture
The human prostate cancer cells, LNCaP (ATCC, Manassas, VA),were grown in improved MEM (IMEM) supplemented with 5% FCS. At70% confluence, the medium was changed to phenol red-free IMEMsupplemented with 5% charcoal-stripped calf serum. Cells were main-tained in hormone-depleted medium for 2 d before treatment withcadmium chloride, zinc chloride, dihydrotestosterone (DHT) (Sigma, St.Louis, MO), methyltrienolone (R1881; NEN Life Science Products, Wil-mington, DE), casodex (Zeneca Pharmaceuticals, Wilmington, DE), andhydroxyflutamide (Sigma).
For anchorage-dependent growth assays, LNCaP cells were plated at105 cells/well into 6-well plates in IMEM supplemented with 10% char-coal-stripped serum. Cells were grown to 40% confluence, and the me-dium was changed to phenol red-free IMEM supplemented with 5%charcoal-stripped serum. After 2 d in this medium, cells were treatedwith either 10�9 m DHT or 10�6 m cadmium chloride. Cells weretrypsinized at the specific time points and counted with a CoulterCounter (Coulter Electronics Inc., Hialeah, FL).
Cell cycle phase analysis
For cell cycle analysis, LNCaP cells were grown as described aboveand treated with either 10�9 m DHT or 10�6 m cadmium chloride. Thecells were trypsinized to obtain a single cell suspension and verifiedmicroscopically after neutralizing with medium containing serum. Thecell number was adjusted to 2 � 106 cells for each treatment. The cellswere washed twice with PBS, centrifuged at 1000 rpm for 5 min, andsuspended in 0.1 ml citrate/dimethylsulfoxide buffer. The DNA contentwas measured by Vindelov staining (29).
Western blot analysis
LNCaP cells were grown as described above and treated with either10�9 m DHT, or 10�10–10�6 m cadmium chloride for 24 h in the presenceor absence of the antiandrogen hydroxyflutamide (10�6 m). Cells werelysed in a buffer containing 20 nm Tris HCl (pH 9), 150 nm sodiumchloride, 1% Nonidet P-40, 20 mm EDTA, 1 mm dithiothreitol, and theprotease inhibitors pefabloc, aprotinin, and leupeptin. Lysates wereloaded onto SDS-polyacrylamide gels. Gels were electrotransferred ontonitrocellulose membranes, washed in PBS five times at room tempera-ture. Membranes were kept in blocking buffer overnight at 4 C andincubated for 1 h with 4 �g/ml of the polyclonal rabbit anti-AR antibody(Affinity BioReagents, Inc., Golden, CO) at room temperature. Afterthree additional washes in PBS, membranes were incubated with thehorseradish peroxidase-conjugated secondary antibody (1:2000) inblocking buffer for 1 h at room temperature. Detection was performedby chemiluminescence using SuperSignal chemiluminescent substrate(Pierce Chemical Co., Rockford, IL).
AR hormone-binding assay
The ability of cadmium to compete with R1881 for binding to the ARwas determined in extracts from LNCaP cells by a multiple-dose ligand-binding assay. LNCaP cells were lysed by sonication in a high-salt buffercontaining 10 mm Tris (pH 7.5), 1.5 mm EDTA, 5 mm sodium molybdate,0.4 m KCl, 1 mm monothioglycerol, and 2 mm leupeptin. The homog-enate was incubated on ice for 30 min and centrifuged at 100,000 � g for1 h at 4 C (30). The protein concentration of the cell extract was deter-mined by the Bradford method. Cell extracts were then pretreated with10�6 m cadmium chloride for 1 h at 4 C; and concentrations of 17�-methyl-[3H]R1881, ranging from 3 � 10�16 m to 3 � 10�9 m were addedin the presence (nonspecific binding, BN) or absence (total binding, BT)of a 1000-fold molar excess of cold R1881. Two hours after incubatingat 37 C, unbound steroid was removed by the addition of 5% dextran-coated charcoal. The amount of radioactivity was measured by scintil-lation counting. Total radioactivity was measured for each [3H]R1881concentration, and specific binding (BS) was determined as the differ-ence between BT and BN (BS � BT � BN). Data were plotted accordingto the method of Scatchard (31). The dissociation constant (Kd) andbinding capacity were determined. The AR concentration was calculated
as binding capacity divided by the total protein concentration and wasexpressed as femtomoles per milligram protein.
The ability of cadmium to bind to the AR was determined in cellextracts from LNCaP cells or COS-1 cells transiently transfected with theAR gene, by a multiple-dose ligand-binding assay. Cell extracts wereincubated with various concentrations of 109Cd (10�12–10�6 m), in thepresence or absence of a 1000-fold molar excess of R1881, for 2 h at 37C. Free radioactivity was removed by the addition of 5% dextran-coatedcharcoal, and the amount of 109Cd bound to the AR was measured byscintillation counting. The data were analyzed by the method of Scat-chard (31).
Measurement of AR, prostate-specific antigen (PSA), andNKX3.1 mRNA
Total cellular RNA was extracted using the RNazol method. Theamounts of AR, PSA, and NKX3.1 mRNA were determined by a ribo-nuclease (RNase) protection assay. 32P-Labeled antisense RNA (cRNA)was synthesized in vitro from AR-2 (32), PSA (32), and acidic ribosomalphosphoprotein PO (p36B4) (33) using T7 polymerase, and from thehuman homeobox gene NKX3.1 (34) using T3 polymerase. The AR probeused for hybridization was a 713-bp EcoRI/HindII fragment coveringnucleotides 1850–2563 of the human AR cDNA (32). Sixty microgramsof total RNA were hybridized, for 16 h at 50 C, to the 32P-labeled cRNAs.The samples were digested with RNase A for 30 min at 25 C. Theprotected cRNA probes were resolved on 6% polyacrylamide gels. Thebands were visualized by autoradiography and quantified by phosphorimaging (Molecular Dynamics, Inc., Sunnyvale, CA). The amounts ofAR, PSA, and NKX3.1 mRNAs were normalized to the amount of acidicribosomal phosphoprotein PO mRNA.
Transient transfections and chloramphenicol acetyltransferase (CAT) assays
A low-temperature and low-pH calcium phosphate method was em-ployed to transfect COS-1 cells (35). COS-1 cells were plated, at a densityof 3 � 106 cells/150-mm dish, in phenol red-free IMEM containing 10%charcoal-treated calf serum, for 24 h. The cells were transfected with 120�g DNA containing 15 �g of an AR expression vector or the chimericreceptor, GAL-AR (amino acids 624–918) (36), 75 �g of the reporterconstruct MMTV-CAT or a GAL4-CAT reporter construct (17m2GCAT),respectively, 6 �g of �-galactosidase, and salmon sperm DNA. TheLipofectAMINE (Life Technologies, Inc./BRL, Rockville, MD) methodwas used to cotransfect COS-1 cells with wild-type AR or the chimericreceptor GAL-AR and MMTV-CAT or 17M2GCAT, respectively (37), inthe presence or absence of the coactivator ARA70 (AR-associated protein70). Cells (106) were plated into 100-mm dishes and transfected with 2�g of the AR expression vector, wild-type or GAL-AR; 10 �g of thereporter construct MMTV-CAT or 17m2GCAT, respectively; and 2, 4, 5,6, or 10 �g of the coactivator AR-associated protein 70 (ARA70). Sixteento 18 h after transfection, the precipitate was washed off, and the cellswere replenished with phenol red-free IMEM containing 10% charcoal-stripped serum in the presence or absence of 10�9 m DHT, 10�6 mcadmium chloride, or 10�5 m zinc chloride. The cells were harvested, 24 hlater, and CAT activity was measured as described previously (38). CATactivity was expressed as the percent conversion of chloramphenicol toits acetylated forms and was normalized to the activity of �-galactosi-dase. The increase in CAT activity, in response to treatment, was ex-pressed relative to untreated controls.
Stable transfections and luciferase assays
Mouse L cells, L929, were cultured in DMEM supplemented with 3%calf serum. The day before transfection, 106 cells were plated on 100-mmtissue culture dishes. The calcium phosphate method (35) was used totransfect 20 �g MMTV-luciferase and 0.5 �g pSV2-neo into the L cells.After transfection, the cells were washed using PBS, and the mediumwas replaced with DMEM containing 3% calf serum for 24 h. Stablytransfected cells were selected in DMEM supplemented with 3% calfserum and 500 �g/ml G418 for about 1 month. Colonies were isolatedand treated with 10�9 m DHT, 10�6 m cadmium chloride, and 10�5 m zincchloride. The amount of luciferase activity was determined using the
264 Endocrinology, January 2002, 143(1):263–275 Martin et al. • Cadmium Regulation of AR
luciferase assay system kit (Promega Corp., Madison, WI) as describedby the manufacturer.
Animal studies
Male Wistar [Hanover and Hsd: (WI) BR] rats and C57BL/6-TgNmice were obtained from Harlan Inc. (Indianapolis, IN) and The JacksonLaboratory (Bar Harbor, MN), respectively. Animals were housed, threeto a cage, in a conventional, well ventilated animal room kept at 22 �2 C and 40–70% relative humidity on a 12-h light, 12-h dark cycle. Theanimals had free access to water and Purina diet (Quality Lab Products,Elkridge, MD). At approximately 8 wk of age, the rats or mice werecastrated either surgically or chemically using cyproterone acetate (CPA,Sigma). CPA was dissolved in 20% benzoylbenzoate in peanut oil at aconcentration of 85 mg/ml. Animals were given ip injections of CPA (50mg/kg body weight) daily for 18 d.
To determine the effect of cadmium chloride on the wet weight of theprostate and the seminal vesicle complex (seminal vesicles with coag-ulating glands), castrated rats or mice were randomly placed into eightgroups. Each group contained five to eight animals. After a 7-d recoveryfrom castration, each group was treated as indicated below. One groupof animals did not receive treatment (control). A testosterone propionatepellet (100 mg/pellet, 112-d release (Innovative Research of America,Sarasota, FL) was sc implanted into the intrascapular region of twogroups of animals under methoxyflurane anesthesia. Two groups ofanimals received a single dose of cadmium chloride (10 �g/kg; ipinjection) prepared in PBS, whereas two additional groups received twodoses of cadmium chloride (2 � 10 �g/kg) over 2 d (ip injection). Groupsthat were treated with an antiandrogen received ip injections of CPA (50mg/kg body weight) daily for 10 d. Ten days after the start of treatment,the rats or mice, as well as four intact animals, were killed using CO2.The prostate, seminal vesicles with the coagulating glands, testes, livers,and kidneys were removed, freed from fat tissue, and weighed. Allorgans were fixed and prepared for histological examination. All animalstudies were conducted in accord with the Guidelines for Care and Useof Experimental Animals.
Real-time PCR assay
Total RNA was isolated from the prostate glands with RNA STAT-60(Tel-Test, Friendswood, TX). Twenty micrograms of RNA were sus-pended in 10 �l of 20 mm Tris-HCl (pH 8.4), 50 mm KCl, and 20 mmMgCl2 containing 2 U deoxyribonuclease and were incubated at 37 C for45 min. Deoxyribonuclease was heat inactivated before the addition of50 U MuLV reverse transcriptase, 20 pmol random hexamers, 20 URNase inhibitor, 10 nm deoxynucleotide triphosphate, and 2 �l RTbuffer. The samples were incubated for 10 min at 25 C, 60 min at 37 C,and 5 min at 95 C. The RT product was diluted to a final vol of 200 �lin sterile water. For real time PCR, the iCycler iQ Detection System(Bio-Rad Laboratories, Inc., Hercules, CA) was used. Amplification wasperformed in a 25-�l final vol containing 1� reaction buffer (SYBR GreenPCR core reagent kit; PE Applied Biosystems, Foster City, CA), 3 mmMgCl2, 0.25 �l Platinum Taq polymerase (Life Technologies, Inc., Carls-bad, CA), 0.2 mm deoxynucleotide triphosphate, 2 �l cDNA, and 0.5 �mprimers. The oligonucleotide primers used to detect rat probasin were5�-GGACCTACTTCCGTCGCATT-3� and 5�-ACGTCTTGGGATCTC-CTTCC-3�. The primers used to detect rat glyceraldehyde 3-phosphatedehydrogenase were 5�-TCCTGCACCACCAACTGCTTAG-3� and 5�-CAGATCCACAACGGATACATTGG-3�. The PCR reaction conditionswere: 10 min at 95 C, followed by 50 cycles of 20 sec at 95 C, 30 sec at58 C, and 30 sec at 72 C. Fluorescent data were collected during the 72-Cstep.
ResultsEffect of cadmium on the growth of LNCaP cells
Previous studies from our laboratory demonstrated thatthe heavy-metal cadmium binds to and activates ER-� (28,38). The goal of the present study was to determine whetherthe metal binds to and activates the AR. To examine theandrogen-like effects of cadmium, the human prostate cancer
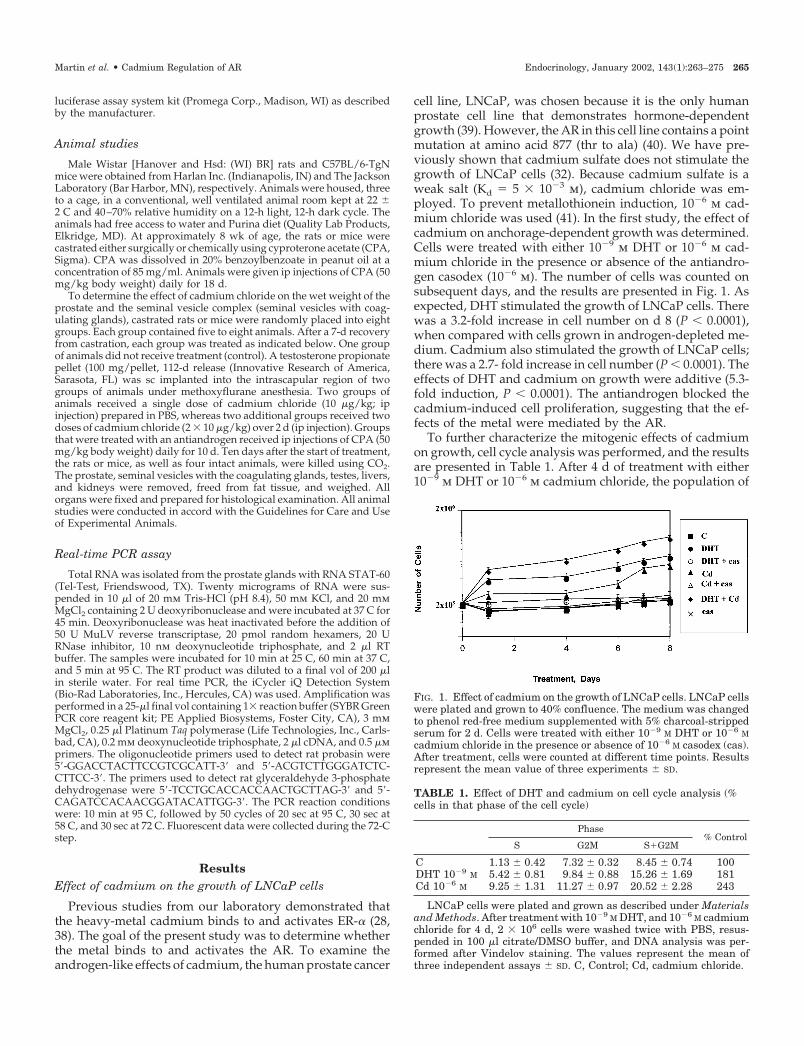
cell line, LNCaP, was chosen because it is the only humanprostate cell line that demonstrates hormone-dependentgrowth (39). However, the AR in this cell line contains a pointmutation at amino acid 877 (thr to ala) (40). We have pre-viously shown that cadmium sulfate does not stimulate thegrowth of LNCaP cells (32). Because cadmium sulfate is aweak salt (Kd � 5 � 10�3 m), cadmium chloride was em-ployed. To prevent metallothionein induction, 10�6 m cad-mium chloride was used (41). In the first study, the effect ofcadmium on anchorage-dependent growth was determined.Cells were treated with either 10�9 m DHT or 10�6 m cad-mium chloride in the presence or absence of the antiandro-gen casodex (10�6 m). The number of cells was counted onsubsequent days, and the results are presented in Fig. 1. Asexpected, DHT stimulated the growth of LNCaP cells. Therewas a 3.2-fold increase in cell number on d 8 (P � 0.0001),when compared with cells grown in androgen-depleted me-dium. Cadmium also stimulated the growth of LNCaP cells;there was a 2.7- fold increase in cell number (P � 0.0001). Theeffects of DHT and cadmium on growth were additive (5.3-fold induction, P � 0.0001). The antiandrogen blocked thecadmium-induced cell proliferation, suggesting that the ef-fects of the metal were mediated by the AR.
To further characterize the mitogenic effects of cadmiumon growth, cell cycle analysis was performed, and the resultsare presented in Table 1. After 4 d of treatment with either10�9 m DHT or 10�6 m cadmium chloride, the population of
FIG. 1. Effect of cadmium on the growth of LNCaP cells. LNCaP cellswere plated and grown to 40% confluence. The medium was changedto phenol red-free medium supplemented with 5% charcoal-strippedserum for 2 d. Cells were treated with either 10�9 M DHT or 10�6 Mcadmium chloride in the presence or absence of 10�6 M casodex (cas).After treatment, cells were counted at different time points. Resultsrepresent the mean value of three experiments � SD.
TABLE 1. Effect of DHT and cadmium on cell cycle analysis (%cells in that phase of the cell cycle)
Phase% Control
S G2M S�G2M
C 1.13 � 0.42 7.32 � 0.32 8.45 � 0.74 100DHT 10�9 M 5.42 � 0.81 9.84 � 0.88 15.26 � 1.69 181Cd 10�6 M 9.25 � 1.31 11.27 � 0.97 20.52 � 2.28 243
LNCaP cells were plated and grown as described under Materialsand Methods. After treatment with 10�9 M DHT, and 10�6 M cadmiumchloride for 4 d, 2 � 106 cells were washed twice with PBS, resus-pended in 100 �l citrate/DMSO buffer, and DNA analysis was per-formed after Vindelov staining. The values represent the mean ofthree independent assays � SD. C, Control; Cd, cadmium chloride.
Martin et al. • Cadmium Regulation of AR Endocrinology, January 2002, 143(1):263–275 265
cells in the S � G2M phases of the cell cycle increased by1.8-fold in DHT-treated cells (P � 0.0031) and by 2.4-fold incadmium treated cells (P � 0.001), suggesting that both DHTand cadmium induced significant proliferation.
Effect of cadmium on AR protein concentration
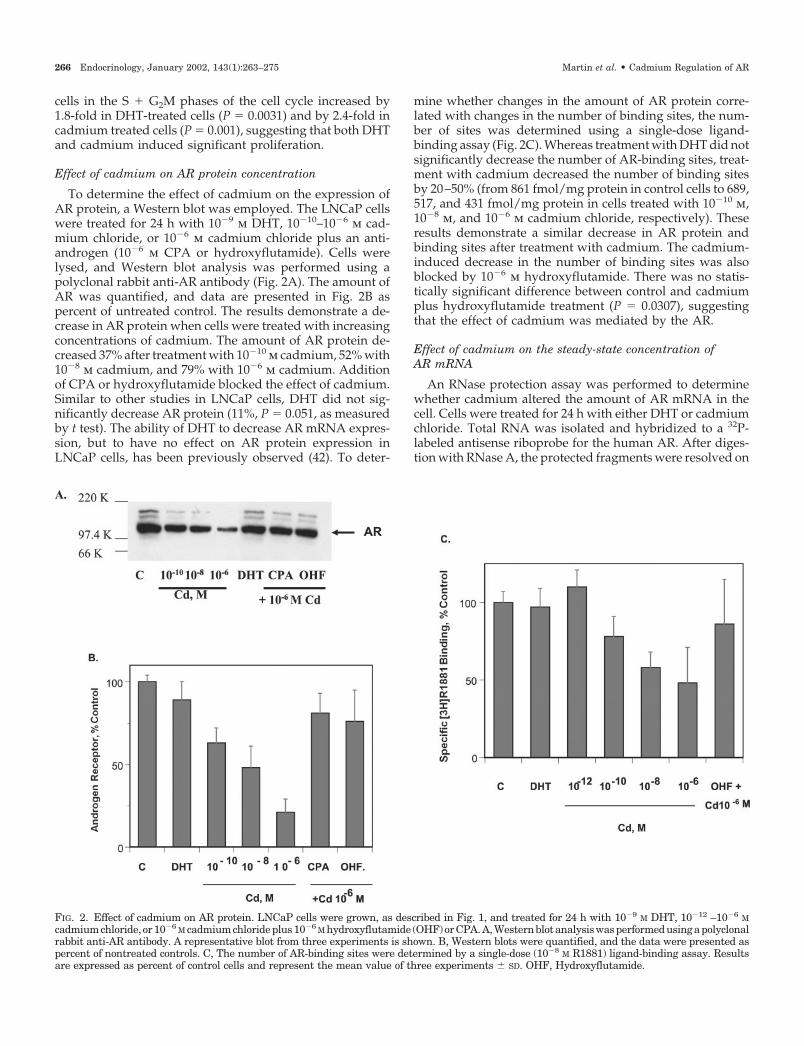
To determine the effect of cadmium on the expression ofAR protein, a Western blot was employed. The LNCaP cellswere treated for 24 h with 10�9 m DHT, 10�10–10�6 m cad-mium chloride, or 10�6 m cadmium chloride plus an anti-androgen (10�6 m CPA or hydroxyflutamide). Cells werelysed, and Western blot analysis was performed using apolyclonal rabbit anti-AR antibody (Fig. 2A). The amount ofAR was quantified, and data are presented in Fig. 2B aspercent of untreated control. The results demonstrate a de-crease in AR protein when cells were treated with increasingconcentrations of cadmium. The amount of AR protein de-creased 37% after treatment with 10�10 m cadmium, 52% with10�8 m cadmium, and 79% with 10�6 m cadmium. Additionof CPA or hydroxyflutamide blocked the effect of cadmium.Similar to other studies in LNCaP cells, DHT did not sig-nificantly decrease AR protein (11%, P � 0.051, as measuredby t test). The ability of DHT to decrease AR mRNA expres-sion, but to have no effect on AR protein expression inLNCaP cells, has been previously observed (42). To deter-
mine whether changes in the amount of AR protein corre-lated with changes in the number of binding sites, the num-ber of sites was determined using a single-dose ligand-binding assay (Fig. 2C). Whereas treatment with DHT did notsignificantly decrease the number of AR-binding sites, treat-ment with cadmium decreased the number of binding sitesby 20–50% (from 861 fmol/mg protein in control cells to 689,517, and 431 fmol/mg protein in cells treated with 10�10 m,10�8 m, and 10�6 m cadmium chloride, respectively). Theseresults demonstrate a similar decrease in AR protein andbinding sites after treatment with cadmium. The cadmium-induced decrease in the number of binding sites was alsoblocked by 10�6 m hydroxyflutamide. There was no statis-tically significant difference between control and cadmiumplus hydroxyflutamide treatment (P � 0.0307), suggestingthat the effect of cadmium was mediated by the AR.
Effect of cadmium on the steady-state concentration ofAR mRNA
An RNase protection assay was performed to determinewhether cadmium altered the amount of AR mRNA in thecell. Cells were treated for 24 h with either DHT or cadmiumchloride. Total RNA was isolated and hybridized to a 32P-labeled antisense riboprobe for the human AR. After diges-tion with RNase A, the protected fragments were resolved on
FIG. 2. Effect of cadmium on AR protein. LNCaP cells were grown, as described in Fig. 1, and treated for 24 h with 10�9 M DHT, 10�12 –10�6 Mcadmiumchloride, or10�6 M cadmiumchlorideplus10�6 M hydroxyflutamide (OHF)orCPA.A,Westernblotanalysiswasperformedusingapolyclonalrabbit anti-AR antibody. A representative blot from three experiments is shown. B, Western blots were quantified, and the data were presented aspercent of nontreated controls. C, The number of AR-binding sites were determined by a single-dose (10�8 M R1881) ligand-binding assay. Resultsare expressed as percent of control cells and represent the mean value of three experiments � SD. OHF, Hydroxyflutamide.
266 Endocrinology, January 2002, 143(1):263–275 Martin et al. • Cadmium Regulation of AR

a 6% polyacrylamide gel. The bands were quantified byscanning densitometry and normalized to the amount ofacidic ribosomal protein PO, which is constitutively ex-pressed in the presence of cadmium (38). In contrast to theeffects of the androgen on the amount of AR protein, treat-ment with 10�9 m DHT produced a decrease of approxi-mately 60% in the amount of AR mRNA that was blocked by10�6 m casodex (Fig. 3A). Treatment for 24 h with 10�10–10�6
m cadmium produced a 50–60% decrease in the amount ofAR mRNA (Fig. 3A), which correlates with the effect of themetal on AR protein concentration. Similar to the effects onAR protein, the effects of cadmium on AR mRNA were alsoblocked by the antiandrogens casodex and hydroxyflut-amide. A time course of the response to 10�6 m cadmium ispresented in Fig. 3B. Cadmium produced a rapid decrease inAR mRNA; by 1 h, a 40% decrease was observed. The max-imum decrease of approximately 60% was observed at 18 h,and the amount of AR mRNA remained suppressed for 48 h.Taken together, these results provide additional support thatthe effects of cadmium are mediated by the AR.
Effect of cadmium on the activity of the AR
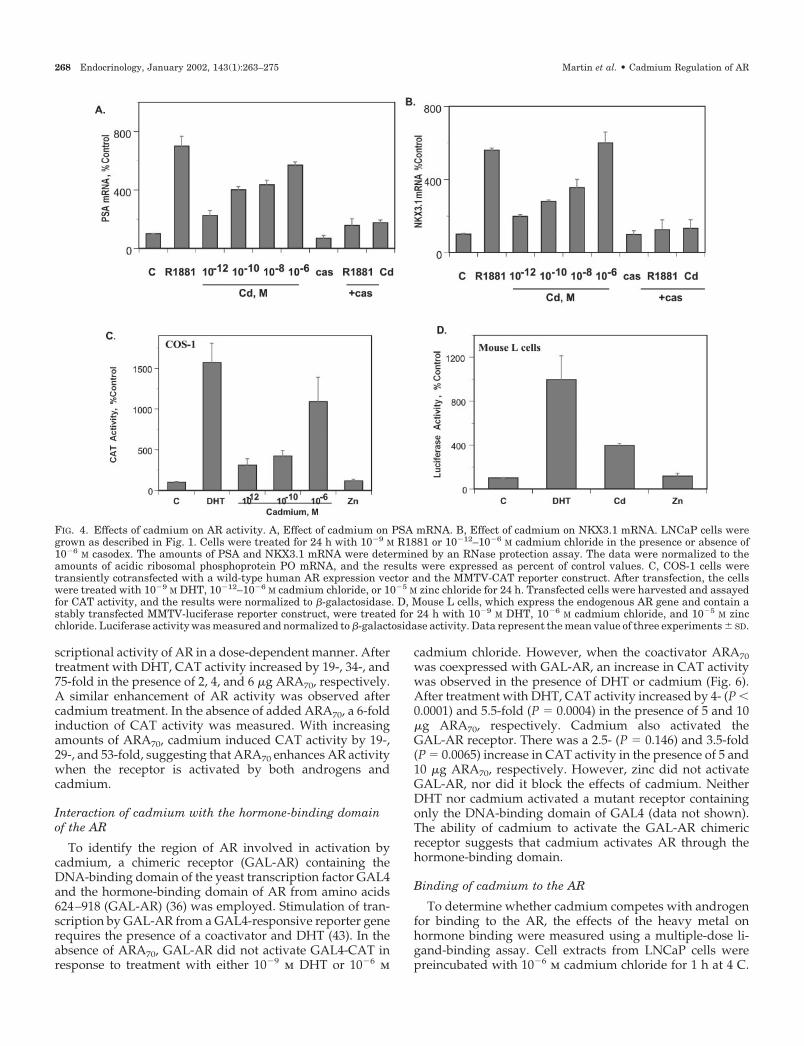
To further characterize the effects of cadmium on the ac-tivity of the AR, the ability of the metal to induce two an-drogen-regulated genes, PSA (32) and the human homeoboxgene NKX3.1 (34), was measured. The LNCaP cells weretreated for 24 h with cadmium chloride concentrations from10�12–10�6 m in the presence or absence of 10�6 m casodex.The synthetic androgen R1881 (10�9 m) was used as a positivecontrol. After treatment, an RNase protection assay was per-formed, and the results are presented in Fig. 4, A and B. Asexpected, R1881 increased PSA and NKX3.1 mRNA by 7- and5.7-fold, respectively. Cadmium increased PSA mRNA andNKX 3.1 mRNA by 2- to 6-fold in a concentration-dependentmanner. Casodex blocked the induction by either R1881 or
cadmium, providing additional evidence that the effects ofthe metal were mediated by the AR.
Because the AR in LNCaP cells contains a mutation atamino acid 877 in the hormone-binding domain that alterssteroid specificity and antiandrogen sensitivity, the effects ofcadmium on wild-type AR activity were tested in two dif-ferent transfection assays. In the first assay system, COS-1cells were transiently cotransfected with a wild-type humanAR expression vector and the mouse mammary tumor virus-CAT reporter gene. In the second assay system, mouse Lcells, which express wild-type AR, were stably transfectedwith the mouse mammary tumor virus-luciferase reporterconstruct. Transfected cells were treated with 10�9 m DHT,10�12–10�6 m cadmium chloride, or 10�6–10�5 m zinc chlo-ride for 24 h, and CAT or luciferase activity was measured(Fig. 4, C and D). In COS-1 cells transfected with the wild-type human AR, cadmium increased the amount of CATactivity by 3- to 10-fold (Fig. 4C). In cells that express theendogenous mouse AR, cadmium chloride treatment re-sulted in a 4-fold increase in luciferase activity (Fig. 4D).Treatment with DHT increased CAT activity by 15-fold, andluciferase activity by 10-fold. However, zinc [10�6 m (data notshown) or 10�5 m] was not capable of increasing either CATor luciferase activity. The ability of cadmium to transactivateAR in these assays demonstrates that the metal also activateswild-type human and mouse AR, and this effect is specific forcadmium and not for zinc.
One of the AR-associated proteins that contributes to theactivation of AR is ARA70 (36). To determine whether ARA70
can enhance the transcriptional activity of AR in the presenceof cadmium, COS-1 cells were transiently cotransfected withwild-type human AR, the mouse mammary tumor virus-CAT reporter, and increasing amounts of ARA70 from 2–6 �g(Fig. 5). ARA70 did not change the baseline CAT activity.However, the coactivator enhanced the DHT-induced tran-
FIG. 3. Effect of cadmium on AR mRNA. LNCaP cells were grown, as described in Fig. 1, and treated with 10�9 M DHT or 10�12–10�6 M cadmiumchloride in the presence or absence of 10�6 M casodex or hydroxyflutamide. Total mRNA was prepared, and AR mRNA was determined by anRNase protection assay as described in Materials and Methods. The effects of cadmium on AR mRNA were normalized to the amount of acidicribosomal phosphoprotein PO mRNA and expressed as percent of control values. Data represent the mean value of four experiments �SD. A,Effect of cadmium concentration on AR mRNA; B, time course of the effect of cadmium on AR mRNA.
Martin et al. • Cadmium Regulation of AR Endocrinology, January 2002, 143(1):263–275 267
scriptional activity of AR in a dose-dependent manner. Aftertreatment with DHT, CAT activity increased by 19-, 34-, and75-fold in the presence of 2, 4, and 6 �g ARA70, respectively.A similar enhancement of AR activity was observed aftercadmium treatment. In the absence of added ARA70, a 6-foldinduction of CAT activity was measured. With increasingamounts of ARA70, cadmium induced CAT activity by 19-,29-, and 53-fold, suggesting that ARA70 enhances AR activitywhen the receptor is activated by both androgens andcadmium.
Interaction of cadmium with the hormone-binding domainof the AR
To identify the region of AR involved in activation bycadmium, a chimeric receptor (GAL-AR) containing theDNA-binding domain of the yeast transcription factor GAL4and the hormone-binding domain of AR from amino acids624–918 (GAL-AR) (36) was employed. Stimulation of tran-scription by GAL-AR from a GAL4-responsive reporter generequires the presence of a coactivator and DHT (43). In theabsence of ARA70, GAL-AR did not activate GAL4-CAT inresponse to treatment with either 10�9 m DHT or 10�6 m
cadmium chloride. However, when the coactivator ARA70was coexpressed with GAL-AR, an increase in CAT activitywas observed in the presence of DHT or cadmium (Fig. 6).After treatment with DHT, CAT activity increased by 4- (P �0.0001) and 5.5-fold (P � 0.0004) in the presence of 5 and 10�g ARA70, respectively. Cadmium also activated theGAL-AR receptor. There was a 2.5- (P � 0.146) and 3.5-fold(P � 0.0065) increase in CAT activity in the presence of 5 and10 �g ARA70, respectively. However, zinc did not activateGAL-AR, nor did it block the effects of cadmium. NeitherDHT nor cadmium activated a mutant receptor containingonly the DNA-binding domain of GAL4 (data not shown).The ability of cadmium to activate the GAL-AR chimericreceptor suggests that cadmium activates AR through thehormone-binding domain.
Binding of cadmium to the AR
To determine whether cadmium competes with androgenfor binding to the AR, the effects of the heavy metal onhormone binding were measured using a multiple-dose li-gand-binding assay. Cell extracts from LNCaP cells werepreincubated with 10�6 m cadmium chloride for 1 h at 4 C.
FIG. 4. Effects of cadmium on AR activity. A, Effect of cadmium on PSA mRNA. B, Effect of cadmium on NKX3.1 mRNA. LNCaP cells weregrown as described in Fig. 1. Cells were treated for 24 h with 10�9 M R1881 or 10�12–10�6 M cadmium chloride in the presence or absence of10�6 M casodex. The amounts of PSA and NKX3.1 mRNA were determined by an RNase protection assay. The data were normalized to theamounts of acidic ribosomal phosphoprotein PO mRNA, and the results were expressed as percent of control values. C, COS-1 cells weretransiently cotransfected with a wild-type human AR expression vector and the MMTV-CAT reporter construct. After transfection, the cellswere treated with 10�9 M DHT, 10�12–10�6 M cadmium chloride, or 10�5 M zinc chloride for 24 h. Transfected cells were harvested and assayedfor CAT activity, and the results were normalized to �-galactosidase. D, Mouse L cells, which express the endogenous AR gene and contain astably transfected MMTV-luciferase reporter construct, were treated for 24 h with 10�9 M DHT, 10�6 M cadmium chloride, and 10�5 M zincchloride. Luciferase activity was measured and normalized to �-galactosidase activity. Data represent the mean value of three experiments � SD.
268 Endocrinology, January 2002, 143(1):263–275 Martin et al. • Cadmium Regulation of AR

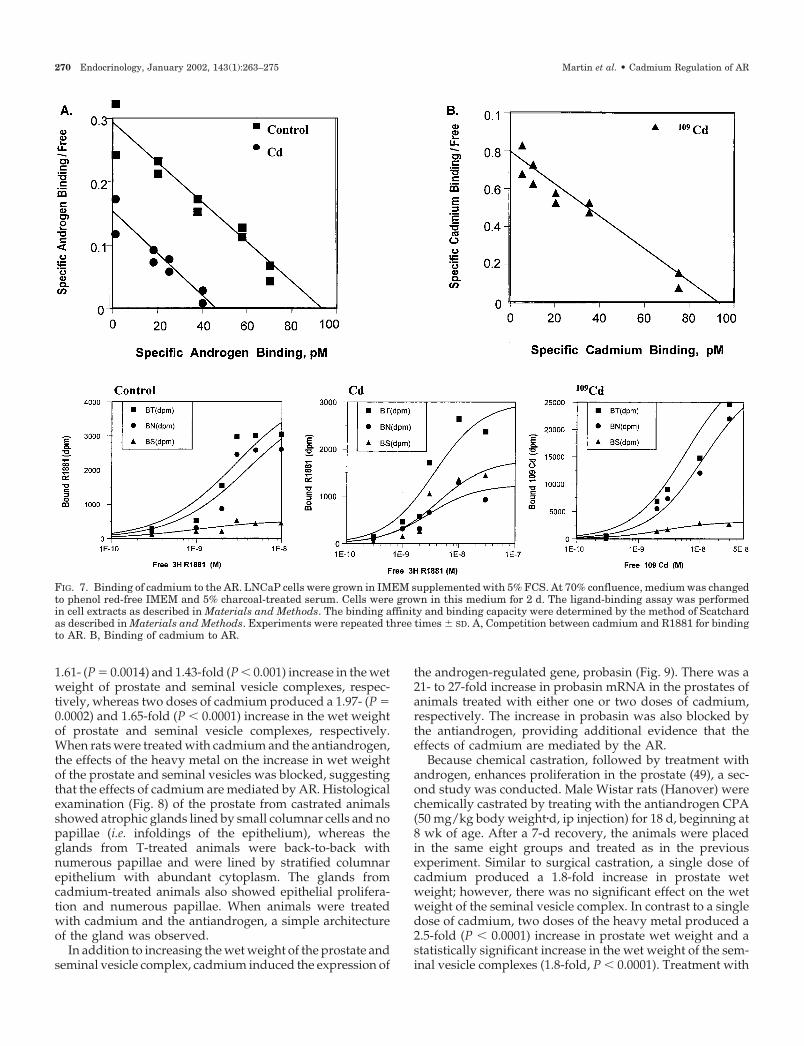
Radiolabeled R1881 was added in the presence or absence ofa 1000-fold molar excess of cold R1881 for 2 h at 37 C. Theaffinity and binding capacity of the receptor were deter-mined according to the method of Scatchard (Fig. 7A). A 45%decrease in the number of androgen-binding sites from 835
to 459 fmol/mg protein was observed after addition of 10�6
m cadmium chloride. The Kd of the [3H]R1881-AR complexwas (2.43 � 0.35) � 10�10 m (n � 3, r � 0.87) and did not showany significant change in the presence of cadmium, (2.75 �0.43) x 10�10 m (n � 3, r � 0.73). These results indicate thatthe metal does not alter the binding affinity of R1881 to thereceptor but blocks hormone binding to the AR.
To determine the binding affinity of cadmium, a multiple-dose ligand-binding assay using 109Cd was employed. Cellextracts from LNCaP cells were incubated with 109Cd (10�12–10�6 m) in the presence or absence of a 1000-fold molar excessof R1881 for 2 h at 37 C. The Scatchard plot is represented inFig. 7B. Cadmium bound AR with high affinity, Kd � (1.19 �0.55) � 10�10 m (n � 3, r � 0.90) and a binding capacity of655 � 81 fmol/mg protein, suggesting the existence of ahigh-affinity binding site for cadmium in the LNCaP cellextracts. To show that the high-affinity site was the AR,COS-1 cells were transiently transfected with the AR gene,and the ability of 109Cd to bind to extracts from control andtransfected cells was measured. High-affinity binding (Kd �2 � 10�10 m, data not shown) was observed in COS-1 cells,whereas no specific binding was observed in control cells,suggesting that cadmium binds with high affinity to the AR.
In vivo effects of cadmium
Both the functional and the structural maintenance ofprostate are dependent on androgens, and the prostate rap-idly regresses after withdrawal of androgen support, such aswith castration (14, 44, 45). To determine whether cadmiummimics androgenic responses in animals, the effects of themetal on the wet weight of the prostate and seminal vesiclecomplex (seminal vesicles and coagulating glands) weretested in castrated animals. The total dose of cadmium chlo-ride was either 10 or 20 �g/kg body weight (�54 or 108nmol/kg, respectively). These doses are 1/250 and 1/500 ofthe LD50 of the metal and are equivalent to the daily exposure(23 �g/kg body weight per day) from food and drinkingwater (46, 47). T propionate, a mitogen in rat prostate and aneffective promoter of prostate cancer in rodents (48), wasemployed as a positive control. In the first study, male Wistarrats (Hanover) were surgically castrated at 8 wk of age. Aftera 7-d recovery, the animals received either one or two ipinjections of cadmium chloride (10 �g/kg body weight) or Tpropionate (1 mg/kg�d). In addition to cadmium and T, someexperimental animals received CPA (50 mg/kg�d for 10 d, ipinjection). The effects of cadmium on the average weight ofprostate and seminal vesicle complexes are shown in Table2. Although a small decrease (10–15%, P � 0.05) in theaverage body weight was observed when the animals werecastrated, there was no statistical difference in body weightbetween treatments. Atrophy of the prostate and seminalvesicle complexes was observed in castrated animals, com-pared with intact rats. As expected, T increased the wetweight of the prostate by 10.8-fold (P � 0.0001), and theweight of the seminal vesicles by 9.7-fold (P � 0.0001), whencompared with nontreated castrated animals (Table 2). Theseresults are in agreement with an earlier report (45). As ex-pected, treatment with the antiandrogen blocked the effectsof the androgen. A single dose of cadmium also induced a
FIG. 5. Effect of cadmium on ARA70 stimulation of AR transcriptionalactivity. COS-1 cells were transiently cotransfected with 2 �g of an ARexpression vector and 10 �g of the MMTV-CAT reporter construct inthe presence or absence of 2, 4, or 6 �g of the coactivator ARA70.Transfected cells were treated for 24 h with either 10�9 M DHT or 10�6
M cadmium chloride, and CAT activity was determined as describedin Materials and Methods. The amount of CAT activity was normal-ized to the amount of �-galactosidase activity. Results are expressedrelative to CAT activity of cells in the absence of ARA70. The resultsare the mean value of three experiments � SD.
FIG. 6. Interaction of cadmium with the hormone-binding domain ofthe AR. COS-1 cells were transiently transfected with GAL-AR anda GAL4-CAT reporter plasmid in the presence or absence of thecoactivator ARA70. The transfected cells were treated with 10�9 MDHT, 10�6 M cadmium chloride, or 10�5 M zinc chloride in the presenceor absence of the antiandrogen casodex (10�6 M). CAT activity wasmeasured as described in Materials and Methods. The results werenormalized to the �-galactosidase activity and expressed as percentof CAT activity in untreated cells. The results represent the meanvalue of three experiments � SD.
Martin et al. • Cadmium Regulation of AR Endocrinology, January 2002, 143(1):263–275 269
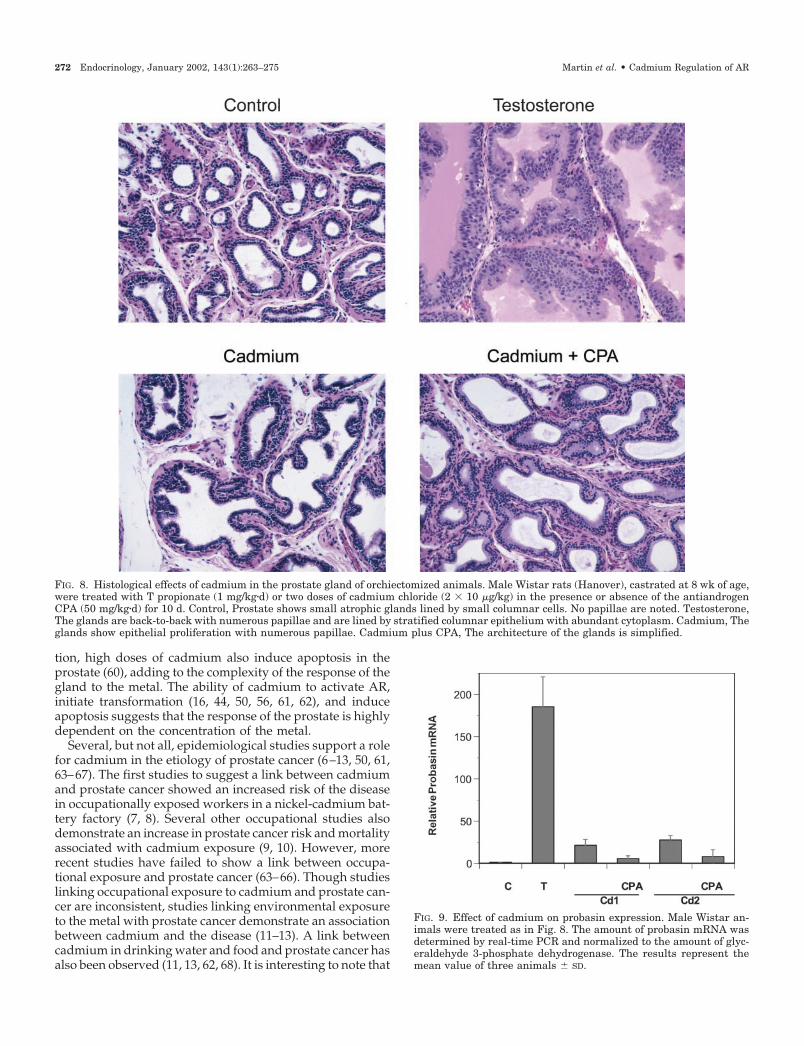
1.61- (P � 0.0014) and 1.43-fold (P � 0.001) increase in the wetweight of prostate and seminal vesicle complexes, respec-tively, whereas two doses of cadmium produced a 1.97- (P �0.0002) and 1.65-fold (P � 0.0001) increase in the wet weightof prostate and seminal vesicle complexes, respectively.When rats were treated with cadmium and the antiandrogen,the effects of the heavy metal on the increase in wet weightof the prostate and seminal vesicles was blocked, suggestingthat the effects of cadmium are mediated by AR. Histologicalexamination (Fig. 8) of the prostate from castrated animalsshowed atrophic glands lined by small columnar cells and nopapillae (i.e. infoldings of the epithelium), whereas theglands from T-treated animals were back-to-back withnumerous papillae and were lined by stratified columnarepithelium with abundant cytoplasm. The glands fromcadmium-treated animals also showed epithelial prolifera-tion and numerous papillae. When animals were treatedwith cadmium and the antiandrogen, a simple architectureof the gland was observed.
In addition to increasing the wet weight of the prostate andseminal vesicle complex, cadmium induced the expression of
the androgen-regulated gene, probasin (Fig. 9). There was a21- to 27-fold increase in probasin mRNA in the prostates ofanimals treated with either one or two doses of cadmium,respectively. The increase in probasin was also blocked bythe antiandrogen, providing additional evidence that theeffects of cadmium are mediated by the AR.
Because chemical castration, followed by treatment withandrogen, enhances proliferation in the prostate (49), a sec-ond study was conducted. Male Wistar rats (Hanover) werechemically castrated by treating with the antiandrogen CPA(50 mg/kg body weight�d, ip injection) for 18 d, beginning at8 wk of age. After a 7-d recovery, the animals were placedin the same eight groups and treated as in the previousexperiment. Similar to surgical castration, a single dose ofcadmium produced a 1.8-fold increase in prostate wetweight; however, there was no significant effect on the wetweight of the seminal vesicle complex. In contrast to a singledose of cadmium, two doses of the heavy metal produced a2.5-fold (P � 0.0001) increase in prostate wet weight and astatistically significant increase in the wet weight of the sem-inal vesicle complexes (1.8-fold, P � 0.0001). Treatment with
FIG. 7. Binding of cadmium to the AR. LNCaP cells were grown in IMEM supplemented with 5% FCS. At 70% confluence, medium was changedto phenol red-free IMEM and 5% charcoal-treated serum. Cells were grown in this medium for 2 d. The ligand-binding assay was performedin cell extracts as described in Materials and Methods. The binding affinity and binding capacity were determined by the method of Scatchardas described in Materials and Methods. Experiments were repeated three times � SD. A, Competition between cadmium and R1881 for bindingto AR. B, Binding of cadmium to AR.
270 Endocrinology, January 2002, 143(1):263–275 Martin et al. • Cadmium Regulation of AR
the antiandrogen also blocked the effects of either androgenor cadmium, suggesting that the effects of the metal aremediated by AR. The weights of the testes were similar in allgroups, independent of treatment. Histological examinationof the prostate, seminal vesicles, coagulating glands, testes,kidney, and liver demonstrated no metal toxicity (data notshown). Because animal strains differ in their response tocastration and subsequent androgen treatment (16, 44, 50),the effects of cadmium in the prostate of surgically castratedWistar rats [Hsd: (WI) BR] was examined. No strain-specificdifferences were observed upon androgen or heavy-metaltreatment (data not shown). In addition to examination ofrats, the androgen-like effects of cadmium in surgically cas-trated mice (C57BL/6-TgN) were examined. After treatmentwith T, the wet weight of the prostate and seminal vesiclecomplex increased 4- and 3-fold, respectively. Cadmium (20�g/kg body weight) produced a 3- and 1.5-fold increase inthe wet weight of the prostate and seminal vesicle complex,respectively. The effects of the androgen and of the heavymetal were blocked by the antiandrogen CPA, suggesting arole for AR. Taken together, these results demonstrate thatcadmium has androgen-like effects in target organs of cas-trated animals. These effects are independent of the strainand species and are mediated by the AR.
Discussion
The goal of this study was to determine whether the heavymetal cadmium activates the AR. The results presentedherein demonstrate that the metal has androgen-like activityin cultured cells and in castrated animals and that the effectsare mediated by the AR. In LNCaP cells, the metal mimics theeffects of androgens on cell growth and gene expression. Theability of an antiandrogen to block these effects suggests thatthe effects of cadmium are mediated through the AR. In
transfection assays, the metal activates the wild-type recep-tor and a chimera containing the hormone-binding domainof AR. In addition, cadmium binds with high affinity to thehormone-binding domain and inhibits the binding of hor-mone to the receptor. The latter observation is also seen in theanterior prostate of the mouse, where cadmium blocks thebinding of hormone to the AR (51). In castrated rats and mice,cadmium mimics the effects of androgens on the wet weightof the prostate gland and the seminal vesicle complex andinduces the expression of an androgen-regulated gene. Thein vivo effects of cadmium are also blocked by an antiandro-gen, suggesting that cadmium activates AR in target tissue.
Although several earlier studies also demonstrate a mi-togenic effect of low doses of cadmium in vitro and in vivo(52–55), these studies did not address the mechanism ofaction of the metal. In human prostate epithelial cells, lowconcentrations of cadmium were shown to stimulate prolif-eration (52–54); whereas, in intact animals, addition of cad-mium to the drinking water significantly increased theweight of the prostate and seminal vesicles but did not re-verse the response of the gland to castration (55). In contrastto the small number of low-dose studies, numerous reportshave examined the toxic and carcinogenic effects of highdoses of the metal (0.3–2 mg/kg). In rodents, high doses ofcadmium induce tumors at several sites, including the site ofinjection, lungs, testes, adrenals, the hematopoietic system,and the prostate (56, 57). The ability of cadmium to initiateprostate tumors is linked to its toxicity in the testes. Exposureof rats to doses of cadmium (approximately 0.2–0.5 mg/kgbody weight), which are nontoxic to the testes, produces adose-related increase in prostate tumors (58, 59); whereasexposure to higher doses of cadmium (0.5 mg/kg bodyweight) produces testicular atrophy and reduces the numberof tumors (58, 59). In addition to disrupting testicular func-
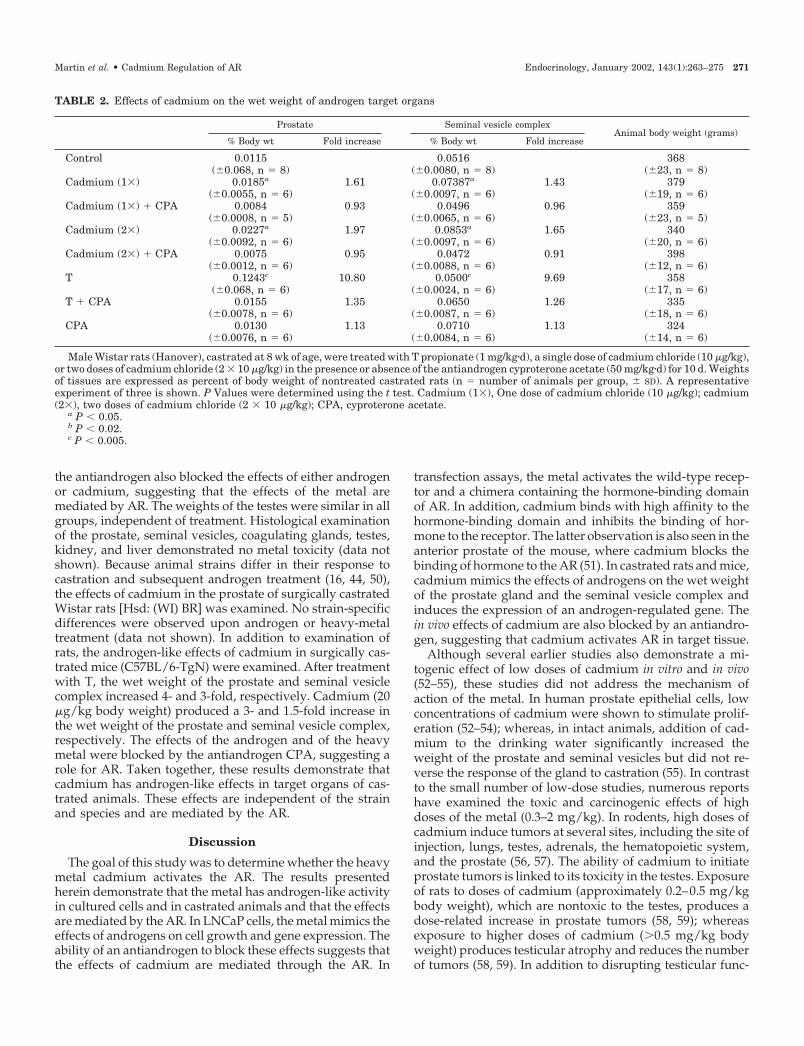
TABLE 2. Effects of cadmium on the wet weight of androgen target organs
Prostate Seminal vesicle complexAnimal body weight (grams)
% Body wt Fold increase % Body wt Fold increase
Control 0.0115 0.0516 368(�0.068, n � 8) (�0.0080, n � 8) (�23, n � 8)
Cadmium (1�) 0.0185a 1.61 0.07387a 1.43 379(�0.0055, n � 6) (�0.0097, n � 6) (�19, n � 6)
Cadmium (1�) � CPA 0.0084 0.93 0.0496 0.96 359(�0.0008, n � 5) (�0.0065, n � 6) (�23, n � 5)
Cadmium (2�) 0.0227a 1.97 0.0853a 1.65 340(�0.0092, n � 6) (�0.0097, n � 6) (�20, n � 6)
Cadmium (2�) � CPA 0.0075 0.95 0.0472 0.91 398(�0.0012, n � 6) (�0.0088, n � 6) (�12, n � 6)
T 0.1243c 10.80 0.0500c 9.69 358(�0.068, n � 6) (�0.0024, n � 6) (�17, n � 6)
T � CPA 0.0155 1.35 0.0650 1.26 335(�0.0078, n � 6) (�0.0087, n � 6) (�18, n � 6)
CPA 0.0130 1.13 0.0710 1.13 324(�0.0076, n � 6) (�0.0084, n � 6) (�14, n � 6)
Male Wistar rats (Hanover), castrated at 8 wk of age, were treated with T propionate (1 mg/kg�d), a single dose of cadmium chloride (10 �g/kg),or two doses of cadmium chloride (2 � 10 �g/kg) in the presence or absence of the antiandrogen cyproterone acetate (50 mg/kg�d) for 10 d. Weightsof tissues are expressed as percent of body weight of nontreated castrated rats (n � number of animals per group, � SD). A representativeexperiment of three is shown. P Values were determined using the t test. Cadmium (1�), One dose of cadmium chloride (10 �g/kg); cadmium(2�), two doses of cadmium chloride (2 � 10 �g/kg); CPA, cyproterone acetate.
a P � 0.05.b P � 0.02.c P � 0.005.
Martin et al. • Cadmium Regulation of AR Endocrinology, January 2002, 143(1):263–275 271
tion, high doses of cadmium also induce apoptosis in theprostate (60), adding to the complexity of the response of thegland to the metal. The ability of cadmium to activate AR,initiate transformation (16, 44, 50, 56, 61, 62), and induceapoptosis suggests that the response of the prostate is highlydependent on the concentration of the metal.
Several, but not all, epidemiological studies support a rolefor cadmium in the etiology of prostate cancer (6–13, 50, 61,63–67). The first studies to suggest a link between cadmiumand prostate cancer showed an increased risk of the diseasein occupationally exposed workers in a nickel-cadmium bat-tery factory (7, 8). Several other occupational studies alsodemonstrate an increase in prostate cancer risk and mortalityassociated with cadmium exposure (9, 10). However, morerecent studies have failed to show a link between occupa-tional exposure and prostate cancer (63–66). Though studieslinking occupational exposure to cadmium and prostate can-cer are inconsistent, studies linking environmental exposureto the metal with prostate cancer demonstrate an associationbetween cadmium and the disease (11–13). A link betweencadmium in drinking water and food and prostate cancer hasalso been observed (11, 13, 62, 68). It is interesting to note that
FIG. 9. Effect of cadmium on probasin expression. Male Wistar an-imals were treated as in Fig. 8. The amount of probasin mRNA wasdetermined by real-time PCR and normalized to the amount of glyc-eraldehyde 3-phosphate dehydrogenase. The results represent themean value of three animals � SD.
FIG. 8. Histological effects of cadmium in the prostate gland of orchiectomized animals. Male Wistar rats (Hanover), castrated at 8 wk of age,were treated with T propionate (1 mg/kg�d) or two doses of cadmium chloride (2 � 10 �g/kg) in the presence or absence of the antiandrogenCPA (50 mg/kg�d) for 10 d. Control, Prostate shows small atrophic glands lined by small columnar cells. No papillae are noted. Testosterone,The glands are back-to-back with numerous papillae and are lined by stratified columnar epithelium with abundant cytoplasm. Cadmium, Theglands show epithelial proliferation with numerous papillae. Cadmium plus CPA, The architecture of the glands is simplified.
272 Endocrinology, January 2002, 143(1):263–275 Martin et al. • Cadmium Regulation of AR

the androgen-like effects of low doses of cadmium found inthis study are consistent with exposure to environmentalconcentrations of cadmium.
Human exposure to cadmium occurs primarily throughdietary sources, cigarette smoking, and occupational expo-sure (46, 47). The average daily intake is estimated to be10–60 �g/d (46, 61). The metal has a biological half-liferanging from 10–30 yr (69), which may account for the sig-nificant accumulation in organs such as the prostate. Accu-mulation in the gland also seems to be associated with themalignant progression of prostate cancer (70, 71). In normalprostate tissue, the amount of cadmium in the nuclear frac-tion was 0.7 �g/g dry weight; and in benign hyperplasia, theamount of metal was 0.98 �g/g dry weight. In prostatetumors, the amount increased to 8.8 �g/g dry weight. InNigerian black men, the amount of cadmium in the prostategland is 8-fold higher in the cancerous tissue, compared withnormal tissue (72); and in Caucasians residing in Great Brit-ain, cadmium levels in resected prostates from patients withprostate cancer were 25 times greater than in normal pros-tates (73). However, no differences in cadmium concentra-tions between malignant and normal prostate tissue wereobserved in Finnish men (74). The concentrations of cad-mium found in human prostate are higher than the concen-trations that mimic the effects of androgens in the presentstudy, supporting a role for the androgen-like effects of themetal in the development of prostate cancer.
Although the mechanism by which cadmium activates theAR remains to be defined, a direct (as well as an indirect)mechanism of action may be proposed. Phosphorylation, asa consequence of the activation of signal transduction path-ways, has been shown to alter the activity of the AR (forreview, see Ref. 75). In response to signaling pathways, theAR is phosphorylated on several serines (76), including 2proline-directed serine phosphorylation sites in the N-terminal region (S81 and S94) and one site in the hinge region(S650) (76). Cadmium also activates protein kinase C (77, 78)and may activate AR, in part, through its effect on this path-way. However, most studies, to date, have shown that ac-tivation of protein kinase C only enhances the activity of ARin the presence of hormone (79–81). Alternatively, cadmiummay activate the AR through a direct interaction with thehormone-binding domain of the receptor. Although the hor-mone-binding domains of nuclear receptors share less than20% sequence homology, they have a similar structure (20–27). They are composed of up to 12 �-helices (H1-H12),folded into a three-layered antiparallel helical sandwich (23,25, 82). The central core layer usually contains three �-helices(H5/6, H9, and H10) sandwiched between 2 additional lay-ers of helices composed of H1-4, H7, H8, and H11. The centralcore of the hormone-binding domain is flanked by helix H12.Upon binding, the ligand induces a conformational change,resulting in the repositioning of helix H12 over the centralcore (20). We have recently shown that cadmium binds toand activates ER-� through a high-affinity interaction withamino acids located on helices H4, H8, H11, and at theinterface of the loop and H12 (28), suggesting that cadmiummay reposition H12 in a manner similar to the repositioningobserved upon ligand binding. Sequence alignment demon-strates that many of the amino acids identified as important
in the interaction of cadmium with ER-� are conserved in thehormone-binding domain of the AR (26, 82). The ability of themetal to bind to and activate the hormone-binding domainof AR suggests that cadmium may activate both receptors bya similar mechanism. However, further studies are requiredto define the precise mechanism by which cadmium activatesthe AR.
Androgens play an important role in prostate cancer pro-motion and progression (15). Consequently, molecules thatcan bind to and activate the AR can potentially affect thepathogenesis of the disease. The results of this study suggestthat cadmium is a candidate for such a role. At environmen-tally relevant concentrations, the metal mimics the effects ofandrogens by a mechanism that involves the hormone-bind-ing domain of the receptor. The ability of cadmium to mimicthe effects of androgens may, in part, explain the risk ofprostate cancer associated with exposure to the metal.
Acknowledgments
We thank Dr. Chawnshang Chang for ARA70; Dr. Robert J. Matusikfor probasin; Drs. Marc E. Lippman and Anna T. Riegel for helpfuldiscussions; and Ann Murray, Tina Wilson, Ignovie Onojafe, AaronFoxworth, Amy Durham, Christine Johnson, Michelle Marriot, TalithaRamey, Laura Rutter-Call, and Winsome West for technical assistance.
Received April 4, 2001. Accepted September 14, 2001.Address all correspondence and requests for reprints to: Mary
Beth Martin, Lombardi Cancer Center, E411 Research Building, 3970Reservoir Road NW, Washington, D.C. 20007. E-mail: [email protected].
This work was supported by the National Institutes of Health GrantCA-70708, American Cancer Society, and an anonymous donor. Supportfor tissue culture, animal, histopathology, and cell cycle analysis corefacilities was provided by P50-CA-58185 and P30-CA-51008.
References
1. Boulikas T 1997 Gene therapy of prostate cancer. Anticancer Res 17:1471–15062. Ingles SA, Ross RK, Yu M, Irvine RA, LaPera G, Haile RW, Coetzee GA 1997
Association of prostate cancer risk with genetic polymorphisms in vitamin Dreceptor and androgen receptor. J Natl Cancer Inst 89:166–170
3. Dhom G 1983 Epidemiologic aspects of latent and clinically manifest carci-noma of the prostate. J Cancer Res Clin Oncol 106:210–218
4. Breslow N, Chan CW, Dhom G, Drury RA, Franks LM, Gellei B, Lee YS,Lundberg S, Sparke B, Sternby NH, Tulinius H 1977 Latent carcinoma ofprostate at autopsy in seven areas. The International Agency for Research onCancer, Lyons, France. Int J Cancer 20:680–688
5. Yatani R, Chigusa I, Akazaki K, Stemmermann GN, Welsh RA, Correa P 1982Geographic pathology of latent prostatic carcinoma. Int J Cancer 29:611–616
6. Lemen RA, Lee JS, Wagoner JK, Blejer HP 1976 Cancer mortality amongcadmium production workers. Ann NY Acad Sci 271:273–279
7. Potts CL 1965 Cadmium proteinuria—the health of battery workers exposedto cadmium oxide dust. Ann Occup Hyg 8:55–61
8. Kipling MD, Waterhouse JAH 1967 Cadmium and prostatic carcinoma. Lan-cet 28:199–204
9. Dubrow R, Wegman DH 1984 Cancer and occupation in Massachusetts: adeath certificate study. Am J Ind Med 6:207–230
10. Blair A, Fraumeni JF 1979 Geographic patterns of prostate cancer in the UnitedStates. J Natl Cancer Inst 61:1379–1384
11. Elghany NA, Schumacher MC, Slattery ML, West DW, Lee JS 1990 Occu-pation, cadmium exposure, and prostate cancer. Epidemiology 1:107–115
12. Bako G, Smith ESO, Hanson J, Dewar R 1980 The geographical distributionof high cadmium concentrations in the environment and prostate cancer inAlberta. Can J Public Health 73:92–94
13. West DW, Slattery ML, Robinson LM, French TK, Mahoney AW 1991 Adultdietary intake and prostate cancer risk in Utah. A case-control study withspecial emphasis on aggressive tumors. Cancer Causes Control 2:85–94
14. Coffey DS, Isaacs JT 1983 Control of prostate growth. Urology 3(Suppl):1715. Bosland MC 2000 The role of steroid hormones in prostate carcinogenesis.
J Natl Cancer Inst Monogr 27:39–6616. Waalkes MP, Rehm S 1994 Cadmium and prostate cancer. J Toxicol Environ
Health 43:251–269
Martin et al. • Cadmium Regulation of AR Endocrinology, January 2002, 143(1):263–275 273

17. Dai JL, Burnstein KL 1996 Two androgen response elements in the androgenreceptor coding region are required for cell-specific up-regulation of receptormessenger RNA. Mol Endocrinol 10:1582–1594
18. Noble RC 1977 The development of prostatic adenocarcinoma in N6 ratsfollowing prolonged sex hormone administration. Cancer Res 37:1929–1933
19. Tsai MJ, O’Malley BW 1994 Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63:451–486
20. Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Grone-meyer H 1996 A canonical structure for the ligand-binding domain of nuclearreceptors. Nat Struct Biol 3:87–94
21. Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, MorasD 1996 Crystal structure of the RAR-� ligand-binding domain bound to all-trans retinoic acid. Nature 378:681–689
22. Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D 1995 Crystalstructure of the ligand-binding domain of the human nuclear receptor RXR-�.Nature 375:377–382
23. Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O,Ohman L, Greene GL, Gustafsson J, Carlquist M 1997 Molecular basis ofagonism and antagonism in the estrogen receptor. Nature 389:753–758
24. Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ1995 A structural role for hormone in the thyroid hormone receptor. Nature378:690–697
25. Tanenbaum DM, Wang Y, Williams SP, Sigler PB 1998 Crystallographiccomparison of the estrogen and progesterone receptor’s ligand binding do-main. Proc Natl Acad Sci USA 95:5998–6003
26. Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N,Joschko S, Scholz P, Wegg A, Basler S, Schafer M, Egner U, Carrondo MA2000 Structural evidence for ligand specificity in the binding domain of thehuman androgen receptor. Implications for pathogenic gene mutations. J BiolChem 275:26164–26171
27. Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE,Salvati ME, Krystek Jr SR, Weinmann R, Einspahr HM 2001 Crystallographicstructures of the ligand-binding domains of the androgen receptor and itsT877A mutant complexed with the natural agonist dihydrotestosterone. ProcNatl Acad Sci USA 98:4904–4909
28. Stoica A, Katzenellenbogen BS, Martin MB 2000 Activation of estrogenreceptor-alpha by the heavy metal cadmium. Mol Endocrinol 14:545–553
29. Vindelov LL, Christensen IJ, Nissen NI 1983 A detergent-trypsin method forthe preparation of nuclei for flow cytometric DNA analysis. Cytometry 3:323–327
30. Stoica A, Saceda M, Fakhro A, Solomon HB, Fenster BD, Martin MB 1997 Therole of transforming growth factor-B in regulation of estrogen receptor ex-pression in the MCF-7 breast cancer cell line. Endocrinology 138:1498–1505
31. Scatchard G 1949 The attractions of protein for small molecules and ions. AnnNY Acad Sci 51:660–672
32. Voeller HJ, Wilding G, Gelmann EP 1991 v-rasH expression confers hormone-independence in vitro growth to LNCaP prostate carcinoma cells. Mol Endo-crinol 5:209–216
33. Saceda M, Lippman ME, Chambon P, Lindsey RK, Pongliktmongkol M,Puente M, Martin MB 1988 Regulation of the estrogen receptor in MCF-7 cellsby estradiol. Mol Endocrinol 2:1157–1162
34. Voeller HJ, Augustus M, Mandike V, Bova GS, Carter KC, Gelmann EP 1997Coding region of NKX3.1, a prostate-specific homeobox gene on 8p21 is notmutated in human prostate cancers. Cancer Res 57:4455–4459
35. Chen C, Okayama H 1987 High-efficiency transformation of mammalian cellsby plasmid DNA. Mol Cell Biol 7:2745–2752
36. Yeh S, Chang C 1996 Cloning and characterization of a specific coactivator,ARA70, for the androgen receptor in human prostate cells. Proc Natl Acad SciUSA 93:5517–5521
37. Webster NJG, Green S, Jin JR, Chambon P 1988 The hormone-binding do-mains of the estrogen and glucocorticoid receptors contain an inducible tran-scriptional activation function. Cell 54:199–207
38. Garcia-Morales P, Saceda M, Kenney N, Kim N, Salomon DS, Gottardis MM,Solomon HB, Sholler PF, Jordan VC, Martin MB 1994 Effect of cadmium onestrogen receptor levels and estrogen-induced responses in human breastcancer cells. J Biol Chem 269:16896–16901
39. Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM,Mirand EA, Murphy GP 1983 LNCaP model of human prostatic carcinoma.Cancer Res 43:1809–1818
40. Veldsholte J, Ris SC, Kuiper GG, Jenster G, Berrevoets C, van Claassen E,Trapman J, Brinkman AO, Mulder E 1990 A mutation in the ligand bindingdomain of the androgen receptor of human LNCaP cells affects steroid bindingcharacteristics and response to anti-androgens. Biochem Biophys Res Com-mun 173:534–540
41. Klaassen CD, Liu J, Chondhuri S 1999 Metallothionein: an intracellular pro-tein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol 39:267–294
42. Wolf DA, Herzinger T, Hermeking H, Blaschke D, Horz W 1993 Transcrip-tional and posttranscriptional regulation of human androgen receptor expres-sion by androgen. Mol Endocrinol 7:924–936
43. Slagsvold T, Kraus I, Bentzen T, Palvimo J, Saatcioglu F 2000 Mutationalanalysis of the androgen receptor AF-2 (activation function 2) core domain
reveals functional and mechanistic differences of conserved residues com-pared with other nuclear receptors. Mol Endocrinol 14:1603–1617
44. Waalkes MP, Rehm S, Devor DE 1997 The effects of continuous testosteroneexposure on spontaneous and cadmium induced tumors in the male Fischer(F344/NG) rat: loss of testicular response. Toxicol Appl Pharmacol 142:40–46
45. Bosland MC 1988 The etiopathogenesis of prostatic cancer with special ref-erence to environmental factors. Adv Cancer Res 51:1–106
46. Gartell MJ, Craun JC, Podrebarae DS, Gunderson ER 1986 Pesticides, selectedelements and other chemicals in adult total diet samples. October 1980–March1982. J Assoc Off Anal Chem 69:146–161
47. Gartell MJ, Craun JC, Podrebarae DS, Gunderson ER 1986 Pesticides, selectedelements and other chemicals in infant and toddler total diet samples. October1980-March 1982. J Assoc Off Anal Chem 69:123–145
48. Bosland MC 1996 Hormonal factors in carcinogenesis of the prostate and testesin humans and in animal models. Prog Clin Biol Res 394:309–352
49. Bosland MC, Prinsen MK 1990 Induction of dorsolateral prostate adenocar-cinomas and other accessory sex gland lesions in male Wistar rats by a singleadministration of N-methyl-N-nitrosourea, 7,12-dimethylbenz(a)anthracene,and 3,2�-dimethyl-4-aminobiphenyl after sequential treatment with cyprot-erone acetate and testosterone propionate. Cancer Res 50:691–699
50. Waalkes MP 2000 Cadmium carcinogenesis in review. J Inorg Biochem 79:241–244
51. Donovan MP, Schein LG, Thomas JA 1979 Inhibition of AR interaction inmouse prostate gland cytosol by divalent metal ions. Mol Pharmacol 17:156–162
52. Dally H, Hartwig A 1997 Induction and repair inhibition of oxidative DNAdamage by nickel(II) and cadmium(II) in mammalian cells. Carcinogenesis18:1021–1026
53. Webber MM 1985 Selenium prevents the growth stimulatory effects of cad-mium on human prostate epithelium. Biochem Biophy Res Commun 127:871–877
54. Holt D, Webb M 1987 Teratogenicity of ionic cadmium in the Wistar rat. ArchToxicol 59:443–447
55. Vissner AJ, Deklerk JN 1979 The effect of dietary cadmium on prostategrowth. Trans Am Assoc Genito-Urinary Surg 70:66–68
56. Waalkes MP, Anver MR, Diwan BA 1999 Chronic toxic and carcinogeniceffects of oral cadmium in the Noble (NBL/Cr) rat: induction of neoplastic andproliferative lesions of the adrenal, kidney, prostate, and testes. J ToxicolEnviron Health A 58:199–214
57. Klaassen CD 1990 Heavy metals and heavy metal antagonists. In: Gilman AG,Rall TW, Nies AS, Taylor P, eds. Goodman and Gilman’s the pharmacologicalbasis of therapeutics. New York: Pergamon Press; 1592–1614
58. Waalkes MP, Rehm S, Riggs CW, Bare RM, Devor DE, Poirier LA, Wenk ML,Hennenman JR, Balaschak MS 1988 Cadmium carcinogenesis in male Wistar[Crl:(WI)BR] rats: dose-response analysis of tumor induction in the prostateand testes and at the injection site. Cancer Res 48:4656–4663
59. Waalkes MP, Anver M, Diwan BA 1999 Carcinogenic effects of cadmium inthe Noble (NBL/Cr) rat: induction of pituitary, testicular, and injection sitetumors and intraepithelial proliferative lesions of the dorsolateral prostate.Toxicol Sci 52:154–161
60. Yan H, Carter CE, Xu C, Singh PK, Jones MM, Johnson JE, Dietrich MS 1997Cadmium-induced apoptosis in the urogenital organs of the male rat and itssuppression by chelation. J Toxicol Environ Health 52:149–168
61. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans 1993Beryllium, cadmium, mercury, and exposures in the glass manufacturingindustry, vol 58, p 119, IARC, Lyon, France
62. Waalkes MP, Rehm S, Coogan TP, Ward JM 1997 Role of cadmium in theetiology of cancer of the prostate. In: Thomas JA, Colby HD, eds. Target organtoxicology series. New York: Raven Press; 277–244
63. Andersson K, Elinder C-G, Hogstedt C, Kjellstrom T, Spang G 1984 Mortalityamong cadmium and nickel exposed workers in a Swedish battery factory.Toxicol Environ Chem 9:53–62
64. Kazantzis G, Lam T-H, Sullivan KR 1988 The mortality of cadmium-exposedworkers; A five-year update. Scand J Work Environ Health 14:220–223
65. Kolonel L, Winkelstein Jr W 1977 Cadmium and prostate cancer. Lancet10:730–731
66. Ross RK, Shimizu H, Paganini-Hill A, Honda G, Henderson BE 1987 Case-control studies of prostate cancer in blacks and whites in southern California.J Natl Cancer Inst 78:869–874
67. Waalkes MP, Oberdorster G 1990 Cadmium Carcinogenesis. In: Foulkes ED,ed. Biological effects of heavy metals. Boca Raton: CRC Press; 129–158
68. Garcia Sanchez A, Antona JF, Urrutia M 1992 Geochemical prospection ofcadmium in a high incidence area of prostate cancer, Sierra de Gata, Salamanca,Spain. Sci Total Environ 116:243–251
69. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans 1976Cadmium, nickel, some expoxides, miscellaneous industrial chemicals andgeneral considerations on volatile anesthetics, vol 11, p 39, IARC, Lyon, France
70. Feustel A, Wennrich R, Steiniger D, Klauss P 1982 Zinc and cadmium con-centration in prostatic carcinoma of different histological grading in compar-ison to normal prostate tissue and adenofibromyomatosis (BPH). Urol Res10:301–303
71. Feustel A, Wennrich R 1984 Determination of the distribution of zinc and
274 Endocrinology, January 2002, 143(1):263–275 Martin et al. • Cadmium Regulation of AR

cadmium in cellular fractions of BPH, normal prostate and prostatic cancersof different histologies by atomic and laser absorption spectrometry in tissueslices. Urol Res 12:253–256
72. Ogunlewe JO, Osegbe DN 1989 Zinc and cadmium concentrations in indig-enous blacks with normal, hypertrophic, and malignant prostate. Cancer 63:1388–1392
73. Habib FK, Hammond GL, Lee IR, Dawson JB, Mason MK, Smith PH, StitchSR 1976 Metal-androgen interrelationships in carcinoma and hyperplasia ofthe human prostate. J Endocrinol 71:133–141
74. Lahtonen R 1985 Zinc and cadmium concentrations in whole tissue and inseparated epithelium and stroma from human benign prostatic hypertrophicglands. Prostate 6:177–183
75. Weigel NL 1996 Steroid hormone receptors and their regulation by phos-phorylation. Biochem J 319:657–667
76. Zhou ZX, Kemppainen JA, Wilson EM 1995 Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol Endo-crinol 9:605–615
77. Mazzei GJ, Girard PR, Kuo JF 1984 Environmental pollutant Cd2� biphasi-cally and differentially regulates myosin light chain kinase and phospholipid/Ca2�-dependent protein kinase. FEBS Lett 173:124–128
78. Bagchi D, Bagchi M, Tamg L, Stohs SJ 1997 Comparative in vitro and in vivoprotein kinase C activation by selected pesticides and transition metal salts.Toxicol Lett 91:31–37
79. deRuiter PE, Teuwen R, Trapman J, Dijkema R, Brinkman AO 1995 Syn-ergism between androgens and protein kinase C on androgen-regulated geneexpression. Mol Cell Endocrinol 110:R1–R6
80. Darne C, Veyssiere G, Jean C 1998 Phorbol ester causes ligand-independentactivation of the androgen receptor. Eur J Biochem 256:541–549
81. Ikonen T, Palvimo JJ, Kallio PJ, Reinikainen P, Janne OA 1994 Stimulationof androgen-regulated transactivation by modulators of protein phosphory-lation. Endocrinology 35:1359–1366
82. Poujol N, Wurtz JM, Tahiri B, Lumbroso S, Nicolas JC, Moras D, Sultan C2000 Specific recognition of androgens by their nuclear receptor. A structure-function study. J Biol Chem 275:24022–24031
Martin et al. • Cadmium Regulation of AR Endocrinology, January 2002, 143(1):263–275 275




















