Risk Considerations for Installation of a New Autoclave in a Pharmaceutical Manufacturing Facility
27
Reference: Sandle, T. (2015): Risk Considerations for Installation of a New Autoclave in a Pharmaceutical Manufacturing Facility, Journal of Validation Technology, 21(1): 1-10 Online: http://www.ivtnetwork.com/article/risk-considerations-installation-new-autoclave- pharmaceutical-manufacturing-facility Risk Considerations for Installation of a New Autoclave in a Pharmaceutical Manufacturing Facility | IVT By Tim Sandle, Ph.D. May 6, 2015 11:00 pm PDT ABSTRACT This paper addresses some of the risk considerations that must be evaluated when replacing a steam sterilizing autoclave within a pharmaceutical processing facility. It demonstrates the application of Failure Modes and Effects Analysis (FMEA) for assessment of risk as part of quality risk management. Formal risk approaches normally share four basic concepts including risk assessment, risk control, risk review, and risk communication. Risk management is fundamentally about understanding what is most important for the control of equipment or design quality and then focusing resources on managing and controlling these aspects. Before risks can be managed, they need to be assessed. FMEA is a widely used risk assessment tools. Steps to perform FMEA are identified; questions to be asked are listed. Criteria for risk assessment must be defined. Either a numerical scoring system or marker phrases such as “high,” “medium,” “low” may be utilized. Three areas are evaluated: Severity of the hazard, likelihood of occurrence, and likelihood of detection. Tables for classifying these aspects are provided. A team approach to risk assessment is recommended. The risk assessment process is demonstrated using the example of a steam sterilizing autoclave replacement. INTRODUCTION This paper addresses some of the risk considerations that must be evaluated when replacing a steam sterilization autoclave within a pharmaceutical processing facility. A cast study format is utilized. In outlining the key risk considerations, the paper demonstrates a risk assessment approach – Failure Modes and Effects Analysis (FMEA).
-
Upload
manchester -
Category
Documents
-
view
0 -
download
0
Transcript of Risk Considerations for Installation of a New Autoclave in a Pharmaceutical Manufacturing Facility

Reference: Sandle, T. (2015): Risk Considerations for Installation of a New Autoclave in a
Pharmaceutical Manufacturing Facility, Journal of Validation Technology, 21(1): 1-10
Online: http://www.ivtnetwork.com/article/risk-considerations-installation-new-autoclave-
pharmaceutical-manufacturing-facility
Risk Considerations for Installation of a New Autoclave in a Pharmaceutical Manufacturing Facility | IVT By
Tim Sandle, Ph.D.
May 6, 2015 11:00 pm PDT
ABSTRACT
This paper addresses some of the risk considerations that must be evaluated when replacing a steam sterilizing autoclave within a pharmaceutical processing facility. It demonstrates the application of Failure Modes and Effects Analysis (FMEA) for assessment of risk as part of quality risk management. Formal risk approaches normally share four basic concepts including risk assessment, risk control, risk review, and risk communication. Risk management is fundamentally about understanding what is most important for the control of equipment or design quality and then focusing resources on managing and controlling these aspects. Before risks can be managed, they need to be assessed. FMEA is a widely used risk assessment tools. Steps to perform FMEA are identified; questions to be asked are listed. Criteria for risk assessment must be defined. Either a numerical scoring system or marker phrases such as “high,” “medium,” “low” may be utilized. Three areas are evaluated: Severity of the hazard, likelihood of occurrence, and likelihood of detection. Tables for classifying these aspects are provided. A team approach to risk assessment is recommended. The risk assessment process is demonstrated using the example of a steam sterilizing autoclave replacement.
INTRODUCTION
This paper addresses some of the risk considerations that must be evaluated when replacing a steam sterilization autoclave within a pharmaceutical processing facility. A cast study format is utilized. In outlining the key risk considerations, the paper demonstrates a risk assessment approach – Failure Modes and Effects Analysis (FMEA).

An autoclave is a pressure chamber used to sterilize equipment and supplies by subjecting them to high pressure saturated steam (1). Autoclaves are used within pharmaceutical facilities to eliminate microbial cells and spores from within a given device. Autoclaves commonly use steam heated to 115–134°C (250 273°F). To achieve sterility, a holding time of at least 30 minutes at 115°C, 15 minutes at 121°C (250°F) or 3 minutes at 134°C (273°F) is required (2).
The validation and verification of the sterilization process is well monitored in the pharmaceutical industry. Operators must ensure that autoclaves comply with various regulatory guidances and regulations. Nonetheless, the purchase of a new autoclave and the assessment requires evaluation. An additional dimension is added when one autoclave is being used to replace another, as with the case study discussed here. To make such an evaluation, a formal risk assessment is required under the auspices of quality risk management.
Risk management and risk assessment principles should be applied as early as possible during the design and construction of steam sterilization devices. The most critical functions in a steam sterilization device are the steam sterilization of direct and indirect the product contact parts. A second important aspect relates to air removal. All of the trapped air must be removed from the autoclave before activation. Trapped air is a very poor medium for achieving sterility during the sterilization cycle.
Formal risk approaches normally share four basic concepts, which are listed below:
Risk assessment Risk control Risk review Risk communication.
This paper considers the application of FMEA, failure modes and effects analysis, to the replacement of a steam-sterilizing autoclave in a pharmaceutical manufacturing facility.
FAILURE MODES AND EFFECTS ANALYSIS (FMEA)
Risk management is fundamentally about understanding what is most important for the control of equipment or design quality and then focusing resources on managing and controlling these aspects to ensure that risks are reduced and contained. Risks relate to a situation, event or scenario where a recognized hazard may result in harm. Before risks can be managed, they need to be assessed (3).
Risk assessment involves identifying risk scenarios. In relation to cleanroom and clean air design, this should ideally be a prospective exercise rather than in reaction to a failure. This process involves determining what can go wrong in the system and all the associated consequences and likelihoods. To achieve this, some kind of risk assessment tool is required (4).

Three key definitions are outlined in ICH Q9(5). These help to contextualize what is meant by “risks.”
Risk: The combination of the probability of occurrence of harm and the severity of that harm
Harm: Damage to health, including the damage that can occur from loss of product quality or availability
Hazard: The potential source of harm.
For engineering systems, one of the most widely used tools for risk assessment is Failure Modes and Effects Analysis (FMEA) (6).
FMEA is a highly structured approach and can be undertaken through the following steps:
a) Setting the scope
b) Defining the problem
c) Setting scales for factors of severity, occurrence and detection (see below)
d) Process mapping
e) Defining failure modes
f) Listing the potential effects of each failure mode
g) Assigning severity ratings to each process step
h) Listing potential causes of each failure mode
i) Assigning and occurrence rating for each failure mode
j) Examining current controls
k) Examining mechanisms for detection
l) Calculating the risk
m) Examining outcomes and proposing actions to minimize risks.
STARTING THE RISK ASSESSMENT PROCESS
Before commencing a risk assessment, it is important to define the size and the scope of the assessment while remaining focused on what is to be achieved, to select the appropriate team (often an interdisciplinary team is best); selecting and reviewing the appropriate risk management tool; deciding upon any numerical scale to be used, and prioritizing the different problems to be addressed.
These steps can be broke down as follows:

Gathering data through an audit and analysis Constructing diagrams of work flows Pin-pointing areas of greatest risk Examining potential sources of contamination Deciding on the most appropriate sample methods Helping to establish alert and action levels Taking into account changes to the work process / seasonal activities Using some type of scoring system so that the risk can be ranked and the level of risk
determined.
In doing so the following questions should be asked:
What is the function of the equipment? What are its performance requirements? How can it fail to fulfill these functions? What can cause each failure? What happens when each failure occurs? How much does each failure matter? What are its consequences? What can be done to predict or prevent each failure? What should be done if a suitable proactive task cannot be found?
These reflective questions help to structure the risk assessment activity.
RISK CRITERIA
It is important to establish the risk assessment criteria as part of the risk assessment exercise. This is necessary in order to place risks in proportion to one another and against a universal scale. Without this, it cannot be determined whether one risk is a greater or lesser problem compared with another, or if a given risk could potentially result in patient harm.
FMEA may utilitze either a numerical scoring system or marker phrases such as “high,” “medium,” “low’ for quantitation. Marker terms are used in the following case study.
These terms are applied to three aspects. These are the severity of the hazard; how likely the hazard is to occur; and whether there are any mechanisms in place to detect the hazard should it occur. Here:
Severity is the consequence of a failure, should it occur Occurrence is the likelihood of the failure happening based on past experience Detection is based on the monitoring systems in place and on how likely a failure can
be detected. Sometimes, a good detection system is described as one that can detect a failure before it occurs.
When these three are cross-compared, the overall risk can be established. The best means to do this is through the use of risk filters. These are illustrated in Tables 1 and 2 below.

Before looking at the risk filter tables, it is necessary to define each of the terms used to establish the overall risk.
Severity of Impact
High = Patients safety will be impacted Medium = Potential patient safety issues Low = No impact on patient
Likelihood of Occurrence
High = 1 failure per year Medium = 1 failure every 5 years Low = 1 failure every 10 years
For documentation, the likelihood is as follows:
High = No Documentation Medium = Incorrect Documentation Low = Documentation present and correct
Probability of Detection
Low = Will not be detected by in-place systems Medium = Only one mechanism for detection by existing systems High = More than one mechanism for detection by existing systems
RISK CLASSIFICATION AND RISK FILTERING
Record the risk classification based upon the impact and likelihood from the table (Table 1). This is established through the use of a filter (or matrix).
TABLE 1: CLASSIFICATION OF RISK
Risk Priority
Record the risk priority based upon the risk classification and probability of detection from the table.

TABLE 2: RISK PRIORITY
Risk Process
In formulating the risk assessment a group was assembled consisting of engineering, validation, microbiology, and the end-users. The process began by identifying the key risk factors and potential failure modes. These were described as “functional details.” Once these were defined, these primary categories were broken down into sub-steps that are termed “sub-functional details.”
Once these were selected and agreed, the steps were grouped together. The group then proceeded to assess the risk for each step, using the FMEA schema outlined above. This consisted of:
a) Considering the relevance of each sub-functional detail. For example, whether it could potentially impact upon product quality b) The possible risk scenarios were considered c) The severity of impact was then assessed as either “high”, “medium” or “low”, using the definitions outlined above d) The likelihood of impact was then assessed, using the same descriptors e) With the severity and likelihood assessed, the risk class was determined using Table 1 above. f) The probability of detection was assessed next g) Knowing the probability of detection allowed the overall risk priority to be determined using Table 2 above. h) The penultimate step was to consider whether any controls or risk mitigation factors were in place i) The final step was to reach a conclusion in relation to risk management.
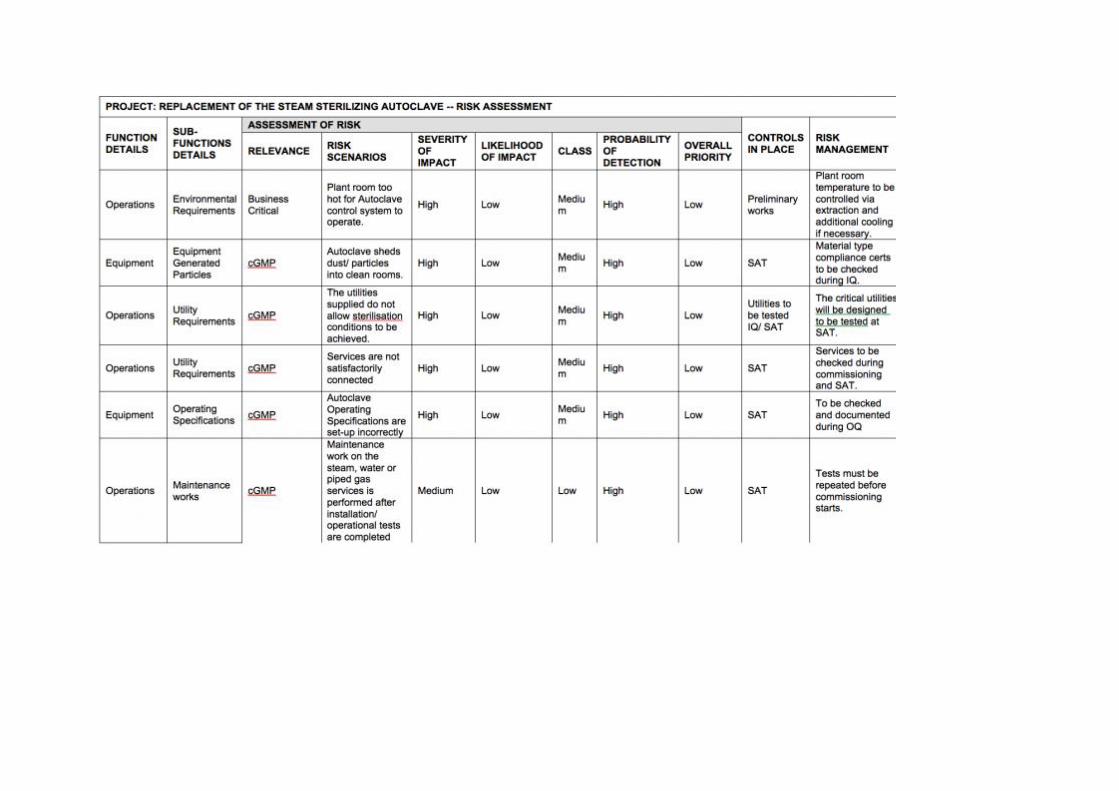
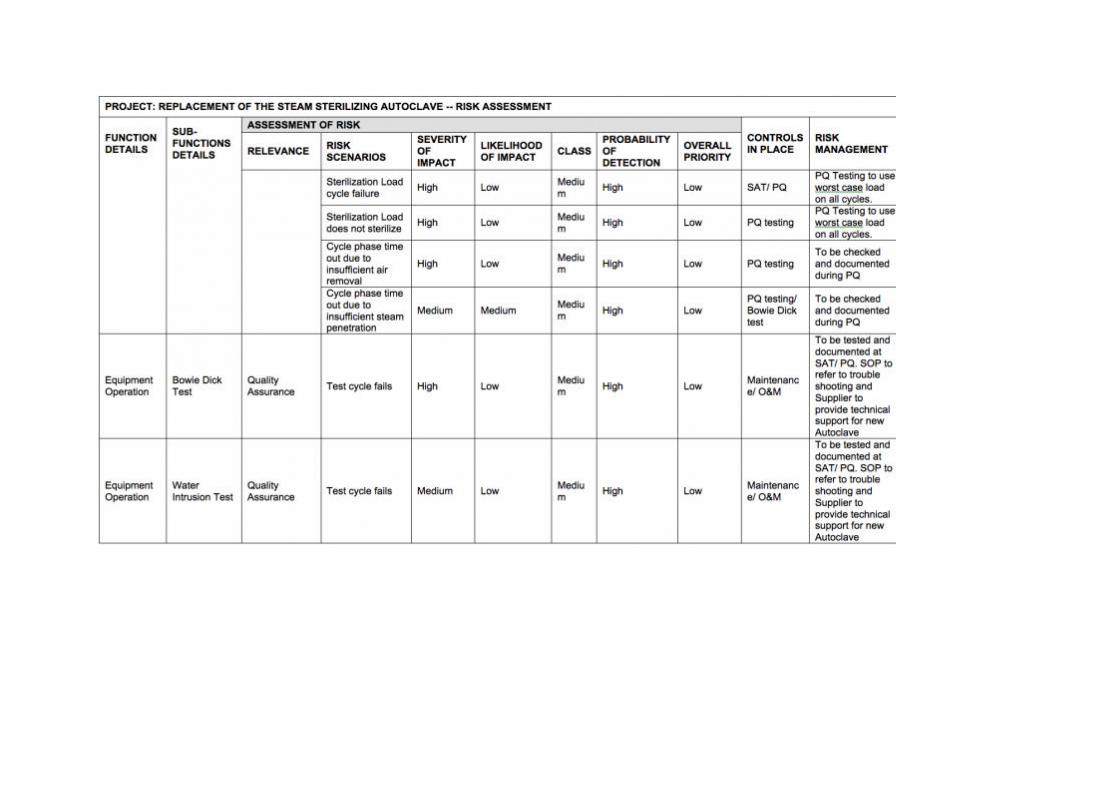
The risk assessment process is outlined in Appendix I using the example of a steam sterilizing autoclave replacement.
Equipment preparation
The defined loading of the autoclave with equipment to be sterilized in an important sub-functional detail is in autoclave operation. The relevance of loading an autoclave incorrectly significantly impacts product quality. Here the risk scenario is an incorrect sterilization of the load. In this case:

a) The severity of impact would be high; b) The likelihood of impact could be medium.
This produces, according to Table 1, a high risk class.
The probability of detection would be low, because the activity is operator dependent and there are no system aspects that prevent an incorrect load from being prepared. Therefore, using Table 2, the risk priority is high.
However, in terms of risk mitigation and risk management, a clear SOP can be put in place and operators can be trained in order to lower the possibility of an incorrect load being prepared. In addition:
Photos of the loads can be prepared during the Operational Qualification Schematics may be included in the SOP Worst-case loads can be verified during both the Operational Qualification and
Performance Qualification.
This example is included in Appendix I. Other sub-functional steps were derived using similar processes.
RISK EXAMPLE
Appendix I contains an example of risk assessment for a replacement steam-sterilizing autoclave. The table is arranged in a fashion to facilitate the risk assessment steps described above where functional and sub-functional steps are outlined. The case study was designed around a particular device and should be regarded as illustrative. Users embarking on a similar process may take note of the items covered. However, they should construct their own risk schematic and reach conclusions that are relevant to their own particular circumstances.
CONCLUSION
The final assessment of the FMEA risk exercise was that the new autoclave can be fitted and that the risks are adequately controlled. It was noted that although the new autoclave has newer control systems technology, the steam sterilization principles together with the load/ test cycles remain in line with the autoclave that is being replaced.
The FMEA risk assessment methodology was appropriate to the task. An alternative approach could have been to use a numerical risk assessment. However, the use of descriptor words like “high,” “medium,” and “low” proved adequate in relation to the activity.

REFERENCES
1. Sandle, T. (2013). Sterility, Sterilisation and Sterility Assurance for Pharmaceuticals: Technology, Validation and Current Regulations, Woodhead Publishing Ltd.: Cambridge, UK, pp93-110
2. Hugo WB (1991). A brief history of heat and chemical preservation and disinfection. J. Appl. Bacteriol. 71 (1): 9–18
3. Sandle, T. (2011): Risk Management in Pharmaceutical Microbiology. In Saghee, M.R., Sandle, T. and Tidswell, E.C. (Eds.) (2011): Microbiology and Sterility Assurance in Pharmaceuticals and Medical Devices, New Delhi: Business Horizons, pp553-588
4. Sandle, T. and Lamba, S. S. (2012) Effectively Incorporating Quality Risk Management into Quality Systems. In Saghee, M.R. Achieving Quality and Compliance Excellence in Pharmaceuticals: A Master Class GMP Guide, New Delhi: Business Horizons, pp89-128
5. ICH Q9: Quality risk management. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH, Geneva (November 2005)
6. Sandle, T. (2003). The use of a risk assessment in the pharmaceutical industry – the application of FMEA to a sterility testing isolator: a case study, European Journal of Parenteral and Pharmaceutical Sciences; 8(2): 43-49
FMEA KEY
BI - Biological Indicator
IQ - Installation Qualification
O&M - Operation and Maintenance
OQ - Operational Qualification
PQ - Performance Qualification
PC - Personal Computer
SOP - Standard Operating Procedure
SAT - Site Acceptance Test
UPS - Uninterruptable Power Supply

APPENDIX 1