
The drift-retention dichotomy on Browns Bank: a model study of interannual variability
Upload
jagiellonianCategory
view
0download
0
DOI: 10.1002/chem.200901805
Resolving Conformation Dichotomy for Y- and T-Shaped Three-CoordinateNiI Carbonyl Complexes with Relativistic DFT Analysis of EPR ACHTUNGTRENNUNGFingerprints
Piotr Pietrzyk,* Katarzyna Podolska, and Zbigniew Sojka[a]
The chemistry of paramagnetic NiI ions has received con-siderable interest, particularly owing to the involvement ofthis species (whether in a ground state, transition state or asthe intermediates) in many important heterogeneous andhomogeneous catalytic systems including organometallic,enzymatic and industrial processes.[1,2] For instance, nickel-exchanged zeolites play an important role in studying themechanistic aspects of selective catalytic reduction of NOx
[2]
or selective hydrogenation of nitro aromatic compounds.[3]
Phosphanylphenolate nickel complexes are active in olefinpolymerization,[4] whereas the methanogenesis cycle is cata-lyzed by methylcoenzyme M (methyl-CoM) reductase withthe cofactor F430 comprising NiI hydrocorphinoid.[5] NiI-car-bonyls of biologically relevant complexes can mimic the ac-tivity of acetylcoenzyme A synthases (acetyl-CoA)[6] or di-hydrogen oxidation by hydrogenases.[7] Another commongroup of NiI�CO complexes is exemplified by silica support-ed and intrazeolitic polycarbonyls with variable number ofCO ligands.[8]
NiI complexes can be obtained by metal-cantered reduc-tion of NiII complexes, whereas the opposite ligand-centredreduction leads to the p-anion radical NiII complexes.[9]
However, the number of known monovalent nickel com-pounds is rather low as compared to other isoelectronic d9
transition-metal complexes. NiI is usually stabilized in theform of robust macrocyclic complexes[10] or other heterolep-tic complexes with polydentate ligands[9] and CO as a coli-gand.[6,8,11] In some cases the presence of hindered ligandswith large steric demand enforces an unusual highly reactivethree-coordinate environment of the nickel core, denotedhereafter as NiI
3c. Depending on the nature of the pliantmonodentate ligand, the NiI
3c complexes can adopt a Y-
shaped or a T-shaped geometry,[6,12] the later observed fa-vourably for d8 transition-metal complexes.[13] The Y or Tconformation entails a significant difference in both theelectronic structure and reactivity of the three-coordinatecomplexes in ligand substitution and transmetalation reac-tions,[12] therefore it appears to be an important factor instructural and kinetic studies. The actual geometry for agiven electron configuration of a metal centre can be ana-lyzed on the basis of the Walsh diagrams showing theenergy change of the metal valence orbitals along the Y toT transformation coordinate. The relative stability of the Yand T geometries was found to be controlled by the groundstate of a transition metal, electron acceptor/donor proper-ties of chelate ligands, presence of agostic interactions, andpredominantly by the steric hindrance of the bulky ligan-d.[12a] However, for small pliant coligands such as CO bothconformations are plausible and the electronic effectsshould be of prime importance.
The structure of the first isolated three-coordinate mono-carbonyl diketiminate-NiI complex (LMeNiI
3cCO) exhibitingthe T-shaped geometry[6a] is shown in Figure 1 a. It has beenassigned based on X-ray crystallography supported by initialEPR and IR investigations. Apart from a spectroscopic char-acterization, the carbonyl complexes of NiI have also beenexamined by density functional theory (DFT) calcula-ACHTUNGTRENNUNGtions[6a,c,14] aiming at elucidation of the influence of electron-ic and steric effects on the T and Y geometry preference.However, despite the substantial effort, diagnostic spectro-scopic signatures allowing for straightforward and quick dis-tinction between Y and T conformation are still lacking.Herein, we provide a DFT-based spectroscopic rationalethat allows for assigning definitively the Y or T conforma-tion based on its EPR characteristics. By relativistic DFTcalculations of g and A(61Ni, 13C) tensors for the LMeNiI
3cCOcomplex and its truncated analogue, and comparison of thecalculated parameters with the available experimentaldata[6a] we demonstrate extreme structural sensitivity of theEPR parameters to the changes of a N-Ni-C angle. The ob-served behaviour was traced back to the changes in the spindensity repartition upon transformation from the Y to the T
[a] Dr. P. Pietrzyk, K. Podolska, Prof. Dr. Z. SojkaFaculty of Chemistry, Jagiellonian Universityul. Ingardena 3, 30-060 Krakow (Poland)Fax: (+48) 12-634-05-15E-mail : [email protected]
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.200901805.
� 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 11802 – 1180711802

geometry, and the associated pronounced rearrangement ofthe d-orbital manifold.
Alignment of CO ligand in NiI complexes has been exam-ined with EPR technique based on rudimentary semiempiri-cal analysis of the g tensor anisotropy[6a,c,8a] and the structureof the 13C hyperfine pattern.[6c,8a] However, observation ofthe metal 61Ni (I=3/2) hyperfine splitting requires isotopi-cally enriched samples, and for three-coordinate complexeshas not been reported yet. Taking the available experimen-tal data as a computational target, we performed DFT cal-culations for the experimental geometry of the LMeNiI
3cCOcomplex in the T conformation (Figure 1 a). The Y geometrywas obtained by decreasing the N-Ni-C angle from its initialvalue of 1598 to the final value of 1318 (Figure 1 b). In addi-tion, spin-unrestricted BP/TZP geometry optimization bymeans of ADF program[15a–c] of the truncated diketiminatemodel was carried out leading to the T-shaped (Figure 1 c)ground-state geometry (C1 symmetry) or, when imposing aC2v symmetry, to the Y-shaped structure (Figure 1 d). Thelater conformation was higher in energy by 8.1 kcal mol�1
with respect to the T geometry. The optimised structures arein a good agreement with the experimental crystallographicdata (Table S1 in the Supporting Information).
The spectroscopic DFT calculations of the g, hyperfine A-ACHTUNGTRENNUNG(61Ni), and superhyperfine A ACHTUNGTRENNUNG(13C) tensors, as well as theirrelative orientation with respect to the molecular frame-work, for both conformations of the LMeNiI
3cCO complexwere performed with ORCA using B3LYP density function-al and the spin-orbit mean field (SOMF) approximation.[15d]
The obtained results are collected in Table 1 (and in TableS2, Supporting Information). For the Y geometry gz>gy>gx
and jAz j> jAy j@ jAx j , whereas in the case of the T struc-
ture gy>gz @gx and jAy j> jAz j> jAx j . Based on the ob-tained results and by assuming typical line-widths, the corre-sponding powder X-band (9.5 GHz) EPR spectra were si-mulated with EPRsim32 software.[16] The resultant plots areshown in Figure 2a,d. The spectrum of the Y-shaped struc-
ture is characterised by bigger g tensor anisotropy and anapparent rhombic symmetry, whereas the spectrum of the T-shaped structure exhibits a pseudo-axial symmetry. Howev-er, the enhanced anisotropy in the gy–gz plane in some par-ticular complexes with strongly disparate ligands may resultin the appreciable rhombicity of the corresponding EPRspectrum, yet still preserving the diagnostic gx !gz, gy pat-tern of the T shape. The same effect may result in a de-crease of the anisotropy in the gx–gy plane for the Y geome-try, preserving the characteristic pattern gz @gx, gy of the Yshape, as it was observed for the LCuIISCPh3 complex.[17a]
The change of the spectrum symmetry is connected with adifferent shape of the spin density contours of both con-
Figure 1. a) Experimental T-shaped structure of the LMeNiI3cCO complex
and b) its Y-shaped conformation along with DFT-optimized diketiminateanalogues of the c) T-shaped (C1) and d) Y-shaped (C2v) structures.
Table 1. B3LYP-SOMF calculation results of the g and hyperfine A ACHTUNGTRENNUNG(61Ni)tensor components for the LMeNiCO complex in Y and T geometry.
Structure gx gy gz Ax ACHTUNGTRENNUNG(61Ni)[MHz]
Ay ACHTUNGTRENNUNG(61Ni)[MHz]
Az ACHTUNGTRENNUNG(61Ni)[MHz]
Y geometry 2.104 2.181 2.350 �17 63 �74T geometry 2.025 2.174 2.158 �46 �113 95experimental[a] 2.014 2.193 2.166 – – –
[a] experimental values taken from Ref. [6].
Figure 2. The X-band (9.5 GHz) EPR spectra simulated based on DFT-calculated g and A ACHTUNGTRENNUNG(61Ni) tensors for the a) Y and d) T geometry of theLMeNiCO complex. Below, the orientation of the principal axes of the gtensor, b) and e), and the spin density contours, c) and f), are shown.
Chem. Eur. J. 2009, 15, 11802 – 11807 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 11803
COMMUNICATION
formers. Dramatic difference in the shape between the EPRspectra for both Y and T structures reveals their high diag-nostic value. As a result, by comparison of the calculatedand experimental parameters listed in Table 1, it is clearthat these results can only reconciled with the T-shapedLMeNiI
3cCO structure. Using the established fingerprints, theunusual rhombic EPR spectra of previously reported isoe-lectronic CuII silica-supported complexes[17b] can be attribut-ed to the three-coordinate structure that was confirmed fur-ther by DFT calculations[17c] (see also the Supporting Infor-mation). Furthermore, other molecular three-coordinateCuII complex exhibiting the Y conformation shows an EPRsignal with gz @gx, gy as expected for the Y geometry.[17a]
However, despite that the local Cs symmetry at the Cu coreimplies a monoclinic spectrum, the anisotropy in the gx–gy
plane is rather small giving rise to an apparent axial shape(as explained above).
The population analysis for the LMeNiI3cCO complex indi-
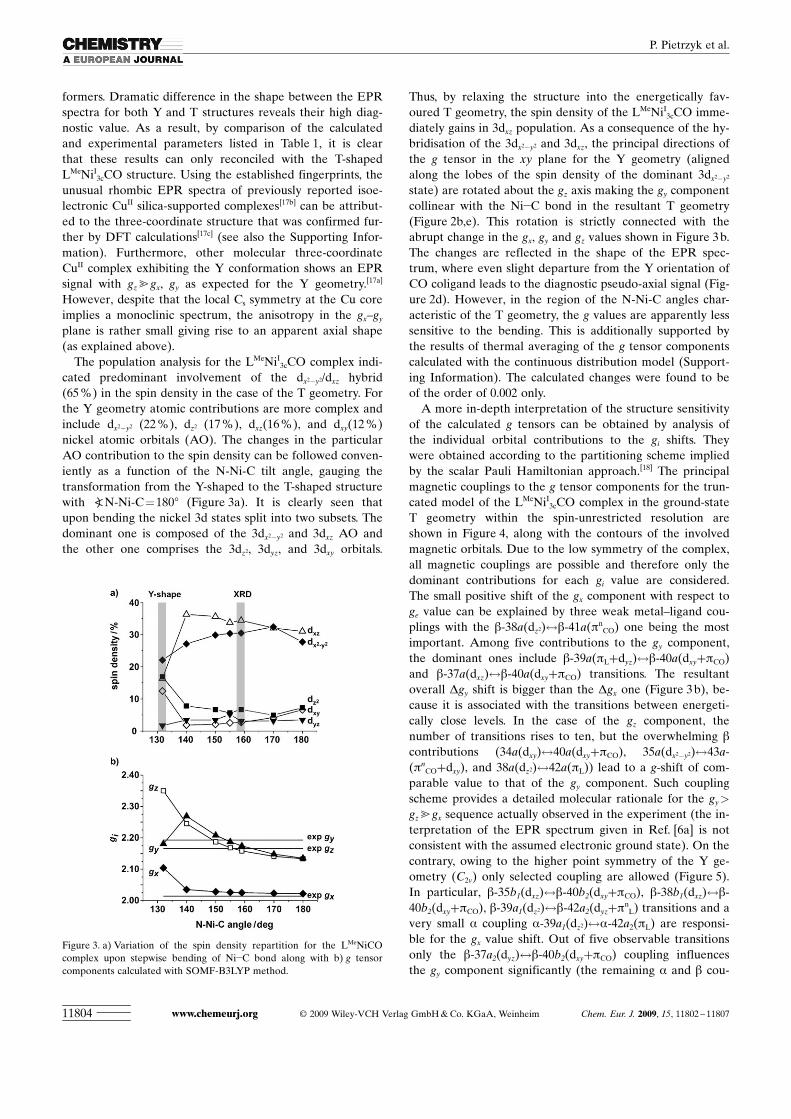
cated predominant involvement of the dx2�y2/dxz hybrid(65 %) in the spin density in the case of the T geometry. Forthe Y geometry atomic contributions are more complex andinclude dx2�y2 (22 %), dz2 (17 %), dxz ACHTUNGTRENNUNG(16%), and dxyACHTUNGTRENNUNG(12 %)nickel atomic orbitals (AO). The changes in the particularAO contribution to the spin density can be followed conven-iently as a function of the N-Ni-C tilt angle, gauging thetransformation from the Y-shaped to the T-shaped structurewith aN-Ni-C =1808 (Figure 3a). It is clearly seen thatupon bending the nickel 3d states split into two subsets. Thedominant one is composed of the 3dx2�y2 and 3dxz AO andthe other one comprises the 3dz2, 3dyz, and 3dxy orbitals.
Thus, by relaxing the structure into the energetically fav-oured T geometry, the spin density of the LMeNiI
3cCO imme-diately gains in 3dxz population. As a consequence of the hy-bridisation of the 3dx2�y2 and 3dxz, the principal directions ofthe g tensor in the xy plane for the Y geometry (alignedalong the lobes of the spin density of the dominant 3dx2�y2
state) are rotated about the gz axis making the gy componentcollinear with the Ni�C bond in the resultant T geometry(Figure 2b,e). This rotation is strictly connected with theabrupt change in the gx, gy and gz values shown in Figure 3 b.The changes are reflected in the shape of the EPR spec-trum, where even slight departure from the Y orientation ofCO coligand leads to the diagnostic pseudo-axial signal (Fig-ure 2d). However, in the region of the N-Ni-C angles char-acteristic of the T geometry, the g values are apparently lesssensitive to the bending. This is additionally supported bythe results of thermal averaging of the g tensor componentscalculated with the continuous distribution model (Support-ing Information). The calculated changes were found to beof the order of 0.002 only.
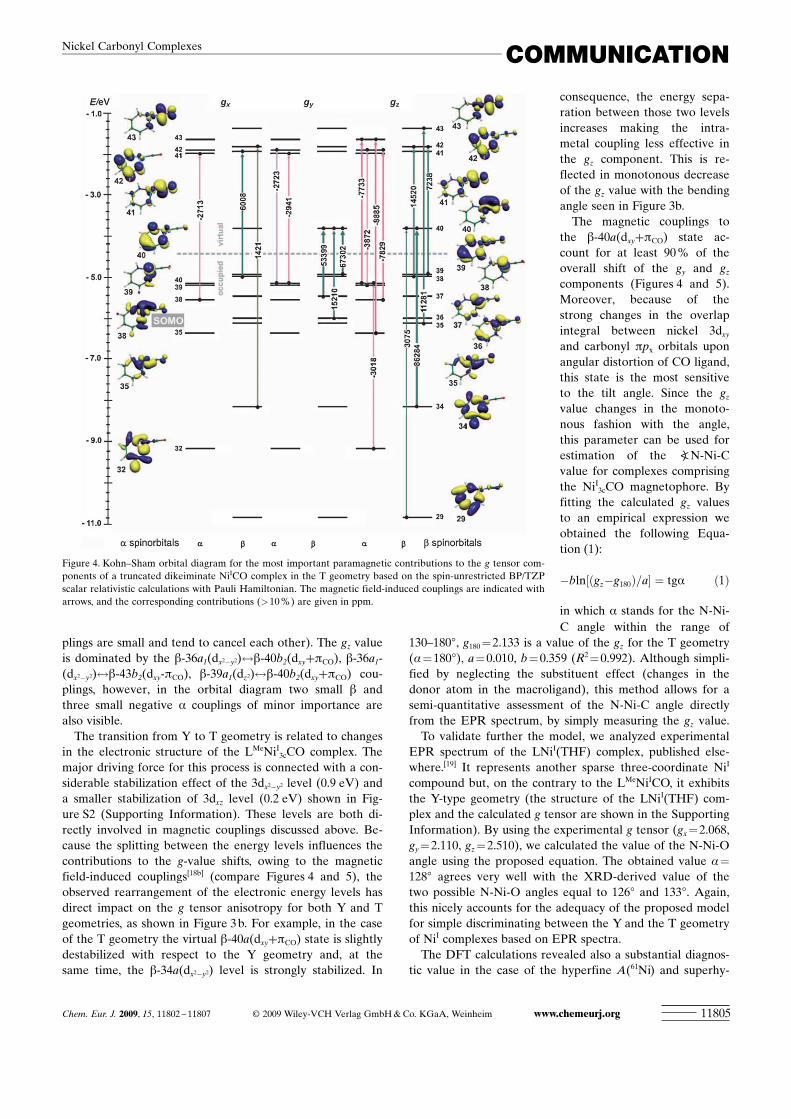
A more in-depth interpretation of the structure sensitivityof the calculated g tensors can be obtained by analysis ofthe individual orbital contributions to the gi shifts. Theywere obtained according to the partitioning scheme impliedby the scalar Pauli Hamiltonian approach.[18] The principalmagnetic couplings to the g tensor components for the trun-cated model of the LMeNiI
3cCO complex in the ground-stateT geometry within the spin-unrestricted resolution areshown in Figure 4, along with the contours of the involvedmagnetic orbitals. Due to the low symmetry of the complex,all magnetic couplings are possible and therefore only thedominant contributions for each gi value are considered.The small positive shift of the gx component with respect toge value can be explained by three weak metal–ligand cou-plings with the b-38a ACHTUNGTRENNUNG(dz2)$b-41a ACHTUNGTRENNUNG(pn
CO) one being the mostimportant. Among five contributions to the gy component,the dominant ones include b-39a ACHTUNGTRENNUNG(pL+dyz)$b-40a ACHTUNGTRENNUNG(dxy+pCO)and b-37a ACHTUNGTRENNUNG(dxz)$b-40a ACHTUNGTRENNUNG(dxy+pCO) transitions. The resultantoverall Dgy shift is bigger than the Dgx one (Figure 3 b), be-cause it is associated with the transitions between energeti-cally close levels. In the case of the gz component, thenumber of transitions rises to ten, but the overwhelming b
contributions (34a ACHTUNGTRENNUNG(dxy)$40a ACHTUNGTRENNUNG(dxy+pCO), 35a ACHTUNGTRENNUNG(dx2�y2)$43a-ACHTUNGTRENNUNG(pnCO+dxy), and 38a ACHTUNGTRENNUNG(dz2)$42a(pL)) lead to a g-shift of com-
parable value to that of the gy component. Such couplingscheme provides a detailed molecular rationale for the gy>
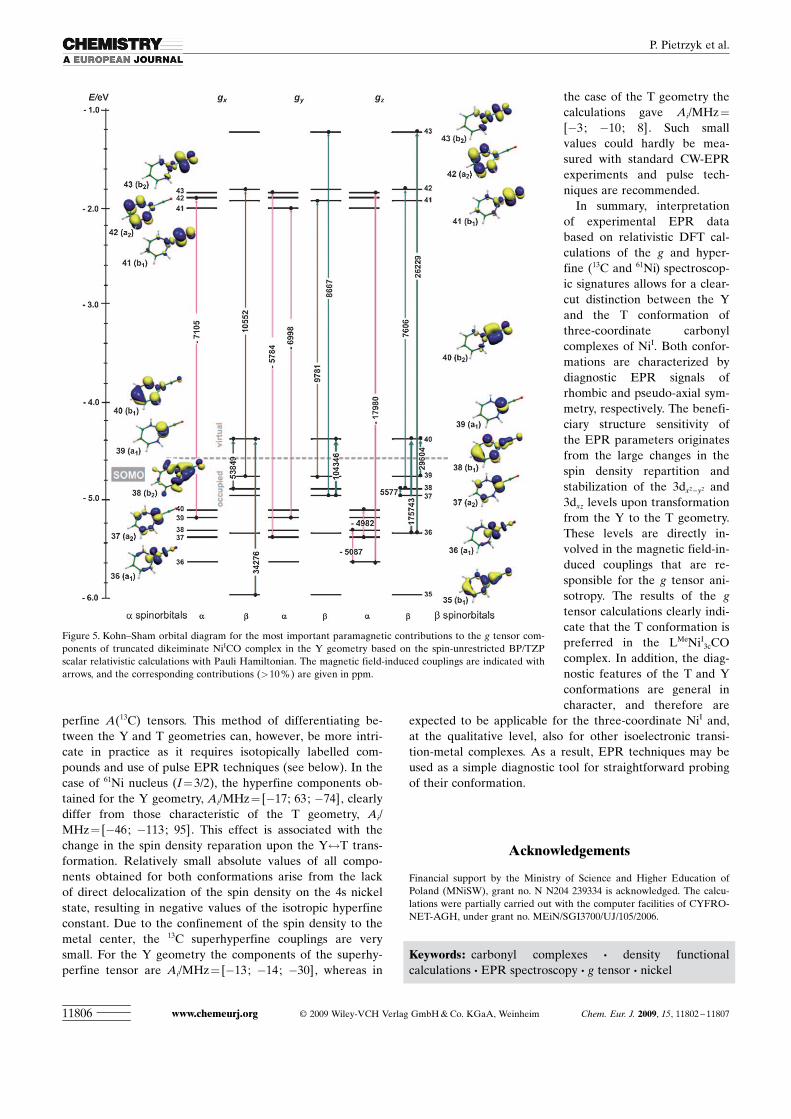
gz @ gx sequence actually observed in the experiment (the in-terpretation of the EPR spectrum given in Ref. [6a] is notconsistent with the assumed electronic ground state). On thecontrary, owing to the higher point symmetry of the Y ge-ometry (C2v) only selected coupling are allowed (Figure 5).In particular, b-35b1ACHTUNGTRENNUNG(dxz)$b-40b2 ACHTUNGTRENNUNG(dxy+pCO), b-38b1 ACHTUNGTRENNUNG(dxz)$b-40b2 ACHTUNGTRENNUNG(dxy+pCO), b-39a1ACHTUNGTRENNUNG(dz2)$b-42a2 ACHTUNGTRENNUNG(dyz+pn
L) transitions and avery small a coupling a-39a1ACHTUNGTRENNUNG(dz2)$a-42a2(pL) are responsi-ble for the gx value shift. Out of five observable transitionsonly the b-37a2ACHTUNGTRENNUNG(dyz)$b-40b2ACHTUNGTRENNUNG(dxy+pCO) coupling influencesthe gy component significantly (the remaining a and b cou-
Figure 3. a) Variation of the spin density repartition for the LMeNiCOcomplex upon stepwise bending of Ni�C bond along with b) g tensorcomponents calculated with SOMF-B3LYP method.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 11802 – 1180711804
P. Pietrzyk et al.
plings are small and tend to cancel each other). The gz valueis dominated by the b-36a1ACHTUNGTRENNUNG(dx2�y2)$b-40b2 ACHTUNGTRENNUNG(dxy+pCO), b-36a1-ACHTUNGTRENNUNG(dx2�y2)$b-43b2ACHTUNGTRENNUNG(dxy-pCO), b-39a1ACHTUNGTRENNUNG(dz2)$b-40b2ACHTUNGTRENNUNG(dxy+pCO) cou-plings, however, in the orbital diagram two small b andthree small negative a couplings of minor importance arealso visible.
The transition from Y to T geometry is related to changesin the electronic structure of the LMeNiI
3cCO complex. Themajor driving force for this process is connected with a con-siderable stabilization effect of the 3dx2�y2 level (0.9 eV) anda smaller stabilization of 3dxz level (0.2 eV) shown in Fig-ure S2 (Supporting Information). These levels are both di-rectly involved in magnetic couplings discussed above. Be-cause the splitting between the energy levels influences thecontributions to the g-value shifts, owing to the magneticfield-induced couplings[18b] (compare Figures 4 and 5), theobserved rearrangement of the electronic energy levels hasdirect impact on the g tensor anisotropy for both Y and Tgeometries, as shown in Figure 3 b. For example, in the caseof the T geometry the virtual b-40a ACHTUNGTRENNUNG(dxy+pCO) state is slightlydestabilized with respect to the Y geometry and, at thesame time, the b-34a ACHTUNGTRENNUNG(dx2�y2) level is strongly stabilized. In
consequence, the energy sepa-ration between those two levelsincreases making the intra-metal coupling less effective inthe gz component. This is re-flected in monotonous decreaseof the gz value with the bendingangle seen in Figure 3b.
The magnetic couplings tothe b-40a ACHTUNGTRENNUNG(dxy+pCO) state ac-count for at least 90 % of theoverall shift of the gy and gz
components (Figures 4 and 5).Moreover, because of thestrong changes in the overlapintegral between nickel 3dxy
and carbonyl ppx orbitals uponangular distortion of CO ligand,this state is the most sensitiveto the tilt angle. Since the gz
value changes in the monoto-nous fashion with the angle,this parameter can be used forestimation of the aN-Ni-Cvalue for complexes comprisingthe NiI
3cCO magnetophore. Byfitting the calculated gz valuesto an empirical expression weobtained the following Equa-tion (1):
�bln½ðgz�g180Þ=a� ¼ tga ð1Þ
in which a stands for the N-Ni-C angle within the range of
130–1808, g180 =2.133 is a value of the gz for the T geometry(a=1808), a=0.010, b= 0.359 (R2 =0.992). Although simpli-fied by neglecting the substituent effect (changes in thedonor atom in the macroligand), this method allows for asemi-quantitative assessment of the N-Ni-C angle directlyfrom the EPR spectrum, by simply measuring the gz value.
To validate further the model, we analyzed experimentalEPR spectrum of the LNiI ACHTUNGTRENNUNG(THF) complex, published else-where.[19] It represents another sparse three-coordinate NiI
compound but, on the contrary to the LMeNiICO, it exhibitsthe Y-type geometry (the structure of the LNiIACHTUNGTRENNUNG(THF) com-plex and the calculated g tensor are shown in the SupportingInformation). By using the experimental g tensor (gx = 2.068,gy = 2.110, gz = 2.510), we calculated the value of the N-Ni-Oangle using the proposed equation. The obtained value a=
1288 agrees very well with the XRD-derived value of thetwo possible N-Ni-O angles equal to 1268 and 1338. Again,this nicely accounts for the adequacy of the proposed modelfor simple discriminating between the Y and the T geometryof NiI complexes based on EPR spectra.
The DFT calculations revealed also a substantial diagnos-tic value in the case of the hyperfine A ACHTUNGTRENNUNG(61Ni) and superhy-
Figure 4. Kohn–Sham orbital diagram for the most important paramagnetic contributions to the g tensor com-ponents of a truncated dikeiminate NiICO complex in the T geometry based on the spin-unrestricted BP/TZPscalar relativistic calculations with Pauli Hamiltonian. The magnetic field-induced couplings are indicated witharrows, and the corresponding contributions (>10%) are given in ppm.
Chem. Eur. J. 2009, 15, 11802 – 11807 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 11805
COMMUNICATIONNickel Carbonyl Complexes
perfine A ACHTUNGTRENNUNG(13C) tensors. This method of differentiating be-tween the Y and T geometries can, however, be more intri-cate in practice as it requires isotopically labelled com-pounds and use of pulse EPR techniques (see below). In thecase of 61Ni nucleus (I=3/2), the hyperfine components ob-tained for the Y geometry, Ai/MHz= [�17; 63; �74], clearlydiffer from those characteristic of the T geometry, Ai/MHz = [�46; �113; 95]. This effect is associated with thechange in the spin density reparation upon the Y$T trans-formation. Relatively small absolute values of all compo-nents obtained for both conformations arise from the lackof direct delocalization of the spin density on the 4s nickelstate, resulting in negative values of the isotropic hyperfineconstant. Due to the confinement of the spin density to themetal center, the 13C superhyperfine couplings are verysmall. For the Y geometry the components of the superhy-perfine tensor are Ai/MHz = [�13; �14; �30], whereas in
the case of the T geometry thecalculations gave Ai/MHz =
[�3; �10; 8]. Such smallvalues could hardly be mea-sured with standard CW-EPRexperiments and pulse tech-niques are recommended.
In summary, interpretationof experimental EPR databased on relativistic DFT cal-culations of the g and hyper-fine (13C and 61Ni) spectroscop-ic signatures allows for a clear-cut distinction between the Yand the T conformation ofthree-coordinate carbonylcomplexes of NiI. Both confor-mations are characterized bydiagnostic EPR signals ofrhombic and pseudo-axial sym-metry, respectively. The benefi-ciary structure sensitivity ofthe EPR parameters originatesfrom the large changes in thespin density repartition andstabilization of the 3dx2�y2 and3dxz levels upon transformationfrom the Y to the T geometry.These levels are directly in-volved in the magnetic field-in-duced couplings that are re-sponsible for the g tensor ani-sotropy. The results of the gtensor calculations clearly indi-cate that the T conformation ispreferred in the LMeNiI
3cCOcomplex. In addition, the diag-nostic features of the T and Yconformations are general incharacter, and therefore are
expected to be applicable for the three-coordinate NiI and,at the qualitative level, also for other isoelectronic transi-tion-metal complexes. As a result, EPR techniques may beused as a simple diagnostic tool for straightforward probingof their conformation.
Acknowledgements
Financial support by the Ministry of Science and Higher Education ofPoland (MNiSW), grant no. N N204 239334 is acknowledged. The calcu-lations were partially carried out with the computer facilities of CYFRO-NET-AGH, under grant no. MEiN/SGI3700/UJ/105/2006.
Keywords: carbonyl complexes · density functionalcalculations · EPR spectroscopy · g tensor · nickel
Figure 5. Kohn–Sham orbital diagram for the most important paramagnetic contributions to the g tensor com-ponents of truncated dikeiminate NiICO complex in the Y geometry based on the spin-unrestricted BP/TZPscalar relativistic calculations with Pauli Hamiltonian. The magnetic field-induced couplings are indicated witharrows, and the corresponding contributions (>10%) are given in ppm.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 11802 – 1180711806
P. Pietrzyk et al.

[1] a) T. C. Harrop, P. K. Mascharak, Coord. Chem. Rev. 2005, 249,3007 – 3024; b) J. Torres-Nieto, W. W. Brennessel, W. D. Jones, J. J.Garcia, J. Am. Chem. Soc. 2009, 131, 4120 – 4126.
[2] a) B. I. Mosqueda-Jim�nez, A. Jentys, K. Seshan, J. A. Lercher, J.Catal. 2003, 218, 375 –385; b) Z. Sojka, P. Pietrzyk, G. Martra, M.Kermarec, M. Che, Catal. Today 2006, 114, 154 – 161.
[3] S.-J. Liu, H.-Y. Cheng, F.-Y. Zhao, J.-Y. Gong, S.-H. Yu, Chem. Eur.J. 2008, 14, 4074 –4081.
[4] J. Heinicke, M. Kçhler, N. Peulecke, M. He, M. K. Kindermann, W.Keim, G. Fink, Chem. Eur. J. 2003, 9, 6093 –6107.
[5] J. Telser, Struct. Bonding (Berlin) 1998, 91, 31 –63.[6] a) N. A. Eckert, A. Dinescu, T. R. Cundari, P. L. Holland, Inorg.
Chem. 2005, 44, 7702 – 7704; b) T. C. Harrop, M. M. Olmstead, P. K.Mascharak, Inorg. Chem. 2006, 45, 3424 –3436; c) J. L. Craft, B. S.Mandimutsira, K. Fujita, C. G. Riordan, T. C. Brunold, Inorg. Chem.2003, 42, 859 –867.
[7] G. J. Kubas, Chem. Rev. 2007, 107, 4152 – 4205.[8] a) L. Bonneviot, D. Olivier, M. Che, J. Mol. Catal. 1993, 21, 415 –
430; b) K. Hadjiivanov, H. Knçzinger, M. Mihaylov, J. Phys. Chem.B 2002, 106, 2618 – 2624.
[9] M. Valente, C. Freire, B. de Castro, J. Chem. Soc. Dalton Trans.1998, 1557 – 1562.
[10] a) I. Zilbermann, M. Winnik, D. Sagiv, A. Rotman, H. Cohen, D.Mayerstein, Inorg. Chim. Acta 1995, 240, 503 – 514; b) M. P. Suh,K. Y. Oh, J. W. Lee, Y. Y. Bae, J. Am. Chem. Soc. 1996, 118, 777 –783.
[11] E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau, T. H. Warren, J.Am. Chem. Soc. 2005, 127, 11248 – 11249.
[12] a) S. Moncho, G. Ujaque, A. Lled�s, P. Espinet, Chem. Eur. J. 2008,14, 8986 – 8994; b) S. Alvarez, Coord. Chem. Rev. 1999, 193–195, 13–41.
[13] a) V. W. Yared, S. L. Miles, R. Bau, C. A. Reed, J. Am. Chem. Soc.1977, 99, 7076 – 7078; b) A. Dedieu, I. Hyla-Kryspin, J. Organomet.Chem. 1981, 220, 115 –123.
[14] a) P. Pietrzyk, K. Podolska, Z. Sojka, J. Phys. Chem. A 2008, 112,12208 – 12219; b) M. Stein, E. van Lenthe, E. J. Baerends, W. Lubitz,J. Phys. Chem. A 2001, 105, 416 –425.
[15] Geometry optimization was carried out with ADF 2007.01 program:a) G. te Velde, F. M. Bickelhaupt, S. J. A. van Gisbergen, C. FonsecaGuerra, E. J. Baerends, J. G. Snijders, T. Ziegler, J. Comput. Chem.2001, 22, 931 – 967; b) C. Fonseca Guerra, J. G. Snijders, G. te Velde,E. J. Baerends, Theor. Chem. Acc. 1998, 99, 391 –403;c) ADF2007.01, SCM, Theoretical Chemistry, Vrije Universiteit,Amsterdam, http://www.scm.com. Spectroscopic calculations wereperformed with ORCA program, d) ORCA—An ab initio, DensityFunctional and Semiempirical Program Package, Version 2.6–00, F.Neese, Lehrstuhl f�r Theoretische Chemie, Bonn, 2007. See detailsin the Supporting Information.
[16] T. Spalek, P. Pietrzyk, Z. Sojka, J. Chem. Inf. Model. 2005, 45, 18–29.
[17] a) P. L. Holland, W. B. Tolman, J. Am. Chem. Soc. 1999, 121, 7270 –7271; b) L. Trouillet, T. Toupance, F. Villain, C. Louis, Phys. Chem.Chem. Phys. 2000, 2, 2005 –2014; c) P. Pietrzyk, J. Phys. Chem. B2005, 109, 10291 –10303.
[18] a) G. Schreckenbach, T. Ziegler, J. Phys. Chem. A 1997, 101, 3388 –3399; b) G. Schreckenbach, T. Ziegler, J. Phys. Chem. 1995, 99, 606 –611.
[19] P. L. Holland, T. R. Cundari, L. L. Perez, N. A. Eckert, R. J. Lachi-cotte, J. Am. Chem. Soc. 2002, 124, 14416 – 14424.
Received: June 30, 2009Published online: September 29, 2009
Chem. Eur. J. 2009, 15, 11802 – 11807 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 11807
COMMUNICATIONNickel Carbonyl Complexes
Copyright © 2022 FDOKUMEN




















