
Redalyc. As Pulp Magazines. Babilónia. Revista Lusófona de ...
Upload
independentCategory
view
1download
0
lable at ScienceDirect
Biomaterials 31 (2010) 2964–2975
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
Resin monomer-induced differential activation of MAP kinasesand apoptosis in mouse macrophages and human pulp cells
Stephanie Krifka a, Christine Petzel a, Karl-Anton Hiller a, Eva-Maria Frank a, Claudia Bosl a,Gianrico Spagnuolo b, Franz-Xaver Reichl c, Gottfried Schmalz a, Helmut Schweikl a,*
a Department of Operative Dentistry and Periodontology, University of Regensburg, University Medical Center, D-93042 Regensburg, Germanyb Department of Oral and Maxillofacial Sciences and Interdisciplinary Research Centre on Biomaterials (CRIB), University of Naples ‘‘Federico II’’, Naples, Italyc Walther-Straub-Institute of Pharmacology and Toxicology, Ludwig-Maximilians-University of Munich, Goethestr. 33, 80336 Munich, Germany
a r t i c l e i n f o
Article history:Received 28 October 2009Accepted 3 January 2010Available online 22 January 2010
Keywords:Resin monomerTEGDMAMAPKCytokineApoptosis
* Corresponding author. Fax: þ49 941 944 6025.E-mail address: [email protected]
0142-9612/$ – see front matter � 2010 Elsevier Ltd. Adoi:10.1016/j.biomaterials.2010.01.005
a b s t r a c t
Triethylene glycol dimethacrylate (TEGDMA) is a resin monomer which is released from polymerizeddental composite materials. It induced apoptosis in various target cells or inhibition of LPS-inducedcytokine production in cells of the immune system after prolonged exposure. In these tissues, mitogen-activated protein kinases (MAPK) regulate signal transduction pathways that support cell survival andcytokine synthesis. The time-dependent regulation of MAPK as well as their linkage to the induction ofapoptosis and cytokine release under the influence of resin monomers is unknown. It was the aim of thepresent study to investigate the kinetics of the up- or down-regulation of the MAPK p38, JNK, and ERK1/2, the induction of apoptosis and cytokine release in RAW264.7 mouse macrophages and human pulp-derived cells. ERK1/2, p38 and JNK were differentially activated by phosphorylation in the presence oflipopolysaccharide (0.1 mg/ml; LPS), a known inducer of MAPK activity, and TEGDMA (3 mM) as detectedby Western blotting. In macrophages, ERK1/2 was activated about 6-fold by LPS, while no activation wasobserved in the presence of TEGDMA after 15 and 30 min. A slight activation of p38 was detected in cellcultures after short exposure to TEGDMA (30 min), but activated JNK was identified after LPS stimulationonly. After a long 24 h exposure period, ERK1/2 and p38 were strongly activated by LPS, a combination ofLPS/TEGDMA, and TEGDMA alone (15–20-fold). In human pulp-derived cells, ERK1/2 was phosphory-lated after exposure to TEGDMA up to 2 h, and sustained activation of ERK1/2 as well as p38 (12–15-fold)was detected after prolonged exposure for 24 h. The LPS-induced, time-related increase in the secretionof the pro-inflammatory cytokines tumor necrosis factor-a (TNF-a) and interleukin-6 (IL-6) as well as theanti-inflammatory IL-10 was instantaneously inhibited by TEGDMA in mouse macrophages. In parallel,the percentage of cells in macrophage cultures in the stage of apoptosis and necrosis increased withexposure period. Yet, in contrast to the inhibition of cytokine release, apoptosis and necrosis caused byLPS and TEGDMA was a late response in both mouse macrophages and human pulp-derived cells. Fromthese data it appears as if MAPK activation, inhibition of cytokine release and the induction of apoptosisand necrosis by TEGDMA are tightly related. The direct causal correlation of these phenomena, however,requires further investigation.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
The adaptive response of eukaryotic cells to environmentalstimuli is controlled by a variety of cellular signal transductionpathways. Among these, the evolutionarily preserved mitogen-acti-vated protein kinases (MAPK) ERK1/2 (extracellular signal-regulatedkinase), p38 and JNK (c-Jun N-terminal kinase) coordinate cellularresponses as diverse as cell proliferation, the release of cytokines or
sburg.de (H. Schweikl).
ll rights reserved.
the induction of apoptosis in case of cell damage [1,2]. While themode of activation through phosphorylation is identical, the functionof MAPK is heterogeneous. Activation of ERK1/2 by, for instance,growth factors is primarily linked with control of cell proliferationand cell survival [3]. Since p38 and JNK pathways are mainly activatedby environmental stresses including radiation, heat shock, thebacterial endotoxin lipopolysaccharide (LPS) or oxidative stress, theseproteins are often classified as SAPK (stress-activated protein kinases)[2,4].
Recent progress in the field of dental research shows thatcompounds of resin materials which are released after polymeri-zation interfere with tissues and cells [5–7]. For instance, the

S. Krifka et al. / Biomaterials 31 (2010) 2964–2975 2965
hydrophilic monomers triethylene glycol dimethacrylate(TEGDMA) and 2-hydroxy ethyl methacrylate (HEMA) may disturbtightly regulated networks of pathways which control cellhomeostasis, thus leading to cell death via apoptosis [8]. Dependingon the exposure period, eluates of composite materials, poly-methacrylates, and resin monomers like TEGDMA and HEMAinduced apoptosis in several cell lines in a concentration-depen-dent manner. The production of reactive oxygen species (ROS) iscurrently considered a key event in the immediate cell responseafter exposure to resin monomers and seems to be associated withthe induction of apoptosis [9–11]. In contrast, it has also beendemonstrated that cells were protected from resin monomer-induced damage by non-enzymatic antioxidants such as N-ace-tylcysteine (NAC) [10–14].
In addition to the observed capability of resin monomers toinitiate cell programs which cause cell death via apoptosis, thesesubstances also interfere with signaling pathways triggered by LPS.In cells of the innate immune system, LPS leads to the activation ofa CD14 and TLR4 complex, and a cascade of signal proteins finallyactivates MAPK and the transcription factor NF-kB. Consequently,only trace amounts of LPS immediately activate the release of pro-and anti-inflammatory mediators such as the cytokines tumornecrosis factor-a (TNF-a), interleukin-6 (IL-6), and interleukin-10(IL-10) [15,16]. It has been shown, however, that the release of TNF-a, IL-6, and IL-10 from LPS-stimulated mouse macrophages wassignificantly reduced by TEGDMA after long exposure periods.Thus, the ability of macrophages to induce an appropriate immuneresponse may be inhibited by this resin compound [17].
These findings demonstrate that resin monomers are able tointerfere with the regulation of cytokine production and apoptosis,and it is well established that MAPK play a crucial role in theregulation of these basic cell functions [2]. Yet, information on theactivation or inhibition of MAPK by dental resin monomers isincomplete. Thus far it has been reported that ERK1/2 is activatedby HEMA and TEGDMA in a salivary gland cell line after longexposure periods, and it has been suggested that p38 is involved inthe apoptotic response to HEMA [18]. In addition, it appears as ifsustained activation of both ERK1/2 and p38 kinases by TEGDMA inTHP-1 cells is associated with TEGDMA-induced cytotoxicity [19].On the other hand, phosphorylation of ERK1/2 was reported inprimary human pulp cells after very short exposure to HEMA, andreduced cell viability after the inhibition of ERK1/2 suggested a pro-survival role of ERK1/2 [20]. Such varying reports illustrate thecomplexity of the multifaceted influence of monomers on thesesignaling pathways.
Although highly desirable, precise information concerning thekinetics of the activation of MAPK by resin monomers in any targettissue is still unavailable. Moreover, a strong correlation betweenMAPK activation, induction of apoptosis and inhibition of therelease of cytokines from LPS-treated macrophages by resinmonomers is also unknown. Thus, we hypothesized that theinduction of apoptosis, as well as the inhibition of LPS-inducedcytokine production in the presence of monomers, may depend ona differential activation of various MAPK. Furthermore, such diversecell responses in cells derived from different target tissues may alsodepend on the exposure period. It is of particular importance tolearn whether a short exposure of cells and tissues is sufficient toinduce apoptosis or whether the stimulation of apoptosis bymonomers requires long and permanent exposure. Therefore, westudied the influence of TEGDMA on the kinetics of MAPK activa-tion, as well as apoptosis and cytokine release in two relevant targetcell lines. TEGDMA served as a model resin monomer because ofthe demonstrated relationship of monomer structure and cyto-toxicity [21]. RAW264.7 mouse macrophages were used as a modelcell line to further analyze the effects of a dental resin monomer on
cells of the innate immune system. In these cells, the release ofcytokines was modified in the presence of TEGDMA [17]. In addi-tion, human pulp-derived cells were utilized as a model for theanalysis of the response of pulp tissue. Although monomer-inducedapoptosis has been shown earlier, a possible relation to the acti-vation of MAPK remains to be elucidated in these cells.
2. Materials and methods
2.1. Chemicals and reagents
Triethylene glycol dimethacrylate (TEGDMA; CAS-No. 109-16-0), lipopolysac-charide (LPS; E. coli, serotype 055:B5), and decaethylene glycol monododecyl ether(C12E10) were purchased from Sigma–Aldrich (Taufkirchen, Germany). Camptothecinwas obtained from BioVision (Mountain View, CA, USA), and Accutase came fromPAA Laboratories GmbH (Colbe, Germany). RPMI 1640 medium containing L-gluta-mine and 2.0 g/l NaHCO3 was purchased from PAN Biotech (Aidenbach, Germany).Fetal bovine serum (FBS), penicillin/streptomycin, and phosphate-buffered salinesupplemented with 5 mM EDTA (PBS–EDTA) came from Life Technologies, Gibco BRL(Eggenstein, Germany). Antiphospho-specific p38 (pT180/pY182) (clone 30/p38MAPK), and anti-p38a (clone 27/p38a/SAPK2a) were purchased from BD Biosciences(Heidelberg, Germany), phospho-specific JNK (anti-ACTIVE JNK, pTPpY) wasobtained from Promega (Madison, WI, USA), antiphospho-specific p44/42 (ERK1/2)(Thr202/Tyr204), anti-p44/42 (ERK1/2), and anti-rabbit IgG HRP-linked antibodywere obtained from Cell Signaling (NEB Frankfurt, Germany), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (clone 6C5) waspurchased from Millipore GmbH (Schwalbach, Germany), goat anti-mouse IgG(H þ L)-HRP conjugate was obtained form Bio-Rad Laboratories (Munich, Germany),and Amersham hyperfilm ECL came from GE Healthcare (Munich, Germany). Aprotease inhibitor cocktail (complete mini) was purchased from Roche Diagnostics(Mannheim, Germany), and a TACS Annexin V-FITC apoptosis detection kit camefrom R&D Systems (Minneapolis, MN, USA). All other chemicals used in the presentstudy were at least chemical grade.
2.2. Cell culture
RAW264.7 mouse macrophages (ATCC TIB71) were maintained in RPMI 1640medium containing L-glutamine, sodium-pyruvate and 2.0 g/l NaHCO3 supple-mented with 10% fetal bovine serum (FBS) and penicillin–streptomycin followingstandard procedures. Human pulp-derived cells (tHPC) were routinely cultivated inMEMa supplemented with 10% FBS, penicillin (100 U/ml), geneticin (50 mg/ml) andstreptomycin (100 mg/ml) at 37 �C and 5% CO2 as described in the literature [22].
2.3. Western blot analysis of MAP kinases
RAW264.7 mouse macrophages (1.5 � 106 cells) and human pulp-derived cells(tHPC) (3� 105 cells) were incubated in cell culture flasks at 37 �C for 48 h. Then, thecell cultures were exposed to 0.1 mg/ml LPS, 3 mM TEGDMA, or a combination of LPS/TEGDMA (3 mM) for 15 min, 30 min, 1 h, 2 h, 6 h, and 24 h. LPS was used as a positivecontrol substance [15]. Exposure was stopped by discarding the exposure media, andthe amount of cytokine present in these supernatants was determined as describedbelow (see 2.4.). Cell cultures were washed with ice-cold PBS. Cells were treated withPBS–EDTA (mouse macrophages) or Accutase (tHPC) and collected by centrifugation.The cell pellet was resuspended and washed twice in ice-cold PBS. Next, the cells werelysed in 50 mM Tris–Cl (pH 7.4), 0.1% Nonidet NP-40, 0.1% C12E10, 150 mM NaCl, 2 mM
Na–EDTA, 5 mM NaF, 1 mM NaVO3, and a protease inhibitor cocktail (complete mini)for 10 min on ice. After centrifugation at 16,000g for 4 min, the supernatant wascollected. The amount of protein present in each cell lysate was determined by a BCAprotein assay (Sigma) using bovine serum albumin as a standard.
For immunoblots, proteins (20 mg per lane) were separated on a 10–12% sodiumdodecyl sulfate-polyacrylamide gel by electorophoresis (SDS-PAGE), and transferredto a nitrocellulose membrane in SDS–electroblot buffer (25 mM Tris–Cl, 192 mM
glycin, 20% methanol, pH 8.3) at 350 mA for 60 min. The membrane was thenwashed twice in TBS (25 mM Tris–Cl, 150 mM NaCl, pH 7.4). After blocking with 5%nonfat milk in TBST (TBS plus 0.1% Tween 20, pH 7.4) at room temperature for60 min, the membrane was incubated with phospho-specific p38, phospho-specificp44/42 (ERK1/2), or phospho-specific JNK primary antibodies over night at coldtemperatures. The membrane was then washed with TBST three times at roomtemperature for 10 min. Primary antibodies were detected by horseradish peroxi-dase-conjugated secondary antibodies in TBST at room temperature for 60 min. Afterwashing for 20 min in TBST and 10 min in PBS, bound secondary antibodies werevisualized by enhanced chemiluminescence (ECL). For reprobing, the membraneswere first stripped at room temperature for 15 min using a CHEMICON re-blot plusmild antibody stripping solution (Millipore, Schwalbach, Germany). Then, themembranes were washed in PBS at room temperature for 30 min and reprobed withcorresponding anti-p38a, anti-p44/42 (ERK1/2), or anti-glyceraldehyde-3-phos-phate dehydrogenase (GAPDH) antibodies. Bound secondary antibodies were again

S. Krifka et al. / Biomaterials 31 (2010) 2964–29752966

15min 30min 15min 30min 15min 30min
Fo
ld
C
ha
ng
e
2
4
6
8
10
12
UC
LPS
LPS/TEGDMA
TEGDMA
pERK1/2 / ERK1/2 pp38 / p38 pJNK / GAPDH
b
c
b
cb
c
a
a
b
c b
c
Fig. 2. Reproducibility of the activation of MAP kinases in RAW264.7 mouse macro-phages after short exposure periods. Mouse macrophage cultures were exposed tolipopolysaccharide (LPS; 0.1 mg/ml), 3 mM TEGDMA and a combination of LPS/TEGDMAfor short exposure periods (15 and 30 min). Bars represent medians (25% and 75%percentiles) obtained from five (n ¼ 5) independent experiments. Statistically signif-icant differences between median fold changes of the ratios of phospho-p38 and p38,phospho-ERK1/2 and ERK1/2, and phospho-JNK and GAPDH were calculated as indi-cated in the text. UC ¼ untreated control cultures. a ¼ significant differences betweenmedian values obtained in cell cultures exposed to LPS and cultures exposed to LPSplus TEGDMA; b ¼ significant differences between median values obtained in cellcultures exposed to LPS and cultures exposed to TEGDMA; c ¼ significant differencesbetween median values obtained in cell cultures exposed to TEGDMA and culturesexposed to LPS plus TEGDMA.
S. Krifka et al. / Biomaterials 31 (2010) 2964–2975 2967
visualized by ECL. The ratio between positive signals for phospho-p38 and p38,phospho-ERK1/2 and ERK1/2, and phospho-JNK and GAPDH was then calculatedafter densitometry using Optimas Corp. version 6.2 image analysis software (Bothell,WA, USA).
2.4. Cytokine release from mouse macrophages and human pulp-derived cells
The amount of tumor necrosis factor-a (TNF-a), interleukin-6 (IL-6), and inter-leukin-10 (IL-10) was determined in the supernatants of cell cultures exposed to LPS,TEGDMA, or a combination of LPS/TEGDMA (as described for Western blot analysis)using ELISA kits (BD Pharmingen, Heidelberg, Germany) according to the manu-facturer’s instructions. All plates were read in a multi-well spectrophotometer(Infinite F200, TECAN) using Magellan version 6.2 software. The lower detection limitsusing standard curves were 15.7 pg/ml for IL-6 and TNF-a, and 31.3 pg/ml for IL-10.
2.5. Determination of apoptosis
RAW264.7 mouse macrophages (1.0 � 105 cells) and human pulp-derived cells(tHPC) (1.5 � 105 cells) were plated in each well of a 6-well plate and incubated at37 �C for 24 h. Then, the cell cultures were exposed to 0.1 mg/ml LPS, 1 or 3 mM
TEGDMA, or a combination of LPS/TEGDMA (3 mM) for 15, 30 min, 1 h, 2 h, 6 h, and24 h. Camptothecin (1 mM) was used as a positive control substance [23]. Theexposure was stopped by discarding the exposure media, and cell cultures whichwere not exposed for 24 h were further incubated in fresh culture medium withouta test substance up to 24 h. Then, the cell culture medium or exposure medium (24 hexposure) was discarded and the cells were washed with PBS at room temperature.Next, the cell cultures were treated with Accutase (tHPC) or PBS–EDTA (macro-phages) on ice, washed in PBS, and finally collected by centrifugation.
Phosphatidyl serine exposure on the cell surface as a marker of apoptosis wasmeasured by the binding of annexin V-FITC. For the differentiation of apoptosis andnecrosis, the cells were also stained with propidium iodide (PI) [24]. About 1.5 � 105
to 1 � 106 cells per cell culture were incubated in 100 ml binding buffer containingannexin V-FITC and PI as recommended by the manufacturer. These samples wereanalyzed on a FACSCanto flow cytometer (Becton Dickinson, San Jose, CA, USA). FITCfluorescence (FL-1) was collected through a 530/30 band pass filter, and PI fluo-rescence (Fl-3) through a 650 nm long pass filter. Data acquisition (at least 104
events for each sample) was performed with FACSDiva� 5.0.2 software, and thesame software was used for analysis. Numbers of viable (annexin V�; PI�) cells werecounted in the lower left quadrant (Q3) of density plots, and the percentages of cellsin apoptosis (annexin Vþ; PI�; lower right quadrant Q4), late apoptosis (annexin Vþ;PIþ; upper right quadrant Q2), and necrosis (annexin V�; PIþ; upper left quadrantQ1) were determined accordingly [24].
2.6. Statistical analyses
Data are presented as medians (25–75% quartiles) from at least four indepen-dent experiments as specified in figure legends, and differences between medianvalues were statistically analyzed using the Mann–Whitney U-test (SPSS 15.0, SPSS,Chicago, IL, USA) for pairwise comparisons among groups at the 0.05 level ofsignificance. The values of the medians, and the upper and lower quartiles were alsoplotted (SigmaPlot 8.0, Systat Software, San Jose, CA, USA).
3. Results
3.1. Phosphorylation of MAP kinases in RAW macrophages
The level of phosphorylated ERK1/2 (phospho-ERK1/2) wasincreased by LPS in mouse macrophages about 6-fold compared tototal amounts of ERK1/2 after 15 and 30 min exposure periods, anda similar effect on ERK1/2 was detected after a co-stimulation of cellcultures with a combination of LPS/TEGDMA (Fig. 1A). Activation ofERK1/2, however, was not detected after exposure to TEGDMAalone. The ratio of phosphorylated p38 (phospho-p38) to total p38was also increased by LPS (3.8-fold) and LPS/TEGDMA (3.0-fold). Noincrease in phospho-p38, however, was detected in macrophagesexposed to TEGDMA for 15 min (Fig. 1A). In contrast,
Fig. 1. Activation of MAP kinases by the resin monomer TEGDMA in RAW264.7 mouse mapolysaccharide (LPS; 0.1 mg/ml), 3 mM TEGDMA and a combination of LPS/TEGDMA for varioumacrophages for 15 and 30 min, (B) for 1 and 2 h, and (C) for 6 and 24 h. (D) ¼ exposure of hand 24 h. Phosphorylation of ERK1/2 (phospho-ERK1/2), p38 (phospho-p38) and JNK (phosamount of ERK1/2, p38 and GAPDH was also monitored on the same membranes after stripratio between positive signals for phospho-p38 and total p38, phospho-ERK1/2 and totalcultures as calculated after densitometry. Control¼ cell lysates of RAW264.7 mouse macrophof phospho-p38. Typical results from at least three independent experiments are shown.
phosphorylation of p38 by LPS was absent after 30 min, but p38was still activated by LPS/TEGDMA (2.9-fold). A slight increase inphospho-p38 (1.8-fold) was even detected in cell cultures treatedwith TEGDMA alone compared to untreated controls (Fig. 1A).Activated JNK was hardly detectable in unstimulated macrophages,but the levels of phospho-JNK increased in the presence of LPS (6.8-fold) and after co-stimulation with LPS/TEGDMA (4.0-fold)compared to the constitutively expressed GAPDH after a 15 minexposure. Although phosphorylation of JNK decreased in thepresence of LPS (2.4-fold) after 30 min, high levels of phospho-JNK(3.7-fold) were still detected in macrophage cultures exposed toLPS/TEGDMA. An increase in the level of phospho-JNK was notdetectable in the presence of TEGDMA (Fig. 1A).
Early activation or inhibition of signal transduction pathwaysthrough MAP kinases is crucial for cell responses like production ofcytokines or the initiation of apoptosis. Therefore, the influence ofLPS, LPS/TEGDMA, and TEGDMA alone on the ratio of phosphory-lated to total amounts of MAP kinases after short exposure periods(15 min and 30 min) was studied in a large number of repeatedexperiments to allow for the analysis of statistically significantdifferences (Fig. 2). It appeared as if TEGDMA could slightly reduceLPS-stimulated phosphorylation ERK1/2 (p ¼ 0.056) compared toan activation observed in the presence of LPS alone (6.6-fold) aftera 15 min exposure period. These differences, however, were notdetected after a 30 min exposure. The increase in ERK1/2 phos-phorylation in the presence of each of the three stimulants was not
crophages and human pulp-derived cells (tHPC). Cell cultures were exposed to lipo-s time periods. Medium ¼ untreated cell cultures. (A) ¼ exposure of RAW264.7 mouseuman pulp-derived fibroblasts (tHPC) for 15 and 30 min, (E) for 1 and 2 h, and (F) for 6pho-JNK) in cell lysates was analyzed by immunoblotting. The expression of the totalping and reprobing with specific antibodies. Numbers indicate the fold change of the
ERK1/2, and phospho-JNK and GAPDH between untreated (medium) and treated cellages exposed to LPS for 15 min in control experiments. The arrow indicates the position

A
TNF-αα
15min 1h 2h 6h 24h
Co
ncen
tratio
n [p
g/m
l]
10-1
100
101
102
103
104
105
UC
LPS
LPS/TEGDMA
TEGDMA
a
a
aa a
b
b
bb
b
BIL-6
15min 1h
Co
ncen
tratio
n [p
g/m
l]
a
a a
b b
C
IL-10
Exposure period [h]
15min
30min
30min
30min 1h
6h
6h
2h 24h
2h 24h
Co
ncen
tratio
n [p
g/m
l]
aa
b b
10-1
100
101
102
103
104
105
10-1
100
101
102
103
104
Fig. 3. Absolute concentrations of TNF-a, IL-6, and IL-10 in RAW264.7 macrophages.The cells were exposed to LPS (0.1 mg/ml), 3 mM TEGDMA and a combination of LPS/TEGDMA for the same time periods used for immunoblot analyses. (A) Amounts ofTNF-a, (B) amounts of IL-6, (C) amounts of IL-10 detected in cell culture supernatants.UC ¼ untreated control. Bars represent medians calculated from triplicates in 12individual values (n) in four independent experiments. a ¼ significant differencesbetween median values obtained in untreated controls (medium) and cell culturesexposed to LPS; b ¼ significant differences between median values obtained inuntreated controls (medium) and cell cultures exposed to LPS plus TEGDMA.
S. Krifka et al. / Biomaterials 31 (2010) 2964–29752968
significantly different after 15 and 30 min exposure periods (Fig. 2).Noteworthy was that LPS-induced activation of p38 was signifi-cantly (p ¼ 0.008) reduced after 30 min compared to a 15 minexposure period. Moreover, TEGDMA significantly (p ¼ 0.008)
increased the ratio of phospho-p38 to total p38 in the presence ofLPS compared to the effect of LPS alone after a 30 min exposure.Such an influence by TEGDMA was not detected after 15 min(Fig. 2). A very similar pattern of activation was found with JNK(Fig. 2). The increase in p38 activation in the presence of TEGDMAalone after a 30 min exposure was only slightly (p¼ 0.056) differentfrom the influence of LPS/TEGDMA on p38 phosphorylation.
The ratio of phospho-ERK1/2 to ERK1/2 in mouse macrophageswas still increased in the presence of LPS compared to untreatedcultures after a 1 h (3.0-fold) and 2 h (1.8-fold) exposure (Fig. 1B).Yet, activation of ERK1/2 in the presence of LPS/TEGDMA was lower(3.0-fold) or even absent (0.8-fold) compared with 15 and 30 minexposure periods. ERK1/2 phosphorylation in macrophages waseven inhibited (0.4-fold) in the presence of TEGDMA alone (Fig. 1B).The amounts of phospho-p38, on the other hand, were about thesame in untreated cell cultures and cultures treated with LPS, LPS/TEGDMA or TEGDMA after 1 h and 2 h exposure periods. Phos-phorylated JNK was not detected in any cell culture (Fig. 1B).
A slight activation of ERK1/2 was detected in macrophages afterexposure to LPS (2.2-fold) and LPS/TEGDMA (1.4-fold) for 6 h, butTEGDMA alone (0.9-fold) was not effective. However, the ratio ofphospho-ERK1/2 to total ERK1/2 in macrophages was furtherincreased by LPS (7.0-fold) after 24 h (Fig. 1C). A combination of LPS/TEGDMA (18.8-fold) and TEGDMA alone (19.1-fold) increasedphosphorylation of ERK1/2 even more than LPS (Fig. 1C). Remark-ably, p38 was not activated by LPS (1.2-fold) after a 6 h exposureperiod, but LPS/TEGDMA (1.8-fold) and TEGDMA alone (1.9-fold)slightly induced p38 phosphorylation. MAPK p38 was again acti-vated by LPS (4.5-fold) after 24 h, but LPS/TEGDMA (15.6-fold) andTEGDMA (15.1-fold) caused an even stronger increase in the ratio ofphospho-p38 to total p38 (Fig. 1C). It appeared as if this increase inERK1/2 and p38 activation was caused, at least in part, by a reducedexpression of total amounts of ERK1/2 and p38 (Fig. 1C). In contrast,activation of JNK in the presence of LPS, LPS/TEGDMA and TEGDMAwas not detected under the current experimental conditions aftera 6 h or 24 h exposure (Fig. 1C).
3.2. Phosphorylation of MAP kinases in human pulp-derived cells
The activation of p38, ERK1/2 and JNK by LPS and TEGDMA inhuman pulp-derived cells (tHPC) was completely different from thepattern detected in mouse macrophages. Most remarkable, none ofthe MAP kinases were activated in the presence of LPS in tHPC aftershort and long exposure periods (Fig. 1D–F). ERK1/2 was foundphosphorylated in unstimulated cells, and the phosphorylation ofERK1/2 was activated in the presence of TEGDMA after a 15 min and30 min (1.9-fold) exposure (Fig. 1D). Similar effects were detectedafter 1 h and 2 h exposure periods (Fig. 1E). The activation of ERK1/2in the presence of TEGDMA was considerably enhanced in cellcultures treated for 6 h (4-fold) and 24 h (16-fold) (Fig. 1F).Although LPS alone did not activate ERK1/2 in tHPC, it appeared asif LPS inhibited the activation of ERK1/2 in cultures treated withTEGDMA. The strong activation of ERK1/2 after a 24 h exposure toTEGDMA alone was, at least in part, a consequence of the lowerexpression of total ERK1/2 (Fig. 1F). Activation of the stress-acti-vated kinases p38 and JNK was not detected in untreated tHPC, orcultures exposed to LPS, LPS/TEGDMA and TEGDMA alone for 15and 30 min as well as 1 h and 2 h. However, total p38 was clearlyidentified in all cell cultures (Fig. 1D and E). Although phosphory-lation of p38 was still absent after 6 h, phospho-p38 was increasedin cultures treated with LPS/TEGDMA (2-fold) for 24 h. An evenstronger activation was detected in cultures exposed to TEGDMAalone (12-fold) (Fig. 1F). JNK was activated similarly to p38, since anincrease in phospho-JNK was detected in cell cultures treated withTEGDMA for 24 h (Fig. 1F).

S. Krifka et al. / Biomaterials 31 (2010) 2964–2975 2969
3.3. Time-dependent secretion of cytokines by macrophages andhuman pulp-derived cells
The exposure of mouse macrophages to LPS-induced a large andtime-related increase in the secretion of all cytokines tested here. Asignificant increase in the amounts of TNF-a in cell cultures treatedwith LPS compared to untreated controls was observed after30 min (Fig. 3A). The release of TNF-a in LPS-treated cultures wasfurther increased more than 20-fold after a 1 h exposure period toabout 2000 pg/ml (Fig. 3A). Maximum levels of TNF-a of 5000–7500 pg/ml were then detected after 2–24 h exposure periods. Therelease of IL-6 and IL-10 from LPS-treated cell cultures was delayedfor about 1–2 h compared with the production of TNF-a butfollowed the same pattern (Fig. 3B and C). The amounts of IL-6 andIL-10 were detectable after a 2 h exposure and further increasedover time. No release of any of the cytokines tested here wasobserved in cell cultures treated with 3 mM TEGDMA (Fig. 3A–C).However, TEGDMA immediately and strongly inhibited the releaseof TNF-a, IL-6, and IL-10 from LPS-treated cell cultures (Fig. 3A–C).Notably, the extent of this inhibition was different depending onthe cytokine. The release of TNF-a from LPS-induced cell cultureswas inhibited about 20–60-fold, but considerable amounts ofTNF-a were still detectable in LPS and TEGDMA co-treated cultures.In contrast, the inhibition of the LPS-induced production of IL-6and IL-10 by TEGDMA was complete even after long exposureperiods (Fig. 3A–C). No release of cytokines by human pulp-derivedcells (tHPC) was detected after exposure to LPS, LPS/TEGDMA orTEGDMA alone (not shown).
3.4. Induction of apoptosis in mouse macrophages and humanpulp-derived cells
The induction of apoptosis in mouse macrophages and humanpulp-derived cells treated with LPS and TEGDMA was analyzed inparallel to the activation of MAPK using flow cytometry. First,representative density blots of macrophage cultures exposed to1 mM camptothecin (positive control), 1 mg/ml LPS and 3 mM
TEGDMA for 24 h illustrate how cells in apoptosis, late apoptosis ornecrosis were identified (Fig. 4). Almost no cells in untreatedcontrol cultures stained with annexin (Vþ) to indicate cells inapoptosis (Q4; 0.4%) or were identified as cells in necrosis (Q1;2.0%) (Fig. 4). However, about 6% of the cells were detected in lateapoptosis (Q2) because of the simultaneous staining with annexinVþ and PIþ. Camptothecin increased the percentage of cells inapoptosis (Q4; 7.2%), late apoptosis (Q2; 47.0%) and necrosis (Q1;31.3%), and a similar pattern of this distribution of cells wasdetected in cultures exposed to 3 mM TEGDMA. It appeared as if celldeath induced by LPS primarily occurred through apoptosis underthe current conditions since an increase in the number of cellsstained with annexin Vþ and PIþ (Q2; 16.4%) was detected (Fig. 4).
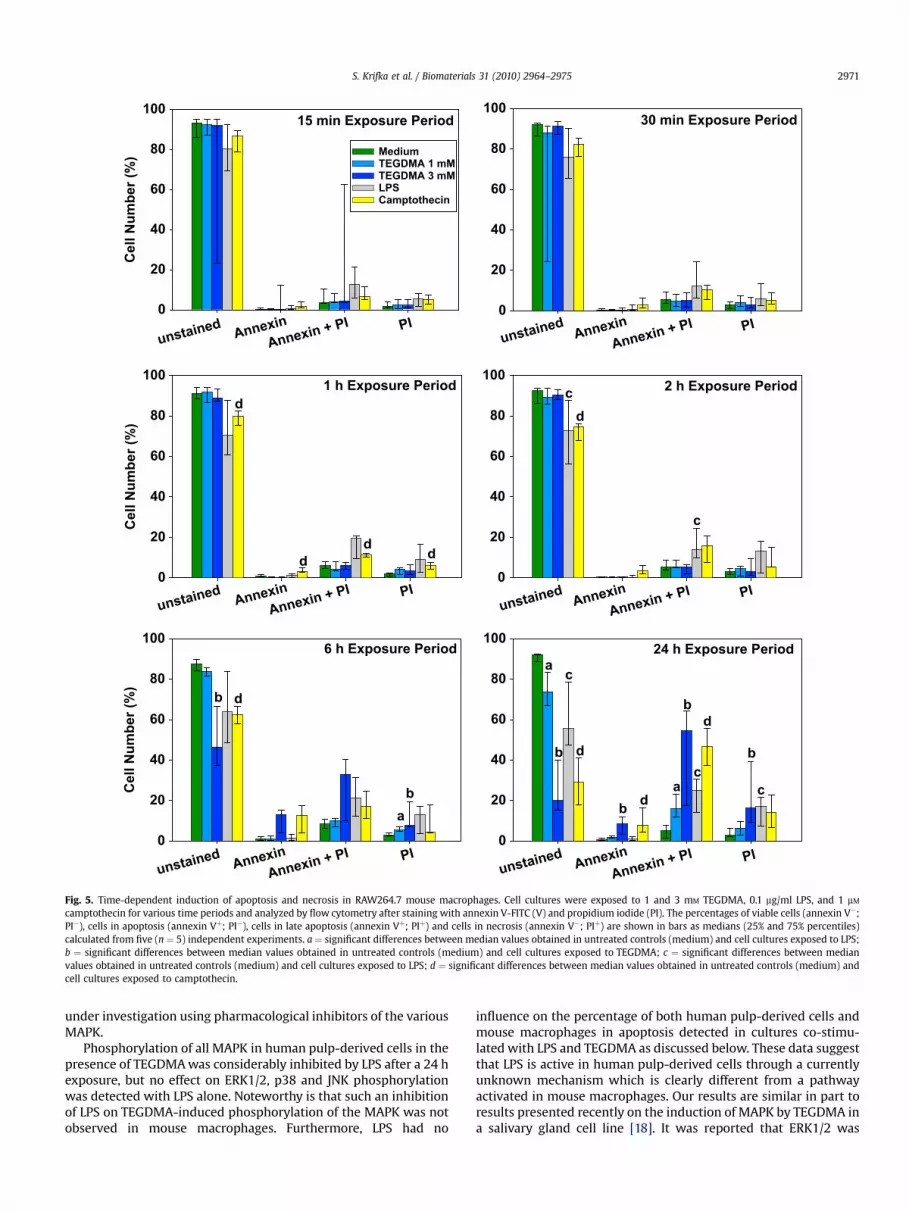
The percentage of cells in apoptosis and necrosis in mousemacrophages differentially induced by camptothecin, LPS, andTEGDMA increased according to the length of the exposure period(Fig. 5). Camptothecin continuously increased the percentage ofcells in apoptosis, late apoptosis, and necrosis after a 1 h to 24 hexposure. In contrast to the effects of camptothecin and TEGDMA,the percentage of cells in late apoptosis was increased in thepresence of LPS after short exposure periods of 15 and 30 min,although the reduction of viable cells to 75–80% was not significantcompared with about 93% viable cells detected in untreatedcultures. The onset of apoptosis caused by TEGDMA, however, wasnot detectable before a 6 h exposure. At this time period, 3 mM
TEGDMA significantly increased the percentage of cells whichstained with annexin (Vþ) to 13% compared with 1% found inuntreated cell cultures (Fig. 5). In addition, 33% of the cells in
TEGDMA-treated cultures were identified in late apoptosiscompared to 8% in untreated controls. Consequently, thepercentage of viable cells significantly decreased from 88% incontrol cultures to 46% in cultures exposed to 3 mM TEGDMA. Aftera 24 h exposure, a significant increase in the number of cells in lateapoptosis was also detected in cultures exposed to 1 mM TEGDMA.In addition, the percentage of cells in late apoptosis and necrosiswas further increased in cell cultures exposed to 3 mM TEGDMA,and the amount of viable cells dramatically decreased by about 20%(Fig. 5).
Apoptosis and necrosis in human pulp-derived cells (tHPC) wasnot detected with any of the substances tested here after exposureperiods up to 2 h (Fig. 6). LPS, and even camptothecin, did notsignificantly decrease cell viability even after long exposure periodsof 6 h and 24 h, and no significant increase in the number of cells inany phase of cell death was observed. In contrast, 1 and 3 mM
TEGDMA caused a concentration-dependent increase in thepercentage of cells in apoptosis and late apoptosis but such anincrease was not detected with cells in necrosis after a 6 h exposure.The percentage of viable cells in cultures treated with 3 mM
TEGDMA was significantly decreased to 65% compared to untreatedcontrols in parallel (Fig. 6). A concentration-dependent shift froma relatively high number of cells in apoptosis towards highernumbers of cells in late apoptosis and necrosis was then detectedafter a 24 h exposure to TEGDMA. About 20% of the cells were foundin late apoptosis and 25% of the cells were identified as necrotic incultures exposed to 3 mM TEGDMA. The percentage of viable cellssignificantly decreased from 90% found in untreated cultures to 75%and 51% detected in cell cultures exposed to 1 and 3 mM TEGDMA,respectively (Fig. 6). Noteworthy is that no significant differencesbetween cells in apoptosis or necrosis were detected in mousemacrophage or tHPC cultures co-treated with LPS and TEGDMA orTEGDMA alone (Fig. 7).
4. Discussion
4.1. Phosphorylation of MAP kinases in mouse macrophages andhuman pulp-derived cells
ERK1/2 and the stress kinases p38 and JNK were differentiallyphosphorylated in mouse macrophages depending on the expo-sure period. At present, we can only speculate about the reasonfor the differential induction of MAPK in mouse macrophagesafter short exposure periods. It is known that the generation ofreactive oxygen species (ROS) or the fluctuation of the cellularredox state lead to the stimulation of various signaling systemsincluding MAP kinases [2]. Recent studies have provided experi-mental evidence on the generation of ROS by dental resinmonomers after a short exposure of cell cultures [18,20,26]. Thus,it is possible that the slight increase in p38 activation aftera 30 min exposure to TEGDMA was initiated by ROS production.On the other hand, the generation of ROS could be the cause ofa slight activation of ERK1/2 in human pulp-derived cells. Thisfinding is very similar to our recent observation regarding theslight phosphorylation of ERK1/2 in pulp cells in the presence ofHEMA after short exposure periods [20]. The reason for thedifferences between the phosphorylation of each MAPK in mousemacrophages and human pulp-derived cells after short exposureperiods remains unclear at the moment. Details relating to therole of oxidative stress in the differential activation of the variousMAPK in the two different cell lines are currently under investi-gation using ROS scavengers.
TEGDMA alone even inhibited the phosphorylation of ERK1/2 inmouse macrophages after 1 h and 2 h exposure periods. Thisobservation suggests that down-regulation of ERK1/2 might be

Fig. 4. Induction of apoptosis and necrosis in RAW264.7 mouse macrophages. Cell cultures were stained with annexin V-FITC (V) and propidium iodide (PI) after exposure andanalyzed by flow cytometry. The representative dual-parameter fluorescence density blots were derived from untreated cell cultures (untreated control) or cell cultures exposed toLPS (0.1 mg/ml), camptothecin (1 mM), and TEGDMA (3 mM) for 24 h. The numbers indicate the percentages of viable cells (Q3; annexin V�; PI�), cells in apoptosis (Q4; annexin Vþ;PI�), cells in late apoptosis (Q2; annexin Vþ; PIþ) and cells in necrosis (Q1; annexin V�; PIþ) in one typical experiment.
S. Krifka et al. / Biomaterials 31 (2010) 2964–29752970
involved in the TEGDMA-induced delay of the cell cycle as previ-ously reported for other cell lines [25]. It is well established thatERK1/2 activation drives cell proliferation in differentiatedeukaryotic cells. There is growing evidence that ERK1/2 regulatescyclin D1 transcription, the assembly of cyclin/CDK complexes andrepresses the transcription of anti-proliferative genes [3]. Activa-tion or inhibition of p38 or JNK was not detected after a 1 h and 2 hexposure period, pointing to a clear difference in the function ofERK1/2 and the stress kinases p38/JNK in response to TEGDMAalone or in combination with LPS.
The large increase in the phosphorylation of both ERK1/2 andp38 after 24 h in cell cultures exposed to TEGDMA alone suggesteda similar mechanism for both MAPK. How this phosphorylationmay possibly be associated with apoptosis is discussed below. Theidentical increase in phosphorylation of ERK1/2 and p38 in mousemacrophages caused by TEGDMA and after co-stimulation with LPSand TEGDMA is most likely caused by TEGDMA since LPS alone wasnot as effective. Moreover, phosphorylation of JNK was not detectedafter long exposure periods suggesting that phosphorylation of the
stress kinases p38 and JNK was independently regulated. Thisfinding also indicates that JNK may not play a role in mousemacrophages if MAPK are involved in the process of the regulationof resin monomer-induced apoptosis. However, it is likely that thisassumption is closely related to the cell type under investigationsince it was reported that JNK was associated with apoptosis inother cell lines [27].
The activation of ERK1/2 and p38 by LPS, LPS/TEGDMA andTEGDMA alone was largely increased after a 24 h exposure inmouse macrophages. It is likely that MAPK were activated by LPSthrough a different mechanism than detected after very shortexposure periods. The short exposure of cell cultures to LPS anda subsequent strong activation of all MAPK was probably related tothe production of cytokines [15]. On the other hand, the phos-phorylation of ERK1/2 and p38 in cultures treated with LPS for 24 hwas related to apoptosis [28–30]. We speculate that a late andsustained activation of p38 in the presence of TEGDMA ispresumably associated with the onset of apoptosis, while ERK1/2was activated to support cell survival. This assumption is currently
15 min Exposure Period
unstainedAnnexin
Annexin + PI PI
Ce
ll N
um
be
r (%
)
0
20
40
60
80
100
Medium
TEGDMA 1 mM
TEGDMA 3 mM
LPS
Camptothecin
30 min Exposure Period
unstained
Annexin
Annexin + PI PI
0
20
40
60
80
100
1 h Exposure Period
unstainedAnnexin
Annexin + PI PI
Cell N
um
ber (%
)
0
20
40
60
80
100
d
d
d
d
2 h Exposure Period
unstained
Annexin
Annexin + PI PI
0
20
40
60
80
100
c
c
d
6 h Exposure Period
unstainedAnnexin
Annexin + PI PI
Cell N
um
ber (%
)
0
20
40
60
80
100
a
b
b
d
24 h Exposure Period
unstained
Annexin
Annexin + PI PI
0
20
40
60
80
100
a
a
b
b
b
b
c
c
c
d
d
d
Fig. 5. Time-dependent induction of apoptosis and necrosis in RAW264.7 mouse macrophages. Cell cultures were exposed to 1 and 3 mM TEGDMA, 0.1 mg/ml LPS, and 1 mM
camptothecin for various time periods and analyzed by flow cytometry after staining with annexin V-FITC (V) and propidium iodide (PI). The percentages of viable cells (annexin V�;PI�), cells in apoptosis (annexin Vþ; PI�), cells in late apoptosis (annexin Vþ; PIþ) and cells in necrosis (annexin V�; PIþ) are shown in bars as medians (25% and 75% percentiles)calculated from five (n ¼ 5) independent experiments. a ¼ significant differences between median values obtained in untreated controls (medium) and cell cultures exposed to LPS;b ¼ significant differences between median values obtained in untreated controls (medium) and cell cultures exposed to TEGDMA; c ¼ significant differences between medianvalues obtained in untreated controls (medium) and cell cultures exposed to LPS; d ¼ significant differences between median values obtained in untreated controls (medium) andcell cultures exposed to camptothecin.
S. Krifka et al. / Biomaterials 31 (2010) 2964–2975 2971
under investigation using pharmacological inhibitors of the variousMAPK.
Phosphorylation of all MAPK in human pulp-derived cells in thepresence of TEGDMA was considerably inhibited by LPS after a 24 hexposure, but no effect on ERK1/2, p38 and JNK phosphorylationwas detected with LPS alone. Noteworthy is that such an inhibitionof LPS on TEGDMA-induced phosphorylation of the MAPK was notobserved in mouse macrophages. Furthermore, LPS had no
influence on the percentage of both human pulp-derived cells andmouse macrophages in apoptosis detected in cultures co-stimu-lated with LPS and TEGDMA as discussed below. These data suggestthat LPS is active in human pulp-derived cells through a currentlyunknown mechanism which is clearly different from a pathwayactivated in mouse macrophages. Our results are similar in part toresults presented recently on the induction of MAPK by TEGDMA ina salivary gland cell line [18]. It was reported that ERK1/2 was

15 min Exposure Period
unstainedAnnexin
Annexin + PI PI
Ce
ll N
um
be
r (%
)
0
20
40
60
80
100
Medium
TEGDMA 1 mM
TEGDMA 3 mM
LPS
Camptothecin
30 min Exposure Period
unstained
Annexin
Annexin + PI PI
0
20
40
60
80
100
1 h Exposure Period
unstainedAnnexin
Annexin + PI PI
Cell N
um
ber (%
)
0
20
40
60
80
100
2 h Exposure Period
unstainedAnnexin
Annexin + PI PI
0
20
40
60
80
100
6 h Exposure Period
unstainedAnnexin
Annexin + PI PI
Cell N
um
ber (%
)
0
20
40
60
80
100
b
24 h Exposure Period
unstained
Annexin
Annexin + PI PI
0
20
40
60
80
100
a
a
b
b
Fig. 6. Time-dependent induction of apoptosis and necrosis in human pulp-derived cells (tHPC). Cell cultures were exposed to 1 and 3 mM TEGDMA, 0.1 mg/ml LPS, and 1 mM
camptothecin for various time periods and analyzed by flow cytometry after staining with annexin V-FITC (V) and PI. The percentages of viable cells (annexin V�; PI�), cells inapoptosis (annexin Vþ; PI�), cells in late apoptosis (annexin Vþ; PIþ) and cells in necrosis (annexin V�; PIþ) are shown in bars as medians (25% and 75% percentiles) calculated fromfive (n ¼ 5) independent experiments. a ¼ significant differences between median values obtained in untreated controls (medium) and cell cultures exposed to 1 mM TEGDMA;b ¼ significant differences between median values obtained in untreated controls (medium) and cell cultures exposed to 3 mM TEGDMA.
S. Krifka et al. / Biomaterials 31 (2010) 2964–29752972
activated by 2 mM TEGDMA after a 6 h exposure period, butexposure to TEGDMA did not result in large alterations of phospho-p38 or phospho-JNK levels. However, the expression of totalamounts of MAPK was not shown. More importantly, a clear andcausal correlation between the phosphorylation of MAPK and theinduction of apoptosis by TEGDMA or any other monomer stillawaits detailed analysis. We observed in a recent study that ERK1/2and p38 were activated in THP-1 cells after exposure to TEGDMA for24 h and 48 h. This activation was reduced in the presence of theROS scavenger NAC [19]. Thus, it appears as if activation of MAPK by
TEGDMA is possible through the generation of oxidative stress incells of the innate immune system.
4.2. Release of cytokines from macrophages
Cells of the innate immune system including macrophagesrespond to harmful foreign substances like the bacterial endotoxinLPS through the activation of MAPK or transcription factors likeNF-kB [15,16]. Small amounts of LPS stimulate these cells via theToll-like receptor (TLR4) and lead to the production of pro-

unstainedAnnexin
Annexin + PI PI
Cell N
um
ber (%
)
0
20
40
60
80
100 Medium
TEGDMA
LPS
LPS+TEGDMA
Camptothecin
24 h Exposure Period
RAW 264.7 mouse macrophages
a
a
a
a
b
b
b
c
c
c
c
d
d
d
d
tHPC
24 h Exposure Period
unstained
Annexin
Annexin + PI PI
Cell N
um
ber (%
)
0
20
40
60
80
100 Medium
TEGDMA
LPS
LPS + TEGDMA
Camptothecina
a
c
c
c
Fig. 7. Time-dependent induction of apoptosis and necrosis in mouse macrophages and human pulp-derived cells (tHPC). Cell cultures were exposed to 3 mM TEGDMA, 0.1 mg/mlLPS, a combination of 0.1 mg/ml LPS and 3 mM TEGDMA (LPS/TEGDMA), and 1 mM camptothecin for 24 h. Bars represent medians (25% and 75% percentiles) calculated from 5 to 6 (n)independent experiments. a ¼ significant differences between median values obtained in untreated controls (medium) and cell cultures exposed to TEGDMA; b ¼ significantdifferences between median values obtained in untreated controls (medium) and cell cultures exposed to LPS; c ¼ significant differences between median values obtained inuntreated controls (medium) and cell cultures exposed to LPS plus TEGDMA; d ¼ significant differences between median values obtained in untreated controls (medium) and cellcultures exposed to camptothecin.
S. Krifka et al. / Biomaterials 31 (2010) 2964–2975 2973
inflammatory mediators, for instance, TNF-a and IL-6 as well asanti-inflammatory cytokines like IL-10 which inhibit the uncon-trolled up-regulated release of TNF-a. Activation of the MAPK p38 isnecessary for production of TNF-a [31,32].
The present study provides the first experimental evidence forthe immediate onset of monomer-induced inhibition of LPS-stim-ulated cytokine release. Exposure of macrophages to LPS-induceda large and time-dependent increase in the secretion of TNF-a identical to earlier observations [33,34]. Consequently, themaximum release of TNF-a between 2 h and 6 h correlated witha delayed increase in its antagonist IL-10 in the present study. Theincrease in LPS-induced cytokine production was inhibited withoutany detectable delay in the presence of TEGDMA after shortexposure periods. This discovery is in agreement with and supportsour recent findings after long exposure periods, including theobservation that TEGDMA alone failed to cause any cytokineproduction [17]. Notable is that the LPS-induced release of TNF-a was inhibited about 1000-fold by TEGDMA at all time pointsanalyzed here, which indicated a continuous mechanism typical fora physiological cell response. At present it remains unclear whetherTEGDMA might interfere with signal transduction pathwaysleading to cytokine release or whether it may inhibit proteinsynthesis. Interestingly, however, the inhibition of the release ofcytokines after exposure periods as short as 15 min was not asso-ciated with cytotoxicity of TEGDMA via apoptosis since thepercentage of viable cells in these cell cultures was as high as inuntreated controls. Thus, it is more likely that the inhibition of anLPS-induced cytokine release in the presence of TEGDMA occurs viathe interference with fast signal transduction pathways leading tothe release of cytokines rather than with the relatively slow processof protein synthesis.
Yet, the mechanism behind these pathways leading to a down-regulation of the LPS-stimulated cytokine release by TEGDMAremains unclear. It has been previously reported that inhibition ofp38 MAPK decreased the release of TNF-a [35]. Since TEGDMA didnot strongly inhibit LPS-stimulated phosphorylation of any MAPKtested here, TEGDMA probably interferes with pathways down-stream of MAPK. It is possible that the resin monomer acts indi-rectly through the generation of ROS as discussed earlier [17]. Theimmediate and strong inhibition of the LPS-stimulated expressionof the pro-inflammatory cytokines TNF-a and IL-6 as well as theanti-inflammatory IL-10 possibly implies physiological tissue
responses. The generation of too much TNF-a may lead to theclinical manifestation of sepsis, but too little TNF-a will negativelyeffect bacterial clearance [36]. Thus, it appears as if TEGDMA hasthe potential to considerably disturb the complex regulationprocess of an appropriate cell response in the innate immunesystem.
4.3. Induction of apoptosis by the resin monomer TEGDMA
Cell death via apoptosis occurred in the presence of TEGDMA inmouse macrophages and human pulp-derived cells. This inductionof apoptosis was time-dependent and followed the same pattern inboth cell lines. LPS was only active in mouse macrophages. Thepercentage of LPS-induced cell deaths remained almost constantduring the entire observation period, and the percentage ofannexin V-positive was low. These findings are very similar to thosedetected by other authors in the same cell line [28,37]. Campto-thecin was similarly effective as TEGDMA in mouse macrophagesafter long exposure periods. It inhibits DNA synthesis via strandscission, thus causing cell death during the S-phase of the cell cycle[23,38].
To our knowledge, a similarly detailed analysis of the kinetics ofa resin monomer-induced cell death via apoptosis in differentrelevant model cell lines has not been shown before. Yet, thefindings presented here correspond with results obtained withprimary human pulp-derived cells and gingival fibroblasts aftersustained exposure [9,39]. Moreover, the concentration-dependentdecrease in the number of vital cells after long exposure periodsprecisely correlated with the activation of MAPK in the presence ofTEGDMA in both mouse macrophages and human pulp-derivedcells. Noteworthy is that LPS appeared to inhibit the activation ofMAPK in pulp cells in the presence of TEGDMA, but the percentageof cells in apoptosis remained the same in cultures treated withTEGDMA alone or with a combination of TEGDMA and LPS. Theseobservations show that LPS is active in human pulp-derived cells,although its inhibitory function on TEGDMA-induced phosphory-lation of the MAPK remains unknown at present. These findingsalso indicate that phosphorylation of MAPK in the presence ofTEGDMA might not be directly related to resin-induced apoptosis,and a clear causal relationship between the activation of MAPK andapoptosis in monomer-treated cells needs to be established. It hasbeen previously reported that ERK1/2 is probably not involved in

S. Krifka et al. / Biomaterials 31 (2010) 2964–29752974
TEGDMA-induced apoptosis in primary pulp cells because thepharmacological inhibition of this pathway only slightly changedthe percentage of cells in apoptosis [39]. It was observed that thenumber of apoptotic salivary gland cells increased after exposure toHEMA and TEGDMA when ERK1/2 activity was inhibited, andinhibition of JNK also reduced the percentage of apoptotic cells [18].These findings as well as our observations suggest that ERK1/2might be activated as a pro-survival factor in cells exposed toTEGDMA, and the activation of the stress kinases p38 and JNK isrelated to TEGDMA-induced apoptosis.
Despite the various findings on the activation of MAPK, a precisetime-related and causal relationship to the induction of apoptosis islacking for any environmental stressor investigated thus far. Sus-tained activation of MAPK was reported, but the regulation of cellsurvival and apoptosis was diversely discussed. For instance, rapidphosphorylation of p38 was observed in the presence of cadmiumchloride, however, phosphorylation of ERK1/2 was detected onlyafter long exposure periods. It appeared that the onset of apoptosiswas related to p38 activated by oxidative stress. In contrast, theactivation of ERK1/2 was inversely correlated with the induction ofapoptosis in some cell models, and the use of a specific inhibitorsuggested a protective effect of sustained ERK1/2 activation uponcell survival [40,41]. Similarly to our observations, treatment ofHeLa cells with hydrogen peroxide caused a time- and dose-dependent induction of apoptosis in parallel with sustained acti-vation of ERK1/2, JNK and p38 kinases. Inhibition of ERK1/2 and JNKactivation suggested that ERK1/2 and JNK inversely influenced cellsurvival in response to oxidative stress, and no effect was detectedwith p38 [42]. However, it was also reported that p38 MAPK wasactivated by various types of oxidative stress followed by apoptosis[43,44].
5. Conclusion
The present study provides experimental evidence thatTEGDMA interferes with the regulation of cellular pathwaysthrough mitogen-activated protein kinases (MAPK) in cells of theinnate immune system and human pulp tissue. The differentialtime-dependent activation of MAPK observed in both target organsof dental resin monomers was unknown thus far. The early andtime-dependent inhibition of LPS-induced cytokines release bymouse macrophages in the presence of TEGDMA is also a novelfinding, although the causal correlation of this inhibition to theactivation or inhibition of MAPK is still under further investigation.The present study demonstrates the obviously high level ofcomplexity between factors leading to the activation of MAPK,inhibition of cytokine release and induction of apoptosis in thepresence of monomers like TEGDMA.
Acknowledgment
This study was supported by the Deutsche For-schungsgemeinschaft DFG (grant number Schw 431/11-1).
Appendix
Figures with essential color discrimination. Figs. 1–7 in thisarticle are difficult to interpret in black and white. The full colorimages can be found in the on-line version, at doi:10.1016/j.biomaterials.2010.01.005.
References
[1] Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. CellMol Life Sci 1999;55:1230–54.
[2] Matsuzawa A, Ichijo H. Stress-responsive protein kinases in redox-regulatedapoptosis signaling. Antioxid Redox Signal 2005;7:472–81.
[3] Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cellcycle regulation. Biochim Biophys Acta 2007;1773:1299–310.
[4] Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellularsignal-related kinase positively regulates ataxia telangiectasia mutated,homologous recombination repair, and the DNA damage response. Cancer Res2007;67:1046–53.
[5] Hanks CT, Strawn SE, Wataha JC, Craig RG. Cytotoxic effects of resin compo-nents on cultured mammalian fibroblasts. J Dent Res 1991;70:1450–5.
[6] Geurtsen W, Spahl W, Muller K, Leyhausen G. Aqueous extracts from dentinadhesives contain cytotoxic chemicals. J Biomed Mater Res 1999;48:772–7.
[7] Reichl FX, Seiss M, Marquardt W, Kleinsasser N, Schweikl H, Kehe K, et al.Toxicity potentiation by H2O2 with components of dental restorative materialson human oral cells. Arch Toxicol 2008;82:21–8.
[8] Schweikl H, Spagnuolo G, Schmalz G. Genetic and cellular toxicology of dentalresin monomers. J Dent Res 2006;85:870–7.
[9] Janke V, von Neuhoff N, Schlegelberger B, Leyhausen G, Geurtsen W. TEGDMAcauses apoptosis in primary human gingival fibroblasts. J Dent Res2003;82:814–8.
[10] Spagnuolo G, D’Anto V, Cosentino C, Schmalz G, Schweikl H, Rengo S. Effect ofN-acetyl-L-cysteine on ROS production and cell death caused by HEMA inhuman primary gingival fibroblasts. Biomaterials 2006:1803–9.
[11] Paranjpe A, Cacalano NA, Hume WR, Jewett A. Mechanisms of N-acetylcysteine-mediated protection from 2-hydroxyethyl methacrylate-inducedapoptosis. J Endod 2008;34:1191–7.
[12] Schweikl H, Hartmann A, Hiller KA, Spagnuolo G, Bolay C, Brockhoff G, et al.Inhibition of TEGDMA and HEMA-induced genotoxicity and cell cycle arrest byN-acetylcysteine. Dent Mater 2007;23:688–95.
[13] Paranjpe A, Cacalano NA, Hume WR, Jewett A. N-acetyl cysteine mediatesprotection from 2-hydroxyethyl methacrylate induced apoptosis via nuclearfactor kappa B-dependent and independent pathways: potential involvementof JNK. Toxicol Sci 2009;108:356–66.
[14] Tsukimura N, Yamada M, Aita H, Hori N, Yoshino F, Chang-Il Lee, et al. N-acetylcysteine (NAC)-mediated detoxification and functionalization of poly(methylmethacrylate) bone cement. Biomaterials 2009;30:3378–89.
[15] Ulevitch RJ. Molecular mechanisms of innate immunity. Immunol Res2000;21:49–54.
[16] West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-likereceptors. Annu Rev Cell Dev Biol 2006;22:409–37.
[17] Eckhardt A, Harorli T, Limtanyakul J, Hiller KA, Bosl C, Bolay C, et al. Inhibitionof cytokines and surface antigen expression in LPS-stimulated murinemacrophages by triethylene glycol dimethacrylate. Biomaterials2009;30:1665–74.
[18] Samuelsen JT, Dahl JE, Karlsson S, Morisbak E, Becher R. Apoptosis induced bythe monomers HEMA and TEGDMA involves formation of ROS and differentialactivation of the MAP-kinases p38, JNK and ERK. Dent Mater 2007;23:34–9.
[19] Eckhardt A, Gerstmayr N, Hiller KA, Bolay C, Waha C, Spagnuolo G, et al.TEGDMA-induced oxidative DNA damage and activation of ATM and MAPKinases. Biomaterials 2009;30:2006–14.
[20] Spagnuolo G, D’Anto V, Valletta R, Strisciuglio C, Schmalz G, Schweikl H, et al.Effect of HEMA on human pulp cells survival pathways ERK and Akt. J Endod2008;34:684–8.
[21] Yoshii E. Cytotoxic effects of acrylates and methacrylates: relationships ofmonomer structures and cytotoxicity. J Biomed Mater Res 1997;37:517–24.
[22] Galler K, Schweikl H, Thonemann B, Schmalz G. Human pulp-derived cellsimmortalized with SV40. Eur J Oral Sci 2006;114:138–46.
[23] Legarza K, Yang LX. New molecular mechanisms of action of camptothecin-type drugs. Anticancer Res 2006;26(5A):3301–5.
[24] Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay forapoptosis. Flow cytometric detection of phosphatidylserine expression onearly apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods1995;184:39–51.
[25] Schweikl H, Altmannsberger I, Hanser N, Hiller KA, Bolay C, Brockhoff G, et al.The effect of triethylene glycol dimethacrylate on the cell cycle of mammaliancells. Biomaterials 2005;26:4111–8.
[26] Engelmann J, Volk J, Leyhausen G, Geurtsen W. ROS formation and glutathionelevels in human oral fibroblasts exposed to TEGDMA and camphorquinone.J Biomed Mater Res B Appl Biomater 2005;75:272–6.
[27] Bode AM, Dong Z. The functional contrariety of JNK. Mol Carcinog2007;46:591–8.
[28] Xaus J, Comalada M, Valledor AF, Lloberas J, Lopez-Soriano F, Argiles JM, et al.LPS induces apoptosis in macrophages mostly through the autocrineproduction of TNF-alpha. Blood 2000;95:3823–31.
[29] Xaus J, Comalada M, Valledor AF, Cardo M, Herrero C, Soler C, et al. Molecularmechanisms involved in macrophage survival, proliferation, activation orapoptosis. Immunobiology 2001;204:543–50.
[30] Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, et al. CD14regulates the dendritic cell life cycle after LPS exposure through NFAT acti-vation. Nature 2009;460:264–8.
[31] Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. Aprotein kinase involved in the regulation of inflammatory cytokine biosyn-thesis. Nature 1994;372:739–46.
[32] Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognitionreceptors and their cross talk. Annu Rev Biochem 2007;76:447–80.

S. Krifka et al. / Biomaterials 31 (2010) 2964–2975 2975
[33] Hsi ED, Remick DG. Rapid determination of cell-associated tumor necrosisfactor production by flow cytometry. Lab Invest 1993;68:740–5.
[34] Bueno C, Rodriguez-Caballero A, Garcıa-Montero A, Pandiella A, Almeida J,Orfao A. A new method for detecting TNF-alpha-secreting cells using direct-immunofluorescence surface membrane stainings. J Immunol Methods2002;264:77–87.
[35] Van den Blink B, Juffermans NP, ten Hove T, Schultz MJ, van Deventer SJ, vander Poll T, et al. p38 mitogen-activated protein kinase inhibition increasescytokine release by macrophages in vitro and during infection in vivo. JImmunol 2001;166:582–7.
[36] Hehlgans T, Pfeffer K. Immunology the intriguing biology of the tumournecrosis factor/tumour necrosis factor receptor superfamily: players, rules andthe games. Immunology 2005;115:1–20.
[37] Seminara AR, Ruvolo PP, Murad F. LPS/IFNgamma-induced RAW 264.7apoptosis is regulated by both nitric oxide-dependent and -independentpathways involving JNK and the Bcl-2 family. Cell Cycle 2007;6:1772–8.
[38] Gallo RC, Whang-Peng J, Adamson RH. Studies on the antitumor activity,mechanism of action, and cell cycle effects of camptothecin. J Natl Cancer Inst1971;46:789–95.
[39] Spagnuolo G, Galler K, Schmalz G, Cosentino C, Rengo S, Schweikl H. Inhibitionof phosphatidylinositol 3-kinase amplifies TEGDMA-induced apoptosis inprimary human pulp cells. J Dent Res 2004:703–7.
[40] Galan A, Garcia-Bermejo ML, Troyano A, Vilaboa NE, de Blas E, Kazanietz MG,et al. Stimulation of p38 mitogen-activated protein kinase is an early regula-tory event for the cadmium-induced apoptosis in human promonocytic cells.J Biol Chem 2000;275:11418–24.
[41] Tong Z, Singh G, Rainbow AJ. Sustained activation of the extracellular signal-regulated kinase pathway protects cells from photofrin-mediated photody-namic therapy. Cancer Res 2002;62:5528–35.
[42] Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidativestress: influences of mitogen-activated protein kinase signalling pathways oncell survival. Biochem J 1998;333(Pt. 2):291–300.
[43] Cai B, Chang SH, Becker EB, Bonni A, Xia Z. p38 MAP kinase mediatesapoptosis through phosphorylation of BimEL at Ser-65. J Biol Chem2006;281:25215–22.
[44] Cao C, Lu S, Kivlin R, Wallin B, Card E, Bagdasarian A, et al. AMP-activatedprotein kinase contributes to UV- and H2O2-induced apoptosis in human skinkeratinocytes. J Biol Chem 2008;283:28897–908.
Copyright © 2022 FDOKUMEN




















