
Optimized water-based ATRP of an anionic monomer: Comprehension and properties characterization
Upload
independentCategory
view
1download
0
ORIGINAL CONTRIBUTION
Influence of the third monomer on lauryl methacrylate–methylmethacrylate emulsion terpolymerization
Rukhsana Shabnam & Abu M. I. Ali &Muhammad A. J. Miah & Klaus Tauer & Hasan Ahmad
Received: 9 February 2013 /Revised: 23 March 2013 /Accepted: 27 March 2013 /Published online: 11 April 2013# Springer-Verlag Berlin Heidelberg 2013
Abstract An experimental study shows how the emul-sion terpolymerization of lauryl methacrylate (LMA)and methyl methacrylate is influenced by the nature ofthe third monomer. The third monomer is either glycidylmethacrylate, hydroxyethyl methacrylate, hydroxypropylmethacrylate, or styrene. We report the synthesis ofterpolymer particles with an appreciably high contentof the very hydrophobic LMA (between 0.2515 and0.238 molar fraction in the monomer mixture) in60:40 weight water/ethanol mixture as the continuousphase, poly(vinyl pyrrolidone) as a polymeric stericstabilizer, and potassium peroxodisulfate as the initiator.The emulsion terpolymerization proceeds smoothly with-out the formation of coagulum and leads to particleswith an average diameter clearly below 1 μm. Wediscuss the overall polymerization behavior regardingconversion–time curves, particle morphology, and glasstransition temperature of the terpolymers in dependenceof the lyophilicity/lyophobicity of the monomer mixture.
Keywords Emulsion copolymerization . Laurylmethacrylate . Rate of polymerization . Particle shape
Introduction
For several decades, emulsion polymerization is a matter ofgreat attention and attraction in both the academia and theindustry [1] because it is a convenient, effective, and simplemethod of preparing sub-micron-sized microspheres with var-iable surface characteristics evenly dispersed in the aqueouscontinuous phase. The simplest recipe for this kind of poly-merization includes water, monomers whose polymers arehydrophobic, initiators generating radicals with certain hydro-philicity, and, in many cases, surfactants. In ab initio batchpolymerizations with lyophilic initiators, as considered here,moderately hydrophobic monomers such as styrene or methylmethacrylate are present as monomer drops which act as amonomer reservoir. After an initial period where polymeriza-tion is restricted to the continuous phase and particle forma-tion takes place, the main polymerization locus shifts to theinterior of the latex particles. In the further course of polymer-ization, the monomer drops supply monomer to the monomer-swollen polymer particles by diffusion through the continuousphase. This brief description already illustrates the importantrole of both the solubility of the monomers in the continuousphase and the solubility of the polymers in the monomers. It isa matter of fact that emulsion polymerization cannot proceedsmoothly if the solubility of the monomer(s) in the continuousphase is below a certain threshold [2, 3]. This prerequisiteoften limits the synthesis of extremely hydrophobic polymersvia aqueous emulsion polymerization. However, for certainspecial applications such as resins for chromatographic sepa-ration, oil absorbency agents, viscosity modifiers, and oil-soluble drag reducers, it is highly desirable to increase thehydrophobicity of latex particles [4, 5]. But also for water-borne coatings, the hydrophobicity of the polymer particles isa crucial application property. Lauryl methacrylate(LMA) is one of the industrially important hydrophobic
R. Shabnam :M. A. J. Miah :H. Ahmad (*)Department of Chemistry, University of Rajshahi,Rajshahi 6205, Bangladeshe-mail: [email protected]
A. M. I. AliDepartment of Chemistry, University of Liverpool,Liverpool L69 7ZD, UK
K. Tauer (*)Max Planck Institute of Colloid and Interfaces, 14424 Potsdam,Germanye-mail: [email protected]
Colloid Polym Sci (2013) 291:2111–2120DOI 10.1007/s00396-013-2952-7

monomers which, in addition to hydrophobicity, also providespolymers with high flexibility (low glass transition tempera-ture, Tg). The very low water solubility (<<0.01 g/100 g at25 °C) of LMA renders this monomer unsuitable for emulsionpolymerization in pure aqueous continuous phases. Conven-tional emulsion polymerization of LMA results in poor con-version, coagulum formation, and poor compositional controlin copolymers [3]. These drawbacks happen because thepolymerization locus is shifted from the latex particles to themonomer droplets as the monomer’s solubility in water is solow that swelling of the latex particles practically not happens.
But the need to increase the hydrophobicity of aqueousemulsion polymers for certain applications initiated intenseactivities in industrial research departments in order to solvethe above-sketched problems. The search for possibilities toincorporate LMA in aqueous emulsion polymers led to thediscovery that β-cyclodextrin (β-CD), a cyclic hepta-amylose which is able to from inclusion complexes withmolecules fitting in the inner cavity [6], is an extremelyuseful additive to facilitate its incorporation [7, 8]. Subse-quently, a few papers appeared in the open literature regard-ing the application of β-CD in the aqueous emulsioncopolymerization of hydrophobic monomers [9–14]. β-CDacts as a kind of quite effective cargo transporter due to thecomplexation or solubilization of LMA in its hydrophobicinterior. The β-CD/LMA complex possessing a hydrophilicouter shell diffuses across the aqueous medium to the poly-mer particles, where the LMA is released. However, themechanism of this shuttle-like behavior of β-CD, particu-larly the question of what triggers the uptake and release ofthe hydrophobic monomers, is not yet fully understood.
Another approach to facilitate the incorporation of LMAin emulsion polymers is the modification of the solvency ofthe continuous phase for this kind of hydrophobic mono-mers. You et al. [15] studied the free radical emulsioncopolymerization of LMA and ethyl methacrylate in propyl-ene glycol which is a good solvent for both monomers. Thekinetics is very similar to conventional aqueous emulsioncopolymerization of monomers with high water solubility.
Additionally, a few reports are available on the solutionfree radical polymerization/copolymerization of LMA [16,17]. Increasing the solvency of water for hydrophobic mol-ecules is also possible with the addition of water-solubleorganic solvents, particularly alcohols. This approach datesback to the early 1940s of the last century when radicalprecipitation polymerization was established in pure alco-hols [18, 19] and in water–alcohol mixtures [20].
Recently, we reported the preparation of sub-micron-sized P(LMA–methyl methacrylate) latex particles via dis-persion polymerization in a water–ethanol mixture as thecontinuous phase using a water-soluble initiator [21]. Thepolymerization rate, average particle size, and morphologyare quite strongly influenced by the ethanol content in the
continuous phase and the type of stabilizer. In a subsequentpaper, the preparation of homo-PLMA particles by suspen-sion polymerization with 2,2-azobis(isobutyronitrile) asmainly the monomer-soluble initiator and poly(vinyl alco-hol) (PVA) as the steric stabilizer is described. The experi-mental data regarding suspension yield, particle diameter,and amount of coagulum prove that the amount of PVAplays a vital role in obtaining a stable suspension [22].PVA content as high as 30 wt% relative to the amount ofmonomer is necessary to reduce the quantity of coagulumbelow 20 %. This result is in accordance with the idea thatthe heterophase polymerization of LMA in a purely aqueouscontinuous phase proceeds almost exclusively inside themonomer droplets. Within this scenario, the coagulum rep-resents the fraction of the oversized monomer droplets dueto poor emulsification by mechanical stirring.
In the present study, we report how the addition of a thirdmonomer influences the heterogeneous copolymerization ofLMA andmethyl methacrylate (MMA). The third monomer isvaried regarding the hydrophilicity and the nature of thepolymerizing group. The following monomers were studied:glycidyl methacrylate (GMA), hydroxyethyl methacrylate(HEMA), hydroxypropyl methacrylate (HPMA), and styrene.The continuous phase is again a water/ethanol mixture (60:40,weight by weight), poly(vinyl pyrrolidone) (PVP) is the stericstabilizer, and potassium peroxodisulfate is the initiator.
Experimental
Materials and instruments
LMA from Fluka, Chemika (Switzerland) was washed with5 % NaOH aqueous solution to remove any inhibitor. StyrenefromAcros Organics (USA) andMMA from Fluka, Chemika,were distilled under reduced pressure and preserved in arefrigerator until use. HEMA and GMA from Fluka, Chemikaand HPMA from Aldrich (USA), all of monomer grade, wereused without purification. Potassium peroxodisulfate (KPS)purchased from LOBA Chem. (India) was recrystallized fromdistilled water and preserved in a refrigerator before use. PVPfrom Fluka, Chemika with a molecular weight of 3.6×105 g mol−1 was used as a polymeric stabilizer. Ethanol wasdehydrated by treating with active CaO and distilled beforeuse. Other chemicals used were of reagent grade. Deionizedwater was distilled using a glass (Pyrex) distillation apparatus.
NICOMP 380 (USA) particle sizer, scanning electronmicroscope (SEM; LEO Electron Microscopy Ltd., UK),Bruker Avance 400-MHz NMR spectrometer, differentialscanning calorimetry (DSC; Q2000, TA Instruments Ltd.,UK), Fourier transform infrared (FTIR; 8044 Shimadzu,Japan), and a TG16-WS high-speed centrifuge machinewere used for the characterization of copolymer latexes.
2112 Colloid Polym Sci (2013) 291:2111–2120

Polymerization procedure
For all polymerizations, the overall amount of monomerswas fixed to 4 g, the amount of the continuous phase to 36 gcomposed of 21.6 and 14.4 g of water and ethanol, respec-tively, the amount of PVP stabilizer was 0.1 g, and theamount of KPS initiator was 0.04 g. Five monomer mixtureswere investigated comprising the copolymerization LMA(2 g)/MMA (2 g) and four terpolymerizations withadditional HEMA, GMA, HPMA, and styrene. For theterpolymerizations, 0.5 g of LMAwas replaced with 0.5 g ofthe corresponding third monomer. All polymerizations werecarried out under nitrogen atmosphere at 70 °C for 12 h in athree-necked, round bottom flask equipped with a mechanicalstirrer (stirrer speed, 95 rpm). To maintain the polymerizationtemperature throughout the reaction, the reactor was placed ina thermostat water bath.
Latex and polymer characterization
Polymer samples were withdrawn from the reactor at definitetime intervals and placed in a pre-weighed dried ceramic Petridish. To prevent further polymerization, the ceramic dishcontained a known amount of 0.1 % hydroquinone solutionand was quenched rapidly in an ice water bath. Samples werekept in an oven at around 90 °C until a constant weight wasreached. It is to be mentioned that the samples were complete-ly dried under these conditions. Then, the percentage of over-all monomer conversion was calculated from the solidscontent by taking into account the amount of auxiliary mate-rials. All the measurements were carried out in duplicate. Theprocedure as applied allowed the determination of conversiondata with an accuracy of ±2–4 %.
Intensity-weighted average hydrodynamic diameterswere measured using dynamic laser light scattering with aNICOMP particle sizer. Before the measurement, the coag-ulum was removed by filtration through cellulose filters andthe sample diluted to around 0.1 % solids content usingdistilled ethanol. Each measurement was repeated twiceand the average is reported. The deviation in the averagesize measurement was less than ±10 %.
The copolymer latexes for FTIR, 1H NMR, and DSC anal-yses were purified by replacing the continuous phase with afresh water–ethanol mixture using a centrifugemachine.Wash-ing was repeated three times before drying the samples for therespective analysis. The FTIR spectra were taken in KBrpellets and the 1H NMR spectra taken in CDCl3 as the solvent.
The glass transition temperatures were measured with DSCaccording to the procedure reported elsewhere [23]. Driedpolymer samples were cooled to −90 °C and then heated upto 150 °C at a heating rate of 20 °C/min under nitrogenatmosphere and quench-cooled at a maximum cooling rate,then reheated for a second time. The glass transition
temperature was collected from the midpoint of the transitionregion in the second heating.
Results and discussion
The addition of a third monomer to the LMA/MMA co-monomer mixture has a profound influence on the course ofpolymerization (conversion and average particle size–timecurves) and on the properties of the terpolymer particles.However, before discussing these experimental data, a briefcharacterization of the reaction system before the start of thepolymerization is necessary. Solubility tests with the singlemonomers reveal that MMA, HEMA, HPMA, and GMA arecompletely miscible with the continuous phase. In contrast,LMA and styrene are not miscible and form droplets in thewater/ethanol mixture. Before starting the polymerization,the reaction system is in the state of an emulsion obviouslydriven by the insolubility of LMA, and also styrene whenapplied. As the monomers during the reaction distributebetween all phases, the situation is quite complex. Determi-nation of the monomer concentration in each of the phases isa complicated task which needs, in order to be solved,comprehensive investigation with sophisticated procedures,which are, however, beyond the scope of this contribution.
The conversion—time curves as summarized in Fig. 1show quite a strong influence of the third monomer on thecourse of the terpolymerization.
The conversion—time curves can be evaluated regardingthe rate of polymerization in each interval, which is deter-mined by differentiation of the corresponding conversionintervals with respect to time, as indicated by the regressionlines in the graphs of Fig. 1. The rates of polymerization, rp1for the lower and rp2 for the higher conversion ranges, aresummarized, together with the other data used for the fol-lowing considerations, in Table 1.
The conversion–time curves possess the characteristicshape with two almost linear intervals, as recently discussedfor LMA copolymerization with MMA [21, 22]. The thirdmonomer influences both the rate of polymerization in eachinterval and the transition between the intervals. GMA asthe third monomer has the strongest influence on the transi-tion between both intervals (cf. graph c of Fig. 1); in fact,the conversion—time curve is actually characterized bythree intervals with distinctly different slopes. Despite thispeculiarity, we subsequently discuss also for GMA only tworates of polymerization, as indicated in Fig. 1.
Obviously, the nature of the third monomer has a specificinfluence on rp1 and rp2 as compared by the bar chart of Fig. 2.
For all monomer combinations, rp1 is expectedly muchgreater than rp2 (cf. Table 2), which is mainly due to the largermonomer concentration during interval 1. The data of Fig. 2also reveal the special position of GMA among the third
Colloid Polym Sci (2013) 291:2111–2120 2113

monomers as it is the only example leading to a higher rate ofpolymerization than the LMA–MMA copolymerization (firstinterval). In all other cases, the rp1 and rp2 of theterpolymerizations are lower than that of the LMA–MMAcopolymerization. Interestingly, the highest rate during thesecond interval for the terpolymerizations is observed forstyrene. Among the methacrylates, GMA leads to the highestrp2 value.
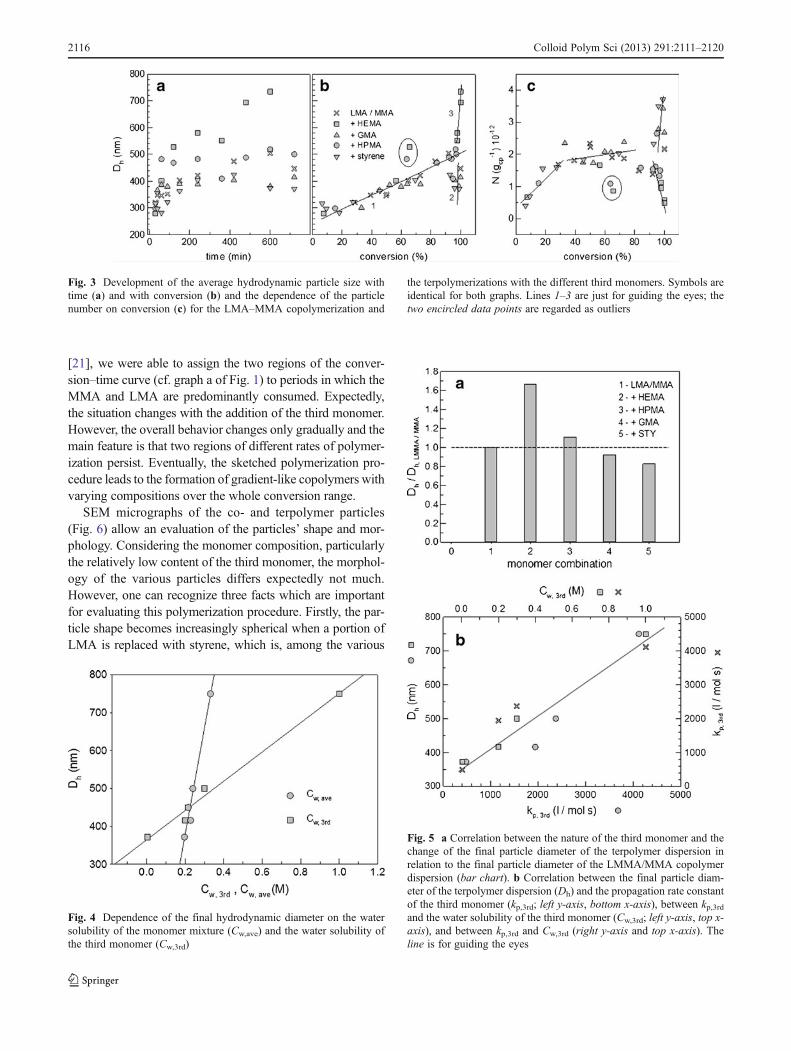
The development of the average particle size as puttogether in the graphs of Fig. 3 shows interesting featuresand reveals an interesting specificity regarding thelyophilic/lyophobic properties of the third monomer. Thefirst and most important feature is that for all polymeriza-tions, the average particle size increases linearly with con-version up to values greater than 85 %. For an initial quiteshort nucleation period and a subsequently constant particle
number, a linear relation between D3 and the conversion areexpected (Eq. 1). The linear dependence over quite a broadrange of conversion hints already an extended particle nu-cleation period as the particle growth with conversion isslower. This conclusion is proven by the particle numbersdepicted in graph 3c. The particle number was calculatedaccording to Eq. 1, assuming an average polymer density of1.1 g cm−3, and refers in graph 3c to the gram of continuousphase (gcp). In Eq. 1, mp is the overall mass of the polymerin grams, mm0 the initial amount of monomers in grams, Xthe monomer conversion in percent, N the particle number,ρp the polymer density in grams per cubic meter, and D theparticle diameter in nanometers.
mp ¼ mm0 � X
100¼ N � p
6� ρp � D3 � 10�21 ð1Þ
Fig. 1 Conversion–time curvesof LMA–MMAcopolymerization (a) andterpolymerizations withdifferent third monomers (b–d);the third monomer is mentionedin the corresponding graph.Dotted lines are the wholeconversion–time curves, blackdots mark the data points usedfor the differentiation tocalculate the rate ofpolymerization, and solid linesare the linear regression lines
Table 1 Relevant parameters for the data evaluation of the different monomer mixtures
Monomers Cmon (M) rp1 (%/min) rp2 (%/min) Dh (nm) kp,ave (l/mol s)a Cw,ave (M)b
LMA/MMA 0.0279 0.9270 0.0594 450 1,195.2 0.2154
+HEMA 0.0297 0.576 9.2100e-3 750 1,546.9 0.3310
+HPMA 0.0294 0.747 6.7900e-3 500 1,307.5 0.2398
+GMA 0.0294 1.006 0.0136 416 1,257.8 0.2279
+Styrene 0.0307 0.299 0.0204 372 1,058.3 0.1961
Cmon is the overall monomer concentration in the reaction mixture and Dh is the final hydrodynamic average particle size. The procedure tocalculate the average values is described in the texta For the calculation of the average propagation rate constant, the following values for the individual monomers, in liters/(mole second), for 70 °Chave been used: LMA, 1,590 [24]; MMA, 1,040 [24]; HEMA, 4,120 [25]; GMA, 1,940 [26]; HPMA, 2,370 [26]; styrene, 482 [24]b For the calculation of water solubility, the following values for the individual monomers (in molars) have been used: LMA, 3×10−7 ; MMA, 0.3;HEMA, 1; GMA, 0.2; HPMA, 0.3; styrene, 4×10−3 , cf. [27, 28]
2114 Colloid Polym Sci (2013) 291:2111–2120

The specificity of the monomer composition, particularlythe property of the third monomer, is expressed by thedevelopment of the average particle size in the very highconversion range (indicated by lines 2 and 3 in graph b ofFig. 3). A possible reason for the opposite behavior ofHEMA, and particularly styrene or GMA, is the differentinteraction of the corresponding terpolymer with the contin-uous phase. The HEMA-rich parts are soluble in the water–ethanol mixture and, hence, cause extensive swelling of theparticles. In contrast, the styrene- and GMA-containingterpolymers are lyophobic and try to minimize the contactwith the continuous phase by shrinking. The interaction ofthe terpolymer particles with the continuous phase becomesworse for these two third monomers with increasing con-version, which causes deswelling and shrinking of the par-ticles. Therefore, the change in the particle numbers atconversion >85 % is the consequence of this effect andcannot be compared with the increase in the particle num-bers at a lower conversion.
The best way to evaluate the polymerization behaviorquantitatively is compositional analysis of the co- and ter-polymers, which, however, is complicated by the fact that
due to the heterogeneous nature of the polymerization amixture of gradient-like co- and terpolymers is obtained[21]. Nevertheless, a kind of quantitative considerationwas tried using an average propagation rate constant(kp,ave) and solubility in water (Cw,ave) for each monomermixture. These averages (cf. Table 1) were calculated con-sidering the values of the corresponding monomers and theinitial molar composition of the monomer mixture.
Interestingly, the only (simple) clear correlation that ex-ists between the experimental data and the kinetically rele-vant parameters put together in Table 1 is the one betweenthe final average hydrodynamic particle size and the averagewater solubility or the water solubility of the third monomer,as depicted in Fig. 4.
Here, we use water solubility as the relevant parameterbecause it is a value which allows the easiest discriminationbetween the third monomers. Moreover, Cw is an expressionof the influence of the third monomer on the hydrophilicityof the resulting polymer, which is an important propertydetermining the dispersed state of the particles at the endof the polymerization (cf. also Fig. 3).
Neither the rate of polymerization nor the average particlesize shows a clear correlation with the propagation rate con-stants. This is not really surprising because under heteroge-neous conditions the polymerization kinetics is governed byan interplay of effects stemming from both the radical poly-merization behavior of the monomers and colloid chemistry.The bar chart diagram of Fig. 5a suggests a clear influence ofthe nature of the third monomer on the change of the finaldiameter in relation to that of the LMA/MMA copolymerdispersion. However, the data summarized in Fig. 5b showthat this correlation is not unambiguous. The final particle sizeof the terpolymer dispersions correlates with both the propa-gation rate and water solubility of the third monomer; more-over, also kp,3rd correlates nicely with Cw,3rd.
Therefore, the correlations between the kinetically andcolloidally important parameters displayed in Fig. 5b supportthe above idea that the scene is dominated by a combination ofpolymerization kinetics and colloid chemistry effects.
Based on the data summarized in Figs. 1 ,2, 3, 4, and 5,the polymerization behavior can be summarized in the fol-lowing way. The reaction starts in the continuous phasewhere the monomers react according to their initiation andpropagation frequency, both of which are controlled by thesolubility in the water–ethanol mixture. After particle nucle-ation, the polymerization is shifted with increasing overallconversion more and more into the particles where themonomer concentration is controlled by swelling. For theco- and terpolymerizations considered here, the swellingcapacity of the particles and the swelling rate differ for thedifferent monomers. For the LMA/MMA copolymerization,this situation is comparatively straightforward because bothmonomers possess distinctly different properties. Recently
Fig. 2 Bar chart illustrating the influence of the third monomer on therate of polymerization in both intervals; the rate of polymerization ineach interval is normalized by the corresponding rate of LMA–MMAcopolymerization (rp1,mon1 and rp2,mon1)
Table 2 Glass transition temperatures (Tg1 and Tg2) measured from themidpoint temperature of the glass transition range during the secondheating of the co- and terpolymers
Copolymers Tg1 (°C) Tg2 (°C)
LMA/MMA copolymer −48.9 98.08
+HEMA −55.23 103.13
+GMA −55.93 100.66
+HPMA −56.57 84.55
+Styrene – 79.51
Colloid Polym Sci (2013) 291:2111–2120 2115
[21], we were able to assign the two regions of the conver-sion–time curve (cf. graph a of Fig. 1) to periods in which theMMA and LMA are predominantly consumed. Expectedly,the situation changes with the addition of the third monomer.However, the overall behavior changes only gradually and themain feature is that two regions of different rates of polymer-ization persist. Eventually, the sketched polymerization pro-cedure leads to the formation of gradient-like copolymers withvarying compositions over the whole conversion range.
SEM micrographs of the co- and terpolymer particles(Fig. 6) allow an evaluation of the particles’ shape and mor-phology. Considering the monomer composition, particularlythe relatively low content of the third monomer, the morphol-ogy of the various particles differs expectedly not much.However, one can recognize three facts which are importantfor evaluating this polymerization procedure. Firstly, the par-ticle shape becomes increasingly spherical when a portion ofLMA is replaced with styrene, which is, among the various
Fig. 3 Development of the average hydrodynamic particle size withtime (a) and with conversion (b) and the dependence of the particlenumber on conversion (c) for the LMA–MMA copolymerization and
the terpolymerizations with the different third monomers. Symbols areidentical for both graphs. Lines 1–3 are just for guiding the eyes; thetwo encircled data points are regarded as outliers
Fig. 4 Dependence of the final hydrodynamic diameter on the watersolubility of the monomer mixture (Cw,ave) and the water solubility ofthe third monomer (Cw,3rd)
Fig. 5 a Correlation between the nature of the third monomer and thechange of the final particle diameter of the terpolymer dispersion inrelation to the final particle diameter of the LMMA/MMA copolymerdispersion (bar chart). b Correlation between the final particle diam-eter of the terpolymer dispersion (Dh) and the propagation rate constantof the third monomer (kp,3rd; left y-axis, bottom x-axis), between kp,3rdand the water solubility of the third monomer (Cw,3rd; left y-axis, top x-axis), and between kp,3rd and Cw,3rd (right y-axis and top x-axis). Theline is for guiding the eyes
2116 Colloid Polym Sci (2013) 291:2111–2120

third monomers, the one with the highest glass transitiontemperature (micrograph e of Fig. 6). A similar but weakenedeffect is observed for HPMA (micrograph d of Fig. 6).
Secondly, the particle size distribution due to the extendedparticle nucleation period is quite broad. Thirdly, estimatingthe particle size from the SEM micrographs and comparingthe largest particles visible with the average hydrodynamicdiameters given in Table 1 supports the above-discussed ideathat the terpolymer particles with HEMA are quite heavilyswollen with the continuous phase. TheDh of about 750 nm atthe end of the polymerization is about a factor of 2.5 largerthan the dimension of the largest dried terpolymer particles.
For the co- and terpolymers considered here, FTIRspectroscopy is not the analytical method which allows reallya clear identification of the composition because of the simi-larity of the monomers and the overwhelming influence ofester groups. However, the spectra of the terpolymer withstyrene prove the presence of the styrene units in the polymerclearly with the δ(C–C) and δ-(C–H) out-of-plane vibration ofthe phenyl ring at about 700 and 795 cm−1, respectively.
The better way to check the composition is NMR spec-troscopy (Fig. 7). In all spectra, the –CH2–O–CO– protonsignal of LMA and the CH3–O–CO– proton signal of MMAappeared at 3.927and 3.60 ppm, respectively. Also, multiple
proton signals at 0.852–1.900 ppm indicate the lauryl partCH3–(CH2)11– of LMA, and the signal at 1.943 ppm repre-sents the C–CH3 proton signal of both LMA and MMA. Inthe terpolymer with styrene, multiple signals at 7.268–6.784 ppm indicate the presence of styrene units. In thespectra of the terpolymers with both HEMA and HPMA,the signal of the –OH protons is visible at 3.930 ppm. Theprotons of the epoxy ring in the case of the terpolymer withGMA appear at 3.644 ppm (inset of Fig. 7). These resultsunambiguously confirm the presence of all monomer unitsin the terpolymers.
Knowing the glass transition temperature (Tg) of a co- orterpolymer allows some more or less rough conclusionsregarding both the composition of the sample and the over-all arrangements of the different monomer units in the co- orterpolymer chain. Figure 8a summarizes the heat flow–tem-perature curves for the various polymers under consider-ation. The LMA/MMA copolymer possesses two distinctTg values, one at about −48.9 °C (Tg1), which is close to thatof pure PLMA, and another at about 98.1 °C (Tg2), which isclose to that of pure PMMA [29, 30]. We believe that thisfinding shows that the copolymer sample is rather a mixtureof two gradient-like copolymers than a mixture of twohomopolymers. One portion of the copolymer is rich in
Fig. 6 SEM micrographs ofLMA−/−MMA copolymerparticles (a) and terpolymerparticles with HEMA (b), GMA(c), HPMA (d), and styrene (e)as third monomers
Colloid Polym Sci (2013) 291:2111–2120 2117

LMA (Tg1) and another rich in MMA units (Tg2). Thisinterpretation is mainly based on two facts. First, comparedwith the glass transition temperatures of the pure homopoly-mers (−65 °C for poly-LMA, 105 °C for poly-MMA) [29, 30],the Tg1 and Tg2 of the copolymer are considerably higher andlower, respectively. Second, for LMA–MMA copolymersprepared via comparable emulsion polymerization procedure
as described [21], conversion dependence of the glass transi-tion was observed. Obviously, the polymerization mechanismof this kind of heterophase polymerization is similar to thatdescribed by Melville et al. for vinyl acetate–methyl methac-rylate emulsion copolymerization [31]. Accordingly, gradient-like structures are obtained when growing radical crosses be-tween phases with drastically different monomer compositions.
Fig. 7 1H NMR spectra ofLMA/MMA copolymers andterpolymers (a) with styrene(b), HEMA (c), HPMA (d), andGMA (e)
Fig. 8 a DSC spectra of the different copolymers prepared by emul-sion copolymerization of LMA in 40 w% ethanol containing thewater–ethanol dispersion media in the presence of 0.1 g PVP withdifferent vinyl co-monomers: MMA (a), MMA and HEMA (b), MMA
and GMA (c), MMA and HPMA (d), and MMA and styrene (e). bCorrelation between the glass transition temperature and the averagesolubility of the monomer mixture; the line is just for guiding the eyes
2118 Colloid Polym Sci (2013) 291:2111–2120

The terpolymers with the methacrylates also show two glasstransition temperatures with, however, slightly shifted valuescompared to those of the LMA/MMA copolymer (cf. Table 2and Fig. 8b). The influence of the third monomer on thearrangement of the monomer units in the terpolymers can beroughly appraised from the shift of theTg values relative to theLMA/MMA copolymer. Tg1 and Tg2 show a tendency tohigher and lower values with decreasing average water solu-bility of the monomer mixture (or the water solubility of thethird monomer; Fig. 8b). The lack of Tg1 for the terpolymerwith styrene points to a considerable shift in the arrangementof the monomer segments along the polymer chain. Themonomer mixture with styrene is the most lyophobic one,and obviously, the main locus of the polymerization is shiftedmore to the monomer-swollen latex particles, leading to arandomized or statistical arrangement of the monomer unitsin the terpolymer chain.
Conclusions
Emulsion co- and terpolymerization of LMA/MMA and athird copolymer can be carried out in water–ethanol mixture(40 wt% ethanol), PVP as the steric stabilizer, and KPS as theinitiator. The rate of polymerization, the latex particle proper-ties, and the polymer properties depend strongly on the natureof the third monomer. The experimental results reveal a com-plex of kinetic and colloid chemical parameters expressed bythe average propagation rate constant and the average solubil-ity in water. These values take the molar composition of themonomer mixture into consideration, and the latter one alsocharacterizes its lyophilicity/lyophobicity. Obviously, thelyophilicity/lyophobicity of the monomer mixtures plays acrucial role as it correlates with the final particle size, particlemorphology, and the arrangement of the monomer units in theterpolymer chain.
Acknowledgments The authors thank Mrs. Rona Pitschke and Mrs.Heike Runge of MPI of Colloid and Interfaces for providing SEMmicrographs.
References
1. Hernandez HF, Tauer K (2002) In: Schlüter AD, Hawker CJ,Sakamoto J (eds) Synthesis of polymers. Wiley-VCH, Weinheim,pp 741–773
2. Lau W (2002) Emulsion polymerization of hydrophobic mono-mers. Macromol Symp 182:283–289
3. Tauer K, Ali AMI, Yildiz U, Sedlak M (2005) On the role ofhydrophilicity and hydrophobicity in aqueous heterophase poly-merization. Polymer 46:1003–1015
4. Xu WJ, Zhu XL, Cheng ZP, Chen JY (2003) Atom transfer radicalpolymerization of lauryl methacrylate. J Appl Polym Sci 90:1117–1125
5. Xu YY, Becker H, Yuan JY, Burkhardt M, Zhang Y, Walther A,Bolisetty S, Ballauff M, Muller AHE (2007) Double-grafted cy-lindrical brushes: synthesis and characterization of poly(laurylmethacrylate) brushes. Macromol Chem Phys 208:1666–1675
6. Lindner K, Saenger W (1982) Crystal and molecular-structure ofcyclohepta-amylose dodecahydrate. Carbohydrate Res 99:103–115
7. Schneiderman E, Stalcup AM (2000) Cyclodextrins: a versatiletool in separation science. J Chromatogr B 745:83–102
8. Loftsson T, Brewster ME (1996) Pharmaceutical applications ofcyclodextrins. 1. Drug solubilization and stabilization. J Pharm Sci85:1017–1025
9. Islam MF, Jenkins RD, Bassett DR, Lau W, Ou-Yang HD (2000)Single chain characterization of hydrophobically modified poly-electrolytes using cyclodextrin/hydrophobe complexes.Macromolecules 33:2480–2485
10. Leyrer RJ, Machtle W (2000) Emulsion polymerization of hydro-phobic monomers like stearyl acrylate with cyclodextrin as a phasetransfer agent. Macromol Chem Phys 201:1235–1243
11. Meyer EE, Islam MF, Lau W, Ou-Yang HD (2000) Complexationkinetics of cyclodextrin with hydrophobic molecules confined inan isolated droplet in water. Langmuir 16:5519–5525
12. Rimmer S, Tattersall PI (1999) The inclusion of beta cyclodextrinprovides a supramolecular solution to the problem of polymeriza-tion of dodecyl and octadecyl methacrylates in aqueous emulsion.Polymer 40:5729–5731
13. Rimmer S, Tattersall PI (1999) Emulsion polymerizations in thepresence of beta-cyclodextrin. Polymer 40:6673–6677
14. Rimmer S (2000) Cyclodextrins in the emulsion polymerization ofvinyl monomers. Macromol Symp 150:149–154
15. You X, Dimonie VL, Klein A (2001) Kinetic study of emulsioncopolymerization methacrylate/lauryl methacrylate in propyleneglycol, stabilized with poly(ethylene oxide)-block-polystyrene-block-poly(ethylene oxide) triblock copolymer. J Appl Polym Sci82:1691–1704
16. Habibi A, Vasheghani-Farahani E, Semsarzadeh MA, SadaghianiK (2004) Estimation of monomer reactivity ratios in free radicalsolution copolymerization of lauryl methacrylate–isobutyl methac-rylate. J Polym Sci Part A Polym Chem Ed 42:112–129
17. Choi EM, Shin KS, Hwang TS (2010) Synthesis and electrochem-ical properties of ion exchange membrane based on crosslinkedstyrene-(2-hydroxyethyl acrylate)-lauryl methacrylate copolymer.Macromol Res 18:1209–1217
18. Abere J, Goldfinger G, Naidus H, Mark H (1945) Polymerizationof styrene under various experimental conditions. J Phys Chem49:211–226
19. Yosida T, Titani T (1941) Polymerisation von Styrol in schweremAlkohot. Bull Chemi Soc Japan 16:125–136
20. Chern C-S, Yu T-C (2005) Effect of 1-pentanol on the styreneemulsion polymerization. Polymer 46:1899–1904
21. Ahmad H, HasanMK,MiahMAJ, Ali AMI, Tauer K (2011) Solventeffect on the emulsion copolymerization of methyl methacrylate andlauryl methacrylate in aqueous media. Polymer 52:3925–3932
22. Ahmad H, Abu-Waesmin M, Rahman MM, Miah MAJ, Tauer K(2013) Preparation of hydrophobic polymer particles by radicalpolymerization and subsequent modification into magneticallydoped particles. J Appl Polym Sci 127:620–627
23. Jin JM, Lee JM, Ha MH, Lee K, Choe S (2007) Highly crosslinkedpoly(glycidyl methacrylate-co-divinyl benzene) particles by pre-cipitation polymerization. Polymer 48:3107–3115
24. Asua JM, Beuermann S, Buback M, Castignolles P, Charleux B,Gilbert RG, Hutchinson RA, Leiza JR, Nikitin AN, Vairon JP, vanHerk AM (2004) Critically evaluated rate coefficients for free-radical polymerization. 5. Propagation rate coefficient for butylacrylate. Macromol Chem Phys 205:2151–2160
25. Buback M, Kurz CH (1998) Free-radical propagation rate coeffi-cients for cyclohexyl methacrylate, glycidyl methacrylate and 2-
Colloid Polym Sci (2013) 291:2111–2120 2119

hydroxyethyl methacrylate homopolymerizations. MacromolChem Phys 199:2301–2310
26. Hutchinson RA, Beuermann S, Paquet DA, McMinn JH, JacksonC (1998) Determination of free-radical propagation rate coeffi-cients for cycloalkyl and functional methacrylates by pulsed-laserpolymerization. Macromolecules 31:1542–1547
27. Daubert TE, Danner RP, Sibul HM, Stebbins CC (1998) Physicaland thermodynamic properties of pure chemicals: data compila-tion. Taylor and Francis, Washington
28. Tauer K (2005) Stability of monomer emulsion droplets and im-plications for polymerizations therein. Polymer 46:1385–1394
29. Andrews RJ, Grulke EA (1999) In: Brandrup J, Immergut EH, GrulkeEA (eds) Polymer handbook (chapter 6). Wiley, New York, p 193
30. Peyser P (1989) In: Brandrup J., Immergut EH (eds) Polymerhandbook (chapter 6). Wiley, New York, p 209
31. Allen PEM, Downer JM, Hastings GW, Melville HW, Molyneux P,Urwin JR (1956) New methods of preparing block copolymers.Nature 177:910–912
2120 Colloid Polym Sci (2013) 291:2111–2120
Copyright © 2022 FDOKUMEN




















