
Rescue of ∆F508-CFTR by Kinase Inhibitors - TSpace
134
Rescue of ∆F508-CFTR by Kinase Inhibitors by Duy (Leo) Nguyen A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Biochemistry University of Toronto © Copyright by Duy (Leo) Nguyen (2013)
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Rescue of ∆F508-CFTR by Kinase Inhibitors - TSpace

Rescue of ∆F508-CFTR by Kinase Inhibitors
by
Duy (Leo) Nguyen
A thesis submitted in conformity with the requirements
for the degree of Master of Science
Graduate Department of Biochemistry
University of Toronto
© Copyright by Duy (Leo) Nguyen (2013)

ii
Rescue of ∆F508-CFTR by Kinase Inhibitors
Duy (Leo) Nguyen
Master of Science
Department of Biochemistry
University of Toronto
2013
Abstract
ΔF508-CFTR is a trafficking mutant that is retained in the ER, unable to reach the plasma
membrane. To identify corrector of this mutant, we screened a kinase inhibitor library enriched
for compounds clinically available or in clinical trials for the treatment of other diseases, using
our recently developed high-content functional screen. Several inhibitors of receptor tyrosine
kinases exhibited strong rescue of ∆F508-CFTR. Moreover, prominent rescue was also observed
with inhibitors of four major pathways: Ras/Raf/MEK/ERK, TAK1/p38, Wnt/GSK-3β, and
PI3K/Akt/mTOR. A complimentary siRNA screen was also performed to identify pathways
involved in the rescue. FGFR1 and several proteins downstream of FGFRs were identified
suggesting a possible role of these receptors in regulating ΔF508- CFTR trafficking. Moreover,
the use of compounds clinically available or in clinical trials for other diseases can expedite
delivery of treatment for CF patients.

iii
Acknowledgements
First, I would like to thank my supervisor, Dr. Daniela Rotin, not only for giving me an
opportunity to work on this very interesting project, but also for teaching me self-discipline, and
providing me guidance and encouragement through the whole Master program. I also want to
thank my committee members, Dr. Christine Bear and Dr. Walid Houry, for their support and
suggestions during the committee meetings.
Secondly, I want to thank Agata Trzcinska-Daneluti, who is a mentor, a colleague, and a
friend. She not only performed the Cellomics and flow cytometry studies, but also has been
giving me helpful suggestions throughout the project. I want to thank Dr. Chong Jiang for
teaching me all the techniques, especially the Ussing chambers, since I started in the lab, and
Ruth Milkereit for helping me extract the macrophages for my phosphoprotein experiment. I also
want to thank all of lab members, Avi, Chen, Ryan, Phillip, Wioletta, and Yunan, who made my
journey much more fun and pleasant.
Lastly, I want to thank my wife, An, and my family for believing and supporting me.
Without them, I would not be able to finish this program.

iv
Table of Contents
Abstract iii
Acknowledgements iv
Table of Contents v
List of Tables viii
List of Figures ix
Abbreviations xi
Chapter 1: Introduction
I. Cystic Fibrosis
1. Overview and pulmonary pathogenesis in CF patients 2
2. Current clinical management of CF 3
II. CFTR and CF-causing mutations
1. CFTR structure 6
2. Channel gating by ATP binding and hydrolysis 9
3. Regulation of CFTR by phosphorylation 12
4. Overview of CF-causing mutations 13
5. ∆F508-CFTR mutation and its defects 15
III. Chaperone systems involved in the processing of CFTR
1. ER-associated and cytosolic chaperone systems 22
a. Hsp70 and its cochaperones 22
b. ER membrane-bound and luminal chaperones 26
c. Hsp90 in the processing of CFTR 27
2. Peripheral chaperone systems 28
IV. Screens for correctors of the ∆F508-CFTR defects
1. High-throughput screens for correctors of ∆F508-CFTR 30
2. Discovery of VX-809 and VX-770 and their clinical trials 32

v
3. Our screens using high-content Cellomics assays 33
V. Project rationale and goals 37
Chapter 2: Materials and Methods
1. Media and reagents 39
2. Small molecules kinase inhibitor library 39
3. Cells 40
4. Cellomics YFP halide exchange screen 41
5. Data analysis 42
6. Immunoblotting 42
7. Flow cytometry 43
8. Short-circuit (Isc) measurements in Ussing chambers 43
9. Isolation of bone marrow-derived macrophages (BDMM) 44
10. Phosphoprotein analysis 44
11. shRNA knockdown and qPCR quantification of knockdown 47
Chapter 3: Results
I. Screen for kinase inhibitors that correct ∆F508-CFTR function using high-content
functional Cellomics assays
II. Validation of the hits
1. Maturation of ∆F508-CFTR
2. Functional analysis of correction of ∆F508-CFTR by the kinase
inhibitors
3. Effect of kinase inhibitors on ∆F508-CFTR chloride channel activity in
primary Human Bronchial Epithelial (HBE) cells harvested from CF
patients
III. Dose response curves of rescue of ∆F508-CFTR in MDCK cells treated with
select kinase inhibitors
IV. Analyses of E6201, a derivative of (5Z)-7-Oxozeaenol
1. E6201 did not rescue ∆F508-CFTR maturation and function
2. Comparison of the effect of E6201 and (5Z)-7-Oxozeaenol on different
signaling pathways
V. Validations of hits of esi-RNA screen
48
57
62
54
66
69
74
80

vi
Chapter 4: Discussion
I. Kinase inhibitor screen 89
II. E6201 and (5Z)-7-Oxozeaenol 94
III. esiRNA screen 95
Future directions
I. Testing the effect of knocking down top hit genes on rescuing ∆F508-CFTR
function using Ussing chamber
II. Elucidate the mechanism of rescuing ∆F508-CFTR by Oxozeaenol 99
III. Elucidate the pathways through which FGFR1 regulates the rescue of ∆F508-CFTR 100
Summary 100
Conclusion 102
References 103
99

vii
List of Tables
Table I. Hit compounds and their validations. 50
Table II. Hit genes of the siRNA screen. 81

viii
List of Figures
Figure 1: Pulmonary pathogenesis in Cystic Fibrosis 4
Figure 2: Schematic diagram and three-dimensional structural model of CFTR. 7
Figure 3: Gating of CFTR channel by ATP hydrolysis. 10
Figure 4: Classes of CFTR mutations. 16
Figure 5: ∆F508-CFTR trafficking and folding defects. 19
Figure 6: ER-associated and peripheral quality control systems involved in the
trafficking of CFTR.
Figure 7: Principles of the Cellomics assays to test rescue of mutant CFTR. 35
Figure 8: Ussing chamber schematic diagram and ∆Isc calculations. 45
Figure 9: Representative hits of the high-content screen. 52
Figure 10: Effect of select kinase inhibitors on ∆F508-CFTR maturation analyzed
by immunoblotting.
Figure 11: Effect of kinase inhibitors on cell surface expression of ∆F508-CFTR
analyzed by flow cytometry.
Figure 12: Effect of compounds’ treatment on ∆F508-CFTR channel activity in
MDCK cells stably expressing ∆F508-CFTR.
Figure 13: Effect of compounds’ treatment on ∆F508-CFTR activity in primary
Human Bronchial Epithelial (HBE) cells harvested from lungs of
∆F508/∆F508 homozygote patients undergoing lung transplant.
Figure 14: Dose response curves of select kinase inhibitors for rescue of ∆F508-
CFTR expressed in MDCK cells.
Figure 15: E6201 does not rescue the function of ∆F508-CFTR. 70
Figure 16: E6201 does not rescue the maturation and function of ∆F508-CFTR
by immunoblotting and Ussing chambers.
Figure 17: Signaling pathways inhibited by (5Z)-7-Oxozeaenol and E6201. 75
23
55
58
60
63
67
72

ix
Figure 18: Both (5Z)-7-Oxozeaenol and E6201 inhibit the phosphorylation of
similar downstream signaling targets.
Figure 19: Validation of hits of the esiRNA screen by Cellomics assays. 82
Figure 20: Validation of hits of the esiRNA screen by immunoblotting. 85
78

x
Abbreviations
∆F508-CFTR deletion of phenylalanine at position 508 in CFTR
∆Isc difference in maximal stimulated current
293MSR genetically engineered HEK 293 cell line expressing the human
macrophage scavenger receptor
4PBA sodium 4-phenylbutyrate
5% Blotto 5% dry milk made with PBST
ABC adenine nucleotide-binding cassette
AMP-PNP adenylyl imidodiphosphate
ASL airway surface liquid
ATP adenosine triphosphate
BHK baby hamster kidney cell
BMM macrophage extracted from mouse bone marrow
CAMK2B calcium/calmodulin-dependent protein kinase II beta
cAMP cyclic adenosine monophosphate
CF cystic fibrosis
CFTR cystic fibrosis transmembrane conductance regulator
CHIP carboxyl terminus of Hsc70 interacting protein

xi
CK2 casein kinase 2
CL4 cytoplasmic loop 4
Corr-4a corrector 4a
CPDR Canadian CF patient registry report
DAG diacylglycerol
DMEM Dulbecco’s Modified Eagle’s Medium
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
DNase deoxyribonuclease
DNDS 4,4-dinitrostilbene-2,2- disulfonic acid
dTDP thymidine diphosphate
dTMP thymidine monophosphate
dTTP deoxythymidine triphosphate
DTYMK deoxythymidylate kinase
EC50 half maximal effective concentration
ECL enhanced chemiluminescence
ENaC epithelial sodium channel

xii
ER endoplasmic reticulum
ERAD ER-associated degradation
ERK extracellular signal-regulated kinase
ESCRT endosomal sorting complex required for transport
esiRNA endonuclease-prepared siRNA
FACS fluorescence-activated cell sorting
FBS fetal bovine serum
FGFR fibroblast growth factor receptor
FIG mixture of forskolin, IBMX, Genistein
FRS2 factor receptor substrate
G551D-CFTR glycine to aspartic acid substitution mutation at position 551 in CFTR
GAB1 Grb2-associated binding protein 1
GFP green fluorescent protein
Gly GlyH-101
GRB2 growth factor receptor-bound protein 2
Gsk3β glycogen synthase kinase 3 beta
HA hemagglutinin
HBE human bronchial epithelial

xiii
HBSS Hank’s balanced salt solution
Hdj human DnaJ homologue
HEK human embryonic kidney cell
HRP horseradish peroxidase
Hsc heat shock cognate protein
HSF1 heat shock factor 1
Hsp heat shock protein
HTS high-throughput screen
IBMX 3-isobutyl-1-methylxanthine
ICD intracellular domain
IP3 inositol triphosphate
IP5 inositol pentakisphosphate
IPMK inositol polyphosphate multikinase
Isc short-circuit current
IκB inhibitor of kappa-B
JNK c-Jun N-terminal kinase
LPS lipopolysaccharide
MAP3K mitogen-activated protein kinase kinase kinase

xiv
MDCK Madin-Darby canine kidney epithelial cell
MEK MAPK/ERK kinase
MSD membrane-spanning domain
mTOR mammalian target or rapamycine
NDB nucleotide-binding domain
NFκB nuclear factor kappa-light-chain-enhancer of activated B cell
NOS2 nitric oxide synthase 2
PAL mixture of pepstatin, aprotinin, and leucine
PBS phosphate buffered saline
PBST phosphate buffered saline with Tween 20
PDGFR platelet derived growth factor receptor
PI3K phosphoinositide 3 kinase
PIAS1 Protein inhibitor of activated STAT1
PIP2 phosphatidylinositol 4, 5-bisphosphate
PKA protein kinase A
PKC protein kinase C
PLC phospholipase C

xv
PMSF phenylmethylsulfonyl fluoride
PRKAR2B cAMP-dependent protein kinase type II-beta regulatory subunit
PRKAR2B cAMP-dependent protein kinase type II-beta regulatory subunit
R domain regulatory domain
RE regulatory extension
RhoA Ras homologue gene family member A
RI regulatory insertion
RIPK receptor-interacting protein kinase
RNA ribonucleic acid
RNAi RNA interference
ROCK Rho-associated, coiled-coil containing protein kinase
RPS6KC1 ribosomal protein S6 kinase delta-1
RSK ribosomal S6 kinase
RTK receptor tyrosine kinase
RT-qPCR quantitative real time polymerase chain reaction
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
shRNA small/short hairpin RNA
siRNA small-interfering RNA

xvi
SOS protein son of sevenless
STAT1 signal transducer and activator of transcription 1
SUMO small ubiquitin-like modifier
TAK TGF-beta activated kinase
TGF transforming growth factor
TMPK thymidylate kinase
TPR tetratricopeptide
USP ubiquitin specific protease
VEGFR vascular endothelial growth factor receptor
WT-CFTR wild-type CFTR
YFP yellow fluorescent protein

1
CHAPTER 1
INTRODUCTION

2
I. CYSTIC FIBROSIS
1. Overview and pulmonary pathogenesis in CF patients:
Cystic Fibrosis (CF) is an autosomal recessive disorder, most common among Caucasian
populations, and affects 1 in 2500 live births (Ratjen and Doring, 2003). It is caused by
mutations in the gene encoding the protein Cystic Fibrosis Transmembrane Conductance
Regulator(CFTR), which is a chloride channel expressed in most secretory and absorptive
epithelial cells. Besides functioning as a chloride channel, CFTR is also known to be permeable
to HCO3- (Devor et al., 1999; Tang et al., 2009) and to regulate other membrane proteins,
especially the sodium channel ENaC. For example, in the sweat glands, CFTR activity is
required for ENaC activation (Reddy et al., 1999), and both channels’ activities are needed to
reabsorb ions back into the sweat ducts. In airways, ENaC activity is elevated in CF, the exact
opposite of what is seen in sweat glands and ducts (Reddy, 2003; Boucher, 2004).
Even though CF is a pleiotropic disease affecting various organs such as the intestine,
liver, pancreas, and vas deferens, the major morbidity and mortality are due to chronic lung
inflammation and disease. CF pulmonary pathogenesis is considered as a failure of the innate
defence mechanisms of the lungs against pathogens. In normal airways, anion secretion is
mediated by CFTR and other alternative chloride channels, coupled with sodium absorption by
ENaC to maintain the fluid homeostasis of the airway surface liquid (ASL). Proper volume
homeostasis of the ASL maintains the viscosity of the mucus layer to promote appropriate cilia
movement and efficient mucocilliary clearance of bacteria. However in the airways of CF
patients, reduced Cl- secretion, due to the lack of functional CFTR on the apical surface, and
hyper-absorption of Na+, due to elevated activity of ENaC, which is negatively regulated by
CFTR in airway cells (Stutts et al., 1995; Stutts et al., 1997; Rubenstein et al., 2010; Gentzsch et

3
al., 2010), lead to dehydration of the ASL. This increases the viscosity of the mucus layer, and
deposition of the thickened mucus collapses the cilia and impairs the mucocilliary clearance
mechanism. Moreover, the deposited layer of mucus creates an environment that promotes
bacterial colonization, commonly by Pseudomonas aeruginosa, and eventually leads to chronic
infection of the lungs (Boucher RC, 2004)(Figure 1). Combined with excessive inflammatory
response due to both infection and dysregulation of inflammatory response in airway cells, CF
patients eventually suffer from irreversible airway damage and respiratory failure (Davies et al.,
2007; Bodas and Vij, 2011).
2. Current clinical management of CF
CF is considered to be a deadly disease of young people (Gadsby DC, 2006). Even
though at birth the airways are uninfected, lung infection and inflammation occur soon after
(Gibson et al., 2003). In 1938, when CF was first recognized as a separate disease, 70% of babies
with CF died within their first year of life due to the inability to absorb nutrients in the intestine
(Garattini et al., 2011). With immense improvements in the treatments of CF, the median
predicted survival age of Canadians with CF in 2011 was estimated to be around 48 years
(Canadian CF patient registry report (CPDR), 2013). However, an ultimate curative treatment for
CF is currently unavailable.
To date, the main goals of the treatments of cystic fibrosis are to relieve the symptoms of
the disease and to enhance the quality of life (Antunovic et al., 2013). The treatments target
nutrition, relief of airway obstruction, and suppression of airway infection and inflammation
(Davis PB, 2006; Garattini et al., 2011). First, long-term nutritional management is very
important for CF patients as about 90% of patients suffer from pancreatic insufficiency, which is

4
Figure 1: Pulmonary pathogenesis in Cystic Fibrosis. In normal airways, the balance between
Na+
absorption (mediated by ENaC in the apical membrane) and anion secretion (mediated by
apical CFTR and alternative anion channels) determines the volume of the fluid on airway
surfaces. This maintains the viscosity of the ASL and allows the proper cilia movement to clear
out bacteria and deposited mucus. In CF airways, lack of functional CFTR leads to reduced
chloride secretion and augments sodium absorption; this, in turn, dehydrates the ASL leading to
the deposition of thicken mucus on the airway surface and impeding the mucocilliary clearance
mechanism. Failure of the clearance mechanism results in bacterial colonization, hyper
inflammation, and chronic infections of the lungs (Modified from Frizzell & Pilewski, 2004).

5

6
caused by the obstruction and damage of the pancreatic duct and results in malabsorption and
deficiency of (especially fat-soluble) nutrients including vitamins (Ooi and Durie, 2012).
Moreover, CF patients also require extra energy to overcome increased work of breathing and
constant battles against infections (http://www.cysticfibrosis.ca), and might experience episodes
of hypochloremia or hyponatremia due to the excessive loss of salt through sweat (Priou-
Guesdon et al., 2010). Therefore, the dietetic management includes very high-caloric intake
(120-150% of the normal recommended daily allowance), daily supplementation of pancreatic
enzymes, fat soluble vitamins (A, D, E, K), and sodium chloride (Antunovic et al., 2013;
Garattini et al., 2012; http://www.cysticfibrosis.ca).
The second main target of CF treatments is to clear out obstruction in the airway. These
therapies include daily airway clearance physiotherapies such as postural drainage, chest
percussion, positive expiratory pressure, and breathing exercises. Complementary treatments to
enhance mucociliary clearance of repiratory secretions may include mucolytics, such as DNase
or hypertonic saline, in combination with bronchodilators to clear out mucus and enlarge the
luminal diameter of the airway. The last main area of treatments is to suppress infection and
inflammation using different antibiotics and anti-inflammatories, including inhaled, oral, or
intravenous medications depending on the type of drug and the severity of the infections
(Antunovis et al., 2013; http://www.cysticfibrosis.ca). In addition to these main areas, CF
patients have to endure additional treatments during an exacerbation or onsets of other
complications such as CF-related diabetes, bone disease that might result in long period of
hospitalization.
Even though these therapies, which have to be carried out across the lifespan of CF
patients, have significantly improved the survivorship and quality of life of the patients, they are

7
still complex and time-consuming processes which greatly affect and become burdens to the
lives of not only the patients but also their family. Eventually, however, lung failure is inevitable,
and lung transplantation is required. Therefore, a drug that could target the basic molecular cause
of the disease is much needed.
II. CFTR AND CF-CAUSING MUTATIONS:
1. CFTR Structure
CFTR was first identified as a member of the adenine nucleotide-binding cassette (ABC)
family of transporters (Riordan et al., 1989). Although it has the core structural architecture of an
ABC transporter, CFTR is the only member of this family functioning as Cl- channel (Kartner et
al., 1991). Like other members of the family, CFTR consists of two symmetrical halves, each
consisting of one nucleotide-binding domain (NDB), which possesses a binding site for ATP,
and one membrane-spanning domain (MSD), which is comprised of six transmembrane
segments (Figure 2). These two halves are linked together by a unique, highly unstructured
regulatory (R) region/domain. While the two MSDs form the anion-selective pore of the channel,
the two NBDs form a head-to-tail dimer with the two ATP binding sites at the interface (Riordan
JR, 2008). To date, no high-resolution structure of the full-length CFTR channel has been solved.
However, models of the structure of the full-length CFTR have been built based on the structure
of the bacterial transporter Savv1866 (Mornon et al., 2008, 2009;

8
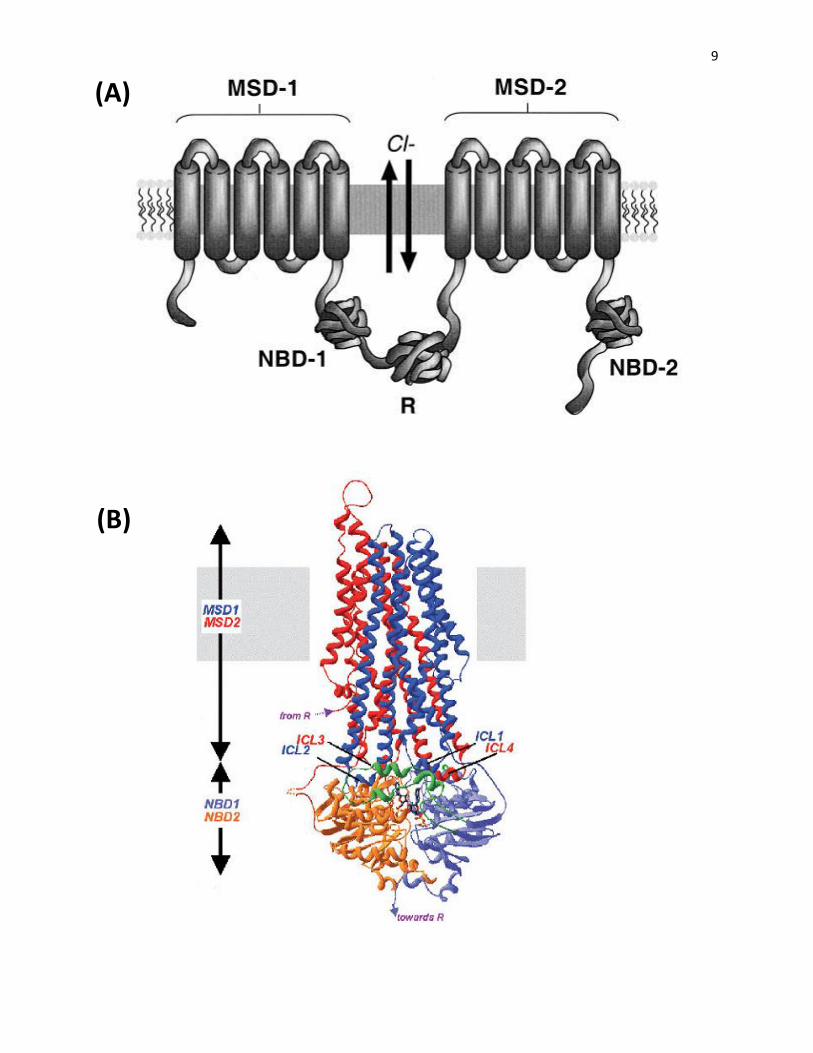
Figure 2: Schematic diagram and three-dimensional structural model of CFTR. A)
Schematic diagram of CFTR with two halves, each consists of one MSD (membrane-spanning
domain) and one NBD (nucleotide binding domain), linked together by the unstructured R region
(domain). B) CFTR three-dimensional built based on the experimental structural model of the
bacterialtransporter Savv1866. The intracellular loops of MSDs (ICL1, ICL2 of MSD1 and
ICL3, ICL4 of MSD2) provide the contacts with NBDs to create the MSD/NBD interfaces
(Lyczak et al., 2002 ; Mornon et al., 2008).
9
(A)
(B)

10
Serohijos et al., 2008).Even though there are some limitations due to the use of Savv1866
structure as a template, this three-dimensional model still permits better insights into the
interactions between the domains of CFTR and the molecular mechanism underlying the activity
and gating of the channel.
2. Channel gating by ATP binding and hydrolysis
The gating mechanism of the channel has been extensively studied. Unlike other
members of the ABC family that use energy from ATP hydrolysis to transport substances against
a concentration gradient, the binding and hydrolysis of ATP in the NBDs of CFTR regulate the
opening and closing of the channel via conformational change caused by the formation and
disruption of the NBD1-NBD2 dimer complex. Like several other members of the ABC family,
the hydrolysis rate of ATP is different between the two domains. The ATP in the binding site of
NBD1 is negligibly slowly hydrolyzed while ATP in the site of NBD2 readily undergoes
hydrolysis (Aleksandrov et al., 2008). Upon binding of ATP to the NBD2 binding site, the two
domains come together to form a dimer complex and open the channel. When hydrolysis of the
ATP at this site occurs, the complex falls apart and the channel closes until the next ATP binding
event (Gadsby et al., 2006).The gating sequence of events are depicted in Figure 3.
The hydrolysis of ATP in the ATP-binding site 2 in NBD2 facilitates the rapid opening
and closing of the channel. However, the hydrolysis of ATP itself is not responsible for closing
and opening of the channel. AMP-PNP, a non-hydrolysable analogue of ATP, can lock the
channel in the open state (Hwang et al., 1994). This suggests that the presence of ATP in the
second binding site is responsible for opening of the channel. This locked open state can also be
observed when the protein is mutated at residue K1250 that abolishes ATP hydrolysis, and leads

11
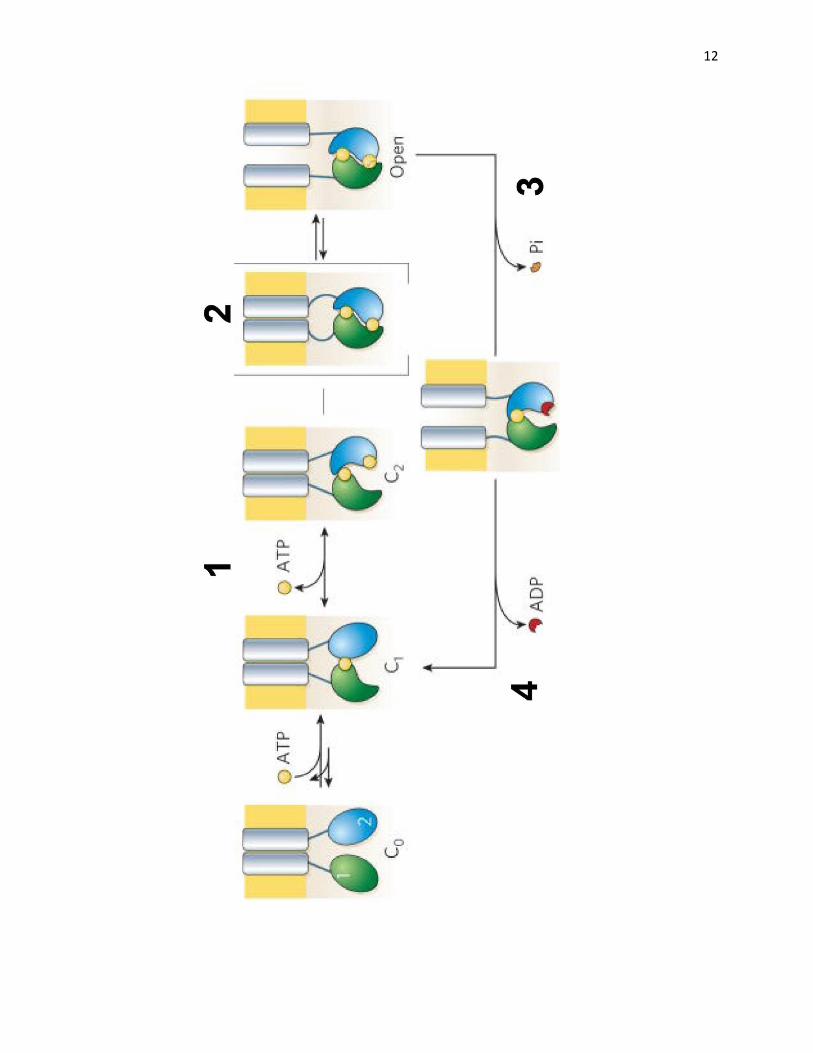
Figure 3: Gating of CFTR channel by ATP hydrolysis. The ATP-binding site in NBD1 has
high affinity and low hydrolysis rate. Thus, the rapid opening of the channel is caused by the
ATP hydrolysis at the second ATP-binding site in NBD2. The opening and closing of the
channel follows 4 steps. Step 1: ATP binds to the NBD2 binding site to initiate the process. Step
2: The conformational change caused by the formation of the NBD dimer causes the channel to
open. Step 3: The dimer is disrupted due to hydrolysis of ATP. Step 4: Pi and ADP release
restores channel to its basal conformation (Gadsby et al., 2006).
12

13
to prolonged binding of ATP (Gunderson and Kopito, 1995). The NBD dimer crystal structure of
other ABC transport reveals that the γ-phosphate of ATP forms a hydrogen bond with the
conserved serine residue (S548 in NBD1, and S1346 in NBD2) and the main chains of the
glycine residues (G550, G551 in NBD1, and G1349 in NBD2) (Hwang TC and Sheppard DN,
2009). In particular, theG551D mutation causes a severe channel gating defect (Bompadreet al.,
2007). The hydrolysis of ATP at the second nucleotide binding site might disrupt these hydrogen
bonds and the dissociation of ADP then closes the channel.
3. Regulation of CFTR by phosphorylation
Another difference between CFTR and other ABC family members is the unique R
region/domain that contains a cluster of dibasic (R-R/K-X-S/T) or monobasic (R-X-S) consensus
sites for phosphorylation, mainly by cAMP-dependent protein kinase A (PKA) (Seibert et al.,
1999). The R domain, until phosphorylated, restrains channel activity. Partial deletion of this
domain produces a constitutively active channel (Ostedgaard et al., 2002). Phosphorylation of
the consensus sites on the R domain provides another level of regulation of CFTR channel
gating. PKA phosphorylation is a pre-requisite for channel opening (Ostedgaard et al., 2001) and
has been shown to increase channel activity by at least 100-fold (Csanady et al., 2005).
Originally, one model for this gating of the channel was that when unphosphorylated, the R
domain blocked the pore of the channel, and thus inhibited any anion transport; upon
phosphorylation by PKA, the accumulation of negative charges created an electrostatic push to
repel the R domain out of the pore and relieved this inhibition (Cheng et al., 1991).This model,
however, was proved to be too simplistic. Later studies have shown that phosphorylation of the
R domain is a very dynamic and complex process, which may cause both stimulatory and
inhibitory effects (Gatsby and Nairn, 1999).

14
Results from site-directed mutation studies (Seibert et al., 1995; Seibert et al., 1999),
combined with evidence of structural rearrangement of the R domain upon phosphorylation
(Dulhanty and Riordan, 1994; Dulhanty et al., 1995), support another theory of the gating
mechanism of the channel. In this model, conformational change of the R domain, rather than the
accumulation of negative charges, is responsible for its regulatory function. Even though it still
remains unstructured and disordered independently of phosphorylation, the R domain has been
shown recently to contain segments of helical structure that most likely interact with other
domains of CFTR.NMR studies performed byte Forman-Kay lab have shown that
phosphorylation reduced not only the helicity of these helical segments but also their interactions
with NBD1. These interactions might play an important role in conferring the regulatory effect
of the R domain on CFTR (Baker et al., 2007).
CFTR is also phosphorylated by other kinases. However, aside from the phosphorylation
of CFTR by PKA and PKC, phosphorylation by other kinases has not been extensively studied.
PKC phosphorylation causes a modest activation of the channel (Dulhanty and Riordan, 1994)
and potentiates the PKA-mediated activation of CFTR, probably by facilitating subsequent PKA
phosphorylation by exposing sites that are otherwise inaccessible (Chang et al., 1993; Jia et al.,
1997). However, the exact mechanism of how PKC directly regulates the channel is unknown.
The R domain does not undergo conformational change when phosphorylated by PKC (Dulhanty
et al., 1994). Yet, PKC does slowly phosphorylate PKA sites (Jia et al., 1997). Recently, another
kinase, Casein kinase2 (CK2), was also shown to regulate CFTR through direct phosphorylation.
Mutating the two CK2 phosphorylation sites diminished both channel conductance and
trafficking of the protein to the plasma membrane (Luz et al., 2011).

15
The R domain is not the only region of CFTR that contains phosphorylation sites.
Mutation of Ser-422, close to the N-terminal of NBD1, in CFTR that already contains nine
mutations in the R domain further reduces channel anion flux and cAMP response (Chang et al.
1993). This phosphorylation site lies in the recently defined regulatory insertion (RI) region of
NBD1 (residues 403-437) (Lewis et al., 2004, 2005). NMR studies have shown that
phosphorylations of the RI region and another region called C-terminus regulatory extension
(RE) at the end of the NBD1 disrupt their binding with NBD1 and expose the binding site for the
first coupling helix of the N-terminal intracellular domain (ICD) of NBD1 (Kanelis et al., 2010).
The helices of ICD are thought to be involved in transmitting the change in conformation during
the formation of NBD1/2 dimer to the MSDs in the regulation of channel opening (Ward et al.
2007). In short, the regulation of CFTR through phosphorylation has been shown to be a
dynamic and complex process which still requires further investigation, and various kinases may
play a role in regulating both channel activity and trafficking.
4. Overview of CF-causing mutations
To date, more than 1800 mutations in the CFTR gene have been identified (CPDR,
2011). However, most of these mutations are rare; and there are only 24 mutations that have been
identified with a frequency of 0.1% or higher (Sick Kids CF mutations database,
www.genet.sickkids.on.ca). These mutations can be classified into six classes depending on their
consequences. Class I mutations produce a stop codon leading to premature transcription
termination signals. These mutations result in truncated or no protein expression. Class II
mutations are usually missense mutations causing the protein to misfold, leading to premature
degradation and failure to reach the apical membrane. The most common CF-causing mutation,
∆F508, belongs to this class. CFTR bearing class III mutations still properly folds and results in

16
Figure 4: Classes of CFTR mutations. Class I mutations produce premature transcription
termination signals resulting in truncated or no protein expression. Class II mutations are
missense mutations causing the protein to misfold, leading to premature degradation. Class III
mutations cause defective channel regulation, resulting in decreased channel activity. Class IV
mutations result in reduced channel conductance due to lower chloride permeability and opening
probability. Class V mutations cause partial defect in producing or processing the protein,
resulting in reduced number of functional channels. Class VI mutations lead to higher rate of
degradation of the channel after biosynthesis (Anderson, 2010).
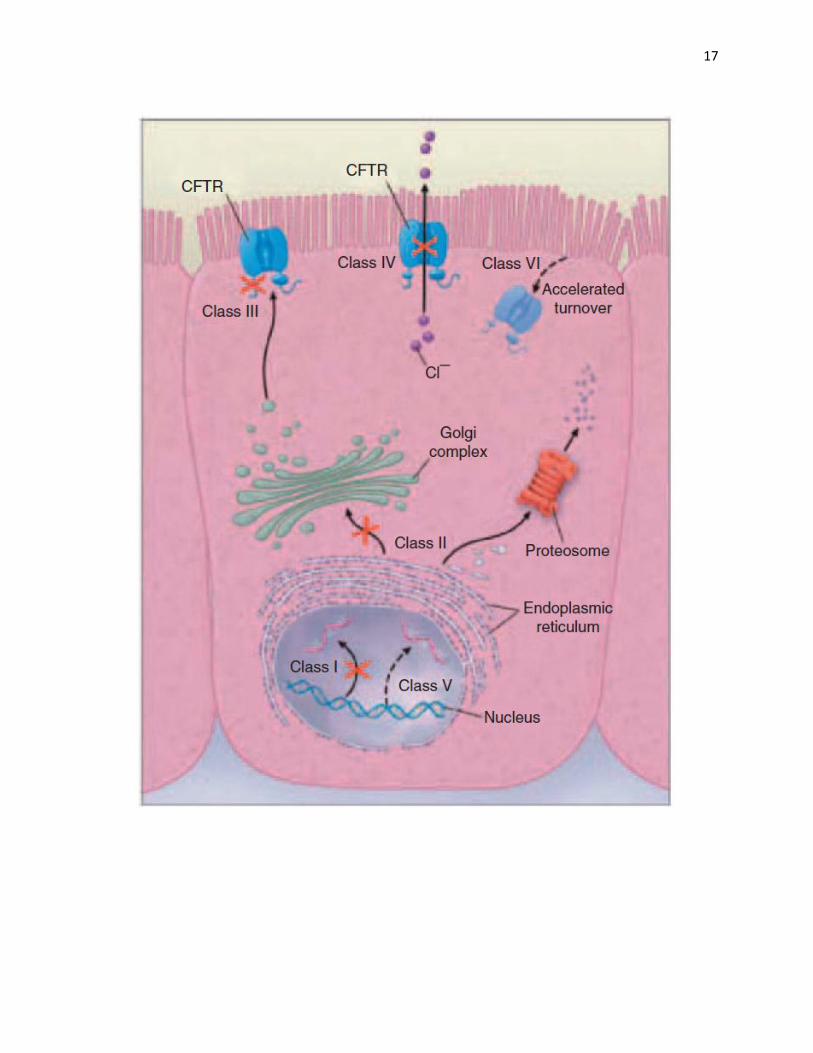
17

18
normal trafficking to the cell surface; however, it suffers a defect in its regulation, resulting in
severely decreased channel activity. A representative from this class is the G551D substitution.
This mutation in the ATP binding site on NBD1 is the third most common CFTR mutation that
results in defects in binding and hydrolysis of ATP (Li et al., 1996). Class IV mutations result in
reduced channel conductance due to lower chloride permeability and opening probability. Class
V mutations cause partly defective production or processing of the protein, resulting in a
reduction in the number of functional channels. Finally, Class VI mutations have only been
characterized recently. Such mutations reduce the channel stability causing an abnormally high
degradation after biosynthesis, and affect CFTR’s regulation of other proteins (Figure 4)(Ratjen
F, 2003; Anderson P, 2010; Okiyoneda and Lukacs 2012).
5. ∆F508-CFTR mutation and its defects
The most common mutation, identified in approximately 90% of CF patients, is the
deletion of phenylalanine at position 508, or ∆F508, a class II mutation. This deletion mutation
lies on the interface between NBD1 and the cytosolic loop 4 (CL4) of the MSD2 domain
(Mornon et al., 2008). It destabilizes the NBD1 thermodynamically and kinetically, and affects
the stability of the NBD1-MSD2interface (Rabeh et al., 2012), and the folding of NBD2 domain
(Du et al., 2005). This consequently disrupts the domain-domain interaction and their assembly
to form the complete channel, and thus causes the protein to be misfolded, kinetically trapped in
the endoplasmic reticulum (ER) and eventually targeted for degradation via ubiquitination by the
ER-associated degradation (ERAD) pathway and the proteasome (Riordan, 2008) (Figure 5).
Normally, the biosynthesis of CFTR starts by being synthesized and core glycosylated in
the ER. After exiting the ER, it is processed through the Golgi and presented on the cell surface

19
as a fully glycosylated mature CFTR (Cheng et al., 1990).CFTR is a large protein, 1480 amino
acid long, which exhibits a very inefficient folding and processing- up to 80% of the wild-type
(WT) CFTR gets degraded during the biosynthesis stage at the ER (Lukacs et al., 1994).This
results from a combination of slow domain assembly and fast degradation by ERAD (Lukacs and
Verkman, 2012). With the ∆F508 mutant, the efficiency is even lower with99% of the mutant
protein targeted for degradation before it reaches the plasma membrane (Ward and Kopito,
1994).This leads to the absence or very low density of the mutant channel on the plasma
membrane, which gives rise to the CF phenotype. This difference can be observed as the very
distinct migration patterns on SDS-PAGE. The WT protein appears as two bands: a prominent
band C represents the fully glycosylated mature form of CFTR (~180kDa), and a less intense
band B represents the core glycosylated immature form (~150kDa). On the other hand, ∆F508-
CFTR shows up predominantly as band B with little or no band C.
In addition, ∆F508 is a temperature-sensitive mutation; some of the mutant channels can
be rescued and reach the plasma membrane by incubation at lower temperature (26-30oC)
(Denning et al., 1992). However, even when rescued to the plasma membrane, ∆F508-CFTRis
quickly internalized by the membrane-associated chaperone system. While WT-CFTR has a half-
life of about 16h on the plasma membrane and is efficiently recycled back to the cell surface
after internalization, the rescued ∆F508 is quickly removed from the plasma membrane with the
half-life of about 2h; misfolding prevents this mutant protein from recycling back to the surface
and promotes its ubiquitination and degradation (Sharma et al., 2004; Swiatecka-Urban et al.,
2005).
Besides reducing the availability of the protein on the apical surface, ∆F508 mutation
also reduces the Cl- permeability of the channel. On the cell surface, ∆F508-CFTR only

20
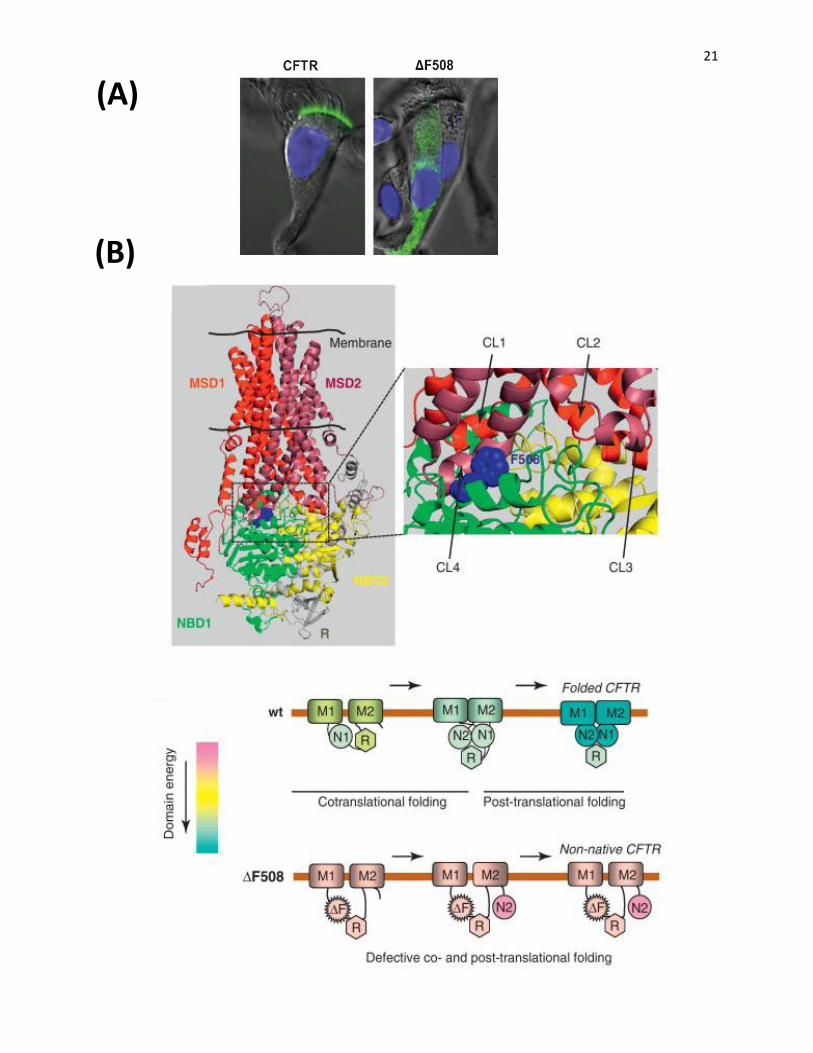
Figure 5: ∆F508-CFTR trafficking and folding defects. A) ∆F508 belongs to class II CF-
causing mutation which facilitates the misfolding of the protein leading to its early degradation at
the ER. The WT CFTR can properly fold, escapes the ER and is further processed in the Golgi
and presented on the plasma membrane as the mature, fully glycosylated channel. Images of
normal and ∆F508 primary airway cells demonstrate the distribution of WT and mutant CFTR,
shown as green fluorescence. WT-CFTR is primarily present on the apical surface while ∆F508-
CFTR mostly resides in the cytosol (Riordan JR, 2008) C) CFTR homology structure shows the
location of ∆F508 at the interface between NBD1and CL4 of MSD2 (upper panel).This mutation
destabilizes the interface and disrupts the assembly of the domains (lower panel) (Lukacs GL
and Verkman AS, 2012).
21
(A)
(B)

22
exhibits partial channel activity in response to PKA (Bear et al., 1992). Therefore, it is very
complicated to find a drug for the treatment of ∆F508. Drugs used to treat ∆F508 have to correct
two main problems in order to achieve near normal lung function in CF patients:
i)prevent premature degradation of the protein and promote its trafficking to the cell surface;
ii) reduce the internalization rate and improve the recycling efficiency at the plasma membrane.
To tackle these problems, a more complex approach is probably required. Recent studies have
shown that stabilizing both NBD1 folding and the NBD1-MSD2 interface are required to fully
reverse the defects of ∆F508-CFTR (Rabeh et al., 2012; Mendoza et al., 2012). Indeed,
suppressant mutations that correct one of these folding defects of ∆F508-CFTR only led to
partial rescue of the channel; while combining mutations that stabilizing both NBD1 and NBD1-
MSD2 interface produced a synergistic rescue (Rabeh et al., 2012).Since CFTR folding and
assembly are monitored by complex systems of chaperones, affecting the chaperones involved in
the processing of CFTR, via a combinational drug therapy, may be one possible approach to
achieve a dual correction effect.
III. CHAPERONE SYSTEMS INVOLVED IN THE PROCESSING OF CFTR
Since the ∆F508 mutant is improperly folded, it is prone to aggregation, and accumulates
in the intracellular compartments (Qu and Thomas, 1996). Therefore, degradation of misfolded
protein is necessary to prevent the formation of large aggregations, which are toxic to cells.
However, when degradation occurs too rapidly, the protein might not have sufficient time for
proper folding. This can be applied to the case of CFTR since the nonubiquinated ∆F508

23
intermediates exist in a folding competent conformation. Inactivating the chaperones, which
target these intermediates for proteasomal degradation via ubiquitination, or treating the cells at
lower temperature, allows the misfolded intermediates to fold properly and reach the cell surface
(Younger et al., 2004). However, in vivo, CFTR biosynthesis is scrutinized at multiple quality
control checkpoints by complex systems of chaperones from the ER-associated chaperones to the
peripheral quality control systems at the plasma membrane (Lukacs and Verkman, 2012). A
summary of the chaperone systems involved in CFTR trafficking and recycling is depicted in
Figure 6.
1. ER-associated and cytosolic chaperone systems
The synthesis of a multi-domain protein, such as CFTR, is a very complex, multi-stage
process controlled by various chaperone systems. It requires not only the proper folding of
individual domains, but also appropriate domain-domain interactions and arrangements. For
CFTR, the first step in the process is the folding of the nascent chain protein which is controlled
by the ER-associated chaperones, both membrane-bound and cytosolic (Chanoux and
Rubenstein, 2012). One of the reasons why CFTR biosynthesis is very inefficient is due to its
uneconomically rigorous early folding steps. The majority of CFTR is degraded in the pre-Golgi
compartments, and thus, never reach the cell surface (Ward and Kopito, 1994).
a. Hsp70 and its cochaperones
One of the first chaperones described to bind to the nascent CFTR chain and mediate the
folding cotranslationally is the heat shock cognate 70 (Hsc70) protein, which belongs to the heat
shock protein 70 (Hsp70) family and is localized to the cytosolic face of the ER. Initially,
Hsc/Hsp70 was thought to be able to distinguish between the mutant and WT protein and have a
prolonged association with the mutant ∆F508 compared to WT-CFTR, and thus retain the mutant

24
Figure 6: ER and peripheral quality control systems involved in the trafficking of CFTR.
A) The ER quality control system involves mainly the complexes of Hsp70 and Hsp90 and their
cochaperones. These complexes sense the folding state of the nascent CFTR chain and target
the misfolded protein for degradation via ubiquitination by E3 ligase such as CHIP. Hsp70 and
Hsp90 can facilitate both degradation and folding of the nascent chain depending on the
cochaperone associated with them. Hdj-2 promotes folding of nascent chain while CHIP
facilitates its degradation when in complex with Hsp70. Cochaperone Aha1 promotes the
degradation pathway of CFTR via Hsp90 (Wang et al., 2006). B) There are overlapping
components between the peripheral and ER quality control. Hsp70/90 and their cochaperones
are also involved in the peripheral machinery that promotes the endocytosis of CFTR from the
plasma membrane and ubiquitination of the protein. Ubiquitinated protein is then removed from
the recycling pool via the ESCRT complex and degraded in the lysosome. Deubiquitination by
USP10 facilitates the recycling of internalized channel back to the plasma membrane (Lukacs
GL and Verkman AS, 2012).
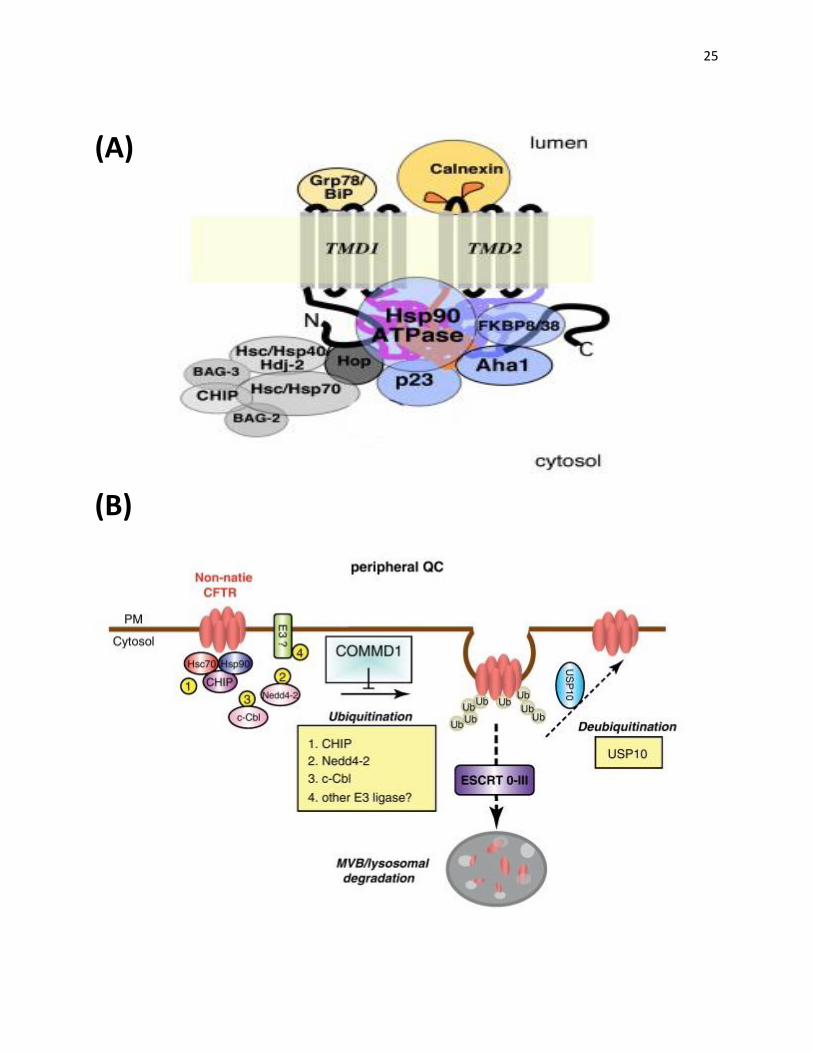
25
(A)
(B)

26
in the ER for later degradation (Yang et al., 1993).However, later studies have shown that
Hsc/Hsp70 can facilitate both the folding and degradation of CFTR nascent chains depending on
the association of other co-chaperones. Meacham et al. showed that Hdj-2 formed a complex
with Hsc/Hsp70, which bound to and promoted the folding of the ribosomal-bound intermediates
protein during the expression of NBD1 (Meacham et al., 1999). This effect dramatically
decreases after the subsequent expression of the R domain and MSDII. TheHdj-2/Hsc70
complex preferentially binds to ∆F508-CFTR and prevents the aggregation of NBD1.
Traditionally, molecular chaperones were thought of as proteins that aided in the folding
of other proteins by promoting their self-assembly (Ellis, 1987). However, in many cases, such
as with CFTR, chaperones can also target the partially folded peptide chain for degradation. The
C-terminus of the Hsc70-Interacting protein (CHIP) is another co-chaperone that can form a
complex with Hsc/Hsp70 (Meacham et al., 2001). However, unlike Hdj-2, CHIP, which is an
ubiquitin ligase, forms a complex with Hsc/Hsp70 to sense the folded state of the nascent chain
of CFTR and targets aberrant proteins for degradation (Meacham et al., 2001).In the case of
CHIP, it acts as an E3 ligase in cooperation with the E2 UbcH5a. These two proteins, when in a
complex with Hsc70, facilitate the degradation of nascent CFTR chains (Younger et al, 2004).
Other cochaperones of Hsc70 have been less well studied. Both HspBP1 and BAG-2
were shown to stimulate the maturation of CFTR by inhibiting the cochaperone CHIP (Alberti et
al., 2004; Arndt et al., 2005). Moreover, Saxena et al. also showed that Hsp105 worked both
independently and as a cochaperone of Hsc70 to stabilize CFTR at an early stage of synthesis,
and to promote the posttranslational folding. In addition, it was also proved to bind preferentially
to ∆F508-CFTR at both the ER and cell periphery and to enhance the expression of the mutant
protein on the cell surface (Saxena et al., 2012).

27
b. ER membrane-bound and luminal chaperones
While the CHIP/Hsc70 complex primarily recognizes the CFTR post-translationally,
another ER membrane-associated ubiquitin ligase complex, consisting of the E3 RMA1, the E2
Ubc6E, and Derlin-1, can recognize folding defects cotranslationally during the synthesis of
MSD1 and target the misfolded nascent CFTR for degradation (Younger et al., 2006).
Overexpression of RMA1, but not CHIP, promoted the degradation of G91R-CFTR, a mutation
that resides in MSD1and prevents proper folding of the protein (Xiong et al., 1997). Moreover,
Derlin-1 coimmunoprecipitated with MSD1. Therefore, in this complex, Younger et al. proposed
that Derlin-1, an ER membrane protein, sensed the folding status of MSD1/2, and formed a
complex with the protein that failed to assemble correctly. Subsequently, Derlin-1 recruited
RMA1 and Ubc6e to facilitate ubiquitination and degradation of CFTR (Younger et al., 2006).
The role of ER luminal chaperones such as calnexin is less well understood(Chanoux and
Rubenstein, 2012). Calnexin was initially thought to bind to immature CFTR and retain ∆F508-
CFTR in the ER due to the fact that it has a prolonged interaction with the mutant compared to
WT-CFTR (Pind et al., 1994). However, other studies have shown that calnexin has a positive
regulatory role in the synthesis of ∆F508-CFTR. Overexpression of calnexin created a pool of
∆F508-CFTR but reduced the degradation and aggregation of the mutant protein (Okiyoneda et
al., 2004). Moreover, knocking down calnexin did not seem to improve the trafficking of ∆F508-
CFTR (Okiyoneda et al. 2008). The role of calnexin is controversial but combined data from
various studies suggests that calnexin alone is not sufficient for the retention of ∆F508-CFTR in
the ER. Other ER luminal chaperones might be a better therapeutic target for CF. A recent study
by Suaud et al. suggested that ERp29 (ER luminal protein of 29 kDa), when overexpressed,
increased both the functional and surface expression of WT and ∆F508-CFTR (Suaud et al.,

28
2011). This ER luminal protein was shown to be upregulated by the compound sodium 4-
phenylbutyrate(4PBA) (Suaud et al., 2011), which was shown to correct ∆F508-CFTR by
altering the expression of chaperones, such as Hsc70 (Rubenstein et al., 1997; Rubenstein and
Zeitlin, 2000).
c. Hsp90 in the processing of CFTR
Another chaperone associated with CFTR maturation that has received considerable
attention is Hsp90, which was shown to stabilize the CFTR folding intermediates (Loo et al.,
1998). However, the activity of Hsp90 depends on the presence of its co-chaperones. Hsp90
cochaperone Aha1 was shown to down-regulate the rescue of misfolding CFTR to the cell
surface. A 50-70% knock down of Aha1led to a significant increase in both band B and band C
of∆F508-CFTR and an increase in halide conductance in CFBE41o- expressing ∆F508-
CFTR(Wang et al., 2006). Aha1 was proposed to increase the binding of Hsp90 to its client
through increasing the ATPase activity of Hsp90 (Wang et al., 2006). Other cochaperones of
Hsp90 were also found to affect Hsp90 in mediating the folding of CFTR. For example,
cochaperone p23was determined to be important in stabilizing and preventing degradation of
∆F508-CFTR (Wang et al., 2006). Recently, Hsp90 was also shown to have a negative impact on
FK506-binding protein 38 (FKBP38), which was localized to the ER membrane and promoted
the posttranslational processing and cell surface expression of CFTR. Mutation on the TPR motif
of this protein, which is required for the binding to Hsp90, uncoupled the two proteins, and
reduced CFTR synthesis but improved the maturation of the channel (Banasavadi-Siddegowda et
al., 2011).

29
2. Peripheral chaperone systems
One of the defects of rescued ∆F508-CFTR is its very rapid removal from the plasma
membrane due to its inability to recycle back to the surface after being internalized(Sharma et
al., 2004; Lukacs et al., 1993; Cholon et al., 2010). This process is the responsibility of the
peripheral chaperone systems. Recent studies done by the Lukacs group have identified
overlapping chaperone systems working as both peripheral and ER control machinery for CFTR
(Okiyoneda et al., 2010). Using siRNA to knock down 33 E3 ligases involved in the down-
regulation of plasma membrane proteins and CFTR ERAD, Okiyoneda et al. determined that
CHIP was the main E3 ligase responsible for the ubiquitination of complex glycosylated
(mature) ∆F508-CFTR. Knocking down CHIP reduced the internalization of mutant CFTR and
partially restored its recycling. Ablation of CHIP also delayed the delivery of internalized
proteins to lysosome for degradation via the endosomal sorting complex required for transport
(ESCRT 0-III) components, which redirected the mutant protein away from the recycling
pathway (Okiyoneda et al., 2010).
Moreover, Hsp70, Hsp90 and a subset of their cochaperones such as Aha1, Hdj-2, and
BAG-1 were also identified to be part of the peripheral quality control machinery. Either
knocking down Hsp70 or Hsp90 or breaking apart their interactions with CHIP via mutating the
TPR domain prevented channel down regulation from the plasma membrane. Ablation of the
cochaperones also reduced the ubiquitination of the mutant channel and its endocytosis
(Okiyoneda et al., 2010). These chaperones and cochaperones are also involved in the quality
control of CFTR at the ER and post-translation (Chanoux and Rubenstein, 2012). Other proteins
have been identified to be a part of the peripheral quality control. C-Cbl functions as an adaptor

30
protein at the plasma membrane promoting the endocytosis of CFTR by a ubiquitin-independent
mechanism. At the early endosomes, however, c-Cbl ubiquitinates and targets CFTR for
degradation (Ye et al., 2010). Moreover, the Ubiquitin Specific Protease-10(USP10)
deubiquitinates CFTR and regulates the recycling back to the plasma membrane of the
internalized channel (Bomberger et al. 2009). Knocking down USP10 reduced both the presence
of CFTR on the apical surface and the channel activity.
The studies of these chaperones systems will shed light on therapeutic approaches to
correct the trafficking defects of ∆F508-CFTR. Targeting the chaperones involved in the
processing of CFTR, rather than CFTR itself, might be a promising approach. However, since
CFTR quality control is such an intricate and complex system with redundant roles, to overcome
the various check points of this network will not be a simple task.
IV. SCREENS FOR CORRECTORS OF THE ∆F508-CFTR DEFECTS
The ∆F508 mutation causes several defects in the processing and function of CFTR. The
protein not only suffers from impaired trafficking from the ER to the plasma membrane, but also
rapid removal from the cell surface due to an impaired recycling mechanism. However, the
∆F508-CFTR trafficking defect is correctable, since treating cells expressing the mutant CFTR at
low temperature or with chemical chaperones such as glycerol can restore the surface expression
of the mutant (Denning et al., 1992; Sato et al., 1996).The fact that ∆F508-CFTR, when it
reaches the plasma membrane, still shows partial channel function (Bear et al., 1993) has
encouraged many groups to screen for small molecules that can rescue ∆F508-CFTR. Moreover,
it was suggested that a rescue of about 10-15% of the ∆F508-CFTR retained in the ER might

31
have therapeutic benefits to the patients (Johnson et al., 1992; Farmen et al. 2005; Zhang et al.,
2009).
1. High-throughput screens for correctors of ∆F508-CFTR
Without any indication of specific drug targets for the rescue of ∆F508-CFTR, high-
throughput screens (HTSs) of large libraries of compounds using functional or biochemical cell-
based assays have become the most practical approach. In these assays, the rescue of ∆F508-
CFTR can be indicated as an increase in the anion transport or through detecting the appearance
of the mutant protein on the cell surface (Pedemonte and Galietta, 2012). With the application of
HTSs, the last decade has witnessed the identifications of a number of compounds that could be
used to correct CF defects. Several of these compounds have shown promising potential. In
general, CF drugs can be divided into two different types: correctors, which correct the
trafficking defect of ΔF508-CFTR, and potentiators, which increase channel activity, e.g. in the
case of G551D-CFTR.
The first HTS was performed by the Verkman group, including an initial screen of a
library of 150,000 chemically diverse compounds and a second screen of 1,500 analogs of active
compounds (Pedemonte et al., 2005). In this screen, they identified the bithiazole corr-4a that
could rescue ∆F508 function in primary human airway epithelial cells obtained from ∆F508
homozygous CF patients (a rescue of about 8% of chloride conductance in non-CF samples, and
at about the same level of conductance as in samples treated at 27oC). Later studies also yielded
bithiazole analogs with improved potency with EC50 as low as 300nM (Yu et al., 2008; Ye et al.,
2010).

32
Since the first screen, several other groups have utilized HTS to identify other correctors
of ∆F508-CFTR. By screening a library of 42,000 compounds using BHK cells stably expressing
∆F508-CFTR bearing three tandem extracellular hemagglutinin (HA) epitote tags and
monitoring the appearance of ∆F508-CFTR on the cell surface as an indicator of the protein
rescue, Robert et al. identified a particular compound, the approved drug sildenafil, that showed
rescue of ∆F508 (Robert et al., 2008). Later on, sildenafil was shown to have a dual effect on
the mutant protein as it worked both as a corrector and potentiator of ∆F508-CFTR (Leier et al.,
2012). However, the author also concluded that the high doses of the drug required for the rescue
of CFTR might limit its application for therapeutic utilization.
The mechanisms of how these correctors work remain mostly unknown. However,
several groups have utilized a method aiming at specific targets based on our current knowledge
of CFTR structure and folding mechanism. The mutation ∆F508 lies on the interface domain
between NBD1 and CL4 of MSD2, and destabilizes the NBD1 and mostly the interface between
NBD1 and MSD1/MSD2 leading to protein misfolding (Mormon et al., 2008; Serohijos et al.,
2008; Lukacs and Verkman, 2012). Based on this conformational information, Sampson et al.,
utilizing a technique called differential scanning fluorimetry which detects ligands that bind and
stabilize purified protein, identified the phenylhydrazone RDR1, which is able to bind and
thermally stabilize purified NBD1 (Niesen et al., 2007; Sampson et al., 2011). Correctors that
bind directly to the protein and stabilize its folding are called pharmaceutical chaperones (PC).
Other correctors that also function as a PC are corr-4a, which was demonstrated to specifically
correct the folding at the ER of ∆F508 but not other mutant CFTR (Pedemonte et al., 2005; Loo
et al., 2008; Grove et al., 2009), and MBP, which was shown to directly bind to NBD1 to
increase CFTR trafficking (Becq et al., 1999; Stratford et al., 2003).

33
The approach to identify pharmaceutical chaperones seems promising since it has less
off-target effects due to the specificity of the binding of these compounds to CFTR and no
alteration of the cellular system (Sampson et al., 2011). Moreover, since it stabilizes the
conformation of the protein, it might correct the processing defects at both the ER and plasma
membrane. Using the homology structure of CFTR, researchers from Epix Pharmaceuticals have
performed structure-based screening of compounds that can increase the stability of the protein
by binding to the interfaces between domains of CFTR thus promoting folding and escape from
the ER (Kalid et al., 2010). Several of these compounds have shown dual activity, working both
as correctors and potentiators. The author suggested that this effect was due to the fact that they
used a model of CFTR in the conducting state, and thus stabilizing this stage not only promoted
the folding but also increased the open probability of the channel.
2. Discovery of VX-809 and VX-770 and their clinical trials
The most successful screen so far was performed by Vertex Pharmaceuticals, which
identified several compounds in the quinazolinone class acting primarily at the ER level to
facilitate folding of the protein (Van Goor et al., 2006). These compounds could rescue Cl-
transport in CF bronchial epithelial cells up to 20% of that in WT cells. This screen has led to
further discovery of some very promising drugs: the corrector VX-809 (Van Goor et al., 2011)
and the potentiator VX-770 (Van Goor et al., 2009). The latter of the two, commercially known
as Ivacaftor or Kalydeco, was recently approved by the FDA for the treatment of CF patients
bearing the mutation G551D, which affects channel gating activity (Ramsey et al., 2011).
Eckford et al., recently showed that VX-770 potentiated the activity of the purified reconstituted
channel in the absence of ATP and had an additive effect with ATP on channel regulation,
suggesting that it bound to a non-canonical site on CFTR (Eckford et al., 2012).

34
Unfortunately, VX-809 was not proven as successful as VX-770. The strong efficacy of
VX-809 in primary cells, exhibited a 25% rescue of ∆F508-CFTR, combined with its safety and
tolerability in vivo, has pushed the compound to a phase II clinical trials (Clancy et al., 2012).
However, except for a significant improvement in the sweat Cl-, the compound did not improve
lung function of ∆F508-CFTR patients. Due to its lower than expected efficacy in patients, VX-
809 is currently going through a phase II studies in combination with VX-770 (Boyle et al.,
2011; Pettit, 2012). Even though this study has shown some promising results, an ideal
combination of the two drugs has not been identified. These results also highlight the difficulty
in correcting the trafficking of ∆F508-CFTR.
3. Our screens using high-content Cellomics assays
So far, most of the HTS performed by various groups have focused on the ability of these
compounds to rescue the phenotype of mutant CFTR without understanding the pathways or
target proteins involved. Moreover, these drugs usually have to go through various clinical trials
stages before approval for clinical use, which usually take a substantial amount of time. Taking a
different approach, our lab had developed a high-content functional screen using Cellomics
KineticScan technology aiming at identifying proteins and small molecules (which are already in
the clinic or in clinical trials for other diseases) that correct the trafficking defect of ∆F508-
CFTR (Trzcinska-Daneluti et al., 2009). In this screen, we utilized human HEK283 MSR
GripTile cells that stably express ∆F508-CFTR and a mutant YFP, YFP(H148Q/I152L) whose
fluorescent signal can be quenched by halide exchange (I- for Cl
-) (Galietta et al., 2001) (Figure
7). Therefore, the quenching of fluorescent signal with time due to the iodide influx into the cells
in exchange for Cl- after exposing the cells to activators of CFTR (Forskolin/IBMX/Genistein)
and a high iodide media acts as an indicator of CFTR activity.

35
Using this approach, our lab identified several proteins that when overexpressed rescue
the function of ∆F508-CFTR. Among the hits were several chaperones, Golgi-associated,
trafficking, signalling proteins and transcription factors. One of the best hits identified was
STAT1 (Signal Transducer and Activator of Transcription 1). Knocking down of PIAS1, an
inhibitor of STAT1, also rescued ∆F508-CFTR, further supporting our findings. PIASI had been
shown to be elevated in CF epithelial cells leading to reduced activation of STAT1 and NOS2,
probably via the activation of the RhoA/ROCK pathway upstream of PIAS1(Kelley and Elmer.,
2000; Kreiselmerier et al., 2003). Recently, RhoA and ROCK were shown to be involved in the
complex with ezrin and actin that, via reorganizing the actin cytoskeleton, tethers ∆F508-CFTR
to the cytoskeleton and stabilizes it at the apical surface (Favia et al, 2010). Increasing the
activity of RhoA can rescue CFTR-dependent chloride efflux.
The above Cellomics is also suitable to identifying small molecules that can rescue
∆F508-CFTR function, or proteins that inhibit ∆F508-CFTR rescue via RNAi screen. Moreover,
one advantage of the screen, focusing on proteins, is that it can identify the pathways that are
involved in the rescue of ∆F508CFTR.

36
Figure 7: Principles of the Cellomics assays to test rescue of mutant CFTR. The mutant YFP
protein (H148Q/I152L), which is expressed in HEK293-GT cells, is halide sensitive, and its
fluorescence is quenched by iodide. The assay is performed with HEK293GT expressing the
mutant YFP and either WT-CFTR or ∆F508-CFTR. In cells expressing WT channel, when
exposed to high iodide/ low chloride media and stimulated with FIG
(Forskolin/IBMX/Genistein), the Cl-/I
- exchange via CFTR leads to quenching of fluorescent
signal. In cells expressing ∆F508-CFTR, the quenching is significantly less due to the absence of
functional channel on the cell surface. The quenching of fluorescent signal works as an indicator
of CFTR activity on the cell surface.

37

38
V) PROJECT RATIONALE AND GOALS:
To identify small molecule correctors of ∆F508-CFTR, our lab performed a Cellomics
screen of a library of 231 kinase inhibitors biased toward drugs that are already in the clinic or
are in clinical trials for the treatment of cancer or inflammation. Such drug repurposing has the
potential to expedite the development of treatment for CF if any of these small molecules can
indeed rescue ∆F508-CFTR. We also performed a complementary siRNA screen for kinases and
related proteins that suppress the rescue of ∆F508-CFTR to help us identify the pathways
involved.
In this thesis, I describe my work done to validate the top hits from the kinase inhibitors
screen and initial validation results of the hits of the siRNA screen. I also present the assessments
of the effect of E6201, a derivative of (5Z)-7-Oxozeaenol (Oxozeaenol), which was one of the
top hits of our kinase inhibitors screen, on the rescue of ∆F508-CFTR, and attempt to elucidate
the pathways involved in the rescue of ∆F508-CFTRby Oxozeaenol.

39
Chapter 2
Materials and Methods

40
1. Media and Reagents
Dulbecco's Modified Eagle's Medium (DMEM), F12 nutrient mixture, Dulbecco's
Phosphate Buffered Saline (D-PBS) with and without calcium or magnesium, fetal bovine serum
(FBS), trypsin, G418, Blasticidin, and Zeocin were obtained from Invitrogen (Carlsbad, CA).
SuperSignal West Femto Maximum Sensitivity kit was from Pierce (Rockford, IL), and
Affinipure goat anti-mouse antibody (Cat.#115005062) was from Jackson ImmunoResearch
(West Grove, PA). The small molecules kinome library was obtained from the Ontario Institute
for Cancer Research (OICR-see below). The mouse M3A7 anti-CFTR monoclonal antibody was
obtained from Millipore (Billerica, MA), and the anti-β-actin monoclonal antibody was from
Sigma (A5441). Mouse anti-HA.11 monoclonal antibody was from Covance (MMS-101R), and
Alexa Fluor 647-labeled goat anti-mouse antibody was from Invitrogen (A21236). The small
molecules kinase inhibitors used for validation of the compound kinome screen were from Tocris
(Bristol, UK), Selleck Chemicals and EMD Chemicals (San Diego, CA). The High Capacity cDNA
Reverse Transcription kit was obtained from Applied Biosystems, and the Platinum® SYBR® Green
qPCR SuperMix-UDG was from Invitrogen. The kinome esiRNA library was obtained from Dr. Laurence
Pelletier (The Samuel Lunenfeld Research Institute – see below). TRC (The RNAi Corsotium) shRNA
clones were a kind gift from Dr. Jason Moffat (University of Toronto), and pGIPZ shRNA clones were
provided by SIDNET (SPARCS), The Hospital for Sick Children.
2. Small Molecules Kinase Inhibitor Library
The OICR (Ontario Institute of Cancer Research) Kinase Inhibitor Cassette that was
screened contains 231 compounds that are reported to inhibit at least 68 kinases. These inhibitors
were purchased from a panel of more than 20 different vendors, or synthesized when not
commercially available. The library was designed to cover as many targets and drug-like

41
compounds as possible. In cases where there are multiple compounds targeting the same primary
kinase, it was anticipated that having multiple chemotypes with different properties and
selectivity profiles would enrich the screening set. Approximately 25% of the library consists of
inhibitors that have made it into the clinic, an additional 25% being compounds in different
phases of discovery (lead generation or optimization), and the remaining 50% are tool
compounds that have not been advanced to the clinic but are known to be active inhibitors
against various kinase targets.
3. Cells
HEK293 MSR GripTite (293MSR-GT) cells stably expressing ΔF508-CFTR or wild type
CFTR (WT-CFTR) protein were stably transfected with eYFP(H148Q/I152L) cDNA in
pcDNA3.1/zeo vector using calcium phosphate (Trzcinska-Daneluti et al., 2009). At 24 h post-
transfection, the cells were seeded onto 5 × 10 cm dishes at various densities (in order to easily
pick individual clones) and selected under 100 μg/ml Zeocin. Individual clones were picked and
expanded. Expression of WT-CFTR or ΔF508-CFTR was validated by immunoblotting using
M3A7 anti-CFTR monoclonal antibodies. Expression of eYFP(H148Q/I152L) was validated by
fluorescent microscopy. 293MSR-GT cells stably co-expressing eYFP(H148Q/I152L) and
ΔF508-CFTR or WT-CFTR protein were cultured in DMEM medium supplemented with 10%
FBS, 1× nonessential amino acids, 0.6 mg/ml G418, 10 μg/ml Blasticidin, and 50 μg/ml Zeocin,
at 37 °C, 5% CO2 in humidified atmosphere. Baby Hamster Kidney (BHK) cells stably
expressing wild type (CFTR-3HA) or mutant (ΔF508-CFTR-3HA) protein with the triple
hemagglutinin (3HA) tag at the ectodomain were a kind gift from D. Y. Thomas (McGill
University, Montreal). The cells were propagated as monolayer cultures in DMEM-F12 medium
(1:1) supplemented with 5% FBS and 0.5 mM Methotrexate at 37 °C, 5% CO2. Madin Darby

42
Canine Kidney (MDCK) cells stably expressing ΔF508-CFTR protein were cultured in DMEM
supplemented with 10% FBS, 1×PenStrep and 5 μg/ml Blasticidin at 37 °C, 5% CO2. Before the
short-circuit current (Isc) studies, MDCK cells were grown on permeable millicell inserts (12
mm, Millipore) for 4 days and then treated with 10 μM kinase inhibitors for 48 h. Primary
human bronchial epithelial (HBE) cells homozygous for ΔF508-CFTR or WT-CFTR were kindly
provided by Dr. P. Karp at the University of Iowa Cell Culture Facility, and propagated on
collagen-coated permeable millicell inserts (12 or 6.5 mm, Millipore) (Zabner et al., 1996). Prior
to the Ussing chamber assay the ΔF508-CFTR inserts were treated with 10 μM kinase inhibitors
or 0.2% DMSO (vehicle control) for 48 h at 37 °C. Parental MDCK and RAW 264.7 cells were
cultured in DMEM supplemented with 5%FBS, 1×PenStrep at 37oC, 5%CO2.
4. Cellomics YFP Halide Exchange Screen
Cellomics halide exchange assay was performed as follow. Briefly, 50,000 293MSR-GT
cells (stably expressing eYFP(H148Q/I152L) and ΔF508-CFTR) per well were seeded in the 96-
well plates. The next day ΔF508-CFTR cells were treated (in triplicate) with 10 μM small
molecule kinome library (separate compound in each well), 0.2% DMSO (vehicle control), or
corr-4a (positive control) at 37 °C, or incubated at 27 °C (positive control). A 10 μM dose was
chosen based on a preliminary screen data (not shown) as a dose that covers a wide range of
inhibiting concentrations but is not toxic to ΔF508-CFTR cells. After 48 h of incubation the
medium was replaced with 152 μL of chloride solution (137 mM NaCl, 2.7 mM KCl, 0.7
mMCaCl2, 1.1 mM MgCl2, 1.5 mM KH2PO4, 8.1 mM Na2HPO4, pH 7.1), in the absence or
presence of FIG (25 μM Forskolin, 45 μM IBMX, 50 μM Genistein) at 37 °C. After 20 min
incubation, 92 μl of iodide buffer (137 mM NaI, 2.7 mM KCl, 0.7 mM CaCl2, 1.1 mM MgCl2,
1.5 mM KH2PO4, 8.1 mM Na2HPO4, pH 7.1) was added (final I−
concentration 52 mM). Using

43
the Cellomics VTI (Thermo Fisher), and a modified target activation algorithm, objects
(individual cells or sometimes clusters of cells) were defined by eYFP(H148Q/I152L)
fluorescence intensity, and the decrease in fluorescence intensity over 24-s time course, at 30 °C,
5% CO2 was recorded. The number of primary objects was used as an indicator of cell toxicity
(cell death). Valid wells contained between 70 and 300 objects per field. After collecting and
analyzing data, a second run of the screen was performed with compounds preselected based on
the first run (∼100 compounds, each in triplicate).
5. Data Analysis
Compounds with a difference in fluorescence intensity between unstimulated (−FIG) and
stimulated (+FIG) samples lower than 0.08 were rejected after the first run of the screen. The rest
of the compounds were subjected to the secondary screen. Only the compounds that exhibited a
difference in average fluorescence intensity between unstimulated and stimulated cells of at least
0.10 were further analyzed. Compounds that displayed a difference in average fluorescence
intensity of at least 0.17 were considered Tier I hits. Compounds that showed a difference in
average fluorescence intensity lower than 0.17 were considered Tier II hits. Representative
compounds of both groups were selected for further validation of the ΔF508-CFTR rescue.
6. Immunoblotting
Prior to immunoblotting ΔF508-CFTR cells were treated with 15 μM kinase inhibitors or
0.3% DMSO (vehicle control) for 48 h at 37 °C, or incubated for 48 h at 27 °C (positive control),
or transfected with shRNA construct as described above. Cells were then rinsed in cold PBS and
lysed in lysis buffer (50 mM Hepes pH7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10%
glycerol (v/v), 1% Triton X-100 (v/v), 2 mM phenylmethylsulfonyl fluoride, 2× PAL inhibitors).

44
Proteins were resolved on SDS-PAGE, transferred to nitrocellulose membranes and
immunoblotted with anti-CFTR monoclonal antibodies (M3A7, 1:1000) or anti-β-actin
antibodies (1:10000). Membranes were washed with 5% Blotto, incubated with HRP-conjugated
goat anti-mouse antibody (1:5000), and washed with PBST. Signal was detected with
SuperSignal West Femto reagent.
7. Flow Cytometry
The rescue of ΔF508-CFTR was also validated by flow cytometry as described
previously. Briefly, at 48 h after adding 10 μM kinase inhibitor or 0.2% DMSO (vehicle control),
BHK cells were trypsinized, washed, and resuspended in ice-cold FACS buffer (PBS
supplemented with 2% FBS). To stain CFTR at the cell surface, cells were incubated with anti-
HA.11 monoclonal antibody (1:25) or AF647-labeled goat anti-mouse antibody (1:200) as a
control, for 1 h at 4 °C. Subsequently, the cells were washed with the cold FACS buffer and
incubated with AF647-conjugated goat anti-mouse antibody (1:200) at 4 °C for 1 h. They were
then washed as above and resuspended in FACS buffer with 1 μg/ml propidium iodide (PI). The
flow-cytometric analysis was performed using LSRII System (BD Biosciences). The data from
10,000 live (propidium iodide negative) cells were collected and analyzed with FlowJo v.7.6.4
software. Cell toxicity, as defined as >10% of cells staining positive for PI, was only observed
for Ki8751 treatment. Alsterpaullone treatment resulted in altered cellular morphology
(increased cell granularity and size) but not toxicity.
8. Short-circuit Current (Isc) Measurements in Ussing Chambers
Cell inserts or Snapwells, seeded with polarized MDCK or HBE cells (expressing ΔF508
or WT -CFTR), were mounted on an Ussing chamber apparatus (Physiological Instruments, San

45
Diego, CA) and studied under voltage clamp conditions. The buffer used in the assay composed
of 1x Hank’s Balanced Salt Solution (HBSS) supplemented with 21mM of NaHCO3, 1.2mM of
CaCl2, and 1.2mM of MgCl2.Prior to stimulation of CFTR, ENaC channels were inhibited with
10 μM amiloride (Sigma), and non-CFTR chloride channels were blocked with 250 μM DNDS
(4,4′-dinitrostilbene-2,2′-disulfonate, Sigma). CFTR currents were then stimulated using FIG,
and after the indicated time (min) inhibited using 15–50 μM GlyH-101 (Gly). Data were
recorded and analyzed using Analyzer 2.1.3. Dose-response analyses (EC50) for the top kinase
inhibitor hits were carried out with increasing inhibitor doses between 1 nM to 10 μM, applied to
MDCK cells stably expressing ΔF508-CFTR. A few of the tested compounds (PKC412,
GDC0941, PD184352, Go6976, Alsterpaullone, Kenpaullone) were toxic to MDCK cells,
resulting in loss of cell monolayer integrity and loss of resistance, detected in the Ussing
chambers. These were thus excluded from the data analysis. Schematic diagram of Ussing
chamber and one sample recorded currents are depicted in figure 8 to demonstrate the principle
of Ussing chamber and how the difference in short-circuit current (ΔIsc) was determined.
9. Isolation of Bone Marrow-derived Macrophages (BDMMs)
BMMs were prepared by culture of bone marrow isolated from femurs and tibias of
C57BL/6 mice (12-14 weeks of age). After red blood cell lysis, the cells were cultured in RPMI
1640 medium supplemented with 20% FBS, 100 units/ml penicillin, 100 µg/ml streptomycin,
and 40 ng/ml macrophage colony-stimulating factor (R & D Systems). LPS stimulations were
performed between 7 and 9 days of cell culture. Experimental and animal care were performed in
accordance with institutional guidelines.
10. Phosphoprotein analysis

46
Figure 8: Ussing chamber schematic diagram and ΔIsc calculations. (A) Schematic
diagram of Ussing chamber. The cell monolayer is mounted in the middle of a U-shaped
chamber to offset any pressure difference. The two halves of the chamber are filled with equal
amount of symmetrical buffer in order to remove any chemical, mechanical and electrical driving
forces. The ion transport across the epithelial monolayer produces a potential difference (voltage
difference). The voltage difference generated is measured using two voltage electrodes placed
near the tissue/epithelium. This voltage is cancelled out by introducing a current via two other
current electrodes. This current is called the Short-circuit current (Isc), and is equal to the net ion
transport across the epithelium (Clarke, 2009). (B) In Ussing chambers analysis, the effect of the
treatment is determined by the difference in stimulated short-circuit current (ΔIsc) between the
treated and control cells. One example of recorded currents is shown. The graph shows currents
recorded between cells treated with (5Z)-7-Oxozeaenol and DMSO control. After the insert is
mounted onto the chamber, amiloride (10 μM ) and DNDS (250 μM ) are added to inhibit the
activity of ENaC and other Cl- channels (not shown on the graph) prior to the stimulation with
FIG (25 μM Forskolin, 25 μM IBMX, and 50 μM Genistein). Then the current is inhibited with
GlyH101 (15µM). To calculate the ΔIsc, currents are normalized to 1 at the point of adding FIG.
ΔIsc is measured as the difference between the maximal current recorded between the two
samples.
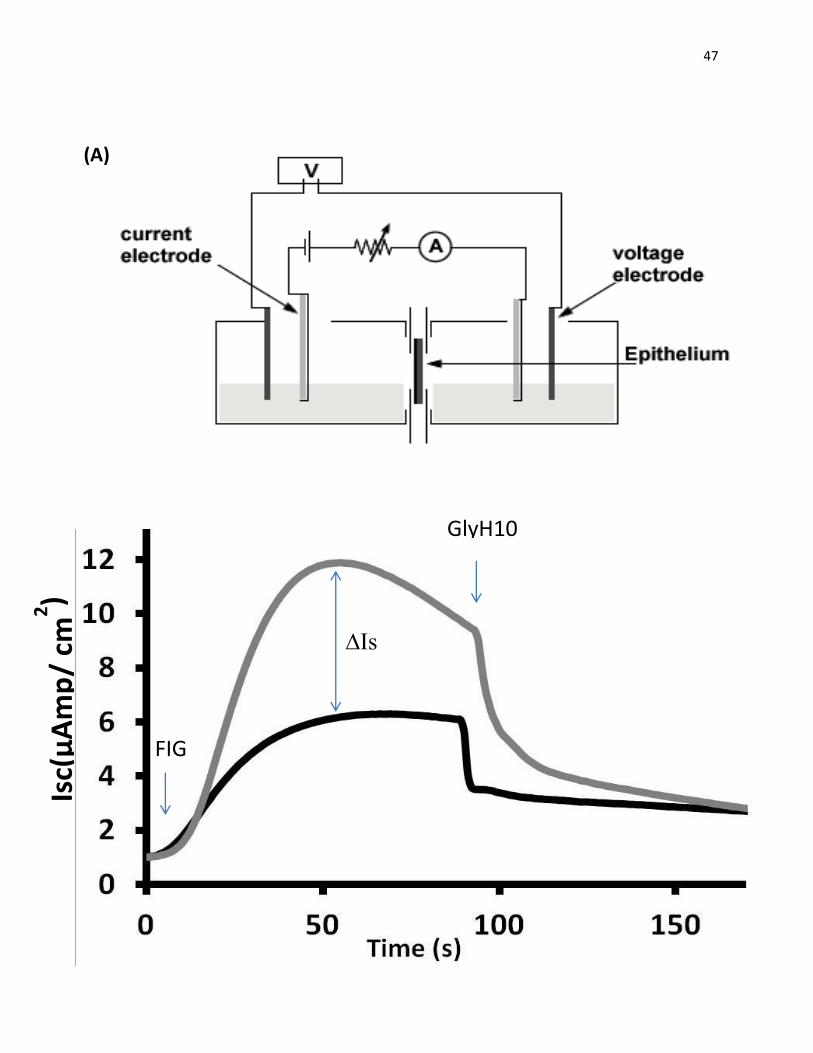
47
Isc(
µA
mp
/ cm
2)
(A)
(A)
GlyH101
∆Is
c
FIG1

48
For analysis of phosphoproteins, RAW264.7 cells or BMMs were treated with indicated
concentrations of Oxozeaenol or E6201 for 1 hour, then stimulated with LPS (100ng/mL) for 5
minute, placed on ice, and washed with ice-cold PBS. The cells were lysed in lysis buffer
(150mMNaCl, 50 mM HEPES, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 10 µg/ml
leupeptin, 10 µg/ml aprotinin, 1 µg/ml pepstatin A, 1 mM PMSF, and 1 mM Na3VO4) and
cleared by centrifugation at 14,000 rpm for 10 min. Equal amounts of proteins were resolved by
SDS-PAGE, transferred to nitrocellulose membrane, and analyzed by immunoblotting with the
indicated antibodies, followed by secondary antibodies and ECL detection (GE Healthcare)
11. shRNA Knockdown and qPCR quantification of knockdown
Prior to the Cellomics halide exchange assay ΔF508-CFTR cells (stably expressing eYFP
(H148Q/I152L) were transfected with target genes or luciferase (nonsilencing control) shRNA
constructs using Lipofectamine 2000, according to the manufacturer's instructions. Medium was
changed 6 h after transfection, and ΔF508-CFTR cells were placed at 37 °C, 5% CO2. 48 h after
transfection the cells were incubated with media containing Puromycin (5 μg/ml, 3 days).
Cellomics halide exchange assay was performed as described above.
Knockdowns were validated by two-step RT-qPCR. Total RNA was isolated using the
RNeasy 96 kit (Qiagen, Dorking, Surrey, UK), and cDNA was prepared using the High Capacity
cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Real time PCR
reactions were performed using Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen)
and CFX96 Real-Time System (BioRad). Primers were obtained from Integrated DNA
Technologies. For standard curves, real time PCR was performed on a fivefold dilution series
DNA.

49
Chapter 3
Results
I performed the majority of the work presented in this thesis, with the exception of the Cellomics
and flow cytometry experiments that were performed by Dr. Agata Trzcinska-Daneluti (Figure
9,11,15B, 19B). The validation results of the kinase inhibitors screen were published in the
journal Molecular & Cellular Proteomics under the title ‘Use of kinase inhibitors to correct
ΔF508-CFTR function’ (Trzcinska-Daneluti, A.M., Nguyen, L. et al., (2012) Mol Cell
Proteomics, 11(9): 745-757).

50
I. Screen for kinase inhibitors that correct ΔF508-CFTR function using high-content
functional Cellomics assays
Using a high-content functional screen our lab previously developed, a library of 231
kinase inhibitors, biased toward FDA-approved drugs or compounds currently in clinical trials
mainly for the treatment of cancer and inflammation, was screened to identify compounds that
could correct ΔF508-CFTR. Several other compounds were also included in the screen such as
(5Z)-7-oxozeaenol, SU5402, and Kenpaullone, which were small molecule kinase inhibitors that
mimicked the effect of knockdown of several of the kinases identified in an independently
performed esiRNA kinome screen. In the current small molecule screen, 293MSR-GT cells
stably co-expressing the Cl− sensitive eYFP (H148Q/I152L) mutant and ΔF508-CFTR (ΔF508-
CFTR cells) were treated with 10µM of each inhibitor from the library for 48h at 37oC. Cells
were then stimulated for 20 min using a mixture of Forskolin (25 μM)/IBMX (45 μM)/Genistein
(50 μM) (FIG) and exposed to low Cl-/high I
- solution. The quenching of fluorescence caused by
Cl-/I
- exchange, presumably by CFTR or its mutant, was recorded and quantified over time. The
list of hit compounds (Table I) consisted of those that exhibited at least 0.1 difference in average
fluorescence intensity between unstimulated (-FIG) and stimulated (+FIG) cells. The hit
compounds included several receptor tyrosine kinase inhibitors (RTKs), especially inhibitors of
FGFR1 (Fibroblast Growth Factor Receptor 1) and its downstream signaling targets (e.g.
SU5402, SU6668, Ki8751), as well as inhibitors of several important cellular signaling pathways
such as Ras/Raf/MEK/ERK (e.g. (5Z)-7-Oxozeaenol, RDEA-119), Wnt/GSK-3β (e.g. GSK-3β
Inhibitor II, Kenpaullone), TAK1/p38 (e.g. (5Z)-7-oxozeaenol, SKF86002), PI3K/Akt/mTOR
(e.g. PI-103, FPA124, 10-DEBC). Figure 9 (B-H) depicts examples of kinase inhibitors that
exhibited rescue of ΔF508-CFTR (e.g. (5-Z)-7-Oxozeaenol, SU5402, GSKInhII, Kenpaullone)

51
Table 1: Hit compounds and their validations. Hits were validated by immunoblotting in
293MSR-GT cells (WB), flow cytometry in BHK cells (Flow), and short circuit current (Isc)
analysis in Ussing chambers on epithelial MDCK cells (MDCK) or on primary Human Bronchial
Epithelial (HBE) cells harvested from lungs of ΔF508/ΔF508 homozygote patients undergoing
lung transplant. 293MSR-GT, BHK, and MDCK cells were stably expressing ΔF508-CFTR. For
MDCK, (+) indicates rescue, (−) no observed rescue and (*) indicates increased toxicity in
MDCK cells. For HBE, (+) indicates rescue observed in a sample from a patient, with (/)
separating between samples from different patients, (−) indicates no observed rescue. For the flow
experiments, (+) indicates >10% rescue, (±) 5–10% rescue, (−) indicates no rescue, (*) indicates
increased cell toxicity and (#) indicates morphological changes observed in the treated BHK cells.
For the immunoblotting experiments, (+) indicates strong rescue of ΔF508-CFTR (manifested as
increase in amount of band C in comparison to vehicle-alone control), (±) poor rescue, (−) no
rescue, and (*) indicates increased toxicity in 293MSR-GT cells. For dose response experiments
(EC50), MDCK cells were treated with increasing concentrations (1 nM to 10 μM range) of select
compounds prior to Isc analysis in Ussing chambers, (§) indicates compounds that rescue ΔF508-
CFTR function at 10 μM only.
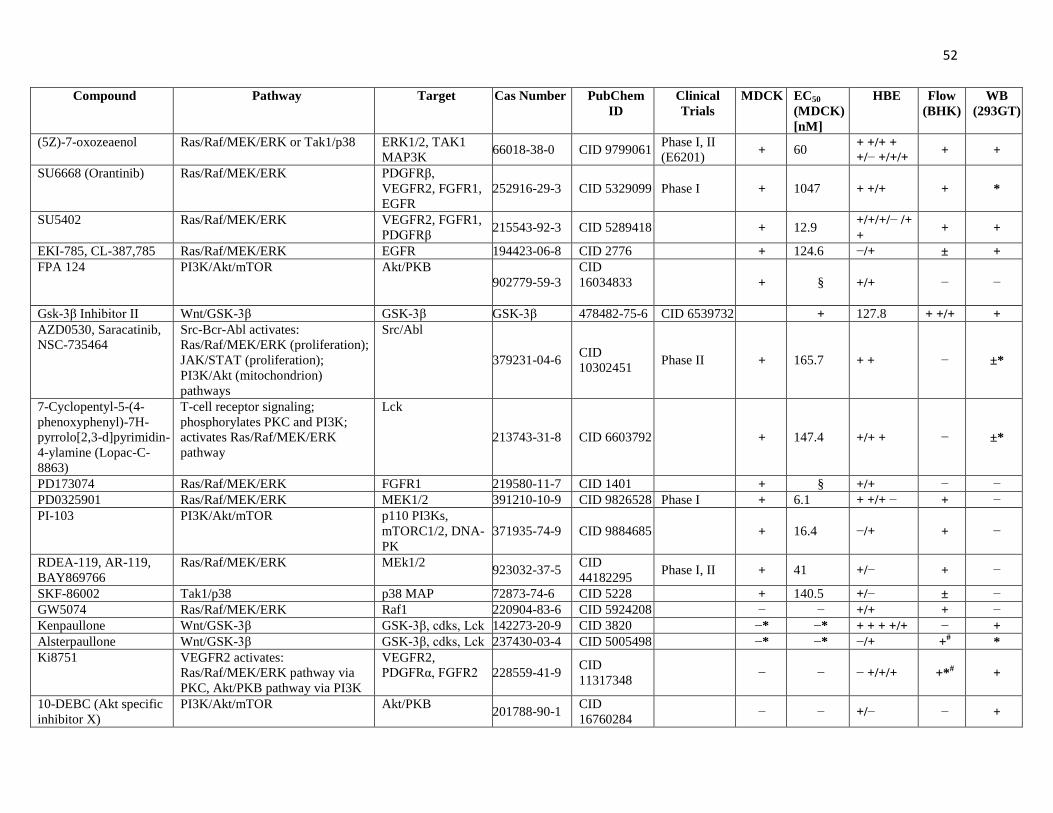
52
Compound Pathway Target Cas Number PubChem
ID
Clinical
Trials
MDCK EC50
(MDCK)
[nM]
HBE Flow
(BHK)
WB
(293GT)
(5Z)-7-oxozeaenol Ras/Raf/MEK/ERK or Tak1/p38 ERK1/2, TAK1
MAP3K 66018-38-0 CID 9799061
Phase I, II
(E6201) + 60
+ +/+ +
+/− +/+/+ + +
SU6668 (Orantinib) Ras/Raf/MEK/ERK PDGFRβ,
VEGFR2, FGFR1,
EGFR
252916-29-3 CID 5329099 Phase I + 1047 + +/+ + *
SU5402 Ras/Raf/MEK/ERK VEGFR2, FGFR1,
PDGFRβ 215543-92-3 CID 5289418 + 12.9
+/+/+/− /+
+ + +
EKI-785, CL-387,785 Ras/Raf/MEK/ERK EGFR 194423-06-8 CID 2776 + 124.6 −/+ ± +
FPA 124 PI3K/Akt/mTOR Akt/PKB
902779-59-3
CID
16034833
+ § +/+ − −
Gsk-3β Inhibitor II Wnt/GSK-3β GSK-3β GSK-3β 478482-75-6 CID 6539732 + 127.8 + +/+ +
AZD0530, Saracatinib,
NSC-735464
Src-Bcr-Abl activates:
Ras/Raf/MEK/ERK (proliferation);
JAK/STAT (proliferation);
PI3K/Akt (mitochondrion)
pathways
Src/Abl
379231-04-6 CID
10302451 Phase II + 165.7 + + − ±*
7-Cyclopentyl-5-(4-
phenoxyphenyl)-7H-
pyrrolo[2,3-d]pyrimidin-
4-ylamine (Lopac-C-
8863)
T-cell receptor signaling;
phosphorylates PKC and PI3K;
activates Ras/Raf/MEK/ERK
pathway
Lck
213743-31-8 CID 6603792 + 147.4 +/+ + − ±*
PD173074 Ras/Raf/MEK/ERK FGFR1 219580-11-7 CID 1401 + § +/+ − −
PD0325901 Ras/Raf/MEK/ERK MEK1/2 391210-10-9 CID 9826528 Phase I + 6.1 + +/+ − + −
PI-103 PI3K/Akt/mTOR p110 PI3Ks,
mTORC1/2, DNA-
PK
371935-74-9 CID 9884685 + 16.4 −/+ + −
RDEA-119, AR-119,
BAY869766
Ras/Raf/MEK/ERK MEk1/2 923032-37-5
CID
44182295 Phase I, II + 41 +/− + −
SKF-86002 Tak1/p38 p38 MAP 72873-74-6 CID 5228 + 140.5 +/− ± −
GW5074 Ras/Raf/MEK/ERK Raf1 220904-83-6 CID 5924208 − − +/+ + −
Kenpaullone Wnt/GSK-3β GSK-3β, cdks, Lck 142273-20-9 CID 3820 −* −* + + + +/+ − +
Alsterpaullone Wnt/GSK-3β GSK-3β, cdks, Lck 237430-03-4 CID 5005498 −* −* −/+ +# *
Ki8751 VEGFR2 activates:
Ras/Raf/MEK/ERK pathway via
PKC, Akt/PKB pathway via PI3K
VEGFR2,
PDGFRα, FGFR2 228559-41-9 CID
11317348 − − − +/+/+ +*
# +
10-DEBC (Akt specific
inhibitor X)
PI3K/Akt/mTOR Akt/PKB 201788-90-1
CID
16760284 − − +/− − +

53
Figure 9: Representative hits of the high-content screen. Average normalized fluorescence
intensity values of ∆F508-CFTR cells (which co-express eYFP(H148Q/I152L) that were (A)
transfected with shRNA for FGFR1, or (B) treated with 10 µM SB431542 (non-corrector), (C)
(5Z)-7-Oxozeaenol, (D) SU5402, (E) GSK-3 Inhibitor II, (F), RDEA119, (G) Kenpaullone, (H),
Ki8751, and grown at 37 °C. After 48 h (5 days in case of shRNA knockdown of FGFR1) cells
were stimulated with FIG (25µM Forskolin, 45µM IBMX and 50µM Genistein), and quenching
of fluorescence during Cl-/I
- exchange of 70–300 cells was quantified simultaneously and re-
corded by the Cellomics. FGFR1 knockdown was 90% (as determined by RT-qPCR)
[Experiments for this figure were carried out by Dr. Agata Trzcinska-Daneluti]
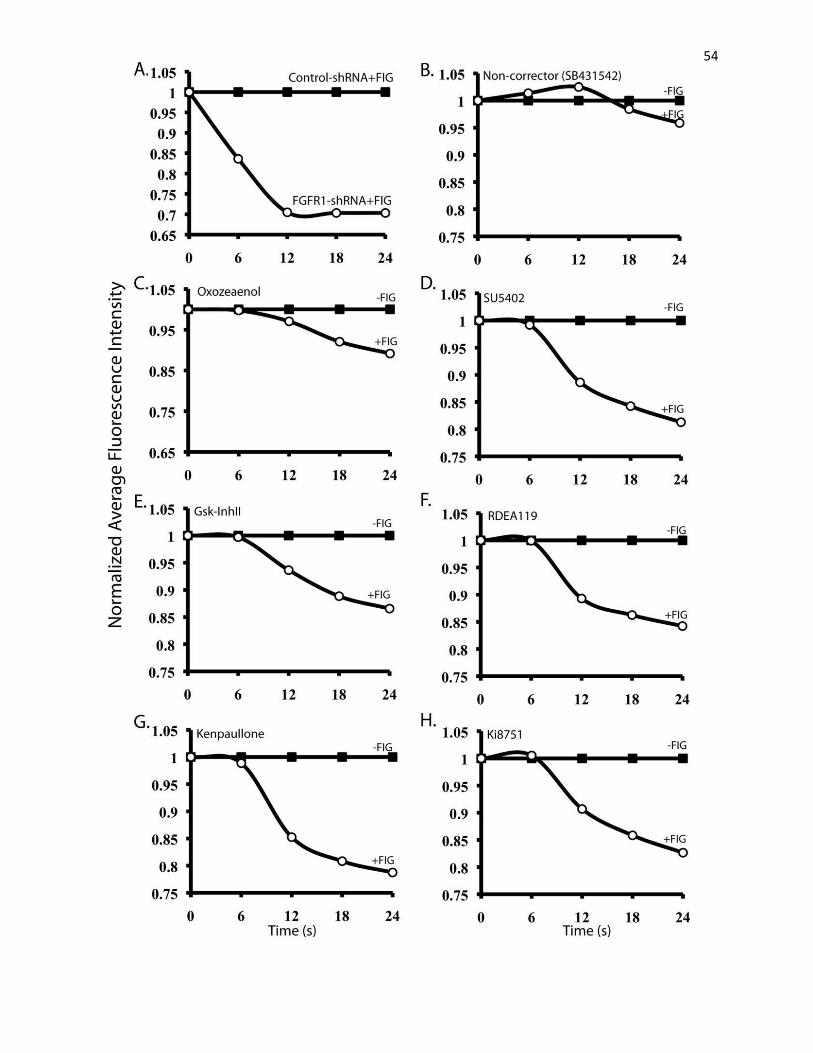
54

55
and one kinase inhibitor that did not rescue the function of mutant CFTR (SB431542). Figure 9A
displays the rescue of ΔF508-CFTR after shRNA knockdown of FGFR1, a target of several
kinase inhibitors identified in the screen.
II. Validation of the Hits
1. Maturation of ΔF508-CFTR
Among the positive hits from the initial screen using Cellomics, 41 representative
compounds were chosen for further analysis and validations of ΔF508-CFTR rescue using
alternative methods. These compounds were selected based on their inhibition of kinases that
participate in four major signaling pathways identified by the screen (e.g. Ras/Raf/MEK/ERK,
Wnt/GSK-3, PI3K/Akt/mTOR and TAK1/p38). First, the effect of the compounds on the
maturation of ΔF508-CFTR was examined by immunoblotting. As previously mentioned, in
SDS-PAGE, WT-CFTR appears as two bands, a prominent band C around 180 kDa representing
the mature, fully glycosylated form of CFTR, and a minor band B around 150 kDa representing
the core glycosylated, immature form. ΔF508-CFTR, which exhibits impaired maturation,
migrates primarily as band B. For this assay, HEK293GT cells stably expressing ΔF508-CFTR
were treated with either DMSO (negative control) or 15µM of selected top hit compounds.
Treatment of the cells at 27oC was used as a positive control, which, as shown in figure 10, led to
a robust increase in band C. Moreover, treatment with some of the indicated compounds (e.g.
(5Z)-7-Oxozeaenol, Kenpaullone) also led to an increase in the maturation of ΔF508-CFTR
(appearance of band C) compared to vehicle control treatment, albeit a less robust rescue than
low temperature. Unfortunately, several compounds exhibited adverse effect on the cells and
thus could not be tested by immunoblotting. In figure 10, TCS2312 illustrates one such
compound. Treatment of cells with this compound abolished both forms of CFTR.

56
Figure 10: Effect of select kinase inhibitors on ∆F508-CFTR maturation analyzed by
immunoblotting.293MSR-GT cells stably expressing ∆F508-CFTR were treated with
15µMkinase inhibitors or 0.3% DMSO (vehicle control), as indicated, grown at37 °C for 48 h,
and the appearance of the mature protein, band C, monitored by immunoblotting with anti-CFTR
antibodies. Band B represents the immature protein. DMSO represents vehicle-alone control,
27oC represents temperature rescue of ∆F508-CFTR at27 °C, 37 °C represents untreated ∆F508-
CFTR control, and WT represents WT-CFTR. Top panels depict the anti-CFTR immunoblot and
bottom panels depict actin (loading) control.**represents cellular toxicity.
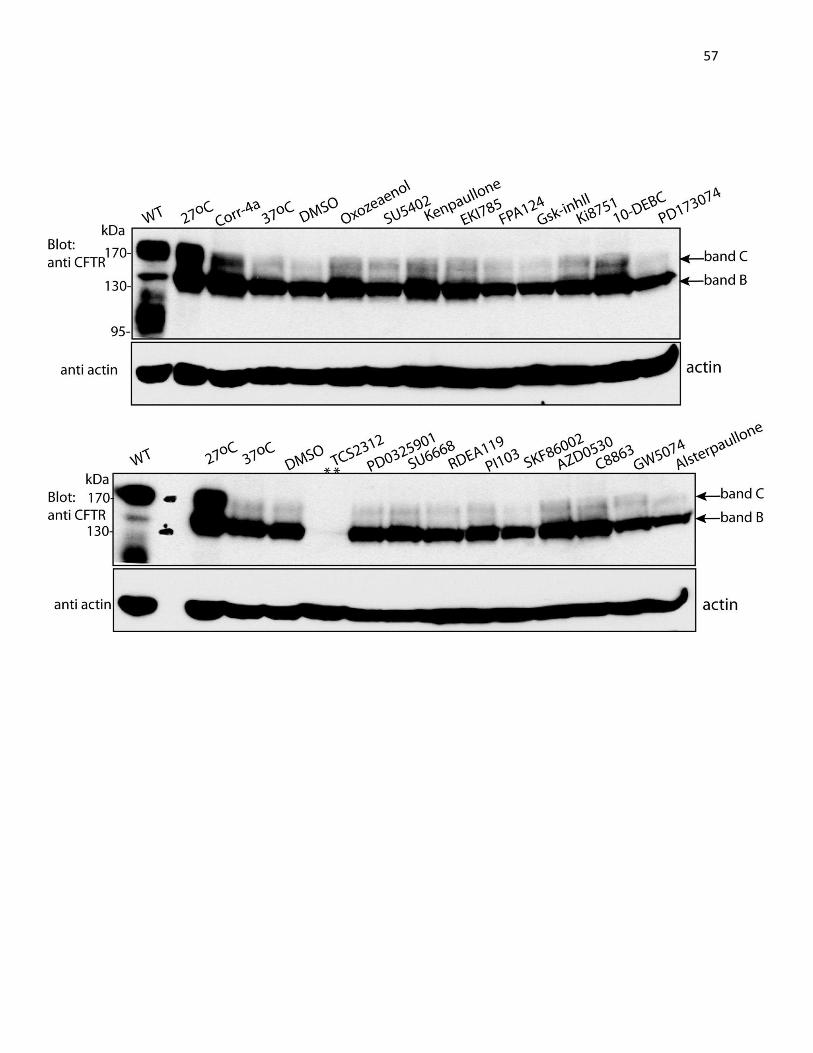
57

58
In addition, flow cytometry was performed to demonstrate the appearance of ΔF508-CFTR at the
plasma membrane. Toward this end, BHK cells stably expressing ΔF508-CFTR bearing a 3HA
tag on the ectodomain of the protein were treated with either 0.2% DMSO (negative control) or
10µM kinase inhibitors; cells grown at 27oC were used as a positive control. To quantify the
amount of ΔF508-CFTR reaching the cell surface, nonpermeabilized BHK cells were then
immunostained with antibodies directed against the 3HA epitote tag before being subjected to
flow cytometry. Figure 11 depicts results of treatment at 27oC (panel A) or with several
compounds (panels B-E), as well as cell surface expression of WT-CFTR (panel F). A summary
of the percentage of increase in cell surface expression of ΔF508-CFTR after treatment with the
kinase inhibitors is shown in panel G. Collectively, these results show that surface expression of
ΔF508-CFTR was rescued by as much as 40-50% after treatment with several inhibitors such as
Oxozeaenol, SU5402, RDEA119 and SU6668. This was about half of the rescue observed by
treatment at lower temperature (about 80% increase in cell surface expression).
2. Functional Analysis of Correction of ΔF508-CFTR by the Kinase Inhibitors
To further analyze the effect of the kinase inhibitors on ∆F508-CFTR, the rescue of
∆F508-CFTR activity was determined by short-circuit current (Isc) analysis in Ussing chambers
using epithelial MDCK cells that stably express ΔF508-CFTR. Cells were grown on inserts with
permeable membrane until they reached confluency, and then treated with 10 μM kinase
inhibitor or 0.2% DMSO (vehicle control) for 48h. Inserts were then mounted in Ussing
chambers for short-circuit current studies. CFTR channel was stimulated with FIG in the
presence of ENaC inhibitor Amiloride and DNDS, an inhibitor of Na+/HCO3
− cotransporters and
Cl−/HCO3
−exchangers; and currents across the layer of cells were recorded. Figure 12A-E

59
Figure 11: Effect of kinase inhibitors on cell surface expression of ∆F508-CFTR analyzed
by flow cytometry. BHK cells stably expressing ∆F508-CFTR-3HA were placed at (A) 27 °C
(positive control) for 48 h, or (B) treated with 10 μM (5Z)-7-oxozeaenol, (C) SU5402, (D)
SU6668, or (E) RDEA-119/AR-119/BAY869766, at 37 °C. (F) BHK cells stably expressing
WT-CFTR. Flow cytometry was then performed on non-permeabilized cells following
immunostaining for the HA epitope located at the ectodomain of ∆F508-CFTR or WT-CFTR, to
quantify the amount of cell-surface CFTR in the analyzed cells. (G) Summary of increase in cell
surface expression of ∆F508-CFTR (%change in fluorescence intensity) of the hits analyzed by
flow cytometry (two independent experiments, 10,000 live cells per treatment per experiment).
[Experiments for this figure were carried out by Dr. Agata Trzcinska-Daneluti]
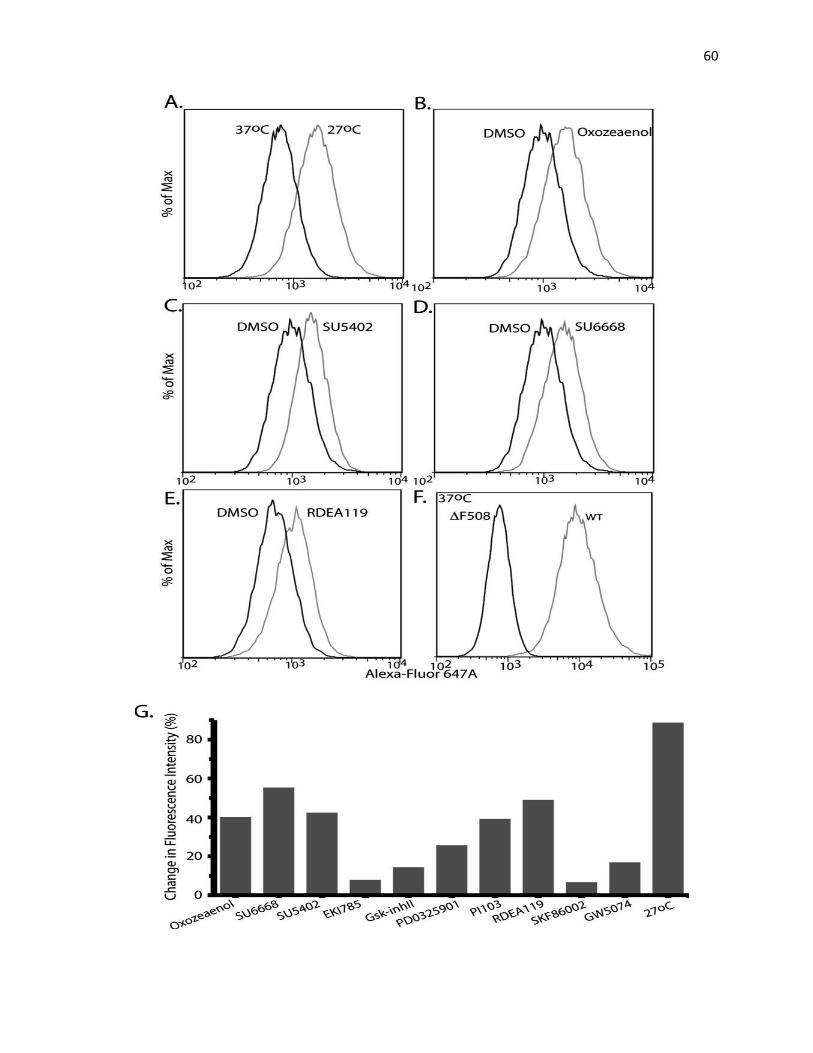
60

61
Figure 12: Effect of compounds treatment on ∆F508-CFTR channel activity in MDCK cells
stably expressing ∆F508-CFTR. Representative short-circuit current (Isc) traces of MDCK
∆F508-CFTR monolayers treated with vehicle (DMSO) alone (black lines), or 10 µM of (A)
(5Z)-7-oxozeaenol, (B) SU5402, (C) SU6668, (D) GSK-3β Inhibitor II, (E) 7-Cyclopentyl-5-(4-
phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamine (C8863) (grey lines), for 48 h prior to
analysis in Ussing chambers. ENaC was inhibited with 10 µM amiloride; non-CFTR chloride
channels were blocked with 250 µM DNDS. CFTR currents were stimulated with FIG (25 µM
Forskolin, 25 µM IBMX and 50 µM Genistein) at time 0 and after the indicated times (arrows)
inhibited using 15 µM GlyH-101 (Gly). (F) Representative short-circuit currents mediated by
untransfected MDCK cells and MDCK cells that stably express WT-CFTR or ΔF508-CFTR. (G)
Summary of the increase in short-circuit currents (∆Isc) in MDCK cells stably expressing
∆F508-CFTR that were treated by the analyzed compounds (relative to DMSO vehicle control
alone). Data are mean ± S.E. (n): number of experiments (*: p<0.05; **: p<0.01, paired one-
tailed Student’s t-test).
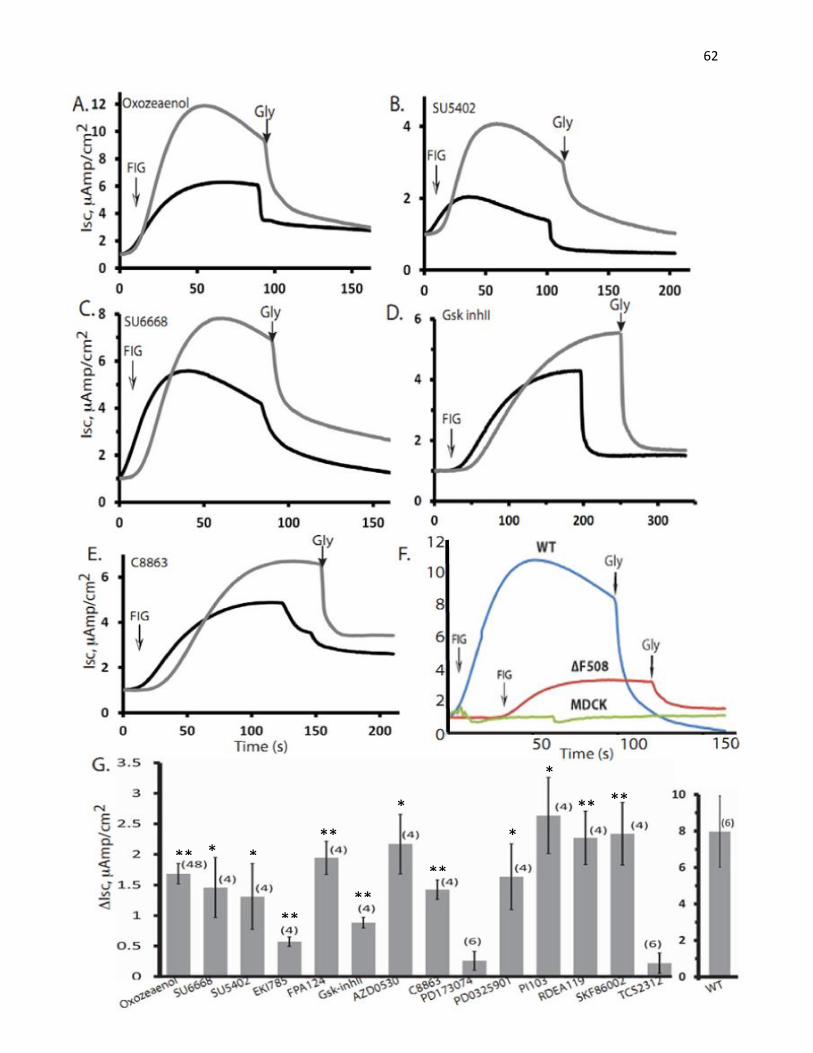
62
**
**
**
**
**
** **
*
*
*
* *

63
displays the effects of treatments with representative compounds (e.g. Oxozeaenol, SU5402,
SU6668, GSK-3β Inhibitor II, 7-Cyclopentyl-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-
4-ylamine/C8863), and others on ΔF508-CFTR function (i.e. chloride channel activity).
Stimulated current in cells stably expressing WT-CFTR is also shown in comparison with that in
cells expressing ΔF508-CFTR and in parental MDCK cells (Figure 12F). Parental MDCK cells
showed no response to either FIG or the CFTR inhibitor, GlyH101. In addition, there was some
channel activity in the untreated ΔF508-CFTR MDCK cells, suggesting some mutant CFTR had
escaped from the ER to the surface in the overexpression system. DMSO control treatment
(0.2%) did not improve this activity. Summary of the difference in maximal stimulated currents
(∆Isc) between cells treated with inhibitors and vehicle control (Figure 12G) shows various
degrees of functional rescue of ΔF508-CFTR with several of these hit compounds. The ∆Isc
between cells expressing WT-CFTR and those expressing the mutant protein was about
8µAmp/cm2. On average, the top compounds (Oxozeaenol, SU5402, SU6668, RDEA-119, PI-
103) exhibited an increase of 20-30% (1.5 to 2.5µAmp/cm2) in stimulated current compared to
what was observed in cells expressing WT-CFTR.
3. Effect of Kinase Inhibitors on ΔF508-CFTR Chloride Channel Activity in
Primary Human Bronchial Epithelial (HBE) Cells Harvested from CF Patients
Even though some of these compounds were proven to be able to rescue the function of
ΔF508-CFTRin vivo in MDCK cells, it was necessary to determine if these inhibitors could
rescue function in bronchial epithelial cells from CF patients. To this end, we examined the
effect of treatment with selected compounds on primary HBE cells obtained from patients that
were homozygous for ΔF508-CFTR and had undergone lung transplant. Since the samples were
limited, only the compounds that rescued the function of ΔF508-CFTR in MDCK cells and/or in

64
Figure13: Effect of compounds treatment on ∆F508-CFTR activity in primary Human
Bronchial Epithelial (HBE) cells harvested from lungs of ∆F508/∆F508 homozygote
patients undergoing lung transplant. Representative short-circuit currents (Isc) mediated by
∆F508-CFTR human bronchial epithelial (HBE) monolayers treated with vehicle (DMSO) alone
(black lines), or 10 µM of (A) (5Z)-7-oxozeaenol, (B) SU5402, (C) SU6668,(D) GSK-3β
Inhibitor II, (E) 7-Cyclopentyl-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-ylamine
(C8863), (F) Kenpaullone (grey lines) for 48 h prior to analysis in Ussing chambers. ENaC
sodium channels were inhibited with 10 µM amiloride; non-CFTR chloride channels were
blocked with 250 µM DNDS. CFTR currents were stimulated with FIG (25 µM Forskolin, 25
µM IBMX and 50 µM Genistein) as indicated, and after the indicated times (black arrows)
inhibited using 15µM (panels A, B, E, F) or 50 µM (panels C, D) GlyH-101 (Gly). In panels C
and D, half of the Gly solution (25 µM) was added twice sequentially, as indicated. (G)
Representative short-circuit currents mediated by HBE cells from non-CF controls (WT-CFTR).
(H) Summary of increase in short-circuit currents (∆Isc) in HBE cells stably expressing ∆F508-
CFTR that were treated with the indicated compounds. Data from individual patients are shown.
Where several replica were tested from the same patient (see Table I), the average value is
shown. Bars represent median values. The baseline currents (before amiloride addition) ranged
between 6 –20 µAmp/cm2 for WT-HBE and 19 – 40 µAmp/cm
2for ∆F508-CFTR HBE. After
adding amiloride, the currents for both WT and ∆F508-CFTR HBE were ~0 –3 µAmp/cm2.
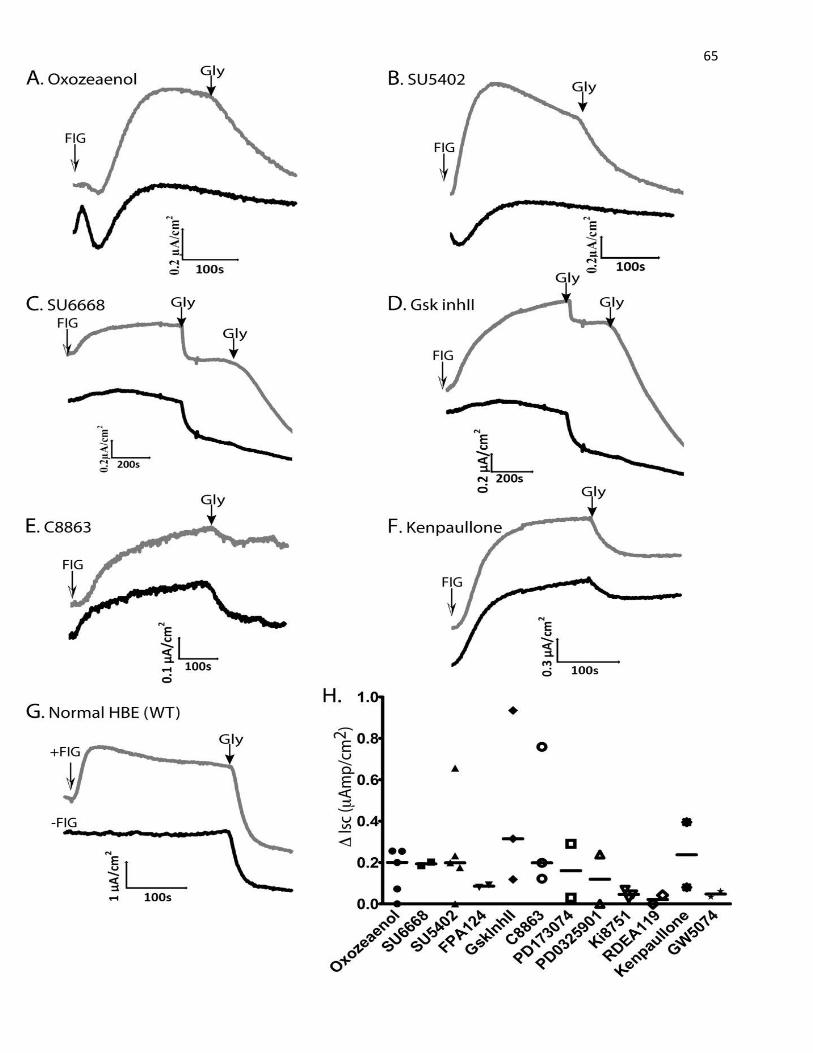
65

66
several other validation assays were chosen to be tested in HBE cells. Because there was
variation between cells obtained from different patients due to differences in the severity of the
disease and conditions of each patient, the effects of the treatments were assessed by comparing
samples from each patient with the control treatment (vehicle alone) from the same patient. This
eliminated patient-to-patient variability.
Like the assays in MDCK cells, primary HBE cells were grown on permeable inserts and
then treated with either 10µM of selected compounds or 0.2% DMSO control. Representative
traces with cells treated with Oxozeaenol, SU5402, SU6668, GSK-3β Inhibitor II, 7-
Cyclopentyl-5-(4-phenoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-ylamine (C8863), and
Kenpaullone are shown in Figure 13A-F. Cells obtained from different patients behaved
differently; and some still exhibited partial activity even with the control treatment. Non-CF
HBE obtained from normal lung was used for comparison (Figure 13G). In non-CF samples, the
stimulated current was about 2µAmp/cm2
compared to unstimulated sample. Most of the
compounds showed variable rescue of ΔF508-CFTR in samples from different patients (Figure
13H), as expected. However, on average, the compounds enhanced the current in HBE samples
to approximately 10% of WT activity (0.2µAmp/cm2). In some cases, the enhancement of the
current was up to 40- 50% of activity of WT HBE.
Combined together, these results suggest that several of these kinase inhibitors can
improve the function of ΔF508-CFTR in both tissue culture cells and in HBE obtained from
patients. This occurs likely by improving the maturation and surface expression of the mutant
channel. However, the possibility that some of these compounds act as potentiators of ΔF508-
CFTR activity (once at the plasma membrane) cannot be excluded based on our functional
analysis. The summary of all the validation results is presented in Table 1. 18 compounds

67
rescued ΔF508-CFTR in at least one of these assays. Several compounds, such as Oxozeaenol or
SU5402, gave consistent rescue of ΔF508-CFTR in all of the assays performed.
III. Dose Response Curves of Rescue of ΔF508-CFTR in MDCK Cells Treated with
Select Kinase Inhibitors
The concentration of the compounds initially used in the screen was 10µM, which was
rather high. Therefore, to further assess the effect of increasing doses of these kinase inhibitors
on rescuing ∆F508-CFTR function, Isc analyses using Ussing chamber were performed in
MDCK cells stably expressing ∆F508-CFTR treated with an increasing concentrations covering
a range from 1nM to 10µM of our top hit inhibitors (Figure 14 and table 1). The estimated half
maximal effective concentrations (EC50) of most of the analyzed compounds were in the
nanomolar range (6 to 125nM) with the exception of SU6668 (EC50~1µM). The effects of two
other compounds, PD173074 and FPA124, could only be seen at concentration of 10µM. Due to
limited amount of HBE samples, dose-response analyses in these cells could not be performed.

68
Figure 14:Dose response curves of select kinase inhibitors for rescue of ∆F508-CFTR
expressed in MDCK cells. Average increase in short-circuit currents (∆Isc) of MDCK cell
monolayers stably expressing ∆F508-CFTR (relative to DMSO vehicle control alone) treated for
48 h with 1, 10, 20, 100, 200, 1000,and 10,000 nM of the top inhibitor compounds, indicated in
panels (A–K). Data are mean ± S.E. of (n) samples.

69
I)

70
IV. Analyses of E6201, a derivative of (5Z)-7-Oxozeaenol
1. E6201 did not rescue ∆F508-CFTR maturation and function
One of our top hits from the screen was a theTAK1/p38 and ERK inhibitor, Oxozeaenol.
However, Oxozeaenol is not useful for therapeutic utilization due to its instability in plasma
(Goto et al., 2009); thus, a derivative of it, E6201, was synthesized by the pharmaceutical
company Eisai. E6201 has now passed phase II clinical trials for the treatment of psoriasis
(Muramoto et al., 2010; Kumar et al., 2012). Therefore, we wanted to determine if E6201 can
rescue ∆F508-CFTR similar to its parental compound, Oxozeaenol. E6201 was provided to us by
Eisai. We thus tested the effect of E6201 on rescue of∆F508-CFTR using the Cellomics assay,
Immunoblotting, and using Ussing chamber as described above. Figure 15A shows structures of
both compounds with the differences encircled. The most prominent change is the N-alkyl
substitution in E6201 (Shen et al. 2010).
Unfortunately, while Oxozeaenol exhibited rescue in all of our validation methods,
treatment of cells with E6201 at several doses did not yield any significant rescue of ∆F508-
CFTR in any of the assays performed. In the Cellomics assays on HEK293GT cells, 48h
treatment with increasing concentrations of E6201 (1nM-50µM) did not yield any significant
rescue of ∆F508-CFTR activity. Lower doses of E6201 (10nM to 10μM) improved the channel
activity, but not significantly, only about 1-2%. The highest increase in activity, about 4-5%, was
observed with the treatment at 10 µM (Figure 15B). Furthermore, treatments at higher doses of
E6201 (20µM to 50µM) seemed to have an adverse effect, resulting in a decreased activity to a
level lower than basal activity. At these concentrations, the quenching of signal was even lower
than that of cells treated with DMSO control. Similar results were observed in Ussing chamber

71
Figure 15: E6201 does not rescue the function of ∆F508-CFTR.(A) Molecular structures of
(5Z)-7-Oxozeaeol (left) and its derivative E6201. The differences are highlighted (circled). The
most important change is the N-akyl substitution at C14 (Shen et al., 2010). (B) E6201 did not
rescue ∆F508-CFTR function in Cellomics assays. Average normalized fluorescence intensity
values of HEK293MSR-GT ΔF508-CFTR cells (which co-express eYFP (H148Q/I152L) that
were treated with either increasing concentrations of E6201 (1nM to 50μM) or 10μM of (5Z)-7-
Oxozeaenol for 48h at 37oC. After 48 h cells were stimulated with FIG and quenching of
fluorescence during Cl−/I
− exchange of minimum 100 cells was quantified simultaneously and
recorded. Data are mean ± SEM of three replicates, *: p<0.05 (unpaired one-tailed Student’s t-
test) [Experiments for this panel were carried out by Dr. Agata Trzcinska-Daneluti].
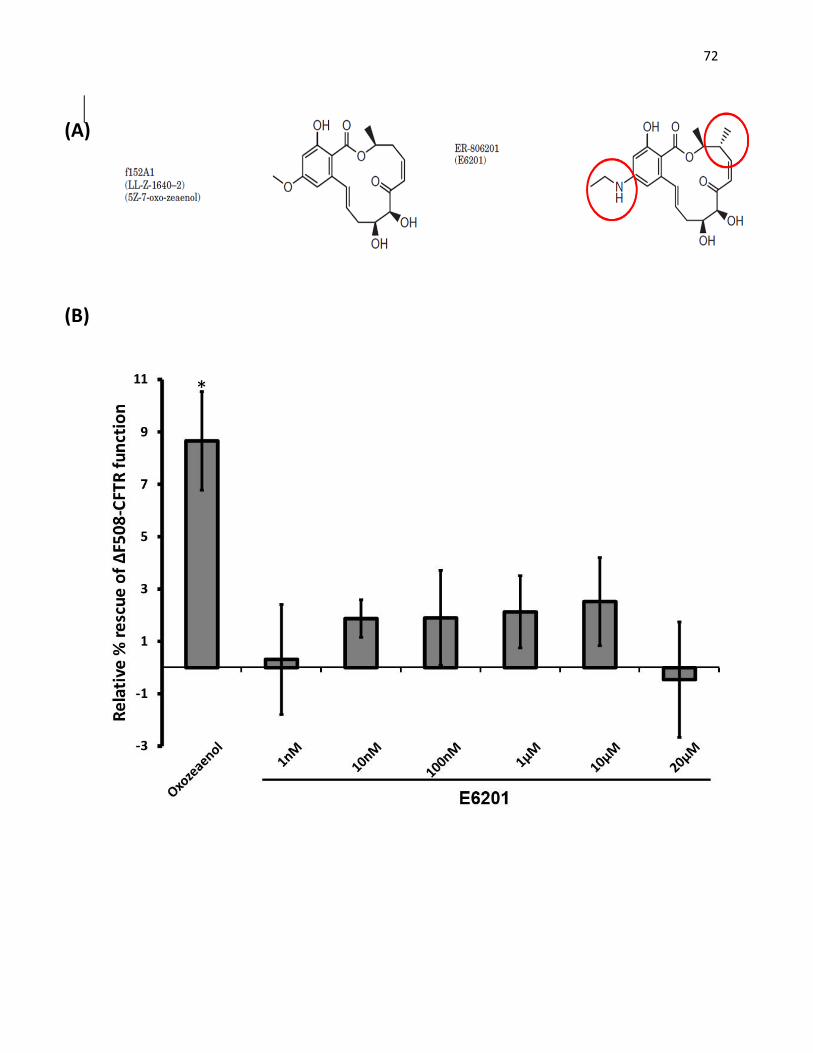
72
(A)
(B)
*

73
Figure 16: E6201 does not rescue the maturation and function of ∆F508-CFTR by
immunoblotting and Ussing chambers.(A) E6201 did not improve the appearance of band C in
immunoblotting. HEK293GT cells expressing ∆F508-CFTR were treated at different
concentrations of E6201 (1nM to 20µM) for 48h at 37oC then lysed and analyzed by
immunoblotting. Treatments at 27oC and (5Z)-7-Oxozeaenol at 15µM were used as positive
controls. Both led to improved appearance of band C. Below is the relative intensity of band B,
band C, and ratio of band B/ band C for each treatment. (B) Summary of the change in short-
circuit currents (ΔIsc) in MDCK cells stably expressing ΔF508-CFTR that were treated by
E6201 (relative to DMSO vehicle control alone). E6201 did not rescue the function of ∆F508-
CFTR in MDCK cells expressing ∆F508-CFTR in Isc analysis by Ussing chambers. MDCK cells
stably expressing ΔF508-CFTR were treated with either increasing concentrations of E6201
(1nM - 20μM) or 10µM (5Z)-7-oxozeaenol, and grown at 37C for 48h prior to analysis in
Ussing chambers. ENaC sodium channels were inhibited with 10 μM amiloride; non-CFTR
chloride channels were blocked with 250 μM DNDS (not shown in the graphs). CFTR currents
were stimulated with FIG at time 0 and after the indicated times (arrows) inhibited using 15 μM
GlyH-101 (Gly). Data are mean ± S.E. (n): number of experiments. *: p<0.05 (paired one-tailed
Student’s t-test)

74
(A)
(B)
* (B)

75
experiments in MDCK cells treated with increasing concentrations of E6201 (1nM-20µM).
Treatments with E6201 at 10 or 20 µM reduced the stimulated current compared to DMSO
control (ΔIsc is below 0), and stimulated currents recorded in cells treated with lower doses
(1nM to 1µM) were not significantly different from DMSO control (ΔIsc~0) (Figure 16B).
Lastly, on Western blot, E6201 also did not improve the appearance of band C in cells
expressing ∆F508-CFTR treated with 1nM to 20µM of E6201 (Figure 16A). Thus, altogether,
these results suggest that unlike the parental compound Oxozeanol, E6201 was not effective at
rescuing ∆F508-CFTR.
2. Comparison of the effect of E6201 and (5Z)-7-Oxozeaenol on different signalling
pathways
Because Oxozeaenol and its derivative, E6201, had different effects on the rescue of
∆F508-CFTR, determining the differences in the effects of both compounds on downstream
signaling pathways might provide us insight into the mechanism/pathways utilized by the
parental compound in the rescue of ∆F508-CFTR. While Oxozeaenol has been shown to inhibit
both TAK1 and the MEK/ERK pathway (even though with much lower potency for the latter
pathway) (Ninomiya-Tsuji et al, 2003), E6201 has only been shown to primarily inhibit the
MEK/ERK pathway (Goto et al, 2009) (Figure 17). This suggests that Oxozeaenol might work
through inhibiting TAK1 and the downstream signaling pathways to rescue ∆F508-CFTR.
To test this hypothesis, inhibition of phosphorylation of signaling targets downstream of
TAK1 was studied. Macrophages extracted from mouse bone marrow (BMM) and RAW264.7
cells were treated with increasing concentrations of either Oxozeaenol or E6201 (100nM to 5 or
10µM) for 1 hour prior to a 20-minute stimulation with LPS. Downstream inhibition of

76
Figure 17: Signaling pathways inhibited by (5Z)-7-Oxozeaenol and E6201. (5Z)-7-
Oxozeaenol is a potent inhibitor of TAK1 (IC50~8nM), which phosphorylates p38, JNK, NFκB
to activate downstream signaling pathways. (5Z)-7-Oxozeaenol also inhibits the MEK/ ERK
pathway, but with a lower potency (IC50~411nM). On the other hand, E6201 has only been
shown to be a strong inhibitor of MEK/ERK pathway with IC50 ~5nM (*Ninomiya-Tsuji et al.,
2003; ** Goto et al., 2009).
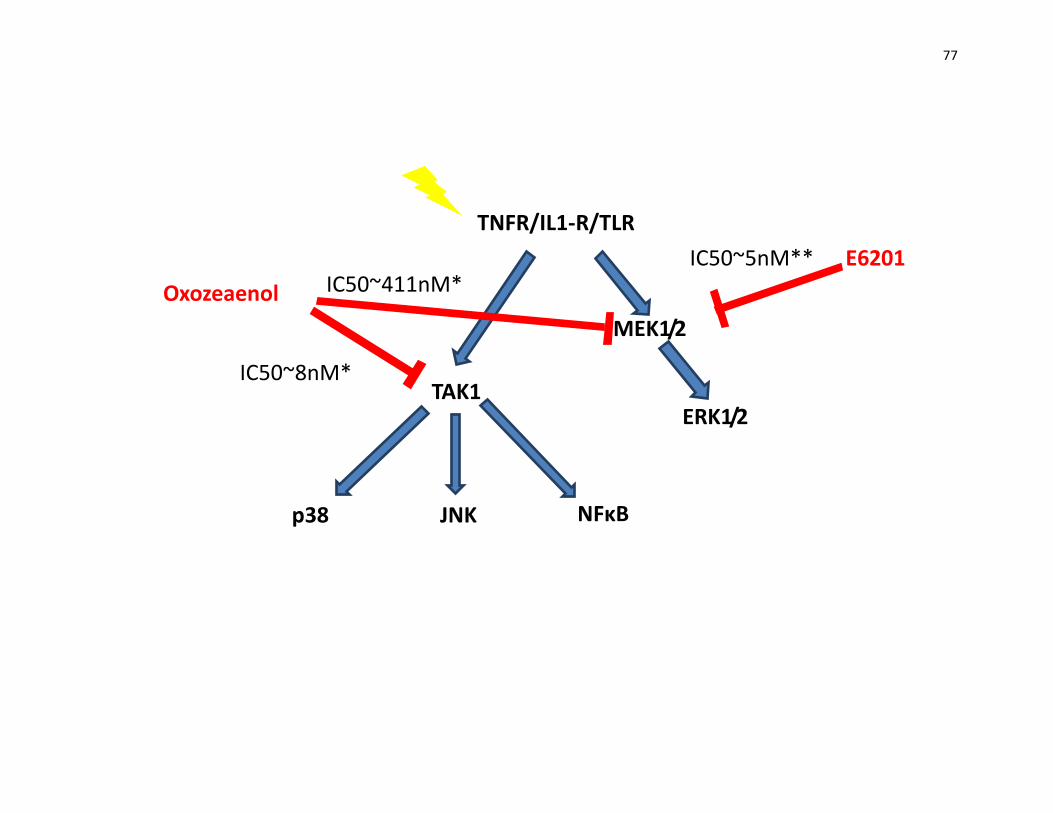
77
E6201
TNFR/IL1-R/TLR
TAK1
MEK1/2
ERK1/2
p38 NFκB
JNK
Oxozeaenol
IC50~411nM*
IC50~8nM*
IC50~5nM**

78
phosphorylation was determined by immunoblotting for phosphorylated p38, JNK, ERK1/2,
NFκB and total level of IκB. Both E6201 and Oxozeaenol were found to inhibit the
phosphorylation of all of the above targets. However, Oxozeaenol exhibited a 5-10 fold better
inhibition of phosphorylation compared to E6201 (Figure 18A, B). For example, Oxozeaenol
abolished phosphorylation of p38 and JNK at 500nM while this could only be observed at 5µM
of E6201. Moreover, Oxozeaenol also exhibited better inhibition of the phosphorylation of
ERK1/2 compared to E6201. This was unexpected as E6201 was proposed to be a more potent
inhibitor of the MEK/ERK pathway (IC50 of 5.1nM compared to 411nM of Oxozeaenol) (Figure
17).
The difference in inhibition of phosphorylation of NFκB was not clear; neither of the two
compounds seemed to completely inhibit NFκB phosphorylation (Figure 18A, B). However, the
re-appearance of the total level of IκB showed a significant difference between the effects of
Oxozeaenol and E6201. IκB is an inhibitor of NFκB that is targeted for degradation during
NFκB activation (Sakurai H, 2012); thus, the restoration of the total level of IκB can be used as
an indicator of the inhibition of NFκB activation. As shown in Figure 18, the level of IκB
restored with the treatment of 1µM of Oxozeaenol was much higher than that with the treatment
of E6201 at 10µM. This indicated a more than 10-fold greater potency for the inhibition of this
pathway. Nevertheless, this difference in potency cannot explain why E6201 did not rescue
∆F508-CFTR because this compound was used at much higher concentrations in both Cellomics
and Ussing chamber assays (20 and 50µM respectively). There might be other different
pathways/mechanism involved, and further investigation is therefore required.

79
Figure 18: Both (5Z)-7-Oxozeaenol and E6201 inhibit the phosphorylation of similar
downstream signaling targets. Both compounds exhibit inhibition of phosphorylation of p38,
ERK1/2, JNK, NFκB in (A) BDMM and (B) RAW264.7 cells stimulated with 100ng/mL LPS
after 1h treatment with the indicated concentrations of either (5Z)-7-Oxozeaenol or E6201. Cells
were lysed 20 min after stimulation and analyzed by immunoblotting with the indicated
antibodies.

80
(B) RAW 264.7
(A) BDMM

81
V. Validation of hits of esi-RNA screen
To complement the kinase inhibitor screen above, an esi-RNA screen was performed to
identify kinases that usually suppressed the rescue of ∆F508-CFTR. Using the same technique as
in the former screen, a library of about 900 esiRNAs targeting primarily kinases and other related
proteins (provided by L. Pelletier, Mt.Sinai, Toronto) was used to knock down target genes.
Among the hits from this screen, 21 genes (Table II) were chosen to be validated by using a
different RNAi technique, shRNA. This would also reduce the chances of having off-target
effects when knocking down target genes.
In the Cellomics assays, since fluorescence-tagged shRNAs could not be used, a panel of
about 200 different untagged TRC-shRNA constructs targeting these 21 genes (multiples clones
for each gene) was used. These constructs were transfected intoHEK293GT cells stably
expressing ∆F508-CFTR and the halide-sensitive YFP mutant, and then subjected to Cellomics
assays to determine their ability to rescue ∆F508-CFTR function. In parallel, qPCR was
performed to determine the knock-down efficiency of these constructs.
To examine the rescue of maturation of ∆F508-CFTR using Western blot analysis,
pGIPZ-shRNA (with GFP tag) constructs were used to knock down target genes in HEK293GT
stably expressing ∆F508-CFTR. The GFP allowed easy confirmation of transfection efficiency.
After transfection, cells were placed in selection media (supplemented with 8µg/mL Puromycin)
for 3 days to obtain the highest transfection efficiency possible (about 80-90% transfection
efficiency).
The TRC clones yielded varied degrees of knock down, but most of the constructs
resulted in more than 50% knock-down of target genes (Figure 19A). In the Cellomics assays, 14

82
Table II: Hit genes of the siRNA screen.
Gene Symbol Description
BRAF v-raf murine sarcoma viral oncogene homolog B1
CAMK2B Calcium/calmodulin-dependent protein kinase II beta
CDK10 Cyclin-dependent kinase 10
CLK3 CDC (cell division cycle)-like kinase 3, dual specificity protein kinase
DTYMK Deoxythymidylate kinase (thymidylate kinase)
DUSP22 Dual specificity phosphatase 22
ERN1 Endoplasmic reticulum to nucleus signaling 1
FGFR1 Fibroblast growth factor receptor 1
FLJ32685 NEK10, NIMA (never in mitosis gene a)-related kinase 10
IPMK Inositol polyphosphate multikinase
MAP3K13 Mitogen-activated protein kinase kinase kinase 13
MET Met proto-oncogene (hepatocyte growth factor receptor)
PANK1 Pantothenate kinase 1
PANK4 Pantothenate kinase 4
PCK2 Phosphoenolpyruvate carboxykinase 2 (mitochondrial)
PRKAR2B Protein kinase, cAMP-dependent, regulatory type II beta
RIPK4 Receptor-interacting serine-threonine kinase 4
RPS6KC1 52 kDa ribosomal protein S6 kinase, S6K-delta-1
SHPK sedoheptulokinase
SOCS1 Suppressor of cytokine signaling 1

83
Figure 19: Validation of hits of the esiRNA screen by Cellomics assays. 200 TRC-shRNA
clones were used to knock down 21 hit proteins of the esiRNA screen. For each target, result of
one representative shRNA clone is presented. (A) For Cellomics assays, average normalized
fluorescence intensity values of HEK293GT cells stably expressing ∆F508-CFTR and mutant
eYFP (H148Q/I152L) that were transfected with TRC-shRNA constructs targeting the indicated
proteins. After transfection, cells were incubated with media containing 5μg/mL Puromycin for 3
days. Cells were then used in Cellomics assays to measure rescue of ∆F508-CFTR [performed
by Agata Trzcinska-Daneluti]. (B) Degrees of knock down of the corresponding shRNA clones
determined by qPCR are shown. Data are means ± S.E. of 3 repeats.
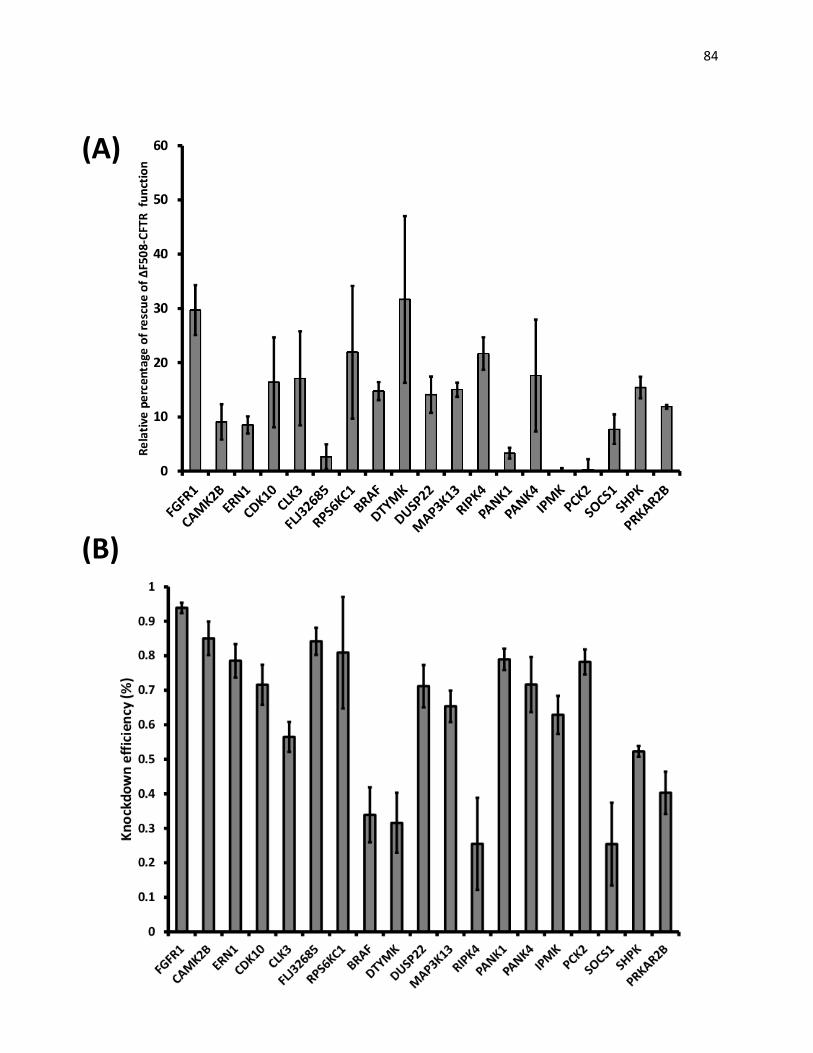
84
(B)
(A)

85
genes, when knocked down by shRNA, produced reproducible rescue of CFTR function (15-
30% increase in channel activity).These included genes encoding for the RTK FGFR1, the Raf
kinase BRAF and the MAPK kinase kinase MAP3K13 of the Ras/Raf/Mek/Erk pathway. Other
hits included genes that encoded for the receptor interacting protein kinase RIPK4, the
thymidylate kinase DTYMK, and the S6 ribosomal protein RPS6KC1 (Figure 19A). The result
for the knock down of MET was not included because the knock down was lethal to the cells,
and the high quenching of fluorescence signal could have been due to the loss of integrity of
cells. In Western blot analysis, most of the pGIPZ constructs had lower degrees of knock down
compared to the representative TRC clones. Most of the PGIPZ constructs resulted in less than
50% of knock down of the target genes (Figure 19B, 20B). However, 9 genes, when knocked
down, still exhibited significant improvements in the appearance of band C (Figure 20A), among
which, several genes such as RPS6KC1, FGFR1, DTYMK, RIPK4, were also confirmed in the
Cellomics assays.
The results from this screen provided complementary data to the kinase inhibitors screen.
For example, from the kinase inhibitor screen, inhibitors of FGFRs showed robust rescue of
∆F508-CFTR function. From the esi-RNA screen, knocking down FGFR1 improved both the
function of CFTR in the Cellomics assays and the appearance of mature band C on Western blot.
Other hits that were confirmed with shRNA were BRAF and MAP3K13, which belonged to the
RAS/RAF/MEK/ERK pathway, one of the four major pathways inhibited by the top hits from
the inhibitors screen (Table I) (Roring and Brummer, 2012; Craig et al, 2008). Other interesting
hits were PRKAR2B gene encoding one of the four regulatory domains of PKA (Yu et al, 2012),
which is a major kinase that phosphorylates and regulate CFTR channel activity, and IPMK,
which is involved in PI3K/Akt pathway downstream of FGFR (Lee et al, 2012).

86
Figure 20: Validation of hits of the esiRNA screen by immunoblotting.HEK293GT cells
expressing ∆F508-CFTR were transfected with either scrambled shRNA or pGIPZ-shRNA
constructs targeting the indicated proteins. 48h after transfection, cells were placed under
Puromycin (8μg/mL) selection for 3 days to obtain the highest transfection efficiency. (A) Cells
were then lysed and analyzed by immunoblotting to detect the appearance of band C. Treatment
at 27oC was used as a positive control. (B) qPCR was performed in parallel to quantify the
degree of knock down of each specific construct. Results are shown as means ± S.E of 3 replica.
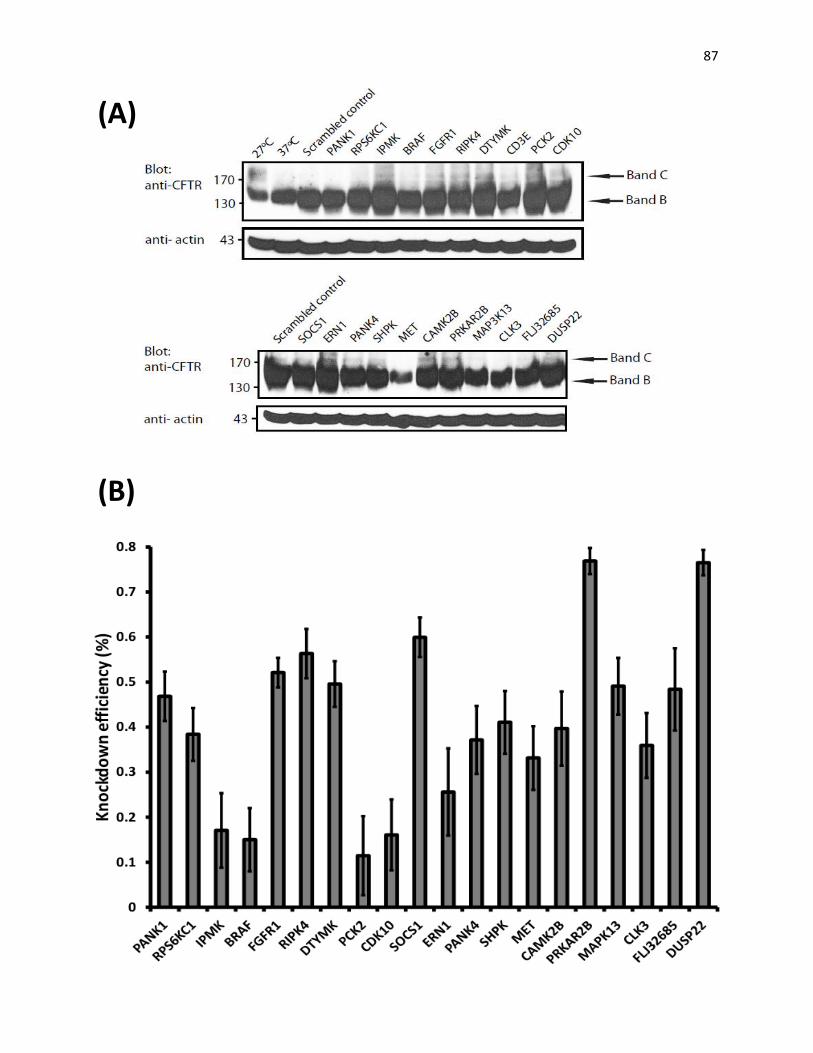
87
(B)
(A)

88
In summary, the work presented in this thesis demonstrates the rescue of ∆F508-CFTR
by several kinase inhibitors in HEK293GT, BHK and MDCK cells stably expressing the mutant
protein. Most importantly, these compounds also facilitated functional rescue of ∆F508-CFTR in
HBE cells obtained from CF patients. Oxozeaenol was one of the top hit compounds;
unfortunately, its derivative, E6201, which has passed phase II clinical trials for the treatment of
psoriasis, did not yield any rescue of ∆F508-CFTR. However, since some of the other top hit
compounds are clinically available or in clinical trials for the treatment of cancer or
inflammation, their use for the treatment of CF can be expedited. The esiRNA screen provided
complimentary data to the kinase inhibitors screen. Results from both screens suggested several
signaling pathways, especially FGFR1 and its downstream signaling pathways, that might be
involved in the trafficking of ∆F508-CFTR. However, the exact mechanism/pathway of the
rescue of ∆F508-CFTR remains to be explored.

89
Chapter 4
Discussion

90
I. Kinase inhibitor screen
In this thesis, I present the results of validations for the hits of a screen of kinase
inhibitors that can correct ∆F508-CFTR. The library of kinase inhibitors used in the screen was
biased towards compounds that are clinically available or in clinical trials for treatment of
cancer, inflammation, or other diseases. This approach provides us two main advantages. First,
once proven effective, these compounds can be quickly moved into clinical trials for the
treatment of ∆F508-CFTR. Secondly, since most of the targets of these inhibitors are known,
signaling pathways involved in the rescue of ∆F508-CFTR can be identified, which might lead to
compounds that yield better rescue or are better suited for treatment in patients.
Different approaches were taken to validate the hits of the kinase inhibitors screen.
Immunoblotting and flow cytometry were used to demonstrate rescues of the maturation and
trafficking of ∆F508-CFTR to the cell surface, respectively. Moreover, to validate the functional
rescue of these inhibitors, functional assays using Isc analyses by Ussing chambers were carried
out in both MDCK cells expressing ∆F508-CFTR and, most importantly, HBE cells obtained
from ∆F508-CFTR patients. The results from HBE cells revealed that the rescue of CFTR
activity (assessed as ∆Isc) in ∆F508-CFTRpatient samples treated with several of the inhibitors
was about 10-30% of that in WT-CFTR cells. This was a significant rescue because it had been
estimated that a partial rescue of ∆F508-CFTR, 10-25%, could restore the airway epithelial
function in patients (Johnson et al., 1992; Farmen et al. 2005; Zhang et al., 2009). From these
assays, however, we could not conclude if any of these inhibitors also act as potentiators of
∆F508-CFTR.

91
In addition, using the data from Isc analyses by Ussing chambers in MDCK cells, the
EC50 of most of the top hit inhibitors identified were determined to be in the nanomolar range.
This suggests that these inhibitors and their derivatives are very promising drugs to be used for
the treatment of CF. However, Rabeh et al. have recently reported that it might require a more
complex approach to give a complete rescue of ∆F508-CFTR. The authors conclude that it is
necessary to correct the stability of both NBD1 and NBD1-MSD2 interface to yield a complete
rescue of ∆F508-CFTR (Rabeh et al., 2012). Therefore, a combination of drugs might be
required to provide a better treatment for ∆F508-CFTR.
The fact that these kinase inhibitors could rescue ∆F508-CFTR suggests that the target
kinases and the downstream signaling pathways commonly inhibit the trafficking of the mutant
CFTR. Several of the compounds, such as SU5402, SU6668, PD173074, Ki8751, are inhibitors
of receptor tyrosine kinase (RTKs) (VEGFR, FGFR, PDGFR). In addition, besides being the
target of several of these inhibitors, FGFR1, when knocked down by shRNA, also led to the
rescue of ∆F508-CFTR. This further supports the possibility that FGFR1, or other RTKs, usually
play a negative role in the maturation of ∆F508-CFTR. However, further investigation is
required to determine the exact mechanism of how inhibiting these RTKs leads to the rescue of
∆F508-CFTR.
Moreover, other compounds, such as (5Z)-7-Oxozeaenol, RDEA-119, PD0325901
specifically target proteins that are downstream of these receptors, such as components of the
MAPK pathways (e.g. Raf, MEK, ERK, p38). Inhibitors of GSK3β in the Wnt and insulin
pathways, such as Kenpaullone and GSK3β Inhibitors II, and inhibitors of Akt in the
PI3K/Akt/mTOR pathway, such as FPA124 and 10-DEBC, were also identified in the screen
(table I). In summary, these inhibitors target four major pathways: 1) Ras/Raf/MEK/ERK, 2)

92
Wnt/GSK-3β, 3) TAK1/p38, and 4) PI3K/Akt/mTOR. Interestingly, the Ras/Raf/MEK/ERK
pathway has been shown recently to be inhibited by STAT1, which was shown to promote the
rescue of ∆F508-CFTR in a previous screen performed by our lab (Trzcinska-Daneluti et al.,
2009). This further supports the conclusion that at least one of these pathways normally inhibits
the trafficking of ∆F508-CFTR. These inhibitors could facilitate the rescue of ∆F508-CFTR
probably by affecting the function or expression level of certain chaperones involved in the
folding and trafficking of ∆F508-CFTR.
The first two pathways, or ERK1/2 and GSK3β to be specific, have been shown to be
involved in suppressing the activity of heat shock factor 1 (HSF1), which is a transcription factor
that induces the expression of heat shock proteins (HSPs) such as Hsp70, Hsp27, CRYAB (Bao
and Liu, 2009; Pirkkala et al., 2001). HSF1 exists in an inactive monomeric form that is
maintained by forming a complex with the chaperone Hsp90 (Zou et al., 1998). During heat
shock, HSF1 exists in a complex with Hsp70 and Hsp40, which act in a negative feedback to
maintain a proper folding state of proteins (Hayashida et al., 2006). Activation of HSF1 is
facilitated through the hyper-phosphorylation of serine residues (Kline and Morimoto, 1997).
However, phosphorylation of specific serine residues represses the transcription activity of HSF1
(Anckarand Sistonen, 2011). In this way, ERK1/2 exhibits its inhibitory role by phosphorylating
HSF1 on Ser307, which primes the transcription factor for the second phosphorylation on Ser303
by GSK3. These simultaneous phosphorylations repress HSF1 function and inhibit subsequent
expression of HSPs (Chu et al., 1996).
Inhibiting ERK1/2 and/or Gsk3β can relieve the inhibition on HSF1 and, as a
consequence, might induce elevated expression levels of HSPs. This event might, in turn, result
in proper folding and rescue of ∆F508-CFTR, since HSPs are chaperones that promote the

93
folding of proteins and prevent the formation of aggregation (Ellis, 1987). For example, Hsp70,
one of the chaperones that are upregulated following activation of HSF1 (Silver and Noble,
2012), has been shown to promote ∆F508-CFTR maturation (Meacham et al., 1999). In our
previous screen, Hsp70- related proteins such as HSPA4 and CRYAB were also identified as
correctors of ∆F508-CFTR (Trzcinska-Daneluti et al., 2009). Moreover, Calamini et al. showed
that small molecules that acted as proteostasis regulators and induced HSF1-dependent
chaperone expression could rescue ∆F508-CFTR (Calamini et al., 2011). These provide further
evidence supporting the notion that the rescue facilitated through inhibiting these two pathways
works through activating HSF1 and upregulating the expression of chaperones.
Furthermore, HSF1 can also be regulated by phosphorylation on other sites. It has been
shown to be directly phosphorylated by the MAPK-activated protein kinase 2 (MK2) on Ser121
(Wang et al., 2005). This phosphorylation inhibits transcriptional activity of HSF1 by reducing
its binding to the promoters of target genes encoded for HSPs and cytokines, and promoting the
binding between HSF1 and its repressor Hsp90. Interestingly, MK2 is one of the downstream
substrates that are directly phosphorylated and activated by p38 in the TAK1/p38 pathway, one
of the pathways identified in the screen (Freshney et al., 1994; Rouse et al., 1994; Rogalla et
al, 1999). Therefore, the rescue of ∆F508-CFTR by inhibitors of this pathway might also be
facilitated through relieving the inhibition on HSF1 by MK2.
However, the first and most notable substrate of p38/MK2 pathway to be identified is the
small heat shock protein Hsp27 (Stokoe et al., 1993), which functions as ATP-independent
chaperone that not only promotes protein folding but also targets unfolded protein for
proteasomal degradation (Jakob et al., 1993; Kostenk and Moens, 2009; Garrido et al., 2012).
The activity and cellular localization of Hsp27 are determined by its ogliomerization state, which

94
is dependent on its phosphorylation (Kostenk and Moens, 2009). Unphosphorylated Hsp27 forms
large ogliomers of up to 40 monomers, while phosphorylation, mainly by MK2, favors the
formation of smaller ogliomers (Rogalla et al., 1999; Parcellier et al., 2005; Kostenk and Moens,
2009). This is a very dynamic process that is crucial for the activity of Hsp27, since the
ogliomerization status modulates the affinity of this chaperone for its substrates (Garrido et al.,
2012).
Inhibiting the p38/MK2 pathway, therefore, terminates the phosphorylations on Hsp27,
and thus favors the large ogliomeric state. Since inhibitors of p38 pathway such as Oxozeaenol
or SKF86002 promote the rescue of ∆F508-CFTR in my study, it suggests that the smaller
ogliomeric states of Hsp27 might have a negative role in the trafficking of the mutant CFTR,
probably by directing the misfolded protein for degradation. Supporting this conclusion is the
fact that it has been shown that large ogliomers of Hsp27 are essential for its chaperone activity,
while the smaller, phosphorylated ogliomers lose their chaperone function and accelerate
degradation of certain proteins (Rogalla et al., 1999; Parcellier et al., 2006). Moreover, Hsp27
was identified as a component of the CFTR interactome (Wang et al., 2006), and recently was
reported to target ∆F508-CFTR for degradation via a SUMO-dependent pathway (Ahner et al.,
2012). In this study, Ahner et al. showed that overexpressed Hsp27, in a complex with the
SUMO E2 conjugating enzyme Ubc9, preferentially promoted SUMOylation of ∆F508-CFTR,
which was then recognized and targeted for degradation by RNF4, a SUMO-targeted ubiquitin
ligase. However, the authors did not assess the effect of different ogliomer sizes and
phosphorylation status of Hsp27 on the maturation of ∆F508. This is an interesting angle to be
further explored.

95
The role of the PI3K/Akt pathway in ∆F508-CFTR trafficking, or how inhibition of this
pathway leads to the rescue of the mutant CFTR, still requires further investigation. Several
reports actually suggest a possible positive role of Akt in CFTR trafficking. For example, Tuo et
al. showed a positive involvement of the PI3K/Akt pathway in trafficking of WT-CFTR to the
plasma membrane (Tuo et al., 2009). Moreover, Akt has been shown to inhibit GSK3β and thus
contribute to the activation of HSF1 (Bijur and Jope, 2000), which, as mentioned earlier, might
induce the expression of chaperones and promote ∆F508-CFTR trafficking and maturation.
However, the PI3K/Akt pathway has also been shown to induce the expression of Hsp27 (Takai
et al., 2006), and, thus, could exhibit its negative regulation of ∆F508-CFTR trafficking through
this mechanism. Moreover, PI3K/Akt actually decreases the plasma membrane availability of
aquaporin-2 and norepinephrine transporter (Jung and Kwon, 2010; Robertson et al., 2010),
suggesting a possible negative role of this pathway in the trafficking of membrane proteins.
Therefore, the exact mechanism for how the inhibitors of PI3K/Akt pathway rescue ∆F508-
CFTR requires further investigation.
II. E6201 and(5Z)-7-Oxozeaenol
Oxozeaenol was one of the top hits of the kinase inhibitors screen. It showed rescue of
ΔF508-CFTR maturation and function in all of our validation approaches (Table 1). However,
since Oxozeaenol is unstable in plasma, its therapeutic utilization is limited (Goto et al., 2009).
In 2009, the Eisai group in Japan identified a derivative of Oxozeaenol, E6201, which now has
gone to phase I trials for the treatment of cancer and phase II trials for the treatment of psoriasis
(Shen Y. et al., 2010; Muramoto K. et al, 2010). We obtained this compound from Eisai and

96
tested it for correction of ΔF508-CFTR using different methods, as in the kinase inhibitors
screen. Unfortunately, it did not yield any significant rescue of ΔF508-CFTR in any of assays
performed, and seemed to have an adverse effect at higher concentrations.
We attempted to determine the possible signaling pathways used by Oxozeaenol in the
rescue of ΔF508-CFTR by examining the different effects of these two compounds on
downstream signaling pathways. Phosphoproteins analysis was performed in BMM and
RAW264.7 cells stimulated with LPS. Using this approach, Oxozeaenol was found to exhibit a
5-10 fold more potent inhibition of phosphorylation of p38, JNK, ERK1/2, and NFκB, compared
to E6201. However, at the higher doses we used, E6201, like its parental compound, should have
inhibited all of the above signaling pathways and hence promote the rescue of ΔF508-CFTR.
One possible explanation for the lack of effect of E6201 might be that at the higher doses at
which it can actually inhibit these signaling pathways, it is also toxic to cells and/or has off-
target effects. In addition, it is possible that Oxozeanol rescued ΔF508-CFTR by inhibiting
pathway(s) downstream of TAK1 that is (are) not inhibited by E6201, which would be important
to investigate. Lastly, there is a possibility that Oxozeanol might have rescued ΔF508-CFTR by
direct binding to and stabilizing this mutant CFTR protein rather than affecting downstream
signaling pathways. This can be tested by using the iodide flux assays on purified CFTR that was
recently developed by the Bear lab (Eckford et al., 2012).
III. esiRNA screen
As a complement to the kinase inhibitor screen, another screen was performed to identify
proteins that suppress the trafficking of ΔF508-CFTR to the plasma membrane. This was done

97
using esiRNA to knock down kinases and related proteins in HEK293GT cells expressing the
mutant eYFP (H148Q/I152L) and ΔF508-CFTR, before subjecting the cells in Cellomics assays
to assess the rescue of ΔF508-CFTR activity in these cells. The top hits (21 genes/proteins, Table
II) were re-tested with another type of RNAi, shRNA (from the TRC collection), to ensure the
observed rescue in the original esiRNA screen was not due to off-target effects. In parallel, GFP-
tagged pGIPZ-shRNA constructs were used to test the effect of knocking down these genes on
the maturation of ΔF508-CFTR by immunoblotting. In both cases, qPCR were performed to
check the degrees of knockdown of the genes of interest. Knocking down of several of these
genes, such as FGFR1, and DTYMK, led to a rescue of up to 30% of CFTR activity in the
Cellomics assays, and an increase in the maturation of CFTR (increase in the appearance of band
C).
The results of the reevaluations performed above lend further support to the results of the
kinase inhibitor screen. Several of the hits are kinases that either are direct targets or are involved
in the signaling pathways inhibited by the kinase inhibitors in the original kinome inhibitors
screen. For example, IPMK (Inositol Polyphosphate Multikinase) is known to act as an important
PI3K that activates Akt (Lee et al., 2012). The fact that knocking down IMPK could rescue
∆F508-CFTR supports the result of the kinase inhibitors screen which suggested a negative role
of the PI3K/Akt pathway in the biogenesis of ∆F508-CFTR. Moreover, FGFR1 is also the target
of several kinase inhibitors we identified, such as SU5402, SU6668, and PD173074(Table I).
FGFR activation leads to the activation of several downstream signaling pathways such as the
Ras/Raf/MEK/ERK cascade, PI3K/Akt and p38 pathways (Javerzat et al., 2002; Turner and
Grose, 2010; Haugsten et al., 2010). As noted previously, these pathways might suppress the
trafficking and maturation of ΔF508-CFTR probably by affecting the activity of HSF1 and

98
Hsp27. Therefore, inhibiting FGFR1 or other downstream effectors involved in these signaling
pathways might relieve this suppression.
Indeed, several hits, such as BRAF and MAP3K13, are directly involved in the
Ras/Raf/MEK/ERK pathways, one of the four main signaling pathways identified (Table 1).
Both of them belong to the MAPK kinase kinase family, which is the first protein to be activated
in the MAPK signaling cascade (Kim and Choi, 2010). Specifically, BRAF is one of the three
isoforms of the Raf kinase family, an important component of the Ras/Raf/MEK/ERK pathway
that acts downstream of Ras and activates ERK through MEK kinase (Craig et al, 2008;
Roskoski , 2010), while MAP3K13 plays a role in activating the JNK and NFκB but not ERK
pathways (Ikeda et al, 2001; Masaki et al., 2003).
Other hits confirmed the involvement of HSF1 and Hsp27 in the rescue of ΔF508-CFTR
by the kinase inhibitors. One notable hit gene is RPS6KC1, which belongs to the S6 ribosomal
kinase family. Another member of this family, the 90-kD ribosomal S6 kinase RSK2, is activated
downstream of ERK and plays a central role in transmitting FGFR-driven signaling pathways
(Anjum and Blenis, 2008). Moreover, RSK2 has been shown to repress the activity of HSF1
(Wang et al., 2000). However, whether RPS6KC1 also inhibits HSF1and suppresses the
maturation of ΔF508-CFTR requires further study. Another hit gene, CAMKIIB, encodes for the
beta subunit of the Ca2+
/calmodulin-dependent protein kinase (CAMKII). Following the
activation of the Wnt pathway and the release of Ca2+
from the intracellular stores, CAMKII is
known to activate TAK1 (Kuhl et al., 2000; Ishitani et al., 2003), and thus leads to
phosphorylation of Hsp27 through the TAK1/p38 pathway (Cai et al., 2008). Therefore, the
rescue of ΔF508-CFTR facilitated by knocking down this gene might work through affecting the
phosphorylation and activity of Hsp27, as discussed earlier.

99
In addition to the four major pathways, results from both screens also suggest a negative
role of the NFκB pathway toward the trafficking and maturation of ΔF508-CFTR. Hsp27 not
only promotes the degradation of ΔF508-CFTR, as mentioned before (Ahner et al., 2012), but
also activates the NFκB pathway via promoting the degradation of phosphorylated IκBα, an
inhibitor protein of NFκB (Parcellier et al., 2003). Moreover, our siRNA screen identified
RIPK4, or RIP4, which belongs to the Receptor Interacting Protein Kinase family that is known
to activate NFκB (Meylan et al., 2002). In CF airways, one factor contributing to the chronic and
hyper-inflammation is the elevated NFκB-mediated IL8 signaling (Tabary et al., 1998; Tabary et
al., 2001). This elevated activity might be due to the lack of inhibitory effect of CFTR on this
pathway in CF cells (Vij et al., 2009). However, whether and how the NFκB pathway inhibits
the trafficking of ΔF508-CFTR requires further investigation.
The results from the siRNA screen not only provide confirmation to the kinase inhibitor
screen, but also suggest new novel pathways that might be involved in the maturation of ΔF508-
CFTR. Other interesting hit genes that do not seem to be involved in any of the signaling
pathways mentioned are PRKAR2B and DTYMK. PRKAR2B encodes one isoform of the
regulatory subunit II of PKA. Upon the binding of cAMP, PKA is activated by the release of its
regulatory subunit, leading to the activation of the catalytic subunit. Therefore, knocking down
one of the regulatory subunits might lead to an increase in PKA activity (Brandon et al., 1997).
Moreover, cAMP/PKA stimulation is known not only to promote CFTR channel activity, but
also to enhance its trafficking to the cell surface and decrease its endocytosis from the plasma
membrane. Whether PRKAR2B promotes rescue of ΔF508-CFTR through this mechanism
remains to be investigated. DTYMK belongs to the family of thymidylate kinase (TMPK). This
kinase family is responsible for catalyzing dTMP to dTDP and dTTP, which is used for DNA

100
synthesis and repair (Huang et al., 1994). Since it plays a very important role in cell growth, it is
often targeted in treatment of cancer (Van Calenberghet al., 2012). However, its role in protein
synthesis and trafficking, if any, is unknown and needs to be explored.
Summary statement
In summary, the work presented in this thesis reveals that several kinase inhibitors can
work as novel correctors of ΔF508-CFTR. Since some of these inhibitors or their derivatives are
clinically available or are in clinical trials for the treatment of cancer or inflammation, their use
for the treatment of ΔF508-CFTR can be accelerated. Moreover, combined with the results of the
siRNA screen, we also identified signaling pathways that might normally suppress the trafficking
and maturation of ΔF508-CFTR. These can be used as targets to design or screen for better
compounds in the future, not only for the treatment of CF but also treatment of other diseases
caused by proteins that exhibit folding defects and are thus stuck in the ER.
FUTURE DIRECTIONS
I. Testing the effect of knocking down top hit genes on rescuing ∆F508-CFTR function
using Ussing Chambers:
To further validate the top hit genes from the siRNA screen, we want to assess the effect of
knocking down these genes in either cultured cells expressing ∆F508-CFTR or, ultimately, HBE
patient cells. Since the shRNA constructs used only target human genes, we are in the process of
generating a human CF cell line by stably expressing ∆F508-CFTR in CFBE410-, a cell line that
was generated by transformation of cystic fibrosis (CF) tracheo-bronchial cells. In the meantime,
since 11 of these genes have higher than 95% identity between human and canine, and thus the

101
shRNAs are compatible to be used in MDCK cells, we will try to knock down these in our
MDCK-∆F508-CFTR cell lines for initial testing.
II. Elucidate the mechanisms of rescuing ∆F508-CFTR by Oxozeaenol:
Both E6201 and Oxozeaenol inhibit similar pathways (at least at higher concentration), but
only Oxozeaenol rescued the mutant CFTR. Therefore, we want to further investigate the
mechanism through which Oxozeaenol rescues ∆F508-CFTR function. One possible explanation
is that Oxozeaenol can exhibit its rescue by directly binding to and promoting the folding of
∆F508-CFTR. This can be tested in collaboration with Christine Bear’s lab using a novel Iodide
flux assay developed recently by that group (Eckford et al, 2012).
III. Elucidate the pathways through which FGFR1 regulates the rescue of ∆F508-CFTR
We have found that inhibiting or knocking down FGFR1 or some of the components of its
downstream signaling pathways (i.e. RAS/RAF/MEK/ERK, PI3K/Akt pathways) can rescue
∆F508-CFTR. We hypothesize that this rescue is facilitated by affecting the chaperones that are
involved in the folding and trafficking of ∆F508-CFTR. Once we obtain HBE cells, we will treat
them for 48 hrs with SU5402, one of the top hits from our kinase inhibitors screen, which
inhibits mainly FGFR1. We will then extract RNA from these cells and use the qPCR array kit
designed to profile the expression levels of heat shock proteins and chaperones to check if there
is any chaperone that is up or down regulated. This will allow us to further investigate the
pathways involved.

102
SUMMARY
To identify compounds and drugs that rescue the trafficking defect of ∆F508-CFTR, our
lab performed a screen of a kinase inhibitor library biased toward small molecules that are
clinically available or in clinical trials for the treatment of cancer and inflammation using a
recently developed high-content Cellomics functional screen. I further validated the top hits of
the screen by: immunoblotting to test the appearance of mature ∆F508-CFTR (band C); flow
cytometry to detect the presence of ∆F508-CFTR at the cell surface; short-circuit current (Isc)
analysis in Ussing chamber to show the increased ∆F508-CFTR activity in MDCK cells stably
expressing the mutant CFTR; and most importantly, Isc analysis to demonstrate improved
∆F508-CFTR activity in Human Bronchial Epithelial (HBE) cells obtained from homozygote
∆F508-CFTR patients who had undergone lung transplant.
The results indicate that several inhibitors of receptor tyrosine kinases such as SU5402
and SU6668, which target FGFRs, VEGFR, and PDGFR, exhibited strong rescue of ∆F508-
CFTR. Moreover, prominent rescue was also observed with inhibitors of four major pathways:
Ras/Raf/MEK/ERK, TAK1/p38, Wnt/GSK-3β, and PI3K/Akt/mTOR. Several of these inhibitors
rescued ∆F508-CFTR in more than one validation approaches, such as (5Z)-7-Oxozeaenol,
SU5402, with EC50 mostly falling in the nanomolar range.
The effect of E6201, one derivative of Oxozeaenol, on the rescue of ∆F508-CFTR was
also examined using similar approaches. E6201 is currently in phase II clinical trials for the
treatment of psoriasis. Unfortunately, it did not exhibit rescue of ∆F508-CFTR in any of the
assays used. The phosphoprotein analysis revealed that E6201 inhibited the phosphorylation of
the same target proteins as Oxozeaenol, but at lower potency. This suggests a possibility that

103
Oxozeaenol could have rescued ∆F508-CFTR using a different mechanism (e.g. direct
binding)or by inhibiting a different signaling pathway that was not tested.
To identify pathways involved in the rescue of ∆F508-CFTR by these inhibitors, we also
performed a complementary siRNA screens for kinases and related proteins that suppress the
rescue. The top hits from this screen were preliminarily validated by immunoblotting. The results
revealed FGFR1, a target of several kinase inhibitors identified in the former screen, and several
kinases such BRAF and MAP3K13, which are involved in the signaling pathways downstream
of FGFRs. This suggests a possible role of FGFRs in regulating ∆F508-CFTR trafficking and
maturation, probably through affecting the expression of chaperones. In addition, other proteins
identified might reveal possible novel pathways involved in ∆F508-CFTR rescue. The exact
mechanisms of the rescue remain to be explored.
CONCLUSION
The results obtained in this study identify several kinase inhibitors that can rescue
ΔF508-CFTR, and suggest that use of compounds or drugs already in the clinic or in clinical
trials for other diseases can expedite delivery of treatment for CF patients.

104
REFERENCES
Ahner, A., Gong, X., Schmidt, B.Z., Peters, K.W., Rabeh, W.M., Thibodeau, P.H., Lukacs,
G.L., Frizzell, R.A. (2012) Small heat shock proteins target mutant CFTR for degradation via a
SUMO-dependent pathway. Mol Biol Cell [Epub ahead of print]
Alberti, S., Bohse, K., Arndt, V., Schmitz, A., Hohfeld, J. (2004) The cochaperone HspBP1
inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis
transmembrane conductance regulator.Mol Biol Cell15(9):4003-4010.
Aleksandrov, L, Aleksandrov, A , Riordan, JR (2008). Mg2+
-dependent ATP occlusion at the
first nucleotide-binding domain (NBD1) of CFTR does not require the second (NBD2). Biochem
J 416: 129–136.
Anckar, J. and Sistonen, L. (2011) Regulation of HSF1 Function in the Heat Stress Response:
Implications in Aging and Disease. Annu Rev Biochem80:1089–1115
Anderson, P. (2010) Emerging therapies in cystic fibrosis.Ther Adv Respir Dis4(3): 177-185
Anjum, R. andBlenis, J. (2008) The RSK family of kinases: emerging roles in cellular
signalling. Nat Rev Mol Cell Biol9(10):747-758.
Antunovic, S.S., Lukac, M., Vujovic, D. (2013) Longitudinal cystic fibrosis care. Clin
Pharmacol Ther 93(1):86-97.
Arndt, V., Daniel, C., Nastainczyk, W., Alberti, S., Hohfeld, J. (2005) BAG-2 acts as an
inhibitor of the chaperone-associated ubiquitin ligase CHIP.Mol Biol Cell16(12):5891-5900.
Baker, J.M., Hudson, R.P., Kanelis, V., Choy, W.Y., Thibodeau, P.H., Thomas,
P.J., Forman-Kay, J.D. (2007) CFTR regulatory region interacts with NBD1 predominantly via
multiple transient helices. Nat Struct Mol Biol14(8): 738-745
Banasavadi-Siddegowda, Y.K., Mai, J., Fan, Y., Bhattacharya, S., Giovannucci,
D.R., Sanchez, E.R., Fischer, G., Wang, X. (2011) FKBP38 peptidylprolyl isomerase promotes
the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum.
J Biol Chem286(50):43071-43080.
Bao, X. Q., and Liu, G. T. (2009) Induction of overexpression of the 27- and 70-kDa heat shock
proteins by bicyclol attenuates concanavalin A-Induced liver injury through suppression of
nuclear factor-kappaB in mice.Mol. Pharm. 75, 1180–1188
Bear, C.E., Li, C.H., Kartner, N., Bridges, R.J., Jensen, T.J., Ramjeesingh, M., Riordan,
J.R.(1992) Purification and functional reconstitution of the cystic fibrosis transmembrane
conductance regulator (CFTR).Cell68(4):809-18.
http://www.ncbi.nlm.nih.gov/pubmed?term=Thibodeau%20PH%5BAuthor%5D&cauthor=true&cauthor_uid=23155000
http://www.ncbi.nlm.nih.gov/pubmed?term=Thibodeau%20PH%5BAuthor%5D&cauthor=true&cauthor_uid=17660831

105
Becq, F., Mettey, Y., Gray, M.A., Galietta, L.J., Dormer, R.L., Merten, M., Metaes, T.,
Chappe, V., Marvingt-Mounir, C., Zegarra-Moran, O., Tarran, R., Bulteau, L., Dérand,
R., Pereira, M.M., McPherson, M.A., Rogier, C., Joffre, M., Argent, B.E., Sarrouilhe, D.,
Kammouni, W., Figarella, C., Verrier, B., Gola, M., Vierfond, J.M. (1999) Development of
substituted Benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride
channel. J Biol Chem274(39):27415-27425.
Bijur, G.N., and Jope, R.S.(2000) Opposing actions of phosphatidylinositol 3-kinase and
glycogen synthase kinase-3beta in the regulation of HSF-1 activity.J Neurochem 75(6):2401-
2408.
Bodas, M. and Vij, N. (2010) The NFκB signaling in cystic fibrosis lung disease:
pathophysiology and therapeutic potential. Discov Med9(47): 346-356
Bomberger, J.M., Barnaby, R.L., Stanton, B.A.(2009) The deubiquitinating enzyme USP10
regulates the post-endocytic sorting of cystic fibrosis transmembrane conductance regulator in
airway epithelial cells. J Biol Chem284(28):18778-89.
Bompadre, S.G., Sohma, Y., Li, M., Hwang, T.C. (2007) G551D and G1349D, two CF-
associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J Gen
Physiol 129(4): 285-298
Boucher, R.C. (2004)New concepts of the pathogenesis of cystic fibrosis lung disease. Eur
Respir J23(1): 146-158
Boyle MP, Bell S, Konstan MW et al. (2011) VX-809, an investigational CFTR corrector, in
combination with VX-770, an investigational CFTR potentiator,in subjects with CF and
homozygous for the F508del-CFTR mutation (abstract). Pediatr Pulmonol(suppl 34):287.
Brandon, E.P., Idzerda, R.L., McKnight, G.S. (1997) PKA isoforms, neural pathways, and
behaviour: making the connection. Curr Opin Neurobiol. 7(3):397-403.
Cai, H., Liu, D., Garcia, J.G. (2008) CaM Kinase II-dependent pathophysiological signalling in
endothelial cells. Cardiovac Res 77:30–34
Calamini, B., Silva, M.C., Madoux, F., Hutt, D.M., Khanna, S., Chalfant, M.A., Saldanha,
S.A., Hodder, P., Tait, B.D., Garza, D., Balch, W.E., Morimoto, R.I.(2011) Small-molecule
proteostasis regulators for protein conformational diseases.Nat Chem Biol 8(2):185-196
Chang, X.B., Tabcharani, J.A., Hou, Y.X., Jensen, T.J., Kartner, N., Alon, N., Hanrahan,
J.W., Riordan, J.R. (1993)Protein kinase A (PKA) still activates CFTR chloride channel after
mutagenesis of all 10 PKA consensus phosphorylation sites. J Biol Chem 268(15): 11304-11311.
Chanoux, R.A and Rubenstein, R.C. (2012) Molecular Chaperones as Targets to Circumvent
the CFTR Defect in Cystic Fibrosis. Front Pharmacol 3:137.

106
Cheng, S.H., Gregory, R.J., Marshall, J., Paul, S., Souza, D.W., White, G.A., O'Riordan,
C.R., Smith, A.E. (1990) Defective intracellular transport and processing of CFTR is the
molecular basis of most cystic fibrosis.Cell 63(4):827-34.
Cheng, S.H., Rich, D.P., Marshall, J., Gregory, R.J., Welsh, M.J., Smith, A.E. (1991)
Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR
chloride channel. Cell 66(5): 1027-1036
Cholon, D.M., O'Neal, W.K., Randell, S.H., Riordan, J.R., Gentzsch, M. (2010) Modulation
of endocytic trafficking and apical stability of CFTR in primary human airway epithelial
cultures.Am J Physiol Lung Cell Mol Physiol 298(3):L304-14.
Chu, B., Soncin, F., Price, B. D., Stevenson, M. A., and Calderwood, S. K. (1996) Sequential
phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses
transcriptional activation by heat shock factor-1. J. Biol. Chem. 271, 30847–30857
Clancy, J.P., Rowe, S.M., Accurso, F.J, Aitken, M.L., Amin, R.S., Ashlock, M.A.,
Ballmann, M., Boyle, M.P., Bronsveld, I., Campbell, P.W., De Boeck, K., Donaldson, S.H.,
Dorkin, H.L., Dunitz, J.M., Durie, P.R., Jain, M., Leonard, A., McCoy, K.S., Moss, R.B.,
Pilewski, J.M., Rosenbluth, D.B., Rubenstein, R.C., Schechter, M.S., Botfield, M., Ordoñez,
C.L., Spencer-Green, G.T., Vernillet, L., Wisseh, S., Yen, K., Konstan, M.W. (2012) Results
of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with
cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67(1):12-18.
Clarke, L.L. (2009) A guide to Ussing chamber studies of mouse intestine. Am J Physiol
Gastrointest Liver Physiol 296(6): G1151-1166.
Craig, E.A., Stevens, M.V., Vaillancourt, R.R., Camenisch, T.D.(2008) MAP3Ks as
centralregulators of cellfate during development.Dev Dyn 237(11):3102-3114.
Csanady, L., Chan, K.W., Nairn, A.C., Gadsby, D.C. (2005) Functional roles of nonconserved
structural segments in CFTR's NH2-terminal nucleotide binding domain. J Gen Physiol 125(1):
43-55
Davies, J.C., Alton, E.W.F.W., Bush, A. (2007) Cystic Fibrosis. BMJ 335(7632): 1255-1259
Davis, P.B. (2006) Cystic fibrosis since 1938.Am J Respir Crit Care Med 173(5): 475-482
Denning, G.M., Anderson, M.P., Amara, J.F., Marshall, J., Smith, A.E., Welsh, M.J. (1992)
Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-
sensitive. Nature 358(6389):761-764.
Devor, D.C., Singh, A.K., Lambert, L.C., DeLuca, A., Frizzell, R.A., Bridges, R.J. (1999)
Biocarbonate and chloride secretion in Calu-3 human airway epithelial cells. J Gen Physiol
113(5): 743-760

107
Du, K., Sharma, M., Lukacs, G.L.(2005) The DeltaF508 cystic fibrosis mutation impairs
domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol
12(1):17-25.
Dulhanty, A.M. and Riordan, J.R. (1994) Phosphorylation by cAMP-dependent protein kinase
causes a conformational change in the R domain of the cystic fibrosis transmembrane
conductance regulator. Biochemistry 33(13): 4072-4079
Dulhanty, A.M., Chang, X.B., Riordan, J.R. (1995). Mutation of potential phosphorylation
sites in the recombinant R domain of the cystic fibrosis transmembrane conductance regulator
has significant effects on domain conformation. Biochem. Biophys. Res. Commun. 206:207–214.
Eckford, P.D., Li, C., Ramjeesingh, M., Bear, C.E.(2012) Cystic fibrosis transmembrane
conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate
of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem
287(44):36639-36649.
Ellis, J. (1987) Proteins as molecular chaperones. Nature 328(6129): 378-379
Farmen, S.L., Karp, P.H., Ng, P., Palmer, D.J., Koehler, D.R., Hu, J., Beaudet, A.L.,
Zabner, J., Welsh, M.J. (2005) Gene transfer of CFTR to airway epithelia: low levels of
expression are sufficient to correct Cl- transport and overexpression can generate basolateral
CFTR. Am J Physiol Lung Cell Mol Physiol 289(6): L1123-1130.
Favia, M., Guerra, L., Fanelli, T., Cardone, R.A., Monterisi, S., Di Sole, F., Castellani, S.,
Chen, M., Seidler, U., Reshkin, S.J., Conese, M., Casavola, V.(2010) Na+/H+ exchanger
regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is
fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in
human airway CFBE41o- cells. Mol Biol Cell 21(1):73-86.
Freshney, N.W., Rawlinson, L., Guesdon, F., Jones, E., Cowley, S., Hsuan, J., Saklatvala,
J.(1994). Interleukin-1 activates a novel protein kinase cascade that results in the
phosphorylation of Hsp27. Cell 78:1039–1049
Gadsby, D.C. and Nairn, A.C. (1999) Control of CFTR channel gating by phosphorylation and
nucleotide hydrolysis.Physiol Rev 79: S77-S107
Gadsby, DC, Vergani, P & Csanady, L (2006). The ABC protein turned chloride channel
whose failure causes cystic fibrosis. Nature 440: 477–483.
Galietta, L.V., Jayaraman, S., Verkman, A.S. (2001) Cell-based assay for high-throughput
quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol
281(5):C1734-1742.

108
Garattini, E., Bilton, D., Cremona, G., Hodson, M. (2011) Adult cystic fibrosis care in the
21st century. Monaldi Arch Chest Dis 75(3):178-184.
Garrido, C., Paul, C., Seigneuric, R., Kampinga, H.H.(2012) The small heat
shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol 44(10):1588-1592.
Gentzsch, M., Dang, H., Dang, Y., Garcia-Caballero, A., Suchindran, H., Boucher, R.C.,
Stutts, M.J. (2010) The cystic fibrosis transmembrane conductance regulator impedes
proteolytic stimulation of the epithelial Na+ channel. J Biol Chem 285(42): 32227-32232
Gibson, R.L., Burns, J.L., Rmasey, B.W. (2003) Pathophysiology and management of
pulmonary infections in cystic fibrosis.Am J Respir Crit Care Med 168(8): 918-951.
Goto, M., Chow, J., Muramoto, K., Chiba, K., Yamamoto, S., Fujita, M., Obaishi, H., Tai,
K., Mizui, Y., Tanaka, I., Young, D., Yang, H., Wang, Y.J., Shirota, H., Gusovsky, F. (2009)
E6201 [(3S,4R,5Z,8S,9S,11E)-14-(ethylamino)-8, 9,16-trihydroxy-3,4-dimethyl-3,4,9,19-
tetrahydro-1H-2-benzoxacyclotetradecine-1,7(8H)-dione], a novel kinase inhibitor of mitogen-
activated protein kinase/extracellular signal-regulated kinase kinase (MEK)-1 and MEK kinase-
1: in vitro characterization of its anti-inflammatory and antihyperproliferative activities. J
Pharmacol Exp Ther 331(2):485-495.
Grove, D.E., Rosser, M.F., Ren, H.Y., Naren, A.P., Cyr, D.M.(2009) Mechanisms for rescue
of correctable folding defects in CFTRDelta F508.Mol Biol Cell20(18):4059-69.
Gunderson, K.L. and Kopito, R.R.. (1995)Conformational states of CFTR associated with
channel gating: the role of ATP binding and hydrolysis. Cell 82: 231-239
Haugsten, E.M., Wiedlocha, A., Olsnes, S., Wesche, J. (2010) Roles of fibroblast growth
factor receptors in carcinogenesis. Mol Cancer Res 8(11):1439-1452.
Hayashida, N., Inouye, S., Fujimoto, M., Tanaka, Y., Izu, H., Takaki, E., Ichikawa, H.,
Rho, J., Nakai, A. (2006) A novel HSF1-mediated death pathway that is suppressed by heat
shock proteins.EMBO J 25:4773–4783
Huang, S.H., Tang, A., Drisco, B., Zhang, S.Q., Seeger, R., Li, C., Jong, A. (1994)
Human dTMP kinase: gene expression and enzymatic activity coinciding with cell cycle
progression and cell growth. DNA Cell Biol 13(5):461-471.
Hwang ,T.C. and Sheppard, D.N. (2009) Gating of the CFTR Cl- channel by ATP-driven
nucleotide-binding domain dimerisation. J Physiol 587(10): 2151-2161
Hwang, T.C., Nagel, G., Nairn, A.C., Gadsby, D.C. (1994) Regulation of the gating of cystic
fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP
hydrolysis. Proc Natl Acad Sci USA 91(11): 4698-4702

109
Ikeda, A., Masaki, M., Kozutsumi, Y., Oka, S., Kawasaki, T.(2001) Identification and
characterization of functional domains in a mixed lineage kinase LZK.FEBS Lett 488(3):190-
195.
Ishitani, T., Kishida, S., Hyodo-Miura, J., Ueno, N., Yasuda, J., Waterman, M., Shibuya,
H., Moon, R.T., Ninomiya-Tsuji, J.,Matsumoto, K. (2003) The TAK1-NLK mitogen-activated
protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin
signaling. Mol Cell Biol 23(1):131-139.
Jakob, U., Gaestel, M., Engel, K., Buchner, J.(1993)Small heat shock proteins are molecular
chaperones.J Biol Chem 268(3): 1517-1520
Javerzat, S., Auguste, P., Bikfalvi, A. (2002) The role of fibroblast growth factors in vascular
development. Trends Mol Med 8(10): 483-489
Jia, Y., Matthews, C.J., Hanrahan, J.W. (1997) Phosphorylation by protein kinase C is
required for acute activation of cystic fibrosis transmembrane conductance regulator by protein
kinase A. J Biol Chem 272(8): 4978-4984
Johnson, L. G., Olsen, J. C., Sarkadi, B., Moore, K. L., Swanstrom, R., and Boucher, R. C.
(1992) Efficiency of gene transfer for restoration of normal airway epithelial function in cystic
fibrosis. Nat Genet 2: 21–25
Jung, H. J., and Kwon, T. H. (2010) Membrane Trafficking of Collecting Duct Water Channel
Protein AQP2 Regulated by Akt/AS160.Electrolyte Blood Press 8:59–65
Kalid, O., Mense, M., Fischman, S., Shitrit, A., Bihler, H., Ben-Zeev, E., Schutz, N.,
Pedemonte, N., Thomas, P.J., Bridges, R.J., Wetmore, D.R., Marantz, Y., Senderowitz,
H.(2010) Small molecule correctors of F508del-CFTR discovered by structure-based virtual
screening.J Comput Aided Mol Des 24(12):971-991.
Kartner, N., Hanrahan, J. W., Jensen, T. J., Naismith, A. L., Sun, S., Ackerley, C. A.,
Reyes, E. F., Tsui, L.-C., Rommens, J. M., Bear, C. E., Riordan, J. R. (1991). Expression of
the cystic fibrosis gene in nonepithelial invertebrate cells produces a regulate anion conductance.
Cell 64: 681-691.
Kelley, T.J.andElmer, H.L.(2000) In vivo alterations of IFN regulatory factor-1 and PIAS1
protein levels in cystic fibrosis epithelium.J Clin Invest 106(3):403-410.
Kim, E.K. and Choi, E.J.(2010) Pathological roles of MAPK signaling pathways in human
diseases. Biochim Biophys Acta 1802(4):396-405.
Kline, M.P. and Morimoto, R.I (1997) Repression of the heat shock factor 1 transcriptional
activation domain is modulated by constitutive phosphorylation.. Mol Cell Biol 17:2107–2115

110
Kostenk, S., and Moens, U. (2009) Heat shock protein 27 phosphorylation: kinases,
phosphatases, functions and pathology. Cell Mol Life Sci 66(20): 3289-3307
Kreiselmeier, N.E., Kraynack, N.C., Corey, D.A., Kelley, T.J.(2003) Statin-mediated
correction of STAT1 signaling and inducible nitric oxide synthase expression in cystic fibrosis
epithelial cells.Am J Physiol Lung Cell Mol Physiol 285(6):L1286-1295.
Kühl, M., Sheldahl, L.C., Park, M., Miller, J.R., Moon, R.T. (2000) The Wnt/Ca2+ pathway:
a new vertebrate Wnt signaling pathway takes shape. Trends Genet 16(7):279-283.
Kumar, V., Schuck, E.L., Pelletier, R.D., Farah, N., Condon, K.B., Ye, M., Rowbottom, C.,
King, B.M., Zhang, Z.Y., Saxton, P.L., Wong, Y.N.(2012) Pharmacokinetic characterization of
a natural product-inspired novel MEK1 inhibitor E6201 in preclinical species.Cancer Chemother
Pharmacol 69(1):229-237.
Lee, JY, Kim, YR, Park, J, Kim, S. (2012) Inositol polyphosphate multikinase signaling in the
regulation of metabolism.Ann N Y Acad Sci 1271:68-74.
Leier, G., Bangel-Ruland, N., Sobczak, K., Knieper, Y., Weber, W.M. (2012) Sildenafil acts
as potentiator and corrector of CFTR but might be not suitable for the treatment of CF lung
disease. Cell Physiol Biochem 29(5-6): 775-790.
Li, C., Ramjeesingh, M., Wang, W., Garami, E., Hewryk, M., Lee, D., Rommens, J.M.,
Galley, K., Bear, C.E.(1996) ATPase activity of the cystic fibrosis transmembrane conductance
regulator.J Biol Chem 271(45):28463-28468
Loo, T.W., Bartlett, M.C., Clarke, D.M. (2008) Correctors promote folding of the CFTR in the
endoplasmic reticulum. Biochem J 413(1):29-36.
Loo, M.A., Jensen, T.J., Cui, L., Hou, Y., Chang, X.B., Riordan, J.R. (1998) Perturbation of
Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by
the proteasome. EMBO J 17(23):6879-6887.
Lukacs, G.L. andVerkman, A.S.(2012) CFTR: folding, misfolding and correcting the ΔF508
conformational defect. Trends Mol Med 18(2):81-91.
Lukacs, G.L., Chang, X.B., Bear, C., Kartner, N., Mohamed, A., Riordan, J.R., Grinstein,
S. (1993) The delta F508 mutation decreases the stability of cystic fibrosis transmembrane
conductance regulator in the plasma membrane. Determination of functional half-lives on
transfected cells. J Biol Chem 268(29):21592-21598.
Lukacs, G.L., Mohamed, A., Kartner, N., Chang, X.B., Riordan, J.R., Grinstein, S.(1994)
Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the
endoplasmic reticulum and requires ATP. EMBO J 13(24):6076-6086.

111
Luz, S., Kongsuphol, P., Mendes, A.I., Romeiras, F., Sousa, M., Schreiber, R., Matos, P.,
Jordan, P., Mehta, A., Amaral, M.D., Kunzelmann, K., Farinha, C.M.(2011) Contribution of
casein kinase 2 and spleen tyrosine kinase to CFTR trafficking and protein kinase A-induced
activity.Mol Cell Biol 31(22): 4392-4404
Lyczak, J.B., Cannon, C.L., Pier, G.B. (2002) Lung Infections Associated with Cystic Fibrosis.
Clin Microbiol Rev 15(2): 194–222.
Masaki, M., Ikeda, A., Shiraki, E., Oka, S., Kawasaki, T. (2003) Mixed lineage kinase LZK
and antioxidant protein-1 activate NF-kappaB synergistically. Eur J Biochem 270(1):76-83.
Meacham, G.C., Lu, Z., King, S., Sorscher, E., Tousson, A., Cyr, D.M. (1999) The Hdj-
2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J 18(6):1492-1505.
Meacham, G.C., Patterson, C., Zhang, W., Younger, J.M., Cyr, D.M. (2001) The Hsc70 co-
chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3(1):100-
105.
Mendoza, J.L., A. Schmidt, Q. Li, E. Nuvaga, T. Barrett, R.J. Bridges, A.P. Feranchak,
C.A. Brautigam, Thomas P.J. (2012)Requirements for efficient correction of ΔF508 CFTR
revealed by analyses of evolved sequences. Cell 148:164–174.
Meylan, E., Martinon, F., Thome, M., Gschwendt, M., Tschopp, J. (2002) RIP4 (DIK/PKK),
a novel member of the RIP kinase family, activates NF-kappa B and is processed during
apoptosis. EMBO Rep 3(12):1201-1208.
Mornon, J.P., Lehn, P., Callebaut, I. (2008)Atomic model of human cystic fibrosis
transmembrane conductance regulator: membrane-spanning domains and coupling interfaces.
Cell Mol Life Sci 65(16): 2594-2612
Mornon, J.P., Lehn, P., Callebaut, I. (2009)Molecular models of the open and closed states of
the whole human CFTRprotein.Cell Mol Life Sci 66(21): 3469-3486
Muramoto, K., Goto, M., Inoue, Y., Ishii, N., Chiba, K., Kuboi, Y., Omae, T., Wang, Y. J.,
Gusovsky, F., and Shirota, H. (2010) E6201, a novel kinase inhibitor of mitogen-activated
protein kinase/extracellular signal-regulated kinase kinase-1 and mitogen-activated protein
kinase/extracellular signal-regulated kinase kinase kinase-1: in vivo effects on cutaneous
inflammatory responses by topical administration. J Pharm Exp Therap 335: 23–31
Niesen,F.H., Berglund, H., Vedadi, M. (2007) The use of differential scanning fluorimetry to
detect ligand interactions that promote protein stability. Nat. Protoc 2: 2212–2221
Ninomiya-Tsuji, J., Kajino, T., Ono, K., Ohtomo, T., Matsumoto, M., Shiina, M., Mihara,
M., Tsuchiya, M., Matsumoto, K. (2003) A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents

112
inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem
278(20):18485-18490.
Okiyoneda, T. andLukacs, G.L.(2012) Fixing cystic fibrosis by correcting CFTR domain
assembly. J Cell Biol 199(2):199-204.
Okiyoneda, T., Barrière, H., Bagdány, M., Rabeh, WM., Du, K., Höhfeld, J., Young, J.C.,
Lukacs, G.L. (2010) Peripheral protein quality control removes unfolded CFTR from the plasma
membrane. Science 329(5993):805-10.
Ooi, C.Y. and Durie, P.R. (2012) Cysticfibrosis transmembrane conductance regulator (CFTR)
gene mutations in pancreatitis. Journal of Cystic Fibrosis 11 (2012): 355–362
Ostedgaard, L.S., Baldusson, O., Welsh, M.J. (2001) Regulation of the cystic fibrosis
transmembrane conductance regulator Cl- channel by its R domain. J Biol Chem 276(11): 7689-
7692
Ostedgaard, L.S., Zabner, J., Vermeer, D.W., Rokhlina, T., Karp, P.H., Stecenko, A.A.,
Randak, C., Welsh, M.J. (2002) CFTR with a partially deleted R domain corrects the cystic
fibrosis chloride transport defect in human airway epithelia in vitro and in mouse nasal mucosa
in vivo. Proc Natl Acad Sci USA 99(5): 3093-3098
Parcellier, A., Brunet, M., Schmitt, E., Col, E., Didelot, C., Hammann, A. Nakayama, K.,
Nakayama, K.I., Khochbin, S., Solary, E., Garrido, C. (2006). HSP27 favors ubiquitination
and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J
20: 1179–1181.
Parcellier, A., Schmitt, E., Gurbuxani, S., Seigneurin-Berny, D., Pance, A., Chantôme, A.,
Plenchette, S., Khochbin, S., Solary, E., Garrido, C. (2003) HSP27 Is a Ubiquitin-Binding
Protein Involved in I-κBα Proteasomal Degradation..Mol Cell Biol 23(16): 5790–5802.
Parcellier, A., Schmitt, E., Brunet, M., Hammann, A., Solary, E., Garrido, C. (2005) Small
heat shock proteins HSP27 and alphaB-crystallin: cytoprotective and oncogenic
functions.Antioxid Redox Signal 7(3-4):404-413.
Pedemonte, N. andGalietta, L.J. (2012) Pharmacological Correctors of Mutant CFTR
Mistrafficking. Front Pharmacol 3:175.
Pedemonte, N., Lukacs, G.L., Du, K., Caci, E., Zegarra-Moran, O., Galietta, L.J.,
Verkman, A.S. (2005) Small-molecule correctors of defective DeltaF508-CFTR cellular
processing identified by high-throughput screening.J Clin Invest 115(9):2564-2571.
Pettit, R.S. (2012) Cystic fibrosis transmembrane conductance regulator-modifying medications:
the future of cystic fibrosis treatment. Ann Pharmacother 46(7-8):1065-75.

113
Pirkkala, L., Nykänen, P., Sistonen, L. (2001) Roles of the heat shock transcription factors in
regulation of the heat shock response and beyond. FASEB J 17(7): 1118-1131
Priou-Guesdon, M., Malinge, M.C., Augusto, J.F., Rodien, P., Subra, J.F., Bonneau, D.,
Rohmer, V. (2010) Hypochloremia and hyponatremia as the initial presentation of cystic fibrosis
in three adults. Ann Enocrinol (Paris) 71(1): 46-50
Qu, B.H. and Thomas, P.J. (1996) Alteration of the cystic fibrosis transmembrane conductance
regulator folding pathway.J Biol Chem. 271(13):7261-7264.
Rabeh, W.M., Bossard, F., Xu H., Okiyoneda, T., Bagdany, M., Mulvihill, C.M., Du, K., di
Bernardo, S., Liu, Y., Konermann, L., Roldan, A., Lukacs, G.L. (2012) Correction of both
NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function.
Cell 148(1-2):150-63.
Ramsey, B.W., Davies, J., McElvaney, N.G., Tullis, E., Bell, S.C., Dřevínek, P., Griese, M.,
McKone, E.F., Wainwright, C.E., Konstan, M.W., Moss, R., Ratjen, F., Sermet-Gaudelus,
I., Rowe, S.M., Dong, Q., Rodriguez, S., Yen, K., Ordoñez, C., Elborn, J.S.; VX08-770-102
Study Group. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D
mutation.N Engl J Med 365(18): 1663-1672.
Ratjen, F. and Doring, G. (2003) Cystic Fibrosis. Lancet 361(9358): 681-689
Reddy, M.M. and Quinton, P.M. (2003) Fucntional interaction of CFTR and ENaC in sweat
glands. Pflugers Arch 445: 499-503
Reddy, M.M., Light, M.J., Quinton, P.M. (1999) Activation of the epithelial Na+ channel
(ENaC) requires CFTR Cl- channel function. Nature 402: 301-304
Riordan, J.R. (2008) CFTR function and prospects for therapy.Annu Rev Biochem 77:701-726
Riordan, J.R., Rommens, J.M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak,
Z., Zielenski, J., Lok, S., Plavsic, N.,Chou, J.L. (1989) Identification of the cystic fibrosis
gene: cloning and characterization of complementary DNA. Science 245(4922): 1066-1073.
Robert, R., Carlile, G.W., Pavel, C., Liu, N., Anjos, S.M., Liao, J., Luo, Y., Zhang, D.,
Thomas, D.Y., Hanrahan, J.W. (2008) Structural analog of sildenafil identified as a novel
corrector of the F508del-CFTR trafficking defect. Mol Pharmacol 73(2):478-489.
Robertson, S. D., Matthies, H. J., Owens, W. A., Sathananthan, V., Chris-tianson, N. S.,
Kennedy, J. P., Lindsley, C. W., Daws, L. C., and Galli, A. (2010) Insulin reveals Akt
signaling as a novel regulator of norepinephrine transporter trafficking and norepinephrine
homeostasis. J Neurosci 30: 11305–11316

114
Rogalla, T., Ehrnsperger, M., Preville, X., Kotlyarov, A., Lutsch, G., Ducasse, C., Paul, C.,
Wieske, M., Arrigo, A.P., Buchner, J., Gaestel M. (1999). Regulation of Hsp27
oligomerization, chaperone function, and protective activity against oxidative stress/tumor
necrosis factor alpha by phosphorylation. J Biol Chem 274:18947-18956.
Roring, M. andBrummer, T. (2012) Aberrant B-Raf signaling in human cancer -- 10 years
from bench to bedside. Crit Rev Oncog 17(1):97-121.
Roskoski, R. Jr. (2010) RAF protein-serine/threonine kinases: structure and regulation. Biochem
Biophys Res Commun 399(3):313-317.
Rouse, J., Cohen, P., Trigon, S., Morange, M., Alonso-Llamazares, A., Zamanillo, D., Hunt,
T., Nebreda, A.R. (1994) A novel kinase cascade triggered by stress and heat shock that
stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell
78:1027–1037.
Rubenstein, R.C., Lockwood, S.R., Lide, E., Bauer, R., Suaud, L., Grumbach, Y. (2001)
Regulation of endogenous ENaC functional expression by CFTR and ∆F508-CFTR in airway
epithelial cells. Am J Physiol Lung Cell Mol Physiol 300(1): L88-L101.
Rubenstein, R. C. and Zeitlin, P. L. (2000) Sodium 4-phenylbutyrate downregulates Hsc70:
implications for intracellular trafficking of ΔF508-CFTR Am J Physiol Cell Physiol 278:
C259–C267
Rubenstein, R. C., Egan, M. E., Zeitlin, P. L. (1997) In vitro pharmacologic restoration of
CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial
cells containing ΔF508-CFTR. J Clin Invest 100:2457–2465.
Sampson, H.M., Robert, R., Liao, J., Matthes, E., Carlile, G.W., Hanrahan, J.W., Thomas,
D.Y. (2011) Identification of a NBD1-binding pharmacological chaperone that corrects the
trafficking defect of F508del-CFTR.Chem Biol 18(2):231-242.
Sato, S., Ward, C.L., Krouse, M.E., Wine, J.J., Kopito, R.R. (1996) Glycerol reverses the
misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 271(2):635-
638.
Saxena, A., Banasavadi-Siddegowda, Y.K., Fan Y., Bhattacharya, S., Roy, G., Giovannucci,
D.R., Frizzell, R.A., Wang, X. (2012) Human heat shock protein 105/110 kDa (Hsp105/110)
regulates biogenesis and quality control of misfolded cystic fibrosis transmembrane conductance
regulator at multiple levels.J Biol Chem 287(23):19158-19170.
Seibert, F.S., Chang, X.B., Aleksandrov, A.A., Clarke, D.M., Hanrahan, J.W., Riordan,
J.R. (1999) Influence of phosphorylation by protein kinase A on CFTR at the cell surface and
endoplasmic reticulum. Biochim Biophys Acta 1461(2): 275-283

115
Seibert,F.S., Tabcharani, J.A., Chang, X.B., Dulhanty, A.M., Mathews, C., Hanrahan,
J.W., Riordan, J.R. (1995) cAMP-dependent protein kinase-mediated phosphorylation of cystic
fibrosis transmembrane conductance regulator residue Ser-753 and its role in channel
activation.J Biol Chem 270(5): 2158-2162
Serohijos, A.W., Hegedus, T., Aleksandrov, A.A., He, L., Cui, L., Dokholyan, N.V.,
Riordan, J.R. (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in
the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S
A105(9): 3256-3261.
Sharma, M., Pampinella, F., Nemes, C., Benharouga, M., So, J., Du, K., Bache, K.G.,
Papsin, B., Zerangue, N., Stenmark, H., Lukacs, G.L. (2004) Misfolding diverts CFTR from
recycling to degradation: quality control at early endosomes. J Cell Biol 164(6):923-933.
Shen, Y., Boivin, R., Yoneda, N., Du, H., Schiller, S., Matsushima, T., Goto, M., Shirota, H.,
Gusovsky, F., Lemelin, C., Jiang, Y., Zhang, Z., Pelletier, R., Ikemori-Kawada, M.,
Kawakami, Y., Inoue, A., Schnaderbeck, M., Wang, Y. (2010) Discovery of anti-
inflammatory clinical candidate E6201, inspired from resorcylic lactone LL-Z1640-2, III. Bioorg
Med Chem Lett 20(10):3155-3157.
Silver, J.T., and Noble, E.G. (2012) Regulation of survival gene hsp70.Cell Stress Chaperon
17(1): 1-9
Stokoe, D., B. Caudwell, P. T. Cohen, Cohen, P. (1993) The substrate specificity and structure
of mitogen-activated protein (MAP) kinase-activated protein kinase-2. Biochem J 296:843-849.
Stratford, F.L., Pereira, M.M., Becq, F., McPherson, M.A., Dormer, R.L. (2003)
Benzo(c)quinolizinium drugs inhibit degradation of Delta F508-CFTR cytoplasmic domain.
Biochem Biophys Res Commun 300(2):524-530.
Stutts, M.J., Canessa, C.M., Olsen, J.C., Hamrick, M, Cohn, J.A., Rossier, B.C., Boucher
R.C (1995) CFTR as a cAMP-dependent regulator of sodium channels. Science 269: 847-850
Stutts, M.J., Rossier, B.C., Boucher, R.C. (1997) Cystic fibrosis transmembrane conductance
regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single
channel kinetics. J Biol Chem 272: 14037-14040.
Suaud, L., Miller, K., Alvey, L., Yan, W., Robay, A., Kebler, C., Kreindler, J.L., Guttentag,
S., Hubbard, M.J., Rubenstein, R.C.(2011) ERp29 regulates DeltaF508 and wild-type cystic
fibrosis transmembrane conductance regulator (CFTR) trafficking to the plasma membrane in
cystic fibrosis (CF) and non-CF epithelial cells. J Biol Chem 286(24):21239-53.
Swiatecka-Urban, A., Brown, A., Moreau-Marquis, S., Renuka, J., Coutermarsh, B.,
Barnaby, R., Karlson, K.H., Flotte, T.R., Fukuda, M., Langford, G.M., Stanton, B.A.
(2005) The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis

116
transmembrane conductance regulator (CFTR) results from accelerated endocytosis of
{Delta}F508-CFTR in polarized human airway epithelial cells. J Biol Chem 280(44):36762-72.
Tabary, O., Escotte, S., Couetil, J.P., Hubert, D., Dusser, D., Puchelle, E., Jacquot, J. (2001)
Relationship between IkappaBalpha deficiency, NFkappaB activity and interleukin-8 production
in CF human airway epithelial cells.Pflugers Arch 443 Suppl 1:S40-44.
Tabary, O., Zahm, J.M., Hinnrasky, J., Couetil, J.P., Cornillet, P., Guenounou,
M., Gaillard, D., Puchelle, E., Jacquot, J. (1998) Selective up-regulation of chemokine IL-8
expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am J Pathol 53(3):921-
930.
Takai, S., Tokuda, H., Matsushima-Nishiwaki, R., Hanai, Y., Kato, K., Kozawa, O. (2006)
Phosphatidylinositol 3-kinase/Akt plays a role in sphingosine 1-phosphate-
stimulated HSP27 induction in osteoblasts. J Cell Biochem 98(5):1249-1256.
Tang, L., Fatehi, M., Linsdell, P. (2009) Mechanism of direct bicarbonate transport by the
CFTR anion channel. J Cyst Fibros 8(2): 115-121.
Trzcinska-Daneluti, A.M., Ly, D., Huynh, L., Jiang, C., Fladd, C., Rotin, D.(2009) High-
content functional screen to identify proteins that correct F508del-CFTR function.Mol Cell
Proteomics 8(4):780-790.
Tuo, B., Wen, G., Zhang, Y., Liu, X., Wang, X., and Dong, H. (2009) Involvement of
phosphatidylinositol 3-kinase in cAMP- and cGMP-induced duodenal epithelial CFTR activation
in mice.Am J Physiol Cell Physiol 297: C503–515
Turner,N. and Grose, R. (2010) Fibroblast growth factor signalling: from development to
cancer. Nat Rev Cancer 10(2):116-129.
Van Calenbergh, S., Pochet, S., Munier-Lehmann, H.(2012) Drug design and identification of
potent leads against mycobacterium tuberculosis thymidine monophosphate kinase.Curr Top
Med Chem 12(7):694-705.
Van Goor, F., Hadida, S., Grootenhuis, P.D., Burton, B., Cao, D., Neuberger, T., Turnbull,
A., Singh, A., Joubran, J., Hazlewood, A., Zhou, J., McCartney, J., Arumugam, V., Decker,
C., Yang, J., Young, C., Olson, E.R., Wine, J.J., Frizzell, R.A., Ashlock, M., Negulescu, P. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770.
Proc Natl Acad Sci U S A 106(44):18825-18830.
Van Goor, F., Hadida, S., Grootenhuis, P.D., Burton, B., Stack, J.H., Straley, K.S., Decker,
C.J., Miller, M., McCartney, J., Olson, E.R., Wine, J.J., Frizzell, R.A., Ashlock, M.,
Negulescu, P.A.(2011) Correction of the F508del-CFTR protein processing defect in vitro by the
investigational drug VX-809.Proc Natl Acad Sci U S A 108(46):18843-18848.

117
Van Goor, F., Straley, K.S., Cao, D., González, J., Hadida, S., Hazlewood, A., Joubran, J.,
Knapp, T., Makings, L.R., Miller, M., Neuberger, T., Olson, E., Panchenko, V., Rader, J.,
Singh, A., Stack, J.H., Tung, R., Grootenhuis, P.D., Negulescu, P. (2006) Rescue of
DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by
small molecules. Am J Physiol Lung Cell Mol Physiol 290(6):L1117-1130.
Vij, N., Mazur, S., Zeitlin, P.L.(2009) CFTR is a negative regulator of NFkappaB mediated
innate immune response. PLoS One 4(2):e4664.
Wang, X., Asea, A., Xie, Y., Kabingu, E., Stevenson, M.A., Calderwood, S.K. (2000) RSK2
represses HSF1 activation during shock. Cell Stress Chaperon 5(5): 432-437
Wang, X., Khaleque, M.A., Zhao, M.J., Zhong, R., Gaestel, M., Calderwood, S.K. (2005)
Phosphorylation of HSF1 by MAPK-activated protein kinase 2 on serine 121, inhibits
transcriptional activity and promotes HSP90 binding. J Biol Chem 281(2): 782-791
Wang, X., Venable, J., LaPointe, P., Hutt, D.M., Koulov, A.V., Coppinger, J., Gurkan,
C., Kellner, W., Matteson, J., Plutner, H., Riordan, J.R., Kelly, J.W., Yates, J.R.
3rd, Balch, W.E. (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding
of CFTR in cystic fibrosis. Cell 127(4):803-815
Ward, C.L. and Kopito, R.R. (1994) Intracellular turnover of cystic fibrosis transmembrane
conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant
proteins.J Biol Chem 269(4): 25710-25718
Xiong, X., Bragin, A., Widdicombe J.H., Cohn J., Skach W.R. (1997) Structural cues
involved in endoplasmic reticulum degradation of G85E and G91R mutant cystic fibrosis
transmembrane conductance regulator.J Clin Invest 100(5):1079-1088.
Yang Y., Janich S., Cohn, J.A., Wilson, J.M. (1993) The common variant of cystic fibrosis
transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi
nonlysosomal compartment.Proc Natl Acad Sci USA 90(20): 9480-9484.
Ye, L., Knapp, J.M., Sangwung, P., Fettinger, J.C., Verkman, A.S., Kurth, M.J. (2010)
Pyrazolylthiazole as DeltaF508-cystic fibrosis transmembrane conductance regulator correctors
with improved hydrophilicity compared to bithiazoles. J Med Chem 53(9):3772-3781.
Ye, S., Cihil, K., Stolz, D.B., Pilewski, J.M., Stanton, B.A., Swiatecka-Urban, A.. (2010) c-
Cbl facilitates endocytosis and lysosomal degradation of cystic fibrosis transmembrane
conductance regulator in human airway epithelial cells. J Biol Chem 285(35):27008-27018.
Younger, J.M., Chen, L., Ren, H.Y., Rosser, M.F., Turnbull, E.L., Fan, C.Y., Patterson,
C., Cyr, D.M. (2006) Sequential quality-control checkpoints triage misfolded cystic fibrosis
transmembrane conductance regulator.Cell 126(3):571-582.

118
Younger, J.M., Ren, H.Y., Chen, L., Fan, C.Y., Fields, A., Patterson, C., Cyr, D.M. (2004)
A foldable CFTR{Delta}F508 biogenic intermediate accumulates upon inhibition of the Hsc70-
CHIP E3 ubiquitin ligase. J Cell Biol 167(6):1075-1085.
Yu, B, Ragazzon, B, Rizk-Rabin, M, Bertherat, J. (2012) Protein kinase A alterations in
endocrine tumors. Horm Metab Res 44(10):741-8.
Yu, G.J., Yoo, C.L., Yang, B., Lodewyk, M.W., Meng, L., El-Idreesy, T.T., Fettinger, J.C.,
Tantillo, D.J., Verkman, A.S., Kurth, M.J. (2008) Potent s-cis-locked bithiazole correctors of
DeltaF508 cystic fibrosis transmembrane conductance regulator cellular processing for cystic
fibrosis therapy. J Med Chem 51(19):6044-6054.
Zhang, L., Button, B., Gabriel, S. E., Burkett, S., Yan, Y., Skiadopoulos, M. H., Dang, Y.
L., Vogel, L. N., McKay, T., Mengos, A., Boucher, R. C., Collins, P. L., and Pickles, R. J. (2009) CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport
to human cystic fibrosis airway epithelium. PLoS Biol 7: e1000155
Zou, J., Guo, Y., Guettouche, T., Smith, D. F., and Voellmy, R. (1998) Repression of heat
shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-
sensitive complex with HSF1. Cell 94: 471–480




















