
Reproductive risks for carriers of complex chromosome rearrangements: Analysis of 25 families
15
American Journal of Medical Genetics 29:247-261 (1988) Reproductive Risks for Carriers of Complex Chromosome Rearrangements: Analysis of 25 Families Jerome L. Gorski, Mildred L. Kistenmacher, Hope H. Punnett, Elaine H. Zackai, and Beverly S. Emanuel Clinical Genetics Center of the Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine (J. L. G., E. H. Z., B. S. E.); Clinical Genetics Center of St. Christopher’s Hospital for Children, Temple University, School of Medicine (M. L. K.. H.H.P.), Philadelphia, Pennsylvania We have determined the empirical reproductive risks for heterozygous carriers of complex chromosome rearrangements (CCRs). CCRs are structural rearrangements involving at least three chromosomes and three or more chromosomal breakpoints. Pregnancy outcome, the frequency and type of chromosomal imbalance in the offspring, and the localization and distribution of chromosome breakpoints were analyzed in 25 CCR families ascertained by the birth of a malformed child or repeated spontaneous abortions. This study included two newly ascertained familial CCRs and a total of 67 informative pregnancies. Analysis of the data, after correction for ascertainment bias, showed that the incidence of spontaneous abortions in CCR families was 48.3%. Approximately one in ten pregnancies and 18.4% of all live births to CCR carriers resulted in phenotypically abnormal offspring. One-half of all CCR carrier liveborn offspring were also CCR carriers. There was a 53.7% incidence of an abnormal pregnancy outcome to CCR carriers. We failed to detect any evidence for a non-random involvement of specific chromosomes in CCRs. However, we did observe a non-random distribution of specific breakpoints at sites lq25,4q13, 6q27, 7p14, 9q12, llpll, llp15,12q21,13q31,and 18q21. Key words chromosomal aberration, chromosomal translocation, aneuploidy, duplication, deficiency, reproductiveoutcome Received for publication September 8, 1986; revision received August 24, 1987. Address reprint requests to Jerome L. Gorski, Division of Pediatric Genetics, Department of Pediatrics, University of Michigan School of Medicine, D1233 Medical Professional Building, Box 0718, Ann Arbor, MI 48109-0010. 0 1988 Alan R. Liss, Inc.
Transcript of Reproductive risks for carriers of complex chromosome rearrangements: Analysis of 25 families

American Journal of Medical Genetics 29:247-261 (1988)
Reproductive Risks for Carriers of Complex Chromosome Rearrangements: Analysis of 25 Families
Jerome L. Gorski, Mildred L. Kistenmacher, Hope H. Punnett, Elaine H. Zackai, and Beverly S. Emanuel
Clinical Genetics Center of the Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine (J. L. G., E. H. Z., B. S. E.); Clinical Genetics Center of St. Christopher’s Hospital for Children, Temple University, School of Medicine (M. L. K.. H.H. P.), Philadelphia, Pennsylvania
We have determined the empirical reproductive risks for heterozygous carriers of complex chromosome rearrangements (CCRs). CCRs are structural rearrangements involving at least three chromosomes and three or more chromosomal breakpoints. Pregnancy outcome, the frequency and type of chromosomal imbalance in the offspring, and the localization and distribution of chromosome breakpoints were analyzed in 25 CCR families ascertained by the birth of a malformed child or repeated spontaneous abortions. This study included two newly ascertained familial CCRs and a total of 67 informative pregnancies. Analysis of the data, after correction for ascertainment bias, showed that the incidence of spontaneous abortions in CCR families was 48.3%. Approximately one in ten pregnancies and 18.4% of all live births to CCR carriers resulted in phenotypically abnormal offspring. One-half of all CCR carrier liveborn offspring were also CCR carriers. There was a 53.7% incidence of an abnormal pregnancy outcome to CCR carriers. We failed to detect any evidence for a non-random involvement of specific chromosomes in CCRs. However, we did observe a non-random distribution of specific breakpoints at sites lq25,4q13, 6q27, 7p14, 9q12, l l p l l , llp15,12q21,13q31,and 18q21.
Key words chromosomal aberration, chromosomal translocation, aneuploidy, duplication, deficiency, reproductive outcome
Received for publication September 8, 1986; revision received August 24, 1987.
Address reprint requests to Jerome L. Gorski, Division of Pediatric Genetics, Department of Pediatrics, University of Michigan School of Medicine, D1233 Medical Professional Building, Box 0718, Ann Arbor, MI 48109-0010.
0 1988 Alan R. Liss, Inc.

248 Gorskietal.
INTRODUCTION
Complex chromosome rearrangements (CCRs) are structural rearrangements involving at least three chromosomes and three or more chromosome breakpoints. CCRs may be balanced or unbalanced, familial or de novo [Pai et al., 1980; Kleczkowska et al., 19821. CCRs are rare. Systematic surveys of liveborn infants [Hamerton et al., 19751, habitual aborters [Jacobs, 1977; Mennuti et al., 19781, and spontaneous abortuses [Carr, 19711 failed to reveal a CCR. Although CCRs are infrequently found in screened populations, balanced CCR carriers are generally phenotypically normal, and the balanced CCR represents a significant reproductive risk. Because of the high probability that chromosomally unbalanced gametes will be produced by these individuals, the expected and observed reproductive outcomes of CCR carriers have included habitual abortions, aneuploid liveborn infants with multiple congenital anomalies, and reduced fertility [Kleczkowska et al., 19821. Although the reproductive risks for the carriers of simple reciprocal translocations have been calculated [Neri et al., 1983; Bou6 and Gallano, 19841, it has remained problematic to define the empirical reproductive risks for individual CCR carriers. In an effort to better define the reproductive risks to CCR carriers, we report a detailed analysis of the reproductive outcomes of CCR carriers consisting of cases personally observed and selected from the literature. The results of this study provide information to assist in assessing reproductive risk for heterozygous CCR carriers of having liveborn infants with unbalanced karyotypes and multiple congenital malforma- tions, spontaneous abortions, or CCR carrier offspring. The distribution of chromosome breakpoints involved in familial CCRs has also been examined.
MATERIALS AND METHODS
Criteria for inclusion into the study included 1) familial segregation of a CCR, 2) completeness of the pedigree, and 3 ) adequate definition of the CCR by chromosome analysis. We define CCRs as complex chromosomal structural rearrangements involving a minimum of three chromosomes and at least three breakpoints. Not included were families carrying two different reciprocal translocations (such as those families described by Hansen et al. [1983], Bass and Sparkes [1979], Bijlsma et al. [1978] or Simoni et al. [ 19791) or complex translocations with multiple rearrangements between two chromo- somes (such a.s Anderson et al. [1981]). Using these criteria, we identified 25 CCR families. Two CCR families were ascertained from our clinical population, and their case reports are included in this study (patients 1 and 2) (Table I). Twenty-three CCR families were ascertained from the literature (cases 3-25). The breakpoints of the CCRs were defined by banding techniques in all but three families. The assigned case numbers, mode of ascertainment, rearranged chromosomes with breakpoints, and references are listed in Table 1. Pedigrees of the CCR families are shown in Figure 1.
Chromosome preparations were made from conventionally phytohemagglutinin- stimulated cultures of peripheral whole blood. Karyotypes were obtained using standard high-resolution trypsinaiemsa banding techniques [Francke and Oliver, 19781.

N A A A * A A N A A A c > A A
2'k% P
Reproductive Risks for CCR Carriers 249
22.) ,Q I
P
Fig. 1. Symbols: 7 , Index carrier; P, proband 0, not analyzed cytogenetically; Q, normal karyotype; O, CCR carrier; A , spontaneous abortus; 0, unbalanced karyotype; @, abnormal phenotype karyotype unavailable.
Pedigrees of CCR families. Case number refers to those of Table I.
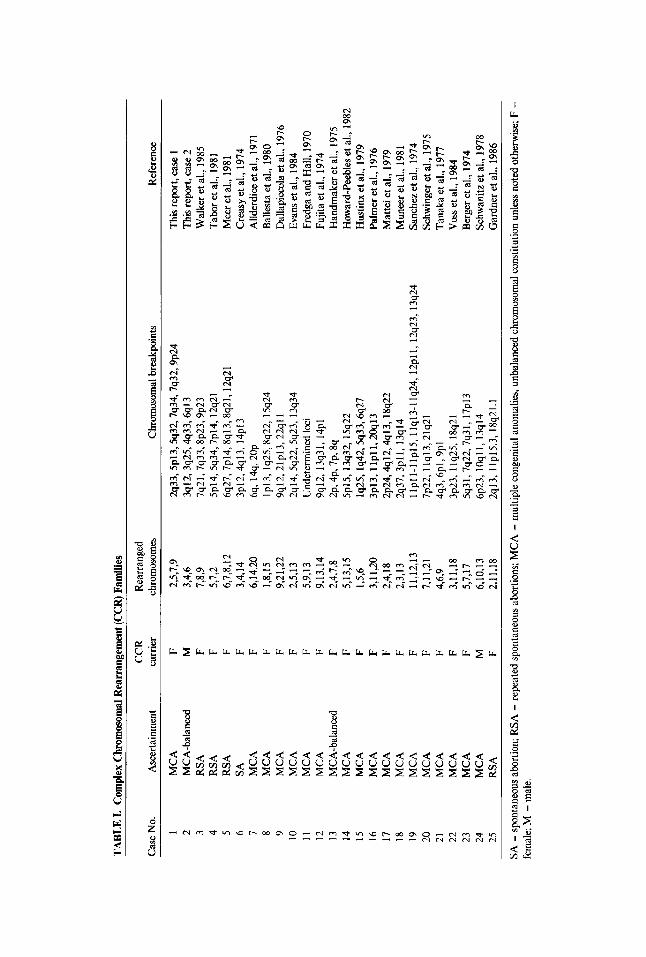
TAB
LE I.
Com
plex
Chr
omos
omal
Rea
rran
gem
ent (
CC
R) F
amili
es
Case
. No.
A
scer
tain
men
t ca
rrie
r ch
rom
osom
es
Chr
omos
omal
bre
akpo
ints
R
efer
ence
C
CR
R
earr
ange
d
1 M
CA
F
2,5,
7,9
%33,5~13,5q32,7q34,7q32,9~24
This
repo
rt, ca
se 1
2
MC
A-b
alan
ced
M
3,4,
6 3q
12,3
q25,
4q33
,6q1
3 T
his r
epor
t. ca
se 2
W
alke
r et a
l., 1
985
3 R
SA
F 7,
8,9
7q21
,7q3
3,8~
23,9
~23
4 R
SA
F 5,
7,2
5p14
,5q3
4,7p
14, 1
2q21
T
abor
et al
., 19
81
5 R
SA
F 6,
7,8,
12
6q27
,7p1
4,8q
13,8
q21,
12q
21
Mee
r et a
l., 1
981
6 SA
F
3,4,
14
3p12
,4q1
3, 1
4~
13
C
reas
y et
al.,
197
4 7
MC
A
F 6,
14,2
0 6%
14%
~O
P
Alld
erdi
ce et
al.,
1971
8
MC
A
F 1,
8,15
lp
13, l
q25,
8q22
,15q
24
Bal
lesta
et a
l., 1
980
9 M
CA
F
9,21
,22
9q12
,21p
13,2
2qll
Dal
lapi
ccol
a et
al.,
197
6 10
M
CA
F
2,5,
13
2q14
,5q2
2,5q
23, 1
3q34
Ev
ans e
t al.,
198
4 11
M
CA
F
5,9,
13
Und
eter
min
ed lo
ci
Fred
ga a
nd H
all,
1970
12
M
CA
F
9,13
,14
9q12
, 13q
31, 1
4pl
Fujit
a et
al.,
1974
13
M
CA
-bal
ance
d F
2,4,
7,8
2p. 4
p, 7
p, 8
q H
andm
aker
et a
l., 1
975
14
MC
A
F 5,
13,1
5 5p
15,1
3q32
, 15
q22
How
ard-
Peeb
les e
t al.,
198
2 15
M
CA
F
1,5,
6 lq
25,lq
42,5
q33,
6q27
H
ustin
x et
al.,
1979
16
M
CA
F
3,
11,2
0 3p
13, l
lpll
, 2O
q13
Palm
er e
t al.,
197
6 17
M
CA
F
2,4,
18
2p24
,4ql
2,*1
3,
38q2
2 M
atte
i et a
l., 1
979
18
MC
A
F 2,
3,13
2q
37,3
pll,
13q
14
Mun
eer e
t al.,
198
1 19
M
CA
F
1 1,1
2,13
ll
pll-
llpl
5, ll
q13-
llq2
4, 1
2~11
, 12q2
3, 1
3q24
Sa
nche
z et a
l., 1
974
20
MC
A
F 7,
11,2
1 7p
22, 1
lq13
,21q
21
Schw
inge
r et a
l., 1
975
21
MC
A
F 4,
6,9
4q3,
6pl,
9~
1
Tana
ka e
t al.,
197
7 22
M
CA
F
3,11
,18
3p23
, 1 lq
25, 1
8q21
23
M
CA
F
5,7,
17
5q31
,7q2
2,7q
31, 1
7~
13
B
erge
r et a
l., 1
974
24
MC
A
M
6,10
,13
6p23
, lO
qll,
13q1
4 Sc
hwan
itz e
t al.,
197
8 25
R
SA
F 2,
11,1
8 2q
13, l
lp15
.3, 1
8q21
.1
Gar
dner
et a
l., 1
986
Vos
s et a
l., 1
984
SA =
spo
ntan
eous
abor
tion;
RSA
= re
peat
ed sp
onta
neou
s abo
rtion
s; M
CA
= m
ultip
le co
ngen
ital a
nom
alie
s, un
bala
nced
chro
mos
omal
cons
titut
ion u
nles
s no
ted
othe
rwise
; F =
fem
ale:
M =
mal
e.

TABL
E II. S
umm
arv of
CC
R F
amilv
Dat
a
No.
of
No.
of
No.
of o
ffsp
ring
No.
of o
ffsp
ring
Sibs
hip
No.
of
spon
tane
ous
No.
of C
CR
ch
rom
osor
nally
ph
enot
ype
and
phen
otyp
e abn
orm
al
Asc
erta
inm
ent
type
pr
egna
ncie
s ab
ortio
ns
carr
iers
ab
norm
al o
ffsp
ring
kary
otyp
e nor
mal
ka
ryot
ype
unst
udie
d
A.
Sum
mar
y of
crud
e da
ta w
ithou
t asc
erta
inm
ent c
orre
ctio
n Pr
oban
d w
ith M
CA
" Pr
imar
y 16
28
15
Pr
oban
d w
ith R
SA"
Prim
ary
31
24
4 Se
cond
ary
6 1
4 To
tal
-
113
53
23
B. S
umm
ary of d
ata
with
asc
erta
inm
ent c
orre
ctio
nb
Prob
and
with
MC
Aa
Prim
ary
56
28
13 4
Seco
ndar
y 4
1 2
RSA
tota
l 11
1
6 To
tal
61
29
19
-
Prob
and
with
RSA
a Pr
imar
y I
~
20
-
20
-
2
8 3 1 12 8 3 1 4 12
5 -
-
5
"MC
A =
mul
tiple
con
geni
tal a
nom
alie
s; R
SA =
repe
ated
spo
ntan
eous
abor
tions
. bA
scer
tain
rnen
t cor
rect
ion
by re
mov
al o
f pro
band
s an
d in
dex
carr
iers
.

252 Gorskiet al.
Fig. 2. Partial G-banded karyotypes of family members of cases 1 and 2. A Partial G-banded karyotype of proband's mother in case 1 showing translocations between 2,5,7, and 9. Case 1 proband's partial karyotype is shown below. Derivative chromosomes are indicated. Extended notation for the balanced carrier is: 46,XX,t(2;5;7;9)(2pter - 2q33::7q34 - 7qter)(2qter - 2q33::7q34 - 7q32::5pl3 - 5q32::9p24 +
9pter)(7pter - 7q32::5p13 - 5pter)(5qter - 5q32::9p24 - 9qter)
der(2) = (2pter - 2q33::7q34 - 7qter) der(5) = (2qter - 2q33::7q34 - 7q32::5p13 - 5q32::9p24 - 9pter) der(7) = (7pter - 7q32::5p13 - Spter) der(9) = (5qter - 5q32::9p24 - 9qter)
Case 1 proband's karyotype - 46,XX,-2,-5,-9, +der(2), +der(5), +der(9), t(2;7)(q33;q34), t[5;der(7)(pl3;q32), t(5;9)(q32;p24) mat. B Partial G-banded karyotype of prospositus in case 2 showing translocations between chromosomes 3,4, and 6. The derivative chromosomes are indicated. Extended notation is 46,XY, t(3;4;6)(3pter - 3q13::6q13 - 6qter)(4ptr - 4q33::3q25 - 3qter)(6qter - 6p13::3q12 - 3q25::4q33 - 4qter).

Reproductive Risks for CCR Carriers 253
CLINICAL REPORTS Patient 1
The proposita was an infant girl, the 2,400-g product of an uncomplicated 40-week gestation. There was no known exposure to recognized teratogens. The mother’s two previous pregnancies resulted in a first-trimester spontaneous abortion and a normal liveborn girl. The patient had a multiple congenital anomaly (MCA) syndrome of hirsutism, three fontanels, wide metopic suture, mongoloid slant of the palpebral fissures, small posteriorly rotated ears, cleft soft palate, short neck, a single simian crease, bilaterally fixed camptodactyly of digits 3, 4, and 5, rugated and partially fused labia majora, unilateral talipes equinovarus, and a single umbilical artery. She exhibited marked hypertonia and abnormally fisted hands. Echocardiography demonstrated an atrial septal defect (ASD) and a ventricular septal defect (VSD). Head CT demonstrated ventricular dilation, partial agenesis of the corpus callosum, and findings consistent with cerebral heterotopias.
Chromosome analysis showed an unbalanced complex rearrangement involving chromosomes 2,5, and 9. A partial karyotype of the proposita and her mother are shown in Figure 2A. Cytogenetic analysis of relatives showed that the patient’s mother and sister are heterozygote carriers of an apparently balanced CCR involving chromosomes 2, 5,7, and 9%46,XX,t(2;7)(q33;q34),t [ 5;der( 7)] (pl3 ,q32),t( 5;9) (q32,p24). The patient’s karyotype was 46,XX, - 2, - 5, - 9, +der(2), +der( 5), +der(s),t(2;7)(q33,q34),t[5;der (7)](~13;q32),t(5;9)(q32;~24)mat. The patient had an apparent duplication of bands 7q32-7q34 and deficiency of 5pl3-+5pter.
Patient 2
The propositus was a 7-year-old boy evaluated for developmental delay (IQ approximately 70) and minor anomalies, including recessed eyes and bilateral fifth digit clinodactyly, with normal growth. His gestational and medical histories were unremark- able. Cytogenetic analysis showed an apparently balanced CCR involving chromosomes 3, 4, and 646,XY,t(3;4;6)(3ql2;4q33;6q13) (Fig, 2B). Cytogenetic analysis of relatives showed that the patient’s healthy and phenotypically normal father and sister are heterozygous carriers of the same CCR.
RESULTS Analysis of Ascertainment
The ascertainment of each of the 25 CCR families was examined. Twenty families were ascertained through a child with multiple congenital anomalies (MCAs); 18 of the index cases had an unbalanced karyotype. In two families the child with MCAs was found to be a balanced CCR carrier (cases 2, 13). While the mildly dysmorphic features of the proposita of case 2 were not observed in the other CCR carriers within his family, similar dysmorphic features were observed in all of the apparently balanced CCR carriers described in case 13 [Handmaker et al., 19751. The remaining five families were ascertained because of fetal wastage.
The parent carrying the balanced CCR was noted in each family (Table I). In two families the carrier parent was the father. Both were ascertained through children with MCAs-ne, a balanced carrier; the other, having unbalanced chromosomes. In 23

254 Gorski et al.
families (92%) the mother was the carrier. In one family the CCR was transmitted through both a male and a female (case 5 ) .
Pregnancy Outcome
The pregnancy of a CCR carrier has four possible outcomes: 1) abortion, 2) liveborn infant with unbalanced chromosomes and variable degrees of phenotypic abnormalities, 3) liveborn infant with a balanced CCR, and 4) chromosomally normal liveborn infant. Data available from the CCR families were used to generate estimates of the average probabilities for each of these possible outcomes. A summary of the CCR family data, with and without ascertainment corrections, is shown in Table 11.
The rate of abortions for both female and male CCR carriers was calculated in exclusion of those primary families ascertained through fetal wastage. The frequency of abortions in female CCR carriers was 23/50 (46%) and in spouses of male carriers was 6/10 (60%). Thus, the total spontaneous abortion frequency was 29/60 (48.3%, SE 12.6). (The SE = f 1.96 d m ) .
Segregation analysis was done using the model for reciprocal translocations developed by Stene and Stengel-Rutkowski [ 19771. This model allows correction for bias related to ascertainment by excluding all probands and translocation carriers with a proband in a direct line of descent. After these corrections, there were 38 liveborn progeny to the 28 CCR carriers available for analysis (Table 11). Seven of these offspring were phenotypically abnormal and presented with multiple congenital anomalies. Two had documented abnormal karyotypes. Five offspring died before investigation and presum- ably had unbalanced chromosomes. Therefore, there was a 7/38 ( 1 8.4%) incidence of abnormal liveborn offspring to CCR carriers. Pregnancies to CCR carriers resulted in a phenotypically abnormal liveborn child in 7 of 67 (10.4%, SE 7.3). This probably represents an underestimate of the actual risk to CCR carriers of having a phenotypically abnormal child, since most families with chromosomally abnormal offspring were ascertained by the birth of the first and only abnormal child (13/19 families). Detailed karyotypes were available on 16 of the chromosomally abnormal probands ascertained on the basis of multiple congenital anomalies. These results provided a compilation of karyotypic abnormalities in offspring of CCR carriers. Five of the probands (31%) had duplication deficiency karyotypes (cases 1 , 7, 14, 15, and 24 of Table I). Ten of the 16 (62.5%) had duplication alone; 3 had dup (9p) (cases 9, 12, and 21), 2 dup(1 lp) (cases 16, 19), and one had dup(l3q) (case 18). A single proband (6.2%) was a CCR carrier trisomic for chromosome 21 and probably resulted from the fertilization of a CCR carrier gamete formed by 4:2 disjunctional segregation (case 20),
Of the 38 recognized pregnancies resulting in liveborn offspring to CCR carriers, 3 1 resulted in phenotypically normal offspring who were studied cytogenetically. These consisted of 12 with normal chromosomes and 19 CCR carriers. Thus, the overall CCR carrier frequency among liveborn progeny to CCR carriers was 19/38 (50%, SE 15.9). The probability for a CCR carrier to produce chromosomally normal liveborn offspring was 12/38 (31.6%, SE * 14.8). There is not a statistically significant difference between these two probabilities.
The frequency of abnormal pregnancy outcome (all malformed liveborns and spontaneous abortions) was calculated. For female CCR carriers this risk was 30/57
Reproductive Risks for CCR Carriers 255
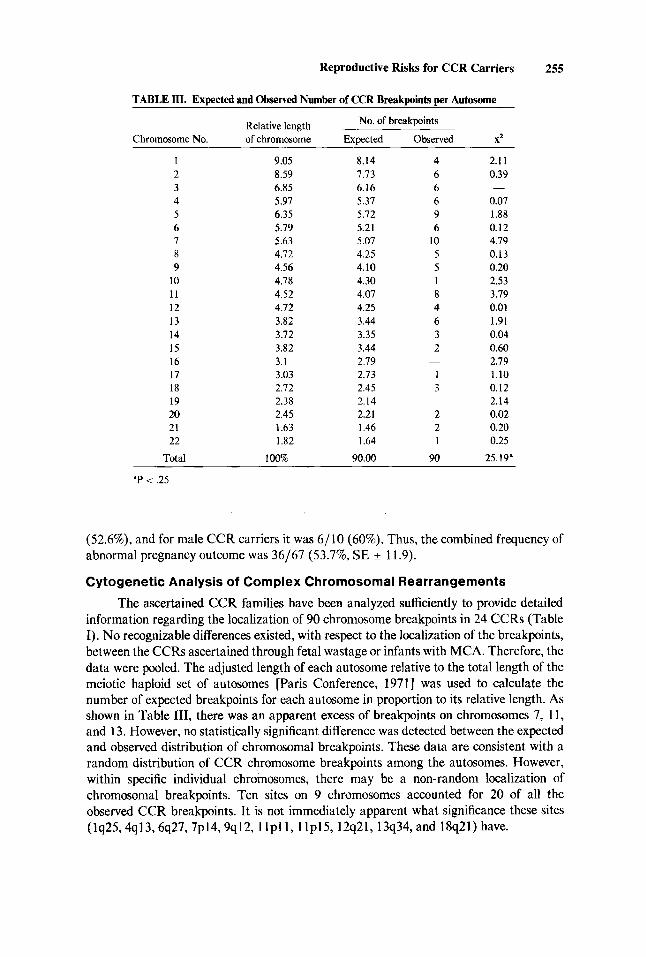
TABLE m. Expected and Observed Number of CCR Breakpoints per Autosome
No. of breakpoints Relative length Chromosome No. of chromosome Expected Observed X2
1 2 3 4 5 6 7 8 9
10 11 12 13 14 15 16 17 18 19 20 21 22
Total
9.05 8.59 6.85 5.97 6.35 5.79 5.63 4.72 4.56 4.78 4.52 4.72 3.82 3.72 3.82 3.1 3.03 2.72 2.38 2.45 1.63 1.82
100%
8.14 7.73 6.16 5.37 5.72 5.21 5.07 4.25 4.10 4.30 4.07 4.25 3.44 3.35 3.44 2.79 2.73 2.45 2.14 2.21 1.46 1.64
90.00
4 6 6 6 9 6
10 5 5 1 8 4 6 3 2
1 3
2 2 1
90
-
-
2.1 I 0.39
0.07 1.88 0.12 4.79 0.13 0.20 2.53 3.79 0.01 1.91 0.04 0.60 2.19 1.10 0.12 2.14 0.02 0.20 0.25
25.19"
-
'P < .25
(52.6%), and for male CCR carriers it was 6/10 (60%). Thus, the combined frequency of abnormal pregnancy outcome was 36/67 (53.796, SE ? 11.9).
Cytogenetic Analysis of Complex Chromosomal Rearrangements
The ascertained CCR families have been analyzed sufficiently to provide detailed information regarding the localization of 90 chromosome breakpoints in 24 CCRs (Table I). No recognizable differences existed, with respect to the localization of the breakpoints, between the CCRs ascertained through fetal wastage or infants with MCA. Therefore, the data were pooled. The adjusted length of each autosome relative to the total length of the meiotic haploid set of autosomes [Paris Conference, 19711 was used to calculate the number of expected breakpoints for each autosome in proportion to its relative length. As shown in Table 111, there was an apparent excess of breakpoints on chromosomes 7, 11, and 13. However, no statistically significant difference was detected between the expected and observed distribution of chromosomal breakpoints. These data are consistent with a random distribution of CCR chromosome breakpoints among the autosomes. However, within specific individual chromosomes, there may be a non-random localization of chromosomal breakpoints. Ten sites on 9 chromosomes accounted for 20 of all the observed CCR breakpoints. It is not immediately apparent what significance these sites (lq25,4q13,6q27,7p14,9q12,1 l p l1 , l lp15,12q21,13q34, and 18q21) have.

256 Gorski et al.
DISCUSSION
We have determined empirical risks for abnormal reproductive outcomes for CCR carriers ascertained by repeated spontaneous abortions or the birth of MCA infants. Although each CCR reported here was unique, we propose that the calculated risks of the reported families represent a good estimate of the risks to CCR carriers, in general. The facts that support this proposition are 1) uniformly, all reported CCR families had a high rate of abnormal pregnancy outcomes (Fig. 1); 2) the CCRs reported involved almost all of the autosomes and not a particular subset; and 3) CCR carriers in other species have similar reproductive risks (61% fetal wastage in CCR carrier chickens) [Bitgood et al., 198 11. There is no evidence that the CCRs or the CCR families presented here represent a distinct subset in chromosome behavior or distinct reproductive risks.
Identification of most CCR carriers by abnormal reproductive outcomes (MCA infants or recurrent abortions) may favor the ascertainment of female CCR carriers and possibly explain, in part, our observed predominance of female CCR carriers in 92% of the families studied. In most reported cases, de novo male CCR carriers are sterile and have been ascertained through infertility evaluations [Joseph and Thomas, 1982; Rodriguez et al., 1985; Saadallah and Hulten, 19851. Testicular biopsies have shown spermatogenic arrest, with the complete absence of spermatozoa with otherwise normal testicular histology in some male CCR carriers [Joseph and Thomas, 1982; Rodriguez, et al., 19851. Meiotic investigations in a subfertile male CCR carrier showed that the hexavalents had a reduced number of chiasmata, possibly representing disturbances in chromosome pairing, although no inadequacies of telomeric synapsis were seen [Saadallah and Hulten, 19851. The identification of three fertile male CCR carriers (cases 2, 5, and 24) within this study demonstrates that not all male CCR carriers are infertile.
Similar but less dramatic inequalities between maternal and paternal carrier rates have been observed in families in whom a structural chromosomal rearrangement was ascertained prenatally through an infant with aneuploidy or recurrent abortions. The mother was the carrier in 68% of the couples in which the anomaly was first detected [BouC and Callano, 19841. These findings in humans may be compared to the results in tobacco mice by Gropp and Winking [1981], who observed that the risk of unbalanced fetuses in almost all of the different types of Robertsonian translocations is significantly higher in female compared with male carriers who have the same chromosomal rearrangement. Together, these findings suggest the possibility of fundamental differences between the sexes in the tolerance of chromosomal rearrangements during meiosis and gametogenesis.
Reproductive risks for CCR carriers can be compared with those of simple reciprocal translocation carriers or to the general population. The calculated 48.3% (SE * 12.6) risk of a CCR carrier’s pregnancy resulting in an early spontaneous abortion is significantly greater than the 15-20% risk in the general population [Warburton and Fraser, 19641 and greater than most of the observed frequencies for reciprocal transloca- tion carriers, which range from 20.4% [Jacobs et al., 19701 to 21.7% [Ford and Clegg, 19691. Neri et al. [1983] reported a 50% incidence of spontaneous abortions in reciprocal translocation carriers, an incidence similar to that reported here. The average risk of spontaneous abortions for Robertsonian translocation carriers has been found to vary between 20 and 25%, depending on the chromosomes involved [Dutrillaux and Lejeune, 1970; Fryns et al., 19821.

Reproductive Risks for CCR Carriers 257
Fig. 3. reciprocal complex translocation with three chromosomal breakpoints.
Pachytene orientation in meiosis of three chromosome pairs (a, b, and c) involved in a hypothetical
The 10.4% risk of a CCR carrier having a phenotypically abnormal liveborn child is close to the predicted risks of malformed offspring of reciprocal or Robertsonian translocation carriers, which range from 6% [Neri et al., 19831 to 14% [Lurie et al., 19781 to the estimated 5-20% of Lejeune et al. [ 19701. BouC and Gallano [ 19841 reported a 1&15% risk for an unbalanced fetus to Robertsonian translocation female carriers, an 11.6% risk to reciprocal translocation carriers (mother or father), and a 5.9% risk to carriers of chromosome inversions. Reproductive risks for reciprocal translocation carriers were found to differ significantly, depending on the mode of ascertainment, with a 20.8% risk for an unbalanced fetus if the carrier was ascertained by the birth of an infant with an unbalanced karyotype, but a 4.9% risk if ascertained by other means [BouC and Gallano, 19841.
The results of this study represent the pooling of CCR families ascertained by either the birth of a MCA infant or recurrent abortions. Insufficient clinical material precludes a separate statistically significant analysis for each family group, based on mode of ascertainment. Additional reports of familial CCR cases will hopefully provide the required clinical material to address the question of possible differences in reproductive risks to CCR carriers ascertained by different means. We strongly suspect, however, that, as demonstrated for reciprocal translocation carrier families [Daniel, 1979; Aurias et al., 19781, the size of the chromosomal abnormality, irrespective of origin, will be the major determinant of survival of an aneuploid CCR fetus to term and, as reported for reciprocal translocation carriers [BouC and Gallano, 19841, the mode of ascertainment of the chromosomal rearrangement will suggest the primary reproductive risk of the couple.
The identification of two families (cases 2, 13) in which an apparently balanced CCR carrier was ascertained by a balanced CCR MCA child suggests that the MCA phenotype is related to the CCR by a mechanism as yet poorly defined. Further reports of similar cases should provide a better sense of whether these cases are exceptional or represent a general phenomenon for which all CCR carriers are at risk.
The high incidence of reproductive abnormalities in CCR carrier offspring can be explained in terms of the chromosomal constitution of the CCR carrier's gametes. Extremely complicated multivalent formation, chromosomal recombination, and meiotic segregation are possible during CCR carrier gametogenesis. The pachytene configuration of a hypothetical CCR with three reciprocal chromosome rearrangements (the least complicated CCR) is shown in Figure 3. Karyotypes of the gametes resulting from the four possible 3:3 disjunctions, karyotypes of the resulting fertilized zygotes, and their predicted phenotypes are shown in Table IV. The existence of hexavalent pachytene configurations similar to that depicted in Figure 3 have been observed in CCR carrier

258 Gorskietal.
TABLE IV. F'redicted Karyotypes and Phenotypes Resulting From the Fertilization of CCR Carrier Gametes After 3 3 Segregation of Pachytene Hexivalents
Gametic Karyotype of zygote after Predicted fertilization with normal gamete phenotype karyotype
1. abc Normal 2. a'b'c' CCR carrier 3. a'bc Partial monosomy a
4. ab'c Partial monosomy b
5 . abc' Partial monosomy c
6 . a'b'c Partial monosomy b
Partial trisomy c
Partial trisomy a
Partial trisomy b
I . a'bc'
8. ab'c'
Partial trisomy c Partial monosomy a Partial trisomy b Partial monosomy c Partial trisomv a
Normal Normal Abnormal or lethal
Abnormal or lethal
Abnormal or lethal
Abnormal or lethal
Abnormal or lethal
Abnormal or lethal
testicular tissue [Saadallah and Hulten, 19851. Only two of the eight possible gametes give rise to phenotypically normal viable offspring. Our observed distribution of CCR carriers (5096, SE 15.9) and karyotypically normal individuals (31.6%, SE k 14.8) among the phenotypically normal CCR carrier offspring are in agreement with the distribution predicted based on the karyotypes of the CCR carrier's gametes. The phenotype and viability of the zygotes resulting from the fertilization of the other six possible gametes would be dependent of the chromosomal breakpoints involved in the CCR. Chromosom- ally abnormal zygotes would be expected to be either phenotypically abnormal or nonviable. Additional chromosomally abnormal gametes could be produced by 4:2 (case 20, for example) or 5:l disjunctional events.
Almost 50% of CCRs ascertained were more complex (Table I) than the hypotheti- cal CCR shown in Figure 3 and involved four or more breakpoints and up to four chromosomes. These more complex rearrangements would be expected to yield still a higher proportion of chromosomally abnormal gametes during meiosis.
Specific chromosomes have been observed to be involved in Robertsonian transloca- tions [Jacobs et al., 19741 and in site-specific familial translocations [Zackai and Emanuel, 19801 in a much higher frequency than would be expected from a random distribution of breakpoints. Stoll [ 19801 identified a statistically significant deviation from random distribution of chromosome breakpoints in 770 reciprocal translocations, with an excess of breakpoints found in chromosomes 4, 9, 13, 18, 21, and 22. Neri et al. [I9831 identified a statistically significant excess of breakpoints in chromosomes 5 , 9, 13, and 15 in 58 reciprocal translocations families. Among liveborn aneuploid infants, excess breakpoints were found in chromosomes 4p, 9p, lOq, 21q, and 22q, with a relative deficiency in chromosomes 1 p, 2p, and 6q [Aurias et al., 19781. Among 545 observed fetal karyotypes, Bout5 and Gallano [ 19841 found that the distribution of the breakpoints did not differ significantly from the expected random distribution when the anomaly was ascertained by a history of recurrent abortions. The distribution of breakpoints did deviate from random, with a relative excess of breakpoints in chromosome 4,9, 18, and 21, when the carrier status of a reciprocal translocation carrier was ascertained through a child with an unbalanced karyotype.

Reproductive Risks for CCR Carriers 259
CCR chromosome breakpoints have been reported previously to exhibit a non- random distribution [Kleczkowska et al., 19821. However, we have found no statistically significant evidence for the non-random involvement of chromosomes in CCRs. The addition of other CCR families will strengthen the statistical power of these analyses.
We recommend that all families containing a person carrying a balanced or unbalanced CCR be fully evaluated cytogenetically. We have presented evidence for the segregation of CCRs through multiple generations. The liveborn offspring of CCR carriers are at significant risk (50%) of being a CCR carrier. The high proportion of pregnancies (53.7%) that results in abnormal outcomes, either spontaneous abnortions or phenotypically abnormal liveborn infants, makes it imperative to identify and counsel all CCR carriers.
ACKNOWLEDGMENTS
We wish to acknowledge the excellent assistance of the Clinical Cytogenetics Laboratory staff. We thank Joanne Owens for the preparation of the manuscript.
J.L.G. was supported in part by National Institutes of Health postdoctoral training grant GM 0751 1. J.L.G. is supported by National Institutes of Health Physician Scientist Award K-1 1-HD 00610. These studies were supported in part by NIH grant GM 32592 to the University of Pennsylvania Genetics Center.
REFERENCES
Allderdice PW, Miller OJ, Miller DA, Breg WR, Gendel E, Zelson C (1971): Familial translocation involving
Anderson 0, Lundsteen C, Niebuhr E (1981): A complex four-break rearrangement between 4 and 13 resulting
Aurias A, Prieur M, Dutrillaux B, Lejeune J (1978): Systematic analysis of 95 reciprocal translocations of
Ballesta F, Fernindez E, Mil6 M (1 980): Translocation t( 1;8;l5) maternelle et trisomie 8qter chez la fille. J
Bass HN, Sparkes RS (1979): Two balanced translocations in three generations of a pedigree: t(7;10)(ql1 ;q22)
Berger R, Derre J, Oritz MA (1974): Les trisomies partielles du bras long du chromosome 7. Nouv Presse Med
Bijlsma JB, deFrance HF, Bleeker-Wagemakers LM, Dijkstra PF (1978): Double translocations t(7;12), t(2;6)
Bitgood JJ, Otis JS, Shoffner RN, Fechheimer NS (1981): A cyclical translocation; t(1;8;5) in the domestic
Bout A, Gallano P (1984): A collaborative study of the segregation of inherited chromosome rearrangements in
Carr DH (1971): Chromosomes and abortions. In Harris € I , Hirschhorn K (eds): “Advances in Human
Creasy MR, Crolla JA, Daker MG (1974): A familial reciprocal translocation between three chromosomes.
Dallapicolla B, Bollea G, Mazzilli C, Gandini E. (1 976): Complex translocation t(9;21)(9;22)(ql2pl3)(p12qll)
Daniel A (1979): Structural differences in reciprocal translocations. Human Genet 51:171-182. Dutrillaux B, Lejeune J (1970): Etude de la dtscendance des porteurs d’une translocation t(2lqDq). Ann Genet
Evans MI, White BJ, Kent SG, Levine MA, Levin SW, Larsen JW Jr (1984): Balanced rearrangement of
Ford CE, Clegg HM (1969): Reciprocal translocation. Br Med Bull 25:11&114.
chromosomes 6, 14 and 20 identified by quinacrine fluorescence. Humangenetik 13:205-209.
in a recombinant chromosome 4. Cytogenet Cell Genet 30:3-10.
autosomes. Hum Genet 45:259-282.
Genet Hum 28:361-366.
and t(14;21)(14qter-.cen-.2lqter). J Med Genet 16:215-218.
3:1801-1804.
heterozygosity in one family. Hum Genet 40:135-147.
chicken Gallus domesticus. Cytogenet Cell Genet 30:243-247.
1356 prenatal diagnoses. Prenatal Diagnosis 4:4547.
Genetics,’’ Vol. 2. New York Plenum, pp 201-249.
Humangenetik 24303-308.
in the family of a child with 9p trisomy syndrome. Hum Genet 33:73-76.
1277-82.
chromosomes 2, 5 and 13 in a family with duplication 5q and fetal loss. Am J Med Genet 19:783-790.

260 Gorskietal.
Francke U, Oliver N (1978): Quantitative analysis of high-resolution trypin-Giemsa bands on human
Fredga K, Hall B (1970): A complex familial translocation involving chromosomes 5, 9, and 13. Cytogenetics
Fryns JP, Kleczkowska A, van den Berghe W (1982): Robertsonian t(Dq;Dq) translocations in man. J Genet
Fujita H, Abe T, Yamamoto K, Furuyama J (1974): Possible complex translocation t(9;14;13)(qI2pl?;q31) in
Gardner RJM, Monk NA, Allen GJ, Parslow MI (1986): A three way translocation in a mother and daughter. J
Gropp A, Winking H (1 98 I): Robertsonian translocations: Cytology, meiosis, segregation patterns and biological
Hamerton JJ, Canning N, Ray M, Smith S (1975): A cytogenetic survey of 14,069 newborn infants. Clin Genet
Handmaker SD, Phil D, Hall BD, Conte FA (1975): A new recognizable syndrome associated with a chromosomal abnormality. In Bergsma D (ed): “New Chromosomal and Malformation Syndromes.” Miami: Symposia Specialists, BDOAS XI(5):181-189.
Hansen RG, Anderson RL, Rary JM (1983): Double balanced chromosomal translocation carrier (6;8)(13;14): A case report. J Hered 74:45G452.
Howard-Pebbles PN, Scarbrough PR, Sharpe J, Finley WH, Finley SC (1982): A complex chromosome rearrangement resulting in trisomy 15q22-qter. J Med Genet 19:224-227.
Hustinx TWJ, Nabben FAE, Scheres JMJC (1979): Partial trisomy of chromosome 1 resulting from a complex maternal rearrangement of chromosomes 1.5, and 6. Am J Med Genet 3:353-358.
Jacobs PA (1977): Structural rearrangements in the chromosomes of man. In Hook EB, Porter 1H (eds): “Population Cytogenetics.” New York Academic, pp 81-97.
Jacobs PA, Aitken J, Frackiewicz A, Law P, Newton MS, Smith PGB (1970): The inheritance of translocations in man: Data from families ascertained through a balanced heterozygote. Ann Hum Genet 34:119-136.
Jacobs PA, Buckton KE, Cunningham C, Newton M (1974): An analysis of the breakpoints of structural rearrangements in man. J Med Genet I 1:5M4.
Joseph A, Thomas IM (1982): A complex rearrangement involving three autosomes in a phenotypically normal male presenting with sterility. J Med Genet 19:375-376.
Kleczkowska A, Fryns JP, van den Berghe H (1982): Complex chromosomal rearrangements (CCR) and their genetic consequences. J Genet Hum 30199-214.
Lejeune J, Dutrillaux B, De Grouchy J (1 970): Reciprocal translocations in human populations (a preliminary analysis). In: “Pfizer Medical Monographs,” No. 5, Edinburgh: University of Edinburgh Press.
Lurie IW, Presman H, Lazjuk GI (1978): Risque gknttique dans les cas de translocations reciprcques. J Genet Hum 26:385-392.
Mattei M-G, Mattei J-F, Bernard R, Giraud F (1979): Partial trisomy 4 resulting from a complex rearrangement of chromosomes 2,4, and 18 with interstitial translocation. Human Genet 515541.
Meer B, Wolff G, Back E (1981): Segregation of a complex rearrangement of chromosomes 6, 7, 8 and 12 through three generations. Hum Genet 58:221-225.
Mennuti MT, Jingeleski S , Schwartz RH, Mellman WH (1978): An evaluation of cytogenetic analysis as a primary tool in the assessment of recurrent pregnancy wastage. Obstet Gynecol52:308-313.
Muneer RS, Donaldson DL, Rennert DM (198 I): Complex balanced translocation of chromosomes 2.3, and 13. Hum Genet 59:182-184.
Neri G, Serra A, Campana M, Tedeschi B (1983): Reproductive risks for translocation carriers: Cytogenetic study and analysis of pregnancy outcome in 58 families. Am J Med Genet 16535-561,
Pai GS, Thomas GH, Mahoney W, Migeon BR (1980): Complex chromosome rearrangements: Report of a new case and literature review. Clin Genet 1 k436-444.
Palmer CG, Poland C, Reed T, Kojetin J (1976): Partial trisomy 11, 46, XX, -3, -20, +der3, +der20, t(3;11;20), resulting from a complex maternal rearrangement of chromosomes 3, 11, 20. Hum Genet 3 1 :219-225.
Paris Conference 1971: Standardization in Human Cytogenetics. (1972): New York The National Founda- tion-March of Dimes. BD:OAS VIII(7):4143.
Rodriguez MT, Martin MJ, Abrisqueta JA (1985): A complex balanced rearrangement involving four chromosomes in an azoospermic man. J Med Genet 22:66-67.
Saadallah N, Hulten M (1985): A complex three breakpoint translocation involving chromosomes 2,4, and 9 identified by meiotic investigations of a human male ascertained for subfertility. Hum Genet 71:312- 320.
prometaphase chromosomes. Hum Genet 45: 137-165.
9:294-306.
Hum 3O:lOl-117.
mother of a child with 9p trisomy syndrome. Humangenetik 25:83-92.
Med Genet 23:90.
consequences of heterozygosity. Symp Zoo1 Soc London 47:141-181.
8:223-243.

Reproductive Risks for CCR Carriers 261
Sanchez 0, Yunis JJ, Escobar JI (1974): Partial trisomy 11 in a child resulting from a complex maternal rearrangement of chromosomes 11, 12, 13. Humangenetik 22:5945.
Schwanitz G, Schmid P, Berthold HJ, Grosse KP (1978): Partial trisomy 13 with clinical signs of Patau syndrome resulting from a complex paternal rearrangement of chromosomes 6, 10 and 13. Ann Genet 21: 10&103.
Schwinger E, Mikkelsen M, Niesen M (1 975): Familial balanced (7;11;21) translocation and Down's syndrome in two siblings. Clin Genet 7:304-307.
Simoni G, Montali E, Rossella F, Dalpra L, Lo Curto F (1979): A woman carrier of two apparently unrelated reciprocal translocations. Hum Genet 46:15%162.
Stene J , Stengel-Rutkowski S (1977): Risk for short arm 10 trisomy: A segregation analysis of eleven families with different translocations. Hum Genet 39:7-13.
Stoll C (1980): Nonrandom distribution of exchange points in patients with reciprocal translocations. Hum Genet 56:89-93.
Tabor A, Jensen LK, Lundsteen C , Niebuhr E (1981): A 5;7, 5;12 double reciprocal translocation in a normal mother and a 5;7 translocation with a recombinant chromosome 5 in her normal child. J Med Genet 18:307-309.
Tanaka N, lkeuchi T, Yara I, Kitahara K (1977): Trisomy 9p due to a maternal complex translocation involving chromosomes 4,6, and 9. Jpn J Hum Genet 21:261-268.
Voss R, Gross-Kieselstein E, Hurvitz H, Dagan J , Kerem E, Zlotogora J (1984): A complex three way translocation resulting in two sibs with partial trisomy 3 p 2 3 ~ 3 p t e r . J Med Genet 21:454459.
Walker S, Howard PJ, Hunter D (1985): Familial complex autosomal translocations involving chromosomes 7, 8, and 9 exhibiting male and female transmission with segregation and recombination. J Med Genet 22484491.
Warburton D, Clarke Fraser F (1964): Spontaneous abortion risks in man: Data from reproductive histories collected in a medical genetics unit. Hum Genet 16:l-25.
Zackai EH, Emanuel BS (1980): Site-specific reciprocal translocation, t(l1;22)(q23;qll) in several unrelated' families with 3:l meiotic disjunction. Am J Med Genet 7507-521.
Edited by John M. Opitz and James F. Reynolds




















