
Relationships between the structure of a zeolite and its adsorption properties
11
ELSEVIER Surface Science 397 (1998) 395-405 surface science Relationships between the structure of a zeolite and its adsorption properties A. Arbuznikov 1, V. Vasilyev, A. Goursot * Laboratotre de Matdrzaux Catalytzques et Catalyse en Chzmw Orgamque, UMR 5618 CNRS, Ecole Natwnale Supdrwure de Chtmw, 8 Rue de l'Ecole Norrnale 34296 Montpelher, Cedex 5, France Receaved 26 June 1997, accepted for pubhcatmn 22 September 1997 Abstract The adsorptmn of N 2 has been stu&ed m different Na-LSX Zeohtes, using an embedded cluster approach. Embedded catmn-molecule systems modeling sates II and sites III of Ideal LSX zeolltes (Si/A1= 1) have been treated quantum chemically, using a method based on density functmnal theory. Two experlmental structures have been compared with two artificially modafied structures The adsorptaon strength of the caUomc sites is correlated with the values of the electrac field and electric field gradaent in the supercages © 1998 Elsevxer Science B V Keywords Clusters. Density functional calculations, Eqmhbrmm thermodynanncs and statistical mechamcs, Nitrogen, Physical adsorption, Sohd gas anterfaces, Zeohtes 1. Introduction Adsorption properties of zeolites are strongly related to the type, number and location of their accessible cations, which are the sites for adsorp- tion. It is not yet clearly established if the frame- work structure also plays a role in the adsorption capacity and selectivity. N2 selective adsorption is an equilibrium process which originates from elec- trostatic interactions with the zeolite cations. In previous theoretical studies, we have shown that N2 adsorption in low sihca Na zeolites (Na-LSX) indeed depends on the positions of the accessible cations [ 1,2]. The comparison of adsorption ener- * Corresponding author Fax (+33) 4 67 14 43 49, e-marl: goursot@rhodmm enscm fr 1Permanent address: Boreskov InsOtute of Catalysis, Prospekt Akademlka Lavrentleva, 5-630090 Novoslblrsk, Russm 0039-6028/98/$19 00 © 1998 Elsevier Science B V All rights reserved. PH S0039-6028 (97) 00760-7 gies calculated at sites II and III in several frame- work geometries of faujasite type zeolite (Fig. 1 ) has revealed that the zeolite structure induces a strong dissymmetry among the cations and modu- lates the adsorption strength at different sites. In these Na-LSX structures, the preferred sites for adsorption are always site III cations in agreement with recent experimental results involving X-zeohtes with monovalent cations [3,4]. Moreover, these previous modeling results have underlined the role of a favorable orientation of the electric field generated by the whole zeolite system m the adsorption selectivity. In the present work, we study in more detail how structural parameters (framework geometry and cation posi- tions) are active factors in the adsorption capacity of Na-LSX zeohtes. To this end, we have compared N2 adsorption energies in two experimental structures which differ by their framework geometry and cation
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Relationships between the structure of a zeolite and its adsorption properties

ELSEVIER Surface Science 397 (1998) 395-405
s u r f a c e s c i e n c e
Relationships between the structure of a zeolite and its adsorption properties
A. Arbuznikov 1, V. Vasilyev, A. Goursot * Laboratotre de Matdrzaux Catalytzques et Catalyse en Chzmw Orgamque, UMR 5618 CNRS, Ecole Natwnale Supdrwure de Chtmw,
8 Rue de l'Ecole Norrnale 34296 Montpelher, Cedex 5, France
Receaved 26 June 1997, accepted for pubhcatmn 22 September 1997
Abstract
The adsorptmn of N 2 has been stu&ed m different Na-LSX Zeohtes, using an embedded cluster approach. Embedded catmn-molecule systems modeling sates II and sites III of Ideal LSX zeolltes (Si/A1 = 1 ) have been treated quantum chemically, using a method based on density functmnal theory. Two experlmental structures have been compared with two artificially modafied structures The adsorptaon strength of the caUomc sites is correlated with the values of the electrac field and electric field gradaent in the supercages © 1998 Elsevxer Science B V
Keywords Clusters. Density functional calculations, Eqmhbrmm thermodynanncs and statistical mechamcs, Nitrogen, Physical adsorption, Sohd gas anterfaces, Zeohtes
1. Introduct ion
A d s o r p t i o n p roper t i e s o f zeoli tes are s t rongly re la ted to the type, n u m b e r and loca t ion o f thei r accessible cat ions , which are the sites for adso rp - t ion. I t is no t yet c lear ly es tab l i shed i f the f rame- w o r k s t ructure also p lays a role in the a d s o r p t i o n capac i ty a n d selectivity. N2 selective a d s o r p t i o n is an equi l ib r ium process which or ig inates f rom elec- t ros ta t i c in te rac t ions wi th the zeoli te cat ions. In prev ious theore t ica l studies, we have shown tha t N2 a d s o r p t i o n in low sihca N a zeoli tes ( N a - L S X ) indeed depends on the pos i t ions o f the accessible ca t ions [ 1,2]. The c o m p a r i s o n o f a d s o r p t i o n ener-
* Corresponding author Fax (+33) 4 67 14 43 49, e-marl: goursot@rhodmm enscm fr
1Permanent address: Boreskov InsOtute of Catalysis, Prospekt Akademlka Lavrentleva, 5-630090 Novoslblrsk, Russm
0039-6028/98/$19 00 © 1998 Elsevier Science B V All rights reserved. PH S0039-6028 (97) 00760-7
gies ca lcu la ted at sites I I and I I I in several f rame- w o r k geometr ies o f faujas i te type zeoli te (F ig . 1 ) has revealed tha t the zeoli te s t ructure induces a s t rong d i s symmet ry a m o n g the ca t ions and m o d u - lates the a d s o r p t i o n s t rength at different sites. In these N a - L S X structures , the p re fe r red sites for a d s o r p t i o n are a lways site I I I ca t ions in agreement with recent exper imenta l results involving X-zeohtes wi th m o n o v a l e n t ca t ions [3,4]. Moreove r , these p rev ious mode l ing results have under l ined the role o f a f avorab le o r i en ta t ion o f the electric field genera ted by the whole zeoli te system m the a d s o r p t i o n selectivity. In the presen t work , we s tudy in m o r e deta i l how s t ruc tura l pa r a me te r s ( f r a me w ork geomet ry and ca t ion posi- t ions) are active fac tors in the a d s o r p t i o n capac i ty o f N a - L S X zeohtes.
To this end, we have c o m p a r e d N2 a dso rp t i on energies in two exper imenta l s t ructures which differ by thei r f r a m e w o r k geomet ry and ca t ion

396 A Arbuzmkov et al / Surface Scwnce 397 (1998) 395-405
Fig 1 Faujaslte type structure with cationic sites (a) "ideal" site III, (b) monocoordmated sate III, and (c) and blcoordlnated sate [[I
distributions and two imaginary structures gener- ated by specific modifications of the experimental structures. In all cases, N2 adsorption energies are evaluated at all the accessible cationic sites. The impact of the structural parameters on the adsorp- tion of N2 is then analyzed in terms of long-range energetic contributions.
2. Model structures
Dehydrated X-zeohtes (Si/A1 < 1 5) contain gen- erally a maximum of 64 cations in sites I ( I ' ) and I I ( I I ' ) , (Fig 1). There are 48 possible sites III , called "1deal", coordinated to two 04 oxygens, whereas 96 other positions are coordinated to O1 and 04 oxygens (192 if A1 and S1 are differenti- ated). Mono-coordinated positions (to O1 or 04) have also been reported.
Assuming a full occupancy of 64 cations in sites I ( I ' ) and I I (II ' ) , 32 cations have thus to be distributed among type I I I sites, for an Ideal LSX zeolite with a Si/A1 ratio equal to 1 For the present study, we have chosen to analyze two recent experi- mental structures, referred to as OLS [5] and ALA [6] because they contain only sodium cations and have comparable composmons with 88 and 86 cations, respectively.
Both contain 32 cations m sites I ' and II. They differ by the location of type I l l cations and also by some structural parameters (Table 1 ). The ALA structure contains type III cations at ideal sites, whereas the OLS structure has 21 N-coordinated and 11 monocoordinated cations. The main struc- tural difference between the two frameworks concern the geometry of the four-membered nngs, with two 04 oxygens much more separated in the ALA structure (4.5 A) than in the OLS structure (3.8 A)

A Arbuzmkov et al / Surface Scwnce 397 (1998) 395-405
Table 1 Structural characterastaces of the studied models (distances in A and angles in degrees)
397
Site III Coordination R(O1-O1) R(O3-O3) (RO4-O4) (SxO4A1) (OIA104) R(Na-O3) R(Na-O4)
Ideal Mono BI
ALA 32 3 66 3 67 4 50 114 5 99 4 2 37 2 28 OLS 11 21 383 366 381 1455 1099 242 ALAM 32 3 66 3 67 3 78 119 3 98 5 3 14 2 28 OLSM 32 3 83 3 66 3 81 145 5 109 9 2 80 2 28
which implies also large differences in the (O1A104) angles. In fact, these f ramework differ- ences m a y be connected with the different ca t ion posit ions. Indeed, a larger 0 4 - 0 4 distance m a y be more suitable for N a ÷ In ideal sites and a larger (O1A104) angle for b t -coord ina ted cat ions This po in t is not essential for our study since our purpose is to delineate how structural characterist ics can be corre la ted with a bet ter adsorp t ion capacity. Start ing f rom these structures, we have also generated two new ad hoc systems: the A L A M mode l is identical to the A L A structure except for the 0 4 - 0 4 distance which has been reduced to 3.78 A, i.e. a value similar to that found in the OLS structure. The (A104Si) angles are kept constant , as is the N a + - O 4 chstance As a consequence, the N a ÷-O3 distance is increased. The second "art i f icial" structure, O L S M , keeps the OLS f ramework structure bu t contains ideal site I I I cations, as in the A L A structure (Table 1) In the four structures, the 32 type I I I cat ions have been dis t r ibuted among the chosen site locat ions using the minimiza t ion of their mutua l Cou lomb repul- sion, all o ther cat ions in sites I ' and I I ' being kept fixed. There are several dis tr ibut ions o f type I I I cat ions which have comparab le stabilities. Fo r the OLS structure, the 32 cat ions (21 b i -coord ina ted and 11 mono-coo rd ina t ed ) are dis t r ibuted among 240 possible sites, which leads to an estmaated error o f about 1 kcal /mol on the N2 adsorp t ion energies (the largest difference ob ta ined f rom three chfferent distr ibutions). This error is reduced to 0.3 kcal /mol when the cat ions are in the ideal sites (only 48 possible locations).
3. Theoretical methodology
The s t ra tegy a d o p t e d for this s tudy IS to mode l N 2 a d s o r p t i o n in tu rn at each o f the 64 accessible
ca t ions in one crysta l uni t cell (32 site I I and 32 site I I I ) . In o rde r to p rope r ly descr ibe the electro- stattc in te rac t ion be tween the molecule and the zeoli te at different sites, an e m b e d d e d cluster a p p r o a c h has been a d o p t e d
A d s o r p t i o n at each site is then descr ibed by a q u a n t u m ca lcu la t ion o f an N a + N 2 system sur- r o u n d e d by its specific ne tw ork o f po in t charges, which s imulate the A1, Si, O and N a ÷ zeoli te ions. In this model , the e lec t ros ta t ic in te rac t ions between the zeoli te ions and the N a ÷ N 2 system are included, as well as thei r effects on its e lec t ron dens i ty and thus on the N 2 adsorp t ion energy ( induc t ion terms). Dispers ive terms between N2 and the f r a m e w o r k ions have been neglected (dis- tances larger than 9 bohrs) .
The ne twork o f ex terna l charges comprises the charges o f a centra l uni t cell (except for the N a ÷ cat ion) , su r rounde d by a full shell o f 26 first ne ighbor cells. Each uni t cell IS neu t ra l and the e r ror o f convergence due to this t runca t ion IS less than 0.4% for the e lec t ros ta t ic po ten t i a l and the electric field, in the centra l uni t cell 2
Ca t ion pos i t ions have been fixed at the experi- men ta l site loca t ions for ideal , m o n o - and b i - coo rd ina t ed ca t ions to faci l i ta te the c o m p a r i s o n be tween site character is t ics . In any case, ca t ion d i sp lacement u p o n N2 a d s o r p t i o n mus t be very small due to the weakness o f this in te rac t ion with respect to the c a t i o n - f r a m e w o r k b ind ing energy,
z This error has been evaluated as the difference between the electrostatic potential (ESP) and electric field (F) values acting on a probe charge situated at the middle of the central umt cell when the embedding is restricted to 26 umt cells and when at reaches its hmlt for a convergence of 10 -3 kcal/mol for the ESP and 10 -6 a u for the field (ESP= - 165 210 kcal/mol and F= 0 045000 a u for the OLS structure)
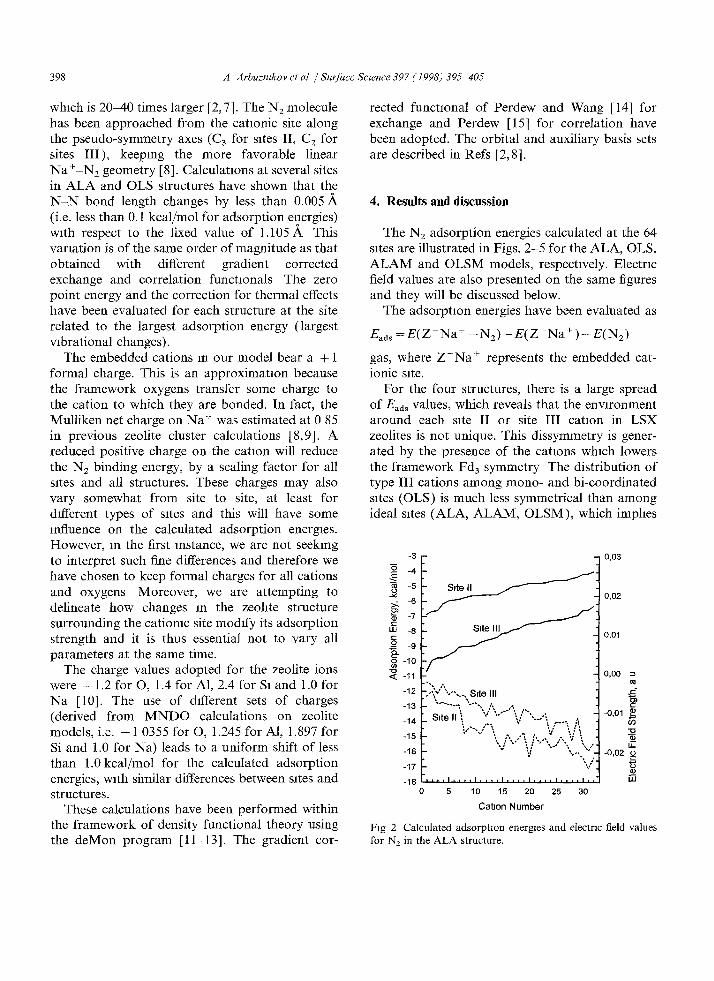
398 A Arbuzmkov et al / Surface Scwnce 397 (1998) 395-405
which is 20-40 times larger [2, 7]. The N 2 molecule has been approached f rom the cationic site along the pseudo-symmetry axes (C 3 for rotes II, C 2 for sites I I I ) , keeping the more favorable linear Na+-N2 geometry [8]. Calculations at several sites in A L A and OLS structures have shown that the N - N bond length changes by less than 0.005 (i.e. less than 0.1 kcal/mol for adsorption energies) with respect to the fixed value of 1.105A This variation is of the same order of magnitude as that obtained with different gradient corrected exchange and correlation functIonals The zero point energy and the correction for thermal effects have been evaluated for each structure at the site related to the largest adsorption energy (largest vibrational changes).
The embedded cations in our model bear a + 1 formal charge. This is an approximation because the f ramework oxygens transfer some charge to the cation to which they are bonded. In fact, the Mulliken net charge on Na ÷ was estimated at 0 85 in previous zeolite cluster calculations [8,9]. A reduced positive charge on the cation will reduce the N2 binding energy, by a scaling factor for all sites and all structures. These charges may also vary somewhat from site to site, at least for different types of sates and this will have some mfluence on the calculated adsorption energies. However, in the first Instance, we are not seeking to interpret such fine differences and therefore we have chosen to keep formal charges for all cations and oxygens Moreover, we are attempting to delineate how changes in the zeohte structure surrounding the catiomc site modify its adsorption strength and it is thus essential not to vary all parameters at the same time.
The charge values adopted for the zeolite ions were - 1.2 for O, 1.4 for A1, 2.4 for S1 and 1.0 for Na [10]. The use of different sets of charges (derived from M N D O calculations on zeolite models, i.e. - 1 0355 for O, 1.245 for A1, 1.897 for Si and 1.0 for Na) leads to a uniform shift of less than 1.0 kcal/mol for the calculated adsorption energies, with similar differences between sites and structures.
These calculations have been performed within the f ramework of density functional theory using the deMon program [11-13]. The gradient cor-
rected functional of Perdew and Wang [14] for exchange and Perdew [15] for correlation have been adopted. The orbital and auxiliary basis sets are described in Refs [2,8].
4. Results and discussion
The N2 adsorption energies calculated at the 64 sites are illustrated in Figs. 2-5 for the ALA, OLS,
A L A M and OLSM models, respectively. Electric field values are also presented on the same figures and they will be discussed below.
The adsorption energies have been evaluated as
Eaas = E ( Z - N a + - - N 2 ) - - E ( Z - N a + ) - - E ( N 2 )
gas, where Z - N a + represents the embedded cat- ionic site.
For the four structures, there is a large spread of Eaa s values, which reveals that the environment around each site II or site I I I cation in LSX zeolites is not unique. This dissymmetry is gener- ated by the presence of the cations which lowers the f ramework Fd3 symmetry The distribution of type I I I cations among mono- and bi-coordinated sites (OLS) is much less symmetrical than among ideal sites (ALA, ALAM, OLSM), which imphes
-3 - 0,03
o~ -5 Site II , i 0 , 0 2 ~, -6
-7
LU -8 O,01
-~ -9 Q. 0 -10 ,~ -11 - 0,00 =
-12 ...~:..; \ . . . ... Site I,1 ,.~ -13 E" -14 Szte II \ 'v" ".-" \ ' " . . . . . \ .,, -0,01 09
-15 ; .", .'~ ~ . . " • V " ~_ 1.1_
-16 . . - . . / -0,02 o
-17 "~ -18 , , , I , , , , I , , , , I , , , , I , , , , I . , , , I , Ill
5 10 15 20 25 30
Cation Number
Fig 2 Calculated adsorptzon energzes and electric field values for N2 in the ALA structure.

A Arbuzmkov et al / Surface Sctence 397 (1998) 395 405 399
-1 - 0,05
-2
0,04 o~ -3 2 ~
-4 0,03 -5
LU ~ 0,02 --~ -7 / Si te III / mo -8 0,01 -o < -9
-10 0,00 .E
-11 ,, . .- . . . , S~te III ~ ) . . . . . , t, , -0,01
-12 ~ ",---"v',,, / i / ' , . ', i" ;, ~ ', i " ',/"-" r-,
-13 ', i " i', i", ":, ' f A : \ ; -0,02
O -14 .t ~ " . t %.'." "'* ; ; " "" -0,03 - -15 S~te II , , j ~ ,t .. - . . . . . . -- . ~m
- 1 6 . , . I . . . . I . . . . I . . . . I . . . . I . . . . I , U J
5 10 15 20 25 30
Cahon N u m b e r
Fig 3 Ca lcu la t ed adso rp t i on energies and electric field values for N 2 in the OLS s t ruc ture
"~ 0,02
>~ -5
-6 ite II 0,01
LU -7
o. -8 0,00 m ° S i t e II -o -9 <
:, , . O5
- 1 0 A -0,01
-11 , ~..'" Y , , . . -v i '~ , . ",.t..t i",, , . . = =
-12 :,: v ..~; " ".';.,~_" il "~ _1 -0 ,02 -o
:' "1 '-f- -13
o E Site Ill
~O ~03
5 10 15 20 25 30 Cat~on N u m b e r
Flg 4 Ca lcu la t ed a d s o r p t m n energies and electric field values for N 2 m the A L A M s t ruc ture
the largest range of adsorption energies for those sites.
The analysis of Figs 2 and 3 shows that in the experimental A L A and OLS structures, adsorption must occur at sites III , which correspond, for the great majority, to larger adsorption energies. This is still the case In the OLSM structure (OLS framework with cations in the ideal sites). However, this is no longer true for the modified
-2 0,04
: -4 0,03
~, -5
-6 0,02 LU O_ ~ -7 / Site III ~ - -8 0,01 o
~< -9 - , , S, te II I 0,00 '~ . E - io 7..,... ..... ==
• , ,, ,,
-11 \ ",i::'~...,, ,,,,,,',, ..',, :, -J -0.01 m ", ~ ; : ' " .~ ,V ' / " , . ,~
-12 t ",. ! k ..'~. ~..i',~,,..,,, ~_~ ft. -13 . . . . • , ; ' ; , ' t ', ; ~ - o ", ", : ¢ ; ' :~ • ' " _n,n'>
-14 S i te II v v "-, : ": L" ",,', ~1 r_ '~ " " . ,1
-15 , , , ~ . . . . i . . . . = . . . . i . . . . = . . . . I , , ] w 0 O3 0 5 10 15 20 25 3O
Cat=on Number
F ig 5 Ca lcu la ted a d s o r p t m n energies and electric field values for N 2 m the O L S M s t ruc ture
A L A M structure, where the adsorption capacity of 1deal site I I I cations is so diminished that site I I cations become preferable. In order to delineate general trends, we have averaged adsorption ener- gies for sites II and III.
These (Eaas)av values, reported m Table 2, lead to the following remarks: • For ALA and OLS structures, site I I I adsorp-
tion is much more favorable, whereas the ten- dency is reversed for the A L A M model. In the OLSM structure, sites I I I are still preferable but the difference with sites II is smaller.
• The A L A structure has the largest adsorption capacity at all sites.
• Keeping the OLS framework and moving site I I I cations f rom mono- and bl-coordlnated posi- tions to ideal posmons (OLSM) increases essen- tially site II adsorption energies.
• Modifying the four-membered ring geometry in the ALA structure (keeping ideal cation posi- tions) degrades considerably site IH adsorption, without affecting site II adsorption. Site occu- pancies at temperature T are gwen by p~=PdPtot, where P, is the Boltzmann factor exp(--Eaas(i)/kT) and Ptot is the sum of the 64 P, values, Eaa~(i) being the adsorption energy at site t

400 A Arbuzmkov et al / SurJace Scwnce 397 (1998) 395 405
Table 2
Average and statasnc adsorption energies and enthalples (kcal/mole)
(E~a~)~ (Ead~) p, values
Site II Site III
//,d,
ALA - 5 1 - 8 2 - 9 9 a OLS - - 2 8 - - 4 8 - 8 6 ~ A L A M - 5 2 - - 4 0 - - 6 3 b
OLSM - 4 2 - 5 4 - 7 3 ~
041, 0 12, 009, 009, 007 --8 7 a
0 67, 0 23, 0.07 --7 6" 0 25, 0 23, 0 06, 0 05, 0 05 b --5 3 b
045, 0 14, 009 - -63"
aSate I l l bSlte II
The statistical adsorption energy ~Eads) is then evaluated as ~[p,Eaas(i)].
The conversion to an enthalpy of adsorption has been obtained using
Haas = <Eaas > + AEt . . . . (T) + AErot(T )
+ A(ZPE) + AE~b (T) + RT,
where AEt . . . . and AEro t are the changes in the translational and rotational energies, A Z P E is the zero point energy contribution to the adsorption energy and AEvl b the contribution for thermal effects on the vibrational energy.
The corresponding (Eaas) and Haa s values are compared In Table 2. We see from these values that the trends indicated by average energies for sites II and I I I are still valid: the adsorption strength of ideal sites I I I in the A L A M structure is so much decreased by the f ramework deforma- tion that sites II become more favorable. The effect of moving site I I I cations In the OLS structure is not so dramatic, although it leads to a decrease of the adsorption energy (OLSM) These results show that there is indeed a clear correlation between structural characteristics and site adsorption strengths.
In an at tempt to dehneate more precisely the physical properties on which this correlation is based, we have used a simplified treatment of intermolecular forces, between the adsorbed N2 molecule and the surrounding zeolite Ions. We can indeed estimate that the adsorption energy at each site can be written as:
Eaas = AE(Na + N 2 ) + Emt ,
where AE(Na+N2) is the binding energy of N2 on
an isolated sodium cation ( - 8 . 5 0 kcal/mol) and gmt corresponds to the energetic modification due to the external charges. Since the zeolite sodium interaction is present in both energies E(Z-Na+N2) and E ( Z - N a + ) , E ~ n t can be approximated as the interaction energy of the surrounding zeolite with a n N 2 molecule at its absorbed position, but already polarized by the sodium cation. The distances between the N 2 mole- cule and the embedding charges being sufficiently large ( > 9 bohrs); therefore, Elnt can be described as a long-range interaction energy [16]. When polarized by the cation, the N: molecule has a non-negligible dipole moment and an increased quadrupole moment For example, a n N 2 molecule polarized by ° a positive point charge located at 2.35 (2.55) A, has dipole and quadrupole moment values of 0.61 (0.52) and - 2 . 0 5 ( - 1 . 8 5 ) a .u , respectively.
Lint can be expressed as a power series in F, iv', etc :
Emt = - - # .g - - ( 1/3)O: g ' - - ( 1/2)#,.F
- ( 1/6)@: F' . . . ,
where the first two terms account for the electro- static interaction between the zeolite and the N2 molecule polarized by the cation only, such that # and O can be considered as its permanent dipole and quadrupole moments The last two terms correspond to the polarization of the N2 molecule by the electric field vector F and electric field gradient tensor F ' due to the embedding charges, which generate Induced dipole (#0 and quadru- pole (03 moments. In this expression, we assume that the multapolar expansion is performed around

A Arbuzntkov et a l / Surface Scwnce 397 (1998) 395-405 401
a single center, i.e. the center of mass of N 2. We believe that this approximation is sufficient to allow qualitative conclusions. The induction terms are always negative and will contribute to increase the N 2 binding energy. In contrast, the electrostatic terms may be positive or negative, depending on the signs of/~, O, F and F'.
The N 2 molecule, polarized by the sodium cation, has a dipole vector directed f rom N1 to N2, N 1 being the end bound to the cation. Depending on the orientation of the electric field vector F, the electrostatic contribution - /~ .F will be negative or positive. In the same way, the quadrupole electrostatic term will contribute to an increase of the adsorption energy if the F ' compo- nents have the same sign as the corresponding O components.
In order to correlate the variations of the calcu- lated adsorption energies with the Emt values, the electric field and its gradient have been evaluated at each cationic site. For this purpose, a local coordinate system is defined at each site, with the origin at the middle of N - N and the OZ axis cohnear with Na +-N1 N2 (positive direction from Na + to NO. The F' tensor is also calculated in this reference system Since the quadrupole tensor of N 2 is diagonal, the off-diagonal elements of F ' do not contribute. Moreover, we use the property that their traces are zero and that the quadrupole tensor has an axial symmetry. Finally, the expres- sion - ( 1 / 3 ) O : F ' is reduced to a single term - - ( 1 / 2 ) O z z F ' z z . The scalar product - /~ . F is also reduced to the single term, - # z F z , in the reference system chosen.
In order to analyze the Fz and F)z components, a grid of points is defined in a plane containing Na+Nz and the two framework oxygens bound to Na +. The values of Fz and F)z are then evaluated at the grid points, leading to contour maps Rotat ing the plane of analysis around the Z-axis allows one to explore the space at each site.
The F)z values at the "best" sites I I and sites I I I are presented in Figs, 6a, b and 7a, b for the ALA, ALAM, OLS and OLSM structures, respec- tively By "best" site is meant the site where the N2 adsorption energy is the largest. All sites I I and all sites I I I for a given structure present very comparable patterns of the F)z component. The
a) Site II
1
t o N 2 o
~ , ,+ ON1 /
-2 "o7'
.3
-1 0
X,A
Site III
1 +
0 .¸
-2
-3 -
-1 0
X, A
b) Site II Site III
<
• N2
o
• N1
-1 X "4" J
f -2
-3 -
-1 0
x,A
-<
1 -
• N2 [
O- I
• N1 -1 l
-1 o I
Fig 6 Contours of the elecmc field gra&ent (F)z component) for the "best" sites II and III m (a) the ALA0 and (b) the ALAM structures
maps presented on Figs. 6 and 7 are thus represen- tative of the gradient patterns at all other cations of the same type in the same structure.
Examination of the maps presented on Fig 6a shows that the whole Na+Nz system is immersed in a region with negative F~zz values, when Na ÷ is a site I I I cation. For sites II, the zero contour is situated before NI, so that the N 2 molecule is not in the negative F)z region but the gradient values around Nz are very small For the A L A M struc-

402 A Arbuzmkov et al / SulJaee Scwnce 397 (1998) 395-405
a)
.q
- 2
-3 z
S~te II
@ N2
~ o4 • N1
J 0
x,A
-1-
M
Site III
N2 •
N1 •
\,
o 1
X,A.
b) S~te II Site III
] Nzo 0
- 3 -
-1 0
X.A
1
• N2 0 -
• N~ -1 4"
-2
-4 , .
-] 0
X.A
Fig 7 Contours of the electric field gradient (F'zz component) for the "'best" sites II and III in (a) the OLS, and (b) the OLSM structures
ture, the two nitrogen atoms are m a posmve F'zz region for both sites I I and sites III .
For both sites I I and I I I of the OLS and OLSM structures (Fig. 7a and b), N2 is in a positive region of the gradient. Only sites I I I of the A L A structure are thus m the favorable situation where the quadrupole electrostatic term ( - 1 / 3 0 : F') is negative, i e. where N2 is stabilized by the zeohte framework.
The OLSM structure which also contains canons at ideal sites I I I has comparable patterns for the
F)z contours with negative wells close to the cations However, the F)z absolute values are much smaller and become zero close to N1. These results show that the shape and values of the F'zz contour lines are mostly determined by the frame- work geometry.
Let us now turn to the examination of the values of the electric field component Fz, evaluated at the middle of the N - N bond, at each canon and for the four structures (Figs. 2-5) . Let us first recall that due to the # direction, a positive Fz compo- nent would favor the N2 adsorption energy (Emt
more negative). Examination of Figs. 2-5 shows that positive Fz values are never found but that, mdeed, for the four structures, the largest adsorp- tion energies correlate with almost zero Fz values Moreover, it is clearly visible that, although there is no one-to-one correspondence between Ea~s and Fz values, there is an overall trend that the adsorp- tion energies decrease when F z values become more negative We can also see that, in the ALAM structure, the reverse order for site I I and site I I I adsorption energies is reflected m the related Fz curves. One striking feature is the very large varia- tion of the Fz values from one site I I to another for the ALAM, OLS and OLSM structures. Since, in contrast, the variations of the electric field component in the ALA structure are comparable at site I I and site III , these characteristics must be related to the f ramework structure.
For all structures, the - # F electrostatic contri- butions are always posmve and must thus contrib- ute to decreasing the Na+-N2 adsorption energy The irregularities which appear in the curves relat- ing Eaas and Fz values show that the other terms of E~nt are also involved in the correlation.
It is thus interesting to analyze the correlation between E a d s and the two terms of the electrostatic contribution, 1.e. Eel~c = - # F-- (1 /3)O: F' To this aim, we have used the dipole and quadrupole moment values induced in an N2 molecule by the presence of a ÷ 1 charge simulating Na +, situated at 2.35 A from N1 for sites I I I in the ALA structure and 2.55 A in all other cases (see above)
The approximate electrostatic contributions to the zeolite-N2 interaction energy are presented on Figs. 8a, b and 9a, b for the ALA, ALAM, OLS and OLSM structures, respectively They are corn-

A Arbuzmkov et al / Surface Scwnce 397 (1998) 395-405 403
-6 E.
II)
ILl
9
8
7
6
5
4
3
2
1
0
-1
-2
-3 -3
i . t "
% (t
/ "
. / , I , I , I , I i I i l i I i I
-2 -1 0 1 2 3 4 5
Earls - Eads(iso), kcal/rnol
a)
m o E
o ~
uJ
13
12
11
10
9
8
7
6
5
4
3
2
1
0
-1 -1
IIiii~ ~. ,~Jl r'1
'
/ ' /
/ J / /
• I , I , I , I , I , I , i , I ,
O 1 2 3 4 5 6 7
Eads-Eads0so), kcal/mol
a)
m o
o" _e
I.U
11
10
9
8
7
6
5
4
3
2
1
! I!
i' .',.i ,# L i
# I
, 4 s ~i
I !'" I
i i! l .'"
b)
I I I , ]
3 4 5 6
Eads - Eads(iso), kcal/mol
Fag 8 Electrostatic contr ibut ion to the N 2 zeohte anteractaon energies compared with calculated adsorption energaes, m (a) the ALA, and (b) the ALAM structures Sates III correspond to sohd hnes and sates II to dashed lanes
11L / 10L Ii
i! ii 'i 8 ~- ,, :,l|~J I - l i i f '
7 f il i, i e
~ 6 I ~ ] -~ ~; " " " 5 I j~, ,...,.
2 ,/"
0 0 1 2 3 4 5 6
Eads-Eads0so), kcal/mol
Fag 9 Electrostatic contrabutton to the N2-zeollte tnteractaon energies compared wath calculated adsorption energies, in. (a) the OLS, and (b) OLSM structures Sates Il l correspond to solid lanes and sates II to dashed lanes
pared with the differences Eads--Eads(iSo), where Eaas(iso) is the Na + N 2 binding energy m the isolated system (--8.50 kcal/mole). Deviation of the curves from the straight line gives a measure of the quality of our approximations.
From these figures, it is clear that the variation
of the N2 adsorption at different sites and in different structures correlates with Its electrostatic interaction with the zeolite The correlation is qmte good for site III and also for site II in the ALA structure. The large oscillations of the elecmc field values at site II in the three other structures still

404 A Arbuzmkov et al / Suiface Sczence 397 (1998) 395-405
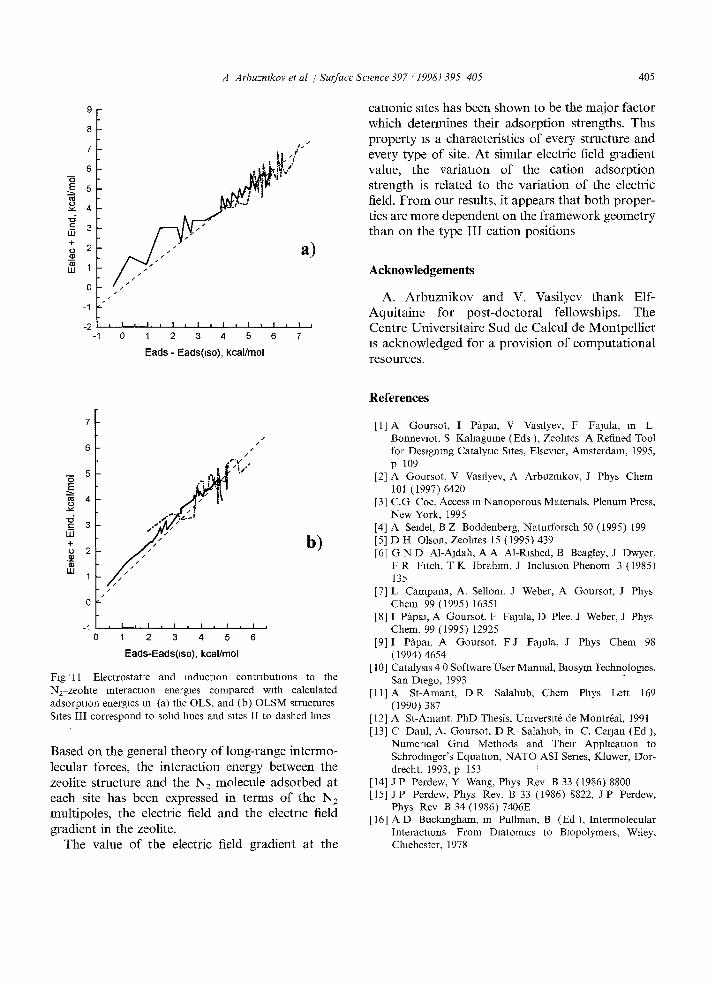
appear on these curves, which means that the effect of the zeolite on the N2 electronic structure is also important. In order to verify that induction inter- actions do, indeed, improve the correlation, the induction term E m a = - - ( 1 / 2 ) l ~ F = - ( 1 / 2 ) ~ : F 2 has been added to the electrostatic contrabution, wath c( /= 15.0 and ~3_ =9.3 a.u. f rom D F T calcula- tions. The induced quadrupole term - (1 /6 )O , : F' is more difficult to evaluate since we have no explicit way to obtain the quadrupole polanzabli ty from our quantum chemical calculations. However, according to the indirect estimations, in our models, the quadrupole induction term never exceeds 0.3 kcal/mol in magnitude and can thus been neglected. 3 The curves corresponding to Eelecq-Emt are presented in Fig 10a (ALA), Fig. 10b (ALAM) , Fig. l l a (OLS) and Fig l l b (OLSM). They show a clear improvement of the correlation with the QM adsorption energies.
These results show that the electrostatic inter- action with the zeolite structure gives the general trend of the N2 adsorption, whereas reduction interactions are necessary for a more quantitative descnptaon Since both the elecmc field and its gradaent play a role, there is a combined influence of the local surrounding of the site (actmg more through F') and of long distance effects (F).
5. Conclusion
Embedded cluster calculations have been used to model N2 adsorption at sites I I and sites II1 of an ideal N a - L S X zeolite. In order to understand how the f ramework structure and cation positions may influence the adsorption strength of these sites, we have studied two experimental structures which display different four-membered geometries and site I I I posmons The study has been com- pleted with two imaginary structures obtained from ad hoc modifications of the two experimental
3 O, was taken as O r - O , where O is the quadrupole moment of the N2 molecule polarized by the point charge located at the same place as its "own" caUon (see above) and Ot is that of the N2 molecule polarized both by its cation and by the whole embedding The maximum 40, zz] obtained m our models is less than 0 3 a u On the other hand, IF)z[ never exceeds 0 006 a u Hence, the upper bound for (1 /6)O,F'=(1/4)O, zzF'z z is 0 0005 a u or 0 3 kcal/mol
3 o
E -~ 2
LI.I +
o 0
LLI -1
-2
-3 -3
/
/ i / / / / / r / / / / /
/ / / / / / i / / I I . I i I i l i I
-2 -1 0 2 3 4
Eads - Eads0so) , kcal /mol
a)
4
8
uJ +
U J
0 0
I /
J
i'n ~ !
. - - : , b) oo ~ ' " , . /
/
/ /
/ 1
/ /
t /
/
, I , I , I , I , I r I
1 2 3 4 5 6
Eads-Eads0so) , kcal /mol
Fig 10 Electrostatic and reduction contributions to the N2-zeohte lnteractmn energies compared with calculated adsorpUon energies, in (a) the ALA, and (b) ALAM structures Sites III correspond to sohd lines and sites II to dashed lines
structures. The analysas of the QM results shows that adsorption essentially occurs at sate I I I cations, in the experimental structures, whichever is the site position, i.e. ideal, mono- or bi-coordlnated Due to the presence of cations, there is no unique sate II or sate III adsorption energies and only few sites should contribute to the initial heat of adsorptaon.
A A r b u z m k o v et al / Sur face Sewnce 397 (1998) 395-405 405
9
8
7
6 0 E 5
S 4
1.1.1 +
Ul ]
0
-1
-2 -1
, !d/ . , :
/ , f ~ . . ' a)
i /
l , I , I I I I I , I ,
0 1 2 3 4 5 6 7
E a d s - E a d s 0 s o ) , k c a l / m o l
cationic sites has been shown to be the major factor which determines their adsorption strengths. This property is a characteristics of every structure and every type of site. At similar electric field gradient value, the variation of the cation adsorption strength is related to the variation of the electric field. From our results, it appears that both proper- ties are more dependent on the framework geometry than on the type III cation positions
Acknowledgements
A. Arbuznikov and V. Vasilyev thank Elf- Aquitaine for post-doctoral fellowships. The Centre Universitaire Sud de Calcul de Montpellier is acknowledged for a provision of computational resources.
7
6
5 E
4 2 ~ ~3 tlA + o 2 ID
ID
U.l 1
/ /
/ /
• / i /
/
_ ~ I , I I I , I , I
0 1 2 3 4 5 6
Eads-Eads0so), kcal/mol
b)
Fig ' l l Electrostatic and reduction contributions to the N2-zeohte interaction energies compared with calculated adsorption energies in (a) the OLS, and (b) OLSM structures Sites III correspond to sohd hnes and sites II to dashed lines
Based on the general theory of long-range intermo- lecular forces, the interaction energy between the zeolite structure and the N2 molecule adsorbed at each site has been expressed in terms of the N2 multipoles, the electric field and the electric field gradient in the zeolite.
The value of the electric field gradient at the
References
[1] A Goursot, I P&pm, V Vasllyev, F Fajula, in L Bonnevlot. S Kahagume (Eds), Zeolltes A Refined Tool for Designing Catalytic Sites, Elsevier, Amsterdam, 1995, p 109
[2] A Goursot, V Vasflyev, A Arbuznmkov, J Phys Chem 101 (1997) 6420
[3] C.G Coe, Access in Nanoporous Materials, Plenum Press, New York, 1995
[4] A Seadel, B Z Boddenberg, Naturforsch 50 (1995) 199 [5] D H Olson, Zeolates 15 (1995) 439 [6] G N D A1-Ajdah, A A A1-Rlshed, B Beagley, J Dwyer,
F R Fitch, T K Ibrahim, J Inclusion Phenom 3 (1985) 135
[7] L Campana, A. Sellom, J Weber, A Goursot, J Phys Chem 99 (1995) 16351
[8] I P~tpal, A Goursot, F Fajula, D Plee, J Weber, J Phys Chem. 99 (1995) 12925
[9] I P&pai, A Goursot, F J Fajula, J Phys Chem 98 (1994) 4654
[ 10] Catalysis 4 0 Software User Manual, Blosym Technologies, San Diego, 1993
[ l l ] A St-Amant, D R Salahnb, Chem Phys Lett 169 (1990) 387
[12] A St-Amant, PhD Thesis. Umversit6 de Montr6al, 1991 [13] C Daul, A. Goursot, D R Salahub, in C. Cerjan (Ed),
Numerical Grid Methods and Their Application to Schrodmger's Equation, NATO ASI Series, Kluwer, Dor- drecht, 1993, p 153
[14] J P Perdew, Y Wang, Phys Rev B 33 (1986) 8800 [15] J P Perdew, Phys Rev. B 33 (1986) 8822, J P Perdew,
Phys Rev B 34 (1986) 7406E [16] A D Buckingham, in Pullman, B (Ed), Intermolecular
Interactions From Dlatomlcs to Blopolymers, Wiley. Chlchester, 1978




















