
Relations between strain and gender dependencies of irinotecan toxicity and UGT1A1, CES2 and TOP1...
7
Toxicology Letters 192 (2010) 395–401 Contents lists available at ScienceDirect Toxicology Letters journal homepage: www.elsevier.com/locate/toxlet Relations between strain and gender dependencies of irinotecan toxicity and UGT1A1, CES2 and TOP1 expressions in mice C. Ahowesso a,b , E. Piccolo c , X.M. Li a,b , S. Dulong a,b , V. Hossard a,b , R. La Sorda c , E. Filipski a,b , N. Tinari c , F. Delaunay d , S. Iacobelli c , F. Lévi a,b,∗ a INSERM, U776 “Rythmes Biologiques et Cancers”, Hôpital Paul Brousse, 14 Avenue PV Couturier, 94800 Villejuif, France b Univ Paris-Sud, SO776, 11, Rue Georges Clemenceau, 91405 Orsay, France c Consorzio Interuniversitario Nazionale per la Bioncologia (CINBO) CE.S.I. - Universita’ “G. d’Annunzio” Via Colle dell’Ara, 66100 Chieti, Italy d Université de Nice-Sophia Antipolis, CNRS FRE3094, 28 Av. Valrose, 06108 Nice, France article info Article history: Received 1 September 2009 Received in revised form 31 October 2009 Accepted 15 November 2009 Available online 29 November 2009 Keywords: Irinotecan Toxicity Personalized medicine Phase II metabolism Gender Cancer abstract Irinotecan hydrochloride (CPT-11) can display severe toxicities in individual cancer patients. CPT-11 is bio-activated through CES, detoxified through UGT1A1 and inhibits TOP1. CPT-11 toxicity and UGT1A1, CES2 and TOP1 mRNAs and UGT1A1 protein were determined in male and female C57BL/6, B6D2F1 and B6CBAF1, as potential models for tailoring CPT-11 delivery. CPT-11 was administered intravenously (40–90 mg/kg/day for 4 days at 7 h after light onset). The relations between dose and lethal toxic- ity or body weight loss were steep and similar in C57BL/6 (lethality, p = 0.001; weight loss, p = 0.002) and B6D2F1 (p = 0.01; p = 0.03, respectively), but weak in B6CBAF1. Females displayed less toxicity than males (p < 0.001). Mean mRNA expression of UGT1A1 was highest in B6CBAF1 (p = 0.039) and in females (p < 0.001). Both CES2 and TOP1 varied according to strain and gender (p < 0.001). The three gene expres- sion data explained the most severe toxicity of CPT-11 in male B6D2F1, but displayed inconsistent relations with toxicity in the other groups. Mean UGT1A1 protein expression was highest in males as compared to females, and so by ∼8-fold in C57BL/6 as compared to B6D2F1 (p < 0.0001). Genetic back- ground and gender significantly altered the molecular prediction of irinotecan toxicity by UGT1A1, CES2 and TOP1 mRNA expressions. © 2009 Elsevier Ireland Ltd. All rights reserved. 1. Introduction Irinotecan, 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbo- nyloxycamptothecin (CPT-11), is a potent and widely used anticancer drug, which can produce severe and unpredictable hematologic, intestinal or systemic toxicities (Innocenti and Ratain, 2003). CPT-11 undergoes bioactivation into SN-38, through a reac- tion catalyzed by carboxylesterase 2 (CES2) that mainly takes place in liver (Rouits et al., 2008). Both CPT-11 and SN-38 inhibit DNA- topoisomerase I (TOP1), thus produce DNA strand breaks during the replicative phase in main toxicity target tissues and in tumor. The potency of SN-38 to produce such lesions is 100–1000-fold greater than CPT-11 (Kawato et al., 1991; Gupta et al., 1994). Liver also rep- resents the main site of CPT-11 and SN-38 detoxification (Rouits et al., 2008). Thus CPT-11 is detoxified by cytochrome P450, CYP3A4 to APC and NPC, whereas SN-38 is conjugated to glucuronic acid by UDP-glycosyltransferase 1 polypeptide A1 (UGT1A1) (Rouits ∗ Corresponding author at: INSERM, U776 “Rythmes Biologiques et Cancers”, Hôpital Paul Brousse, 14 Avenue PV Couturier, 94800 Villejuif, France. E-mail address: [email protected] (F. Lévi). et al., 2008). Both CPT-11 and SN-38 are subject to spontaneous interconversion between a lactone (active) form and a carboxy- late (inactive) form, depending on the pH of the fluid (Rouits et al., 2008). The relevance of decreased glucuronoconjugation for CPT- 11 toxicity was first shown in patients with Gilbert syndrome (Wasserman et al., 1997). This finding led to the identification of UGT1A1 polymorphisms as main predictors of CPT-11 toxicity in cancer patients (Marcuello et al., 2004; Rosner et al., 2008). In turn, the Food and Drug Administration recommended the use of a dedicated test of UGT1A1*28 polymorphism prior to the admin- istration of CPT-11, in order to reduce the risk of undue and sometime fatal toxicity of this drug (Innocenti and Ratain, 2006). However, recent reports regarding the relevance of this test to pre- dict for CPT-11 toxicity have shown mitigated results (Lankisch et al., 2008; Cecchin et al., 2009). Several studies identify gender, age and treatment schedule as possibly interfering with the ability of UGT1A1 polymorphisms to predict for CPT-11 toxicity (Kweekel et al., 2008). The relevance of genetic polymorphisms for predict- ing CPT-11 toxicities has also been emphasized for other UGT1A gene family, CYP3A and CES2 (Lamba et al., 2002; Charasson et al., 2004). 0378-4274/$ – see front matter © 2009 Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.toxlet.2009.11.017
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Relations between strain and gender dependencies of irinotecan toxicity and UGT1A1, CES2 and TOP1...

RU
CNa
b
c
d
a
ARRAA
KITPPGC
1
nah2titrptratb
H
0d
Toxicology Letters 192 (2010) 395–401
Contents lists available at ScienceDirect
Toxicology Letters
journa l homepage: www.e lsev ier .com/ locate / tox le t
elations between strain and gender dependencies of irinotecan toxicity andGT1A1, CES2 and TOP1 expressions in mice
. Ahowessoa,b, E. Piccoloc, X.M. Li a,b, S. Dulonga,b, V. Hossarda,b, R. La Sordac, E. Filipskia,b,. Tinari c, F. Delaunayd, S. Iacobelli c, F. Lévia,b,∗
INSERM, U776 “Rythmes Biologiques et Cancers”, Hôpital Paul Brousse, 14 Avenue PV Couturier, 94800 Villejuif, FranceUniv Paris-Sud, SO776, 11, Rue Georges Clemenceau, 91405 Orsay, FranceConsorzio Interuniversitario Nazionale per la Bioncologia (CINBO) CE.S.I. - Universita’ “G. d’Annunzio” Via Colle dell’Ara, 66100 Chieti, ItalyUniversité de Nice-Sophia Antipolis, CNRS FRE3094, 28 Av. Valrose, 06108 Nice, France
r t i c l e i n f o
rticle history:eceived 1 September 2009eceived in revised form 31 October 2009ccepted 15 November 2009vailable online 29 November 2009
eywords:rinotecan
a b s t r a c t
Irinotecan hydrochloride (CPT-11) can display severe toxicities in individual cancer patients. CPT-11 isbio-activated through CES, detoxified through UGT1A1 and inhibits TOP1. CPT-11 toxicity and UGT1A1,CES2 and TOP1 mRNAs and UGT1A1 protein were determined in male and female C57BL/6, B6D2F1 andB6CBAF1, as potential models for tailoring CPT-11 delivery. CPT-11 was administered intravenously(40–90 mg/kg/day for 4 days at 7 h after light onset). The relations between dose and lethal toxic-ity or body weight loss were steep and similar in C57BL/6 (lethality, p = 0.001; weight loss, p = 0.002)and B6D2F1 (p = 0.01; p = 0.03, respectively), but weak in B6CBAF1. Females displayed less toxicity than
oxicityersonalized medicinehase II metabolismenderancer
males (p < 0.001). Mean mRNA expression of UGT1A1 was highest in B6CBAF1 (p = 0.039) and in females(p < 0.001). Both CES2 and TOP1 varied according to strain and gender (p < 0.001). The three gene expres-sion data explained the most severe toxicity of CPT-11 in male B6D2F1, but displayed inconsistentrelations with toxicity in the other groups. Mean UGT1A1 protein expression was highest in males ascompared to females, and so by ∼8-fold in C57BL/6 as compared to B6D2F1 (p < 0.0001). Genetic back-ground and gender significantly altered the molecular prediction of irinotecan toxicity by UGT1A1, CES2
ons.
and TOP1 mRNA expressi. Introduction
Irinotecan, 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbo-yloxycamptothecin (CPT-11), is a potent and widely usednticancer drug, which can produce severe and unpredictableematologic, intestinal or systemic toxicities (Innocenti and Ratain,003). CPT-11 undergoes bioactivation into SN-38, through a reac-ion catalyzed by carboxylesterase 2 (CES2) that mainly takes placen liver (Rouits et al., 2008). Both CPT-11 and SN-38 inhibit DNA-opoisomerase I (TOP1), thus produce DNA strand breaks during theeplicative phase in main toxicity target tissues and in tumor. Theotency of SN-38 to produce such lesions is 100–1000-fold greaterhan CPT-11 (Kawato et al., 1991; Gupta et al., 1994). Liver also rep-
esents the main site of CPT-11 and SN-38 detoxification (Rouits etl., 2008). Thus CPT-11 is detoxified by cytochrome P450, CYP3A4o APC and NPC, whereas SN-38 is conjugated to glucuronic acidy UDP-glycosyltransferase 1 polypeptide A1 (UGT1A1) (Rouits∗ Corresponding author at: INSERM, U776 “Rythmes Biologiques et Cancers”,ôpital Paul Brousse, 14 Avenue PV Couturier, 94800 Villejuif, France.
E-mail address: [email protected] (F. Lévi).
378-4274/$ – see front matter © 2009 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.toxlet.2009.11.017
© 2009 Elsevier Ireland Ltd. All rights reserved.
et al., 2008). Both CPT-11 and SN-38 are subject to spontaneousinterconversion between a lactone (active) form and a carboxy-late (inactive) form, depending on the pH of the fluid (Rouits et al.,2008).
The relevance of decreased glucuronoconjugation for CPT-11 toxicity was first shown in patients with Gilbert syndrome(Wasserman et al., 1997). This finding led to the identification ofUGT1A1 polymorphisms as main predictors of CPT-11 toxicity incancer patients (Marcuello et al., 2004; Rosner et al., 2008). Inturn, the Food and Drug Administration recommended the use of adedicated test of UGT1A1*28 polymorphism prior to the admin-istration of CPT-11, in order to reduce the risk of undue andsometime fatal toxicity of this drug (Innocenti and Ratain, 2006).However, recent reports regarding the relevance of this test to pre-dict for CPT-11 toxicity have shown mitigated results (Lankisch etal., 2008; Cecchin et al., 2009). Several studies identify gender, ageand treatment schedule as possibly interfering with the ability of
UGT1A1 polymorphisms to predict for CPT-11 toxicity (Kweekelet al., 2008). The relevance of genetic polymorphisms for predict-ing CPT-11 toxicities has also been emphasized for other UGT1Agene family, CYP3A and CES2 (Lamba et al., 2002; Charasson et al.,2004).
3 ogy Le
emt2ivchtWmta
2
2
oClbr
imsEo
iwuma
2
Mts
2
ammn1psa
2
w
2
2
iaaraeio4(c
Mean mortality rates with corresponding standard error of mean (SEM) were
96 C. Ahowesso et al. / Toxicol
Whereas the genetic profile of the tumor may modify drugffects, the genetic background of the patient with respect toetabolizing enzymes and drug transporters plays a critical role in
he relation between drug dose and therapeutic effects (van Schaik,008). Here, we hypothesize that CPT-11 toxicity varies accord-
ng to genetic background and gender, and we seek whether theseariations are predicted by UGT1A1 expression in liver. The pre-linical toxicity of anticancer drugs has been mostly assessed inomozygous C57BL/6 mice. We investigate the modifications inoxic outcome in 3 mouse strains (C57BL/6, B6D2F1 and B6CBAF1).
e further study the relations between CPT-11 toxicity and theRNA expressions of UGT1A1, CES2 and TOP1, as well as the pro-
ein expression of UGT1A1 in the liver as a function of both gendernd genetic background.
. Materials and methods
.1. Study design
Four experiments (Exp) were performed in a total of 260 males and femalesf three mouse strains C57BL/6, B6D2F1 (♀ C57BL/6 ×♂ DBA2) and B6CBAF1 (♀57BL/6 ×♂ CBA). CPT-11 was administered daily for 4 consecutive days at 7 h after
ight onset (also named Zeitgeber 7, ZT 7) (Aschoff, 1965). This time was shown toe close to that of least toxicity and best antitumor efficacy in ♂ B6D2F1 mice, as aesult of circadian rhythms (Granda et al., 2002; Filipski et al., 2004).
Exp 1 and 2 aimed at the determination of relations between dose and toxicityn ♂ and ♀ mice from the 3 strains. The main endpoints were lethal toxicity and
aximum body weight loss following the daily administration of CPT-11 for 4 con-ecutive days. Daily doses of 40, 50, 60 or 70 mg/kg were administered to the mice inxp 1. In Exp 2, daily doses of 70, 80 or 90 mg/kg were injected in ♂ and ♀ B6CBAF1nly.
The physiological mRNA expression of UGT1A1, CES2 and TOP1 were determinedn the liver of 3–5 ♂ and ♀ mice from each of the 3 strains in Exp 3. Mouse livers
ere sampled at ZT 7, immediately frozen into liquid nitrogen, and kept at −80 ◦Cntil analysis. UGT1A1 protein expression was assessed in the right liver lobe of 5ice from each group in Exp 4. All samples were obtained between ZT 6 and ZT 7,
nd immediately put into formalin.
.2. Drug
The clinical solution for intravenous (i.v.) injection of CPT-11 (20 mg/ml; Aventis,ilan, Italy) was diluted in 0.9% sodium chloride on each study day, prior to injec-
ions. The final drug solution was injected i.v. into the right retro-orbital venousinus of the mice (10 ml/kg of body weight) (Filipski et al., 2004).
.3. Animals and synchronization
All procedures were performed in accordance with the French guidelines fornimal care and experimentation (Decree 87-843). C57BL/6, B6D2F1 and B6CBAF1ice, 7 weeks of age, were purchased from Janvier (Le Genest st Isle, France). All theice were housed two or three per cage with water and food ad libitum and synchro-
ized for 3 weeks with 12 h of light (L) in alternation with 12 h of darkness (D) (LD2:12). The cages were randomly allocated to one of the several treatment groupslanned for each experiment. Cages from each group were placed on a differenthelf of an autonomous chronobiological animal facility in temperature controlledt 21–23 ◦C (ESI-Flufrance, Arcueil, France) (Tampellini et al., 1998).
.4. Toxicity assessment
Toxic deaths were recorded over the 2 weeks following treatment onset. Bodyeights were measured daily at ZT 7 over the same time span.
.5. UGT1A1, CES2 and TOP1 mRNA expression
.5.1. RNA extractionEach sample was flushed with 2 ml of PBS. 600 �l of lysis buffer GIT (guanosine
sothiocyanate: sodium citrate and N-lauryl sarcosine 10%, 2-mercaptoethanol) wasdded. The lysate obtained was recovered in a tube for RNA extraction (Chomczynskind Sacchi, 2006). For each sample, were added 1 vol. of phenol and 0.2 vol. of chlo-oform:isoamyl (24:1). After agitation, the samples were incubated 15 min on icend then centrifuged (20 min, 9660 × g, at 4 ◦C). The aqueous phase was then recov-
red. One volume of isopropanol was added and the mixture was obtained throughterative reversals of the solution. Precipitated messenger RNAs were incubatedvernight at −80 ◦C. The next day, samples were centrifuged (30 min, 9660 × g, at◦C) and supernatants were removed. The pellets were rinsed with ethanol 70%500 �l), then centrifuged (20 min, 9660 × g, at 4 ◦C). The supernatants were dis-arded and pellets dried 10 min in ice and then re-suspended in water RNA free. RNA
tters 192 (2010) 395–401
samples were analyzed by formaldehyde-agarose gel electrophoresis, and integritywas confirmed by visualization of 18S and 28S rRNA bands. The RNAs obtained werequantified by spectrometry (Biophotometer Plus Eppendorf, Dominique Dutscher,Brumath, France) and stored at −80 ◦C.
Reverse transcription involved denaturing step of RNA followed by a retro tran-scription step itself (RT Invitrogen, Cergy-Pontoise, France). The thermocycler wasused for all stages of heating and cooling. For each sample, a solution containing5 �g of total RNA and 100 ng random primers (hexamers solution at 100 ng/�l)was prepared. It was then heated in the thermocycler at 65 ◦C for 5 min (dena-turing step), then it was cooled at 4 ◦C for 5 min. Along with the denaturing step,the solution required for the retro-transcription was prepared. It contained desoxy-nucleoside triphosphates (dATP, dCTP, dTTP and DGTP) (10 mM), buffer containingthe enzyme cofactors essential for the functioning of the enzyme (250 mM Tris–HCl,375 mM KCl, 15 mM MgCl2), dithiothreitol (0.1 mM solution), and reverse transcrip-tase (SuperScript II Reverse Transcriptase, Invitogen, Cergy-Pontoise, France). Thereverse transcription step was performed by incubating samples for 50 min to 42 ◦C,followed by incubation for 15 min at 75 ◦C for enzyme denaturation. The sampleswere then diluted to the tenth before the PCR.
PCR was performed with a Light Cycler 3 (Roche Applied Science, Mey-lan, France) using SYBR green I dye detection according to the manufacturer’srecommendations. cDNA was added to a reaction mixture (Faststart DNASYBR Green I, Roche Diagnostics, Meylan, France) with appropriate primersat 0.5 �M each. Relative mRNA abundance was calculated using a standardcurve method. Expression levels were normalized to the levels of the constitu-tively and non-rhythmic expressed 36B4 (acidic ribosomal phosphoprotein P0).The following primers were used for UGT1A1 (Accession number NM 201645.1;forward, 5′-agattaccccaggcccatc-3′ and reverse, 5′-atggctttcttctccggaat-3′), CES2(Accession number NM 145603.1; forward, 5′-ggccatgtgtctgcaaaatc-3′ and reverse,5′-caccatcacaggcaggttag-3′) TOP1, (Accession number NM 009408.2; forward,5′-gttcacgaatcaagggtgag-3′ and reverse, 5′-tccttctcattgcctgctct-3′) and for 36B4(Accession number NM 007475.3; forward, 5′-gctgatgggcaagaacacca-3′ and reverse,5′-cccaaagcctggaagaagga-3′).
2.6. UGT1A1 protein expression
UGT1A1 protein expression was assessed using immunohistochemistry andconfocal microscopy. Livers from mice were fixed in 4% buffered formalin for 24 hat room temperature. The fixed tissues were then dehydrated in ethanol, cleared inBioclear (Bioptica, Milan, Italy) and embedded into paraffin. Section of 5 �m werecut and taken on positively charged slides. Slides were incubated overnight at 37 ◦Cand immersed in xylol in order to remove paraffin. Tissue sections were rehydratedin a graded alcohol series (100%, 96% and 80%) for 5 min in each bath, and rinsed withrunning tap water. Haematoxylin and eosin (H&E) staining was performed accord-ing to a routine protocol in order to estimate the quality of the tissue. Sections wereincubated for 30 min with 1% BSA in PBS to minimize non specific binding, thenovernight at 4 ◦C with rabbit anti-human UGT1A1 antibody (Santa Cruz Biotech-nology, Santa Cruz, CA) diluted 1:30 in PBS, and Pan-Cadherin antibody (Abcam,Cambridge, UK) directly conjugated to Alexa-568 fluorophore diluted 1:50, to visu-alize plasma membrane. After three washes in PBS, sections were co-incubated for60 min at room temperature with an Alexa Flour 488-conjugated goat anti-rabbitsecondary antibody from Molecular Probes (Invitrogen, Milan, Italy) and with thefar-red fluorescent DNA dye DRAQ5 (Alexis, Lausen, Switzerland) to visualize nuclei.As a negative control, tissues were stained with rabbit IgG (Jackson Laboratories, BarHarbor, Maine) at the same concentrations used for the primary antibody. Followingthree brief PBS washes, samples were mounted using Eukitt (Bioptica, Milan, Italy).Slides from the same liver of the same BalbC mouse were included as a control inall the experiments.
Images were acquired with a Zeiss LSM 510 meta-confocal microscope using a488 and 633-nm lasers. Detector gain voltages and pinhole were set at the beginningof the experiment and maintained constant during the acquisition of all samples.For each tissue section, 3 images were collected corresponding to different areasof the section. To quantify UGT1A1 protein expression, images were analyzed withImage ProPlus 6.0 Software (Media Cybernetics, Bethesda, MD). The standard ver-sion of the software was modified in order to measure the fluorescent area in anautomatic mode (Immagini & Computer, Milan, Italy). Briefly, for each image thesoftware estimated the number of nuclei and measured the fluorescent area. Thelatter was divided by the number of nuclei to obtain a mean value of fluorescencearea. To reduce inter-assay variability, mean values were expressed as percentagesof UGT1A1 expression in the control liver from male BalbC mouse.
2.7. Statistical analyses
computed and plotted as a function of dose for each strain, for each gender and foreach gender in each strain. Incidence data were compared by �2 test. Dose–responserelations were studied using linear regression for both mortality and body weightloss. Intergroup differences were statistically validated by multiple-way analysesof variance (ANOVA) and contrast tests using SPSS v.16.0 for Windows software(Paris-La Défense, France).
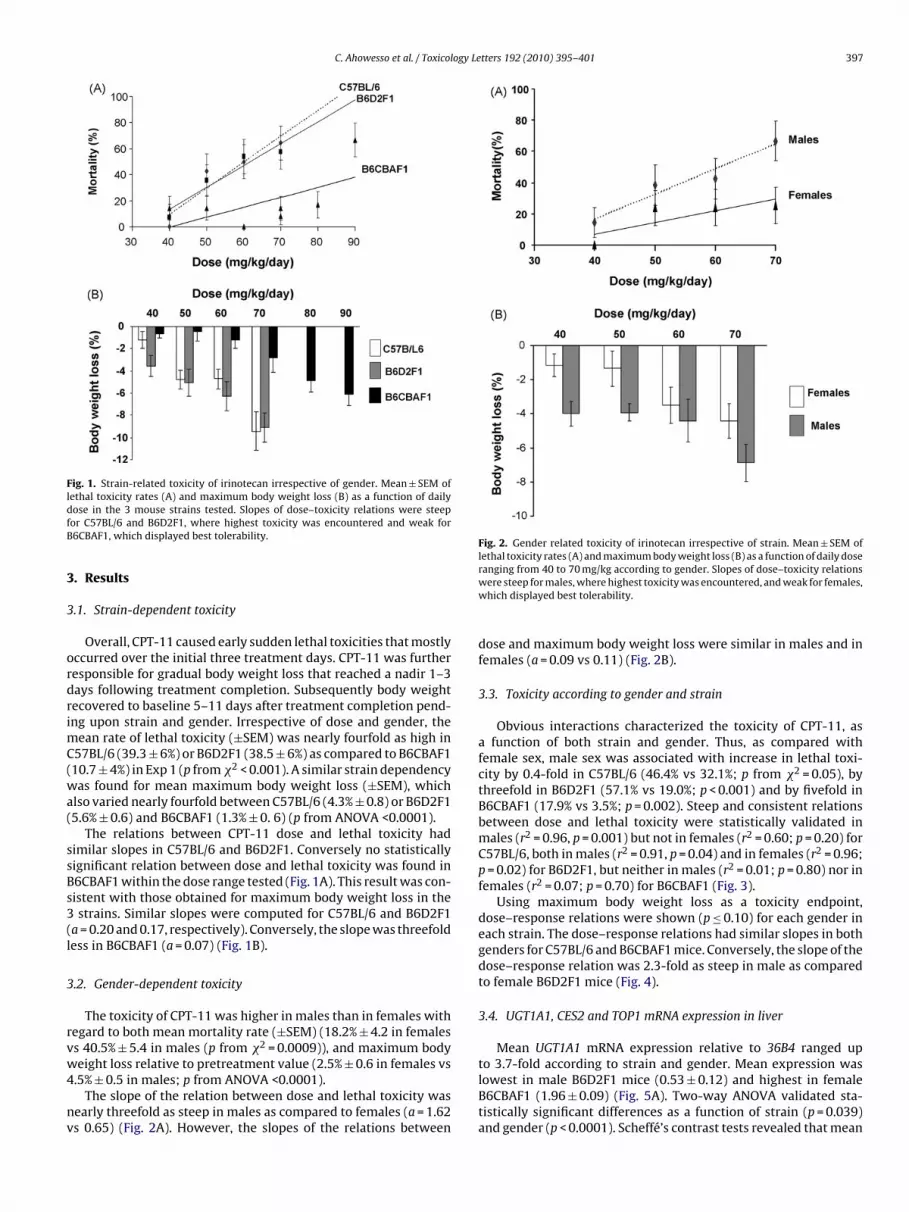
C. Ahowesso et al. / Toxicology Letters 192 (2010) 395–401 397
Fig. 1. Strain-related toxicity of irinotecan irrespective of gender. Mean ± SEM oflethal toxicity rates (A) and maximum body weight loss (B) as a function of dailydfB
3
3
ordrimC(wa(
ssBs3(l
3
rvw4
nv
Fig. 2. Gender related toxicity of irinotecan irrespective of strain. Mean ± SEM oflethal toxicity rates (A) and maximum body weight loss (B) as a function of daily dose
to 3.7-fold according to strain and gender. Mean expression was
ose in the 3 mouse strains tested. Slopes of dose–toxicity relations were steepor C57BL/6 and B6D2F1, where highest toxicity was encountered and weak for6CBAF1, which displayed best tolerability.
. Results
.1. Strain-dependent toxicity
Overall, CPT-11 caused early sudden lethal toxicities that mostlyccurred over the initial three treatment days. CPT-11 was furtheresponsible for gradual body weight loss that reached a nadir 1–3ays following treatment completion. Subsequently body weightecovered to baseline 5–11 days after treatment completion pend-ng upon strain and gender. Irrespective of dose and gender, the
ean rate of lethal toxicity (±SEM) was nearly fourfold as high in57BL/6 (39.3 ± 6%) or B6D2F1 (38.5 ± 6%) as compared to B6CBAF110.7 ± 4%) in Exp 1 (p from �2 < 0.001). A similar strain dependencyas found for mean maximum body weight loss (±SEM), which
lso varied nearly fourfold between C57BL/6 (4.3% ± 0.8) or B6D2F15.6% ± 0.6) and B6CBAF1 (1.3% ± 0. 6) (p from ANOVA <0.0001).
The relations between CPT-11 dose and lethal toxicity hadimilar slopes in C57BL/6 and B6D2F1. Conversely no statisticallyignificant relation between dose and lethal toxicity was found in6CBAF1 within the dose range tested (Fig. 1A). This result was con-istent with those obtained for maximum body weight loss in thestrains. Similar slopes were computed for C57BL/6 and B6D2F1
a = 0.20 and 0.17, respectively). Conversely, the slope was threefoldess in B6CBAF1 (a = 0.07) (Fig. 1B).
.2. Gender-dependent toxicity
The toxicity of CPT-11 was higher in males than in females withegard to both mean mortality rate (±SEM) (18.2% ± 4.2 in femaless 40.5% ± 5.4 in males (p from �2 = 0.0009)), and maximum bodyeight loss relative to pretreatment value (2.5% ± 0.6 in females vs
.5% ± 0.5 in males; p from ANOVA <0.0001).The slope of the relation between dose and lethal toxicity was
early threefold as steep in males as compared to females (a = 1.62s 0.65) (Fig. 2A). However, the slopes of the relations between
ranging from 40 to 70 mg/kg according to gender. Slopes of dose–toxicity relationswere steep for males, where highest toxicity was encountered, and weak for females,which displayed best tolerability.
dose and maximum body weight loss were similar in males and infemales (a = 0.09 vs 0.11) (Fig. 2B).
3.3. Toxicity according to gender and strain
Obvious interactions characterized the toxicity of CPT-11, asa function of both strain and gender. Thus, as compared withfemale sex, male sex was associated with increase in lethal toxi-city by 0.4-fold in C57BL/6 (46.4% vs 32.1%; p from �2 = 0.05), bythreefold in B6D2F1 (57.1% vs 19.0%; p < 0.001) and by fivefold inB6CBAF1 (17.9% vs 3.5%; p = 0.002). Steep and consistent relationsbetween dose and lethal toxicity were statistically validated inmales (r2 = 0.96, p = 0.001) but not in females (r2 = 0.60; p = 0.20) forC57BL/6, both in males (r2 = 0.91, p = 0.04) and in females (r2 = 0.96;p = 0.02) for B6D2F1, but neither in males (r2 = 0.01; p = 0.80) nor infemales (r2 = 0.07; p = 0.70) for B6CBAF1 (Fig. 3).
Using maximum body weight loss as a toxicity endpoint,dose–response relations were shown (p ≤ 0.10) for each gender ineach strain. The dose–response relations had similar slopes in bothgenders for C57BL/6 and B6CBAF1 mice. Conversely, the slope of thedose–response relation was 2.3-fold as steep in male as comparedto female B6D2F1 mice (Fig. 4).
3.4. UGT1A1, CES2 and TOP1 mRNA expression in liver
Mean UGT1A1 mRNA expression relative to 36B4 ranged up
lowest in male B6D2F1 mice (0.53 ± 0.12) and highest in femaleB6CBAF1 (1.96 ± 0.09) (Fig. 5A). Two-way ANOVA validated sta-tistically significant differences as a function of strain (p = 0.039)and gender (p < 0.0001). Scheffé’s contrast tests revealed that mean

398 C. Ahowesso et al. / Toxicology Letters 192 (2010) 395–401
Fig. 3. Strain and gender dependency of the relation between CPT-11 dose and lethaltag
UBo
ae(se(f(
im(vrbo
lfhtAs
protein expressions of UGT1A1, across the 6 groups here stud-ied. Indeed, the highest mRNA expression was found in female
oxicity in three mouse strains. Mean ± SEM of lethal toxicity rates according to dosend gender in C57BL/6 (A), B6D2F1 (B) and B6CBAF1 (C). Note: Major relevance ofender in B6D2F1.
GT1A1 mRNA expression differed significantly between female6CBAF1 and male B6D2F1 (1.96 ± 0.09 vs 0.53 ± 0.12; p = 0.008)r male C57BL/6 (0.76 ± 0.06; p = 0.037).
Mean CES2 mRNA varied up to 5.4-fold according to strainnd gender (Fig. 5B). Once more, statistically significant differ-nces were validated by two-way ANOVA, according to strainp < 0.001) and gender (p < 0.0001). Scheffé’s contrast tests, furtherhowed that mean CES2 mRNA expression was significantly high-st in male B6D2F1 (3.94 ± 0.54) as compared with female C57BL/60.73 ± 0.14; p < 0.0001), female B6D2F1 (1.93 ± 0.14; p = 0.003),emale B6CBAF1 (1.24 ± 0.14; p < 0.0001), as well as male C57BL/61.01 ± 0.18; p < 0.0001) and male B6CBAF1 (1.65 ± 0.08; p = 0.001).
Mean TOP1 mRNA expression ranged up to 3.6-fold accord-ng to strain and gender. Here, mean expression was lowest in
ale C57BL/6 mice (0.72 ± 0.19) and highest in female B6D2F12.58 ± 0.13) (Fig. 5C). Strain-related differences were statisticallyalidated with two-way ANOVA (p < 0.001). Scheffé’s contrast testsevealed that mean TOP1 mRNA expression differed significantlyetween B6D2F1 (2.13 ± 0.25) and C57BL/6 (0.80 ± 0.08; p = 0.001)r B6CBAF1 (1.07 ± 0.12; p < 0.0001).
Overall, male B6D2F1 mice displayed highest CES2 expression,owest UGT1A1 and high TOP1 expression in liver. On the opposite,emale B6CBAF1 had low expressions of both CES2 and TOP1, and
ighest UGT1A1 expression. Noteworthy, a strain * gender interac-ion term was close to statistical significance according to the 2-wayNOVA for CES2 (p = 0.06), UGT1A1 (p = 0.1) and TOP1 (p = 0.07),uggesting that the gender-dependent mRNA expression could dif-Fig. 4. Effects of strain, gender and dose on body weight change at nadir.Mean ± SEM of body weight change according to dose and gender in C57BL/6 (A),B6D2F1 (B) and B6CBAF1 (C). Note: Major relevance of gender in B6D2F1.
fer according to strain. This was obvious in B6D2F1 mice, wheremales displayed higher CES2 expression, lower UGT1A1 expres-sion and higher TOP1 expression as compared to females, while nosuch sex-related difference was apparent in C57BL/6 or B6CBAF1(Fig. 5).
3.5. UGT1A1 protein expression in liver
UGT1A1 protein expression also differed largely as a functionof strain and gender as illustrated for a representative mouse ofeach sex in each strain. Lowest immunofluorescence was foundin the female mice of each strain, and in B6D2F1 mice (Fig. 6A).Mean UGT1A1 protein expression ranged by 12.8-fold, from a low-est mean value (±SEM) in female B6D2F1 (20.9 ± 11.4) to a highestone in male C57BL/6 (266.8 ± 52.7) (Fig. 6B). Two-way ANOVA val-idated statistically significant differences as a function of strain(p < 0.0001) and suggested that male gender could also be asso-ciated with increased UGT1A1 protein expression (p = 0.069). Nostrain * gender interaction was found (p = 0.19).
Thus, no consistent relation was found between mRNA and
B6CBAF1, where protein expression had intermediate values. Con-versely lowest mRNA expressions were detected in male B6D2F1and male C57BL/6 mice; mean protein expressions ranked secondto lowest and highest in these two groups, respectively.

C. Ahowesso et al. / Toxicology Le
Fig. 5. Bar graph of UGT1A1, CE2, Top1 mRNA expression in the liver of female ormsdB
4
lCaig2
BaBiTs
tCociFtHg1mmt
ale mice from three strains. (A) UGT1A1 mRNA expression, (B) CES2 mRNA expres-ion, (C) TOP1 mRNA expression. Mean ± SEM in 3–5 mice per group (triplicateeterminations). Note: Major relevance of gender for all three gene expressions in6D2F1.
. Discussion
CPT-11 toxicity, as assessed with lethal toxicity and body weightoss, varied up to several-fold among three strains of mice with57BL/6 background. In order to minimize variability, CPT-11 wasdministered at a fixed circadian time, associated with least toxicityn male B6D2F1 and male ICR mice, according to prior investi-ations (Ohdo et al., 1997; Granda et al., 2002; Filipski et al.,004).
In the current study, CPT-11 toxicity was least in female6CBAF1 and highest in male B6D2F1 or C57BL/6. Sex and strainlso affected the slope of dose–toxicity, an estimate of drug safety.oth tolerability and safety of CPT-11 were greatest in B6CBAF1
rrespective of gender and in female mice irrespective of strain.he steepest slope was found in male B6D2F1, suggesting poorestafety.
CPT-11 toxicity mostly results from its bioactivation into SN-38hrough CES activities. SN-38 is 100–1000-fold more potent thanPT-11, with regard to inhibition of TOP1, the main target enzymef camptothecins (Kawato et al., 1991; Gupta et al., 1994). SN-38atabolism primarily involves UGT1A1 (Rouits et al., 2008). The crit-cal role of UGT1A1 expression for CPT-11 tolerability has led theDA to recommend UGT1A1*28 polymorphism assessment prioro CPT-11 administration in patients (Innocenti and Ratain, 2006;uang and Ratain, 2009). However gender, drug schedule and other
enetic and lifestyle factors were recently shown to influence CPT-1 toxicities (Kweekel et al., 2008). In the current study, meanRNA expression of UGT1A1 was higher in female than in maleice, in three strains, in good agreement with the sex-dependentolerability of CPT-11. Similarly, increased UGT1A1 mRNA expres-
tters 192 (2010) 395–401 399
sion was reported in the liver of C57BL/6 mice (Buckley andKlaassen, 2007).
The gender-dependent differences in UGT1A1 were recentlyrelated to the male pattern in growth hormone (GH) secretion andto androgens (Buckley and Klaassen, 2009). In turn, the dimorphismin GH secretion pattern was found both to be critical for cytochromeP450 oxidases and to be controlled by the circadian clock (Bur et al.,2009). These findings indicate multiple control mechanisms of theseveral Phase I, II, and III metabolic pathways of CPT-11. Indeed, notonly gender, but also genetic background significantly altered bothCPT-11 toxicity and UGT1A1 mRNA expression, as well as the rela-tion between both endpoints. Thus, despite mean UGT1A1 mRNAexpression were similar in female C57BL/6 (1.05 ± 0.24) and in maleB6CBAF1 (0.91 ± 0.09) (p from Scheffé’s contrast test = 0.99), cor-responding mortality rates differed by nearly twofold (32.1% and17.9% respectively; p from �2 = 0.033).
However mean UGT1A1 protein expression was highest inmales as compared with females, and in C57BL/6 as comparedto B6D2F1, with intermediate mean values in B6CBAF1. Thus noconsistent relation was found between gene and protein UGT1A1expression, as assessed with immunohistochemistry and confocalmicroscopy. This method provided quantified estimate of UGT1A1protein expression in liver cells located around blood vessels andbiliary ducts, while most methods that determine gene expressionand proteins use whole liver extracts (Luquita et al., 2001). Note-worthy, low glucuronidation activity was not related to low mRNAexpression of UGT1A1 in healthy human subjects with P229L sin-gle nucleotide polymorphism (Kaniwa et al., 2005). In a separatehuman study, the mRNA expressions of UGT isoforms, as assessedwith qPCR, were compared to UGT1A1 protein expression, as deter-mined with Western blotting in healthy human livers (Izukawa etal., 2009). A correlation between gene and protein expression wasfound only for UGT1A1, but not for UGT1A4, UGT1A6, or UGT2B7(Izukawa et al., 2009). However, an earlier work had found sig-nificant correlation between UGT1A6 gene and UGT1A6 proteinexpressions in another series of human livers (Krishnaswamy etal., 2005). Taken together, these and our current results support thepossibility of post-translational regulation of UGT1A1 expression, amechanism that remains poorly understood as yet (Lévesque et al.,2007), and could be differentially modulated by genetic backgroundand sex. Moreover, differences in post-translational UGT modi-fication, substrate absorption, cosubstrate abundance, competingmetabolic pathways, or metabolite excretion have been consideredas potential factors that explain sex differences in UGT1A1 activ-ity, beside UGT isoform transcription (Stern et al., 2008). Additionalfactors that reportedly modify UGT1A1 prediction of CPT-11 toxic-ity and display sex- and genetic dependencies include CES (Cecchinet al., 2005), CYP3A4 (Rouits et al., 2008; van Schaik, 2008), TOP1(Moisan et al., 2006) and ABC transporters (de Jong et al., 2007).
In our study, mean mRNA expression of CES2 was higher in malethan in female mice, with highest expression in male B6D2F1 andlowest one in female C57BL/6. CES2 expression levels were corre-lated with different rates of CPT-11 bioactivation into SN38 in livercellular extracts (Xu et al., 2002). In cancer patients, Cecchin et al.(2005) observed a high interindividual variability in CES2 mRNAexpression in peripheral blood mononuclear cells. They furthershowed that CES2 mRNA expression was significantly associatedwith the rate of CPT-11 bioactivation into SN-38, the main cytotoxicmetabolite of CPT-11.
We also document large strain-related differences in TOP1expression, with highest expression in B6D2F1, and lowest one in
C57BL/6. TOP1 mRNA expression also displayed large variabilityamong colorectal cancer specimens from different patients, sug-gesting that such variable expression could account for differenttumor susceptibility for CPT-11 (Takatani et al., 1997). Thus, TOP1silencing is a feature of some CPT-11-resistant cells (Sugimoto et
400 C. Ahowesso et al. / Toxicology Letters 192 (2010) 395–401
Fig. 6. Strain- and gender dependencies of UGT1A1 protein expression in mouse liver (A) Confocal analysis of UGT1A1 expression in liver from C57BL/6, B6D2F1 or B6CBAF1(columns) and male or female sex (rows). Fixed and paraffin-embedded mouse liver sections were stained with antibodies against UGT1A1 (green). Pan-Cadherin antibody(red) was used to visualize plasma membrane of hepatocytes, while DraQ5 (blue) was used to visualize nuclei. Three images from each section were acquired with 40×objective. Pinhole, laser potency and detector gain were kept constant during all the acquisitions. (B) Bar graph of mean ± SEM protein expression of UGT1A1 according tos ale Bar
aC
twUipc
tttewnos
train and gender. Protein expression is expressed relative to a control liver of a meader is referred to the web version of the article.)
l., 1990; Taniguchi et al., 1996), whereas TOP1 up-regulation favorsPT-11 cytotoxicity (Fukuoka et al., 1997).
Taken together, the three gene expression data concur to explainhe most severe toxicity of CPT-11 that is found in male B6D2F1,hose liver displays highest CES2 and TOP1 expressions and lowGT1A1 expression. These findings support largest bioactivation
nto SN-38, highest toxicity through interactions with TOP1, andoor detoxification into SN-38G in male B6D2F1. However, no suchonsistent relations are found in the other groups of mice.
Our results show that the measurements of the mRNA and pro-ein expressions of a single gene such as UGT1A1 are insufficiento predict for CPT-11 toxicity, despite this enzyme critically affectshe metabolism of this drug. We further show that the high mRNA
xpressions of three critical genes of CPT-11 pharmacology accountell for the highest toxicity of CPT-11 in a single mouse strain, butot in the two others. Rather, the validity of a systematic mappingf these critical genes and proteins responsible for CPT-11 toxicityhould further include ABC transporters and Cyp3a4 in multiple pre-lbC mouse. (For interpretation of the references to colour in this figure legend, the
clinical models. This should be integrated within a systems biologyapproach to anticancer drug pharmacology development, so as toguide the clinical assessment of model-based toxicology (Faratianet al., 2009; Wolkenhauer et al., in press). We believe that circa-dian timing should be an integral part of such approach (Lévi andSchibler, 2007; Lévi et al., 2010). Our ultimate goal is to improvetherapy through the identification of genetic variants dictating CPT-11-induced toxicity. To this end, the mechanisms underlying thecircadian rhythm of CPT-11-induced toxicity are investigated fromthe viewpoints of the sensitivity of target tissues and drug phar-macokinetics in both genders of the three strains here studied. Weexpect that this approach contributes to the true individualizationof dosing and timing of this and other anticancer agents, in order
to improve safety, quality of life and efficacy in each patient.Conflict of interest statement
None declared.

ogy Le
A
s(pEtF
R
A
B
B
B
C
C
C
C
F
F
F
G
G
I
H
I
I
d
K
C. Ahowesso et al. / Toxicol
cknowledgements
C. Ahowesso is supported by a PhD fellowship from Univer-ity Paris Sud XI and Association pour la Recherche sur le CancerARC), (9 rue Guy Moquet, 94803 VILLEJUIF Cedex). S. Dulong is aostdoctoral fellow from INSERM. The study is supported by theuropean Union Thought STREP TEMPO (Temporal genomics forailored chronotherapeutic, LSHG-CT-2006-037543), as part of theP6.
eferences
schoff, J., 1965. The phase-angle difference in circadian periodicity. In: Aschoff, J.(Ed.), Circadian Clocks. North Holland Press, Amsterdam, pp. 262–278.
uckley, D.B., Klaassen, C.D., 2007. Tissue- and gender-specific mRNA expression ofUDP glucuronosyltransferases (UGTs) in mice. Drug Metab. Dispos. 35, 121–127.
uckley, D.B., Klaassen, C.D., 2009. Mechanism of gender-divergent UDP-glucuronosyltransferase mRNA expression in mouse liver and kidney. DrugMetab. Dispos. 37, 834–840.
ur, I.M., Cohen-Solal, A.M., Carmignac, D., Abecassis, P.Y., Chauvet, N., Martin, A.O.,van der Horst, G.T., Robinson, I.C., Maurel, P., Mollard, P., Bonnefont, X., 2009.The circadian clock components CRY1 and CRY2 are necessary to sustain sexdimorphism in mouse liver metabolism. J. Biol. Chem. 284, 9066–9073.
ecchin, E., Corona, G., Masier, S., Biason, P., Cattarossi, G., Frustaci, S., Buonadonna,A., Colussi, A., Toffoli, G., 2005. Carboxylesterase isoform 2 mRNA expressionin peripheral blood mononuclear cells is a predictive marker of the irinote-can to SN-38 activation step in colorectal cancer patients. Clin. Cancer Res. 11,6901–6907.
ecchin, E., Innocenti, F., D’Andrea, M., Corona, G., De Mattia, E., Biason, P., Buon-adonna, A., Toffoli, G., 2009. Predictive role of the UGT1A1, UGT1A7, and UGT1A9genetic variants and their haplotypes on the outcome of metastatic colorec-tal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J. Clin.Oncol. 27, 2457–2465.
harasson, V., Bellott, R., Meynard, D., Longy, M., Gorry, P., Robert, J., 2004. Pharma-cogenetics of human carboxylesterase 2, an enzyme involved in the activationof irinotecan into SN-38. Clin. Pharmacol. Ther. 76, 528–535.
homczynski, P., Sacchi, N., 2006. The single-step method of RNA isolation by acidguanidinium thiocyanate–phenol–chloroform extraction: twenty-somethingyears on. Nat. Protoc. 1, 581–585.
aratian, D., Goltsov, A., Lebedeva, G., Sorokin, A., Moodie, S., Mullen, P., Kay, C., Lang-don, S., Goryanin, I., Harrison, D.J., 2009. Systems biology reveals new strategiesfor personalizing cancer medicine and confirms the role of PTEN in resistanceto trastuzumab. Cancer Res. 69, 13–20.
ilipski, E., Lemaigre, G., Liu, X.H., Méry-Mignard, D., Mahjoubi, M., Lévi, F., 2004.Circadian rhythm of irinotecan tolerability in mice. Chronobiol. Int. 21, 3–30.
ukuoka, K., Adachi, J., Nishio, K., Arioka, H., Kurokawa, H., Fukumoto, H., Ishida,T., Nomoto, T., Yokote, H., Tomonari, A., Narita, N., Yokota, J., Saijo, N., 1997.p16INK4 expression is associated with the increased sensitivity of human non-small cell lung cancer cells to DNA topoisomerase I inhibitors. Jpn. J. Cancer Res.88, 1009–1016.
randa, T.G., D’Attino, R.M., Filipski, E., Vrignaud, P., Garufi, C., Terzoli, E., Bissery,M.C., Lévi, F., 2002. Circadian optimisation of irinotecan and oxaliplatin efficacyin mice. Br. J. Cancer 86, 999–1005.
upta, E., Lestingi, T.M., Mick, R., Ramirez, J., Vokes, E.E., Ratain, M.J., 1994. Metabolicfate of irinotecan in humans: correlation of glucuronidation with diarrhea. Can-cer Res. 54, 3723–3725.
nnocenti, F., Ratain, M.J., 2003. Irinotecan treatment in cancer patients with UGT1A1polymorphisms. Oncology 17, 52–55.
uang, R.S., Ratain, M.J., 2009. Pharmacogenetics and pharmacogenomics of anti-cancer agents. CA Cancer J. Clin. 59, 42–55.
nnocenti, F., Ratain, M.J., 2006. Pharmacogenetics of irinotecan: clinical perspectiveson the utility of genotyping. Pharmacogenomics 7, 1211–1221.
zukawa, T., Nakajima, M., Fujiwara, R., Yamanaka, H., Fukami, T., Takamiya, M.,Aoki, Y., Ikushiro, S., Sakaki, T., Yokoi, T., 2009. Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers.Drug Metab. Dispos. 37, 1759–1768.
e Jong, F.A., Scott-Horton, T.J., Kroetz, D.L., McLeod, H.L., Friberg, L.E., Mathijssen,
R.H., Verweij, J., Marsh, S., Sparreboom, A., 2007. Irinotecan-induced diarrhea:functional significance of the polymorphic ABCC2 transporter protein. Clin.Pharmacol. Ther. 81, 42–49.aniwa, N., Kurose, K., Jinno, H., Tanaka-Kagawa, T., Saito, Y., Saeki, M., Sawada,J.I., Tohkin, M., Hasegawa, R., 2005. Racial variability in haplotype frequenciesof ugt1a1 and glucuronidation activity of a novel single nucleotide polymor-
tters 192 (2010) 395–401 401
phism 686C > T (P229L) found in an African-American. Drug Metab. Dispos. 33,458–465.
Kawato, Y., Aonuma, M., Hirota, Y., Kuga, H., Sato, K., 1991. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effectof CPT-11. Cancer Res. 51, 4187–4191.
Krishnaswamy, S., Hao, Q., Al-Rohaimi, A., Hesse, L.M., von Moltke, L.L., Greenblatt,D.J., Court, M.H., 2005. UDP glucuronosyltransferase (UGT) 1A6 pharmacogenet-ics. I. Identification of polymorphisms in the 5′-regulatory and exon 1 regions,and association with human liver UGT1A6 gene expression and glucuronidation.J. Pharmacol. Exp. Ther. 313, 1331–1339.
Kweekel, D., Guchelaar, H.J., Gelderblom, H., 2008. Clinical and pharmacogeneticfactors associated with irinotecan toxicity. Cancer Treat Rev. 34, 656–669.
Lamba, J.K., Lin, Y.S., Schuetz, E.G., Thummel, K.E., 2002. Genetic contribution to vari-able human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 54, 1271–1294.
Lankisch, T.O., Schulz, C., Zwingers, T., Erichsen, T.J., Manns, M.P., Heinemann,V., Strassburg, C.P., 2008. Gilbert’s Syndrome and irinotecan toxicity: combi-nation with UDP-glucuronosyltransferase 1A7 variants increases risk. CancerEpidemiol. Biomarkers Prev. 17, 695–701.
Lévesque, E., Girard, H., Journault, K., Lépine, J., Guillemette, C., 2007. Regulation ofthe UGT1A1 bilirubin-conjugating pathway: role of a new splicing event at theUGT1A locus. Hepatology 45, 128–138.
Lévi, F., Schibler, U., 2007. Circadian rhythms: mechanisms and therapeutic impli-cations. Annu. Rev. Pharmacol. Toxicol. 47, 593–628.
Lévi, F., Okyar, A., Dulong, S., Innominato, P.F., Clairambault, J., 2010. Circadian Timingin Cancer Treatments. Annu. Rev. Pharmacol. Toxicol. 50, 375–419.
Luquita, M.G., Catania, V.A., Pozzi, E.J., Veggi, L.M., Hoffman, T., Pellegrino, J.M.,Ikushiro, S., Emi, Y., Iyanagi, T., Vore, M., Mottino, A.D., 2001. Molecular basisof perinatal changes in UDP-glucuronosyltransferase activity in maternal ratliver. J. Pharmacol. Exp. Ther. 298, 49–56.
Marcuello, E., Altes, A., Menoyo, A., Del Rio, E., Gomez-Pardo, M.B., 2004. UGT1A1gene variations and irinotecan treatment in patients with metastatic colorectalcancer. Br. J. Cancer 91, 678–682.
Moisan, F., Longy, M., Robert, J., Le Morvan, V., 2006. Identification of gene polymor-phisms of human DNA topoisomerase I in the National Cancer Institute panel ofhuman tumour cell lines. Br. J. Cancer. 95, 906–913.
Ohdo, S., Makinosumi, T., Ishizaki, T., Yukawa, E., Higuchi, S., Nakano, S., Ogawa, N.,1997. Cell cycle-dependent chronotoxicity of irinotecan hydrochloride in mice.J. Pharmacol. Exp. Ther. 283, 1383–1388.
Rosner, G.L., Panetta, J.C., Innocenti, F., Ratain, M.J., 2008. Pharmacogenetic pathwayanalysis of irinotecan. Clin. Pharmacol. Ther. 84, 393–402.
Rouits, E., Charasson, V., Pétain, A., Boisdron-Celle, M., Delord, J.P., Fonck, M., Laurand,A., Poirier, A.L., Morel, A., Chatelut, E., Robert, J., Gamelin, E., 2008. Pharma-cokinetic and pharmacogenetic determinants of the activity and toxicity ofirinotecan in metastatic colorectal cancer patients. Br. J. Cancer 99, 1239–1245.
van Schaik, R.H., 2008. CYP450 pharmacogenetics for personalizing cancer therapy.Drug Resist. Updat. 11, 77–98.
Stern, S.T., Tallman, M.N., Miles, K.K., Ritter, J.K., Smith, P.C., 2008. Androgen regula-tion of renal uridine diphosphoglucuronosyltransferase 1A1 in rats. Drug Metab.Dispos. 36, 1737–1739.
Sugimoto, Y., Tsukahara, S., Oh-Hara, T., Isoe, T., Tsuruo, T., 1990. Decreased expres-sion of DNA topoisomerase I in camptothecin-resistant tumor cell lines asdetermined by a monoclonal antibody. Cancer Res. 50, 6925–6930.
Takatani, H., Oka, M., Fukuda, M., Narasaki, F., Nakano, R., Ikeda, K., Terashi, K.,Kinoshita, A., Soda, H., Kanda, T., Schneider, E., Kohno, S., 1997. Gene muta-tion analysis and quantitation of DNA topoisomerase I in previously untreatednon-small cell lung carcinomas. Jpn. J. Cancer Res. 88, 160–165.
Taniguchi, K., Kohno, K., Kawanami, K., Wada, M., Kanematsu, T., Kuwano, M., 1996.Drug-induced down-regulation of topoisomerase I in human epidermoid cancercells resistant to saintopin and camptothecins. Cancer Res. 56, 2348–2354.
Tampellini, M., Filipski, E., Liu, X.H., Lemaigre, G., Li, X.M., Vrignaud, P., Francois, E.,Bissery, M.C., Lévi, F., 1998. Docetaxel chronopharmacology in mice. Cancer Res.58, 3896–3904.
Wasserman, E., Myara, A., Lokiec, F., Goldwasser, F., Trivin, F., Mahjoubi, M., Mis-set, J.L., Cvitkovic, E., 1997. Severe CPT-11 toxicity in patients with Gilbert’ssyndrome: two case reports. Ann. Oncol. 8, 1049–1051.
Wolkenhauer, O., Auffray, C., Baltrusch, S., Blüthgen, N., Byrne, H., Cascante, M.,Ciliberto, A., Dale, T., Drasdo, D., Fell, D., Ferrell, J., Gallahan, D., Gatenby, R.,Guenther, U., Herzel, H., Junghanss, C., Kunz, M., Laplace, F., van Leeuwen, I.,Lenormand, P., Lévi, F., Linnebacher, M., Lowengrub, J., Maini, P.K., Marcus, F.,Malik, A., Rateitschak, K., Sansom, O., Schäfer, R., Schürrle, K., Sers, C., Schnell,
S., Harms, B., Shibata, D., Tyson, J., Vera, J., White, M., Zhivotovsky, B., Jaster, R.,in press. Systems biologists hope for a full integration of their field into cancerresearch. Cancer Res.Xu, G., Zhang, W., Ma, M.K., McLeod, H.L., 2002. Human carboxylesterase 2 is com-monly expressed in tumor tissue and is correlated with activation of irinotecan.Clin. Cancer Res. 8, 2605–2611.




















