
Nitroxyl enhances myocyte Ca2+ transients by exclusively targeting SR Ca2+-cycling

Regulation of cardiac myocyte contractility by phospholemman: Na�/Ca2�
exchange versus Na�-K�-ATPase
Jianliang Song,1,2 Xue-Qian Zhang,1,2 JuFang Wang,1,2 Ellina Cheskis,2 Tung O. Chan,2
Arthur M. Feldman,2 Amy L. Tucker,3 and Joseph Y. Cheung1,2
1Division of Nephrology and 2Center of Translational Medicine, Department of Medicine, Jefferson Medical College,Thomas Jefferson University, Philadelphia, Pennsylvania; and 3Cardiovascular Division, Department of Internal Medicine,University of Virginia Health Sciences Center, Charlottesville, Virginia
Submitted 17 March 2008; accepted in final form 12 August 2008
Song J, Zhang XQ, Wang J, Cheskis E, Chan TO, Feldman AM,Tucker AL, Cheung JY. Regulation of cardiac myocyte contractility byphospholemman: Na�/Ca2� exchange versus Na�-K�-ATPase. Am JPhysiol Heart Circ Physiol 295: H1615–H1625, 2008. First publishedAugust 15, 2008; doi:10.1152/ajpheart.00287.2008.—Phospholemman(PLM) regulates cardiac Na�/Ca2� exchanger (NCX1) and Na�-K�-ATPase in cardiac myocytes. PLM, when phosphorylated at Ser68,disinhibits Na�-K�-ATPase but inhibits NCX1. PLM regulates car-diac contractility by modulating Na�-K�-ATPase and/or NCX1. Inthis study, we first demonstrated that adult mouse cardiac myocytescultured for 48 h had normal surface membrane areas, t-tubules, andNCX1 and sarco(endo)plasmic reticulum Ca2�-ATPase levels, andretained near normal contractility, but �1-subunit of Na�-K�-ATPasewas slightly decreased. Differences in contractility between myocytesisolated from wild-type (WT) and PLM knockout (KO) hearts werepreserved after 48 h of culture. Infection with adenovirus expressinggreen fluorescent protein (GFP) did not affect contractility at 48 h.When WT PLM was overexpressed in PLM KO myocytes, contrac-tility and cytosolic Ca2� concentration ([Ca2�]i) transients revertedback to those observed in cultured WT myocytes. Both Na�-K�-ATPase current (Ipump) and Na�/Ca2� exchange current (INaCa) inPLM KO myocytes rescued with WT PLM were depressed comparedwith PLM KO myocytes. Overexpressing the PLMS68E mutant(phosphomimetic) in PLM KO myocytes resulted in the suppressionof INaCa but had no effect on Ipump. Contractility, [Ca2�]i transientamplitudes, and sarcoplasmic reticulum Ca2� contents in PLM KOmyocytes overexpressing the PLMS68E mutant were depressed com-pared with PLM KO myocytes overexpressing GFP. Overexpressingthe PLMS68A mutant (mimicking unphosphorylated PLM) in PLMKO myocytes had no effect on INaCa but decreased Ipump. Contractil-ity, [Ca2�]i transient amplitudes, and sarcoplasmic reticulum Ca2�
contents in PLM KO myocytes overexpressing the S68A mutant weresimilar to PLM KO myocytes overexpressing GFP. We conclude thatat the single-myocyte level, PLM affects cardiac contractility and[Ca2�]i homeostasis primarily by its direct inhibitory effects onNa�/Ca2� exchange.
adult mouse cardiac myocyte culture; excitation-contraction coupling;FXYD1; �-adrenergic stimulation; phosphorylation
PHOSPHOLEMMAN (PLM) is the first cloned member of the FXYDfamily of regulators of ion transport (19). This small 72-aminoacid integral membrane phosphoprotein with a single trans-membrane domain was initially proposed to be involved in cellvolume regulation in noncardiac tissues (7). In the mammalianheart, PLM regulates the activities of both Na�-K�-ATPase (8,16, 26) and the cardiac Na�/Ca2� exchanger (NCX1) (14, 17).
PKA phosphorylates Ser68, whereas PKC phosphorylates bothSer68 and Ser63 (23), of PLM. PLM, when phosphorylated atSer68, disinhibits Na�-K�-ATPase (8, 16) and actively inhibitsNCX1 (18, 25).
In adult rat myocytes, overexpression of PLM alters con-tractility and cytosolic Ca2� concentration ([Ca2�]i) transientamplitudes (17). Specifically, at low (0.6 mM) extracellularCa2� concentrations ([Ca2�]o), both contraction and [Ca2�]i
transient amplitudes are larger in myocytes overexpressingPLM. At high (5.0 mM) [Ca2�]o, cell shortening and [Ca2�]i
transient amplitudes are smaller in myocytes overexpressingPLM. Na�-K�-ATPase current (Ipump) is inhibited in adult ratmyocytes overexpressing PLM (26). Enhanced inhibition ofNa�-K�-ATPase by overexpressed PLM would be expected toraise the cytosolic Na� concentration ([Na�]i), thereby reduc-ing the thermodynamic driving force for Ca2� efflux via NCX1and result in an increase, rather than the observed decrease, incontractility and [Ca2�]i transient amplitudes. The contractilephenotype observed in myocytes overexpressing PLM is sim-ilar to that observed in myocytes in which NCX1 is downregu-lated (21) and provides circumstantial evidence that PLM maydirectly inhibit Na�/Ca2� exchange. Indeed, in a heterologousexpression model system, PLM inhibits Na�/Ca2� exchangecurrent (INaCa) and Na�-dependent Ca2� uptake (1). In adultrat myocytes, PLM colocalizes with (28) and coimmunopre-cipitates NCX1 (1, 14).
In adult rat myocytes in which PLM has been downregulated(14) and in adult mouse myocytes in which PLM has beenengineered to be absent (25), INaCa is higher than in respectivecontrol myocytes. Compared with their respective controls,contractile and [Ca2�]i transient amplitudes are lower at 0.6but higher at 5.0 mM [Ca2�]o. The contractile phenotype ofPLM downregulated or PLM knockout (KO) myocytes mimicsthat observed in myocytes in which NCX1 is overexpressed(29). These results, when viewed together, support the hypoth-esis that PLM regulates cardiac contractility by regulatingNa�/Ca2� exchange. However, in adult myocytes, Ipump is alsoinhibited by PLM either by lowering Vmax (10, 11, 26) or byraising Km for Na� (8). At present, the mechanism by whichPLM regulates cardiac contractility is being actively investi-gated and is a topic of lively debate (5).
The present study was undertaken to define, at the single-myocyte level, whether inhibition of Na�-K�-ATPase or Na�/Ca2� exchange is the primary mechanism by which PLM
Address for reprint requests and other correspondence: J. Y. Cheung, Div. ofNephrology, Jefferson Medical College, 833 Chestnut St., Suite 700, Phila-delphia, PA 19107 (e-mail: [email protected]).
The costs of publication of this article were defrayed in part by the paymentof page charges. The article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Am J Physiol Heart Circ Physiol 295: H1615–H1625, 2008.First published August 15, 2008; doi:10.1152/ajpheart.00287.2008.
0363-6135/08 $8.00 Copyright © 2008 the American Physiological Societyhttp://www.ajpheart.org H1615

affects cardiac contractility. We examined, in cultured myo-cytes isolated from congenic PLM KO hearts, whether over-expression of PLM mutants that affect only Na�/Ca2� ex-change or Na�-K�-ATPase can bring about contractility and[Ca2�]i transient changes.
METHODS
Generation of PLM-deficient mice and animal care. PLM KO micebackcrossed to a pure congenic C57BL/6 background were generatedas previously described (12, 22). Homozygous adult littermates of �3mo old were used. Mice were housed and fed on a 12:12-h light-darkcycle at the Thomas Jefferson University Animal Facility and weresupervised by veterinary staff members. Standard care was providedto all mice used for experiments. All protocols applied to the mice inthis study were approved and supervised by the Institutional AnimalCare and Use Committees at Thomas Jefferson University and theUniversity of Virginia.
Isolation and culture of adult murine cardiac myocytes. Cardiacmyocytes were isolated from the septum and left ventricular free wallof wild-type (WT) and PLM KO mice (�3 mo old, 25–37g) accordingto the protocol of Zhou et al. (30) and as modified by us (22). Isolatedmyocytes were plated on laminin-coated coverslips, and the Ca2�
concentration of the buffer was progressively increased from 0.05 to0.125 to 0.25 to 0.5 mM (10-min intervals each). The 0.5 mM Ca2�
buffer was then aspirated and replaced with minimal essential medium(MEM) containing 1.2 mM Ca2�, 2.5% FBS, and antibiotics (1%penicillin-streptomycin). The pH was adjusted to 7.0 in 4% CO2 bythe addition of NaHCO3 (0.57 g/l). After 1 h (4% CO2, 37°C), themedium was replaced with FBS-free MEM containing 0.1 mg/mlBSA, antibiotics, and insulin-transferrin-selenium (ITS) supplement.2,3-Butanedione monoxime (BDM; 10 mM) was present throughoutthe heart perfusion and culture. Media were changed every day.Before measurements of contraction, [Ca2�]i transients, INaCa, Ipump,and sarcoplasmic reticulum (SR) Ca2� content, culture medium con-taining BDM was aspirated, and cells were bathed with MEM withoutBDM and returned to the incubator (37°C) for 30 min. Coverslipscontaining cultured myocytes were then taken out of the incubator,mounted in the Dvorak-Stotler chamber, and bathed in fresh mediabefore measurements.
Staining of t-tubules in myocytes. Myocytes isolated from WThearts and cultured for 0, 24, and 48 h were stained with di-8-ANEPPS (10 �M) at 37°C for 10 min. Stained myocytes were washedwith MEM followed by examination using both confocal (ZeissLSM510, excitation and emission wavelengths at 488 and 505 nm,respectively) and conventional wide-field fluorescence microscopy(Olympus IX71, excitation and emission wavelengths at 480 � 20 and535 � 25 nm, respectively). In some experiments, myocytes wereexposed to formamide (1.5 mol/l) at 37°C for 15 min to inducedetubulation before being imaged.
Adenoviral infection of cardiac myocytes. Recombinant, replica-tion-deficient adenovirus (Adv) expressing either green fluorescentprotein (GFP) alone, GFP and WT dog PLM, or GFP and dog PLMmutants (S68A or S68E) were constructed as previously described(17, 18). Two hours after isolation, myocytes were infected withAdv-GFP, Adv-GFP-PLM, or Adv-GFP-PLM mutants at a multiplic-ity of infection of 2 for 3.5 h. Media were then changed, and myocyteswere studied after 48 h. For the sake of brevity, PLM KO myocytesinfected with Adv-GFP, Adv-GFP-PLM, and Adv-GFP-PLM mutantsare referred to as KO-GFP, KO-PLM, and the respective designationfor the PLM mutant (e.g., KO-S68A) myocytes, respectively.
Myocyte shortening measurements. Myocytes adherent to cover-slips were bathed in 0.6 ml of air- and temperature-equilibrated (37°C)HEPES-buffered (20 mM, pH 7.4) medium 199 containing either 1.8or 5.0 mM [Ca2�]o. Measurements of myocyte contraction (1 Hz)were performed as previously described (14, 17, 18, 21, 22, 29).
[Ca2�]i transient measurements. Myocytes loaded with fura-2(0.67 �M, 15 min, 37°C) were field stimulated to contract (1 Hz,37°C) in medium 199 containing either 1.8 or 5.0 mM [Ca2�]o.[Ca2�]i transient measurements, daily calibration of fura-2 fluorescentsignals, and [Ca2�]i transient analyses were performed as previouslydescribed (14, 17, 18, 21, 22, 29).
INaCa, Ipump, and SR Ca2� content measurements. Whole cellpatch-clamp recordings were performed at 30°C as previously de-scribed (21, 22). The pipette diameter was 4–6 �m and the pipetteresistance was 0.8–1.4 M� when filled with standard internal solu-tion. For INaCa measurements, the pipette solution contained (in mM)100 Cs�-glutamate, 7.25 Na�-HEPES, 1 MgCl2, 12.75 HEPES, 2.5Na2ATP, 10 EGTA, and 6 CaCl2 (pH 7.2). Free Ca2� in the pipettesolution was 205 nM. Myocytes were bathed in an external solutioncontaining (in mM) 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5CaCl2, 10 HEPES, 10 Na�-HEPES, and 10 glucose (pH 7.4) (14, 18,25). Verapamil (1 �M), ouabain (1 mM), and niflumic acid (10 �M)were used to block L-type Ca2� currents, Ipump, and Cl� currents,respectively. The myocyte was held at the calculated equilibriumpotential for INaCa (ENaCa) of �73 mV for at least 5 min before currentwas elicited with a descending-ascending voltage ramp (from �100 to�120 and back to �100 mV, 500 mV/s). INaCa was defined as thedifference current in the absence and presence of CdCl2 (1 mM). Ourconditions for measuring INaCa were carefully chosen to minimizecontamination by Na�-K�-ATPase activity (K� free and in thepresence of ouabain) and ion fluxes through NCX1 before the onset ofvoltage ramp (by holding the cell at the calculated ENaCa), therebyallowing [Na�]i and [Ca2�]i to equilibrate with those present in thepipette solution.
For Ipump measurements (26), the standard pipette solution con-tained (in mM) 70 Na�-aspartate, 20 K�-aspartate, 8 CsOH, 7MgSO4, 11 EGTA, 10 TEA.Cl, 1 CaCl2, 5 HEPES, 5 Na2ATP, and0.2 GTP (pH 7.2). To decrease the pipette Na� concentration([Na�]pip) to 10 mM, Cs� was substituted for Na�. The externalsolution contained (in mM) 137.7 NaCl, 18 KCl, 2.3 NaOH, 1 MgCl2,2 BaCl2, 1 CdCl2, 5 HEPES, and 10 glucose (pH 7.4). The holdingpotential was switched from �70 to �40 mV (300 ms) before theapplication of a negative voltage ramp (from �60 to �120 mV, 20mV/s), first in the absence and then in the presence of 1 mM ouabain.Ipump was defined as the difference current before and after theaddition of ouabain.
SR Ca2� content was estimated by integrating forward INaCa
induced by caffeine exposure as previously described (21, 22, 29).The pipette solution consisted of (in mM) 100 Cs�-glutamate, 1MgCl2, 30 HEPES, and 2.5 MgATP (pH 7.2). The external solutioncontained (in mM) 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5CaCl2, 20 HEPES, and 10 glucose (pH 7.4, 30°C). The holdingpotential was �70 mV. At 200 ms after the 11th conditioning pulse(from �70 to 0 mV, 300 ms, 1 Hz), with membrane potential held at�70 mV, caffeine (5 mM, 2.4 s) was applied by puffer superfusion.The resulting inward current was digitized at 0.5 kHz and collected for5 s. To convert the INaCa time integral (in Coulombs) to moles, thecharge was divided by Faraday’s constant of 96,487 coulombs/equivalent, based on 3 Na� being exchanged for each Ca2�. SR Ca2�
content was normalized to cell size (in fmol/fF).Immunoblot analysis. Myocytes were harvested for immunoblot
analysis after 48 h of culture as previously described (14, 17, 18, 21,26). For PLM immunoblot analysis, proteins in myocyte lysates weresubjected to 12% SDS-PAGE under reducing (5% �-mercaptoetha-nol) conditions. Polyclonal C2 antibody (1:10,000) was used to detectthe COOH-termini of both endogenous mouse and WT dog PLM (thepredominantly unphosphorylated form) (16, 17, 26). Monoclonal B8antibody (1:5,000) was used to detect the NH2-termini of WT dogPLM and its Ser68 mutants (18). Polyclonal CP68 antibody (1:500)was used to detect PLM phosphorylated at Ser68 (16, 26).
For the detection of NCX1, sarco(endo)plasmic reticulum Ca2�-ATPase 2 (SERCA2), calsequestrin, and the �1-subunit of Na�-K�-
H1616 PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

ATPase, myocyte homogenates were subjected to 7.5% SDS-PAGEunder nonreducing (10 mM N-ethylmaleimide for NCX1) or reducing(for SERCA2, calsequestrin, and Na�-K�-ATPase) conditions. Theprimary antibodies used were as follows: for NCX1, 11-13 polyclonalantibody (1:700, Swant, Bellinzona, Switzerland); for SERCA2, BL337polyclonal antibody (1:1,000, Bethyl Laboratories, Montgomery,TX); for calsequestrin, rabbit anti-calsequestrin antibody (1:5,000,Swant); and for the �1-subunit of Na�-K�-ATPase (1:1,000,Upstate, Charlottesville, VA). The secondary antibodies used weredonkey anti-rabbit and sheep anti-mouse IgG (Amersham). Immu-noreactive proteins were detected with an enhanced chemilumines-cence Western blot system. Protein band signal intensities werequantitated by scanning autoradiograms of the blots with a phos-phorimager.
Statistics. All results are expressed as means � SE. For the analysisof a parameter (e.g., maximal contraction amplitude) as a function of daysin culture and [Ca2�]o, two-way ANOVA was used. When the parameterwas examined as a function of group (e.g., WT vs. PLM KO), [Ca2�]o,and days in culture, three-way ANOVA was used. For the analysis of ioncurrents as a function of group and voltage or as a function of group and[Na�]pip, two-way ANOVA was used. A commercial software package(JMP version 7, SAS Institute, Cary, NC) was used. In all analyses, P 0.05 was taken to be statistically significant.
RESULTS
Culture and adenoviral infection of adult mouse myocytes.In our hands, adult mouse myocytes survived better in culturewhen plated on laminin- rather than Matrigel-coated cover-
slips. Cultured mouse myocytes maintained their elongated rodshapes when cultured in FBS for 48 h. With serum-free culture,the inclusion of BDM, ITS supplement, and BSA in MEMgave the best results in terms of the maintenance of rod shapes(�65%) after 48 h (Fig. 1). When infected with adenovirusexpressing GFP, green fluorescence started to appear by 18–24h and increased throughout 48 h of culture (Fig. 1). With fewexceptions, GFP fluorescence was much more intense in rod-shaped myocytes compared with rounded and hypercontractedmyocytes. Other agents that inhibited myocyte contraction[blebbistatin (5.7 �M) or cytochalasin D (2 �M)] were not aseffective as BDM in terms of maintaining myocyte viability inculture. The required presence of BDM precluded pacing adultmouse myocytes while in culture.
Effects of culture and adenoviral infection on whole cellmembrane capacitance, t-tubules, and selected proteins in-volved in excitation-contraction coupling. Membrane capaci-tance (Cm) in PLM KO myocytes cultured for 48 h (180 � 5pF, n � 27) was not significantly (P 0.065) different thanthat measured in freshly isolated PLM KO myocytes (164 � 5pF, n � 50), suggesting that myocyte surface membrane area didnot appreciably change with short-term culture. When di-8-ANEPPS was used to stain surface sarcolemma and t-tubules,myocytes cultured for 24 and 48 h retained their t-tubular networkcompared with freshly isolated myocytes (Fig. 2). Pretreatment
Fig. 1. Culture and adenoviral infection of adultmouse myocytes. Cardiac myocytes were isolatedfrom adult congenic phospholemman (PLM)knockout (KO) mice, infected with adenovirusexpressing both green fluorescent protein (GFP)and the PLMS68A mutant, and placed in cultureas described in METHODS. A: transmitted lightimage of adult mouse myocytes after 24 h ofculture. B: fluorescent image of A demonstratingthe expression of GFP. C: transmitted light imageof another adult mouse myocyte culture at 48 h.D: fluorescent image of C. Both B and D are rawimages and were taken at the same time usingidentical excitation light intensities, with thecamera set at the same gain but with autoexpo-sure mode. The higher background in B com-pared with D is due to the longer exposure timebecause of the much lower GFP fluorescenceemission from adenovirus-infected myocytes cul-tured for 24 h. In contrast, GFP fluorescenceemission from infected myocytes cultured for48 h was much stronger and required a shorterexposure time, as indicated by the lowerbackground.
H1617PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org
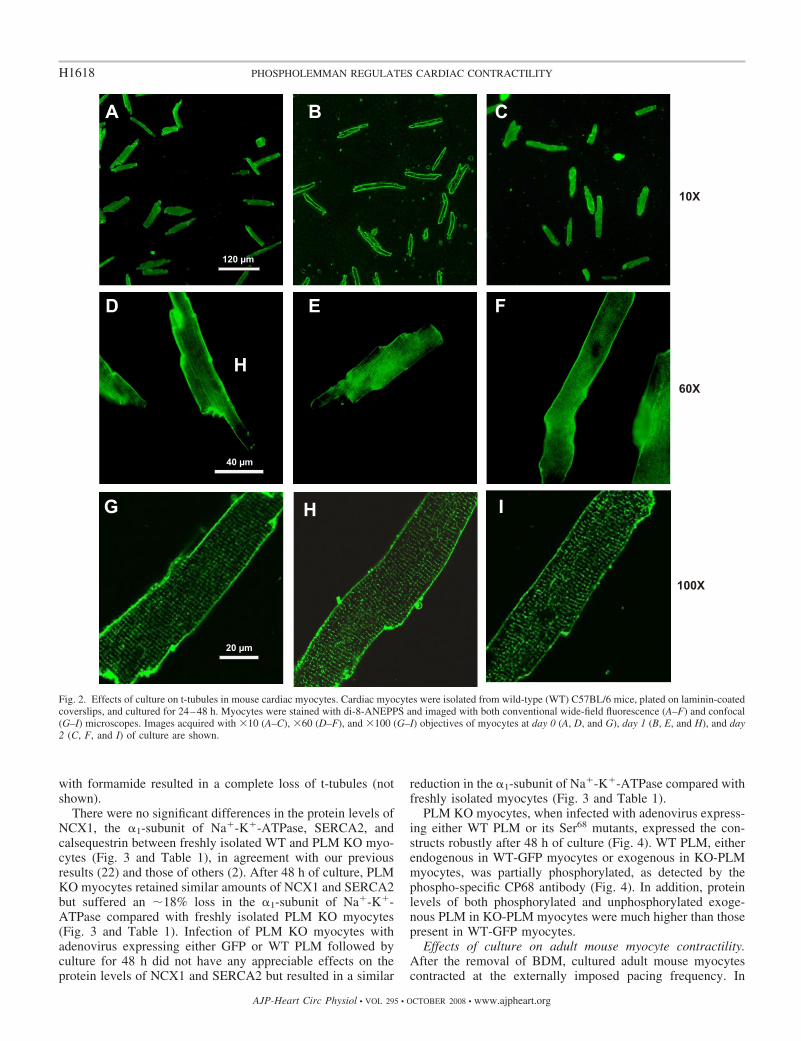
with formamide resulted in a complete loss of t-tubules (notshown).
There were no significant differences in the protein levels ofNCX1, the �1-subunit of Na�-K�-ATPase, SERCA2, andcalsequestrin between freshly isolated WT and PLM KO myo-cytes (Fig. 3 and Table 1), in agreement with our previousresults (22) and those of others (2). After 48 h of culture, PLMKO myocytes retained similar amounts of NCX1 and SERCA2but suffered an �18% loss in the �1-subunit of Na�-K�-ATPase compared with freshly isolated PLM KO myocytes(Fig. 3 and Table 1). Infection of PLM KO myocytes withadenovirus expressing either GFP or WT PLM followed byculture for 48 h did not have any appreciable effects on theprotein levels of NCX1 and SERCA2 but resulted in a similar
reduction in the �1-subunit of Na�-K�-ATPase compared withfreshly isolated myocytes (Fig. 3 and Table 1).
PLM KO myocytes, when infected with adenovirus express-ing either WT PLM or its Ser68 mutants, expressed the con-structs robustly after 48 h of culture (Fig. 4). WT PLM, eitherendogenous in WT-GFP myocytes or exogenous in KO-PLMmyocytes, was partially phosphorylated, as detected by thephospho-specific CP68 antibody (Fig. 4). In addition, proteinlevels of both phosphorylated and unphosphorylated exoge-nous PLM in KO-PLM myocytes were much higher than thosepresent in WT-GFP myocytes.
Effects of culture on adult mouse myocyte contractility.After the removal of BDM, cultured adult mouse myocytescontracted at the externally imposed pacing frequency. In
Fig. 2. Effects of culture on t-tubules in mouse cardiac myocytes. Cardiac myocytes were isolated from wild-type (WT) C57BL/6 mice, plated on laminin-coatedcoverslips, and cultured for 24–48 h. Myocytes were stained with di-8-ANEPPS and imaged with both conventional wide-field fluorescence (A–F) and confocal(G–I) microscopes. Images acquired with �10 (A–C), �60 (D–F), and �100 (G–I) objectives of myocytes at day 0 (A, D, and G), day 1 (B, E, and H), and day2 (C, F, and I) of culture are shown.
H1618 PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

contrast to freshly isolated myocytes, most mouse myocytescultured for 48 h failed to contract when stimulated at 0.6 mM[Ca2�]o. This phenomenon made it very difficult to preciselyquantify contraction amplitudes at 0.6 mM [Ca2�]o. For thisreason, in this study, we only examined contraction and[Ca2�]i transients at 1.8 and 5.0 mM [Ca2�]o.
As shown in Table 2, WT and PLM KO myocytes culturedfor 24 and 48 h in serum-free media maintained contractionamplitudes that were slightly less than those observed forfreshly isolated myocytes (day effect, P 0.035). As expected,elevating [Ca2�]o increased contraction amplitudes ([Ca2�]o
effect, P 0.0001). In addition, contraction amplitudes weredifferent between cultured WT and PLM KO myocytes (groupeffect, P 0.003). Specifically, contraction amplitudes inPLM KO myocytes were significantly higher than WT myo-cytes at 5.0 but not 1.8 mM [Ca2�]o (group � [Ca2�]o
interaction effect, P 0.03). Since freshly isolated myocytesfrom PLM KO hearts exhibited contraction amplitudes thatwere similar at 1.8 mM [Ca2�]o but higher at 5.0 mM [Ca2�]o
compared with WT myocytes (22), our present observationsuggests that the contractile differences between WT and PLMKO myocytes were preserved after 48 h of culture.
Effect of adenoviral infection on cultured myocyte contrac-tility. Infection with recombinant, replication-deficient adeno-virus has been shown to be highly efficient for gene delivery toadult cardiac myocytes (17, 21, 29). Indeed, 95% of rod-shaped adult mouse myocytes exhibited robust GFP fluores-cence after an infection with Adv-GFP (Fig. 1). Compared withuninfected myocytes, PLM KO myocytes infected with Adv-GFP had similar cell shortening amplitudes after 24 and 48 hof culture, regardless of whether contraction was measured
with 1.8 or 5.0 mM [Ca2�]o (Table 3). This conclusion wassupported by three-way ANOVA, which indicated insignificantgroup (PLM KO vs. KO-GFP, P 0.80) and day (P 0.11)effects but significant [Ca2�]o (P 0.0001) effects. Therewere no significant interaction effects.
Rescue of PLM deficiency in PLM KO myocytes. Since PLMKO myocytes maintained their contractile phenotype aftershort-term culture (Table 2), we next determined if the con-tractile phenotype would revert back to that observed for WTmyocytes after “rescue” with PLM. Compared with KO-GFPmyocytes, contraction amplitudes in KO-PLM myocytes weresimilar at 1.8 mM [Ca2�]o but lower at 5.0 mM [Ca2�]o (Fig. 5and Table 4; group � [Ca2�]o interaction effect, P 0.025).There were no differences in contraction amplitudes betweenWT and KO-PLM myocytes, indicating that PLM rescue wassuccessful in restoring the contractile phenotype in PLM KOmyocytes back to that observed in WT myocytes. [Ca2�]i
transients in PLM KO myocytes were similarly affected byPLM overexpression: systolic [Ca2�]i and [Ca2�]i transientamplitudes were similar at 1.8 mM [Ca2�]o but significantly
Fig. 3. Effects of culture on selected proteins involved in excitation-contrac-tion coupling. Myocyte homogenates were prepared from day 0 WT (WT-D0),day 0 PLM KO (KO-D0), day 2 PLM KO (KO-D2), day 2 GFP-expressing KO(KO-GFP), and day 2 PLM-expressing KO (KO-PLM) myocytes and sub-jected to SDS-PAGE followed by Western blot analysis. Primary antibodies(METHODS) were used to detect cardiac Na�/Ca2� exchanger (NCX1), the�1-subunit of Na�-K�-ATPase, sarco(endo)plasmic reticulum Ca2�-ATPase 2 (SERCA2), and calsequestrin (CLSQ). Composite results areshown in Table 1. Fig. 4. Expression of WT PLM and its Ser68 mutants in cultured PLM KO
myocytes. Adult cardiac myocytes isolated from congenic PLM KO mice wereinfected with adenovirus expressing either GFP alone (KO-GFP), GFP � WTdog PLM (KO-PLM), GFP � PLMS68A (KO-S68A), or GFP � PLMS68E(KO-S68E) and cultured for 48 h. Myocytes from WT littermates infected withadenovirus expressing GFP (WT-GFP) and cultured for 48 h were used ascontrols. Proteins in myocyte lysates (50 �g/lane) were separated by gelelectrophoresis and transferred to ImmunBlot polyvinylidene difluoride mem-branes. Endogenous mouse and WT dog PLM, unphosphorylated and phos-phorylated at Ser68, was probed with C2 and CP68 anti-PLM antibodies,respectively, whereas dog PLM Ser68 mutants were detected with B8 anti-PLMantibody. C2 antibody recognizes predominantly the unphosphorylatedCOOH-terminus of both dog and rodent PLM, and CP68 antibody is specificfor PLM phosphorylated at Ser68 (16, 26), whereas B8 antibody recognizes theNH2-terminus of dog but not rodent PLM (18).
Table 1. Effects of culture and adenoviral infection on selected proteins in PLM KO myocytes
WT-D0 KO-D0 KO-D2 KO-GFP KO-PLM
Na�/Ca2� exchanger isoform 1 5.2�0.7 5.3�0.5 4.8�0.2 5.6�0.6 5.9�0.2Sarco(endo)plasmic reticulum Ca2�-ATPase 2 9.0�0.6 8.9�0.2 8.2�1.1 7.9�0.2 8.0�0.3�1-Subunit of Na�-K�-ATPase 9.9�0.5 8.1�1.0 6.6�0.2* 6.4�0.2* 5.5�1.0*
Values are means � SE (in arbitrary units). WT-D0, day 0 wild-type (WT) myoctyes; KO-D0, day 0 phospholemman (PLM) knockout (KO) myocytes;KO-D2, day 2 PLM KO myocytes; KO-GFP, PLM KO myocytes infected with adenovirus expressing green fluorescent protein (GFP); KO-PLM, PLM KOmyocytes infected with adenovirus expressing PLM � GFP. Except for the WT-D0 group (n � 3), all other groups consisted of myocytes prepared from 4different mice. To correct for slight variations in protein loading, the protein band intensities shown in Fig. 3 were normalized to the corresponding calsequestrinlevels. *P 0.001 compared with WT-D0 myocytes.
H1619PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

lower at 5.0 mM [Ca2�]o (Fig. 5 and Table 5). The half-life(t1/2) of [Ca2�]i decline, an estimate of in situ SR Ca2� uptakeactivity (27), was not different between KO-PLM and KO-GFPmyocytes (Table 5), indicating that PLM had no effect onSERCA2 activity. In support of this interpretation, there wereno differences in SERCA2 protein levels between KO-GFPand KO-PLM myocytes (Fig. 3 and Table 1). Importantly, bothINaCa (Fig. 6) and Ipump (Fig. 7) were significantly suppressedin KO-PLM myocytes compared with KO-GFP myocytes.Compared with WT myocytes, both INaCa (Fig. 6) and Ipump
(Fig. 7) were higher in KO-GFP but much lower in KO-PLMmyocytes. Severe suppression of INaCa and Ipump in KO-PLMmyocytes is likely due to the supraphysiological levels of bothphosphorylated and unphosphorylated exogenous PLM in KO-PLM myocytes compared with the endogenous levels of PLMfound in WT myocytes (Fig. 4). Despite no differences in SRCa2� uptake activity and SERCA2 protein levels, SR Ca2�
content measured at 5 mM [Ca2�]o was higher in KO-GFPcompared with KO-PLM myocytes (Table 6).
Effects of the PLMS68E mutant on contractility, [Ca2�]i
transients, SR Ca2� uptake, SR Ca2� content, INaCa, and Ipump
on PLM KO myocytes. Previous studies have indicated thatPLM, when phosphorylated at Ser68, strongly inhibits NCX1(18, 25) while it simultaneously relieves its inhibition onNa�-K�-ATPase (8, 10, 11, 16). To investigate whether theregulation of myocyte contractility by PLM is mediated via itseffects on NCX1 versus Na�-K�-ATPase, we used the phos-phomimetic PLM mutant S68E, which has been shown toinhibit INaCa in adult rat cardiac myocytes (18) and transfectedHEK-293 cells (25). In PLM KO myocytes devoid of endog-enous PLM, overexpression of the PLMS68E mutant resultedin a large suppression of INaCa (Fig. 6). Because phosphory-lated PLM has been reported to either lower the Km for Na� (8)or increase the Vmax of Na�-K�-ATPase (16, 26), we measuredIpump at [Na�]pip of 10 mM (Km effect) and 80 mM (Vmax
effect). Overexpression of the PLMS68E mutant in PLM KOmyocytes did not affect Ipump (Fig. 7; group effect, P 0.35,and group � [Na�]pip interaction effect, P 0.80). At 5.0 mM[Ca2�]o, contraction amplitudes of KO-S68E myocytes weresignificantly (P 0.0004) lower than those of KO-GFP myo-cytes but similar (P 0.99) to those of KO-PLM myocytes(Fig. 5 and Table 6). Similarly, [Ca2�]i transient amplitudes ofKO-S68E myocytes measured at 5.0 mM [Ca2�]o were lowercompared with those of KO-GFP myocytes (Fig. 5 and Table6). There were no differences in SR Ca2� uptake, although SR
Ca2� content was significantly lower in KO-S68E comparedwith KO-GFP myocytes (Table 6).
Effects of the PLMS68A mutant on contractility, [Ca2�]i
transients, SR Ca2� uptake, SR Ca2� content, INaCa, and Ipump
on PLM KO myocytes. The PLMS68A mutant has been shownto have no effect on INaCa in adult rat cardiac myocytes (18)and in transfected HEK-293 cells (25). Overexpression of thePLMS68A mutant in PLM KO myocytes had no effect on INaCa
(Fig. 6) but resulted in significant inhibition of Ipump (Fig. 7),which appeared to be more prominent at [Na�]pip of 80 mM(�66% inhibition) than 10 mM (�51% inhibition). This con-clusion was supported by two-way ANOVA, which indicatedsignificant group (P 0.0001), [Na�]pip (P 0.0001), andgroup � [Na�]pip interaction (P 0.0025) effects. Our resultssuggest that unphosphorylated PLM regulates Na�-K�-ATPase pri-marily by a Vmax effect, although a Km effect cannot beexcluded. Contraction and [Ca2�]i transient amplitudes inKO-S68A myocytes were not different than those observed inKO-GFP myocytes, whether measured at 1.8 or 5.0 mM[Ca2�]o (Fig. 5 and Table 6). Similarly, there were no differ-ences in SR Ca2� uptake and SR Ca2� content (Table 6)between KO-GFP and KO-S68A myocytes.
DISCUSSION
Current research on cardiac pathophysiology frequentlymakes use of genetically manipulated mouse models. Com-pared with adult rat hearts, it is technically more challenging toisolate viable myocytes from adult mouse hearts and maintaintheir viability in culture. In 2000, Zhou et al. (30) made a majorcontribution in establishing the experimental protocols forisolation, culture, and adenoviral infection of adult mousecardiac myocytes. In 2003, O’Connell et al. (15) published amodified method for the isolation and culture of adult mousecardiac myocytes. Although both studies demonstrated successin maintaining rod-shaped morphology in culture for up to 72 h(15, 30), functional studies (contraction, [Ca2�]i transients, ioncurrents, etc.) were only shown for myocytes cultured for18–24 h. Our present study has extended these observations bydemonstrating that myocyte contractility and [Ca2�]i ho-meostasis were well preserved at 48 h. In addition, myocytesurface membrane area, t-tubule organization, and proteinlevels of NCX1, SERCA2, and calsequestrin were not affectedafter 48 h of culture. One caveat is that unlike freshly isolatedmyocytes (22), the majority of adult mouse myocytes culturedfor 48 h failed to contract when [Ca2�]o was lowered to 0.6mM. Another change brought on by short-term culture is the
Table 2. Effects of culture on contractility in WT and PLMKO myocytes
Day 0 Day 1 Day 2
Means � SE n Means � SE n Means � SE n
1.8 mM �Ca2��o
WT 5.28�0.44 16 5.25�0.42 14 5.08�0.48 7PLM KO 5.84�0.58 9 5.55�0.24 23 4.85�0.51 6
5.0 mM �Ca2��o
WT 8.77�0.34 15 8.06�0.41 19 7.59�0.48 15PLM KO 10.43�0.94* 9 9.42�0.33* 19 8.90�0.50* 12
Values are means � SE of maximal contraction amplitude (as a percentageof resting cell length); n, no. of cells. [Ca2�]o, extracellular Ca2� concentra-tion. *P 0.03, WT vs. PLM KO myocytes.
Table 3. Adenoviral infection had no effect on contractilityin PLM KO myocytes
Day 0 Day 1 Day 2
Means � SE n Means � SE n Means � SE n
1.8 mM �Ca2��o
PLM KO 5.84�0.56 9 5.55�0.24 23 4.85�0.51 6KO-GFP 5.34�0.22 14 4.91�0.33 16
5.0 mM �Ca2��o
PLM KO 10.43�0.94 9 9.42�0.33 19 8.90�0.50 12KO-GFP 9.18�0.55 21 8.99�0.22 26
Values are means � SE of maximal contraction amplitude (as a percentageof resting cell length); n, no. of cells.
H1620 PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

small reduction (�18%) in the �1-subunit of Na�-K�-ATPase.[Ca2�]i transients and ion currents, however, were easily mea-surable in adult mouse cardiac myocytes after 48 h of culture.Another important finding is that adenovirus-mediated genetransfer in adult mouse cardiac myocytes resulted in the robustexpression of the exogenous gene with functional conse-quences after 48 h of culture. In our hands, the major factorsfor a successful outcome are gentle and careful isolationprocedures to ensure a maximum yield of healthy rod-shapedmyocytes, lowering of pH in culture from 7.40 to 7.00, inclu-sion of ITS supplement and BSA in the culture medium, andthe absolute requirement of BDM during myocyte isolation andculture. We have tried other inhibitors of excitation-contractioncoupling, such as blebbistatin and cytochalasin D, withoutmuch success.
Overexpression (17) and downregulation (14) or elimination(22) of PLM have profound effects on cardiac myocyte con-
tractility and [Ca2�]i transients. Since PLM regulates bothNa�-K�-ATPase (8, 10, 16, 26) and NCX1 (22, 25, 28) in theheart, it is difficult to dissect which ion transport pathway is theprimary mediator of PLM’s effect on myocyte contractility. Toapproach the issue of whether inhibition of Na�-K�-ATPaseor NCX1 represents the major mechanism by which PLMregulates cardiac contractility, we used myocytes isolated fromPLM KO hearts in which there was no endogenous PLM. Amajor finding of our present study is that contraction differ-ences between PLM KO and WT mouse myocytes werepreserved after 48 h of culture. Another important finding isthat adenovirus infection, by itself, did not affect myocytecontractility examined after 24 and 48 h of culture. These twoobservations made it possible to express PLM or its mutants inPLM KO myocytes to examine the effects on contractility,[Ca2�]i transients, Ipump, and INaCa. The reintroduction of WTPLM into cultured PLM KO myocytes had no effects on the
Fig. 5. Effects of PLM and its Ser68 mutants on contrac-tility and cytosolic Ca2� concentration ([Ca2�]i) tran-sients in cultured PLM KO myocytes. Adult cardiacmyocytes isolated from congenic PLM KO myocyteswere infected with adenovirus expressing either GFPalone (A and E), GFP � WT dog PLM (B and F), GFP �PLMS68A (C and G), or GFP � PLMS68E (D and H)and cultured for 48 h. Myocytes were incubated at 5 mMextracellular Ca2� concentration at 37°C and paced tocontract at 1 Hz. Steady-state myocyte contractions(A–D) and [Ca2�]i transients (E–H) are shown. Compos-ite data are shown in Tables 4–6.
H1621PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

protein levels of NCX1, the �1-subunit of Na�-K�-ATPase,SERCA2, and calsequestrin compared with matched PLM KOmyocytes overexpressing GFP. Contraction and [Ca2�]i tran-sient amplitudes in KO-PLM myocytes, however, were re-stored to levels observed in WT myocytes. In addition, bothIpump and INaCa in PLM KO myocytes were suppressed by WTPLM overexpression. These results indicate that PLM has animportant role in the regulation of cardiac contractility and[Ca2�]i homeostasis in resting myocytes, and the mechanismmay involve modulating Na�-K�-ATPase and/or NCX1 activ-ities. Despite no change in SERCA2 levels and SR Ca2�
uptake activity, SR Ca2� content was lower in KO-PLMcompared with KO-GFP myocytes. This observation suggeststhat altered SR Ca2� content is likely the result of inhibition ofNa�/Ca2� exchange by PLM, either directly or indirectlythrough its action on Na�-K�-ATPase. The lower SR Ca2�
content in KO-PLM myocytes largely accounts for the de-creased [Ca2�]i transient and contraction amplitudes.
To differentiate the effects of PLM on Na�-K�-ATPaseand/or NCX1, we used the PLMS68E mutant, which inhibitedINaCa but not Ipump when overexpressed in PLM KO myocytes.PLMS68E overexpression resulted in a depression of contrac-tion and [Ca2�]i transient amplitudes at 5.0 but not 1.8 mM[Ca2�]o compared with matched PLM KO myocytes express-ing GFP. This phenotype is characteristic of myocytes in whichNCX1 was downregulated (21). In addition, SR Ca2� content
Table 4. Reversion to the WT contractile phenotype in PLMKO myocytes infected with advenovirus containing PLM
Day 0 Day 1 Day 2
Means � SE n Means � SE n Means � SE n
1.8 mM �Ca2��o
WT 5.28�0.44 16 5.25�0.42 14 5.08�0.48 7KO-GFP 5.34�0.22 14 4.91�0.33 16KO-PLM 5.39�0.34 9 5.16�0.40 4
5.0 mM �Ca2��o
WT 8.77�0.34 15 8.06�0.41 19 7.59�0.48 15KO-GFP 9.18�0.55 21 8.99�0.22 26KO-PLM 7.97�0.48* 18 7.35�0.33* 13
Values are means � SE of maximal contraction amplitude (as a percentageof resting cell length); n, no. of cells. *P 0.025, KO-GFP vs. KO-PLMmyocytes.
Table 5. [Ca2�]i transients in PLM KO myocytes infectedwith advenovirus containing PLM and cultured for 48 h
KO-GFP KO-PLM
Means � SE n Means � SE n
Systolic �Ca2��i, nM1.8 mM �Ca2��o 191�6 33 175�16 135.0 mM �Ca2��o 301�93 54 224�12* 23
Diastolic �Ca2��i, nM1.8 mM �Ca2��o 98�4 85�105.0 mM �Ca2��o 95�4 86�7
�Ca2��i transient amplitude,%increase in fluorescence
1.8 mM �Ca2��o 18.2�0.6 18.6�1.05.0 mM �Ca2��o 37.1�0.9 27.1�1.0*
t1/2 of �Ca2��i decline, ms1.8 mM �Ca2��o 191�12 186�95.0 mM �Ca2��o 149�5 163�9
Values are means � SE; n, no. of cells. �Ca2�[]i, cytosolic Ca2� concen-tration; t1/2, half-time. The results of two-way ANOVA were as follows: forsystolic �Ca2��i, group (P 0.0001), �Ca2��o (P 0.0001), and group ��Ca2��o interaction effects (P 0.009); for diastolic �Ca2��i, group (P 0.13), �Ca2��o (P 0.49), and group � �Ca2��o interaction effects (P 0.43);for �Ca2��i transient amplitude, group (P 0.0001), �Ca2��o (P 0.0001),and group � �Ca2��o interaction effects (P 0.0001); and for t1/2 of �Ca2��i
decline, group (P 0.55), �Ca2��o (P 0.002), and group � �Ca2��o
interaction effects (P 0.33). *P 0.01, KO-GFP vs. KO-PLM myocytes.
Fig. 6. Effects of WT PLM and its Ser68 mutants on Na�/Ca2� exchangecurrent (INaCa) in cultured PLM KO myocytes. Adult cardiac myocytes fromcongenic PLM KO mice were infected with adenovirus expressing either GFPalone, GFP � WT PLM, GFP � PLMS68A, or GFP � PLMS68E. Aftermyocytes had been cultured for 48 h, INaCa was measured as described inMETHODS. Current density-voltage relationships of INaCa (means � SE) fromKO-GFP (�; n � 11), KO-PLM (Œ; n � 11), KO-S68A (F; n � 7), andKO-S68E (E; n � 9) myocytes are shown. For comparison, current density-voltage relationships of INaCa from WT (}; n � 20) myocytes are also shown.The reversal potential of INaCa in all myocytes examined was approximately�60 mV. Note that INaCa was lower in WT compared with KO-GFP myocytes.In addition, INaCa was suppressed by WT PLM and the PLMS68E mutant butnot by the PLMS68A mutant. Error bars are not shown if they fall within theboundaries of the symbol.
Fig. 7. Effects of WT PLM and its Ser68 mutants on Na�-K�-ATPase current(Ipump) in cultured PLM KO myocytes. Adult myocytes from congenic PLMKO hearts were infected with adenovirus expressing either GFP alone, GFP �WT PLM (PLM), GFP � PLMS68A (S68A), or GFP � PLMS68E (S68E).After 48 h of culture, Ipump was measured at 18 mM extracellular K�
concentration at 0 mV at 30°C with pipette Na� concentrations ([Na�]pip) ofeither 10 mM (open bars) or 80 mM (solid bars). For comparison, Ipump of WTmyocytes is shown. At 10 mM [Na�]pip, there were 8 WT, 9 KO-GFP, 10KO-PLM, 6 KO-S68A, and 7 KO-S68E myocytes. At 80 mM [Na�]pip, therewere 5 WT, 7 KO-GFP, 6 KO-PLM, 8 KO-S68A, and 8 KO-S68E myocytes.Two-way ANOVA indicated significant differences between WT and KO-GFP(P � 0.05), WT and KO-PLM (P 0.0004), WT and KO-S68A (P 0.001),KO-GFP and KO-PLM (P 0.002), KO-GFP and KO-S68A (P 0.003),KO-PLM and KO-S68E (P 0.003), and KO-S68A and KO-S68E (P 0.004) myocytes, but not between KO-GFP and KO-S68E (P 0.8), KO-PLMand KO-S68A (P 0.8), and WT and KO-S68E (P 0.3) myocytes.
H1622 PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

was lower in KO-S68E myocytes despite no changes in SRCa2� uptake. These observations suggest that direct inhibitionof NCX1 by phosphorylated PLM resulted in decreased SRCa2� content, leading to reduced [Ca2�]i transient and con-traction amplitudes. Conversely, when we overexpressed thePLMS68A mutant, which inhibited Ipump but not INaCa in PLMKO myocytes, there were no differences in SR Ca2� uptake,SR Ca2� content, contraction, and [Ca2�]i transient amplitudescompared with matched PLM KO myocytes infected withAdv-GFP. Our results clearly indicate that in isolated myocytesstudied under our conditions, the effects of PLM on cardiaccontractility were mediated largely by its inhibitory effects onNCX1 rather than Na�-K�-ATPase.
Some insight into the mechanism by which PLM regulatescardiac contractility can also be gleaned from the distributionof the isoforms of Na�-K�-ATPase, NCX1, and PLM in adultheart muscle. Whereas PLM and NCX1 are distributed in bothsarcolemma and t-tubules (28), the �1-isoform (ouabain insen-sitive) and �2-isoform (ouabain sensitive) of Na�-K�-ATPaseare preferentially distributed in the sarcolemma and t-tubules,respectively (3, 20). In rodent hearts, PLM associates with andmodulates the activity of the �1-isoform but not �2-isoform ofNa�-K�-ATPase (16), although both �1- and �2-isoforms ofNa�-K�-ATPase coimmunoprecipitate with PLM in bovine(6) and rabbit (4) cardiac tissues. In adult rat cardiac myocytes,inhibition of the �2-isoform of Na�-K�-ATPase results inincreased INaCa and enhanced cardiac myocyte contractility(20), indicating intimate cross-talk between the �2-isoform ofNa�-K�-ATPase and NCX1 in the t-tubules. Similar intimatecross-talk between NCX1 and the �2-isoform of Na�-K�-ATPase has also recently been demonstrated for mouse aorticsmooth muscle cells (13). The observations that in rodentcardiac myocytes PLM regulates the �1-isoform but not �2-isoform of Na�-K�-ATPase, and that cross-talk occurs pre-dominantly between the �2-isoform of Na�-K�-ATPase andNCX1 in the t-tubules, support our hypothesis that PLMregulates cardiac contractility by mechanisms other than inhi-bition of Na�-K�-ATPase. Our results with the PLMS68Amutant expressed in PLM KO myocytes, showing that itinhibited Ipump but had no effects on contraction and [Ca2�]i
transient amplitudes, can be explained by the concept that inrodent cardiac myocytes, PLM inhibits the �1-isoform but not
�2-isoform of Na�-K�-ATPase (16). Definitive proof of thishypothesis, however, will require further investigation withmouse cardiac myocytes.
In PLM KO myocytes cultured for 48 h and expressing GFP,reducing [Na�]pip from saturating 80 to 10 mM resulted in an�66% decrease in Ipump, suggesting tht the Km for Na� in PLMKO myocytes was �10 mM. This interpretation is consistentwith the findings that the Km for Na� was 14.6 � 1.9 mM infreshly isolated PLM KO myocytes (8) and �10 mM in freshlyisolated adult rat myocytes (26). When the PLMS68A mutantwas expressed in PLM-KO myocytes to simulate maximalinhibition of Na�-K�-ATPase by unphosphorylated WT PLM,the major effect was a decrease in the maximal amplitude ofIpump. This Vmax effect of PLM on Ipump is in agreement withthe observations in adult rat myocytes overexpressing PLM(26) and in freshly isolated guinea pig myocytes stimulatedwith forskolin (16). A comparison of Na�-K�-ATPase activ-ities measured under saturating Na� conditions in heart ho-mogenates obtained from WT and PLM KO mice also indi-cated a Vmax effect of PLM (2). In contrast, when pump activitywas measured as the Na�-K�-ATPase-mediated [Na�]i de-cline in Na�-loaded mouse myocytes, phosphorylating Ser68 ofPLM resulted in a decrease in the Km for Na� with little effecton the Vmax of Na�-K�-ATPase (8). Only when both Ser68 andSer63 of PLM were phosphorylated was an increase in Vmax
evident in mouse myocytes (11). These results, when viewedtogether, suggest that PLM affects both the Km for Na� andVmax of Na�-K�-ATPase in rodent cardiac myocytes.
There are limitations to the present study. The first limitationis that our measurements were exclusively performed in singlecultured myocytes. Therefore, caution must be exercised whenextrapolating our findings to the intact heart. A good exampleof this is that despite evidence of hypertrophy in PLM KOhearts (2, 12), neither we (22) nor Despa et al. (8) could detecthypertrophy in myocytes isolated from PLM KO hearts. In-deed, whether contractile performance in PLM KO hearts wassuperior (12) or depressed (2) in the intact animal remainscontroversial at the present time. The second limitation is thatcontractility of cultured adult mouse myocytes, although pre-served, was not quite “normal,” as evidenced by the highincidence of contractile failure at 0.6 mM [Ca2�]o, and con-traction amplitudes did decline slightly with increasing days in
Table 6. Effects of PLMS68E and PLMS68A mutants on PLM KO myocytes
KO-GFP KO-PLM KO-S68E KO-S68A
Means � SE n Means � SE n Means � SE n Means � SE n
Maximal contraction amplitude, %resting cell length1.8 mM �Ca2��o 4.91�0.33 16 5.16�0.40 4 5.02�0.27 15 4.92�0.28 135.0 mM �Ca2��o 8.99�0.22 26 7.35�0.33* 13 7.19�0.21* 28 8.73�0.41 19
�Ca2��i transient amplitude, %increase in fluorescence1.8 mM �Ca2��o 18.2�0.6 33 18.6�1.0 13 17.9�1.2 16 17.8�1.3 175.0 mM �Ca2��o 37.1�0.9 54 27.1�1.0* 23 26.4�1.3* 22 36.2�1.0 12
t1/2 of �Ca2��i decline, ms1.8 mM �Ca2��o 191�12 186�9 188�10 195�175.0 mM �Ca2��o 149�5 163�9 174�6 159�14
SR Ca2� content, fmol/fF5.0 mM �Ca2��o 12.1�0.2 4 5.4�1.6* 5 6.0�0.9* 8 11.3�0.9 6
Values are means � SE; n, no. of cells. Contractility, �Ca2��i transients, t1/2 of �Ca2��i decline, and sarcoplasmic reticulum (SR) Ca2� contents were measuredafter adenovirus infection and 48 h of culture. For comparison, SR Ca2� content measured at 5 mM �Ca2��o was 14.3 � 0.6 fmol/fF (n � 9) in freshly isolatedPLM KO myocytes (22). KO-S68E and KO-S68A, PLM KO myocytes infected with adenovirus expressing PLMS68E and PLMS68A mutants, respectively.*P 0.025 compared with KO-GFP myocytes.
H1623PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

culture. In addition, protein levels of the �1-subunit of Na�-K�-ATPase were slightly depressed compared with freshlyisolated myocytes. The third limitation is that the reintroduc-tion of WT PLM into PLM KO myocytes by adenovirus-mediated gene transfer resulted in higher than physiologicallevels of PLM. The supraphysiological levels of both phos-phorylated and unphosphorylated PLM in infected myocytes,compared with PLM levels naturally present in WT controls,resulted in a severe suppression of INaCa and Ipump in KO-PLMmyocytes compared with WT myocytes. Finally, the interpre-tation of our results relies on the assumption that PLMS68Aand PLMS68E mutants faithfully mimic the unphosphorylatedand phosphorylated forms of PLM, respectively. This appearsreasonable since in HEK-293 cells coexpressing NCX1 andWT PLM or its mutants, PLMS68E but not PLMS68A mim-icked the inhibitory effects of forskolin on INaCa (25).
In summary, adult mouse myocytes were successfully main-tained in culture for 48 h with near-normal contractility; nochanges in membrane surface area, t-tubular organization,SERCA2, NCX1, and calsequestrin levels; and a slight (�18%)decrease in �1-subunit of Na�-K�-ATPase levels. Infectionwith adenovirus did not affect myocyte contractility. Contrac-tility differences between WT and PLM KO myocytes werepreserved after 48 h in culture. Overexpression of WT PLM inPLM KO myocytes had no effect on NCX1 and SERCA2levels, and SR Ca2� uptake, but INaCa, Ipump, and SR Ca2�
content were decreased, and resulted in contraction amplitudesindistinguishable from those of cultured WT myocytes. Over-expression of the phosphomimetic mutant of PLM (S68E) inPLM KO myocytes inhibited INaCa but not Ipump or SR Ca2�
uptake and resulted in decreased SR Ca2� content, contraction,and [Ca2�]i transient amplitudes compared with PLM KOmyocytes. In contrast, overexpression of the PLMS68A mutant(which cannot be phosphorylated) in PLM KO myocytes in-hibited Ipump but not INaCa or SR Ca2� uptake and had noeffects on SR Ca2� content, contraction, and [Ca2�]i transientamplitudes compared with PLM KO myocytes. We concludethat in single cardiac myocytes, the regulation of contractilityand [Ca2�]i transients by PLM was mediated by its effects onNCX1 rather than Na�-K�-ATPase.
GRANTS
This work was supported in part by National Institutes of Health GrantsRO1-HL-58672 and RO1-HL-74854 (to J. Y. Cheung), RO1-DK-46678 (toJ. Y. Cheung, coinvestigator), RO1-HL-69074 (to A. L. Tucker), and PO1-HL-91799 (to A. M. Feldman) and by an American Heart Association ScientistDevelopment Grant (to T. O. Chan).
REFERENCES
1. Ahlers BA, Zhang XQ, Moorman JR, Rothblum LI, Carl LL, Song J,Wang J, Geddis LM, Tucker AL, Mounsey JP, Cheung JY. Identifi-cation of an endogenous inhibitor of the cardiac Na�/Ca2� exchanger,phospholemman. J Biol Chem 280: 19875–19882, 2005.
2. Bell JR, Kennington E, Fuller W, Dighe K, Donoghue P, Clark JE, JiaLG, Tucker AL, Moorman JR, Marber MS, Eaton P, Dunn MJ,Shattock MJ. Characterization of the phospholemman knockout mouseheart: depressed left ventricular function with increased Na-K-ATPaseactivity. Am J Physiol Heart Circ Physiol 294: H613–H621, 2008.
3. Berry RG, Despa S, Fuller W, Bers DM, Shattock MJ. Differentialdistribution and regulation of mouse cardiac Na�/K�-ATPase alpha1 andalpha2 subunits in t-tubule and surface sarcolemmal membranes. Cardio-vasc Res 73: 92–100, 2007.
4. Bossuyt J, Ai X, Moorman JR, Pogwizd SM, Bers DM. Expression andphosphorylation of the Na-pump regulatory subunit phospholemman inheart failure. Circ Res 97: 558–565, 2005.
5. Cheung JY. Regulation of cardiac contractility: high time for FXYD.Am J Physiol Heart Circ Physiol 294: H584–H585, 2008.
6. Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Phospholem-man (FXYD1) associates with Na,K-ATPase and regulates its transportproperties. Proc Natl Acad Sci USA 99: 11476–11481, 2002.
7. Davis CE, Patel MK, Miller JR, John JE 3rd, Jones LR, Tucker AL,Mounsey JP, Moorman JR. Effects of phospholemman expression onswelling-activated ion currents and volume regulation in embryonic kid-ney cells. Neurochem Res 29: 177–187, 2004.
8. Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, TuckerAL, Bers DM. Phospholemman-phosphorylation mediates the beta-adren-ergic effects on Na/K pump function in cardiac myocytes. Circ Res 97:252–259, 2005.
9. Despa S, Tucker AL, Bers DM. PLM-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during �-adrenergic stimulation inmouse ventricular myocytes. Circulation 117: 1849–1855, 2008.
10. Fuller W, Eaton P, Bell JR, Shattock MJ. Ischemia-induced phosphor-ylation of phospholemman directly activates rat cardiac Na-K-ATPase.FASEB J 18: 197–199, 2004.
11. Han F, Bossuyt J, Despa S, Tucker AL, Bers DM. Phospholemmanphosphorylation mediates the protein kinase C-dependent effects onNa�/K� pump function in cardiac myocytes. Circ Res 99: 1376–1383,2006.
12. Jia LG, Donnet C, Bogaev RC, Blatt RJ, McKinney CE, Day KH, BerrSS, Jones LR, Moorman JR, Sweadner KJ, Tucker AL. Hypertrophy,increased ejection fraction, and reduced Na-K-ATPase activity in phos-pholemman-deficient mice. Am J Physiol Heart Circ Physiol 288: H1982–H1988, 2005.
13. Lynch RM, Weber CS, Nullmeyer KD, Moore ED, Paul RJ. Clearanceof store-released Ca2� by the Na�/Ca2� exchanger is diminished in aorticsmooth muscle from Na�-K�-ATPase �2-isoform gene-ablated mice.Am J Physiol Heart Circ Physiol 294: H1407–H1416, 2008.
14. Mirza MA, Zhang XQ, Ahlers BA, Qureshi A, Carl LL, Song J,Tucker AL, Mounsey JP, Moorman JR, Rothblum LI, Zhang TS,Cheung JY. Effects of phospholemman downregulation on contractilityand [Ca2�]i transients in adult rat cardiac myocytes. Am J Physiol HeartCirc Physiol 286: H1322–H1330, 2004.
15. O’Connell TD, Ni YG, Lin KM, Han H, Yan Z. Isolation and culture ofadult mouse cardiac mouse myocytes for signaling studies. Alliance CellSignal Res Rep 1: 1–9, 2003.
16. Silverman BZ, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY,James AF, Shattock MJ. Serine 68 phosphorylation of phospholemman:acute isoform-specific activation of cardiac Na/K ATPase. Cardiovasc Res65: 93–103, 2005.
17. Song J, Zhang XQ, Carl LL, Qureshi A, Rothblum LI, Cheung JY.Overexpression of phospholemman alter contractility and [Ca2�]i tran-sients in adult rat myocytes. Am J Physiol Heart Circ Physiol 283:H576–H583, 2002.
18. Song J, Zhang XQ, Ahlers BA, Carl LL, Wang J, Rothblum LI, StahlRC, Mounsey JP, Tucker AL, Moorman JR, Cheung JY. Serine 68 ofphospholemman is critical in modulation of contractility, [Ca2�]i tran-sients, and Na�/Ca2� exchange in adult rat cardiac myocytes. Am JPhysiol Heart Circ Physiol 288: H2342–H2354, 2005.
19. Sweadner KJ, Rael E. The FXYD gene family of small ion transportregulators or channels: cDNA sequence, protein signature sequence, andexpression. Genomics 68: 41–56, 2000.
20. Swift FB, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, LouchWE, Sjaastad I, Sejersted OM. Altered Na�/Ca2�-exchanger due todownregulation of Na�-K�-ATPase �2-isoform in heart failure. Cardio-vasc Res 78: 71–78, 2008.
21. Tadros GM, Zhang XQ, Song J, Carl LL, Rothblum LI, Tian Q, DunnJ, Lytton J, Cheung JY. Effects of Na�/Ca2� exchanger downregulationon contractility and [Ca2�]i transients in adult rat myocytes. Am J PhysiolHeart Circ Physiol 283: H1616–H1626, 2002.
22. Tucker AL, Song J, Zhang XQ, Wang J, Ahlers BA, Carl LL,Mounsey JP, Moorman JR, Rothblum LI, Cheung JY. Altered con-tractility and [Ca2�]i homeostasis in phospholemman-deficient murinemyocytes: role of Na�/Ca2� exchange. Am J Physiol Heart Circ Physiol291: H2199–H2209, 2006.
23. Waalas SI, Czernik AJ, Olstad OK, Sletten K, Walaas O. Proteinkinase C and cyclic AMP-dependent protein kinase phosphorylate phos-pholemman, an insulin and adrenaline-regulated membrane phosphopro-tein, at specific sites in the carboxy terminal domain. Biochem J 304:635–640, 1994.
H1624 PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org

24. Wang J, Zhang XQ, Ahlers BA, Carl LL, Song J, Rothblum LI, StahlRC, Carey DJ, Cheung JY. Cytoplasmic tail of phospholemman interactswith the intracellular loop of the cardiac Na�/Ca2� exchanger. J BiolChem 281: 32004–32014, 2006.
25. Zhang XQ, Ahlers BA, Tucker AL, Song J, Wang J, Moorman JR,Mounsey JP, Carl LL, Rothblum LI, Cheung JY. Phospholemmaninhibition of the cardiac Na�/Ca2� exchanger. Role of phosphorylation.J Biol Chem 281: 7784–7792, 2006.
26. Zhang XQ, Moorman JR, Ahlers BA, Carl LL, Lake DE, Song J,Mounsey JP, Tucker AL, Chan YM, Rothblum LI, Stahl RC, CareyDJ, Cheung JY. Phospholemman overexpression inhibits Na�-K�-ATPase in adult rat cardiac myocytes: relevance to decreased Na�
pump activity in postinfarction myocytes. J Appl Physiol 100: 212–220, 2006.
27. Zhang XQ, Ng YC, Moore RL, Musch TI, Cheung JY. In situ SRfunction in postinfarction myocytes. J Appl Physiol 87: 2143–2150, 1999.
28. Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ,Rothblum LI, Cheung JY. Phospholemman modulates Na�/Ca2� ex-change in adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol284: H225–H233, 2003.
29. Zhang XQ, Song J, Rothblum LI, Lun M, Wang X, Ding F, Dunn J,Lytton J, McDermott PJ, Cheung JY. Overexpression of Na�/Ca2�
exchanger alters contractility and SR Ca2� content in adult rat myocytes.Am J Physiol Heart Circ Physiol 281: H2079–H2088, 2001.
30. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B,Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviralinfection of adult mouse cardiac myocytes: methods for cellular geneticphysiology. Am J Physiol Heart Circ Physiol 279: H429–H436, 2000.
H1625PHOSPHOLEMMAN REGULATES CARDIAC CONTRACTILITY
AJP-Heart Circ Physiol • VOL 295 • OCTOBER 2008 • www.ajpheart.org
Copyright © 2022 FDOKUMEN




















