
Overexpression of junctin causes adaptive changes in cardiac myocyte Ca 2+ signaling
12
Cell Calcium 39 (2006) 131–142 Overexpression of junctin causes adaptive changes in cardiac myocyte Ca 2+ signaling Uwe Kirchhefer a,∗ , Gabriela Hanske a , Larry R. Jones b , Isabel Justus a , Lars Kaestner c , Peter Lipp c , Wilhelm Schmitz a , Joachim Neumann d a Institut f ¨ ur Pharmakologie und Toxikologie, Westf¨ alische Wilhelms-Universit¨ at, Domagkstr. 12, 48149 M¨ unster, Germany b Krannert Institute of Cardiology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN 46202, USA c Institut f ¨ ur Molekulare Zellbiologie, Geb¨ aude 61, Universit¨ at des Saarlandes, 66421 Homburg/Saar, Germany d Institut f ¨ ur Pharmakologie und Toxikologie, Martin-Luther-Universit¨ at Halle-Wittenberg, Magdeburger Str. 4, 06112 Halle, Germany Received 7 July 2005; received in revised form 2 October 2005; accepted 5 October 2005 Available online 9 November 2005 Abstract In cardiac muscle, junctin forms a quaternary protein complex with the ryanodine receptor (RyR), calsequestrin, and triadin 1 at the luminal face of the junctional sarcoplasmic reticulum (jSR). By binding directly the RyR and calsequestrin, junctin may mediate the Ca 2+ -dependent regulatory interactions between both proteins. To gain more insight into the underlying mechanisms of impaired contractile relaxation in transgenic mice with cardiac-specific overexpression of junctin (TG), we studied cellular Ca 2+ handling in these mice. We found that the SR Ca 2+ load was reduced by 22% in cardiomyocytes from TG mice. Consistent with this, the frequency of Ca 2+ sparks was diminished by 32%. The decay of spontaneous Ca 2+ sparks was prolonged by 117% in TG. This finding was associated with a lower Na + -Ca 2+ exchanger (NCX) protein expression (by 67%) and a higher basal RyR phosphorylation at Ser 2809 (by 64%) in TG. The shortening- and [Ca] i -frequency relationships (0.5–4 Hz) were flat in TG compared to wild-type (WT) which exhibited a positive staircase for both parameters. Furthermore, increasing stimulation frequencies hastened the time of relaxation and the decay of [Ca] i by a higher percentage in TG. We conclude that the impaired relaxation in TG may result from a reduced NCX expression and/or a higher SR Ca 2+ leak. The altered shortening-frequency relationship in TG seems to be a consequence of an impaired excitation–contraction coupling with depressed SR Ca 2+ release at higher rates of stimulation. Our data suggest that the more prominent frequency-dependent hastening of relaxation in TG results from a stimulation of SR Ca 2+ transport reflected by corresponding changes of [Ca] i . © 2005 Elsevier Ltd. All rights reserved. Keywords: Ca 2+ release units; Junctional sarcoplasmic reticulum; Ca 2+ handling; Excitation–contraction coupling; Ca 2+ sparks; Cardiac function; Transgenic mouse model 1. Introduction The Ca 2+ release units of the jSR are formed by stable uniform complexes of four proteins consisting of the Ca 2+ release channel (i.e., ryanodine receptor, RyR), calsequestrin (CSQ), triadin 1 (TRD), and junctin (JCN) [1,2]. The RyR contains a large cytoplasmic domain that reaches into the space between jSR and the T-tubules and a transmembrane domain that forms the conduction pore of the Ca 2+ release ∗ Corresponding author. Tel.: +49 251 8355510; fax: +49 251 8355501. E-mail address: [email protected] (U. Kirchhefer). channel [3,4]. CSQ is a Ca 2+ storage protein in the lumen of the jSR [5]. It forms an electron-dense matrix in the jSR lumen which was ascribed to the ability of CSQ to sequester Ca 2+ near the Ca 2+ release sites [6–8]. CSQ may act as a Ca 2+ sensor that inhibits the Ca 2+ release channel at low luminal Ca 2+ [9] and is anchored to the RyR either directly or via two auxiliary proteins, TRD and JCN. TRD and JCN are structurally similar transmembrane proteins constituted of short N-terminal cytoplasmic domains and highly charged C-terminal domains projecting into the jSR lumen [10,11]. TRD and JCN may be involved in SR luminal Ca 2+ sensing by mediating the inhibitory interactions between CSQ and 0143-4160/$ – see front matter © 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.ceca.2005.10.004
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Overexpression of junctin causes adaptive changes in cardiac myocyte Ca 2+ signaling

Cell Calcium 39 (2006) 131–142
Overexpression of junctin causes adaptive changes incardiac myocyte Ca2+ signaling
Uwe Kirchhefera,∗, Gabriela Hanskea, Larry R. Jonesb, Isabel Justusa, Lars Kaestnerc,Peter Lippc, Wilhelm Schmitza, Joachim Neumannd
a Institut fur Pharmakologie und Toxikologie, Westfalische Wilhelms-Universitat, Domagkstr. 12, 48149 Munster, Germanyb Krannert Institute of Cardiology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN 46202, USA
c Institut fur Molekulare Zellbiologie, Gebaude 61, Universitat des Saarlandes, 66421 Homburg/Saar, Germanyd Institut fur Pharmakologie und Toxikologie, Martin-Luther-Universitat Halle-Wittenberg, Magdeburger Str. 4, 06112 Halle, Germany
Received 7 July 2005; received in revised form 2 October 2005; accepted 5 October 2005Available online 9 November 2005
Abstract
he luminaltaxation intheyr
thermore,thatncyesation of SR
nic
nSRsterawtly
CNtutedged
and
In cardiac muscle, junctin forms a quaternary protein complex with the ryanodine receptor (RyR), calsequestrin, and triadin 1 at tface of the junctional sarcoplasmic reticulum (jSR). By binding directly the RyR and calsequestrin, junctin may mediate the Ca2+-dependenregulatory interactions between both proteins. To gain more insight into the underlying mechanisms of impaired contractile reltransgenic mice with cardiac-specific overexpression of junctin (TG), we studied cellular Ca2+ handling in these mice. We found thatSR Ca2+ load was reduced by 22% in cardiomyocytes from TG mice. Consistent with this, the frequency of Ca2+ sparks was diminished b32%. The decay of spontaneous Ca2+ sparks was prolonged by 117% in TG. This finding was associated with a lower Na+-Ca2+ exchange(NCX) protein expression (by 67%) and a higher basal RyR phosphorylation at Ser2809(by 64%) in TG. The shortening- and�[Ca]i -frequencyrelationships (0.5–4 Hz) were flat in TG compared to wild-type (WT) which exhibited a positive staircase for both parameters. Furincreasing stimulation frequencies hastened the time of relaxation and the decay of [Ca]i by a higher percentage in TG. We concludethe impaired relaxation in TG may result from a reduced NCX expression and/or a higher SR Ca2+ leak. The altered shortening-frequerelationship in TG seems to be a consequence of an impaired excitation–contraction coupling with depressed SR Ca2+ release at higher ratof stimulation. Our data suggest that the more prominent frequency-dependent hastening of relaxation in TG results from a stimulCa2+ transport reflected by corresponding changes of [Ca]i .© 2005 Elsevier Ltd. All rights reserved.
Keywords: Ca2+ release units; Junctional sarcoplasmic reticulum; Ca2+ handling; Excitation–contraction coupling; Ca2+ sparks; Cardiac function; Transgemouse model
1. Introduction
The Ca2+ release units of the jSR are formed by stableuniform complexes of four proteins consisting of the Ca2+
release channel (i.e., ryanodine receptor, RyR), calsequestrin(CSQ), triadin 1 (TRD), and junctin (JCN)[1,2]. The RyRcontains a large cytoplasmic domain that reaches into thespace between jSR and the T-tubules and a transmembranedomain that forms the conduction pore of the Ca2+ release
∗ Corresponding author. Tel.: +49 251 8355510; fax: +49 251 8355501.E-mail address: [email protected] (U. Kirchhefer).
channel[3,4]. CSQ is a Ca2+ storage protein in the lumeof the jSR[5]. It forms an electron-dense matrix in the jlumen which was ascribed to the ability of CSQ to sequeCa2+ near the Ca2+ release sites[6–8]. CSQ may act asCa2+ sensor that inhibits the Ca2+ release channel at loluminal Ca2+ [9] and is anchored to the RyR either direcor via two auxiliary proteins, TRD and JCN. TRD and Jare structurally similar transmembrane proteins constiof short N-terminal cytoplasmic domains and highly charC-terminal domains projecting into the jSR lumen[10,11].TRD and JCN may be involved in SR luminal Ca2+ sensingby mediating the inhibitory interactions between CSQ
0143-4160/$ – see front matter © 2005 Elsevier Ltd. All rights reserved.doi:10.1016/j.ceca.2005.10.004

132 U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142
the RyR[2]. In addition, a direct modulatory role of TRDhas been proposed by inhibiting the activity of the skeletalmuscle RyR in reconstitutional systems[12].
JCN like TRD has a jSR luminal domain which accountsfor the binding of CSQ and to the RyR. The interaction ofJCN with CSQ is diminished at high Ca2+ concentrations[2]. Furthermore, JCN can also bind to TRD independentlyof different Ca2+ concentrations and to itself[2]. Junctin runsas a 26-kDa protein in SDS-PAGE and is highly enriched inthe jSR in both cardiac and skeletal muscle[11]. Ultrastruc-tural studies have shown that JCN is mainly localized at theperipheral (i.e., junctional) SR[11,13,14]. It has been sug-gested that JCN (and TRD) may play more direct roles in Ca2+
release beyond simply anchoring CSQ to the RyR. In line withthis hypothesis, the application of JCN to the luminal side ofpurified cardiac RyRs increased the open probability of thechannels[9]. To shed light on the functional role of JCN inthe regulation of SR Ca2+ release in the heart, we developeda transgenic mouse model (TG) with cardiac-specific over-expression of JCN[15]. The 10-fold overexpression of JCNwas associated with a tighter packing of CSQ in proximityof the jSR membrane[14]. Moreover, TG mice exhibited amild hypertrophy and a reduced expression of TRD and theRyR. The relaxation was prolonged in papillary muscles andisolated cardiomyocytes of TG mice. Consistently, the decayof Ca2+ transients was prolonged in TG[16].
s bys ntingf CNs sa teine ion oft neda datas of ani byl mala
2
2
inej ribed[ thso pro-t theU ity,U area
2
leso col
as described[17]. Ca2+ATPase in SR vesicles was deter-mined by measuring colorimetrically the release of inorganicphosphate (Pi) from ATP at different CaCl2 concentrations[18,19].
2.3. Measurement of Ca2+ transients, cell shortening,and SR Ca2+ load
Cardiomyocytes were enzymatically isolated from TGand WT hearts by a standard procedure as reported before[17]. Intracellular Ca2+ transients were determined at 0.5 Hzand 2 mM extracellular Ca2+ with Indo-1/AM (Sigma, St.Louis, MO, USA). The fluorescence indicator was excitedat 365 nm. The emitted fluorescence was detected at 405and 495 nm. The cytosolic free Ca2+ concentration was esti-mated by calculating the ratio of Indo-1 signals (405/495 nm).The maximum Ca2+ concentration, the minimum Ca2+ con-centration, and their difference were set as the systolicratio, diastolic ratio, and�[Ca]i , respectively. Where indi-cated, stimulation frequency was increased stepwise from0.5 to 4 Hz. At each frequency steady state was reachedafter 5 min. The shortening of cardiomyocytes was recordedsimultaneously using a video edge detection system[16].Moreover, the SR Ca2+ content was estimated by mea-surement of Ca2+ transients in the presence of 10 mM caf-feine and 5 mM Ni2+, blocking effectively theI current[
2
cedot heN inf offl on-o ec edw d aN is-i as 27 sl Hz.S er Vis-i ired2 rdingf ana-l SA)a go,U -ld ences imalr othp
Here, we have extended these initial observationtudying in depth the physiological mechanisms accouor the impaired relaxation in TG. Overexpression of Jlowed the decay of spontaneous Ca2+ sparks. This effect wassociated with a dramatically down-regulated NCX proxpression in TG and an enhanced basal phosphorylathe RyR at Ser2809. Moreover, relaxation was more hastet increasing stimulation frequencies in TG. Thus, ouruggest that the prolonged relaxation in TG is the resultmpaired global cellular Ca2+ homeostasis which is causedocal structural and functional alterations of the sarcolemnd jSR Ca2+ cycling.
. Methods
.1. Experimental animals
TG mice with cardiac-specific overexpression of canunctin were generated and identified as previously desc15]. All experiments were performed on mice of 4–5 monf age. Mice were handled and maintained according to
ocols approved by the animal welfare committees ofniversity of Munster, Germany, and Indiana UniversSA, which also conform to the NIH Guidelines for the Cnd Use of Laboratory Animals.
.2. Ca2+ATPase assay
SR vesicles were prepared from individual ventricf TG and WT mice according to a modified proto
Na/Ca20].
.4. Detection of Ca2+ sparks by confocal microscopy
Cardiomyocytes of either TG or WT animals were plan poly-l-lysin coated coverslips and loaded with 2�M of
he indicator fluo-4/AM (Molecular Probes, Leiden, Tetherlands) for 30 min followed by a period of 10 m
or de-esterification of the internalised dye. Excitationuo-4 was performed with the 488 nm line of an Argr Argon-Krypton laser. The Ca2+ spark frequency and thharacteristics of individual Ca2+ sparks were measurith laser-scanning confocal microscopes. We useipkow-disk-based confocal microscope (QLC-100, V
Tech International, Sunderland, UK) to estimate the C2+
park frequency in quiescent cardiomyocytes duringong recording periods at a constant frame rate of 19patiotemporal fine details of individual Ca2+ sparks wer
ecorded on an ultra-fast confocal microscope (VTeye;Tech International, Sunderland, UK). For this we acquD-data from resting cardiomyocytes at a constant reco
requency of 120 or 360 Hz. The resulting images wereyzed using ImageJ (Wayne Rasband, NIH, Bethesda, Und IgorPro 4.0 software (WaveMetrics, Lake OsweSA). The amplitude of individual Ca2+ sparks was ana
yzed by the following procedure. A box of 5× 5 pixels wasrawn around each maximal increased fluo-4 fluorescignal (F), which had to be at least 25% above the minesting level (F0). The background was subtracted from barameters and ratio F/F0 was calculated.
U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142 133
2.5. Heart perfusion
To determine the phosphorylation of PLN, hearts fromTG and WT mice were perfused in the anterograde modeusing a working-heart set-up[17]. After a 30-min stabi-lization period, either buffer alone or buffer supplementedwith isoproterenol (Iso, 0.1�M) was applied to the perfusionsystem for further 10 min. For measurement of basal RyRphosphorylation, hearts were perfused using the proceduredescribed above with 1 mM NaF in the perfusate[21].
2.6. Immunoblotting
Aliquots (50�g) of prepared membrane vesicles fromventricles of TG and WT mice were solubilized in 5% SDS-stop solution[22], subjected to 8% SDS-PAGE[23], andtransferred to nitrocellulose. The nitrocellulose was incu-bated with the mouse monoclonal antibody C2C12 raisedto purified canine cardiac NCX (ABR, Golden, CO, USA)[20] and with a rabbit antibody to canine cardiac CSQ[24].Antibody-reacting bands were detected by an anti-mouseantibody conjugated with alkaline phosphatase (Sigma) andby125I-labeled protein A (Perkin-Elmer, Boston, MA, USA).Signals were quantified using a PhosphorImager (Bio Rad,Hercules, CA, USA). In addition, perfused hearts fromTG and WT mice were homogenized in 10 mM NaHCO3,5 ◦ .Ha 0w MT d at ratedo l-l anti-b hnol-o illa,L no-cp tion-sw tifiedu
2
-e ed byA -s
3
3C
S ]
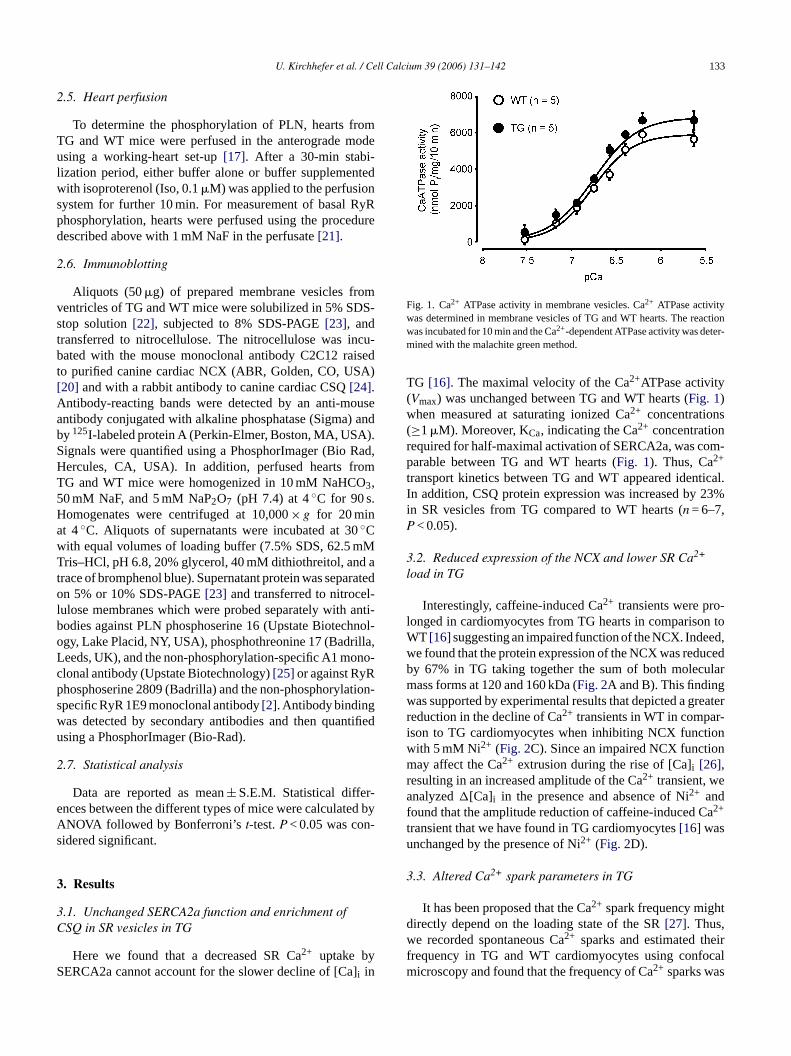
Fig. 1. Ca2+ ATPase activity in membrane vesicles. Ca2+ ATPase activitywas determined in membrane vesicles of TG and WT hearts. The reactionwas incubated for 10 min and the Ca2+-dependent ATPase activity was deter-mined with the malachite green method.
TG [16]. The maximal velocity of the Ca2+ATPase activity(Vmax) was unchanged between TG and WT hearts (Fig. 1)when measured at saturating ionized Ca2+ concentrations(≥1�M). Moreover, KCa, indicating the Ca2+ concentrationrequired for half-maximal activation of SERCA2a, was com-parable between TG and WT hearts (Fig. 1). Thus, Ca2+
transport kinetics between TG and WT appeared identical.In addition, CSQ protein expression was increased by 23%in SR vesicles from TG compared to WT hearts (n = 6–7,P < 0.05).
3.2. Reduced expression of the NCX and lower SR Ca2+
load in TG
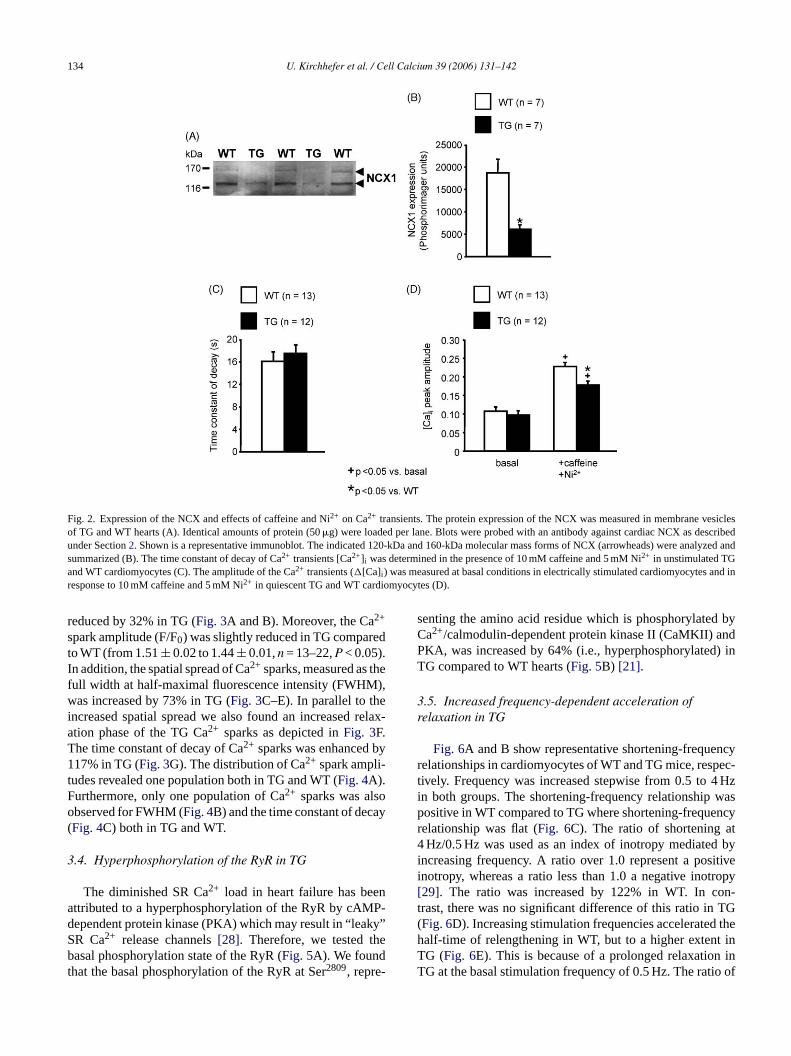
Interestingly, caffeine-induced Ca2+ transients were pro-longed in cardiomyocytes from TG hearts in comparison toWT [16] suggesting an impaired function of the NCX. Indeed,we found that the protein expression of the NCX was reducedby 67% in TG taking together the sum of both molecularmass forms at 120 and 160 kDa (Fig. 2A and B). This findingwas supported by experimental results that depicted a greaterreduction in the decline of Ca2+ transients in WT in compar-ison to TG cardiomyocytes when inhibiting NCX functionwith 5 mM Ni2+ (Fig. 2C). Since an impaired NCX functionmay affect the Ca2+ extrusion during the rise of [Ca]i [26],r 2+
af atu
3
tdw eirf calm s
0 mM NaF, and 5 mM NaP2O7 (pH 7.4) at 4 C for 90 somogenates were centrifuged at 10,000× g for 20 mint 4◦C. Aliquots of supernatants were incubated at 3◦Cith equal volumes of loading buffer (7.5% SDS, 62.5 mris–HCl, pH 6.8, 20% glycerol, 40 mM dithiothreitol, an
race of bromphenol blue). Supernatant protein was sepan 5% or 10% SDS-PAGE[23] and transferred to nitroce
ulose membranes which were probed separately withodies against PLN phosphoserine 16 (Upstate Biotecgy, Lake Placid, NY, USA), phosphothreonine 17 (Badreeds, UK), and the non-phosphorylation-specific A1 molonal antibody (Upstate Biotechnology)[25] or against RyRhosphoserine 2809 (Badrilla) and the non-phosphorylapecific RyR 1E9 monoclonal antibody[2]. Antibody bindingas detected by secondary antibodies and then quansing a PhosphorImager (Bio-Rad).
.7. Statistical analysis
Data are reported as mean± S.E.M. Statistical differnces between the different types of mice were calculatNOVA followed by Bonferroni’st-test.P < 0.05 was conidered significant.
. Results
.1. Unchanged SERCA2a function and enrichment ofSQ in SR vesicles in TG
Here we found that a decreased SR Ca2+ uptake byERCA2a cannot account for the slower decline of [Cai in
esulting in an increased amplitude of the Catransient, wenalyzed�[Ca]i in the presence and absence of Ni2+ and
ound that the amplitude reduction of caffeine-induced C2+
ransient that we have found in TG cardiomyocytes[16] wasnchanged by the presence of Ni2+ (Fig. 2D).
.3. Altered Ca2+ spark parameters in TG
It has been proposed that the Ca2+ spark frequency mighirectly depend on the loading state of the SR[27]. Thus,e recorded spontaneous Ca2+ sparks and estimated th
requency in TG and WT cardiomyocytes using confoicroscopy and found that the frequency of Ca2+ sparks wa
134 U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142
Fig. 2. Expression of the NCX and effects of caffeine and Ni2+ on Ca2+ transients. The protein expression of the NCX was measured in membrane vesiclesof TG and WT hearts (A). Identical amounts of protein (50�g) were loaded per lane. Blots were probed with an antibody against cardiac NCX as describedunder Section2. Shown is a representative immunoblot. The indicated 120-kDa and 160-kDa molecular mass forms of NCX (arrowheads) were analyzed andsummarized (B). The time constant of decay of Ca2+ transients [Ca2+]i was determined in the presence of 10 mM caffeine and 5 mM Ni2+ in unstimulated TGand WT cardiomyocytes (C). The amplitude of the Ca2+ transients (�[Ca]i ) was measured at basal conditions in electrically stimulated cardiomyocytes and inresponse to 10 mM caffeine and 5 mM Ni2+ in quiescent TG and WT cardiomyocytes (D).
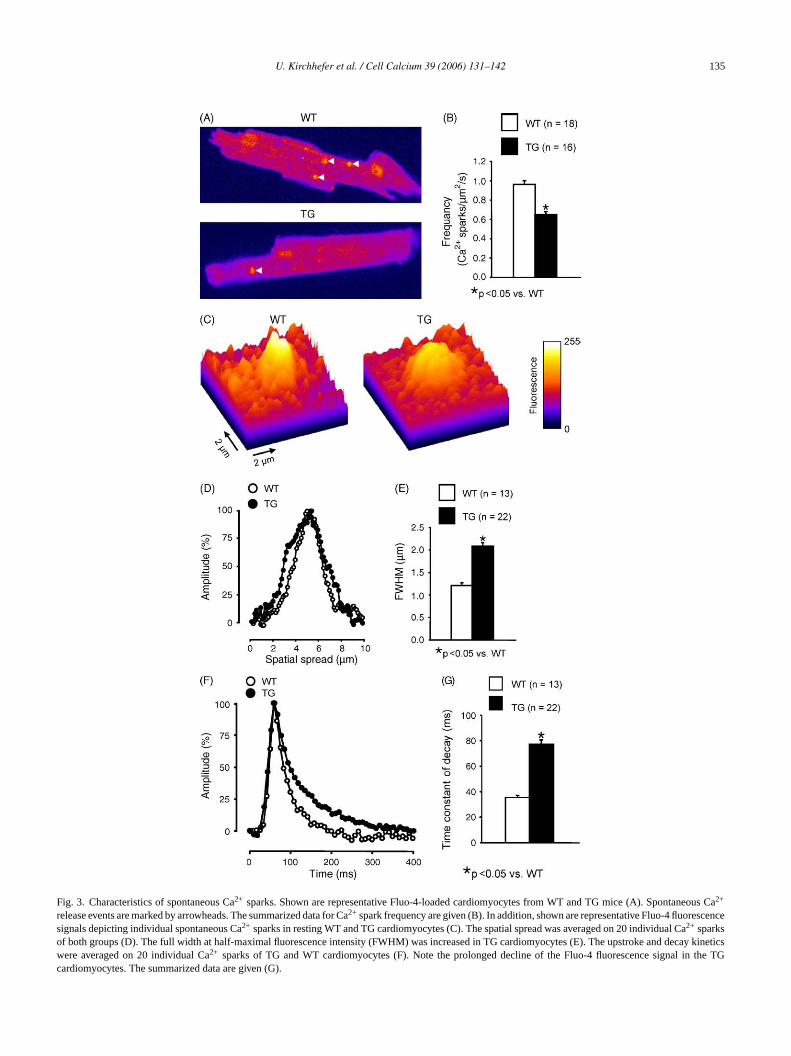
reduced by 32% in TG (Fig. 3A and B). Moreover, the Ca2+
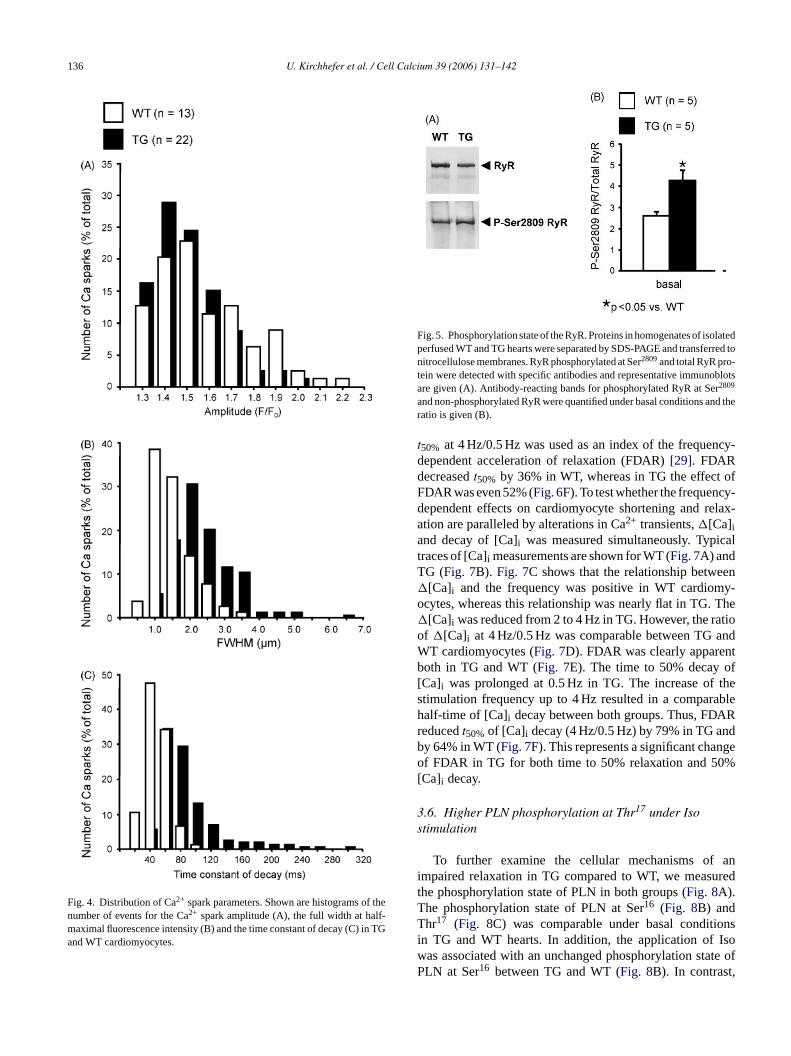
spark amplitude (F/F0) was slightly reduced in TG comparedto WT (from 1.51± 0.02 to 1.44± 0.01,n = 13–22,P < 0.05).In addition, the spatial spread of Ca2+ sparks, measured as thefull width at half-maximal fluorescence intensity (FWHM),was increased by 73% in TG (Fig. 3C–E). In parallel to theincreased spatial spread we also found an increased relax-ation phase of the TG Ca2+ sparks as depicted inFig. 3F.The time constant of decay of Ca2+ sparks was enhanced by117% in TG (Fig. 3G). The distribution of Ca2+ spark ampli-tudes revealed one population both in TG and WT (Fig. 4A).Furthermore, only one population of Ca2+ sparks was alsoobserved for FWHM (Fig. 4B) and the time constant of decay(Fig. 4C) both in TG and WT.
3.4. Hyperphosphorylation of the RyR in TG
The diminished SR Ca2+ load in heart failure has beenattributed to a hyperphosphorylation of the RyR by cAMP-dependent protein kinase (PKA) which may result in “leaky”SR Ca2+ release channels[28]. Therefore, we tested thebasal phosphorylation state of the RyR (Fig. 5A). We foundthat the basal phosphorylation of the RyR at Ser2809, repre-
senting the amino acid residue which is phosphorylated byCa2+/calmodulin-dependent protein kinase II (CaMKII) andPKA, was increased by 64% (i.e., hyperphosphorylated) inTG compared to WT hearts (Fig. 5B) [21].
3.5. Increased frequency-dependent acceleration ofrelaxation in TG
Fig. 6A and B show representative shortening-frequencyrelationships in cardiomyocytes of WT and TG mice, respec-tively. Frequency was increased stepwise from 0.5 to 4 Hzin both groups. The shortening-frequency relationship waspositive in WT compared to TG where shortening-frequencyrelationship was flat (Fig. 6C). The ratio of shortening at4 Hz/0.5 Hz was used as an index of inotropy mediated byincreasing frequency. A ratio over 1.0 represent a positiveinotropy, whereas a ratio less than 1.0 a negative inotropy[29]. The ratio was increased by 122% in WT. In con-trast, there was no significant difference of this ratio in TG(Fig. 6D). Increasing stimulation frequencies accelerated thehalf-time of relengthening in WT, but to a higher extent inTG (Fig. 6E). This is because of a prolonged relaxation inTG at the basal stimulation frequency of 0.5 Hz. The ratio of
U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142 135
Fig. 3. Characteristics of spontaneous Ca2+ sparks. Shown are representative Fluo-4-loaded cardiomyocytes from WT and TG mice (A). Spontaneous Ca2+
release events are marked by arrowheads. The summarized data for Ca2+ spark frequency are given (B). In addition, shown are representative Fluo-4 fluorescencesignals depicting individual spontaneous Ca2+ sparks in resting WT and TG cardiomyocytes (C). The spatial spread was averaged on 20 individual Ca2+ sparksof both groups (D). The full width at half-maximal fluorescence intensity (FWHM) was increased in TG cardiomyocytes (E). The upstroke and decay kineticswere averaged on 20 individual Ca2+ sparks of TG and WT cardiomyocytes (F). Note the prolonged decline of the Fluo-4 fluorescence signal in the TGcardiomyocytes. The summarized data are given (G).
136 U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142
Fig. 4. Distribution of Ca2+ spark parameters. Shown are histograms of thenumber of events for the Ca2+ spark amplitude (A), the full width at half-maximal fluorescence intensity (B) and the time constant of decay (C) in TGand WT cardiomyocytes.
Fig. 5. Phosphorylation state of the RyR. Proteins in homogenates of isolatedperfused WT and TG hearts were separated by SDS-PAGE and transferred tonitrocellulose membranes. RyR phosphorylated at Ser2809and total RyR pro-tein were detected with specific antibodies and representative immunoblotsare given (A). Antibody-reacting bands for phosphorylated RyR at Ser2809
and non-phosphorylated RyR were quantified under basal conditions and theratio is given (B).
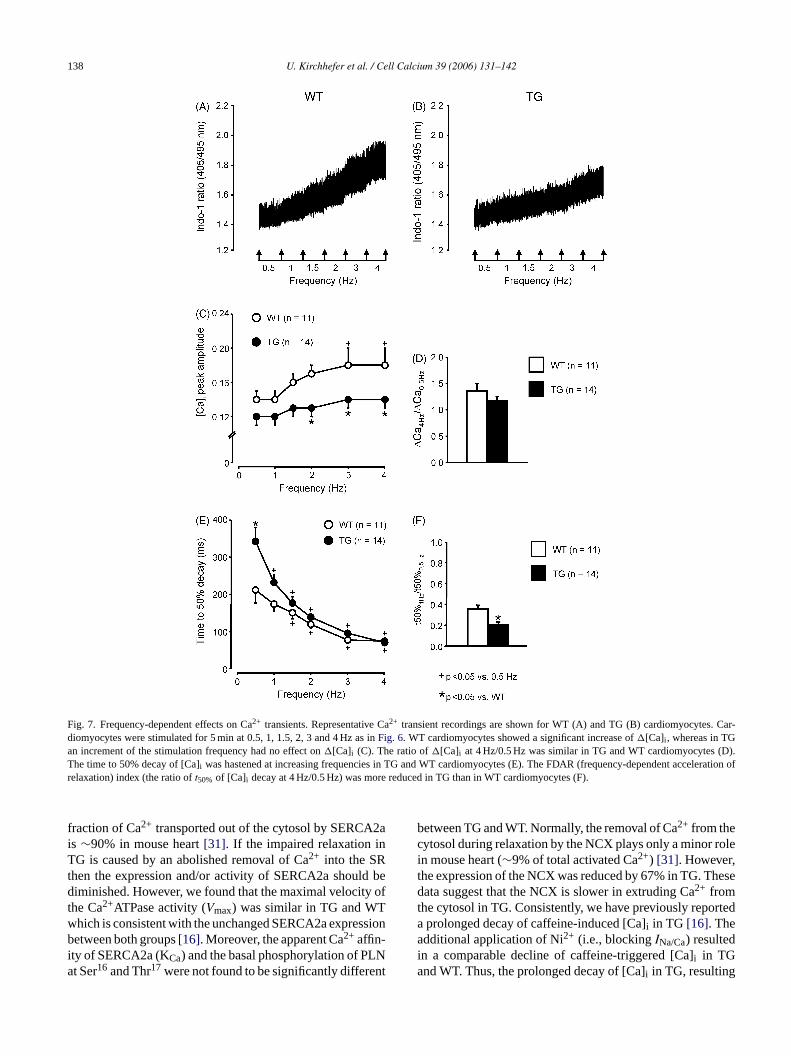
t50% at 4 Hz/0.5 Hz was used as an index of the frequency-dependent acceleration of relaxation (FDAR)[29]. FDARdecreasedt50% by 36% in WT, whereas in TG the effect ofFDAR was even 52% (Fig. 6F). To test whether the frequency-dependent effects on cardiomyocyte shortening and relax-ation are paralleled by alterations in Ca2+ transients,�[Ca]iand decay of [Ca]i was measured simultaneously. Typicaltraces of [Ca]i measurements are shown for WT (Fig. 7A) andTG (Fig. 7B). Fig. 7C shows that the relationship between�[Ca]i and the frequency was positive in WT cardiomy-ocytes, whereas this relationship was nearly flat in TG. The�[Ca]i was reduced from 2 to 4 Hz in TG. However, the ratioof �[Ca]i at 4 Hz/0.5 Hz was comparable between TG andWT cardiomyocytes (Fig. 7D). FDAR was clearly apparentboth in TG and WT (Fig. 7E). The time to 50% decay of[Ca]i was prolonged at 0.5 Hz in TG. The increase of thestimulation frequency up to 4 Hz resulted in a comparablehalf-time of [Ca]i decay between both groups. Thus, FDARreducedt50% of [Ca]i decay (4 Hz/0.5 Hz) by 79% in TG andby 64% in WT (Fig. 7F). This represents a significant changeof FDAR in TG for both time to 50% relaxation and 50%[Ca]i decay.
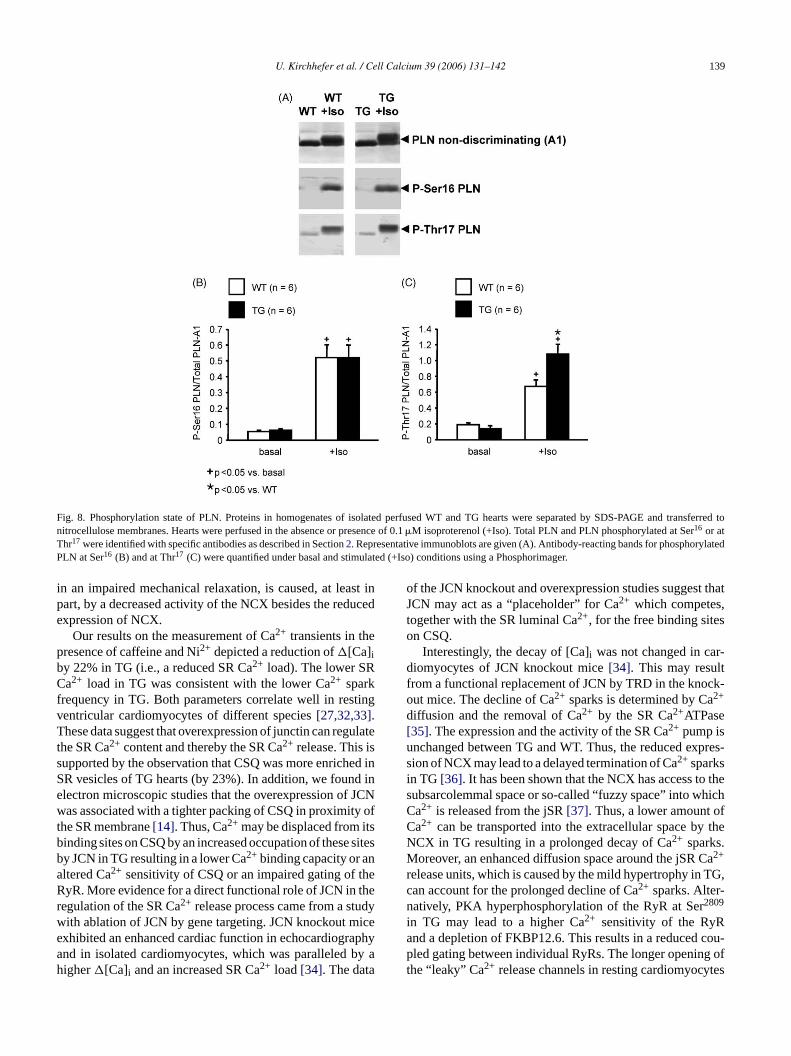
3.6. Higher PLN phosphorylation at Thr17 under Isostimulation
ani redtTT onsi Isow ate ofP ,
To further examine the cellular mechanisms ofmpaired relaxation in TG compared to WT, we measuhe phosphorylation state of PLN in both groups (Fig. 8A).he phosphorylation state of PLN at Ser16 (Fig. 8B) andhr17 (Fig. 8C) was comparable under basal conditi
n TG and WT hearts. In addition, the application ofas associated with an unchanged phosphorylation stLN at Ser16 between TG and WT (Fig. 8B). In contrast

U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142 137
Fig. 6. Frequency-dependent changes of cell shortening and relaxation. Shown are representative traces of edge detection measurements recorded oncardiomy-ocytes of WT (A) and TG mice (B). Frequency was stepwise increased every 5 min from 0.5 to 4 Hz. Average data of experiments are given for cell shortening(C). The frequency-dependent inotropic effect was determined by the ratio of cell shortening (CS) at 4 Hz/0.5 Hz (D). Cardiomyocytes of both groups exhibitedan acceleration of relaxation under increasing frequencies as measured by the time to 50% relengthening from 0.5 to 4 Hz (E). However, those of TG miceshowed a higher reduction of the FDAR (frequency-dependent acceleration of relaxation) index (F, the ratio oft50% at 4 Hz/0.5 Hz).
the overexpression of JCN resulted in a 1.6-fold increasedphosphorylation state of PLN at Thr17 under�-adrenergicreceptor (�-AR) stimulation (Fig. 8C).
4. Discussion
The SR Ca2+ release is maintained by structural unitswhich are organized into quaternary protein complexes con-sisting of the RyR, CSQ, TRD, and JCN[2]. Biochemi-
cal studies revealed that JCN enables the Ca2+-dependenttethering of CSQ to the RyR[2,9]. The use of transgenicmodels with cardiac-specific overexpression of JCN pro-vided new insight into the dynamic processes of SR Ca2+
release[15,30]. The 10-fold JCN overexpression was associ-ated with an impaired relaxation in isolated cardiomyocytesand papillary muscles[16]. However, the underlying cellularmechanisms remained undefined.
The dominant process that determines the decay of [Ca]iis thought to be Ca2+ transport by the SR Ca2+ATPase. The
138 U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142
Fig. 7. Frequency-dependent effects on Ca2+ transients. Representative Ca2+ transient recordings are shown for WT (A) and TG (B) cardiomyocytes. Car-diomyocytes were stimulated for 5 min at 0.5, 1, 1.5, 2, 3 and 4 Hz as inFig. 6. WT cardiomyocytes showed a significant increase of�[Ca]i , whereas in TGan increment of the stimulation frequency had no effect on�[Ca]i (C). The ratio of�[Ca]i at 4 Hz/0.5 Hz was similar in TG and WT cardiomyocytes (D).The time to 50% decay of [Ca]i was hastened at increasing frequencies in TG and WT cardiomyocytes (E). The FDAR (frequency-dependent acceleration ofrelaxation) index (the ratio oft50% of [Ca]i decay at 4 Hz/0.5 Hz) was more reduced in TG than in WT cardiomyocytes (F).
fraction of Ca2+ transported out of the cytosol by SERCA2ais ∼90% in mouse heart[31]. If the impaired relaxation inTG is caused by an abolished removal of Ca2+ into the SRthen the expression and/or activity of SERCA2a should bediminished. However, we found that the maximal velocity ofthe Ca2+ATPase activity (Vmax) was similar in TG and WTwhich is consistent with the unchanged SERCA2a expressionbetween both groups[16]. Moreover, the apparent Ca2+ affin-ity of SERCA2a (KCa) and the basal phosphorylation of PLNat Ser16 and Thr17 were not found to be significantly different
between TG and WT. Normally, the removal of Ca2+ from thecytosol during relaxation by the NCX plays only a minor rolein mouse heart (∼9% of total activated Ca2+) [31]. However,the expression of the NCX was reduced by 67% in TG. Thesedata suggest that the NCX is slower in extruding Ca2+ fromthe cytosol in TG. Consistently, we have previously reporteda prolonged decay of caffeine-induced [Ca]i in TG [16]. Theadditional application of Ni2+ (i.e., blockingINa/Ca) resultedin a comparable decline of caffeine-triggered [Ca]i in TGand WT. Thus, the prolonged decay of [Ca]i in TG, resulting
U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142 139
Fig. 8. Phosphorylation state of PLN. Proteins in homogenates of isolated perfused WT and TG hearts were separated by SDS-PAGE and transferred tonitrocellulose membranes. Hearts were perfused in the absence or presence of 0.1�M isoproterenol (+Iso). Total PLN and PLN phosphorylated at Ser16 or atThr17 were identified with specific antibodies as described in Section2. Representative immunoblots are given (A). Antibody-reacting bands for phosphorylatedPLN at Ser16 (B) and at Thr17 (C) were quantified under basal and stimulated (+Iso) conditions using a Phosphorimager.
in an impaired mechanical relaxation, is caused, at least inpart, by a decreased activity of the NCX besides the reducedexpression of NCX.
Our results on the measurement of Ca2+ transients in thepresence of caffeine and Ni2+ depicted a reduction of�[Ca]iby 22% in TG (i.e., a reduced SR Ca2+ load). The lower SRCa2+ load in TG was consistent with the lower Ca2+ sparkfrequency in TG. Both parameters correlate well in restingventricular cardiomyocytes of different species[27,32,33].These data suggest that overexpression of junctin can regulatethe SR Ca2+ content and thereby the SR Ca2+ release. This issupported by the observation that CSQ was more enriched inSR vesicles of TG hearts (by 23%). In addition, we found inelectron microscopic studies that the overexpression of JCNwas associated with a tighter packing of CSQ in proximity ofthe SR membrane[14]. Thus, Ca2+ may be displaced from itsbinding sites on CSQ by an increased occupation of these sitesby JCN in TG resulting in a lower Ca2+ binding capacity or analtered Ca2+ sensitivity of CSQ or an impaired gating of theRyR. More evidence for a direct functional role of JCN in theregulation of the SR Ca2+ release process came from a studywith ablation of JCN by gene targeting. JCN knockout miceexhibited an enhanced cardiac function in echocardiographyand in isolated cardiomyocytes, which was paralleled by ahigher�[Ca]i and an increased SR Ca2+ load[34]. The data
of the JCN knockout and overexpression studies suggest thatJCN may act as a “placeholder” for Ca2+ which competes,together with the SR luminal Ca2+, for the free binding siteson CSQ.
Interestingly, the decay of [Ca]i was not changed in car-diomyocytes of JCN knockout mice[34]. This may resultfrom a functional replacement of JCN by TRD in the knock-out mice. The decline of Ca2+ sparks is determined by Ca2+
diffusion and the removal of Ca2+ by the SR Ca2+ATPase[35]. The expression and the activity of the SR Ca2+ pump isunchanged between TG and WT. Thus, the reduced expres-sion of NCX may lead to a delayed termination of Ca2+ sparksin TG [36]. It has been shown that the NCX has access to thesubsarcolemmal space or so-called “fuzzy space” into whichCa2+ is released from the jSR[37]. Thus, a lower amount ofCa2+ can be transported into the extracellular space by theNCX in TG resulting in a prolonged decay of Ca2+ sparks.Moreover, an enhanced diffusion space around the jSR Ca2+
release units, which is caused by the mild hypertrophy in TG,can account for the prolonged decline of Ca2+ sparks. Alter-natively, PKA hyperphosphorylation of the RyR at Ser2809
in TG may lead to a higher Ca2+ sensitivity of the RyRand a depletion of FKBP12.6. This results in a reduced cou-pled gating between individual RyRs. The longer opening ofthe “leaky” Ca2+ release channels in resting cardiomyocytes

140 U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142
may then contribute to the depletion of SR Ca2+ stores[38].However, the contribution of a PKA hyperphosphorylationof the RyR to the prolonged decay of Ca2+ sparks and [Ca]iremains controversial[21,28,39,40]. The small reduction ofthe Ca2+ spark amplitude in TG may be attributable to thediminished SR Ca2+ load or the reduced protein expressionof the RyR rather than to an altered activity. Moreover, thehigher spread width of Ca2+ sparks (FWHM) in TG suggestsa higher recruitment of single RyRs in each Ca2+ releaseevent. This conclusion is supported by the fact that the proteinexpression of the RyR is reduced by 32% in TG[16]. Alter-natively, the lower NCX expression may reduce the apparentCa2+ diffusion coefficient.
Frequency is a powerful modulator of excitation–contraction coupling influencing the amplitude and relax-ation of cardiac contraction. The frequency-dependent effectsdepend, in part, on the SR Ca2+ load at the initial frequencyand on the recovery of the RyR from inactivation at thefollowing frequencies. The basal stimulation at 0.5 Hz wasassociated with a similar shortening and�[Ca]i betweenboth groups despite a reduced SR Ca2+ load in TG. A highermaximal-stimulated fractional SR Ca2+ release could havecontributed to that effect in TG. We suggest that the loss ofICaat higher frequencies, which is related to slow recovery ofthe L-type Ca2+ channel from inactivation[41], results in alower fractional SR Ca2+ release in TG and consequently in afll -t inga -q alsor ation( de-pw n ther ngeso ast es intra-c KII[C yla-t sei -o onC y-lw LNk KIIi II-d ireP ofCF PLNp ents hos-
phorylation of SERCA2a by CaMKII[51] may contribute toan acceleration of SR Ca2+ transport and improved relaxationat higher frequencies in TG. However, the study by Valverdeand coworkers[50] found no evidence for an associationof FDAR with changes in the CaMKII-dependent phospho-rylation of SERCA2a. These authors suggested that highintracellular Ca2+ by itself, without CaMKII activation, mayeffect the interaction between PLN and SERCA2a, as demon-strated by in vitro studies[52]. The enhanced phosphorylationof PLN at Thr17 under�-AR stimulation in beating TG heartsdoes not indicate that CaMKII-dependent mechanisms areinvolved in FDAR. It has been shown by use of transgenicmodels with mutation of Ser16 and Thr17 to alanine that thephosphorylation of Ser16 in PLB by PKA is sufficient in medi-ating its maximal contractile response to�-AR stimulation[53,54]. Thus, the phosphorylation of PLN at Ser16 might bea prerequisite for Thr17 phosphorylation of PLN. Moreover,it has been shown more recently that the increase in PLNphosphorylation at Ser16 after Iso application was associ-ated with a relaxant effect, which was further enhanced whenphosphorylation of PLN at Thr17 reached significant levels[50]. This suggests that FDAR and isoproterenol-dependentacceleration of relaxation do not share common underly-ing mechanisms. Thus, our results lead us conclude that thehigher phosphorylation of PLN at Thr17 in Iso-stimulated TGhearts may contribute to the hastened relaxation after�-ARs
del:( as eousC n oft ryla-t Ni ationo stt h all
A
sis-t
(toL SFB5 jectB gerF
R
ineeticu-
at staircase. Indeed, the inactivation kinetics ofICawere pro-onged at basal conditions in TG[16]. A lower phosphorylaion of PLN at Ser16may also account for the lower shortennd�[Ca]i in TG at higher frequencies[42]. Increasing freuencies affect not only the amplitude of contraction butelaxation. Frequency-dependent acceleration of relaxFDAR) occurs in mammalian ventricular preparations inendently of a positive or negative staircase[43–45]. Heree measured a higher effect of increasing frequencies o
elengthening of TG cardiomyocytes paralleled by chaf the decay of [Ca]i . There is controversy in the literature
o whether the SR Ca2+ load contributes to FDAR. On onide, it has been shown that higher frequencies elevateellular [Ca]i leading to an increased activation of CaM29,46], phosphorylation of PLN at Thr17 and higher SRa2+ load [47,48]. This frequency-dependent phosphor
ion of PLN at Thr17 is closely correlated with a decrean the relaxation time (t50%) of rat ventricular cardiomycytes[48]. On the other side, a dependence of FDARaMKII activation[45,49,50]and a higher PLN phosphor
ation after an increase of the stimulation frequency[49,50]ere not observed. Moreover, FDAR also occurred in Pnockout mice and was inhibited by the potent CaMnhibitor KN-93 suggesting that FDAR depends on CaMKependent stimulation of SR Ca2+ release but does not requLN phosphorylation[29]. Thus, a potential involvementaMKII-dependent phosphorylation of PLN at Thr17 duringDAR remains controversial. Here, we did not detect thehosphorylation status or the CaMKII activity at differtimulation frequencies. It is conceivable that a higher p
timulation[16].In summary, here we report novel findings on this mo
i) the lower SR Ca2+ load is associated with a reduced C2+
park frequency; (ii) the prolonged decay of spontana2+ sparks in TG is accompanied by a down-regulatio
he NCX protein expression and an enhanced phosphoion of the RyR at Ser2809; and (iii) the overexpression of JCs paralleled by a higher frequency-dependent accelerf relaxation and a faster decay of [Ca]i . Thus, we sugge
hat the impaired relaxation in TG may result from botower sarcolemmal Ca2+ extrusion and/or a higher SR Ca2+
eak.
cknowledgements
We thank N. Hinsenhofen for excellent technical asance.
This work was supported by NIH Grant HL-28556.R.J.), the Deutsche Forschungsgemeinschaft and the56 projects B1 and Z2 (to J.N.), and SFB 530 pro6 as well as HBFG (to P.L.) and HOMFOR (Homburorschungsforderprogramm) project A2003/C5 (to L.K.).
eferences
[1] W. Guo, K.P. Campbell, Association of triadin with the ryanodreceptor and calsequestrin in the lumen of the sarcoplasmic rlum, J. Biol. Chem. 270 (1995) 9027–9030.

U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142 141
[2] L. Zhang, J. Kelly, G. Schmeisser, Y.M. Kobayashi, L.R. Jones,Complex formation between junctin, triadin, calsequestrin, and theryanodine receptor, J. Biol. Chem. 272 (1997) 23389–23397.
[3] H. Takeshima, S. Nishimura, T. Matsumoto, et al., Primary struc-ture and expression from complementary DNA of skeletal muscleryanodine receptor, Nature 339 (1989) 439–445.
[4] M.B. Bhat, J. Zhao, H. Takeshima, J. Ma, Functional calcium releasechannel formed by the carboxyl-terminal portion of ryanodine recep-tor, Biophys. J. 73 (1997) 1329–1336.
[5] K.P. Campbell, D.H. MacLennan, A.O. Jorgensen, M.C. Mintzer,Purification and characterization of calsequestrin from canine car-diac sarcoplasmic reticulum and identification of the 53,000 daltonglycoprotein, J. Biol. Chem. 258 (1983) 1197–1204.
[6] C. Franzini-Armstrong, L.J. Kenney, E. Varriano-Marston, The struc-ture of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study, J. Cell. Biol. 105 (1987) 49–56.
[7] Y. Sato, D.G. Ferguson, H. Sako, et al., Cardiac-specific overex-pression of mouse cardiac calsequestrin is associated with depressedcardiovascular function and hypertrophy in transgenic mice, J. Biol.Chem. 273 (1998) 28470–28477.
[8] L.R. Jones, Y.J. Suzuki, W. Wang, et al., Regulation of Ca2+ sig-naling in transgenic mouse cardiac myocytes overexpressing calse-questrin, J. Clin. Invest. 101 (1998) 1385–1393.
[9] I. Gyorke, N. Hester, L.R. Jones, S. Gyorke, The role of calse-questrin, triadin, and junctin in conferring cardiac ryanodine receptorresponsiveness to luminal calcium, Biophys. J. 86 (2004) 2121–2128.
[10] W. Guo, A.O. Jorgensen, L.R. Jones, K.P. Campbell, Biochemicalcharacterization and molecular cloning of cardiac triadin, J. Biol.Chem. 271 (1996) 458–465.
[11] L.R. Jones, L. Zhang, K. Sanborn, A.O. Jorgensen, J. Kelley,tion
car-995)
[ ofstrin,
[ sen,s in.
[ alse-and
Mol.
[ turalrex-
[ n in2003)
[ phyin 1,
[ tero-hem.
[ banus-
um
[ in 1n-
004)
[ se Aodine
receptor) in normal and failing hearts, J. Biol. Chem. 278 (2003)444–453.
[22] J. Neumann, P. Boknik, A.A. DePaoli-Roach, et al., Targeted overex-pression of phospholamban to mouse atrium depresses Ca2+ transportand contractility, J. Mol. Cell. Cardiol. 30 (1998) 1991–2002.
[23] M.A. Porzio, A.M. Pearson, Improved resolution of myofibrillar pro-teins with sodium dodecyl sulfate-polyacrylamide gel electrophore-sis, Biochim. Biophys. Acta 490 (1977) 27–34.
[24] L. Mahony, L.R. Jones, Developmental changes in cardiac sarcoplas-mic reticulum in sheep, J. Biol. Chem. 261 (1986) 15257–15265.
[25] T. Suzuki, J.H. Wang, Stimulation of bovine cardiac sarcoplasmicreticulum Ca2+ pump and blocking of phospholamban phospho-rylation and dephosphorylation by a phospholamban monoclonalantibody, J. Biol. Chem. 261 (1986) 7018–7023.
[26] J.W.M. Bassani, R.A. Bassani, D.M. Bers, Relaxation in rabbit andrat cardiac cells: species-dependent differences in cellular mecha-nisms, J. Physiol. 476 (1994) 279–293.
[27] H. Satoh, L.A. Blatter, D.M. Bers, Effects of [Ca2+]i , SR Ca2+ load,and rest on Ca2+ spark frequency in ventricular myocytes, Am. J.Physiol. 272 (1997) H657–H668.
[28] S.O. Marx, S. Reiken, Y. Hisamatsu, et al., PKA phosphorylationdissociates FKBP12.6 from the calcium release channel (ryanodinereceptor): defective regulation in failing hearts, Cell 101 (2000)365–376.
[29] J. DeSantiago, L.S. Maier, D.M. Bers, Frequency-dependent accel-eration of relaxation in the heart depends on CaMKII, but notphospholamban, J. Mol. Cell. Cardiol. 34 (2002) 975–984.
[30] C.S. Hong, M.C. Cho, Y.G. Kwak, et al., Cardiac remodeling andatrial fibrillation in transgenic mice overexpressing junctin, FASEBJ. 16 (2002) 1310–1312.
[31] L. Li, G. Chu, E.G. Kranias, D.M. Bers, Cardiac myocyte calciumdoge-7.
[ ntaryscle,
[ es-a
ar
[ ene
[
96)
[ ku-arksr, J.
[ ofneous995)
[ , K.cium2001)
[ le in
[ Acium309–
[ ismsa
Purification, primary structure, and immunological characterizaof the 26-kDa calsequestrin binding protein (junctin) fromdiac junctional sarcoplasmic reticulum, J. Biol. Chem. 270 (130787–30796.
12] M. Ohkura, K.I. Furukawa, H. Fujimori, et al., Dual regulationthe skeletal muscle ryanodine receptor by triadin and calsequeBiochemistry 37 (1998) 12987–12993.
13] C. Thompson, W. Arnold, A.C.-Y. Shen, L.R. Jones, A.O. JorgenSubcellular distribution of calsequestrin (CSQ) binding proteincardiac sarcoplasmic reticulum, Biophys. J. 72 (1997) 378–386
14] P. Tijskens, L.R. Jones, C. Franzini-Armstrong, Junctin and cquestrin overexpression in cardiac muscle: the role of junctinthe synthetic and delivery pathways for the two proteins, J.Cell. Cardiol. 35 (2003) 961–974.
15] L. Zhang, C. Franzini-Armstrong, V. Ramesh, L.R. Jones, Strucalterations in cardiac calcium release units resulting from ovepression of junctin, J. Mol. Cell. Cardiol. 33 (2001) 233–247.
16] U. Kirchhefer, J. Neumann, D.M. Bers, et al., Impaired relaxatiotransgenic mice overexpressing junctin, Cardiovasc. Res. 59 (369–379.
17] U. Kirchhefer, J. Neumann, H.A. Baba, et al., Cardiac hypertroand impaired relaxation in transgenic mice overexpressing triadJ. Biol. Chem. 276 (2001) 4142–4149.
18] L.R. Jones, S.E. Cala, Biochemical evidence for functional hegeneity of cardiac sarcoplasmic reticulum vesicles, J. Biol. C256 (1981) 11809–11818.
19] F.N. Briggs, K.F. Lee, A.W. Wechsler, L.R. Jones, Phospholamexpressed in slow-twitch and chronically stimulated fast-twitch mcles minimally affects calcium affinity of sarcoplasmic reticulCa2+-ATPase, J. Biol. Chem. 267 (1992) 26056–26061.
20] U. Kirchhefer, L.R. Jones, F. Begrow, et al., Transgenic triadoverexpression alters SR Ca2+ handling and leads to a blunted cotractile response to�-adrenergic agonists, Cardiovasc. Res. 62 (2122–134.
21] S. Reiken, M. Gaburjakova, S. Guatimosim, et al., Protein kinaphosphorylation of the cardiac calcium release channel (ryan
transport in phospholamban knockout mouse: relaxation and ennous CaMKII effects, Am. J. Physiol. 274 (1998) H1335–H134
32] H. Cheng, W.J. Lederer, M.B. Cannell, Calcium sparks: elemeevents underlying excitation-contraction coupling in heart muScience 262 (1993) 740–744.
33] V. Lukyanenko, S. Viatchenko-Karpinski, A. Smirnov, T.F. Winer, S. Gyorke, Dynamic regulation of sarcoplasmic reticulum C2+
content and release by luminal Ca2+-sensitive leak in rat ventriculmyocytes, Biophys. J. 81 (2001) 785–798.
34] Q. Yuan, G.-C. Fan, X. Sun, et al., Ablation of junctin by gtargeting results in enhanced cardiac contractility and Ca2+ kinetics,Circulation 110 (Suppl.) (2004), III-131.
35] A.M. Gomez, H. Cheng, W.J. Lederer, D.M. Bers, Ca2+ diffusion andsarcoplasmic reticulum transport both contribute to [Ca2+]i declineduring Ca2+ sparks in rat ventricular myocytes, J. Physiol. 496 (19575–581.
36] J.I. Goldhaber, S.T. Lamp, D.O. Walter, A. Garfinkel, G.H. Fumoto, J.N. Weiss, Local regulation the threshold for calcium spin rat ventricular myocytes: role of sodium-calcium exchangePhysiol. 520 (1999) 431–438.
37] A.W. Trafford, M.E. Diaz, S.C. O’Neill, D.A. Eisner, Comparisonsubsarcolemmal and bulk calcium concentration during spontacalcium release in rat ventricular myocytes, J. Physiol. 488 (1577–586.
38] S.O. Marx, J. Gaburjakova, M. Gaburjakova, C. HenriksonOndrias, A.R. Marks, Coupled gating between cardiac calrelease channels (ryanodine receptors), Circ. Res. 88 (1151–1158.
39] A.R. Marks, Cardiac intracellular calcium release channels: roheart failure, Circ. Res. 87 (2000) 8–11.
40] Y. Li, E.G. Kranias, G.A. Mignery, D.M. Bers, Protein kinasephosphorylation of the ryanodine receptor does not affect calsparks in mouse ventricular myocytes, Circ. Res. 90 (2002)316.
41] G. Antoons, K. Mubagwa, I. Nevelsteen, K.R. Sipido, Mechanunderlying the frequency dependence of contraction and [C2+]i

142 U. Kirchhefer et al. / Cell Calcium 39 (2006) 131–142
transients in mouse ventricular myocytes, J. Physiol. 543 (2002)889–898.
[42] K. Brixius, A. Wollmer, B. Bolck, W. Mehlhorn, R.H.G. Schwinger,Ser16-, but not Thr17-phosphorylation of phospholamban influ-ences frequency-dependent force generation in human myocyardium,Pflugers Arch. 447 (2003) 150–157.
[43] W.D. Gao, N.G. Perez, E. Marban, Calcium cycling and contractileactivation in intact mouse cardiac muscle, J. Physiol. 507 (1998)175–184.
[44] B. Pieske, B. Kretschmann, M. Meyer, et al., Alterations in intracel-lular calcium handling associated with the inverse force-frequencyrelation in human dilated cardiomyopathy, Circulation 92 (1995)1169–1178.
[45] Z. Kassiri, R. Myers, R. Kaprielian, H.S. Banijamali, P.H. Backx,Rate-dependent changes of twitch force duration in rat cardiac tra-beculae: a property of the contractile system, J. Physiol. 524 (2000)221–231.
[46] R.A. Bassani, A. Mattiazzi, D.M. Bers, CaMKII is responsiblefor activity-dependent acceleration of relaxation in rat ventricularmyocytes, Am. J. Physiol. 268 (1995) H703–H712.
[47] V.J.A. Schouten, Interval dependence of force and twitch duration inrat heart explained by Ca pump inactivation in sarcoplasmic reticu-lum, J. Physiol. 431 (1990) 427–444.
[48] D. Hagemann, M. Kuschel, T. Kuramochi, W. Zhu, H. Cheng, R.P.Xiao, Frequency-encoding Thr17 phospholamban phosphorylation isindependent of Ser16 phosphorylation in cardiac myocytes, J. Biol.Chem. 275 (2000) 22532–22536.
[49] M. Hussain, G.A. Drago, J. Colyer, C.H. Orchard, Rate-dependentabbreviation of Ca2+ transient in rat heart is independent of phos-pholamban phosphorylation, Am. J. Physiol. 273 (1997) H695–H706.
[50] C.A. Valverde, C. Mundina-Weilenmann, M. Said, et al., Frequency-dependent acceleration of relaxation in mammalian heart: a propertynot relying on phospholamban and SERCA2a phosphorylation, J.Physiol. 562 (2005) 801–813.
[51] T. Toyofuku, K. Curotto Kurzydlowski, N. Narayanan, D.H.MacLennan, Identification of Ser38 as the site in cardiac sarcoplasmicreticulum Ca2+-ATPase that is phosphorylated by Ca2+/calmodulin-dependent protein kinase, J. Biol. Chem. 269 (1994) 26492–26496.
[52] M. Asahi, E. McKenna, K. Kurzydlowski, M. Tada, D.H.MacLennan, Physiological interactions between phospholamban andsarco(endo)plasmic reticulum Ca2+-ATPases are dissociated by ele-vated Ca2+, but not by phospholamban phosphorylation, vanadate, orthapsigargin, and are enhanced by ATP, J. Biol. Chem. 275 (2000)15034–15038.
[53] W. Luo, G. Chu, Y. Sato, Z. Zhou, V.J. Kadambi, E.G. Kranias,Transgenic approaches to define the functional role of dual sitephospholamban phosphorylation, J. Biol. Chem. 273 (1998) 4734–4739.
[54] G. Chu, J.W. Lester, K.B. Young, W. Luo, J. Thai, E.G. Kranias, Asingle site (Ser16) phosphorylation in phospholamban is sufficientin mediating its maximal cardiac responses to beta-agonists, J. Biol.Chem. 275 (2000) 38938–38943.




















