
Regulation of arginase pathway in response to wall shear stress
8
Atherosclerosis 210 (2010) 63–70 Contents lists available at ScienceDirect Atherosclerosis journal homepage: www.elsevier.com/locate/atherosclerosis Regulation of arginase pathway in response to wall shear stress Tyler N. Thacher a , Veronica Gambillara a , Fabienne Riche a , Paolo Silacci d , Nikos Stergiopulos a , Rafaela F. da Silva a,b,c,∗ a Swiss Federal Institute of Technology Lausanne, Lausanne, Switzerland b Department of Neurosurgery, Geneva University Medical Center, Switzerland c Department of Fundamental Neurosciences, Faculty of Medicine, University of Geneva, Switzerland d Station de recherche Agroscope Liebefeld, Posieux, Switzerland article info Article history: Received 22 March 2009 Received in revised form 22 October 2009 Accepted 24 October 2009 Available online 5 November 2009 Keywords: Arginase Nitric oxide Hemodynamic forces Wall shear stress Oscillatory shear stress Mechanotransduction Atherosclerosis abstract Objective: Alterations of wall shear stress can predispose the endothelium to the development of atherosclerotic plaques. Ample evidence indicates that arginase expression and/or activity correlates with several risk factors for cardiovascular disease including atherosclerosis. We investigated the regulation of arginase pathway in response to distinct patterns of wall shear stress. Methods: Isolated porcine endothelial cells and carotid arterial segments were perfused under unidirec- tional high shear stress (HSS) or oscillatory shear stress (OSS) for 1 and 3 days. Arginase I and II expression, cellular localization and enzyme activity were, respectively, assessed by Western blot, immunohisto- chemistry and colorimetric determination of urea. The contribution of arginase to the processes of endothelial dysfunction, cell proliferation and arterial remodeling induced by OSS was evaluated by administration of the arginase inhibitor N--hydroxy-nor-l-arginine (nor-Noha). Results: Only arginase II isoform was detected on porcine carotid endothelial cells and on carotid artery. Exposure of arteries to OSS increased arginase II expression and activity as compared to HSS. Inhibition of arginase by nor-Noha improved NO-dependent endothelial function and decreased total vascular ROS formation in arteries submitted to OSS. In addition, inhibition of arginase activity decreased smooth muscle cell proliferation rate with no effect on collagen content after OSS. Conclusions: Exposure of carotid artery to oscillatory flow induced a more pronounced activation of arginase as compared to HSS. Inhibition of arginase in arteries exposed to OSS improved NO-dependent endothelial function and decrease smooth muscle cell proliferation rate, both processes are important for the focal development of atherosclerotic plaque. © 2009 Elsevier Ireland Ltd. All rights reserved. 1. Introduction Alterations of wall shear stress, the frictional force generated by blood flow, can act as a local risk factor for the develop- ment of atherosclerotic plaque. At straight areas of the vasculature blood flows in a laminar and unidirectional fashion with high mean shear stress values (HSS). These areas are relatively resis- tant to the development of early atherosclerosis. In contrast, at vessel bifurcations flow is bi-directional having low mean shear stress values. This flow condition, also termed oscillatory (OSS), is known to affect gene and protein expression, morphology and function of endothelium, which in turn becomes more suscepti- ble to plaque development [1–3].Impairment of endothelial nitric ∗ Corresponding author at: Laboratory of Hemodynamics and Cardiovascular Technology, Building AI1232, Federal Institute of Technology Lausanne (EPFL), 1015 Lausanne, Switzerland. Tel.: +41 21 693 9654; fax: +41 21 693 8083. E-mail address: rafaela.fernandesdasilva@epfl.ch (R.F. da Silva). oxide synthase (eNOS) expression and phosphorylation has been suggested to partially mediate the endothelial dysfunction induced by OSS [4–6]. The activity of eNOS is controlled by multiple factors, including the availability of its substrate l-arginine [7,8]. In that regard, arginases expressed in vascular tissues can compete with eNOS for the common l-arginine substrate resulting in lower nitric oxide (NO) formation [9–11]. Arginase exists as two genetically distinct isoforms named cytosolic arginase I (ArgI) and mitochon- drial arginase II (ArgII). Several key mediators of atherothrombosis such as thrombin [12], inflammatory cytokines [13] and oxidized LDL [14] have been shown to up-regulate the activity of ArgII iso- form in endothelial cells (EC). More recently, vascular increase in arginase activity has been shown to contribute to the mechanism of endothelial dysfunction in atherosclerotic mice [15]. Whether arginase mediates the endothelial dysfunction induced by OSS is not yet known. In addition to the NO pathway, the formation of l-ornithine by arginase present in vascular smooth muscle cells (SMC) and in EC is the only source of polyamines (putrescine, spermidine, and 0021-9150/$ – see front matter © 2009 Elsevier Ireland Ltd. All rights reserved. doi:10.1016/j.atherosclerosis.2009.10.031
-
Upload
anhanguera -
Category
Documents
-
view
2 -
download
0
Transcript of Regulation of arginase pathway in response to wall shear stress

R
TNa
b
c
d
a
ARRAA
KANHWOMA
1
bmbmtvsifb
TL
0d
Atherosclerosis 210 (2010) 63–70
Contents lists available at ScienceDirect
Atherosclerosis
journa l homepage: www.e lsev ier .com/ locate /a therosc leros is
egulation of arginase pathway in response to wall shear stress
yler N. Thachera, Veronica Gambillaraa, Fabienne Richea, Paolo Silaccid,ikos Stergiopulosa, Rafaela F. da Silvaa,b,c,∗
Swiss Federal Institute of Technology Lausanne, Lausanne, SwitzerlandDepartment of Neurosurgery, Geneva University Medical Center, SwitzerlandDepartment of Fundamental Neurosciences, Faculty of Medicine, University of Geneva, SwitzerlandStation de recherche Agroscope Liebefeld, Posieux, Switzerland
r t i c l e i n f o
rticle history:eceived 22 March 2009eceived in revised form 22 October 2009ccepted 24 October 2009vailable online 5 November 2009
eywords:rginaseitric oxideemodynamic forcesall shear stress
scillatory shear stressechanotransduction
therosclerosis
a b s t r a c t
Objective: Alterations of wall shear stress can predispose the endothelium to the development ofatherosclerotic plaques. Ample evidence indicates that arginase expression and/or activity correlates withseveral risk factors for cardiovascular disease including atherosclerosis. We investigated the regulationof arginase pathway in response to distinct patterns of wall shear stress.Methods: Isolated porcine endothelial cells and carotid arterial segments were perfused under unidirec-tional high shear stress (HSS) or oscillatory shear stress (OSS) for 1 and 3 days. Arginase I and II expression,cellular localization and enzyme activity were, respectively, assessed by Western blot, immunohisto-chemistry and colorimetric determination of urea. The contribution of arginase to the processes ofendothelial dysfunction, cell proliferation and arterial remodeling induced by OSS was evaluated byadministration of the arginase inhibitor N-�-hydroxy-nor-l-arginine (nor-Noha).Results: Only arginase II isoform was detected on porcine carotid endothelial cells and on carotid artery.Exposure of arteries to OSS increased arginase II expression and activity as compared to HSS. Inhibition
of arginase by nor-Noha improved NO-dependent endothelial function and decreased total vascular ROSformation in arteries submitted to OSS. In addition, inhibition of arginase activity decreased smoothmuscle cell proliferation rate with no effect on collagen content after OSS.Conclusions: Exposure of carotid artery to oscillatory flow induced a more pronounced activation ofarginase as compared to HSS. Inhibition of arginase in arteries exposed to OSS improved NO-dependentendothelial function and decrease smooth muscle cell proliferation rate, both processes are importantt of a
for the focal developmen. Introduction
Alterations of wall shear stress, the frictional force generatedy blood flow, can act as a local risk factor for the develop-ent of atherosclerotic plaque. At straight areas of the vasculature
lood flows in a laminar and unidirectional fashion with highean shear stress values (HSS). These areas are relatively resis-
ant to the development of early atherosclerosis. In contrast, atessel bifurcations flow is bi-directional having low mean shear
tress values. This flow condition, also termed oscillatory (OSS),s known to affect gene and protein expression, morphology andunction of endothelium, which in turn becomes more suscepti-le to plaque development [1–3].Impairment of endothelial nitric∗ Corresponding author at: Laboratory of Hemodynamics and Cardiovascularechnology, Building AI1232, Federal Institute of Technology Lausanne (EPFL), 1015ausanne, Switzerland. Tel.: +41 21 693 9654; fax: +41 21 693 8083.
E-mail address: [email protected] (R.F. da Silva).
021-9150/$ – see front matter © 2009 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.atherosclerosis.2009.10.031
therosclerotic plaque.© 2009 Elsevier Ireland Ltd. All rights reserved.
oxide synthase (eNOS) expression and phosphorylation has beensuggested to partially mediate the endothelial dysfunction inducedby OSS [4–6]. The activity of eNOS is controlled by multiple factors,including the availability of its substrate l-arginine [7,8]. In thatregard, arginases expressed in vascular tissues can compete witheNOS for the common l-arginine substrate resulting in lower nitricoxide (NO) formation [9–11]. Arginase exists as two geneticallydistinct isoforms named cytosolic arginase I (ArgI) and mitochon-drial arginase II (ArgII). Several key mediators of atherothrombosissuch as thrombin [12], inflammatory cytokines [13] and oxidizedLDL [14] have been shown to up-regulate the activity of ArgII iso-form in endothelial cells (EC). More recently, vascular increase inarginase activity has been shown to contribute to the mechanismof endothelial dysfunction in atherosclerotic mice [15]. Whether
arginase mediates the endothelial dysfunction induced by OSS isnot yet known.In addition to the NO pathway, the formation of l-ornithineby arginase present in vascular smooth muscle cells (SMC) and inEC is the only source of polyamines (putrescine, spermidine, and

6 roscle
s[tptasaamt
sttlaHotO
2
2p
cCcod(Comb1
2
w1m[r(rfleAsedfBeb(
2
s
4 T.N. Thacher et al. / Athe
permine), which are essential for regulation of cell proliferation16]. l-Ornithine can also be metabolized into l-proline, an essen-ial aminoacid for the synthesis of collagen, which is involved in therocess of arterial remodeling [16]. We previously demonstratedhat exposure of porcine carotid for 3 days to OSS increased SMCnd EC proliferation as compared to HSS [17]. Furthermore, in theame study it has been suggested that OSS triggers the initiation ofrterial remodeling as evidenced by the decrease in plasminogenctivator inhibitor-1 (PAI-1) expression and the increase in matrixetalloproteinase expression and activity. Whether arginase par-
icipates in these cellular processes is not yet known.To evaluate the regulation of arginase in response to wall shear
tress in a well-controlled hemodynamic environment and withouthe influence of systemic neuroendocrine factors, we used two dis-inct perfusion systems previously developed and validated in ouraboratory [18,19]. Porcine carotid EC seeded on compliant tubesnd carotid arterial segments were exposed to either unidirectionalSS or bi-directional OSS for 1 or 3 days. Pharmacological inhibitionf arginase by N-�-hydroxy-nor-l-arginine (nor-Noha) was usedo investigate mechanistic aspects of the regulation of arginase bySS.
. Material and methods
.1. Cell culture of isolated porcine carotid EC and in vitro flowerfusion
EC from porcine left common carotid artery were isolated andoated on compliant silicon tubes as previously described [18].ompliant silicon tubes seeded with EC were mounted in spe-ially designed fittings and inserted in a perfusion loop composedf a medium reservoir and a gear pump (Ismatec) as previouslyescribed. In brief, the pump was controlled by a function generatorStandford research system) to produce a 1-Hz sinusoidal flow rate.ells were exposed to either unidirectional HSS (6 ± 3 dynes/cm2)r to a bi-directional OSS values (0.3 ± 3 dynes/cm2). Flow rate waseasured by a Transonic flowmeter and shear stress calculated
y Womersley approximation. Mean pressure was maintained at00 mmHg with a pulsation of ±20 mmHg.
.2. Arterial perfusion system
Segments of left common porcine carotid artery (PCA)ere obtained at a local slaughterhouse (from pigs weighting
20–150 kg, 6 months old). The arteries were harvested andounted on the perfusion system as described in detail elsewhere
17,20]. Additional information can be seen on supplement mate-ial. In brief, perfusion flow was generated by a gear pump systemIsmatec). A function generator producing a 1-Hz sinusoidal flowate controlled the pump. Flow rate was measured with ultrasonicowmeters (Transonic System INC, EMKA). Arterial lumen diam-ters were measured using ultrasonic echotracking device (NIUS,sulab). Perfusion pressure was set at 80 mmHg and pulse pres-ure amplitude was ±10 mmHg. Arterial segments were randomlyxposed to either unidirectional HSS (6 ± 3 dynes/cm2) or to a bi-irectional OSS values (0.3 ± 3 dynes/cm2). Arteries were perfusedor 1 and 3 days. The arginase inhibitor nor-Noha (10−4 mol/L,achem) was added to the medium for the oscillatory shear stressxperiment. From each arterial segment a small ring was excisedefore perfusion to be used as control group. Six to eight arteriesn = 6–8) were included in each group.
.3. Western blot analysis
Total protein from porcine endothelial cells (PEC) and carotidegments were extracted respectively with RIPA and Brij-35 lysis
rosis 210 (2010) 63–70
buffer containing �-mercaptoethanol. Twenty, 50, 100 or 200 �gof total protein was electrophoresed and transferred to a nitro-cellulose membrane (Amersham). Membranes were incubatedovernight at 4 ◦C with the following primary antibodies: rabbitand mouse anti-arginase I (1:200 Santa Cruz and 1:500, BD Bio-sciences, respectively) and rabbit anti-ArgII (1:200, Santa CruzAntibody), mouse anti-GAPDH (1:5000, General Electric) and rab-bit anti-collagen IV (1:100, Fitzgerald). After several washes andblocking with 5% milk, membranes were incubated with either goatanti-rabbit or mouse IgG horseradish peroxidase-linked secondaryantibodies (1:1000, Amersham). Enhanced chemiluminescencewas used for detection (Amersham). Protein expression was quan-tified using a Kodak Image Station 2000R and with Kodak 1D ImageAnalysis Software. ArgI and II protein expression levels were calcu-lated as their ratio with GAPDH house keeping protein level.
2.4. Arginase activity
Arginase activity in the cells and carotid tissue lysates were mea-sured by colorimetric determination of urea formed from l-argininesubstrate. We followed the protocol previously published by Minget al. [12].
2.5. Immunohistochemistry
At the end of the perfusion period, part of the arterial segmentwas rinsed with 0.9% NaCl, snap-frozen in OCT compound (Tissue-Tek) and stored at −80 ◦C for further analysis. For stainings, serialsections of 10 �m were cut, air-dried, and fixed in 100% acetonefor 10 min at −20 ◦C. Sections were incubated successively with0.1% Triton X-100 in PBS for 10 min, then incubated overnight at4 ◦C with rabbit anti-ArgII (1:200, Santa Cruz Antibody), rat anti-CD31 (1:250, BD Pharmagen), mouse anti-�-smooth muscle actin(�-SMA; 1:500, kind gift from Boris Hinz laboratory), mouse anti-ki67 (1:50, Chemicon International) in 10% normal goat serumin PBS. The sections were then incubated with goat anti-rabbitIgG alexa540-conjugated (1:500, BD Biosciences) and goat anti-rat IgG alexa488-conjugated (1:500, BD Biosciences) for 45 min atroom temperature. Sections were examined on a Zeiss Axiovert 135microscope. Collagen fibers were stained with 0.5% picrus–siriusred solution. Collagen type I and III were visualized by polarizedlight microscopy (Olympus AX 70) and semi-quantitative analysiswas performed as previously described [21]. In brief, type I col-lagen appeared very brilliant and strongly birefringent yellow orred fibers. On the other hand, type III collagen displays a weakbirefringence of a greenish color. An image analysis algorithm wasdeveloped on MetaMorph 6.3 (Molecular Devices) to compute therelative proportion for collagen I and III. The signal intensity forthe red, green, and yellow fibers was threshold at 80% of total sig-nal strength for each three fibers to reduce background artifacts.Data were expressed as the fraction of collagen I and III to thetotal signal measured on the birefringence slides, considered onpolarization as the total collagen content. For DHE detection 5 �marterial frozen cryosections were incubated for 30 min at 37 ◦C with5 �mol/L dihydroethidium (DHE) (FluoProbes) in PBS, then rinsedand mounted in PBS. Sections were examined on a Zeiss Axiovert135 microscope at 20× magnification. All DHE staining steps wereperformed in the dark.
2.6. Assessment of vascular function
Vascular function of carotid artery segments was tested beforeand after each perfusion experiment according to previous workperformed on porcine carotid arteries [22]. When resting ten-sion achieved 2 g, arteries were contracted with non-receptordependent KCl (90 mmol/L). To determine the endothelium-

roscle
dwcSvttvncwwwG
2
pn
3
3
ose(p(t
Fp(Lsicai
T.N. Thacher et al. / Athe
ependent relaxation capacity, arterial rings were precontractedith norepinephrine (10−4 mol/L, Sigma) and a cumulative
oncentration–response curve to bradykinin (10−9–10−5 mol/L,igma). The endothelium-independent relaxation capacity waserified by performing a cumulative concentration-response curveo sodium nitroprusside (10−9–10−4 mol/L, Sigma). To verifyhe effects of arginase inhibition on endothelium-dependentasorelaxation capacity, arterial rings were pre-incubated withor-Noha (10−4 mol/L, Bachem) for 1 h and a cumulativeoncentration–response curve to bradykinin (10−9–10−5 mol/L)as performed in the presence of nor-Noha. Tension evolutionas sampled at a rate of 1 Hz and recorded using of IOX Data soft-are, version 1.7.0 (EMKA Technology). Data were analyzed withraphPad Prism (version 5).
.7. Statistics
Data were analyzed using one-way ANOVA and Bonferroni’sost-test or unpaired ‘t’ test. Value of p ≤ 0.05 was considered sig-ificant.
. Results
.1. Arginase expression on porcine EC and carotid artery
Since no data were available about the expression of arginasen PCA, we started our experiments by characterizing the expres-ion and localization of ArgI and ArgII in this vessel. ArgII was
xpressed in fresh carotid artery tissue and EC isolated from PCAFig. 1A; n = 4). Immunohistochemistry analysis revealed a moreronounced localization of ArgII in endothelium than in SMCFig. 1B, ArgII expression represented in red, n = 4). In contrast,he expression of ArgI isoform was not detectable in isolated ECig. 1. Characterization of arginase expression in carotid porcine EC (PEC) andorcine carotid artery (PCA). (A) Immunoblot showing the expression of ArgI35 kDa), ArgII (40 kDa) and GAPDH (38 kDa) for different quantity of protein load.iver and kidney total protein extracts were used as a positive control for the expres-ion of ArgI and II respectively. (B) Immunohistochemistry visualization of ArgII (red)n endothelial cell (EC) and smooth muscle cell (SMC) of PCA tissue. Cell nuclei areolored with DAPI (blue). Green represents autofluorescence signal of elastin. Datare representative of four experiments. (For interpretation of the references to colorn this figure legend, the reader is referred to the web version of the article.)
rosis 210 (2010) 63–70 65
even after loading 150 �g of total protein cell extract. ArgI was alsonot detectable in porcine carotid fibroblasts and SMC, as indirectlyindicated by blotting fresh carotid artery tissue (Fig. 1A).
3.2. Regulation of arginase by wall shear stress in PCA perfused exvivo
As described in Fig. 2A, ArgII expression tended to increase after1 day exposure to HSS condition as compared to fresh tissue. Thisexpression was further enhanced after 3 days of flow exposure(n = 6, p ≤ 0.05). OSS flow condition also increased the expressionof ArgII. In this condition, changes were significant already after1 day of flow exposure (p ≤ 0.05 as compared to fresh tissue)and markedly increased after 3 days perfusion (p ≤ 0.01). Interest-ingly, after 3 days flow exposure OSS significantly increased theexpression of ArgII as compared to HSS (n = 6, p ≤ 0.05). Increaseon arginase protein expression in response to wall shear stresswas accompanied by changes in its enzymatic activity (Fig. 2B).Finally, immunohistochemistry analysis revealed that increase inArgII expression (Red panels in Fig. 2C and D) by OSS strongly co-localized with endothelium (Fig. 2C; double-immunolabelling ofArgII with the EC marker CD31) and only slightly co-localized withSMC (Fig. 2D; double-immunolabelling of ArgII with �-SM actin).
3.3. Endothelial function in arteries perfused under differentshear stress conditions: the role of arginase inhibition
Perfusion of PCA for 3 days under HSS flow condition didnot alter the endothelial-dependent vasorelaxation capacity inresponse to bradykinin as compared to same arteries before perfu-sion (Fig. 3A). In contrast, endothelial function was significantlyreduced after exposure of arteries for 3 days to OSS as com-pared to same arteries before perfusion (Fig. 3B; Emax 29 ± 7.4%and 47.3 ± 4.6%, respectively, n = 6, p ≤ 0.05). Pre-incubation of OSSarteries for 1 h with the arginase inhibitor nor-Noha (10−4 mol/L)significantly improved the NO-dependent relaxation capacity inresponse to bradykinin (Fig. 3B, n = 8). No differences were observedamong the 3 groups on endothelium-independent relaxationcapacity in response to cumulative concentrations of sodium nitro-prusside (data not shown).
3.4. Regulation of arginase by wall shear stress in porcine ECperfused in three-dimensional compliant tubes
Due to the abundant expression of ArgII in EC, the regulation ofarginase II expression and activity by wall shear stress was evalu-ated using an in vitro perfusion system. PEC coated on compliantsilicon tubes were exposed to either OSS or HSS. Similarly to our exvivo system, OSS for 24 h significantly increased the expression andactivity of ArgII (Fig. 4A and B; n = 4, p ≤ 0.05). In contrast, exposureto HSS for 24 h did not alter the expression of ArgII as compared tostatic culture (Fig. 4A and B).
3.5. Potential downstream mechanisms of OSS-induced arginaseexpression and activity
To evaluate the mechanism of shear-induced arginase expres-sion, nor-Noha (10−4 mol/L) was added in the medium whilearteries were perfused under OSS. Nor-Noha effectively decreasedarginase activity in arteries exposed to OSS (20% decrease, data notshown). As revealed by positive DHE staining (Fig. 5A), exposure
of arteries to OSS for 3 days markedly increased total vascular ROSformation as compared to HSS condition (n = 5, p ≤ 0.05). Nor-Nohatreatment decreased DHE staining particularly in the endothelium(n = 5, p ≤ 0.05). OSS increased the proliferation rate of SMC as com-pared to HSS (Fig. 5B; 0.12 and 1.8% of ki67 positive cells in HSS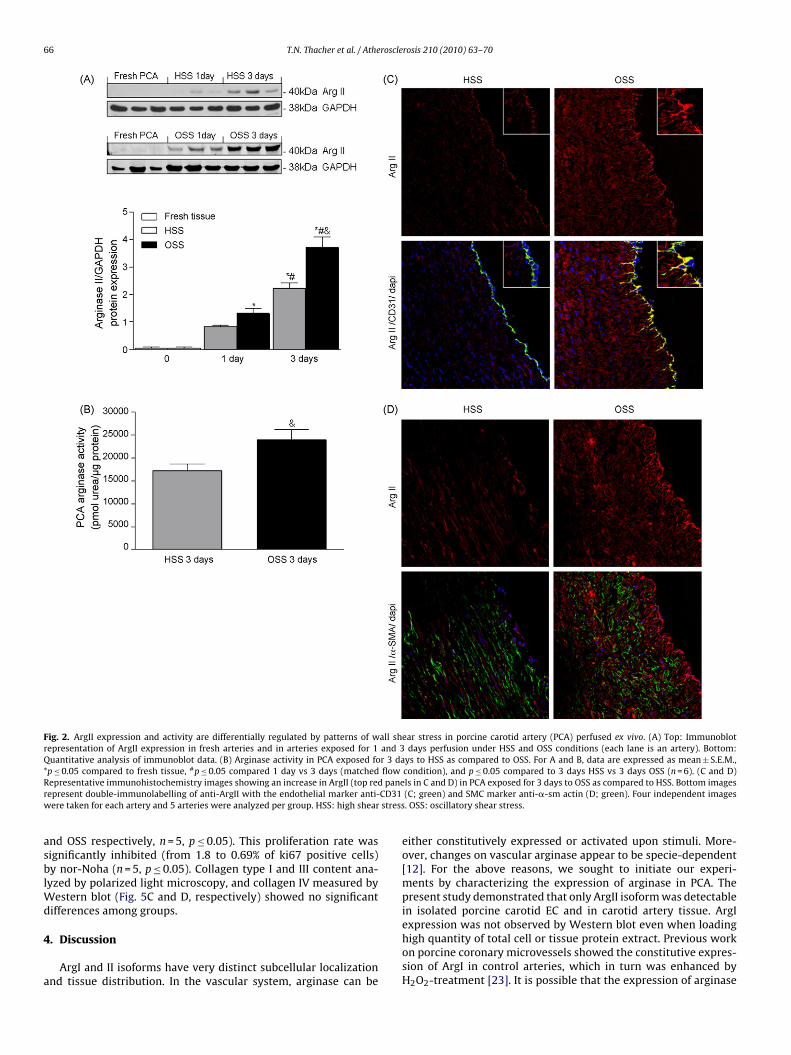
66 T.N. Thacher et al. / Atherosclerosis 210 (2010) 63–70
Fig. 2. ArgII expression and activity are differentially regulated by patterns of wall shear stress in porcine carotid artery (PCA) perfused ex vivo. (A) Top: Immunoblotrepresentation of ArgII expression in fresh arteries and in arteries exposed for 1 and 3 days perfusion under HSS and OSS conditions (each lane is an artery). Bottom:Quantitative analysis of immunoblot data. (B) Arginase activity in PCA exposed for 3 days to HSS as compared to OSS. For A and B, data are expressed as mean ± S.E.M.,* # flowR paner CD31w stress
asblWd
4
a
p ≤ 0.05 compared to fresh tissue, p ≤ 0.05 compared 1 day vs 3 days (matchedepresentative immunohistochemistry images showing an increase in ArgII (top redepresent double-immunolabelling of anti-ArgII with the endothelial marker anti-ere taken for each artery and 5 arteries were analyzed per group. HSS: high shear
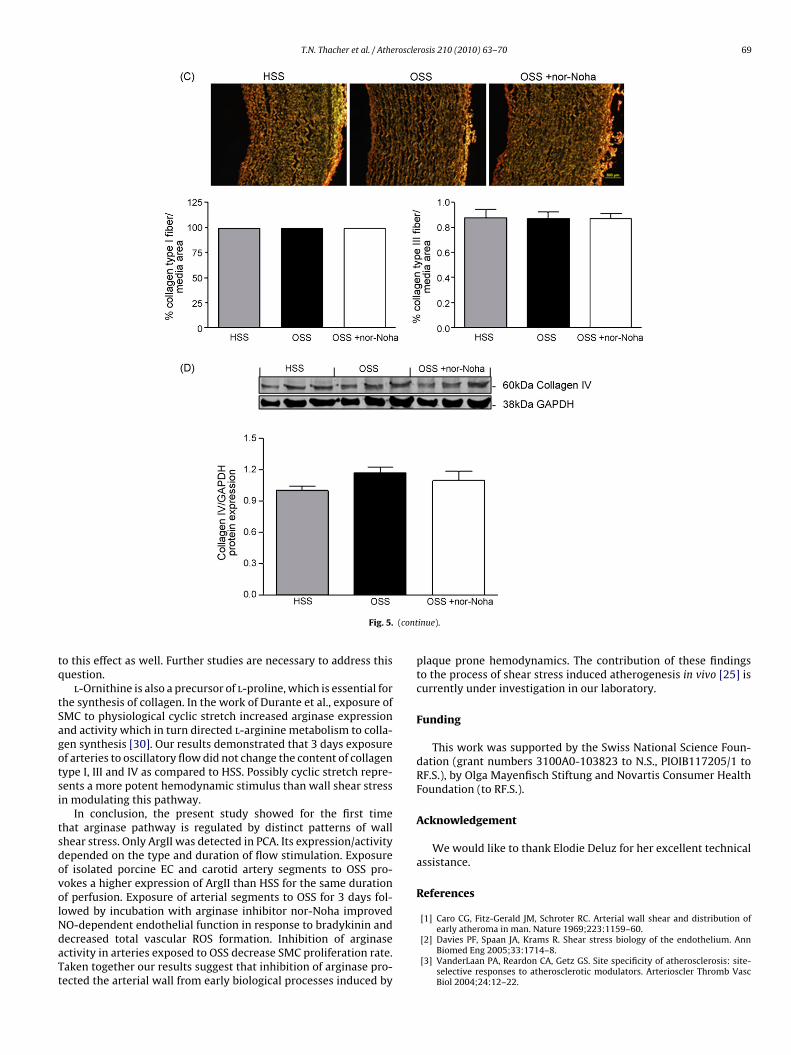
nd OSS respectively, n = 5, p ≤ 0.05). This proliferation rate wasignificantly inhibited (from 1.8 to 0.69% of ki67 positive cells)y nor-Noha (n = 5, p ≤ 0.05). Collagen type I and III content ana-
yzed by polarized light microscopy, and collagen IV measured byestern blot (Fig. 5C and D, respectively) showed no significant
ifferences among groups.
. Discussion
ArgI and II isoforms have very distinct subcellular localizationnd tissue distribution. In the vascular system, arginase can be
condition), and p ≤ 0.05 compared to 3 days HSS vs 3 days OSS (n = 6). (C and D)ls in C and D) in PCA exposed for 3 days to OSS as compared to HSS. Bottom images(C; green) and SMC marker anti-�-sm actin (D; green). Four independent images. OSS: oscillatory shear stress.
either constitutively expressed or activated upon stimuli. More-over, changes on vascular arginase appear to be specie-dependent[12]. For the above reasons, we sought to initiate our experi-ments by characterizing the expression of arginase in PCA. Thepresent study demonstrated that only ArgII isoform was detectablein isolated porcine carotid EC and in carotid artery tissue. ArgI
expression was not observed by Western blot even when loadinghigh quantity of total cell or tissue protein extract. Previous workon porcine coronary microvessels showed the constitutive expres-sion of ArgI in control arteries, which in turn was enhanced byH2O2-treatment [23]. It is possible that the expression of arginase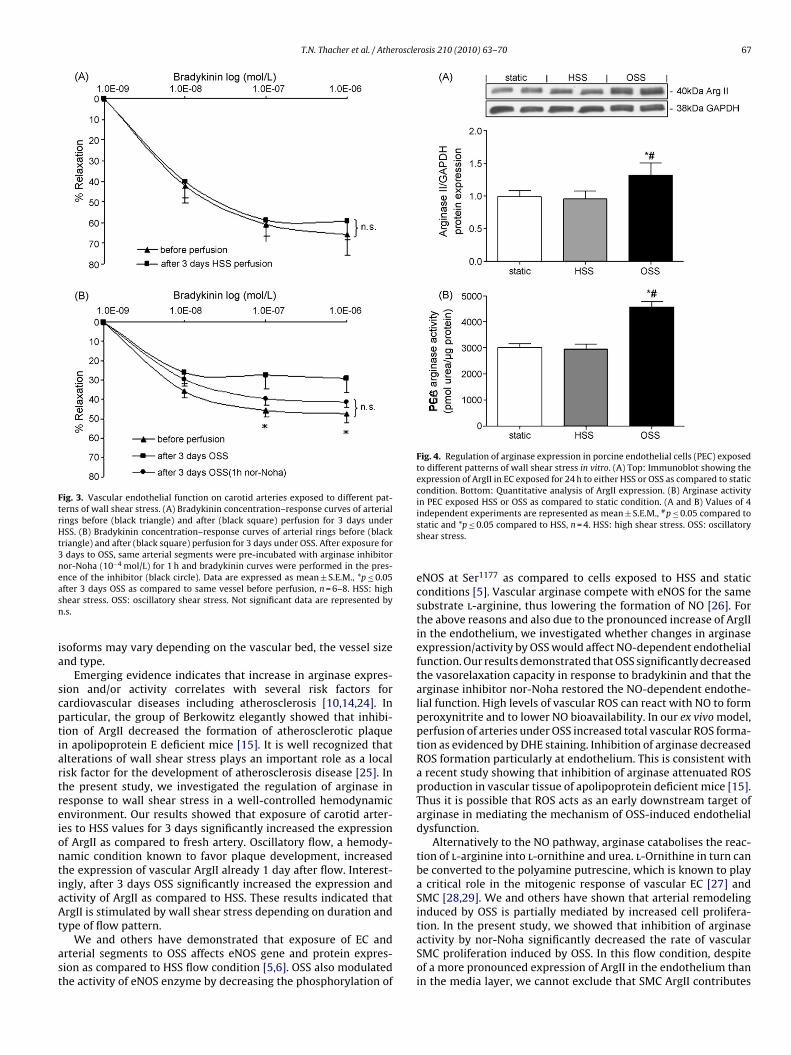
T.N. Thacher et al. / Atherosclerosis 210 (2010) 63–70 67
Fig. 3. Vascular endothelial function on carotid arteries exposed to different pat-terns of wall shear stress. (A) Bradykinin concentration–response curves of arterialrings before (black triangle) and after (black square) perfusion for 3 days underHSS. (B) Bradykinin concentration–response curves of arterial rings before (blacktriangle) and after (black square) perfusion for 3 days under OSS. After exposure for3 days to OSS, same arterial segments were pre-incubated with arginase inhibitornor-Noha (10−4 mol/L) for 1 h and bradykinin curves were performed in the pres-easn
ia
scptiartreiontiaAt
ast
Fig. 4. Regulation of arginase expression in porcine endothelial cells (PEC) exposedto different patterns of wall shear stress in vitro. (A) Top: Immunoblot showing theexpression of ArgII in EC exposed for 24 h to either HSS or OSS as compared to staticcondition. Bottom: Quantitative analysis of ArgII expression. (B) Arginase activityin PEC exposed HSS or OSS as compared to static condition. (A and B) Values of 4
nce of the inhibitor (black circle). Data are expressed as mean ± S.E.M., *p ≤ 0.05fter 3 days OSS as compared to same vessel before perfusion, n = 6–8. HSS: highhear stress. OSS: oscillatory shear stress. Not significant data are represented by.s.
soforms may vary depending on the vascular bed, the vessel sizend type.
Emerging evidence indicates that increase in arginase expres-ion and/or activity correlates with several risk factors forardiovascular diseases including atherosclerosis [10,14,24]. Inarticular, the group of Berkowitz elegantly showed that inhibi-ion of ArgII decreased the formation of atherosclerotic plaquen apolipoprotein E deficient mice [15]. It is well recognized thatlterations of wall shear stress plays an important role as a localisk factor for the development of atherosclerosis disease [25]. Inhe present study, we investigated the regulation of arginase inesponse to wall shear stress in a well-controlled hemodynamicnvironment. Our results showed that exposure of carotid arter-es to HSS values for 3 days significantly increased the expressionf ArgII as compared to fresh artery. Oscillatory flow, a hemody-amic condition known to favor plaque development, increasedhe expression of vascular ArgII already 1 day after flow. Interest-ngly, after 3 days OSS significantly increased the expression andctivity of ArgII as compared to HSS. These results indicated thatrgII is stimulated by wall shear stress depending on duration and
ype of flow pattern.
We and others have demonstrated that exposure of EC andrterial segments to OSS affects eNOS gene and protein expres-ion as compared to HSS flow condition [5,6]. OSS also modulatedhe activity of eNOS enzyme by decreasing the phosphorylation of
independent experiments are represented as mean ± S.E.M., #p ≤ 0.05 compared tostatic and *p ≤ 0.05 compared to HSS, n = 4. HSS: high shear stress. OSS: oscillatoryshear stress.
eNOS at Ser1177 as compared to cells exposed to HSS and staticconditions [5]. Vascular arginase compete with eNOS for the samesubstrate l-arginine, thus lowering the formation of NO [26]. Forthe above reasons and also due to the pronounced increase of ArgIIin the endothelium, we investigated whether changes in arginaseexpression/activity by OSS would affect NO-dependent endothelialfunction. Our results demonstrated that OSS significantly decreasedthe vasorelaxation capacity in response to bradykinin and that thearginase inhibitor nor-Noha restored the NO-dependent endothe-lial function. High levels of vascular ROS can react with NO to formperoxynitrite and to lower NO bioavailability. In our ex vivo model,perfusion of arteries under OSS increased total vascular ROS forma-tion as evidenced by DHE staining. Inhibition of arginase decreasedROS formation particularly at endothelium. This is consistent witha recent study showing that inhibition of arginase attenuated ROSproduction in vascular tissue of apolipoprotein deficient mice [15].Thus it is possible that ROS acts as an early downstream target ofarginase in mediating the mechanism of OSS-induced endothelialdysfunction.
Alternatively to the NO pathway, arginase catabolises the reac-tion of l-arginine into l-ornithine and urea. l-Ornithine in turn canbe converted to the polyamine putrescine, which is known to playa critical role in the mitogenic response of vascular EC [27] andSMC [28,29]. We and others have shown that arterial remodelinginduced by OSS is partially mediated by increased cell prolifera-tion. In the present study, we showed that inhibition of arginase
activity by nor-Noha significantly decreased the rate of vascularSMC proliferation induced by OSS. In this flow condition, despiteof a more pronounced expression of ArgII in the endothelium thanin the media layer, we cannot exclude that SMC ArgII contributes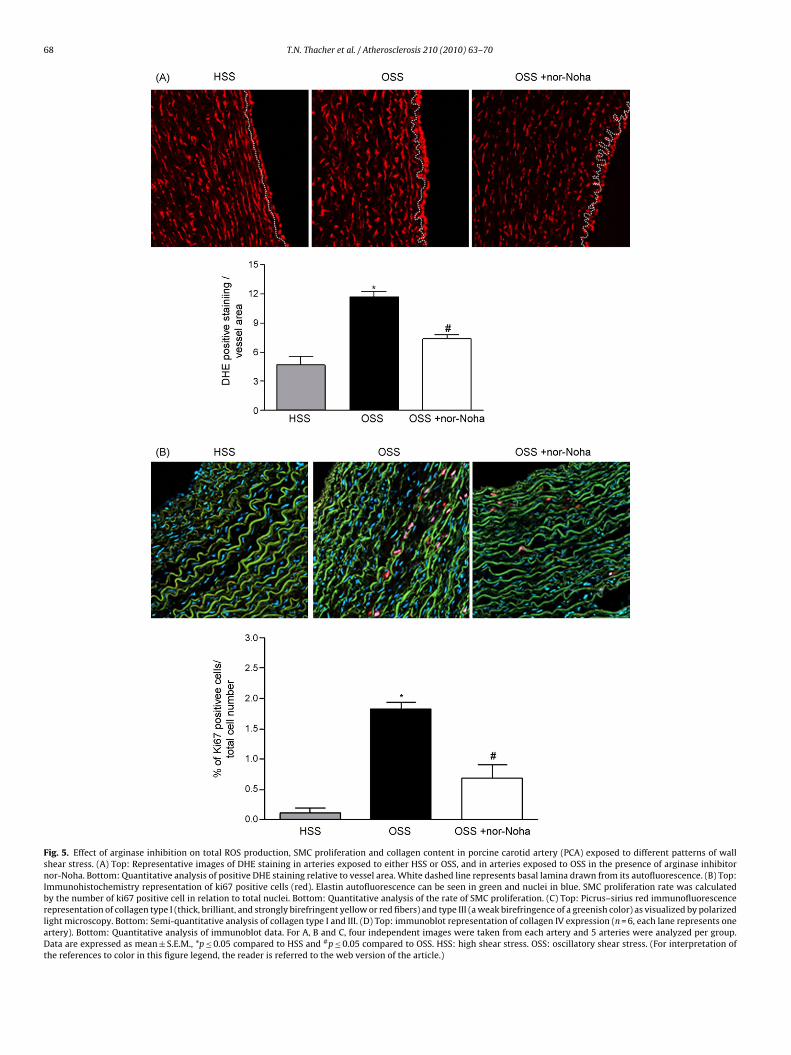
68 T.N. Thacher et al. / Atherosclerosis 210 (2010) 63–70
Fig. 5. Effect of arginase inhibition on total ROS production, SMC proliferation and collagen content in porcine carotid artery (PCA) exposed to different patterns of wallshear stress. (A) Top: Representative images of DHE staining in arteries exposed to either HSS or OSS, and in arteries exposed to OSS in the presence of arginase inhibitornor-Noha. Bottom: Quantitative analysis of positive DHE staining relative to vessel area. White dashed line represents basal lamina drawn from its autofluorescence. (B) Top:Immunohistochemistry representation of ki67 positive cells (red). Elastin autofluorescence can be seen in green and nuclei in blue. SMC proliferation rate was calculatedby the number of ki67 positive cell in relation to total nuclei. Bottom: Quantitative analysis of the rate of SMC proliferation. (C) Top: Picrus–sirius red immunofluorescencerepresentation of collagen type I (thick, brilliant, and strongly birefringent yellow or red fibers) and type III (a weak birefringence of a greenish color) as visualized by polarizedlight microscopy. Bottom: Semi-quantitative analysis of collagen type I and III. (D) Top: immunoblot representation of collagen IV expression (n = 6, each lane represents oneartery). Bottom: Quantitative analysis of immunoblot data. For A, B and C, four independent images were taken from each artery and 5 arteries were analyzed per group.Data are expressed as mean ± S.E.M., *p ≤ 0.05 compared to HSS and #p ≤ 0.05 compared to OSS. HSS: high shear stress. OSS: oscillatory shear stress. (For interpretation ofthe references to color in this figure legend, the reader is referred to the web version of the article.)
T.N. Thacher et al. / Atherosclerosis 210 (2010) 63–70 69
(cont
tq
tSagotsi
tsdovolNdaTt
Fig. 5.
o this effect as well. Further studies are necessary to address thisuestion.l-Ornithine is also a precursor of l-proline, which is essential for
he synthesis of collagen. In the work of Durante et al., exposure ofMC to physiological cyclic stretch increased arginase expressionnd activity which in turn directed l-arginine metabolism to colla-en synthesis [30]. Our results demonstrated that 3 days exposuref arteries to oscillatory flow did not change the content of collagenype I, III and IV as compared to HSS. Possibly cyclic stretch repre-ents a more potent hemodynamic stimulus than wall shear stressn modulating this pathway.
In conclusion, the present study showed for the first timehat arginase pathway is regulated by distinct patterns of wallhear stress. Only ArgII was detected in PCA. Its expression/activityepended on the type and duration of flow stimulation. Exposuref isolated porcine EC and carotid artery segments to OSS pro-okes a higher expression of ArgII than HSS for the same durationf perfusion. Exposure of arterial segments to OSS for 3 days fol-owed by incubation with arginase inhibitor nor-Noha improved
O-dependent endothelial function in response to bradykinin andecreased total vascular ROS formation. Inhibition of arginasectivity in arteries exposed to OSS decrease SMC proliferation rate.aken together our results suggest that inhibition of arginase pro-ected the arterial wall from early biological processes induced byinue).
plaque prone hemodynamics. The contribution of these findingsto the process of shear stress induced atherogenesis in vivo [25] iscurrently under investigation in our laboratory.
Funding
This work was supported by the Swiss National Science Foun-dation (grant numbers 3100A0-103823 to N.S., PIOIB117205/1 toRF.S.), by Olga Mayenfisch Stiftung and Novartis Consumer HealthFoundation (to RF.S.).
Acknowledgement
We would like to thank Elodie Deluz for her excellent technicalassistance.
References
[1] Caro CG, Fitz-Gerald JM, Schroter RC. Arterial wall shear and distribution of
early atheroma in man. Nature 1969;223:1159–60.[2] Davies PF, Spaan JA, Krams R. Shear stress biology of the endothelium. AnnBiomed Eng 2005;33:1714–8.
[3] VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb VascBiol 2004;24:12–22.

7 roscle
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
0 T.N. Thacher et al. / Athe
[4] Cheng C, van Haperen R, de Waard M, et al. Shear stress affects the intracellulardistribution of eNOS: direct demonstration by a novel in vivo technique. Blood2005;106:3691–8.
[5] Gambillara V, Chambaz C, Montorzi G, Roy S, Stergiopulos N, Silacci P. Plaque-prone hemodynamics impair endothelial function in pig carotid arteries. Am JPhysiol Heart Circ Physiol 2006;290:H2320–2328.
[6] Silacci P, Formentin K, Bouzourene K, Daniel F, Brunner HR, Hayoz D. Uni-directional and oscillatory shear stress differentially modulate NOS III geneexpression. Nitric Oxide 2000;4:47–56.
[7] Gornik HL, Creager MA. Arginine and endothelial and vascular health. J Nutr2004;134:2880S–7S [discussion 2895S].
[8] Goumas G, Tentolouris C, Tousoulis D, Stefanadis C, Toutouzas P. Therapeu-tic modification of the l-arginine–eNOS pathway in cardiovascular diseases.Atherosclerosis 2001;154:255–67.
[9] Berkowitz DE, White R, Li D, et al. Arginase reciprocally regulates nitric oxidesynthase activity and contributes to endothelial dysfunction in aging bloodvessels. Circulation 2003;108:2000–6.
10] White AR, Ryoo S, Li D, et al. Knockdown of arginase I restores NO signaling inthe vasculature of old rats. Hypertension 2006;47:245–51.
11] Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginasein microvascular endothelial cells counteracts nitric oxide-mediated vasodila-tory function. Faseb J 2001;15:1264–6.
12] Ming XF, Barandier C, Viswambharan H, et al. Thrombin stimulates humanendothelial arginase enzymatic activity via RhoA/ROCK pathway: implica-tions for atherosclerotic endothelial dysfunction. Circulation 2004;110:3708–14.
13] Satriano J. Arginine pathways and the inflammatory response: interregulationof nitric oxide and polyamines: review article. Amino Acids 2004;26:321–9.
14] Ryoo S, Lemmon CA, Soucy KG, et al. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitricoxide signaling. Circ Res 2006;99:951–60.
15] Ryoo S, Gupta G, Benjo A, et al. Endothelial arginase II: a novel target for thetreatment of atherosclerosis. Circ Res 2008;102:923–32.
16] Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxidesynthesis and vascular function. Clin Exp Pharmacol Physiol 2007;34:906–11.
17] Gambillara V, Montorzi G, Haziza-Pigeon C, Stergiopulos N, Silacci P. Arte-rial wall response to ex vivo exposure to oscillatory shear stress. J Vasc Res2005;42:535–44.
[
[
rosis 210 (2010) 63–70
18] da Silva RF, Chambaz C, Stergiopulos N, Hayoz D, Silacci P. Transcriptional andpost-transcriptional regulation of preproendothelin-1 by plaque-prone hemo-dynamics. Atherosclerosis 2007;194:383–90.
19] Thacher T, Gambillara V, Da Silva R, Montorzi G, Stergiopulos N, Silacci P. Oscil-latory shear stress and reduced compliance impair vascular functions. ClinHemorheol Microcirc 2007;37:121–30.
20] Bardy N, Karillon GJ, Merval R, Samuel JL, Tedgui A. Differential effects of pres-sure and flow on DNA and protein synthesis and on fibronectin expression byarteries in a novel organ culture system. Circ Res 1995;77:684–94.
21] Fonck E, Prod’hom G, Roy S, Augsburger L, Rufenacht DA, Stergiopulos N.Effect of elastin degradation on carotid wall mechanics as assessed by aconstituent-based biomechanical model. Am J Physiol Heart Circ Physiol2007;292:H2754–2763.
22] Corriu C, Feletou M, Puybasset L, Bea ML, Berdeaux A, Vanhoutte PM.Endothelium-dependent hyperpolarization in isolated arteries taken fromanimals treated with NO-synthase inhibitors. J Cardiovasc Pharmacol1998;32:944–50.
23] Thengchaisri N, Hein TW, Wang W, et al. Upregulation of arginase by H2O2
impairs endothelium-dependent nitric oxide-mediated dilation of coronaryarterioles. Arterioscler Thromb Vasc Biol 2006;26:2035–42.
24] Zhang C, Hein TW, Wang W, et al. Upregulation of vascular arginase inhypertension decreases nitric oxide-mediated dilation of coronary arterioles.Hypertension 2004;44:935–43.
25] Cheng C, Tempel D, van Haperen R, et al. Atherosclerotic lesion size andvulnerability are determined by patterns of fluid shear stress. Circulation2006;113:2744–53.
26] Ash DE. Structure and function of arginases. J Nutr 2004;134:2760S–4S [dis-cussion 2765S–2767S].
27] Li H, Meininger CJ, Kelly KA, Hawker Jr JR, Morris Jr SM, Wu G. Activities ofarginase I and II are limiting for endothelial cell proliferation. Am J PhysiolRegul Integr Comp Physiol 2002;282:R64–69.
28] Peyton KJ, Ensenat D, Azam MA, et al. Arginase promotes neointima formationin rat injured carotid arteries. Arterioscler Thromb Vasc Biol 2009;29:488–94.
29] Wei LH, Wu G, Morris Jr SM, Ignarro LJ. Elevated arginase I expression in rataortic smooth muscle cells increases cell proliferation. Proc Natl Acad Sci U S A2001;98:9260–4.
30] Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Physiological cyclic stretchdirects l-arginine transport and metabolism to collagen synthesis in vascularsmooth muscle. Faseb J 2000;14:1775–83.




















