![1] Material classification: There are different ways of classify](https://static.fdokumen.com/doc/165x107/6335324ed2b728420307cfa1/1-material-classification-there-are-different-ways-of-classify.jpg)
1] Material classification: There are different ways of classify
Upload
independentCategory
view
0download
0
Pergamon
0145-2126(93)E0012-K
Leukemia Research Vol 18, No 3. pp. 149 1611. 1994 Elsevier Science Ltd
Printed in Great Britain All rights reserved 0145 2126/04 $6.1X) + (),(X}
REGROWTH RESISTANCE IN LEUKEMIA AND LYMPHOMA: THE NEED FOR A NEW SYSTEM TO CLASSIFY TREATMENT FAILURE
AND FOR NEW APPROACHES TO TREATMENT
H. D. PREISLER and V. GOPAL
Division of Hematology/Oncology, Rush Cancer Institute. 1725 West Harrison Street. Chicago, IL 60612, U.S.A.
(Received 1 August 1993. Accepted 11 November 1993)
Abstract--Treatment failure in drug sensitive malignancies is a complex phenomenon resulting from both drug resistance and from the rapid regrowth of malignant cells ('regrowth resistance'). Attempts to overcome regrowth resistance during the treatment of the aggressive lymphomas by increasing the frequency of cytotoxic therapy appears to have failed. An alternative approach of significant potential would be to administer biologically active agents to directly slow the regrowth of neoplastic cells between courses of full dose cytotoxic therapy. To facilitate this approach a new system for classifying treatment failure in the leukemias and lymphomas is needed so that the extent of regrowth resistance and the effects of treatment on regrowth resistance can be directly assessed. Accordingly, a new classification system is proposed.
Key words: Leukemia, lymphoma, regrowth resistance.
Introduction
THE TREATMENT o f neoplastic diseases has long been based on the use of therapies designed to kill neo- plastic cells. Since resistance to treatment is generally assumed to be synonymous with 'classical resistance', that is the ability of neoplastic cells to survive cyto- toxic therapy, current approaches to improve treat- ment outcome have focused on increasing the intensity of cytotoxic therapy. While the logic behind this approach is unassailable, there is a second and potentially highly effective strategy for increasing the effectiveness of cytotoxic agents, the inhibition of neoplastic cell regrowth between courses of cytotoxic therapy. As illustrated in Fig. l(a), since neoplastic regrowth between courses of cytotoxic therapy can offset the effects of cytotoxic therapy, the inhibition of regrowth has the potential of significantly increas- ing the efficacy of cytotoxic therapy in those neo- plastic diseases which are drug sensitive and which have a high regrowth rate [1].
Correspondence to: Harvey D. Preisler, Director, Rush Cancer Institute, Division of Hematology/Oncology, Rush-Presbyterian-St. Luke's Medical Center, 1725 West Harrison Street, Professional Building III-Suite 855, Chicago, IL 60612, U.S.A. (Tel: (312) 563-2190; Fax: (312) 455-9635).
149
While Fig. l(a) illustrates regrowth subsequent to a course of chemotherapy, regrowth may be so rapid as to even occur between individual doses of radiation therapy [2,3]. There is reason to believe that regrowth resistance is a widespread phenomenon affecting the outcome of the treatment of head and neck cancers [4], testicular cancer [5] and breast cancer [6] as well as the neoplastic diseases which are the focus of this paper. Further, the drug sensitivities of other neoplastic diseases such as small cell car- cinoma of the lung, acute lymphocytic leukemia, and many pediatric neoplasms are sufficiently high for regrowth resistance to be considered when the clini- cal resistance of these diseases to treatment is being evaluated. Despite the fact that regrowth resistance is a common impediment to treatment, this phenom- enon has not been systematically studied either at the biological or at the clinical level.
To develop the means to overcome regrowth resist- ance one must first be able to define the extent of the problem in different neoplastic diseases and identify those patients in whom regrowth resistance contributes to treatment failure. Acute and chronic myelogenous leukemia and the aggressive lym- phomas will be used to illustrate the problem posed by regrowth resistance and to explore possible approaches to reduce this type of resistance.
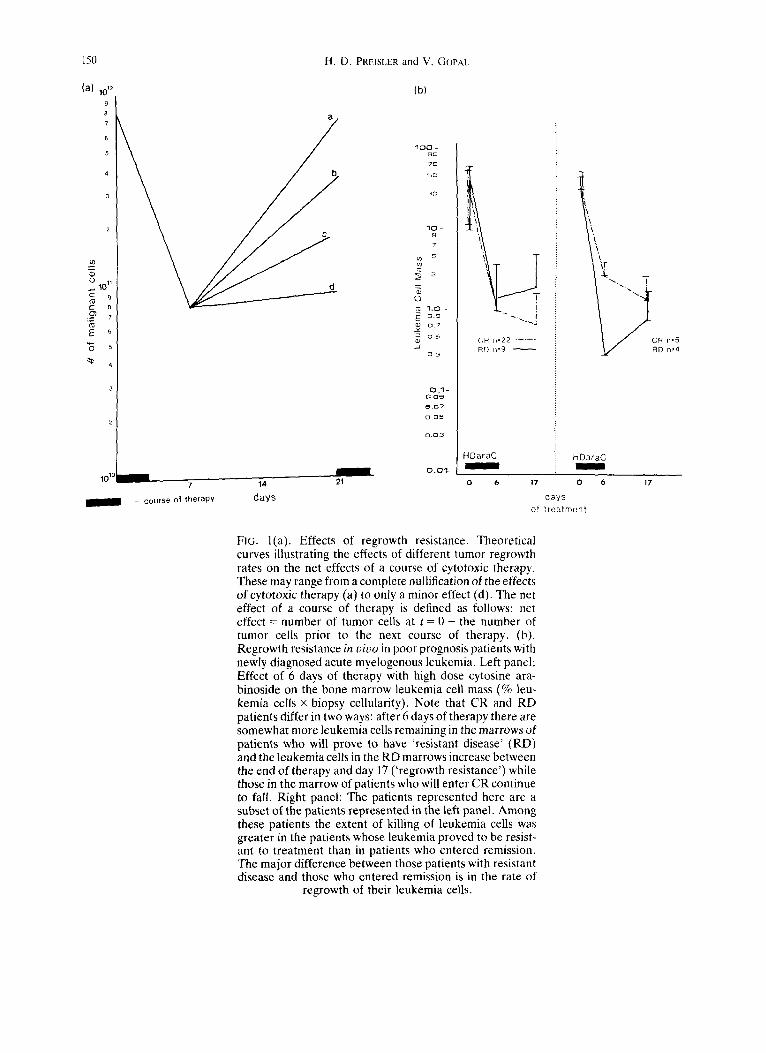
150 H.D. PREISLER and V, GOPAI.
(a) 1012 9 8 7
6
5
1011
c 8 o) • - 7
E
a
b
C
m 101° ~ 7 14 21
= course o | t h e r a p y days
(b)
100 ~o 7(3
%{3
1 0 - B 7 S
~ o 7
~) o 5 _/
o 3
0 1 0.09
0.05
O.O1
CR n=22 . . . . . RD n=9
HDaraC
~ C R n,5 RD n,4
HDaraC
o 6 17 0
days o[ t reatment
6 17
FIG. l(a). Effects of regrowth resistance. Theoretical curves illustrating the effects of different tumor regrowth rates on the net effects of a course of cytotoxic therapy. These may range from a complete nullification of the effects of cytotoxic therapy (a) to only a minor effect (d). The net effect of a course of therapy is defined as follows: net effect = number of tumor cells at t = 0 - the number of tumor cells prior to the next course of therapy. (b). Regrowth resistance in vivo in poor prognosis patients with newly diagnosed acute myelogenous leukemia. Left panel: Effect of 6 days of therapy with high dose cytosine ara- binoside on the bone marrow leukemia cell mass (% leu- kemia ceils x biopsy cellularity). Note that CR and RD patients differ in two ways: after 6 days of therapy there are somewhat more leukemia cells remaining in the marrows of patients who will prove to have 'resistant disease' (RD) and the leukemia cells in the RD marrows increase between the end of therapy and day 17 ('regrowth resistance') while those in the marrow of patients who will enter CR continue to fall. Right panel: The patients represented here are a subset of the patients represented in the left panel. Among these patients the extent of killing of leukemia cells was greater in the patients whose leukemia proved to be resist- ant to treatment than in patients who entered remission, The major difference between those patients with resistant disease and those who entered remission is in the rate of
regrowth of their leukemia cells.

Classification of treatment failure 151
C l i n i c a l
/ signif icant drug++
resi stance
Resi s tance
+ + +
presumptive regrowth
resistance
to T r e a t m e n t .
2 patient expires before a determination can be made as
to whether the patient wi l l
enter complete remission or
wi l l prove to have "resistant"
disease.
FIG. 2. Classification of treatment failure in the acute leukemias and aggressive lymphomas.
+Clinical resistance to a treatment regimen is present when the treatment regimen fails to produce the desired result, in this case a complete remission, regardless of the reason for treatment failure.
++Significant drug resistance--the therapeutic regimen fails to produce a significant reduction in the extent of leukemic infiltration in a marrow or a reduction in the size of lymphomatous masses.
+++Presumptive regrowth resistance--the therapeutic regimen produces a significant reduction in leukemia cells or in the size of lymphomatous masses but the leukemia cells begin to reappear or the size of the lymphomatous masses begins to increase prior to the next course of ther-
apy.
The leukemias
Classification of treatment failure
The system currently being used to classify treat- ment failure in the leukemias provides the means to identify patients in whom there is a high likelihood of regrowth resistance. As originally described, the system identified two types of documented drug resistance: absolute or significant drug resistance and relative drug resistance [7, 8]. These categories differ in that in the former, cytotoxic chemotherapy fails to produce a significant reduction in marrow cellu- larity. In the latter, severe marrow hypocellularity is produced but leukemia cells rather than normal cells repopulate the marrow. As far as one can measure, the effects of remission induction therapy on the bone marrows of many patients in the relative drug resistance category are similar to the effects on the marrows of patients who enter remission. Regrowth resistance makes a major contribution to remission induction failure in patients with relative drug resist- ance. This has proved to be the case in as many as 30-50% of patients with poor prognosis acute myelogenous leukemia [9, 10]. Figure l(b) illustrates this problem.
In current practice the two categories of docu-
mented drug resistance have been merged into a single category of resistant disease. To help define the contribution of regrowth resistance to remission induction failure in acute leukemia, treatment fail- ures should once again be subdivided into 'significant' drug resistance and 'relative' drug resistance as in the original classification system [7, 8]. However, to focus attention on the problem of regrowth resist- ance, we suggest that the term 'relative drug resist- ance' should be replaced by 'presumptive regrowth resistance' since in these patients leukemia cells reappeared after severe marrow hypocellularity was produced. A proposed scheme for this classification system is provided in Fig. 2. The identification of patients in whom regrowth resistance is a likely prob- lem will facilitate both the study of this problem as well as the identification of patients for whom the inhibition of regrowth resistance should be a thera- peutic goal.
Proliferatioe rates and regrowth resistance in acute myelogenous leukemia
The relationship between the proliferative charac- teristics of leukemia cells, the course of AML, and treatment outcome has long been a focus of clinical investigation. Until recently the relationships have

152 H.D. PREISLER and V. GOPAL
been equivocal with some studies pointing to a relationship between proliferative characteristics and treatment outcome [14, 15[ and others finding no relationship [16, 17]. The underlying reasons for the conflicting and confusing information are related to the methods used and the designs of the studies.
The fundamental problem with these studies is the fact that the vast majority utilized either the in vitro labeling or flow cytometric analysis of marrow aspir- ates or peripheral blood cells [14-22] to assess the proliferative characteristics of these cell populations. The problems associated with the use of marrow aspirates or peripheral blood cells are two-fold: (1) the peripheral blood is not the site of significant proliferation of leukemia cells; and (2) marrow aspir- ates are invariably diluted with peripheral blood cells [18, 20, 22]. Attempts to overcome this problem by using spicules derived from marrow aspirates or by crushing marrow biopsies can only partially correct this problem [18, 20-22]. Additionally, labeling cells or performing DNA histogram analysis can, at best, provide only an estimate of the percent cells syn- thesizing DNA (the labeling index or Li). These methods cannot provide an estimate of the most important cell cycle measurement, the cell cycle time (L).
Conceptual problems also abound in the studies described above. Almost all were designed to deter- mine if there was a relationship between the Li and remission induction outcome. Until recently these studies did not distinguish between treatment failure due to patient death and treatment failure resulting from drug resistant disease [7, 8]. Hence any relation- ship between the latter and the Li would have been masked by the noise in the system resulting from the inclusion of patients whose treatment failure was not the result of resistant disease [23, 24]. A second problem in study design is that many studies attempted to relate the Li to treatment outcome when S-phase-specific agents were not used or were not administered alone [14-21]. Clearly, such studies asked more from this proliferative measurement than was reasonable.
A recent study, not hampered by the above design problems, explored the relationship between the kill- ing of leukemia cells in vivo by single agent high dose cytosine arabinoside (araC) therapy and the proliferative characteristics of AML cells [10]. This study demonstrated that proliferative characteristics do play a role in determining the sensitivity of leu- kemia cells to this cytotoxic agent, but only in those leukemias which are metabolically sensitive to araC. These studies demonstrated that in and of themselves the proliferative characteristics of leukemia cells do not determine the extent of killing even by S-phase-
specific cytotoxic agents. Hence the rate at which leukemia cells are multiplying is not a primary deter- minant of sensitivity to cytotoxic agents but rather it plays a permissive role under appropriate cir- cumstances.
The first large study of the relationship between the pre-therapy proliferative rate of leukemia cells and treatment outcome yielded somewhat unex- pected results: only a weak relationship between remission induction outcome and the cell cycle time [25] but a strong relationship between remission dur- ation and cell cycle time [26]. Long remission dur- ations were most common in patients whose leukemia cells were cycling slowly. It appears likely that this relationship is reflective of the slow regrowth rate of leukemia cells between courses of therapy and also once therapy has concluded [26]. The short remissions of patients with rapidly proliferating disease appear to be due to regrowth resistance. Similar relationships between proliferative charac- teristics and treatment outcome have been reported for the lymphomas [27], head and neck cancers [28] and breast cancer [29, 30]. There is suggested evi- dence that this is true for pediatric acute lymphocytic leukemia as well [31].
Chronic myelogenous leukemia (CML)
The chronic and blastic phases of CML are usually viewed as being resistant to cytotoxic therapy. This conclusion is based upon the inability of cytotoxic therapy to suppress the chronic phase of the disease [32, 33] and by the low response rate of myeloid blastic crisis to this type of therapy [34, 35]. As pointed out elsewhere, the conclusion that CML is resistant to cytotoxic therapy is simultaneously cor- rect and inaccurate. When evaluating the efficacy of cytotoxic therapy with respect to the chronic phase of the disease, it is correct to conclude that at the clinical level the disease is resistant to such an approach. However, this does not necessarily mean that CML cells are resistant to the cytotoxic effects of chemotherapy. The basis for this statement is the fact that the trials of cytotoxic therapy referenced above did produce reversions to at least partially normal hematopoiesis in a fair proportion of patients [32, 33]. However, these responses were incomplete and transient in nature leading to the conclusion that the disease is 'drug resistant'.
When viewed from this perspective it appears that the disease is 'sensitive' to cytotoxic therapy since if it were resistant in the 'classical' sense even transient responses would not have been produced. Responses can occur only if the CML cells are, in fact, more sensitive than normal hematopoietic cells to cytotoxic therapy. It is likely that the responses were incom-

Classification of treatment failure 153
plete and transient because of the proliferative advantage which CML cells enjoy over their normal counterparts [36]. This advantage permitted the CML cells to rapidly return to the marrow sup- pressing and replacing the normal marrow elements thereby abrogating the effects produced by cytotoxic therapy.
Support for this explanation can be found in recent observations that in chronic phase CML, subsequent to cytotoxic therapy, Phi-negative cells appear in the peripheral blood soon after the conclusion of such therapy but by the time the white blood cell count reaches 4000-5000/~tl the Phi-negative cells have been replaced by PhI-positive cells [17, 38]. The most cogent interpretation of these observations is that cytotoxic therapy selectively killed PhI-positive cells permitting the reappearance of Phi-negative cells. Unfortunately, the Phi-positive cells reappeared before a normal cell count could be achieved because of their growth advantage. The observation that interferon requires 1-1.5 years to induce cytogenetic responses suggests that this agent probably sup- presses the proliferative advantage of CML cells per- mitting normal hematopoietic elements to slowly replace the CML elements [36]. Hence the adminis- tration of this agent subsequent to courses of cyto- toxic therapy has the potential for increasing the efficacy of cytotoxic agents by reducing regrowth resistance.
Similarly, while the leukemia cells present in myeloid blastic crisis are certainly relatively drug resistant, this characteristic alone cannot account for the difficulty in treating this disease. Clinical experience has demonstrated that while it is possible to produce the same degree of marrow hypoplasia in blastic crisis as is produced in AML the remission rate is far lower in blastic crisis [34, 35]. The frequently observed rapid repopulation of the marrow by blastic phase cells subsequent to intensive cytotoxic therapy strongly suggests that regrowth resistance makes a significant contribution to the refractory nature of this disease.
These data suggest that cytotoxic agents could play a more significant role in the treatment of all phases of CML if a means were developed to reduce the regrowth of the CML cells between courses of cyto- toxic therapy. Such an effect, though not the reason for the study design, may account for the apparent effectiveness of the combination of low dose cytosine arabinoside/o~-interferon in chronic phase CML [39].
Regrowth resistance in the lymphomas
Clinical observations and the classification of treat- mentfailure. Clinicians often state that the high pro- liferative rates of the aggressive lymphomas present
an obstacle to effective treatment because they are associated with the regrowth of the lymphomas between courses of cytotoxic therapy [3, 11]. These statements are based on observations that lym- phomas may respond early in treatment only to have the tumor regrow before completion of the treatment regimen [12, 13]. Despite these statements the actual extent of regrowth resistance in this disease is unknown since tumor reappearance prior to the con- clusion of an entire treatment regimen could also result from the appearance of drug resistant neo- plastic cells. Additionally, the relationship of this type of treatment failure to the proliferative rate of the lymphoma is unknown since studies relating these phenomena have not been performed.
To develop information regarding the contribution which regrowth resistance makes to remission induc- tion failure in the lymphomas, a system to classify treatment failures similar to that used for the acute leukemias could be employed (Fig. 2). To this end, measurement of tumor masses should be performed midway between the courses of therapy as well as prior to the administration of each course of cytotoxic therapy as is usually done. In this way the sensitivity of the lymphoma to the cytotoxic agents being admin- istered can be quantitated together with the extent of regrowth of the lymphoma prior to the next course of cytotoxic therapy.
The use of this system will increase our under- standing of the biology of the response of the lym- phomas to cytotoxic therapy and will also help to distinguish between two categories of patients: those who might benefit from more intensive cytotoxic therapy and those for whom attempts to reduce tumor regrowth between courses of therapy would be a more appropriate approach to improving treatment outcome. This information is not currently available because treatment response is evaluated and quan- titated only immediately prior to the administration of the next course of therapy.
Third generation regimens vs CHOP. As noted, most hematologists and medical oncoiogists are aware of the problem of regrowth resistance in the lymphomas, although they may not think of it in the same manner in which it is being discussed here. In fact, the third generation lymphoma treatment protocols were designed to administer as many courses of therapy in as short a time as possible at least in part so as to reduce the extent of lymphoma regrowth between courses of chemotherapy [40-43]. Unfortunately, a recently completed large ran- domized intergroup trial comparing three third gen- eration protocols with the standard first generation CHOP regimen failed to demonstrate that any of these new regimens were superior to CHOP [44].
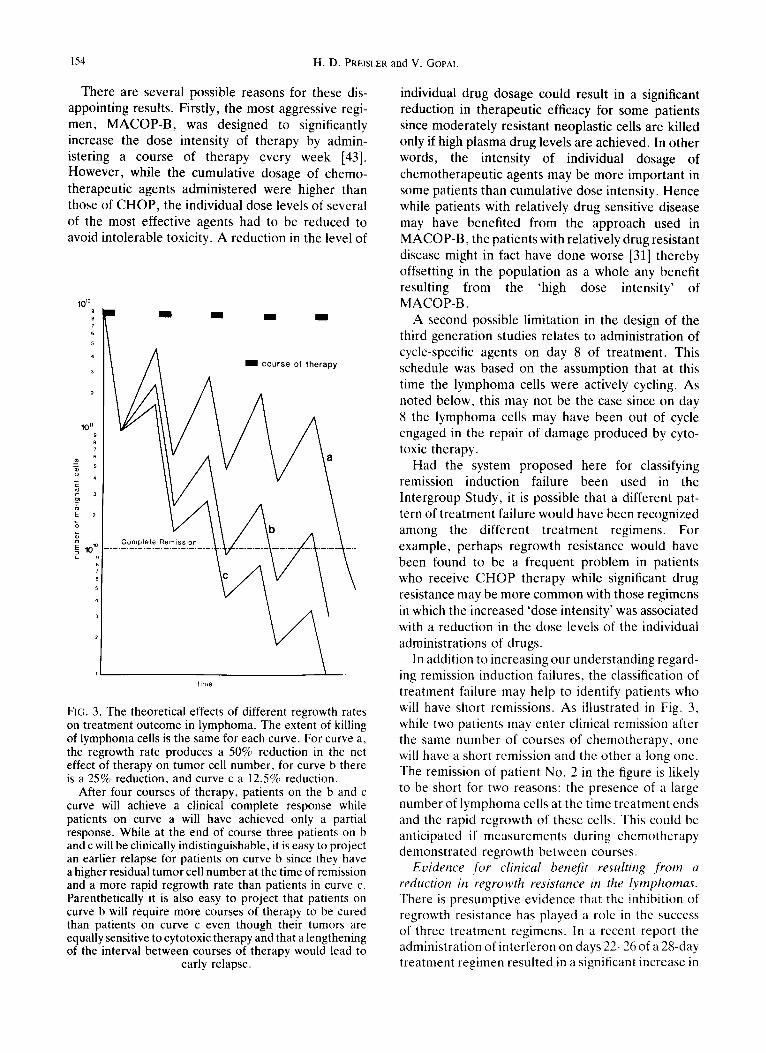
154 H . D . PREISLER and V. GOPAL
There are several possible reasons for these dis- appointing results. Firstly, the most aggressive regi- men, MACOP-B, was designed to significantly increase the dose intensity of therapy by admin- istering a course of therapy every week [43]. However, while the cumulative dosage of chemo- therapeutic agents administered were higher than those of CHOP, the individual dose levels of several of the most effective agents had to be reduced to avoid intolerable toxicity. A reduction in the level of
1012
101
m
E m
-&
f i
"6
E= 1~ E
t i m e
FIG. 3. The theoretical effects of different regrowth rates on treatment outcome in lymphoma. The extent of killing of lymphoma cells is the same for each curve. For curve a, the regrowth rate produces a 50% reduction in the net effect of therapy on tumor cell number, for curve b there is a 25% reduction, and curve c a 12.5% reduction.
After four courses of therapy, patients on the b and c curve will achieve a clinical complete response while patients on curve a will have achieved only a partial response. While at the end of course three patients on b and c will be clinically indistinguishable, it is easy to project an earlier relapse for patients on curve b since they have a higher residual tumor cell number at the time of remission and a more rapid regrowth rate than patients in curve c. Parenthetically it is also easy to project that patients on curve b will require more courses of therapy to be cured than patients on curve c even though their tumors are equally sensitive to cytotoxic therapy and that a lengthening of the interval between courses of therapy would lead to
early relapse.
individual drug dosage could result in a significant reduction in therapeutic efficacy for some patients since moderately resistant neoplastic cells are killed only if high plasma drug levels are achieved. In other words, the intensity of individual dosage of chemotherapeutic agents may be more important in some patients than cumulative dose intensity. Hence while patients with relatively drug sensitive disease may have benefited f rom the approach used in MACOP-B, the patients with relatively drug resistant disease might in fact have done worse [31] thereby offsetting in the population as a whole any benefit resulting from the 'high dose intensity' of MACOP-B.
A second possible limitation in the design of the third generation studies relates to administration of cycle-specific agents on day 8 of treatment. This schedule was based on the assumption that at this time the lymphoma cells were actively cycling. As noted below, this may not be the case since on day 8 the lymphoma cells may have been out of cycle engaged in the repair of damage produced by cyto- toxic therapy.
Had the system proposed here for classifying remission induction failure been used in the Intergroup Study, it is possible that a different pat- tern of t reatment failure would have been recognized among the different t reatment regimens. For example, perhaps regrowth resistance would have been found to be a frequent problem in patients who receive C H O P therapy while significant drug resistance may be more common with those regimens in which the increased 'dose intensity' was associated with a reduction in the dose levels of the individual administrations of drugs.
In addition to increasing our understanding regard- ing remission induction failures, the classification of treatment failure may help to identify patients who will have short remissions. As illustrated in Fig. 3, while two patients may enter clinical remission after the same number of courses of chemotherapy, one will have a short remission and the other a long one. The remission of patient No. 2 in the figure is likely to be short for two reasons: the presence of a large number of lymphoma cells at the time treatment ends and the rapid regrowth of these cells. This could be anticipated if measurements during chemotherapy demonstrated regrowth between courses.
Evidence +for clinical benefit resulting .from a reduction in regrowth resistance in the lymphomas. There is presumptive evidence that the inhibition of regrowth resistance has played a role in the success of three t reatment regimens. In a recent report the administration of interferon on days 22-26 of a 28-day treatment regimen resulted in a significant increase in
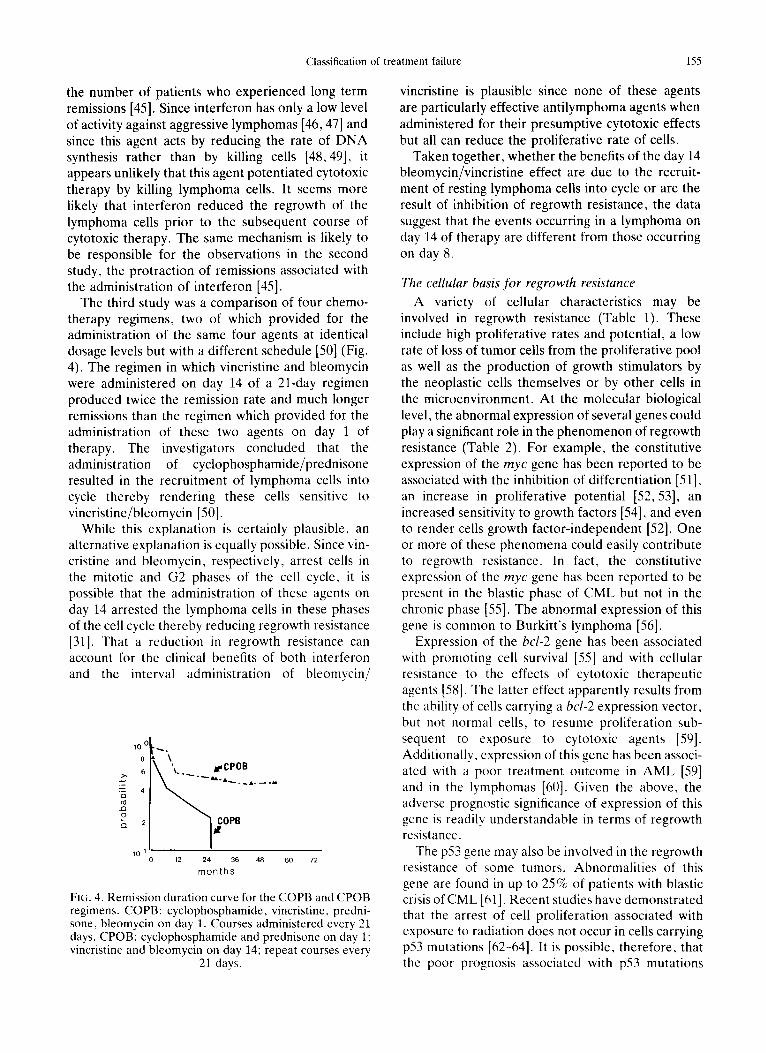
Classification of t reatment failure 155
the number of patients who experienced long term remissions [45]. Since interferon has only a low level of activity against aggressive lymphomas [46, 47] and since this agent acts by reducing the rate of DNA synthesis rather than by killing cells [48,49], it appears unlikely that this agent potentiated cytotoxic therapy by killing lymphoma cells. It seems more likely that interferon reduced the regrowth of the lymphoma cells prior to the subsequent course of cytotoxic therapy. The same mechanism is likely to be responsible for the observations in the second study, the protraction of remissions associated with the administration of interferon [45].
The third study was a comparison of four chemo- therapy regimens, two of which provided for the administration of the same four agents at identical dosage levels but with a different schedule [50] (Fig. 4). The regimen in which vincristine and bleomycin were administered on day 14 of a 21-day regimen produced twice the remission rate and much longer remissions than the regimen which provided for the administration of these two agents on day 1 of therapy. The investigators concluded that the administration of cyclophosphamide/prednisone resulted in the recruitment of lymphoma cells into cycle thereby rendering these cells sensitive to vincristine/bleomycin [50].
While this explanation is certainly plausible, an alternative explanation is equally possible. Since vin- cristine and bleomycin, respectively, arrest cells in the mitotic and G2 phases of the cell cycle, it is possible that the administration of these agents on day 14 arrested the lymphoma cells in these phases of the cell cycle thereby reducing regrowth resistance [31]. That a reduction in regrowth resistance can account for the clinical benefits of both interferon and the interval administration of bleomycin/
10 0
8
6
25 4 r~ c~
o
o . 2
10 1
~,A'\'~.. fCPOB
~ OPB
12 24 36 48 60 72
months
FIG. 4. Remission duration curve for the COPB and CPOB regimens. COPB: cyclophosphamide, vincristine, predni- sone, bleomycin on day 1. Courses administered every 21 days. CPOB: cyclophosphamide and prednisone on day 1 ; vincristine and bleomycin on day 14; repeat courses every
21 days.
vincristine is plausible since none of these agents are particularly effective antilymphoma agents when administered for their presumptive cytotoxic effects but all can reduce the proliferative rate of cells.
Taken together, whether the benefits of the day 14 bleomycin/vincristine effect are due to the recruit- ment of resting lymphoma cells into cycle or are the result of inhibition of regrowth resistance, the data suggest that the events occurring in a lymphoma on day 14 of therapy are different from those occurring on day 8.
The cellular basis for regrowth resistance
A variety of cellular characteristics may be involved in regrowth resistance (Table 1). These include high proliferative rates and potential, a low rate of loss of tumor cells from the proliferative pool as well as the production of growth stimulators by the neoplastic cells themselves or by other cells in the microenvironment. At the molecular biological level, the abnormal expression of several genes could play a significant role in the phenomenon of regrowth resistance (Table 2). For example, the constitutive expression of the myc gene has been reported to be associated with the inhibition of differentiation [51], an increase in proliferative potential [52, 53], an increased sensitivity to growth factors [54], and even to render cells growth factor-independent [52]. One or more of these phenomena could easily contribute to regrowth resistance. In fact, the constitutive expression of the myc gene has been reported to be present in the blastic phase of CML but not in the chronic phase I55]. The abnormal expression of this gene is common to Burkitt's lymphoma [56].
Expression of the bcl-2 gene has been associated with promoting cell survival [55] and with cellular resistance to the effects of cytotoxic therapeutic agents [58]. The latter effect apparently results from the ability of cells carrying a bcl-2 expression vector, but not normal cells, to resume proliferation sub- sequent to exposure to cytotoxic agents [59]. Additionally, expression of this gene has been associ- ated with a poor treatment outcome in AML [59] and in the lymphomas [60]. Given the above, the adverse prognostic significance of expression of this gene is readily understandable in terms of regrowth resistance.
The p53 gene may also be involved in the regrowth resistance of some tumors. Abnormalities of this gene are found in up to 25% of patients with blastic crisis of CML [61]. Recent studies have demonstrated that the arrest of cell proliferation associated with exposure to radiation does not occur in cells carrying p53 mutations [62-64]. It is possible, therefore, that the poor prognosis associated with p53 mutations

156 H. D. PREISLER and V. GOPAL
TABLE l . CELLULAR CHARACTERISTICS WHICH PLAY A ROLE IN DETER- MINING THE EXTENT OF REGROWTH RESISTANCE
No. Characteristic
1 Number of malignant cells which survive the prior course of therapy. The proliferative rate of the malignant cells. Proportion of malignant cells in the proliferative pool. Rate of cell loss from the proliferative pool. Sensitivity to growth stimulators and inhibitors. Production of cytokines by the malignant cell population or by cells in the microenvironment.
TABLE 2. GENES WHICH MAY INFLUENCE REGROWTII RATE
Gene Effect
myc Expression is associated with cell proliferation and with the failure of cells to differentiate. The myc gene can also play a role in the immortalization of cells. Can prevent apoptosis when cells are deprived of necessary growth factors, can confer resistance to cytotoxic agents, can cooperate with c-myc to immortalize cells. Normal expression is necessary for the pause in cell proliferation subsequent to DNA damage. Cells bearing mutant genes continue to proliferate despite exposure to radiation.
bcl-2
p53
[65-68] is a consequence of the ability of tumor cells bearing mutant p53 genes to actively proliferate during cytotoxic therapy thereby replacing the cells being killed.
Phase II Regimens For Treating Aggressive Lymphomas
cyc lophosphamide rn i toxant rone V M - 2 6 bleomycin prednisone [d l -5] [araC] v inc r is t ine
I 1 14 15
. - o ( IFN ,--]
repeat cycle
22 26
FIG. 5. Two regimens designed to inhibit the regrowth of lymphoma cells between courses of cytotoxic therapy. The regimen is the same for newly diagnosed and relapsed patients except that the cytosine arabinoside (araC) is administered only for the latter patients. The regimen for newly diagnosed patients has been designed to evaluate, in a pilot study, the combined effects of two approaches designed to reduce growth resistance. The regimen for relapsed disease evaluates, in pilot fashion, the effect of adding an S-phase specific agent at a time when accelerated proliferation and/or recruitment of lymphoma cells into
cycle is likely to occur.
Strategies to overcome regrowth resistance
The current strategy being employed to reduce regrowth resistance is to administer cytotoxic therapy as frequently as possible. This strategy has been employed without significant benefit in the lym- phomas [44] and has recently been recommended for the treatment of carcinoma of the breast [6]. As noted above, the necessary reduction in individual dose levels of cytotoxic agents is a serious potential problem with this approach. A more effective approach would be to administer maximal individual dosages of cytotoxic therapy and then to reduce the regrowth of the tumor cells between courses of cytotoxic therapy by administering noncytotoxic agents.
Studies in acute myelogenous leukemia have demonstrated that the administration of 13-cis reti- noic acid and or-interferon can slow the proliferation of AML cells in vivo in patients [69] and can reduce the level of myc and myb expression [70, 71]. Initial studies in chronic myelogenous leukemia have demonstrated that these two agents can alter the behavior of these myeloid leukemia cells in vivo in patients as well (Table 3). Hence, in AML and CML, a reasonable approach would be to test the prop-
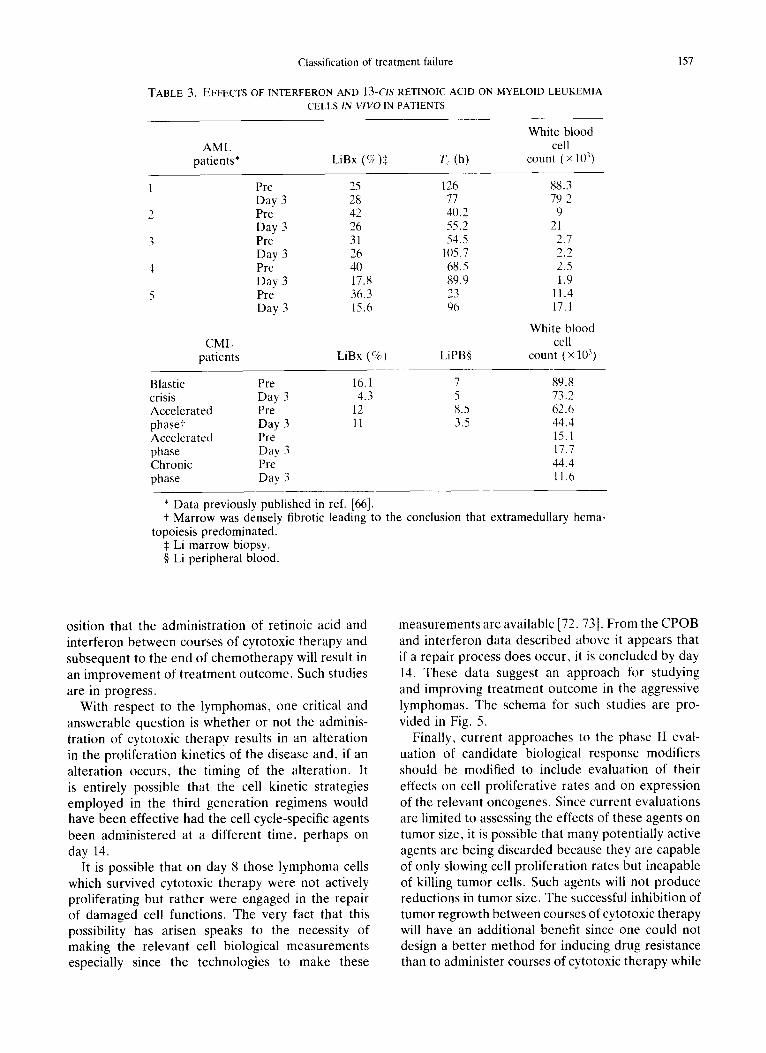
Classification of treatment failure
TABLE 3. EFFECTS OF INTERFERON AND 13-CIS RETINOIC ACID ON MYELOID LEUKEMIA CELLS IN VIVO IN PATIENTS
White blood AML cell
patients* LiBx (%)$ T c (h) count (x 10 ))
CML patients
Pre 25 126 88.3 Day 3 28 77 79.2 Pre 42 4//.2 9 Day 3 26 55.2 21 Pre 31 54.5 2.7 Day 3 26 1/)5.7 2.2 Pre 40 68.5 2.5 Day 3 17.8 89.9 1.9 Pre 36.3 23 11.4 Day 3 15.6 96 17.1
White blood cell
LiBx (%) LiPB§ count (× 1113)
Blastic Pre 16.1 7 89.8 crisis Day 3 4.3 5 73.2 Accelerated Pre 12 8.5 62.6 phase+ Day 3 11 3.5 44.4 Accelerated Pre 15.1 phase Day 3 17.7 Chronic Pre 44.4 phase Day 3 11.6
* Data previously published in ref. [66]. + Marrow was densely fibrotic leading to the conclusion that extramedullary
topoiesis predominated. $ Li marrow biopsy. § Li peripheral blood.
hema-
157
osition that the administration of retinoic acid and interferon between courses of cytotoxic therapy and subsequent to the end of chemotherapy will result in an improvement of treatment outcome. Such studies are in progress.
With respect to the lymphomas, one critical and answerable question is whether or not the adminis- tration of cytotoxic therapy results in an alteration in the proliferation kinetics of the disease and, if an alteration occurs, the timing of the alteration. It is entirely possible that the cell kinetic strategies employed in the third generation regimens would have been effective had the cell cycle-specific agents been administered at a different time, perhaps on day 14.
It is possible that on day 8 those lymphoma cells which survived cytotoxic therapy were not actively proliferating but rather were engaged in the repair of damaged cell functions. The very fact that this possibility has arisen speaks to the necessity of making the relevant cell biological measurements especially since the technologies to make these
measurements are available [72, 73]. From the CPOB and interferon data described above it appears that if a repair process does occur, it is concluded by day 14. These data suggest an approach for studying and improving treatment outcome in the aggressive lymphomas. The schema for such studies are pro- vided in Fig. 5.
Finally, current approaches to the phase II eval- uation of candidate biological response modifiers should be modified to include evaluation of their effects on cell proliferative rates and on expression of the relevant oncogenes. Since current evaluations are limited to assessing the effects of these agents on tumor size, it is possible that many potentially active agents are being discarded because they are capable of only slowing cell proliferation rates but incapable of killing tumor cells. Such agents will not produce reductions in tumor size. The successful inhibition of tumor regrowth between courses of cytotoxic therapy will have an additional benefit since one could not design a better method for inducing drug resistance than to administer courses of cytotoxic therapy while

158 H.D. PREISLER and V. GOPAL
permitt ing the regrowth of the neoplastic cells between each course. The inhibition of tumor regrowth should reduce the rate of appearance of drug resistant cells.
Acknowledgement - -This work was supported by the National Cancer Institute grant Nos CA60085 and CA60086.
References
1. Preisler H. D. & Raza A. (1993) Regrowth resistance, a frequent but neglected cause of treatment failure. Contemp. Oncol. 3, 12-23.
2. Trott K. R. & Kummermehr J. (1985) What is known about tumor proliferative rates to choose between accelerated fractionation or hyperfractionation? Radiother. Oncol. 3, 1-9.
3. Yahalom J., Gulati S. C., Toia M. et al. (1993) Accel- erated hyperfractionated, total-lymphoid irradiation, high-dose chemotherapy, and autologous marrow transplantation for refractory and relapsing patients with Hodgkin's disease. J. clin. Oncol. 11, 1062-1071.
4. Begg A. C., Hofland I., Van Globekke G. etal. (1992) Predictive value of potential doubling time for radio- therapy of head and neck tumor patients: results from the EORTC cooperative trial 22851. Semin. Rad. Oncol. 2, 22-26.
5. Bajorin D. F., Sarosdy M. F., Pfister D. G. etal. (1993) Randomized trial of etoposide and cisplatin versus etoposide and carboplatin in patients with good-risk germ cell tumors. J. clin. Oncol. 11,598-606.
6. Crown J., Kritz A., Vahdat L. et al. (1993) Rapid administration of multiple cycles of high-dose myelo- suppressive chemotherapy in patients with metastatic breast cancer. J. clin. Oncol. 11, 1144-1150.
7. Preisler H. D. (1978) Failure of remission induction in acute myelocytic leukemia. Med. Ped. Oncol. 4, 275- 276.
8. Preisler H. D. (1982) Treatment failure in AML. Blood Cells 8, 585-602.
9. Raza A., Maheshwari Y., Brereton W. et al. (1987) An analysis of cell cycle characteristics and course of the disease in ANLL. A m . J. Hemat. 24, 66-75.
10. Preisler H. D., Raza A., Larson R. et al. (1991) Some reasons for the lack of progress in the treatment of acute myelogenous leukemia. Leukemia Res. 15, 773- 780.
11. Armitage J. O. (1992) Treatment of non-Hodgkin's lymphoma. New Engl. J. Med. 328, 1023-1030.
12. Schein P. S., Chabner B. A., Canellos G. P. et al. (1974) Potential for prolonged disease-free survival following combination chemotherapy of non- Hodgkin's lymphoma. Blood 43, 181-189.
13. Schein P. S., DeVita V. T., Hubbard S. et al. (1976) Bleomycin, adriamycin, cyclophosphamide, vincristine and prednisone (BACOP) combination chemotherapy in the treatment of advanced lymphoma. Ann. Intern. Med. 85, 417-422.
14. Hillen H., Wessels J. & Haanen C. (1975) Bone mar- row proliferation patterns in acute myeloblastic leu- kemia determined by pulse cytophotometry. Lancet 1, 509-511.
15. Hart J., George A. L., Frei E. et al. (1977) Prognostic significance of pretreatment proliferative activity in adult acute leukemia. Cancer 39, 1603-1617.
16. Crowther D., Beard M. E. J., Bateman C. J. T. et al. (1975) Factors influencing prognosis in adults with acute myelogenous leukaemia. Br. J. Cancer 32, 456- 464.
17. Murphy S. B., Aur R. J. A., Simone J. V. etal. (1977) Pretreatment cytokinetic studies in 94 children with acute leukemia, relationship to other variables at diag- nosis and to outcome of standard treatment. Blood 49, 683-691.
18. Hiddemann W., Buchner T., Andreef M. et al. (1982) Cell kinetics in acute leukemia. A critical reevaluation based on new data. Cancer 50, 250-258.
19. Amadori S., Petti C., Mastrovincenzo M. G. T. et al. (1980) Blast cell kinetics and prognosis in acute nonlymphocytic leukemia. Leukemia Res. 4, 239-244.
20. Colly L. P., Peter W. G., Hermans J. et al. (1987) Percentage of S-phase cells in bone marrow aspirates, biopsy specimens and bone marrow aspirates corrected for blood dilutions from patients with acute leukemia. Leukemia Res. 11,209-213.
21. Riccardi A., Danova M., Montecucco C. et al. (1986) Adult acute nonlymphoblastic leukemia: reliability and prognostic significance of bone marrow S-phase size determined by flow cytometry. Scand. J. Haemat. 36, 11-17.
22. Raza A., Maheswari Y. & Preisler H. D. (1987) Dif- ferences in cell cycle characteristics amongst patients with acute nonlymphocytic leukemia. Blood 69, 1647- 1653.
23. Preisler H. D., Epstein J., Barcos M. et al. (1984) Prediction of response of acute nonlymphocytic leu- kaemia to therapy with high dose cytosine arabinoside. Br. J. Haemat. 58, 19-32.
24. Preisler H. D., Azarnia N., Raza A. et al. (1984) Relationship between the per cent of marrow cells in S-phase and the outcome of remission-induction therapy for acute nonlymphocytic leukaemia. Br. J. Haemat. 56, 399-407.
25. Raza A., Bohokari J., Yousuf N. et al. (1991) Cell kinetic studies in human cancers: development of three DNA-specific labels in three decades. Arch. Path. Lab. Med. 115, 873-879.
26. Raza A., Preisler H. D., Day R. et al. (1990) Direct relationship between remission duration in acute myeloid leukemia and cell cycle kinetics. A leukemia intergroup study. Blood 76, 2191-2197.
27. Christensson B., Tribukaint B., Linden I. L. et al. (1986) Cell proliferation and DNA content in non- Hodgkin's lymphoma. Cancer 58, 1295-1304.
28. Begg A. C., Moonen L., Holland I. etal. (1988) Human tumor cell kinetics using a monoclonal antibody against iododeoxyuridine: intratumor sampling variations. Radiother. Oncol. 11,337-347.
29. Meyer J. S. & Lee J. Y. (1980) Relationships of S-phase fraction of breast carcinoma in relapse to duration of remission, estrogen receptor content, therapeutic responsiveness and duration of survival. Cancer Res. 40, 1890-1896.
30. Silvestrini R., Daidone M. G., Valagressa P. et al. (1987) Cell kinetics as a prognostic marker in locally advanced breast cancer. Cancer Treat. Rep. 71, 375- 380.

Classification of treatment failure 159
31. Preisler H. D., Raza A., Bonomi P. et al. (1993) Regrowth resistance as a major contributor to treat- ment failure in neoplastic disease. J. clin. Oncol.
32. Tura S. et al. (1975) A clinical trial of early splen- ectomy, hydroxyurea and cyclic cytosine arabinoside, vincristine, and prednisone in chronic myelogenous leukemia. Italian cooperative study group in chronic myeloid leukemia. Ser. Hemat. 8, 121-125.
33. Cunningham I., Gee T., Dowling M. et al. (1979) Results of treatment of Phi+ chronic myelogenous leukemia with an intensive treatment regimen (L5 Pro- tocol). Blood 53, 375-385.
34. Preisler H. D., Raza A., Hibgy D. et al. (1984) Treat- ment of myeloid blastic crisis of chronic myelogenous leukemia. Cancer Treat. Rep. 58, 1251-1255.
35. Lambertenghi-Deillers G., Annaloro C., Cortellaro M. et al. (1991) Idarubicin in blastic crisis of chronic myelogenous leukemia. Haematology 76, 406-408.
36. Preisler H. D., Raza A. & Baccarani M. 119931 Pro- liferative advantage rather than classical drug resist- ance as the cause of treatment failure in chronic myelogenous leukemia. Leukemia Lymphoma 10, Suppl. 2.
37. Carelli A. E., Pollicardo N., Pungolino E. et al. (1993) Mobilization of cytogenetically normal blood pro- genitor cells by intensive conventional chemotherapy for chronic myeloid and acute lymphoblastic leukemia. Leukemia L y m p h o m a 9, 477-483.
38. Deisseroth A. Unpublished observations. 39. Kantarjian H. M., Keating M. J., Estey E. H. et al.
(1992) Treatment of advanced stages of Philadelphia- positive chronic myelogenous leukemia with interferon cr and low dose cytarabine. J. clin. Oncol. 10, 772- 778.
41). Inwards D. J. & Armitage J. O. (1991) Modern chemo- therapeutic regimens in the management of aggressive non-Hodgkin lymphoma: can they be improved? Eur. J. Cancer 27, 510-513.
41. Skarin A. T., Canellos G. P., Rosenthal D. S. et al. (19831 Improved prognosis of diffuse histiocytic and undifferentiated lymphoma by use of high dose metho- trexate alternating with standard agents (M-BACOD). J clin. Oncol. 1, 91-98.
42. Fisher R. l., DeVita R. V. Jr, Hubbard S. M. et al. (1984) Randomized trial of ProMACE-MOPP vs ProMACE-CytaBOM in previously untreated, advanced stage diffuse aggressive lymphoma. Proc. Am. Soc. Clin. Oncol. 3, 242, Abstract no. C-945.
43. Kilmo P. & Connors J. M. (1985) MACOP-B chemo- therapy for the treatment of diffuse large-cell lymphoma. Ann. Intern Med. 102, 596-602.
44. Fisher R. I., Gaynor E. R., Dahlberg S. et al. (1993) Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non- Hodgkin's lymphoma. New Engl. J. Med. 328, 1002- 1006.
45. Smalley R. V., Andersen J. W., Hawkins M. J. et al. (1992) Interferon alfa combined with cytotoxic therapy for patients with non-Hodgkin's lymphoma. New Engl. J. Med. 327, 1336-1341.
46. Louie A. C., Gallagher J. G., Sikora K. et al. (1981) Follow-up observations on the effect of human leu- kocyte interferon in non-Hodgkin's lymphoma. Blood 58, 712-718.
47. Foon K. A., Sherwin S. A., Abrams P. G. etal. (19841 Treatment of advanced non-Hodgkin's lymphoma with
recombinant leukocyte A-interferon. New Engl. J. Med. 311, 1148-1152.
48. Baron S., Tyring A. K., Fleischmann R. Jr etal. (1991) The interferons. Mechanisms of action and clinical applications. J A M A 266, 1375-1383.
49. Preisler H. D., Gopal V., Banavali S. et al. (1992) Multiparameter assessment of the cell cycle effects of bioactive and cytotoxic agents. Cancer Res. 52, 1-7.
50. Johnson G. J., Costello W. G., Oken M. M. et al. 11983) Sequential cyclophosphamide-prednisone and vincristine-bteomycin (CPOB): an effective schedule dependent treatment for advanced diffuse histiocytic lymphoma. Cancer 52, 1133-1141.
51. Coppola J. A. & Cole M. D. (1986) Constitutive c-myc gene expression blocks erythroleukemia-cell differenti- ation but not commitment. Nature 320, 760-762.
52. Mourgneau E., Lemieux L., Rassoulzadegan M. et al. (1984) Biological activities of v-myc and rearranged c- mvc oncogenes in rat fibroblast cells in culture. Proc. natn. Acad. Sci. U.S.A. 81, 5758-5762.
53. Gionti E., Pontarelli G. & Cancedda R. (1985) Avian myelocytomatosis virus immortalizes differentiated quail chondrocytes. Proc. natn. Acad. Sci. U.S.A. 82, 2756-2760.
54. Sorrentino V., Drozdoff V., McKinney M. D. et al. (1986) Potentiation of growth factor activity by exogen- ous c-myc expression. Proc. natn. Acad. Sci. U.S.A. 83, 8167-8171.
55. Gopal V., Kadam P., Preisler H. D. et al. 11992) Abnormal regulation of the myc gene in myeloid leu- kemia. Med. Oncol. Tumor Pharmac. 9, 1-9.
56. Leda P., Barley J., Lenoin G. et al. (1983). Trans- locations among antibody genes in human cancer. Science 222, 765-771.
57. Vaux D. L., Cory S. & Adams J. M. (1988) bcl-2 geue promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B-cells. Nature 335,440- 442.
58. Miyashita T. & Reed J. C. 119931 bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood 81, 151-157.
59. Campos L., Rouault J. P., Abido O. et al. (1993) High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81, 3091-3096.
60. Yunis J. J., Mayer M. G., Arnesen M. A. et al. (1989) bcl-2 and other genomic alterations in the prognosis of large-cell lymphoma. New Engl. J. Med. 320, 11147- 1054.
61. Marshal R., Shtalrid M., Talpaz M. et al. (1990) Rearrangement and expression of p53 in the chronic phase and blast crisis of chronic myelogenous leukemia. Blood 75, 180-189.
62. Kastan M. B., Onyekwere O., Sidransky D. et al. (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res. Sl, 6304- 6311.
63. Kuerbitz S. J., Plunkett B. S., Walsh W. V. et al. (1992) Wild-type p53 is a cell cycle check point determinant following irradiation. Proc. natn. Aead. Sci. U.S.A. 89, 7491-7495.
64. Livingstone L. R., White A., Sprouse J. et al. (1992) Altered cell cycle arrest and gene amplification poten- tial accompany loss of wild-type p53. Cell 70, 923-935.
65. Thor A. D., Moore D. H. II, Edgerton S. M. et al. 119921 Accumulation of p53 tumor suppressor gene

160 H.D. PREISLER and V. GOPAL
protein: an independent marker of prognosis in breast cancers. J. Natl Cancer Inst. 84, 845-855.
66. Isola J., Visakopi T., Holli K. et al. (1992) Association of overexpression of tumor suppression protein with rapid cell proliferation and poor prognosis in none- negative breast cancer patients. J Natl Cancer Inst. 84, 1109-1114.
67. Visakorpi T., Kallioniemi O. P., Heikkinen A. et al. (1992) Small subgroup of aggressive highly proliferative prostatic carcinomas defined by p53 accumulation. J. Natl Cancer lnst. 84, 883-887.
68. Sun X. F., Castensen J. M., Zhang H. et al. (1992) Prognostic significance of cytoplasmic p53 oncoprotein in colorectal adenocarcinoma. Lancet 340, 1369- 1373.
69. Preisler H. D., Raza A. & Larson R. A. (1992) Alter- ation of the proliferative rate of acute myelogenous
leukemia cells in vivo in patients. Blood 80, 2600- 2603.
7(1. Banavali S., Pancoast J., Tricot G. et al. (1993) Serial studies of c-myc expression in bone marrow biopsies: effects of cytotoxic agents, rhGM-CSF, and retinoic acid and interferon. Eur. J. Cancer (in press).
71. Preisler H. D., Gopal V. & Raza A. (1993) The effects of rhGM-CSF on myc and myb expression in acute myelogenous leukemia cells in vil~o. Br. J. Haernat. (in press).
72. Yanik G., Yousuf N., Miller M. A. et al. (1992) In t~ioo determination of cell cycle kinetics of non-Hodgkin's lymphomas using iododeoxyuridine and bromode- oxyuridine. J. Histochem. Cytochem. 40, 723-728.
73. Pavelic K., Pavelic Z. P., Denton D. et al. (1990) Immunohistochemical detection of c-myc oncoprotein in paraffin embedded tissues. J. exp. Path. 5, 143 153.
Copyright © 2022 FDOKUMEN