
Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication
10
JOURNAL OF VIROLOGY, Mar. 2011, p. 2818–2827 Vol. 85, No. 6 0022-538X/11/$12.00 doi:10.1128/JVI.01969-10 Copyright © 2011, American Society for Microbiology. All Rights Reserved. Receptor Tyrosine Kinase Inhibitors Block Multiple Steps of Influenza A Virus Replication ‡ Naveen Kumar, Yuhong Liang,† Tristram G. Parslow, and Yuying Liang* Department of Pathology and Laboratory Medicine, Emory University, Atlanta, Georgia 30322 Received 16 September 2010/Accepted 22 December 2010 Host signaling pathways play important roles in the replication of influenza virus, but their functional effects remain to be characterized at the molecular level. Here we identify two receptor tyrosine kinase inhibitors (RTKIs) of the tyrphostin class that exhibit robust antiviral activity against influenza A virus replication in cultured cells. One of these (AG879) is a selective inhibitor of the nerve growth factor receptor and human epidermal growth factor receptor 2 (TrkA/HER2) signaling; the other, tyrphostin A9 (A9), inhibits the platelet- derived growth factor receptor (PDGFR) pathway. We find that each inhibits at least three postentry steps of the influenza virus life cycle: AG879 and A9 both strongly inhibit the synthesis of all three influenza virus RNA species, block Crm1-dependent nuclear export, and also prevent the release of viral particles through a pathway that is modulated by the lipid biosynthesis enzyme farnesyl diphosphate synthase (FPPS). Tests of short hairpin RNA (shRNA) knockdown and additional small-molecule inhibitors confirmed that interventions targeting TrkA can suppress influenza virus replication. Our study suggests that host cell receptor tyrosine kinase signaling is required for maximal influenza virus RNA synthesis, viral ribonucleoprotein (vRNP) nuclear export, and virus release and that specific RTKIs hold promise as novel anti-influenza virus therapeutics. Influenza virus imposes substantial burdens on public health by causing annual epidemics and occasional pandemics of acute respiratory disease that may lead to potentially severe and deadly complications, such as pneumonia. Antiviral ther- apeutics are a critical tool in combating influenza virus infec- tions, especially in years when the vaccine strain does not match well with the circulating virus, when vaccines are un- available at the early pandemic stage, or when vaccines are in short supply. Development of novel anti-influenza virus drugs is urgent, as variant strains resistant to all currently available drugs have been isolated and are expected to evolve rapidly (7, 8, 26, 44, 59). Targeting host cell signaling pathways or other host factors required for influenza virus replication offers an alternative strategy for antiviral drug development. Recent proteomic screening using small interfering RNA (siRNA) libraries has identified hundreds of host factors that may pro- mote influenza virus replication (3, 16, 22, 24, 49), but the challenge of validating, characterizing, and interdicting their respective activities through pharmacological means remains. Influenza A virus is an enveloped, negative-strand RNA virus with a segmented RNA genome (38). Influenza virus enters cells through receptor-mediated endocytosis after bind- ing to sialylated receptors (50). After internalization, the low-pH environment in endosomes triggers fusion of viral and endosomal membranes and facilitates the release of viral ribo- nucleoprotein (vRNP) complexes into the cell cytoplasm (58). The released vRNPs then enter the nucleus, where viral RNA (vRNA) replication and transcription occur (38). Newly syn- thesized vRNPs are exported from the nucleus via the cellular Crm1-mediated nuclear export pathway (1, 12, 28, 55). Virus budding is mediated mainly by the viral M1 protein, which interacts with viral integral membrane proteins (HA, NA, and M2) and vRNP complexes at the plasma membrane (5, 33). The final release of virions from the cell surface requires the neuraminidase activity of viral NA protein (37, 39). Despite extensive studies, many aspects of influenza virus replication are incompletely understood, including the roles of host sig- naling pathways and cellular factors at each step of the virus life cycle. Identification of small-molecule compounds target- ing any of these processes can yield biological insights as well as potential new therapies. For example, amantadine was found to block virus uncoating (4, 29), and viruses resistant to amantadine were found to harbor mutations in the ion channel region of the M2 transmembrane domain, suggesting both that the viral M2 protein is the target of amantadine (17) and that M2 ion channel activity is essential for virus uncoating. Viral HA protein was also found to influence amantadine sensitivity, implying an interaction between HA and M2 (17). Receptor tyrosine kinases (RTKs) are a group of growth factor receptors that, upon ligand binding, undergo autophos- phorylation at Tyr residues (18, 48, 52). These phosphorylated tyrosines then recruit Src homology 2 (SH2)- and phosphoty- rosine-binding (PTB) domain-containing proteins that activate or link to downstream signaling pathways, such as the Ras/ ERK/MAPK, PI3K/Akt, and JAK/STAT pathways (40, 48). Together, the complex signaling network triggered by RTKs leads to regulation of cell growth, migration, metabolism, and differentiation. Due to their critical roles in the development * Corresponding author. Mailing address: Department of Pathology and Laboratory Medicine, 615 Michael St., Ste. 177, Rm. 175, White- head Biomedical Research Bldg., Emory University, Atlanta, GA 30322. Phone: (404) 727-3243. Fax: (404) 727-8538. E-mail: yliang5 @emory.edu. † Present address: Department of Microbiology, Mount Sinai School of Medicine, New York, NY 10029. ‡ Supplemental material for this article may be found at http://jvi .asm.org/. Published ahead of print on 5 January 2011. 2818 on June 6, 2016 by guest http://jvi.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication

JOURNAL OF VIROLOGY, Mar. 2011, p. 2818–2827 Vol. 85, No. 60022-538X/11/$12.00 doi:10.1128/JVI.01969-10Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Receptor Tyrosine Kinase Inhibitors Block Multiple Steps of InfluenzaA Virus Replication�‡
Naveen Kumar, Yuhong Liang,† Tristram G. Parslow, and Yuying Liang*Department of Pathology and Laboratory Medicine, Emory University, Atlanta, Georgia 30322
Received 16 September 2010/Accepted 22 December 2010
Host signaling pathways play important roles in the replication of influenza virus, but their functional effectsremain to be characterized at the molecular level. Here we identify two receptor tyrosine kinase inhibitors(RTKIs) of the tyrphostin class that exhibit robust antiviral activity against influenza A virus replication incultured cells. One of these (AG879) is a selective inhibitor of the nerve growth factor receptor and humanepidermal growth factor receptor 2 (TrkA/HER2) signaling; the other, tyrphostin A9 (A9), inhibits the platelet-derived growth factor receptor (PDGFR) pathway. We find that each inhibits at least three postentry steps ofthe influenza virus life cycle: AG879 and A9 both strongly inhibit the synthesis of all three influenza virus RNAspecies, block Crm1-dependent nuclear export, and also prevent the release of viral particles through apathway that is modulated by the lipid biosynthesis enzyme farnesyl diphosphate synthase (FPPS). Tests ofshort hairpin RNA (shRNA) knockdown and additional small-molecule inhibitors confirmed that interventionstargeting TrkA can suppress influenza virus replication. Our study suggests that host cell receptor tyrosinekinase signaling is required for maximal influenza virus RNA synthesis, viral ribonucleoprotein (vRNP)nuclear export, and virus release and that specific RTKIs hold promise as novel anti-influenza virustherapeutics.
Influenza virus imposes substantial burdens on public healthby causing annual epidemics and occasional pandemics ofacute respiratory disease that may lead to potentially severeand deadly complications, such as pneumonia. Antiviral ther-apeutics are a critical tool in combating influenza virus infec-tions, especially in years when the vaccine strain does notmatch well with the circulating virus, when vaccines are un-available at the early pandemic stage, or when vaccines are inshort supply. Development of novel anti-influenza virus drugsis urgent, as variant strains resistant to all currently availabledrugs have been isolated and are expected to evolve rapidly (7,8, 26, 44, 59). Targeting host cell signaling pathways or otherhost factors required for influenza virus replication offers analternative strategy for antiviral drug development. Recentproteomic screening using small interfering RNA (siRNA)libraries has identified hundreds of host factors that may pro-mote influenza virus replication (3, 16, 22, 24, 49), but thechallenge of validating, characterizing, and interdicting theirrespective activities through pharmacological means remains.
Influenza A virus is an enveloped, negative-strand RNAvirus with a segmented RNA genome (38). Influenza virusenters cells through receptor-mediated endocytosis after bind-ing to sialylated receptors (50). After internalization, thelow-pH environment in endosomes triggers fusion of viral and
endosomal membranes and facilitates the release of viral ribo-nucleoprotein (vRNP) complexes into the cell cytoplasm (58).The released vRNPs then enter the nucleus, where viral RNA(vRNA) replication and transcription occur (38). Newly syn-thesized vRNPs are exported from the nucleus via the cellularCrm1-mediated nuclear export pathway (1, 12, 28, 55). Virusbudding is mediated mainly by the viral M1 protein, whichinteracts with viral integral membrane proteins (HA, NA, andM2) and vRNP complexes at the plasma membrane (5, 33).The final release of virions from the cell surface requires theneuraminidase activity of viral NA protein (37, 39). Despiteextensive studies, many aspects of influenza virus replicationare incompletely understood, including the roles of host sig-naling pathways and cellular factors at each step of the viruslife cycle. Identification of small-molecule compounds target-ing any of these processes can yield biological insights as wellas potential new therapies. For example, amantadine wasfound to block virus uncoating (4, 29), and viruses resistant toamantadine were found to harbor mutations in the ion channelregion of the M2 transmembrane domain, suggesting both thatthe viral M2 protein is the target of amantadine (17) and thatM2 ion channel activity is essential for virus uncoating. ViralHA protein was also found to influence amantadine sensitivity,implying an interaction between HA and M2 (17).
Receptor tyrosine kinases (RTKs) are a group of growthfactor receptors that, upon ligand binding, undergo autophos-phorylation at Tyr residues (18, 48, 52). These phosphorylatedtyrosines then recruit Src homology 2 (SH2)- and phosphoty-rosine-binding (PTB) domain-containing proteins that activateor link to downstream signaling pathways, such as the Ras/ERK/MAPK, PI3K/Akt, and JAK/STAT pathways (40, 48).Together, the complex signaling network triggered by RTKsleads to regulation of cell growth, migration, metabolism, anddifferentiation. Due to their critical roles in the development
* Corresponding author. Mailing address: Department of Pathologyand Laboratory Medicine, 615 Michael St., Ste. 177, Rm. 175, White-head Biomedical Research Bldg., Emory University, Atlanta, GA30322. Phone: (404) 727-3243. Fax: (404) 727-8538. E-mail: [email protected].
† Present address: Department of Microbiology, Mount SinaiSchool of Medicine, New York, NY 10029.
‡ Supplemental material for this article may be found at http://jvi.asm.org/.
� Published ahead of print on 5 January 2011.
2818
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from

and progression of various cancers, RTKs have recently beenstudied extensively as targets for anticancer therapeutics. Hostsignaling through RTKs and other tyrosine kinases has alsobeen shown to play important roles in virus replication. Thetyrosine kinase inhibitor genistein was found to block replica-tion of HIV-1, herpes simplex virus type 1 (HSV-1), and arena-virus (51, 53, 61), for example, and Src family kinases areknown to be important for assembly and maturation of denguevirus and West Nile virus (6, 19). The Raf/MEK/ERK (42) andPI3K/Akt (9, 10, 15) pathways downstream of RTKs play im-portant roles in influenza virus replication. It has been shownthat Raf/MEK/ERK signaling is required for the nuclear ex-port of influenza vRNPs (42). The functional mechanism bywhich the PI3K pathway affects influenza virus replication isunclear, however. One recent report indicates that epidermalgrowth factor receptor (EGFR) signaling promotes influenzaA virus uptake by cells (11).
In this study, we identify two specific RTK inhibitors(RTKIs), known as AG879 and tyrphostin A9 (A9), that havestrong antiviral activity against influenza A virus, and we dem-onstrate that they both inhibit the Crm1-dependent nuclearexport of the vRNP complex, viral RNA synthesis, and virusrelease. We show that diverse interventions targeting TrkA canimpede influenza virus replication, thus validating this specificRTK as a candidate drug target. Our findings provide mecha-nistic insights into the potential roles of host RTK signaling infacilitating influenza virus replication, and they also suggestthat specific RTKIs could be developed as potential anti-influ-enza virus therapeutics.
MATERIALS AND METHODS
Cells and viruses. 293T cells (human kidney epithelial cells) and A549 cells(human lung epithelial cells) were grown in Dulbecco’s modified Eagle’s mediumsupplemented with 10% heat-inactivated fetal bovine serum. Madin-Darby ca-nine kidney (MDCK) cells were maintained in Eagle’s minimal essential mediumsupplemented with 5% fetal bovine serum. After infection with influenza A virus,MDCK cells were grown in L-15 medium containing 15 mM HEPES (pH 7.5),nonessential amino acids, 0.75 g of NaHCO3 per liter, and 0.125% (wt/vol)bovine serum albumin. Influenza virus strains A/WSN/33 (A/WSN) andA/PR8/34 (A/PR8) were grown in 10-day-old embryonated chicken eggs, andtheir titers were determined by plaque assay on MDCK cells. The WSN-LUCreporter viruses were generated as previously described (25).
Plasmids, antibodies, and inhibitors. Plasmid expressing farnesyl diphosphatesynthase (FPPS) was provided by P. Creswell (Yale University). Plasmid express-ing NF-�B molecule p65 was obtained from W. Greene (UCSF). Plasmid ex-pressing the Rev-green fluorescent protein (GFP) fusion protein was provided byA. Mergia (University of Florida). Generation of the luciferase (LUC)-encodingreporter constructs vNA-LUC and cNA-LUC was described previously (45).Anti-FPPS antibody was obtained from P. Edward (UCLA). Anti-NP monoclo-nal antibody was purchased from Serotec. The library of kinase inhibitors waspurchased from BIOMOL and includes 80 kinase inhibitors. TyrphostinAG879, tyrphostin A9, tyrphostin AG494, ammonium pyrrolidinedithiocar-bamate (PDTC), Bay11-7082 (Bay11), ribavirin, and AG1296 were purchased fromSigma. U0126 was purchased from Promega. GW441756 was purchased from SantaCruz. K252a, TAK-165, ZD1839, and SKI-606 were purchased from LC Lab.
MTT assay. The MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazo-lium bromide] assay was used to measure the effect of selected compounds oncell viability. MDCK or A549 cells in 96-well plates were treated with sequential10-fold dilutions of either a given compound or dimethyl sulfoxide (DMSO), intriplicates, in a total of 100 �l growth medium for 48 h. Freshly made 5 mg/mlMTT solution (20 �l) was added to each well, and the cells were incubated at37°C for 5 h before the medium was replaced with 200 �l DMSO to dissolve thecrystals. The plates were further incubated at 37°C for another 5 min to dissolveany air bubbles before the MTT signal was measured at an absorbance of 550 nm.The MTT assay was performed on each compound, and no cytotoxicity wasobserved for A9 at 5 �M, AG879 at 81 �M, AG494 at 27 �M, Bay11-7082
(Bay11) at 10 �M, U0126 at 50 �M, or PDTC at 100 �M. Accordingly, through-out this work, except where noted, A9 was used at 4 �M, AG879 at 10 �M,AG494 at 10 �M, Bay11 at 10 �M, U0126 at 50 �M, and PDTC at 50 �M.
Virus attachment assay. A549 cells were preincubated with DMSO or selectedcompounds for 30 min and then infected with A/WSN (multiplicity of infection[MOI] of 2) at 4°C for 2 h. After cells were washed 6 times with phosphate-buffered saline (PBS), cell lysates were prepared by rapid freeze-thaw. Virus titerwas determined by plaque assay.
Inhibition of nucleocytoplasmic trafficking. A549 cells were infected withA/WSN virus at an MOI of 5 for 1 h and replaced with fresh medium containingvehicle control or respective inhibitors. Intracellular localization of influenzavRNPs in the virus-infected cells at various times postinfection was detected byimmunofluorescence assay as described previously (25). To examine the effectsof the inhibitors on the nucleocytoplasmic trafficking of HIV rev protein, A549cells grown on coverslips were transfected with a plasmid expressing rev-GFPfusion protein and, 18 h later, treated with DMSO or respective inhibitors for 4 hprior to observation under a fluorescence microscope. The intensity of fluores-cein isothiocyanate (FITC)-NP or rev-GFP signals in both nucleus (defined byDAPI [4�,6-diamidino-2-phenylindole] staining) and cytoplasm of individual cellswas quantified using an image analysis program, and the percentage of nuclearsignal (nuc%) was calculated for each cell. The mean percentage of nuclearsignal was then calculated for 40 cells per drug treatment per time point, andstatistical comparisons among groups were conducted using Student’s t test.
Quantitative real-time RT-PCR. The levels of influenza vRNA, cRNA, andmRNA in the virus-infected cells were quantified by real-time reverse transcrip-tion PCR (RT-PCR) as described previously (25).
Reporter-based influenza virus RNA transcription assay. The luciferase(LUC)-based five-plasmid RNA transcription assay was conducted as describedpreviously (25). In brief, A549 cells grown in serum-free medium were trans-fected with 5 plasmids, four encoding the PA, PB1, PB2, and NP proteins,respectively, and one expressing the viral RNA promoter-directed LUC gene(vNA-LUC or cNA-LUC). DMSO or inhibitors were added at 8 h posttransfec-tion, and LUC activity was measured at 24 h posttransfection.
Virus release assay. A549 cells were infected with influenza A virus at an MOIof 2. At 8 h postinfection (hpi), cells were washed 5 times with PBS and replacedwith fresh medium containing vehicle control or respective inhibitors. At differ-ent time points (15, 30, 45, and 60 min) following addition of inhibitors, super-natants were collected and cell pellets were lysed by rapid freeze-thaw on dryice-ethanol and a 37°C water bath. Virus titers in the supernatant (extracellularvirus) and cell lysate (membrane-associated virus) were then quantified byplaque assay on MDCK cells.
shRNA knockdown. We used two different short hairpin RNAs (shRNAs) toknock down human TrkA expression. Two different pairs of oligonucleotides, 5�CCAGTGACCTCAACAGGAAGAttcaagagaTCTTCCTGTTGAGGTCACTGG 3� and 5� TCATCGAGAACCCACAATACTttcaagagaAGTATTGTGGGTTCTCGATGA 3� (only forward sequences are shown), were each cloned intothe psiRNA-hH1 vector (InvivoGen) between the Acc65I and HindIII sitesdownstream of the human H1 RNA promoter. The resulting plasmids, psiRNA-TrkA1 and psiRNA-TrkA3, were verified by sequence analysis. For shRNAknockdown, A549 cells were transfected twice in 48 h using Lipofectamine(Invitrogen) with either the psiRNA-hH1Luc control plasmid, which expressesluciferase-specific shRNA, or the combined psiRNA-TrkA1 and -TrkA3 plas-mids and then, at 72 h posttransfection, infected with A/WSN virus at an MOI of1. Virus titer was determined at 12 hpi.
RESULTS
Identification of two RTKIs, AG879 and A9, with anti-influ-enza virus activity. Using the WSN-LUC reporter virus systemthat we have previously described (25), we screened a smalllibrary of 80 kinase inhibitors for anti-influenza virus activity.MDCK cells on 96-well plates were infected with WSN-LUCvirus at an MOI of 0.5 for 1 h, washed with PBS, and treatedwith DMSO vehicle control, respective compounds at 10 �m,or ribavirin at various concentrations as a positive control. TheLUC activity in the infected MDCK cells was measured andnormalized against that for the DMSO control (Fig. 1A, lane1). As shown in a representative screen (Fig. 1A), most of thetested compounds showed no inhibitory effects on LUC activ-ity in the WSN-LUC-infected cells, whereas the nonspecific
VOL. 85, 2011 RTK SIGNALING AND INFLUENZA VIRUS 2819
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from
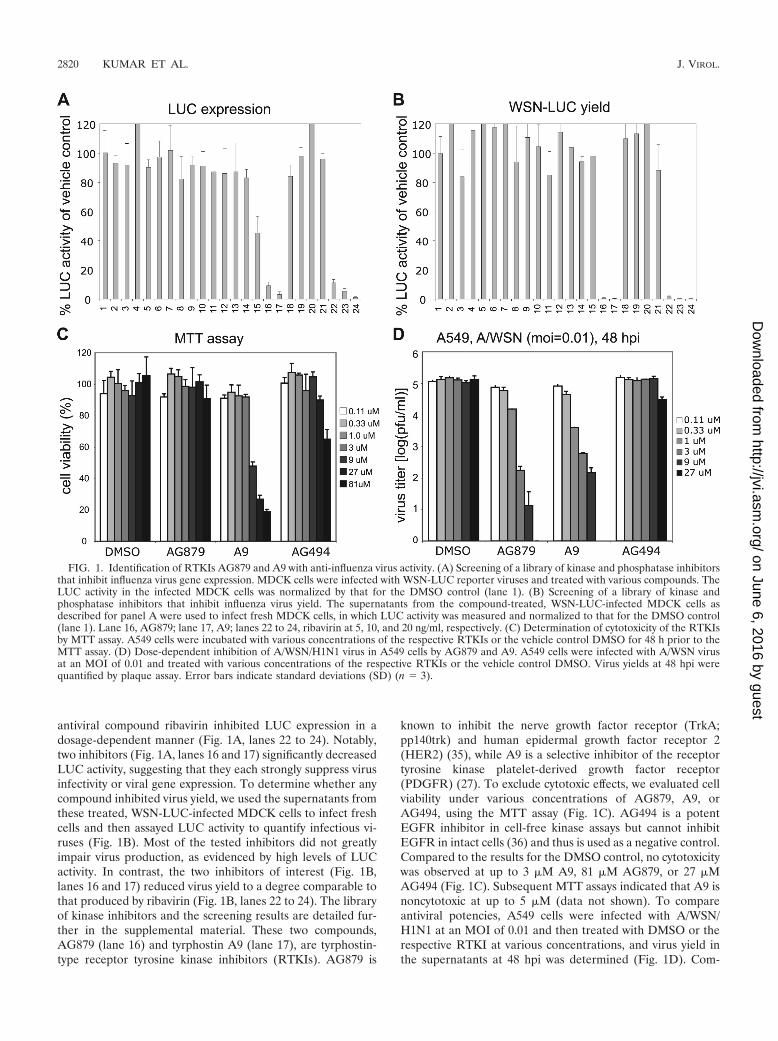
antiviral compound ribavirin inhibited LUC expression in adosage-dependent manner (Fig. 1A, lanes 22 to 24). Notably,two inhibitors (Fig. 1A, lanes 16 and 17) significantly decreasedLUC activity, suggesting that they each strongly suppress virusinfectivity or viral gene expression. To determine whether anycompound inhibited virus yield, we used the supernatants fromthese treated, WSN-LUC-infected MDCK cells to infect freshcells and then assayed LUC activity to quantify infectious vi-ruses (Fig. 1B). Most of the tested inhibitors did not greatlyimpair virus production, as evidenced by high levels of LUCactivity. In contrast, the two inhibitors of interest (Fig. 1B,lanes 16 and 17) reduced virus yield to a degree comparable tothat produced by ribavirin (Fig. 1B, lanes 22 to 24). The libraryof kinase inhibitors and the screening results are detailed fur-ther in the supplemental material. These two compounds,AG879 (lane 16) and tyrphostin A9 (lane 17), are tyrphostin-type receptor tyrosine kinase inhibitors (RTKIs). AG879 is
known to inhibit the nerve growth factor receptor (TrkA;pp140trk) and human epidermal growth factor receptor 2(HER2) (35), while A9 is a selective inhibitor of the receptortyrosine kinase platelet-derived growth factor receptor(PDGFR) (27). To exclude cytotoxic effects, we evaluated cellviability under various concentrations of AG879, A9, orAG494, using the MTT assay (Fig. 1C). AG494 is a potentEGFR inhibitor in cell-free kinase assays but cannot inhibitEGFR in intact cells (36) and thus is used as a negative control.Compared to the results for the DMSO control, no cytotoxicitywas observed at up to 3 �M A9, 81 �M AG879, or 27 �MAG494 (Fig. 1C). Subsequent MTT assays indicated that A9 isnoncytotoxic at up to 5 �M (data not shown). To compareantiviral potencies, A549 cells were infected with A/WSN/H1N1 at an MOI of 0.01 and then treated with DMSO or therespective RTKI at various concentrations, and virus yield inthe supernatants at 48 hpi was determined (Fig. 1D). Com-
FIG. 1. Identification of RTKIs AG879 and A9 with anti-influenza virus activity. (A) Screening of a library of kinase and phosphatase inhibitorsthat inhibit influenza virus gene expression. MDCK cells were infected with WSN-LUC reporter viruses and treated with various compounds. TheLUC activity in the infected MDCK cells was normalized by that for the DMSO control (lane 1). (B) Screening of a library of kinase andphosphatase inhibitors that inhibit influenza virus yield. The supernatants from the compound-treated, WSN-LUC-infected MDCK cells asdescribed for panel A were used to infect fresh MDCK cells, in which LUC activity was measured and normalized to that for the DMSO control(lane 1). Lane 16, AG879; lane 17, A9; lanes 22 to 24, ribavirin at 5, 10, and 20 ng/ml, respectively. (C) Determination of cytotoxicity of the RTKIsby MTT assay. A549 cells were incubated with various concentrations of the respective RTKIs or the vehicle control DMSO for 48 h prior to theMTT assay. (D) Dose-dependent inhibition of A/WSN/H1N1 virus in A549 cells by AG879 and A9. A549 cells were infected with A/WSN virusat an MOI of 0.01 and treated with various concentrations of the respective RTKIs or the vehicle control DMSO. Virus yields at 48 hpi werequantified by plaque assay. Error bars indicate standard deviations (SD) (n � 3).
2820 KUMAR ET AL. J. VIROL.
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from
pared to results for vehicle control DMSO and negative con-trol AG494, both AG879 and A9 showed dose-dependent in-hibition of influenza virus yield in A549 cells. Note that thevirus yield with AG879 at 27 �M was �0 PFU and is thereforeundetectable in Fig. 1D and that the strong virus inhibitionproduced by A9 at 9 �M could partly be due to cytotoxicity atthat concentration (Fig. 1C). Based on these studies of cyto-toxicity and antiviral efficacy, we used A9 at 4 �M and AG879and AG494 at 10 �M in all subsequent work. Taken together,these initial studies identified two specific RTKIs that canstrongly inhibit both influenza A viral gene expression andvirus yield.
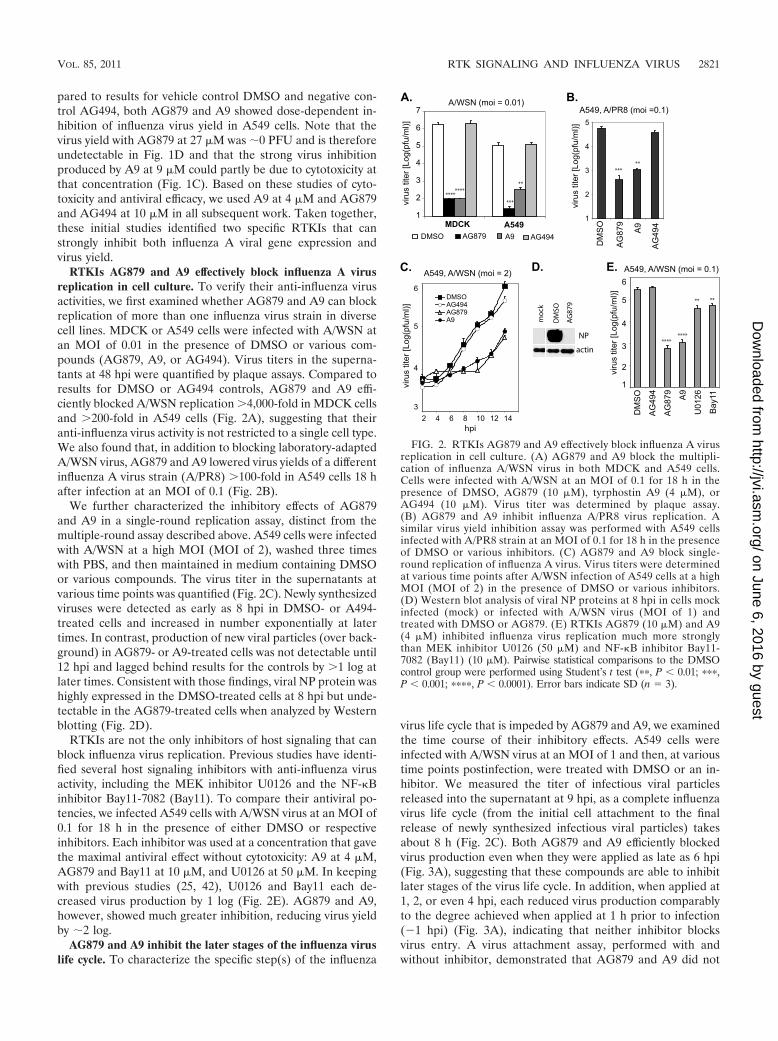
RTKIs AG879 and A9 effectively block influenza A virusreplication in cell culture. To verify their anti-influenza virusactivities, we first examined whether AG879 and A9 can blockreplication of more than one influenza virus strain in diversecell lines. MDCK or A549 cells were infected with A/WSN atan MOI of 0.01 in the presence of DMSO or various com-pounds (AG879, A9, or AG494). Virus titers in the superna-tants at 48 hpi were quantified by plaque assays. Compared toresults for DMSO or AG494 controls, AG879 and A9 effi-ciently blocked A/WSN replication �4,000-fold in MDCK cellsand �200-fold in A549 cells (Fig. 2A), suggesting that theiranti-influenza virus activity is not restricted to a single cell type.We also found that, in addition to blocking laboratory-adaptedA/WSN virus, AG879 and A9 lowered virus yields of a differentinfluenza A virus strain (A/PR8) �100-fold in A549 cells 18 hafter infection at an MOI of 0.1 (Fig. 2B).
We further characterized the inhibitory effects of AG879and A9 in a single-round replication assay, distinct from themultiple-round assay described above. A549 cells were infectedwith A/WSN at a high MOI (MOI of 2), washed three timeswith PBS, and then maintained in medium containing DMSOor various compounds. The virus titer in the supernatants atvarious time points was quantified (Fig. 2C). Newly synthesizedviruses were detected as early as 8 hpi in DMSO- or A494-treated cells and increased in number exponentially at latertimes. In contrast, production of new viral particles (over back-ground) in AG879- or A9-treated cells was not detectable until12 hpi and lagged behind results for the controls by �1 log atlater times. Consistent with those findings, viral NP protein washighly expressed in the DMSO-treated cells at 8 hpi but unde-tectable in the AG879-treated cells when analyzed by Westernblotting (Fig. 2D).
RTKIs are not the only inhibitors of host signaling that canblock influenza virus replication. Previous studies have identi-fied several host signaling inhibitors with anti-influenza virusactivity, including the MEK inhibitor U0126 and the NF-�Binhibitor Bay11-7082 (Bay11). To compare their antiviral po-tencies, we infected A549 cells with A/WSN virus at an MOI of0.1 for 18 h in the presence of either DMSO or respectiveinhibitors. Each inhibitor was used at a concentration that gavethe maximal antiviral effect without cytotoxicity: A9 at 4 �M,AG879 and Bay11 at 10 �M, and U0126 at 50 �M. In keepingwith previous studies (25, 42), U0126 and Bay11 each de-creased virus production by 1 log (Fig. 2E). AG879 and A9,however, showed much greater inhibition, reducing virus yieldby �2 log.
AG879 and A9 inhibit the later stages of the influenza viruslife cycle. To characterize the specific step(s) of the influenza
virus life cycle that is impeded by AG879 and A9, we examinedthe time course of their inhibitory effects. A549 cells wereinfected with A/WSN virus at an MOI of 1 and then, at varioustime points postinfection, were treated with DMSO or an in-hibitor. We measured the titer of infectious viral particlesreleased into the supernatant at 9 hpi, as a complete influenzavirus life cycle (from the initial cell attachment to the finalrelease of newly synthesized infectious viral particles) takesabout 8 h (Fig. 2C). Both AG879 and A9 efficiently blockedvirus production even when they were applied as late as 6 hpi(Fig. 3A), suggesting that these compounds are able to inhibitlater stages of the virus life cycle. In addition, when applied at1, 2, or even 4 hpi, each reduced virus production comparablyto the degree achieved when applied at 1 h prior to infection(�1 hpi) (Fig. 3A), indicating that neither inhibitor blocksvirus entry. A virus attachment assay, performed with andwithout inhibitor, demonstrated that AG879 and A9 did not
FIG. 2. RTKIs AG879 and A9 effectively block influenza A virusreplication in cell culture. (A) AG879 and A9 block the multipli-cation of influenza A/WSN virus in both MDCK and A549 cells.Cells were infected with A/WSN at an MOI of 0.1 for 18 h in thepresence of DMSO, AG879 (10 �M), tyrphostin A9 (4 �M), orAG494 (10 �M). Virus titer was determined by plaque assay.(B) AG879 and A9 inhibit influenza A/PR8 virus replication. Asimilar virus yield inhibition assay was performed with A549 cellsinfected with A/PR8 strain at an MOI of 0.1 for 18 h in the presenceof DMSO or various inhibitors. (C) AG879 and A9 block single-round replication of influenza A virus. Virus titers were determinedat various time points after A/WSN infection of A549 cells at a highMOI (MOI of 2) in the presence of DMSO or various inhibitors.(D) Western blot analysis of viral NP proteins at 8 hpi in cells mockinfected (mock) or infected with A/WSN virus (MOI of 1) andtreated with DMSO or AG879. (E) RTKIs AG879 (10 �M) and A9(4 �M) inhibited influenza virus replication much more stronglythan MEK inhibitor U0126 (50 �M) and NF-�B inhibitor Bay11-7082 (Bay11) (10 �M). Pairwise statistical comparisons to the DMSOcontrol group were performed using Student’s t test (��, P � 0.01; ���,P � 0.001; ����, P � 0.0001). Error bars indicate SD (n � 3).
VOL. 85, 2011 RTK SIGNALING AND INFLUENZA VIRUS 2821
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from
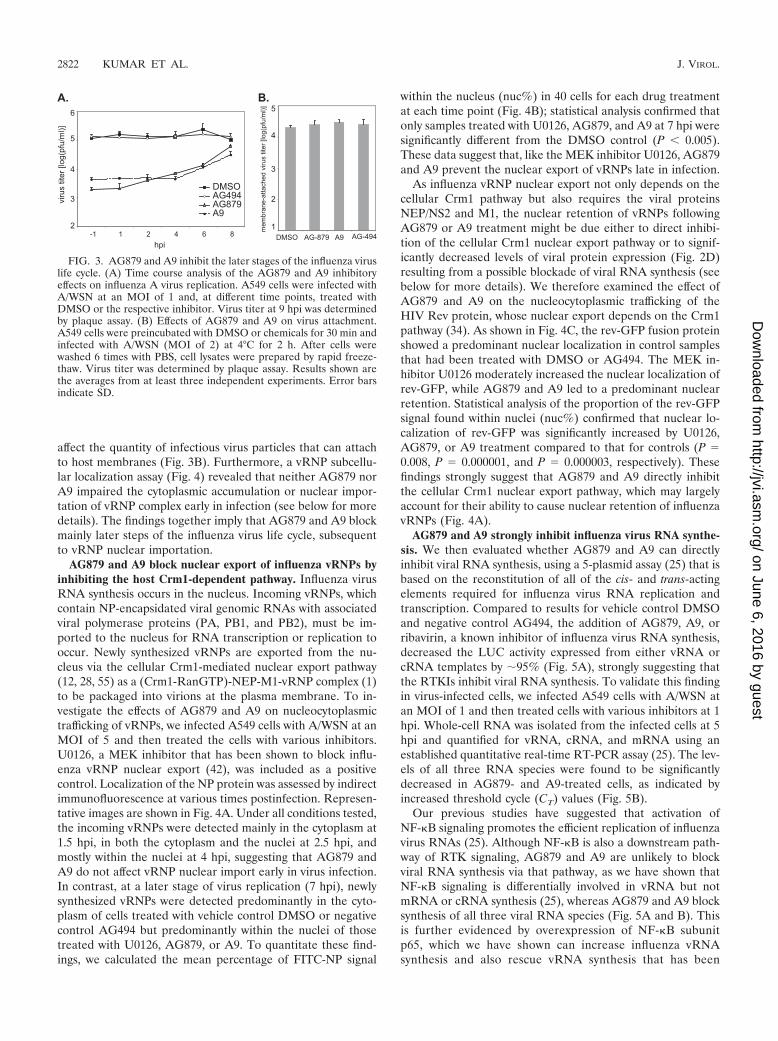
affect the quantity of infectious virus particles that can attachto host membranes (Fig. 3B). Furthermore, a vRNP subcellu-lar localization assay (Fig. 4) revealed that neither AG879 norA9 impaired the cytoplasmic accumulation or nuclear impor-tation of vRNP complex early in infection (see below for moredetails). The findings together imply that AG879 and A9 blockmainly later steps of the influenza virus life cycle, subsequentto vRNP nuclear importation.
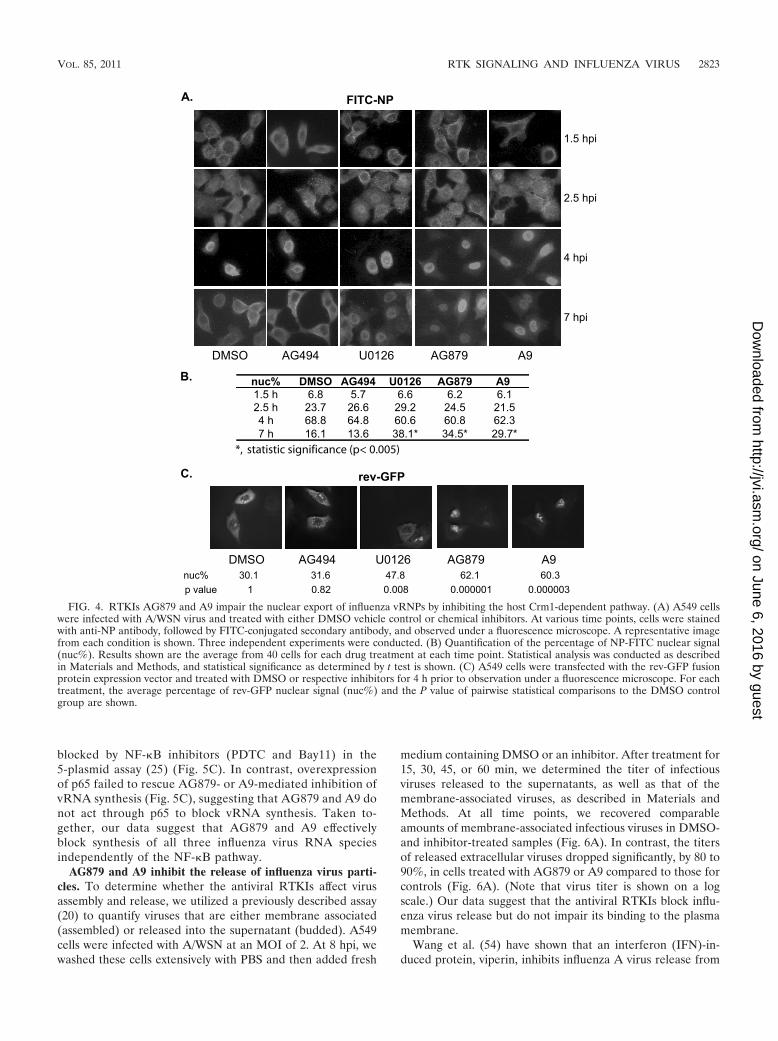
AG879 and A9 block nuclear export of influenza vRNPs byinhibiting the host Crm1-dependent pathway. Influenza virusRNA synthesis occurs in the nucleus. Incoming vRNPs, whichcontain NP-encapsidated viral genomic RNAs with associatedviral polymerase proteins (PA, PB1, and PB2), must be im-ported to the nucleus for RNA transcription or replication tooccur. Newly synthesized vRNPs are exported from the nu-cleus via the cellular Crm1-mediated nuclear export pathway(12, 28, 55) as a (Crm1-RanGTP)-NEP-M1-vRNP complex (1)to be packaged into virions at the plasma membrane. To in-vestigate the effects of AG879 and A9 on nucleocytoplasmictrafficking of vRNPs, we infected A549 cells with A/WSN at anMOI of 5 and then treated the cells with various inhibitors.U0126, a MEK inhibitor that has been shown to block influ-enza vRNP nuclear export (42), was included as a positivecontrol. Localization of the NP protein was assessed by indirectimmunofluorescence at various times postinfection. Represen-tative images are shown in Fig. 4A. Under all conditions tested,the incoming vRNPs were detected mainly in the cytoplasm at1.5 hpi, in both the cytoplasm and the nuclei at 2.5 hpi, andmostly within the nuclei at 4 hpi, suggesting that AG879 andA9 do not affect vRNP nuclear import early in virus infection.In contrast, at a later stage of virus replication (7 hpi), newlysynthesized vRNPs were detected predominantly in the cyto-plasm of cells treated with vehicle control DMSO or negativecontrol AG494 but predominantly within the nuclei of thosetreated with U0126, AG879, or A9. To quantitate these find-ings, we calculated the mean percentage of FITC-NP signal
within the nucleus (nuc%) in 40 cells for each drug treatmentat each time point (Fig. 4B); statistical analysis confirmed thatonly samples treated with U0126, AG879, and A9 at 7 hpi weresignificantly different from the DMSO control (P � 0.005).These data suggest that, like the MEK inhibitor U0126, AG879and A9 prevent the nuclear export of vRNPs late in infection.
As influenza vRNP nuclear export not only depends on thecellular Crm1 pathway but also requires the viral proteinsNEP/NS2 and M1, the nuclear retention of vRNPs followingAG879 or A9 treatment might be due either to direct inhibi-tion of the cellular Crm1 nuclear export pathway or to signif-icantly decreased levels of viral protein expression (Fig. 2D)resulting from a possible blockade of viral RNA synthesis (seebelow for more details). We therefore examined the effect ofAG879 and A9 on the nucleocytoplasmic trafficking of theHIV Rev protein, whose nuclear export depends on the Crm1pathway (34). As shown in Fig. 4C, the rev-GFP fusion proteinshowed a predominant nuclear localization in control samplesthat had been treated with DMSO or AG494. The MEK in-hibitor U0126 moderately increased the nuclear localization ofrev-GFP, while AG879 and A9 led to a predominant nuclearretention. Statistical analysis of the proportion of the rev-GFPsignal found within nuclei (nuc%) confirmed that nuclear lo-calization of rev-GFP was significantly increased by U0126,AG879, or A9 treatment compared to that for controls (P �0.008, P � 0.000001, and P � 0.000003, respectively). Thesefindings strongly suggest that AG879 and A9 directly inhibitthe cellular Crm1 nuclear export pathway, which may largelyaccount for their ability to cause nuclear retention of influenzavRNPs (Fig. 4A).
AG879 and A9 strongly inhibit influenza virus RNA synthe-sis. We then evaluated whether AG879 and A9 can directlyinhibit viral RNA synthesis, using a 5-plasmid assay (25) that isbased on the reconstitution of all of the cis- and trans-actingelements required for influenza virus RNA replication andtranscription. Compared to results for vehicle control DMSOand negative control AG494, the addition of AG879, A9, orribavirin, a known inhibitor of influenza virus RNA synthesis,decreased the LUC activity expressed from either vRNA orcRNA templates by �95% (Fig. 5A), strongly suggesting thatthe RTKIs inhibit viral RNA synthesis. To validate this findingin virus-infected cells, we infected A549 cells with A/WSN atan MOI of 1 and then treated cells with various inhibitors at 1hpi. Whole-cell RNA was isolated from the infected cells at 5hpi and quantified for vRNA, cRNA, and mRNA using anestablished quantitative real-time RT-PCR assay (25). The lev-els of all three RNA species were found to be significantlydecreased in AG879- and A9-treated cells, as indicated byincreased threshold cycle (CT) values (Fig. 5B).
Our previous studies have suggested that activation ofNF-�B signaling promotes the efficient replication of influenzavirus RNAs (25). Although NF-�B is also a downstream path-way of RTK signaling, AG879 and A9 are unlikely to blockviral RNA synthesis via that pathway, as we have shown thatNF-�B signaling is differentially involved in vRNA but notmRNA or cRNA synthesis (25), whereas AG879 and A9 blocksynthesis of all three viral RNA species (Fig. 5A and B). Thisis further evidenced by overexpression of NF-�B subunitp65, which we have shown can increase influenza vRNAsynthesis and also rescue vRNA synthesis that has been
FIG. 3. AG879 and A9 inhibit the later stages of the influenza viruslife cycle. (A) Time course analysis of the AG879 and A9 inhibitoryeffects on influenza A virus replication. A549 cells were infected withA/WSN at an MOI of 1 and, at different time points, treated withDMSO or the respective inhibitor. Virus titer at 9 hpi was determinedby plaque assay. (B) Effects of AG879 and A9 on virus attachment.A549 cells were preincubated with DMSO or chemicals for 30 min andinfected with A/WSN (MOI of 2) at 4°C for 2 h. After cells werewashed 6 times with PBS, cell lysates were prepared by rapid freeze-thaw. Virus titer was determined by plaque assay. Results shown arethe averages from at least three independent experiments. Error barsindicate SD.
2822 KUMAR ET AL. J. VIROL.
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from
blocked by NF-�B inhibitors (PDTC and Bay11) in the5-plasmid assay (25) (Fig. 5C). In contrast, overexpressionof p65 failed to rescue AG879- or A9-mediated inhibition ofvRNA synthesis (Fig. 5C), suggesting that AG879 and A9 donot act through p65 to block vRNA synthesis. Taken to-gether, our data suggest that AG879 and A9 effectivelyblock synthesis of all three influenza virus RNA speciesindependently of the NF-�B pathway.
AG879 and A9 inhibit the release of influenza virus parti-cles. To determine whether the antiviral RTKIs affect virusassembly and release, we utilized a previously described assay(20) to quantify viruses that are either membrane associated(assembled) or released into the supernatant (budded). A549cells were infected with A/WSN at an MOI of 2. At 8 hpi, wewashed these cells extensively with PBS and then added fresh
medium containing DMSO or an inhibitor. After treatment for15, 30, 45, or 60 min, we determined the titer of infectiousviruses released to the supernatants, as well as that of themembrane-associated viruses, as described in Materials andMethods. At all time points, we recovered comparableamounts of membrane-associated infectious viruses in DMSO-and inhibitor-treated samples (Fig. 6A). In contrast, the titersof released extracellular viruses dropped significantly, by 80 to90%, in cells treated with AG879 or A9 compared to those forcontrols (Fig. 6A). (Note that virus titer is shown on a logscale.) Our data suggest that the antiviral RTKIs block influ-enza virus release but do not impair its binding to the plasmamembrane.
Wang et al. (54) have shown that an interferon (IFN)-in-duced protein, viperin, inhibits influenza A virus release from
FIG. 4. RTKIs AG879 and A9 impair the nuclear export of influenza vRNPs by inhibiting the host Crm1-dependent pathway. (A) A549 cellswere infected with A/WSN virus and treated with either DMSO vehicle control or chemical inhibitors. At various time points, cells were stainedwith anti-NP antibody, followed by FITC-conjugated secondary antibody, and observed under a fluorescence microscope. A representative imagefrom each condition is shown. Three independent experiments were conducted. (B) Quantification of the percentage of NP-FITC nuclear signal(nuc%). Results shown are the average from 40 cells for each drug treatment at each time point. Statistical analysis was conducted as describedin Materials and Methods, and statistical significance as determined by t test is shown. (C) A549 cells were transfected with the rev-GFP fusionprotein expression vector and treated with DMSO or respective inhibitors for 4 h prior to observation under a fluorescence microscope. For eachtreatment, the average percentage of rev-GFP nuclear signal (nuc%) and the P value of pairwise statistical comparisons to the DMSO controlgroup are shown.
VOL. 85, 2011 RTK SIGNALING AND INFLUENZA VIRUS 2823
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from
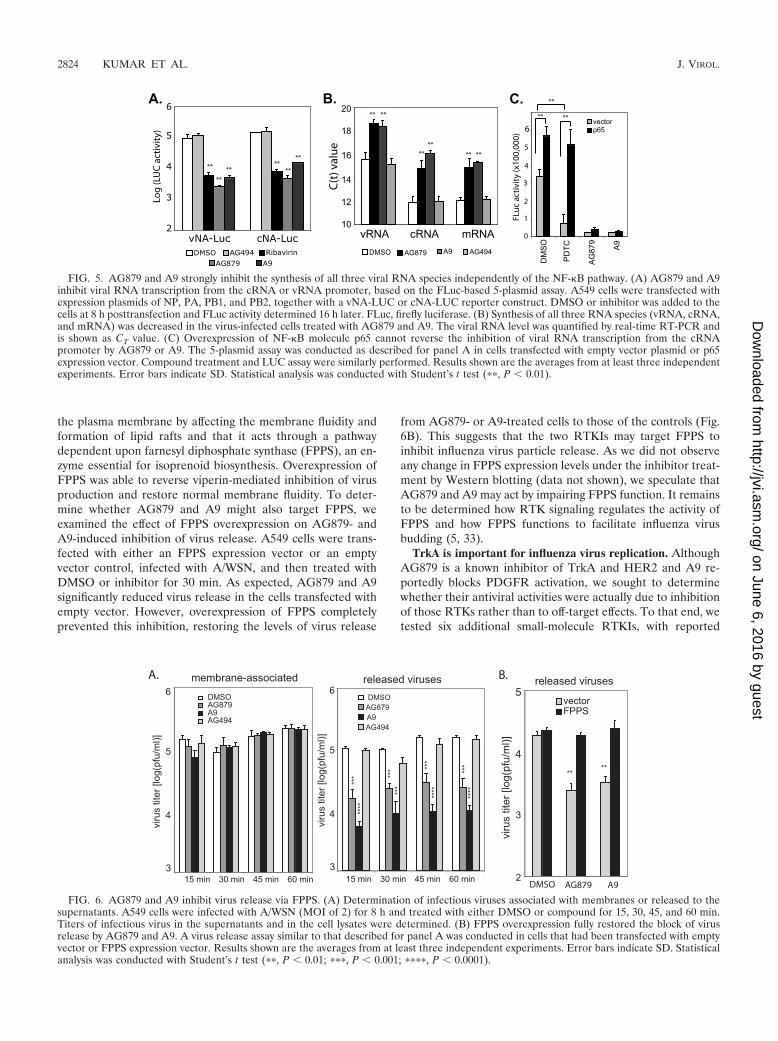
the plasma membrane by affecting the membrane fluidity andformation of lipid rafts and that it acts through a pathwaydependent upon farnesyl diphosphate synthase (FPPS), an en-zyme essential for isoprenoid biosynthesis. Overexpression ofFPPS was able to reverse viperin-mediated inhibition of virusproduction and restore normal membrane fluidity. To deter-mine whether AG879 and A9 might also target FPPS, weexamined the effect of FPPS overexpression on AG879- andA9-induced inhibition of virus release. A549 cells were trans-fected with either an FPPS expression vector or an emptyvector control, infected with A/WSN, and then treated withDMSO or inhibitor for 30 min. As expected, AG879 and A9significantly reduced virus release in the cells transfected withempty vector. However, overexpression of FPPS completelyprevented this inhibition, restoring the levels of virus release
from AG879- or A9-treated cells to those of the controls (Fig.6B). This suggests that the two RTKIs may target FPPS toinhibit influenza virus particle release. As we did not observeany change in FPPS expression levels under the inhibitor treat-ment by Western blotting (data not shown), we speculate thatAG879 and A9 may act by impairing FPPS function. It remainsto be determined how RTK signaling regulates the activity ofFPPS and how FPPS functions to facilitate influenza virusbudding (5, 33).
TrkA is important for influenza virus replication. AlthoughAG879 is a known inhibitor of TrkA and HER2 and A9 re-portedly blocks PDGFR activation, we sought to determinewhether their antiviral activities were actually due to inhibitionof those RTKs rather than to off-target effects. To that end, wetested six additional small-molecule RTKIs, with reported
FIG. 5. AG879 and A9 strongly inhibit the synthesis of all three viral RNA species independently of the NF-�B pathway. (A) AG879 and A9inhibit viral RNA transcription from the cRNA or vRNA promoter, based on the FLuc-based 5-plasmid assay. A549 cells were transfected withexpression plasmids of NP, PA, PB1, and PB2, together with a vNA-LUC or cNA-LUC reporter construct. DMSO or inhibitor was added to thecells at 8 h posttransfection and FLuc activity determined 16 h later. FLuc, firefly luciferase. (B) Synthesis of all three RNA species (vRNA, cRNA,and mRNA) was decreased in the virus-infected cells treated with AG879 and A9. The viral RNA level was quantified by real-time RT-PCR andis shown as CT value. (C) Overexpression of NF-�B molecule p65 cannot reverse the inhibition of viral RNA transcription from the cRNApromoter by AG879 or A9. The 5-plasmid assay was conducted as described for panel A in cells transfected with empty vector plasmid or p65expression vector. Compound treatment and LUC assay were similarly performed. Results shown are the averages from at least three independentexperiments. Error bars indicate SD. Statistical analysis was conducted with Student’s t test (��, P � 0.01).
FIG. 6. AG879 and A9 inhibit virus release via FPPS. (A) Determination of infectious viruses associated with membranes or released to thesupernatants. A549 cells were infected with A/WSN (MOI of 2) for 8 h and treated with either DMSO or compound for 15, 30, 45, and 60 min.Titers of infectious virus in the supernatants and in the cell lysates were determined. (B) FPPS overexpression fully restored the block of virusrelease by AG879 and A9. A virus release assay similar to that described for panel A was conducted in cells that had been transfected with emptyvector or FPPS expression vector. Results shown are the averages from at least three independent experiments. Error bars indicate SD. Statisticalanalysis was conducted with Student’s t test (��, P � 0.01; ���, P � 0.001; ����, P � 0.0001).
2824 KUMAR ET AL. J. VIROL.
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from
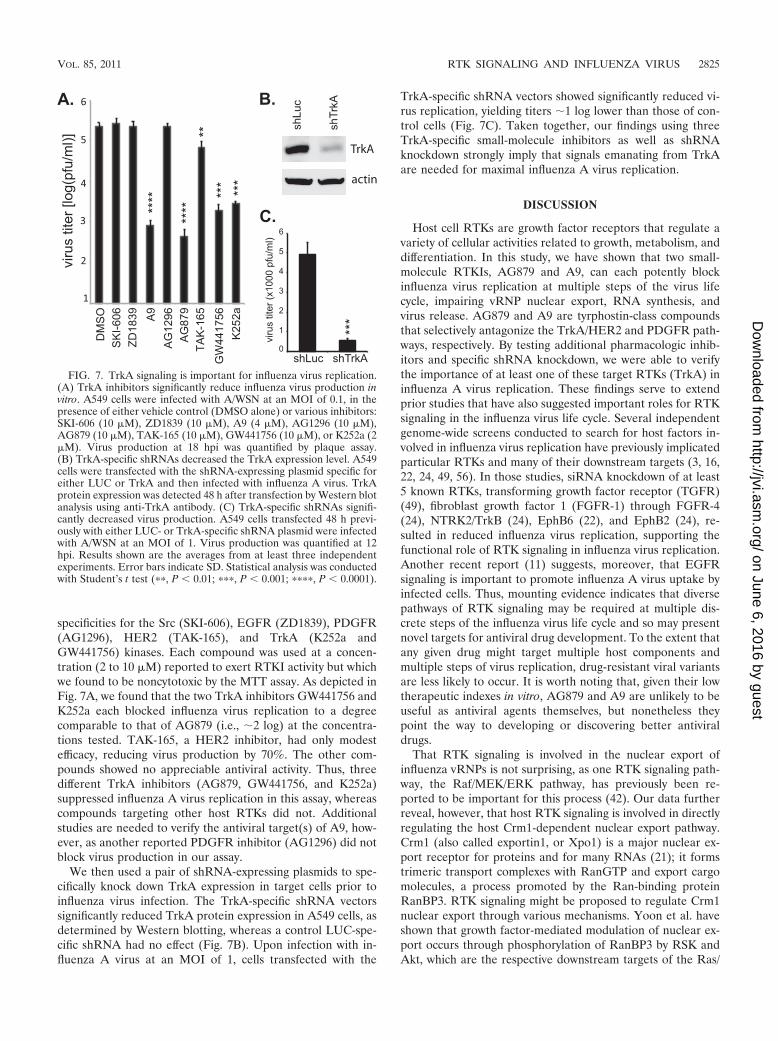
specificities for the Src (SKI-606), EGFR (ZD1839), PDGFR(AG1296), HER2 (TAK-165), and TrkA (K252a andGW441756) kinases. Each compound was used at a concen-tration (2 to 10 �M) reported to exert RTKI activity but whichwe found to be noncytotoxic by the MTT assay. As depicted inFig. 7A, we found that the two TrkA inhibitors GW441756 andK252a each blocked influenza virus replication to a degreecomparable to that of AG879 (i.e., �2 log) at the concentra-tions tested. TAK-165, a HER2 inhibitor, had only modestefficacy, reducing virus production by 70%. The other com-pounds showed no appreciable antiviral activity. Thus, threedifferent TrkA inhibitors (AG879, GW441756, and K252a)suppressed influenza A virus replication in this assay, whereascompounds targeting other host RTKs did not. Additionalstudies are needed to verify the antiviral target(s) of A9, how-ever, as another reported PDGFR inhibitor (AG1296) did notblock virus production in our assay.
We then used a pair of shRNA-expressing plasmids to spe-cifically knock down TrkA expression in target cells prior toinfluenza virus infection. The TrkA-specific shRNA vectorssignificantly reduced TrkA protein expression in A549 cells, asdetermined by Western blotting, whereas a control LUC-spe-cific shRNA had no effect (Fig. 7B). Upon infection with in-fluenza A virus at an MOI of 1, cells transfected with the
TrkA-specific shRNA vectors showed significantly reduced vi-rus replication, yielding titers �1 log lower than those of con-trol cells (Fig. 7C). Taken together, our findings using threeTrkA-specific small-molecule inhibitors as well as shRNAknockdown strongly imply that signals emanating from TrkAare needed for maximal influenza A virus replication.
DISCUSSION
Host cell RTKs are growth factor receptors that regulate avariety of cellular activities related to growth, metabolism, anddifferentiation. In this study, we have shown that two small-molecule RTKIs, AG879 and A9, can each potently blockinfluenza virus replication at multiple steps of the virus lifecycle, impairing vRNP nuclear export, RNA synthesis, andvirus release. AG879 and A9 are tyrphostin-class compoundsthat selectively antagonize the TrkA/HER2 and PDGFR path-ways, respectively. By testing additional pharmacologic inhib-itors and specific shRNA knockdown, we were able to verifythe importance of at least one of these target RTKs (TrkA) ininfluenza A virus replication. These findings serve to extendprior studies that have also suggested important roles for RTKsignaling in the influenza virus life cycle. Several independentgenome-wide screens conducted to search for host factors in-volved in influenza virus replication have previously implicatedparticular RTKs and many of their downstream targets (3, 16,22, 24, 49, 56). In those studies, siRNA knockdown of at least5 known RTKs, transforming growth factor receptor (TGFR)(49), fibroblast growth factor 1 (FGFR-1) through FGFR-4(24), NTRK2/TrkB (24), EphB6 (22), and EphB2 (24), re-sulted in reduced influenza virus replication, supporting thefunctional role of RTK signaling in influenza virus replication.Another recent report (11) suggests, moreover, that EGFRsignaling is important to promote influenza A virus uptake byinfected cells. Thus, mounting evidence indicates that diversepathways of RTK signaling may be required at multiple dis-crete steps of the influenza virus life cycle and so may presentnovel targets for antiviral drug development. To the extent thatany given drug might target multiple host components andmultiple steps of virus replication, drug-resistant viral variantsare less likely to occur. It is worth noting that, given their lowtherapeutic indexes in vitro, AG879 and A9 are unlikely to beuseful as antiviral agents themselves, but nonetheless theypoint the way to developing or discovering better antiviraldrugs.
That RTK signaling is involved in the nuclear export ofinfluenza vRNPs is not surprising, as one RTK signaling path-way, the Raf/MEK/ERK pathway, has previously been re-ported to be important for this process (42). Our data furtherreveal, however, that host RTK signaling is involved in directlyregulating the host Crm1-dependent nuclear export pathway.Crm1 (also called exportin1, or Xpo1) is a major nuclear ex-port receptor for proteins and for many RNAs (21); it formstrimeric transport complexes with RanGTP and export cargomolecules, a process promoted by the Ran-binding proteinRanBP3. RTK signaling might be proposed to regulate Crm1nuclear export through various mechanisms. Yoon et al. haveshown that growth factor-mediated modulation of nuclear ex-port occurs through phosphorylation of RanBP3 by RSK andAkt, which are the respective downstream targets of the Ras/
FIG. 7. TrkA signaling is important for influenza virus replication.(A) TrkA inhibitors significantly reduce influenza virus production invitro. A549 cells were infected with A/WSN at an MOI of 0.1, in thepresence of either vehicle control (DMSO alone) or various inhibitors:SKI-606 (10 �M), ZD1839 (10 �M), A9 (4 �M), AG1296 (10 �M),AG879 (10 �M), TAK-165 (10 �M), GW441756 (10 �M), or K252a (2�M). Virus production at 18 hpi was quantified by plaque assay.(B) TrkA-specific shRNAs decreased the TrkA expression level. A549cells were transfected with the shRNA-expressing plasmid specific foreither LUC or TrkA and then infected with influenza A virus. TrkAprotein expression was detected 48 h after transfection by Western blotanalysis using anti-TrkA antibody. (C) TrkA-specific shRNAs signifi-cantly decreased virus production. A549 cells transfected 48 h previ-ously with either LUC- or TrkA-specific shRNA plasmid were infectedwith A/WSN at an MOI of 1. Virus production was quantified at 12hpi. Results shown are the averages from at least three independentexperiments. Error bars indicate SD. Statistical analysis was conductedwith Student’s t test (��, P � 0.01; ���, P � 0.001; ����, P � 0.0001).
VOL. 85, 2011 RTK SIGNALING AND INFLUENZA VIRUS 2825
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from

ERK/RSK and PI3K/Akt pathways (60). We have not yet eval-uated whether AG879 or A9 can block the phosphorylation ofRanBP3 or affect other components of the Crm1 nuclear ex-port complex. In addition, interaction of Crm1 with cargoproteins can be regulated by cargo phosphorylation (21). Sev-eral viral protein components of influenza vRNPs are known tobe phosphoproteins, including PA (47), NP (23, 43), M1 (13,14, 23), and NEP/NS2 (46), and hyperphosphorylation of amutant M1 protein has been shown to cause its aberrant nu-clear retention (57). Conflicting results concerning the rela-tionship between NP phosphorylation and nuclear export havebeen reported: one study reported that phosphorylated NPaccumulated in the nucleus and cytoplasm with similar kinetics(41), suggesting that phosphorylation did not affect NP nucleo-cytoplasmic trafficking, whereas another study found thatvRNPs isolated from the nucleus contained much more phos-phorylated NP than those from the cytoplasm, consistent withdifferential nuclear export (2). Whether phosphorylation ofvRNP components regulates nuclear export and whetherAG879 and A9 cause nuclear retention of vRNPs by specifi-cally blocking that process require further investigation.
A variety of host signaling pathways and other host factorshave been implicated in regulating influenza virus RNA syn-thesis, but their underlying mechanisms are largely unknown.Several cellular factors that stimulate influenza virus RNAsynthesis have been identified, including Hsp90 (31), the splic-ing-related factor AP56/BAT1 (30), and the chaperone Tat-SF1 (32). Recent proteomic screens using siRNA librarieshave identified hundreds of candidate host factors that affectinfluenza virus replication (3, 16, 22, 24, 49), but exactly whichhost factors are functionally required for viral RNA synthesisand how they function await further research. We have previ-ously shown that NF-�B signaling can differentially regulateinfluenza virus RNA synthesis by promoting vRNA but notmRNA or cRNA synthesis (25). Here we present evidence thathost RTK signaling, by contrast, is important for the synthesisof all three influenza virus RNA species, through mechanismsthat remain to be characterized.
Influenza virus particles are assembled and bud at theplasma membrane at sites that are enriched in cholesterol andglycosphingolipids, forming lipid raft microdomains (5, 33).The eventual release of virus from the plasma membrane re-quires closure of the bud and separation of the virus particlefrom the host membrane, processes that are influenced by viralcomponents as well as host factors. It has been shown thatinhibitors of certain G proteins and protein kinases can inhibitinfluenza virus budding (20), suggesting an important role forhost signaling in this process. An enzyme essential for iso-prenoid biosynthesis, FPPS, appears to be critically involved ininfluenza virus budding, possibly owing to its role in the for-mation of lipid rafts (54). At least two different classes ofinfluenza virus inhibitors, the IFN response protein viperin(54) and the antiviral RTKIs AG879 and A9 (Fig. 6B), blockinfluenza virus release via FPPS. Moreover, siRNA-mediatedknockdown of FPPS significantly reduces influenza virus rep-lication (54), confirming that FPPS is a potential target fordeveloping anti-influenza virus drugs. Further studies will castlight on the functional mechanisms by which FPPS, as well asother host factors, affects influenza virus budding and release.
ACKNOWLEDGMENTS
We thank Y. Kawaoka (University of Wisconsin—Madison) for theinfluenza virus protein plasmids, P. Creswell (Yale University) for theFPPS expression plasmid, W. Greene (UCSF) for the plasmid express-ing NF-�B molecule p65, A. Mergia (University of Florida) for therev-GFP expression plasmid, and P. Edward (UCLA) for the anti-FPPS antibody.
This work was supported by NIH grants AI067704 to Tristram G.Parslow and AI083409 to Yuying Liang.
REFERENCES
1. Akarsu, H., et al. 2003. Crystal structure of the M1 protein-binding domainof the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 22:4646–4655.
2. Almond, J. W., and V. Felsenreich. 1982. Phosphorylation of the nucleopro-tein of an avian influenza virus. J. Gen. Virol. 60:295–305.
3. Brass, A. L., et al. 2009. The IFITM proteins mediate cellular resistance toinfluenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254.
4. Bukrinskaya, A. G., N. K. Vorkunova, G. V. Kornilayeva, R. A. Narmanbe-tova, and G. K. Vorkunova. 1982. Influenza virus uncoating in infected cellsand effect of rimantadine. J. Gen. Virol. 60:49–59.
5. Chen, B. J., and R. A. Lamb. 2008. Mechanisms for enveloped virus budding:can some viruses do without an ESCRT? Virology 372:221–232.
6. Chu, J. J., and P. L. Yang. 2007. c-Src protein kinase inhibitors blockassembly and maturation of dengue virus. Proc. Natl. Acad. Sci. U. S. A.104:3520–3525.
7. Cox, N. J., and K. Subbarao. 1999. Influenza. Lancet 354:1277–1282.8. de Jong, M. D., et al. 2005. Oseltamivir resistance during treatment of
influenza A (H5N1) infection. N. Engl. J. Med. 353:2667–2672.9. Ehrhardt, C., et al. 2006. Bivalent role of the phosphatidylinositol-3-kinase
(PI3K) during influenza virus infection and host cell defence. Cell. Micro-biol. 8:1336–1348.
10. Ehrhardt, C., et al. 2007. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 81:3058–3067.
11. Eierhoff, T., E. R. Hrincius, U. Rescher, S. Ludwig, and C. Ehrhardt. 2010.The epidermal growth factor receptor (EGFR) promotes uptake of influenzaA viruses (IAV) into host cells. PLoS Pathog. 6:e1001099.
12. Elton, D., et al. 2001. Interaction of the influenza virus nucleoprotein withthe cellular CRM1-mediated nuclear export pathway. J. Virol. 75:408–419.
13. Gregoriades, A., T. Christie, and K. Markarian. 1984. The membrane (M1)protein of influenza virus occurs in two forms and is a phosphoprotein.J. Virol. 49:229–235.
14. Gregoriades, A., G. G. Guzman, and E. Paoletti. 1990. The phosphory-lation of the integral membrane (M1) protein of influenza virus. VirusRes. 16:27–41.
15. Hale, B. G., D. Jackson, Y. H. Chen, R. A. Lamb, and R. E. Randall. 2006.Influenza A virus NS1 protein binds p85beta and activates phosphatidylino-sitol-3-kinase signaling. Proc. Natl. Acad. Sci. U. S. A. 103:14194–14199.
16. Hao, L., et al. 2008. Drosophila RNAi screen identifies host genes importantfor influenza virus replication. Nature 454:890–893.
17. Hay, A. J., A. J. Wolstenholme, J. J. Skehel, and M. H. Smith. 1985. Themolecular basis of the specific anti-influenza action of amantadine. EMBO J.4:3021–3024.
18. Heldin, C. H. 1995. Dimerization of cell surface receptors in signal trans-duction. Cell 80:213–223.
19. Hirsch, A. J., et al. 2005. The Src family kinase c-Yes is required for matu-ration of West Nile virus particles. J. Virol. 79:11943–11951.
20. Hui, E. K., and D. P. Nayak. 2002. Role of G protein and protein kinasesignalling in influenza virus budding in MDCK cells. J. Gen. Virol. 83:3055–3066.
21. Hutten, S., and R. H. Kehlenbach. 2007. CRM1-mediated nuclear export: tothe pore and beyond. Trends Cell Biol. 17:193–201.
22. Karlas, A., et al. 2010. Genome-wide RNAi screen identifies human hostfactors crucial for influenza virus replication. Nature 463:818–822.
23. Kistner, O., K. Muller, and C. Scholtissek. 1989. Differential phosphoryla-tion of the nucleoprotein of influenza A viruses. J. Gen. Virol. 70:2421–2431.
24. Konig, R., et al. 2010. Human host factors required for influenza virusreplication. Nature 463:813–817.
25. Kumar, N., Z. T. Xin, Y. Liang, H. Ly, and Y. Liang. 2008. NF-kappaBsignaling differentially regulates influenza virus RNA synthesis. J. Virol.82:9880–9889.
26. Le, Q. M., et al. 2005. Avian flu: isolation of drug-resistant H5N1 virus.Nature 437:1108.
27. Levitzki, A., and C. Gilon. 1991. Tyrphostins as molecular tools and potentialantiproliferative drugs. Trends Pharmacol. Sci. 12:171–174.
28. Ma, K., A. M. Roy, and G. R. Whittaker. 2001. Nuclear export of influenzavirus ribonucleoproteins: identification of an export intermediate at thenuclear periphery. Virology 282:215–220.
2826 KUMAR ET AL. J. VIROL.
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from

29. Martin, K., and A. Helenius. 1991. Nuclear transport of influenza virusribonucleoproteins: the viral matrix protein (M1) promotes export and in-hibits import. Cell 67:117–130.
30. Momose, F., et al. 2001. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viralRNA synthesis. J. Virol. 75:1899–1908.
31. Momose, F., et al. 2002. Identification of Hsp90 as a stimulatory host factorinvolved in influenza virus RNA synthesis. J. Biol. Chem. 277:45306–45314.
32. Naito, T., et al. 2007. An influenza virus replicon system in yeast identifiedTat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc. Natl.Acad. Sci. U. S. A. 104:18235–18240.
33. Nayak, D. P., E. K. Hui, and S. Barman. 2004. Assembly and budding ofinfluenza virus. Virus Res. 106:147–165.
34. Neville, M., F. Stutz, L. Lee, L. I. Davis, and M. Rosbash. 1997. The impor-tin-beta family member Crm1p bridges the interaction between Rev and thenuclear pore complex during nuclear export. Curr. Biol. 7:767–775.
35. Ohmichi, M., et al. 1993. The tyrosine kinase inhibitor tyrphostin blocks thecellular actions of nerve growth factor. Biochemistry 32:4650–4658.
36. Osherov, N., A. Gazit, C. Gilon, and A. Levitzki. 1993. Selective inhibition ofthe epidermal growth factor and HER2/neu receptors by tyrphostins. J. Biol.Chem. 268:11134–11142.
37. Palese, P., and R. W. Compans. 1976. Inhibition of influenza virus replicationin tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid(FANA): mechanism of action. J. Gen. Virol. 33:159–163.
38. Palese, P., and M. L. Shaw. 2007. Orthomyxoviridae: the viruses and theirreplication, p. 1647–1689. In D. M. Knipe and P. M. Howley (ed.), Fieldsvirology, 5th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA.
39. Palese, P., K. Tobita, M. Ueda, and R. W. Compans. 1974. Characterizationof temperature sensitive influenza virus mutants defective in neuraminidase.Virology 61:397–410.
40. Pawson, T. 1995. Protein modules and signalling networks. Nature 373:573–580.
41. Petri, T., and N. J. Dimmock. 1981. Phosphorylation of influenza virusnucleoprotein in vivo. J. Gen. Virol. 57:185–190.
42. Pleschka, S., et al. 2001. Influenza virus propagation is impaired by inhibitionof the Raf/MEK/ERK signalling cascade. Nat. Cell. Biol. 3:301–305.
43. Privalsky, M. L., and E. E. Penhoet. 1978. Influenza virus proteins: identity,synthesis, and modification analyzed by two-dimensional gel electrophoresis.Proc. Natl. Acad. Sci. U. S. A. 75:3625–3629.
44. Puthavathana, P., et al. 2005. Molecular characterization of the completegenome of human influenza H5N1 virus isolates from Thailand. J. Gen.Virol. 86:423–433.
45. Regan, J. F., Y. Liang, and T. G. Parslow. 2006. Defective assembly of
influenza A virus due to a mutation in the polymerase subunit PA. J. Virol.80:252–261.
46. Richardson, J. C., and R. K. Akkina. 1991. NS2 protein of influenza virus isfound in purified virus and phosphorylated in infected cells. Arch. Virol.116:69–80.
47. Sanz-Ezquerro, J. J., et al. 1998. The PA influenza virus polymerase subunitis a phosphorylated protein. J. Gen. Virol. 79:471–478.
48. Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell 103:211–225.
49. Shapira, S. D., et al. 2009. A physical and regulatory map of host-influenzainteractions reveals pathways in H1N1 infection. Cell 139:1255–1267.
50. Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusionin virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569.
51. Stantchev, T. S., I. Markovic, W. G. Telford, K. A. Clouse, and C. C. Broder.2007. The tyrosine kinase inhibitor genistein blocks HIV-1 infection in pri-mary human macrophages. Virus Res. 123:178–189.
52. Ullrich, A., and J. Schlessinger. 1990. Signal transduction by receptors withtyrosine kinase activity. Cell 61:203–212.
53. Vela, E. M., G. C. Bowick, N. K. Herzog, and J. F. Aronson. 2008. Genisteintreatment of cells inhibits arenavirus infection. Antiviral Res. 77:153–156.
54. Wang, X., E. R. Hinson, and P. Cresswell. 2007. The interferon-inducibleprotein viperin inhibits influenza virus release by perturbing lipid rafts. CellHost Microbe 2:96–105.
55. Watanabe, K., et al. 2001. Inhibition of nuclear export of ribonucleoproteincomplexes of influenza virus by leptomycin B. Virus Res. 77:31–42.
56. Watanabe, T., S. Watanabe, and Y. Kawaoka. 2010. Cellular networks in-volved in the influenza virus life cycle. Cell Host Microbe 7:427–439.
57. Whittaker, G., I. Kemler, and A. Helenius. 1995. Hyperphosphorylation ofmutant influenza virus matrix protein, M1, causes its retention in the nu-cleus. J. Virol. 69:439–445.
58. Whittaker, G. R., and P. Digard. 2006. Entry and intracellular transport ofinfluenza virus, p. 37–64. In Y. Kawaoka (ed.), Influenza virology—currenttopics. Caister Academic Press, Norwich, United Kingdom.
59. Winquist, A. G., K. Fukuda, C. B. Bridges, and N. Cox. 1999. Neuraminidaseinhibitors for treatment of influenza A and B infections. MMWR Surveill.Summ. 48:1–9.
60. Yoon, S. O., et al. 2008. Ran-binding protein 3 phosphorylation links the Rasand PI3-kinase pathways to nucleocytoplasmic transport. Mol. Cell 29:362–375.
61. Yura, Y., H. Yoshida, and M. Sato. 1993. Inhibition of herpes simplex virusreplication by genistein, an inhibitor of protein-tyrosine kinase. Arch. Virol.132:451–461.
VOL. 85, 2011 RTK SIGNALING AND INFLUENZA VIRUS 2827
on June 6, 2016 by guesthttp://jvi.asm
.org/D
ownloaded from




















