
Recent developments in the analysis of brominated flame retardants and brominated natural compounds
27
Journal of Chromatography A, 1153 (2007) 145–171 Review Recent developments in the analysis of brominated flame retardants and brominated natural compounds Adrian Covaci a,b,∗ , Stefan Voorspoels a , Lourdes Ramos c , Hugo Neels a , Ronny Blust b a Toxicological Centre, Department of Pharmaceutical Sciences, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk, Belgium b Ecophysiology, Biochemistry and Toxicology Group, Department of Biology, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium c CSIC, IQOG, Department of Instrumental Analysis and Environmental Chemistry, Juan Cierva 3, E-28006 Madrid, Spain Available online 6 December 2006 Abstract This article reviews recent literature on the analysis of brominated flame retardants (BFRs) and brominated natural compounds (BNCs). The main literature sources are reviews from the last five years and research articles reporting new analytical developments published between 2003 and 2006. Sample pretreatment, extraction, clean-up and fractionation, injection techniques, chromatographic separation, detection methods, quality control and method validation are discussed. Only few new techniques, such as solid-phase microextraction (SPME) or pressurized liquid extraction (PLE), have been investigated for their ability of combining the extraction and clean-up steps. With respect to the separation of BFRs, the most important developments were the use of comprehensive two-dimensional gas chromatography for polybrominated diphenyl ethers (PBDEs) and the growing tendency for liquid-chromatographic techniques for hexabromocyclododecane (HBCD) stereoisomers and of tetrabromobisphenol-A (TBBP-A). At the detection stage, mass spectrometry (MS) has been developed as well-established and reliable technology in the identification and quantification of BFRs. A growing attention has been paid to quality assurance. Interlaboratory exercises directed towards BFRs have grown in popularity and have enabled laboratories to validate analytical methods and to guarantee the quality of their results. The analytical procedures used for the identification and characterization of several classes of BNCs, such as methoxylated polybrominated diphenyl ethers (MeO-PBDEs) (also metabolites of PBDEs), halogenated methyl or dimethyl bipyrroles (DBPs), are reviewed here for the first time. These compounds were generally identified during the routine analysis of BFRs and have received little attention until recently. For each topic, an overview is presented of its current status. © 2006 Elsevier B.V. All rights reserved. Keywords: Brominated flame retardants; Brominated natural compounds; Analysis; Review Contents 1. Introduction ............................................................................................................ 146 2. Brominated flame retardants .............................................................................................. 146 2.1. Advances in sample preparation .................................................................................... 146 2.1.1. Extraction ................................................................................................ 146 2.1.2. Clean-up and fractionation ................................................................................. 153 2.1.3. Quality assurance/quality control ........................................................................... 154 2.2. Advances in GC–MS and LC–MS analysis .......................................................................... 155 2.2.1. Polybrominated diphenyl ethers ............................................................................ 155 2.2.2. Hexabromocyclododecanes ................................................................................ 160 2.2.3. Tetrabromobisphenol-A ................................................................................... 161 2.2.4. Brominated flame retardants in polymers .................................................................... 162 2.3. Interlaboratory studies on brominated flame retardants ................................................................ 162 ∗ Corresponding author. Tel.: +32 3 820 2704; fax: +32 3 820 2722. E-mail address: [email protected] (A. Covaci). 0021-9673/$ – see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.chroma.2006.11.060
Transcript of Recent developments in the analysis of brominated flame retardants and brominated natural compounds

Journal of Chromatography A, 1153 (2007) 145–171
Review
Recent developments in the analysis of brominated flame retardantsand brominated natural compounds
Adrian Covaci a,b,∗, Stefan Voorspoels a, Lourdes Ramos c, Hugo Neels a, Ronny Blust b
a Toxicological Centre, Department of Pharmaceutical Sciences, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk, Belgiumb Ecophysiology, Biochemistry and Toxicology Group, Department of Biology, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium
c CSIC, IQOG, Department of Instrumental Analysis and Environmental Chemistry, Juan Cierva 3, E-28006 Madrid, Spain
Available online 6 December 2006
Abstract
This article reviews recent literature on the analysis of brominated flame retardants (BFRs) and brominated natural compounds (BNCs). Themain literature sources are reviews from the last five years and research articles reporting new analytical developments published between 2003 and2006. Sample pretreatment, extraction, clean-up and fractionation, injection techniques, chromatographic separation, detection methods, qualitycontrol and method validation are discussed. Only few new techniques, such as solid-phase microextraction (SPME) or pressurized liquid extraction(PLE), have been investigated for their ability of combining the extraction and clean-up steps. With respect to the separation of BFRs, the mostimportant developments were the use of comprehensive two-dimensional gas chromatography for polybrominated diphenyl ethers (PBDEs) andthe growing tendency for liquid-chromatographic techniques for hexabromocyclododecane (HBCD) stereoisomers and of tetrabromobisphenol-A(TBBP-A). At the detection stage, mass spectrometry (MS) has been developed as well-established and reliable technology in the identificationand quantification of BFRs. A growing attention has been paid to quality assurance. Interlaboratory exercises directed towards BFRs have grown inpopularity and have enabled laboratories to validate analytical methods and to guarantee the quality of their results. The analytical procedures usedfor the identification and characterization of several classes of BNCs, such as methoxylated polybrominated diphenyl ethers (MeO-PBDEs) (alsometabolites of PBDEs), halogenated methyl or dimethyl bipyrroles (DBPs), are reviewed here for the first time. These compounds were generallyidentified during the routine analysis of BFRs and have received little attention until recently. For each topic, an overview is presented of its currentstatus.© 2006 Elsevier B.V. All rights reserved.
Keywords: Brominated flame retardants; Brominated natural compounds; Analysis; Review
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1462. Brominated flame retardants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 146
2.1. Advances in sample preparation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1462.1.1. Extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1462.1.2. Clean-up and fractionation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1532.1.3. Quality assurance/quality control . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
2.2. Advances in GC–MS and LC–MS analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1552.2.1. Polybrominated diphenyl ethers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
2.2.2. Hexabromocyclododecanes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1602.2.3. Tetrabromobisphenol-A . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1612.2.4. Brominated flame retardants in polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1622.3. Interlaboratory studies on brominated flame retardants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
∗ Corresponding author. Tel.: +32 3 820 2704; fax: +32 3 820 2722.E-mail address: [email protected] (A. Covaci).
0021-9673/$ – see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.chroma.2006.11.060

146 A. Covaci et al. / J. Chromatogr. A 1153 (2007) 145–171
3. Brominated natural compounds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1643.1. Methoxylated and hydroxylated brominated diphenyl ethers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1663.2. Brominated bipyrroles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1663.3. Brominated dibenzo-p-dioxins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1673.4. Other brominated natural compounds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
4. Future perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168. . . .
. . . . .
1
n(hwipeaAhonti
Bmapl
dwBTnqBiosPTaFn
2
2
ne
pia[cppdepotbsfbofn(ftddos
2
egTts
2wa
2aH
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction
Brominated flame retardants (BFRs), including polybromi-ated diphenyl ethers (PBDEs), hexabromocyclododecaneHBCD) stereoisomers and tetrabromobisphenol-A (TBBP-A),ave attracted tremendous attention over the past decade. Theiridespread production and use, together with strong evidence of
ncreasing contamination of the environment, wildlife, and peo-le and limited knowledge of potential biological/physiologicalffects highlights the importance of identifying emerging issuesssociated with the use of BFRs. These BFRs (except TBBP-) appear to be lipophilic and bioaccumulate in wildlife andumans [1]. Furthermore, BFRs are linked to adverse physi-logical effects both in vitro and in vivo (e.g. interference ineurobehavioural development, foetal health and thyroid func-ion). However, adequate data on the effects are currently stillnsufficient to fully understand their toxicology [2].
Due to the exponential growth in the literature on analysis ofFRs, the basis for this review had to be defined adequately. Theain literature sources are reviews from the last five years [3–5]
nd research articles reporting new analytical developmentsublished in the period 2003–2006. However, when required,iterature published before 2003 has also been used.
The present review is divided into two sections. The firsteals with new trends and advances in the sequence of analysisith respect to sample treatment, separation and detection ofFRs. Specific advice on the analysis of some BFRs, such asBBP-A, BDE 209 and HBCDs, will be given separately wheneeded. A special emphasis was given to the need of adequateuality control, which is necessary for the reliable analysis ofFRs. The second section reviews for the first time the analyt-
cal procedures used for the identification and characterizationf several classes of brominated natural compounds (BNCs),uch as methoxylated polybrominated diphenyl ethers (MeO-BDEs), halogenated methyl or dimethyl bipyrroles (DBPs).hese compounds were generally identified during the routinenalysis of BFRs and have received little attention until recently.inally, future perspectives with regard to the analysis of bromi-ated compounds are discussed.
. Brominated flame retardants
.1. Advances in sample preparation
Despite the tremendous growth during recent years in theumber of papers dealing with the determination of the BFR lev-ls in different environmental matrices, no standard analytical
T(i7
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
rocedures have yet been set for these analytes. This has resultedn a variety of analytical approaches for both sample preparationnd instrumental analysis, as shown in several previous reviews3–6]. For PBDEs, sample treatment procedures have typi-ally been based on protocols previously established for traceersistent organic pollutants (POPs), such as organochlorineesticides, polychlorinated biphenyls (PCBs) or polychlorinatedioxins and furans (PCDD/Fs). Because of the complexity ofnvironmental matrices and the low levels at which these com-ounds are present, such sample treatments include a numberf steps for exhaustive extraction and preconcentration of thearget compounds, followed by purification and fractionationefore final chromatographic separation and detection. Table 1ummarises relevant data on selected analytical procedures usedor the determination of BFRs in a wide variety of abiotic andiotic samples. The similarities among the analytical meth-ds have allowed the simultaneous determination of severalamilies of microcontaminants, typically PBDEs, polybromi-ated biphenyls (PBBs) and polybrominated dioxins and furansPBDD/Fs), after a common sample preparation protocol as usedor POPs [11,46]. In most instances, the need for additional frac-ionation usually depends on the selected chromatographic andetection systems and/or on the specific study goal. Furthermore,ue to particular physico-chemical properties, the determinationf individual HBCD diastereomers and TBBP-A may requirepecific analytical approaches.
.1.1. ExtractionFor both abiotic and biotic samples, the selection of the
xtraction technique depends on the nature of the matrix investi-ated; different procedures are used for solid and liquid samples.he amount of sample required varies largely depending on
he contamination level anticipated in the sample and on theensitivity provided by the detection technique.
.1.1.1. Abiotic samples. Abiotic matrices reviewed include (i)ater; (ii) air and dust; (iii) soil, sediment and sewage sludge;
nd (iv) polymers.
.1.1.2. Water. Liquid–liquid extraction (LLE) is a simplepproach for the determination of BFRs in aqueous samples.owever, because of their hydrophobic character (except for
BBP-A) and thus low concentrations in water, large volumesup to 1000 mL) are typically required to ensure detectabil-ty [7,10]. Suzuki and Hasegawa [7] reported recoveries above7% for �-, �- and �-HBCD, after two sequential LLE with

A.C
ovacietal./J.Chrom
atogr.A1153
(2007)145–171
147Table 1Overview of typical analytical procedures used for the determination of PBDEs, HBCDs and TBBP-A in selected matrices
BFRs (# of congeners) Sample type (mL)—water Pretreatment Extraction procedure Clean-up Instrumentalanalysis
Recovery (%) RSD (%) LOD (ng/mL, pg/g)
Refs.
�- ,�- ,�-HBCD Landfill leachate (250) – LLE (DCM, 2 mL × 25 mL) – LC–MS/MS 77–92 9–10 0.002 [7]�- ,�- ,�-HBCD and TBBP-A Landfill leachate (1000) Filtration Abselut Nexus SPE (Acet, 5 mL) – LC–MS/MS 54–85 and 103 8–16 and 13 0.002 and
0.0002[7]
Tri-hexa-BDEs (9) Sewage treatment plant water – Semipermeable membrane(20 h) + MAE (DCM:C6, 60 mL,85 ◦C)
GPC LVI–GC–ITD-MS/MS 72–91 6–13 <0.03 [8]
Tetra-hexa-BDEs (6) Tap water + wastewater (10) Filtration HS-SPME (PDMS, 100 ◦C,stirring, 30 min)
– GC–ITD-MS/MS Tetra-penta-BDEs:87–109
1–19 0.008–0.2 [9]
Hexa-BDEs: ND-7
Tri-hexa-BDEs (9) Effluent from plastic company(100)
20% MeOH SBSE (25 h) – TD–GC–MS (scan) 94–103 1–5 0.4–10 [10]
BFRs (# of congeners) Sample type (g)—air and dust
Pretreatment Extraction procedure Clean-up Instrumentalanalysis
Recovery (%) RSD (%) LOD (ng/mL, pg/g)
Refs.
Tri-hexa-BDEs (9) Indoor air (PUF disksamplers)
– Soxhlet (C6, 12 h) H2SO4 + LLE(DMSO) + Florisil®
GC–EI-MS 75 to −95 1–6 0.1 pg/m3 [11]
Mono-deca-BDEs (42) Indoor dust (3.4) Sieve (2 mm) Soxhlet (DCM:C6, 8 h) SiO2 + Al2O3 GC–ECNI-MS 70–84 5–10 32–305 [12]TBBP-A Air samples Glass fibre and
PUFSonication (2 × 5 mL,2 × 20 min, AcN)
Filtration LC–ESI-MS/MS 75–93 – – [13]
Tri-deca-BDEs (26) + �- ,�-,�-HBCD
Air samples PUF and XAD-2resin
Soxhlet (Acet:C6, 1:1,24 h)
deact SiO2 (1–3.5%H2O)
GC–ECNI-MS Tri-hexa-BDEs: 85–110 – PBDEs:0.1–0.3 �g/m3
[14]
LC–MS/MS Hepta-deca-BDEs,HBCDs: 50–60
HBCDs:0.07–0.1 �g/m3
Tri-hexa-BDEs (9) Indoor dust (1) Sieve (0.5 mm) PLE (Florisil®; C6,150 ◦C, 10 MPa)
H2SO4 + LLE(DMSO) + Florisil®
GC–EI-MS 45–67 1–6 30 [11]
Tri-deca-BDEs (22) Indoor dust (0.1–0.5) Sieve (1 mm) PLE (DCM, 100 ◦C,13.8 MPa)
SiO2 GC–ECNI-MS – <25 1000–6000 [15]
BFRs (# of congeners) Sample type (g)—sediment and sewagesludge
Pretreatment Extraction procedure Clean-up Instrumental analysis Recovery(%)
RSD (%) LOD(ng/mL,pg/g)
Refs.
Mono-hepta-BDEs + deca-BDE (20)
Spiked sea sand (4) Na2SO4 (20 g) + Cu (15 g) Soxhlet (Acet:C6, 1:1,18 h)
SiO2 − H2SO4 + Al2O3 GC–ITD-MS/MS 46–88 11–26 13–250 [16]
�-, �-, �-HBCD and TBBP-A Sediments Ground with Na2SO4 Soxhlet (Acet:C6, 3:1,6 h or 1:1, 12 h)
LLE withH2SO4 + GPC + SiO2
LC–MS HBCD:47–104
HBCD: 5 0.2–330 and0.1–97
[17]
Tri-hexa-BDEs+ deca-BDE(10)
River sediment cores (1) Air-dry + Cu (3 g) Hot Soxhlet (Acet:C6,1:3, 2 h)
Cu + SiO2 − H2SO4 GC–ECNI-MS 82–93 <16 50–200 [18]
�- ,�- ,�-HBCD and TBBP-A Marine sediment (1) Air-dry Sonication (Acet, 10 min) – LC–MS/MS 104–108and 101
3–5 and 4 0.002 and0.0002
[7]
Tri-deca-BDEs (22) Sediment (20) Homogenization + drying withdiatom. earth
PLE (DCM, 100 ◦C,14 MPa)
Cu + Al2O3 GC–EI-MS >50 <35 0.1–1.5 [19]
TBBP-A Sediment (10), sewage sludge(1.0)
Freeze-dry + homogenization PLE (DCM, 100 ◦C,12.7 MPa)
Derivatisation(CH2N2) + SiO2
+ deact SiO2 + SiO2
− H2SO4 + SiO2
− AgNO3
GC-HRMS >80 – – [20]
Tri-hexa-BDEs (9) River sediment (20) Air-dry + grind + sieve PLE (DCM:C6, 1:1,100 ◦C, 10 Mpa)
Al2O3 GC–EI-MS 70–100 – 500–1400 [21]
Mono-hepta-BDEs (39) Marine and river sediments(1)
Freeze-dry + grind + sieve(2 mm)
PLE (Cu + Al2O3, 1:2;DCM:C6, 1:1, 100 ◦C,10 Mpa)
– GC–ECNI-MS 22–88 1–14 1–46 [22]

148A
.Covacietal./J.C
hromatogr.A
1153(2007)
145–171Table 1 (Continued )
BFRs (# of congeners) Sample type (g)—sediment and sewagesludge
Pretreatment Extraction procedure Clean-up Instrumental analysis Recovery(%)
RSD (%) LOD(ng/mL,pg/g)
Refs.
Di-hexa-BDEs + deca-BDE(14)
Sewage sludge (2.5) Air-dry + grind PLE (DCM:C6, 1:1,100 ◦C, 10 Mpa)
H2SO4 + SiO2
− H2SO4 + Al2O3
Di-hexa-BDEs:GC–MS/MS
– – – [23]
Deca-BDE:GC–ECNI-MS
Tri-hexa-BDEs (9) Marine sediment (5) Freeze-dry + grind + sieve MAE (Acet:C6, 1:1,150 ◦C)
Filtration + GPC (DCM) LVI–GC–MS/MS 81–96 4–13 4–20 [24]
Tetra-hexa-BDEs (6) Soil, sediment, sewage sludge(0.5)
Air-dry + homog + sieve(0.3 mm) + H2O (2 mL)
HS-SPME (100 ◦C,60 min)
– GC–ITD-MS/MS 82–105 4–16 5–625 [25]
Mono-deca-BDEs Polymers Pyrolysis – – GC–EI-MS (scan) – – – [26]
BFRs (# ofcongeners)
Sample type (g,mL)—biologicalsamples
Pretreatment Extraction procedure Clean-up Instrumental analysis Recovery (%) RSD (%) LOD (ng/mL,pg/g)
Refs.
Tri-deca-BDEs (27) Fish serum Heparinized,centrifugate
LLE (HCl, 2-propanol,C6 + MTBE)
GPC GC–ECNI-MS – – – [27]
Tri-deca-BDEs (12) Human serum (5) Formic acid Oasis® HLB SPE SiO2 − H2SO4 GC–ECNI-MS 64–95 <17 0.2–25 (wetweight)
[28]
TBBP-A Human serum (5) Formic acid Abselut Nexus SPE SiO2 LC–ESI-MS/MS 83–104 <8 0.8 (wet weight) [29]Tri-deca-BDEs (8) Human serum (4) Formic acid Oasis® HLB SPE SiO2 − KOH + SiO2 − H2SO4 GC-HRMS 69–95 19–27 1–6 (wet weight) [30]Tri-hepta-BDEs (10) Human serum (8) Formic
acid + 2-propanol inwater (3%)
Isolute® ENV+ SiO2 + SiO2 − H2SO4 GC–ECNI-MS 48–126 2–18 0.01–0.12 (lipidweight)
[31]
Tri-deca-BDEs (8) Human milk (1) Pasteurization Diatomaceous earth (elutionwith DCM)
SiO2+SiO2 − H2SO4 GC-HRMS 60–89 4–9 0.1–1.2 (lipidweight)
[32]
Tri-deca-BDEs (30), Blood serum (10) – LLE (EtOAc,12 + 8 mL) + LLE (AcN,3 mL + C6, 3 mL × 3 mL)
C6 layer (PBDEs): Oasis® HLBSPE + SiO2 + SiO2 − H2SO4
PBDEs: GC-HRMS Tri-hepta-BDEs:60–110
Tri-hepta-BDEs:3–22
Tri-hepta-BDEs:<0.5–10
[33]
�- ,�- ,�-HBCD,TBBP-A
Adipose tissue (0.5) – LLE (AcN, 3 mL + C6,3 mL × 3 mL)
ACN layer (HBCD + TBBP-A):enzymatic hydrolysis (50 ◦C,4 h) + Oasis® HLB + silica
�- ,�- ,�-HBCD:LC–MS/MS
Deca-BDE: 80–110 Deca-BDE: 3–4 Deca-BDE:30–500
Human milk (1) Freeze-dry,homogenization
SLE (Acet:DCM, 1:1,12 + 6 mL) + LLE (AcN,3 mL + C6, 3 mL × 3 mL)
SPE TBBP-A (silylated):GC-HRMS
13C-�-HBCD: 40 HBCD: 8–25 HBCD: 30–500
TBBP-A: 40 TBBP-A: 4–7 TBBP-A: 0.2–4
Tetra-hexa-BDEs (3) Seal plasma (1–4) Heparinized,centrifuged, Na2SO4
Column elution (DCM:C6) SiO2 − NaOH + SiO2
+ SiO2 − H2SO4
GC–EI-MS 46–139 – – [34]
Mono-deca-BDEs(47)
Tree bark (15–25) Cut (1 cm) Soxhlet (Acet:C6, 1:1) LLE (H2SO4 + C6) + Al2O3 GC–ECNI-MS >80 (30–60 fordeca-BDE)
– 10–1500 [35]
Tri-hepta-BDEs (7) Hair (0.3–0.5) Water wash (1 h,40 ◦C) + drying + cut(2 mm)
LLE (HCl 4 M + DCM:C6,4:1, 40 ◦C, overnight)
SiO2 − H2SO4 GC–ECNI-MS – – <50 [36]
Tetra-penta-BDEs(3)
Fish feed (1.5) – MSPD (C18,1 g) + SiO2 − H2SO4 + Al2O3
(C6, 30 mL)
– GC–ITD-MS/MS 70–96 5–9 300–900 [37]
Tetra-penta-BDEs(3) + PBBs (2)
Turbot feed (1) Grind + freeze-dry SFE (in-cell basicAl2O3 + SiO2 − H2SO4; CO2,60 ◦C) + C18 trap (25 ◦C)
Desorption from C18 trap (C6,2 mL) + HS-SPME (75 ◦C,60 min)
GC–ITD-MS/MS 82–101 14–16 2–9 [38]
Mono-deca-BDEs(40)
Chicken fat (1) – SLE (DCM) + filtration SiO2 − H2SO4 + SiO2
+ SiO2 − KOH + Al2O3
GC-HRMS 76–114 14–20 – [39]
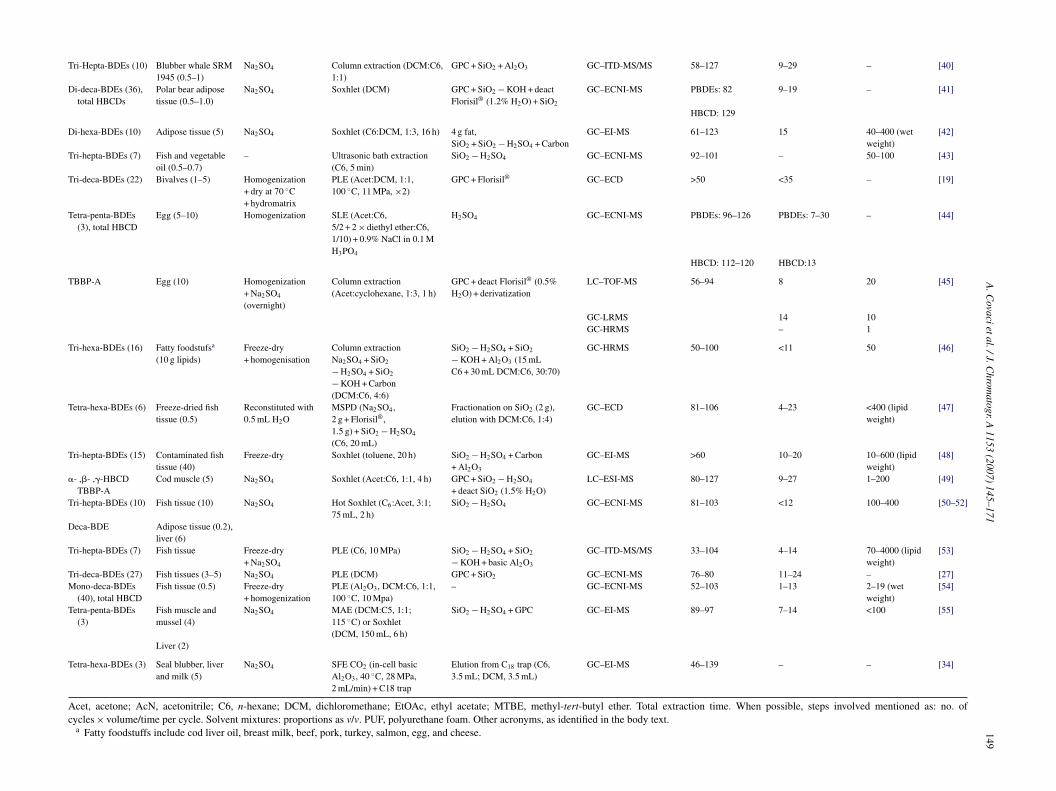
A.C
ovacietal./J.Chrom
atogr.A1153
(2007)145–171
149Tri-Hepta-BDEs (10) Blubber whale SRM
1945 (0.5–1)Na2SO4 Column extraction (DCM:C6,
1:1)GPC + SiO2 + Al2O3 GC–ITD-MS/MS 58–127 9–29 – [40]
Di-deca-BDEs (36),total HBCDs
Polar bear adiposetissue (0.5–1.0)
Na2SO4 Soxhlet (DCM) GPC + SiO2 − KOH + deactFlorisil® (1.2% H2O) + SiO2
GC–ECNI-MS PBDEs: 82 9–19 – [41]
HBCD: 129
Di-hexa-BDEs (10) Adipose tissue (5) Na2SO4 Soxhlet (C6:DCM, 1:3, 16 h) 4 g fat,SiO2 + SiO2 − H2SO4 + Carbon
GC–EI-MS 61–123 15 40–400 (wetweight)
[42]
Tri-hepta-BDEs (7) Fish and vegetableoil (0.5–0.7)
– Ultrasonic bath extraction(C6, 5 min)
SiO2 − H2SO4 GC–ECNI-MS 92–101 – 50–100 [43]
Tri-deca-BDEs (22) Bivalves (1–5) Homogenization+ dry at 70 ◦C+ hydromatrix
PLE (Acet:DCM, 1:1,100 ◦C, 11 MPa, ×2)
GPC + Florisil® GC–ECD >50 <35 – [19]
Tetra-penta-BDEs(3), total HBCD
Egg (5–10) Homogenization SLE (Acet:C6,5/2 + 2 × diethyl ether:C6,1/10) + 0.9% NaCl in 0.1 MH3PO4
H2SO4 GC–ECNI-MS PBDEs: 96–126 PBDEs: 7–30 – [44]
HBCD: 112–120 HBCD:13
TBBP-A Egg (10) Homogenization+ Na2SO4
(overnight)
Column extraction(Acet:cyclohexane, 1:3, 1 h)
GPC + deact Florisil® (0.5%H2O) + derivatization
LC–TOF-MS 56–94 8 20 [45]
GC-LRMS 14 10GC-HRMS – 1
Tri-hexa-BDEs (16) Fatty foodstufsa
(10 g lipids)Freeze-dry+ homogenisation
Column extractionNa2SO4 + SiO2
− H2SO4 + SiO2
− KOH + Carbon(DCM:C6, 4:6)
SiO2 − H2SO4 + SiO2
− KOH + Al2O3 (15 mLC6 + 30 mL DCM:C6, 30:70)
GC-HRMS 50–100 <11 50 [46]
Tetra-hexa-BDEs (6) Freeze-dried fishtissue (0.5)
Reconstituted with0.5 mL H2O
MSPD (Na2SO4,2 g + Florisil®,1.5 g) + SiO2 − H2SO4
(C6, 20 mL)
Fractionation on SiO2 (2 g),elution with DCM:C6, 1:4)
GC–ECD 81–106 4–23 <400 (lipidweight)
[47]
Tri-hepta-BDEs (15) Contaminated fishtissue (40)
Freeze-dry Soxhlet (toluene, 20 h) SiO2 − H2SO4 + Carbon+ Al2O3
GC–EI-MS >60 10–20 10–600 (lipidweight)
[48]
�- ,�- ,�-HBCDTBBP-A
Cod muscle (5) Na2SO4 Soxhlet (Acet:C6, 1:1, 4 h) GPC + SiO2 − H2SO4
+ deact SiO2 (1.5% H2O)LC–ESI-MS 80–127 9–27 1–200 [49]
Tri-hepta-BDEs (10) Fish tissue (10) Na2SO4 Hot Soxhlet (C6:Acet, 3:1;75 mL, 2 h)
SiO2 − H2SO4 GC–ECNI-MS 81–103 <12 100–400 [50–52]
Deca-BDE Adipose tissue (0.2),liver (6)
Tri-hepta-BDEs (7) Fish tissue Freeze-dry+ Na2SO4
PLE (C6, 10 MPa) SiO2 − H2SO4 + SiO2
− KOH + basic Al2O3
GC–ITD-MS/MS 33–104 4–14 70–4000 (lipidweight)
[53]
Tri-deca-BDEs (27) Fish tissues (3–5) Na2SO4 PLE (DCM) GPC + SiO2 GC–ECNI-MS 76–80 11–24 – [27]Mono-deca-BDEs
(40), total HBCDFish tissue (0.5) Freeze-dry
+ homogenizationPLE (Al2O3, DCM:C6, 1:1,100 ◦C, 10 Mpa)
– GC–ECNI-MS 52–103 1–13 2–19 (wetweight)
[54]
Tetra-penta-BDEs(3)
Fish muscle andmussel (4)
Na2SO4 MAE (DCM:C5, 1:1;115 ◦C) or Soxhlet(DCM, 150 mL, 6 h)
SiO2 − H2SO4 + GPC GC–EI-MS 89–97 7–14 <100 [55]
Liver (2)
Tetra-hexa-BDEs (3) Seal blubber, liverand milk (5)
Na2SO4 SFE CO2 (in-cell basicAl2O3, 40 ◦C, 28 MPa,2 mL/min) + C18 trap
Elution from C18 trap (C6,3.5 mL; DCM, 3.5 mL)
GC–EI-MS 46–139 – – [34]
Acet, acetone; AcN, acetonitrile; C6, n-hexane; DCM, dichloromethane; EtOAc, ethyl acetate; MTBE, methyl-tert-butyl ether. Total extraction time. When possible, steps involved mentioned as: no. ofcycles × volume/time per cycle. Solvent mixtures: proportions as v/v. PUF, polyurethane foam. Other acronyms, as identified in the body text.
a Fatty foodstuffs include cod liver oil, breast milk, beef, pork, turkey, salmon, egg, and cheese.

1 atogr
dtNsa(a
mthql(TewrtilStmat
auauaau[Bppweipwa1cc
(f1pms(to
iltos
2wedcTbRmwa(
ecnPiestsri1uLwm
2iadv[mpt(ttiiahw
50 A. Covaci et al. / J. Chrom
ichloromethane (DCM) of a spiked landfill leachate. However,he authors suggested solid-phase extraction (SPE) on Abselutexus cartridges as a faster and valuable alternative allowing the
imultaneous determination of TBBP-A (recovery 103 ± 16%)nd a significant reduction in the organic solvent consumptionfrom 50 mL DCM to 5 mL acetone) that still provided accept-ble recoveries (54–85%) of the three HBCD diastereomers.
New membrane-based techniques may also overcome theain limitations of the large scale and highly manipula-
ive LLE approaches. Microwave assisted extraction (MAE)as been demonstrated to be an interesting alternative foruantitative recovery of tri- to hexa-BDEs preconcentrated inow-density polyethylene semipermeable membrane devicesSPMDs) deployed for 20 h in a sewage treatment plant [8].he MAE method provided similar results to dialysis (recov-ries of 72–91% and 96–103% at the 2 ng/membrane levelith RSDs <13% and 11%, respectively), but allowed the
eduction of solvent consumption to one-third and shortenedhe extraction time from 24 h to 5 min. A potential shortcom-ng of MAE as compared to dialysis is the co-extraction ofarger amounts of potential interfering components from thePMDs, which can complicate the subsequent clean-up of
he extracts. To overcome this problem, gel permeation chro-atography (GPC) combined with a selective detection of the
nalytes by gas chromatography–ion-trap-tandem mass spec-rometry (GC–ITD-MS/MS) was used [8].
Solid-phase microextraction (SPME), a rapid, solvent-freend low-cost analytical technique, has been increasingly eval-ated for several environmental determinations, including thenalysis of PBDEs. Since PBDEs have relatively high molec-lar weights and low vapour pressures, one would expect tochieve higher recoveries by direct immersion of the fibre inliquid sample than by sampling the headspace (HS) when
sing SPME as preconcentration step. Nevertheless, Polo et al.9] observed the opposite trend when analysing tetra- to penta-DEs in tap water and wastewater from an urban treatmentlant (spiking level, 0.02–1.0 ng/L). Beside filtration, no otherretreatment was required before direct HS-SPME of 10 mLater heated at 100 ◦C for 30 min. Apart from the enhanced
xtraction efficiencies, the use of HS-SPME instead of directmmersion prevented the contamination with non-volatile com-ounds and prolonged the lifetime of the fibre. Linear responsesere observed in the 0.2–500 ng/L range, with recoveries >87%
nd RSDs <19%. However, since hexa-BDEs (BDE 153 and54), could not be recovered under the proposed experimentalonditions, the method seems to be limited up to penta-BDEongeners [9].
A closely related technique, stir bar sorptive extractionSBSE) on polydimethylsiloxane (PDMS) stir bar, has been usedor the quantitative preconcentration of tri- to hexa-BDEs from00 mL of surface water contaminated by the effluents from alastic-production company [10]. The analytes were then ther-ally desorbed from the stir bar in a GC injection port. The only
ample pretreatment required was the addition of 20% methanolv/v) to the water sample. The long preconcentration time used inhe experiments (25 h) resulted in low limits of detection (LODs)f 0.4–10 ng/L, although GC–MS with electron ionization (EI)
RtBd
. A 1153 (2007) 145–171
n scan mode was used for the final detection. However, a ratherong preconditioning of the stir bar (300 ◦C, 4 h) was requiredo avoid cross contamination. Under these conditions, the carry-ver was <2% for tri- to hexa-BDEs after the analysis of wateramples spiked at the 600 ng/L level [10].
.1.1.3. Air and dust. For abiotic solid samples, Soxhlet isidely accepted as a robust, efficient and low-cost solid–liquid
xtraction (SLE) technique. Soxhlet has been used for theetermination of PBDEs and HBCDs in indoor air (after pre-oncentration on polyurethane foam [11,14]) and dust [12].ypical solvents include n-hexane, DCM, acetone and theirinary mixtures. Gevao et al. [12] reported recoveries >70%,SDs <10% and LODs in the range of 32–305 pg/g for 42ono- to deca-BDEs after Soxhlet extraction of 3.4 g house dustith DCM:n-hexane (1:1, v/v), clean-up on silica and alumina
nd GC–MS operated in electron-capture negative ionizationECNI).
The main drawbacks of the Soxhlet extraction, i.e. longxtraction times (typically >8 h) and large solvent consumption,an be at least partially avoided by alternative extraction tech-iques, such as pressurized liquid extraction (PLE). Recently, aLE-based procedure was validated for the PBDE analysis using
ndoor dust standard reference material (SRM 2585) [15]. Thextraction was carried out with DCM (20 min at 100 ◦C) and aimple clean-up on silica SPE cartridges sufficed to obtain ready-o-analyse extracts. The use of n-hexane at 150 ◦C as extractionolvent for PLE of nine tri- to hexa-BDEs from indoor dustesulted in somehow lower recoveries (45–67%) [11]. Interest-ngly, Harrad et al. [11] included an in-cell purification using.5 g Florisil® in the extraction cell under the sample. Evensing this approach, the extracts had to be further purified byLE extraction with concentrated H2SO4 and back-extractionith dimethyl sulfoxide (DMSO), followed by column chro-atography on Florisil® before instrumental analysis.
.1.1.4. Soil, sediment and sewage sludge. Soxhlet extractions also a primary option for the determination of BFRs in soilsnd sediments. In general, mixtures of acetone and n-hexane inifferent proportions (1:1 or 1:3, v/v) have been found to pro-ide the best recoveries for PBDEs [6,56], HBCDs and TBBP-A17]. However, wet samples require the use of additional water-iscible solvents, such as acetone or methanol, to facilitate the
enetration of the solvent in the matrix and thus to ensure quan-itative extraction of BFRs [57]. The Soxhlet extraction times6–24 h) can be substantially shortened by replacing the tradi-ional Soxhlet system by a semi-automated hot Soxhlet [18]. Inhis technique, the solvent distilled into the extraction chambers also heated below the boiling point of the solvent. The samples consequently in permanent contact with hot solvent, whichccelerates the analyte desorption from the matrix. The use ofot Soxhlet allows a significant reduction in the extraction timeithout affecting the final results. High recoveries (82–93%,
SDs <16%) have been achieved after 2 h hot Soxhlet extrac-ion with acetone:n-hexane (1:3, v/v) of 1 g sediment used in theROC interlaboratory study [58]. Such a rapid analytical proce-ure was subsequently applied to the analysis of river sediment

atogr
cias(Hpa
dbtPPDiapattawswi1lPrimperbr(or
oedatwaew(isiuo1
o3
2bPttail
dpHpbf
Biceavmaa[[oh
2rippiEasacp
2siieho
A. Covaci et al. / J. Chrom
ores [18] and was also used as reference method when evaluat-ng the feasibility of MAE [24]. Although GPC instead of Cu andcidified silica was used for clean-up in the latter case, similaratisfactory recoveries (81–96%) and comparable repeatabilityRSDs <13%) were reported for MAE compared to hot Soxhlet.owever, the use of large volume injection (LVI) in the MAErocedure, combined with a larger sample intake (5 g versus 1 gs in [18]) resulted in a 10-fold reduction of LODs.
PLE has also been evaluated for the analysis of BFRs inried soils, sediments and sewage sludge. DCM at 100 ◦C haseen found to provide quantitative recoveries (∼80%) of tetra-o deca-BDEs [59] and TBBP-A [20], providing that severalLE cycles (e.g. 2 min × 5 min cycle) instead of a single longerLE cycle [19] were carried out. As an alternative, mixtures ofCM and n-hexane at 100 ◦C have been used [21,22]. Interest-
ngly, de la Cal et al. [22] proposed a selective PLE approachllowing complete sample preparation in a single step by dis-ersion of 1 g sediment in 2 g Cu powder and activated aluminand packing the resulting homogenized mixture in the extrac-ion cell on top of 6 g alumina. The sample was then subjectedo 2 min × 10 min static cycles with DCM:n-hexane (1:1, v/v)nd the concentrated extracts were analysed by GC–ECNI-MSithout any further treatment. Soxhlet extraction with the same
olvent mixture followed by clean-up on SPE alumina cartridgeas used as reference method. The PLE procedure provided sim-
lar results for 29 tri- to hepta-BDEs (recoveries 47–82%; RSDs–10%) to those found using the reference method, but somehowower recoveries (22–41%) for the mono- and di-BDEs. TheseBDE congeners are however found at very low levels in envi-onmental samples. The authors suspected the co-extraction ofnterfering compounds which eluted in the first part of the chro-
atogram. These interferences were probably eliminated by theurification scheme used in the reference method [22]. Nev-rtheless, it should be noted that low recoveries were alreadyeported for mono- and di-BDEs when alumina was used as sor-ent for in-cell PLE in preliminary experiments [22]. The higherecoveries reported by Samara et al. [21] for tri- to hexa-BDEs70–100%) using a similar analytical procedure, but includingff-line alumina purification of the PLE extracts obtained fromiver sediments, seem to confirm this observation.
The use of HS-SPME with PDMS fibres for the extractionf PBDEs from soil, sediment and sewage sludge slurries wasvaluated by Salgado-Petinal et al. [25]. The experimental con-itions were set according to those previously optimized forqueous samples [10] but, because of the higher complexity ofhe sample matrix, longer extraction times (typically 60 min)ere used. This resulted in higher recoveries of tetra-BDEs
nd, to a minor extent, penta-BDEs, as compared to recov-ries obtained by MAE (2 g extracted for 15 min at 120 ◦Cith 8 mL of acetone:n-hexane, 4:1) or by ultrasonic extraction
8 mL solvent, 15 min). However, differences between recover-es decreased with the increasing bromination level and thereforeimilar recoveries were obtained for hexa-BDEs with the three
nvestigated techniques. The recoveries of the spiked compoundssing HS-SPME decreased dramatically as the organic contentf the sample increased from 0.18% (sandy marine sediment) to.0–3.5% (river sediment and soil), and very low recoveries wereohip
. A 1153 (2007) 145–171 151
btained for the sewage sludge slurry (average organic content,5%).
.1.1.5. Polymers. The European directive 2002/95/EC [60]ans the use of PentaBDE and OctaBDE and restricts the use ofBBs in the electrical and electronic equipment (EEE), while
he European directive 2003/11/EC [61] prohibits the distribu-ion of products that contain levels of PentaBDE or OctaBDEbove 0.1%. Since the levels at which BFRs have to be measuredn polymers are high, simplified methods were described in theiterature.
A fast qualitative screening method consisting in the thermalesorption of analytes at temperatures below polymer decom-osition (i.e. 300–400 ◦C) and GC–MS has been proposed [26].owever, quantitative determination required a complete samplereparation including LLE of the dissolved sample (for solu-le polymers) or Soxhlet extraction (for non-soluble polymers)ollowed by GPC clean-up before instrumental analysis [26].
Altwaiq et al. [62] examined different procedures to extractFRs from various polymer materials. These procedures
ncluded supercritical carbon dioxide (CO2), modified super-ritical CO2, solvent and Soxhlet extraction. The extractionfficiency of the investigated BFRs varied according to thepplied methods. The results proved the high capacity of sol-ents such as toluene, tetrahydrofuran and acetonitrile to extractany BFRs, whereas extraction by using a combination of CO2
nd organic solvents overcomes the lower extraction efficiencychieved with only CO2. Fast extraction techniques, such as PLE63] and ultrasonic solvent extraction (USE) with 2-propanol64] have been developed for the extraction and identificationf BFRs in polymers used in EEE, such as TV and PC monitorousings.
.1.1.6. Biotic samples. Table 1 summarises relevant methodselated to BFRs in biotic samples. The reviewed categoriesnclude (i) biological fluids (i.e. serum and plasma); (ii) feed andlant samples; and (iii) fatty foodstuffs and animal tissue sam-les. Similar to abiotic samples, only drying and homogenizations usually carried out before extraction of biological samples.xcept for serum and plasma, (semi)liquid (e.g. eggs) samplesre usually freeze-dried and then treated as any other solid bioticample. In general, similar extraction techniques and solventsre used for BFR analysis in abiotic and fat-containing matri-es, and the main differences between both sets of analyticalrotocols refer only to the subsequent clean-up steps.
.1.1.7. Serum and plasma. BFRs are usually extracted fromerum by successive treatment with solvents of different polar-ty. In some cases, a treatment with a HCl:2-propanol mixtures carried out for protein denaturation before LLE [27]. How-ver, direct solvent shaking with ethyl acetate and acetonitrileas also been demonstrated to provide quantitative recoveriesf PBDEs (60–110%, RSDs 2–22%), despite the high number
f manipulation steps [33]. Lower, but reproducible recoveriesave been obtained for �-HBCD and TBBP-A using this analyt-cal approach. One of the main limitations of these LLE-basedrocedures is the long waiting time or centrifugation required
1 atogr
ftth(aoNe(ttrii
2dnrd1yltpbscsDbrasto
oHvaB
2sa9StNl1P
aw
saPAprluiruWapob
hlt[fas[
Mtiaowelosoboprt(1mMcaal
ie
52 A. Covaci et al. / J. Chrom
or phase separation. To avoid this, Wolkers et al. [34] driedhe serum with anhydrous Na2SO4, loaded the resulting mix-ure onto a glass column and extracted the analytes (tetra- toexa-BDEs) with DCM:n-hexane. A wide range of recoveries46–139%) were obtained using this approach, which also hadlarger solvent consumption. Alternatively, SPE-based meth-
ds using Oasis® HLB [28,30], Isolute® ENV+ [31] or Abselutexus [29] sorbents have been proposed. Relatively good recov-
ries (>60%) were obtained in most cases, including BDE 209Table 1). A similar SPE-based procedure has been proposed forhe determination of PBDEs in human milk [32]. Furthermore,he SPE-based methods proved to be less laborious and allowededuced solvent consumption and processing time, possibil-ty of miniaturization, and parallel sample preparation, whichncreases throughput.
.1.1.8. Feedstuffs and plant samples. Due to the high interestevoted to the presence of BFRs in the aquatic environment, aumber of studies dealing with the analysis of fish feed have beeneported. A simple and fast method based on matrix solid-phaseispersion (MSPD) of 1.5 g aquaculture feedstuffs dispersed ong C18-modified silica was proposed for the simultaneous anal-sis of several POP classes [37]. The resulting mixture was thenoaded on top of acidified silica and alumina in a glass SPE car-ridge and eluted with 30 mL n-hexane. No additional extracturification was required. The acidified silica and alumina sor-ents were found to be efficient for effective fat removal duringupercritical fluid extraction (SFE) of turbot feed [38]. In thisase, the sample was loaded in the extraction cell on top of theorbent layers and extracted with CO2 at 60 ◦C and 16.5 MPa.espite the limited number of PBDE congeners included inoth studies (BDEs 47, 99 and 100), promising recoveries in theange 70–96% and 82–101% were obtained with these MSPD-nd SFE-based procedures, respectively. MSPD provided alsoatisfactory repeatability (RSDs 5–9% versus 14–16%, respec-ively). To the best of our knowledge, the presence of HBCDsr TBBP-A in animal feed has not yet been investigated.
Up to now, BFR levels in plant samples have been evaluatednly in a limited number of studies. As an example, Zhu andites [35] used Soxhlet extraction with acetone:n-hexane (1:1,
/v) for the determination of PBDEs in tree bark. Recoveriesbove 80% were reported for 46 mono- to nona-BDEs, whileDE 209 had a lower extraction efficiency (30–60%).
.1.1.9. Biological tissues and fatty foodstuffs. For fat and oilamples, the first sample treatment is to dissolve the lipids in anppropriate solvent. This has be achieved by melting the fat at0 ◦C followed by LLE with an apolar solvent [33], by directLE with an apolar solvent [39], column extraction with a mix-
ure of apolar solvents after drying of the sample with anhydrousa2SO4 [40], Soxhlet extraction [41,42] or sonication with apo-
ar solvents [43]. Typically sample intake was between 0.5 g andg and quantitative recoveries >60% have been reported for
BDEs and HBCDs.Column extraction using a multi-layer column containingppropriate sorbents for a preliminary purification has beenidely used for biological tissues. In this case, the initial sample
tuam
. A 1153 (2007) 145–171
ize determines the volume of solvents to be used. Fernandes etl. [46] needed 400 mL of DCM:n-hexane (2:3, v/v) to eluteBDEs from a multi-layer column containing 10 g of food.lternatively, the intimate contact between the sample com-onents and the sorbent particles achieved by MSPD usuallyesults in a more efficient retention of impurities. Moreover,ower solvent consumption and cleaner extracts can be expectedsing MSPD compared to the alternative, in which the samples packed above the sorbent in a column. Martınez et al. [47]eported high recoveries (81–106%) for six tetra- to hexa-BDEssing only 0.5 g naturally contaminated fish tissue (WELL-MF-01) dispersed on 1.5 g Florisil® and packed on top of
cidified silica. Only 20 mL n-hexane was needed for the com-lete elution of analytes. Except for a subsequent fractionationf PBDEs from PCBs on silica, no other treatment was requiredefore GC with electron-capture detection (ECD).
The same extraction techniques as used in abiotic samplesave been used for BFR determination in more complex bio-ogical tissues and fatty foodstuffs. Binary solvent mixturesypically containing acetone:n-hexane [6,44,49], acetone:DCM19], DCM:n-hexane [45,54] or toluene [48] have been preferredor Soxhlet-based extractions. This technique has a number ofdvantages, such as minimum sample pretreatment required,implicity, and high recoveries obtained for PBDEs (>60%,48,65]) and for HBCDs and TBBP-A (>80%, [17]).
Alternative enhanced extraction techniques, such as PLE orAE, have also been used. Eljarrat et al. [54] adapted a selec-
ive PLE, previously validated for the determination of PBDEsn sediments [22], for the simultaneous analysis of PBDEsnd total HBCDs in fish tissue. Ready-to-analyse extracts werebtained also here, but, slightly higher recoveries (52–103%)ere reported as compared to those found for sediments. The
fficiency of this particular PLE procedure proved to be simi-ar to other PLE-based methods in which clean-up was carriedut off-line by manual [27] or semi-automated [53] multi-tep procedures. The use of PLE-based extraction methods forrganic pollutants, including BFRs, has recently been reviewedy Bjorklund et al. [66]. For MAE, purification should be carriedut off-line after separation of the solvent from the matrix com-onents, but its high extraction efficiency allowed a significanteduction of the extraction time [55] compared to conven-ional extraction procedures. Recoveries in the range of 89–97%RSDs <14%) have been reported for MAE of BDEs 47, 99 and00 from spiked marine biological tissues with variable fat andoisture contents (range 1.2–38% and 48–78%, respectively).oreover, the use of MAE provided only slightly lower con-
entrations (difference <15%) than the Soxhlet procedure useds reference method (6 h extraction with 150 mL DCM) for thenalysis of two certified reference materials (SRM 1588a – codiver oil and SRM 2978 – mussel tissue).
SFE has been used to a lesser extent for the BFR analysisn biological samples. Wolkers et al. [34] achieved a selectivelution of organic pollutants, including PCBs, chlordane and
hree PBDE congeners, from seal blubber, liver and milk (5 g)sing CO2 at 40 ◦C and 28 MPa, alumina as in-cell fat retainernd C18 as solid trap. Although low blank levels (<10% of theeasured concentrations) and short extraction time (25 min)
atogr
wp
2
ccigtehclsswpbOe
2epst[apo
2[bedThdmtslscufo
atBh[bi
Atasss
2osodapttpoitptioiwmtBafioaciALt
2tpd
pPbPmseit
A. Covaci et al. / J. Chrom
ere obtained, variable recoveries (46–139%) were reported,robably due to the matrix-sensitive character of SFE.
.1.2. Clean-up and fractionationThe non-selective nature of the exhaustive extraction pro-
edures and the complexity of the sample matrices result inomplex extracts that require further purification. Despite thenherent advantages derived from partial [23] or complete inte-ration [53] of these tedious and time-consuming purificationreatments, or by combining the clean-up treatment with thextraction [22,34], up to now, the development in this fieldas been rather limited and the analytical steps involved inlean-up protocols for BFRs have usually been carried out off-ine. Typical purification and fractionation procedures have beenummarized in Table 1. For abiotic samples (sediment, soil, andewage sludge), the clean-up should ensure sulphur removal,hile, for biotic samples, lipid elimination should be accom-lished before chromatographic analysis. Lipid elimination cane accomplished by destructive or non-destructive methods.therwise, similar protocols can be used for purification of the
xtracts almost irrespective of the matrix nature.
.1.2.1. Sulphur removal. Sediment, soil and sewage sludgextracts often contain relatively large amounts of elemental sul-hur, which may hamper the determination of BFRs even ifelective separation and detection techniques are used. Simplereatments of either the sample [6,16,22,67] or the sample extract18,19] with Cu powder or the use of GPC [6,24,49] are efficientpproaches for sulphur elimination. Other less frequently usedrocedures included treatment with AgNO3-modified silica [20]r with tetrabutylammoniumsulphite [57].
.1.2.2. Non-destructive methods for lipid removal. GPC24,27] and adsorption chromatography on selected sor-ents [12,21] are non-destructive treatments applied for lipidlimination. GPC is mainly carried out using polystyrene-ivinylbenzene columns and DCM as eluent [8,19,24,27,59].he current use of prepacked high performance GPC columnsas resulted in higher separation efficiencies, improved repro-ucibility and lower solvent consumption as compared toanually packed columns [19]. Satisfactory isolation of the
arget compounds from the co-extracted organic material afteringle GPC analysis has been achieved for samples containingimited amounts of lipids, such as sewage sludge [24] or fisherum [27]. However, for more complex matrices, two seriallyonnected GPC columns [17] or GPC followed by further clean-p by adsorption chromatography [6,40,59] are often requiredor complete fat removal and/or isolation of BFRs from otherrganohalogenated compounds.
Silica gel, alumina and Florisil® with different degrees ofctivation have been widely used for lipid removal by adsorp-ion chromatography under atmospheric conditions (Table 1).ecause of its limited capacity for retention of lipids, silica
as been used in combination with alumina [6,12] or Florisil®41]. Alumina and Florisil® have been preferred as fat retainersecause of their higher lipid retaining capacity in proceduresnvolving MSPD [47], in-cell PLE [11,22,54] and SFE [34,38].
ePPw
. A 1153 (2007) 145–171 153
s an alternative to these sorbents, Carro et al. [37] proposedhe use of apolar sorbents, such as C18-modified silica, for
preliminary fat retention during MSPD. For obvious rea-ons, when extraction and clean-up are combined in a singletep, the total lipid content determination should be carried outeparately.
.1.2.3. Destructive methods for lipid removal. Similarly tother organohalogenated compounds, BFRs are stable undertrong acid conditions [3,6,17]. Due to its efficient removal ofrganic matter, sulphuric acid treatment is one of the most usedestructive treatments in BFR analysis (Table 1). The simplestpproach consists of direct addition of the acid to the sam-le extract dissolved in n-hexane [6,11,23,35,44,76]. However,his treatment requires several sequential LLE and centrifuga-ion steps, which results in a multi-step and time-consumingrocedure. The dispersion of sulphuric acid onto the surfacef activated silica gel results in a sorbent which can be eas-ly loaded into a column. The use of acidified silica avoidshe emulsion problems of the LLE approach, reduces the sam-le handling and solvent consumption and increases samplehroughput. Only 5 g of acidified silica (40%, w/w) prepackedn an SPE cartridge was sufficient for the purification of 0.7 gf fish or vegetable oil samples [43]. The target analytes, whichncluded seven tri- to hepta-BDEs, were quantitatively elutedith 15 mL n-hexane and 10 mL DCM. Silica gel can also beodified with alcoholic NaOH [34] or KOH [41,46], but such
reatment may cause losses of Br atoms from highly brominatedFRs, such as HBCDs, PBBs or PBDEs [6]. Although in manypplications the use of acidified silica is enough to yield suf-ciently clean extracts, several studies have described the usef acidified silica in combination with neutral silica [20,33,42]nd/or base-modified silica [34,39,41,46,53,67] in multi-layerolumns for improved purification. All approaches provide sim-lar satisfactory results concerning recovery and reproducibility.lternative procedures for destructive lipid removal, such asLE with dimethyl sulfoxide [11] are less frequently used than
hose quoted above.
.1.2.4. Fractionation. For specific applications, isolation ofhe target analytes from other organohalogenated compoundsresent in the extract can be mandatory to avoid interferencesuring final determination.
Due to their higher polarity than other organohalogenatedollutants typically present in the extract, e.g PCBs or PCDD/Fs,BDEs can be isolated in a separate fraction using classical sor-ents, such as silica gel, alumina and Florisil®. On silica gel,BDEs elute with mixtures of DCM and n-hexane [47,68,69],ixtures of diethyl ether and n-hexane [6] or with DCM [68] in a
econd, more polar, fraction after PCBs have been quantitativelyluted with n-hexane. However, BDE 209 elutes quantitativelyn the first fraction [17]. Alumina and Florisil® show less selec-ivity for PBDEs than silica gel and, using similar solvents as
xtractants, some planar PCBs were found to elute together withBDEs in the second fraction [47]. In addition, lower brominatedBDEs, such as BDE 47, partially eluted in the first fractionhen using Florisil® columns [41]. Deactivated silica gel has
1 atogr
aPisedi
tsap(vp
Hdhct
PPs4qTbPbn[ooE2ptisPaatfwt[
Gepi2ir
2
ttddssolcanns
sSpttPtd
asuDoTvasWgtB
TcrmopbiimBioB
54 A. Covaci et al. / J. Chrom
lso been successfully applied for the quantitative isolation ofBDEs from HBCD diasteroisomers and TBBP-A. In this case,
so-octane was used for the elution of PBDEs, while a more polarolvent, i.e. 15% diethyl ether:iso-octane (v/v), was required tolute HBCDs and TBBP-A [17,49]. The use of the semi-polariethyl ether was necessary to recover the late eluting �-HBCDsomer [49].
Florisil® (activated at 450 ◦C for 12 h and subsequently deac-ivated with 0.5% H2O, w/w) has been successfully used toeparate neutral organohalogenated compounds from phenolicnalytes, including TBBP-A [45]. In this case, neutral com-ounds were firstly eluted with mixtures of DCM and n-hexane1:3, v/v), while polar mixtures of acetone and n-hexane (15:85,/v) and methanol and DCM (12:88, v/v) were needed to elutehenolic analytes.
Polystyrene divinyl benzene-based sorbents, such as OasisLB®, are a valuable alternative for the fast separation of HBCDiasteroisomers from TBBP-A. Only 7 mL of a mixture DCM:n-exane (1:1, v/v) was required to elute HBCDs from the SPEartridge, while 8 mL of DCM sufficed for subsequent quantita-ive elution of TBBP-A [33].
Activated carbon has been used to separate the bulk ofCBs and PBDEs from the planar non-ortho-PCBs, PCDDs andCDFs [46,48,70]. Although this sorbent provided an efficienteparation, large solvent volumes, e.g. 100 mL of n-hexane and00 mL of DCM:n-hexane (2:3, v/v), were required to ensureuantitative elution of ortho-substituted PCBs and PBDEs [46].he higher efficiency provided by the use of porous graphitic car-on (Hypercarb)-HPLC resulted in a better separation betweenBBs and less brominated PBDEs, on the one hand, and highlyrominated congeners (including BDE 209) and planar bromi-ated compounds (PBDDs and PBDFs), on the other hand71]. However, the separation between the two latter classesf brominated compounds required an extra fractionation stepn a 2-(1-pyrenyl)ethyldimethylsilylated (PYE) HPLC column.ven in these conditions, BDE 209 was not separated from,3,7,8-BDD/Fs [71] using DCM:n-hexane (1:10, v/v) as mobilehase. In another application, Gomara et al. [72] have shownhat the addition of 2% toluene to iso-octane and the increasen the temperature of the PYE column to 45 ◦C resulted in aignificant reduction of the elution time of the more retainedBDEs and PBBs. Under these experimental conditions, ancceptable separation between PCBs and PBDEs + PBBs waschieved within 10 min. Only the less brominated PBDEs inves-igated (BDEs 17 and 28) were found to elute in the bulk PCBraction, while CBs 170 + 194, which were strongly retained,ere eluted in the second collected fraction containing the eight
etra- to hepta-BDEs and nine penta- to hepta-PBBs studied72].
Finally, in a rather different approach, Bayen et al. [55] usedPC for the isolation of PBDEs from selected OCPs classes in
xtracts of marine biological tissues obtained by MAE. Onlyartial separation was achieved as the BDEs 47, 99 and 100
ncluded in the study were found to elute between 18 mL and5 mL together with OCPs such as DDTs and chlordanes but,n principle, this approach should provide simultaneous lipidemoval within the first 16 mL.vat[
. A 1153 (2007) 145–171
.1.3. Quality assurance/quality controlDespite the profound influence that the measurement of
otal lipid content has on the lipid-normalized BFR concen-rations, up to now, no universal procedure for lipid contentetermination in biota has been adopted. Although separateetermination of the total lipid content using non-chlorinatedolvents has been proposed [73,74], most laboratories performimple lipid gravimetrical measurements on the same extractr on an aliquot of the extract used for determination of pol-utants. As a consequence, high lipid content in the sample, inombination with low analyte levels, has also been identifieds an additional source of variability in the results in inter-ational testing programs [75], also because laboratories doot adapt their standard purification protocols for such specificamples.
However, the lipid determination is not the only potentialource of error during sample preparation for BFR analysis.ome treatments should be avoided to preserve the integrity ofarticular BFRs, e.g. HBCDs are not stable under basic condi-ions, while high temperatures and/or extensive saponificationimes can also result in decomposition of highly brominatedBDEs and PBBs [6]. Especially for BDE 209, high tempera-
ures should be avoided during sample treatment to prevent itsebromination.
Complete evaporation of the extracts to dryness should bevoided as PBDEs and PBBs tend to adsorb to glass even moretrongly than PCBs, which can result in incomplete dissolutionpon reconstitution. For BDE 209, solvents such as toluene,CM or acetone:n-hexane mixtures should be preferred becausef its limited solubility in other organic solvents [3]. Similarly,BBP-A tends to adsorb to glass when using n-hexane as sol-ent, while it remains in solution with methanol [76]. BDE 209nd possible other higher brominated BDE congeners are photo-ensitive and thus direct exposure to UV light should be avoided.
rapping the containers with aluminium foil or the use of amberlassware are probably the simplest preventive measures. Addi-ional recommendations regarding this issue can be found in deoer and Wells [75].
Reported data for PBDE and, especially for HBCDs andBBP-A, should be supported by appropriate QA/QC proto-ols. In certain laboratories, QA and QC have been estimated toepresent up to 30% of the whole analytical effort [77]. Internaleasures to be performed in a routine basis include the analysis
f reagent blank and at least one procedural blank per set of sam-les and sample type analysed [77]. Procedural blanks shoulde analysed under the same conditions as the samples and takennto consideration for quantification. PBDEs have been reportedn procedural blanks during analysis of house dust [12], sedi-
ent [18] and serum [28,33]. According to Papke et al. [77],FR values in samples should only be reported when exceed-
ng the levels found in procedural blanks by a minimum factorf two. Alternatively, it was proposed that reported levels ofFRs should be above a value equal to the procedural blankalue plus 10 times [53], 5 times [76] or 3 times [50] the associ-
ted SD of the procedural blanks. Some analytical alternativeso reduce blank values have been recommended by Papke et al.77].
atogr
mrodrrnHeh1no
dtdaiscn
d[GidAb
pdfiatd
2
easvoaia
2
Gsw
btdtataGbcdgi
td[isaanddsg
odtawtlrt[a
giPfeCtisc4ia[
A. Covaci et al. / J. Chrom
The best procedure to evaluate the accuracy of analyticalethods used for BFR determination is the analysis of certified
eference materials. However, there are only a limited numberf reference materials certified for BFRs (SRM 2585 – indoorust; SRM 1589a – lyophilized human serum) and more mate-ials are needed. Furthermore, changes in the EU system foreference material preparation have reduced the rate at whichew materials are produced and exacerbated this problem [58].owever, indicative values are already available for several ref-
rence materials [78–80] and some BNCs, such as MeO-BDEs,ave also been measured in some of these materials, e.g. SRM945. These materials have the advantage of being homoge-eous and are probably used in many labs for the QA/QC ofther classes of halogenated pollutants.
When certified reference materials are not available, the stan-ard addition method may still be accepted as a valid approacho evaluate the accuracy and precision of the analytical proce-ures [55,77]. Preferably, validation should be performed usingQC sample spiked at least at two different (realistic) spik-
ng levels. When possible, proper incubation and aging of thepiked samples should be carried out in order that the spikedompounds mimic as closely as possible the behaviour of theaturally incurred analytes [47].
[13C]-labelled PBDEs should be preferred as internal stan-ards for quantitation based on the isotopic dilution method3,75]. When this approach is not possible, e.g. duringC–ECNI-MS analysis, PBDE congeners which are not present
n technical mixtures or environmental samples or fluorinatederivatives (F-PBDEs, [81]), can be used as internal standards.dditional recommendations regarding internal standards cane found in Papke et al. [77].
Shewhart plots for target compound data in selected QC sam-les allow an efficient detection of special causes of variationuring the analysis and are considered as a valuable approachor internal QC [77]. External QC should include regular partic-pation in interlaboratory studies and proficiency tests coveringwide variety of matrices (see Section 2.3). Additional sugges-
ions and recommendations related to QC/QA can be found inedicated literature [75,77].
.2. Advances in GC–MS and LC–MS analysis
Separation, identification and quantification of BFRs are gen-rally performed by means of GC–MS and/or LC–MS. Thenalytical methodology for the determination of BFRs, with apecial emphasis on polybrominated diphenyl ethers, was pre-iously reviewed by Covaci et al. [3]. The present section buildsn the previous review and highlights advances in the separationnd detection of BFRs in the last years, which are summarisedn Tables 2 and 3. Special attention was given to the numerousdvances concerning HBCDs and TBBP-A.
.2.1. Polybrominated diphenyl ethers
The most applied analytical method for PBDEs is stillC–MS. However, seeing the degradation problems that areometimes experienced for certain congeners, more insightas generated regarding this issue and the methodology has
adma
. A 1153 (2007) 145–171 155
een further optimised. Bjorklund et al. [97] described howhe characteristics of the GC system significantly influence theetermination of PBDEs by GC–MS. If not selected properly,he column brand, type of retention gap, press-fit connector,nd stationary phase, as well as column length and injectionechnique may have a very strong influence on the accuracynd precision of the PBDE analysis. By selecting an erroneousC set-up, the yield of nona- and deca-brominated BDEs cane reduced to zero and the precision of the determination ofongeners with more than five bromine atoms can be stronglyecreased. Bjorklund et al. [97] also concluded that the pro-rammable temperature vaporizer (PTV) injector is a suitablenjector that, if used properly, did not degrade labile PBDEs.
To overcome quality problems associated with the degrada-ion of BDE 209, the use of [13C]-labelled BDE 209 for isotopicilution in the ECNI-MS mode as proposed by Bjorklund et al.98] has found a broad application. Due to the overlap of theon clusters of native and labelled BDE 209, the ion-selectionhould be done with care. A combination of the [Br]− (m/z 79nd 81) and [C6Br5O]− (m/z 486.7 and 488.7 for BDE 209nd m/z 494.7 and 496.7 for [13C]-BDE 209) in ECNI-MS sig-ificantly increases selectivity, sensitivity and accuracy in theetermination of BDE 209. Apart from using adequate stan-ards, Bjorklund et al. [98] also highlighted the need to optimiseeveral other parameters, such as ionisation energy, moderatingas pressure, ion source temperature, and analyser temperature.
Furthermore, Bjorklund et al. [99] used a rotary-valven-column injector, which has a number of advantages: (i)ecreased discrimination of high molecular PBDEs; (ii) nohermal degradation in the injector; (iii) no liner needed; (iv)vailability of large volume injection; and (v) easy hyphenationith a preceding HPLC. A good precision was obtained for injec-
ion volumes up to 50 �l. For samples with low contaminationevels, such as air, injection of 50 �l might not be satisfactory inegard to LODs. Therefore, other types of injection, such as loopype injection with an early solvent vapour exit was investigated100]. Up to 500 �l could be injected with high reproducibilitynd low carry-over.
Several potential chromatographic interferences can hamperood quality data [3,101]. When working in EI-MS, potentialnterferences originate from chlorinated compounds, such asCBs; the nominal masses corresponding to ions monitoredor di-BDEs and penta-CBs are the same (m/z = 326). Theirxact masses are 325.8942 for di-BDEs and 325.8804 for penta-Bs and a resolution power of 24,000 is needed to separate
hem. However, this is not recommended due to the signif-cant loss of sensitivity at this elevated resolution [101]. Aimilar interference occurs for tetra-BDEs and hepta-CBs. Spe-ial attention should be paid to the co-elution between BDE7 and CB 180 on 30 m DB-5 type columns [3] and therefore,dentification criteria should be very restrictive (retention timend relative isotopic peak ratios). For ECNI-MS, where onlyBr]− ions are monitored, other brominated compounds, such
s MeO-PBDEs or PBBs, could also interfere with the PBDEeterminations. One MeO-tetra-BDE congener and one MeO-onochloro-tetra-BDE were found to coelute with BDE 100nd BDE 99, respectively, using a DB-5 column [102], while
156A
.Covacietal./J.C
hromatogr.A
1153(2007)
145–171
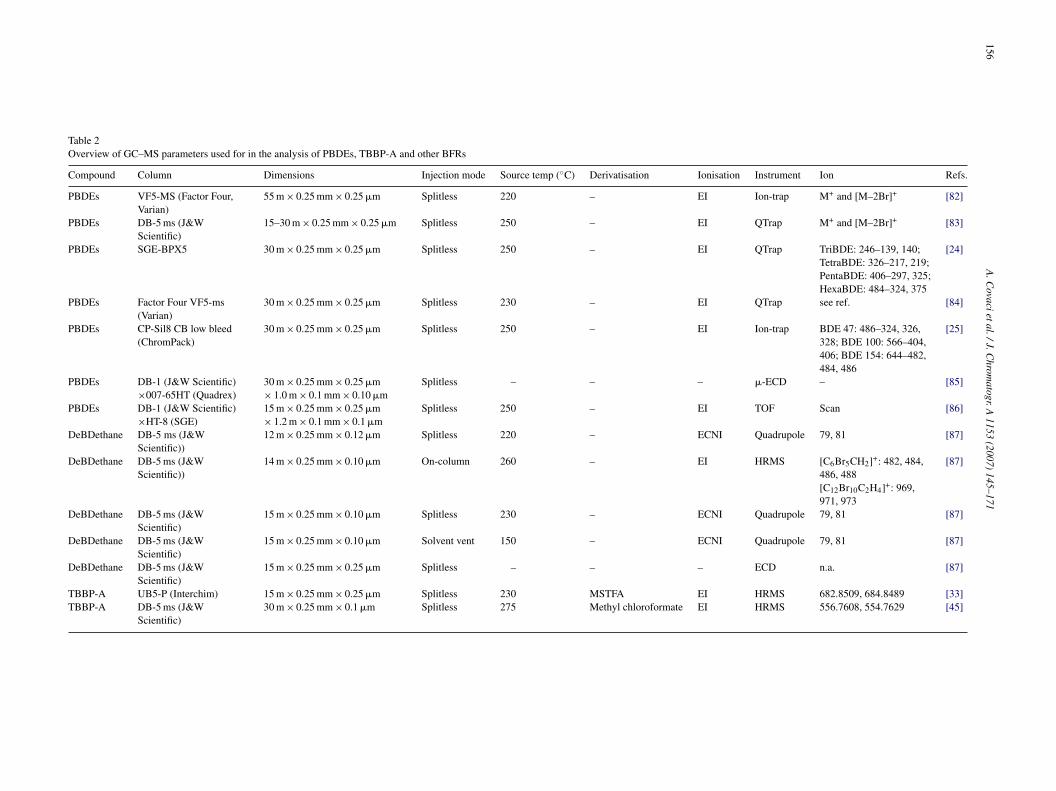
Table 2Overview of GC–MS parameters used for in the analysis of PBDEs, TBBP-A and other BFRs
Compound Column Dimensions Injection mode Source temp (◦C) Derivatisation Ionisation Instrument Ion Refs.
PBDEs VF5-MS (Factor Four,Varian)
55 m × 0.25 mm × 0.25 �m Splitless 220 – EI Ion-trap M+ and [M–2Br]+ [82]
PBDEs DB-5 ms (J&WScientific)
15–30 m × 0.25 mm × 0.25 �m Splitless 250 – EI QTrap M+ and [M–2Br]+ [83]
PBDEs SGE-BPX5 30 m × 0.25 mm × 0.25 �m Splitless 250 – EI QTrap TriBDE: 246–139, 140;TetraBDE: 326–217, 219;PentaBDE: 406–297, 325;HexaBDE: 484–324, 375
[24]
PBDEs Factor Four VF5-ms(Varian)
30 m × 0.25 mm × 0.25 �m Splitless 230 – EI QTrap see ref. [84]
PBDEs CP-Sil8 CB low bleed(ChromPack)
30 m × 0.25 mm × 0.25 �m Splitless 250 – EI Ion-trap BDE 47: 486–324, 326,328; BDE 100: 566–404,406; BDE 154: 644–482,484, 486
[25]
PBDEs DB-1 (J&W Scientific)×007-65HT (Quadrex)
30 m × 0.25 mm × 0.25 �m× 1.0 m × 0.1 mm × 0.10 �m
Splitless – – – �-ECD – [85]
PBDEs DB-1 (J&W Scientific)×HT-8 (SGE)
15 m × 0.25 mm × 0.25 �m× 1.2 m × 0.1 mm × 0.1 �m
Splitless 250 – EI TOF Scan [86]
DeBDethane DB-5 ms (J&WScientific))
12 m × 0.25 mm × 0.12 �m Splitless 220 – ECNI Quadrupole 79, 81 [87]
DeBDethane DB-5 ms (J&WScientific))
14 m × 0.25 mm × 0.10 �m On-column 260 – EI HRMS [C6Br5CH2]+: 482, 484,486, 488
[87]
[C12Br10C2H4]+: 969,971, 973
DeBDethane DB-5 ms (J&WScientific)
15 m × 0.25 mm × 0.10 �m Splitless 230 – ECNI Quadrupole 79, 81 [87]
DeBDethane DB-5 ms (J&WScientific)
15 m × 0.25 mm × 0.10 �m Solvent vent 150 – ECNI Quadrupole 79, 81 [87]
DeBDethane DB-5 ms (J&WScientific)
15 m × 0.25 mm × 0.25 �m Splitless – – – ECD n.a. [87]
TBBP-A UB5-P (Interchim) 15 m × 0.25 mm × 0.25 �m Splitless 230 MSTFA EI HRMS 682.8509, 684.8489 [33]TBBP-A DB-5 ms (J&W
Scientific)30 m × 0.25 mm × 0.1 �m Splitless 275 Methyl chloroformate EI HRMS 556.7608, 554.7629 [45]

A.C
ovacietal./J.Chrom
atogr.A1153
(2007)145–171
157Table 3Overview of LC–MS parameters used for in the analysis of HBCDs, TBBP-A, PBDEs and other BFRsa
Compound Column Dimensions Mobile phase (gradient) Flow(mL/min)
Mobile phasemodifiers
Ionisation Instrument Ion Sourcetemp (◦C)
Ref.
(±) HBCDs Nucleodex(Machery-Nagel)
200 mm × 4 mm; 5 �m AcN:MeOH:H2O (y) 0.5 – ESI Triplequadrupole
MRM(640.6 => 79,81)
– [88]
HBCDs Luna C18
(Phenomenex)150 mm × 2 mm; 5 �m AcN:MeOH:H2O (y) 0.2 Ammonium
acetateESI Ion-
trap/quadrupole640.7 160 [17,49]
HBCDs Vydac 218MS5(Mandel Scientific)
150 mm × 2.1 mm; 5 �m AcN:MeOH:H2O (y) – Ammoniumacetate
ESI Triplequadrupole
MRM(640.6 => 79)
125 [89]
HBCDs Genesis C18
(Chromatogr.Specialties)
50 mm × 2.1 mm; 4 �m MeOH:H2O (y) 0.3 – ESI Triplequadrupole
MRM(640.6 => 79,81)
500 [90]
HBCDs Symmetry C18
(Waters)150 × 2.1; 5 �m AcN:MeOH:H2O (y) 0.25 – ESI Triple
quadrupoleMRM(640.6 => 79,81)
– [88]
HBCDs Zorbax Stable BondC18 (Agilent)
150 mm × 2.1 mm; 3.5 �m MeOH:toluene:H2O (y) 0.3 – APPI Triplequadrupole
MRM(640.6 => 79)
n.a. [91]
HBCDs Develosil C30
(Nomura)150 mm × 2 mm; 5 �m AcN:H2O (y) 0.2 – APPI Triple
quadrupoleMRM 250 [7]
TBBP-A:542.7 => 445.8HBCDs:640.6 => 79
HBCDs Luna C18
(Phenomenex)150 mm × 2 mm; 5 �m AcN:H2O (y) 0.25 – ESI Quadrupole 640.7 150 [49]
HBCDs Vydac 218MS5(Mandel Scientific)
150 mm × 2.1 mm; 5 �m AcN:MeOH:H2O (y) 0.15–0.2 Ammoniumacetate
ESI Triplequadrupole
MRM(640.6 => 79,81)
125 [92]
HBCDs Symmetry C18
(Waters)150 mm × 2.1 mm; 3.5 �m AcN:MeOH:H2O (y) 0.25 Acetic acid ESI Triple
quadrupoleMRM(640.6 => 79,81)
– [33]
PBDE, TBBP-A,TBPE
Luna 5 �
Phenyl-Hexyl(Phenomenex)
150 mm × 4.6 mm; 5 �m(50 ◦C)
MeOH:2-aminoethanolbuffered water (n)
2.5 – – UV(200–400 nm)
– – [64]
PBDEs Ultrabase RP18
(SFCC)250 mm × 2 mm; 5 �m MeOH:toluene:H2O (n) 0.2 – APPI QTrap Scan n.a. [93]
PBDEs, HBCDs,TBBP-A
Hypersil ODS C18
(Thermo Electron)250 mm × 4.6 mm; 5 �m(40 ◦C)
MeOH:buffer(ammonium acetate) (n)
1 – – UV(203 nm)/triplequad
Scan(150–1000)
n.a. [63]
TBBP-A Nucleodur 100-C8
(Interchim)250 mm × 4 mm; 5 �m AcN:H2O (y) 1b Acetic acid APPI QTrap Scan n.a. [93]
TBBP-A Develosil C30
(Nomura)150 mm × 2 mm; 5 �m AcN:H2O (y) 0.2 – APCI Triple
quadrupoleMRM 250 [7]
TBBP-A:542.7 => 445.8HBCDs:640.6 => 79
TBBP-A Mightysil (Kanto) 150 mm × 2 mm; 3 �m AcN:MeOH:H2O (y) 0.2 – ESI Triplequadrupole
MRM(542.7 => 445.8)
– [29]

158 A. Covaci et al. / J. ChromatogrTa
ble
3(C
onti
nued
)
Com
poun
dC
olum
nD
imen
sion
sM
obile
phas
e(g
radi
ent)
Flow
(mL
/min
)M
obile
phas
em
odifi
ers
Ioni
satio
nIn
stru
men
tIo
nSo
urce
tem
p(◦
C)
Ref
.
TB
BP-
AX
terr
aC
18(W
ater
s)15
0m
m×
2.1
mm
;3.5
�m
MeO
H:H
2O
(y)
0.2
Am
mon
ium
acet
ate
APC
I,E
SIT
ripl
equ
adru
pole
SIM
(542
.7,
544.
7)sc
an70
–550
135
[13]
TB
BP-
AD
isco
very
C18
(Sup
elco
)—se
dim
ent
50m
m×
2.1
mm
;5�
mM
eOH
:H2O
(y)
0.3
–E
SIIo
n-tr
apSc
an(1
45–5
43)
–[9
4]
TB
BP-
ASy
mm
etry
C8
(Wat
ers)
—se
wag
esl
udge
150
mm
×4.
6m
m;3
.5�
mM
eOH
:H2O
(y)
0.3
–E
SIIo
n-tr
apSc
an(1
45–5
43)
–[9
4]
TB
BP-
AL
una
C18
(Phe
nom
enex
)15
0m
m×
2m
m;5
�m
AcN
:H2O
(y)
0.25
Am
mon
ium
acet
ate
ESI
Qua
drup
ole
540.
915
0[1
7]
TB
BP-
AA
ce3
C18
(Adv
ance
dC
hrom
atog
raph
yTe
chno
logi
es)
150
mm
×2.
1m
m;3
.0�
mM
eOH
:H2O
(y)
0.2
Am
mon
ium
acet
ate
ESI
TO
FSc
an(2
30–5
50)
130
[45]
TB
PP-A
Gen
esis
C18
(Chr
omat
ogra
phic
Spec
ialti
es)
150
mm
×2.
1m
m;4
�m
MeO
H:H
2O
(y)
0.2
–E
SIT
ripl
equ
adru
pole
MR
M(5
43=
>81
)13
0[9
5]
TB
BP-
A-d
bpe
Zor
bax
XD
B-C
18
(Agi
lent
)15
0m
m×
4.6
mm
;5�
m(4
0◦ C
)A
cN:H
2O
(y)
1–
–D
AD
––
[96]
aFo
rac
rony
ms,
see
Tabl
e1
orte
xt.
bPo
st-c
olum
nsp
littin
g1:
10.
Bos
PddtrCdtsndcitnemtwol
KlPewtGTioftrsael0
[h[caiamtpbs
. A 1153 (2007) 145–171
DE 154 and PBB 153 were also co-eluting on the same typef column [3]. Therefore, alternative stationary phases that canolve these co-elutions have been suggested [3,102].
Korytar et al. [103] investigated the elution order of 126BDEs on seven different GC stationary phases. The resultingatabase facilitates selection of the most suitable GC columns foreveloping a quantitative, congener-specific PBDE analysis andhe testing of retention prediction algorithms based on structureelationships of GC phases and congener substitution patterns.ompared to PCBs, less co-elutions for PBDE congeners withifferent bromine numbers were observed (Table 4). This meanshat the use of MS detection in the EI mode, which enables aeparate quantification of co-eluting homologues with a differentumber of Br substituents, is not of primary importance and thatetection in the ECNI mode will yield reliable results in manyases. Stationary phase dependent degradation was observed,ndicating that column equivalency is not always a suitable cri-eria for column selection [97,103]. Based on three criteria; (i)umber of PBDEs involved in co-elutions; (ii) number of co-luting groups; and (iii) number of co-elutions observed forajor PBDEs, a DB-XLB (J&W Scientific) column was found
o be the most efficient for PBDE congener-specific separation,ith a DB-1 (J&W Scientific) column as runner-up. The latterne is, however, preferred for routine analysis because it causesess degradation of hepta- and higher brominated congeners.
The experimental retention time (tR) database produced byorytar et al. [103] was used by Wang et al. [104] to calcu-
ate quantitative structure GC relative tR models for all 209BDE congeners on seven stationary phases. Based on param-ters such as the branching of the molecules, the interactionith the stationary phase could be predicted. Strong correla-
ions were obtained between relative tR obtained using differentC configurations and under different operation conditions.hese correlation equations can be used to predict the chem-
cal identities of unknown peaks. Using the same tR databasef Korytar et al. [103], Zhao et al. [105] developed a methodor GC peak recognition of PBDEs under different tempera-ure programs. On the basis of an identification database ofetention parameters of gas chromatography and tR of theelected internal standards, the relative tR of PBDEs can beccurately estimated. These predicted relative tR matched thexperimental relative tR under similar conditions reported ear-ier by Korytar et al. [85] to a great extent (highest deviation was.043).
Recently, new analytical approaches, such as ITD-MS25,82,83] or quadrupole ion-storage MS (QTrapMS, [24,84])ave been evaluated for the analysis of PBDEs. Gomara et al.82] have established a method for the determination of 10 PBDEongeners by means of GC–ITD-MS operated in EI. Parametersffecting isolation and fragmentation of precursor ions in theon-trap were optimized in order to achieve the best robustnessnd sensitivity for all PBDE congeners investigated. The frag-ent ions monitored correspond to loss of two Br atoms from
he molecular ion. The feasibility of the overall method (sam-le preparations plus instrumental detection) has been evaluatedy participating in interlaboratory comparison exercises withatisfactory results.

A. Covaci et al. / J. Chromatogr. A 1153 (2007) 145–171 159
Table 4Co-elution of PBDE congeners on seven GC columns according to three criteria
Major PBDEs
DB-1 DB-5 HT-5 DB-17 DB-XLB HT-8 CP-Sil 1962a 63a 66a 67a 56a 62a 72a
24b 26b 27b 30b 22b 26b 29b
Co-elution with major PBDEs28 16, 33 16, 33 16, 33, 38 – – 16, 33, 38 16, 3347 – – – – – – –49 68,80 68 68 62 46, 48, 68, 71 68 51, 7599 – – 116 127 – – –100 – – 109 101 – 109, 120 –153 – – HBCD 168 – – –154 MeTBBP-A, BB 153 MeTBBP-A, BB 153 – 105 – 126 BB 153183 BB 169 BB 169 – – – – –
M
fcsttapofsuittaBais1ebstbpo
togpMQnf(
([wpP5eassassct
TC
T
EEEIQ
M
odified from reference [103].a No. of co-eluting PBDEs.b No. of co-elutions.
Using GC-QTrapMS for the analysis of 20 PBDEs, dif-erences in isolation and fragmentation patterns for PBDEsongeners with degree of bromination were observed [84]. Theensitivity obtained was similar to that of ECNI and greaterhan that of EI quadrupole MS. The applicability of QTrapMSo real samples and matrix effects were evaluated through thenalysis of sewage sludge sample from a waste-water treatmentlant [84]. Similar characteristics of the QTrapMS system werebserved by Wang et al. [83], allowing the monitoring of dif-erent ions for congeners with the same number of bromineubstituents. For the mono-, di- and tri-PBDEs, the molec-lar ions ([M]+ or [M + 2]+) were observed to be the mostntense peaks and thus were selected as the precursor ions forhe subsequent MS/MS analysis. Different fragmentation pat-erns, however, were obtained for tetra- to deca-BDEs. Forll ortho-substituted tetra- through hepta-BDEs and for deca-DE, predominant ion clusters resulting from the loss of two Brtoms (i.e. [M–2Br + 2]+ or [M–2Br + 4]+) were seen, while thentense molecular ion clusters were observed for the non-ortho-ubstituted tetra- through hepta-PBDEs (e.g. BDE 77 and BDE26). This is in agreement with the data published by Alaeet al. [106], who reported that PBDEs with ortho-substitutedromines favoured the formation of the [M–2Br]+ over the [M]+
pecies when analyzed by EI-MS. Wang et al. [83] demonstratedhat MS/MS using QTrapMS is a highly specific method that can
e applied to the determination of PBDEs in environmental sam-les. The optimisation of the dissociation processes by variationf the experimental parameters was found to be important forpmh
able 5omparison of the principal mass spectrometric techniques used for the analysis of P
echnique Sensitivity Identity confirmati
CNI-MS ++ + (bromine ion)I-MS + ++ (nominal molecI-HRMS +++ ++ (exact molecul
TD-MS/MS + ++ (nominal molecTrap-MS/MS ++ ++ (nominal molec
odified from reference [84]. +: low; ++: medium; +++: high.
he MS/MS analysis. However, despite the detection specificitybtained by MS/MS, it is still possible that co-eluting con-eners with higher bromine content could give rise to isobaricrecursor and fragment ions that would both contribute to theS/MS channel monitored for a targeted PBDE. Nevertheless,TrapMS may constitute a low-cost, rapid and reliable alter-ative to high-resolution mass spectrometry (HRMS) devicesor the analysis of selected PBDEs in environmental samplesTable 5).
Comprehensive two-dimensional gas chromatographyGC × GC), a technique that offers excellent separation power107] has been evaluated also for the analysis of PBDEs. Itas previously shown that even on the most efficient stationaryhases, single dimension GC cannot separate all or nearly allBDE congeners; from the 125 BDE congeners that were tested,5 congeners were involved in 22 co-elutions for the mostfficient phase, DB-XLB [103]. As a consequence, Korytar etl. [85] evaluated six column combinations for the GC × GCeparation of PBDEs with �-ECD or time of flight-masspectrometry (TOF-MS) as detectors. They concluded that
DB-1 × 007-65HT (Quadrex) combination was the mostuitable because of: (i) the highest number of PBDE congenerseparated; (ii) the least decomposition of higher brominatedongeners; and (iii) the most suitable maximum operatingemperature. With this set-up, there were only 17 co-eluting
airs involving 35 congeners. Since other BFRs or MeO-BDEsay be present in environmental samples, Korytar et al. [85]ave shown that the second dimension column improves the
BDEs
on Accuracy Cost
++ (internal standard) +ular mass) +++ (isotope dilution) +
ar mass) +++ (isotope dilution) +++ular mass) +++ (isotope dilution) +ular mass) +++ (isotope dilution) +

1 atogr
saa
itadsnGbtBatwD
PuIGaasd[
waaoaEtctshois(iDratedtneDft
hechi
cotTtooioFtffobcott
2
a[hsitsy[
iAtattta6h4aris
60 A. Covaci et al. / J. Chrom
eparation of Me-TBBP-A, TBBP-A, BB 169, 6-OH-BDE 47nd 6-MeO-BDE 47, while most of other compounds werelready separated in the first dimension.
Korytar et al. [85] also found that F-BDEs were valuablenternal standards for the determination of PBDEs because ofheir very similar physico-chemical properties and excellent sep-ration from the corresponding PBDEs, mainly in the secondimension. GC × GC–TOF-MS and GC × GC–�-ECD werehown to be useful tools to identify decomposition products ofona- and deca-substituted BDEs, which are formed during theC run. All three nona-BDEs (206, 207 and 208) were shown toe the major decomposition products of BDE 209. The intensi-ies of the decomposition curves are seen to decrease in the orderDE 207 > BDE 208 > BDE 206. This suggests that the loss ofbromine atom in the meta position is the most, and that from
he ortho position the least-favoured elimination. Some columnsere found to lead to a complete degradation of BDE 209 (e.g.B-XLB column).Focant et al. [86] have set-up a multi-residue method for
BDEs, PBBs, PCBs and OCPs in human serum and milksing comprehensive GC × GC–TOF-MS and isotope dilution.n contrast to the reference methods based on single dimensionC–MS, a single injection resulted in accurate identification
nd quantification with method LODs ranging between 1 pg/gnd 15 pg/g. While the GC × GC ensured the chromatographiceparation of most compounds, TOF-MS allowed mass spectraleconvolution of co-eluting compounds, as well as the use of13C]-labelled internal standards.
Only few reports on new or previously un-investigated BFRsere published. Since these new BFRs have similar structures as
lready known BFRs, the existing analytical methodology waspplied. Kierkegaard et al. [87] have investigated the presencef decabrominated diphenyl ethane (DeBDethane), marketed asn alternative to BDE 209, in sewage sludge, sediment and air.I-MS proved to be the most appropriate ionisation mode for
he identification of DeBDethane (Table 2). Although identifi-ation was carried out using HRMS by monitoring the ions ofhe most abundant fragment [C6Br5CH2]+, quantitative mea-urement are preferably done by means of ECNI due to itsigher sensitivity. Similar to BDE 209, a rising baseline wasbserved before the DeBDethane peak, pointing at degradationn the GC system. Because DeBDethane has an extremely lowolubility in most organic solvents, a combination of solventsacetone, tetrahydrofurane and toluene) together with ultrason-cation was needed to dissolve a small amount of DeBDethane.ue to the lack of an appropriate reference standard, the levels
eported by Kierkegaard et al. [87] are only indicative becausemounts were roughly estimated using a relative response fac-or between DeBDethane and BDE 209. Later, Konstantinovt al. [108] investigated the differences regarding recovery andetector response between DeBDethane and BDE 209. Seeinghe similarities between BDE 209 and DeBDethane, Konstanti-ov et al. [108] emphasised that similar measures are taken to
nsure good quality data (e.g. the use of corresponding [13C]-eBDethane as internal standard, now commercially availablerom Wellington Labs). Using similar techniques as used forhe analysis of PBDEs, e.g. GC–ECNI-MS, Hoh et al. [109]
dgs
. A 1153 (2007) 145–171
ave reported the presence of 1,2-bis(2,4,6-tribromophenoxy)-thane (TBE) in air and sediment samples. Unexpectedly, TBEoncentrations in air were comparable to those of tetra- throughexa-BDEs and often higher than those of BDE 209, reflectingts increasing usage.
Recently, Debrauwer et al. [93] have investigated the appli-ability for LC techniques to the analysis of PBDEs. The usef atmospheric pressure photo ionisation (APPI) may facili-ate the analysis of PBDEs and phenolic compounds, such asBBP-A, in the same run. Negative APPI gave better sensi-
ivity than positive APPI, but due to the low resolving powerf LC compared to GC, this technique has not been furtherptimised for PBDE analysis. The use of APPI seems promis-ng for the identification of more polar Phase II metabolitesf PBDEs, such as glucuronic acid or glutathione conjugates.urthermore, LC–APPI-MS could be a potential alternative for
he analysis of BDE 209, seeing the difficulties encounteredor this congener during GC–MS analysis [93]. Without per-orming full optimisation, LODs were estimated in the rangef 200–1500 pg on-column. Further optimisation is necessaryefore this methodology can be taken into consideration to beomplementary to GC–MS analysis of PBDEs. This methodol-gy opens a way to the use of LC–MS/MS based methods forhe identification of BFR biotransformation products for BFRshat can be formed in vitro [110].
.2.2. HexabromocyclododecanesTraditionally, HBCD has been analysed using GC–MS, usu-
lly operated in ECNI-mode for which the monitoring of theBr]− ions allows a higher sensitivity. However, this techniqueas a number of serious limitations, such as the interconver-ion of HBCD diastereomers above 160 ◦C and therefore thempossibility of separating them, decomposition of HBCDs atemperatures above 240 ◦C and partial breakdown in dirty GCystems [3]. Detailed information regarding the GC–MS anal-sis of HBCDs can be found in recent reviews by Covaci et al.111] and Law et al. [112].
In contrast to GC, reversed-phase LC, coupled to electrosprayonisation (ESI) or atmospheric pressure chemical ionizationPCI-MS, is a versatile tool for the isomer-specific determina-
ion of HBCDs in environmental samples. Although the structurend chemical properties of HBCD suggest the use of APCI forhis molecule, Budakowski and Tomy [90] have shown that thisechnique does not meet the expectations and that the ion intensi-ies using ESI were significantly higher. Using LC–ESI-MS/MSnd single reaction monitoring for the transition [M–H]− (m/z40.6) → [Br]− (m/z 79 and 81), Budakowski and Tomy [90]ave developed a selective and sensitive method with LODs of–6 pg on-column for a standard solution of �-HBCD. Janak etl. [88] have further optimized both the LC and MS conditionsesulting in baseline separation of the HBCD diastereomers andn LODs of 0.5 pg and 5 pg on-column for �-HBCD in standardolutions and fish extracts, respectively.
Although LC–MS/MS is now the method of choice for theetermination of HBCDs, a lot of pitfalls can hamper results ofood quality. Tomy et al. [89] have studied crucial parameters,uch as co-elution with other analytes or with matrix compo-

atogr
nc((mlHpbssom
macdamf
tpHco7ltwHwMTwtoa6er
o[safEcb[swesl
b�abcsdry
2
rdPtmoaLBabL
cmpbcdbsoAfib
mumssafaFmfa
t
A. Covaci et al. / J. Chrom
ents, which can lead to ion-suppression. This effect can beompensated for by means of: (i) standard addition method;ii) dilution of the sample; (iii) improved sample clean-up;iv) improved chromatographic separation; (v) use of a matrix-atched external standards; or (vi) spiking the sample with
abelled internal standards prior to injection (e.g. deuteratedBCDs). This latter standard performs a dual role: to com-ensate for instrumental fluctuations of the mass spectrometeretween injections and to compensate for matrix related ionuppression or enhancement effects that can occur in the ionource. Tomy et al. [89] concluded that accurate measurementsf native HBCD isomers in environmental matrices are compro-ised without the use of labelled standards.In order to enhance the signal or sensitivity, mobile phase
odifiers, such as ammonium acetate [13,17,45,49,89,92] andcetic acid [33] are commonly used. Tomy et al. [89] were alsooncerned by the apparent differences in the stability of HBCDiastereomers in various solvents. This was explained by the rel-tively lower solubility of �-HBCD in acetonitrile compared toethanol. Therefore, the authors suggested the use of methanol
or final extracts before LC–MS analysis.Although triple quadrupole instruments are preferred due to
heir high selectivity and sensitivity, Morris et al. [49] com-ared a single quadrupole MS and an ITD-MS for the analysis ofBCDs. Using a single quadrupole MS, collision induced disso-
iation is not possible. Although ITD-MS allows the monitoringf daughter ions, the HBCD-specific transition (m/z 640.6 → m/z9 and 81) cannot be used because the m/z of the daughter-ion isower than the cut-off value of the instrument (typically 25% ofhe parent ion). Differences in the sensitivity of different HBCDsere observed between the two MS instruments [49]. The �-BCD isomer was the most sensitive on the quadrupole MS,hile the �-HBCD isomer was the most sensitive on the ITD-S. The linearity of the response was limited (20–250 ng/�L).
he decreased response in ESI-MS at high analyte concentrationas attributed to “analyte saturation”, where charge competi-
ion occurs on the ESI probe [49]. LOQs were around 150 pgn-column for both quadrupole and ITD instruments. Morris etl. [49] also reported the presence of a weaker MS signal at m/z76.7 [M + Cl–H]− originating from a chlorine adduct. How-ver, this fragment showed variable presence during analyses ofeal samples.
Although Budakowski and Tomy [90] favoured the use of ESIver APCI for the ionisation of HBCDs, Suzuki and Hasegawa7] reported on the analysis of HBCDs and TBBP-A in leachateamples by LC–APCI-MS. In this case, ionisation by APCI led to2–5 times higher S/N ratio compared to ESI for HBCDs, while
or TBBP-A the S/N ratio using APCI was almost half that ofSI. They also concluded that ESI was more sensitive to matrixomponents [7]. Other ionisation modes, such as APPI, can alsoe used, but rather as complementary technique to ESI or APCI91]. While the common problems for ESI and APCI, such asuppression of the analyte ionisation efficiency by compounds
ith a higher ionisation potential or compounds present in largexcess, had a lower influence on the ionization by APPI, theensitivity obtained using APPI was approximately 10 timesower compared to ESI [91].
rauL
. A 1153 (2007) 145–171 161
HPLC on chiral, permethylated �-cyclodextrin columns haseen successfully employed to separate enantiomers of the (+/−)-, (+/−) �- and (+/−) �-HBCD stereomers [88,113]. The sep-ration was achieved in a single analysis, while LODs variedetween 10 pg and 20 pg injected on-column. However, matrixomponents co-eluting with HBCDs may lead to serious ionuppression of the primary ion [M–H]−, resulting in stronglyecreased sensitivity [114]. This problem may be (partially)esolved through extensive sample clean-up prior to HPLC anal-sis.
.2.3. Tetrabromobisphenol-AAlthough TBBP-A is the most widely used brominated flame
etardant, this compound is not frequently measured, probablyue to its presence at lower concentrations in biota compared toBDEs and HBCDs and due to its lower bioaccumulation poten-
ial. Among the predominant BFRs, TBBP-A is the most polarolecule, which demands therefore more complicated meth-
ds for a proper determination. Acidification and derivatisationre compulsory before analysis by GC can be carried out, whileC has the advantage that no derivatisation step is required [75].oth the derivatisation and acidification step can introduce errorsnd/or losses [75]. In terms of sensitivity, LC–ESI-MS/MS cane competitive with published GC–EI-MS/MS techniques, withODs in the ppt-range [29].
A GC-HRMS method requiring derivatisation with methyl-hloroformate was developed by Berger et al. [45]. However, thisethod suffered from a rather restricted linear range and incom-
lete derivatisation, leading to lower recoveries. This might alsoe explained by the presence of bulky bromine substituents adja-ent to the two hydroxyl groups, resulting in an incompleteouble derivatisation. Although the GC–MS method showedetter separation properties and was more sensitive for standardolutions, LC–ESI-TOF-MS was superior for the quantificationf egg sample extracts, with a satisfying LOQ of 3 pg TBBP-
on-column [45]. This was explained by the high-resolutionltering potential of the TOF-MS which eliminates the matrixackground from the analyte’s mass chromatograms.
The LC separation of TBBP-A from other compounds andatrix components is greatly dependent on the mobile phase
sed. Chu et al. [95] reported one-third higher response withethanol instead of acetonitrile. For optimized chromatographic
eparation and/or ionisation response, mobile phase additives,uch as formic acid, tris(hydroxymethyl)aminomethane andmmonium acetate, are often used. However, while the twoormer products were found to give a decreased ESI-response,mmonium acetate significantly increased the response [95].urthermore, using a methanol mobile phase alone resulted in aore stable detector baseline and thus a lower LOQ [95]. There-
ore, using methanol and water as a mobile phase can be moredvantageous for the quantitative analysis of TBBP-A.
The commercial availability of [13C]-TBBP-A has improvedhe quality of the LC–MS analysis to a great extent. The occur-
ence of matrix effects, which can affect analyte ion intensity,ccuracy and reproducibility, can be much diminished by these of labelled standards. Furthermore, the soft ionisation inC–ESI-MS makes possible to monitor the intact molecule [13].
1 atogr
Ta
o[rl
fsmmreiTp
cafooLsfSs[s
bmpoestwbsvst2
Uoetwttp
ea
aAnfttCti
2
p[PQwtgamdeKRwBarta
2
bTptv
NAiEeP2(aswe
62 A. Covaci et al. / J. Chrom
ogether with sample clean-up, these two features improve bothccuracy and precision of the analyses.
Hayama et al. [29] have set-up a method for the determinationf TBBP-A in human serum using SPE and LC–ESI-MS/MS.13C]-TBBP-A was found suitable as an internal standard for theeproducible determination of TBBP-A. Method LOQs were asow as 4 pg/g serum.
For the determination of TBBP-A in air, Tollback et al. [13]ound that ESI in negative ionisation mode was optimal, withensitivity up to 40 times higher compared to APCI. The use ofultiple reaction monitoring (MRM) produced mainly ions with/z ratios of 79 and 81. Other fragments could not significantly
educe the LOD, because they were only formed in low amounts,ven at high collision energy. The most sensitive method was runn SIM (m/z 542.7, 544.7). Tollback et al. [13] also noted thatBBP-A standard solution can be debrominated if not properlyrotected from light.
Apart from triple quadrupole MS, ITD-MS was also suc-essfully used for the determination of TBBP-A in sedimentnd sewage sludge [94]. Sensitivity of the system clearly suf-ered after running a batch of sewage sludge extracts, but with anptimised and clean system, an LOD of 5 pg on-column could bebtained for standard solutions. This value is comparable to theOD reported for GC–MS after derivatisation [115]. The ITDcan range was set from m/z 145–543, with mass transitionsrom m/z 543–528 and from m/z 543–448 being also useful.urprisingly, little ion suppression of the TBBP-A signal fromediment matrix components in the ESI process was observed94]. However, sewage sludge extracts greatly influenced the ionuppression, but extensive clean-up minimised this effect.
Another TBBP-A related product, such as tetrabromo-isphenol A bis(2,3-dibromopropylether) (TBBP-A-dbpe), isarketed as an additive for polymers, including polypropylene,
olyethylene and high-impact polystyrene (HIPS). As a resultf its poor water solubility, the TBBP-A-dbpe released into thenvironment is expected to accumulate in sediments and sewageludges. Attempts to use LC–ESI-MS or LC–APCI-MS in nega-ive or positive ion mode for the determination of TBBP-A-dbpeere unsuccessful [96]. The samples were therefore quantifiedy LC–DAD using external calibration with purified substanceince no native or isotopic labelled standards were available. Thealidation of the method was accomplished using sediment andewage sludge samples spiked with defined amounts of authen-ic TBBP-A-dbpe. The LODs using LC–DAD were 10 ng/g and2 ng/g in sediment and sewage sludge, respectively.
In a study of degradation products of TBBP-A formed afterV-exposure, ESI did not prove very useful, while the ionisationf TBBP-A degradation products by means of APPI was veryfficient [93]. A potential drawback of APPI was its suscep-ibility towards the mobile phase composition. Higher signalsere seen at the end of the gradient elution, close to 100% ace-
onitrile. This particular feature could constitute a limitation forhe quantitative analysis of mixtures of BFRs, their degradation
roducts or metabolites [93].Next to LC techniques, capillary electrophoresis (CE), anfficient technique for the separation of charged species, waslso found useful for the analysis of TBBP-A [116]. Blanco et
acat
. A 1153 (2007) 145–171
l. [116] developed a method for the determination of TBBP-and other phenolic compounds in environmental samples by
on-aqueous capillary electrophoresis coupled to DAD (210 nmor TBBP-A), with MeOH as solvent. Some parameters affectinghe electrophoretic separation were studied, such as salt concen-ration, electrolyte pH, and capillary and solution temperature.alibration curves were linear from 0.5 ng/�L to 10 ng/�L. This
echnique was successfully applied for the analysis of TBBP-An water samples. Method LOQs were in the range of 12 pg/�L.
.2.4. Brominated flame retardants in polymersFor polymers containing high concentrations of BFRs, sim-
le analysis techniques have been proposed. Schlummer et al.63] used LC–UV/MS to identify and quantify HBCD, TBBP-A,BDEs and PBBs in post-consumer plastics from waste of EEE.uantification proved to be more reproducible by UV, whichas probably due to the co-extraction of polymer components
hat accumulate in the APCI source. The authors therefore sug-est using UV for the quantification and MS for identificationnd validation purposes. A similar approach based on a rapidethod for the extraction of BFRs followed by LC–UV was
eveloped by Pohlein et al. [64]. The overall runtime required forxtraction and chromatographic analysis was less than 10 min.ikuchi et al. [117] analysed PBDEs in matrix polymers byaman spectroscopy without any sample pretreatment. LODas approximately 100 �g/g and analysis time took only 1 min.ased on the distinctive bands, the DecaBDE technical mixturend TBBP-A could be identified. Energy dispersive X-ray fluo-escence analysis, LC–UV, GC–MS and infrared spectroscopyechniques have also been evaluated for the analysis of polymersfter various extraction procedures [62].
.3. Interlaboratory studies on brominated flame retardants
Since 1999, several international interlaboratory studies haveeen organized to improve the quality in the analysis of BFRs.able 6 summarizes the most important exercises that have takenlace until now. A wide range of matrices have been used duringhese exercises and this has enabled researchers to adequatelyalidate their methods and to implement reliable QC procedures.
Four interlaboratory studies were organized by Theetherlands Institute for Fisheries Research (RIVO), Qualityssurance of Information for Marine Environmental Monitor-
ng in Europe (QUASIMEME) and the Bromine Science andnvironmental Forum (BSEF). Detailed descriptions of thesexercises were presented in several articles [75,118]. All majorBDE congeners (BDE 28, 47, 99, 100, 153, 154, 183, and09), as well as HBCDs, TBBP-A and dimethylated TBBP-AMe-TBBP-A) were targeted. The test materials included fishnd marine mammal tissue, fish oil, shellfish, sediments, sewageludge, human milk and standard solutions (Table 6). In parallelith the increasing number of participating laboratories, a gen-
ral tendency for qualitative improvements of results and thus of
nalytical methods was observed. This was possible due to theontinuous advice given by the organisers after each exercisend to the increased availability of (labelled) standards. Never-heless, these interlaboratory studies have shown that most labo-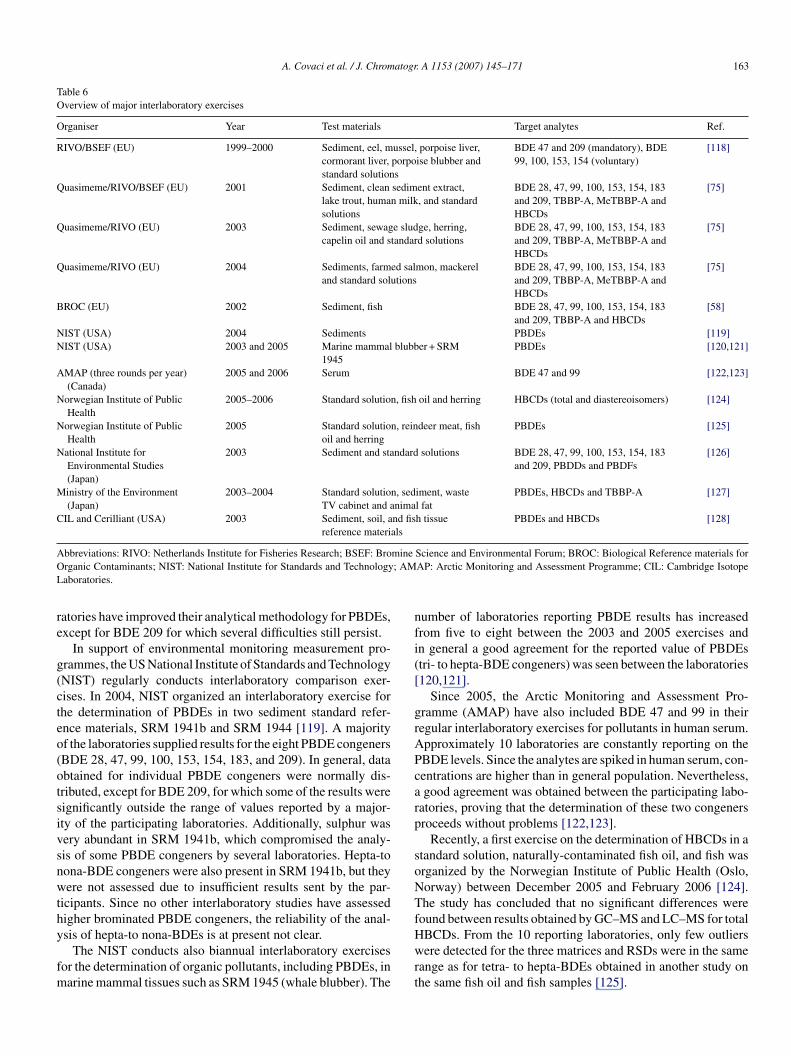
A. Covaci et al. / J. Chromatogr. A 1153 (2007) 145–171 163
Table 6Overview of major interlaboratory exercises
Organiser Year Test materials Target analytes Ref.
RIVO/BSEF (EU) 1999–2000 Sediment, eel, mussel, porpoise liver,cormorant liver, porpoise blubber andstandard solutions
BDE 47 and 209 (mandatory), BDE99, 100, 153, 154 (voluntary)
[118]
Quasimeme/RIVO/BSEF (EU) 2001 Sediment, clean sediment extract,lake trout, human milk, and standardsolutions
BDE 28, 47, 99, 100, 153, 154, 183and 209, TBBP-A, MeTBBP-A andHBCDs
[75]
Quasimeme/RIVO (EU) 2003 Sediment, sewage sludge, herring,capelin oil and standard solutions
BDE 28, 47, 99, 100, 153, 154, 183and 209, TBBP-A, MeTBBP-A andHBCDs
[75]
Quasimeme/RIVO (EU) 2004 Sediments, farmed salmon, mackereland standard solutions
BDE 28, 47, 99, 100, 153, 154, 183and 209, TBBP-A, MeTBBP-A andHBCDs
[75]
BROC (EU) 2002 Sediment, fish BDE 28, 47, 99, 100, 153, 154, 183and 209, TBBP-A and HBCDs
[58]
NIST (USA) 2004 Sediments PBDEs [119]NIST (USA) 2003 and 2005 Marine mammal blubber + SRM
1945PBDEs [120,121]
AMAP (three rounds per year)(Canada)
2005 and 2006 Serum BDE 47 and 99 [122,123]
Norwegian Institute of PublicHealth
2005–2006 Standard solution, fish oil and herring HBCDs (total and diastereoisomers) [124]
Norwegian Institute of PublicHealth
2005 Standard solution, reindeer meat, fishoil and herring
PBDEs [125]
National Institute forEnvironmental Studies(Japan)
2003 Sediment and standard solutions BDE 28, 47, 99, 100, 153, 154, 183and 209, PBDDs and PBDFs
[126]
Ministry of the Environment(Japan)
2003–2004 Standard solution, sediment, wasteTV cabinet and animal fat
PBDEs, HBCDs and TBBP-A [127]
CIL and Cerilliant (USA) 2003 Sediment, soil, and fish tissuereference materials
PBDEs and HBCDs [128]
A mineO ; AML
re
g(cteo(otsivsnwthy
fm
nfi([
grAPcarp
soNTf
bbreviations: RIVO: Netherlands Institute for Fisheries Research; BSEF: Brorganic Contaminants; NIST: National Institute for Standards and Technologyaboratories.
atories have improved their analytical methodology for PBDEs,xcept for BDE 209 for which several difficulties still persist.
In support of environmental monitoring measurement pro-rammes, the US National Institute of Standards and TechnologyNIST) regularly conducts interlaboratory comparison exer-ises. In 2004, NIST organized an interlaboratory exercise forhe determination of PBDEs in two sediment standard refer-nce materials, SRM 1941b and SRM 1944 [119]. A majorityf the laboratories supplied results for the eight PBDE congenersBDE 28, 47, 99, 100, 153, 154, 183, and 209). In general, databtained for individual PBDE congeners were normally dis-ributed, except for BDE 209, for which some of the results wereignificantly outside the range of values reported by a major-ty of the participating laboratories. Additionally, sulphur wasery abundant in SRM 1941b, which compromised the analy-is of some PBDE congeners by several laboratories. Hepta-toona-BDE congeners were also present in SRM 1941b, but theyere not assessed due to insufficient results sent by the par-
icipants. Since no other interlaboratory studies have assessedigher brominated PBDE congeners, the reliability of the anal-
sis of hepta-to nona-BDEs is at present not clear.The NIST conducts also biannual interlaboratory exercisesor the determination of organic pollutants, including PBDEs, inarine mammal tissues such as SRM 1945 (whale blubber). The
Hwrt
Science and Environmental Forum; BROC: Biological Reference materials forAP: Arctic Monitoring and Assessment Programme; CIL: Cambridge Isotope
umber of laboratories reporting PBDE results has increasedrom five to eight between the 2003 and 2005 exercises andn general a good agreement for the reported value of PBDEstri- to hepta-BDE congeners) was seen between the laboratories120,121].
Since 2005, the Arctic Monitoring and Assessment Pro-ramme (AMAP) have also included BDE 47 and 99 in theiregular interlaboratory exercises for pollutants in human serum.pproximately 10 laboratories are constantly reporting on theBDE levels. Since the analytes are spiked in human serum, con-entrations are higher than in general population. Nevertheless,good agreement was obtained between the participating labo-
atories, proving that the determination of these two congenersroceeds without problems [122,123].
Recently, a first exercise on the determination of HBCDs in atandard solution, naturally-contaminated fish oil, and fish wasrganized by the Norwegian Institute of Public Health (Oslo,orway) between December 2005 and February 2006 [124].he study has concluded that no significant differences were
ound between results obtained by GC–MS and LC–MS for total
BCDs. From the 10 reporting laboratories, only few outliersere detected for the three matrices and RSDs were in the sameange as for tetra- to hepta-BDEs obtained in another study onhe same fish oil and fish samples [125].

1 atogr
3
ibwaofba[mpm
b
aTvbleB
f(aBoA
64 A. Covaci et al. / J. Chrom
. Brominated natural compounds
To date, over 3600 halogenated natural compounds have beendentified and the largest proportion are BNCs [129]. BNCs maye produced by marine species (sponges, corals, sea slugs, seaorms), seaweed, plants, fungi, lichen, algae, bacteria, microbes
nd some mammals [129,130]. In the past few years, the rangef non-polar halogenated compounds has been further expandedollowing the identification of several BNCs, such as the tetra-romo phenoxyanisole BC-2 and related compounds (BC-10nd BC-11) [131] or halogenated 1,1′-dimethyl-2,2′-bipyrroles132]. Fig. 1 presents the chemical structures of the most com-only found BNCs, most of which present also bioaccumulative
roperties. These BNCs have already been identified in fish,
arine mammals and birds, human milk, air, and other matrices.The analytical chemistry of the described BNCs has not yeteen reviewed because only in a few cases, their identification
f
f
Fig. 1. Chemical structures of the most commonl
. A 1153 (2007) 145–171
nd quantification has been the principal scope of research [133].he use of ECNI-MS and the monitoring of the [Br]− ion pro-ide a sensitive and selective approach to the determination ofrominated compounds by GC–MS [133]. Due to this pecu-iarity, the presence of additional peaks in chromatograms ofnvironmental and food samples during the routine analysis ofFRs has led to the identification of several BNCs [133].
In most cases, a comparison of retention times and ECNI or EIull scans on various GC columns between reference compoundswhen available) and the corresponding unknown compoundsre necessary for correct identification and characterization ofNCs. A major drawback in the analysis of the BNCs is that mostf them are not commercially available as reference standards.s a consequence, concentrations cannot always be determined
or all individual BNCs.Table 7 summarises a selection of analytical methods used
or the determination of BNCs in a wide variety of biological
y detected brominated natural compounds.

A.C
ovacietal./J.Chrom
atogr.A1153
(2007)145–171
165
Table 7Overview of typical analytical procedures used for the determination of brominated natural compounds in selected matricesa
Compounds Matrices Extraction Clean-up Analysis Ref.
MeO-BDEs (2′-MeO-BDE 68 + 6-MeO-BDE47 + MeO-triBDEs)
Cod liver, mussel, marine mammals MAE GPC + deactivated andactivated SiO2
GC–ECNI-MS, GC–EI-MS [131]
MeO-BDEs + Cl-MeO-BDEs Salmon blood C6:MTBE KOH partitioning, GPC,activated SiO2 + acidifiedSiO2
GC–ECNI-MS, GC–EI-MS [102]
HO-BDEs + MeO-BDEs Red algae, blue mussels Acet:C6 + C6:MTBE KOH partitioning,SiO2 + acidified SiO2
GC–ECNI-MS [134]
MeO-BDEs Whale and dolphin liver SFE Basic Al2O3 and C18 (as fatretainers)
GC–ECNI-MS [135]
MeO-BDEs (2′-MeO-BDE 68 + 6-MeO-BDE47)
Pike Acet:C6 + C6:MTBE H2SO4 + acidified SiO2 GC–ECNI-MS, GC–EI-MS [136]
MeO-BDEs (2′-MeO-BDE 68 + 6-MeO-BDE47)
Fish, guillemot Soxhlet (toluene) H2SO4 + Florisil® + carboncolumn
GC–EI-MS [137]
Sea lion blubber
MeO-BDEs (2′-MeO-BDE 68 + 6-MeO-BDE47)
European shag eggs, fish liver, bluemussels
Acet:cyclohexane (3/1) GPC + Florisil® GC–ECNI-MS [138]
MeO-BDEs (2′-MeO-BDE 68 + 6-MeO-BDE47)
Fish oil supplements Soxhlet (Acet:C6) Acidified SiO2 GC–ECNI-MS, GC–EI-MS [139]
MeO-BDEs (2′-MeO-BDE 68 + 6-MeO-BDE47) + DBP-Br4Cl2
Sea lion blubber PLE (DCM) GPC + SiO2 GC–ECNI-MS, GC–EI-MS [140]
DBP-Br3Cl3, DBP-Br4Cl2, DBP-Br5Cl,DBP-Br6
Fish and seafood, seabird liver andeggs, marine mammals
PLE (Acet:C6, 1:2), Soxhlet,column extraction
GPC + Florisil® GC–ECNI-MS [132,141,142]
DBP-Br3Cl2, DBP-Br3Cl3, DBP-Br4Cl2,DBP-Br5Cl, DBP-Br6
Whale and dolphin blubber Column extraction DCM:C6(1/1)
GPC + activated SiO2 GC–EI-MS [143]
MBP-HBr5Cl, MBP-Br6Cl and MBP-Br7 Blubber marine mammals + squid Column extraction, DCM:C6(1:1)
GPC + SiO2/Al2O3
fractionationGC–EI-MS, GC–ECNI-MS [144]
TriBDDs + tetraBDDs Blue mussels Acet:C6 + C6:MTBE H2SO4, SiO2, aminopropylLC column, PYE column
GC-HRMS [145]
Di- and tribromoindoles Common oysters Column extraction (DCM) Florisil® GC–ITD-MS, GC–ECNI-MS [146]Di- and tribromoindoles Water and sediment LLE or SLE (Acet:C6, 1:1) GPC + cyanopropyl LC
column (sediment),SiO2 + GPC
GC–ITD-MS, GC–ECNI-MS [147]
2,2′-diMeO-BB80 Blubber and liver marine mammals SLE (Acet:C6, 1:1) H2SO4, acidified SiO2, SiO2 GC–PICI-MS,GC–ECNI-MS GC–EI-MS
[148]
TriBHD and tetraBHD Fish and seal blubber SLE (Acet:C6, 1:2) GPC, H2SO4, SiO2 GC–ECNI-MS GC–EI-MS [149]
a For acronyms, see Table 1 and text.
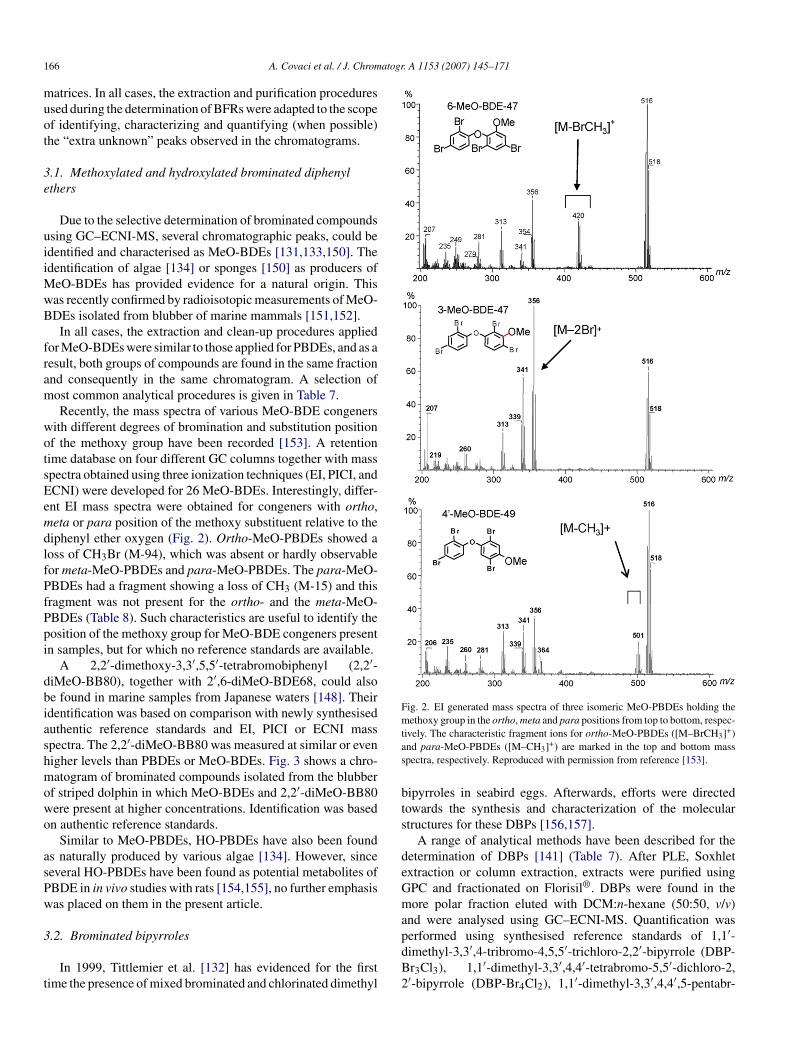
1 atogr. A 1153 (2007) 145–171
muot
3e
uiiMwB
fram
wotsEemdlfPfPpi
dbiashmowo
asPw
3
t
Fig. 2. EI generated mass spectra of three isomeric MeO-PBDEs holding themethoxy group in the ortho, meta and para positions from top to bottom, respec-tas
bts
deGma
66 A. Covaci et al. / J. Chrom
atrices. In all cases, the extraction and purification proceduressed during the determination of BFRs were adapted to the scopef identifying, characterizing and quantifying (when possible)he “extra unknown” peaks observed in the chromatograms.
.1. Methoxylated and hydroxylated brominated diphenylthers
Due to the selective determination of brominated compoundssing GC–ECNI-MS, several chromatographic peaks, could bedentified and characterised as MeO-BDEs [131,133,150]. Thedentification of algae [134] or sponges [150] as producers of
eO-BDEs has provided evidence for a natural origin. Thisas recently confirmed by radioisotopic measurements of MeO-DEs isolated from blubber of marine mammals [151,152].
In all cases, the extraction and clean-up procedures appliedor MeO-BDEs were similar to those applied for PBDEs, and as aesult, both groups of compounds are found in the same fractionnd consequently in the same chromatogram. A selection ofost common analytical procedures is given in Table 7.Recently, the mass spectra of various MeO-BDE congeners
ith different degrees of bromination and substitution positionf the methoxy group have been recorded [153]. A retentionime database on four different GC columns together with masspectra obtained using three ionization techniques (EI, PICI, andCNI) were developed for 26 MeO-BDEs. Interestingly, differ-nt EI mass spectra were obtained for congeners with ortho,eta or para position of the methoxy substituent relative to theiphenyl ether oxygen (Fig. 2). Ortho-MeO-PBDEs showed aoss of CH3Br (M-94), which was absent or hardly observableor meta-MeO-PBDEs and para-MeO-PBDEs. The para-MeO-BDEs had a fragment showing a loss of CH3 (M-15) and thisragment was not present for the ortho- and the meta-MeO-BDEs (Table 8). Such characteristics are useful to identify theosition of the methoxy group for MeO-BDE congeners presentn samples, but for which no reference standards are available.
A 2,2′-dimethoxy-3,3′,5,5′-tetrabromobiphenyl (2,2′-iMeO-BB80), together with 2′,6-diMeO-BDE68, could alsoe found in marine samples from Japanese waters [148]. Theirdentification was based on comparison with newly synthesiseduthentic reference standards and EI, PICI or ECNI masspectra. The 2,2′-diMeO-BB80 was measured at similar or evenigher levels than PBDEs or MeO-BDEs. Fig. 3 shows a chro-atogram of brominated compounds isolated from the blubber
f striped dolphin in which MeO-BDEs and 2,2′-diMeO-BB80ere present at higher concentrations. Identification was basedn authentic reference standards.
Similar to MeO-PBDEs, HO-PBDEs have also been founds naturally produced by various algae [134]. However, sinceeveral HO-PBDEs have been found as potential metabolites ofBDE in in vivo studies with rats [154,155], no further emphasisas placed on them in the present article.
.2. Brominated bipyrroles
In 1999, Tittlemier et al. [132] has evidenced for the firstime the presence of mixed brominated and chlorinated dimethyl
pdB2
ively. The characteristic fragment ions for ortho-MeO-PBDEs ([M–BrCH3]+)nd para-MeO-PBDEs ([M–CH3]+) are marked in the top and bottom masspectra, respectively. Reproduced with permission from reference [153].
ipyrroles in seabird eggs. Afterwards, efforts were directedowards the synthesis and characterization of the moleculartructures for these DBPs [156,157].
A range of analytical methods have been described for theetermination of DBPs [141] (Table 7). After PLE, Soxhletxtraction or column extraction, extracts were purified usingPC and fractionated on Florisil®. DBPs were found in theore polar fraction eluted with DCM:n-hexane (50:50, v/v)
nd were analysed using GC–ECNI-MS. Quantification was
erformed using synthesised reference standards of 1,1′-imethyl-3,3′,4-tribromo-4,5,5′-trichloro-2,2′-bipyrrole (DBP-r3Cl3), 1,1′-dimethyl-3,3′,4,4′-tetrabromo-5,5′-dichloro-2,′-bipyrrole (DBP-Br4Cl2), 1,1′-dimethyl-3,3′,4,4′,5-pentabr-
A. Covaci et al. / J. Chromatogr. A 1153 (2007) 145–171 167
Table 8Ion fragments obtained in GC–EI-MS and GC–ECNI-MS for brominated natural compounds
Compounds Ionization Ions Ref
6-MeO-BDE 47 ECNI [M–Br]− (434), [M–2Br]− (354), [C7H6Br2O]− (264) [131,138,151,152]6-MeO-BDE 47 EI [M + 4]+ (516), [M–BrCH3]+ (418), [M–2Br]+ (354) [131,138,151,152]2′-MeO-BDE 68 ECNI [M–Br]− (434), [M–2Br]− (354) [138,151,152]2′-MeO-BDE 68 EI [M + 4]+ (516), [M–BrCH3]+ (418), [M–2Br]+ (354) [138,151,152]6′-Cl-2′-MeO-BDE 68 EI [M + 4]+ (550), [M–BrCH3]+ (456), [M–BrCl]+ (436), [M–2Br]+ (390) [102]6′-Cl-2′-MeO-BDE 68 ECNI [M + 4]− (550), [M–Br]− (470), [M–2Br]− (390), [M–C6H3Br2CH3]− (298) [102]Dibromoindole EI (ITD) [M]+ (273), [M–Br]+ (194) [146]Tribromoindole EI (ITD) M+ (351), [M–Br]+ (272), [M–HBr2]+ (192) [146]DBP-Br4Cl2 EI M+ (544), [M–C2H3NBr]+ (424), [M–2Br]+ (384) [140]DBP-Br3Cl2 EI M+ (462), [M–C2H3NBr]+ (344), [M–2Br]+ (306) [143]MBP-HBr5Cl EI [M]+ (576), [M–Cl]+ (541), [M–Br]+ (495), [M–BrCl]+ (460), [M–2Br]+ (416), [M–3Br]+ (335) [144]M + + + (575 + + +
MM ]+ (53
o3
aAbtboMtft[nCa
Fbi
eb
setnDccp
3
BP-Br6Cl EI [M] (654), [M–Cl] (619), [M–Br]BP-Br6Cl ECNI [M]− (654), [M–Br]− (576)BP-Br7 EI [M]+ (697), [M–Br]+ (619), [M–2Br
mo-5′-chloro-2,2′-bipyrrole (DBP-Br5Cl) and 1,1′-dimethyl-,3′,4,4′,5,5′-hexabromo-2,2′-bipyrrole (DBP-Br6) [141].
Recently, mixed halogenated methyl bipyrroles (MBPs) havelso been evidenced in samples of marine mammals [144,158].lthough structures of polybrominated naphthols have initiallyeen proposed [159], subsequent investigations have shownhat the correct structures had a 1′-methyl-1,2′-bipyrrole back-one (Fig. 1). Further investigations have shown the presencef higher brominated MBPs (MBP-HBr5Cl, MBP-Br6Cl andBP-Br7) in marine mammal species [144]. Similar to DBPs,
he analytical procedures for MBPs involved column extractionollowed by GPC purification and silica and alumina fractiona-ion. Again, MBPs were eluted in a second, more polar fraction144]. Since reference standards for these MBP derivatives were
ot available, quantification was done using a standard of DBP-l6 [144]. It is important to mention that both mixed brominatednd chlorinated DBP and MBP derivatives degrade in the pres-ig. 3. GC–ECNI-MS chromatogram (m/z 79 and 81 monitored) from blub-er of a striped dolphin using a non-polar Equity-5 column (30 m × 0.25 mm.d. × 0.1 �m film thickness). Reproduced with permission from reference [148].
imfpnecFaPgosbatbpbS
3
c
), [M–BrCl] (538), [M–2Br] (494), [M–3Br] (415) [144][144]
8), [M–3Br]+ (459), [M–4Br]+ (380), [144]
nce of concentrated H2SO4 and therefore acid treatment shoulde avoided during lipid elimination from extracts.
In an Arctic marine food web (zooplankton – Arctic cod –eabirds), DBP-Br4Cl2 was found to biomagnify to a greaterxtent than DBP-Br3Cl3, DBP-Br5Cl and DBP-Br6, even thoughhe Kow of DBP-Br4Cl2 is lower than that of DBPs with higherumber of bromines [160]. The biomagnification potential ofBPs, which is in the same range as the recalcitrant PCB 153,
an have serious consequences regarding toxic effects of suchompounds at relatively higher levels, such as found in top-redators [160].
.3. Brominated dibenzo-p-dioxins
The presence of polybrominated dibenzo-p-dioxins (PBDDs)n the marine environment has been recently evidenced in blue
ussels from the Baltic Sea [145] and their natural formationrom cyanobacteria has been demonstrated [161]. The analyticalrocedures were similar to those routinely used for the determi-ation of PBDEs in biological samples and typically consisted inxtraction with acetone:n-hexane followed by MTBE:n-hexane,lean-up with concentrated H2SO4 and fractionation on silica.urther clean-up was done by the use of an amino LC columnnd a PYE column for the isolation of planar compounds, such asBDDs [161]. Identification and characterization of PBDD con-eners was carried out using GC-HRMS and three GC columnsf different polarity, including a column with enantioselectivetationary phase. The identity of several PBDD congeners coulde unequivocally confirmed since some congeners were avail-ble as individual reference standards. A plausible hypothesis forhe origin of the PBDDs isolated from the blue mussels coulde the biogenic formation by biotransformation of naturally-roduced precursor molecules such as HO-PBDEs, which haveeen evidenced in red algae and blue mussels from the Balticea [134].
.4. Other brominated natural compounds
Di- and tribromoindoles (BIs) have been evidenced in theommon oyster during the investigation of anthropogenic con-

1 atogr
t[wutot(Gaa1tbfLms
dtctHdnbsdmd
m[mpra
4
ir(arctr
liyrit
tmila
tnrmhei
tidapetwEpt
A
Cst
R
68 A. Covaci et al. / J. Chrom
aminants (e.g. pesticides) using a common analytical procedure146]. An aliquot (5–7 g) oyster wet tissue was homogenizedith sodium sulphate and the mixture was column extractedsing 200 mL DCM. After concentration, the extract was frac-ionated on 18 g 1% water-deactivated Florisil® using fractionsf increasing eluant polarity. Two di- and one tri-BIs dominatedhe second fraction eluted with 100 mL 20% DCM in n-hexanev/v) and they were identified using full scan GC–ECNI-MS andC–EI-MS. In both cases, the molecular ions were the most
bundant ions (Table 8). Since reference standards were notvailable, samples were quantified using the response factor of,4-dibromonaphthalene. A more recent study [147] has shownhe presence of di- and tri-BIs in water and sediment samples. Foroth matrices, samples were extracted using acetone:n-hexane,urther cleaned-up using GPC and fractionated on a cyanopropylC column (sediment) or silica (water). BIs were quantitativelyeasured by GC–ECNI-MS using commercially available or
ynthesized reference standards [147].Recently, tri- and tetrabrominated hexahydroxanthene
erivatives (PBHDs) were detected for the first time duringhe routine analysis of commercial fish for PBDEs [149]. Afterolumn extraction of ∼20 g fish and purification using GPC,he obtained extract was further purified using concentrated
2SO4 and fractionated on silica. The brominated xantheneerivatives were present in the more polar fraction eluted with-hexane:toluene (65:35, v/v), which contained also other poly-rominated compounds. Identification was carried out using fullcan GC–ECNI-MS and GC–EI-MS. Interestingly, the sameerivatives were tentatively identified in fish oil dietary supple-ents [139], but the correct structure could not be yet attributed
ue to the lack of reference standards.Other BNCs that were less frequently detected in
arine samples are bromophenols [138,162], tribromoanisole131,138,162], brominated carbazoles [163] and a dibro-otrichloro monoterpene (MHC-1) [131,164]. These com-
ounds were always present in the more polar fraction whichesults from silica fractionation and could be structurally char-cterized using GC–EI-MS and/or GC–ECNI-MS.
. Future perspectives
The ongoing international interlaboratory studies on BFRsn environmental matrices have resulted in an improved compa-ability of laboratory results for a number of PBDE congenersBDE 47, 99, 100, 153, and 154). However, other PBDEs (183nd 209) are still a cause of serious concern for most laborato-ies. Only a few laboratories have demonstrated that they canarry out reliable HBCD and TBBP-A analyses based on LCechniques. For these two analytes, LC–MS based methods areecommended over the traditional GC–MS methods.
A general observation concerns the extensive time taken byaboratories to install and validate analytical methods for emerg-ng contaminants, such as BFRs. With a few exceptions, several
ears are needed to pass before a sufficient number of labo-atories can produce reliable results for a new contaminant. Its important to mention the relatively low number of labora-ories participating in interlaboratory studies compared to the. A 1153 (2007) 145–171
remendous number of publications and reports which suggests auch larger number of analytical laboratories actually perform-
ng analyses of BFRs. It is still uncertain how many of theseaboratories participate regularly in intercomparison exercisesnd therefore how reliable is the reported data.
Another issue that may probably receive more attention inhe future is the identification and quantification of bromi-ated compounds released from the use of polymeric flameetardants [165]. A number of compounds, such as pentabro-oethylbenzene, pentabromotoluene, pentabromobenzene or
exabromobenzene, have already been identified during thexperimental thermal stress of brominated polymers [165] orn the air [109].
Lastly, more efforts should be directed towards the iden-ification and characterization of “unknown” peaks occurringn chromatograms. Most of the peaks identified to date wereescribed as natural compounds. More data is needed to learnbout their sources, environmental occurrence, bioaccumulativeroperties, exposure to humans and toxic effects. In view of sev-ral open questions, the variety of unknown compounds, andhe fact that there are no regulatory limits for BNCs in food,e are convinced that research on this topic is an urgent matter.stablishing yet-unknown structures and determining the toxicotential of natural products is an important task in environmen-al chemistry and toxicology.
cknowledgements
A.C. acknowledges financial support from the Researchouncil of the University of Antwerp and a postdoctoral fellow-
hip of the Funds of Scientific Research Flanders (FWO). L.R.hanks MEC for financial support (CTQ2006-14993/BQU).
eferences
[1] C.A. de Wit, Chemosphere 46 (2002) 583.[2] L.S. Birnbaum, D.F. Staskal, Environ. Health Perspect. 112 (2004) 9.[3] A. Covaci, S. Voorspoels, J. de Boer, Environ. Int. 29 (2003) 735.[4] E. Eljarrat, D. Barcelo, Trends Anal. Chem. 23 (2004) 727.[5] K. D’Silva, A. Fernandes, M. Rose, Crit. Rev. Environ. Sci. Technol. 34
(2004) 141.[6] J. de Boer, C. Allchin, R. Law, B. Zegers, J.P. Boon, Trends Anal. Chem.
20 (2001) 591.[7] S. Suzuki, A. Hasegawa, Anal. Sci. 22 (2006) 469.[8] V. Yusa, A. Pastro, M. de la Guardia, Anal. Chim. Acta 565 (2006) 103.[9] M. Polo, G. Gomez-Noya, J.B. Quintana, M. Llompart, C. Garcia-Jares,
R. Cela, Anal. Chem. 76 (2004) 1054.[10] J. Llorca-Porcel, G. Martinez-Sanchez, B. Alvarez, M.A. Cobollo, I.
Valor, Anal. Chim. Acta 569 (2006) 113.[11] S. Harrad, S. Hazrati, C. Ibarra, Environ. Sci. Technol. 40 (2006) 4638.[12] B. Gevao, M. Al-Bahloul, A.N. Al-Ghadban, Al Al-Omair, L. Ali, J.
Zafar, M. Helaleh, Chemosphere 64 (2006) 603.[13] J. Tollback, C. Crescenzi, E. Dyremark, J. Chromatogr. A 1104 (2006)
106.[14] E. Hoh, R.A. Hites, Environ. Sci. Technol. 39 (2005) 7794.[15] H.M. Stapleton, N.G. Dodder, J.H. Offenberg, M.M. Schantz, S.A. Wise,
Environ. Sci. Technol. 39 (2005) 925.
[16] D. Wang, Z. Cai, G. Jiang, M.H. Wong, W.K. Wong, Rapid Commun.Mass Spectrom. 19 (2005) 83.[17] S. Morris, C.R. Allchin, B.N. Zegers, J.J.H. Haftka, J.P. Boon, C. Belpaire,
P.E.G. Leonards, S.P.J. van Leeuwen, J. de Boer, Environ. Sci. Technol.38 (2004) 5497.

atogr
A. Covaci et al. / J. Chrom[18] A. Covaci, A. Gheorghe, S. Voorspoels, J. Maervoet, E. Steen-Redeker,R. Blust, P. Schepens, Environ. Int. 31 (2005) 367.
[19] D.R. Oros, D. Hoover, F. Rodigari, D. Crane, J. Sericano, Environ. Sci.Technol. 39 (2005) 33.
[20] S.C. Quade, M. Alaee, C. Marvin, R. Hale, K.R. Solomon, N.J. Bunce,A.T. Fisk, Organohalogen Compd. 62 (2003) 327.
[21] F. Samara, Ch.W. Tsai, D.S. Aga, Environ. Int. 139 (2006) 489.[22] A. de la Cal, E. Eljarrat, D. Barcelo, J. Chromatogr. A 1021 (2003) 165.[23] B. Fabrellas, D. Larrazabal, M.A. Martinez, E. Eljarrat, D. Barcelo,
Organohalogen Compd. 66 (2004) 3755.[24] V. Yusa, O. Pardo, A. Pastro, M. de la Guardia, Anal. Chim. Acta 557
(2006) 304.[25] C. Salgado-Petinel, M. Garcia-Chao, M. Llompart, C. Garcia-Jares, R.
Cela, Anal. Bioanal. Chem. 385 (2006) 637.[26] M. Geissler, The Applications Book, 2006, p. 34.[27] H.M. Stapleton, B. Brazil, R.D. Holbrook, C.L. Mitchelmore, R. Bene-
dict, A. Konstantinov, D. Potter, Environ. Sci. Technol. 40 (2006) 4653.[28] A. Covaci, S. Voorspoels, J. Chromatogr. B 827 (2005) 216.[29] T. Hayama, H. Yoshida, S. Onimaru, S. Yonekura, H. Kuroki, K. Todoroki,
H. Nohta, M. Yamaguchi, J. Chromatogr. B 809 (2004) 131.[30] A. Sjodin, R.S. Jones, C.R. Lapeza, E.E. McGahee III, J.F. Focant, D.G.
Patterson Jr., Anal. Chem. 76 (2004) 1921.[31] M. Karlson, A. Julander, B. van Bavel, G. Lindstrom, Chromatographia
61 (2005) 67.[32] A. Sjodin, E.E. McGahee III, J.F. Focant, R.S. Jones, C.R. Lapeza, Y.
Zhang, D.G. Patterson Jr., Anal. Chem. 76 (2004) 4508.[33] R. Cariou, J.P. Antignac, P. Marchand, A. Berrebi, D. Zalko, F. Andre, B.
Le Bizec, J. Chromatogr. A 1100 (2005) 144.[34] H. Wolkers, M.O. Hammill, B. van Bavel, Environ. Pollut. 142 (2006)
476.[35] L. Zhu, R.A. Hites, Environ. Sci. Technol. 40 (2006) 3711.[36] H. D’Have, A. Covaci, J. Scheirs, P. Schepens, R. Verhagen, W. de Coen,
Environ. Sci. Technol. 39 (2005) 6016.[37] A.M. Carro, R.A. Lorenzo, F. Fernandez, R. Rodil, R. Cela, J. Chromatogr.
A 1071 (2005) 93.[38] R. Rodil, A.M. Carro, R.A. Lorenzo, R. Cela-Torrijos, Anal. Chem. 77
(2005) 2259.[39] J.K. Huwe, M. Lorentzsen, K. Thuresson, A. Bergman, Chemosphere 46
(2002) 635.[40] M. Lebeuf, B. Gouteux, L. Measures, S. Trottier, Environ. Sci. Technol.
38 (2004) 2971.[41] D.C.G. Muir, S. Backus, A.E. Derocher, R. Dietz, T.J. Evans, G.W.
Gabrielsen, J. Nagy, R.J. Norstrom, C. Sonne, I. Stirling, M.K. Taylor,R.J. Letcher, Environ. Sci. Technol. 40 (2006) 449.
[42] B. Johnson-Restrepo, K. Kannan, D.P. Rapaport, B.D. Rodan, Environ.Sci. Technol. 39 (2005) 5177.
[43] M.N. Jacobs, A. Covaci, A. Gheorghe, P. Schepens, J. Agric. Food Chem.52 (2004) 1780.
[44] U. Sellstrom, A. Bignert, A. Kierkegaard, L. Haggberg, C.A. de Wit, M.Olsson, B. Jansson, Environ. Sci. Technol. 37 (2003) 5496.
[45] U. Berger, D. Herzke, T.M. Sandanger, Anal. Chem. 76 (2004) 441.[46] A. Fernandes, S. White, K. D’Silva, M. Rose, Talanta 63 (2004) 1147.[47] A. Martınez, M. Ramil, R. Montes, D. Hernanz, E. Rubı, I. Rodrıguez,
R. Cela-Torrijos, J. Chromatogr. A 1072 (2005) 83.[48] P. Isosaari, A.K. Lundebye, G. Ritchie, Ø. Lie, H. Kiviranta, T. Vartiainen,
Food Addit. Contam. 22 (2005) 829.[49] S. Morris, P. Bersuder, C.R. Allchin, B. Zegers, J.P. Boon, P.E.G.
Leonards, J. de Boer, Trends Anal. Chem. 25 (2006) 343.[50] S. Voorspoels, A. Covaci, P. Schepens, Environ. Sci. Technol. 37 (2003)
4348.[51] V.L.B. Jaspers, A. Covaci, S. Voorspoels, T. Dauwe, M. Eens, P. Schepens,
Environ. Pollut. 139 (2006) 340.[52] S. Voorspoels, A. Covaci, P. Lepom, S. Escutenaire, P. Schepens, Environ.
Sci. Technol. 40 (2006) 2937.[53] C. Pirard, E. de Pauw, J.F. Focant, J. Chromatogr. A 1115 (2006) 125.[54] E. Eljarrat, A. de la Cal, D. Raldua, C. Duran, D. Barcelo, Environ. Sci.
Technol. 38 (2004) 2603.[55] S. Bayen, H.K. Lee, J.Ph. Obbard, J. Chromatogr. A 1035 (2004) 291.
. A 1153 (2007) 145–171 169
[56] M.D. Scrimshaw, K.H. Langford, J.N. Lester, Environ. Technol. 25 (2004)967.
[57] K. Nylund, L. Asplund, B. Janssen, P. Jonsson, K. Litzen, U. Sellstrom,Chemosphere 24 (1992) 1721.
[58] S.P.J. van Leeuwen, R. Van Cleuvenbergen, M. Abalos, A.L. Pasini, U.Eriksson, M. Cleemann, J. Hajslova, J. de Boer, Trends Anal. Chem. 25(2006) 397.
[59] R.C. Hale, M.J. la Guardia, E. Harvey, T.M. Mainor, Chemosphere 46(2002) 729.
[60] Directive 2002/95/EC of the European Parliament and the Council of 27January 2003 on the restriction of the use of certain hazardous substancesin electrical and electronic equipment, Official Journal L037, 13/02/2003.
[61] Directive 2003/11/EC of the European Parliament and of the Coun-cil of 6 Feburary 2003 amending for the 24th time Council directive76/769/EEC relating to restrictions on the marketing and use of certaindangerous substances and preparations (pentabromodiphenyl ether andoctabromodiphenyl ether), Official Journal L042, 15/02/2003.
[62] A.M. Altwaiq, M. Wolf, R. van Eldik, Anal. Chim. Acta 491 (2003)111.
[63] M. Schlummer, F. Brandl, A. Maurer, R. van Eldik, J. Chromatogr. A1064 (2005) 39.
[64] M. Pohlein, A. Segura Llopis, M. Wolf, R. van Eldik, J. Chromatogr. A1066 (2005) 111.
[65] M.N. Jacobs, A. Covaci, P. Schepens, Environ. Sci. Technol. 36 (2002)2797.
[66] E. Bjorklund, S. Sporring, K. Wiberg, P. Haglund, C. von Holst, TrendsAnal. Chem. 25 (2005) 318.
[67] S. Voorspoels, A. Covaci, J. Maervoet, P. Schepens, J. Environ. Monit. 6(2006) 914.
[68] B. Fangstrom, M. Athanasiadou, I. Athanassiadis, A. Bignert, P. Grand-jean, P. Weihe, A. Bergman, Chemosphere 60 (2005) 836.
[69] H. Liu, Q. Zhang, Z. Cai, A. Li, Y. Wang, G. Jiang, Anal. Chim. Acta 557(2006) 314.
[70] J.W. Choi, J. Onodera, K. Kitamura, S. Hashimoto, H. Ito, N. Suzuki, S.Sakai, M. Morita, Chemosphere 53 (2003) 637.
[71] N. Hanari, K. Kannan, Y. Miyake, T. Okazawa, P.R.S. Kodavanti, K.M.Aldous, N. Yamashita, Environ. Sci. Technol. 40 (2006) 4400.
[72] B. Gomara, C. Garcıa-Ruiz, M.J. Gonzalez, M.L. Marina, Anal. Chim.Acta 565 (2006) 208.
[73] S. Jensen, L. Haggberg, H. Jorunsdottir, G. Odham, J. Agric. Food Chem.51 (2003) 5607.
[74] F. Smedes, Analyst 124 (1999) 1711.[75] J. de Boer, D.E. Wells, Trends Anal. Chem. 25 (2006) 364.[76] A. Sjodin, Occupational and dietary exposure to organohalogenated sub-
stances with special emphasis on polybrominated flame retardants, Ph.D.thesis, Stockholm University, Sweden, 2000.
[77] O. Papke, P. Furst, T. Herrmann, Talanta 63 (2004) 1203.[78] H.M. Stapleton, T. Harner, M. Shoeib, J.M. Keller, M.M. Schantz, S.D.
Leigh, S.A. Wise, Anal. Bioanal. Chem. 384 (2006) 791.[79] J.R. Kucklick, K.J.S. Tuerk, S.S. van der Pol, M.M. Schantz, S.A. Wise,
Anal. Bioanal. Chem. 378 (2004) 1147.[80] L.Y. Zhu, R.A. Hites, Anal. Chem. 75 (2003) 6696.[81] G. Luthe, P.E.G. Leonards, H. Liu, J.E. Johansen, Organohalogen Compd.
66 (2004) 3715.[82] B. Gomara, L. Herrero, L.R. Bordajandi, M.J. Gonzalez, Rapid Commun.
Mass Spectrom. 20 (2006) 69.[83] D. Wang, Z. Cai, G. Jiang, M.H. Wong, W.K. Wong, Rapid Commun.
Mass Spectrom. 19 (2005) 83.[84] D. Larrazabal, M.A. Martınez, E. Eljarrat, D. Barcelo, B. Fabrellas, J.
Mass Spectrom. 39 (2004) 1168.[85] P. Korytar, A. Covaci, P.E.G. Leonards, J. de Boer, U.A.Th. Brinkman, J.
Chromatogr. A 1100 (2005) 20.[86] J.F. Focant, A. Sjodin, W.E. Turner, D.G. Patterson Jr., Anal. Chem. 76
(2004) 6313.[87] A. Kierkegaard, J. Bjorklund, U. Friden, Environ. Sci. Technol. 38 (2004)
3247.[88] K. Janak, A. Covaci, S. Voorspoels, G. Becher, Environ. Sci. Technol. 39
(2005) 1987.

1 atogr
70 A. Covaci et al. / J. Chrom[89] G.T. Tomy, T. Halldorson, R. Danell, K. Law, G. Arsenault, M. Alaee, G.MacInnis, C.H. Marvin, Rapid Commun. Mass Spectrom. 19 (2005) 1.
[90] W. Budakowski, G. Tomy, Rapid Commun. Mass Spectrom. 17 (2003)1399.
[91] S. Backus, S. Batchelor, M. Alaee, D. Ueno, L.M. Hewitt, OrganohalogenCompd. 67 (2005) 240.
[92] C.H. Marvin, G.T. Tomy, M. Alaee, G. MacInnis, Chemosphere 64 (2006)268.
[93] L. Debrauwer, A. Riu, M. Jouahri, E. Rathahao, I. Jouanin, J.P. Antignac,R. Cariou, B. Le Bizec, D. Zalko, J. Chromatogr. A 1082 (2005) 98.
[94] R. Saint-Louis, E. Pelletier, Analyst 129 (2004) 724.[95] S. Chu, G.D. Haffner, R.J. Letcher, J. Chromatogr. A 1097 (2005) 25.[96] R. Koppen, R. Becker, C. Jung, C. Piechotta, I. Nehls, Anal. Bioanal.
Chem. 384 (2006) 1485.[97] J. Bjorklund, P. Tollback, C. Hiarne, E. Dyremarck, C. Ostman, J. Chro-
matogr. A 1041 (2004) 201.[98] J. Bjorklund, P. Tollback, C. Ostman, J. Mass Spectrom. 38 (2003) 394.[99] J. Bjorklund, P. Tollback, E. Dyremark, C. Ostman, J. Sep. Sci. 26 (2003)
594.[100] J. Bjorklund, P. Tollback, C. Ostman, J. Sep. Sci. 26 (2003) 1104.[101] E. Eljarrat, A. de la Cal, D. Barcelo, J. Chromatogr. A 1008 (2003) 181.[102] G. Marsh, M. Athanasiadou, A. Bergman, L. Asplund, Environ. Sci.
Technol. 38 (2004) 10.[103] P. Korytar, A. Covaci, J. de Boer, A. Gelbin, U.A.Th. Brinkman, J. Chro-
matogr. A 1065 (2005) 239.[104] Y. Wang, A. Li, H. Liu, Q. Zhang, W. Ma, W. Song, G. Jiang, J. Chro-
matogr. A 1103 (2006) 314.[105] H. Zhao, X. Xue, Q. Xu, F. Zhang, X. Liang, J. Chromatogr. A 1107
(2006) 248.[106] M. Alaee, D.B. Sergeant, M.G. Ikonomou, J.M. Luross, Chemosphere 44
(2001) 1489.[107] J. Dalluge, J. Beens, U.A.Th. Brinkman, J. Chromatogr. A 1000 (2003)
69.[108] A. Konstantinov, G. Arsenault, B. Chittim, T. Loic, K. MacPherson,
A. McAlees, R. McCridle, D. Potter, E.J. Reiner, C. Tashiro, B. Yeo,Chemosphere 64 (2006) 245.
[109] E. Hoh, L.Y. Zhu, R.A. Hites, Environ. Sci. Technol. 39 (2005) 2472.[110] D. Zalko, C. Prouillac, A. Riu, E. Perdu, L. Dolo, I. Jouanin, C. Canlet,
L. Debrauwer, J.P. Cravedi, Chemosphere 64 (2006) 318.[111] A. Covaci, A.C. Gerecke, R.J. Law, S. Voorspoels, M. Kohler, N.V. Heeb,
H. Leslie, C.R. Allchin, J. de Boer, Environ. Sci. Technol. 40 (2006) 3679.[112] R.J. Law, M. Kohler, N.V. Heeb, A.C. Gerecke, P. Schmid, S. Voorspoels,
A. Covaci, G. Becher, K. Janak, C. Thomsen, Environ. Sci. Technol. 39(2005) 281A.
[113] N.V. Heeb, W.B. Schweizer, M. Kohler, A.C. Gerecke, Chemosphere 61(2005) 65.
[114] N.G. Dodder, A.M. Peck, J.R. Kucklick, L.C. Sander, J. Chromatogr. A1135 (2006) 36.
[115] C. Thomsen, E. Lundanes, G. Becher, J. Sep. Sci. 24 (2001) 282.[116] E. Blanco, M.C. Casais, M.C. Mejuto, R. Cela, J. Chromatogr. A 1071
(2005) 205.[117] S. Kikuchi, K. Kawauchi, S. Ooki, M. Kurosawa, H. Honjo, T. Yagishita,
Anal. Sci. 20 (2004) 1111.[118] J. de Boer, W.P. Cofino, Chemosphere 46 (2002) 625.[119] H.M. Stapleton, J.M. Keller, M.M. Schantz, J.R. Kucklick, S.A. Wise,
NISTIR 7278 (2005).[120] J.R. Kucklick, R.S. Pugh, P.R. Becker, M.M. Schantz, S.A. Wise, T.K.
Rowles, NISTIR 7269 (2005).[121] J.R. Kucklick, R.S. Pugh, P.R. Becker, M.M. Schantz, S.A. Wise, T.K.
Rowles, Description and results of the 2005 NIST/NOAA interlabora-tory comparison exercise program for organic contaminants in marinemammal tissues, in press.
[122] AMAP ring test for persistent organic pollutants in human serum, Rounds
2005-1, 2005-2 and 2005-3, Institute National de Sante Publique, Quebec,Canada, 2005.[123] AMAP ring test for persistent organic pollutants in human serum,Rounds 2006-1 and 2006-2, Institute National de Sante Publique, Quebec,Canada, 2006.
. A 1153 (2007) 145–171
[124] L.S. Haug, C. Thomsen, G. Becher, First Interlaboratory Comparisonon HBCD in Biological Samples, Norwegian Institute of Public Health,Oslo, Norway, 2006.
[125] L.S. Haug, G. Becher, Interlaboratory comparison on dioxins in food2005, 6th round of an International study, Report 2005:7, NorwegianInstitute of Public Health, Oslo, Norway, ISBN 82-8082-145-7.
[126] S. Takahashi, S.I. Sakai, I. Watanabe, Chemosphere 64 (2006) 234.[127] S. Takahashi, S. Sakai, I. Watanabe, Organohalogen Compd. 67 (2005)
430.[128] The Standard (CIL), Cambridge Isotope Laboratories/Cerilliant Corpo-
ration 2003 International Interlaboratory Study on Sediment, Soil, andFish Tissue Reference Materials, vol. 9 (1), August 2003.
[129] G.W. Gribble, Chem. Soc. Rev. 28 (1999) 335.[130] G.W. Gribble, Environ. Sci. Pollut. Res. 7 (2000) 37.[131] W. Vetter, W. Jun, Chemosphere 52 (2003) 423.[132] S.A. Tittlemier, M. Simon, W.M. Jarman, J.E. Elliott, R.J. Norstrom,
Environ. Sci. Technol. 33 (1999) 26.[133] W. Vetter, Anal. Chem. 73 (2001) 4951.[134] A. Malmvarn, G. Marsh, L. Kautsky, M. Athanasiadou, A. Bergman, L.
Asplund, Environ. Sci. Technol. 39 (2005) 2990.[135] A. Petterson, B. van Bavel, M. Engwall, B. Jimenez, Arch. Environ.
Contam. Toxicol. 47 (2004) 542.[136] A. Kierkegaard, A. Bignert, U. Sellstrom, M. Olsson, L. Asplund, B.
Jansson, C.A. de Wit, Environ. Pollut. 130 (2004) 187.[137] S. Sinkkonen, A.L. Rantalainen, J. Paasivirta, M. Lahtipera, Chemosphere
56 (2004) 767.[138] W. Vetter, R. von der Recke, D. Herzke, T. Nygard. Environ. Int., in press
(http://dx.doi.org/10.1016/j.envint.2006.06.019).[139] A. Covaci, S. Voorspoels, J. Wyckmans, A. Gelbin, H. Neels, Organohalo-
gen Compd. 68, in press (https://www5.shocklogic.com/scripts/JMEvent/abstracts/FCC-2602-376675-2-376675.pdf).
[140] H.M. Stapleton, N.G. Dodder, J.R. Kucklick, C.M. Reddy, M.M. Schantz,P.R. Becker, F. Gulland, B.J. Porter, S.A. Wise, Mar. Pollut. Bull. 52(2006) 522.
[141] S. Tittlemier, A. Borrell, J. Duffe, P.J. Duignan, P. Fair, A. Hall, P. Hoek-stra, K.M. Kovacs, M.M. Krahn, M. Lebeuf, C. Lydersen, D. Muir, T.O’Hara, M. Olsson, J. Pranschke, P. Ross, U. Siebert, G. Stern, S. Tanabe,R. Norstrom, Arch. Environ. Contam. Toxicol. 43 (2002) 244.
[142] S.A. Tittlemier, J. Agric. Food. Chem. 52 (2004) 2010.[143] K. Haraguchi, Y. Hisamichi, T. Endo, Arch. Environ. Contam. Toxicol.
49 (2006) 1.[144] E.L. Teuten, B.E. Pedler, A.N. Hangsterfer, C.M. Reddy, Environ. Pollut.
144 (2006) 336.[145] A. Malmvarn, Y. Zebuhr, S. Jensen, L. Kautsky, E. Greyerz, T. Nakano,
L. Asplund, Environ. Sci. Technol. 39 (2005) 8235.[146] K.A. Maruya, Chemosphere 52 (2003) 409.[147] N. Reineke, S. Biselli, S. Franke, W. Francke, N. Heinzel, H. Huhnerfuss,
H. Iznaguen, U. Kammann, N. Theobald, M. Vobach, W. Wosniok, Arch.Environ. Contam. Toxicol. 51 (2006) 186.
[148] G. Marsh, M. Athanasiadou, I. Athanassiadis, A. Bergman, T. Endo, K.Haraguchi, Environ. Sci. Technol. 39 (2005) 8684.
[149] J. Hiebl, J. Melcher, H. Gundersen, M. Schlabach, W. Vetter, J. Agric.Food Chem. 54 (2006) 2652.
[150] W. Vetter, E. Stoll, M.J. Garson, S.J. Fahey, C. Gaus, J.F. Muller, Environ.Toxicol. Chem. 21 (2002) 2014.
[151] E.L. Teuten, L. Xu, C.M. Reddy, Science 307 (2005) 917.[152] E.L. Teuten, C.G. Johnson, M. Mandalakis, L. Asplund, O. Gustafs-
son, M. Unger, G. Marsh, C.M. Reddy, Chemosphere 62 (2006)197.
[153] M. Athanasiadou, G. Marsh, I. Athanassiadis, L. Asplund, A. Bergman,J. Mass Spectrom. 41 (2006) 790.
[154] T. Malmberg, M. Athanasiadou, G. Marsh, I. Brandt, A. Bergman, Envi-ron. Sci. Technol. 39 (2005) 5342.
[155] G. Marsh, M. Athanasiadou, I. Athanassiadis, A. Sandholm, Chemo-sphere 63 (2006) 690.
[156] G.W. Gribble, D.H. Blank, J.P. Jasinski, Chem. Commun. 21 (1999) 2195.[157] S.A. Tittlemier, D.H. Blank, G.W. Gribble, R.J. Norstrom, Chemosphere
46 (2002) 511.

atogr
A. Covaci et al. / J. Chrom[158] E.L. Teuten, R. Saint-Louis, B.E. Pedler, L. Xu, E. Pelletier, C.M. Reddy,Mar. Pollut. Bull. 52 (2006) 572.
[159] R. Saint-Louis, E. Pelletier, Mar. Pollut. Bull. 50 (2005) 889.[160] S.A. Tittlemier, A.T. Fisk, K.A. Hobson, R.J. Nostrom, Environ. Pollut.
116 (2002) 85.[161] A. Malmvarn, U. Eriksson, L. Kautsky, L. Asplund, Organohalogen
Compd. 67 (2005) 1380.
. A 1153 (2007) 145–171 171
[162] W. Vetter, D. Janussen, Environ. Sci. Technol. 39 (2005) 3889.[163] L.Y. Zhu, R.A. Hites, Environ. Sci. Technol. 39 (2005) 9446.
[164] W. Vetter, J. Hiebl, N.J. Oldham, Environ. Sci. Technol. 35 (2001)4157.[165] B. Gouteux, M. Alaee, S. Mabury, D. Muir, Organohalogen Compd. 68,
in press (https://www5.shocklogic.com/scripts/JMEvent/abstracts/FCC-2602-379201-1-Gouteux%20et%20al.pdf).




















