
Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis of Natural Products
25
Send Orders of Reprints at [email protected] 2206 Current Organic Chemistry, 2012, 16, 2206-2230 1385-2728/12 $58.00+.00 © 2012 Bentham Science Publishers Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis of Natural Products Juan. Á. Bisceglia and Liliana R. Orelli* Departamento de Química Orgánica. Facultad de Farmacia y Bioquímica. Universidad de Buenos Aires. CONICET. Junín 956, (1113) Buenos Aires, Argentina Abstract: The Horner-Wadsworth-Emmons reaction has evolved in the last years as one of the most powerful and reliable method for stereocontrolled olefin synthesis. The reaction has become a widespread standard tool in natural products total syntheses, providing ac- cess to relatively simple as well as highly complex synthetic targets. In this work we present the most representative applications within the field of natural product synthesis from 2000 to the present year. The examples comprise the synthesis of macrocycles, 5 to 7 mem- bered rings, lipids and related compounds, alkaloids and cyclic ethers. They were chosen from a large amount of literature reports and il- lustrate the use of the HWE reaction in the synthesis and elaboration of key intermediates and in the crucial assembly of highly function- alized synthetic precursors. Keywords: Horner-Wadsworth-Emmons (HWE) reaction, Carbonyl olefinations, Stabilized phosphonate carbanions, Natural products, Total synthesis, ,-Unsaturated carbonyl compounds. 1. INTRODUCTION In 1958, L. Horner published a novel Wittig reaction between phosphine-oxide stabilized carbanions and carbonyl compounds [1], which was further modified by W.S. Wadsworth and W. D. Em- mons by employing phosphonates [2]. Since then, the so called “Horner-Wadsworth-Emmons (HWE) olefination reaction” gained popularity and has become a widespread tool for de novo C=C bond formation [3-7]. Although the HWE reaction has not been reviewed in the last years, some general aspects are discussed in two more recent publications devoted to carbonyl olefinations [8,9]. Among them, some generally known data will be briefly mentioned only if they are of critical importance in order to settle down the context of the present review. The classical HWE reaction is predominantly an E-alkene for- mation tool. In spite of this, it has been demonstrated that the stere- ochemical outcome of the reaction depends both on the structure of the reactants as well as on the reaction medium. This includes the base and solvent but also the use of certain additives, such as metal salts, crown ethers, or even the deliberate exclusion of salts (result- ing in the so-called “salt-free conditions”). Regarding the structure of the reagents, Z-selectivity can be achieved using either bis(2,2,2- trifluorethyl)phosphonoacetates developed by Still and Gennari [10] or bis(O-aryl)phosphonates proposed by Ando [11]. In addition to the standard conditions (i. e. NaH/THF), different well estab- lished protocols are now available in order to perform the reaction. In the field of natural product synthesis, the HWE reaction is ubiquitous. It is used not only for the elaboration of more or less complex synthetic precursors but quite often as a key step, to cou- ple preformed fragments of the target molecule or to introduce a newly formed olefin with a defined stereochemistry. Many features *Address correspondence to the author at the Departamento de Química Orgánica. Facultad de Farmacia y Bioquímica. Universidad de Buenos Aires. CONICET. Junín 956, (1113) Buenos Aires, Argentina; Tel: -----------------------; Fax: ---------------------; E-mail: [email protected] make the HWE reaction the best choice to rely on during the crucial steps of a natural product synthesis. First of all, it is a robust reac- tion, with a predictable outcome, since it has been put to test by many groups in quite different phosphonate and carbonyl combina- tions. Moreover, since the first papers were published, a number of modifications to the original protocol were developed that broaden the scope of the reaction in terms of substrate tolerance to the reac- tion conditions. Furthermore, in-depth mechanistic studies have revealed details of the influence of base, solvent and the presence (or absence) of salt additives on the stereochemical outcome of the reaction. Such important body of knowledge gives the synthetic chemist “freedom of choice”, allowing for the reaction to be se- lected as a key step to assemble advanced synthetic precursors. Since the HWE reaction has become a standard method for double bond formation, or more precisely for the introduction of ,unsaturated ketones and esters, listing each and every use of it in a natural product synthesis would be both an overwhelming and pointless task. This review is devoted to such examples where the olefination reaction is a key step, where its use was unavoidable due to the target molecule characteristics or because other methods failed to achieve the desired transformation. When available, a comparison with alternative methodologies is included. In some cases the reaction is chosen because it paves the way for further synthetic modifications, as the resulting enones and ,unsaturated esters are suitable substrates for further function- alization. Such examples will also be highlighted. This work is organized in sections considering the general structure of the targeted natural product. This subdivision, however, cannot be stringent, due to the structural complexity of the natural products targeted by modern synthetic chemists. The chosen exam- ples are limited to stabilized ylides as HWE reagents, and cover the literature from 2000 to 2012. A previous microreview by K. C. Nicolaou et al. (1997) [12] contains a useful introduction on some selected aspects of the Wittig and HWE reactions, together with an account on representative total syntheses of natural products from their groups employing such reactions.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis of Natural Products

Send Orders of Reprints at [email protected]
2206 Current Organic Chemistry, 2012, 16, 2206-2230
1385-2728/12 $58.00+.00 © 2012 Bentham Science Publishers
Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis of
Natural Products
Juan. Á. Bisceglia and Liliana R. Orelli*
Departamento de Química Orgánica. Facultad de Farmacia y Bioquímica. Universidad de Buenos Aires. CONICET. Junín 956,
(1113) Buenos Aires, Argentina
Abstract: The Horner-Wadsworth-Emmons reaction has evolved in the last years as one of the most powerful and reliable method for
stereocontrolled olefin synthesis. The reaction has become a widespread standard tool in natural products total syntheses, providing ac-cess to relatively simple as well as highly complex synthetic targets. In this work we present the most representative applications within
the field of natural product synthesis from 2000 to the present year. The examples comprise the synthesis of macrocycles, 5 to 7 mem-
bered rings, lipids and related compounds, alkaloids and cyclic ethers. They were chosen from a large amount of literature reports and il-
lustrate the use of the HWE reaction in the synthesis and elaboration of key intermediates and in the crucial assembly of highly function-
alized synthetic precursors.
Keywords: Horner-Wadsworth-Emmons (HWE) reaction, Carbonyl olefinations, Stabilized phosphonate carbanions, Natural products, Total synthesis, , -Unsaturated carbonyl compounds.
1. INTRODUCTION
In 1958, L. Horner published a novel Wittig reaction between phosphine-oxide stabilized carbanions and carbonyl compounds [1], which was further modified by W.S. Wadsworth and W. D. Em-mons by employing phosphonates [2]. Since then, the so called “Horner-Wadsworth-Emmons (HWE) olefination reaction” gained popularity and has become a widespread tool for de novo C=C bond formation [3-7]. Although the HWE reaction has not been reviewed in the last years, some general aspects are discussed in two more recent publications devoted to carbonyl olefinations [8,9]. Among them, some generally known data will be briefly mentioned only if they are of critical importance in order to settle down the context of the present review.
The classical HWE reaction is predominantly an E-alkene for-mation tool. In spite of this, it has been demonstrated that the stere-ochemical outcome of the reaction depends both on the structure of the reactants as well as on the reaction medium. This includes the base and solvent but also the use of certain additives, such as metal salts, crown ethers, or even the deliberate exclusion of salts (result-ing in the so-called “salt-free conditions”). Regarding the structure of the reagents, Z-selectivity can be achieved using either bis(2,2,2-trifluorethyl)phosphonoacetates developed by Still and Gennari [10] or bis(O-aryl)phosphonates proposed by Ando [11]. In addition to the standard conditions (i. e. NaH/THF), different well estab-lished protocols are now available in order to perform the reaction.
In the field of natural product synthesis, the HWE reaction is ubiquitous. It is used not only for the elaboration of more or less complex synthetic precursors but quite often as a key step, to cou-ple preformed fragments of the target molecule or to introduce a newly formed olefin with a defined stereochemistry. Many features
*Address correspondence to the author at the Departamento de Química Orgánica. Facultad de Farmacia y Bioquímica. Universidad de Buenos Aires. CONICET. Junín 956, (1113) Buenos Aires, Argentina; Tel: -----------------------; Fax: ---------------------; E-mail: [email protected]
make the HWE reaction the best choice to rely on during the crucial steps of a natural product synthesis. First of all, it is a robust reac-tion, with a predictable outcome, since it has been put to test by many groups in quite different phosphonate and carbonyl combina-tions. Moreover, since the first papers were published, a number of modifications to the original protocol were developed that broaden the scope of the reaction in terms of substrate tolerance to the reac-tion conditions. Furthermore, in-depth mechanistic studies have revealed details of the influence of base, solvent and the presence (or absence) of salt additives on the stereochemical outcome of the reaction. Such important body of knowledge gives the synthetic chemist “freedom of choice”, allowing for the reaction to be se-lected as a key step to assemble advanced synthetic precursors.
Since the HWE reaction has become a standard method for double bond formation, or more precisely for the introduction of , unsaturated ketones and esters, listing each and every use of it
in a natural product synthesis would be both an overwhelming and pointless task. This review is devoted to such examples where the olefination reaction is a key step, where its use was unavoidable due to the target molecule characteristics or because other methods failed to achieve the desired transformation. When available, a comparison with alternative methodologies is included. In some cases the reaction is chosen because it paves the way for further synthetic modifications, as the resulting enones and , unsaturated esters are suitable substrates for further function-
alization. Such examples will also be highlighted.
This work is organized in sections considering the general structure of the targeted natural product. This subdivision, however, cannot be stringent, due to the structural complexity of the natural products targeted by modern synthetic chemists. The chosen exam-ples are limited to stabilized ylides as HWE reagents, and cover the literature from 2000 to 2012. A previous microreview by K. C. Nicolaou et al. (1997) [12] contains a useful introduction on some selected aspects of the Wittig and HWE reactions, together with an account on representative total syntheses of natural products from their groups employing such reactions.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2207
2. SYNTHESIS OF MACROCYCLES
2.1. Ring Closing Reactions
HWE olefination has proven very useful for large size ring clos-ing reactions, as in the synthesis of macrolactones and related mac-rolides, together with acylations of the Yamaguchi type, organopal-ladium couplings and RCMs. It has also been widely used in the assembly of elaborated fragments in early steps of the synthetic plan. In this context, the reaction provides not only the possibility of highly convergent synthesis, but also leaves behind valuable scaffolds for further transformations.
Since phosphonoacetic acid and its derivatives are commer-cially available, O-phosphonoacetylation of suitable advanced pre-cursors having also a latent aldehyde moiety is often used for the construction of the C2-C3 bond in macrolactones by an in-tramolecular HWE olefination. An example of this approach can be seen in White’s total synthesis of Phorboxazole A2, in which the advanced precursor of C3-C46 (1a) was oxidatively deprotected at C24 and immediately phosphonoacetylated by means of DCC acti-vation of the acid [13] to give 1b. After selective fluoride unmask-ing of C3 hydroxyl followed by DMP generation of the aldehyde, an intramolecular HWE olefination yielded the desired intermediate 1c. The high Z- selectivity observed might be a consequence of both the use of a potassium base and geometric constraints imposed by the molecule’s structure. Removal of silyl and ketal protecting groups completed the synthesis of the target molecule (Scheme 1) in a way analogous to that previously used in William’s synthesis of Phorboxazole A [14].
A complementary strategy to construct unsaturated macrolac-tones employs phosphonoketones bearing a carboxylic acid group which reacts with an alcohol containing an aldehyde precur-sor moiety. This is exemplified in the total synthesis of (-)-5,6-Dihydrocineromycin B proposed by Li [15]. Firstly, C9-C13 and C1-C8 fragments (2a,b, respectively) were assembled by esterifica-tion. Oxidative cleavage of the terminal alkene then generated the aldehyde moiety of 2c, which by intramolecular HWE olefination under Masamune-Roush conditions [16], originated the C8-C9 double bond (Scheme 2). The resulting enone 2d was then sub-jected to stereoelective reduction, and subsequent deprotection, delivering the desired allylic alcohol 2e with a diastereomeric ratio of 2.5:1.
The same strategy was followed in Jiang’s synthesis of Palmer-olide A, a cytotoxin isolated from the antarctic marine tunicate Synoicum adareanum (Scheme 3) [17]. Esterification of 3b with 3a was performed under Yamaguchi’s conditions and, after acidic deprotection of both TES ethers, the primary alcohol 3c was con-verted to the requisite aldehyde. In this case, HWE olefination was brought about by use of potassium carbonate/18-c-6 conditions to give 3d.
Palmerolide A was also targeted by Nicolaou’s group. In their hands, the Masamune-Roush protocol applied to the C19 phos-phonoacetylated precursor 4a gave the best results after screening many base/solvent systems [18]. In this work, the authors also compare various macrocyclization strategies using appropriate syn-thetic precursors (Scheme 4). This valuable study highlights the effectiveness of the HWE reaction, which is comparable, in terms
O
N
O
Br
H3CO
OTBDPSOCH3
OOPMB
N
OO O
TBDPSO
OTBDPS
3
24
46
1) DDQ, DCM
2) (H3CO)2P(O)CH2CO2H,
DCC, DCM, 91%
O
N
O
Br
H3CO
OTBDPSOCH3
OO
N
OO O
TBDPSO
OTBDPS
O PO(OMe)2
O
N
O
BrH3CO
R1O
OCH3
R2O
OO
N
O
O
O
OR1
1) NH4F, MeOH
2) DMP, DCM
3) K2CO3, 18-c-6,
toluene, 81% (Z/E 4:1)
R1: TBDPS
R2: Me
1) TBAF, THF
2) HCl(6%), THFPhorboxazole A2
R1 = R2 = H
O
1a
1b
1c
Scheme 1.

2208 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
of chemical yield to RCM and only surpassed by the Yamaguchi approach. Mitsunobu reaction delivered lower quantities of the desired macrocycle, and Stille coupling yielded 45% of the desired product that the authors claim to be accompanied by an unidenti-fied, and non separable, geometric isomer (22%).
Macrolactones can also be useful as synthetic intermediates. This is exemplified in Suzuki’s first total synthesis of (+)-Phomopsidin, a microtubule assembly inhibitor [19]. The tricyclic precursor 5d of this interesting secondary metabolite is obtained from a polyunsaturated macrocycle (5c) by means of a transannular
O O
O
OTES
HO O
O
OTES
(EtO)2OP
OH
+ 1) DCC, DMAP, DCM
2) OsO4, NMO
3) NaIO4
O O
O
OTES
(EtO)2OP
OHC
DIPEA, LiCl, MeCN, 65%
O O
OH
OH1) S-BINAL, THF
2) TBAF
(-)-5,6-Dihydrocineromycin B
2a 2b 2c
2d2e
Scheme 2.
1) PhI(OAc)2, TEMPO, CH2Cl2/H2O
2) K2CO3, 18-c-6, PhMe, 60 °C
(70%, two steps)
O HOO
MeO2C
O
OTIPS
O HOO
MeO2C
O
OTIPS
PO(OMe)2
HO
OH TESO
MeO2C
OTIPSTESO
CO2H
O
PO(OMe)2
+1) 2,4,6-Cl3BzCl, NEt3, DMAP, toluene
2) PPTS, MeOH
O OOHN
OH
OH
O
H2NOCPalmerolide A
3a
3b
3c
3d
Scheme 3.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2209
Diels Alder reaction (TADA) (Scheme 5). After bromothriphenyl-phosphonium bromide mediated phosphonoacetylation of 5a fol-lowed by mild acid hydrolysis of the ethoxyethyl ether protecting group, the precursor phosphonoaldehyde was prepared from the resulting alcohol 5b upon DMP oxidation and submitted to HWE cyclization without purification, yielding the intermediate mac-rolactone 5c.
Closely related to macrolactones but less often targeted are macrolactams and cyclic polypeptides, where aminophosphonates and phosphonoacetamides are found to be very useful synthons. As it can be seen in the total synthesis of macrodilactams Syringolin A and B (Scheme 6) [20], N-( formyl)substituted diethyl phos-phonoacetamides obtained by oxidation of 6a are cyclized in very mild conditions by means of the Helquist protocol, which uses zinc triflate and an organic base to achieve olefination [21]. The reaction proved to be selective towards the E-enamides 6b.
2.2. Elaboration of Advanced Precursors
The HWE reaction is also used in the setting up of functional-ized open chain precursors towards macrolide elaboration. An in-teresting example of this methodology is found in Müller’s synthe-sis of a Pladienolide B analogue (Scheme 7) [22]. Aldehyde 7a and
phosphonomethylketone 7b are coupled with the aid of BaO to the early synthetic intermediate 7c. After macrolactonization was achieved through acylation, the allylic alcohol 7d was produced diasteroselectively by NaBH4/CeCl3 treatment.
Uenishi reported in 2009 a closely related approach to the cyto-toxic macrolide (-)-Zampanolide, isolated from the marine sponge Fasciospongia rimosa. The open chain precursor 8c was obtained via a cesium carbonate promoted HWE between the highly func-tionalized aldehyde 8a and phosphonoketone 8b, using iso-propanol as the solvent. Intramolecular esterification followed by functional group interconversion led to (-)-Dactylolide, which upon treatment with unsaturated amide 8d afforded the target compound (Scheme 8) [23].
It would seem, at this point, that the order of olefination and acylation reactions leading to a macrocycle could be selected at will. In certain cases, however, the most convenient macrocycliza-tion strategy is determined by the chemical properties of the sub-strates. This is illustrated in the construction of the epoxide-containing macrolactone nucleus of Oximidine I [24]. Such com-pound accommodates seven consecutive sp2 carbon atoms, includ-ing an o-phenylene and a Z-olefin adjacent to a fused oxirane moi-ety, in a 12-membered ring. The first synthetic approach, consisting
O
O
O
R
OR3
OR2
R1
O
O
O
R
OH
OH
CONH2
RCM. Grubbs II
73%
PO(OEt)2O
O
O
R
OR3
OR2
R1
CHO
HWE. Masamune-Roush
73%
O
OI
O
R
OR3
OR2
R1
Bu3Sn
Stille. Pd(dba)2, AsPh3
45%
HO2C
HO
O
R
OR3
OR2
R1
Yamaguchi
81%
HO2C
HO
O
R
OR3
OR2
R1
Mitsunobu (DEAD, Ph3P, toluene)
31%
4a Palmerolide A
Scheme 4.

2210 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
O
OTIPS
O
O
OTIPS
O
(EtO)2OP
O
OTIPSH
H
O
BHT, toluene
reflux
OH
OTIPS
OOEt
OH
O
(EtO)2OP
+
1) DMP, DCM
2) K2CO3, 18-c-6,
toluene, 0.005 M
78%, 2 steps
1) CBr4, PPh3, Py, DCM
2) PPTS, EtOH
HO
OHH
H
CO2H
(+)-phomopsidin
5a
5b 5c
5d
Scheme 5.
(EtO)2OP
HN
NH
OH
HNO
O
RHN
NH
O
HN
R
O1) DMP
2) Zn(OTf)2, TMEDA, Et3N, 5mM
65%, R=Boc
81%, R=Cbz
2 steps
Syringolin A and B
common precursors6a 6b
Scheme 6.
MeO2COTES
H
(MeO)2OP
O
OTBS
OMEM
I
O
+
BaO, Et2O, H2O
82%
CO2Me
OTES
O
OTBS
OMEM
I
O
OH
OTBS
OMEM
I
O
7a 7b
7c
7d
Pladienolide B analogue
Scheme 7.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2211
O
O
PMBOOH
H H
19
(-)-Dactylolide
(EtO)2OP
OOH
O2
3
+
O
OPMBO
OH
H H
HO2C
Cs2CO3, i-PrOH, 89%
(-)-Zampanolide
O
OOHC O
H H
O
O
OO
H H
O
NH
OHONH2
O
H+
8a 8b 8c
8d
Scheme 8.
CO2R
PO(OEt)2CO2R
O
OTBDPS
OH OH
O
O
OAc
O
HO
CHO
O
OTBDPS
KOtBu, THF,
36% (R=Me),
77% (R=(CH2)2SiMe3);
Deprotection
2,4,6-Cl3BzCl,
NEt3, THF, DMAP, toluene, 70%;
R=H
O
O
O1) Cs2CO3, NEt3, MeOH, 0°C
2) Swern
3) KOtBu, 18-crown-6, toluene, 14–25%.
OAc
O
O
PO(OEt)2
O
9a
9b
9c 9d
9e
9f
Oximidine I
Scheme 9.
in a HWE olefination of aldehyde 9b with phosphonate 9a (R H) followed by deprotection of ester 9c and Yamaguchi lactonization failed to give any product but benzofuranone 9d (Scheme 9). The resulting acid 9c (R=H) reacted in a SN2´ fashion regardless of the
deprotection method used, i.e. basic (R=Me) or fluorous (R=TMS-ethyl), leading to epoxide ring opening. On the other hand, acyla-tion of the epoxide-containing alcohol 9e with phosphonoacid 9a (R=H) followed by functional group manipulation of diester 9f and

2212 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
HWE olefination, delivered the desired macrolactone, albeit in low yield.
2.3. Synthesis of Building Blocks
One pot sequential aminoxylation-HWE-deamination is a well established protocol for the synthesis of , unsaturated
hydroxyesters [25]. This procedure was employed in the stereo-selective total synthesis of macrodiolide Clonostachydiol, where the C1-C5 building block 10b was prepared from aldehyde 10a (Scheme 10) [26].
Unsaturated and polyunsaturated phosphonoacids (vinylogous to phosphonoacetic) can be used to introduce conjugated acid or acid derivative moieties in the synthetic target. For example, in the synthesis of the C1-C27 fragment of Aplyronine A, two different HWE reactions were used in a highly convergent way to establish
the C1-C20 fragment of the 24-membered lactone (Scheme 11) [27].
Firstly, C5-C14 phosphonomethylketone 11c was constructed by selective alkylation of the dianion of diethyl 3-oxo-2-butyl phosphonate 11b with iodide 11a, followed by HWE coupling with a suitably functionalized aldehyde (11d). The TBSO group was then removed and, after Dess-Martin oxidation of the resulting alcohol, treatment of aldehyde 11e with triethyl fosfonocrotonate led to the fully elaborated C1-C20 , , , unsaturated ester 11f. This approach was further extended to sorbic acid derived phos-phonates. In the synthesis of the macrocyclic core of Ansatrienins, ethyl-6-(diethoxyphosphoryl)sorbate 12a was used to create the E,E,E triene system present in this antibiotic (Scheme 12) [28].
Another example of this principle can be found in Aman’s syn-thesis of the monomeric counterpart of Marinomycin A, a polyene
CHO
OTBS OTBS
CO2Et
1) PhNO, L-proline, DMSO,
2) (EtO)2POCH2CO2Et, DBU, LiCl,
3) MeOH, CuSO4, H2O
60% (one pot)
OH10a 10b
Scheme 10.
I OTBS
OMTMOTBS
(EtO)2OP
O
+NaH, BuLi
THF, 86%
OTBS
OMTMOTBS
PO(OEt)2
O
1) Ba(OH)2, THF:H2O(40:1), 67%
2) CSA, MeOH, 91%
3) DMP, DCM, quant
OPMB
O OMe
OPMB
OMe
O
OMTMOTBS
OOPMB
OMe
OMTMOTBS
O
CO2Et
(EtO)2POCH2CHCHCO2Et
LHMDS, THF, 77%
11a 11b 11c
11d
11e11f
Scheme 11.
O
OCHO
OTBDPS
(EtO)2OP CO2Et+ LHMDSO
O
OTBDPS
CO2Et(E:Z 6:1)
12a
Scheme 12.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2213
macrodiolide [29,30]. The phosphonate group was instaurated to the ester moiety in 13a and then coupled with stannylated aldehyde 13b, yielding a highly unsaturated stannane (13c) proposed as the key synthon for the C1-C13 fragment of Marinomycin A (Scheme 13). Other stannylated synthetic precursors have been prepared by means of the HWE reaction as in the synthesis of key fragments to Liodelide A by Chellat [31].
In some cases, the enone product of a HWE olefination is used for the elaboration of densely functionalized synthetic precursors, as it delivers three consecutive reactive centers. Paterson’s synthe-
sis of the differentially protected C20-C38 fragment of Brasili-nolides is an example of this concept [32]. After the C20-C38 chain was assembled by means of a Ba(OH)2 mediated HWE between 14a and 14b, the resulting enone 14c was submitted to asymmetric reduction followed by Sharpless epoxidation, leading to the intrin-cate anti hydroxy-oxirane arrangement present in the natural prod-uct (Scheme 14).
The same principle was applied by Nicolaou as a part of the to-tal synthesis of Apoptolidin [33]. In this case, after Ba(OH)2 pro-moted HWE reaction of 15a and 15b, the resulting enone 15c har-
O O
OH
O
1
O O
PO(OEt)2
O
1
O O
O
1
SnBu3
O SnBu3
13NaH, THF, 53%
1) PBr3, Et2O
2) P(OEt)3, toluene, reflux
95%, 2 steps
13a
13b
13c
Scheme 13.
PO(OMe)2
O
Ba(OH)2, THF, H2O (40:1)
91%ODMB
OODMB
1) CH3PO(OMe)2, BuLi
2) DMP, DCM
73% 2 steps
+
O
OTES O O ODEIPS
PMP
OODMB OTES O O ODEIPS
PMP
OHODMB OTES O O ODEIPS
PMP
O
1) (R)-Me-CBS cat., BH3.SMe2, THF
2) (+)-DIPT, TBHP, Ti(OiPr)4, DCM
21 37
14a 14b 14c
Scheme 14.
(MeO)2OP OMe
O
OMe
OTBS
O O O OPMB
TBSTBS
+
Ba(OH)2, THF, H2O (40:1), 80%
OMe
OMe
OTBS
O O O OPMBTBSTBS
OOMe
OTBS
OMe
OH
HO
OH
OPMB
OMe H
1) AD-MIX-
2) TBAF, SiO2
3) MeOH, pTsOH
15a
15b
15c
15d
Scheme 15.

2214 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
nesses the construction of a pyrane ring by ketalization with a latent -alcohol, while the double bond is used for the introduction of the syn vicinal diol present in 15d (Scheme 15).
Some authors exploit the robustness of the HWE reaction to prepare building blocks of related natural products. In Roy’s syn-theses of the C18-C34 fragment of Amphidinolide C (16d) and the C18-C29 fragment of Amphidinolide F (16e), the same functional-ized phosphonate reagent 16a is coupled with two different alde-hydes (16b and 16c, respectively), thus paving the way for closely related synthetic targets [34]. The generation of this interesting HWE reagent and its use in the preparation of allylic alcohols pre-cursors is depicted in Scheme 16. It is to be noted that the genera-tion of the keto group to the existing phosphonate could not be achieved by means of a Wacker reaction. The substrate had to be carefully manipulated, first to an inconsequent diastereomeric mix-ture of borinated products and then to the alcohol and ketone. Ce-sium carbonate-promoted HWE followed by stereoselective reduc-tion of the carbonyl group gave access to the desired allylic alco-hols 16d,e.
Although HWE chemistry is compatible with base resistant pro-tecting groups such as ketals and silyl ethers, certain structural fea-tures in the substrates can lead to unexpected reactivity. An exam-ple of this can be found in Hillier’s study on the total synthesis of Disorazole C1, where HWE reaction conditions sufficed to bring about concomitant 1,5-O-silyl group transfer, aldehyde deprotection and the proposed olefination. The author’s rationalization of this behavior is depicted in Scheme 17 [35].
HWE with vinylogous phosphonoacetates is also a versatile synthetic tool when highly unsaturated materials are to be prepared. In Srinivasarao’s synthesis of the Apoptolidin D core [36]. Phos-phonate 18a was prepared by Arbuzov reaction of tert-butyl-6-bromo-2,4-dimethylsorbate and then coupled with a functionalized butanal derivative (18b) to afford the E,E,E-triene ester 18c which, after functional group manipulation, was converted into aldehyde 18d. Subsequent reaction of 18d with a vinylsulfone derived phos-phonate (18e) [37] delivered the desired pentaunsaturated ester 18f (Scheme 18).
OCO2Me
(MeO)2OP
OCO2Me
PO(OMe)2PMBO
OH
PMBO
OH
PO(OMe)2
PMBO O
PO(OMe)2PMBOO
O
PMBOO
O
PMBO
O
O
TBSO
O
OTBS
O
PMBO
O
OH
TBSO
PMBO
OOHH H
Grubbs II, CuI, DCM, 78%
Pd(Ph3P)4, DIPEA,
THF, 88%
1) B2pin2, CuI, t-BuONa, MeOH, DPEPhos, THF
2) NaBO3, H2O, THF
3) TPAP, NMO, DCM
58%, 3 steps
24
CsCO3, i-PrOH, 93% CsCO3, i-PrOH, 93%
L-Selectride, THF, 88%L-Selectride, THF, 94%
+
C18-C34 fragment of Amphidinolide C C18-C29 fragment of Amphidinolide F
16a16b
16c
16d16e
Scheme 16.
OEt
OTBSOH
OEt
OO
TBS
OEt
OOTBS
OOTBS
EtO-
OTBS
CO2EtNaH
(EtO)2P(O)CH2CO2Et
NaH
Scheme 17.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2215
Incednine is a macrolactam antibiotic that exhibits significant inhibitory activity against some anti-apoptotic oncoproteins [38]. It contains a methoxy- , unsaturated amide motif as part of a conjugated pentaene skeleton. Its precursor, i.e. carboxylic acid 19b, was prepared in preliminary studies towards the total synthesis of the natural product by HWE reaction of trimethyl 2-methoxy phosphonoacetate and aldehyde 19a (Scheme 19).
The HWE reaction also proved useful in the total synthesis of Stephanotic acid, a cyclic peptide from the moroidin family (Scheme 20) [39]. Compound 20a, in which formyl group reactivity is enhanced by Boc protection of the indole NH, was reacted with the phosphorylated valyl-glycine synthon 20b in salt free conditions using Schmidt’s Z-selective DBU protocol [40]. Although Z-
enamide 20c was obtained in good chemical yield, the basic me-dium caused epimerization of the isopropyl and benzylcarbamate groups, delivering a 1:1 diastereomeric mixture of the desired prod-uct, which was separated chromatographycally before asymmetric reduction of the double bond.
2.4. Other Examples
In Sanchez’s synthesis of (+)-Dactylolide, a cytotoxic metabo-lite from the sponge Dactylospongia spp., HWE reaction is used for chain elongation as well as in the ring closing step [41]. In this work, enone 21c is generated by means of a HWE reaction between aldehyde 21a and phosphonoketone 21b using Paterson’s Ba(OH)2 procedure [42]. After functional group manipulation to produce alcohol 21d, phosphonoacetylation of the 19 OH group and aldehyde
generation, NaHMDS promoted intramolecular HWE olefination of 21e yields the C2-C5 E,E-diene system in 21f (Scheme 21). It is note-
worthy that the authors use polymer supported DCC to achieve phosphonoacetic acid activation in order to simplify the purification of this highly polar intermediate.
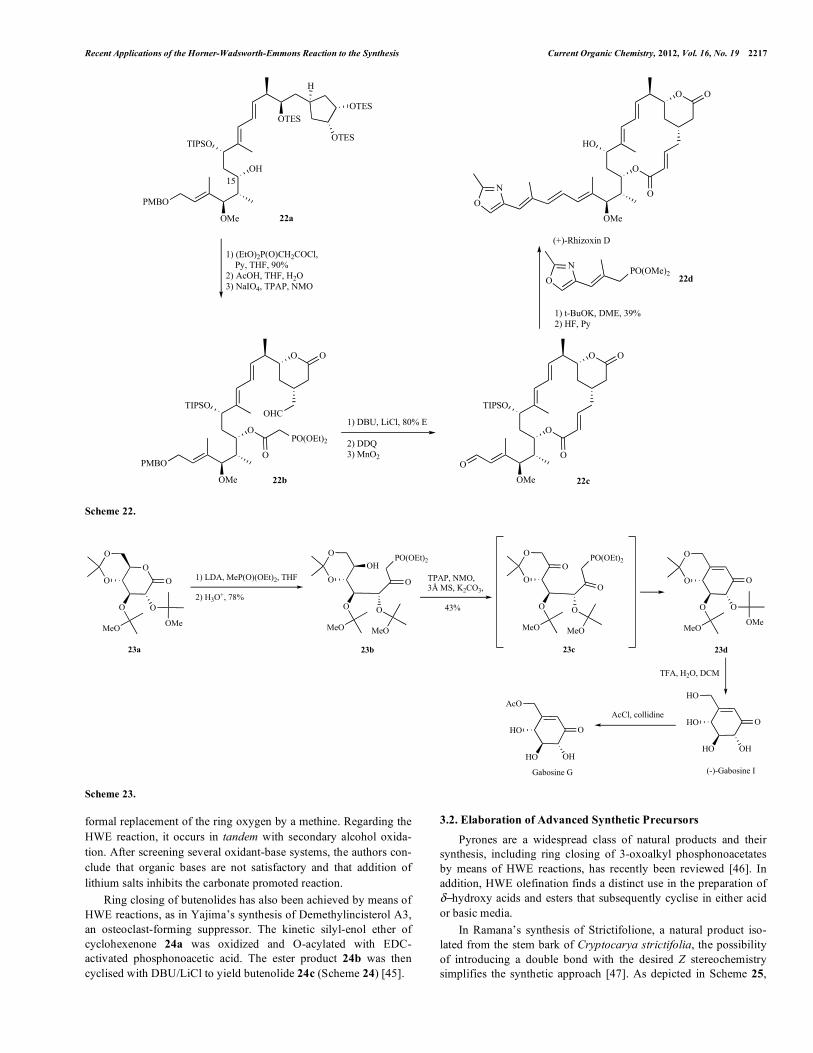
HWE olefination has also been used for side chain annexation after the macrocyclic core had been assembled, as in the total syn-thesis of the antitumor macrolide (+)-Rhizoxin D reported by Jiang [43]. In this case, after phosphonoacetylation of 22a using the cor-responding acyl chloride, the aldehyde moiety in 22b is generated by means of vicinal diol cleavage. Intramolecular E-selective HWE olefination at high dilution (0,0001M) is used as the key step to form the 16-membered , unsaturated macrolactone 22c. A sec-ond intermolecular HWE reaction then establishes the side chain E,E,E-triene by means of phosphonomethyl vinyl oxazole 22d. Unwanted ring opening of the tetrahydropyranone moiety would account for the modest yield of this step (Scheme 22).
3. SYNTHESIS OF FIVE TO SEVEN-MEMBERED RINGS
3.1. Ring Closing Reactions
An interesting application of the HWE reaction is found in the synthesis of carbasugars Gabosine I and Gabosine G [44].
Gluconolactone, protected as the mixed acetal 23a, was treated with diethyl methylphosphonate and LDA to afford hydroxy phos-phonate 23b (Scheme 23). Oxidation of the latter, with TPAP-NMO in the presence of K2CO3, generated ketone 23c that collapsed to the Z-alkene 23d within the reaction medium. Deprotection of the enone led to Gabosine I, and selective acetylation of the resulting primary alcohol provided Gabosine G. This sequence has some remarkable features: from the synthetic standpoint, it represents the
O
TBDPSO
PMBO
Br
O-tBu
O
PO(OEt)2
O-tBu
O
P(OEt)3
toluene,
reflux,
91%
BuLi, THF, 90%
O-tBu
O
TBDPSO
PMBO
1) NH4F
2) Swern
O-tBu
O
O
PMBOSO2Ph
PO(OEt)2
BuLi, THF, 90%
O-tBu
O
PMBO
SO2Ph
18a
18b
18c
18d
18e
18f
Scheme 18.
I
CHOTESO
OTES
1) (MeO)2P(O)CH(OMe)CO2Me,
KHMDS, 18-c-6, THF, 82%
2) 1.0 M KOH aq,dioxane
I
TESO
OTES
CO2H
OMe19a 19b
Scheme 19.

2216 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
N
Boc
CHO
CbzHN
OHN
CO2-t-Bu BocHN
HN CO2Me
PO(OMe)2
N
Boc
CbzHN
O
NHt-Bu-O2C
BocHNNH
CO2Me
DBU, DCM, 73%
BocHNNH2
N2 CO2Me
PO(OMe)2
O
O
+
cat. Rh2(OCOC7H15)4
CHCl3, reflux, 64%
O
NH
HN
O HN
NHN
H
O
HN
O
O
CO2Me
Stephanotic acid methyl ester
O
20a
20b
20c
Scheme 20.
O
O
PMBOPMBO
O
H H
O
PO(OEt)2
O
OTBDPS
PMBOPMBO
OH
H H
1) PS.DCC, DMAP,
DMAP.HCl, 100%
2) HF,Py, 74%
3) TPAP, NMO, 86%
HO
O
PO(OEt)2
O
OPMBPMBO
O
H H
O
19
19
19
2
3
NaHMDS
60%
(+)-Dactylolide
O
OTBDPS
OPMBO
OTBS
H H
(MeO)2OP
OTBDPS
O
O
O
PMBOOTBS
H H+
Ba(OH)2, THF, H2O (40:1)
79%
FGI
21a
21b
21c
21d21e
21f
Scheme 21.
Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2217
O
OHC
O O
O
OMe
TIPSO
PMBO
PO(OEt)2
O
O O
O
OMe
TIPSO
O
O
NPO(OMe)2
O
N
O
O O
O
OMe
HO
1) DBU, LiCl, 80% E
2) DDQ
3) MnO2
1) t-BuOK, DME, 39%
2) HF, Py
(+)-Rhizoxin D
OH
OMe
TIPSO
PMBO
H
OTES
OTES
OTES
15
1) (EtO)2P(O)CH2COCl,
Py, THF, 90%
2) AcOH, THF, H2O
3) NaIO4, TPAP, NMO
22a
22b 22c
22d
Scheme 22.
O
O
O O
O
O
MeOOMe
PO(OEt)2
1) LDA, MeP(O)(OEt)2, THF
2) H3O+, 78%
OH
O
O O
O
O
MeO MeO
PO(OEt)2O
O
O O
O
O
MeO MeO
O
O O
O
O
MeOOMe
TPAP, NMO,3Å MS, K2CO3,
43%
HO
HO OH
O
HO
HO
HO OH
O
AcO
TFA, H2O, DCM
(-)-Gabosine I
AcCl, collidine
Gabosine G
23a 23b 23c 23d
Scheme 23.
formal replacement of the ring oxygen by a methine. Regarding the HWE reaction, it occurs in tandem with secondary alcohol oxida-tion. After screening several oxidant-base systems, the authors con-clude that organic bases are not satisfactory and that addition of lithium salts inhibits the carbonate promoted reaction.
Ring closing of butenolides has also been achieved by means of HWE reactions, as in Yajima’s synthesis of Demethylincisterol A3, an osteoclast-forming suppressor. The kinetic silyl-enol ether of cyclohexenone 24a was oxidized and O-acylated with EDC-activated phosphonoacetic acid. The ester product 24b was then cyclised with DBU/LiCl to yield butenolide 24c (Scheme 24) [45].
3.2. Elaboration of Advanced Synthetic Precursors
Pyrones are a widespread class of natural products and their synthesis, including ring closing of 3-oxoalkyl phosphonoacetates by means of HWE reactions, has recently been reviewed [46]. In addition, HWE olefination finds a distinct use in the preparation of
hydroxy acids and esters that subsequently cyclise in either acid or basic media.
In Ramana’s synthesis of Strictifolione, a natural product iso-lated from the stem bark of Cryptocarya strictifolia, the possibility of introducing a double bond with the desired Z stereochemistry simplifies the synthetic approach [47]. As depicted in Scheme 25,

2218 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
after establishment of the Z-unsaturated ethyl ester by submitting alcohol 25a oxidation product to Ando´s conditions, mild exposure of 25b to PPTS suffices to bring about not only acetonide, silyl ether and ester deprotection, but also lactonization.
A similar approach is used in Srihari’s synthesis of closely re-lated (+)-Dodoneine [48], a natural product isolated from the methanolic extract of the hemiplant parasite Tapinanthus dodonei-
folius. In this case, bis(2,2,2-trifluoromethyl)(methoxycarbonyl-methyl)phosphonate was employed to establish the , unsaturated ester with Z stereochemistry using the Still and Gennari protocol.
Other dihydropyranones have been synthesized using slight variations of this strategy. In 2004, Singh reported the one step preparation of the methoxy lactone moiety present in (+)-Dihydrokawain-5-ol (Scheme 26). The intermediate Z-unsaturated ester 26b was obtained by HWE olefination of aldehyde 26a under Ando’s conditions [49]. Subsequent treatment of 26b with K2CO3/methanol was used to achieve ester cleavage, lactonization
and methoxide conjugate addition in a single step, yielding the pre-cursor 26c.
Z-selective HWE reaction is not necessary when tetrahydro-pyranones are the synthetic target, since the resulting double bond can be hydrogenated before the cyclization step. The stereoselective total synthesis of (+)-Garvensintriol [50], isolated from the stem bark of Goniothulamus arvensis, provides an illustrative example. The sequence starts with a one pot aminoxylation-HWE-deamination sequence of aldehyde 27a, followed by protection of the resulting allylic alcohol (27b). Subsequent hydrogenation and deprotection-cyclization led to the desired hydroxy- lactone (Scheme 27).
This strategy was taken even further in the synthesis of Passi-floricin A reported by Kumar [51], where three aminoxylation-HWE-deamination cycles were employed to elaborate the polyhy-droxylated side chain of this dihydropyranone (Scheme 28). Start-ing from palmitaldehyde, a first aminoxylation-HWE-reduction-
H
HO
H
HO
O
O(EtO)2OPH
H
O
O
1) LDA, TMSCl, THF,
2) OsO4, NMO, THF, H2O, t-BuOH,
3) (EtO)2POCH2COOH,
EDC, DMAP, DCM
LiCl, DBU, THF,82%
H
H
O
O
OH
demethylincisterol A3
24a 24b 24c
Scheme 24.
Ph OH
O OTBSO
Ph
O OTBSO
CO2EtPh
OH OOH
O
1) Swern
2) ethyl (di-o-tolyl
phosphono)acetate,
NaH, THF
(2 steps, 81%)
PPTS
EtOH, 55˚
67% Strictifolione25a 25b
Scheme 25.
Ph
OHCO
CO2Et
ethyl (di-o-tolyl
phosphono)acetate,
NaH, THF,
77%OPMB
Ph
OOHCO
OPMB
K2CO3, MeOH, 0˚
77% Ph
O
OPMB
O
OMe Ph
O
OH
O
OMe
(+)-Dihydrokawain-5-ol26a 26b 26c
Scheme 26.
O O
Ph
MOMO
HO
CO2EtO O
Ph
MOMO
O
1) MOMCl, DIPEA;
2) 10% Pd/C, H2,
3) PTSA, MeOH, reflux
1) PhNO, L-proline, DMSO,
2) (EtO)2P(O)CH2CO2Et, DBU, LiCl,
3) MeOH, NH4Cl, Cu(OAc)2,
45% (one-pot)
OOH
Ph
OHOH
O
(+)-Garvensintriol
27a 27b
Scheme 27.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2219
protection sequence leads to the silyloxyaldehyde 28a, which is then submitted to a four steps-one carbon homologation to obtain 28b. A second aminoxylation-HWE-reduction-protection cycle delivers the bis(silyloxy) ester 28c, which upon reduction and a third aminoxylation-HWE-reduction-protection step yields the de-sired acyclic precursor 28d.
3.3. Functionalization
HWE olefination is also useful for the introduction of meth-ylidene and exocyclic alkylidene groups. The reaction finds twofold use in the straightforward and elegant synthesis of the antitumour agent (±)-bis-Homosarkomycin ethyl ester depicted in Scheme 29 [52]. Alkylation of triethyl phosphonoacetate with bromide 29a
leads to tetraethyl-2-phosphonopimelate, which is subjected to HWE olefination in heterogeneous conditions to deliver the desired
methylenepimelate 29b. A phospha-Michael reaction of the lat-ter followed by base promoted cyclization produced the desired phosphorylated cycloheptanone substrate for the second HWE reac-tion (29c), allowing for the introduction of the exocyclic methylene group found in this natural product (Scheme 29). It is noteworthy that the last reaction occurs in mild basic aqueous medium without hydrolysis of the ester.
The introduction of an exocyclic double bond with a defined stereochemistry is sometimes difficult when the carbonyl partner of the HWE reaction is a cyclic ketone devoid of substituents. In such cases, chiral phosphonates are to be used in order to obtain
O
12OH
OHOH
O
Passifloricin A
12OTBS
OTBSOTBS
CO2Et12
OTBS
OTBS
CO2Et
12OTBS
CHO
12
1) PhNO, D-proline, DCM,
(EtO)2POCH2CO2Et, DBU, LiCl
2) H2, Pd/C, AcOEt
3) TBSCl, DMF, imidazole
75%, 3 steps
ee>93%
4 steps
12OTBS
CHO
1) PhNO, D-proline, DCM,
(EtO)2POCH2CO2Et, DBU, LiCl
2) H2, Pd/C, AcOEt
3) TBSCl, DMF, imidazole
65%, 3 steps
de>92%
1) DIBAL, DCM, -78˚ C
2) PhNO, D-proline, DCM,
(EtO)2POCH2CO2Et, DBU, LiCl
3) H2, Pd/C, AcOEt
4) TBSCl, DMF, imidazole
65%, 4 steps
de>92%
CO2Et
28a
28b28c
28d
Scheme 28.
(EtO)2OP CO2Et
BrCO2Et
CO2Et
CO2Et
CO2Et
PO(OEt)2
O
CO2Et
O
+
1) NaH, THF, 70%
2) (HCHO)n, K2CO3,
THF, 96%
1) (EtO)2P(O)H, K2CO3,
HSTBA, THF, 86%
2) NaH, DME, 78%
HCHO (30%aq),
K2CO3, THF/H2O,
56%
(±)-bis-homosarkomycin
ethyl ester29a 29b 29c
Scheme 29.
O
H
Me
H
TBDPSOO
PO(OR)2O
OR'
base, 76% O
H
Me
H
TBDPSO
O
O
R'
R=R'=Et, base=LHMDS, de=20%
R=Me, R'=nor-8-phenylmenthyl, base=BuLi, de=63%
(-)-Platensimycin
Scheme 30.

2220 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
high diasteroselectivities. One example of this is the synthesis of a precursor of the novel antibacterial agent (-)-Platensimycin, in which the use of a nor-8-phenylmenthyl ester clearly improves E-diasteroselectivity (Scheme 30) [53].
The same methodology allowed for the diasteroselective prepa-ration of an advanced intermediate in the synthesis of 3-Oxacarbacyclin (Scheme 31) [54].
In a striking example of HWE’s reaction versatility, the exocyc-lic allylic alcohol of Zoapatanol was introduced by simple HWE olefination of the requisite 3-oxepanone 32b followed by LAH reduction of the ester function to give 32c. Such precursor, in turn, was prepared by intramolecular HWE olefination of 32a (Scheme 32), after the original synthetic strategy (which relayed on a RCM-hydroboration-oxidation sequence) had failed [55].
4. LIPIDS
HWE reaction is also a powerful and widespread two carbon homologation tool. An example of this is the synthesis of (2E,6Z)-farnesol and (E,E,E)-geranylgeraniol reported by Yu [56], in which polyprenylated methyl ketones undergo HWE olefination with triethylphosphonoacetate in the presence of NaH/15-c-5 with high E-diasteroselectivity.
The HWE reaction has also been used to prepare putative lyco-pene metabolites (Scheme 33) by desymmetrization of unsaturated dialdehydes [57].
Two-carbon homologation of aldehydes is a straightforward method for the stepwise construction of extended polyene systems. However, the classic reaction with phosphonoacetate derivatives necessarily involves the introduction of a reduction step of the ester product to the aldehyde group needed for further elongation. The best yields for this transformation are obtained by sequential LAH reduction to the corresponding alcohol followed by selective allylic oxidation. Interestingly, a reagent that maintains the oxidation state of the aldehyde throughout the two carbon homologation sequence has been developed [58]. Replacement of the ester group in phos-phonoacetates directly by formyl is obviously not possible, due to its inherent incompatibility, but the formylhydrazone group is stable enough to be used in standard HWE reaction protocols both as ylide stabilizing as well as protective group. The methodology derived from the use of this reagent was compared with the classical reduc-tion-oxidation sequence in Petrosky’s preparation of a conjugated tetraene insect pheromone (Scheme 34). This resulted in a shorter (by 2 steps after 2 HWE/deprotection cycles) and higher yielding (37% vs. 20%) synthesis than the one previously reported [59]. Removal of the hydrazone group to deliver the sensitive unsaturated
O
OTBS
OTBS
O
Ph
LiO PO(OMe)2
THF, 88%
de=90%
OTBSOTBS
OPh
O
3-Oxacarbacyclin
Scheme 31.
O
O
BnO
TBDPSOO
BnO
TBDPSO
1)LHMDS, 97%, E/Z=7:3
2) LiAlH4, Et2O
CH2OH
O
O
BnO
TBDPSO
O
BnO
TBDPSO
PO(OMe)2
O
BnO
TBDPSO
EtO2CCH2P(O)(OEt)21) PDC, DCM
2) NaH, THF
53%, 2 steps
3) H2, Pd/C, EtOH
Grubbs, toluene
70%
Various conditions
(+)-Zoapatanol
OH
32a32b 32c
Scheme 32.
O
O(EtO)2OPCH2COOEt, t-BuOK, THF, 38% O
OEt
O
Scheme 33.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2221
aldehyde is accomplished in very mild aqueous hydrochloric acid/petroleum ether biphasic system.
P-gp (permeability glycoprotein) is an ATP dependent efflux pump for xenobiotic compounds, whose activity is known to be related to multidrug resistance in cancer cells [60]. Diterpenes of the jatrophane family modulate glycoprotein expression [61], and their de novo synthesis has been a subject of investigation in order to determine structure-activity relationships. In this context, (-)-15-acetyl-3-propionyl-17-norcharaciol was synthesized using two HWE olefinations among the key steps [62]. A first HWE reaction between acetoxyphosphonate 35a and aldehyde 35b with N,N-
dimethylformamidine as base led to 35c, which was then used for the elaboration of the cyclic precursor 35d. After the ketophospho-nate moiety had been established, a second HWE reaction per-formed with racemic aldehyde 35e gave the E-alkene 35f, which was then elaborated to the target diterpene (Scheme 35).
The precursor of the side chain of Arieianal (36d), an ant repel-lent prenylated benzoic acid isolated from Piper arieianum, was synthesized using a HWE olefination as the key step. Halide dis-placement of bromide 36a with triethylphosphonoacetate in basic medium afforded isoprenoid phosphonate 36b, which was then coupled with terpenoid aldehyde 36c, yielding the synthetic inter-mediate 36d (Scheme 36) [63].
HWE reaction has also been used in Durand’s synthesis of phy-toprostanes and isoprostanes. In a first report (2004), the eight di-astereoisomers of the Syn-Anti-Syn Phytoprostanes F1 Types I and II were prepared from a common precursor and its enantiomer (not shown). As depicted in Scheme 37, both cyclopentylmethyl alco-hols (37a,c) were DMP oxidized and subjected to HWE protocol without purification (due to the unstable nature of the aldehyde), using dimethyl 2-oxobutylphosphonate for the Phytoprostanes F1 type I, and dimethyl 9-(ethoxycarbonyl)-2-oxononylphosphonate
CHO
EtO2C
(EtO)2OP
(EtO)2OPN
NN
N
CO2Et CH2OH
CHO
LDA
BuLi
HCl / petr. ether
78%, 2 steps
LiAlH4
MnO2
45%, 3 steps
Scheme 34.
TMSOO
PO(OEt)2
TBSO
O
OTES
+
TMSOO
TBSO
OTES
O
OH
AcO
AcO
(-)-15-Acetyl-3-propionyl-17-norcharaciol
BuLi, THF
58%
O
OTBSEtO2C
OAc
PO(OEt)2
OTBS
CO2Et
AcO
+
LiCl, (Me2N)2CNH, THF, 91% E/Z 4:1
35a 35b
35c
35d 35e 35f
Scheme 35.

2222 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
for the Phytoprostanes F1 type II. The resulting enones (37b,d) were selectively reduced with S or R BINAL-H, providing for the natural products and their epimers, respectively [64].
The same approach was reported in 2008, when the synthesis of the four enantiomerically pure diastereoisomers of phytoprostanes E1 type II and of 15-E2t-isoprostanes was achieved. In this work, dimethylacetals were used as precursors of the aldehyde moiety. The yields over two steps were comparable, and the resulting enones were manipulated in a similar fashion (Scheme 37) [65].
HWE reaction has also been used to synthesize glycosylthio Lipid II phosphonate analogues 38d [66]. Conjugate addition of tri-O-acetyl-1-thio-N-acetyl-D-glucosamine 38a to 1,1-bis-(diethyl-phosphono)ethene (38b) led to the corresponding glycosylthioethyl-diphosphonate 38c, which reacted with octanal and decanal in the presence of NaH (Scheme 38). As frequently occurs with long chain aldehydes, the products were obtained only in moderate yield and with poor diasteroselectivity. The reaction can also be per-formed as a one-pot procedure, with analogous results.
TBSO CHO
Br
CO2Et
PO(OEt)2KO-t-Bu
91%
36b, KO-t-Bu, toluene
64% E alkene
CO2EtTBSO
Arieianal
triethylphosphonoacetate
36a 36b
36c 36d
CHO
OH
OH
HO2C
Scheme 36.
BzO
BzO
OH
1) DMP
2) dimethyl 9-(ethoxycarbonyl)-
2-oxononylphosphonate,
NaHMDS, THF
79%, 2 steps
BzO
BzO
CO2Et
O
Phytoprostanes F1
Type II
BzO
BzO
OH
1) DMP
2) dimethyl 2-oxobutyl
phosphonate,
NaHMDS, THF
60%, 2 steps
Phytoprostanes F1
Type I
CO2Et
6
BzO
BzO
CO2Et
6
O
EOMO
TBSO
CH(OMe)2
1) TsOH
2) dimethyl 9-(ethoxycarbonyl)-
2-oxononylphosphonate,
NaHMDS, THF
70%, 2 steps
EOMO
TBSO
CO2Et
O
Phytoprostanes E1
Type II
EOMO
TBSO
CH(OMe)2
(CH2)3CO2Et
1) TsOH
2) (MeO)2P(O)CH2C(O)C5H11
NaH, THF
72%, 2 steps
EOMO
TBSO
(CH2)3CO2Et
OIsoprostanes 15-E2t
37a 37b
37c 37d
37e 37f
37g 37h Scheme 37.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2223
However, in the synthesis of a C-analogue of suphated galacto-sylceramide [67], HWE reaction between sugar derived dimethyl-phosphonoketone 39a and tetradecanal gave exclusively the E-alkene 39b in 71% yield, showing that substrate structure and reac-tion conditions may in some cases overcome poor reactivity of long chain aldehydes (Scheme 39).
In 2002, Lee and co-workers developed a method for the prepa-ration of various substituted dimethyl 2-oxo-4-aminobutylphospho-nates (40b) via nucleophilic ring opening of activated azetidinones 40a, and investigated their application in the synthesis of -amino-’, ’-unsaturated ketones 40c (Scheme 40) [68]. This transforma-
tion is interesting since it transfers the chirality of a heterocyclic nucleus, readily available through a variety of stereoselective meth-
ods, to a valuable chiral synthon in a single step. The authors then applied this methodology to the formal synthesis of L-erythro-sphingosine and D-lyxo-phytosphingosine. Azetidinone 40d was reacted with lithium dimethoxy methylphosphonate and then cou-pled with tridecanal, yielding , -unsaturated ketone 40e. Reduc-tion of the latter provided 40f, whose enantiomer had already been used for the preparation of D-erythro-sphingosine and L-lyxo-phytosphingosine.
5. ALKALOIDS
A HWE reaction of aldehyde 41b with phosphonomethylketone 41a was used to introduce the polyene side chain as a key step to-wards the total synthesis of (+)-Torrubiellone C, an insecticidal
PO(OEt)2
PO(OEt)2
O SH
OAc
NHAcAcO
OAcPO(OEt)2
PO(OEt)2
O S
OAc
NHAcAcO
OAc PO(OEt)2
O S
OAc
NHAcAcO
OAc
n
n=6, 33%, E:Z=1:1.2
n=8, 33%, E:Z=1:1.4
DCM, Et3N
90% NaH, dioxane
CHO
n+
38a
38b
38c 38d
Scheme 38.
O
OBnBnO
BnO
OBn
PO(OMe)2
NHBoc
O
C13H27CHO
K2CO3, MeCN
71%
O
OBnBnO
BnO
OBn
NHBoc
O
C13H27
39a 39b
Scheme 39.
N
TBDPSO
O
1) CH3P(O)(OMe)2,
BuLi, (2 equiv.), THF, 95%
2) C12H25CHO, K2CO3,
EtOH, 70%BocHN
OTBDPS
O
Boc
C12H25
N
R1 R2
O
CH3P(O)(OMe)2,
BuLi, THF
59-95%
RCHO
K2CO3, EtOH or
KO-t-Bu, THF
45-89%HN
R1
R2
O
BocBoc
(MeO)2OP
HN
R1
R2
O
Boc
R
R3
R3 R3
R1= OTBDPS, OBn, Et, i-Pr
R2= H, Ph
R3= CH2OTBS, CH2OTBDPS, H
R= 4-ClPhCHO,
5-Cl, 2NO2PhCHO, Ph(CH2)2CHO
EtCHO40a 40b 40c
40d 40e 40f
TBSO
OTBSHN
OTBDPS
O
Boc
C12H25
OTBS
H2, Pd/C, EtOAc
L-erythro-sphingosine
and
D-lyxo-phytosphingosine
Scheme 40.

2224 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
substance extracted from entomopathogenic fungi isolates (Scheme 41). The authors explicit that a mixture of THF/water and LiOH as base were found to be essential to suppress by-product formation [69]. Removal of silyl and benzyl ethers in 41c led directly to the polyene-pyridone natural product.
In 2010, Cui reported the total synthesis of phenantroindolizine (R)-Antofine and phenantroquinolizine (R)-Cryptopleurine alka- loids using an aminoxylation-HWE-reduction sequence (Scheme 42) [70]. Starting from 3,6,7-trimethoxy-phenantrene-9-carbalde- hyde (42a), the propanal derivative 42c was produced in 2 synthetic steps, comprising HWE followed by hydrogenation to produce ester 42b, and reduction followed by selective oxidation. L-Proline cata-lyzed aminoxylation generated the chiral aminoxyaldehyde 42d
which was directly submitted to a second HWE reaction and imme-diate reduction without purification. The resulting 5-aryl-4-hydroxypentanoate 42e served then as common precursor for both alkaloids.
A different and quite expeditious approach to a series of indol-izidine alkaloids was reported recently, [71] using aldehyde 43b as a chiral synthon and three different phosphonate reagents (Scheme 43). Compound 43b was prepared in a 5 gram scale from ester 43a (derived in four synthetic steps from L-aspartic acid). Phosphonate 43d was obtained by nucleophilic ring opening of butyrolactone 43c with dimethoxy methylphosphonate lithium salt. Phosphonate 43g was prepared from hexanal using an aminoxylation-HWE-reduction sequence followed by treatment of the resulting ester 43f with lithiated dimethoxy methylphosphonate. The use of L-proline
in the aminoxylation step gave access to ent-43g (not shown). HWE coupling of 43b and 43d under Paterson’s conditions led to 43h, which upon reductive cyclization, MsCl mediated intramolecular nucleophilic substitution and deprotection provided 43i, an already known precursor of (-)-indolizidines 167B and 209D. Reaction of 43b with 43g or ent-43g under the same conditions allowed for the formation of 43j and 43k, respectively. Compound 43j was ma-nipulated in the same manner as 43h in order to establish the indol-izidine nucleus. The product was then elaborated onto (-)-indolizidines 239AB and 195B, which is just 3-epi-monomorine. The same sequence performed on compound 43k led to (-)-Monomorine.
Radicamines belong to the rare alkaloid family of aryl substi-tuted iminosugars. In 2011, Shankaraiah developed a high yielding (9.75% overall) total synthesis of (+)-Radicamine B relying on HWE olefination and Sharpless epoxidation as the key steps (Scheme 44) [72]. IBX oxidation of 44a followed by carbonyl ole-fination with triethylphosphonoacetate sodium salt led to ethyl 5-aryl-2-pentenoate derivative 44b. Reduction of the ester moiety followed by asymmetric epoxidation, deprotection and intramolecu-lar ring opening of the oxirane yielded the pyrrolidine nucleus pre-sent in the natural product with the desired stereochemistry.
6. CYCLIC ETHERS
Cyclic ethers are isolated mainly from marine natural sources, some of them being toxic or displaying other biological activities.
NH
OMe
O
PMBOO
PO(OMe)2 OHC
TBDPSO
LiOH, THF/H2O, 3 days, 45%NH
OMe
O
PMBOO
OTBDPS
E/Z = 12:141a
41b
41c
Deprotection
(+)-Torrubiellone C
Scheme 41.
CHO
OMe
MeO
MeO
OMe
MeO
MeO
CO2Et
OMe
MeO
MeO
CHO
OMe
MeO
MeO
CHO
ONHPh
OMe
MeO
MeO
OH CO2Et
1) (EtO)2P(O)CH2CO2Et, NaH
2) H2, Pd/C, 98%, 2 steps
1) LAH
2) Swern
PhNO
L-prolineDMSO
1) (EtO)2P(O)CH2CO2Et, DBU, LiCl, MeCN
2) H2, Pd/C,
40%, 3 steps
OMe
MeO
MeO
N
H
n=1, R-Antofine
n=2, R-Cryptopleurinen
42a 42b 42c 42d
42e
Scheme 42.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2225
Their intricate array of different ring size saturated oxacycles, stereogenic elements and polyfunctionality renders some of them amongst the most overwhelming synthetic targets available.
In the synthesis of Amphidinol developed by Crimmins (Scheme 45) [73], the Ba(OH)2 promoted HWE olefination between
phosphonate 45a and aldehyde 45b was used to assemble the polyoxygenated bis-tetrahydropyran core (45c) present in this natu-ral product.
Brevisamide, a monocyclic ether isolated from the red tide dinoflagellate Karenia brevis, was prepared using a HWE reaction
43b 43d+
1) PhNO, D-proline, DMSO
2) (EtO)2P(O)CH2CO2Et,
DBU, LiCl, MeCN
3) H2, Pd/C, AcOEt
62%, 3 steps
TBSO CO2Me
NHCbz
TBSO CHO
NHCbz
43a 43b
O
O
(MeO)2P(O)CH3,
BuLi, THF
80%
43c
OH
O
PO(OMe)2
43d
DIBAL-H
toluene
CHO
OH
CO2Et
(MeO)2P(O)CH3,
BuLi, THF
63% OH
O
PO(OMe)2
43e 43f 43g
TBSO
NHCbz
OH
O
Ba(OH)2.H2O,
THF,H2O, 89%
43h
43b 43g+ TBSO
NHCbz OBa(OH)2.H2O,
THF,H2O, 87%
43j
N
H
n-C3H7
N
H
n-C5H11
(-)-indolizidine 167B (-)-indolizidine 209D
OH
43b ent-43g+ TBSO
NHCbz OBa(OH)2.H2O,
THF,H2O, 86%
43k
OH
1) H2, 10% Pd/C, EtOH
2) MsCl, Et3N, DCM,
3) 0.1M HCl, EtOH
N
H
CH2OH
and
43i
N
H
N
H
Me
(-)-indolizidine 239AB (-)-indolizidine 195B
and
n-C4H9 n-C4H9
HO
N
H
Me(-)-monomorine
n-C4H9
Scheme 43.
OTs
OH
OBnBocHN
OTs
OBnBocHN
CO2Et
OH
HONH
HO
HO
(+) Radicamine B
1) IBX, DMSO, THF
2) (OEt)2PO(CH2COOEt), NaH, THF
90%, 2 steps
44a 44b
Scheme 44.
OO
O
O
OTBSO
OMOM
OHC
H
H
O
O
OBn PO(OMe)2
O
O
O
BnO
OH
OTBSH
OO
O
O
OTBSO
OMOM
H
H
O
O
OBn
O
O
O
BnO
OH
OTBSHBa(OH)2, THF, H2O, 74%+
45a 45b 45c
Scheme 45.

2226 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
to couple phosphonocrotonate 46a with propanal derivative 46b, in order to introduce the unsaturated side chain aldehyde, which was obtained after redox manipulation of ester 46c (Scheme 46) [74].
In 2010, Wilson reported a synthesis of the benzannulated spi-roketal core (47g, Scheme 47) of Berkelic acid (a natural product with activity against ovarian cancer) by means of a HWE/oxa-Michael cascade [75]. Benzyl 4-benzyloxybutyrate 47b was pre-pared in a single step from butyrolactone 47a. Phosphonoketone 47c was synthesized by treatment of 47b with lithiated dimethoxy methylphosphonate. The requisite aldehyde was prepared in situ by reduction of isochromanone 47d and reacted with 47c in the pres-ence of NaH. The resulting enone 47e underwent an intramolecular oxa-Michael addition, thus affording isochroman 47f as a 1:1 mix-ture of diastereoisomers. Chromatographic separation followed by spiroketalization of the cis epimer, led to 47g.
The HWE reaction is also found as a key step in many synthe-ses of some natural products known as ladder toxins, due to the
alternating disposition of the oxygen atom in consecutive rings. The main applications of the reaction are the linkage of preformed ring synthons and ring construction, as the enone product can be further manipulated through oxidation, conjugated addition and ketaliza-tion. A generally used disconnection is shown in Scheme 48.
In Crimmins synthesis of the GHIJ fragment of Brevetoxin A, a neurotoxic ladder toxin produced by K. brevis [76], the oxocane phosphonate precursor of ring G (49a) was coupled with the formyltetrahydropyrane precursor of ring J (49b) by means of a HWE reaction. The resulting four carbon linker in 49c was then elaborated in various steps (including hydrogenation of the double bond, ketalization-dehydration, hydroboration-oxidation, ketaliza-tion and reduction) to the H and I tetrahydropyranyl rings present in precursor 49d (Scheme 49). The same author had previously re-ported a synthesis of the BCDE fragment of the toxin by a similar approach [77].
PO(OEt)2EtO2C OO
HN
OTBS
O
+BuLi, THF
80% O
HN
OTBS
O
EtO2C
1) DIBAL-H
2) MnO2
O
HN
OTBS
O
OHC
46a 46b 46c
Brevisamide
Scheme 46.
O
O
OBn
O
OBn
OBn
O
PO(OMe)2
47a 47b 47c
KOH, BnBr,
toluene, DS
LiCH2P(O)(OMe)2,
THF, 88%
O
C5H11
O OBn
OBn
1) DIBAL-H, toluene
2) 47c, NaH, THF, 80% HO
C5H11
BnO OBn
BnO
O
O
C5H11
BnO OBn
BnO
O
d.r. 1:1
47d 47e
47f
O
C5H11
O OHO
47g
Pd(OH)2, HCl
THF, H2
Scheme 47.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2227
In 2010 Crimmins reported the synthesis of a putative precursor for (-)-Brevenal [78], which paradoxically behaves as a brevitoxine antidote and as a potential therapeutic agent for cystic fibrosis. A barium hydroxide promoted HWE reaction between phosphono-methylketone 50a and aldehyde 50b was used to establish the C and D ring precursor tether in 50c (Scheme 50). At variance with the previous example, a substituted acetaldehyde precursor of ring E was used, as in this case the resulting enone would be further elabo-rated into a pyrane and an oxepane.
Maitotoxin, isolated from the dinoflagellate Gambierdiscus
toxicus, is the most toxic and the largest among ladder toxins known to date. Such molecule probably represents a landmark for modern organic synthesis, containing the impressive number of 32 fused alternating ether rings, 98 chiral centers, 28 hydroxyl groups and two sulphated positions among various challenging structural features. Some fragments of this molecule have been prepared us-
ing a HWE reaction as a key disconnection (Scheme 51). In 2008, Morita used a HWE olefination to synthesize the C’D’E’F’- side chain fragment (51d) of the toxin [79]. An aldehyde precursor of the C’D’E’ fragment was obtained by ozonolysis of the appropriate vinyl precursor 51a and then subjected to olefination with a large excess (14 equiv.) of phosphonate 51b, which provided both the side chain and the precursor of the F’ ring in 51c.
In 2010, Nicolaou reported the synthesis of the ABCDEFG segment of Maitotoxin (51h), lacking the side chain [80]. In this case, phosphonomethylketone 51e was coupled under Masamune-Roush conditions with the aldehyde precursor of ring G (51f). The resulting enone tether in 51g was then manipulated onto F ring and 1,2-syn-propylenediol moietiy present in 51h. In both syntheses, the carbonyl group of the enone was exploited to create a pyrane ring trough ketalization (Scheme 51).
O CHO
OPG O
OPG
(RO)2OP
O
O
OPG
OPGO
O
O
O O
OOR
HWE
+A
D
B C AA
D
D
O
O O
PGO
BA D
Scheme 48.
O
O
O
O
OBn
OPO(OMe)2
OPiv
TIPSO
OPMB
OO
O
OO
OBn
OPiv
TIPSO
OPMB
O
O
O
OBn
O
OBn
OPiv
TIPSO
O
H H
H H H
+
Ba(OH)2, THF, H2O
80%G
H I
J
G
G
J
J
49a 49b
49c
49d
Scheme 49.
O
O
O
O
PO(OMe)2TIPSO
O
MeH
Me
O
TESOOBn
Me
BnO
O
O
O
O
O
OTIPS
OMe
H
Me
O
TESO
BnO Me
OBn
O
O
OH
O
TIPSO
Me
H
Me
OO
TBSO
Me
OTBS
+
Me
HH
H
H
A
B
C
D
EA
AB
B
E
E
Ba(OH)2, THF
90%
50a
50b 50c
Scheme 50.

2228 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
CONCLUDING REMARKS
Since its inception and after more than 50 years, the HWE reac-tion has become one of the most powerful methods for C=C bond formation. Modifications regarding novel phosphonate reagents and reaction conditions, as well as the development of intramolecular HWE reactions have widened the scope of the original method. Further developments have led to novel sequential reactions that are standard tools in modern synthetic organic chemistry. Additional features like robustness of the reaction, wide functional group toler-ance, availability of a large variety of phosphonate reagents, ease of introduction of the phosphonate moiety in advanced synthetic pre-cursors and predictable stereochemical outcome have established the HWE reaction as one of the most reliable tools in the field of natural product synthesis. The vast quantity of recent applications reflects its fundamental importance in this area in the preparation of building blocks, of advanced synthetic precursors and very often as a key disconnection to assemble highly complex molecular frame-works.
CONFLICT OF INTEREST
The author(s) confirm that this article has no conflicts of inter-est.
ACKNOWLEDGEMENTS
This work was supported by the University of Buenos Aires (20020100100935) and by CONICET (PIP 286).
ABBREVIATIONS
15-c-5 = 15-crown-5
18-c-6 = 18-crown-6
2,4,6-Cl3BzCl = 2,4,6-trichlorobenzoyl chloride
Ac = acetyl
B2pin2 = bis(pinacolato)diborane
BHT = 2,6-di-tert-butyl-4-methylphenol
Bn = benzyl
Boc = tert-butyloxycarbonyl
Bz = benzoyl
Cbz = benzyloxycarbonyl
CSA = camphorsulfonic acid
dba = dibenzylideneacetone
DDQ = 2,3-dichloro-5,6-dicyanobenzoquinone
DEAD = diethyl azodicarboxylate
O
O
O
O
O
BnO
PO(OMe)2
H
HBnO
BnO
H
H
HH
BnO
BnO
OBn
OTES
O
O
O
O
O
O
BnO
H
H
BnO
BnO
H
H
HH
BnO
BnO
BnO
OTES
O
OOOBn
BnO
OBn
OBn
DIPEA, LiCl, MeCN, 91%
O
OBn
BnO OBn
OBn
O
O
O
O
O
HOH
HOH
OHHHH
H
HO
HO OH
O OOH
HO
OH
OHH
HOH
OH
A B
C
D
E
F
G
G
A B
C
D
E
A
B C D E G
OTBDPS
PO(OMe)2
O
O
O
O
O
O
H HTBSO OBn
OBn
OH
OHO
O
O
H HO OBn
OBn
HH
TBDPSO
O
OO O
O
O
H
H
TBSO
OBn
BnO
1) O3, DCM; then Me2S
2) 51b, NaH, THF-DMF,
84%, 2 steps
C'D'E'F'
C'D'E'
C'
D'
E'
51a
51b
51c
51d
51e
51f
51g
51h Scheme 51.

Recent Applications of the Horner-Wadsworth-Emmons Reaction to the Synthesis Current Organic Chemistry, 2012, Vol. 16, No. 19 2229
DEIPS = diethyl isopropylsilyl
DIBAL = diisobutylalane
DIPEA = diisopropyl ethylamine
DMAP = 4-(dimethylamino)pyridine
DMB = 3,4-dimethoxybenzyl
DMP = Dess-martin periodinane (1,1,1-Triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one)
DPEPhos = bis(2-diphenylphosphinophenyl)ether
DS = Dean-Stark trap
EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodi-imide
EOM = ethoxymethyl
FGI = functional group interconversion
Grubbs I, II = Grubbs type I or II catalyst
HSTBA = tetrabutylammonium hydrogen sulphate
IBX = 2-iodoxybenzoic acid
KHMDS = potassium hexamethyldisilylamide
LHMDS = lithium hexamethyldisilylamide
MEM = -methoxyethoxymethyl ether
MOM = methoxymethyl
MTM = methylthiomethyl
MsCl = trifluoromethanesulfonyl chloride
NHMDS = sodium hexamethyldisilylamide
NMO = N-methylmorpholine-N-oxide
PDC = pyridinium dichromate
PG = protecting group
Piv = pivaloyl
PMB = 4-methoxybenzyl
PMP = 4-methoxyphenyl
PPTS = pyridinium 4-toluenesulphonate
PS.DCC = polymer supported dicyclohexylcarbodi-imide
PTSA, TsOH = 4-toluenesulphonic acid
Py = pyridine
S- and R-BINAL= chiral binaphtol-aluminium hydride reagent
Selectride = tri-sec-butylhydroborate
Swern = DMSO based oxidation
TBAF = tetrabutylammonium fluoride
TBDPS = tert-butyldiphenylsilyl
TBS = tert-butyldimethylsilyl
TEMPO = 2,2,6,6-tetramethylpiperidine-N-oxide
TES = triethylsilyl
TFA = trifluoracetic acid
TIPS = triisopropylsilyl
TMEDA = N,N,N’,N’-tetramethyl ethylenediamine
TPAP = tetrapropylammonium perruthenate
REFERENCES
[1] (a) Horner, L.; Hoffmann, H. M. R.; Wippel, H. G. Phosphororganische Verbindungen, XII. Phosphinoxyde als Olefinierungsreagenzien. Ber. 1958, 91, 61-63. (b) Horner, L.; Hoffmann, H. M. R.; Wippel, H. G.; Klahre, G. Phosphororganische Verbindungen, XX. Phosphinoxyde als Ole-finierungsreagenzien. Ber. 1959, 92, 2499-2505.
[2] Wadsworth W. S, Emmons W. D. The Utility of Phosphonate Carbanions in Olefin Synthesis. J. Am. Chem. Soc. 1961, 83, 1733-1738.
[3] Williams, J.M.J. (Ed.) Preparation of Alkenes (Oxford: Oxford University Press, 1996).
[4] Trost, B.M.; Fleming, I. (Eds). Comprehensive Organic Synthesis, Vol. 1 (Oxford: Pergamon Press, 1999).
[5] Murphy, P.J.; Brennan, J. The Wittig olefination reaction with carbonyl compounds other than aldehydes and ketones . Chem. Soc. Rev. 1988, 17, 1-30.
[6] Maryanoff, B.E.; Reitz, A.B. The Wittig Olefination Reaction and Modifica-tions Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mecha-nism, and Selected Synthetic Aspects. Chem. Rev. 1989, 89, 863-927.
[7] Boutagy, J.; Thomas, R. Olefin Synthesis with Organic Phosphonate Carban-ions. Chem. Rev. 1974, 74, 87-99.
[8] Korotchenko, V.N.; Nenajdenko,V.G.; Balenkova, E.S.; Shastin, A.V. Olefi-nation of carbonyl compounds: modern and classical methods. Russ. Chem.
Rev. 2004, 73 (10), 957-989. [9] Edmonds, M; Abell, A. "The Wittig and related reactions", in Modern Car-
bonyl Olefination. Takeda, T. (Ed.), 2004, Chapter 1. WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
[10] Still, W.C.; Gennari, C. Direct synthesis of Z-unsaturated esters. A useful modification of the Horner-Emmons olefination. Tetrahedron Lett. 1983, 24 (41), 4405–4408.
[11] (a) Ando, K. Highly Selective Synthesis of Z-Unsaturated Esters by Using New Horner Emmons Reagents, Ethyl (Diarylphosphono)acetates. J. Org.
Chem. 1997, 62, 1934-1939. (b) Ando, K. Z-Selective Hor-ner Wadsworth Emmons Reaction of -Substituted Ethyl (Diarylphos-phono)acetates with Aldehydes. J. Org. Chem. 1998, 63, 8411-8416. (c) Ando, K. Convenient Preparations of (Diphenylphosphono)acetic Acid Es-ters and the Comparison of the Z-Selectivities of Their Hor-ner Wadsworth Emmons Reaction with Aldehydes Depending on the Ester Moiety. J. Org. Chem. 1999, 64, 8406-8408. (d) Ando, K.; Oishi, T.; Hirama, M.; Ibuka, T. Z-Selective Horner-Wadsworth-Emmons Reaction of Ethyl (Diarylphosphono)acetates Using Sodium Iodide and DBU. J. Org. Chem. 2000, 65, 4745-4749.
[12] Nicolaou, K.C.; Harter, M.W.; Gunzner, J.L.; Nadin, A. The Wittig and Related Reactions in Natural Product Synthesis, Liebigs Ann./Recueil, 1997,
1283- 1301. [13] White, J.D; Lee, T.H; Kuntiyong, P. Total Synthesis of Phorboxazole A. 2.
Org. Lett., 2006, 8 (26), 6043-6046. [14] Williams, D.R; Kiryanov, A.A.; Emde, U., Clark, M.P.; Berliner, M.A.;
Reeves, J.T. Total Synthesis of Phorboxazole A. Angew. Chem. Int. Ed., 2003, 42 (11), 1258-1262.
[15] Li, G.; Yang, X.; Zhai, H. Total Synthesis of (-)-5,6-Dihydrocineromycin B. J. Org. Chem., 2009, 74, 1356-1359.
[16] Blanchette, M.A.; Choy, W.; Davis, J.T; Essenfeld, A. P., Masamune, S.; Roush, W.R.; Sakai, T. Horner-Wadsworth-Emmons reaction: Use of lithium chloride and an amine for base-sensitive compounds. Tetrahedron Lett., 1984, 25 (21), 2183-2186.
[17] Jiang, X.; Liu, B.; Lebreton, S; De Brabander, J.K. Total Synthesis and Structure Revision of the Marine Metabolite Palmerolide A. J. Am. Chem.
Soc., 2007, 129, 6386-6387. [18] Nicolaou, K.C.; Sun, Y.-P.; Guduru, R.; Banerji, B.; Chen, D.Y.-K. Total
Synthesis of the Originally Proposed and Revised Structures of Palmerolide A and Isomers Thereof. J. Am. Chem. Soc., 2008, 130, (11) 3633-3644.
[19] Suzuki, T.; Usui, K.; Miyake, Y.; Namikoshi, M.; Nakada, M. First Total Synthesis of Antimitotic Compound, (+)-Phomopsidin. Org. Lett., 2004, 6 (4), 553-556.
[20] Pirrung, M.C.; Biswas, G.; Ibarra-Rivera, T.R. Total Synthesis of Syringolin A and B. Org. Lett., 2010, 2 (10), 2402-2405.
[21] Schauer, D. J.; Helquist, P. Mild Zinc-Promoted Horner-Wadsworth-Emmons Reactions of Diprotic Phosphonate Reagents. Synthesis, 2006, 3654–3660.
[22] Müller, S.; Mayer, T.; Sasse, F.; Maier, M.E. Synthesis of a Pladienolide B Analogue with the Fully Functionalized Core Structure. Org. Lett., 2011, 13 (15), 3940-3943.
[23] Uenishi, J.; Iwamoto, T.; Tanaka, J. Total Synthesis of (-)-Zampanolide and Questionable Existence of (-)-Dactylolide as the Elusive Biosynthetic Pre-cursor of (-)-Zampanolide in an Okinawan Sponge. Org. Lett., 2009, 11 (15), 3262-3265.
[24] Harvey, J.E.; Raw, S.A.; Taylor, R.J.K. The first synthesis of the epoxide-containing macrolactone nucleus of oximidine I. Tetrahedron Lett., 2003, 44 (38) 7209-7212.
[25] Zhong, G.; Yu, Y. Enantioselective Synthesis of Allylic Alcohols by the Sequential Aminoxylation Olefination Reactions of Aldehydes under Ambi-ent Conditions. Org. Lett. 2004, 6, 1637-1639.
[26] Ramulu, U.; Ramesh, D.; Rajaram, S.; Reddy, S.P.; Venkatesham, K.; Venkateswarlu, Y. Stereoselective total synthesis of clonostachydiol. Tetra-
hedron: Asymmetry, 2012, 23, 117–123. [27] Hong, W.P.; Noshi, M.N.; El-Awa, A.; Fuchs, P.L. Synthesis of the C1C20
and C15C27 Segments of Aplyronine A. Org. Lett., 2011, 13, (24) 6342-6345.
[28] Wrona, I.E.; Agouridas, V.; Panek, J.S. Design and synthesis of ansamycin antibiotics. C. R. Chimie, 2008, 11, 1483-1522.

2230 Current Organic Chemistry, 2012, Vol. 16, No. 19 Bisceglia and Orelli
[29] Amans, D.; Bellosta, V.; Cossy, J. An Efficient and Stereoselective Synthesis of the Monomeric Counterpart of Marinomycin A. Org. Lett., 2007, 9 (8), 1453-1456.
[30] Amans, D.; Bareille, L.; Bellosta, V.; Cossy, J. Synthesis of the Monomeric Counterpart of Marinomycin A. J. Org. Chem., 2009, 74, 7665-7674.
[31] Chellat, M.F.; Proust, N.; Lauer, M.G.; Stambuli, J.P. Synthesis of Key Fragments of Leiodelide A. Org. Lett., 2011, 13, (12) 3246–3249.
[32] Paterson, I.; Burton, P.M.; Cordier, C.J.; Housden, M.P.; Mühlthau, F.A.; Loiseleur, O. Toward the Total Synthesis of the Brasilinolides: Construction of a Differentially Protected C20-C38. Org. Lett., 2009, 11 (3), 693-696.
[33] Nicolaou, K.C.; Li, Y.; Sugita, K.; Monenschein, H.; Guntupalli, P.; Mitchell, H.J.; Fylaktakidou, K.C.; Vourloumis, D.; Giannakakou, P. O’Brate, A. Total Synthesis of Apoptolidin: Completion of the Synthesis and Analogue Synthesis and Evaluation. J. Am. Chem. Soc., 2003, 125, 15443-15454.
[34] Roy, S.; Spilling, C.D. Synthesis of the C(18)-C(34) Fragment of Amphidi-nolide C and the C(18)-C(29) Fragment of Amphidinolide F. Org. Lett., 2010, 12 (22), 5326-5329.
[35] Hillier, M.C.; Price, A.T.; Meyers, A. I. Studies on the Total Synthesis of Disorazole C1. An Advanced Macrocycle Intermediate. J. Org. Chem. 2001, 66, 6037-6045.
[36] Srinivasarao, M.; Kim, Y.; Li, X.H.; Robbins, D.W.; Fuchs, P.L. Studies on the Synthesis of Apoptolidin: Synthesis of a C1C27 Fragment of Apoptolidin D. J. Org. Chem., 2011, 76, 7834-7841.
[37] Li, X.; Lantrip, D.; Fuchs, P.L. -Allyl Phosphinoyl Phenyl Sulfone (GAPPS): A Conjunctive Reagent for the Synthesis of EE, EZ, and ET 1,3-Dienes. J. Am. Chem. Soc., 2003, 125 (47) 14262–14263.
[38] Ohtani, T.; Kanda, H.; Misawa, K.; Urakawa, Y.; Toshima, K. Synthetic studies of incednine: synthesis of C1–C13 pentaenoic acid segment. Tetrahe-
dron Lett. 2009, 50, 2270–2273. [39] Bentley, D.J.; Slawin, A.M.Z.; Moody, C.J. Total Synthesis of Stephanotic
Acid Methyl Ester. Org. Lett., 2006, 8 (10) 1975-1978. [40] Schmidt, U.; Griesser, H.; Leitenberger, V.; Lieberknecht, A.; Mangold, R.;
Meyer, R.; Riedl, B. Amino Acids and Peptides. Part 81. Diastereoselective Formation of Z- Didehydroamino Acid Esters. Synthesis, 1992, 487-490.
[41] Sánchez, C.C.; Keck, G.E. Total Synthesis of (+)-Dactylolide. Org. Lett., 2005, 7 (14), 3053-3056.
[42] (a) Paterson, I.; Yeung, K-S. Studies in marine macrolide synthesis: A stere-ocontrolled synthesis of a C17-C32 subunit of scytophycin C. Tetrahedron
Lett., 1993, 34, 5347-5350. (b) Paterson, I.; Yeung, K.-S.; Smaill, J.B. The Horner-Wadsworth-Emmons Reaction in Natural Products Synthesis: Expe-dient Construction of Complex (E)-Enones Using Barium Hydroxide. Syn-
lett, 1993, 774-776. [43] Jiang, Y.; Hong, J.; Burke, S.D. Stereoselective Total Synthesis of Antitumor
Macrolide (+)-Rhizoxin D. Org. Lett., 2004, 6, (9), 1445-1448. [44] Shing, T.K.M.; Cheng, H.M. Short Syntheses of Gabosine I and Gabosine G
from d-D-Gluconolactone. J. Org. Chem., 2007, 72, 6610-6613. [45] Yajima, A.; Kagohara, Y.; Shikai, K.; Katsuta, R.; Nukada, T. Synthesis of
two osteoclast-forming suppressors, demethylincisterol A3 and chaxine A. Tetrahedron, 2012, 68, 1729-1735.
[46] Marco, J.A.; Carda, M.; Murga, J.; Falomir, E. Stereoselective syntheses of naturally occurring 5,6-dihydropyran-2-ones. Tetrahedron, 2007, 63, 2929–2958.
[47] Ramana, C.V.; Raghupathi, N.; Gurjar, M.K.; Chorghade, M.S. A carbohy-drate-based approach for the total synthesis of strictifolione. Tetrahedron
Lett., 2005, 46 (23), 4073-4075. [48] Srihari, P.; Rajendar, G.; Srinivasa Rao, R.; Yadav, J.S. First stereoselective
total synthesis of (+)-dodoneine. Tetrahedron Lett., 2008, 49, 5590-5592. [49] Singh, R.P.; Singh, V.K. Facile One-Step Synthesis of b-Alkoxy Lactone via
Sequential Lactonization and 1,4-Addition of Alkoxide Group: Total Synthe-sis of All Stereoisomers of Dihydrokawain-5-ol. J. Org. Chem. 2004, 69, 3425-3430.
[50] Yadav, J.S.; Subba Reddy, U.V.; Anusha, B.; Subba Reddy, B. V. The stereoselective total synthesis of (+)-garvensintriol. Tetrahedron Lett., 2010, 51 (42) 5529-5531.
[51] Kumar, P.; Pandey, M.; Gupta, P.; Dhavale, D.D. Organocatalytic stereose-lective synthesis of passifloricin A. Org. Biomol. Chem., 2012, 10, 1820-1825.
[52] Samarat, A.; Landais, Y.; Amri, H. First synthesis of (±)-bis-homosarkomycin ethyl ester. Tetrahedron Lett., 2004, 45 (10), 2049-2050.
[53] Ghosh, A.K.; Xi, K. Total Synthesis of (-)-Platensimycin, a Novel Antibacte-rial Agent. J. Org. Chem., 2009, 74, 1163-1170.
[54] Kim, M.; Gais, H.-J. Fully Stereocontrolled Syntheses of 3-Oxacarbacyclin and Carbacyclin by the Conjugate Addition-Azoalkene-Asymmetric Olefina-tion Strategy. J. Org. Chem., 2006, 71, 4642-4650.
[55] Taillier, C.; Gille, B.; Bellosta,V.; Cossy, J. Synthetic Approaches and Total Synthesis of Natural Zoapatanol. J. Org. Chem., 2005, 70, 2097-2108.
[56] J.S. Yu, T.S. Kleckley, D.F. Wiemer. Synthesis of Farnesol Isomers via a Modified Wittig Procedure. Org. Lett. 2005, 7 (22), 4803-4806.
[57] Reynaud, E.; Aydemir, G.; Rühl, R.; Dangles, O.; Caris-Veyrat, C. Organic Synthesis of New Putative Lycopene Metabolites and Preliminary Investiga-tion of Their Cell-Signaling Effects. J. Agric. Food Chem. 2011, 59, 1457–1463.
[58] Petroski, R.J.; Bartelt, R.J. Direct Aldehyde Homologation Utilized To Construct a Conjugated-Tetraene Hydrocarbon Insect Pheromone. J. Agric.
Food Chem., 2007, 55, 2282-2287. [59] Bartelt, R.J.; Weisleder, D.; Plattner, R.D. Synthesis of nitidulid beetle
pheromones: alkyl-branched tetraene hydrocarbons. J. Agric. Food Chem., 1990, 38, 2192-2196.
[60] Borst, P.; Elferink, R. O. Mammalian ABC transporters in health and dis-ease. Annu. Rev. Biochem. 2002, 71, 537-592.
[61] Hohmann, J.; Molnar, J.; Redei, D.; Evanics, F.; Forgo, P.; Kalman,A.; Argay, G.; Szabo, P. Discovery and Biological Evaluation of a New Family of Potent Modulators of Multidrug Resistance: Reversal of Multidrug Resis-tance of Mouse Lymphoma Cells by New Natural Jatrophane Diterpenoids Isolated from Euphorbia Species. J. Med. Chem. 2002, 45 (12), 2425-2431.
[62] Helmboldt, H.; Köhler, D.; Hiersemann, M. Synthesis of the Norjatrophane Diterpene (-)-15-Acetyl-3-propionyl-17-norcharaciol. Org. Lett., 2006, 8 (8), 1573-1576.
[63] Odejinmi, S.I.; Wiemer, D.F. Synthesis of arieianal, a prenylated benzoic acid from Piper arieianum. J. Nat. Prod., 2005, 68 (9), 1375-1379.
[64] El Fangour, S.; Guy, A.; Despres, V.; Vidal, J.-P.; Rossi, J.-C.; Durand, T. Total Synthesis of the Eight Diastereomers of the Syn-Anti-Syn Phyto-prostanes F1 Types I and II. J. Org. Chem., 2004, 69, 2498-2503.
[65] Pinot, E.; Guy, A.; Fournial, A.; Balas, L.; Rossi, J.-C.; Durand, T. Total Synthesis of the Four Enantiomerically Pure Diasteroisomers of the Phyto-prostanes E1 Type II and of the 15-E2t-Isoprostanes. J. Org. Chem., 2008, 73, 3063-3069.
[66] Borbás, A.; Herczegh; P. Synthesis of lipid II phosphonate analogues. Car-
bohydrate Research, 2011, 346 (12) 1628-1632. [67] Modica, E.; Compostella, F.; Colombo, D.; Franchini, L.; Cavallari, M.;
Mori, L.; De Libero, G.; Panza, L.; Ronchetti, F. Stereoselective Synthesis and Immunogenic Activity of the C-Analogue of Sulfatide. Org. Lett., 2006, 8 (15), 3255-3258.
[68] Lee, H.K Kim, E.-K.; Pak, Ch.S. Facile transformation of 2-azetidinones to unsaturated ketones: application to the formal synthesis of sphingosine and phytosphingosine. Tetrahedron Lett., 2002, 43 (52), 9641-9644.
[69] Jessen, H.J.; Schumacher, A.; Schmid, F.; Pfaltz, A.; Gademann, K. Catalytic Enantioselective Total Synthesis of (+)-Torrubiellone C. Org. Lett., 2011, 13 (16), 4368-4370.
[70] Cui, M.; Song, H.; Feng, A.; Wang, Z.; Wang, Q. Asymmetric Synthesis of (R)-Antofine and (R)-Cryptopleurine via Proline-Catalyzed Sequential a-Aminoxylation and Horner-Wadsworth-Emmons Olefination of Aldehyde. J.
Org. Chem., 2010, 75, 7018-7021. [71] Reddy, Ch.R.; Latha, B.; Rao, N.N. Enantioselective access to (-)-
indolizidines 167B, 209D, 239AB, 195B and (-)-monomorine from a com-mon chiral synthon. Tetrahedron, 2012, 68, 145-151.
[72] Shankaraiah, G.; Sateesh Chandra Kumar, R.; Poornima, B.; Suresh Babu, K. Stereoselective synthesis of (+)-radicamine B. Tetrahedron Lett., 2011, 52 (38), 4885-4887.
[73] Crimmins, M.T.; Martin, T.J.; Martinot, T.A. Synthesis of the Bis-tetrahydropyran Core of Amphidinol 3. Org. Lett, 2010, 12 (17), 3890-3893.
[74] Fadeyi, O.O.; Lindsley, C.W. Total synthesis of brevisamide. Org. Lett., 2009, 11 (17), 3950-3952.
[75] Wilson, Z.E.; Brimble, M.A. A flexible asymmetric synthesis of the tetra-cyclic core of berkelic acid using a Horner–Wadsworth–Emmons/oxa-Michael cascade. Org. Biomol. Chem., 2010, 8, 1284–1286.
[76] Crimmins, M.T; Zuccarello, J.L.; Cleary, P.A.; Parrish, J.D. Convergent, Stereoselective Synthesis of the GHIJ Fragment of Brevetoxin A. Org. Lett., 2006, 8 (1), 159-162.
[77] Crimmins, M.T.; McDougall, P.J.; Emmitte, K.A. A Convergent Coupling Strategy for the Formation of Polycyclic Ethers: Stereoselective Synthesis of the BCDE Fragment of Brevetoxin A. Org. Lett., 2005, 7 (18), 4033-4036.
[78] Crimmins, M.T.; Shamszad, M.; Mattson, A.E. A Highly Convergent Ap-proach toward (-)-Brevenal. Org. Lett., 2010, 12 (11), 2614-2617.
[79] Morita, M.; Ishiyama, S.; Koshino, H.; Nakata, T. Synthetic Studies on Maitotoxin. 1. Stereoselective Synthesis of the C D E F -Ring System Hav-ing a Side Chain. Org. Lett., 2008, 10 (9), 1675-1678.
[80] Nicolaou, K.C.; Aversa, R.J.; Jin, J.; Rivas, F. Synthesis of the ABCDEFG Ring System of Maitotoxin. J. Am. Chem.Soc., 2010, 132, 6855-6861.




















