
Injection Moulding Tool Design Manufacturing, Estimation and ...
Upload
khangminh22Category
view
10download
0
Reaction Injection Moulding of Syndiotactic Polystyrene: The Effect of Reaction Parameters on Monomer
Conversion and Polymer Properties
Colin Li Pi Shan
A ehesis subrnitted to the Department oÇChemistry in conformity with the
requirements for the degree of Mas ter of Science (Engineering)
Queen's University
Kingston, Ontario, Canada
September 1997
copyright O Colin Li Pi Shan, 1997

National Library I*I of Canada Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques
395 Wellington Street 395. rue Wellington OttawON K1AON4 Ottawa ON K1A ON4 Canada Canada
The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sel1 reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la fome de microfiche/^ de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur consenre la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fkom it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

Abstract
The primyy objective of this research w u to investigate the narure of the conversion
limitation in the developrnent of a novel syndionctic polystyrene (sPS) rel-ctioii injection mouldiiig
@Il i ) pmcess. By v q i n g the reaction p*Lu;uIietee of the estent of mising, reaction rime w d mould
wall tempenture, the effects on the monorner conversion ünd polymer properties were determinrd.
fi , 3, styrene Cdiztng a two component medlocene cataiyst system comprised of Cp*TiuMe~ and B(C F-)
monomer \vas polmerized in bulk in a specially constructed RIM app;irJtus to produce syndior~ctic
polysryrene. I r \vas obsemed diat the reÿction wÿs highly esothecmic md that hi& conversions to
hi$? syndiotactic polymer occurred.
It w:is found tliat r-ing the mould w;Jl tempenture Ii:id the largest effecr on tlic
polynerization. .At the lower mould tempemures, increases ù i the conversion were fouiid. At Liotli
the highest and lowest rempenture estrernes snidied, it wvs discovered that die polymer proprrries
detecior~ted. In studying different reaction times, it was found t1i:ir the catalysr activity ç:ui I)c
sustaîned for longer duntion to increase the conversion while m~iiitaiiiilg the polyner propertics.
.it short reaction times the polyrnerizarion occurs quite mpidly -and hi& convenioiis c m be diievcd
within minutes. Vsqing the estent of misuig had little to rio effect on die coiiversion and polymrr
properries.
In dl die conditions snidied to reduce an- difhsion or temperature limirritions, a limitirig
conversion w;is sd l reached. Investigations into the cystdlirie iiamre of the polymer revealed thac
the coiivenion limitation of the polymerïzation may be due to the enrrapmenr of styrene monorner
widiin die c~srailùie regioiis of sPS. This entrapmenr is hypodiesized to occur via a styrene-sPS
rnolecuhr cornples which is related to the hel ical conformations of s yiidio tactic pol ystyrene.

Acknowledgements
1 would like to thank Dr. Vi.E. Baker for his guidance and supervision, riot onlv in the
aspects of the project but as educator in the aJLiing of Engmeering Cliernists. 1 dso esrend my
thanks CO both Dr. M.F. Cunningham and Dr. K.E. Russell whose researcli esperieiice ïiid
suggestions were induable. -Yso, 1 musr acknowledge Dr. MC. Baird a i d his Iab g o u p fr)r
providing the srarting materials and technicd expertise for the cntalysc sydieses.
My thmks, dso spreads out to the members of the Baker L;lb who suppocted my
frustrarions ÿnd triumphs while making the esperience enjoyable. Specid diÿnks so to D. (Iuk :uid
S. Hojabr for helpfLl discussions and to Dr. T. Liu for his previous esperieiice -and assistance witli
the ariaiyticd TG.% work.
Lastly, 1 would like to express my sincere gr~titude co rny Eimily h r alwïdys supporurig my
endeavours. -4 special tvord is dso bestowed ro my friends, for the friendships 1 have treasured at
Queen's.

Table Of Contents
Absuact Acknowledgements Table of Contents List of Figures List of Tables List of Symbols
1.0. Introduction
Z . I . Syndiotaak Polysîyrene 1.1.1 Background to sPS
1.1.1.1 Properties of Polystyrene 1.1.1.2 Structud Polyrnorphism of sPS
1.1.2 Syndiospedic Polymerization O f Styrene 1.1.2.1 Cadysa and Mechanisms 1.1.2.2 Effect O t Po1ymeriz;ition Conditions on Catalyst ;ictivity ;ind
Material Properties 1.1 . î .3 High Conversion Styrene Polymerizations
1.2 Con ven ti'onai Sryrene PoI'erÙatio~ 1.2.1 Thermÿl and Bulk Polymerization of Styrenr 1.2.2 Difhsion ControIIed Phenomena
3.3 MM Processing 1.3.1 Introduction to RIhI 1.3.2 Wh1 Processing Requirements 1.3.3 Previous studies of RIhl Processing of sPS
1.4 Auns of diis Study
2.0 Expetimental
2.2 M a t e d s 2.2 Metdocene Cacllyst Synthesis
2.2.1 Prepmtion O t Cp0Tihfe, Catdyst 2.2.2 Prepmtion of B(C,Fd, Cocatdyst
2.3 Reaction Injecrion MouidUIg PolymerUations 2.3.1 RliLl hlising =\pparatus 2.3.2 Rihl Technique 2.3.3 Temperature M o n i t o ~ g 2.3.4 Esperimennl Studies
2.3.4.1 Prepolymerization o f Styrene by B o r n e Coçaralys t 2.3.1.2 Benchmark Control Study 2.3.4.3 M.sing Study 2.3.4.4 Reaction Time Study 2.3.4.5 .\lould \Xd Tempemture Study
2.3.5 Rcsidual LIonomcr Loss/(:onversion Estimate

Table of Contents Cont'd.
2.3.5.1 \Txuum Oven drying 2.3.5.2 Thermogravime t ic rùidilysis
2.4 Charactenkarion of PS Polymers 2.4.1 Deterrnination of sPS/aPS Frxtions 2.4.2 Product Identification and Txticity Analysis 2.4.3 Determination of T h e d Properties 2.4.4 Deterrnination of Molenilar Weight 2.4.5 Estimaaon ofsPS Crystdlinity and Identification of Crysnlline Forms
3.0 Results and Discussion
3.1. EstUnating the Monomer Conversion ofBulX- Pof'enZed RIM Samples 3.2 Control Smdy of RIM Polperizations
3.2.1 Benchmark Reproducibility 3.2.2 Characteristics of RIhl Polystyrene
3.3 Effect of ilhkihg on the MM Poiymerization 3.3.1 Effect of Mising on Conversion 3.3.2 Eifect of hllxing on Material Properties
3.1 Effect of T h e on the RIM Polymerization 3.4.1 Ef'fect of Retiction Tirne on Conversion 3.4.2 Effect of Reaction Tirne on Marerixi Pro perties
3.5 Effecr of Mouid Temperature on the MM Polymnkatio~ 3-51 Effect of Mould \Vail Temperature on Conversion 3.5.2 Effect of Mould W'dI Temperature on Materid Properties
3.6 Conversion Limirations of the Polymenkation
4.0 Conclusions
5.0 Recommendations and Future Work
References

Table of Contents Cont'd.
Appendices and Curriculum Vitae
Typicd TGA Therrnogm of sPS RIM Smple Randomized Experimentd Run List Borane Prepolymerization Smdy =H N&fR ofsPS 'H YhIR ofaPS 13c U i R of sPS
NMR of aPS DSC Thermal Trace ofsPS Sample lilxing Study
G: Reacuon Time Study H: Mould \Val1 Temperature S ~ d y 1: Crysnllinity Estimates of sPS Samples
Curriculum Vitae

List of Figures
Fig. 1.1: Fig. 1.2.1: Fig. 1.2.2: Fig. 1.2.3: Fig. 1.2.4:
Fig. 1.3.1: Fig. 1.3.7: Fig. lA.1: Fig. 1 -42 Fig. 1.4.3: Fig. 1.4.4: Fig. 1.4.5:
Fig. L.5.1: Fig. 1.5.2: Fig. 1.6: Fig. 1.7:
Strucninl configurations of polysryrene sPS zigzag and helical represenntions Methods of obnining sPS ~xystdline forms Structurai representations of sPS-roluene molecular complrs Representation of the entrapment of benzene molecules within sPS moie~vles MeMi-coordinated insertion mechanism of styrene P-hydrogen elimination reaction in styrene polymerizations Styrene polymerimtion canlyzed by Cp'TiiLIe,/,LLiO EFfect ofstyrene concentration on the monomer conversion Effeect of increasing polymerizi~tion time on the monomer conversion EEeçt of increasing temperature on the monomer conversion Variation of syndiotactic yield and melting point of sPS versus pol y merization temperature Thermal Initiation O ï Styrene Chain Growth Propagation of Styrenr Effect of Diffusion Controlled Temination Schematic of a RIM process
Fig.2.1: RIM hlishead Schematic Fig 2.2: RI,\;[ :lppariltus Setup Fig 2 3 : Flowchart of the Ch~mcrerization of RIM S:unples Fig 2.4: ' H S M R of mehine and methylene C region of polystyrenes
13 Fig 2.5: C S M R of phenyl C region of polystyrenes Fig 2.6: Eupanded inkued specaa of sPS smples in three differerent regions Fig 2.7: FTIR IR s p e c n of sPS dis tinguis hing a and P çrystalline t o m s Fig 2.8: FTIR IR specm ot'sPS distinguishing 6 and y crystdline forms
Fig. 3.1: Typicd sPS R N polymerization reaction temperature pro file Fig. 3.3- 1: Effect of mixing method on the monorner conversion Fig 3.3.2: Effect of the mking method on the sPS fraction Fig. 3.4.1: EfFect of reaction time on the monomer conversion

List Of Figures Cont'd.
Fig. 3.4.2: Effect of reaction time on the sPS Enchon Fig. 3.4.3: Effect of reaction time on sPS ~vt. avg. molecular weight Fig. 3.4.4: Effect of reaction time on sPS num. iivg. rnolecular weight Fig. 3.5.1: 60 OC mould wdl temperature pro tile Fig. 3.5.2: 110 OC mould wdvall tempenture profile Fig. 3.5.3: O OC mould wdl temperature profile Fig. 3.5.4: -20 O C mould wdl tempenture profile F i . 3 . 5 : Effect oE mould 4 1 temperature on the monomer conversion Fig. 3.5.6: Effect of mould d l temperature on the sPS h a i o n Fig. 3.5.7: Effect of mould w;dI tempenture on the sPS lvt. avg. moleculi~ weighr Fig. 3.6.1: FTIR - Identification of sPS helicd forms in the 400-650 cm ' region Fig. 3.6.2: FTIR - Identification oisPS helicd forms in the 660-940 cm.' regton Fig. 3.6.3: FTIR - Suggested presence of zig-mg forms in the 1 100-1400 cm ' region Fig. 3.6.4 FTIR - Distinguishing the S-form from the y-form in rhr 940-1020 cm '
regton Fig 3.6.5: FTIR - Identitication of the morphous f o m in the 820-860 cm ' region

List Of Tables
Table 1.0: DCP RIM Formulation 29 Table 3.1: Cornparison of Conversion Estimates for Vacuum Oven Drying and TG.\ 53
Wt. LOSS Table 3.2: sPS RIhf Benchmark Consenion Reproducibility 56 Table 3.3: sPS RIM Benchmark Polymer Properties 37 Table 3.4: Mould \Val1 Average Reaction Tempemtures -!
/ 2 Table 3.5: DSC Crysnllinity Estimates of sPS Samples 81 Table 3.6: Predicted Conversions due to Monomer Entmpmenr by sPS Crysrals s 1

List of Syrnbols
aPS - anctic polysqrene . U S - poIy(acqloniuile-bundiene-sqrene) .UBX - azoisobutyronitnle BM - benchmark Cp - qclopentadienyl Cp' - pei~omediylqclopen~dienyl d - chamber diameter (cm) DCP - dicyclopemadiene DOF - degree of freedorn DSC - differentid scanning caloturieuy ESR - elecuon spin resonance spectroscopy F I I R - Fourier uansfom infmed specuoscopy GPC - gel pemeation chromaropphy HTGPC - high temperature gel permeauon chrom;itognphy iPS - isotactic polystyrene HIPS - tiigh impact polystyrene LDhl - large diametei rnising LLAO - methylduminosane hlEK - methyl ediyl ketone SMR - nucle-ar mgnetic resonance spectroscopy S S l I - no sratic misiiig PDCP - polydiqcloperitadiene PE - polyethyiene PET - polyethyiene terephdialate PP - polypropyleric PS - polyscyrene Q - tlowr~te in cm3/s Re - Reynolds number RIM - re.~ction injection rnoulding RRIM - reiriforced reaction injection rnoulding sPS - syiidior,icric polysryrene S.<\ - poly(syrene-acryioni trile) SE\ [ - scanning electron microscopy SINC - solvent induced cqsdlization TCB - trichlorobenzene Tg - g h s s tmisition TG .A - thermogr~vimetric mal ysis TI,\.[ - thennoplastic injection rnoulding Thl\ - trünethylduminum
- test cube mising Z-S - Ziegler-Sam cadysts
p - zigzag plaiiÿr (onhorhombic) crysdline form ofsPS 6 - Iielicd crysrdine form of sPS y - Iielicd crysdine form ofsPS q - riscosity (poise)

Chapter 1

1.0 Introduction
1.1 Syndioractic Polystyrene (sPS)
1S.l Background to sPS
1.1.1.1 Properties of Polystyrene
The monomeric precursor, styrene, is the simplest m m t i c compound haviiy :ui
wamrated side-ch&. Uniquely, the polynerization of styrene proceeds readily, using dI mediods
of poly-nerization, under the influence of heat alone md/or m initiator. It is one of die &w
monomers thar c m be polynerized by the four disthicr mectiiuiisms of free r ~ d i d , the ioiiic
mechanisms of mionic and cationic ünd coordination polymerizatioii. 50 yecirs ilgo, Dow Chttmtcd
\vas die first cornpany to cornmercialize polysvrene successtl1ly.l Tr~dïy, polys-rene P S ) is ;L
wmmodity polmer aiid maiy different grades are produced by a wriety of differeiit processer ibr :L
varie- of applications. r\ very large business hÿs developed for polystyeiie a i d its copolymers suc11
u mbber-modified polystyene (HIPS), ac-loiiitrile-sqreiie çopolymrrs (S.AS) and rubber modified
licqlonitrile-scyrerie copol ymers (.%Bq.
n i e most distinguishitig characreristic of generai purpose polystyene is diiit ir is :ui
amorphous, bntrie, glas-like solid below 100OC.I \.el1 above its &us-transition rernpemture (Tg),
the polymer is tluid-like whicli allows it to be e:dy shaped into mai- usehl forms. n i e comrnrrci:il
success of polystyrene is largely due to its uanspareiiq, escellent stiffiiess, go«d prc~cessabili- iuid
low çost. Typicd applications for generd-purpose polystyreiie resiiis iiicludr pacbigng produçrs.
disposable medical wue, roys etc. F o m applications iiiclude eg: c:irtoris, meat-packaging tciys.
"clamsheiis" for k t - food packagine; and espdiided polystyreiie cusliioriinç mirterials for packagng.
The only restriction is in use at hi& tempenture where it loses dimeiisioiiai snbility above its Tg.
Stereoreguliinty is important in coiitrollirig the properties of pd!.mer molecules. Durilig rlic
polyrnerizÿtioii of rinyl monomers (CHz=CHR) depeiiding on the iiisertimi iirrmh~meiir. ;~t:icric.
isor~ctic :md syndionçtic polyiners c m be formed. In the c:w o f styrciie moriomer, ;i ciiidoni

arrangement of the pheriyl groups alortg the backbone result in ar~çriç polystyrerie. \Shen dl the
phenyi groups are on the sarne side, the structure is isoracUc and wheri the phen. groups dternare
positions above and below the backbone, the structure Ïs named syiidior~ctic polyswrene. Plese
cefer to Figure 1.1 for the structural represenmtions of polystyrene.
.\tactic Pol ystyrene (iiPS)
Isotactic Polyscyrene ( 9 s )
Figure 1.1: Structural configurations of polys tyrene
Cp to diis point, essentially dl of die commercial polystyreric in use today is ar~ctic in nature.
In theory, stereoregul*~ polysqrenes were predicted to esist ;md to have Iiigii rneltiiig points duc to
die rigd pheiiyl groups ;ittaciied ro die polymer biickhrie. In 105.5, Litrd discovered isoocric
polys-relie iuid i t did i~ideed Iiaw :i hidi me1 ring point of 2-11 1.2 Lnfc)rni~ii~tcI~, rhc txtc ( ) f

qsrdlization of this polystyrene was too slow for practicd use and it \v:is iiever comrnercidizrd. I r
was not u n d 1985, that Ishihara and the Idernitsu Kosaii Cu. discovered syidiotactic polystyreiir
with the use of a medocene canlyst.3 -4s predicted, sPS has a higli melting point of ?70°C and ;i
npid crystallization rate that is pncticd for industry. Due to in crystdluie nature, sPS results ixi a
polymer with high heat resistance. This feature coupled wirh irs hydrocirboii backbone result in
escellent resistance towards moisture, s t e m and various chernical solvetits. sPS has an uriusudly f i s r
crystallization rate which reaches a ma.uimum around lGO°C.= At this temperature, the rate is so fiasr.
d ix cold crysdliution can occur s i d a r to polyethyleiie tereplithdate (PET). This cipid
qsrdlizatioii makes sPS pncticd for a nurnber of ÇomiLig openuoiis iricludiiig injection mouldi~iç,
extrusion and therrnoforrning. Compared to odier engineering diemoplastics, it ediibits l o ~ w
moisture uprke, lower shrinkdge a id i higher degree of dimeiisioiid ; I C L U ~ J ~ m d sr~biliy \vheti
rnoulded.=
For sPS to be used ;is ÿn engineering thermoplüstic, it musc he reiiiforced wirli tiber&;üs,
mineral fillers and/or rubber elastomen. Fibergiass reinforced sPS lias p o d dyriÿmic :uid tlienno-
mechmicd properties ediibibng a high load heat distoriioii temperature of 7jO0C.= Tliese Iiigfi
temperature propercies malie fibergiass reinforced sPS just ÿs effective as con\~entioriÿl engiiieeriiig
tliermoplas tics.
.-ùiodier beiiefit of sPS is its specific gravity advaiinge of 35O;0 [)ver odier erigmeeriiig resins.
sPS dso surpasses many other engineering resins with a higher e1ectric:d resisrmce md lowrr
dielecuic dissipation putting sPS ui direct cornpetition with teffoii.= .YI of die above mmeriuoned
properties, combtned with its esceprioiid electricai perfomÿiice, low specific gpv iv nid tougli~iess
d e s it competirive widi other hidi heat c~snll ir ie engineering tliermoplastics. ;\s a result a iiumher
of applications are espected in the :uas such ;is electricd, ilutomotive, tilms :uid fibres.
In esploriiig the potential applications, the developmerit of :i re;ictioii injection mouldiii~
(RIA[) process for syndiotactic polystyreiie could e rpmd die pncticd uses of die polymrr :uid the
RISI process. In the upcomiiig sectioiis, details of the c~stdl i i ie minire of sPS, iiisrghr mto rlic

cÿdytic chernisuy, a look at conventional styrenr polynerization ;uid reÿçtiori inirctioii mouldirig
pcocess operations w d be discussed to gain a berter understanding of some of the concems tn tlic
development of this novel sPS RIiLl process.
1.1.1.2 Stnictural Polymorphism of sPS
Since the discoveq of sPS, there have been many snidies uito tlie nature of the crysrdliiie
structure and in formation. Oiguiaiiy, Isliihua et al. repocted a zigzag piairar structure based on s-
ray diffraction data.3 Today, syndionctic polystyrene has been shown to have very comples
polynorpliic structures dependent on the c~sdlizatioii conditions. Csiiig die ii»mriiçl;inirc
proposed bu G u e m et al., four different cqsralline fomis exist.4 .Ci a aiid P iorm a~iioiniiig
planïr zigz;lg c i i i ~ i ~ 0 with fibre identity periods of 5.0-5.1 -4 whtle nvo others, the 8 ;uid 7,
conriniiig (7/1)75 Iieliciil chahs (ITGG) with fibre identity periods of 7.5-7.7 -4. The molecu1;ir
structure represenntions -are shown in Fig 1-21. The generd pattern is tiirdier complicated Li! the
fact diat botli the a (trigond) ;uid P
T (odiortiombic) fimns cul esist in di fferei~t 4 in r~ modifications ch;mcterized by different degees
of structurai order, whcre a' and p' are die nvci
modifications and U" ;uid P.'
ordered moditicauons. \[arc
limiting disordercd
.are the nvo limiting
recently, the presence of a mesarn«cphic fonn,
coritaiiiing ch;iiiis in the t~uis-pl;u~x
conformation of srniill ;md imperfect crysr;ds of
Figure 1-2-19 sPS zigzag and heiical represen tations the trigond (a) hiis ;dso been rept~rtcd.~
For simplici~, only the four major crystdliiir forrns will be discussed ;uid the coiidiriotis
required to obtiiiii each f o m are sfiown in Figure 1.7.2. Snrriiig frcim die i~morphous gl;issy kinn,

the a fom cm be obnlied by annealhg above die Tg or by aniiedi~ig the helical 7 f o m abore 181 1
OC. From melt cryscillization, puce a and P or a mixture of both c m be obrained dependuig on the
coolîng rate. For rapid cooling from the meit, the a form is obained whiie for low cooling mes or
i s o t h e d crysdlization, die crysnlline form which is obnined (a, P or mised) depends on die
sarting m~tenai. If the starting material is in the P forrn, P €om crysrds are iilways obrained. If die
srutbig mnterial is in the a or y f o n the produced material is a or j3 drpendiiig on die mi~~ imum
temperature reached in the melr -and on the residence tirne at tiiat temperature. This cm br
esplÿined by the fact that the a f o m crystals c m eshibit a memory effect, wliicti promores self-
nuclention during melt crysdlir~tion. Wheti diis memory e&ct is absent or delered ;it Iiigh
rempenmres, the octhorhornbic P f o m is obrined. The J3 form c m dso be made frorn soluririii
casting at high rempentures benveen 130-170°C. hlost of the transfom;itions discussed 1i:ire liren
from die a form to the p f o m suggesting rhnt the P t o m is therrnodyimiadly hroured. C ie rg
confnrmation andysis have Lidicated that uideed die P f o m is lower in energy than die a
hlore iirerestuigiy, foomiatioii of the Iielicd il pliase cm oiily l x obtairied in die preserice OC ;L
solvenr or esposure to solvent vapour. This process is hiown ÿs solreiit induced crysrai1iz:itioii
(SISC) wliicli aids cqsdizarion due to tlie eiihmcemeiit of segment mobility as a result of the
presence of the solvent. In essence, diffusion is enhmiced Li? a redisuiburioii of the free volume tr)
allow large-scaie structud re:umigemerits and penemtioii of solreiit molecules. During tlie
crysnllization process, the solvenr molecules ;ire espelied from the cryst;dlized zones.' Tco nid rliis
SINC process, polymer solvenr interactions c m dso occur nid the degree of crystd1iz;itioii is
dependent oii die nature of the solvent. There lias beeti much debate as to the iiÿture of these
polymer solrenr interactions and there is evidence for the Fom~t ion of solvent-rnu1rcui;ir
çompounds with a-xious solvents sucli as cldoroform, decalin, o-yieiie. toluesie, berizriie :uid
cfilorobenzenes.'.8,9-10 m k e s tlie solution behaviour of sPS quitc cornpies and ;it rrmn
remperxture, sPS is iior soluble in good solvents çap:il)le of dissolvins ar;ictic prilystyeiie. Oiily upoii


he~ring w d th= polyner dissolve ;tnd this results in therrnoreversible gels. These tlirrmorewrsit~lt.
sels have been observed for many soivents and cm be esplained by the theos. that rhe solvent
moledes xts as helicd stabilizers, preventing the c h a h from faldirig ;uid this resulrs in enhim~ed
chain rigidity of the swoiieii polymer." This hel id snbdintion has beeii esphined bu the
entnpment of solvent molecules within die helical ch.hs. The position of the phenyl groups causes
the creation of cavities that cari house the solveiit molecules. C-scd structures have been reponed
bu Charani" and Guenet'? for toluene ;wid benzeiie sPS moIecular complexes and represeriutive
structures of their possible entrapment -are sliown in Fkqres 1-23 ;uid 1.2.4. These solveiir
moledes -are bound in hvo differerit ways, loosely bound arid tiglitly bouiid. For berizerie it Ii;is
been reported that 4 molecules of benzene per sryrene monomer c;ui be tcipped. Tliret. of dit.
moledes have been found ro be loosely bound and 1 tightiy bound. Chly at die boiliiis point of the
solvent is die tightly bound solvent molecule releied. This has beeri determined bu the preserictt of
nvo solvent rciporation p e k duting DSC scans of medium concenrr~tioii sPS/benzeiic gels ilii-
4(.)0h).E Due to die diffidty of rernoving the tolueiie molecules, C1i;it;uii hypodiesized tlic
eiimpment of toluene moIecuIes. Jliis eritrxpmelit w;is fouiid dso in ;i 4: 1 r;itio vid the w&gfit Ioss
by t h e m ~ ~ ~ v i m e t r i c analysis was 14.1%. Cpori removal of the solveiir, it w;ü: huiid rlint die 6 forni
is convetted to die y form.
I t is important to discuss each of these forrns because it h;is beeii sliowii rhat tliesc
structures di eshibit diffèrent morphologies, wliich as a consequerice affects die mecii:uiic;ii
properties of die polymer. Based on the he l id coiifomiatio1is of sPS, porentiiil ;ipplicatiotis of sPS
being used i i s inciusion compouiids (clarhrates) have been iiivestigi~ted. Typicdy these cl;~dir,ites
rnainly involve zeolites but helical stmcmr;il polymers like sPS, tliat c m include @est moledes, ;ire
being considered for applicatioris for cliernicai sep;il..~tiori, purifictrioii of p e s %id liquids ;uid
câtaiysis.lJ

Figure 1.2.3:U Structural represenation of sPS-toluene m .otecular complex
&k
Figure 1.2.4:u Representation of the eouapment of benze~e molecules within sPS molecules.

1.1.2.1 Cataiysts and Mechanisms
In the earlv 1980's, I;aminshyy's discoveq of homogeneous carÿlytic systerns of rne~docerir
ÿnd m e t h ~ u m i n o x m e (hL\O) capable of producing highly linearl5 :uid stereoregul:u16 polyoleiuis,
represented an imporrmt break-through in dkene polyrnerization. Since then, mera.llocenrs Ii;w
become the hottest area Li catalyst chemistry, being considered the most v e r s d e ~iu~siriot i merd
cadysts for the stereospecific polyrnerization of 0lefuis.1~ These catdysts are capable of prepiirriig
polymen widi ntionally nilored properties by dieir ability ro control molecular weight, r~cuciy :uid
melriiig point. Metÿllocenes have beeii showii to prepare IUiear low density polyediylene, ethyfeiie
propylene diene monomer rubber, isotactic and syidioc~cuc polypropylene, syndionctic polystytttic
and polycycloolefins. These are in tact new miiteriiils utilizing inespensive commodiy moiiomcrs
diat ;LI ready have es tablis lied teclinoiogies for processiiig.18
In 1983, Ishihan. et d. tvere the first ro synthesize syndi«r~ctic polystyrerie by iitiliziiig ;i
homogeneous orgmornenllic catdytic sys tem based on tirmium cornpouiids ;ind .Ll.\0.3 .\[os t
catdyst systerns such as Zieder-Natta (2--T) catdysts bi die past have beeri Iieterogeneous. I t i ordcr
to obtain uriifom activity and particle size witli 2 -N c;italysts, the c;it-dysts are ofteii placed oti solid
supports such üs MgC12. The most effective citalyss for syidiospecific styreiie polymtirizatioiis t1i;it
have beeri repolred üre bsed on ritaniurn.' Other group IV mecds such as zirconium ;md hatiiium
have been shown to cariilyze sPS but in cornparison with tiraiiiurn compourids the); show lowr
accivity and Iower ~tereoregular i ty .~~3~ Metallocenes originaily were based on die structure of
CpLSM2 wliere Cp = qclopenedienyl liguid, S = mecd ceiitcr ;uid 11 = ligand, rmgirig h m
haiogens to dl+ groups. =Uthougii the terrn metdlocene is now used loosely for catalysts widi
organometallic nature, for titanium cornpleses, titanocenes refer tci cornpleses widi hvo Cp iigiiids
(CP:TL\[~) and half titanocenes to cornpleses with one Cp liguid (CpTiLh). 1 t is these 1i:ilf
titanocenes widi one qclopenndienyl ligand that yield the highesr :ictirity hr sPS. -4ldiougli the

merhod of conuolling mcricity is nor well undecstood. the basic pruiciplr is d ix die irisenio~i
orientation of the monomer group is conuolled by the constrained geomeq of the bulky ligands.
The topic of cadvst design and the substitution of various ligands is quire extensive. For
purposes of diis review, only the cataiyst used u i this smdy qj-penr~mediyl~lope~itadienyltit~iiurn
uimerhvl (CpTiLled wili be discussed in denil. r\s mentioned, tirmium merdlocene compleses \ d i
one q-clopeiindienyl ligand yield the highest activity for sPS. \Vieri die substinienn on die
cyclopentadienyl ligmd are electron donor groups, higher polymerizatiori activities have been fouiid.=
Rj-Cp Iigmds sudi *as Cp' widi 5 methyl groups, provide an iiicreased electroii derisity ori tlie
tirani- tiius snbilizing the active species. This increased electroii deiisity or steric hindraiçc
around the active species dso cause the polymeritatioiis ro be more stereospecitic. \Yirliour die R=-
Cp ligand, the moledar weights produced by Cp-met.illoceiie polymerizarions terid to be lo-.vrr ;uid
when polymerizuig over a rmge of remperanires, a greater elfect on the xctivity c m lx olisen-çd.
This h;is been cxplÿuied by the Rs-Cp ligand's bener ability ro sribilize the active ceiiter ÿnd retard P-
h~dro~gen el~mination.'~
For die systern to be active, n c o c d y s t is required. LIost systems h:we beeii developed wirh
.\l-\O cocatdvsr but more recentiy, novel merhydurninosaiie free cocmlysn such as H(C6Fi)i
@orme) have been shown to be effective."- Tlir cocatalyst is 1iecess:iry tOr the activatioii of rlie
caralyst cornples. ;Uthough die nature and Fornition of the true active species are nor yer h l l y
elucidamd, it k genenlly accepted thar liomogeneous cardysts b;ised on group IV.-\ medloceiics
consisr of catiotiic compleses. For die hLIO sysrem, the following mive species formation Ii:u beeii
suggested L i equations 1.1 - 1 .3:z5
where S = halogens or dli7.1 ligands

. \ L i 0 connvls mounts of free uimetliyldumtnurn mLi) wliicli k important u i the dkylylïtioii (if
the cataivst. The timiurn cornplex is initidy in a T i09 osidation srite a i d reduces to a Ti(iIl) luid
rernains in this state *as m active species. This mechanism has been estetisively studied and ESR
spectroscopy kas demonsuated that this mechÿnism is highly pl:cu~iLile.~~8zJ~~ It sliould br
mentioned thar the rnethyl radicals fomed c m dso initiate the ndicd polymerization of styrene CO
atactic p~lymer.-~ Due to the low value of the equilibriurn consratit Üi equatiotis 12-13, the use of il
I;uge escess of MI0 is often required and it has been found espehnennlly thit large r~tios of A/Ti
are required for high activity.
For the borme, tt has been discovered that the active comples begiiis witli metliul
abstr~ction from the catalyst .as sliown below in equütiori 1.4:=.'
CpTiMe3 + B(GF;), + [CpTiMez]+ P(GF,),Me]- (1-4)
In this case, die active species cm Lie formed ordy using ÿn d h i l tsmium cornpouiid. The rxio o f
the cadyst to cocatdyst is 1:l md no escess is required ,as cornparcd to die hl.\( 1 susrem. T'lie
active species for this system is dso sdl in debate. ESR meiisuremerits, 1i;ive demcinsrnited rh;it rlit.
active species is still a Ti(I1I) cornples but from die above comples fornation a Ti(l\) systern sliould
be preseiit."J I t lias been suggested by Zamhelli d u t in the presrtice [if styreiie or solveiir, dic
Ti(l\') cationic comples c m stepwise decompose, passibly letduig to ;i TiflII) comples [CpTi.l[rl-
which is consistent widi the active species of the .ILW systern." Duriiig the comples h m t i o n , rlic
titanium catalyst is stabilized by the uitenction of the monomer or solvetit.
It lias heen gcncrally iccepted diat tlie rnecliaiism of polyrnerizatioii is ai insertion
mechanism is shown in Ficgure 1.3.1 Initiation and propagauon occur together, in die seiise diar once
the [CpWTiMel+ P ( C ~ F S ) ~ ~ [ ~ ] - has been formed, the styene monomer coordin;ites wirh the Ti centcr
and tliis auses cis-openhg of die double bond side (side \vith the wo 1iydrr)gens) ;uid rlieri tlic
methyl group amches ro die P-cxbon. The nest monomer rliar c«rmliwtes t» tlie tiruiium cciiter
dso hÿs cis-opeiiing of its double bond :uid d~en die polymer segneiir anaclics to rlie P-cirbon. -nit

syndionctic contigur~tion uises from the phenyl-plienyl repulsivr inreractiori bçnvçeii die lasr
inserted unit of the growing chah and the incotning rnonomer.
In rnedlocene catalyzed polymerizations, terminatioil ractions arc geriecdly coiisidered
absenr." If the polymerization mecliïnisrn was d y living, molenilÿr weiglit distributions of 1
would be found. This is the not the case, since P-hydro~n m s f e r reactions do esist which limit the
rnolecuiar weight. The existence of these reactions have beexi detected by gas ccliromtognpliy afrrr
queiiching a reaction with methmol.' The preseiice of ethylbenzeiie suggested thar retniriariori ri:i :i
second-. insertion occurs after a P-hydrogen elimiiiation. The preserice of ri-propyl beiizeiie dso
sugges ted a second,q mechanism, 2,l head to nil insertion mech;uiism. The P-h ydrogen eliinixiirioii
scheme is showii in Figure 1 - 3 2 Broidenuig of the mole~.ul;u weight distributions a u i idsu occur
due to different caraiytic cenrers, esistence of diain t r a d e r and fornirion of aPS due to r.idid or
ionic initiation. hIechanisms that wouid ause temiiiiation, c;üi occur by iiiclusion of car.dyst
p,micles within precipitated pol !mer and deactivatioii of the active c;it;tiytic si tes.=.'
h[ainlv we have been discussing the c a t d ~ i c iictivity of the CpTiMes. Tlie B(C:hF5)5 ;dotic.
in the preserice of a countetion (probably a trace imouiic of wter) h:u ; h o beeri sliowri to be ;i good
c;irbocationic initiator for ethyl vinyl ether cmd sryrene.- The bonne itself will slowly prilyrrierrizt.
sryrene to low rnoIecular weight ;iPS. In most of die reported Iite~iture po1ymeriz:itioris u i the
presence of a soIverit, the anctic palymerizatson was rio t sigriificm t. Hawever, w i d ~ aiiy devia tioiis
from a 1:l cataiysr/cocataIyst ratio with additioiiid borme, increases die frxtiori of aPS formed.'
Interestingiy, for the Cp'TiAIes aiid borme system, it lm beeri reported bu Baird et al., rliat a
dud nature to die cadysr esists. The system c m polymerize die siune monorner by wo differciir
rnechanisrns, a carbocatioriic polyrnerizatiori mechmism ririd il Ziegler-Satta mechanism. For
styrene polymerizatioiis irivolving toluerie as a solveiit it w;is showii th;it ;uiy polymeriz;irioti r i f
styrene below O°C resulted in the formation of iio syidiotactic polymer md only iitmic." This
phenornelia t v s iitrribured to the Iow concentrarian of the sPS actlvc cornples iit tfiese tempecitures
.uid conttnuatioii of die po1ymeriz;itioii t ~ y ;i c*xt~oc;itioriic mechiuiisrn.

Figure 13.1:27 Me tal-coordina ted insertion mechanism of s tyrene
Figure 13.29 P-bydrogen elirninaaon reaction in styrene poly~nerizauons

In the developmenr of die sPS RIhI process, it is evideiit di:ir tlie cadysr cliemistry will p l + -
a significant role. Having uisight uito the fomxion of the active species md its polperizÿtloii
mechanisms will help to esplah and understand in canlyric behaviour under differenr conditiuiis.
The observed resulo from some o f the different polymetization coridiùoiis will be presenad in the
nest section.
1.1.2.2 Effect of Pofymerizatioa Conditions on Catalyst Activity and Matehl Properties
Tliere have been mm? studies reported regarding a vÿriety of different cardysr systems m d
as the active species and mech;inism are being elucidated, bener and more eficierit cadysrs are beirig
discovered." For die purpose of this review, climcreris tics of tlie CpTilLe3 / B(C6Fï)s the sys rem
used in tliis study wdl be focussed on but other similar cataivsr s'terns will be discussed. I r must Lie
noted that most of the dari reported are for solution polynerizxioiis iii rolueiie ÿiid tirnt very litde
buik polymerization d a n is a d a b l e . Alrhough die dan is reported :it :i varie? of monomer ;uid
cataiyst concentrations die effect of die polymerization conditions and b~nerxl treiids sliould t ~ e
comp-xible.
Tlie oti@nal discovery of sPS udized AL+(-> ;LS a cocariyst. .\LA(> is curreiitly Lieiiig widely
used but its activiv tends to depend on the .LM0 composition wirli a 1;rrge escess of . \ L i 0 required
for optimum activity." The use o f B(GF~))J requires a 1:l ~ ~ t d ~ ~ t / ~ o ~ i l t d y ~ r ratio. For c~mparisoti.
ChieiiLJ lias reported tlie effect of .il/Ti ratios oii die sPS yield utilizirig die Cp*T~\[e3 cacll~st ;is
shown in Figure 1.4.1. It c m be seeii that a n t io of500:l iU/Ti is required br optimum acrivity I~ur
in geneni die yields are quite Ion- for the reported coiiditions. For the oprimized C~'TL!I~J:L\( )
nrio, the acriMty wïs reported to be 1.22 r 1 0 6 g I>S/rnol(Tï) wliile in die s + m e smdy, 3 R((J,Fi)3
çatal~*zed rwction, had n calculated activity of 3.83 r 106 g PS/mol(iÏ) \vliicli is 3 rimes Iiislier in
actiriry. The reason for the lower hLI0 ncuvi? is due tri the lower efticieiicy «i esrrrictirin of rlic
methide ion. The efficienq of the esuüction eshibits ;ui optimum ;irouiid 500:1 .-U/Ti ratio but
inconsis t enq occurs at Iiidier .il /Ti r~tios. 1 t lias tieen siiLggested rh:it çr>rnples;itiriii of the ;icrire

Figure 1.4.1:a Styrene polyrnerization catalyzed by CpTiMe,/MAO Conditions: [CpTiMc:j = 1.0 r IO--' M, [styrene] = 0.8 hl, 50 t11L tcilucuc
b = 60 uiiii. Tp = 50°C
ions c m occur ,and, due to stetic reasons, d e die catalyst system less active.3
Tlie higli activiq of the CP 'TL\ I~~/B(GF~)~ has beeii reported by odiers and diese d i o r s
have fourid the syndiocictic yield (fraction uisoluble in ,LlEkJ, to be higti, Lietweeri 97-Ocl wt. ",O. Ilic
resdting sPS dso found to be sterically pure by NhCR wliich w;is continned bu ;i higti meItiiig
Zmbelli, recentiy preserited ai in depdi kuietic study of die Cp'Tihk3/B(C6F+ sustem ici
coluene solutiori, in an attempt to eIucidate the nature of die active spe~ies.~' .-1s CO the proposcd
mech;mism of reduction of (CpTiXie2]- to [Cp'TLLleI-, it was found t1i:it iipon the addition o f
syrene, die estent of reduction depended on the concentratiori of moriomer. I t was hund di;ir rhc
concentration dependence on the monomer was first order. .\c prescrit. it is the oiily snidy itir.nl\-ing
die CPTL\I~~/R(G,F~)~ cacdyst svstem under a range of conditions of low +-relie coriceiicr~tioiis.
short polynerization cimes and a& room tcrnpermtre. The effect of styrerie concentrxior~ or1
die polyrnerization is showti u i Figure 1.4.2. -4s espected, incre;isiiig die styreiie coriccnmtinii resulrs
in ;i higher cunversion. For this 5 nun room temperature polymeriz;itiori, weight iivcragc moleculx
weiglits produced were around 2(.)0,(J00 g/mol widi polydispersi tics migirig h m 3 ) - 2.6.

(3p'TLL les / B (G F3)3
O 0.2 O -4 O -6 0 .8 1 1 .7
Sty rene Conceiitration (mol/L)
Figure 1.4.2:31 Effect of styrene concentration on the monomer conversion Coriditions: [CpT&Ie,l = 1.1 a 10-3 hi. T i B = 1: 1, 35 uil. of toiucric,
tp = 300 s, Tp = 25 OC
Tlie eEect o f increasing polymerization rime wÿs dso considered ÿtid die results are sho\tm
Li Figure 1.4.3. .ils0 as espected, increÿsing polymerimtion tirne resul rs in Uicre-ased amversion but i t
O 100 100 300 400 500 600 700
Time (s)
Figure 1.4.3:Jl Effect of increasing polymerization cime on the monomer conversion Coriditioris: [CpTi\le:l = 1.1 x !')-: LM!, TiiB = 1 : 1, [Styrcuel = 1.09 tiiol/L, 35 iiL o f tolucrie,
T, = 35 OC
Campbell et d. at Dow Chernical Co., liave reponed data for i l cirkty of complcscs
results show diat catdvst actiritv cm be miritained h r mm: haurs, dthougli thet rate o f
polymerization does decreatie widi tirnc.

Tlie effect of increasing polperizatio~i temperature uns dso snidied bu Zmtielli I i i i r
unfominately, it was for a, C p T i ( C ) B ( C F ) catdysr sy~tern.~l Tlie gened r ffeçt of
ampenture on the conversion is showi in Figure 1.4.4. .Uthougli diis cadyst system is sli@id>-
differenr thui die CpTLVe, systern, it mi be seeii that die highest acriviq is reported around 70°C
Temperature (C)
Figure 1.4.4:" Effect of increasiag temperature on the monomer conversion Conditious: [C~TI(CH~P~~)~J = 1.1 x IO-! hl . Ti:B = 1:l , [Styrriiel = 1.09 riiol/L. 35 tiL d toliierrr-.
tp = 600 s
This suggesrs a temperature dependence o C the active pal ymerizarion speçies, widi dr~ct i~i t io i i
occurring at Iiiglier rempentures due ro thermal decompos irion of the org..uiornerdlic comples a i d :it
lower temperxures, a lower conceiimtiori of active species due to slower tmisitioii of tlie Ti(l1) rri
Ti(III) species. The effecr of molecdar weight with temperature showed ï drmiitic decrease in
molecuiar \veighrs to 30,UOO g/mol atier 70°C but eshibired no c1i:uigs in the IWD. Sever.d
remperaure but a sligl~dy lower optimum tempennire for Cp systems ;uouiid *OC. presumdily due
CO lack of methyl groups to snbilire die Cp rÜig.s'JOJ= Ir was iilso ol~sen-ed d m tlie nmlecul:ir
weigtirs tend ro drop as the ce~ction temperature is incrc'ased. The effeçt of molecul;ir weighc ;uid
temperature \vas tiirther studied at a varie? of tempecitures for a
(ten-bu~l~clopen~idie~~~l)tir~i~um cÿrdyst \vit11 .LL40.J3 Tl ie resiilts ;it (i°C, ?i°C ;uid 0-5O(:

ÿrourtd U°C. .At higher tempenmres, they found that the stenc purity of the syidiomctic fnctioii
decreases with increasing reaction ternpenmre as contimed by the decrease in melring point. It w-as
also observed that the tempenture dependence of die '/O syidioractiç yield was Cowes ui iiarurr
e s h i b i ~ g a masimum ît -lj°C. The temperature dependence of borh die melting point ÿrid ' 0
syndiotactic yield are shown in Figure 1.45. It w u suggested that at hi& rreÿction temperatures, rlie
srndiospecific acti1.e sires change into ÿspecific sites which causes a deçrease in die syiidiocictic +Id
and lowen the melting point due to an increased -mount of monomer insertion errors. Tlir rise i t i
activity from lower tempentures was amibured to die gredter conceiitcition of syidiotactiç active
Figure 1.4.5:" Variation of syndiotactic yield (0) and melting point (a) of sPS versus poiymetization temperature

1.1.2.3 Nigh Convenioo Sryrene Polymeriza Lions
For most of the studies reported so far, very few have reported the conveniori of styeiir ro
tiigh d u e s Li the dilute solutions used. Low catrlyst and monomer conceiimtions in roluene
solution were used to improve heat tnnsfer for bener temperxure control .md to increÿse [lie
Huidity of the reaction mistuce due to die tremeridous viscosity iiicrme at Iiigfier conversioiis.
However, some artempa have been described. Ishîlim reported for CpTiCI3 and CeTiCl3 /.\LI( 1
sustem, nearly 100 wt. o'. conmnioti, at a styrenr çoncentratioii of 1.7 .L[ and catdyst conceiimitioii
of 4.2 s 104 M, when the reaction wris left for 2 lics.3u Chien dso reported up ru '14% coiiversioii
using a CpTi(0Bu)j -and hLAO.32 ;\t a styrene concentration of 2.9 hI at 6iJ°C mid ï cadysr
concentration of 8.3 s IO-' .LI it was reported die stir bar in die re~crioii ressel ceïsed tliictir~iiiiig
f t e r 5 min. Iiiterestingiy, the highest conversion reported by zunhelli is 107°.'~ in 8 miriurrs for :i
reaction ï t 9i.i°C for Cp.TiCl)/hLi\O system, at a c;miyst cot~centration of 9.1 P 1 0 - ~ .II iuid inrelie
concentr~tiori of 3.3 M ; ~ t 9O0C.=I This 1 U 2 O h m;iy be due to weighuig errors, but ir would be
mteresting to deduce whether it was due to entr~pped residud moiiomrr or solverit. For rlic
Cp-TL\Le3/bome s ys tem, Baird er d., report pol ymerizütion dat;i ï r die Iiighes t catid !*sr md
monorner concentr~tioris.~ The? utilized styreiie coricetitr~tioris of 4.3 ht ;uid c;it;dysr
concentrations of 7.5 s 10-3 ht whicfi is 10 to 1(.i0 fold the -mouiit uscd by odiers. Theu o b s e n d
tliat the reaction mismres solidified withiri seconds vid coiitained 15-20 wt. "'0 trapped moriomer.
Cnder these conditions, the cmde material coritained relatively smdI wnouiits of ;iPS, uid the sPS
had melting points of 27Z0C, with moleculv weights raiging from 100,000 g/mol at 7iPC md
3,üoo,0~1Ci g/mol at ii°C with polydispersities ;irourid 2.3.

î.2 Con ventionai Sryrene Po~vmerizarlun
1.2.1 Thermal and Bulk Polymetization of Styrene
During the d y comrnercidization years of polystyrene, tlie rdre of polymerization of
styrene w;ls fcightenlig. The high rates and the resulting esothermicity delayed some of die eïrly
development because of apparent uncontrollable rmctivity. Irihibitors such as t-butylcatechol werc
necessq to prevent high yield losses during distillation and storage. Tlie Tint commercializîtioti \cis
by a themal bulk polymeL-ization using Dow Chernicd's "Gui" process wliich uiwli-ed tillitig 10-
g d o n cms with monorner and hating the cans at progressively h&er temperatures for seved days
CO rerch 99O6 c o n ~ e r s i o n . ~ ~
The styreiie F d y of monomers is unique because of dieir abiliv to undergo spoimmous
or thermal polvmeriz~tion merely by heating to 100°C or above. Ciider appropriate polymeriz:iririti
conditions, styrene cvi act -as its own initiator. Styrene monomer c m geiier;ite eliougli free r~dicds
during heatitig tliat high conversion and liigli moleculxr weigtit polymer c m be prepared widiour rlie
use of chernical initiators. This mechariism of thermal polymeriz-atiori 11;s beeti proposed ri] proceed
by a dow Diels-.Uder dimerization ceaction, wliich is then able to hm mriiior~dic;lls cip;ible of
in~tiüting the styrene polumerization. The hvo steps -are shown in Figurcs 1-51.
CH,
Fig. 1.5.194 Thermal Initiation of Styrene

The n t e of rhis themial polynetization has beeti showii to follow die relatioiisliip:
Initial rate (WL O/O poiymer/hr) = 10['uj-a170/T(v]
.it lower temperatures below l0iIoC, the rate is quxte slow and has a zero order of ceaction. For
60°C, Russeil has repocted a m e of 0.0070 mol/L/hr in die absence of oqgeri ÿnd inhibitors wliicli
is approximately 0.073 O h (wt/vol.) per hour of anctic polystyreiir.J5 Today, die comrnercid use of
the t h e d polymerization of styrene is quite uncornmon and insteüd, free radical initiaton sucli as
72-azobisisobutyronitnlr (rUB;'I) are often used. The propagation mechaiiism for ch in gniwdi of
polystyrene occurs predoininandy by head to rd addition :uid the iiiirinririg sclirme is slir)wii 111
Figure 1.5.3. The additioii occurs L i a Iiead to rad addtioii because of die seÿter snbility of die
benzylic r ad id over the methylene radical.J4
Figure 15.2:3' Chain Growth Propagation of Styrene
Tvpicall y, the bulk pol ynerization of styreiie hivolves die process of formirig pol ystyeiic
from pure, uiidiluted monomer. It usually involves the processiiig of ven viscous tluids brç:iusc
polystyrene is soluble in its own monomer forming :i single continuous liquid pliase. Oiice ti1gti
molecular weiglit polymer is fomed, viscosity iiicreases up to lu4 or more c m occur. In iiidusrcy,
solution poly-nerizarions are often used by the addition of j-lj?r solvent as ;i processirg üid t o
reduce die riscosin ÿnd to provide chah uyiskr agents. .Utliough the use of large volumes of
solvent mi- seem attractive to :moid high viscosities ÿnd ru colirrd the tempenmre, ve? téw
successtiil processes have beeii developed in diis manner. hi interesriiig feiture of biilk
po1yneriz:ition is the Iiigh esotherrnicity. The typic;d lieat of polymerizatioii of styeiie is 70 kJ/mol
aid a cd~uhtioii of the adabatic tempemure cise results in ;i t1ieoretiç:il temperdnire rise o f 3 3 i0(:.3f1
Durttipj pn~cessiiig, 1n;idequ:ire hcit remov:d resuits in :m sccelecircd rc;icrioii ;uid iiiçrmsc III

temperature. This large generation of heat, coupled with the lower themd difisivit). of the rwctirig
miunire, often leads to t h e d runaway. This rnakes the process very diffi~ulr tu control since the
rise in temperimre lowers the degree of po[ymecïzation. This causes the molecuiar weigiir
distribution to broaden during the course of a reacuon which l ads to a deterior~tion of the
mechmical propertïes of die polymer. I t is undesirable to have thermal niriaway since the rmctiori
misture usudly approaches a ceiling temperdture where monomers vid polymers are in equihbrium.
For styrene die calculated c e h g temper-ature is 310°C." The polynen recovered after thermd
niri;nvay have low moIecular weight ;uid are of linte comrnercid value. For practicd resons, m;uiy
reactors have autorefrigeration systems rhat limit the redctioir tempeciture below the boilirig poitir of
the monomer (for styrenc 14S°C).
1.2.2 Diffusion Controiied Phenornena
-\s ;i continu;ition of the previous discussion, the lknitirig fe:itures of bu1 k pol~meriz;iriclii
gves rise to the serious problem -ssociated wttti high viscosity systems rliat quickly becorne diffusiciii
controlled.
Tfie most cornmon cliw~cteristic of buik polymerization 1s die preseiice [if hi& moleci11;ir
weighr polyrner in die conrinuous phase whicli gves rise to l;niri;ir-tlow systerns witli m;iss vid he;ir
transfer limitritioiis. It is well known for the free-r~dicd polynerizatiori of triiiyl moiiomers such ils
me thyl methacrylate and sqretie, thüt c1;issical pol ymerization kinetics do iio t appl y at Iiigli
conversioris.38 In die moderate coiicentration cmge wi th coiiversioiis greater thui Gi PO, die cite r )f
propagation Lg-adually decreases as the reaction proceeds and die concentrations of monomer ;uid
initiator are slowly depleted. However, for systems witli Iiigti moriomer coticent~irioiis, ;ui
acceler~tioii in rate is usually observed. \Chen the polymer coriceiitr,itioii becomes tiigh enou&, dit-
growing polymer chairis wiii become erimgied witli segments o€ orher polymer ch;iiris. .\s ;i resulr,
the mobility of the polymeric mdic;lls decreases ;uid the probability o f them ericouiitering rnoriomcr
decrc;ises. The prop;igatioti rc;ictiori invol\-es the re;iction of ;i 1;irgc r;ulicd ivirli a sm;iiI rnc~iic)n.icr

molecule whose diffusion is nor changed significmdy whereas the rermuiarioii process involves WU
rnacroradicals whose ends have reduced rnobility, because motion of tlieir ceiiten of rnass lias
become restrained. The net result is an effective increase in the m e of polymerization. This
;iutoacceleration phenomenon has been termed the 'gel effect' or Norrish-Trornsdorff effect. Tliis
gel effecr causes a fàsrer tempenmre rise and fasrer initiator decomposition. In addition to this pl
effecr, anorher diffusion conuolled phenomena cm dso occur cdled rhe '$;iss effecr'. This occurs
when the polymerization mixture vitrifies and die propagation step becornes subiecr to difhsioii
conuol. This usudly ocnin below the &as trmsition of die system (misture cif monomtx xid
polyner) ;uid causes poly-nerizations to case before dl the monomer in the system I i ÿ s k e n
consumed.38
Esperimeiital evidence for die gel effect cm be seeii -as a rise in siope in a plot of moriomer
conversion versus time. At low conversion, wiiere classicd kirietics (steady-mte ;ipprc~simarioii)
applies, rhe conversion increases p d d y . .At sorne coiivenion, the mqnitude of which drpeiids on
the monomer and other factors, die polymerizatioii rate bepis to iiicrease ro A mucti tiigher levcl.
resulring in 2 tiigher dope (see Figure 1.6). The iiicrased conversion rare usudly leads to ;i
Figure 1.6:39
500 1 O00 Timc (hrs)
Effect of Diffusion Connolled Termination Bulk Polyneriziition of Styrene ~t 60°C witli .URS (0.09 I[)

temperature cise, which causes a Fer higher coriversion rate d u t dso muses a higher tempwinire
etc.l8
described so hr, there are muy cornplesities in the polymerizatioti of styrene. Klien
de-ding with sPS polymetizations the hctors to consider are the metailocetie cataiyst chernis- vici
charxtersstics, dieand uiitia tion, and di f ision control and the type of cqsnlliiie polmer t'omrd.
hlmv of rhese hctors m y play a role when uivestigating and developinç a MAC process.

1.3 Ri1MProcesshg
1.1 Introduction to RIM
The following is a summan, of some of the mÿin highliglirs :uid kitures of RIh[ processiiig
:ti isutfàied by the extensive compilation wrinen by C. blacosko."
Reaction injection modduig (MM) is a p o l p e r process for the npid production of complrs
plastic purs. The process involves the impuigement of nvo reactïve liquid cornponeiits iusr brOm
rhey are injeçred üito a mould, die shÿpe of the finished part. Durliig inisirig iuid füling tlir rnould
the reacrioii misnire usudy p«lymenzes r~pidly. Once the po1yrneriz;irioii is cornplrre die polymer ts
cooled md the p m cm ofteii be demoulded in less dian one m i i i ~ r e . ~ ~ *
-1 tvpiç:il MM schem~tic is siiov.m ui Figure 1.7. Tlie R I l l prucess is riirirely :L b:ircli
operation. The process involves the delivery of nvo or more liquid rextuirs from sror+r,e raiks.
From diese r:uih, rlie liquids *are purnped at hi* pressure iiito a misbig cliÿmber. n i e Row cirio
brnveeri die nvo s r r e m 1s caretLIIy metered to maiirain die stoichiometric bdmce rif the re:icraiirs.
Line from cornponent
storage tank
F
Dry air
Hydraulic cylinders
I htet tine
Lance metering zylinders
7ecirculation line
I 1
Figure 1.7:1' Schematic of n RIM process

Irnpingernetit in the midiead then ocmrs which causes intensive misuig o f die rextioii srreÿms. Tlir
materid polymerizes as it flows out in to the mould cavity, which typicdly tkes about 5 seconds-
Once the polperization is near completioii or solid enough to withscuid the stresses of demoddirig,
the part is ejected. This demouldlig usually ocmrs beweeii 0.5 - 4 min depending on the s!-stem.
The part then undergoes fiiiishhg processes to flash off the volatiles and postcure the mrerid.
jlfter the pan is dned, it is cleaned *md then ~ainted.~O~
One of the main advanmges of MM is chat it k not a veq eriergy intensive process. The kev
to MM processing is the activation of the reactioti by impüigemeiit mising of low viscosi~ liquids-
This use of low viscosity liquids avoids the high remper-tures rind pressures required by convcritiuiid
themioplastic injection moulding 0. In TIN higti temper-tures ;ire required to iriiecr the
viscous molten polymer into moulds usirig pressures of 100(.1 bar and 31iOO ton c1;unping hrces. For
RL\L, the mould tempemures are usually benvreii 50-80°C, die siunr temperature ;ü die st;irririg
rcactmts, and pressures for irnpingement mising are only 10r 1 bar and the sliimpiiig forces oiil!- 3 1
tons. .\fter the rnisuig, pressures of less tliaii 1iJ bar are required to fil1 tiie niould sirice die rniitcd 1s
still ~ O W in viscosity. These low pressures dlow smdler mould cI;imps to Lie used. Tliis 1e;ids to Icss
espensive tooling and opention especiall y when produciiig large p;i~-ts.~'~
One of die major disadvanrages of RIA[ is that longer cyclirig cimes ;ire required wlittii
compüred to Tihf. Iri TIM, parts c m ofreri be demciulded in 30 seconds ;uid rio specd materid
h;indling is required. Besides the toxicity ofsome of RIM reageiits, diere are difficulties in seding rlw
svstems *and in some cases, the atmosphere musc be kept free of oq-geii and water. -hotlier
disadwitage Lc that specid mmould relme agents are required suice the re;ictivc materid sometirncs
adlieres to the merdic mouIds?0c
Xinetv five percent of die m;iterids processed by Rlhb are typicdly polyuret1i;uies \vlitle
others include pol yes rets, eposies, tiylotis iuid dic).cIope~it.,idieties. 1 los t r) f the polyurcrli;uic
mateciais ;ire e1;iscomeric and structurd foams whicli Iii~ve hund tIieir bigest ripplicacioii in dic.
.lutoinorive iridustry for bumpers ;uid h~cia.~M Poly~rerli;u~e cliemisrq- i~ivolves the cr)iidcns;itioii

reacuon of diols ÿnd diisocyutes. The chemisq is quite suir.ible fiir RI.11 silice die kinrrics :ire
npid -and nead? 100°h convenion c m be xhieved. The focmularimis of polyurediÿiie are quite
versaule in that crosslinken, oligomen ÿiid cliYn estenden c m be added to rdor the proprrtirs of
die polymer. To hrther irnpmve the dimensioiid snbility and mech;uiic:il properties of the pms.
the option of adding minenl fiers or giass fibers into the reactmt feedstreams is used, ladbig to ii
process called reinforced MM ~ i L L ) . 4 0 e
.is meiitioned above, the key to a Rihl process is the impiiigemeiir mising of die reictivc
Iiquids. ;Utiioub,$ there ;ire mi. mishe-ad designs, it hÿs been fouiid tliat n 'T' miser, as sli«wii iii
die Wh[ schematic, is efficient-JOf To mesure the effectiveness of the impirigemerit misirig iui
rstim;ition of the Reynolds number (Re) is often used. The Re is a dirneiisionless vdue o f rlir ~ i r i o oI
the inertial forces to the viscous forces which include die effects [if dow rxe (0, derisity (p).
viscosity(q) -and cliÿmber diamerer (d) -as sliown iii tlie equation below:
For polyurethÿiie cliemistry, Ahcosko lias fouiid thnt die vdues of' Re should br grexter dim 300 i i i
order to gve good impingemerit misirig for a R N pr~cess.-'~g
.Liother factor dependent on good misiiig is tlie masimum adiabiiric remperxurr nse. In
gened, the €!aster the mising, the higher the peïk temperature wliicli :ho correspoiids to a ge;iter
Reynolds numbrr. The irnpomice of rnislig is to reduce the s triatioii rhickiess. S triarion rliichies';
is i rnpomt, süice the lmellar mode1 of rnisiiig geiientes a disrribution of thichesses. Tiiiii
sui;itions ;dlow monomer to difiise m d react to t o m polyrner wliile 1;irger striations cause
stoichiometric imbdances, which d o w monomer to becorne tnpped benveen tlie polyrner 1;iyers.
This is usudy chnrxterized by slower misirig rates and leads to a lower peak tempecintre. Brner
mising, leads to Iiigher molecdar weights ÿrid ;i higher tempermm rise wliicli increase die re:içtioii
m e to drive the reaction to complete c o n ~ e r s i o r i . ~ ~ ~ ~

2 RIM Processing Requirements
-4s rnentioned previously, most of the RIhl producn have Lieen bÿsed on polyuredivie
chemisv. This is a condensation polyrnerizatiori method uid is vsrly differenr h m dic
coordination polymerization method of syndiotactic polystyrene since iio c ~ d y s r is required md rlie
monomer is consumed irnmediately upon miwing. However, Li 1983 Hercules iriuoduced die iirst
polymer which \vas invented pÿmcui~xlirly for RIiLL.dhC Polydiqclopeiir~diene (PDCP) is ;i
crosslinlied polymer which forms by a menthesis ractioii of the iiorbomeiie ring of the
di-lopentadiene (DCP). This resuls ui a p o i ~ e r with hidi modulus aid hi& impact strengh.
t\h:it is interesting iibout this rextioii is that i r utiIizes î coordination npe of catdyst. h v c ~ p ~ r t
RI.\[ system hns been developed, in which a tuiigsten chloride c~tÿlpt dissolved in DCP is miscd
with a diettiylduninum chloride cocardyst in î 1:l r~tio. The reactioti of tliese nvn liquids is
estrcmely ripid ;ind esotiiemüc, to die point diat inhibitors such as di-si-butyl etiier are d d e d r o
dela! the torrnation of the catalytic comples. The cidiab& temperature rise for DCP is above 20O
OC. For die successfiii, DCP RIhI fomul;ition, ii few additions 1i;ive Liecn miide. The ti,miul;ittoii is
shown ui Table 1.iJ. Tlie catdysts are sensitive and must be protected frcim risygeri a id wter . l l ic
94 wt. O/o
5 wt. ",'a
0.67 mol O'O
1.0 mol O/O
DCP 94 wt. O "
h t o n 1103 S \m. O %
\Y/'CIa/pheiiol 0.1 mol "'O
toluene 1.5 \m. O,'O
benzorii trile o. 11 mol O ; ' ~
Table 1.0:w DCP RIM Formulation
reacrants ;ire kept at 3 5 O C . h t o n 1102 a syrerie-buudierie-screne uitilock copolymer ts ;tiso
dissolved in the reactants, not otilp to imprcnve impact resisr~ice but ro incresse die rcacr;uir
viscostty. This increase in reacrant vkcosit)r reduces the air entmpmeiir duriiig filling ;uid the ;unount
of t1;isiiing th;ir ocairs. The orher companents suc11 as die tr~luetie. pliencil ;uid I~ciizonitrilc ;ire

required to help solubilize the \%'CIo cadyst ÿiid ro preverit prepolyrneriziriori. For tlie most p m ,
there -are man. sda r i t i e s ui cornparison with the mentioned syitliesis mrthod of syndioaçric
po l~yrene . -4ithough PDCP differs €rom sPS in that it is non-crysrdluie -and crosslinked, i r dors
have a hi& Tg of llo°C. It is also prep-xed by coordination polyrnerization, which is simpler diai :i
urediane polymecization. Msing should be easier because of the cliatri polymerization mechmism
chat does noc require perfect stoichiometric Liiilan~e.~Ol
In the development o f ï non-urethane sPS MM process, hl:icosko40* lias outliiied some of
rhe requiremena for ï material to be successhi of which some ÿre listed beluw:
Reacmts must be stable for weeks at room tempemture
Condition of m;ichuie should operare ï t less diaii 70°C or 15U°C for Iiigli ternpccinire
machines
hlising of nvo ceactive components but ;i diird ma- be added
Low viscosiy to dlow good irnpirigemeiit rnising
hIust have ai increase in riscosity after inisirig but dlow tirne for mould fiiliris, s o as ro
preverit bubble entnprnerit
Khile ~ f i n g , a low mould rempermm should Lie uscd wirli ri« polymer deq.idxioii biir
compensation for shrinkage should be allowed
Demouldirig rimes sliould be less tlim 3 minutes or 45 seconds for higti production
95% conversion should be achieved vid have sufficient green streriL@i for demouldiiig
Little or no flashing of the volatiles
n i e post cure should Lie miiiimized and the product sliould 11e pïirit~ble
Csirig the basic fuiidamentais of RIXl and noting die corisider~tïoiis outiiried bu .\ Iiici~sko.
the development o f a RIA[ process for syndiotactic polystyetie seems feÿsitile. Givrn the simi1:irirics
in tlie DCP polvmerization, the adaptation to a merdlocerie RIAI process sliould be possible :ü \vil1
lie esplairied in the followiiig section.

1.33 Previous studies of RIM Processing of sPS
Observations of the mpid kinetics of a sPS polymerizatioii, gwe rise to die tdea -id
possibility of developuig an sPS RI&[ process. The npid production of engiieering parts hariris the
liigh crystailine me1 ting point of sPS is seen s having great potentid and cornmerciai d u e . l [ÿny of
the elements of a RI&[ process, such as iow viscosity monorner, nvo reactant s t r e m , npid chernical
h e t i c s and quick soliditication are preseiit and the adaptatioti seerns fiisible. The motivltioii
behind the worh is CO produce high value, etigineering thermopl;istic p u t s from a cnmrnodin-
monorner, s tyrene. Baker et al. pioneered the sPS FüM process in 1994, discoveritig somc
shortcomings but dso considenble promise, meeting m i y of die requirerneiits h r ;L RIXL p r o c t t s ~ . ~ ~
The initid investigations into the sPS RIhI process b e p utilizirig a bencli scde R N device.
The ripp;iratus was senip under inert aunosphere conditions ;uid d l materid hriridlirig w u ins~dt. ;i
$ovebox. .\ CpTiiiIe3 c;ztaiyst vid B((>F+ cocataiyst sysrem w;ti used ;uid the niaterd w;is
dissolved either in sryrene/toluene or styreiie/styretie. The solutiotis were irnpiriged in LI miskici
and the styrene polymetized r~pidly to t o m crystdlirie sPS. It w;is reported diat moiiomer
conversions varied from 72 to 83°:'o. These coiiversions were well lielow die requiremciit of '15" 4,
conversion as outlined by Maco~ko.'~k h h i y uivestigirtioiis were irito iticrefiiritr, the
conversion. -\t first, they believed die crystailinity to be die limitiiig Eictnr with monomer being
mpped widiiii die crystdline chaim. Copolymetizatioiis with 3(4) mediyl-styrerie were made uid r i o
significant inprovernent in the conversion occurred. However, polymeriziiig 3(4)-rnethyl styeric
done did result in a Iiigher conversion of 93Vo but resultcd in the Ioss of o~yt;ilIiiiity. Sest, dic
effecr of c;itd ys t coricentratioris was uivestig~ted in die i~icreasirig cuige of 1 :57( 1 to 1 :3 c;italvst to
monomer ratios (mol/mol) vid it wu tound, rit best, that 8 9 O , / 0 convcrsiori could be acliieved. ,-\ few
novel techniques were dso attempted, to iiicretise the ccin\*ersioii by impartiiig energ to the rt.;icttiig
s y ~ r e r n . ~ ~ HF subrnergmg the mould in ;ui ultrasotiic water bxth ;ui iricre;ise in airit.ersiori rn '1 l"e w:is
tolind. .As a p s t curirig technique, bomtxirdment by neutron r;idiatioii W;LS artcmpred tr) polymcrixc.

the residuai monomer. It wris found rhat 93% conversion wis acllieved lifter 124 1 min of Ïrr-diatioii
time. A more f a i b l e process investig~ted was themai posr mring. By seding the plug UI rpo?
m d placing them in a vacuum oven at temperatures of 180°C and above, it \vas found, ;ir besr, 5'' 1 1
residual monomer was left for the 10 min trials but afier 5 lirs, 1.5"'~ residual monomer remairied
indic;iting that significuit anctic polymerization had ocnirred.
h t l y to deal with the issue of sPS bcirdeness, &ton Cl652 mbber succrsshlly
dissolved in the monomer catdyst streams in the range of 10 ro ?O wr. S'O ;uid polymerized r o hm
solid plup. I t wvs fouiid rhat o d v the 10-15 wt. ?O results were promising siiice gmss pliase
sepanrion occurred ar higher weight percenrages of b t o n . From SE.\[, ir was observrd diït g m d
dispersion wm achieved with ÿn werdge size of die dispersed p1i:ise beitis 0.15 p.43
It \vas concluded the sPS RIM process bad corisidenble poreiitid. However, die! did iiorc
that the maior problems of iiicomplete coiivenioti of the monomer aid die st~bility of the c:irdysr
would have to be irwestigated h d i e r .

1.4 Aims of tltu's Sm*
In the punuit of achieving higher convenions in the sPS Rihl process, it was fel t that ceniiii
hndamentai questions should be investigated. A smdy of the effect of rextion parmeters sucli ;s
rime, temperature aiid the estent of mising on die conmrsioii and dso on die sPS material properties
wouid give insight into the nature of die ceacting system.
To obnin :i bener understmding inro die nature of the coiiversioii limicition of the RIAI SI'S
pcilymerizations, the following objectives were outliiied:
ro derermine if die styene rnooomer coiivenion of the RIAI sPS polymecizatioii a m bc
dtered by the estent of misiiig,
ro determine if the monomer conversion of the RIhI sPS polymeriz;itioii am be iricre~sed II-
longer reaction timcs,
ro smdy the effect of the mould wdl tempenture on the moiiomer cotivenioii of tlic RI.\[
sPS polymerization
to de termine die effect of die above re~ctioii p'xameters on the polymer properties sucii ;is
the sPS fr~ction, melcing point, ucticity ;uid molecular wei&t,
to discuss die rianire of die cotiversioii limitation in ternis of eidier beirig difhsicjli Iimircd.
tempemture limited or intluenced by ;inorher factor such as die cryrdliiie iiature of sPS.

Chapter 2
Experimental

2.1 Ma tenkls
Spirene monomer ( 9 9 % ~ ~ ,LW IN, adrich) uihibited wirh 10-15 ppm Ctert-butylc~tecliol
was dned over calcium hydnde (<>50:0, r\ldrich) ;ind disded under reduced pressure tu remove dit.
inhibitor. The styrene was kept dry over activated M m o l e d u sieres (BDH) under a blaiiket d
nitrogen. To prevent aiiy themial polymerization, the monomer mas retiiger~ted ;uid stored ui die
dar k.
Polperizatioiis were carried out under nitrogeii (Liquid (:;trlioiiic, prepurtfied) rhat w:is
h d e r dried by püssing tiirou@i ;i columi of dry 4-A rnoleculilr sieves.
- R e starting materials for the canlyst syritheses were purchÿsed from .ildricli a id wwre used
without hrdier puritication. CpbTiC13 precunor w a s obniiied h m n stock supply, preriously
svndirsized bv Dr. II--\. Koeslag (lab of ILC. Baird, Queen's Chemistry Dept.) usiiiç liter.inire
procedures. .ifter the sviitlieses, the catdysts were stored nid refrlger~red iriside :i \-miiirn
.\trnospheres giovebos.
hlethyl edyl ketone (hlEKJ +and methmol used in die puritic;itioris were re;igerir g-cide.
q5-Penrmediylylopenndien$ tiniiiumrrimethyl (CpBTi.\ le,) c i td ys t :uid
tris (pe~itiifluorop heryl) b o m (B(GF5)3) cocatalys t were syithestzed iii-linuse, under puri fied
nitropn, usiiig standard Shlenk line tediriiques, a V;lcuum ..\trnospheres glovebos m d
d n e d / d e o n ~ n a t e d solvents.

2.2.1 Preparaaon of Cp(TiMe3 Catalys t
The prepÿration of CpWTii\.1e, wÿs moditied from that of .\.letin ;uid Ruya45 2 g of Cp'Ti(:l i
precursor (6.91 rnmol) was dissolved in 80 mL of hexanes ÿnd cooled to -U°C (~cetonitrile slusli).
20.73 mL of rnechyiithium (14.81 mm01 of 1.4 mol/L solution L i etlier) t v : ~ ïdded dropwise ovrr :i
period of 30 miri to the above sucred solution . The resulaiig suspeiisicin \vas duk green in colour.
.\fier 3 Iirs die slush bath w;is removed m d sùrrüig \vas continued. .\fier aiother 7 Iirs the solutiori
wxs tïdtered over Celite and evaporared to dryness urider vacuum. The producr cryscals were d;uk
green in colour ruid the yield was approsim;itely 75% (1.18 g). 'H ShCR iridicared iI6.GU'~ puri? l ~ y
inteption *and comparison with the neigliboruig p e i . The cacalyst producr wils used widiout
h d i e r purification and the same batch wüs used t'or dl the po1ymeriz:itÏciris.
IF1 NlCR (ppm in GD6), 1.74 (sr 15 H, Cp'), 0.99 (s, ?Fi, Ti-&le)
2.2.2 Preparation of B(GFj)3 Cocatalyst
The prepamtion of S(C>Fj)3 \V;S moditied froni diat o f .LI:tisey ;uid P;irk.46 10-0 g of
bromopentafluorobenzeiie (5.04 mi,, 40.4 m m l ) w;is dissolved in 350 mL 1ies;iiies. The soluriciii
tvas theri freeze-&;LW degxssed arid cooied to - 7 8 T (isoprc~pariol slusfi) . \K hile s tirriiig the ;ib( n-c
solution, 41.5 mm01 of buslithium (35.3 mL of 1.6 mol/L solution iii tiesaiies) wws added dropwisc.
white suspension resulted ;md it was stirred for 2 hours. Nesr, 133 mmol of horai tric1ilc)ridc
(13.3 mL of 1.0 mol /L solution in Iieprane) was ridded dropwise. Tiie reactioii was theri s rirred fijr 1
hr more at -78OC: before rhe slush bath was removed. =\fter stirring 7 hrs hrtlier, wliile dlowirig dit.
solution to w a m to room temperamre die misture w:is Ieft to sertle for I ' L iirs. n i e supcni;iraiit
was tïdtered rlirougti Celite imd then evapor~ted tr, dryiess under m x u m . The producr c-srds tvcrc
brownrsh-white in coIour and a yieId of 47°'o (3.3 g) was obtaiiied. Furtiier piirific~riori \cx ciirricd
out bv sublimation under vacuum ;it 8j°C onto water-crloled cold firiger, n i e resulrliig B(Ct,F5)3
crystds were white i r i colour.

23.1 RIM MMng Apparatus
.UI of the syndiospecific styrene po1yneriz;ltions were carried out widi n miiii RI.\[
apparatus. The RLLl apparatus consisted of a specially consuucred & s s mishr~d a1d ;i
pol yxop ylene Kenics sntic mixer (Chemineer- Kenics) combination (Fi y re 2.1). The knpingemrrir
midie~d had injection porr bore diameters of 1.4 mm ;uid a 0.50 cm c~vity dWmerer (8 mm, sp d l ) .
To mesure die effectiveness of the impingemen t misirig ;ui es timiitioii o Ç rlir Rc yiolds
number (Re) was used. .b mentioned in section 1.3.1, Macosko has outlined fhat a vdue of Re > 3 H i
provides good impingement rnising for a RIM process based on polyurediaiie cl~emistry.~" -;\ simi1:ir
cdda t ion for the sustem, based on 1 ml/s tlowrares, die room ternper.iture deiisity :md viscosi~ o f
srvrene and the iniecrion port diameter resulted in ;i Re of 4 1070 wliich sliouid provide sutticieiir
mising. To enhmce the rnising from the impingrneiit sectioii, a 74 elemeiit ii.5 cm OD Kenics
Impingement n r l
M ixhead
Cocatalys r + +
Styreiie
Static Mixer
Note. =@am not to s c d c Iiito the AIouId
Figure 2.1: RIM Mixhead Schematic

satic miser wÿs dso udized. The Kenics mixer design çorisisad of a series of 18U0 nvisred leti
handed and cight handed elements digned at 90° to complete die rnising by tlow division, tloiv
reversai and radial rni,~i.ng.~'
2.3.2 RIM Technique
.\ typicai polymerization cun was cirried out as follows. . i l1 apparatus materials were d d
ui a vacuum oven at 60°C before use. The appmtus was assembled as diapmmed in Figpre 7.2.
The mi-shead ports were s d e d with mbber sepn (8mm OD, .Udricli) and fitred to the Krtiics st:itic
miser by PE tubing (l/C' I.D., Fisherbrand). Both the rnisheÿd/sr:iric miser iuid the thrmocouplc
were fitted through a rubber septum (20.5 mm, .Udricli) ;md s d e d iiiside tlic test tube mould (25 s
150 mm, Comuig). .ifter seiiiis the tictings with p;irxfdrn Eipe, the i i p p a ~ i t u s wis rvacu:itrd fiir 1
Iir. .ifter evacuatioii, the apparatus atmosphece was back flled widi iiitroseii :uid ;i coiista~it tlo\v
was liept. .hi- escess pressure wvas re1e;lsed dirougii a needle valve looited ar the eiitr.uice to the
mould. To proride a consisteiit mould \;il1 remperxure, the dass mouid KLS submerscd i i i ;i l:irgc
silicone oil biith.
Both die c;itdyst ;uid cocat;ilysc were weighed iuid recrieved from ;i ir;icuum .imosplicrcs
giovebos. 1 1 mg of Cp'TiiMe3 m s weigiied m d seded iii a test nihe witli ii rubber sepn/p:ir~tïdm.
78 mg of B(CbF5), was also weighed ÿnd seded in a test tube wirli ;i rulilier septdpar~filrn dong wirli
a 1/Y" Tetlon magnetic stir bar. n i e srir bar m d stir plate were utilized to promore lierter misiiig o t
the borme :uid styrerie. PE/PP syringes (10 mL, Forruna) were titred with saiiiless steel iierdles
(PT:, Y, Hamilton) and backfdled wirh S. ùi a separate sealed test tube. Prerir>usly distilled styeiic
was removed from tlie refrigentor and ailowed to w r n i to room temperature.
Iri the following order, 2 mL of styreiie was iidded to the Cp'TihIc3 to il~liicvc .i
coiiceritratioii of (i.341 AI. Sest, 3.25 d of styerie was added to tlie stirred test nibe of B((:aF;j)> rc J
achieve a coizceritr~tion of (1.343 LI. T l l~ e s t n ( 1.25 mL of moriomer \v:is ;iddecl to die B(C,,Fjjs ri,
prereiit tlir uptalie of insolul>le st~lids if preseiit. 2 mL of die (:pTi.\lr, solution \v;is t;Lcii up . i i d

then the needle tip was insened into die rïght seded port of the mishrïd. Sest, oidy 3 rnL of rlie
B(C&)J solution was taken up and insened into the left srded port of die mixhad- T h
prep-mtion tirne of the canlyst solutions was esumated to be benvrçn 45-60 seconds. Fidly , borli
syringes were simulrmeously depressed slowly -and metered to keep the tlow and impingemerir
uniform. The materid could be viewed while flowing through die mislied, formuig a blecli
coloured comples m d then pÿssuig through the Kenics miser. In die bottom of the mould, rlie
polymer plug solidified benveen 13-10 seconds and it was estirnired that the injection proçcss
occurred widiin 4 seconds. Due ro the mpid solidificxtion time, ir w;is obsemed tliat up t o n ppni of
the reacnnt material b r c m e trapped in the rnishead, mosdy iii die sr~tic miser. 031 :nVerdge rlie ror:il
time for prepmtion -and complere injection into the mould was ;ipprtxirn~tely 1 mirute.
RIhI Irnpiugeriierit Sectioti
Stirrcd B (C. Fs) 2
Cocdyst Solution
Figure 2.2: RIM Apparatus Setup

2.3 3 Temperature Monitoring
.A Est response 1 / 16" type K tliemocouple supplied by O m e p Eiigiiieeriiig was posi tioiied
precisely in die center of the &ss mould arid elevared slighdy above die botrom. Just prior tu tlir
injection of the reaccuit rnaterials, n dan acquisition compter progra.cn was initiared to record dit.
temperature *and tirne via rn Omega E n g i n e e ~ g DP-41 Temperature b [eter.
2.3.4.1 Polymerization of Styrene by Borane Cocatalys t
Tc) determine die m o u n t of styrene polynerized by the homie c o c l r d y duriiig rlic
premising step of die borane aiid the sryrene, 36 mg of borarie w:o mised widi 3 mL of styreiic
(NO23 59 at room tempenmre. .Uter 45 seconds, 10 mL of :iciditied merhaiiol w;ü: iiiiected to
terminate the pol~er izat ion. Tiie precipinted polymer was then dried in the v:icuum oïeii nid
weighed. To determine the moui i r of styrene polymetized :it a Iiigher rempecimre, rlic sunc
rmction was wried out using sryreiie preheated tbr 10 miii at l(.)O°C.
2.3.4.2 Benchmark Controt Study
Ili order to est-blish a weU defilied bericf;.n-~rk condition ;uid ro test die reproducibili' o t
the reacrion procedure, four polymerizations were carried out ÿs desctibed nbove widi the sniid:irci
cataiyst recipe, room temperature reaccmts and ;i one Iiour reactiori rime. Tlie moriomer çunversioii
wru deterr-nined ;is described in 23-51 and the polymer product c1iür;icterizcd as described in 2.4.
The results of these polymerizatioris were statisticall~ taliulnted as the bencfim;irk ii~Giisr \vliicli ;dl
odier polymerlzatioii coiiditioiis would be compared.
2.3.4.3 Mucing Smdy
Tc> determirie wliedier or iiar die polymerizatioris were misiiig limited, kiur differeiit misirig
ïciiidittons were s tudied. Firstl y, ;i no mlsiiig coiiditiori \GIS memp red in wliich die rc:icr;inrs nwc

uijrçred direçdy Uito the g l s s test tube mould cotinuiing ody the dit.moçouplr. n i e secolid KU i i
no sutic miring condition in which the Kenics misiig elemeiits were removed nid the Liipiiigemeiir
rnishead outiet wu: fitted direcdy Lito the mould. The third ÿnd rnarked as the sniidard rnisiiig
condition was the use of the RIIL[ mi-shead and smtic miser cornbuiatioii as outliried in section 3.3.3.
The I s r condition was the use of a Iarger dimeter rni.uhead. .A polypropflene T-ioiiir from Sdgeiir
with larger bore diameten o f 4 mm on die injection pom uid cavity oudet wxs araclied in place o t
the glass MM misfiead.
23.4.4 Reaction Time Study
To d e t e d e the tirne to complete the reÿction, RIM pol~meriz;lrioiis wcre carrird out \r.itli
reaction cime iritewals of 2 min, 10 min, 1 hour, 6 hours smd 34 hours.
23.4.5 Mould Wail Temperature Study
To determinr the effect of mould rempenture on the RI.\.[ polyrneriz;itioii, mould \ d l
temper~tures above and below r m m temperature were evdu-ated iii ;i r m ~ e Irom -X°C tci 1 li IO( :.
Room temperm.tre polymerizations were cvried our in a silicoiie oil txidi as a I i a t tmisfer rncdiiim.
Runs perfomed at elevated temperatures were rhemosr~ticdly coiitrnlled usirig :i CI:i;Ae D ( 3
Tempenture Conuoller. For these runs, the mould appÿrxus was pre1ie;ired b r ' i Iiour itiside tlic
bath. The ceactants were kept :it room rempenture and nor prelieated, tu prevetir atiy sigiitiç:uir
tliermÿl polymerizauoii of styreiie. Runs perfomed at o°C were çooled usirig a srirred ice-wircr
bath. Bodi the mould and reactvir styrene were cliilled for at l a t 15 minutes prior tr) the mising of
die catalyst solutioiis and injection into die RIM appmrus. Ruris perfomed ;it ccygtiriic
temperatures were cooled using a temciil«roedi;uie/liquid iiitrogii cold b:itii. Roth the mould :uid
reactant screne were dso chilled prior tu die m~sirig of die catdyst solutioiis ;uid irilect~oii ~ i i t r j dit.
RI,\ [ appiintus.

2.3.5.1 Vacuum Oven Drying
To mesure the polyrnerizatioii yield, tiie eritire üpp;ir~nis a id @;fis rnould were wei#tiecl
prior to and üfrer the compleced reaction to determine the -mount of polymer iii the iipp;ir.tus ;uid
giass mould. To temuiiate die reactioti, the polymer plug was powdered i r i a coffee prider. Tlic
tirne elapsed brtween weighuig ÿnd grinding of the plugs w-as appmsirnately one minute. .-Ifter
powdering, a noticeable coIour chuige from d d green/brown to ;i bright orange WJS obsen-ed.
signi5hg die climge in the Ti caraiyst osidatioii state. The sample w;is weigtied oii a +pl;ice
S ~ ~ O U S bdaiice and the residual monomer wris removed by drying in a vacuum overi at Sii°C untd
constant weight v a s acfiieved. It was observed diat die dqiiig rime to ;~cliieve corist;uit \veiglir
ocmrred benveen 3-4 weeks which is unusudly long when compared to the d ~ i r i g of ;ir;ictic
polystyrene smples. The cdcuiated residwd monomer loss was then rakeri ;s ai estim;ite of rlic
ovecd1 moriomer conversion.
2.3 5.2 Thermogravimetric Anabsis (TGA)
Ta veri. die vacuum oven dqirig merhod, the moriomer weight loss bu TG.-\ nhds ;ho
detemüried, using a Metder T-'UOOi:, system with TG30 thermnbdmce. The TG=\ iuidysis W;IS
carried out by Iieauiig the smple Çrom 3U°C to JOO°C at 3 m e nf lfi°C/miii. The mass loss ;ir 311
O C : was used as ;in indication to the mount of residud monomer lost by die sample.

2.1 CharacteI-izarion of PS Polymers Please refer to Figure 2.3 For ;r flow chan of the ch-mcterization procedure used.
2.4.1 Detemination of SPS/aPS Fractions
During the polytnerirition, both a~ictic ÿnd syndiotactic polystyrene -are brmed. To
derecmine the -mount of syndiorictic and anctic polymer produced, the fractions were separited b p
solvent estraction with MEK. A Soshlet estraction apparatus was setup. Ground polymer w;w
weiglied ;uid placed inside an estraction diimble and reflused for 48 Iiours. .ifter esrrxtiun. die
thirnble wvas dried in the vacuum oven at 60uC and the . L U X insotuble polymer weiglied (SIS). Tlic
X E K solubie polmer (.aPS) \vas precipinted by dissolutiori ui 10 times die solution volume of
medianol. The resulrîng polymer wvs chen dried in die vacuum oveii :it 6ii°C and weiglied. Duniig
die esrnctioii process, it was caiculated h r the material losses were arourid 10 '/O. This is belirved
to have occurred duriiig the reprecipintion -and tiltentig process for rlic .\EL soluble polymer.
2.4.2 Product Identification and Tacticity Analysis
The purified aPS and sPS polymer \vas cliÿracterized by ShLR specrroscopy. 1H-SI[R \v:ü
performed on a Bniker .kM-700 spectrometer oper~tiiig at 200.137 I M z while the sPS lJ(:-SIIR
were obrined on a B d e r C\'P-300 miming at 50.306 hLHL . The aPS 13C ShCR spertmm was
me;isured ar room temperature in tolune-da while tlie aPS IH, sPS 1H ;uid 13C ?;.\IR were masurrd
at 393K in 1,122,-tetrachloroethvie-d,. For che hi& temperature spectri, the s-amples wcrc
equilibrited in a he~ter block to d o w swelling and dissolution o f rlie polymer. To differeiiti:itc
benveen die ar~ctic imd die syidionctic polymer, 1H NMR peaks iii the 1-3 ppm regai wcrc
analyzed. Figure 7.4 depica the CH and CHr proton signais for rlie diree types of poly-yreiies.
Since the CH and CH2 protons are split equivalently in the dtenintiris structure o f sPS, ;i tripler 1s
detected ;it 1.4 ppm instead of a broad resorimce or a multiplet ;is in aPS ;uid iPS resprcrivcly. -nit
O O sytidioraxicin of die polystyreiie \t.ifi detemiriied by 13C ShlR where by tlie plieriyl Cl

MEK Insoluble Polymer
Polymer plug powdercd in CO fke grinde r 1
Powdered sarnple is weighed and chen dried
in Vacuum O h e n
sPS hlolecular Wei& t DetemÜnatiori bu
HTGPC
3
Determination of " 'o Syxiiotacticity f-
Deteminarion of Residud Monomer and Conversion
MEK Soluble Polyrner
sPS Identification bu 1H ‘i;LCR, "(3
N &,IR
aPS A [olecular Weigh t De temination 11y
HTGP c:
aPS Ideriti ficatioii by 'H XMR, 'SC
NhDi
Figure 2.3: Flowchan of the characterization of RIM samples

carbon resonmce gdve *an indication to the stereoregulÿnty of die peiinds L i rhr polyrner. Fib~re 2.5
shows the Cl phenyl resonmces and a s h q singiet at 145.2 ppm corresponds ro the rrrr peiicid
configuration while odier pentad combinations are shifred downfield.
Figure 2.4: 1H NMR of methine and melhylene C region of polystyrenes
Chernical Shi fr (ppm )
Figure 2.5: 2 *C NMR of phenyl C repion of polystyrenes
2.43 Determination of Thermai Properties
>leIrkg point, dass tmisition and uidothermd crysraiiit~rioii temperxures were determitietl
for the unpurificd -and dried sPS samples in a Metder T.\3OU(.) Differeiiti;il Scumirig Ciilorimtiter.
Two scaiis were performed. The firsr scm was at a heatuig rxe of I(!OC/miri h m 3c37(ioC tu
ense die themai histocy of the sarnple by the DSC cycle of melting followed by qurrichtii~ wrli
liquid nitrogen. The second scan was at a heating nte of 10°C/miii from 50-34 )()OC and from tiiis
scan, die tempemures were recorded.
2-4.4 Detemination of MoIecular Weight
>hlcmlur weigiirs of the sPS and aBc; materid were meüsured b y IiigIi temper.irurc gcl
pemcatioii chr«m;itopphy (HTGPQ on ;i \Yaters 150-C GPC ;ir l-lj°C iii rriclilorol~eiizeiie ff-(:RI.

Solurions o f 0 . l NT- ''O sPS polymer ui TCB were prepared aiid bared in a 145 OC: silicone oil biidi ro
obnui çomplere visible dissolution of the polymer. This dissolution procedure wÿs necessa? stricr
penerration of the TCB was difimit due to the crysnllinity of the sPS samples. ÿDS polymers u i :i
0.1 W. solution were readily soluble in TCB. .i cdibntion cunre coiistructed ushg TSK TOSOH
polystyrene standards in a range of 9100 g/mol ro 1,0911,OM) g/mol w u used to esrimire rlic
molecular weight and polydispersity index of the sarnple using hkxirna's Baseliiie so fnv'are.
2.4.5 Estimation of sPS Crystallinity and Identification of Crystdline Forms
To estimate the degree of cqsnllinity of the sPS smple, the eiithdpy of meIrin3 \c:ü
meüsured i t i a .\letder TrUOOO Differentid Scuuiirig Cdocimerer. n i e undried polyner siimpie wü
scanned at 10°C/min in n range of 5i1°C to 3(10°C. Tlie melting eiidorherm char occurred miund
?7iJ°C \vas ititegxed to estimate the eritlidpy of rneltiiig of the cryst;dliz:ible ponioii of die smple.
This endidpu cornp:md to die enthÿlpy of hsioii of liHl0'o crystdliiie sPS (AH' 53.2 J/pjtn to
esrinwte die "10 ~ r y d h i i t y of the sample. Since sPS c m i s o d i e d l v crysrilize, it is possible rli:ir
these crysdluiity esrimates are higli, beiiig panid- represetir~tive of die urigirid :imouiit :uid rlic
ctycdlizariori during die DSC heatuig cycle.
To determine die crystalline form produced during the sPS polymerizauori, FI'IR idys i s
ut;ü carried out on an undried polymer sample. For cornparison, a specrrum was ais» obrairied from
n dned m d pun tied smpie. nie specm were obraüied €rom a BO,\.E.\ [ .\LR Scries IR instrumeiir III
FT mode with a resolution of 4 cm-'. The sarnple \vas powdered :mi pressed Lienveeri D r dists
behre scm~iüig benveen 400-4UOO cm-'. To ideritifj- die sPS c~s rd l inc fmn, the sample \\+:fi
mdyzed in die regions benveeii ll(.)O-lXlO cm-', 811(.)-lOOiI cm-' ;uid 400-6j0 cm1 :uid tvcrc
compared to die lirerature specrra shown in Figure 2.6 depictiiig the differeiices betweeii :unorpla~us.
6-helicd, P - z i p g and a-zigzag structural (omis. Specific buids h r identieing the fiirms c n u r ;ir

6 ünd y forms and in the -IUT)-63.0 cm-' regiori, p& at SiiO m d 570 cm-' cle~rly depÎct ;L Iiclid
structure. The y t om spectnim looks very similiir to the 6 t o m escept Los ;i slut't in inrensicies.
shifr in 1-xger intensity for the 975 cm-' when compared to 965 cm-' is indicative of the 7 t o m ;uid
this is shown in Figure 3.8.

1400 1350 lm i 2 M 1200 150 Wa..nr i ikr in cm"
Warmrrnkr h cm"
Figure 2.6: Expanded infrared spectra of sPS crystalline forms (Guerra et al.)l (A) 1100-1400 cm-' (B) 860-940 cm-' (C) 400-650 cm-'

Figure 2.7: FTIR spectra of sPS distinguishing a and P crystalline forms (Guena et al.)'
Wovenurnber in cm-'
Figure 2.8: FTIR spectra of sPS distinguishing 6 and y crystalline forms (Musto et al.)'
4'1

Chapter 3
Results and Discussion

3.0 Results and Discussion
3.1. Estbnathg the Monomer Conversion of BuU- Poi'erized H M Samples
In rn indusuial RL\.[ process, the pam produced ;ire usually demoulded in their fuid use
fom. When dealing with a bulk polymerized RLLf s-ample, difficuly ÿnses in estimÿwig die
conversion of n solid polymer plug since quick termination of the rwcrion ÿrid accurmare remo~id of
the residud monomer is important. Dunng the re~ctioii, it was observed diat the polymeriziiig
rnisture wodd se1 ÿnd solidifv widiin 13-30 secoiids. .-\fter 2 miiiutes. die resultiiig pdyner plug 1 ~ : ~ s
liard and dmost d~ to the touch. Tliis npid solidifie-~tion çmses diffi~ulty in estimihiig dit.
conversion. h[osr methods to estimate the conversion require thar the sample Lie dissolved in
solution. -4s mentioned previousl y, sPS has escellent solmnt resistmçe. sPS will on1 y dissolve ;it
higli temperature in a few selective solvents such i s uichiorobeiizene and tolueiie. Tlie usc of :i
solvenr ÿr high temperature to dissolve the polyner was considered but at the +10i.l0C tempecirures
required. d i e d polymerlzition of die residud sryreiie would occur whicii would rcsulr tii higticr
polymer yields from the additional atactic polymer. ;\nother disadcuit~ge o f usiiig a precipir~tioii
method would be €rom the normal ilmaterial losses iricurred duririg die rniuiiid m;uiipul;itiori ;uid
tilteriiig of tlie smple. If the residual styrene monomer was erduated by a c l i r ~ i m t o ~ ~ : i p l i ~
mediod, the s m e themai polymenzation coiicems would be ~ d i d bur more pmblemïtiç would be
the dissolution of the sPS polymer ui the curreiit chromatopphic solverits in use. TIius rlic
methods of dissolutioii ;uid precipintion and p / l iqu id chromatog,ipliy were nor used ro rstirn;irr
the conversion.
For metdlocene solution polymerizatioiis, die addiriori of acidified meth;uiol will sufficieiitly
Ml dl the active catalytic sites by coordination with the ;icid iuid idcolml goups. 1ietli;riir)I iidditioii
also aids in die precipicition and separation of die polyrner from the escess m«iiomer/solrrnt. For
die i l I I I plug. the use of a queiicliing solreiit w;is deemed uiifeÿsilile silice tlie high miirersioiis I t x i
ni :i solid polymer plus widi ;L 7.5 cm di:merer. Diftiision iuid peiiercitioii o f :L solïriir iiiro rliis solid

plug would be very slow m d result in pooc tetmiiatioii of the reÿctiori. .-\ more feadie medicd oi
termination was by grinding the polymer plug uito powder usirig n converitionii cdfee <ginder.
Cnfominatelr, during this process the shape of die moulded polyrner was lost but by esposing rhe
grearer mount of surface a r a the catalyst was deactivated by the oygen/moisnire nmosphere. This
canlyst deactivation was noted by the change in osidation snte of die residud Ti caralyst u i die
polymer From 3 Ti(l1I) dark green colour to a bright orange. At rhis s q e , the addition of licidifird
methmol was considered but it was felt that any esm sample rnanipul;itioii would wli ;~wiy wmc
of die residual monomer -and hinder the accume weighing of die smple.
To quiclily m d effectively remove the residud styrene monomer, ciiily r i remperimre nbovc
the polyscyrene Tg ar 110 OC would provide die mobility of the polymec c1i;iii.i~ CO -dow dit.
udiindered diffusion of the moriomer fiom the ;morphous regioris. Cnfornrriately, at 1 IOOC, thc
rate of styrene themal polymerizatioii would become signifiant, so iiiste;id, die polymer w;is d d
below die Tg in a vamum oven. .-Uthou$ the Tg t m s unaffected bu the w m u m pressure, rlie
ev;ipor~tioii of the moiiorner vas considembly erihaiiced suice die styene vapour pressure escceds
die vacuum pressure (30 iniig below atmospheric) ;it a temperature of 33.(J°CJ9 To 1xil:uice tlic
effect of die reduced monomer mobility below d-ie Tg m d tiie d iemd po1yneriz;itioti of residiid
styrene, die polymer powder was dried nt BO0C so as to be liigli ericiugli h r d i f i s h i rri occur btir
low enough for the rate of themid polymerizatiori to be low. As meiitioiied 1n section 23-51, rlic
srunples took 3-4 weeks to reacli a consrnt weight. Duruig diis lerigdiy period, it w;is h~urid t l i ~ t
mosr of die residual styrene was removed within the first ~ ~ v o houfi ;uid oi i ly smdl weiglir losscs
resulted aftenvards. If the m e of thermal polymerizatioti w;is sigiificiuit, it would be espected thrit
die coiistmt weight loss would be acliieved more quickly. Looking ;tt the mech;u~ism for styreiic
themil polynerization as outlined in section 1.7.1, it is possible tli;it the preseiice of residd Ti
catdyst prevented die formation of styrerie cadicals by binding to thern -as the? were frmned.
-Uthougii tiie Ti catdyst is preserit in very smdl quantiries, tliis mecli;uiism mil? he sufficieiit ro

prevent low rates of themxd polynecintion. If this were m e , tliis would esplain why :i shortcr
dqing penod for the residual monomer removal was not observed.
-4s mentioned previously, it would have been desinble to obmin a conversion esrimm o f
the enrire polymer sample. Uniomnately, rhis w u not possible since material wÿs often a-pped in
rhe R h [ mising apparatus and lost d u ~ g the gniiding process. It was also iiecessary to sa\*e scimr
of the widried polymer for hriher -mdysis. To estimate die conversion r>f the polymer, "a relative
calculacion" was used. The O4o residual monomer removed dutitig die dqirig of a test portion \vas
considered represenr~tive of the eritire polyner sample produced. The ''0 polymer cernaking atier
die residud moriomer was removed was then takeii -as the conversiori estimate under die mumpttori
diat the sample was Iiomogeneous.
To verle the method of vacuum oven dryiiig, themuFivimetric ;ui;tiysis W-JS carned out r o
determine die weiglit loss of the simple durüig a temperature tise of 1( ,OC/miri. Duriiig rliis he:iriiig
procrss, a monorner evapor~tion peak was detected benveeii 14~-15i)~C: and a polymer deg-~id;irioti
p r k ÿrouiid -IIii°C. Please rehr to .-\ppendis .A €or the TG.\ tliermowm. Tlie ("O siimple wei~lir
remaining after the weight loss sr~bilized (arourid 3(HJ0q was nkeii iis the estimate of die startiiig
m a s of the smple that h;id polymecized. The conversion estim~tes t'rom die vacuum oven dqiiig
-md TG.\ W. loss frorn nvo of the samples are showii u i Table 3.1:
Conversion Estimation ,Lietliod
-- -
TabIe 3.1: Cornparison of Conversion Estimates for Vacuum Oven Drying and TGA Wt. Loss
(Ca: ssOc)
On cornparisoii of the vacuum oveii d+g and die TC;..\ weidit loss From smples .A K. R, the TCi.4
conversion estimites were higher dian the vacuum oven estimaccs. -Udiough it is difficult to ;ir91t.
th;it these results are physiciilly differeiit, die diffemice rnq be ui die measuremeiir irself. In the
TG:\ rneiisuremeiit, milliLgram smple w-eiglits were uscd wliicli m;iy iiot hiive becri rcpreseiitritivc O t
rlie ~4iole s;unple. \Yhile lie:itirig ;ir ;i lU°C/rniii cite, tliemd p ~ l ~ m c ~ i ~ : i t i ~ t i r)f syrciie m:iy ti:ivc
(@; 31 ioOq I
-A (BhI #3) 78.2 9'0 82.1 "/O ---- B (3 min #2) 78.8 ?O #J.I ?'O

ocmrred at the Iiigher temperitures, at a gredter nte tlmi die caraiyric buiding mecliuiism rneiitioiicd
previously. This extra polymecizaaon wouid account for the Iiigtier conversioii estirnates h m rtie
TG-\ measurements. Regardless of the small ciifferences, both mediods contirmed thar arowid 2iIU.
residual monomer was present in the ~ndned smples and thar bodi conversion estimrioii
techniques u e üi general agreement and consistent wirh previous srudie~.~'
3.2.1 Benchmark Reproducibility
One of the important objectives in c q u i g out this study wrs tr) esr~biish 2 well-defiiicd
polyneriz~tion procedure chat would provide reproducible results. In dediiig with die ;Gr-seiisirivc.
organometdlic compounds, reproducibiliry ws ;rl\v~ys a conceni. The ç;it;dytic xtivity of dirse
cornpounds c m vary depending on die ÿmount of impurities iii;idvertetitly ;ilIowed iiito die systern.
These impurities cm be l inhd to a iiwnber of sources, such as isx?geii/moisnire coiiretir in dic
amosplirre, idsorbed moismre on the giÿsswxe and tiiuidliiig aiid t~uisfer techniques of the c ~ i l y s t
miiterids ;ind moiiomer.
Some of the reproducible ch-aracteristics of the rextioii were ol)sened quiiiicirively. Duriris
the initiai sr;iges of die reaction, observations of the npid solidifimticiii ;uid hish remperdrure riscs;
were utdized as criteria for reproducibility. -\ s~milar remark from Baird et ;il., menrioricd tliar
"ri~orous exclusion of moismre \vas i i rcessq for npid aiid reprodiicible polymerizatioii ro hi@
moleculÿr weiglit product to occuri'.i 1 t \s later nohced rhat lower temperdture rises utcf slower
solidification times riften dtered the balance of syndiotactic/at;ictic polymer ciusing a decrezise in rlir
s yndiotactic fmctioii while some times, but iio t dwys , affectirig the overall convers ioti. .-\ t q~ icd
tempemture rise versus tirne curve is showii in Figure 3.1. Ir c;m be seeii tliat ;L 1( 1 - 3 1 secoiid
induction period w s eshibited during the formation of the iictive comples. .iftenvards a cipid
tempecinire rise w i s obsemed widi the peak temperature occurriii~ ;ifter 1 minute. This r.~pid

remperdcure rise, dthough similar to a 'iorrish-Trwnsdorff gel effect is probalil- not dur ro .l
reduction in the tennination rate since coordination polymerizatioiis Iiwe 110 defiitive terminauon
mechiuiisrns unlike free radical polymerizations. Instead this rapid tempenture cise I: artributeci ro
the acceleration of die reaction rJte as the rempenture of the reaction rnisnire incrrrses. Mrer die
peak tempenrure, the reaction temperature dropped npidly back to the initial silicone barh
remperanire after 10 minutes. This tempenture profile was faidy reprt~duçible ui most of the rmm
remperanire polymerizations perfomed.
Figure 3.1: Typical sPS EUM polyrnerization reactioo temperature profile
It was noted in section 3.3.3 that m estra mising srep wis used tci dissolve die brxuic
cocanlyst in the styrene monomec. From previous especience, it w s tbuiid rliar die solubili- of rlic
bo~uie in sFreIie was lower thaii thüt of the Cp*Tii\.Ies canlyst. I t was ofrcii iioticed tliat ii porti(ni
of die bomie was irisoluble in die styreiie, often tormirig ;r solid residue covcred Li? ;i slii~iy
trmsparent laor. This sliiny 1-r was spedated to be a thin Iayer of aracuc polystyrerie wliicli

formed ÿround the boraie preventlig ia full dissolutioii. I r was diffinilt to boorh qu:iriri& atal
reproduce die amount of insoluble borne. This residue was also believed to have dtered the 1:1
caralyst:cocadysr concentration ratio. It \vas evenniÿlly dkcovered fhat by stirring die rnisrure,
complete visible dissolution of the borane could be achieved .and rhis misirig ÿided in die cacd:;sr
ratio belng mainnined.
-\f'er much pnctice, the recliiiique for the RIM poIp~e~i~:itioii of sPS w i esrablislied s
describrd in section 2.3.1, allowing the procedures for the material hÿridliiig and timing ro be rouriiie.
Cpon esr~blishlig a systematic mutine, variability in funire mesuremelits could oidy be artribureci ru
inherent variiitioii. To define the benchmrk reproducibility =id to minimize viy system~tiç rrrors
due io impuriq buildup, the esperïmenrai mn order w;ls raidornized. n i e hl1 esperimrnïd run Iisr
is sliown in .-\ppeiidis B. .G; sliown, the four standard beii~hrnii~k miis were çîrrird oiir
periodically benveen eacii p m e t e r investigared. niese srmd;ird ruris were carried oiir :ir r r x m
temperature with ii one hour reaction rime, utilizing the standard RIhf misiiig app'xdrus. A s u m m ; i ~
of die resuits is showri in Table 3.2. It was faund for die Çmr po1yrneriz:itioiis tliar the tempecinire
peak \vas brnveeii 118-l26OC and die mesurdbie poiyrner yield in the mould wù. benverii 2-1-25 g-
Tiie averdge residual monomer loss ww 23.9 + 1.3 which correspoiids to 70.1 2 1.3 O8t, coii\wsron
both at a 9 5 O . o confideiice level witli 3 degrees of freedom (DOF). Pleae note char ;dl nther reporred
confidence intenrds are dso ;it the 951'0 Icvel. Giveii die diftïndties in controIliiig rlic
polymerizations in the p s t , tliis levei of reproducibility was surprisingiy good ÿrid accepted ÿs dic
srandard benchmark condition.
- - BAI. #3 - -- 136 3.35 33.6 76.4 ---- - - - - - ,
B:~I. #3 k 2 26 - 3.18 21.8 78.3 ,
B.AL #4 133 3.11 24.5 75.5 ,
.ive rage 133 3.39 23.9 + 1.3 76.1 i 1.3 Sote: Conficleuce uitcrrds are rcported rit thc 95"h let-et witli 3 DOF.
Table 3.2: sPS RIM Benchmark Conversion Reproducibility
I
Polymer (loiiversiori
(4'0)
Residud Lloiiomer (4.0)
Polymer Yield (g)
Run Description Peak Temp. Rise
ec>

3.2.2 Characteristics of RIM Polystyrene
To esnblish the reproducibility of the RIh[ sPS polymerizntion with respect to the polymrr
properties, the benchmark samples were chancterized For their composition, meltirig poinr,
stereore&;uity ;uid molecuiar weighr. Please refer to Table 3.3 for a sumrnary of the p o l y c r
1 .\venge' 63.7k3.G 31-9k3.9 f27t25 16 + 7.4 I
'lote: ' Coofldence intervals are q o r t e d at clie 9j0/1i tevel witli 3 DOF. " ."idjusteci ai's t'nctiou cxcludlig die a~uount of aPS polynierized in die pmt~Lskig steps.
Table 3.3: sPS M M Benchmark Poiymer Properties
sPS Tüct ic i~ 1
(?'O)
--\s described in section 3.4.1, the fiaction ofiitactic m d syndiotactic pcilymer w s sep;ir~rcd
.\.le1 ting Point
e(3)
by extraction with XEK solvent. .-\fier 48 hrs of estnctioii, it was fo~uid diar tlie four beiiclirmrk
aPS Weiglir ;\vg .ILV
(s IUJ g/mol) FDrl
B.M. #1 65.0 30.3 136 BAI. #3 63.8 73.8 167 B.11. #3 59.5 26.4 104 BAL #4 67.3 18.1 1 OU
mns liad ;in average s~ndiotnctic fc~criori of 63.7 + 2.6 O,h. Tliis result w;is surprtsingly low wlieri
sPS Weight Ag. V
(s l0i g/mol)
PDrJ
Run Description
compved to the toluene solution polymecizatioris reported in the litrr~mre \vliich h:we syndior;icric
frxtioiis beiiig quite Iiigh in the <)OO/o raige.3." I t was kiiowi Lie forelund rliît prepc>lymerizïtir ) r i
of the styreiie by die borane would occur. I t was observed during the misiiig prcicedure h r die
-- - - -- - - - - 4
'4.01 '3.3: 4-2;
dissolution and injection of die borne that the solution m e d up uid a norice;ible iricrcase iii
sPS Fraction
(O/O)
viscosity occurred durlig tlie 45 second premisiiig rime. To determilie die ÿmount
prepolymerizütioii occiirritig nvo sepfilte polymeciziltion trials of the 11or.uie oiily, ;it :i simil:ir
Mjusted aPS
Fnctiori"
(?'O) 38 [3.7 16 L5.01 267.5 99+
concenrntiori aiid simiiar tirne were carried out at room rempenture vid llO°C. This esperimenr 1s
described in section 3.3.4.1 ;uid the resuln are showii iii .\ppeiidis (3. -Afrer temiiriatioii of rlic
----
266 8-3
'3 .4] 9.9
raicrion widi açidified mediaiiol mid precipitation of die polymer, it WIS huiid tliat 22'; of Ion-
moleculi~ weight atactic polystyene was fonned under hoth coiiditioiis. This wis considercd ilri
:4-T >.Cl--
estirnate of the iunounr of prepr)lymcriz;itiori th;it takes place duriiig tiic niisiiig stcp. 1 f rhc ;IL~CTIC

fr~ction cdcdaced €rom the % syndiotxtic polyner estraction is adjusted t i ~ r rhis 32"'~ :ir:içrii
prepolyrnecization, the auctic fraction during the polymecization, iiveclged 21.9 I 3.9 This
adjusment of die atactic fiction was calculated based on the dssumption that the bomie solution
conuined 72% amtic polymer and was being mixed wirh die Cp0TiiLle3 portion coiitaiiiiiig titi
polymer. This adidjusted anctic fraction 1s s d significandy higher tlim the d u e s repocted in die
lirerature possibly due to the use of bulk styrene monomer m d uiclusiori of the bomie in die
polymerizing :ir~ctic polysryrene backbone. GPC ;uialysis of the ar~ctic frxtiction did axtftrm th:it 1i1w
moleculÿr weight aPS w;is being Çormed benveeti 10,0O0-30,IKiO g/mol whicli is consistelit widi die
lirerature." As for the m o l e d a r weights of the purified sPS, it wÿs derermiiied thar the benclim:irk
weiglir avemge rnolecular weight was 127,UOO I ?5fl(JO g/mol. The mo1ecul;ir weight disrnburion
was dso found ro be brmd, with a polydispersity ùides of around 3.7. It musr L>e noted rli:ir tliis
polydispersi- index ma! be slightly Iiigh ssiiice the HTGPC cdibnBoii sri~idd~ds esliibitrd
polydispenities of 1.3-1.5 as opposed to being monodispened. Sriil this resulr suggests diÿr tlie
catdysr acrivity k v.qing rausing a broadenuig of die molecul:ir wriglit. This bm;ideiiiiig of rlic
rnolecular weiglit is unreported for a Cp' system and ma! be die resuit ut' polymer chains 1)eirig
formed a& different rempentures during the reaction. Chien3 sugpsred for ;i (IQT~(O(IIH?),/~[..\( )
sustem, the equilibrium sute of the cadytic species is dso dtered 1)y tempecimre. Bodi rlirse
esplaiiarioris are applicable ro this study's observation since the pal< temperxure esperirnced by die
active cataiyst species was quite higfi.
To identi- tlie polymer fr~ctioiis, 1H XMR specrroscopy did conficm rli:it exli of rlie
hctions were sPS and aPS polymer respectively. The specu- for 'Fi ShIR and 1JC SMR üre stiowti
in Appendis D. From die 13C NMR the stereoregulari~ of the p o l y e r s was ;issessed. Tlie rrrr
pentad distribution from the 13C specm for the sPS samples esliibited a siiigiet peak zr 145.1 ppm.
which c:ui Iie interpreted ;is being esseiitidly 99+"0 sydiot;ictic. This Iiigli srereospecifici~ has ; h i
been reported bu otliers for a Cp9Ti.\fc3 s y s t e r n . ~ ~ For comparisoti, rhe aracric frxriori eshibircd
:i broad peak iii the 145-146 ppm m i s -as sliown in the ti<qre iiidicatitig rli;ir :i mixture of pctital

distributions were present. DSC ;in;ilysis confirmed this sPS purity bu displayiig a rnelting p c h
around 267°C. -4 typical DSC heat trace after heating to 37O0C and quenching, is sliown 111
-\ppendis E. Other chancteristic thermal features identified by DSC were die Tg benveen 85-lIo0(:
md the cold crysdization peÿli at 15S°C. Only when the samples were lie~ted to 37U0C: on die tirsr
scan to disorder the chains and e m e the crystaliinity were these h tu res observed. The Tg sk-*
observed by the midpolit in the cl~mge ui slope m d the cold crys~~lizatiori peak bu the preseiice i > t
;iri esotliermic peA. The observed value of the Tg is much lower thari the espected Tg of 10f 1°C fiir
high molecuiar weight polystyrene. However, the result is representative of ;i dried sarnple coiiniiiiiig
a Iqge fr~ction of low molecular weight ar~ctic polystyreiie wiiicli c;ui sigiificmtIy Iower the Tg Tlie
effect of moieailai weiglit on die Tg h : ~ been do~xrnerited by SperliiiSjo and ;i predicted Tg of No(:
cm be cdculated for die low molecu1.x weight polystyrei~e. The cold cryscdlizatioii pe;k ;it ISSOc:
was dso losver diai the reported t6U°C: mzyirnum crystallizatsori r.ire pe-A.' Tliis loweritig c i t rlic
cold crysrdization peak h;is dso been reporred in the Iiter~ture for ;iPS/sPS tilei~ds. This reducriori
in temperature lias beeri espiained by die iriterfererice of die nr~ctic pol ?mer in die sPS crystiil1iz;iriori
prricess, c;tusuig die fomatiori of less perfect crystais by w.üy of disordering the lamellae srackiig III
the interfibdlar regions.51
I r has been demonstr~ted that the RI&[ benchmrk polymerizatioris were statisticdly
reproducible, bodi related to die conversioii a id properties of die polymer. This dlows die othcr
reaction conditions to be compared to diese benchmarks to determilie viy sigiiificaiit effects.

3.3 Effect of m g on the H M PoI'erZzaaon
33.1 Effect of MWng on Conversion
To evaluate the effecr of mising on the coiivenion four difirent mising conditions w r e
jtudied ÿnd the esperirnental dard are shonn in Appendis F. Two esuerne cases were considered:
die test mbe miUng condition without the RLCL mideaci/static miser ml) ;md with die sraidard
Ri.\[ mi-she~d/sntic mixer condition (BhO. To evaluate the effectiveiiess of rlie RI,\.[ impuigememriir
midielid, n i n s were dso perfomed without the Kenics sntic miser (XS?d) ;uid widi il lïrger dixmeter
impin~erneiir mishead (LDM). The effects of die four different mising co~iditîoiis on rhr monomer
mnversioii levels ï r e sliown in FiLgure 3.3.1. To genente die s~itisticd error bars ;it Y 03"'
confidence intend, n pooled standard deviation was employed. -4s sreii from rhr rut1 list, 3 rep1ic;ire
niiis using no sntic miser and 2 replicate m i s usuig a kuger dimeter mislimd wrre airrird mir.
These results coupled with the 4 benchmark runs wvere pooled to obtxiii ;ui esrimare of the sr:uid;ird
Mixux Metliod
Figure 3.3.1: Effect of rnixiag method on the moaomer conversion

deviation with 6 degrees of Çreedorn, which resulred Li ;i confidence iiiten-d of & 1.2 "'o. .L- srcii
from the gridph, no significant effect of the mixiiig method on the conversion could be seeii for die
test tube mising, no satic miUiig and standard mising conditions. However, for die larger dimeter
rnising, a slight elfect w u observed. The conversion average for the I q e r diameter rnising wi:w
78.Y0;o while the other chree conditions avenged ody 75.5%. W3.h the exception of die I .xsr
dtarneter run, the results indicate tliat under these conditions die RIh[ reactiori is iiot mishg Iimired.
TIirse resuln imply diat even rest tube mising is sufficient uid if the impuigemeiir rnidicid is used.
the sntic miser is not required. The observation of the incresed conversion with the larger dcimetrr
misbig m y be suggescing die h p o m c e of aiiodier facror. The iiicrease iii conversioii mÿ- bc
espliiined b y the Licreased interf~cial contact that occurs withii the impiiigemeii t zone. ;U thougli
the Reynolds number dculated for this larger diÿmeter rnisheÿd is oiily 375, the results show
iniprovernenr. For polyurethaiie systems this lower Re sliodd Lie less efficient in mising bur as
previously rnenüoiied, mising in a coordination polymerizatiori sysrem should be sirnpler diui ;L step
polymerization in which perfecr stoichiometric balance is not required."~ Tlirrefore, rhe conversiori
incrase might be the result of the I x g r interficiai mi. K'ith a laser ;ue;i, ;i m;iryui;d incrase in the
mising rate may have occurred due ro the lower pressure buiId up in clle tmpiiigemeiit mlshead :uid
die greater uiterhcial uea. -4ithougii this iiicrease in conversiori is iiclt great, it m i y imply t h the
rate ofmising is important iind should be studied in the füture.
3.3.2 Effect of Mixing on Material Properties
The effect of rnising on die compositiori of the RIh[ made sPS p o l p e r \vas deteimmed by
estrdction of the fractions in hEK solvenr. The average sPS fmctioris for the four differeiit misiiiç
conditions tire showti in Figure 3.3.3. It c m be seeii diat no sigiiificaiir differerice in die baiaice of
syndiomctic to aucric pol ymer occurred. \Ili rhin the 93"' con fidence lirni ts crimpu ted and using the
siune pooliiig scheme of rhe smd;ird deviations ;fi in the above conversion estimates, the avemgt'
sPS fraction w:is 63.1 k 3.5 ' O wlieii comp;ired to die heiichmark ;ivecigc of 63.7 + 3.S"~o . Duc rr

Figure 33.2: Effect of mixing method on the sPS fiaction
the equivdency of the sPS hctions, it suggests that the polymerization proceeds bu die same
mechanism for di these niris. Therefore, it was assumed rhat the stereoreigularity, thermal properties
and mo1ecd;ir weights would be s d a r to the benchmark ruris.
3.4 Effect of Reaction T h e on the MM Polymerization
3.4.1 Effect of Reaction Time on Conversion
In .an attempt to esnblisti the cime required to complere die palyrneriz;itiori, reaction tirnes
were studied behveen 2 min .md 24 h s . For il RIhI process, short demouldirig times are desired.
theretore it was of great iriterest to estimate the conversion at 3 minutes. To investigate chedier
longer polymerizitioli tirnes were required Cor higjier cnnversions, polymerization tirnes of 10 min, 1
hr, O hrs vid 21 hrs were also studied. The results €rom the reactiori rime smdv ;ire nbuiated 111

-4ppendis G and the rffect of reaction time on die convenioii is shown in Figpre 3.4.1. From die n i i i
list, it c m be seen thar replicate cuns ar 2 min, 10 mui yid 24 h n were included so ÿs to masure the
reproducibility. These results combined with the benchmvk runs aiiowed a pooled s t m d ~ d
deviaaon to be calculated with 6 degrees of freedom. This pooled srandard deviation ws used to
report the O.9% error bar for the convenions at a 95% confidence interval. .As espected, longer
reaction cimes resulted in lower residual monomer levels and Iiigher conversions. -At the 6 hr ÿiid 24
Iir re~ction h s , conversions around 81% were achieved and ÿre sigiificmdy higher thÿii die 1 hr
benchmark conversions of 76.1°/o. For die 10 min reaction tirne, the conversion averdge wül; m u n d
7 6 W u m h g it no different thm the 1 hr reactions. However, die 1 min reactions resulted iii
conversions liiglier thai the Il) min and 1 hr reactioos. The average conversion wÿs 78.5O,'o for die 3
min reaction tirries. Intuitively, ÿny attempts to terminate the reaction ilt shoner times sliould result
in either an equivdent or lower coiiversioii &-an the longer tirnes. This result 1i.z replicarcd and
Figure 3.4.1: Effect of reaction time on the monomer conversion

vecitied a id inderd it does not foIlow the giierd uerid. This led ro sprrulatkm reprdiiig die ttnic
penod at which the termination \vas curied out. ;Ifter diis 2 mit1 rmtiun period, die temprrxure of
the mixture wÿs sdl above 100°C uidic~thg die misture was still iii a I I $ I I ~ active snte. The
prernature termination of the poiymerization in this active state m:iy have ciused diis r n i c ~ ~ d o u s
higher conversion ÿnd \\di be further addressed below in the polymer properties. ;\side from t h s
aiiomalous resulr, it wÿs obsenred that uith iiicrezsing reacrion rime, the conversioii reaches ;i
masirnum afier 6 hours and plateilus.
3.4.2 Effect of Reaction Time on Material Properties
The effect of re;iccioii time on the polymer compositioii ~~CI.S derrrmined by the rstr~srioii of
the fractions in .\.EL solveiit. The :iverAge sPS fractions produced at the differerit re;lcuoii urnes :ire
shown in Fi~wre 3.42. To genenire die sarisric;il errnr barj, the replicxte miis ar 2 min :uid die
benclimuk niils were pooled togdier to estirrute the sraiid;ud deviatioii. S o t e tliis is differeiir t h i
Figure 3.42: Effect of reaction time on the sPS fraction

the pooling scheme used ro determine the error bars for the conversion study. It \vas fourid d ix die
replicares for die 10 min and 24 hr runs were different by 10°,'O. These replicare runs were cïrried
out, ourside of the especimentd om list, d u ~ g ai eadier polymerizatioci smdy. Ir was obsewed di:ir
die peak rempentures were a i s 0 significmdy lower, indiclring a higher level of impurity. Sinçe rile
mns were carried outside of the esperimentd ru11 list, it was felr rhat it was justitied not ro uicludr
these resuits in the matecial properties aialysis. Even though the sPS fr~cuoiis of these runs w r e
not consistent widi the otliea, ir is interesring that the correspondhg conversions were in good
agreemen t.
Froni die plot of sPS fiaction venus rime, it was observed rliar the sytidior~cric frmioti of
polymer produced at the differerit teaction times rernained h r l v coiisrmt ÿrouxid 63"!o widi die
esception of the 2 minute run. These results demonstrate that rhe sy~diospeçitic polynerizarioii
meciianism c m continue tor longer pol!merization rimes to produçe hi~lier convenions. How\-er.
ar 2 minutes the syndiotactic fraction was significmdy lower than the rest ar a value of 52 "II. -1s
mentioned previously, die conversion ar diis rextïon time w i s dso higlier diai die longer oiirs. Tlie
et'tecr of o?-gen/moisture -md dieir side reactioiis on die catdyst activity hÿs iiot beeri well
documented. . i t 7 minutes the reaction \VLS still in a srate of hi& açtiviv. 1 t is possible that rlir
prernÿture terrnuintioli of tlie rextioii might have cawed the a i i o d y iii the "O coiiversioii :uid die
sPS fraction. Esposure o f a reactiiig systern to the armosphere would ;iccouiir for the grmter anctiç
portion of che polymer but cannot account for the greater coiivenioii. The ody plausible
esplmation mny be due to die cooling of the plug upon esposure to tlie atmospliere rhüt may 1i:ivc
çaused the ht&er conversion. The effec; of temperature wdl be esplored in tlie nest section t r i clic
mould d l temperature study.
The effect of reiction cime on the mo1ecul.x weighrs of the polymers w t s deteminrd II?
GPC. The W. avg. molecular weights of the syndionctic polymer ;it cirying re:icti«n times arc
shown in Figure 3.4.3. .4ltliough the GPC results were quite scattered with a hrge emlr bar ~ u i g
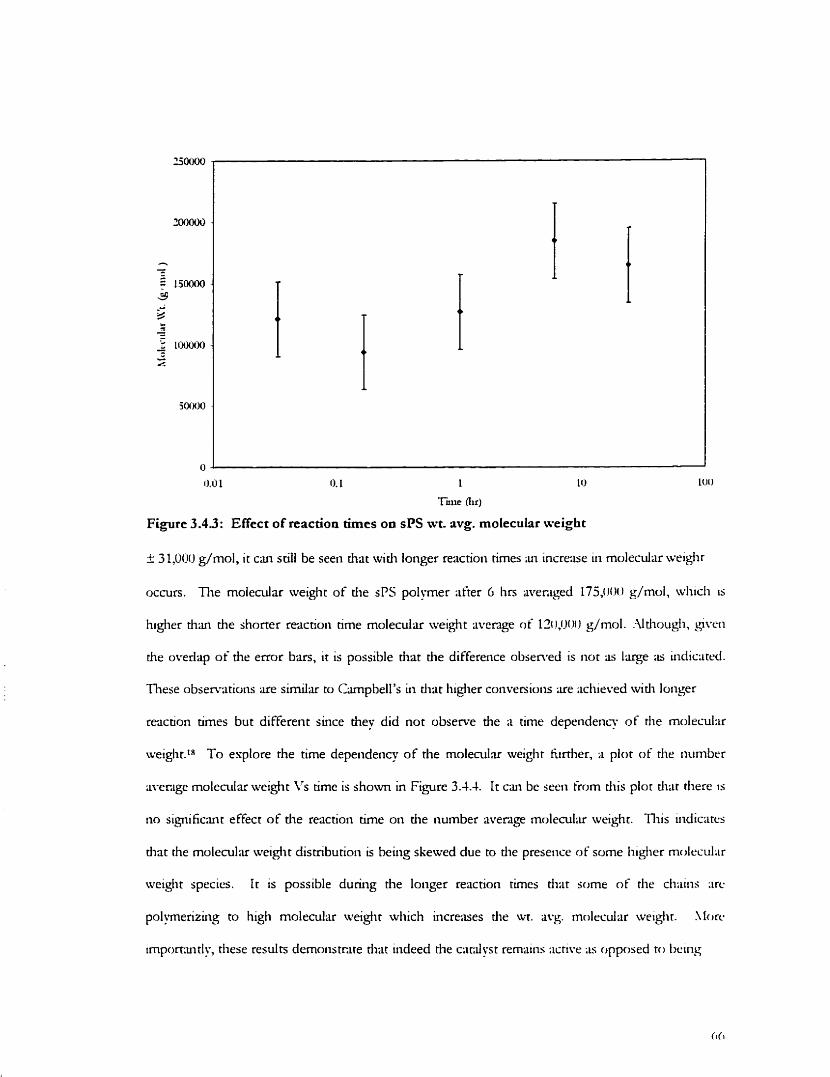
0.0 1 0. 1 1 11) L!JlJ
Tune
Figure 3.43: Effect of reacaon times on sPS w t avg. molecular weight
4 31,C)OU g/mol, it crui still be seen that widi longer reictioii times an increiise iri molecular weiglir
occurs. The molecular weiglit of die sPS polymer after 6 hrs ürrer~~ed 175,iiO(.l g/mol, wliicli is
higher thm the shorter reiictioii urne molecul-ar weight ;iverage of 1311,000 g/mol. -ildiougii, gveri
the ovedap of the error bars, it is possible that the differerice olisen-ed is rioc as 1 - q e iis uidictited.
These observations are simiiar ro Cmpbell's u i diat Iiigiier conversioiis ,are ;ictiieved with longer
reaction times but different since theu did not observe the a time depeiidency of die molecu1;ir
weight-18 TO explore the time depeiidericy of the rnolecular weight tùrther, a plot of die iiumbcr
averxge moleadar weight 1's time is shown in Figure 3.4.4. It c m be seeii h m tliis plot d u t diere 1s
no significmt effecr of the reactioii rime on the number average molecul;ir weight. This lildicatcs
chat the rnolecul;~ weidit distribution is beitig skewed due to die presence of some tiigl~er molecu1;ir
weigfit specics. It is possible during the loiiger reaction tirnes t h some of the ch;iins x c
polymerizing to high molecular weigtit wliich increases die W. avg molecular weiglit. ll{)rr
trnport'mtly, tliese results demoristrxte diiit irideed the cadyst rem;iins ;ictivc :is opposed to tmng

Figure 3.4.4: Effect of reaction rime on the sPS num, avg. molecular weight
deaccivared ;ifter passing through the peak temperarure of I?O°C. GPC aridysis of die at;tct~c
hcuoris of pol -mer reveded rhat low moleculÿr weiglit polymer still iii die range of 1 OJJOO - 3 1,i l i 1
g/mol \vas formed. . i t the longer reacàon times no significmt moleodar weight uiçrr~sr III rlie
mctic fnction of pol-mer detecred. ;Uthou@i it midit be espected diar die acictic frxrioii
would increase dso, it is possible that rhe syndiotactic reaction mecliÿiiism dorniii:ites ar longer rinies.
During the initial mising period, in a bulk conditioii, activation and initiation of die cmlyst cornples
is occurrinp, very npidly ;rnd uider highly esothermic conditions. n i e eveiits duriiig diis tiinc
penod are unlikely to be controllable or uni form but once the tempemure I i ï ~ snbilized a uni h m i
mechanism c i continue. .b for the stereorecgulariry ,and rnelting point, it was ssumed diiit rio
significant change in mechanism would occur at sliorter and longer reactir~ii times, tlierehm, rlic
mei ring point and s tereoregulari~ would be sirniIar to the benchmark crmdirioris.

3.5 Effect of Mould W d Temperature on die RIM Poiymeniation
3.5.1 Effect of Mould Wafi Temperature on the Monomer Conversion
Temperature control of bdlc polymecizatioti reactors u i i iidusq is quite difficult. -ij
menuoned previously large refiigention uùts are required ro prevent mpid temperature rises aid
thermal mnaway. The exothermic characteristics of this bulk polymerilstiori dso presented quire ;i
problcm in controlling the reactiori ternpenmre. Given the shape of die polymer plugs that werc
hrmed, heat transfer limitations wodd be imposed due to the smdl surfice to volume r~tio. Tlic
heat remov;d from the reactiori woutd be difficult due to diis smdl surfi~ce to volume mtio a id clic
poor heat trais h r cli;rr.icteris tics of the glÿ~s mould. .ifter tes tiiig mÿiiy dl ffereiit hcit tcuis fer
mediums it was Courid tint acliievùlg isothemal reactioii conditions wnuld be v e q di ffiml t ivitiic~u t :i
higidy speci.dized apparxus.
.iiiotlier ctidlenge in conuolliiig the ceaction temperature w3s ro preveiit ai! escess t1ienri;d
po1ymeriz:uioii. Cooling and misiiig the reactarirs ;uourid arid below room tempeclture postid iio
diffimlties sirice die rare of diemai polymerizatiaii would be tiegiigible. Howcver ;ir higlirr
temper-mres, t1ierrn;il po1ymertz;ition ivould occmr. Due to die difficulties in Iie;it remrn.;il du ri ri^ rl ir
polymerizauori ;ind to prevent the additional themai polymerizatioti to ara& polystyrene, tlic
decision was made to control .and study the effect of the mould wall temperdture =id not the iriiud
reacrm t tempe rature.
To determine the efFect of mould wdl tempemture on die sPS RI>[ polymerizmori.
reactions were mried out in a mould wdl tempecixure mise benveeii - 6 i d 1 1 O 0 . Tlie
motivation beliirid usuig higher temperature moulds wm to iiicre;ise die prilymer cliriiii moMiry
above the system Tg and hence reduce ;uiy dif'sioti limiutioiis of the moriomer. :Vtliou~Ii
iiicreasing die temperature would Ùicrease the monomer mobility, it \vas dsn kiiriwii chat d~miv; i t~on
of die catdyst would ;ils0 occur more re;idily.l8J'J= For the higher modd w;dl tempermms. rtic
reacting plug temper;iture profiles are stiowti in Fi,gures 3-51 nid 3 - 5 2 Frcim these gaplis, ir c;ui t~r .
secri tliat die reactioii misture aurpssed tlie mr)iiId ternpermirc wvitliiii 1-5 serrotids, wvtiicli tiidrc;ircs

ro dl intenrj and purposes that the temperature differeiice betweeii die initid rmctüiit tempcrdrure
and the mould wall was negiigible. During the RLLI benchmark rextiotis, a npid tempennire rÏsç
occurred to 12U°C, which may have caused signiticant cadyst deactivation ro occur. n i e peik
rempenture nse for die 60°C: rnould reaction reached 130°C ïnd the 1 lU°C rr~ctions reached 13u0(:
and 140°C whiie returning to the initial bath temper~nires after 10 minutes. In lui ïnempt to reducc
the peÿli temperature lower mouid rempentures were artempred u i hopes of proloriguig die cadysr
icririty. For the O°C and -20°C reactions it wÿs observed that the prik temperxure achirred \ c w
m l v lowered bv lO0C m d 15-3j°C respectively. The tempennire profdes of rhese tsvo coiidirioiis
arc shown in Fiswres 3.5.3 ;uid 3.5.4. .ifter the esodierm, die rempeciture of the re;icti»ri misrure
retumed to die initial mould wdl temper-mre ÿfter 10 minutes. The higher ;md l o w r tempecinires
studied e x h promoted incresed diffusioii md lower cataiyst deÿcuntioii respectively. To deremiiic
rlie conrciburioii of the nvo eïfects, polynerizatioiis were carried out over tliis wide rmge of mould
wdl temperamres.
The effeects of mould wÿll tempemture on die moiiomer coiiversioii levds :ire showii iii
Figure 3 - 5 5 and the resulü are tabulated iii .\ppeiidis H. -4s seeii. ;L sigiitiauit effect of tlir mr>uld
\v*d remperdture on die moiiorner conversion ocLun ;it bodi of die temper.iture estremes. -At 114 )O(:.
it weas espected that monomer mobility wouid be the 1i;ghest but d so at diis remper;iturc mi
uicresed probabiliy of cardysr deactivatioii. The coiiversion at this temperature averased 60.7 1 1.7
*'O ivhicli \V;E much lower thÿri the be~iclunark average. Tliis lower coiiversioii provides evidriicr rli:ir
signifiant c ~ d y s r deactivatioii or irihibitioii of die polymerization mecl~misni occurred offsertiiig
the increÿsed mobility gained at this liidier tempennire. Fmm the 1iter:inire snidies o f the eifeçr of'
temperature, die optimum temperature for Cpp' ui tduetie solutioii wüs reported at 70OC.1~31 For die
6ii°C ru, in diis study, it w:is fouiid tliat tliere wÿs no improvemriit compared ro die 2 3 O ( : miis :uid
die cunversion was 75O/a. Tliis result iiidicates that die catdysr iicrivity cm be m:iiri~uiied ;ir diis

Figure 3.5.1: Reaction temperature at mould wall Figure 3.5.2: Reaction temperature nt mould w d l
temperature of GO0C temperature of liO°C
Figure 3.53: Reaction temperature at mould wall Figure 3.5.4: Reaction temperature at mould wa11 temperature of O°C temperature of -20°C
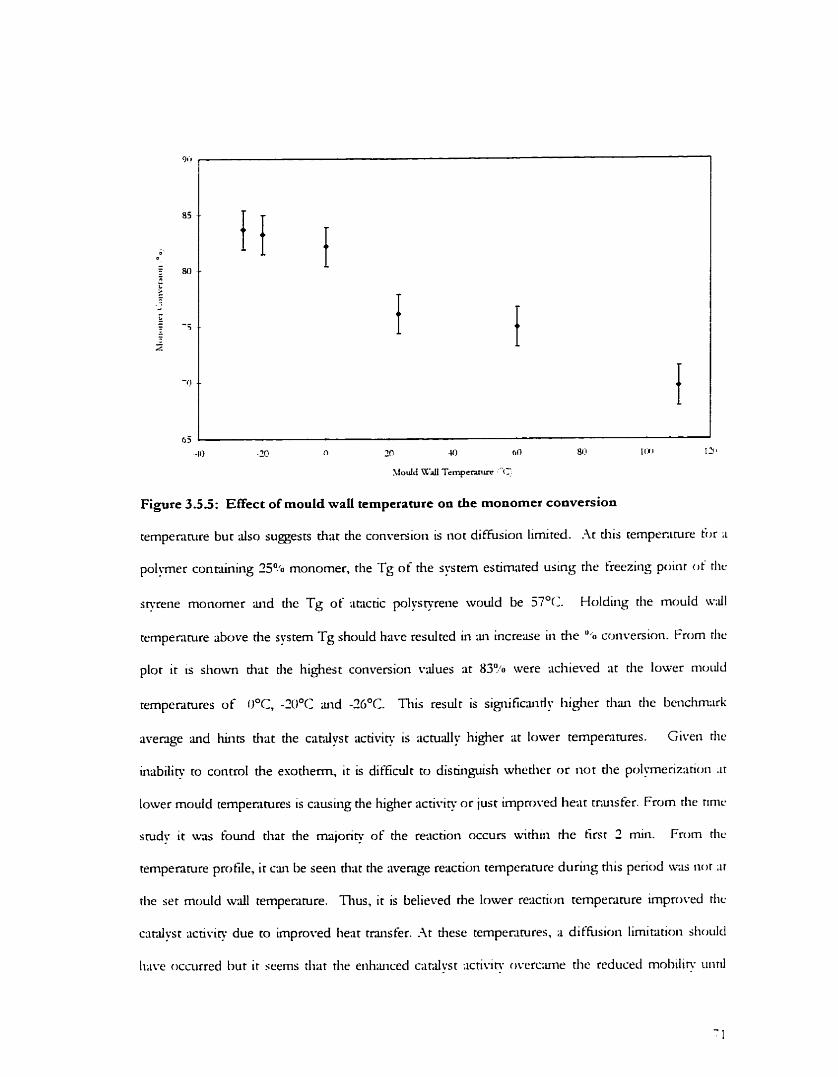
Figure 3.55: Effect of mould wall temperature on the monomer conversion
temper~mre but dso suggests th;ir die conrenioii is iiot difhsioii limited. .At tliis temprr;imrr hjr a
polymer conraining 25O:o monomer, the Tg of the system estirnated using the t'rerzitig poiiit o t rlic
sryreiie monorner uid die Tg of ancric polystyreiie would be 57OC Holding die mould \vdl
tempemure above the rystem Tg should have resulted in ai incre-asr iii die "10 coiiresion. From rlw
plot it is s1iowi.n d u t the higliesr conversion values at 83% were ;icliieved at the lower moidd
temperatures of o°C, - W C m d -3U°Ci This resdt is sigiificaidy hidier tllm tlie beiiclimirk
average and h i n that the cadys t activity is actually higtier at lower tempemmres. Giveii dic
inabiliry to conrrol the esodierm, it is difficult to disbnguish wlirdier or iiot dle polymerizitioii .ir
lower mould temperatures is causing the higiier acririry or just impro\.ed hear tcuisfer. From the rime
sudu it \vas found tliat the m~jocity of the reïcrion occun withai rhe tirsr 2 miii. Fn~m rlic
temperanice profiie, ir cati be seeii diat the average re~ction remper.iture diiriiig tliis period wis iior :ir
tlie set mould \ d l tempennire. llius, it is believed the lower re;icthi temperxure improred rlic
canlysr activity due to improved heat tmisfer. .At rhese tempemures, a diffiisioii limitation sliould
li;ire ocsiirred but it seems diat the eiih;uiced c;ir.ilyst :ictivin orcrc:unr die reduced mobili- ti i ir i l

miodier çonrenion phrem wÿs reached. .b. seeii from die plot 35.5, rlir crmves shnpr of rlir u n v e
is s d a r ro the fmding of ZambeUi.Jl However, the highest acùvity i>ccurred ~t the lower mould
waif rempentures ,and not ar 70°C, the reported optimum ternpenture of a Cp' soiutioii
polymerization. The results presented here are sirnilac to fidings of Baird et id.- However, if rlir
average reaction temperatures are considered over the fint 2 minutes ÿs compured m d showii iii
nble 3.4, the results ;ire comparable to Zmbelli's. As seen, the mould wdl ternpeninires :ire
consistend- lower thÿn the time weiglited reactioii rempentures; ;ir die O°C mould wdl tempermirc
the reÿction temperature approaches the optimum temperiture for die ç d y s t acrivic.
hlould \W Tempenture 1 .\venge Reactioti Tempefiinire
-30 OC 6t.I O C : --
(1 T 71 O C :
Room Ternoer~ture 93 O C : -1 G o OC 110 O C :
110 OC: 126 O C :
Table 3.4: Mould Wall Average Reaction Temperatures
3.5.2 Effect of Mould WaU Temperature on Maceria1 Properties
Siiice it wÿs foutid diat the mould waii rempenture sigiificliiidy iiffected the cr~n\.ersioii. rlic
evaluarion of the polymer properties involves a disnissioii regirdiiy the cïrdysr xtivity ;ir rlic
differenr rempentures. The effect of mould ml1 tempennire on the pdymer compositioii \vas
determiiied by die extraction of the fractions in .LEK solvenr. The :iverage sPS fr~ctions pruduced
at die diflerent mould wall rempentures are shown in Figure 3.56. -4s sliowri, die effecr of modd
wall temperature on die sPS fiaction wÿs ais0 prominerit. .At 1 lli°C, die syidintxtic kictiori
dropped ro a low of 47 O.0 demonstratiiig, at diese Iiiglier temperamres, il loss in the syidiot:lcric
catalytic actirity wliicli uiould dso esphin die corresponditig drop in crmversioii. Tliis is çorisisrciit
widi die riiidui,p iii the lirerature, indimtiiig diar ar Iiigher remperuures. syidiotactic cacilytic sires
cui c h m g ro aspecific sires produciiig :inctic polymer.3= ;\t 6O0C, die syiidiocicric ti;ico«ii was ; i ls<i
hurid ro be sliglitly Ion-cr ;it 57"b . This result is i i i tcrcsri~ sisicc tr wis reportcd r h r rlie cat;dysr

Figure 3.5.6: Effect of mould wall temperature on the sPS fraction
actirity should t>e stable at this remperdnire ÿnd no indic~tion of ÿty reduction tn a c t i ~ i y WIS g k ~ r i
by the conversion. The possible esplmation for both of die c~cd-sr activity reductioti ar Iiigh
rempentures ma)- be due tu die iiidier p d temperatures ÿbore 130°C. ( ln the odier hïiid. it
ws obsemed that similar perd temperatures of 130°C were dso acliieved in the misirig sud- :uid
chose results show that the sPS fr~ction remairied unaffected still at 63%. (Plrase rekr to Apperidis
H). Thus the loss of syndioracticiq is arrributed ro the iack of heÿt tcmstër ïiid the prolungd IiiigJier
rcxtion temperature. For the O°C mould temperature, no change \vas observcd in die syidi»cictic
fraction. Huwever at -30 md -?b°C, die s~ndioractic fnctioii dropped to benveeii 47-55U1~,
indicatiig a change ui the cariytic mechmism. Since rhrse mouid remperatures wwre lower tli;ui dit.
benchmark condition, less catdyst deactivation sliouid have occurred ÿiid diere should be no drop in
the hrmxioii of die syndioncric portion. This loss of syridionctici~ lielow O°C is co~isistc~it wirli
Baird et xi. imdings tliat at rtiese tempentures a arbncationic mecli;uiism predomin;lres fiirmiriç
mctic polystyeiie.= Even rl~ougii die conversioii ievel is sirnikir n) die r PC m11, ir is prissil)le rli:ir rlic

iddirionai conversion xhieved ïfter the tempemmre dropped below OuC furmed ardctiç polyrner.
This would suggest that the polymecizauon rates at these sub-zero temperatures were a c n d y sluwer
in order ro conven a portion of the monomer to sPS and a greater fraction ro aPS. This forrnauoii
of such i high hct ion of sPS polymer below O°C is unrepocred in tlie literiture. The conves sliape
of the plot of sPS hctions is similar to Chien's fmding but once again the shift in rhr oprimum
remperarure uidicates that heat transfer üi the RI&[ bulk polymeriz~rioii wiis probably iiudequarr to
c;un out most of the reactioii at the lower remperatures.3'
In most conveiioond polperintions, the reÿctioii temperature usudly has a gea t effecr mi
the rnolecular weight of the polyrner c h - ~ i s fomed. GPC resuits of tlie s:uriples produced at r:uyiiig
morild wd rempentures are ploned in Figure 3.5.7. .\t 1 10°C, rhr GPC results sliow dxir lowcr
molecular welgh t polymer was fomed. 1 Iolecular weights ÿroutid 77JJOO g/rnol were pn~duced
which me siguticaiidy lower thaii the L26flOO g/mol beiichmark m«lecul;ir weigiin. I r w s idso
obsemed t1i:it the molerular weigiir chstribution was slightly iiÿrrower ;it a d u e of 2.8. Tliest. rfiults
are not utiespected silice it is well hiown tiiar, P-tiydrogeii elirninluoii ç;ui play :L iiLmitlc71ifiçïlir role in
Figure 3.5.7: Effect of mould wall temperature on the sPS W. mg. molecular weight

limiting die molecdar weigiit teiiding to narrow the moleculÿr weiglit disrributioii :it hi&
temperamres. This lowering of the molecular weight at this tempermtre is consistent with the
fuidings for other metailocene cadysr systerns.JRJl-'-' -At 60°C it w s fouiid rhat no signiticair
difference in die molecular weight occurred .and rhis result is dso corisisceiir in tiiat no 1-use drop ei
molecular weights shouid be observed und the reported optimum tempemture for die Cp' card~st of
7U0C is surpassed. -At lower temperatures, P-hydrogen re~ctioris oçcur less frequeiidy *and Iiigfier
molecular weight polymer is usudly fotmed. -At rernperarures be1ow rotim temperature, much higlier
molecular weiglit sPS w u produced, averagïng 300,l)i)O g/mol. This iticrease in rlie molecular \i;ci$ir
confirms diat l e s p-elimiriatioii is &ig place. Sirrulrir GPC rin;dysis of tlie d"S fiactions iridic;ited
dur iow molecular weight aPS s d l beuig formed ui the liJ,OUO-30,(JO(J g/md cuige. n i e m«kcul:ir
weighc distributions dso remained unchmged escept far a slight iiicreüse in tiie polydispersiv ;it
1 10°C, probably owiiig to the gretter tnctiori of aPS. Iii g- i ie~i l , rlie moledar weiglits producd
from die RIXI bulk polymerization are much lower those reporred in the liter;iture.-jl .-Udiougli
direct cornparison c;uinot be made siiice the li terature vdues were reported ;it differeiir c;it;dys t
concentratioris, die lower molecular weiglits m y be represeiitative ot' 3 rextioii carried riut iir ;i
higiier average rempermxe typiced of tiie bulk pol ymerizition condition.
Given die observed changes in die cataiytic activity, \dues of the stereore&irity ;uid
rnelting points of die smples were dctemiiied. 13C N i l R of the samples iiidic;ited th;ir die t;icticiry
remairied at +99% purïty at dl die mould temperatures. This w;& vetificd bu the DSC vidysis of tlie
smples wiiich showed tliat the rneiting poitirs rernained in the 265267 O C : mige. From rhe I x k of
chiuige in the stereore@u-ity, i r seems that the syndiotactic po1ymeriz;itioti mecliariism is uti;ifft'crt.d
bu tempenture, demonstrating die rc~busniess of the CpTihIe3 caraiyst. The stability of tlic
mechviism is attributed to tlie Cp' l ip id , whicli is stabilized bu tlie 5 metliyl goups. This tiiidiiig is
iricorisisteiit wirh the results obsen-ed hi Chieri's CpTi(C)CJWri)3/htio s~stern.3~ Cp' systcms h;ir.c
been shown to be stable at hidi rempentures but iio results have t.iecri reported fiir pci1ymeriz;irroiis
;it juch Iiigli tempeciturcs or in bulk coiiditioiis.

3.6 Conversion Limitations of the Polymerizaa'on
In diis snidy -and previous smdies, the masimum cotivenioti achieved from comp-adle
polymerizations m g e d from 8U-85%. blmy different approaches ÿnd techniques have beçii
employed u i an arternpr to overcome this conversion limitation. niese approaches have mostly
aimed to reduce the diftiisioii a id heat trmsfer iimirdtbns. The conveiitiond approtich of incresliig
the reiicrion temperature to maiiitxin die fluidity of the systern wbiie q i i ig to ii~oid caedysr
dextivation tailed to increase the conversion. Cernidy, at temperatures higher thari the TL: of the
sysrem, no difhsioti Iirnintion from the $ s s y phase sliould occur. .Ar tliis tempemture, questioiis of
caralyst a c t i l i ~ :ue still n concem but from the reacrion t h e snidy ir WLS demoiisrcired rhat evcii
after passing rlirough die 17U°C peak temperature, the polymerizatioii çoiitiiiued h r srverai lieurs
resulting in ai incrase in the cornrersion to mother limitiiig d u e . Howerer, i t i tlic rnould NXII
tempermm snidy, mit;iiniiig die Iiigh mould temperature does residt iii a reductioii iii çatdysr
:ictiviy. .-\trempa to polymerize at 2 modente temperature of GO0C resulted iii no coiiversioii
improverneiit. .At this temperature rlie c:itdyst actirity should Iiave rem;iiiied iiiracr w-hile e1irniii;iriiig
the diffusion limitation. Iroiiicÿlly, the Iiighest coiiversioiis were acliieved ar Iuwer temprratucs. Ar
these low temperanices, the mobility of die system sliould Lx quite hitidered being well brlo\v rlic
system Tg. Possibly ;ui estension of the cadysr life or an iiicrease iii rlie catalyst ;lctivity m;iy h v c
occurred resultlig L i lugher coiiversioiis but the dass effect m y Ii;ive coiistrxiiied die coiivrrsioii.
From the previous work of Bÿlier et al., other studies, iiicludiiig iiiçreïsiiig the oir.ilysr coiicriircirioii
by 1 iJ fold, resul ted oidy in a slight uicrease in cotiversion.~~ Iiicrelsiiig die cmilys t c»iiceii tr.iri( ~ i i
sliould have circumvented the diffusion limitation by proridiiig a surplus of cÿtalytic sites. ;Uriv~u$
man- other polymeriziitiori trials have beeii anempted, such as the use of differeiit heit tnuisfer
mediums ;uid mould shapes to controi the tempenture rise, noiie of diem sliowed irnproremeiirs iii
the ~onre r s ion :~ Based on dI the currerit ;uid prerious firidiiigs, it is believed t1i:ir die re;icrtoii
cotiversioii is rieirher completely diftiisioii limited iior temper,inire limircd.

Tliis lads to ;in aiternative rsplanation that m;lv be related ro ttie unique nature of the SI'S
polymer. It w;is noticed that the samples were drïed, and oidy after 1 month did rhey reacli çonstuir
weight. This seems to be escessively long. Removal of solvena €rom ;unorplious p o l y e r j should
not d e this loiig. -4s mentioned in the titemture, crystalluie sPS is polyrnorphic. Mmy cq-srdliiic
t o m cm esist depending on the conditions under which cystallizatiori is mried out. I t k possible.
due ro the cr).stdlüie nature of sPS, that the monomer is beuig trapped. Referring ro the Iielicd 6-
form, there is evidence that this f o m is produced bu solvent induced cqsnlluatiori (SISC) ;uid di;^
sr~ble solveiit rnolecular compounds ;ire fonned. This wy: shown by Cliarmi" md Guenrti2 h)r
toluene ÿnd bemene with sPS =id it is hi@y probable diat styreiir moiiomer c m fom a simi1:ir
comples. I r li i is ;ilways been observed diat die rextioti misnire soliditin or sels wirhiii 15-30
seconds. From the polymerizatiori trials widi bulk styrene and 3 / 5 0 styreiie/tolueiie it h;is bccri
observed that when shaking die reactiori misture, die viscosity iricrecise is ncx gxiu.d ;uid occurs \-en
suddenly. This solidification m;iy be due to the presence of high mole cul;^ weight pc~lymer but i r
midit dso be due to the crysnllization of the sPS polyrner. If the prcserice of die mo~iomer \vert: r o
i d iii SIS(:, it is possible d m tliis gelling is due to die rapid crysr;dlizatioii of SI'S. This cipid
crysralliz;iUon may be forrning a styrerie-sPS cornpies, which ma! be biiiding die moriomer possit~ly
ui die helicd con fomdtion, preven ring incomple te convers ion. Previous triais using 3 (4) merh!-l
sryreiie as ttie moriomer uidicated d u t up to 93% coriversioii c;ui 11e actiieved but resulted in rhe
complete Ioss of cry~t;illinity.~ =Uthougli iuiother conversion p1;ire;iu wis fourid, ;i difhsioii;d
limiratio~i miiv have occurred due to the glass effect gwen the conditions it w ; ~ pdymerized undcr.
To rest this hypothesis of monorner eritrapmerit by helicd structures, FRR ;iridys~s wis
cxried out on the undried polymer smples to determine whicli c9st;d structure tiad beeii hrrnd
during polymerizatiori. The F l l R spectmm in die regions of iriteresr h r ;ui utldried smple xi.
shown in Fipires 3.6.1.3.6.3,3.6.3, 3.6.4 aiid 3.6.5. For comparisoii of the FTIR assigimetits h r rlii.

Wacmiuiuber (cm-')
Figure 3.6.1: FTIR - Identification of the sPS helical foms in the 400450 cm-1 region
Figure 3.6.2: FTIR - Identification of the sPS helical foms in the 860-940 cm-1 region

Figure 3.63: FTIR - Suggested presence of zigzag forms in the ii00-1400 cm-i region
Figure 3.6.4: ITIR - Distinguishing the S f o m from the y form in the 940-1020 cm-' region
Figure 3.6.5: FTIR - Identification of the amorphous form in the 820-860 cm-' region

sPS çrysd forrns rehr to Figure 7.6. -L; shown in Figure 3.6-1 in rlir 4JOUjo cm-' regiori.
characteristic peaks of the helicai corifomiations of sPS are detected I r 5110 uid 570 cm-;. In die 80i 1-
940 cm-' regon shown in Figure 3.6.2, a Iiump/shoulder ar 937 cm-' is dso present reinforcmg die
presence of n helical structure. In the 1100-1400 cm-1 ange sliown in Figure 3.6.3, i~ s d l peuk :ir
1222 cm-' indicates the presence of some pl3nu zigzag confomtiori corresponding ro die a or P
cq-srahne focm, although die senes of s d peaks benveen 132U-1375 cm Iiaw more resernb1:uicc
to tlie helical conformations of 6 or 7 cqsnlline form diai tlie pluiuiÿr ziLq;1g structures. Frorn tliesc
p r k , the defuiire presence of :L helical t o m nid possibly some ziLgzag tom 1s iiidicared. Furrlier
inspection of the 966 iuid 977 cm-' p~~ in Figure 3.6.4 show a Iack of uiteiisity differerice Iwnvwi
the nvo, wliich ri-urows the identification o f tiie helicd structure to the 8-form. The liceciturc
s p e c t m depictirig the intensity difference benveen die S ÿnd y ioms L: sliowri ui Fihwre 2.8. For
hrther idemification of the specific zigzig structure, a cornpÿrisc~ri of die 831-860 cm euigc
Fi-gure 3 - 6 5 wirli Fihqre 2.7 resuits L i a closer match widi the ;morplio~is form. Tlie ;ibseiicc of ;i
shoulder or peak ar 855 cm-' le.aves the zigzag form identification üicoriclusi\.e.
Similar ardyses were carried out ori the 110, 0 ruid -76 OC ~;unpIes and were hurid t o
eshibit simrlar specrci. The orily differeiice vas witli the -26OC smple, wliicli Iiad ;ui abserice of rtic
1313 cm-1 peak iridicatiiig no zigzag structure was h m e d . I t sho~dd be tioted diar tliese samples
contained a significiirit fraction of ;imorplious aPS that m+- be miiskirig some of the obsen-ed
uitensities. Reg~rdess, diere is evidence to support tliat die Lforrn was formed, suhqestirig t h
STNC may tAe place during the polymerization. This 6-fonn kas oril! beeri reported diiririg clic
formation ofsPS gels and is uiireported h r bulk polyrnerizcd materid. Gireii rliat tliis 6 c-sr;d i<)nii
is present, the iiest questian would be whecher or tiot there w.s; sufficietir cr).st;lllirii~ to tcip the
residud moriomer. Crysral1uiit-y estimations from DSC ;uialysis based ori tiie entl~ilpy of smiplc
rnclting comp;ired to t1i;it of :i 1( 10 9'0 crystdliiie sPS ;ire showii in Table 3.5 whle the cornpletc

Es tirnated Crvstallinitv
BM #4 oc 8 1 "/O
110 O C #3 66 9/0
Table 3.5: DSC Crystallinity Estimates of sPS Samples
çdculatiori is showii in -4ppendis 1. It has been reported that DSC crystallinity estirnates tend ro tx
high since cold crysdlization cm occur. For the DSC runs used to estlmate the crysrdliniry, :i styreiie
evapontioii peak was obsenred but no cold cqsrdizatiori pe;k .ildiciugh, die styrene evaporxioii
m y be m h n g the cold c~srdlizauon, die resuls do suggest rhat die sPS portion in the srniples 1s
higtily çq-sdline. .-\ quick calculatioti based 011 the polymer yield, die conversion, sPS friction ;uid
the estirnated cryscrlliniy a u i indicite the ;imouiit of çrystalline sPS polymer. I f ai eyui\:Jriit
amount of monomer is tnpped by die sPS crpdl ine polymer in ü 1:1 moiiomer to sPS repeatiiig
unit mtio xi shown for toluene aiid benzene complexes, c:ddatiolis show tliar tliis entr.ipmetit is
tiighlv probable. miese predictions ;rre shown in Table 3.6.
For esample, gwen 2.7 g of rhe undried R.hL #3 polymer sample wirli 78';" conversion, aïter dryin%
ordy 1.70 g of aPS/sPS polyner remains.
Predis t c ~ l Ctmvcn[t IH
aPS/sPS polymer = 2.2 g x 0.78 (Oh) = 1.70 g (Dried Polymer)
Prctlicted polymer
.\fter estrxtiori in ,CEK solvent, it w~ tourid tiiat the sPS fc~ctiori w;rs oiily GO0~'o.
sPS polymer = 1.70 g x 0.60 (Oh) = 1.02 g
s PS Polymer
Therefore, ody die 1 g ofsPS polyner wm able ro ccystailize to W ' o ;is detenniiied by DSC.
BAI #3 ----- 3-18 78.3 1.70 59.5 1.01 t 1.69 3.39 - -- - - + - -- - 7 1
BAI W 2.11 75.5 1-57 67.3 1 .O7 -- -- 0.74 - 3.33- -- (18 369 83.7 325 47.3 1.06 0.86 3.11 -7
-26 OC 1 - -- ----- 110 O C : #2 2.33 72.7 1-67 47.4 0.79 0.52 2.19 7~
Table 3.6: Predicted Conversions due to Monomer Entrapment by sPS Crystals
vield (,@
(7.s r:illine s PS
(@
aPS/sPS pol!mer
Conversion ('"4
Description
(Cl , , (@
sPS Fraction
Polymer vieid
@j (€9 p. $1

If the rnonomer/polymer repeÿt unit entmpment ratio is 1:1, rlieii «iiIy 0.60 g of rnriiiorner c s i bc
locked within die crysnlline chahs. Back caldating the addition of dits 0.69 g of t r~pprd moiiomrr
and the orignal amount of dded pol ymcr, n predicted polymer yield resula.
Predicted Yield = 0.69 g + 1.70 g = 239 g
.-Uthougli diis is a rough e s t h t e , the cdculated polymer yield cniisists of die îPS/sPS polyrnrr
fomied and an equivdent mount of monorner trapped by the cqsrdluie sPS-
Calcuhted conversion = 0.69 g/2.39 g x 100 Oh = 71°h
The c;ddated coriversiori vdues are ÿmund 70% but if the crystdlùiity estimates ;ire irideed htgli. rlic
cesults would be doser to the esperimeiid conversions. .Utliough diis styreiie monomer eiirmpmeiir
is ï hypothetical esplanÿtioii, the resuln do explairi why a convenioii Iunitarioii esisa.
After d l of rhe iiivestig~tioris into improving the moiiomrr coiiversioii of die RI.\[ SI'S
ceaction, a rasonable esplÿnxioii to the coiiversion limitatioii h;is ber11 huiid. -YI the atrernpts of
adjusrui~ die reïctioii conditions to improve the rnoiiomer mobility or rn;iirieiriiig the cacdysr
;ictivity cilrd ro uicrease die conversion sigiiificmdy. This sugests thar rlie polymerizitiori IS
neirher difhsion limited nor temperature limired. Iiistead, it is belirved rhar SIS(: is occurriii~ by
rr~ppuig rlie monomer within sPS's helicd cliaiiis. It is this entnpmrnr of rnoiiomer rliiit is c~usiiig
the polymeriz~tion to c m e preminirely before cnmplete coriversiori c m be reaclied. Furtlier snidics
into controlluig die type of sPS cq-snlliiie fom produced during die po1yrneriz;~rion srem w~ircmred.

Chapter 4 & 5
Conclusions and Recomrnendations

4.0 Conclusions
In this study of the sPS RIM polymetiz;ition, che effects of die esreiit of misuig, reacrioii
Ume .and mould 4 rempenrure on the monomer convesion a id m t e d properties w r e
mves tigated.
For the benchmark polpecizations at room tempenmre a id with a reaction tirne of oiie
hour it \vas found that die average conversion was 76%. n i e rcsulùiig sPS fraction \vas 63% mid
exhibited a m o l e d x weight of 126,000 g/rnol with 2 polydispersity of 3.7. The ;iPS furmeci u;is
luw moiecdar weigfit between 1U,i)U(J-3U,1)00 g/mol. The sPS polymer wiis steriçdly puft. ;la
deterrnined by NXIR, wliich was dso contirmed by die corresporidixig meltitig poiiit of 267OC.
I r was fouiid d u t v q i i i g the mould wall tempenture Iind die most promuieiit rffeçt oii tlic
RI.\[ polynerizition. Controlling the modd wd rernpemtuce had the overdl e f k t of moder.itiiig
die average reactiori temperature. Iiicreasirig the mould wÿll rempemture to I loO(: decreiüed tlic
conversion ro 70?0 and lowered die sPS fcictioii to 470/0. Decreasiiig the mould \ d l tempeciturr
below room remperxure increiued die coiivesioii to 81% but rempecinires belmv 0°(: ciecre;i';ed rlic
sPS fr~ctioii to 47Oh. I t wiis dso fouiid diat die moleculx weidit of die sPS polymrr ctiultl Iie
iricreased to 20( ),Ui )(.) g/mol the inould tempeciture was lowered, iiidicitiiig tliat Iess P-liydrogm
elirmnarioii occurj. Tlie stereoregul:irity ;uid me1 ting point of the sPS wece uiiaffected bu the mould
w - d temperature sliowiiig the syndiouctic insertion mectimism for die Cp'Ti\[e, systern \v:is
uiiaffected.
I t wifi Found that longer polymerization times mi iiicrease the moriomer coiiversioii up ici
t31"/0 but it w u discnvered tliat the majoriry of die po1ymeriz;itiori is romplete (76%) ;ifter 2 m~iiures-
However widi re;iction times g a r e r di-m 6 lm, it was fouiid tliat ilii iiicre;ise in tlic sPS mcilec~il:ir
weight to 3U0,OW g/mol occurred. The sPS fmctioii was uriaffected Liy the re;ictioti time itidic;itiiig
the catdyst mechviism rmiiiniris the bdrince of syndinnctic ;uid ;icictic polymer. llicse rcsdrs slio\v

diat the cataiyst system cemains active ÿfter passing through a pe.A tempeclnire of + 130°(: 31d
conwiued polymerization c m occur.
The estent of rnising of the RihI polymerization showed litde effect un the conversion iuid
sPS fr~ction indicatkg that the process is not rnixuig Iimited. Hoivever, a sliglit uicresr in rlir
crmvrrsion to 789!0 wÿr observed with a I.xger diÿmeter mishead, suggesring die :unouiit of interfKi:il
ue~/cotitact may be of importance.
Giveii rhe inability to increase the conversion significmtly uiider the variety of coiidiriciiis
anempted, it is believed diat the sPS polymerizaaon is neither diffusion lirnired nor remprrJnire
limited. In~esti~qtions of the cq-stailine nature of the sPS polymer produceci ceraird that r l i r SI'S
polyner assumes ï tielical confomÿtioii, nÿmely die Gform, diat c m oidy be h m e d by the proccss
of solvent uiduced crystdlization. it is hupodiesized diat during tliis SISC prticess, tlic moiiomrr l s
bring rr~pped widiiii die crystduie regioiis in 3 sryrene-sPS complcs. DSC ÿiidysis lias iiidicared tlic
w-produçed sPS is I i i f l y crystdine wliicli suggests tliat this cryscd eiirclprnetit 1s Iiigtily p1:tudilc.
Tli~is ~r is believed diat the rianire of die conversion limitanori u i die sPS R b 1 prcxxss is liriked r o
the eiitnpment of styene monomer widiin the sPS Iielical chains.
In grierd, it w a s fouiid diar by v q i ~ i g the misuig medind, reactiuii time iuid moiilci \v:iII
temperature, oidy ï rni~urnurn conversion of 8 1 O 4 1 /avs achieved. Ciider bulk conditions, ir \v:ü
discovered diat die m a j o r i ~ of the raction is complered iii 3 miiiutes imd t h r rtic
CpTi!IeJB(GFs), catalys t system was faidy robusr in producing sPS poly-mer uiider die varie- o f
conditions investigted. r\ conversion limitatioii to the sPS polymerizatioii dors esist ;uid ~r ic,
believed to be mhereiit to the niiture of the crys~dliiie polymer.

5.0 Recommendations and Future Work
This study and previous studies have revded that die developmeiit of the sPS RIlI proçrss
is quite complicated and the high conversions required are stdl unattaïn;ible.
In the rnixuig study, it was found chat a larger diamerer rnislir~d promoted E-ter misuis
which m i have incrased the conversion slightly. During scde-up of this pnicess, diis Eicror
become impohuit ;md hence, a snidy inro die effect of interfacial coiiracr widiui diifereiir midirïd
desigis mi? be IieIptL1.
It wu evidenr in die mould nid rempemture study, rhat coritrdlhg the rractioii tempeciturc
1 challenge. I r was mentioned chat a higjdy specialized apparatus would be required for rfficictir
heat tram fer. Investigations uito usbis a mould constructed out of metai witli a 1;ug.e sururt:.içr ;uei
m q dlow berrer coiitrol over die ceaction temperature. ;Utliougli it is doubttLl diat Ixge iiicre:ism
in the conversion would occur, the polymer properties would benefit.
I t is uiihowri whether die coiiversioii limiratioli is catrilytic. It would be iritere~tirig ro
invesripte tlie e Eectiveness of die iiewer, more s nl~le, higlier tempenicure ç:it;Jys ts d ix liai-e becti
recendy discovered. Higher polymeriz;itioii remperdtures m ~ y prevriit/del:iy the tiirmari<iii of rlic
ityreiie-sPS cornples. .A more stable cardyst will dso preveiir the rn;iterid property deteriomrioii ;ir
higtier reiction temperirures.
O v e d , die most esciting coiiclusion w:ls the possible eiitmpmeiit of die s-rene moiiomrr
widiiii die crysrdliiie structure of sPS. Studies iiito the staliility of rliis s~reiic-sPS çomples rii;iy
proride :m undenrmding for its fomi~tiori which m y , ui tuni provide insislit for its preveiirioii. Tlic
use ofditTerent additives to modi. the crystalliriity miy have a sigiif?c;ult impiict on tlie cririvttrsioii.
Iiivestigations into the use of :i volatile solvent diat is pretërentid tr) ti)miiri~ ;i more stable comples
dian die styrene-sPS comples ma. have tlie desired effecr of iiicre;c;uig die conversicxi wliilr ilo ou-
e-ay remord of the solvent. Ciiforniiiarely, the choice and use of I;u-se quiuitities of iidditi\+çs in A
RI11 process is uridersir;it->Ie ;uid lirnited by the orgmomctdlic nmire of tlie c;it;dysr systcm.

References
1. .\Iark, H., Biliales, S., Overberger, C., hfenges, G., Encvc1opedi;i of Polyrner Scieiice iuid
EnPineerïn~ LTÏiey and Sons tnc., Toronto, 16, 1987, p. 1
2 Ishihara, N.; hramoto, M.; Smd. Surc Sci. Catal., 1994, 89,339-3.
3. Ishth-xa, S.; Seimiya, T.; Kuramoto, M.; Uoi, M.; .Ilacromolecules, 1986, 19,34652466.
4 Guem, G.; .\Lusto, P.; b z , F. E.; .LIachight, WJ.; bfaliromol. Chem., 1990, 19 1.21 11-21 19.
5 Musto, P.; Tavone, S.; Guerm, G.; De Rosa, C.; J. Polym. Sci., Pm B, Polym. Phys., I1197,3j, 7.
lO55- 1U66.
6. Sapolitano, R.; Ptrozzi, B.; Macromolecules, 1993,26,7325-7338.
7. Vittoria, V.; De Caridia, F.: Caroteriuto, hl.; Guadiigno, L.; J. hhcrcmol. Sci. Phvs R lL196. 35, 2.
365373.
Y. I'ittocia, V.; Russo, R.; Polymer, 1991,32, 18,3371-3375.
1- Deberdt, F.; Bergturims, K.; Polymer, 1993,35, 10,3193-3201.
li 1. Roels, T.; Deberdt, F.; Ber@irnaris, H.; .L[acromolecules, 1994, 27,G216-032iL
1 1. Deberdt, F.; Bergtu-nms, H.; Polymer, 1994,35,8, 1694-17W.
12. Guenet, J.11.; Deluca. MD.; Daniel, C.; Polymer, 1996,37,7, 1373- l28( i .
13. Chat:rili, 1-.; Shimme, Y.; Itugki, T.; Polymer, 1993,34, 8, 1620-11134.
14. lI.mfiedi, C.; Del Sobile, hI..A.; hlensitieri, G.; Guerra, G.; Rap;iccruolo, M.; J. Polym. Sa., Part
R, Polym. Phys., 1997,35, 133-140.
15. Sinri, H.; I;;imu~sLy, K.'.; .-\dv. Orcgaiomer. Cllem., 1980, 18, 09.
16. Kmuisky, W.; Mri, AI.; Sinn, H.; VUoldt. R; hLakrornol. Cliern., Rapid. Crimmuii., 1083,-t, 417.
17. Ewen, J .; Sci. .&II., 1997, My, 86-91.
18. Campbell, R.E.; Newman,T.H.; Malang, MT.; .Lfacromol. Symp., 1997,97, 151-160.
1'1. Zmbelli, .A., Pellecchia, (1.; Proro, ri.; 1IacromoI. Surrip., 1795, 89, 373-383.
3?,. Pellecdiia, C.; Longo, P.; Proto, -A.; Z4unbelli, .A.; ,Lhkromol. Ctiern., Rapid Commun., 1002, 13,
365-768.
31. Longo, P.; Proto, -A.; Zambelli, -\.; Macromol. Cliem. Pliqrs., 1995, 196,30153029.
23. Ki;mg, Q.; Quyoum, R.; Gillis, D.; Tudoret, A1.J.; Jeremic, D.; Hutirer, B.; Biurd, >K.;
Orguiomet;illics, 1996, lj,693-7( 13.
33. Kuclit, H.; Kucht, -1.; Chieri, J.; Rausch, LI.; -\ppl. Organomet. Cliem., 1994, Y, 393-3911.
34. PO', R.; Cardi, S.; Prog. Polvm. Sci., 1996,21,47-88.
25. Chien, J.; Sdajka, 2.; Dong, S.; hhcromolecues, 1992,35,3199-3203.
20. Gcissi, .A.; Pellecchia, (1.; Oliva, L.; lhcromol. (:liem. Phys, 1095, 100, li 103- 1 1( i t 1.
27. Gclssi, -4.: zarni~ell~, -A.; ( k~u~c)me~dI ic s , 1906, 15, 2, -+si 1, 482-

28. \..mg Q.; Baird, .1I.C.; .Llacromolecules, 1995,38,8021-8037.
29. Foster, P.; Chien, J.; Rausch, U.; OrpomerJlics. 1996, 15, 24(KZ4OL).
30. Ishhara, 'I .;Kumoto, hi.; Uoi, !d.; Macromolecules, 1988,21,3336-33Gt).
31. Gmsi, A.; Lmberti, C.; Zmbelli, A.; Llinpzzi, 1.; Xlacrornolecules, 1997, 30, 7, 188C1880.
32. Chien, J.; Salajka, 2.; J. Polym. Sci., Part .\, Polym. Chem., 1991,39, 133-1263.
33. Duncdf; D.; Kiade, H.; Watersori, C.; Derrick, P.; Haddleton, D.; . I lcCdey, -A.;
,L~acromolecules, 1996,39,6399-6-103.
34. I lark, F i . ; Bikales, 3.; Overberger, C.; Menges, G .: Encyclopedia of Pol -mer Science ;uid
Enpineerin~ Jolm Wdey & Sons., 16, 1987, pp 1-36.
33. Russell, K.E.; Tobolsky, A.V.; J. ;\m. Cliem. Soc., 1953,755052-5( 154.
36. llark, H.; BiMes, S.; Cherberger, (3.; Menges, G.: Enqclo
Envineerinc John mile? & Sons., 7,1987, pp 501-514.
37. Odiai, G.; Pririci~les of Pol mrrintioii, 3" Ed., Jolin \Wey Kr Som Iiic., Toronto, 199 1, p. 28-5.
38. Rudin. A.; R i e Elemerits of Polvmer Science aici Er rieetirlg, - k d e m i c Press, Toroiito, 1983.
pp. 93( 1-73 1.
30. .LImeii, F.L., Hamielec, .\.E.; J. -\ppl. Polym. Sci., 1983, 27,489-505.
40. htacosko, C., F~iiid;unenr,ds nf Reictinn Iniection .LIci\ildiiig, Hanser, Sew York, 1989, (a) pg. 1.
@) Pa. 6, (c) pg- 7, (d) pg. 3, (e) pg. 709, (t) pg. 87, @ pg. 93, (11) pp. 93-L10, (3 pp. 193-200, (1) pg.
199 0;) pp. 177 - 182
41. llark, H.; Bikales, S.; (Sverberger, (1.; hienges, G.; Eric
Engirieeriw, John Wley & Sons., 14, 1987, pp 73-74.
43. Geer, R.P.; Stoutiand, R.D.; Proc. of the . h ~ u ; i l (lotit of the Socle' of Plastic Gigineers, 198-5,
1232-1235.
43. Liu, T'Al.; Baker, W.E.; Scliyn, \-.;Jolies, T.: Baird, MC.; J. 'ippl. Pol-m. Sc;., 1 W G , 62. 1 1,
1807-1828.
U. Cnpublished work in W.E. Baker's labontory, 1995.
5 . .\lena, 11.; Royo, P.; Serrmo, R.; Pellinghelli, M.A.; Tiripiccliio. :\.; (>rguiomer;dlics, 1089, 8,
476482.
16. llassey, -i.G.; Park, A.J.4. Orpiomet. Chem., 19W, 2,245.
47. Harnbv, S.; Edwards, 11.F.; Nienow, .4.W.; ,Ilisiiw in the Prncess Iridustrie5, Rutrenvortli ,uid
(30. Ltd., 1985, p. 228.
48- Evans, .-\..\.I.; Keiiar, E.J.C., G~owles, J.; Galiotis, C.; Cxriere, C.J.; .-ùidrews, E.H., Polym. Lis.
Sci., 1997,37, 1.
49. Rrmdup,].; Polvmer H;uidt~otk, 3d Ed., Joliii \Y'iley & Srms Iiic., 1075, p.

3. Sperluig, L.H.; Inrrodtiction to Phvsical Polvmer Science, 2nd Ed., J r h i K ï l e y J: Sons Iiic.. ILIL)?.
p. 353.
51. Park, J.Y.; Lee, H.S.; Park, 0.0.; Conf. Proceedùig of the 13" .Guiual hleering of die Polymer
Processing Society, 6B, Secaucus, SJ., June 1997.

Appendices and Curriculum Vitae

Appendix A - Typical TGA Therrn~~parn of sPS RIM Sample

Appendix B - Randornized Experimenml Run List
1 Run # Run Description 1 Peak Temperature (OC) 1 1 Benchmark (Bhn #1 118
3 - - No Static iCLs.in,q (NSiLI) #1 65 - 3 Luger Dimeter ALsing &DM) #l 126 4 No Static iCLising (NSiLI) #3 131 - 3 Test Tube Xliuing (rrlcr> 128 6 Benchrnd (BM) #2 126 7 24 hr #1 124 8 10 mui #1 133 9 2 min #1 102
2 min ff3 I
Additional Replicare Runs frcm i I
Preno~s SN& Run # Run Description Peak Temperature (OC') 9 9 -- IO min #2 100

Appendix C - Borane Prepolymerization Study
l[olecuiar K'eight @/mol)
Styrene Conversion
\Veight of aPS Polymer Ce;)
Run Condition Initial Wt.
Styrene


Appendix D Cont'd - 1H NMR of aPS

Appendix D - Cont'd U C NMR of sPS

Appendk D - Cont'd U C NMR of aPS

Appendtr E - DSC Thermal Trace of sPS Sample
SCAN P A R A M E T E R S S T A R T TEHP. OC R A T E K / H I N . END T E M P - OC T f M E ISO, M I N . P L O T CM R A N G E F S m U OFFSET .Y / O
P A N T Y P E 1/2 L I M I T ' mW SCREEN D Y N / I S O 1 1 2
F I L E N O . EDENT. N O . WE 1 GHT mG
TEHPERa TURE JC
5 0 10 ZOO O 20 20 8 0
HEAT FLOW EXOTHERMAL-->
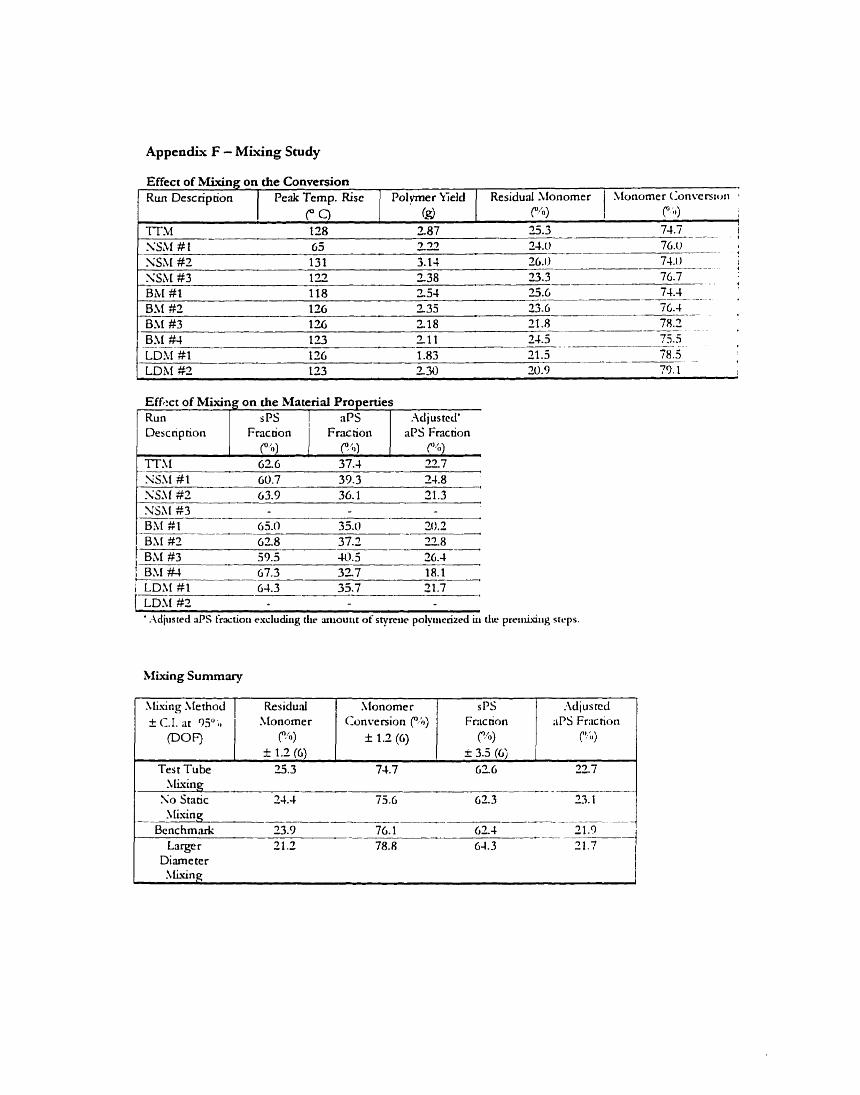
Effect of Mixine: on the Material Prouercies
Effect of Mwng on the Conversion
' :\djiisted aPS fraction c.ucludiiig die miouiit o f s ~ r e i i e polviirerized Li die prrriisiig srrps.
1 Run Descnp rion
e q ce;) e/4 e 4 l +
'ITM 128 3 8 7 35.3 74.7 - -- I
SSM #l 63 -- 7 77 24.1) 76.U I --P -- ------- t
74.1 1 S S N #2 131 3.14 26.1) -- 1 I
SSM #3 139 3 3 8 23.3 -- 76.7 - BM #l 118 754 25.6 - 74.4 - --
t t
B M #3 136 335 23.6 -- 76.4 -- ?
B M #3 126 7 1 8 21 -8 78.2 - +
BAL . 123 2.11 24.5 --- -- 75..5
LDM $1 1 2G 1.83 21 -5 78.;- -
LDM $2 123 330 20.9 79.1
Polpner kïeld Residud Monorner Run Description
Large r 31.2 78.8 64.3 31.7 Diame ter LLuing
.Clonorner Conversiori ! Peak Temp. Rise
TT1 1 63.6 37.4 937
ALixing !dethoJ + (7.1. ar 95" ;,
P O FI
-Adjus tcd' aPS Fraction
e.3
sPS 1 aPS Fraction
Test Tube 25.3 74.7 6 3 6 --- 73 7
Fraction ml
:\ci j us rcd ;iPS Fr;iction
(?q
Residud Monomer
(".'#,) t 1.2 (6)
Monomer Conversion (?!O)
k 1.2 (6)
s PS Fraction
("/O)
+ 3.3 (Gj

Appendk G - Reaction Time Study
.---
2 & # 2 128 22.2 10 min #1 133 349 93.6 76.4
Effect of Reaction Time on the Conversion
Effect of Reaction Time on the ~Mnterid Properties Run s PS =\diustedg sPS Molecdar ;PS Description Fraction aPS WC. Mo1ecul:ir Wt-
Fciction [x IV g/mo[) Ç s 20' g/mol)
Monorner Conversion
?./O)
Reaction Erne S u m q
2 min $1 102 349 21.8 78.2
Reac tion T i c
Pol!mer \r'idd
(3 Peak Temp.
Rise
eci
--. 39 L3.7 - -- 16 (3.ilJ
8.3 k4.31 9.9-~4.15] -- 3?- [3 - -__
10 min ft3 53.7 32.0 129 I hr #1 65.0 20.3 136 1 hr #2 62.8 23.8 167 I hr #3 59.5 3G.4 104
Residual lionorner
PO>
3.1 '4.0 '3.3 ,4.2
A Lising .Ilcthod
f C.I. rlr 930, O
@OF)
1 hr #4 67.3 18.1 I i JO 6 hr 65.0 21 -4 185
-3.4 4.5:
sPS Molecular N' t .
(s 1(Y g/mol)
i 22 (4) PDrI
Residud 1 Monomer sPS Fraction
ea) i 3.4 (4)
;rPS h11~Iccul;~r \K.;. ( s I W . q r n l ~ ~ ;
PD11
.Vonorner P :))
i U.3 (6)
.idjustecl <u)S Fmction
(%)
Conversion c '4
i 0.9 (6)

Appendix H - Mould Wall Temperature Study
Effect of Mould Temperature on the Conversion Run Description 1 Peak TemD. Rise 1 Polymer k'ield 1 Residud Monomer 1 !vfonorner
I I cj 0 e,o> 1 Conversion (" :,)
- - ,-- -- - ---
23 O C : #2 126 335 13.6 - 76.4
33 O(: #3 12G 2.18 21.8 78.3 --- -- 9- C
33 OC A 4 133 2.1 1 3-35 13.3 - -----
GO "C A-
130 2-60 5.0 75.0 -----
110 O C #1 140 205 373 -+ 67.7
1 10 OC- #7 136 333 28.3 71.7
MouId Temperature Summary
Effect of Mould Temperature on the Material Propenies s PS
T m m ~ 1 r' 11) 1
1 -'6 - 47.3 39.6 201 [3.91 365
t--- * - + -
VI + -30 OC: 55.5 31.3 186 [ 3 4 i l [-!-Cl 26 -5 .------- - - - -- - . - -. - - - . iJ O C #l 67.3 19.3 171 [3.9] - 10 [4.9] 266 .- 99t . -- i l O C #2 60.6 36.3 337 [3.4 .- ---- - - --- - - - - - -- - -
33 O C ##I 65.0 20.2 ---- 136 [4.0] - 29[3.q
A--.------ -- -.-- -
33 =c #2 --
62.8 32.8 167 [3.3] 16 [5.01 367..5 9% - - - - - -- . - . -- - -
33 O(: $3 65.0 20.9 10-3 [421 8.3 [4.,51 , - - .- - . -- - -- --- A---p- - - - - - . - .
23 O C , +#4 67.3 18.1 l( IO [3.41 9.8 [-!.GI 266 .--- - -* - -
60 O C : 56.3 38.8 143 (3.61 14 [4-6] 267 --- -- -* ---- -
110 "C #I 47.3 36.5 82 [381 363 99 + .--PT - p- --4- - .
110 O C #2 47.4 37.3 71 [2.91 24 15.81 ' =\djilsted &'S frxtiori cxcludiiig die miourit oistyrcrie polytiierized in die preiiii.uirig steps.
.\ Iould Temperature
.id jus ted' riPS
Faction V f n )
s PS Fraction
? 4
sPS Molecular Wr. ( X I @ Q / ~ O I ) PD11
a PS Molecuiru K't.
( K I W ~ ~ / ~ O ~ ]
FDrJ
' sPS l k l a n g Point

Appendix I - Crystaiiinity Estimates of sPS Samples
The "k crystdlinity h a k e n adjus ted based on the 'mount of crystdlizablc sPS
sPS Friction
S;imple Description
Monomer Conversion
Bk[ #3 (Cridried) 14.50 243.31 16.8 6.75 36.1 31.3 6 7 3 ------ Blf #3 (Dried) 5 . 0 109.83 22.0 3.98 36.9 41.3 -- - . -. ---
* 69.1 1 BA[ #4 (t ndried) l(J-5( 1 187.98 17.9 5.33 33.3 33.7
- - - 66.3
-26 OC Wndried) 1O.0O 170.4 17.0 3-95 43.1 32.( 1 -- - 81.0 j 110 O C Kndried) 1(!-25 1 23 .O6 17.0 3 -48 35.3 32.6 66.1 j
S-rdliiiity
(O O )
hIelt Exirhdpy
(r /&
-\dpistcd~ 1 S - ~ d i m n ~ 1
! II 'a
DSC Sÿmple Endothem Enthdpy
cmn
Ssample LVeigtir
h p ; )
hfelt Enthdpy
O&
Crvsnllizable Weight (mg)
Copyright © 2022 FDOKUMEN




















