
Devices and systems targeted towards augmented robotic radical prostatectomy

Prostate Cancer: Screening, Prevention and Therapy –Lessons Learnt from Current Trials
Proceedings of an International ConsultationOrganized by the WHO Collaborating Center for Urologic Tumors
Stockholm, SwedenSeptember 8-10, 2010

Prostate Cancer: Screening, Prevention and Therapy –Lessons Learnt from Current Trials
WHO International Consultation September 8-10, 2010, Stockholm
Hasselbacken Conference Center, Stockholm
This conference is EACCME accredited
Supporting Organizations
Acta OncologicaAnonymous donorAstellas PharmaAstraZenecaBristol-Myers SquibbBayer HealthcareCancer Society in StockholmGlaxoSmithKlineJanssen-CilagNCI, National Institutes of HealthOrion PharmaSanofi -AventisSwedish Cancer Society
Organizing Committee
Sten Nilsson Chairman, WHO Collaborating Center for Urologic Tumors Karolinska University Hospital, Stockholm
Lennart Andersson Co-chairman,WHO Collaborating Center for Urologic TumorsJan Adolfsson Oncologic Center, Karolinska University HospitalOlof Akre Dept. of Clin. Epidemiology, Karolinska University HospitalMarianne Brehmer Dept. of Urology, Karolinska University HospitalGabriella Cohn-Cedermark Dept. of Oncology, Karolinska University HospitalPeter Ekman Dept. of Urology, Karolinska University HospitalPer-Uno Malmström Dept. of Urology, Uppsala University Hospital, UppsalaMartin Schumacher Dept. of Urology, Karolinska University HospitalBernhard Tribukait Dept. of Med. Radiobiology, Karolinska University HospitalAnders Ullén Dept. of Oncology, Karolinska University HospitalPeter Wiklund Dept. of Urology, Karolinska University Hospital
Secretariat
Diana Eriksson WHO Collaborating Center for Urologic Tumors Karolinska University Hospital, Stockholm
Conference agency Meetagain Konferens AB [email protected] www.meetagain.se
Additional scientifi c contributions from:Øyvind Bruland, Oslo, Jan-Erik Damber, Gothenburg, Bo Lennernäs, Gothenburg,Seymour H. Levitt, Minneapolis, Oliver Sartor, New Orleans,the WHO Center is under re-designation.

Contents
Introduction
1Introduction from the chairmen of the WHO International Consultation on Prostate cancer
Nilsson S. & Andersson L.
Editorial
2Screening for prostate cancer: Defi ning critical issues
Holmberg L. & Akre O.
Review Articles
4Screening for prostate cancer – The controversy continues, but can it be resolved?
Bul M. & Schröder F. H. 12
The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial: The prostate cancer screening results in context
Berg C. D. 18
Early detection of prostate cancer with emphasis on genetic markersAly M., Wiklund F. & Grönberg H.
24 Introduction
Joniau S.
Original Articles
25State-of-the-art uroradiologic imaging in the diagnosis of prostate cancer
Heijmink S. W. T. P. J., Fütterer J. J., Strum S. S., Oyen W. J. G., Frauscher F., Witjes J. A. & Barentsz J. O.
39Developing imaging strategies for castration resistant prostate cancer
Fox J. J., Morris M. J., Larson S. M., Schöder H. & Scher H. I.
Review Articles
49Pathology in prostate research: Optimizing the pathological data
Berney D. M., Montironi R. & Egevad L. 53
Pathology in prostate research: Optimizing tissue quality Berney D. M., Montironi R. & Egevad L.
56Role of histopathology and molecular markers in the active surveillance of prostate cancer
Montironi R., Egevad L., Bjartell A. & Berney D. M. 61
Tumor markers in prostate cancer I: Blood-based markers Shariat S. F., Semjonow A., Lilja H., Savage C., Vickers A. J. & Bjartell A.
76Tumour markers in prostate cancer II: Diagnostic and prognostic cellular biomarkers
Bjartell A., Montironi R., Berney D. M. & Egevad L. 85
Tumour markers in prostate cancer III: Biomarkers in urine Roobol M. J., Haese A. & Bjartell A.

Introduction
90Introduction: Therapy with curative intent
Nilsson S., Cohn-Cedermark G. & Wiklund P.
Review Article
92Radical retropubic prostatectomy: A review of outcomes and side-effects
Hugosson J., Stranne J. & Carlsson S. V.
Original Articles
98Curative radiation therapy in prostate cancer
Harmenberg U., Hamdy F. C., Widmark A., Lennernäs B. & Nilsson S. 104
Four and fi ve dimensional radiotherapy with reference to prostate cancer – defi nitions, state of the art and further directions – an overview
Lennernäs B., Nilsson S. & Levitt S. 111
Hypofractionation for radiotherapy of prostate cancer using a low alfa/beta ratio – possible reasons for concerns? An example of fi ve dimensional radiotherapy
Lennernäs B., Nilsson S. & Levitt S.
Editorial
116Natural history of prostate cancer, chemoprevention and active surveillance
Bratt O. & Schumacher M. C.
Review Articles
120When is active surveillance the appropriate treatment for prostate cancer?
Albertsen P. C. 127
Chemoprevention of prostate cancer Rittmaster R. S.
Editorial
137Management of advanced prostate cancer – new drugs
Ullén A., Shah C.-H. & Kirkali Z.
Review Article
141Broadening horizons in medical management of prostate cancer
Niraula S. & Tannock I. F.
Original Article
148Prostate cancer from the horizon of the patientDenis L. J., Roobol M. & Dourcy-Belle-Rose B.
Commentary
155WHO International Consultation on Prostate Cancer: A summary
Albertsen P. C.

INTRODUCTION
Introduction from the chairmen of the WHO International Consultation on Prostate cancer
The reports from the two large PSA-based screening studies of prostate cancer reported in 2009, ERCP in Europe and PLCO in USA and Canada, supple-mented by a Scandinavian trial, based in Gothen-burg, have widened our understanding of the value of systemic screening and, at the same time, thrown light upon the shortcoming, or rather imperfection, inherited in studies based upon PSA values. These data inspired us to undertake a more wide front updating of not only screening bus as well diagnostic procedures and therapy of prostate cancer. What has emerged in modern research with respect to prog-nostic markers, evaluation of tumor extent and pro-gressive character, treatment modalities, etc? When is active surveillance justifi ed? Feasibility of prophy-lactic measures? Are some of the drugs in the big fl ora of substances with a presumed cytostatic prop-erty of any value in the treatment of prostate cancer? And – not least – how can the profession collaborate with our patients and their organizations to arrive at optimal solutions in situations, many times of a dif-fi cult character – when it comes to the selection of treatment modality.
With all this in view, we decided to invite a faculty of scientists representing a manifold expertise and all
of them selected among those world-leading at present, to collaborate with us to focus on these issues in the light of modern research. After a period of pre-paration we came together for an open conference in Stockholm, entitled ” Prostate Cancer: Screening, Prevention and Therapy – Lessons Learnt from Cur-rent Trials. WHO International Consultation ” . This book presents those conclusions we arrived at. In case at least some of these of our conclusions will stand the test of time, our common efforts will be worthwhile. We even hope our reports will open a window to new research. And – like in all our inter-national consultations – the ultimate aim of our efforts is to serve our patients.
We wish to convey our warm thanks to all our Faculty members for your devoted contributions at non-profi t conditions and for our stimulating debates. We also wish to convey our sincere thanks to our supporting organizations. Without your fi nancial and idealistic support this endeavor would never had become a reality.
Sten Nilsson Lennart Andersson
Acta Oncologica, 2011; 50(Suppl 1): 1
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.542176

Correspondence: [email protected]
(Received 6 September 2010 ; accepted 17 September 2010 )
EDITORIAL
Screening for prostate cancer: Defi ning critical issues
LARS HOLMBERG & OLOF AKRE
King’s College London, Medical School, Division of Cancer Studies, London, UK and Karolinska Institutet, Department of Clinical Epidemiology, Stockholm, Sweden
Several facts point towards that prostate cancer in theory is a suitable goal for secondary prevention with screening. It is a common disease in many coun-tries with often a malignant course when detected clinically. Clinical symptoms are unspecifi c or few and clinical diagnosis often means late diagnosis in a stage where medical interventions cannot today claim to be curative. There is presently no clearly identifi able life style risk factor that could be changed to infl uence the incidence substantially. There is a long preclinically identifi able phase where the disease is limited and with a low probability of having produced clinically viable micrometastases during that time. For the last 5 – 10 years there has been increasing evidence that locally radical intervention at a stage where the disease is limited can save lives [1,2].
However, we already knew when the randomised studies to evaluate the effi cacy of prostate cancer screening [3,4] started that two major obstacles stand in our way to realise expectations of a successful screening program: First, the current means to detect prostate cancer in a symptomatic stage – PSA testing followed by biopsies – will diagnose a substantial number of men with cancer that would not have sur-faced clinically during their lifetime, thus causing overdiagnosis [5,6]. Our hope to fi nd a reliable method to distinguish the patients that benefi t from treatment from those that do not remains unfulfi lled. Second, a local radical intervention for prostate can-cer is still despite many remarkable improvements in surgical technique associated with a clinically sig-nifi cant risk of side-effects that may be unacceptable given the potentially indolent course of disease. These two obstacles combine in an unfortunate way to risk of overtreatment with severe consequences that then
threatens the cost-benefi t balance certainly for small, but even for modest and clearly clinically relevant eventual mortality reductions by screening.
We now have the fi rst evidence from two large randomised trials in prostate cancer screening, the ERSPC [4] and the PLCO [3] trial. In this section of the proceedings of the WHO consultation on pros-tate cancer, further evidence than those already pub-lished in the New England Journal of Medicine will be presented and discussed. Furthermore, the trialists set the empirical fi ndings in a context of the discus-sions emerging from the fi rst publications. Given what we know hitherto from the publications, a fi rst priority is to understand the very basic fact: is there a mortality reduction to expect from PSA screening or not? This task is not trivial. Mass screening trials are extremely complex to undertake, analyse and interpret. A multiprofessional, multidisciplinary, care-ful, realistic and sincere approach free from irrational confl icts of interest – be they commercial, political, academic or any – is needed from all stakeholders. We need to understand that we are only in the beginning of the follow-up and of this discussion. It is sobering to think that the fi rst randomised data in breast screening now came some 40 years ago, and still, though a majority of experts and reviews are very clear about a mortality reduction following mammog-raphy screening, new debates pops up from time to time and health care providers many times get con-fused. The same is true for screening for colorectal cancer, where as in mammography screening the ran-domised data are so much richer and older than for prostate cancer screening. The medical community should show that we have a collective memory and learning so that we in the prostate cancer debate avoid the sometimes irrational and emotional overtones in
Acta Oncologica, 2011; 50(Suppl 1): 2–3
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.529459

Editorial 3
cancer screening history, which have hindered people ’ s understanding of the screening issues.
A second task is to defi ne the most important and pressing questions that rise from the current problem situation. We suggest that rather than using a number of critical questions only as criticisms and dismissal of trial data, they should be brought forward as impor-tant research issues to be solved either with data from the current trials or in other research in existing datasets or in prospective trials. For such an approach to give answers sooner than later, the ERSPC and PLCO trialists need support for their efforts and oth-ers have to collaborate constructively in focused efforts. Some such questions are obvious and are already on the research agenda for many groups, i.e. fi nding a way to distinguish potentially lethal cancer from the very slow growing or improving the screen-ing test itself. However, such a magic bullet may not be found for many years and if screening continues, we need to engage in large studies of immediate clin-ical problems: Is active surveillance a safe option? Is there a low-toxic intervention that could be offered for men with low volume disease with benefi cial prog-nostic markers? Such studies need to be done in large, collaborative networks. In this section of the proceed-ings there is an important discussion about the pos-sible contribution from genetic biomarkers to solve these problems.
If the results of these two tasks can be clearly formulated it will go a long way to help us with the ultimate goal: to have science inform men and health care providers what the current state of affairs tells us about prostate cancer screening, what is known,
which are the great enigmas and what are the pos-sible clinical implications. We cannot in the long run shy away from trying to solve one of the most impor-tant medical and ethical dilemmas today: should screening with PSA for prostate cancer be stopped, should it be encouraged on a large population scale, or should it only be offered after a very careful indi-vidual information including an informed consent? This problem solving requires a lot of very good, honest collaborative teamwork.
References
Bill-Axelson A, Holmberg L, Filen F, Ruutu M, Garmo H, [1] Busch C, et al. Radical prostatectomy versus watchful wait-ing in localized prostate cancer: The Scandinavian prostate cancer group-4 randomized trial. J Natl Cancer Inst 2008; 100:1144 – 54. Widmark A, Klepp O, Solberg A, Damber JE, Angelsen A, [2] Fransson P, et al. Endocrine treatment, with or without radiotherapy, in locally advanced prostate cancer (SPCG-7/SFUO-3): An open randomised phase III trial. Lancet 2009; 373(9660):301 – 8. Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia [3] D, Church TR, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009;360:1310 – 9. Schroder FH, Hugosson J, Roobol MJ, Tammela TL, [4] Ciatto S, Nelen V, et al. Screening and prostate-cancer mor-tality in a randomized European study. N Engl J Med 2009; 360:1320 – 8. Heinsdijk EAM, der Kinderen A, Wever EM, Draisma G, [5] Roobol MJ, de Koning DJ. Overdetection, overtreatment and costs in prostate-specifi c antigen screening for prostate cancer. Br J Cancer 2009;101:1833 – 8. Stark JR, Mucci L, Rothman KJ, Adami HO. Screening [6] for prostate cancer remains controversial. BMJ 2009;339: b3601.

Correspondence: Fritz H. Schr ö der, Erasmus MC, University Medical Center Rotterdam, Department of Urology, P.O. Box 2040, 3000 CA Rotterdam, The Netherlands. E-mail: [email protected]
(Received 16 August 2010 ; accepted 6 September 2010 )
REVIEW ARTICLE
Screening for prostate cancer – The controversy continues, but can it be resolved?
MEELAN BUL & FRITZ H. SCHR Ö DER
Department of Urology, Erasmus MC, University Medical Center, Rotterdam, The Netherlands
Abstract Background. In 2009, the European Randomized Study of Screening for Prostate Cancer (ERSPC) was one of two studies to report interim data on the effect of screening for prostate cancer (PC) on the disease specifi c mortality. Contradictory results caused considerable discussion and misunderstanding in secondary literature. Methods. This document is based on a non systematic review of recent evidence for and against screening for PC, specifi cally considering three recently published randomized screening trials [1 – 3]. Results . The ERSPC data are based on a core age group of 162 387 men, aged 55 – 69 years, who were identifi ed through population registries in seven European countries. Men were randomized between a screening group that received screening at an average of once every four years and a control group. After a median follow-up of nine years, a reduction in the rate of death from PC by 20% was shown which increased to 31% after adjusting for non-compliance and contamination. Overdetection and subsequent overtreatment (with a number needed to treat (NNT) of 48) are considered to be the major down sides of screening. The recently published 14-year results have shown that these down sides strongly depend on the duration of follow-up. In response to the outcomes of the ERSPC, several points of discussion have been brought up by various authors concerning the usefulness of screening considering benefi ts, harms and costs, the methodology of the ERSPC and the interpretation of its outcomes. Important issues to address regarding PC screening are addressed. Conclusions . This paper sheds a light on the controversial points of the ERSPC as well as on the priority issues of PC screening. On July 2, 2010 the Swedish section of ERSPC (G ö teborg screening trial) published their results with a median follow-up of 14 years. With longer follow-up the data confi rm the trend seen in improvement of PC mortality and suggest much more favorable future outcomes also with respect to the NNT to prevent one PC death.
In 2009, two large, randomized, controlled trials [1,2] reported their interim data on prostate cancer specifi c mortality, addressing the matter of screening for prostate cancer (PC). The two studies have appar-ently contradictory results, which created consider-able discussion and misunderstanding in secondary literature. Recent literature profoundly outlined the differences between the two trials and the underlying causes explaining the contradictory outcomes [4]. However, as one editorial stated [5], the main point regarding present prostate cancer screening studies, is to avoid meaningless controversy over which study is right or wrong, thus hiding the real issues behind a smoke screen.
In this paper we would like to defi ne the results of the ERSPC as well as its supposed controversial points as they were mentioned in various communi-cations, in order to throw light on different sides of
the study and to report the priority issues and the way they should be taken on. In addition, the recently published results of the G ö teborg screening trial [3], which is part of the ERSPC, will be considered in the context of the ERSPC trial as a whole.
Evidence from the European Randomized Study of Screening for Prostate Cancer
The European Randomized Study of Screening for Prostate Cancer (ERSPC) [1] was initiated in 1991 as a randomized, multicenter trial of screening for PC, with PC mortality being the main endpoint. In eight different countries men, aged 50 – 74 years, were ran-domized between either a screening group or control group. The core age group consisted of men aged 55 – 69 years (N � 162 387), who were screened with a four-year (87%) or two-year (13%) interval. Screening
Acta Oncologica, 2011; 50(Suppl 1): 4–11
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.522197

Controversies in Screening for Prostate Cancer 5
was performed by means of a prostate-specifi c anti-gen (PSA) test, with a cut-off value of 3.0 ng/ml as an indication for lateralized sextant biopsy. Never-theless, slightly differing PSA cut-offs of 2.5 – 4.0 ng/ml with ancillary tests as digital rectal examination, free to total PSA ratio and transrectal ultrasound have been used by different centers during the study period as was all clearly reported earlier in the orig-inal paper (Table I).
After randomization, a total of 72 890 men were assigned to the screening group and 89 353 to the control group, with a mean age of 60.8 years. Of all PSA-tests performed, 20 437 (16.2%) were positive and led to biopsy recommendation, which 17 543 (85.5%) of men complied with. In 24.1% of men biopsied, results turned out positive (positive predic-tive value or PPV), amounting to 75.9% of false positive results. PC was detected in 5 990 (8.2%) men in the screening group and in 4 307 (4.8%) men in the control group.
After a mean and median follow-up of 8.8 and 9.0 years respectively, 214 PC deaths occurred in the screening group and 326 PC deaths in the control group (Table II). This corresponded to a signifi cant reduction of 20% fewer men dying of PC in the screening group (p � 0.04). The cumulative risk of death from PC over time for both groups is shown in Figure 1, with diverging rates of death from seven to eight years on and a trend suggesting larger effects with longer follow-up.
The intention-to-screen (ITS) analysis showed an absolute risk reduction of seven per 10 000 screened men, with a number of 1 410 men who
Table I. Methods used for the ERSPC.
Recruitment Men identifi ed from population registries
Randomization- Before consent Finland, Sweden, Italy- After consent Netherlands, Belgium, Switzerland,
SpainCore age group 55 – 69 years (range 50 – 74 years)Biopsy indication
- Netherlands PSA � 4 or � ve DRE or TRUS at PSA � 3; since 1997 PSA � 3
- Finland PSA � 4 or � ve DRE or TRUS at PSA � 3
- Sweden PSA � 3- Italy PSA � 4 or � ve DRE or TRUS at
PSA � 2.5- Spain PSA � 4- Belgium PSA � 4 or � ve DRE or TRUS- Switzerland PSA � 3
Biopsy procedure (lateralized) sextant biopsiesScreening interval 4-years (87%)
2-years (13%)
PSA � prostate specifi c antigen (ng/ml), DRE � digital rectal examination, TRUS � transrectal ultrasound.
Table II. Results of the ERSPC.
Screening group Control group
Number of participants 72 890 89 353Mean age (years) 60.9 60.7Mean follow-up (years) 8.8 9.0PC detected (%) 5 990 (8.2%) 4 307 (4.8%)PC deaths 214 326
ITS analysis Secondary analysis
Mortality reduction 20% 30%Metastatic disease
reduction25% 32%
PSA � prostate specifi c antigen, PPV � positive predictive value, PC � prostate cancer, ITS � intention to screen.
need to be screened (NNS) and another 48 men who need to be treated (NNT) in order to prevent one PC death. When adjusted for non-compliance, a 27% reduction in PC mortality was seen in men who were actually screened. After adjustment for the differ-ences in stage distribution between the two arms, no difference was seen in treatment, which makes a mortality reduction solely caused by a treatment effect very improbable [6].
One of the major drawbacks of PC screening in general is detecting PC in men who would not have clinical symptoms during their lifetime if it was not for screening, with an over detection rate in the ERSPC screening group that has been estimated to be as high as 50% [7].
A secondary analysis [8] was carried out accord-ing to the method described by Cuzick et al. [9] to assess the impact of adjustment for men assigned to the screening arm, who did no undergo screening ( non-compliance ) and for men assigned to the control arm, who sought opportunistic PSA-based screen-ing ( contamination ). This analysis showed a further
Figure 1. Cumulative risk of death from prostate cancer.

6 M. Bul & F. H. Schröder
enhancement of the PC-specifi c mortality reduction after adjustment for both non-compliance and con-tamination by up to 31%.
The effect of this secondary analysis on the rate of metastatic PC was analyzed by Kerkhof et al. [10]. The ITS analysis resulted in a signifi cant reduction of 25% (RR 0.75, 95%CI 0.59 – 0.95, p � 0.02) for developing metastatic PC in the screening arm, with an even more distinct reduction of 32% (RR 0.68, 95%CI 0.49 – 0.94, p � 0.02) in the adjusted analysis reckoning non-compliance and contamination. This hence leads to an improvement in the relative risk of 7% in the secondary analysis. While the ITS analysis is diluted by non-compliance and contamination it shows the effect on the study population as a whole, where the secondary analysis has the ability to give a better adjusted estimate of the effect of screening at the individual level for those men who are actually screened.
In the meantime the results of the G ö teborg screening trial have been published [3]. This trial was initiated as an independent study in 1994 but joined the ERSPC trial shortly thereafter by signing the ‘ agreement of participation ’ , a research contract. The adjusted power calculation is based on the random-ization of 20 000 men, aged 50 – 64 years, identifi ed in the population registry in 1994. A power of 80% was predicted with a follow-up of 14 years and a participation rate of 76% to show a difference of 40% in prostate cancer mortality by screening. These con-ditions were met when the follow-up was complete up to the end of 2008. The results show a rate ratio for PC death of 0.56 (95%CI 0.39 – 0.82, p � 0.002) in the ITS analysis and of 0.44 (95%CI 028 – 068, p � 0.0002) after adjustment for non-compliance. This resulted in a NNS of 293 and a NNT of 12, and for attendees after adjustment for non-compliance a NNS of 234 and NNT of 15. The main differences with the ERSPC as a whole are the type of random-ization, younger age, a shorter screen interval, and, most importantly, a longer follow-up due to the simul-taneous randomization of all participants in 1994.
What is true and what is not true in the controversy around ERPSC?
So what are we to believe? This year, the ‘ inventor ’ of PSA, Richard Ablin, called PSA-based screening a “ public health disaster ” and pictured the idea of one man being saved by screening while 47 other men might experience loss of sexual function or urine leakage for no good reason [11]. The numbers in this example clearly relate back to the NNT out-come of the ERSPC. Are the future prospects really that bad and, if so, why should we try to decrease PC mortality at all?
Prostate cancer is a major public health problem being the most numerous cancer among men and the second most important cause of cancer related deaths with 192 280 and 27 360 incident cases respectively in the US in 2009 [12]. Worldwide numbers of PC, show an incidence of 679 023 and a mortality of 221 002 in 2002 [13]. With a seemingly achievable 35% mortality reduction, this could lead to the prevention of 77 350 men dying from PC, provided that the price we have to pay to do this is acceptable. In the US, a mortality reduction of 35%, would lead to PC going from a second to a fi fth place in leading cancer related deaths [12].
Despite these large incidence numbers and a sub-stantial disease-specifi c mortality reduction in the ERSPC, the effect on overall mortality will be mini-mal even if the PC mortality reduction would double, as seen in the G ö teborg trial. Some critics [14] pro-mote the view that overall mortality would be the appropriate end point. A trial addressing overall mor-tality as an end point would need several million participants to create enough statistical power to show an overall mortality reduction and this would thus be very unlikely to ever be accomplished. At present, the aim of health care systems worldwide is to gain improvements in mortality of different types of cancer that will in the end hopefully decrease the total burden of cancer mortality. The ERPSC study contributes to this process.
Some authors have suggested that ERSPC is not a coherent study and that pooled analysis is not jus-tifi ed, but should be replaced by a meta-analysis. These authors suggest that the ERSPC actually is a collection of seven studies with differing screening protocols and consider this to be a weakness [15,16]. Truth is, that agreement on a common data set with central data collection and agreements on mutually accepted small differences per country including the core age group (55 – 69 years), were already agreed upon in 1994 and 1995, at the time European coop-eration was initiated. National regulations caused randomization protocols to differ among countries, which led to a population-based effectiveness trial in Finland, Sweden and Italy, where randomization took place before informed consent. In the Nether-lands, Belgium, Switzerland, Spain and the 2005 late comer France, men were randomized after providing informed consent, also known as an effi cacy trial. Irrespective of the way of randomization, population registries were used to identify trial subjects [1]. Validation of randomization to test for heterogeneity of outcomes between (groups of) centers was pre-planned as was defi ned in the published ERSPC monitoring plan [17] and was carried out with suc-cessful results [1]. Since in all centres a trend toward mortality reduction by screening can be observed,

Controversies in Screening for Prostate Cancer 7
the agreed differences in national protocols appar-ently do not exert major effects on the outcome. The described heterogeneity must be considered as a strength rather than a weakness of ERPSC.
Critical remarks have been made in various com-munications [15,18] about the effect on PC mortal-ity being due to treatment instead of being due to screening. In a screening study cases in both arms should be treated similarly to rule out treatment as a confounder in mortality reduction. If treatment is reported to be more aggressive in the screening arm then inequalities in treatment choices between the two groups could be causing the mortality reduction instead of screening. Within ERSPC however, after correcting for stage and grade, no difference favor-ing more aggressive treatment in the screening arm could be found. The observed mortality reduction is therefore very unlikely to be solely caused by treat-ment effect [6]. This study also shows that the only difference in treatment that was found, was the com-bination of radiotherapy and endocrine treatment which is superior in high-risk patients and was applied more often in the control arm, so if any effect was expected it might be in favor of the control arm. Noteworthy is, that treatment decisions in PC cases in both arms were left to regional care provid-ers, as was recorded in the study protocol (www.erspc.org; publications). In practice, general practi-tioners were encouraged to refer patients to regional urologists and both were contacted in this respect beforehand. Also the G ö teborg trial did not report treatment difference which could impact on screen-ing as the determinant of outcomes.
Then, there is the issue of α -spending that could cause future analyses to lack power and become sta-tistically invalid, because of the interim analyses that were performed previously. When sequential testing is performed in interim monitoring of clinical trials, the α -value should be adjusted accordingly at each look to preserve the overall type-I error (i.e. stating an event is signifi cant when it is not). This was already anticipated in the monitoring plan [17] and described in the 2009 publication [1]; an α -spending curve (O ’ Brian-Fleming rule) of ≈1% each time was used, with a division of uneven weights and higher weight at the end. All three interim analyses of the ERSPC had their power adjusted for α -spending.
Others stated that the re-assurance of men with negative test results is not appropriate [16,19]. This is indeed a statement that embodies a continuous worry. Studies show that there is no PSA cut-off below which a man can be reassured that he has no PC. As was shown in the only empirical analysis of the performance characteristics of PSA, a cut-off value of 3.0 ng/ml misses 67.8% of biopsy detectable PC and 42.4% of potentially aggressive ones [20].
Low PSA levels can therefore not even rule out the presence of high-grade PC, although both risks of fi nding a PC on biopsy are directly related to PSA levels, also in the lower range.
For the ERSPC it was calculated that, of 48 867 men in six of the participating countries with an ini-tial PSA � 3.0 ng/ml, 5% was diagnosed with PC after a mean follow-up of nine years, of which 4.6% was confi rmed to be high-grade [21]. Substantial numbers of cancers must be therefore assumed to be missed. Only long-term follow-up, as it is planned within the ERSPC, can shed light on their natural history and on the rate of their detection at subse-quent screens. In addition to this, results of ERSPC Rotterdam [22] showed 9.4% of men who had initial negative biopsies to develop PC over an 11-year fol-low-up period, with the number of potentially missed cancers with a poor outcome in terms of progression-free survival (9%) and deaths from PC (2.4%) being very low. More aggressive screening may result in the detection of additional aggressive PC, but this would be at the cost of detecting a lot of potentially indolent PC, which will increase overdiagnosis, overtreatment and the NNT.
Does the benefi t shown in the ERSPC match the damage? We expressed this as an important downside in our 2009 paper [1], when we addressed overdiag-nosis and overtreatment as the most important adverse effects induced by screening. Overdiagnosis in the ERSPC was estimated to be no less than 50% in the screening group [7] and although this is a major concern, it is also one of the main achieve-ments of the ERSPC to quantify and report on these events. Furthermore, the NNS and the NNT are time dependent and can be expected to become more favorable with longer follow-up, as the mortal-ity reduction increases.
Unfortunately, PSA lacks the specifi city to be a solid tool in determining a biopsy indication, result-ing in large numbers of unnecessary biopsies and, at the same time, missing PC diagnoses in men with PSA values � 3.0 ng/ml. The ideal screening tool for PC, which could identify potentially aggressive can-cers in an early, curative stage, but could also avoid detection of cancers that would never cause any symptoms, let alone deaths, unfortunately does not yet exist. However, the unnecessary treatments can with some as yet unidentifi ed risk for the patient, be delayed by offering “ active surveillance ” . About 25% of men in ERSPC have made this choice.
Since PSA testing can not distinguish lethal PC from indolent disease and a lowering of the PSA threshold to proceed to prostate biopsy would imply detecting more and more PC that would be over-diagnosed and potentially overtreated. Addressing this issue, Roobol et al. [23] have applied the PC

8 M. Bul & F. H. Schröder
Riskcalculator (www.prostatecancer-riskcalculator.com) to reduce the number of unnecessary biopsies, while still detecting most clinically important PC cases. Including ultrasound volume, digital rectal exam and transrectal ultrasound together with the PSA value in a model, while applying an additional probability cut-off value of, for example, 12.5% to trigger a biopsy, resulted in a substantial increase of PPV in initial (from 29 to 38%), as well as repeat screening (from 19 to 25%). The predictive value for PSA alone showed an AUC of 0.64, which increased to 0.77 when the Riskcalculator was used. With this method, cancer diagnoses would be missed, but this would predominantly concern indolent PC (70 – 81%) and only a very small proportion of potentially important PC. Just increasing the PSA cut-off to � 4.0 ng/ml for example, would also result in a con-siderable decrease in biopsies, but considerably higher numbers of missed PC diagnoses (1.8 – 2.6 times higher). This individualized screening algo-rithm might contribute to counteract two of the most negative side-effects of screening, namely unneces-sary invasive testing and overdiagnosis with the related overtreatment. The fact that some cancers will always be missed by increasing test specifi city must however be considered. It is unknown at pres-ent what proportion of cancers will escape all efforts and which cancers can be safely detected during a
subsequent round of screening. Besides this, hope-fully in the future, new markers and improved nomo-grams will become available to lead to a more selective detection of aggressive PC.
What are the “ real issues ” and how can they be dealt with?
As incidence rates are high and PC is one of the most frequent causes of cancer related deaths, it is a rel-evant public health problem to decrease the PC mor-tality provided that this can be done at an acceptable price. This ‘ price ’ should comprise of a number of considerations.
First, the quality-adjusted life years (QALY ’ s) and other costs and benefi ts of screening should be matched against the achievable decrease of PC mor-tality. A paper on this subject addressing the cost effectiveness and quality of life in the ERSPC is in preparation and will be updated with increasing follow-up.
Then, testing for PC should become more selec-tive, resulting in detecting less non-aggressive can-cers and decreasing the amount of overdiagnosis and resulting overtreatment. As discussed earlier, PSA alone is not an optimal marker for PC screening, but individualized screening by means of the ERSPC based Risk Calculator is a step in the right direction.
Figure 2. Riskcalculator predicting the risk of positive biopsy based on solely PSA.This fi gure shows the predicted risk of having a positive biopsy, solely based on the PSA-value, irrespective of any other values of ancillary tests. In this example a man with a PSA-value of 4.0 ng/ml has a predicted risk of 21%. The estimation is based on data from the fi rst 6288 participants in the Rotterdam arm of the ERSPC.

Controversies in Screening for Prostate Cancer 9
follow-up in the ERSPC study is still too short to draw defi nite conclusions and further follow-up has to be awaited. If longer follow-up shows a larger difference in PC mortality – which can be expected from the 2009 mortality curves – the NNS and NNT will decrease. With an assumed 35% relative risk reduction in the intention to screen analysis, the NNS would decrease to 806 and the NNT to 27. If a 40% reduc-tion would be accomplished, this would lead to a NNS of 256 and NNT of 14. For those men with PSA values � 3.0 ng/ml, longer follow-up will help quantify the predictive value of a negative result and to better be able to reassure men with certain characteristics.
These estimates are similar to the results of the G ö teborg screening trial. If effects of similar size were shown in ERSPC as a whole, these fi ndings are likely to be more relevant because of the size of the trial, the European multiple country setting, the fact that small protocol differences still produce identical trends, the possibility to study multiple aspect on how to screen best for PC and the ongoing analysis of QALY ’ s based on ERSPC fi ndings.
Another important issue to study in this context, will be the screen detected PC that escape all efforts
These decision tools use risk modifying techniques to identify the cancers that will most likely be indo-lent and can be managed with active surveillance. An example of the application of the Riskcalculator (step 3) to three different men all presenting with a PSA value of 4.0 ng/ml is presented in Figures 2 – 4. The ERSPC based Riskcalculator among others, calcu-lates the probability of having a positive biopsy (step 3) and of indolent cancer (step 6). The calculator was updated and validated [24] and used for the study by Roobol et al. [23] mentioned above. Nevertheless, better markers and more accurate, advanced nomo-grams should be developed in order to decrease over-diagnosis even more.
Next, continued follow-up of the ERSPC is needed, since until now about 22% of participants died, there are still many more events to be expected. In the Scan-dinavian SPG-4 study of clinically diagnosed locally confi ned PC [25], the difference in cumulative inci-dence of death due to PC between groups assigned to radical prostatectomy or watchful waiting, remained stable only after about ten years. In the G ö teborg trial, even after 14 years of follow-up, the mortality curves still continue to diverge. This illustrates that the
Figure 3. Riskcalculator step 3. Prediction of the chance of a positive sextant biopsy in a man who was never screened before; PSA � 4.0 ng/ml, low risk.This fi gure shows the prediction of a positive biopsy, making use of the results of transrectal ultrasound (TRUS), digital rectal examination (DRE), prostate volume at ultrasonography and PSA-value. In this example a man with a PSA-value of 4.0 ng/ml, normal TRUS and DRE and prostate volume of 60cc were used, which results in a 7% chance.

10 M. Bul & F. H. Schröder
in spite of early detection and treatment. We are co-operating internationally to identify the characteris-tics of such patients and the mechanism that causes these “ escapes ” . Again, the resulting knowledge can be used to improve testing and screening. There will always be a group of unavoidable “ escapes ” which, when defi ned and quantifi ed, will contribute to resolving many controversies.
Finally, validated mechanisms for shared decision taking should be established. Results of the ERSPC have been adopted in several guidelines, including those of the European Association of Urologists [26], but so far no commonly accepted information mate-rial exists about the proper interpretation of the trial results and for men who wish to be informed about the possible risks and uncertainties. Effort should be taken at an international level to create validated decision aids.
So, what should we tell patients who wish to be screened, taking the contemporary evidence into account? This message has changed dramatically by the results of recent studies and should include the following statements. In the case of PC detected with
screening, the chances of dying of the disease are decreased by at least 31%. The downside remains though, as long as we have to deal with a high chance of being diagnosed and treated for disease which oth-erwise may not harm you within a period of nine years or longer. However, when non aggressive dis-ease is suspected, treatment can be avoided at least for some time.
Conclusion
In conclusion, we can say that the ERSPC shows a reduction of PC mortality for screened men of 20 – 31% at nine years of follow-up. In the G ö teborg screening trial these fi gures amount to 44 – 56% at 14 years. The resulting overdiagnosis and overtreatment are major worries, but can be decreased by screening less aggressively and more selectively using available risk modifying calculators. Establishing population based screening for PC as a health policy, will depend on lowering the NNT, while focussing on methods for more selective screening and achieving an accept-able risk-benefi t ratio.
Figure 4. Riskcalculator step 3. Prediction of the chance of a positive sextant biopsy in a man who was never screened before; PSA � 4.0 ng/ml, high risk.This fi gure shows the prediction of a positive biopsy, making use of the results of transrectal ultrasound (TRUS), digital rectal examination (DRE), prostate volume at ultrasonography and PSA-value. In this example a man with a PSA-value of 4.0 ng/ml, abnormal TRUS and DRE and prostate volume of 25cc were used, which results in a 65% chance.

Controversies in Screening for Prostate Cancer 11
We think screening will become an accepted health policy, but it will take time and work to be done. Key developments in the fi eld of screening show, that PSA should be used as a marker within an algorithm instead of a single biopsy indicator, to achieve better predictions of positive biopsies. Nev-ertheless, we have to work hard to fi nd new, better and more selective markers for screening purposes. It is important however to be aware of the fact that we will always miss cancers with screening and we have to learn to selectively miss the ones that would otherwise stay indolent and those that will always escape all efforts in spite of screening.
Funding statement
The ERSPC is supported by grants from the Dutch Cancer Society (KWF 94-869, 98-1657, 2002-277, 2006-3518), The Netherlands Organization for Health Research and Development (002822820, 22000106, 50-50110-98-311), 6 th Framework Pro-gram of the EU: P-Mark: LSHC-CT-2004-503011, Beckman Coulter Hybritech Inc and of Europe against Cancer (SOC 95 35109, SOC 96 201869 05F02, SOC 97 201329, SOC 98 32241). The ERSPC received Erasmus MC and Ministry of Health institutional review board approval.
Declaration of interest: The authors report no confl icts of interest. The authors alone are responsible for the content and writing of the paper.
References
Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, [1] Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009;360:1320 – 8. Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, [2] Chia D, Church TR, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009;360:1310 – 9. Hugosson J, Carlsson S, Aus G, Bergdahl S, Khatami A, [3] Lodding P, et al. Mortality results from the Goteborg ran-domised population-based prostate-cancer screening trial. Lancet Oncol 2010. Schroder FH, Roobol MJ. ERSPC and PLCO prostate can-[4] cer screening studies: What are the differences? Eur Urol 2010;doi:10.1016/j.eururo.2010.03.033. Holmberg L. Prostate cancer screening: The need for prob-[5] lem-solving that puts men’s interests fi rst. Eur Urol 2009;56:34 – 7. Wolters T, Roobol MJ, Steyerberg EW, van den Bergh RC, [6] Bangma CH, Hugosson J, et al. The effect of study arm on prostate cancer treatment in the large screening trial ERSPC. Int J Cancer 2010;126:2387 – 93. Draisma G, Boer R, Otto SJ, van der Cruijsen IW, Damhuis [7] RA, Schroder FH, et al. Lead times and overdetection due to prostate-specifi c antigen screening: Estimates from the
European Randomized Study of Screening for Prostate Cancer. J Natl Cancer Inst 2003;95:868 – 78. Roobol MJ, Kerkhof M, Schroder FH, Cuzick J, Sasieni P, [8] Hakama M, et al. Prostate cancer mortality reduction by prostate-specifi c antigen-based screening adjusted for nonat-tendance and contamination in the European Randomised Study of Screening for Prostate Cancer (ERSPC). Eur Urol 2009;56:584 – 91. Cuzick J, Edwards R, Segnan N. Adjusting for non-compliance [9] and contamination in randomized clinical trials. Stat Med 1997;16:1017 – 29. Kerkhof M, Roobol MJ, Cuzick J, Sasieni P, Roemeling S, [10] Schroder FH, et al. Effect of the correction for non-compliance and contamination on the estimated reduction of metastatic prostate cancer within a randomized screening trial (ERSPC section rotterdam). Int J Cancer 2010; Epub 2010 Mar 1. Ablin RJ. The great prostate mistake. New York Times. 2010 [11] March 10. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer [12] statistics, 2009. CA Cancer J Clin 2009;59:225 – 49. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statis-[13] tics, 2002. CA Cancer J Clin 2005;55:74 – 108. Dubben HH. Trials of prostate-cancer screening are not [14] worthwhile. Lancet Oncol 2009;10:294 – 8. Boyle P, Brawley OW. Prostate cancer: Current evidence [15] weighs against population screening. CA Cancer J Clin 2009;59:220 – 4. Brawley OW, Ankerst DP, Thompson IM. Screening for [16] prostate cancer. CA Cancer J Clin 2009;59:264 – 73. De Koning HJ, Hakulinen T, Moss SM, Adolfsson J, Smith [17] PH, Alexander FE, et al. Monitoring the ERSPC trial. BJU Int 2003;92(Suppl 2):112 – 4. Barry MJ. Screening for prostate cancer – the controversy [18] that refuses to die. N Engl J Med 2009;360:1351 – 4. Stark JR, Mucci L, Rothman KJ, Adami HO. Screening for [19] prostate cancer remains controversial. BMJ 2009;339:784 – 6. Thompson IM, Ankerst DP, Chi C, Lucia MS, Goodman PJ, [20] Crowley JJ, et al. Operating characteristics of prostate-specifi c antigen in men with an initial PSA level of 3.0 ng/ml or lower. JAMA 2005;294:66 – 70. Roobol MJ, Aus G, Auvinen A, Bangma CH, Berenguer A, [21] Ciatto S, et al. How to screen for prostate cancer after 2008? PSA as a biopsy indicator, part II. Eur Urol Suppl 2009;9:191. Schroder FH, van den Bergh RC, Wolters T, van Leeuwen PJ, [22] Bangma CH, van der Kwast TH, et al. Eleven-year outcome of patients with prostate cancers diagnosed during screening after initial negative sextant biopsies. Eur Urol 2010;57:256 – 66. Roobol MJ, Steyerberg EW, Kranse R, Wolters T, van den [23] Bergh RC, Bangma CH, et al. A risk-based strategy improves prostate-specifi c antigen-driven detection of prostate cancer. Eur Urol 2010;57:79 – 85. Steyerberg EW, Roobol MJ, Kattan MW, van der Kwast TH, [24] de Koning HJ, Schroder FH. Prediction of indolent prostate cancer: Validation and updating of a prognostic nomogram. J Urol 2007;177:107 – 12. Bill-Axelson A, Holmberg L, Filen F, Ruutu M, Garmo H, [25] Busch C, et al. Radical prostatectomy versus watchful wait-ing in localized prostate cancer: The Scandinavian prostate cancer group-4 randomized trial. J Natl Cancer Inst 2008;100:1144 – 54. Heidenreich A, Aus G, Bolla M, Joniau S, Matveev VB, [26] Schmid HP, et al. EAU guidelines on prostate cancer. Eur Urol 2008;53:68 – 80.

Correspondence: Christine D. Berg, Early Detection Research Group, Division of Cancer Prevention, National Cancer Institute, Bethesda, MD 20852, USA. E-mail: [email protected]
(Received 27 September 2010 ; accepted 28 September 2010 )
REVIEW ARTICLE
The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial: The prostate cancer screening results in context
CHRISTINE D. BERG
Early Detection Research Group, Division of Cancer Prevention, National Cancer Institute, Bethesda, USA
Abstract Background . The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) was conducted in sites around USA during a period of marked secular changes in the use of prostate specifi c antigen (PSA) screening for prostate cancer. Material and methods . Trends in prostate cancer incidence, stage at presentation and mortality are useful when interpreting the results from a screening trial that commenced in 1993 and enrolled participants through 2001. The last participants com-pleted active screening in 2006. Incidence and mortality data published to date on PLCO need to be placed into the context of the secular trends. Additional data analyses have been conducted on subsets of the participants and these results can also enhance the interpretation of the trial. Additionally, the accompanying biospecimen repository has served as a rich research resource yielding informative fi ndings. Results . The PLCO is best viewed as a trial comparing a regimented active annual screening program of PSA screening for six rounds, four of which had accompanying digital rectal examination (DRE) to patterns of screening that were occurring in the population in many academic and community settings across the USA. The epidemiology and molecular genetics of prostate cancer is becoming better understood and analyses of the PLCO resource have contributed. One approach to risk assessment utilizing genetic markers from selected members of the PLCO prostate cancer cohort has been developed. A modeling effort with CISNET-ERSPC-PLCO is underway to compare and contrast fi ndings such as effects of different PSA thresholds and screening intervals. Conclusions . The information emerging from PLCO is useful to inform the debate around prostate cancer screening. An understanding of the biologic differences underpinning indolent and aggressive prostate cancer will better guide the future development of screening and treatment strategies.
Background
Prostate cancer has been the leading cause of cancer in males in USA for decades [1]. In the late 1970s near the inception of the Surveillance Epidemiology and End Results (SEER) Registry (http://seer.cancer.gov/), pros-tate cancer incidence was similar to lung cancer. The decline in smoking in males has led to a gradual decline in incidence in lung cancer. Prostate cancer screen-ing appears to have contributed to the marked rise in incidence witnessed in the late 1980s and early 1990s. Although, prostate cancer incidence has fallen from its peak, it remains elevated compared to the pre-prostate specifi c antigen (PSA) era. Mortality from pros-tate cancer fell relatively rapidly after the introduction of PSA testing. Debate has raged as to the extent to which treatment advances and/or widespread PSA testing have contributed to this decline.
The Food and Drug Administration approved the PSA test in 1986 as a way to monitor men after treat-ment. It was then studied as a means of detection of prostate cancer [2]. The medical community recog-nized at the time that interest and uptake would be generated particularly as it was a non-invasive, rela-tively inexpensive, blood test. Many prominent mem-bers of the urologic community and the National Can cer Institute (NCI) met to discuss how best to assess the effect of the emergence of PSA testing. A prospective, randomized trial with a prostate cancer specifi c mortality endpoint was judged as the most defi nitive approach. Other outstanding screening questions for lung, colorectal and ovarian cancer screening were identifi ed. A multi-modal screening trial was thought to have cost-effi ciencies. Also, mature adults when receiving their medical care are
Acta Oncologica, 2011; 50(Suppl 1): 12–17
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.531283

PLCO Prostate Cancer Screening 13
Table II. Tumor stage and Gleason score for all prostate cancers at 10 years.
Screening Group n � 3 452
Control Group n � 2 974
Stage I 2 (0.1 %) 15 (0.5 %) II 1458 (97.2 %) 2790 (93.8 %) III 22 (1.5%) 56 (1.9 %) IV 15 (1.0 %) 79 (2.7 %) Unknown 3 (0.2 %) 34 (1.1 %)Gleason score on biopsy 2–4 94 (6.3 %) 137 (4.6 %) 5–6 963 (64.2 %) 1656 (55.7%) 7 318 (21.2 %) 779 (26.2 %) 8–10 98 (6.5 %) 341 (11.5 %) Unknown 27 (1.8%) 61 (2.1 %)
Table I. Demographics of male enrollees in the PLCO .
percent
Screening Group
n � 38 343
Control Group
n � 38 350
Age 55 – 59 yr 32.3 32.3 60 – 64 yr 31.3 31.3 65 – 69 yr 23.2 23.2 70 – 74 yr 13.2 13.2Race or ethnic group; self-reported Non-Hispanic white 86.2 83.8 Non-Hispanic black 4.5 4.3 Hispanic 2.1 2.1 Asian 4.0 3.9 Other 0.8 0.9 Missing data 2.4 5.0Family history of prostate cancer 7.1 6.7PSA test within past 3 years Once 32.8 31.9 Two or more times 9.4 9.8
evaluated for their risk of cancer as it continues to be the second leading cause of death in the United States. Their clinicians would benefi t from informa-tion to guide screening choices and a multi-modal approach to assessment would better refl ect the clin-ical reality [3].
Material and methods
The design of the PLCO focused on developing a randomized trial in males and females that would be adequately powered to assess the impact on cancer specifi c mortality of screening for lung and colorectal cancer in males and females, ovarian cancer screen-ing in females and prostate cancer screening in males [4]. The trial initially set out to enroll individuals 60 to 75, reasoning that these were ages of high incidence of cancer. More elderly individuals had and still have higher rates of competing mortality and therefore it is more diffi cult to assess the effect of screening in this group and they were excluded from the study. As the trial progressed it became clear that the enrollment that was occurring, although more rapid in the younger age groups, was slow and to expand the numbers the age for entry was lowered to 55.
To refl ect the geographic, racial and ethnic diver-sity of the US population a Request for Proposals was broadly solicited. The proposals were reviewed for cap-abilities of the sites to perform the recruitment, screen-ing, retention and follow-up needed. The resulting sites were distributed throughout the continental US and Hawaii (http://prevention.cancer.gov/plco/centers). One poorly performing site was replaced early in the trial by the University of Alabama which has a strong emphasis on recruiting African-Americans from sur-rounding communities into trials. Efforts at another site, the University of Colorado Health Systems focused on Hispanic recruitment. A location at the Pacifi c Health Research Institute (now Pacifi c Health Research and Education Institute) was aimed at recruiting Asian and Pacifi c-Islanders. All sites including the NCI had the study approved by their local Institutional Review Boards. The PLCO participants signed an informed consent detailing the nature and risks from the screen-ing interventions. The control participants were encour-aged to continue to receive care from their usual health care providers. Screening was neither encouraged nor discouraged for them.
The resulting demographics of the enrolled male population can be seen in Table I. An analysis of the characteristics of the participants confi rmed that the PLCO enrollees like many in clinical trials are “ healthy volunteers ” [5]. They are of higher socio-economic and educational attainment than the population at large and tend to have a better health profi le. This needs to be recognized as one analyzes the results of the
trial. Enrollment occurred from 1993 to 2001. Pros-tate cancer screening was with annual PSA testing for six years (T0 – T5) and digital rectal examination (DRE) annually for four years (T0 – T3). The thresh-old for an abnormal serum PSA was set at � 4 nano-grams per milliliter (ng/ml). A reference laboratory was established at UCLA where all specimens were shipped on dry ice after centrifugation and serum sep-aration. PSA tests were analyzed with the Tandem-R PSA assay until January 1, 2004 and with the Access Hybritech PSA after that (both assays were manu-factured by Beckman Coulter). Additional blood specimens were drawn from screening arm partici-pants and buccal cells were collected from control arm participants to establish a biospecimen repository [6]. Tissue samples were also collected from participants in both arms. Tissue microarrays were constructed and cores obtained from formalin-fi xed paraffi n embedded specimens.

14 C. D. Berg
At enrollment, all participants completed a base-line questionnaire that inquired about screening prac-tices in the three year period prior to enrollment, demographic characteristics and potential risk fac-tors for malignancy. Separate dietary questionnaires were also administered. The participants were asked if they had had prior PSA or DRE testing and the number of tests. After 1995, those participants who had more than one PSA blood test in the previous three years were excluded. During the conduct of the screening portion of the trial, participants in the con-trol arm were also assessed for screening test uptake. A randomly selected group was queried every one to two years. For prostate cancer screening, the par-ticipants were asked if they had ever had a PSA blood test for prostate cancer or a digital rectal examination of the prostate. Those who answered yes were then asked when the most recent test was. The categories for response were “ within the past year ” , “ 1 – 2 years ago ” , “ 2 – 3 years ago ” and “ more than three years ago. ” The reasons for the test were also queried to determine if it was routine or for evaluation of a specifi c health problem. Those participants who had repeated PSA testing prior to entry were assumed to continue screening annually. This comprised 9.8% of the con-trol group. A weighted average, of the percent respond-ing both “ within the past year ” and routine, and the 100% estimate in the group screened frequently prior to enrollment, was used to provide an estimate of over-all contamination. Assessment of compliance in the screening arm was determined by attendance at the annually scheduled screening appointments and an estimate of compliance was calculated by dividing that by the number expected.
Results
The trial was monitored from inception by an inde-pendent Data and Safety Monitoring Board. Reviews of the accumulating data were done every six months with regular planned interim analyses. Publication of prostate cancer specifi c mortality results for up to ten years from recruitment occurred in March 2009 [7]. Median follow-up was 11.5 years and vital status was known for 98% of participants at seven years and 67% at ten years. The decision to report was made as no evidence of difference between arms was emerging at that point but evidence of harms from diagnostic evaluations following screening and after treatment was noticed. Earlier publications had pre-sented results from the baseline [8] and all screening rounds [9].
Six annual rounds of screening were conducted and at seven years, 2 820 prostate cancers and 50 prostate cancer specifi c deaths were noted in the screened group. In the control group, 2 322 cancers and 44 deaths
were noted. The data at ten years (67% complete) showed 3 452 screened versus 2 974 control group cancers and 92 compared to 82 deaths. Of note, the excess number of prostate cancer cases persisted after completion of screening. Also, although 25% more prostate cancers were diagnosed in the active screen-ing arm at seven years, mortality rates through seven to ten years were the same in each arm. Whether or not the small differences in stage and Gleason ’ s score between arms will result in differential survival in the future remains to be seen.
The percentage of patients having late stage dis-ease {AJCC Stages III and IV, [10]} Table II, at diag-nosis was low in both arms. At ten years, 3.5% of all screening arm subjects were clinical stage III and IV compared to 4.6% in the control arm. This compares with a 25% incidence of presentation of late stage disease prior to the advent of PSA testing and a rate of 4% in the population as a whole in 2002. A com-parison of Gleason ’ s score on biopsy, revealed aggres-sive Gleason ’ s 8 to 10 in 8.4% of screened arm participants and 11.5% in the control arm partici-pants. Additional follow-up on the entire cohort is continuing. Whether these differences in stage at diagnosis and in Gleason ’ s grade will translate into differences in prostate cancer specifi c mortality will be seen.
A separate analysis of the effect of the contamina-tion on rates of prostate cancer has been conducted [11]. The rates of reported test usage increase if one includes any testing within the year compared with rou-tine testing. Rates increased from 33 to 40% at study year 0 and from 46% at study year 5 to 54 – 55%. At year 0, 38% of men reported no history of PSA test-ing while at year 5, 15% did. Also, at year fi ve, 18% reported testing one to two years earlier. Compliance with the screening protocol overall was 85% for PSA testing and 86% for DRE, lower than the study design estimate of 90%. Clearly, the men in the control arm were being screened but at a lower frequency and inten-sity than men in the screened arm.
To estimate what would have occurred in the absence of screening, SEER rates from 1985 – 1987 prior to the onset of the PSA era were utilized. Five year age groups and race (white, black, other) were constructed and the SEER rates were applied to the person-years at risk for control and screened arm men during the screening period of the trial (the fi rst six years). A separate calculation was done utilizing SEER rates contemporaneous to the screening period of the trial. During the six screening years of the trial, 2 538 prostate cancers were identifi ed in the screened arm and 1 958 in the control arm. In the screened arm there were an excess of 1 589 and in the control arm, 1 024 com pared to the pre-PSA screening era. When one compares with the contemporaneous

PLCO Prostate Cancer Screening 15
SEER rates, 927 and 354 (screening: control) excess cancers were diagnosed. For the control arm, this refl ects a 22% excess over the expected number if screening had been conducted in the control arm as in the populations covered by SEER.
Ancillary data analyses
The information available from the PLCO prostate cancer screening trial can be analyzed to improve the understanding of the role of other factors that infl u-ence the conduct of screening and evaluation for pro state cancer. A large fraction of screened men, initially have low PSA levels ( � 2 ng/ml). In the PLCO, in men with baseline PSAs less than 1 ng/ml, 1.5% were found to have a PSA of more than 4 ng/ml by year 5, while in those with PSAs between 1.0 and 1.99 ng/ml the rate of progression was somewhat higher with 7.4% progressing by year fi ve [12]. A total of 33.5% and 79% of men with initial PSA of 2.0 to 2.99 and 3.0 to 4.0 converted by year 5. This information could help to inform thresholds for pos-itivity and screening frequency. Information on PSA velocity (PSAV) may also help to inform decisions as to when to biopsy. In 1 441 men enrolled in the PLCO who received � 2 PSA screens, and were diagnosed with prostate cancer within one year of the last screen, PSAV was calculated using all available PSA levels [13]. Both PSA and PSAV were related to biopsy Gleason score. The median PSAV was 0.60 ng/ml per year for men with Gleason scores from 2 to 6 versus 0.84 ng/ml for men with Gleason scores from 7 to 10 (p � 0.0001). Information such as this can inform watchful waiting and active sur-veillance approaches as well. Another issue is what happens after an initial negative prostate biopsy. The probability of having a repeat biopsy within three years of initial biopsy was 43% for 1 736 men with suspicious PSA levels after an additional round of screening [14]. An analysis of men who had an initial false positive result was done to assess the impact of this result on subsequent screening behavior. Given the subsequent high risk of repeat biopsy it was wor-risome to note that in a multi-variable model being African American (p � 0.016 and having a high school education or less p � 0.007) were predictive of not returning for prostate cancer screening within the fol-lowing year [15]. Additional effort may be warranted to facilitate compliance with screening in this group.
The impact of associated diseases and conditions can also be assessed in the PLCO cohort. Analysis within the PLCO has confi rmed an inverse relation-ship between PSA concentration and body mass index [16]. Dietary factors and supplement use have been analyzed within the cohort. There was no overall asso-ciation between dietary or supplemental intake of
vitamin E, beta-carotene or vitamin C and prostate cancer risk [17, 18]. Also, no evidence was found sub-stantiating the hypothesis that lycopene and tomato product intake affected risk of prostate cancer [19].
The PLCO biospecimen repository with high qual-ity DNA specimens has contributed to our under-standing of the molecular genetic factors that are associated with increased risk of prostate cancer. A locus within the 8q24 chromosome, rs6983267, was identifi ed separate from the initially reported locus at rs1447295 [20]. Additional SNP analyses done in second stage replication scans confi rmed three previ-ously reported loci, two in 8q24 and one in HNF1B [21] and loci on chromosomes 7, 10 (2 loci) and 11 were highly signifi cant. The loci on chromosome 10 include MSMB which encodes β -microseminoprotein, a primary constituent of semen and CTBP2, a gene with antiapoptotic activity. Additional fi ne mapping and functional analysis confi rmed the strong associa-tion with prostate cancer risk of the rs10993994 locus in MSMB and gene expression was higher in cell lines with a CC or a CT genotype than with a TT geno-type. [22].
Utilizing knowledge of SNPs to assess risk of malignancy is a developing endeavor. A project utiliz-ing a population-based case-control study in Sweden and a nested case-control study from the PLCO developed a risk-prediction model utilizing SNPs and family history [23]. Men with 11 risk alleles (mode) and negative family history were considered at base-line risk and those who had � 14 risk alleles and a positive family history had an odds ratio of 4.92 (95% CI: 3.64 – 6.64) for prostate cancer in the Swedish study and this was confi rmed in the PLCO. This could be utilized to calculate a man ’ s absolute risk of pros-tate cancer. For example, a 65-year-old man in the US with a family history and � 14 risk alleles, has a 41% risk of being diagnosed with prostate cancer compared with a population average of 13%. The utility of these types of assessments for screening and consideration of chemoprevention still need to be determined.
Discussion
The PLCO trial was conceived at the beginning of utilization of the PSA test for screening for prostate cancer. Rapid dissemination and widespread initial use of PSA testing for prostate cancer screening occurred in the years leading up to the launch of the trial. Sub-sequent to launch, regular PSA screening remained common in the community. The peak in prostate cancer incidence in males in the US coincided with the launch of the trial in the early 1990s [1]. Also, with the increasing incidence in the disease, a decrease in advanced stage disease was noted. Subsequent modeling was consistent with much of this decline in late stage

16 C. D. Berg
disease being a consequence of screening [24]. Several years after the trial launch, prostate cancer mortality in the US fell, going below pre-PSA rates by 2003. The results of the trial must be placed into this context.
When one looks at the incidence rates in the PLCO the control group actually has higher rates than compared with contemporaneous results from the SEER registry. This can be explained by the char-acteristics of the enrollees. As mentioned earlier, they represent a “ healthy volunteer ” who is of higher socio-economic status and better educated than aver-age. This demographic undergoes screening more frequently. The prostate cancer specifi c mortality in both arms of the study is low.
A limitation of the PLCO could be the cut-off of � 4 ng/ml chosen as the threshold for referral for fur-ther evaluation. This was the commonly accepted threshold at the time of trial initiation. Subsequently, a trial of fi nasteride in prostate cancer prevention had as part of the design an exit biopsy in all men in the placebo arm regardless of PSA level. This demon-strated that overall, men with PSAs of less than 4 ng/ml had a 15% incidence of prostate cancer of which 14.9% was Gleason ’ s 7 � [25]. The prevalence of pro-state cancer in men with PSAs of � 0.5 ng/ml was 6% of which 12.5% were high grade. If PSA screening were to be implemented as an organized program, the ideal threshold value is unclear. Also, the PLCO investigators did not prescribe evaluations or therapy. Participants and their physicians decided on a course of evaluation of an elevated PSA and if prostate can-cer were diagnosed the treatments were also determined in the same manner. It should be noted that these same diagnostic and treatment approaches were the ones employed during the period of rapid increase in pros-tate cancer incidence in SEER and also in the decrease in mortality seen in the US.
An analysis under the auspices of the Cancer Inter-vention and Surveillance Modeling Network {(CIS-NET) http://cisnet.cancer.gov/} will compare results across ERSPC and PLCO. Differences in the screen-ing approaches such as PSA threshold and screening interval, and populations will have to be noted when these comparisons are presented.
Conclusion and next steps
Continued collection of endpoint data in the PLCO is critical. An impact of the differential in Gleason ’ s grade between arms and the very small difference in stage may emerge. The analyses of CISNET-ERSPC-PLCO will be revealing. The molecular genetics of prostate cancer risk are better understood through contributions of the PLCO. Much more remains to be accomplished and the prostate tumor cores and TMAs from the PLCO
and the matched pre-diagnostic biospecimens are available to the research community. Interesting under standings of the behaviors of indolent and aggressive disease may emerge.
The goal of all is to minimize the adverse impact of prostate cancer in our ageing society. Clearly, the mor-tality reductions achieved to date are to be applauded. However, they do come with a high rate of overdiagno-sis. Treatment side effects are also substantial. Knowl-edge gained from watchful waiting and active surveillance approaches will be important. As discussed elsewhere in this monograph advances in the chemoprevention of prostate cancer have also been made.
The PLCO trial has contributed to this emerging database. However, it is important when analyzing this trial to place the results in the context of the times. When designing screening trials for the future these issues need to be considered.
Acknowledgements
The PLCO trial was funded by contracts from the National Cancer Institute. The ClinicalTrials.gov identifi er for PLCO is NCT00002540. The author acknowledges the contributions to this work made by the co-authors on the other prostate cancer manu-scripts emanating from the PLCO. She thanks the study subjects for their contributions in making this study possible. There are no confl icts of interest to report for this article.
Declaration of interest: The author reports no con-fl icts of interest. The author alone is responsible for the content and writing of the paper.
References
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA [1] Cancer Clin J 2010;60:277 – 300. Epub 2010 Jul 7. Catalona WJ, Smith DS, Ratliff TL, Dodds KM, Coplen DE, [2] Yuan JJJ, et al. Measurement of prostate-specifi c antigen in serum as a screening test for prostate cancer. N Engl J Med 1991;324:1156 – 61. Gohagan JK, Prorok PC, Hayes RB, Kramer BS. The Prostate, [3] Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial of the National Cancer Institute: History, organization, and status. Control Clin Trials 2000;21(Suppl):251S – 272S. Prorok PC, Andriole GL, Bresalier RS, Buys SS, Chia D, [4] Crawford ED, et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials 2000;21(Suppl):273S – 309S. Pinsky PF, Miller A, Kramer BS, Church T, Reding D, [5] Prorok P, et al. Evidence of a healthy volunteer effect in the Prostate, Lung, Colorectal and Ovarian cancer screening trial. Am J Epidemiol 2007;165:874 – 81. Hayes RB, Reding D, Kopp W, Subar A, Bhat N, Rothman R, [6] et al. Etiologic and early marker studies in the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials 2000;21(Suppl):349S – 355S.

PLCO Prostate Cancer Screening 17
Grubb RL, Black A, Izmirlian G, Hickey TP, Pinsky PF, [16] Mabie JE, et al. Serum prostate-specifi c antigen hemodilu-tion among obese men undergoing screening in the Prostate, Lung, Colorectal, and Ovarian cancer screening trial. Cancer Epidemiol Biomarkers Prev 2009;18:748 – 51. Koralek DO, Peters U, Andriole G, Reding D, Krish V, Subar A, [17] et al. A prospective study of dietary alpha-linoleic acid and the risk of prostate cancer (United States). Cancer Causes Control 2006;17:783 – 91. Kirsh VA, Hayes RB, Mayne ST, Chatterjee N, Subar AF, [18] Dixon LB, et al. Supplemental and dietary vitamin E, beta-carotene, and vitamin C intakes and prostate cancer risk. J Natl Cancer Inst 2006;98:245 – 54. Kirsh VA, Mayne ST, Peters U, Chatterjee N, Leitzmann MF, [19] Dixon LB, et al. A prospective study of lycopene and tomato product intake and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev 2006;15:92 – 8. Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, [20] et al. Genome-wide association study of prostate cancer iden-tifi es a second risk locus at 8q24. Nat Genet 2007;39:645 – 9. Epub 2007 Apr 1. Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, [21] et al. Multiple loci identifi ed in a genome-wide association study of prostate cancer. Nat Genet 2008;40:310 – 5. Lou H, Yeager M, Li H, Bosquet JG, Hayes RB, Orr N, et al. [22] Fine mapping and functional analysis of a common variant in MSMB on chromosome 10q11.2 associated with prostate can-cer susceptibility. Proc Natl Acad Sci USA 2009;106:7933 – 8. Xu J, Sun J, Kader AK, Lindstr ö m S, Wiklund F, Hsu FC, [23] et al. Estimation of absolute risk for prostate cancer using genetic markers and family history. Prostate 2009;69:1565 – 72. Etzioni R, Gulati R, Falcon S, Penson DF. Impact of PSA [24] screening on the incidence of advanced stage prostate cancer in the United States: A surveillance modeling approach. Med Decis Making 2008;28:323 – 31. Thompson I, Pauler D, Goodman P, Tangen C, Lucia M, [25] Parnes H, et al. Prevalence of prostate cancer among men with a prostate-specifi c antigen level of � 4.0 ng per milliliter. N Engl J Med 2004;350:2239 – 46.
Andriole GL, Crawford ED, Grubb RL, Buys SS, Chia D, [7] Church TR, et al. Mortality results from a randomized pros-tate-cancer screening trial. N Engl J Med 2009;360:1310 – 9. Andriole GL, Levin DL, Crawford ED, et al. Prostate cancer [8] screening in the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial: Findings from the initial screening round of a randomized trial. J Natl Cancer Inst 2005;97:433 – 8. Grubb RL III, Pinsky PF, Greenlee RT, et al. Prostate cancer [9] screening in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial: Update on fi ndings from the initial four rounds of screening in a randomized trial. BJU Int 2008;102:1524 – 30. Fleming JD, Cooper JS, Henson DE, et al., editors. AJCC [10] cancer staging manual. 5 th ed. Philadelphia: Lippincott-Raven; 1997. Pinsky PF, Black A, Kramer BS, Miller A, Prorok PC, [11] Berg C. Assessing contamination and compliance in the prostate component of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Clin Trials 2010;7:303 – 11. Crawford ED, Pinsky PF, Chia D, Kramer BS, Fagerstrom RM, [12] Andriole G, et al. Prostate specifi c antigen changes as related to the initial prostate specifi c antigen: Data from the prostate, lung, colorectal and ovarian cancer screening trial. J Urol 2006;175:1286 – 90. Pinsky PF, Andriole G, Crawford ED, Chia D, Kramer BS, [13] Grubb R, et al. Prostate-specifi c antigen velocity and pros-tate cancer Gleason grade and stage. Cancer 2007;109:1689 – 95. Pinsky PF, Crawford ED, Kramer BS, Andriole GL, [14] Gelmann EP, Grubb R, et al. Repeat biopsy in the prostate, lung, colorectal and ovarian cancer screening trial. BJU Int 2007;99:775 – 9. Ford ME, Havstad SL, Demers R, Cole-Johnson C. Effects [15] of false-positive prostate cancer screening results on subse-quent prostate cancer screening behavior. Cancer Epidemiol Biomarkers Prev 2005;14:190 – 4.

Correspondence: Henrik Gr ö nberg, Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, PO Box 281, SE-171 77 Stockholm, Sweden. Tel: � 46 8 52482347. Fax: � 46 8 314975. E-mail: [email protected]
(Received 22 September 2010 ; accepted 4 October 2010 )
REVIEW ARTICLE
Early detection of prostate cancer with emphasis on genetic markers
MARKUS ALY 1,2 , FREDRIK WIKLUND 1 & HENRIK GR Ö NBERG 1
1 Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden and 2 Division of Urology, Department of Clinical Sciences, Danderyd Hospital, Karolinska Institutet, Stockholm, Sweden
Abstract Background. The recent advances in genomic research have made it possible to identify several new genomic-based biomar-kers for prostate cancer. In this review we evaluate these new markers and speculate about future scenarios. Results. Today 35 single nucleotide polymorphisms (SNPs) have been identifi ed and independently validated to associate with prostate cancer. These SNPs are common in the population ( � 5%) but the effect of these SNPs in these regions on prostate cancer risk is modest with odds ratios typically ranging between 1.1 and 1.3. It is estimated that these markers explain 25% of the familial risk of prostate cancer. However, it is anticipated that additional 50 – 75 prostate cancer SNPs will be identifi ed in the near future. The SNPs associated with prostate cancer so far are not associated with disease stage or outcome. There are several efforts to identify germline genetic markers that can be used as prognostic markers. There are also tumor-based methods that are promising in identifying new genetic markers that can be easily measured in plasma or urine. Conclusion. There are several new “ genetic ” markers that in the near future might be used in clinical routine. These markers are easy to measure and stable over time. However the challenge is not only to identify new biomarkers but the real test is to vali-date new biomarkers in several large well-characterized patient populations. This validation must be done together will all other known biomarkers at the same time as it not likely that one single marker is enough, but a panel of different markers. Today 2010 there are over 19 000 publications in the area of biomarkers and prostate cancer, but only one biomarker, PSA, is used in the clinic today!
Since the late 1980s Prostate Specifi c Antigen (PSA) has been used as a clinical biomarker initially used to follow treatment outcome but later as a tool to identify patients at a high risk of being diagnosed with prostate cancer.
Two recent studies have presented different out-comes as to whether PSA can serve as a tool for screening or not. The European study, ERPC, showed a 20% reduction in prostate cancer mortality com-pared to the non-intervention arm. The PLCO study did not show any reduction in mortality, probably due to contamination of the control group and rela-tively few events in the intervention group. Although it seems as if screening would affect the prostate can-cer related deaths it has not led to general screening in most western countries mostly due to the fact that screening inevitably leads to overdiagnosis and over-treatment which also was shown in the ERCP study
where 1 400 men had to be screened and 48 men had to be treated to avoid one death related to prostate cancer [1,2]. In an updated analysis of the G ö teborg part of the ERPC, a 44% reduction in prostate cancer mortality was reported. This results reduces the numbers needed to screen to 293 and number needed to treat to only 12 [3].
Still this calls for novel biomarkers with a better specifi city in order to better predict which men would benefi t from further diagnostic interventions and eventually treatment. Biological markers associated with aggressiveness would be very valuable in order to better be able to individualize treatment.
In this review we are focusing on genetic markers that can be measured in blood, either from DNA or plasma/serum. Genetic markers measured in urine or tumors are covered in other articles. We have the following defi nitions of a genetic marker:
Acta Oncologica, 2011; 50(Suppl 1): 18–23
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.529824

Genetic Markers in Prostate Cancer 19
Germline (inherited) genetic markers
Point mutations/copy number variation in the human genome. A single nucleotide polymorphism (SNP) is an inherited mutation that is present in more than 1% of the population. These markers can be analyzed from any normal tissue in the body but in most cases DNA from white blood cells are used.
Germline genetic markers and prostate cancer risk
A family history of prostate cancer is one of the strongest risk factors and twin studies suggest that as much as 42% of the disease risk is explained by her-itable factors [4]. Attempts to decipher the heritable component of prostate cancer based on candidate gene association studies and genome-wide linkage studies in multiple case families have suggested numerous prostate cancer susceptibility genes and loci. However, an inability to replicate reported link-age and association fi ndings suggest that prostate cancer is genetically complex with multiple common low-penetrance genes involved in prostate cancer predisposition [5].
Recently, genome-wide association studies (GWAS) have emerged as a powerful method to identify genomic low-risk susceptibility regions for complex diseases including cancer [6]. Through genotyping platforms that explore hundreds of thousands of single nucleotide polymorphisms (SNPs) simultaneously it is possible to screen the complete genome for common genetic variation associated with the disease of interest. In 2006 the fi rst prostate cancer susceptibility region was identi-fi ed at chromosome 8q24. Subsequent GWAS and region-focused studies have revealed fi ve distinct linkage disequilibrium blocks harbouring prostate cancer susceptibility alleles at 8q24 [7 – 13]. The 8q24 region has also been shown to harbour sus-ceptibility alleles for breast cancer [14], colorectal cancer [15], bladder cancer [16], and ovarian can-cer [10]. The 1.2-Mb sequence at 8q24 containing all observed risk alleles does not code for any known genes and the biologic mechanisms underlying these associations are unknown. The oncogene c-MYC is the closest distal gene to this region and it has been suggested that the observed associations refl ect long-range control of myc expression. To date, 38 distinct genetic loci harbouring prostate cancer risk alleles have been identifi ed and consistently repli-cated (Table I). In general, the effect of variants in these regions on prostate cancer risk is modest with odds ratios typically ranging between 1.1 and 1.3. It has been estimated 17 that hitherto identifi ed variants together explain approximately 22% of the familial risk of prostate cancer and it is anticipated
that many more prostate cancer susceptibility vari-ants will be identifi ed in the future. All these studies have been conducted on European/North American populations making the translation to other ethnic groups uncertain. However, a recent Japanese study showed that x/x of known prostate cancer suscepti-bility loci was confi rmed in Japanese men [18]. In addition fi ve new loci was reported and it is unknown if these loci are specifi c to the Japanese population or not.
Germline genetic markers and disease aggressiveness
To date there is no reliable way of predicting whether prostate cancer will be an aggressive, fast-growing disease or a non-aggressive, slow-growing type of cancer. In general, a combination of tumor staging (using the tumor-node-metastasis staging system [19]), tumor grading (using the Gleason scoring sys-tem [20]) and diagnostic PSA serum levels are used to classify patients into different prognostic risk groups to guide clinicians in treatment decisions. In genetic association studies, prostate cancer patients are commonly classifi ed as having a more aggressive form of the disease if they fulfi ll any of the following criteria: 1) disease spread outside of the prostate gland, or presence of cancer in the lymph nodes or other metastatic sites; 2) presence of poorly differen-tiated cancer as indicated by a high Gleason score (i.e. 4 � 3 � 7 or higher); or 3) a serum PSA level associated with a high likelihood of extensive disease (i.e. � 20 ng/ml).
Several studies have explored the capacity of established prostate cancer risk variants to distinguish between less aggressive and more aggressive disease [7 – 9,21 – 43]. Overall, results are inconclusive, with some studies reporting stronger associations for some of these variants among more aggressive prostate can-cer patients, while others did not. In a large replication study from the PRACTICAL (Prostate cancer asso-ciation group to Investigate cancer associated altera-tions in the genome) consortium, which evaluated genetic variants at chromosome 3p12, 6q25, 7q21, 10q11, 11q13, 19q13 and Xp11 among 7 370 pros-tate cancer cases and 5 742 controls, no association with tumor grade was observed for any of the explored variants [42]. Fitzgerald and coworkers assessed the same seven variants and an additional six variants at chromosome 7p15, 8q24, 10q26, and 17q12 in a pop-ulation-based study comprising 1 308 cases and 1 267 controls for association with family history and clinical features of more aggressive disease [43]. No associa-tion was observed between any of the evaluated risk variants and a composite measure of disease aggres-siveness; however, two variants, rs10993994 at 10q11

20 M. Aly et al.
(p � 0.02) and rs5945619 at Xp11 (p � 0.03) were nominally signifi cantly associated with Gleason score.
Most of the published studies exploring estab-lished risk variants with respect to prostate cancer aggressiveness had several limitations including small sample size, heterogeneous defi nition of aggressive disease across multiple study populations, and reli-ance on clinical grading and staging of tumors. To address these limitations Xu and coworkers evalu-ated 20 established risk variants in 17 distinct genomic regions among 5 895 prostate cancer patients of European descent who underwent radical prostatectomy for treatment of prostate cancer. Based on the entire prostate gland each tumor was uni-
formly graded and staged using the same protocol. For 18 of the 20 variants explored no signifi cant dif-ference was observed in risk allele frequencies between patients with more aggressive and less aggressive disease. Two variants were signifi cantly asso-ciated with disease aggressiveness; SNP rs2735839 downstream of the kallikrein 3 gene ( KLK3 , p � 8.4�10 �7 ), the gene coding for PSA, and SNP rs10993994 in the microseminoprotein beta gene ( MSMB , p � 0.046). Since these risk alleles have been shown to strongly associate with higher PSA levels among population controls [25,44,45], it is possible that the observed association with aggressive disease may partly refl ect a PSA detection bias.
Table I. Validity SNPs and chromosomal loci associated with prostate cancer risk.
dbSNP No. Chromosome Gene ∗ Risk Allele † Per Allele OR Study
rs1465618 2p21 THADA A 1.08 Eeles et al. 2009 [17]rs721048 2p15 EHBP1 A 1.15 Gudmundsson et al. 2008 [24]rs12621278 2q31.1 ITGA6 A 1.33 Eeles et al. 2009 [17]rs4857841 3q21.3 EEFSEC A 1.12 Gudmundsson et al. 2009 [55]rs12500426 4q22.3 PDLIM5 A 1.08 Eeles et al. 2009 [17]rs17021918 4q22.3 PDLIM5 C 1.11 Eeles et al. 2009 [17]rs7679673 4q24 FLJ20032 C 1.10 Eeles et al. 2009 [17]rs9364554 6q25.3 SLC22A3 T 1.17 Eeles et al. 2008 [25]rs10486567 7p15.2 JAZF1 G 1.35 Thomas et al. 2008 [23]rs6465657 7q21.3 LMTK2 C 1.12 Eeles et al. 2008 [25]rs1512268 8p21.2 NKX3-1 T 1.18 Eeles et al. 2009 [17]rs12543663 8q24.21 C 1.08 Al Olama et al. 2009 [12]rs10086908 8q24.21 T 1.15 Al Olama et al. 2009 [12]rs1016343 8q24.21 T 1.21 Al Olama et al. 2009 [12]rs13252298 8q24.21 A 1.19 Al Olama et al. 2009 [12]rs6983561 8q24.21 C 1,47 Al Olama et al. 2009 [12]rs16901979 8q24.21 A 1,79 Gudmundsson et al. 2007 [7]rs16902094 8q24.21 G 1.21 Gudmundsson et al. 2009 [55]rs445114 8q24.21 T 1.14 Gudmundsson et al. 2009 [55]rs620861 8q24.21 C 1.11 Al Olama et al. 2009 [12]rs6983267 8q24.21 G 1.26 Al Olama et al. 2009 [12]rs1447295 8q24.21 A 1.29 Amundadottir et al. 2006 [56]rs10993994 10q11.23 MSMB T 1.25 Eeles et al. 2008 [25]rs4962416 10q26.13 CTBP2 C 1.20 Thomas et al. 2008 [23]rs7127900 11p15.5 A 1.22 Eeles et al. 2009 [17]rs12418451 11q13.2 A 1.15 Zheng et al. 2009 [31]rs11228565 11q13.2 A 1.23 Gudmundsson et al. 2009 [55]rs10896449 11q13.2 G 1.28 Thomas et al. 2008 [23]rs11649743 17q12 HNF1B G 1.28 Sun et al. 2008 [27]rs4430796 17q12 HNF1B A 1.22 Gudmundsson et al. 2007 [22]rs1859962 17q24.3 G 1.20 Gudmundsson et al. 2007 [22]rs8102476 19q13.2 PPP1R14A C 1.12 Gudmundsson et al. 2009 [55]rs2735839 19q13.33 KLK3 A 1.20 Eeles et al. 2008 [25]rs9623117 22q13.1 TNRC6B C 1.11 Sun et al. 2009 [28]rs5759167 22q13.2 BIK G 1.16 Eeles et al. 2009 [17]rs5945619 Xp11.22 NUDT11 C 1.19 Eeles et al. 2008 [25]
∗ These genes are within the linkage-disequilibrium block defi ned by the associated variant. THADA denotes thyroid adenoma associated isoform 1, EHBP1 the EH domain binding protein 1, ITGA6 the integrin alpha chain 6, EEFSEC the elongation factor for selenoprotein translation, PDLIM5 the PDZ and LIM domain 5 isoform d, FLJ20032 the hypothetical protein LOC54790, SLC22A3 the solute carrier family 22 member 3, JAZF1 the juxtaposed with another zinc fi nger gene 1, LMTK2 the lemur tyrosine kinase 2, SLC25A37 the mitochondrial solute carrier protein, NKX3-1 the NK3 transcription factor related locus 1, MSMB the beta-microseminoprotein isoform a precursor, CTBP2 the C-terminal binding protein 2 isoform 2, HNF1B hepatocyte nuclear factor 1 homeobox B, PPP1R14A the protein phosphatase 1, regulatory inhibitor, KLK3 the kallikrein 3 gene, TNRC6B the trinucleotide repeat containing 6B isoform 2, BIK the BCL2-interacting killer, NUDT11 the nudix-type motif 11. † Risk alleles as defi ned from published data cited in the column.

Genetic Markers in Prostate Cancer 21
Somatic (tumor) genetic marker
Acquired genetic mutations/rearrangement in a tumor during the development of the tumor. These changes can be detected from DNA or RNA from the specifi c tumor. New techniques make it possible to detect these somatic mutations (DNA) or copies of gene expression (RNA) directly in blood plasma.
Since the 1970s it has been known that elevated levels of free circulating DNA can be detected in blood in patients with malignant disease [46]. It was also shown in the same paper that the levels correlated with metastatic status and that the levels decreased after therapy. Chun et al. described 2006 that plasma DNA level is predictive and highly accurate to assess the risk of positive biopsy outcome in prostate cancer. Increased levels of cDNA have also been shown in patients with metastatic disease, although they also found elevated levels in patients with benign prostatic hyperplasia [47]. Papadopoulou et al. showed that cell-free DNA could be used to distinguish between patients with prostate cancer and those not diagnosed with the disease [48]. Cherepanova et al. has also shown that patients with prostate cancers have elevated levels in blood in com-parison to patients with benign diseases of the prostate [49]. The utility of detecting tumor-specifi c rearrange-ments in plasma is currently limited by the heterogene-ity of solid tumors. In an attempt to map structural variation in breast cancer, none of 24 tumors harbored any identical rearrangements [50]. Therefore, for most cancer types, the primary tumor needs to be investi-gated fi rst, which limits the possibility to use circulating DNA for early detection. Although it would be possible to detect individual-specifi c tumor rearrangements directly in plasma or serum it is not practically feasible due to costs since tumor DNA constitutes � 30% of the total amount of circulating nucleic acids [51]. Unlike other adenocarcinomas, prostate tumors har-bors a rearrangement (TMPRSS2-ERG gene fusion) that occur in 50% of cases [52]. These rearrangements may be detectable in blood and could be of diagnostic and perhaps also of prognostic value. Further studies are needed to assess this as a clinically useful marker.
miRNA
MikroRNAs are small, up to 22 nucleotides long, non-coding functional RNAs. It is estimated that 1 000 such small RNAs exists and they are all expressed in a tissue specifi c manner suggesting that they could be used as markers for various conditions. miRNAs are known to regulate gene expression. Porrka et al. identifi ed 51 dif-ferent miRNA being either up- or down from an expres-sion profi le of 319 genes encoding miRNA in prostatic cancer tissue as compared to normal prostatic tissue. Twenty two of these were downregulated in all prostate cancer samples and 15 were only downregulated in
castration resistant specimens. It has also been demon-strated that miRNA profi les were consistent with prostate cancer disease process [53]. miRNA plays a biological role that may imply their correlation with diagnosis and therapeutic outcome and miRNA are possible to detect in plasma samples.
Identifi cation of somatic mutations in prostate tumors
With the introduction of massive parallel sequencing it is now possible to screen for somatic mutation in the entire coding parts of the genome (all genes) or even the entire genome in 10 – 100 of tumors. This has been demonstrated to be successful in several other tumors as breast cancer, colon cancer and lung cancer in which several new key genes and pathways have been identifi ed. If protein products from these altered genes are secreted in either the urine or the bloodstream new biomarkers might be identifi ed.
Conclusion
There are several promising new “ genetic ” markers that in the near future might be used in clinical routine. These markers are easy to measure and stabile over time. We foresee the following areas as most promising:
1. Identifying high-risk populations using a combination of prostate cancer susceptibility alleles (SNPs).
Individually, each risk variant has a modest effect on disease risk and they will clearly not be useful for individualized risk prediction. However, risk profi les based on a combination of risk variants lead to an appreciable increased risk of disease [32] and there is potential for the predictive power to increase considerably as more risk variants are detected [54]. Combining the fi rst 28 prostate cancer SNPs in the Swedish CAPS study, the top 8% of the population had three times of more increased lifetime risk of prostate cancer. These high-risk men can be selected for targeted screening or chemoprevention.
2. Using prostate cancer susceptibility alleles (SNPs) in current risk calculators for prostate cancer.
These risk variants might be used in combination with current risk calculation aiding clinicians when to do prostate biopsy or not.
3. The identifi cation of new prognostic markers both germline genetic variants and other biomarkers based somatic mutations in the tumors are highly warranted.
However the challenge is not only to identify new biomarkers but the real test is to validate

22 M. Aly et al.
new biomarkers in several large well-characterized patient populations. This validation must be done together will all other known biomarkers at the same time as it not likely that one single marker is enough, but a panel of different markers. Today 2010 there are over 19 000 publications in the area of biomarkers and prostate cancer, but only one biomarker, PSA, is used in the clinic today!
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto [1] S, Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009;360:1320 – 8. Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia [2] D, Church TR, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009;360:1310 – 9. Hugosson J, Carlsson S, Aus G, Bergdahl S, Khatami A, [3] Lodding P, et al. Mortality results from the Goteborg ran-domised population-based prostate-cancer screening trial. Lancet Oncol 2010;11:725 – 32. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, [4] Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000;343:78 – 85. Schaid DJ. The complex genetic epidemiology of prostate [5] cancer. Hum Mol Genet 2004;13(Spec No. 1):R103 – 21. Chung CC, Magalhaes WC, Gonzalez-Bosquet J, Chanock [6] SJ. Genome-wide association studies in cancer – current and future directions. Carcinogenesis 2010;31:111 – 20. Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, [7] Gudbjartsson D, Helgason A, et al. Genome-wide associa-tion study identifi es a second prostate cancer susceptibility variant at 8q24. Nat Genet 2007;39:631 – 7. Haiman CA, Patterson N, Freedman ML, Myers SR, [8] Pike MC, Waliszewska A, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 2007;39:638 – 44. Zheng SL, Sun J, Cheng Y, Li G, Hsu FC, Zhu Y, et al. [9] Association between two unlinked loci at 8q24 and prostate cancer risk among European Americans. J Natl Cancer Inst 2007;99:1525 – 33. Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai [10] Z, Driver KE, et al. Multiple loci with different cancer spe-cifi cities within the 8q24 gene desert. J Natl Cancer Inst 2008;100:962 – 6. Yeager M, Xiao N, Hayes RB, Bouffard P, Desany B, Burdett [11] L, et al. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum Genet 2008;124:161 – 70. Al Olama AA, Kote-Jarai Z, Giles GG, Guy M, Morrison J, [12] Severi G, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet 2009;41:1058 – 60. Yeager M, Chatterjee N, Ciampa J, Jacobs KB, Gonzalez-[13] Bosquet J, Hayes RB, et al. Identifi cation of a new prostate cancer susceptibility locus on chromosome 8q24. Nat Genet 2009;41:1055 – 7. Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thomp-[14] son D, Ballinger DG, et al. Genome-wide association study
identifi es novel breast cancer susceptibility loci. Nature 2007;447:1087 – 93. Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, [15] Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifi es a susceptibility variant for colorectal can-cer at 8q24.21. Nat Genet 2007;39:984 – 8. Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, [16] Stacey SN, et al. Sequence variant on 8q24 confers suscep-tibility to urinary bladder cancer. Nat Genet 2008;40:1307 – 12. Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG, Guy M, [17] Severi G, et al. Identifi cation of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet 2009;41:1116 – 21. Takata R Akamatsu S, Kubo M, Takahashi A, Hosono N, [18] Kawaguchi T, et al. Genome-wide association study identifi es fi ve new susceptibility loci for prostate cancer in the Japanese population. Nat Genet 2010;42:751 – 4. Hoedemaeker RF, Vis AN, Van Der Kwast TH. Staging pros-[19] tate cancer. Microsc Res Tech 2000;51:423 – 9. Epstein JI, Allsbrook WC, Jr., Amin MB, Egevad LL. The [20] 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Car-cinoma. Am J Surg Pathol 2005;29:1228 – 42. Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder [21] S, et al. Genome-wide association study of prostate cancer identifi es a second risk locus at 8q24. Nat Genet 2007;39:645 – 9. Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson [22] JT, Thorleifsson G, Manolescu A, et al. Two variants on chro-mosome 17 confer prostate cancer risk and the one in TCF2 protects against type 2 diabetes. Nat Genet 2007;39:977 – 83. Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr [23] N, et al. Multiple loci identifi ed in a genome-wide association study of prostate cancer. Nat Genet 2008;40:310 – 5. Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Mano-[24] lescu A, Gudbjartsson D, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate can-cer. Nat Genet 2008;40:281 – 3. Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugur-[25] nauth SK, et al. Multiple newly identifi ed loci associated with prostate cancer susceptibility. Nat Genet 2008;40:316 – 21. Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, [26] Wiklund F, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst 2007;99:1836 – 44. Sun J, Zheng SL, Wiklund F, Isaacs SD, Purcell LD, Gao Z, [27] et al. Evidence for two independent prostate cancer risk-as-sociated loci in the HNF1B gene at 17q12. Nat Genet 2008;40:1153 – 5. Sun J, Zheng SL, Wiklund F, Isaacs SD, Li G, Wiley KE, [28] et al. Sequence variants at 22q13 are associated with prostate cancer risk. Cancer Res 2009;69:10 – 5. Chang BL, Cramer SD, Wiklund F, Isaacs SD, Stevens VL, [29] Sun J, et al. Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum Mol Genet 2009;18:1368 – 75. Hsu FC, Sun J, Wiklund F, Isaacs SD, Wiley KE, Purcell LD, [30] et al. A novel prostate cancer susceptibility locus at 19q13. Cancer Res 2009;69:2720 – 3. Zheng SL, Stevens VL, Wiklund F, Isaacs SD, Sun J, Smith [31] S, et al. Two independent prostate cancer risk-associated Loci at 11q13. Cancer Epidemiol Biomarkers Prev 2009;18:1815 – 20.

Genetic Markers in Prostate Cancer 23
Ahn J, Berndt SI, Wacholder S, Kraft P, Kibel AS, Yeager M, [44] et al. Variation in KLK genes, prostate-specifi c antigen and risk of prostate cancer. Nat Genet 2008;40:1032 – 4; Author reply 35 – 6. Wiklund F, Zheng SL, Sun J, Adami HO, Lilja H, Hsu FC, [45] et al. Association of reported prostate cancer risk alleles with PSA levels among men without a diagnosis of prostate can-cer. Prostate 2009;69:419 – 27. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in [46] the serum of cancer patients and the effect of therapy. Can-cer Res 1977;37:646 – 50. Jung K, Stephan C, Lewandowski M, Klotzek S, Jung M, [47] Kristiansen G, et al. Increased cell-free DNA in plasma of patients with metastatic spread in prostate cancer. Cancer Lett 2004;205:173 – 80. Papadopoulou E, Davilas E, Sotiriou V, Koliopanos A, [48] Aggelakis F, Dardoufas K, et al. Cell-free DNA and RNA in plasma as a new molecular marker for prostate cancer. Oncol Res 2004;14:439 – 45. Cherepanova AV, Tamkovich SN, Bryzgunova OE, Vlassov [49] VV, Laktionov PP. Deoxyribonuclease activity and circulat-ing DNA concentration in blood plasma of patients with prostate tumors. Ann N Y Acad Sci 2008;1137:218 – 21. Stephens PJ, McBride DJ, Lin ML, Varela, I, Pleasance ED, [50] Simpson JT, et al. Complex landscapes of somatic rearrange-ment in human breast cancer genomes. Nature 2009;462:1005 – 10. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. [51] Detection and quantifi cation of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 2005;102:16368 – 73. Perner S, Svensson MA, Hossain RR, Day JR, Groskopf J, [52] Slaughter RC, et al. ERG rearrangement metastasis patterns in locally advanced prostate cancer. Urology 2010;75:762 – 7. Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread [53] deregulation of microRNA expression in human prostate cancer. Oncogene 2008;27:1788 – 93. Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Poly-[54] genes, risk prediction, and targeted prevention of breast cancer. N Engl J Med 2008;358:2796 – 803. Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, [55] Gylfason A, Agnarsson BA, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet 2009;41:1122 – 6. Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, [56] Baker A, Agnarsson BA, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet 2006;38:652 – 8.
Zheng SL, Sun J, Wiklund F, Smith S, Stattin P, Li G, et al. [32] Cumulative association of fi ve genetic variants with prostate cancer. N Engl J Med 2008;358:910 – 9. Sun J, Chang BL, Isaacs SD, Wiley KE, Wiklund F, Stattin [33] P, et al. Cumulative effect of fi ve genetic variants on prostate cancer risk in multiple study populations. Prostate 2008;68:1257 – 62. Xu J, Isaacs SD, Sun J, Li G, Wiley KE, Zhu Y, et al. Asso-[34] ciation of prostate cancer risk variants with clinicopathologic characteristics of the disease. Clin Cancer Res 2008;14:5819 – 24. Wang L, McDonnell SK, Slusser JP, Hebbring SJ, Cunning-[35] ham JM, Jacobsen SJ, et al. Two common chromosome 8q24 variants are associated with increased risk for prostate cancer. Cancer Res 2007;67:2944 – 50. Suuriniemi M, Agalliu I, Schaid DJ, Johanneson B, McDon-[36] nell SK, Iwasaki L, et al. Confi rmation of a positive associa-tion between prostate cancer risk and a locus at chromosome 8q24. Cancer Epidemiol Biomarkers Prev 2007;16:809 – 14. Cheng I, Plummer SJ, Jorgenson E, Liu X, Rybicki BA, [37] Casey G, et al. 8q24 and prostate cancer: Association with advanced disease and meta-analysis. Eur J Hum Genet 2008;16:496 – 505. Helfand BT, Loeb S, Cashy J, Meeks JJ, Thaxton CS, Han [38] M, et al. Tumor characteristics of carriers and noncarriers of the deCODE 8q24 prostate cancer susceptibility alleles. J Urol 2008;179:2197 – 201; Discussion 202. Tan YC, Zeigler-Johnson C, Mittal RD, Mandhani A, Mital [39] B, Rebbeck TR, et al. Common 8q24 sequence variations are associated with Asian Indian advanced prostate cancer risk. Cancer Epidemiol Biomarkers Prev 2008;17:2431 – 5. Terada N, Tsuchiya N, Ma Z, Shimizu Y, Kobayashi T, Naka-[40] mura E, et al. Association of genetic polymorphisms at 8q24 with the risk of prostate cancer in a Japanese population. Prostate 2008;68:1689 – 95. Robbins C, Torres JB, Hooker S, Bonilla C, Hernandez W, [41] Candreva A, et al. Confi rmation study of prostate cancer risk variants at 8q24 in African Americans identifi es a novel risk locus. Genome Res 2007;17:1717 – 22. Kote-Jarai Z, Easton DF, Stanford JL, Ostrander EA, Sch-[42] leutker J, Ingles SA, et al. Multiple novel prostate cancer predisposition loci confi rmed by an international study: The PRACTICAL Consortium. Cancer Epidemiol Biomarkers Prev 2008;17:2052 – 61. Fitzgerald LM, Kwon EM, Koopmeiners JS, Salinas CA, [43] Stanford JL, Ostrander EA. Analysis of recently identifi ed prostate cancer susceptibility loci in a population-based study: Associations with family history and clinical features. Clin Cancer Res 2009;15:3231 – 7.

Correspondence: Steven Joniau, Department of Urology, University Hospitals Leuven, Herestraat 49, 3000 Leuven, Belgium. Tel: � 32 16 346930. Fax: � 32 16 346931. E-mail: [email protected]
Introduction
STEVEN JONIAU
Department of Urology, University Hospitals Leuven, Leuven, Belgium
We are facing tremendous dilemmas in the fi eld of prostate cancer detection and treatment. Of all prostate cancers diagnosed, many do not need treatment while others are lethal, even when found and treated in a localized clinical stage. The main challenge today is not so much how to treat, but whom to treat.
Can we reliably identify patients who do not need aggressive treatment? Do we have the right tools at hand to identify lethal prostate cancer? As clinicians, we rely on clinical factors such as prostate-specifi c antigen (PSA), clinical stage and biopsy Gleason score. Based upon those, patients can be classifi ed using risk stratifi cation models of whom the D ’ Amico risk groups are amongst the most frequently used. Such models are convenient for physicians, as treat-ment guidelines are built around them. Nevertheless, these risk stratifi cation models fail to provide accu-rate risk predictions. Improved, individual risk pre-dictions can be achieved using nomograms. However, no matter how refi ned these tools are, they only pro-vide statistical estimations with a certain degree of uncertainty. What we really need is individualized patient care using patient-specifi c tools.
Ways to overcome these hurdles have emerged in the last couple of years.
Imaging modalities have improved tremendously. Multiparametric magnetic resonance imaging (MRI) not only delivers extremely detailed anatomical images, but also provides tissue-specifi c information by using magnetic resonance (MR) spectroscopy, dynamic contrast enhanced (DCE) MRI and diffu-sion weighted (DW) MRI. These modalities help us to discern benign from malignant tissues. Further-more, our understanding of prostate cancer biology has improved tremendously. Based upon this knowl-edge, novel positron emission tomography (PET)
tracers like 18 F-FDHT, probing the androgen (AR) signaling axis, have been developed. The role of MRI and PET will be discussed in detail in the papers by Heijmink and Fox.
Besides imaging, even more individualized infor-mation can be gained from the tumor itself. Serum PSA still is the most important biomarker for the detection and follow-up of prostate cancer. PSA based screening can reduce disease specifi c mortality but coinciding unnecessary testing and over diagnosis warrant further research for more specifi c biomarkers. Attractive, because non-invasive, are urine-based tests. Numerous, very interesting candidate tumor markers have been identifi ed and are presently being tested. The prostate cancer antigen 3 (PCA3) test, the TMPRSS2 – ERG fusion gene and their combination have been subject of many studies showing encour-aging results. Finally, most of the information can be derived from the cancer tissue itself. On one side, correct tissue handling and reporting after biopsy taking or surgical removal is vital, as outcome pre-diction and thus tumor marker validation is fully dependent on this. On the other side, careful tissue collection and internal quality control of the tissues is of extreme importance in the development of tumor marker development. Those issues will be further elaborated in the contributions by Roobol, Berney and Montironi.
Better identifi cation of those patients who do not need treatment and those who harbor a potentially lethal form of prostate cancer have a far-reaching impact on global healthcare. Undoubtedly, further development of modern imaging modalities and tumor markers are key in this process and will change the face of prostate cancer management forever. Truly individualized prostate cancer care is on the horizon.
Acta Oncologica, 2011; 50(Suppl 1): 24
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.578378

Correspondence: Stijn W.T.P.J. Heijmink, Department of Radiology, Radboud University Nijmegen Medical Center, PO Box 9101, NL-6500 HB Nijmegen, The Netherlands. E-mail: [email protected]
(Received 12 October 2010; accepted 10 November 2010)
ORIGINAL ARTICLE
State-of-the-art uroradiologic imaging in the diagnosis of prostate cancer
STIJN W. T. P. J. HEIJMINK1 , JURGEN J. F Ü TTERER1 , STEPHEN S. STRUM2 , WIM J. G. OYEN3 , FERDINAND FRAUSCHER4 , J. ALFRED WITJES5 & JELLE O. BARENTSZ1
1Department of Radiology, Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands, 2Medical Oncologist, Ashland, OR 97520, USA, 3Department of Nuclear Medicine, Radboud University Nijmegen Med. Center, Nijmegen, The Netherlands, 4Department of Radiology II, University Hospital Innsbruck, A-6020 Innsbruck, Austria and 5Department of Urology, Radboud University Nijmegen Medical Center, Nijmegen, The Netherlands
Abstract In the diagnostic process of prostate cancer, several radiologic imaging modalities signifi cantly contribute to the detection and localization of the disease. These range from transrectal ultrasound (TRUS) and magnetic resonance imaging (MRI) to positron emission tomography (PET). Within this review, after evaluation of the literature, we will discuss the advantages and disadvantages of these imaging modalities in clarifying the patient ’ s clinical status as to whether he has prostate cancer or not and if so, where it is located, so that therapy appropriate to the patient ’ s disease may be administered. TRUS, specifi cally with the usage of intravenous contrast agents, provides an excellent way of directing biopsy towards suspicious areas within the prostate in the general (screening) population. MRI using functional imaging techniques allows for highly accurate detection and localization, particularly in patients with prior negative ultrasound guided biopsies. A promising new development is the performance of biopsy within the magnetic resonance scanner. Subsequently, a proposal for optimal use of radiologic imaging is presented and compared with the European and American urological guidelines on prostate cancer.
With a total of 217 730 new cases estimated for 2010, prostate cancer (PC) now accounts for 28% of all new male cancers diagnosed in the USA [1]. In their lifetime, one in six men will be clinically diag-nosed with having PC, although many more men are found to have histological evidence of PC at autopsy [2 – 4]. Presently, approximately 1 in 10 men will die of PC [5,6]. The ever-aging population and wider spread use of the prostate-specifi c antigen (PSA) test [7,8], as well as the tendency to apply lower cut-off levels for this test [9], will further increase the diag-nosis of this disease [10].
An elevated PSA level, abnormal changes in PSA level (i.e. PSA dynamics) such as PSA velocity or doubling time, or an abnormal digital rectal exami-nation are biologic indicators signaling an increased risk of PC. With the improvement and wider range of curative therapies, detection and subsequent exact
localization of PC have become increasingly important because of their infl uence on treatment strategy [11,12]. Two such affected treatments are laparo-scopic (robotic) radical prostatectomy and intensity-modulated radiation therapy (IMRT [13]). The urologist ’ s inability to palpate the operating fi eld dur-ing laparoscopic surgery makes it even more crucial to know where the cancer is located. Similarly, the urologist must know whether the cancer is near a neurovascular bundle since this affects the decision of whether or not to perform nerve-sparing prostate-ctomy [14]. IMRT also necessitates accurate PC localization. While giving a standard dose to the pros-tate, a higher (i.e. boost) dose can be given to any dominant intraprostatic lesion(s) as these lesions regularly appear to be the sites of recurrent disease [15]. Furthermore, precision radiation dosimetry will decrease radiation complications, particularly
Acta Oncologica, 2011; 50(Suppl 1): 25–38
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.578369

26 S. W. T. P. J. Heijmink et al.
rectal wall toxicity [16], thereby likely dimini-shing the development of post-radiation rectal cancer [17].
In order to determine the optimal treatment for the individual patient, it is necessary to evaluate all patient and cancer characteristics. Most often used for this purpose are laboratory values (PSA level and dynamics), the results of digital rectal examina-tion (clinical staging), and histopathological pros-tatic biopsy fi ndings (Gleason score). However, imaging may play an important role in detecting and localizing areas most refl ective of the actual aggressiveness of the cancer. This directly infl uences the assessment of the patient and may lead to important changes in treatment strategy which can mean the difference between treatment success and failure.
Currently, a spectrum of imaging modalities is available to clinicians for tackling detection- and localization-related problems. To provide optimal and cost effi cient patient care, these techniques should be used in the appropriate clinical context to aid clinicians in detecting and localizing PC.
This review 1) presents an overview of the currently available imaging methods to aid in PC detection and localization. 2) Additionally, a scheme is proposed for optimal evidence-based use of imaging in detecting and localizing prostate cancer and a critical comparison is made between this scheme and the most recent guidelines as put forward by the American Urological Association (AUA) and European Association of Urology (EAU).
Literature search
Relevant articles were retrieved using combinations of both Medical Subject Headings (MeSH) and free search terms in the MedLine ® (WebSPIRS Version 5.12, Build 20060224, Ovid Technologies) and Pubmed (U.S. National Library of Medicine) online search engines.
MeSH terms included: “ Prostate ” , “ Anatomy ” , “ Prostatic Neoplasms ” , “ Neoplasm Staging ” , “ Ultra-sonography ” , “ Tomography, X-Ray Computed ” , “ Magnetic Resonance Imaging ” , “ Magnetic Reso-nance Spectroscopy ” , “ Diffusion Magnetic Resonance Imaging ” , “ Radionuclide Imaging ” , and “ Positron-Emission Tomography ” .
Free search terms included: “ prost * ” , “ cancer ” , “ detect * ” , “ localization ” , “ localisation ” , “ biops * ” , “ stag * ” , “ capsul * ” , “ extracapsular penetration ” , “ ext-racapsular extension ” , “ seminal vesicle invasion ” , “ transrectal ultraso * ” , “ TRUS ” , “ computed tomogra-phy ” , “ CT ” , “ magnetic resonance imaging ” , “ MRI ” , “ ferumoxtran-10 ” , “ magnetic resonance spectroscopy ” ,
“ spectroscopy ” , “ MRS ” , “ bone scan * ” , “ bone scintig-raphy ” , “ positron-emission tomography ” , “ PET ” , and “ ProstaScint ” .
Reference lists of selected articles were further analyzed for relevant articles.
Prostate cancer detection and localization in reference to prostate anatomy and essential prostate cancer characteristics
In order to effectively apply the various imaging modalities, it is important to fi rst understand both the normal prostate anatomy and the distribution and intrinsic characteristics of PC.
Normal anatomy as related to PC localization
On the basis of its embryological origins, the pro-state is anatomically divided into three zones that are eccentrically located around the urethra: the innermost transition zone (TZ), the central zone, and the outermost peripheral zone (PZ) [18,19]. In older patients, the former two cannot be distin-guished radiologically due to compression of the central zone by benign prostatic hyperplasia (BPH) in the TZ and together they are referred to as the central gland; this as opposed to the outer gland, which comprises the PZ. Furthermore, the prostate is craniocaudally divided into apex (the caudal one-third), mid-gland, and base (the cranial one-third).
Anatomical distribution of PC
Up to 70 – 80% of PC is located in the PZ [20] and overall analysis of these cancers has shown homoge-neous distribution across the entire PZ [21], with over half of the prostates containing two or more distinct cancer foci [22]. Nevertheless, while up to 20 – 52% of all PC originate in the TZ, only a small (3.6 – 25%) percentage of these cancers [21,23] occur solely in the TZ as many will have concurrent PZ cancer foci [20,24,25].
Pathological grading of PC aggressiveness
Presently, the most widely used histological scaling system for PC aggressiveness is the Gleason score [26,27], which consists of two numbers: a primary and secondary Gleason grade refl ecting the two grades most frequent in the specimen. Each Gleason grade is assigned a value between 1 and 5, the higher numbers indicating a more aggressive cancer. The prognostic value of the Gleason grading system is well-documented [28,29].

State-of-the-art radiologic imaging in prostate cancer 27
Patients clinically at risk for prostate cancer: Radiological imaging to detect and localize primary PC
Transrectal ultrasound (TRUS): Reliable, although not perfect, daily-use modality
Grayscale TRUS. Today, in regular clinical prac-tice, prostate biopsies are performed under TRUS guidance. Even though the traditional ultrasound appearance of PC is a PZ hypoechoic lesion (Figure 1A, B), other conditions such as prostatitis and prostatic intraepithelial neoplasia may also pres-ent as hypoechoic lesions [30,31]. It is important to note that over 40% of PC lesions are isoechoic (Figure 1C, D) while only 5% are hyperechoic [32]. Therefore, targeting only hypoechoic areas is not an optimal approach for successful PC detec-tion [33] and various biopsy protocols that sample tissue at standard locations (i.e. systematic biopsy) within the prostate have become the most common biopsy technique [34]. The number of cores taken per session varies across institutions. Recently, how-ever, emphasis has been put on adequate tissue sam-pling from more laterally located PZ regions [35,36] and on the relative unimportance of biopsying the
central gland [37]. Despite the use of extended systematic biopsy protocols, there is still an approxi-mately 20% chance that the Gleason score at pros-tatectomy will differ from that at biopsy to a clinically relevant degree [38]. Recently, it was observed that biopsies performed with an endfi re probe obtained a signifi cantly higher biopsy rate compared with side-fi re probes [39]. PC detection rates have varied from 19 – 40% [40,41] and repeat biopsy sessions are often necessary [42]. Localization sensitivity varied widely between 39 – 75% (Table I).
Doppler TRUS. Because increased blood fl ow due to neovascularity is one of the characteristics of PC, this is a means of targeting lesions. In a study of 96 patients with lower urinary tract symptoms and PSA levels over 4 ng/ml [43], the degree of Doppler signal correlated with the microvessel density and Gleason score of a lesion. One study achieved Doppler imaging-based de-tection rates of 40% [44]. Power Doppler TRUS could improve the localization specifi city [45]. However, a drawback of Doppler imaging is the high inter-observer variability [46,47], refl ected in the widely spread sen-sitivity and specifi city fi gures in the literature (27 – 98% and 46 – 84%, respectively) (Table I).
Figure 1. (A) Axial gray-scale transrectal ultrasound image (Aplio™, Toshiba) of the prostate of a 65-year-old man (PSA level 19.02 ng/ml, biopsy Gleason score 6, normal digital rectal examination). A hypoechoic lesion was observed in the right peripheral zone (arrows). (B) Histopathology of the prostatectomy specimen confi rmed the presence of a Gleason 3 � 4 adenocarcinoma (T). (C) In another patient (69 years, PSA level 3.59 ng/ml, biopsy Gleason score 7, normal digital rectal examination), the axial gray-scale image did no show any echogenic abnormality while at histopathology (D) a number of cancer foci (T) were reported.

28 S. W. T. P. J. Heijmink et al.
Contrast-enhanced TRUS. An innovation is the application of gas-fi lled microbubble contrast agents, such as Levovist ® (Schering, Berlin, Ger-many) and SonoVue ® (Bracco, Milan, Italy) [48]. These microbubbles remain intravascular, thereby enhancing the visibility of the vascular tree in and around the prostate. This improves the ability to de-tect PC and to thus target areas more representative of the aggressiveness of PC. In experienced hands, it is reported that compared with systematic biopsy, targeting only lesions with pathological enhance-ment after contrast administration requires less than half the number of biopsy cores to obtain the same diagnostic yield [49 – 51]. A recent random-ized clinical trial comparing systematic biopsy and contrast-enhancement targeted biopsy confi rmed these fi ndings [52]. In addition, contrast-enhanced TRUS biopsies on average detected signifi cantly more aggressive cancers compared with systematic biopsy. Therefore, we can speculate that by using this technique the difference in Gleason score be-tween biopsy and prostatectomy specimens would most likely diminish. If the latter is confi rmed by future studies, pre-therapeutic risk assessment of patients will increase in accuracy. Disadvantages of using contrast agents are the longer duration and higher degree of invasiveness of the examination; however, the risk of hypersensitivity to the substance is rare and most adverse events are minor and self-resolving [53]. Sensitivities and specifi cities of PC detection using contrast-enhanced TRUS varied between 48 – 94% and 46 – 88%, respectively (Table I). A preliminary study suggests that a 14-day pre-biopsy course of dutasteride, a dual 5 α -reductase inhibitor, causes a relative high reduction in blood fl ow in healthy prostate tissue compared with can-cer tissue and could increase the diagnostic yield of contrast-enhanced TRUS targeted biopsy [54].
Sonoelastography. Transrectal sonoelastography is a new non-invasive technique that analyzes the com-pression characteristics of prostate tissue. A study by K ö nig et al. of 404 men undergoing biopsies based on real-time sonoelastography revealed a detection rate of 37.4% [55]. In a comparative study, Pallwein et al. found a signifi cantly higher per core detection rate for sonoelastography-targeted biopsy compared with systematic biopsy. Sonoelastography-targeted biopsy was 2.9 times more likely to detect cancer [56]. A drawback of the latter study was the hetero-geneity of the population since more than half of the patients had already undergone one or more nega-tive biopsy sessions. A study comparing real-time sonoelastography with radical prostatectomy re-ported a localization sensitivity of 88% [57]. While T
able
I.
Ove
rvie
w o
f st
udie
s th
at e
xam
ined
the
dia
gnos
tic
perf
orm
ance
of
loca
lizin
g pr
osta
te c
ance
r by
im
agin
g m
odal
ity.
Imag
ing
mod
alit
y R
efer
ence
s
Per
iods
of
publ
icat
ion,
ra
nge
Num
ber
of p
atie
nts,
ra
nge
Acc
urac
y (%
) S
ensi
tivi
ty (
%)
Spe
cifi c
ity
(%)
PP
V (
%)
NP
V (
%)
Tra
nsre
ctal
ult
raso
und
Gra
y-sc
ale
[47,
75,1
30]
1998
– 200
047
– 265
52 – 6
239
– 75
40 – 8
245
– 84
36 – 7
2D
oppl
er[4
4,13
1 – 13
5]19
97 – 2
004
136 –
591
54 – 8
327
– 98
46 – 8
416
– 84
69 – 9
9C
ontr
ast-
enha
nced
[33,
49,5
0,13
6,13
7]20
00 – 2
005
59 – 3
8058
– 91
48 – 9
446
– 88
33 – 8
855
– 93
End
orec
tal
MR
im
agin
g at
1.5
T
T2-
wei
ghte
d im
agin
g[6
7,73
– 75,
138,
139]
1999
– 200
724
– 53
69 – 8
252
– 83
46 – 8
838
– 80
54 – 8
9D
iffu
sion
-wei
ghte
d im
agin
g[8
7]20
1048
86 – 8
971
– 88
Pro
ton
MR
SI
[73 –
75,1
38,1
40,1
41]
1999
– 200
724
– 94
67 – 8
757
– 92
57 – 8
848
– 83
51 – 9
9 E
ndor
ecta
l M
R
imag
ing
at 3
T
T2-
wei
ghte
d M
R i
mag
ing
[102
]20
0746
76 – 8
053
– 58
83 – 8
648
– 56
85 – 8
7 P
ET
sca
nnin
g F
DG
11
C-c
holin
e[1
07,1
10]
[115
,116
]19
99 – 2
001
2005
– 200
624
– 44
41 – 4
3N
/A71
– 72
4 – 64 66
100
81 – 8
410
087
– 88
2555
– 59
PP
V, p
osit
ive
pred
icti
ve v
alue
; NP
V, n
egat
ive
pred
icti
ve v
alue
; MR
SI,
mag
neti
c re
sona
nce
spec
tros
copi
c im
agin
g; P
ET
, pos
itro
n em
issi
on to
mog
raph
y, F
DG
, 18-
fl uor
ine-
labe
lled
fl uor
odeo
xygl
ucos
e,
11 C
-cho
line,
car
bon-
11 l
abel
ed c
holin
e.
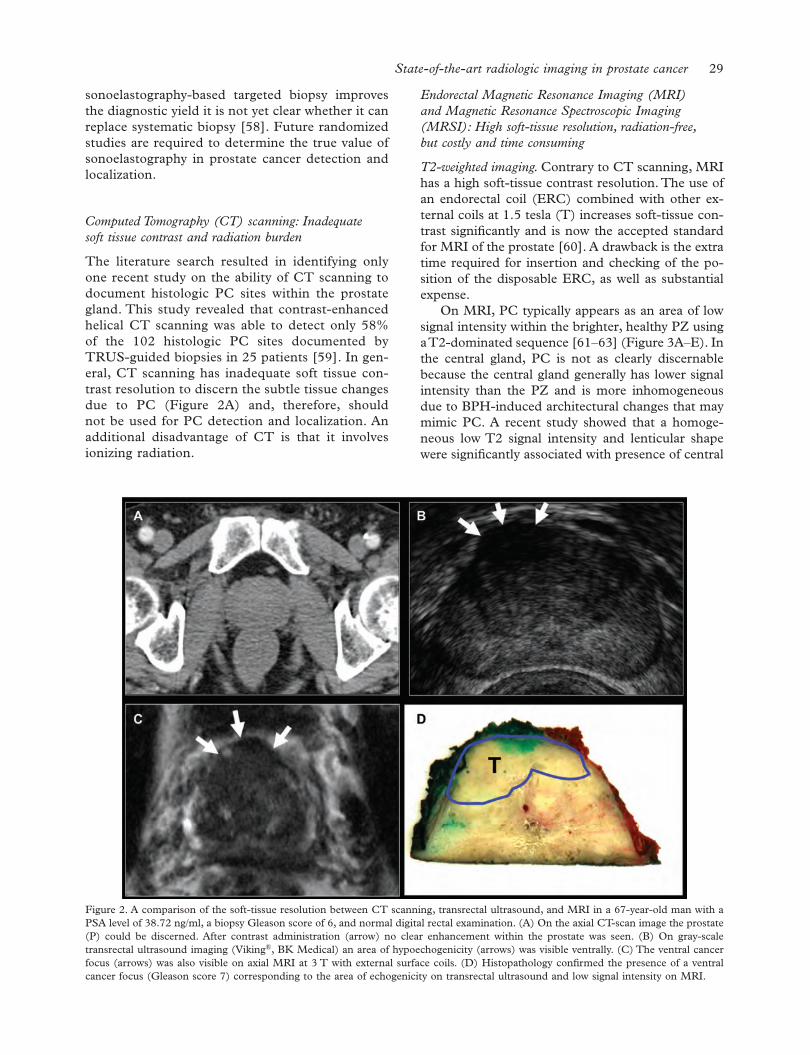
State-of-the-art radiologic imaging in prostate cancer 29
sonoelastography-based targeted biopsy improves the diagnostic yield it is not yet clear whether it can replace systematic biopsy [58]. Future randomized studies are required to determine the true value of sonoelastography in prostate cancer detection and localization.
Computed Tomography (CT) scanning: Inadequate soft tissue contrast and radiation burden
The literature search resulted in identifying only one recent study on the ability of CT scanning to document histologic PC sites within the prostate gland. This study revealed that contrast-enhanced helical CT scanning was able to detect only 58% of the 102 histologic PC sites documented by TRUS-guided biopsies in 25 patients [59]. In gen-eral, CT scanning has inadequate soft tissue con-trast resolution to discern the subtle tissue changes due to PC (Figure 2A) and, therefore, should not be used for PC detection and localization. An additional disadvantage of CT is that it involves ionizing radiation.
Endorectal Magnetic Resonance Imaging (MRI) and Magnetic Resonance Spectroscopic Imaging (MRSI): High soft-tissue resolution, radiation-free, but costly and time consuming
T2-weighted imaging. Contrary to CT scanning, MRI has a high soft-tissue contrast resolution. The use of an endorectal coil (ERC) combined with other ex-ternal coils at 1.5 tesla (T) increases soft-tissue con-trast signifi cantly and is now the accepted standard for MRI of the prostate [60]. A drawback is the extra time required for insertion and checking of the po-sition of the disposable ERC, as well as substantial expense.
On MRI, PC typically appears as an area of low signal intensity within the brighter, healthy PZ using a T2-dominated sequence [61 – 63] (Figure 3A – E). In the central gland, PC is not as clearly discernable because the central gland generally has lower signal intensity than the PZ and is more inhomogeneous due to BPH-induced architectural changes that may mimic PC. A recent study showed that a homoge-neous low T2 signal intensity and lenticular shape were signifi cantly associated with presence of central
Figure 2. A comparison of the soft-tissue resolution between CT scanning, transrectal ultrasound, and MRI in a 67-year-old man with a PSA level of 38.72 ng/ml, a biopsy Gleason score of 6, and normal digital rectal examination. (A) On the axial CT-scan image the prostate (P) could be discerned. After contrast administration (arrow) no clear enhancement within the prostate was seen. (B) On gray-scale transrectal ultrasound imaging (Viking®, BK Medical) an area of hypoechogenicity (arrows) was visible ventrally. (C) The ventral cancer focus (arrows) was also visible on axial MRI at 3 T with external surface coils. (D) Histopathology confi rmed the presence of a ventral cancer focus (Gleason score 7) corresponding to the area of echogenicity on transrectal ultrasound and low signal intensity on MRI.

30 S. W. T. P. J. Heijmink et al.
informative since the ratio between choline and citrate alters during the transformation from healthy to malignant prostatic cells [69,70] and an increas-ing choline � creatine/citrate ratio was correlated with higher Gleason scores [71]. Presently, 3D MRSI of the entire prostate can be performed [72], thereby aiding in the diagnosis of central gland PC. The addition of 3D MRSI to MRI has increased lo-calization accuracy, particularly by raising specifi c-ity up to 91% [73]. However, a limitation of MRSI is its low spatial resolution. Compared to systematic biopsy, PC localization by means of MRI and MRSI was found to be more sensitive (67% and 76% ver-sus 50%) but less specifi c (69% and 57% versus 82%) than systematic biopsy [74]. With whole-mount section histopathology as standard of refer-ence, 3D MRSI had a signifi cantly larger area under the receiver operating curve (AUC) of 0.80 in local-izing cancer, compared with 0.68 with T2-weighted MRI [75]. Adding the combination of T2-weighted imaging and MRSI to clinical data was shown to have the highest accuracy (AUC 0.85) in predict-ing the probability of a patient having insignifi cant prostate cancer [76], signifi cantly higher than that of clinical nomograms. A recent multi-institutional American College of Radiology Imaging Network study raised doubts on the additive value of MRSI over T2-weighted imaging alone [77]. However, potential factors for this result were the selected
gland PC [64]. It was reported that higher Gleason score cancers had lower signal intensities (relative to muscle) compared with low Gleason score cancers [63]. T2-weighted imaging can be performed in mul-tiple planes or as a three dimension (3D) volume acquisition [65]. Comparing T2-weighted MRI with prostatectomy specimens, MR attained high (52 – 83%) sensitivities in PC localization, while specifi ci-ties were somewhat lower (46 – 88%) (Table I).
A study that directly compared endorectal MRI with digital rectal and TRUS-guided biopsy localiza-tion revealed signifi cant incremental value from MRI [66]. In patients subjected to multiple prior negative TRUS-guided biopsies, anatomical MRI by means of T2-dominated acquisition plays an important role. In this patient population, an 83% sensitivity and 50% positive predictive value for MRI have been established [67].
Postbiopsy hemorrhage causes areas of low signal intensity on T2-weighted imaging, thereby making prostate cancer detection more diffi cult. However, it was shown recently that the amount of hemorrhage was signifi cantly lower in areas of cancer compared with healthy tissue [68].
MRSI. Additionally, MRSI (Figure 3G) can be add-ed to the protocol to provide metabolic information based on the citrate, choline, and creatine levels, and their ratios within the prostate. This is highly
Figure 3. 59-year-old patient with PSA 12, Gleason, eight tumor (circle) right PZ, stage T3a (arrows) at prostatectomy. (A) T2-w axial image, (B) DCE image showing wash-out at the tumor site, (C) ADC-map tumor shows marked restriction, which argues for high Gleason grade, (D) DWI image (b 800) tumor has high signal, (E) sagital T2-weighted image, (F) concentration time curve shows fast wash-in and fast wash-out. G. MR spectrum shows high choline (arrow). This patient scores for all modalities fi ve points (20/20). Scale: 1 no tumor, 5 defi nitely tumor.

State-of-the-art radiologic imaging in prostate cancer 31
prostatectomy population, the small average cancer focus size, and the inclusion of centers without any previous MRSI experience.
Diffusion weighted imaging (DWI). DWI is a novel non invasive technique that measures the fractional anisotropy of water molecules within the prostate which is expressed in apparent diffusion coeffi cient (ADC) mapping. Thereby, cancer tissue is deemed to result in a more restricted movement of water molecules and thus producing lower ADC values (Figure 3C, D) [78,79]. A recent study in 38 patients, performed at 1.5T with an ERC observed that the mean ADC values of regions of interest placed within prostate cancer tissue was signifi cantly lower than those placed within healthy prostate tissue [80]. In preliminary studies, combining this technique with MRSI [80] or T2-weighted imaging [81] signifi cantly improved the localization accuracy. A recent study in
37 patients revealed a signifi cant increase in sensitiv-ity from 51% for T2-weighted imaging to 71% for combined T2-weighted and DWI reading [82]. In a recent multiparametric analysis, DWI was the best-performing parameter [83]. Preliminary studies at 3T show promising results [84 – 86]. The b value used appears to affect the PC localization accuracy, as in a preliminary study imaging with a b value of 2000 s/mm 2 was shown to have a signifi cantly higher accu-racy compared with 1000 s/mm 2 [87], possibly due to a fall in the signal-to-noise ratio. At biopsy, DWI may aid in differentiating between low-risk and high-risk patients [88].
Dynamic contrast-enhanced MRI [83,89]. To further enhance localization accuracy of MRI, one may use contrast agents. Dynamic contrast-enhanced endorectal MRI, in which the contrast agent concentration is followed in time [90], is able to
Figure 4. A comparison of the image quality between axial endorectal coil (ERC) MRI at 1.5 T (A) and 3 T (B) in the same patient (age: 58 years, PSA level: 2.7 ng/ml, Gleason biopsy score: 6, normal digital rectal examination). The visibility of the internal architecture of the central gland (*) increased and the capsule (arrowheads) is better delineated at a fi eld strength of 3 T and the tumor (T) as outlined by the histopathology (C) is also better appreciated (arrows).

32 S. W. T. P. J. Heijmink et al.
discriminate between healthy prostatic tissue and PC [91]. Early contrast enhancement and high (relative) peak enhancement are the most accurate predictors of PC of the PZ, while fast washout of contrast agent and high permeability of the blood vessels (Figure 3B, F) are most sensitive for central gland PC [92,93]. A recent study showed that the AUC for localizing PC increased signifi cantly from 0.68 with regular anatomical MRI to 0.91 by ap-plying contrast agent [75]. However, limitations of using contrast agents are the higher costs and pos-sible adverse reactions, of which the most serious, anaphylaxis, is rare [94,95].
Multiparametric imaging. Combining any number of these techniques ( ‘ multiparametric imaging ’ ) has shown to increase the ability of MRI to detect and localize prostate cancer (Figure 3A – G) [96 – 98].
High-fi eld imaging. An important future direction is the use of higher magnetic fi eld strengths (e.g. 3T) [99 – 101] (Figure 4). Compared with body array coil MRI, the higher resolution obtained with ERC MRI at 3T signifi cantly improved PC localization accuracy [102].
Biopsy. Another development is to directly biopsy the prostate by means of MRI [103]. Preliminary results of direct MR-guided transrectal biopsy of suspicious lesions on pre-biopsy MRI in patients with prior negative or inconclusive TRUS-guided biopsy results demonstrated the feasibility of MR prostate biopsy without complications. Neverthe-less, disadvantages of the biopsy device are its limit-ed reach, particularly towards the base of the pros-tate, and procedure duration [104]. In a study of 68 patients with at least two prior negative TRUS biopsy sessions, MR guided biopsy established cancer in 59% [105].
Positron Emission Tomography (PET) Scanning: Metabolic information yet not suffi ciently discriminatory from benign disease
FDG. The utility of PET scanning with fl uorine-18-labelled deoxyglucose (FDG) in detecting PC is compromised by the relatively low uptake of FDG by prostate cancer cells [106] and signifi cant over-lap with marker uptake by BPH. Moreover, reports of FDG uptake correlating with PC aggressive-ness have been confl icting, although FDG uptake was substantially higher in metastasized compared to organ-confi ned primary cancers [107]. A
further drawback is that the normal urinary FDG excretion results in high bladder activity which ob-scures pathological FDG uptake in the prostate. Generally, FDG PET is not recommended for evaluation of the prostate as sensitivities are as low as of 4 – 64% with a specifi city in the order of 50% [108 – 110].
11 C-choline. Another tracer, carbon-11-labelled choline ( 11 C-choline), accumulates in prostatic cells and has the advantage that, unlike FDG, it is not excreted via the urinary tract, and thereby does not infl uence the visualization of the pros-tate [111]. Furthermore, the prostate is the only organ in the pelvis to accumulate 11 C-choline. The 11 C-choline uptake was higher in PC compared with BPH, but the difference was not signifi cant [112]. In a direct comparison between 11 C-choline PET and MRSI, a signifi cant linear correlation was observed between the maximum standardized uptake value (SUV) of 11 C-choline and the MRSI metabolite ratios. Also, 11 C-choline PET was more accurate than MRSI in accurately predicting the laterality (i.e. left- or right-sidedness) of the cancer: 81% (13/16) versus 50% (8/16), respectively [113]. Recently, it was shown that 11 C-choline preferen-tially detected more aggressive prostate cancer foci [114]. Drawbacks are the high costs of 11 C-choline and the short half-life of 11 C-choline (20 minutes). This latter precludes application of 11 C-choline in centers without cyclotrons. Nevertheless, the re-sults of the fi rst two studies combining 11 C-choline PET/CT scanning were encouraging, with a sensi-tivity of 66% and specifi city between 81 – 84% on a sextant basis [115,116]. However, the high rate of false negative fi ndings was a concern. A direct preoperative comparison between 11C-choline PET, FDG PET, and MRI in 43 patients showed that 11C-choline outperformed FDG PET in localizing prostate cancer but that MRI was superior to both [117].
Other radiopharmaceuticals. A preliminary PET/CT study using fl uoro-18-choline ( 18 F-choline) dem-onstrated its feasibility, but reported its inability to distinguish cancer from BPH [118]. In a small popu-lation of both primary and recurrent disease, dual-phase 18 F-choline showed that areas of malignancy had stably high or increasing uptake while benign areas had decreasing uptake [119]. Thereby, this technique may aid in differentiating malignant from benign prostatic tissue. In a double-tracer study, 11 C-acetate PET was more sensitive than FDG, showing consistently increased uptake in PC lesions [120]. A further advantage was that 11 C-acetate did not

State-of-the-art radiologic imaging in prostate cancer 33
accumulate in the urine. Again, a considerable uptake overlap was described between normal pros-tatic tissue, BPH and PC [121].
ProstaScint ® scanning: No place in regular clinical practice
ProstaScint ® (Cytogen, Princeton, NJ) is an Indium-111 labeled monoclonal mouse antibody specifi c for prostate-specifi c membrane antigen. A signifi cant association between the PSA level and detection of ProstaScint ® activity in the prostate was reported [122]. A recent study revealed sensi-tivities between 37 – 87% and specifi cities between 0 – 50%, concluding that the scan could not be used to reliably localize prostate cancer foci within the prostate [123]. In a single study of only seven patients in which the results of ProstaScint ® fusion with CT scanning were correlated with systematic biopsy a sensitivity and specifi city of 79% and 80%, respectively, were found [124]. In 47 of 51 (92%) preoperative patients at high risk of meta-static disease an increased ProstaScint ® activity in the prostate was observed [125].
A drawback is that the antibodies clear slowly from the vasculature and muscle. Blood and bone
marrow activity may cause false-positive fi ndings. In regular clinical practice, this modality has no place in primary prostate cancer detection and localization.
Conclusions and discussion
In summary, new developments in ultrasound imag-ing (Doppler imaging and particularly the applica-tion of contrast agents) have proved capable of increasing the PC detection rate with fewer biopsy cores necessary as well as detecting relatively more aggressive cancer foci. This is a substantial improve-ment for the patient, who will have to undergo fewer biopsies. In addition, treatment guidance is improved since more representative areas are discovered at biopsy and thus the subsequent diagnostic process can be more accurately performed. TRUS remains the primary imaging tool because of its ease-of-use and its role in guiding prostate biopsy. However, TRUS accuracies varied widely among studies, in part due to the inherent high inter-observer varia-tion, particularly in Doppler imaging.
MRI achieves high accuracy rates, particularly when functional information from dynamic
Figure 5. Proposed scheme for optimal use of imaging in patients at risk of prostate having prostate cancer. Abbreviations: PSA, prostate-specifi c antigen; DRE, digital rectal examination; TRUS, transrectal ultrasound; PPA, pelvic phased-array coil.

34 S. W. T. P. J. Heijmink et al.
contrast-enhanced MR and MRSI are added. A multiparametric approach was shown to optimize the diagnostic accuracy. This compensates for the longer examination time and the discomfort of the use of an ERC. Nevertheless, on a cost-effectiveness basis, MRI cannot be performed in all patients at risk of PC [126]. In patients with one or more prior negative TRUS-guided biopsy sessions and con-tinuing suspicion of PC, MRI can provide valuable additional information for PC detection and local-ization and thereby reduce the future number of biopsies the patient must undergo. Direct MRI guided biopsy is a novel method of performing tar-geted prostate biopsy.
CT scanning does not play a role in PC detec-tion or localization because of its low soft-tissue resolution and radiation burden. This also applies to PET scanning due to its high costs and invasive nature, as well as the availability of alternative imaging modalities. PET scanning may possibly be used in instances in which TRUS-guided biopsies are negative and absence of evidence of PC on MRI. In addition, combining or fusing PET scanning with, for instance, MRI may be of additional value.
Proposals for optimal usage of imaging
Scheme
Based on the abovementioned, the authors propose the following scheme for patient care in patients at risk for prostate cancer (Figure 5).
Comparison with AUA and EAU guidelines [127 – 129]
Both associations recognize that the stage migration during the PSA era necessitates more accurate tech-niques in detecting and localizing prostate cancer. Use of TRUS to guide biopsy is regarded the stan-dard of reference. The EAU ’ s guideline, however, does not mention any contrast-enhanced Doppler imaging based biopsy strategies. This is in contrast to the data presented in our review. Neither CT scanning nor MRI is recommended or mentioned in relation to prostate cancer diagnosis. The latter is in contrast with our proposal in which MRI is used in patients in whom no cancer was found on fi rst TRUS biopsy but with persistently high or rising PSA levels.
Acknowledgements
The authors would like to thank Yvonne L. Hoogeveen for her assistance in preparing the
manuscript and Pieter H. M. de Mulder for his guiding recommendations while editing the manuscript.
Declaration of interest: The authors report no confl icts of interest. The authors alone are responsible for the content and writing of the paper.
References
Jemal AJ, Siegel R, Xu J, Ward E. CA Cancer J Clin 2010;[1] 60(5):277 – 300. Carter HB, Piantadosi S, Isaacs JT. Clinical evidence for [2] and implications of the multistep development of prostate cancer. J Urol 1990;143:742 – 6. Parkin DM, Bray FI, Devesa SS. Cancer burden in the year [3] 2000. The global picture. Eur J Cancer 2001;37(Suppl 8): S4 – 66. Konety BR, Bird VY, Deorah S, Dahmoush L. Comparison [4] of the incidence of latent prostate cancer detected at autopsy before and after the prostate specifi c antigen era. J Urol 2005;174:1785 – 8. Crawford ED. Epidemiology of prostate cancer. Urology [5] 2003;62(Suppl 1):3 – 12. Stewart SL, King JB, Thompson TD, Friedman C, Wingo PA, [6] et al. Cancer mortality surveillance – United States, 1990 – 2000. MMWR Surveill Summ 2004;53:1 – 108. Catalona WJ, Loeb S, Han M. Viewpoint: Expanding pros-[7] tate cancer screening. Ann Intern Med 2006;144:441 – 3. Hoffman RM. Viewpoint: Limiting prostate cancer screen-[8] ing. Ann Intern Med 2006;144:438 – 40. Graif T, Loeb S, Roehl KA, Gashti SN, Griffi n C, Yu X, [9] et al. Under diagnosis and over diagnosis of prostate cancer. J Urol 2007;178(1):88 – 92. Max W, Rice DP, Sung H, Michel M, Breuer W, Zhang X, [10] et al. The economic burden of prostate cancer, California, 1998. Cancer 2002;94:2906 – 13. Mangar SA, Huddart RA, Parker CC, Dearnaley DP, Khoo [11] VS, Horwich A. Technological advances in radiotherapy for the treatment of localised prostate cancer. Eur J Cancer 2005;41:908 – 21. Meraney AM, Haese A, Palisaar J, Graefen M, Steuber T, [12] Huland H, et al. Surgical management of prostate cancer: Advances based on a rational approach to the data. Eur J Cancer 2005;41:888 – 907. Bucci MK, Bevan A, Roach M, III. Advances in radiation [13] therapy: Conventional to 3D, to IMRT, to 4D, and beyond. CA Cancer J Clin 2005;55:117 – 34. Hricak H, Wang L, Wei DC, Coakley FV, Akin O, Reuter [14] VE, et al. The role of preoperative endorectal magnetic resonance imaging in the decision regarding whether to preserve or resect neurovascular bundles during radical ret-ropubic prostatectomy. Cancer 2004;100:2655 – 63. Cellini N, Morganti AG, Mattiucci GC, Valentini V, Leone [15] M, Luzi S, et al. Analysis of intraprostatic failures in patients treated with hormonal therapy and radiotherapy: Implica-tions for conformal therapy planning. Int J Radiat Oncol Biol Phys 2002;53:595 – 9. Roach M, III. Reducing the toxicity associated with the use [16] of radiotherapy in men with localized prostate cancer. Urol Clin North Am 2004;31:353 – 66. Baxter NN, Tepper JE, Durham SB, Rothenberger DA, [17] Virnig BA. Increased risk of rectal cancer after prostate radiation: A population-based study. Gastroenterology 2005;128: 819 – 24.

State-of-the-art radiologic imaging in prostate cancer 35
McNeal JE. Normal anatomy of the prostate and changes [18] in benign prostatic hypertrophy and carcinoma. Semin Ultrasound CT MR 1988;9:329 – 34. Coakley FV, Hricak H. Radiologic anatomy of the prostate [19] gland: A clinical approach. Radiol Clin North Am 2000;38: 15 – 30. Chen ME, Johnston DA, Tang K, Babaian RJ, Troncoso P. [20] Detailed mapping of prostate carcinoma foci: Biopsy strat-egy implications. Cancer 2000;89:1800 – 9. McNeal JE, Redwine EA, Freiha FS, Stamey TA. Zonal [21] distribution of prostatic adenocarcinoma. Correlation with histologic pattern and direction of spread. Am J Surg Pathol 1988;12:897 – 906. Miller GJ, Cygan JM. Morphology of prostate cancer: The [22] effects of multifocality on histological grade, tumor volume and capsule penetration. J Urol 1994;152(Pt 2):1709 – 13. Brossner C, Winterholer A, Roehlich M, Diouhy-Schutz E, [23] Serra V, Sonnleithner M, et al. Distribution of prostate car-cinoma foci within the peripheral zone: Analysis of 8,062 prostate biopsy cores. World J Urol 2003;21:163 – 6. Horninger W, Reissigl A, Rogatsch H, Volgger H, Studen M, [24] Klocker H, et al. Prostate cancer screening in the Tyrol, Austria: Experience and results. Eur J Cancer 2000;36:1322 – 35. Ohori M, Kattan M, Scardino PT, Wheeler TM. Radical [25] prostatectomy for carcinoma of the prostate. Mod Pathol 2004;17:349 – 59. Gleason DF. Histologic grade, clinical stage, and patient [26] age in prostate cancer. NCI Monogr 1988;7:15 – 8. Gleason DF. Histologic grading of prostate cancer: A per-[27] spective. Hum Pathol 1992;23:273 – 9. Egevad L, Granfors T, Karlberg L, Bergh A, Stattin P. Prog-[28] nostic value of the Gleason score in prostate cancer. BJU Int 2002;89:538 – 42. Rasiah KK, Stricker PD, Haynes AM, Delprado W, Turner [29] JJ, Golovsky D, et al. Prognostic signifi cance of Gleason pattern in patients with Gleason score 7 prostate carci-noma. Cancer 2003;98:2560 – 5. Lee F, Gray JM, McLeary RD, McHugh TA, Solomon MH, [30] Kumasaka GH, et al. Prostatic evaluation by transrectal sonography: Criteria for diagnosis of early carcinoma. Radi-ology 1986;158:91 – 5. Meirelles LR, Billis A, Cotta AC, Nakamura RT, Caserta [31] NMG, Prando A. Prostatic atrophy: Evidence for a possible role of local ischemia in its pathogenesis. Int Urol Nephrol 2002;34:345 – 50. Ellis WJ, Brawer MK. The signifi cance of isoechoic prostatic [32] carcinoma. J Urol 1994;152(Pt 2):2304 – 7. Frauscher F, Klauser A, Berger AP, Halpern EJ, Feuchtner [33] G, Koppelstaetter F, et al. The value of ultrasound (US) in the diagnosis of prostate cancer. Radiologie. 2003;43:455 – 63. Hodge KK, McNeal JE, Terris MK, Stamey TA. Random [34] systematic versus directed ultrasound guided transrectal core biopsies of the prostate. J Urol 1989;142:71 – 4. Presti JC, Jr. Prostate biopsy: How many cores are enough? [35] Urol Oncol 2003;21:135 – 40. Eskicorapci SY, Baydar DE, Akbal C, Sofi kerim M, Gunay [36] M, Ekici S, et al. An extended 10-core transrectal ultra-sonography guided prostate biopsy protocol improves the detection of prostate cancer. Eur Urol 2004;45:444 – 8. Pelzer AE, Bektic J, Berger AP, Halpern EJ, Koppelst ä tter [37] F, Klauser A, et al. Are transition zone biopsies still neces-sary to improve prostate cancer detection? Results from the Tyrol screening project. Eur Urol 2005;48(6):916 – 21. King CR, McNeal JE, Gill H, Presti JC, Jr. Extended pros-[38] tate biopsy scheme improves reliability of Gleason grading:
Implications for radiotherapy patients. Int J Radiat Oncol Biol Phys 2004;59:386 – 91. Ching CB, Moussa AS, Li J, Lane BR, Zippe C, Jones JS, [39] et al. Does transrectal ultrasound probe confi guration really matter? End fi re versus side fi re probe prostate cancer detection rates. J Urol 2009;181:2077 – 82. Gore JL, Shariat SF, Miles BJ, Kadmon D, Jiang N, Wheeler [40] TM, et al. Optimal combinations of systematic sextant and laterally directed biopsies for the detection of prostate can-cer. J Urol 2001;165:1554 – 9. Scherr DS, Eastham J, Ohori M, Scardino PT. Prostate [41] biopsy techniques and indications: When, where, and how? Semin Urol Oncol 2002;20:18 – 31. Djavan B, Remzi M, Schulman CC, Marberger M, Zlotta [42] AR, et al. Repeat prostate biopsy: Who, how and when? A review. Eur Urol 2002;42:93 – 103. Wilson NM, Masoud AM, Barsoum HB, Refaat MM, [43] Moustafa MI, Kamal TA, et al. Correlation of power Dop-pler with microvessel density in assessing prostate needle biopsy. Clin Radiol 2004;59:946 – 50. Okihara K, Kojima M, Nakanouchi T, Okada K, Miki T. [44] Transrectal power Doppler imaging in the detection of prostate cancer. BJU Int 2000;85:1053 – 7. Eisenberg ML, Cowan JE, Carroll PR, Shinohara K. The [45] adjunctive use of power Doppler imaging in the preoperative assessment of prostate cancer. BJU Int 2010;105(9):1237 – 41. Rifkin MD, Sudakoff GS, Alexander AA. Prostate: Tech-[46] niques, results, and potential applications of color Doppler US scanning. Radiology 1993;186:509 – 13. Lavoipierre AM, Snow RM, Frydenberg M, Gunter D, [47] Reisner G, Royce PL, et al. Prostatic cancer: Role of color Doppler imaging in transrectal sonography. AJR Am J Roentgenol 1998;171:205 – 10. Jakobsen JA, Correas JM. Ultrasound contrast agents and [48] their use in urogenital radiology: Status and prospects. Eur Radiol 2001;11:2082 – 91. Frauscher F, Klauser A, Volgger H, Halpern EJ, Pallwein L, [49] Steiner H, et al. Comparison of contrast enhanced color Doppler targeted biopsy with conventional systematic biopsy: Impact on prostate cancer detection. J Urol 2002;167:1648 – 52. Pelzer A, Bektic J, Berger AP, Pallwein L, Halpern EJ, [50] Horninger W, et al. Prostate cancer detection in men with prostate specifi c antigen 4 to 10 ng/ml using a combined approach of contrast enhanced color Doppler targeted and systematic biopsy. J Urol 2005;173:1926 – 9. Mitterberger M, Pinggera GM, Horninger W, Bartsch G, [51] Strasser H, Sch ä fer G, et al. Comparison of contrast enhanced color doppler targeted biopsy to conventional systematic biopsy: Impact on Gleason score. J Urol 2007;178(2):464 – 8. Mitterberger M, Horninger W, Pelzer A, Strasser H, Bartsch [52] G, Moser P, et al. A prospective randomized trial comparing contrast-enhanced targeted versus systematic ultrasound guided biopsies: Impact on prostate cancer detection. Pros-tate 2007;67:1537 – 42. Jakobsen JA, Oyen R, Thomsen HS, Morcos SK. Safety of [53] ultrasound contrast agents. Eur Radiol 2005;15:941 – 5. Mitterberger M, Pinggera G, Horninger W, Hannes [54] Strasser, Halpern E, Pallwein L, et al. Dutasteride prior to contrast-enhanced colour Doppler ultrasound prostate biopsy increases prostate cancer detection. Eur Urol 2008;53: 112 – 7. Konig K, Scheipers U, Pesavento A, Lorentz A, Ermert H, [55] Senge T. Initial experiences with real-time elastography guided biopsies of the prostate. J Urol 2005;174:115 – 7.

36 S. W. T. P. J. Heijmink et al.
Pallwein L, Mitterberger M, Struve P, Horninger W, Aigner [56] F, Bartsch B, et al. Comparison of sonoelastography guided biopsy with systematic biopsy: Impact on prostate cancer detection. Eur Radiol 2007;17(9):2278 – 85. Frauscher F, Klauser A, Koppelstaetter F, Berger AP, [57] Horninger W, Bartsch G, et al. Real-time elastography for prostate cancer detection: Preliminary experience. Eur Radiol 2004; 14(S2):150. Nelson ED, Slotoroff CB, Gomella LG, Halpern EJ. Tar-[58] geted biopsy of the prostate: The impact of color Doppler imaging and elastography on prostate cancer detection and Gleason score. Urology 2007;70:1136 – 40. Prando A, Wallace S. Helical CT of prostate cancer: Early [59] clinical experience. AJR Am J Roentgenol 2000;175:343 – 6. Tempany CM, Zhou X, Zerhouni EA, Rifkin MD, Quint [60] LE, Piccoli CW, et al. Staging of prostate cancer: Results of Radiology Diagnostic Oncology Group project compar-ison of three MR imaging techniques. Radiology 1994;192:47 – 54. Cruz M, Tsuda K, Narumi Y, Kuroiwa Y, Nose T, Kojima [61] Y, et al. Characterization of low-intensity lesions in the peripheral zone of prostate on pre-biopsy endorectal coil MR imaging. Eur Radiol 2002;12:357 – 65. Claus FG, Hricak H, Hattery RR. Pretreatment evaluation [62] of prostate cancer: Role of MR imaging and 1H MR spec-troscopy. Radiographics 2004;24(Suppl 1):S167 – 80. Wang L, Mazaheri Y, Zhang J, Ishill NM, Kuroiwa K, [63] Hricak H. Assessment of biologic aggressiveness of prostate cancer: Correlation of MR signal intensity with Gleason grade after radical prostatectomy. Radiology 2008;246:168 – 76. Akin O, Sala E, Moskowitz CS, Kuroiwa K, Ishill NM, [64] Pucar D, et al. Transition zone prostate cancers: Features, detection, localization, and staging at endorectal MR imag-ing. Radiology 2006;239:784 – 92. Rosenkrantz AB, Neil J, Kong X, Melamed J, Babb JS, Taneja [65] SS, et al. Prostate cancer: Comparison of 3D T2-weighted with conventional 2D T2-weighted imaging for image quality and tumor detection. AJR Am J Roentgenol 2010;194:446 – 52. Mullerad M, Hricak H, Kuroiwa K, Pucar D, Chen H, [66] Kattan MW, et al. Comparison of endorectal magnetic resonance imaging, guided prostate biopsy and digital rec-tal examination in the preoperative anatomical localization of prostate cancer. J Urol 2005;174:2158 – 63. Beyersdorff D, Taupitz M, Winkelmann B, Fischer T, Lenk [67] S, Loening SA, et al. Patients with a history of elevated prostate-specifi c antigen levels and negative transrectal US-guided quadrant or sextant biopsy results: Value of MR imaging. Radiology 2002;224:701 – 6. Tamada T, Sone T, Jo Y, Yamamoto A, Yamashita T, Egashira [68] N, et al. Prostate cancer: Relationships between postbiopsy hemorrhage and tumor detectability at MR diagnosis. Radi-ology 2008;248:531 – 9. Kurhanewicz J, Vigneron DB, Hricak H, Narayan P, Carroll [69] P, Nelson SJ. Three-dimensional H-1 MR spectroscopic imaging of the in situ human prostate with high [0.24-0.7 cm 3 ] spatial resolution. Radiology 1996;198:795 – 805. Coakley FV, Qayyum A, Kurhanewicz J. Magnetic reso-[70] nance imaging and spectroscopic imaging of prostate can-cer. J Urol 2003;170(Pt 2):S69 – 75. Zakian KL, Sircar K, Hricak H, Chen H, Shukla-Dave A, [71] Eberhardt S, et al. Correlation of proton MR spectroscopic imaging with Gleason score based on step-section pathologic analysis after radical prostatectomy. Radiology 2005;234:804 – 14.
Scheenen TW, Klomp DW, Roll SA, F ü tterer JJ, Barentsz [72] JO, Heerschap A. Fast acquisition-weighted three-dimen-sional proton MR spectroscopic imaging of the human prostate. Magn Reson Med 2004;52:80 – 8. Scheidler J, Hricak H, Vigneron DB, Kyle K, Yu KK , Sokolov [73] DL, et al. Prostate cancer: Localization with three-dimen-sional proton MR spectroscopic imaging – clinicopathologic study. Radiology 1999;213:473 – 80. Wefer AE, Hricak H, Vigneron DB, Coakley FV, Lu Y, Wefer [74] J, et al. Sextant localization of prostate cancer: Comparison of sextant biopsy, magnetic resonance imaging and mag-netic resonance spectroscopic imaging with step section histology. J Urol 2000;164:400 – 4. F ü tterer JJ, Heijmink SWTPJ, Scheenen TWJ, Veltman J, [75] Huisman HJ, Vos P, et al. Prostate cancer localization with dynamic contrast-enhanced MR imaging and proton MR spectroscopic imaging. Radiology 2006;241:449 – 58. Shukla-Dave A, Hricak H, Kattan MW, Pucar D, Kuroiwa [76] K, Chen H, et al. The utility of magnetic resonance imaging and spectroscopy for predicting insignifi cant prostate can-cer: An initial analysis. BJU Int 2007;99:786 – 93. Weinreb JC, Blume JD, Coakley FV, Wheeler T, Cormack [77] JB, Sotto CK, et al. Prostate cancer: Sextant localization at MR imaging and MR spectroscopic imaging before prosta-tectomy – results of ACRIN prospective multi-institutional clinicopathologic study. Radiology 2009;251:122 – 33. Sato C, Naganawa S, Nakamura T, Kumada H, Miura S, [78] Takizawa O, et al. Differentiation of noncancerous tissue and cancer lesions by apparent diffusion coeffi cient values in transition and peripheral zones of the prostate. J Magn Reson Imaging 2005;21:258 – 62. Zelhof B, Pickles M, Liney G, Peter Gibbs P, Rodrigues G, [79] Kraus S, et al. Correlation of diffusion-weighted magnetic resonance data with cellularity in prostate cancer. BJU Int 2009;103:883 – 8. Mazaheri Y, Shukla-Dave A, Hricak H, Fine SW, Zhang, J, [80] Inurrigarro G, et al. Prostate cancer: Identifi cation with combined diffusion-weighted MR imaging and 3D 1H MR spectroscopic imaging – correlation with pathologic fi nd-ings. Radiology 2008;246:480 – 8. Haider MA, Van Der Kwast TH, Tanguay J, Evans AJ, [81] Hashmi A, Lockwood G, et al. Combined T2-weighted and diffusion-weighted MRI for localization of prostate cancer. AJR Am J Roentgenol 2007;189:323 – 8. Yoshimitsu K, Kiyoshima K, Irie H, Tajima T, Asayama Y, [82] Hirakawa M, et al. Usefulness of apparent diffusion coeffi -cient map in diagnosing prostate carcinoma: Correlation with stepwise histopathology. J Magn Reson Imaging 2008; 27:132 – 9. Langer DL, Van Der Kwast TH, Evans AJ, Trachtenberg J, [83] Wilson BC, Haider A. Prostate cancer detection with mul-ti-parametric MRI: Logistic regression analysis of quantita-tive T2, diffusion-weighted imaging, and dynamic contrast-enhanced MRI. J Magn Reson Imaging 2009;30:327 – 34. Gibbs P, Pickles MD, Turnbull LW. Diffusion imaging of [84] the prostate at 3.0 tesla. Invest Radiol 2006;41:185 – 8. Pickles MD, Gibbs P, Sreenivas M, Turnbull LW. Diffusion-[85] weighted imaging of normal and malignant prostate tissue at 3.0T. J Magn Reson Imaging 2006;23:130 – 4. Miao H, Fukatsu H, Ishigaki T. Prostate cancer detection [86] with 3-T MRI: Comparison of diffusion-weighted and T2-weighted imaging. Eur J Radiol 2007;61:297 – 302. Kim CK, Park BK, Kim B. High-b-Value diffusion-weighted [87] imaging at 3 T to detect prostate cancer: Comparisons between b values of 1,000 and 2,000 s/mm 2 . AJR Am J Roentgenol 2010;194:W33 – 7.

State-of-the-art radiologic imaging in prostate cancer 37
Woodfi eld CA, Tung GA, Grand DJ, Pezzullo JA, Machan [88] JT, Renzulli JF, et al. Diffusion-weighted MRI of peripheral zone prostate cancer: Comparison of tumor apparent dif-fusion coeffi cient with Gleason score and percentage of tumor on core biopsy. AJR Am J Roentgenol 2010;194:W316 – 22. Langer DL, Van Der Kwast TH, Evans AJ, Plotkin A, [89] Trachtenberg J, Wilson BC, et al. Prostate tissue composi-tion and MR measurements: Investigating the relationships between ADC, T2, K[trans], v[e], and corresponding histologic features. Radiology 2010;255:485 – 94. Barentsz JO, Engelbrecht M, Jager GJ, de LaRosette J, van [90] Der Sanden BP, Huisman HJ, et al. Fast dynamic gadolin-ium-enhanced MR imaging of urinary bladder and prostate cancer. J Magn Reson Imaging 1999;10:295 – 304. Padhani AR, Gapinski CJ, Macvicar DA, Parker GJ, [91] Suckling J, Revell PB, et al. Dynamic contrast enhanced MRI of prostate cancer: Correlation with morphology and tumour stage, histological grade and PSA. Clin Radiol 2000;55:99 – 109. Engelbrecht MR, Huisman HJ, Laheij RJ, Jager GJ, van [92] Leenders GJLH, Hulsbergen-Van De Kaa CA, et al. Dis-crimination of prostate cancer from normal peripheral zone and central gland tissue by using dynamic contrast-en-hanced MR imaging. Radiology 2003;229:248 – 54. Van Dorsten FA, Van Der Graaf M, Engelbrecht MR, van [93] Leenders GJ, Verhofstad A, Rijpkema M, et al. Combined quantitative dynamic contrast-enhanced MR imaging and [1]H MR spectroscopic imaging of human prostate cancer. J Magn Reson Imaging 2004;20:279 – 87. Runge VM. Safety of approved MR contrast media [94] for intravenous injection. J Magn Reson Imaging 2000;12: 205 – 13. Lin SP, Brown JJ. MR contrast agents: Physical and phar-[95] macologic basics. J Magn Reson Imaging 2007;25:884 – 99. Kitajima K, Kaji Y, Fukabori Y, Yoshida K, Suganuma N, [96] Sugimura K. Prostate cancer detection with 3 T MRI: Comparison of diffusion-weighted imaging and dynamic contrast-enhanced MRI in combination with T2-weighted imaging. J Magn Reson Imaging 2010;31:625 – 31. Sciarra A, Panebianco V, Ciccariello M, Salciccia S, [97] Cattarino S, Lisi D, et al. Value of magnetic resonance spec-troscopy imaging and dynamic contrast-enhanced imaging for detecting prostate cancer foci in men with prior negative biopsy. Clin Cancer Res 2010;16:1875 – 83. Turkbey B, Pinto PA, Mani H, Bernardo M, Pang Y, [98] McKinney YL, et al. Prostate cancer: Value of multipara-metric MR imaging at 3 T for detection- – histopathologic correlation. Radiology 2010;255:89 – 99. Futterer JJ, Scheenen TW, Huisman HJ, Klomp DW J, van [99] Dorsten, FA, Hulsbergen-van de Kaa, CA, et al. Initial experience of 3 Tesla endorectal coil magnetic resonance imaging and 1H-spectroscopic imaging of the prostate. Invest Radiol 2004;39:671 – 80. Beyersdorff D, Taymoorian K, Knosel T, Schnorr D, Felix [100] R, Hamm B, et al. MRI of prostate cancer at 1.5 and 3.0 T: Comparison of image quality in tumor detection and staging. AJR Am J Roentgenol 2005;185:1214 – 20. Scheenen TW, Heijmink SW, Roell SA, Hulsbergen-Van de [101] Kaa CA, Knipscheer BC, Alfred Witjes JA, et al. Three-dimensional proton MR spectroscopy of human prostate at 3 T without endorectal coil: Feasibility. Radiology 2007;245: 507 – 16. Heijmink SWTPJ, Futterer JJ, Hambrock T, Takahashi S, [102] Scheenen TWJ, Huisman HJ, et al. Prostate cancer: Body-array versus endorectal coil MR imaging at 3 T –
Comparison of image quality, localization, and staging performance. Radiology 2007;244:184 – 95. Yakar D, Hambrock T, Hoeks C, Barentsz JO, F ü tterer, JJ. [103] Magnetic resonance-guided biopsy of the prostate: Feasibility, technique, and clinical applications. Top Magn Reson Imaging 2008;19:291 – 5. Beyersdorff D, Winkel A, Hamm B, Severin Lenk S, [104] Loening SA, Taupitz M. MR imaging-guided prostate biopsy with a closed MR unit at 1.5 T: Initial results. Radi-ology 2005;234:576 – 81. Hambrock T, Somford DM, Hoeks C, Bouwense SA, [105] Huisman H, Yakar D, et al. Magnetic resonance imaging guided prostate biopsy in men with repeat negative biopsies and increased prostate specifi c antigen. J Urol 2010;183: 520 – 7. Shreve PD, Grossman HB, Gross MD, Wahl RL. Metastatic [106] prostate cancer: Initial fi ndings of PET with 2-deoxy-2-[F-18]fl uoro-D-glucose. Radiology 1996;199:751 – 6. Oyama N, Akino H, Suzuki Y, Kanamaru H, Sadato N, [107] Yonekura Y, et al. The increased accumulation of [18F]fl uorodeoxyglucose in untreated prostate cancer. Jpn J Clin Oncol 1999;29:623 – 9. Effert PJ, Bares R, Handt S, Wolff JM, Bull U, Jakse G. [108] Metabolic imaging of untreated prostate cancer by positron emission tomography with 18fl uorine-labeled deoxyglu-cose. J Urol 1996;155:994 – 8. Haseman MK, Reed NL, Rosenthal SA. Monoclonal anti-[109] body imaging of occult prostate cancer in patients with elevated prostate-specifi c antigen. Positron emission tom-ography and biopsy correlation. Clin Nucl Med 1996;21:704 – 13. Liu IJ, Zafar MB, Lai YH, Segall GM, Terris MK. Fluoro-[110] deoxyglucose positron emission tomography studies in diagnosis and staging of clinically organ-confi ned prostate cancer. Urology 2001;57:108 – 11. Hara T, Kosaka N, Kishi H. PET imaging of prostate can-[111] cer using carbon-11-choline. J Nucl Med 1998;39:990 – 5. Sutinen E, Nurmi M, Roivainen A, Varpula M, Tolvanen T, [112] Lehikoinen P, et al. Kinetics of [[11]C]choline uptake in prostate cancer: A PET study [correction for stydy]. Eur J Nucl Med Mol Imaging 2004;31:317 – 24. Yamaguchi T, Lee J, Uemura H, Sasaki T, Takahashi N, [113] Oka T, et al. Prostate cancer: A comparative study of [11]C-choline PET and MR imaging combined with proton MR spectroscopy. Eur J Nucl Med Mol Imaging 2005; 32:742 – 8. Piert M, Park H, Khan A, Siddiqui J, Hussain H, [114] Chenevert T, et al. Detection of aggressive primary prostate cancer with 11C-choline PET/CT using multimodality fusion techniques. J Nucl Med 2009;50:1585 – 93. Farsad M, Schiavina R, Castellucci P, Nanni C, Barbara [115] Corti B, Martorana G, et al. Detection and localization of prostate cancer: Correlation of [11]C-choline PET/CT with histopathologic step-section analysis. J Nucl Med 2005;46:1642 – 9. Martorana G, Schiavina R, Corti B, Farsad M, Salizzoni E, [116] Brunocilla E, et al. 11C-Choline positron emission tomog-raphy/computerized tomography for tumor localization of primary prostate cancer in comparison with 12-core biopsy. J Urol 2006;176:954 – 60. Watanabe H, Kanematsu M, Kondo H, Kako N, Yamamoto [117] N, Yamada T, et al. Preoperative detection of prostate can-cer: A comparison with 11C-choline PET, 18F-fl uorodeox-yglucose PET and MR imaging. J Magn Reson Imaging 2010;31:1151 – 6. Schmid DT, John H, Zweifel R, Cservenyak T, Westera G, [118] Goerres GW, et al. Fluorocholine PET/CT in patients with

38 S. W. T. P. J. Heijmink et al.
prostate cancer: Initial experience. Radiology 2005;235:623 – 8. Sandblom G, S ö rensen J, Lundin N, H ä ggman M, Malm-[119] str ö m PU. Positron emission tomography with c11-acetate for tumor detection and localization in patients with pros-tate-specifi c antigen relapse after radical prostatectomy. Urology 2006;67:996 – 1000. Oyama N, Akino H, Kanamaru H, Suzuki Y, Muramoto S, [120] Yonekura Y, et al. 11C-acetate PET imaging of prostate cancer. J Nucl Med 2002;43:181 – 6. Kato T, Tsukamoto E, Kuge Y, Takei T, Shiga T, Shinohara [121] N, et al. Accumulation of [11C]acetate in normal prostate and benign prostatic hyperplasia: Comparison with prostate cancer. Eur J Nucl Med Mol Imaging 2002;29:1492 – 5. Sodee DB, Malguria N, Faulhaber P, Resnick MI , Albert [122] J, Bakale G, et al. Multicenter ProstaScint imaging fi ndings in 2154 patients with prostate cancer. The ProstaScint Imaging Centers. Urology 2000;56:988 – 93. Mouraviev V, Madden JF, Broadwater G, Mayes JM, [123] Burchette JL Schneider F, et al. Use of 111in-capromab pendetide immunoscintigraphy to image localized prostate cancer foci within the prostate gland. J Urol 2009;182:938 – 47. Ellis RJ, Kim EY, Conant R, Sodee DB, Spirnak JP, Dinch-[124] man KH, et al. Radioimmunoguided imaging of prostate cancer foci with histopathological correlation. Int J Radiat Oncol Biol Phys 2001;49:1281 – 6. Burgers JK, Hinkle GH, Haseman MK. Monoclonal anti-[125] body imaging of recurrent and metastatic prostate cancer. Semin Urol 1995;13:103 – 12. Jager GJ, Severens JL, Thornbury JR, de la Rosette JJMCH, [126] Ruijs SHJ, Barentsz JO. Prostate cancer staging: Should MR imaging be used? – A decision analytic approach. Radi-ology 2000;215:445 – 51. Report on the management of clinically localized prostate [127] cancer. Available from: http://www.auanet.org/guidelines/proscan07.cfm. 2007. Heidenreich A, Aus G, Abbou CC, Bolla M, Joniau S, [128] Matveev V, Schmid HP, et al. Guidelines on prostate cancer 2007. Available from: http://www.uroweb.org/fi leadmin/user_upload/Guidelines/Prostate%20Cancer.pdf. 2007. Thompson I, Thrasher JB, Aus G, Burnett AL, Canby-[129] Hagino ED, Cookson MS, et al. Guideline for the manage-ment of clinically localized prostate cancer: 2007 update. J Urol 2007;177:2106 – 31. Salomon L, Colombel M, Patard JJ, Lefr è re-Belda MA, [130] Bellot J, Chopin D, et al. Value of ultrasound-guided sys-tematic sextant biopsies in prostate tumor mapping. Eur Urol 1999;35:289 – 93.
Cornud F, Belin X, Piron D, Chr é tien Y, Flam T, Casanova [131] JM, et al. Color Doppler-guided prostate biopsies in 591 patients with an elevated serum PSA level: Impact on Gleason score for nonpalpable lesions. Urology 1997;49:709 – 15. Halpern EJ, Strup SE. Using gray-scale and color and [132] power Doppler sonography to detect prostatic cancer. AJR Am J Roentgenol 2000;174:623 – 7. Kuligowska E, Barish MA, Fenlon HM, Blake M. [133] Predictors of prostate carcinoma: Accuracy of gray-scale and color Doppler US and serum markers. Radiology 2001; 220:757 – 64. Sauvain JL, Palascak P, Bourscheid D, Chabi C, Atassi A, [134] Bremon JM, et al. Value of power doppler and 3D vascular sonography as a method for diagnosis and staging of pros-tate cancer. Eur Urol 2003;44:21 – 30. Remzi M, Dobrovits M, Reissigl A, Ravery V, Waldert M, [135] Wiunig C, et al. Can Power Doppler enhanced transrectal ultrasound guided biopsy improve prostate cancer detec-tion on fi rst and repeat prostate biopsy? Eur Urol 2004; 46:451 – 6. Unal D, Sedelaar JP, Aarnink RG, van Leenders GJ, Wijk-[136] stra H, Debruyne FM, et al. Three-dimensional contrast-enhanced power Doppler ultrasonography and conventional examination methods: The value of diagnostic predictors of prostate cancer. BJU Int 2000;86:58 – 64. Halpern EJ, Rosenberg M, Gomella LG. Prostate cancer: [137] Contrast-enhanced us for detection. Radiology 2001; 219:219 – 25. Yuen JS, Thng CH, Tan PH, Khin LW, Phee SJ, Xiao D, [138] et al. Endorectal magnetic resonance imaging and spectros-copy for the detection of tumor foci in men with prior negative transrectal ultrasound prostate biopsy. J Urol 2004;171:1482 – 6. Graser A, Heuck A, Sommer B, Massmann J , Scheidler J, [139] Maximillian Reiser M, et al. Per-sextant localization and staging of prostate cancer: Correlation of imaging fi ndings with whole-mount step section histopathology. AJR Am J Roentgenol 2007;188:84 – 90. Zakian KL, Sircar K, Hricak H, Chen H, Shukla-Dave A, [140] Eberhardt S, et al. Correlation of proton MR spectroscopic imaging with gleason score based on step-section patho-logic analysis after radical prostatectomy. Radiology 2005;234:804 – 14. F ü tterer JJ, Scheenen TWJ, Heijmink SWTPJ, Huisman [141] HJ, Hulsbergen-van de Kaa CA, Witjes JA, et al. Standard-ized threshold approach using three-dimensional proton magnetic resonance spectroscopic imaging in prostate can-cer localization of the entire prostate. Invest Radiol 2007; 42:116 – 22.

Correspondence: Josef J. Fox, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York 10065, USA. Tel: � 1 212 6297371. Fax: � 1 212 7173263. E-mail: [email protected]
(Received 14 December 2010 ; accepted 25 February 2011)
ORIGINAL ARTICLE
Developing imaging strategies for castration resistant prostate cancer
JOSEF J. FOX 1 , MICHAEL J. MORRIS 2,3 , STEVEN M. LARSON 1 , HEIKO SCH Ö DER 1 & HOWARD I. SCHER 2,3
1 Nuclear Medicine Service, Department of Radiology, Memorial Sloan-Kettering Cancer Center, New York, New York, USA; 2 Sidney Kimmel Center for Prostate and Urologic Cancer, Genitourinary Oncology Service, Department of Medicine, Memorial Sloan-Kettering Cancer Center, New York, New York, USA; 3 Department of Medicine, Joan and Sanford E. Weill College of Medicine of Cornell University, New York, New York, USA
Abstract Recent advances in the understanding of castrate-resistant prostate cancer (CRPC) have lead to a growing number of experimental therapies, many of which are directed against the androgen-receptor (AR) signaling axis. These advances generate the need for reliable molecular imaging biomarkers to non-invasively determine effi cacy, and to better guide treatment selection of these promising AR-targeted drugs. Methods. We draw on our own experience, supplemented by review of the current literature, to discuss the systematic development of imaging biomarkers for use in the context of CRPC, with a focus on bone scintigraphy, F-18 fl uorodeoxyglucose (FDG)-positron emission tomography (PET) and PET imaging of the AR signaling axis. Results. The roadmap to biomarker development mandates rigorous standardiza-tion and analytic validation of an assay before it can be qualifi ed successfully for use in an appropriate clinical context. The Prostate Cancer Working Group 2 (PCWG2) criteria for “ radiographic ” progression by bone scintigraphy serve as a paradigm of this process. Implemented by the Prostate Cancer Clinical Trials Consortium (PCCTC), these con-sensus criteria may ultimately enable the co-development of more potent and versatile molecular imaging biomarkers. Purported to be superior to single-photon bone scanning, the added value of Na 18 F-PET for imaging of bone metas-tases is still uncertain. FDG-PET already plays an integral role in the management of many diseases, but requires further evaluation before being qualifi ed in the context of CRPC. PET tracers that probe the AR signaling axis, such as 18 F-FDHT and 89 Zr-591, are now under development as pharmacodynamic markers, and as markers of effi cacy, in tandem with FDG-PET. Semi-automated analysis programs for facilitating PET interpretation may serve as a valuable tool to help navigate the biomarker roadmap. Conclusions. Molecular imaging strategies, particularly those that probe the AR signaling axis, have the potential to accelerate drug development in CRPC. The development and use of ana-lytically valid imaging biomarkers will increase the likelihood of clinical qualifi cation, and ultimately lead to improved patient outcomes.
About 32,050 men will die of prostate cancer in the United States in 2010 [1], the majority after the tran-sition to a castration resistant state, the invariably lethal form of the disease [2]. The hallmark of castra-tion resistant prostate cancer (CRPC) is evidence of tumor growth despite castrate levels of serum andro-gens [3]. Several important insights into the molecu-lar pathogenesis of CRPC, including mechanisms of androgen receptor (AR) signaling, have now been elucidated, leading to the development of new and
more potent therapies targeting the AR pathway [4]. These novel agents include inhibitors of CYP17, an enzyme required for androgen synthesis; direct AR-antagonists that prevent nuclear translocation; inhibi-tors of HSP90 which protects AR from degradation; inhibitors of histone deacetylases which is required for optimal AR mediated transcription, and tyrosine kinase inhibitors. A number of these drugs appear to durably repress disease growth, even after numerous prior hormonal treatments and chemotherapy [5–7].
Acta Oncologica, 2011; 50(Suppl 1): 39–48
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2011.572914

40 J. J. Fox et al.
Despite these advances there remains a pressing need for continued drug development in this arena. The diffi culty in introducing new prostate cancer drugs to the market is well documented. Among the challenges confronted is the disease ’ s heterogeneous clinical course, which complicates eligibility and response cri-teria for clinical trials. This problem has been addressed in part through the use of the “ clinical-states ” frame-work for organizing the natural history of the disease [8], but substantial impediments persist. Notably, the chief manifestation of metastatic disease (i.e., bone metastases) is notoriously diffi cult to monitor. Further-more, multiple mechanisms have been implicated in AR signaling reactivation, which in part accounts for the non-uniform response to AR directed therapy [9]. These obstacles highlight the need for biomarker development in CRPC. Molecular imaging with PET has the potential to address this need through its ver-satility, non-invasiveness and quantitative capabilities.
The biomarker roadmap
A biomarker is “ a factor that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention ” [10]. Biomarkers can be sub-classifi ed into various categories, including: (1) predic-tive/risk markers, (2) phar macodynamic markers, and (3) biologic response/progression markers. The road-map for biomarker qualifi cation requires rigorous stan-dards for trial design, data capture, reporting, and analysis [11]. Imaging biomarkers, like any assay, should be measured in an analytic test system with well-estab-lished performance characteristics. The validated imag-ing test must then undergo qualifi cation, which is the evidentiary process of linking a biomarker with a bio-logical process or clinical endpoint, in other words, establishing “ fi tness for purpose ” [12]. A validated imaging test qualifi ed for use in one disease entity, or cancer-type, may not qualify for use in other settings. Similarly, an assay that is validated for predicting risk may not be useful as a marker of progression.
Imaging biomarkers of response/progression are often used as endpoints in oncologic trials. Overall sur-vival (OS) is regarded as the gold standard of clinical endpoints, defi ned as the time from the start of study treatment to the date of death of any cause. Because survival endpoints can take years to reach, regulatory authorities, such as the Federal Drug Administration (FDA) in the United States, allow for accelerated approval in certain life-threatening diseases. Acceler-ated approval is usually based on an endpoint “ reason-ably likely to predict ” clinical bene fi t. It is distinct from a surrogate endpoint which, as defi ned by Prentice, must meet the following criteria: 1) treatment has a statistically signifi cant impact on the true endpoint;
2) treatment has a statistically signifi cant impact on the surrogate endpoint; 3) the surrogate endpoint has a statistically signifi cant impact on the true endpoint; and 4) the full effect of the treatment on the true end-point should be captured by surrogate endpoint [13]. Surrogate endpoints commonly incorporated in onco-logic clinical trials include objective response rate (ORR), time to progression (TTP) and progression free survival (PFS) [14]. ORR is defi ned as the propor-tion of subjects with a predefi ned amount of reduction in tumor burden, often assessed on the basis of radio-logic criteria, as in Response Evaluation Criteria in Solid Tumors (RECIST) [15]. Unfortunately, RECIST has a limited role in CRPC drug development, and is only applicable to the assessment of soft-tissue disease [16]. TTP is often a composite endpoint defi ned as the time from the start of study treatment to the date of the fi rst documentation of objective tumor progres-sion/relapse, initiation of other cancer therapy, or death as a result of the tumor, whichever comes fi rst. PFS is similar to TTP, but also includes death from any cause. These composite endpoints face diffi culties in terms of data collection and analysis, and are susceptible to sub-jective bias. The precise defi nition of TTP and PFS, specifi cally, the measurement of progression or relapse, continues to evolve as new biomarkers for progression are established.
The broader oncology community in the United States is attempting to address the need for qualifi ed biomarkers through collaborative efforts between the FDA, the National Cancer Institute (NCI) and other regulatory bodies, with the formulation of consortia such as the Oncology Biomarkers Qualifi cation Ini-tiative (OBQI) and the AACR-FDA-NCI Cancer Biomarkers Collaborative [17]. Meanwhile, tangible measures to improve patient outcomes have been undertaken by the prostate cancer clinical trials com-munity. Specifi cally, the Prostate Cancer Clinical Tri-als Consortium (PCCTC) was created in 2006, supported by the Prostate Cancer Foundation and the US Department of Defense, for the purpose of carefully designing and conducting phase 1 and 2 multicenter clinical trials. Memorial Sloan-Kettering Cancer Center (MSKCC) is the coordinating center for the consortium, which is currently comprised of 13 prostate cancer research centers.
Morris MJ, Basch EM, Wilding G, Hussain M, Carducci MA, Higano C, et al. Department of Defense prostate cancer clinical trials consortium: a new instrument for prostate cancer clinical research. Clin Genitourin Cancer. 2009 Jan;7(1):51-7.. The PCCTC has already successfully conducted studies controlling for patient population and prior treat-ment, as well as for scanning algorithm, imaging end-points and interpretation. The consortium’s effort to redefi ne endpoints in prostate cancer clinical trials
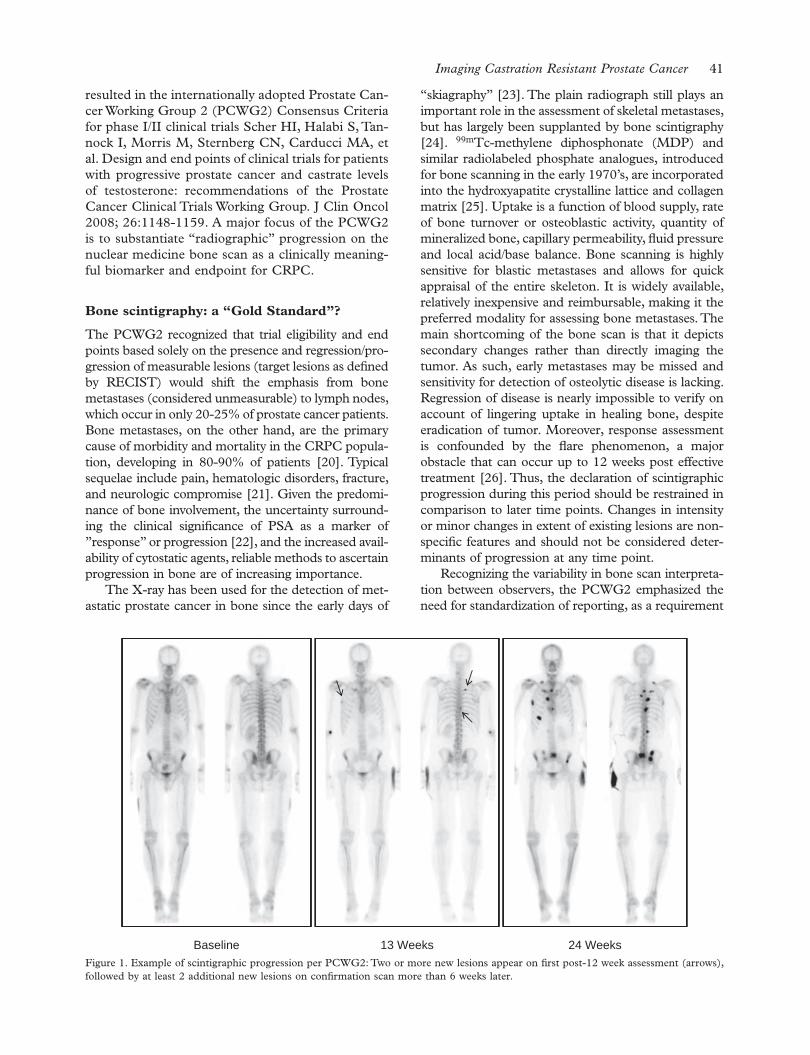
Imaging Castration Resistant Prostate Cancer 41
resulted in the internationally adopted Prostate Can-cer Working Group 2 (PCWG2) Consensus Criteria for phase I/II clinical trials Scher HI, Halabi S, Tan-nock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008; 26:1148-1159. A major focus of the PCWG2 is to substantiate “radiographic” progression on the nuclear medicine bone scan as a clinically meaning-ful biomarker and endpoint for CRPC.
Bone scintigraphy: a “ Gold Standard ” ?
The PCWG2 recognized that trial eligibility and end points based solely on the presence and regression/pro-gression of measurable lesions (target lesions as defi ned by RECIST) would shift the emphasis from bone metastases (considered unmeasurable) to lymph nodes, which occur in only 20-25% of prostate cancer patients. Bone metastases, on the other hand, are the primary cause of morbidity and mortality in the CRPC popula-tion, developing in 80-90% of patients [20]. Typical sequelae include pain, hematologic disorders, fracture, and neurologic compromise [21]. Given the predomi-nance of bone involvement, the uncertainty surround-ing the clinical signifi cance of PSA as a marker of ” response ” or progression [22], and the increased avail-ability of cytostatic agents, reliable methods to ascertain progression in bone are of increasing importance.
The X-ray has been used for the detection of met-astatic prostate cancer in bone since the early days of
“ skiagraphy ” [23]. The plain radiograph still plays an important role in the assessment of skeletal metastases, but has largely been supplanted by bone scintigraphy [24]. 99m Tc-methylene diphosphonate (MDP) and similar radiolabeled phosphate analogues, introduced for bone scanning in the early 1970 ’ s, are incorporated into the hydroxyapatite crystalline lattice and collagen matrix [25]. Uptake is a function of blood supply, rate of bone turnover or osteoblastic activity, quantity of mineralized bone, capillary permeability, fl uid pressure and local acid/base balance. Bone scanning is highly sensitive for blastic metastases and allows for quick appraisal of the entire skeleton. It is widely available, relatively inexpensive and reimbursable, making it the preferred modality for assessing bone metastases. The main shortcoming of the bone scan is that it depicts secondary changes rather than directly imaging the tumor. As such, early metastases may be missed and sensitivity for detection of osteolytic disease is lacking. Regression of disease is nearly impossible to verify on account of lingering uptake in healing bone, despite eradication of tumor. Moreover, response assessment is confounded by the fl are phenomenon, a major obstacle that can occur up to 12 weeks post effective treatment [26]. Thus, the declaration of scintigraphic progression during this period should be restrained in comparison to later time points. Changes in intensity or minor changes in extent of existing lesions are non-specifi c features and should not be considered deter-minants of progression at any time point.
Recognizing the variability in bone scan interpreta-tion between observers, the PCWG2 emphasized the need for standardization of reporting, as a requirement
Baseline 13 Weeks 24 Weeks Figure 1. Example of scintigraphic progression per PCWG2: Two or more new lesions appear on fi rst post-12 week assessment (arrows), followed by at least 2 additional new lesions on confi rmation scan more than 6 weeks later.
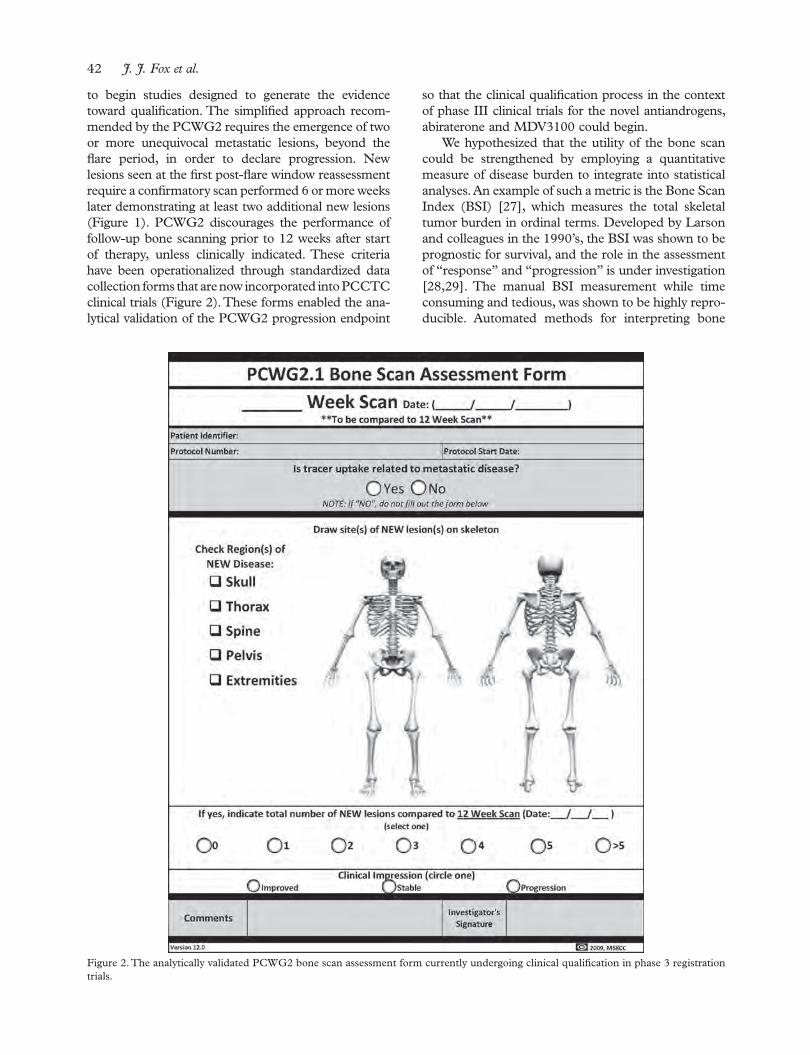
42 J. J. Fox et al.
to begin studies designed to generate the evidence toward qualifi cation. The simplifi ed approach recom-mended by the PCWG2 requires the emergence of two or more unequivocal metastatic lesions, beyond the fl are period, in order to declare progression. New lesions seen at the fi rst post-fl are window reassessment require a confi rmatory scan performed 6 or more weeks later demonstrating at least two additional new lesions (Figure 1). PCWG2 discourages the performance of follow-up bone scanning prior to 12 weeks after start of therapy, unless clinically indicated. These criteria have been operationalized through standardized data collection forms that are now incorporated into PCCTC clinical trials (Figure 2). These forms enabled the ana-lytical validation of the PCWG2 progression endpoint
so that the clinical qualifi cation process in the context of phase III clinical trials for the novel antiandrogens, abiraterone and MDV3100 could begin.
We hypothesized that the utility of the bone scan could be strengthened by employing a quantitative measure of disease burden to integrate into statistical analyses. An example of such a metric is the Bone Scan Index (BSI) [27], which measures the total skeletal tumor burden in ordinal terms. Developed by Larson and colleagues in the 1990 ’ s, the BSI was shown to be prognostic for survival, and the role in the assessment of “ response ” and “ progression ” is under investigation [28,29]. The manual BSI measurement while time consuming and tedious, was shown to be highly repro-ducible. Automated methods for interpreting bone
Figure 2. The analytically validated PCWG2 bone scan assessment form currently undergoing clinical qualifi cation in phase 3 registration trials.

Imaging Castration Resistant Prostate Cancer 43
scans that can produce results within seconds and with 100% reproducibility are under development and may prove useful for calculating the BSI [30 – 32]. Though promising, the automated platform itself must fi rst be validated against the manual technique before its prog-nostic value can be determined.
Additional efforts to standardize the assessment of bone disease include the proposed criteria from MD Anderson (MDA), the PET Response Criteria in Solid Tumors (PERCIST) by Wahl and colleagues, and the updated RECIST 1.1 publication [33 – 35]. The latter only addresses osteolytic lesions with soft tissue components that are infrequent in prostate can-cer. The MDA report proposes a multimodality approach that incorporates bone scan, X-ray, CT and MRI. The recently proposed PERCIST is presumed to apply equally to bone and soft tissue lesions alike, given that the functional information derived from PET is largely independent of tissue type. As was the case with the proposed PCWG2 bone scan progres-sion criteria, both the MDA and PERCIST proposed criteria will require analytical validation before they can begin qualifi cation testing. Until PET and other molecular imaging biomarkers are qualifi ed in the context of CRPC, rigorously standardized bone scan-ning may for now serve as the groundwork by which these other modalities are tested.
Positron Emission Tomography (PET)
Several PET tracers have shown promise as potential biomarkers in CRPC [36]. 18 F-Sodium Fluoride (NaF) is a high affi nity bone seeking agent that if employed in lieu of the single photon 99m Tc agents could enhance traditional bone scanning. 18 F-FDG-PET is a marker of tumor glycolytic rate (Warburg effect [37]) with established benefi ts in several contexts [38–40], while the role in prostate cancer management is still consid-ered “ investigational ” . Additional metabolic agents such as 18 F-FACBC, 18 F-Choline, and 11 C-methionine have been studied extensively in prostate cancer, but are beyond the scope of this review. Finally, 18 F-FDHT and novel tracers such as 89 Zr-J591 are under develop-ment for probing the AR signaling axis.
18F-sodium fl ouride (NaF) PET
Ironically, NaF was developed for bone scanning as a single photon agent prior to the advent of the 99m Tc phosphonates [41], but is being reinvestigated as a PET tracer. Possible advantages of NaF over 99m Tc-agents are attributable to the higher affi nity for osteoblastic activity, and the superior imaging charac-teristics of PET. Several studies have suggested that NaF performs better than 99m Tc-agents for the detec-tion of metastases, particularly when combined with
the anatomic information derived from CT. Even-Sapir et al. compared planar bone scintigraphy, bone SPECT, NaF PET, and NaF PET/CT in patients with localized high-risk or metastatic prostate cancer. The reported sensitivity and specifi city for detection of bone lesions was higher for NaF PET/CT (100% and 100%, respectively) than for planar bone scanning (70% and 57%), bone SPECT (92% and 82%) or NaF PET (100% and 62%) [42]. These results seem to favor NaF PET/CT over traditional bone scanning; however, the study included a mixed population and did not include a standard comparator. Further inves-tigation of NaF in the context of rigorously controlled prospective trials is needed before it can be recom-mended to replace the single photon bone scan, which is less expensive and more widely available.
18 F-FDG: Imaging tumor glycolysis
Despite the apparent advantage of NaF-PET over single photon bone scanning, it remains an indirect
Figure 3. (A). Kaplan-Meier survival curves for 22 patients with low ( � 6.10) and 21 patients with high ( � 6.10) SUVmax, p � 0.002. Reprinted with permission from Meirelles et al. [29]. (B). 69-year-old male with CRPC. MIP and axial images shows markedly FDG-avid bone lesions in the thoracic spine, SUVmax 16.8. The patient died within 18 months after the scan.

44 J. J. Fox et al.
with survival was also shown for BSI (14.7 vs. 28.2 months if BSI � 1.27 vs. � 1.27; p � 0.004); however, only SUVmax was an independent factor in multivariate analysis.
Paralleling the approach to develop the BSI, our group evaluated a single quantitative measure of tumor burden on PET, termed the SUV max-avg , which is an average of the 5 lesions (bone, lymph node or soft tissue) with the highest SUV max . We showed the value of SUV max-avg as a prognostic factor for survival and treatment response [46, 47] . Nevertheless, while a RECIST-like target lesion analysis has merit, it is feasible and perhaps desirable to perform a total lesion analysis that better estimates the true tumor burden and has potential for capturing inter-lesional heterogeneity. This daunting task could be mitigated by semi-automated applications of acquisitions and for interpretation that are now in clinical testing. PET VCAR (Volume Computer Assisted Reading) is one such program that was co-developed by Larson and colleagues in collaboration with GE Healthcare Systems. PET-VCAR is based on the fi duciary marker of the skeleton [48], a count-based edge recognition program [49], and introduction of novel parameters to associate with clinical outcomes (total lesion gly-colysis or gross metabolic volume) [50]. The applica-tion bookmarks regions of interest and propagates them from one time point to another, improving analysis and workfl ow. These features, in addition to accelerating the interpretation process, may also
method of imaging bone metastases. FDG, on the other hand, has the benefi t of directly assessing tumor metabolism, and is useful for both bone lesions and soft tissue lesions. These characteristics raise the pos-sibility of developing a “ response ” biomarker that occurs earlier than TTP or OS, without sacrifi cing the emphasis on bone metastases. This in turn could lead to shorter approval times for novel CRPC ther-apies. Initial studies of FDG-PET in prostate cancer examined heterogeneous patient populations [43, 44]. The less-than-favorable results highlighted the need to study patient populations controlled for clin-ical state, disease progression, therapy, scanning algorithm and clinical endpoints. Subsequent studies adhering to this approach have suggested a role for FDG-PET as an outcome measure in CRPC [45, 46]. With respect to the context of prognosis, one study [29] found an inverse relationship between FDG-PET SUVmax and survival of CRPC patients (median survival 14.4 vs. 32.8 months if SUVmax � 6.10 vs. � 6.10, p � 0.002) (Figure 3). A combination of SUVmax and a nomogram for progressive prostate cancer dichotomized patients into a high versus low risk group (median survival 14.4 vs. 34.6 months, p � .015) that was more prognostic than either alone. In this same study, of 105 FDG-positive bone lesions that were negative on bone scan, 84 (80%) lesions eventually turned positive on follow-up bone scan, indicating that FDG-PET bone fi ndings are clinically relevant. A similar inverse relationship
BA
FDHT FDHT
FDG FDG
Figure 4. (A). CRPC patient with multiple osteoblastic metastases. Sagittal fused PET/CT and PET images (FDG top row, FDHT bottom row) demonstrate prominent FDG and FDHT uptake, consistent with a “ Glycolysis/AR Concordant ” phenotype. (B). Second CRPC patient with multiple osteoblastic metastases. Sagittal fused PET/CT and PET images (FDG top row, FDHT bottom row) demonstrate intense FDHT uptake and relatively low level FDG uptake, consistent with an “ AR Predominant ” phenotype.

Imaging Castration Resistant Prostate Cancer 45
increase inter and intra observer agreement in assess-ment of lesions. The ability to compare two separate PET tracers with high precision and accuracy is par-ticularly relevant to drug development in CRPC, where probing of the AR signaling axis is likely to require a multi-tracer strategy.
Imaging the Androgen Receptor Signaling Axis
FDHT is an analog of the primary ligand of AR, dihydrotestosterone (DHT), and is thus a rational candidate for imaging of the AR [51]. The feasibility of FDHT-PET imaging in CRPC patients and dis-placement of FDHT by antiandrogens are already established [52,53]. Quantitative kinetic models of FDHT uptake as a measure of AR expression in human tumors, in vivo, were recently elucidated [54]. These studies were the foundation for FDHT-PET as a vital component of broader AR imaging strate-gies in CRPC; however, continued investigation is needed to further determine its optimal context of use. As a step towards this goal, we have studied more than 100 CRPC patients with baseline
FDHT-PET and FDG-PET assessments prior to entry into clinical trials, and the majority of these patients have undergone early and late post-treat-ment scans. Facilitated by the PET-VCAR software, this imaging strategy has suggested diverse metabolic phenotypes of CRPC on both a patient and lesional basis, which we prefer to classify as either “ AR Pre-dominant ” , “ Glycolysis Predominant ” or “ AR/Gly-colysis Concordant ” [55] (Figures 4 and 5). This classifi cation scheme may have prognostic and treat-ment predictive implications, as associations with specifi c molecular determinants in the tumor itself are being explored.
One specifi c context for FDHT-PET was its use as a pharmacodynamic response indicator in the phase 1 trial of the next generation anti-androgen MDV3100 [5] . The study demonstrated antitumor effects in patients with CRPC, with PSA declines by � 50% in 56%, soft tissue regression in 22%, bone disease stabilization in 56%, and conversion from unfavorable to favorable circulating tumor cell counts in 49% of patients. The pharmacodynam-ics of MDV3100 was evaluated by measuring the change in FDHT uptake after start of treatment
SUVm
ax
16.0
14.0
12.0
10.0
8.0
6.0
4.0
2.0
0.01 3 5 7 9 11 13 15 17 19 21 23
FDGFDHT
25 27 29
Lesion
A B
C
Figure 5. (A) Axial fused PET/CT and CT images show an example of a discordantly positive nodal mass on 18 F-FDG (top, crosshairs), negative on 18 F-FDHT (bottom). (B) In the same patient, axial images show an example of a discordantly positive bone lesion on 18 F-FDHT (bottom, crosshairs), negative on 18 F-FDG (top). (C) VCAR derived bar graph of total lesional SUVmax for the same patient demonstrating substantial heterogeneity in lesion avidity.

46 J. J. Fox et al.
(displacement) in a subset (n � 22) of patients receiving dosages ranging from 60 mg to 480 mg per day. A clear reduction in FDHT uptake was seen in all patients ( ∼ 20-100%), with some indication that those receiving lower dosages had a smaller reduc-tion (mean decrease � 50%) than those receiving dosages in the higher range (mean decrease � 50%). No appreciable difference in FDHT uptake was seen in the higher dose range, despite substantial differ-ences in serum levels of MDV3100. This suggests that the maximal effect of the drug may be reached before reaching the maximum tolerated dose (Figure 6). Based on the fi ndings of this pilot study, FDHT-PET appears useful as a pharmacodynamic marker in discrete contexts. Further optimization of the imaging with a form of background correction is ongoing. Interestingly, these same 22 patients also underwent FDG-PET scans to assess tumor glycoly-sis. Modulation of tumor glycolytic rate was evidenced by reductions in FDG metabolism with declines of SUVmax-avg of 25% or more in 45% of cases. The fact that the across-the-board reduction of FDHT uptake did not parallel the FDG-PET response seems consistent with FDHT-PET as a pharmacodynamic
marker, as opposed to a response indicator. Never-theless, it is possible that FDHT-PET may prove to be a response-indicator in other settings. For instance, with drugs that decrease androgen synthesis (e.g. CYP17 inhibitors), but do not directly target the AR, a reduction of FDHT uptake may signal a true cytostatic or cytotoxic response.
While changes in FDHT uptake do refl ect AR ligand-receptor interaction, there remains a discon-nect between the detection of AR occupancy and the effects of the drug on downstream AR signaling. An investigational agent, 89 Zr-J591 [56], is hypothesized to distinguish between these two entities. J591 is an antibody that binds to an external epitope on pros-tate specifi c membrane antigen (PSMA) that has been studied extensively for both imaging and radio-immunotherapy purposes [57]. Androgen depriva-tion has been shown to upregulate PSMA expression. As such, changes in PSMA expression detected by 89 Zr-J591 are proposed to refl ect the downstream effects of AR inhibition [58,59]. 89 Zr-J591 and simi-lar downstream imaging agents will hopefully improve our understanding of the biology of CRPC and its escape mechanisms. If successful, these
Figure 6. Pharmacodynamics of MDV3100. (A) Sagittal fused PET/CT and PET images 1 h after administration of FDHT at baseline and 4 weeks after start of MDV3100 therapy show a reduction in FDHT accumulation in tumor within the vertebrae, compared with the cardiac and aortic blood pool, in which FDHT metabolites circulate bound to serum proteins. (B) Percentage change in FDHT average maximum standard uptake value (SUVmax) from baseline to 4 weeks by dose. At baseline, all 22 patients had at least one FDHT-avid lesion that could serve as index lesions: 17 patients had fi ve index lesions, three had three index lesions, and two had one index lesion. At baseline, the median FDHT SUVmax average was 7.81 (IQR 4.9 – 9.6). Reprinted with permission and modifi cation from Scher et al. [5].

Imaging Castration Resistant Prostate Cancer 47
non-invasive AR probes, in tandem, will give us the capacity to discern four key aspects of targeted ther-apy: the presence of the target, the ability of the drug to localize to the target, the ability of the drug to inhibit downstream target effects, and the ability of the drug to modulate tumor viability.
CONCLUSION
Recent advances in the understanding of prostate cancer biology have lead to the development of much needed experimental therapies for CRPC with proven effi cacy, several of which target the AR signaling axis. Qualifi ed imaging biomarkers are sorely needed to facilitate the continued development and approval of these drugs. The bone-tropic nature of metastatic CRPC justifi es the emphasis on rigorously standard-ized bone scanning as the gatekeeper through which more potent and versatile imaging biomarkers are co-developed. In concert with the metabolic infor-mation derived from FDG, molecular imaging of the AR signaling axis promises to reveal important insights into CRPC that will, hopefully, result in improved patient outcomes.
Acknowledgements
Support for this work came from: Department of Defense grant PC071610, ICMIC grant P50 CA086438-08, and the Memorial Sloan-Kettering Cancer Center Specialized Program of Research Excellence (SPORE) Grant in Prostate Cancer (P50 CA92629).
Declaration of interest: The authors report no confl icts of interest. The authors alone are responsible for the content and writing of the paper.
References
Cancer.org [internet]. American Cancer Society; 2011. Avail-[1] able from: http://www.cancer.org/Cancer/ProstateCancer/DetailedGuide/prostate-cancer-key-statistics. Scher H. Prostate carcinoma: Defi ning therapeutic objectives [2] and improving overall outcomes. Cancer Suppl 2003;97:758 – 71. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, [3] et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33 – 9. Chen Y, Sawyers CL, Scher HI. Targeting the androgen [4] receptor pathway in prostate cancer. Curr Opin Pharmacol 2008;8:440 – 8. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, [5] Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1-2 study. Lancet 2010;375:1437 – 46. Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, [6] Smith MR, et al. Phase II multicenter study of abiraterone
acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol 2010;28:1496 – 501. Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, [7] Fong PC, et al. Signifi cant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol 2010;28:1489 – 95. Scher HI, Heller G. Clinical states in prostate cancer: [8] Toward a dynamic model of disease progression. Urology 2000;55:323 – 7. Feldman BJ, Feldman D. The development of androgen- [9] independent prostate cancer. Nat Rev Cancer 2001;1:34 – 45. Atkinson Jr AJ, Colburn WA, DeGruttola VG, DeMets DL, [10] Downing GJ, Hoth DF, et al. Biomarkers Defi nitions Work-ing Group. Biomarkers and surrogate endpoints: Preferred defi nitions and conceptual framework. Clin Pharmacol Ther 2001;69:89 – 95. Goodsaid FM, Frueh FW, Mattes W. Strategic paths for [11] biomarker qualifi cation. Toxicology 2008;245:219 – 23. Wagner JA, Williams SA, Webster CJ. Biomarkers and sur-[12] rogate end points for fi t-for-purpose development and regu-latory evaluation of new drugs. Clin Pharmacol Ther 2007;81:104 – 7. Prentice RL. Surrogate and mediating endpoints: Current [13] status and future directions. J Natl Cancer Inst 2009;101:216 – 7. WHO handbook for reporting results of cancer treatment. [14] Geneva: World Health Organization Offset Publication; 1979. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan [15] RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. J Nat Cancer Inst 2000;92:205 – 16. Scher HI, Morris MJ, Kelly WK, Schwartz LH, Heller G. [16] Prostate cancer clinical trial end points: “RECIST”ing a step backwards. Clin Cancer Res. 2005;11(14):5223 – 32. Khleif SN, Doroshow JH, Hait WN. AACR-FDA-NCI [17] Cancer Biomarkers Collaborative consensus report: Advanc-ing the use of biomarkers in cancer drug development. Clin Cancer Res 2010;16:3299 – 318. Morris MJ, Basch EM, Wilding G, Hussain M, Carducci [18] MA, Higano C, et al. Department of Defense prostate cancer clinical trials consortium: A new instrument for prostate can-cer clinical research. Clin Genitourin Cancer 2009;7:51 – 7. Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, [19] Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148 – 59. Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Wili [20] N, et al. Metastatic patterns of prostate cancer: An autopsy study of 1589 patients. Hum Path 2000;31:578 – 83. Carlin BI, Andriole GL. The natural history, skeletal compli-[21] cations and management of bone metastases in patients with prostate carcinoma. Cancer 2000;88:2989 – 94. Mulders PF, Schalken JA. Measuring therapeutic effi cacy in [22] the changing paradigm of castrate-resistant prostate cancer. Prostate Cancer Prostatic Dis 2009;12:241 – 6. Evans TC. Two cases of cancer of prostate with bony metas-[23] tases. Proc R Soc Med 1929;22:1383 – 6. Levenson RM, Sauerbrunn BJ, Bates HR, Newman RD, [24] Eddy JL, Ihde DC. Comparative value of bone scintigraphy and radiography in monitoring tumour response in sys-temically treated prostatic carcinoma. Radiology 1983;146:513 – 8. Subramanian G, McAfee JG. A new complex of 99mTc for [25] skeletal imaging. Radiology 1971;99:192 – 6.

48 J. J. Fox et al.
Pollen JJ, Witztum KF, Ashburn WL. The fl are phenomenon [26] on radionuclide bone scan in metastatic prostate cancer. Am J Roentgenol 1984;142:773 – 6. Imbriaco M, Larson SM, Yeung HW, Mawlawi OR, Erdi Y, [27] Venkatraman ES, et al. A new parameter for measuring met-astatic bone involvement by prostate cancer: The bone scan index. Clin Cancer Res 1998;4:1765 – 72. Sabbatini P, Larson SM, Kremer A, Zhang ZF, Sun M, Yeung [28] H, et al. Prognostic signifi cance of extent of disease in bone in patients with androgen-independent prostate cancer. J Clin Oncol 1999;17:948 – 57. Meirelles GS, Schoder H, Ravizzini GC, Gonen M, Fox JJ, [29] Humm J, et al. Prognostic value of baseline [18F] fl uorode-oxyglucose positron emission tomography and 99mTc-MDP bone scan in progressing metastatic prostate cancer. Clin Cancer Res 2010;16(24):6039–9. Erdi YE, Humm JL, Imbriaco M, Yeung H, Larson SM. [30] Quantitative bone metastases analysis based on image seg-mentation. J Nucl Med 1997;38:1401 – 6. Sadik M, Hamadeh I, Nordblom P, Suurkula M, Hoglund [31] P, Ohlsson M, et al. Computer-assisted interpretation of pla-nar whole-body bone scans. J Nucl Med 2008;49:1958 – 65. Sadik M, Suurkula M, H ö glund P, J ä rund A, Edenbrandt L. [32] Improved classifi cations of planar whole-body bone scans using a computer-assisted diagnosis system: A multicenter, multiple-reader, multiple-case study. J Nucl Med 2009;50:368 – 75. Costelloe CM, Chuang HH, Madewell JE, Ueno NT. Cancer [33] response criteria and bone metastases: RECIST 1.1, MDA and PERCIST. J Cancer 2010;1:80 – 92. Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST [34] to PERCIST: Evolving considerations for PET response crite-ria in solid tumors. J Nucl Med 2009;50(Suppl 1):S122 – 50. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sar-[35] gent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228 – 47. Apolo A, Pandit-Taskar N, Morris MJ. Novel tracers and [36] their development for the imaging of metastatic prostate can-cer. J Nucl Med 2008;49:2031 – 41. Vander Heiden MG, Cantley LC, Thompson CB. Under-[37] standing the Warburg effect: The metabolic requirements of cell proliferation. Science 2009;324:1029 – 33. Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, [38] Cheng EY, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug develop-ment. Clin Cancer Res 2005;11:2785 – 808. Shankar LK, Hoffman JM, Bacharach S, et al. Guidelines for [39] the use of 18 F-FDG PET as an indicator of therapeutic response in patients in National Cancer Institute trials. J Nucl Med 2006;47:1059 – 66. Larson SM, Schwartz LH. 18F-FDG PET as a candidate for [40] “qualifi ed biomarker”: Functional assessment of treatment response in oncology. J Nucl Med 2006;47:901 – 3. Blau M, Nagler W, Bender MA. Fluorine-18: A new isotope [41] for bone scanning. J Nucl Med 1962;3:332 – 4. Even-Sapir E, Metser U, Mishani E, Mishani E, Lievshitz G, [42] Lerman H, et al. The detection of bone metastases in patients with high risk prostate cancer: 99mTc-MDP planar bone scin-tigraphy, single and multifi eld of view SPECT, 18F- fl uoride PET and 18F-fl uoride PET/CT. J Nuc Med 2006;47:287 – 97. Yeh SD, Imbriaco M, Larson SM, Garza D, Zhang JJ, [43] Kalaigian H, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol 1996;23:693 – 7. Shreve PD, Grossman HB, Gross MD, Wahl RL. Metastatic [44] prostate cancer: Initial fi ndings of PET with 2-deoxy-2-[F-18]fl uoro-D-glucose. Radiology 1996;199:751 – 6.
Morris MJ, Akhurst T, Osman I, Nunez R, Macapinlac H, [45] Siedlecki K, et al. Fluorinated deoxyglucose positron emis-sion tomography imaging in progressive metastatic prostate cancer. Urology 2002;59:913 – 8. Morris MJ, Akhurst T, Larson SM, Ditullio M, Chu E, Sie-[46] dlecki K, et al. Fluorodeoxyglucose positron emission tom-ography as an outcome measure for castrate metastatic prostate cancer treated with antimicrotubule chemotherapy. Clin Cancer Res 2005;11:3210 – 6. Morris MJ, Fox JJ, Dennis ER, Tse K, Stephenson RD, [47] Flatts E, et al. Pretreatment fl uorodeoxyglucose (FDG) PET and survival for castration-resistant metastatic prostate cancer (CRMPC). 2010 Genitourinary Cancers Symposium; March 5–7, 2010; San Francisco, CA; Abstract 108. Erdi YE, Srivastava NC, Humm JL, Larson SM. A coordinate [48] system for tumor identifi cation in positron emission tomo-graphy (PET) imaging. Clin Positron Imaging 2000;3:131 – 6. Erdi YE, Mawlawi O, Larson SM, Imbriaco M, Yeung H, [49] Finn R, et al. Segmentation of lung lesion volume by adaptive positron emission tomography image thresholding. Cancer 1997;80:2505 – 9. Larson SM, Erdi Y, Akhurst T, Mazumdar M, Macapinlac [50] HA, Finn RD, Casilla C, et al. Tumor treatment response based on visual and quantitative changes in global tumor glycolysis using PET-FDG imaging: The visual response score and the change in the total lesion glycolysis. Clin Posi-tron Imaging 1999;3:159 – 71. Liu A, Dence CS, Welch MJ, Katzenellenbogen JA. Fluorine-[51] 18-labeled androgens: Radiochemical synthesis and tissue distribution studies on six fl uorine-substituted androgens, potential imaging agents for prostatic cancer. J Nucl Med 1992;33:724 – 34. Larson SM, Morris M, Gunther I, Beattie B, Humm JL, [52] Akhurst TA, et al. Tumor localization of 16beta-18F-fl uoro-5alpha-dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J Nucl Med 2004;45:366 – 73. Dehdashti F, Picus J, Michalski JM, Dence CS, Siegel BA, [53] Katzenellenbogen JA, et al. Positron tomographic assessment of androgen receptors in prostatic carcinoma. Eur J Nucl Med Mol Imaging 2005;32:344 – 50. Beattie BJ, Smith-Jones PM, Jhanwar YS, Schoder H, [54] Schmidtlein CR, Morris MJ, et al. Pharmacokinetic assess-ment of the uptake of 16beta-18F-fl uoro-5alpha-dihydrotes-tosterone (FDHT) in prostate tumors as measured by PET. J Nucl Med 2010;51:183 – 92. Fox J, Blanc E, Schoder H, Morris M, Scher H, Larson S, [55] et al. Diversity of biology in castrate resistant prostate cancer. J Nucl Med 2009;50(Suppl 2):523. Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson [56] SM, Lewis JS. 89Zr-DFO-J591 for immunoPET of prostate-specifi c membrane antigen expression in vivo. J Nucl Med. 2010;51(8):1293 – 300. Pandit-Taskar N, O’Donoghue JA, Morris MJ, Wills EA, [57] Schwartz LH, Gonen M, et al. Antibody mass escalation study in patients with castration-resistant prostate cancer using 111In-J591: Lesion detectability and dosimetric projections for 90Y radioimmunotherapy. J Nucl Med 2008;49:1066 – 74. Wright GL Jr, Grob BM, Haley C, Grossman K, Newhall K, [58] Petrylak D, et al. Upregulation of prostate-specifi c membrane antigen after androgen-deprivation therapy. Urology 1996;48:326 – 34. Kuroda K, Liu H, Kim S, Guo M, Navarro V, Bander NH. [59] Docetaxel down-regulates the expression of androgen recep-tor and prostate-specifi c antigen but not prostate-specifi c membrane antigen in prostate cancer cell lines: Implications for PSA surrogacy. Prostate 2009;69:1579 – 85.

Correspondence: Daniel M. Berney, Department of Molecular Oncology and Imaging, Institute of Cancer, Queen Mary University of London, London, UK. E-mail: [email protected]
(Received 3 August 2010 ; accepted 16 September 2010 )
REVIEW ARTICLE
Pathology in prostate research: Optimizing the pathological data
DANIEL M BERNEY 1 , RODOLFO MONTIRONI 2 & LARS EGEVAD 3
1 Department of Molecular Oncology and Imaging, Institute of Cancer, Queen Mary University of London, London, UK, 2 Institute of Pathological Anatomy and Histopathology, School of Medicine, Polytechnic University of the Marche Region, Ancona, Italy and 3 Department of Pathology, Karolinska University Hospital, Stockholm, Sweden
Abstract Pathology remains the gold standard for the diagnosis and local staging and grading of prostate cancer. However, as in any discipline, there are variations in national standards and protocols leading to possible signifi cant intra-observer variations. This can signifi cantly impact on the data supplied to clinical trials. Diagnostic and grading criteria . Error rates in the diag-nosis of prostate cancer have improved but the possibility that diagnostic error may be discovered has to be addressed in any research series. Major changes in Gleason grading have occurred in the past 40 years and this may lead to suboptimal application of grades in research cohorts, falsely raising the prognostic power of new biomarkers. Tumor measurements and staging criteria. Further information that may provide additional prognostic information include various measures of tumor extent and peri-neural invasion in biopsy specimens. Standardization of measures of tumor extent is necessary to give more useful assessments of prognosis. In radical prostatectomy specimens there are a number of other staging measurements which might be applied, including tumor volume, margin status, extra-capsular extension and nodal positivity though many of these variables are interdependent. Conclusion. Appropriate utilization of such pathological material will produce improved cohorts in which it will be possible to test new biomarkers with increased rigor.
Pathological data in clinical research
Validation of the diagnosis
The pathological diagnosis of prostate cancer on needle biopsy is still recognized to be a challeng-ing area. Retrospective analyses of cases has revealed error rates of between 7 – 20% [1,2] though modern immunohistochemical methods may have reduced this [3]. Nevertheless, recent series still reveal sig-nifi cant errors in both the under-diagnosis and over-diagnosis of cancer [4]. Specialist uropathologists have been shown to have lower error rates than non-specialists. Error rates will also probably increase in older series. However it should be realized that review of old cases may lead to ethical questions of whether the patient should be informed of such changes if cases are reviewed rather than case notes abstracted. This may be handled in a number of ways. In one ret-rospective series, the patients were informed, after 20 years if their diagnosis of prostate cancer was incor-rect [1]. No legal action resulted from this (personal communication from the author). A second similar
series had built-in anonymization to their results and was given ethical approval that the patients would not be informed of such changes [2]. These issues have to be addressed before the commencement of any review.
Gleason scoring
Gleason scoring, devised over 40 years ago, remains the most powerful predictor of prostate cancer behav-ior in any biopsy or resection specimen [5,6]. How-ever, signifi cant changes have occurred in Gleason scoring since it was devised in the late 1960s and early 1970s [7]. This has resulted in signifi cant shifts in Gleason scoring, invariably to higher scores. The reasons for this are many, but include the recognition that the lowest Gleason scores probably do not rep-resent cancer, meaning they are no longer diagnosed. The ISUP 2005 consensus conference made a num-ber of signifi cant changes to Gleason scoring. Unfor-tunately these were not fully validated, and it remains uncertain whether traditional or ‘ revised ’ Gleason
Acta Oncologica, 2011; 50(Suppl 1): 49–52
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.525223

50 D. M. Berney et al.
scoring is a better predictor of behavior [8], espe-cially for the crucial cut-off between Gleason pattern 3 and Gleason pattern 4. It should also be recognized that the modern criteria are a ‘ consensus ’ and there-fore there may be variations between different labo-ratories and different pathologists in how Gleason grading is calculated [9]. The results of this Gleason drift is the so called ‘ Will Rogers ’ phenomenon, where survival curves appear to show improvement over time due to upgrading of the disease [10].
Examples of this include the calculation of Gleason score from a biopsy series. Some pathologists will calculate a composite score, taking into account the appearances in every core with cancer. Other groups report the ‘ worst ’ score seen in any of the cores sam-pled. A second example is the calculation of tertiary grade [11]. There was no consensus at the meeting in radical prostatectomy specimens as to whether the worst pattern seen, if it was a tertiary element should be incorporated into the main Gleason score.
Unfortunately it is not yet possible to state on the optimal method. However there is evidence that abstraction of notes from a retrospective series will provide much poorer information than a single pathologist reviewing to modern criteria [12]. This is important in any biomarker assessment since merely using abstracted information from multiple cen-ters will considerably under-estimate the power of Gleason scoring and therefore lead to a falsely more powerful assessment of a given tumor biomarker.
Therefore for both retrospective and prospective studies there is considerable variation how Gleason score has been and is calculated, and any utilization of Gleason score should include a description of the methodologies used.
Tumor extent and staging in diagnostic samples
Estimates of tumor extent are being increasingly used in estimation of recurrence and progression risk and should therefore be used in multivariate analy-ses. Unfortunately multiple methods of assessment are available leading to non-comparability of large series. Tumor extent in TURP specimens may be measured counting the number of positive chips or by visual estimates of involved tissue though one paper suggests these are comparable [13].
Measurements of tumor extent are prognosti-cally more important in prostate biopsies, and most series have shown that tumor content of prostate biopsies yields prognostic information on multi-variate analysis. A systematic review [14] has shown that multiple methodologies for core measurement and assessment preclude proper analyses. However, tumor extent does appear in nomograms and tables and some assessment of tumor extent should be
regarded as standard practice in the evaluation of new biomarkers on needle biopsies [15].
Perineural invasion
The importance of reporting perineural invasion prostate cancer is still controversial. A recent review concluded that variations in study design precluded meta-analysis [16] but that the weight of evidence suggested that in localized disease, the presence of perineural invasion might favor the use of more radical therapy.
Pathologic data in radical prostatectomy specimens
As in biopsy specimens, details of the methods by which Gleason grade is calculated should be regarded as an essential criterion in any multivariate assess-ment of disease progression after RP in clinical tri-als. However there are other important assessments of disease extent which are not routinely of impor-tance in biopsy specimens. The International Society of Urological Pathologists has recently published guidelines to standardize the acceptable approaches to radical prostatectomy processing [17 – 21].
a. TNM Stage. The major decision in radical pro-statectomy (RP) specimens is to distinguish between tumors limited to the prostate (organ confi ned, pT2) or involving extra-prostatic tissues (pT3) [22]. In radical prostatectomy specimens tumor volume measurements and sub dividing the category of organ confi ned tumors (pT2) does not appear to provide use-ful independent prognostic information as it is less informative than other prognostic para-meters such as stage, capsular involvement, mar gin positivity and lymph node status. Assess-ments of tumor volume in this scenario have yielded contradictory information [23,24].
b. Margin status . Many studies have reported on the prognostic signifi cance of involved margins and the extent of positivity is also of prognostic signifi cance [25,26].
c. Seminal vesicle involvement (SVI, pT3b) is a poor prognostic factor after radical prostatec-tomy [27].
d. Vascular invasion. The presence of vascu-lar invasion is an independent predictor of biochemical recurrence following radical prosta tec tomy [28].
e. Nodal positivity is a poor prognostic factor and the diameter of the largest metastasis appears to be more predictive of cancer-specifi c sur-vival than the number of positive nodes alone. However the presence of extranodal extension was not prognostic [29,30].

Prostate pathology data in research 51
Conclusions
There is an urgent need for new biomarkers to pre-dict outcome in prostatic carcinoma. However the search for such markers must be backed up with full and thorough assessment of all pathological criteria which might compete with a novel assess-ment. There is a concern that novel markers may appear more promising if measured in series with suboptimal or incomplete pathological assessments. Utilization of the maximum amount of pathologi-cal data will obviate these concerns and hopefully lead to biomarkers progressing from the research laboratory into clinical practice.
Declaration of interest: The authors state no con-fl ict of interest and have received no payment in the preparation of this manuscript.
References
Bostwick DG, Chang L. Overdiagnosis of prostatic adeno-[1] carcinoma. Semin Urol Oncol 1999;17:199 – 205. Berney DM, Fisher G, Kattan MW, Oliver RT, Moller H, [2] Fearn P, et al. Pitfalls in the diagnosis of prostatic cancer: Retrospective review of 1791 cases with clinical outcome. Histopathology 2007;51:452 – 7. Wojno KJ, Epstein JI. The utility of basal cell-specifi c anti-[3] cytokeratin antibody (34 beta E12) in the diagnosis of prostate cancer. A review of 228 cases. Am J Surg Pathol 1995;19:251 – 60. Epstein JI, Walsh PC, Sanfi lippo F. Clinical and cost impact [4] of second-opinion pathology. Review of prostate biopsies prior to radical prostatectomy. Am J Surg Pathol 1996;20:851 – 7. Kattan MW, Wheeler TM, Scardino PT. Postoperative nom-[5] ogram for disease recurrence after radical prostatectomy for prostate cancer. J Clin Oncol 1999;17:1499 – 507. Albertsen PC, Hanley JA, Fine J. 20-year outcomes following [6] conservative management of clinically localized prostate cancer. JAMA 2005;293:2095 – 101. Epstein JI, Allsbrook WC, Jr., Amin MB, Egevad LL. The [7] 2005 International Society of Urological Pathology (ISUP) consensus conference on Gleason grading of prostatic carcinoma. Am J Surg Pathol 2005;29:1228 – 42. Zareba P, Zhang J, Yilmaz A, Trpkov K. The impact of the [8] 2005 International Society of Urological Pathology (ISUP) consensus on Gleason grading in contemporary practice. Histopathology 2009;55:384 – 91. Melia J, Moseley R, Ball RY, Griffi ths DF, Grigor K, Harnden P, [9] et al. A UK-based investigation of inter- and intra-observer reproducibility of Gleason grading of prostatic biopsies. Histopathology 2006;48:644 – 54. Albertsen PC, Hanley JA, Barrows GH, Penson DF, [10] Kowalczyk PD, Sanders MM, et al. Prostate cancer and the Will Rogers phenomenon. J Natl Cancer Inst 2005;97:1248 – 53. Harnden P, Shelley MD, Coles B, Staffurth J, Mason MD. [11] Should the Gleason grading system for prostate cancer be modifi ed to account for high-grade tertiary components? A systematic review and meta-analysis. Lancet Oncol 2007;8:411 – 9. Berney DM, Fisher G, Kattan MW, Oliver RT, Moller H, [12] Fearn P, et al. Major shifts in the treatment and prognosis of
prostate cancer due to changes in pathological diagnosis and grading. BJU Int 2007;100:1240 – 4. Foucar E, Haake G, Dalton L, Pathak DR, Lujan JP. The [13] area of cancer in transurethral resection specimens as a prog-nostic indicator in carcinoma of the prostate: A computer-assisted morphometric study. Hum Pathol 1990;21:586 – 92. Harnden P, Shelley MD, Naylor B, Coles B, Mason MD. [14] Does the extent of carcinoma in prostatic biopsies predict prostate-specifi c antigen recurrence? A systematic review. Eur Urol 2008;54:728 – 39. Amin M, Boccon-Gibod L, Egevad L, Epstein JI, Humphrey PA, [15] Mikuz G, et al. Prognostic and predictive factors and report-ing of prostate carcinoma in prostate needle biopsy speci-mens. Scand J Urol Nephrol Suppl 2005;216:20 – 33. Harnden P, Shelley MD, Clements H, Coles B, Tyndale-Biscoe [16] RS, Naylor B, et al. The prognostic signifi cance of perineural invasion in prostatic cancer biopsies: A systematic review. Cancer 2007;109:13 – 24. van der Kwast TH, Amin MB, Billis A, Epstein JI, Griffi ths D, [17] Humphrey PA, et al. International Society of Urological Pathology (ISUP) consensus conference on handling and staging of radical prostatectomy specimens. Working group 2: T2 substaging and prostate cancer volume. Mod Pathol 2010 Sep 3. Tan PH, Cheng L, Srigley JR, Griffi ths D, Humphrey PA, [18] van der Kwast TH, et al. International Society of Urological Pathology (ISUP) consensus conference on handling and staging of radical prostatectomy specimens. Working group 5: Surgical margins. Mod Pathol 2010 Aug 20. Magi-Galluzzi C, Evans AJ, Delahunt B, Epstein JI, Griffi ths DF, [19] van der Kwast TH, et al. International Society of Urological Pathology (ISUP) consensus conference on handling and staging of radical prostatectomy specimens. Working group 3: Extraprostatic extension, lymphovascular invasion and locally advanced disease. Mod Pathol 2010 Aug 27. Egevad L, Srigley JR, Delahunt B. International Society of [20] Urological Pathology (ISUP) consensus conference on han-dling and staging of radical prostatectomy specimens: Rationale and organization. Mod Pathol 2010 Aug 27. Berney DM, Wheeler TM, Grignon DJ, Epstein JI, Griffi ths DF, [21] Humphrey PA, et al. International Society of Urological Pathology (ISUP) consensus conference on handling and staging of radical prostatectomy specimens. Working group 4: Seminal vesicles and lymph nodes. Mod Pathol 2010 Sep 3. Epstein JI, Amin M, Boccon-Gibod L, Egevad L, Humphrey PA, [22] Mikuz G, et al. Prognostic factors and reporting of prostate carcinoma in radical prostatectomy and pelvic lymphadenec-tomy specimens. Scand J Urol Nephrol Suppl 2005;216:34 – 63. Nelson BA, Shappell SB, Chang SS, Wells N, Farnham SB, [23] Smith JA, Jr., et al. Tumour volume is an independent predic-tor of prostate-specifi c antigen recurrence in patients under-going radical prostatectomy for clinically localized prostate cancer. BJU Int 2006;97:1169 – 72. Kikuchi E, Scardino PT, Wheeler TM, Slawin KM, Ohori M. [24] Is tumor volume an independent prognostic factor in clini-cally localized prostate cancer? J Urol 2004;172:508 – 11. Marks RA, Koch MO, Lopez-Beltran A, Montironi R, [25] Juliar BE, Cheng L. The relationship between the extent of surgical margin positivity and prostate specifi c antigen recurrence in radical prostatectomy specimens. Hum Pathol 2007;38:1207 – 11. Babaian RJ, Troncoso P, Bhadkamkar VA, Johnston DA. [26] Analysis of clinicopathologic factors predicting outcome after radical prostatectomy. Cancer 2001;91:1414 – 22. Tefi lli MV, Gheiler EL, Tiguert R, Banerjee M, Sakr W, [27] Grignon DJ, et al. Prognostic indicators in patients with

52 D. M. Berney et al.
seminal vesicle involvement following radical prostatectomy for clinically localized prostate cancer. J Urol 1998;160(3 Pt 1):802 – 6. Ferrari MK, McNeal JE, Malhotra SM, Brooks JD. Vascular [28] invasion predicts recurrence after radical prostatectomy: Stratifi cation of risk based on pathologic variables. Urology 2004;64:749 – 53.
Cheng L, Bergstralh EJ, Cheville JC, Slezak J, Corica FA, [29] Zincke H, et al. Cancer volume of lymph node metastasis predicts progression in prostate cancer. Am J Surg Pathol 1998;22:1491 – 500. Cheng L, Pisansky TM, Ramnani DM, Leibovich BC, [30] Cheville JC, Slezak J, et al. Extranodal extension in lymph node-positive prostate cancer. Mod Pathol 2000;13:113 – 8.

Correspondence: Daniel M. Berney, Department of Molecular Oncology and Imaging, Institute of Cancer, Queen Mary University of London, London, UK. E-mail: [email protected]
(Received 3 August 2010 ; accepted 16 September 2010 )
REVIEW ARTICLE
Pathology in prostate research: Optimizing tissue quality
DANIEL M. BERNEY 1 , RODOLFO MONTIRONI 2 & LARS EGEVAD 3
1 Department of Molecular Oncology and Imaging, Institute of Cancer, Queen Mary University of London, London, UK, 2 Institute of Pathological Anatomy and Histopathology, School of Medicine, Polytechnic University of the Marche Region, Ancona, Italy and 3 Department of Pathology, Karolinska University Hospital, Stockholm, Sweden
Abstract The collection of tissue from the prostate gland for research creates unique challenges in the identifi cation of cancer and in preserving pathological material. Value and uses of formalin fi xed tissue. Formalin fi xed paraffi n embedded (FFPE) tissue is often available in abundance after pathological processing and reporting of specimens but is limited in value for detailed molecular tests. Tissue micro-array if carefully performed is a helpful technique for examining many FFPE specimens with immunohistochemical or fl uorescence in situ hybridization tests. Value and uses of frozen tissue. The collection of fresh tissue prior to formalin fi xation and later validation samples of fresh prostate cancer is diffi cult as prostate cancer is very diffi cult to identify macroscopically on cut prostate specimens. Also, the act of manipulation and dissection of the gland while fresh and without compromising surgical margins is challenging. Methods which have been used to dissect the fresh prostate gland and also collect fresh tissue from other prostatic specimens are discussed. The ethical challenges of collecting research tissue without compromising patient care are discussed. Conclusions. Prostate cancer tissue banks, particularly of frozen tissue are still relatively few in number. Enhanced collection methods which do not prohibit full pathological examination are available but require expertise to maximize their potential.
Formalin fi xed paraffi n embedded tissue
Formalin fi xed and paraffi n embedded tissue (FFPE) allows a variety of molecular techniques to be assayed after histological diagnosis. Storage can be under-taken at room temperature and the vast majority of epitopes will remain stable even after 10 – 20 years or more in paraffi n blocks. A number of factors may affect this. Firstly if another fi xative such as Bouin ’ s is used, this may cause loss or different expression of markers [1]. Secondly, after FFPE blocks have been cut onto slides there is a loss of strength of signal with some antigens. For instance, HER-2 is known to degrade within a few weeks of sections being cut [2]. It has been suggested that if unstained slides are stored then they are preserved from degradation by storing at a low temperature or an oxidation free environment [3].
Tissue microarrays (TMA) allow multiple speci-mens to be examined on a single slide while preserv-ing their identity and also allowing a single immunohistochemical experiment to be performed on the same tissue [4,5]. It has proven a valuable
technique for high throughput of multiple tumors but nevertheless requires careful technical prepara-tion. Areas for microarray need to be identifi ed on adjacent Haematoxalin and Eosin sections, prefera-bly by a pathologist. This is especially true for pros-tate carcinoma where diagnosis is often challenging, especially in low grade disease. Secondly the hetero-geneity of tumor needs to be refl ected in sampling. This will include sampling of the index tumor. As Gleason scoring has shown, prostate cancer shows unique heterogeneity. At least three areas of tumor should be sampled and all grades of tumor, if differ-ent grades are present [6]. TMA is a technique fre-quently used in TURP and radical prostatectomy specimens. However recently it has been shown to be possible to create TMAs from biopsy material as well as TURP and RP specimens [7].
Fresh tissue
Molecular profi ling techniques such as real-time PCR and microarrays require the harvesting of high quality tissue, yet the process of formalin fi xation
Acta Oncologica, 2011; 50(Suppl 1): 53–55
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.525224

54 D. M. Berney et al.
results in non-specifi c degradation of delicate intra-cellular molecules, particularly RNA [8,9]. Therefore although essential for cell culture studies, the collec-tion of fresh tissue is also greatly preferable for DNA and especially RNA studies. However, the collection of fresh prostatic tissue, especially tumor tissue is far more challenging than tissue collection of most other tumors. A major challenge is to identify tumor at gross examination which is often very diffi cult or impossible in prostate cancer. Thus, morphological verifi cation is absolutely mandatory. It must in no way compromise the diagnostic or prognostic infor-mation than can be extracted from the tissue. There-fore the ability to extract fresh tissue depends upon the surgical procedure undertaken.
TRUS biopsy
18 gauge needle biopsies are too small to identify cancer macroscopically, and inevitably the whole of the core is needed for histopathological processing. Due to the insensitivity of PSA screening and lack of precision of needle biopsies, only about 30 – 40% of TRUS biopsy procedures lead to the identifi cation of cancer [10]. Methods of collecting fresh tissue from TRUS include the collection of extra needle cores for research. However this may pose ethical problems, especially if cancer is identifi ed in the cores, while the specimens taken for routine FFPE processing are negative. It is likely to be useful only in extensive or high grade tumors. Some groups have attempted to use tissue imprints from biopsy cores [11] by transferring the superfi cial cells onto a sur-face. However, this also will tend to identify only high grade tumors.
TURP collection
As few pathology departments process all the tissue removed at TURP, there is usually scope to collect fresh tissue from these operations. However this is usually hyperplastic tissue from the transition zone or non-neoplastic central zone tissue. Although it might be considered an adequate ‘ normal ’ control tissue, it must be remembered that most of the tissue is not from the peripheral zone, where the majority of prostatic cancers arise. Techniques for sampling fresh tissue from TURP specimens include sampling the middle of a TURP chip while processing the outer portions routinely to correlate the morphology with the fresh tissue [12]. Occasionally prostatic can-cers are operated upon using this method, especially in fast growing tumors which are not amenable to radical therapy, in cases of bladder outfl ow obstruc-tion. In these cases, abundant tumor tissue is avail-able. However, it should be remembered that these
tumors are unlikely to be representative of the full spectrum of prostate cancer. They are usually hor-mone pre-treated and occupy an unusual part of the prostate cancer spectrum, being tumors that have spread rapidly locally but have not yet resulted in death by metastatic spread.
Radical prostatectomy specimens
The harvesting of validated prostate tissue from rad-ical prostatectomy specimens, probably represents the optimal method of the accrual of tissue from low risk prostate cancer. However the macroscopic dis-section of the prostate gland is diffi cult and critical attention needs to be paid to margin status and cap-sular invasion. Therefore any dissection should always be conducted by a pathologist according to an agreed protocol.
A number of methods are currently in use to col-lect fresh tissue. Some centres use 18 gauge biopsies on the fresh gland before fi xation in formalin [13]. This has the advantage of less distortion of the gland and minimal disruption of the surgical margin. How-ever disadvantages are that little tissue is sampled and immediate correlation with macroscopic internal appearances of the gland are not possible. Further-more, it is very diffi cult to localize the tumor as the majority of prostate cancers at radical prostatectomy are currently T1c tumors, i.e. non-palpable disease.
Another technique includes immediate section-ing of the gland with the dissection of suspicious areas of cancer from the center of the specimen where there is less danger of the disruption of surgi-cal margins [14,15]. Disadvantages include the sub-sequent potential distortion of the specimen in formalin rendering assessment challenging, and the fact that increasingly prostate cancers are too small to identify macroscopically. A method has been described for harvesting tissue from cut surfaces without compromising surgical margins [16].
Radical cystectomy specimens removed for urothelial carcinomas are also a potential source of prostate tissue. Although small prostate cancers may be found in many cases, they represent a poten-tial source of ‘ normal ’ controls and do not require such rigorous assessment of margin status of the prostate.
Storage of fresh prostate tissue for research
Due to rapid autolysis, tissue should be stabilized as soon as possible after removal from the patient. Com-mon methods include snap freezing of tissue and stor-age at �70 ° C. Cooling the specimen immediately in theater by placing it on ice may be helpful. However for transport this is often cumbersome and the

Prostate pathology data in research 55
development of an RNA stabilization solution RNAlater may used if RNA is to be analysed [15]. A disadvantage of RNAlater is that it impairs the inter-pretation of morphology. Correlation of all tissue with either frozen sections or adjacent FFPE tissue is important if cancer is to be properly validated [17].
When harvesting for cell culture, morphological verifi cation with frozen section or RNAlater fi xed material is not possible as the harvested tissue needs to remain fresh. This may also be the case with tissue intended for proteomic analysis. A morphological validation can then be carried out using cytological sampling technique [18].
Ethical issues
The ethical issues involved in the collection and use of fresh tissue will vary greatly between countries [19 – 21]. However they are underpinned by an ethos that no patient should suffer harm from the research, and that their confi dentiality must be ensured. Ethi-cal approval in the country of origin should always be obtained, and depending on the procedure and country, consent by the patient is required. Harm is conceivable if the patient undergoes unnecessary procedures: for instance research orientated TRUS biopsies with no diagnostic use or intent. The patient may undergo indirect harm if research manipulation of tissue renders a specimen diffi cult to interpret for margin status.
Conclusions
Harvesting the prostate, especially cancer tissue, for research creates problems not encountered in other organs and cancers, mostly secondary to the unique methods by which prostate cancer is diagnosed and sampled. However by using the variety of techniques suggested above and with cooperation of local pathol-ogists it is possible to collect both frozen and FFPE tissue banks which can be used for future research programs.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Ananthanarayanan V, Pins MR, Meyer RE, Gann PH. Immu-[1] nohistochemical assays in prostatic biopsies processed in Bouin’s fi xative. J Clin Pathol 2005;58:322 – 4. Jacobs TW, Prioleau JE, Stillman IE, Schnitt SJ. Loss of [2] tumor marker-immunostaining intensity on stored paraffi n slides of breast cancer. J Natl Cancer Inst 1996;88:1054 – 9.
DiVito KA, Charette LA, Rimm DL, Camp RL. Long-term [3] preservation of antigenicity on tissue microarrays. Lab Invest 2004;84:1071 – 8. Battifora H. The multitumor (sausage) tissue block: Novel [4] method for immunohistochemical antibody testing. Lab Invest 1986;55:244 – 8. Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml [5] P, Leighton S, et al. Tissue microarrays for high-throughput molecular profi ling of tumor specimens. Nat Med 1998;4:844 – 7. Rubin MA, Dunn R, Strawderman M, Pienta KJ. Tissue [6] microarray sampling strategy for prostate cancer biomarker analysis. Am J Surg Pathol 2002;26:312 – 9. Jhavar S, Bartlett J, Kovacs G, Corbishley C, Dearnaley D, [7] Eeles R, et al. Biopsy tissue microarray study of Ki-67 expres-sion in untreated, localized prostate cancer managed by active surveillance. Prostate Cancer Prostatic Dis 2009;12:143 – 7. Haselkorn R, Doty PJ. The reaction of formaldehyde with [8] polynucleotides. J Biol Chem. 1961;236:2738 – 45. Ben-Ezra J, Johnson DA, Rossi J, Cook N, Wu A. Effect of [9] fi xation on the amplifi cation of nucleic acids from paraffi n-embedded material by the polymerase chain reaction. J His-tochem Cytochem 1991;39:351 – 4. Roddam AW, Hamdy FC, Allen NE, Price CP. The impact [10] of reducing the prostate-specifi c antigen threshold and including isoform refl ex tests on the performance character-istics of a prostate-cancer detection programme. BJU Int 2007;100:514 – 7. Gaston SM, Soares MA, Siddiqui MM, Vu D, Lee JM, [11] Goldner DL, et al. Tissue-print and print-phoresis as plat-form technologies for the molecular analysis of human surgical specimens: Mapping tumor invasion of the prostate capsule. Nat Med 2005;11:95 – 101. Riddick AC, Barker C, Sheriffs I, Bass R, Ellis V, Sethia KK, [12] et al. Banking of fresh-frozen prostate tissue: Methods, vali-dation and use. BJU Int 2003;91:315 – 23; discussion 23 – 4. Walton TJ, McCulloch TA, Rees RC, Bishop MC. Obtaining [13] fresh prostate cancer tissue for research: A novel biopsy needle and sampling technique for radical prostatectomy specimens. Prostate 2005;64:382 – 6. Wheeler TM, Lebovitz RM. Fresh tissue harvest for research [14] from prostatectomy specimens. Prostate 1994;25:274 – 9. Furman J, Murphy WM, Rice L, Drew PA, Narayan P. Pros-[15] tatectomy tissue for research: Balancing patient care and discovery. Am J Clin Pathol 1998;110:4 – 9. Egevad L, Engstrom K, Busch C. A new method for handling [16] radical prostatectomies enabling fresh tissue harvesting, whole mount sections and landmarks for alignment of sections. J Urol Pathol 1998;9:17 – 28. Grotzer MA, Patti R, Geoerger B, Eggert A, Chou TT, [17] Phillips PC. Biological stability of RNA isolated from RNAlater-treated brain tumor and neuroblastoma xenografts. Med Pediatr Oncol 2000;34:438 – 42. Lexander H, Hellman U, Palmberg C, Auer G, Hellstrom M, [18] Franzen B, et al. Evaluation of two sample preparation methods for prostate proteome analysis. Proteomics 2006;6:3918 – 25. European Group on Ethics in Science and New Technolo-[19] gies; human tissue banks; human embryo research. Hum Reprod Genet Ethics 1999;5:1. Ashcroft R. The ethics of reusing archived tissue for research. [20] Neuropathol Appl Neurobiol 2000;26:408 – 11. Clark BJ. Tissue banking in a regulated environment—does [21] this help the patient? Part 1--Legislation, regulation and eth-ics in the UK. Pathobiology 2007;74:218 – 22.

Correspondence: Rodolfo Montironi, Pathological Anatomy, Polytechnic University of the Marche Region, School of Medicine, United Hospitals, Via Conca 71, I � 60126 Torrette, Ancona, Italy. E-mail: [email protected]
(Received 3 August 2010 ; accepted 25 August 2010 )
REVIEW ARTICLE
Role of histopathology and molecular markers in the active surveillance of prostate cancer
RODOLFO MONTIRONI 1 , LARS EGEVAD 2 , ANDERS BJARTELL 3 & DANIEL M. BERNEY 4
1 Institute of Pathological Anatomy and Histopathology, School of Medicine, Polytechnic University of the Marche Region, Ancona, Italy, 2 Department of Pathology, Karolinska University Hospital, Stockholm, Sweden, 3 Department of Urology, Skåne University Hospital, Malmö, Sweden and 4 Department of Molecular Oncology and Imaging, Institute of Cancer, Queen Mary University of London, London, UK
Abstract Surgery or radiation therapy remain the standard curative treatments for newly diagnosed prostate cancer patients. Nonetheless, these aggressive treatments are associated with decreased quality of life with altered sexual and urinary func-tions. The objective was a systematic review of active surveillance protocols to investigate the role of histopathology and molecular markers in the active surveillance of prostate cancer. Medline was searched using the following terms: prostate cancer, active surveillance and expectant management. Selection criteria, follow-up strategies and outcomes . Using modern risk stratifi cation, several centres have gained signifi cant experience in identifying patients with a low risk of prostate cancer progression and have adopted an active surveillance program with delayed curative therapy. Interestingly, only limited numbers of patients under active surveillance require additional treatment. Recent data suggest that delayed treatment does not appear to alter the clinical outcome among those highly selected patients. The future and conclusions . A better under-standing of the molecular determinants of prostate cancer behaviour would not only enable healthcare professionals to identify which cases need aggressive treatment but, perhaps more importantly, would also indicate potential targets for the development of novel therapeutic strategies.
The aim of active surveillance (AS) of early prostate cancer is to individualise therapy by selecting for curative therapy only patients with signifi cant can-cers. Patients with favourable tumour characteris-tics in terms of clinical T stage, Gleason score and serum prostate specifi c antigen (PSA) testing are closely monitored using serum PSA kinetics and repeat prostate biopsies. The choice between con-tinued observation and radical treatment is based on evidence of disease progression, with progres-sion defi ned in terms of ‘ upgrading ’ at repeat biopsy and PSA doubling time (PSADT). The aim is to identify cases for treatment long before any symp-toms or overt clinical signs of tumour progression are evident [1].
AS should be distinguished from ‘ watchful wait-ing ’ . The latter involves relatively lax observation with late, palliative treatment for those who develop symptoms of progressive disease, whereas AS involves
close monitoring with early, radical treatment in those with signs of progression [1 – 3].
Selection criteria
A critical factor for successful AS is the best possible selection of patients with prostate cancer with low risk of progression (Table I). Patients with an identifi able low risk of progression are most likely to be safely observed and treated only when necessary. Epstein et al. introduced prostate biopsy criteria to predict insignifi cant prostate cancer (PCa) in the radical prostatectomy (RP) specimens [4]. In addition to the original Epstein criteria, multiple selection or entry criteria, based on preference or on experience and not always obtained on hard data, have been pub-lished [1,4 – 9] (Table II). The most common clinical data used to defi ne low-risk prostate cancer include a Gleason score � 6 (no pattern 4 or 5 disease), PSA
Acta Oncologica, 2011; 50(Suppl 1): 56–60
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.522199

Active surveillance 57
Table I. Prostate cancer aggressiveness risk strata and related care options.
Early Stage Prostate Cancer Aggressiveness Category
Measures of prostate cancer severity
Prostate Cancer Care Options Biopsy Gleason Score Clinical Stage Serum PSA (ng/ml)
Low risk � 6 T1 or T2a � 10 Active surveillance, prostatectomy, brachytherapy, or external radiotherapy
Intermediate risk � 6 T1 or T2a or b 10 – 20 Prostatectomy, external radiotherapy with adjuvant androgen suppressive therapy, or brachytherapy
7 T1 or T2a or b � 20
High risk � 7 T1 or T2a, b, or c � 20 External radiotherapy with adjuvant androgen suppressive therapy or prostatectomy
8 – 10 T1 or T2a, b, or c Any PSA
Table II. Entry criteria for active surveillance (authors in alphabetic order).
Source Entry criteria
Dall ’ Era et al. [1] (Most common clinical criteria)
Gleason score 6 No Gleason pattern 4 or 5 PSA level � 10 ng/ml and stable PSA kinetics � 50% single core involvement � 33% positive cores
D ’ Amico et al. [5] PSA level � 10 ng/ml No Gleason pattern 4 or 5 Clinical stage T2a or lower
Epstein et al. [4] Clinical stage T1c PSA density � 0.15 ng/ml/cm3 No Gleason pattern 4 or 5 � 3 positive cores � 50% cancer per core
Patel et al. [6] Clinical stage T3 or lower Gleason sum � 7
PRIAS ∗ (Van den Bergh et al.) [9]
Clinical stage T1c – T2b No Gleason pattern 4 or 5 PSA density � 0.20 ng/ml/cm3 PSA level � 10 ng/ml Fewer than three positive cores
Soloway et al. [7] Clinical stage T2 or lower PSA level � 15 ng/ml No Gleason pattern 4 or 5 � 50% cancer per two positive cores
Van As et al. [8] Clinical stage T1 – T2a Gleason sum � 7 (3 � 4) PSA level � 15 ng/ml � 50% of biopsy cores positive
∗ PRIAS, Prostate Cancer Research International Active Surveillance.
level � 10 ng/ml, and clinical stage T1 to T2a disease. Other characteristics which have been used include PSA kinetics (stable) before diagnosis, PSA density (PSAD) � 0.15 ng/ml/cm3, percent positive cores at biopsy � 33%, and the extent of cancer in any core � 50% [1]. The percentage of core involvement and percentage positivity of the biopsies are both depen-dent on the length and number of cores respectively. Measurements of cancer length may be more helpful. Prospective studies comparing entry criteria for AS protocols with subsequent disease progression and
treatment patterns are needed to clarify the best candidates for AS.
Issues on the role of prostate biopsies in patient selection
Some authors have shown that a non-negligible pro-portion of patients who meet the entry criteria for AS actually harbour aggressive or locally advanced disease if they are submitted to RP [10]. Ploussard et al. pro-vided a detailed analysis evaluating, for the fi rst time, the misclassifi cation rate in patients who could be suit-able for an AS program according to different biopsy schemes [10]. All of their patients were submitted to a 21-core fi rst biopsy mapped by location. The authors found that patients who could have been selected for AS programs based on a 12-core biopsy scheme showed higher rates of unfavourable prostate cancer character-istics at RP compared to patients who would have been included only in a 21-core biopsy scheme (overall unfa-vourable prostate cancer: 28.6 – 35.9% vs . 14.0 – 17.6%, respectively). Interestingly, among patients without cancer evidence in the 12-core scheme but with cancer diagnosed only at the 21-core biopsy, roughly 16% showed unfavourable disease at RP, defi ned as Gleason score � 8 or category pT3 or higher. These data showed that a certain proportion of patients initially submitted to AS actually harbour aggressive disease at the time of diagnosis. The data by Ploussard et al. [10] could help reduce the misclassifi cation risk by introducing high density biopsy strategies in the initial management of patients submitted to AS.
Follow-up strategies to detect prostate cancer progression
Even though different AS follow-up strategies have been adopted (Table III) [6 – 8,11 – 15], the criteria are somewhat similar. Besides a regular repeat biopsy, regular PSA level testing, digital rectal examination (DRE) and optional transrectal ultrasound studies are warranted [15].

58 R. Montironi et al.
Table III. Predicting progression during active surveillance (authors in alphabetic order).
Source PSA DRE TRUS Rebiopsy
Carter et al. [11,12] Every 6 months Every 6 months No mention YearlyDall ’ Era et al. [13] Every 3 months Every 3 months 6 – 12-month interval Every 12 – 24 monthsHardie et al. [14] Every 3 – 6 months for 2 years,
then every 6 months if PSA is stable
Every 3 – 6 months for 2 years, then every 6 months
Not routine Not routine
Klotz et al. [15] Every 3 months for 2 years, then every 6 months if PSA level is stable
Every 3 months for 2 years, then every 6 months if PSA level is stable
Optional At 12 – 18 months
Patel et al. [6] Every 3 months for 1 year, then every 6 months
Every 3 months for 1 year, then every 6 months
At 6 months At 6 months
Soloway et al. [7] Every 3 months for 2 years Every 3 months No mention At 6 – 12 months, afterwards when indicated
Van As et al. [8] Year 1: monthly Year 2: every 3 months Afterwards: every 6 months
Every 3 months for 2 years, then every 6 months
No mention At 18 – 24 months, then biannually
DRE, digital rectal examination; TRUS, transrectal ultrasound; PSA, prostate-specifi c antigen.
The detection of prostate cancer progression in a patient selected for AS remains a continuing chal-lenge. What will serve as the best parameter to cor-rectly identify patients that progress to more aggressive cancer in order not to miss the window of curability is still a matter of debate. At present, the choice between radical treatment and continued observa-tion is based on evidence of disease progression, with progression defi ned in terms of ‘ upgrading ’ at repeat biopsy and PSADT.
Prostate cancer ‘ upgrading ’ at repeat biopsy is a major criterion for active treatment [6 – 8,11 – 17]. The study by van As et al. used PSA kinetics profi les, progression of Gleason grade, and increased percent-age of cancer per core as indicators to stop AS in patients with low-risk prostate cancer [8]. Interest-ingly, in the cohort of Klotz et al., only 4% of patients were treated because of progression of Gleason grade alone [16]. The greatest trigger for intervention in the Toronto cohort remained the PSADT, with 21% of the cohort having a PSADT � 3 years [17].
Outcomes
Multiple studies have reported their experience with AS, but the value of most studies is limited by a relatively short follow-up time (Table IV) [6 – 8,11 – 15,17 – 19]. However, recent data suggest that delayed treatment does not appear to alter the clini-cal outcome among those highly selected patients. In a recent study by van As et al. it was found that 20% of patients received delayed radical treatment after a median follow-up of 22 months. Within this time frame no patient developed metastatic disease or died of prostate cancer [8]. Hardie et al. reported similar fi ndings at a median follow-up of 42 months [14]. Approximately 91% of the patients had a Gleason
score � 6 and 73% a PSA level � 10 ng/ml. All patients revealed organ-confi ned disease in the RP specimen.
The future
AS provides an ideal opportunity for healthcare pro-fessionals to improve their understanding of the basis for the extraordinary variation in prostate cancer behaviour. A better understanding of the molecular markers of prostate cancer behaviour would not only enable healthcare professional to identify which cases need treatment but, perhaps more importantly, would also indicate potential targets for the development of novel therapeutic strategies.
Molecular markers
Multiple susceptibility genes and many additional mechanisms involved in carcinogenesis and cancer progression have been discovered [20 – 23]. However, no single biomarker capable of improving the com-mon clinical parameters included in the currently used predicting models has yet been identifi ed in any prospective active surveillance series.
The Prostate CAncer gene 3 (PCA3) assay is a novel tool that might aid in the diagnosis of prostate cancer and that might indicate the signifi cance of the disease [24,25]. The PCA3 urinary assay might be used to guide biopsy decisions in: (i) men with an elevated serum total prostate specifi c antigen (tPSA) level and one or more previous negative biopsies; (ii) men with a normal tPSA level and a family history of prostate cancer; (iii) men with an elevated tPSA level (2.5 – 10 ng/mL) and no previous biopsy; (iv) men with an elevated tPSA level and a concomitant urinary condition. In addition, in men diagnosed

Active surveillance 59
Table IV. Treatment criteria (authors in alphabetic order).
Source Treatment criteria Median follow-up,
months Percentage of patients
with treatment
Carter et al. [11,12] Gleason score � 7on rebiopsy, any pattern 4/5, � 2 cores involved, � 50% any single core involved
23 31
Dall ’ Era et al. [13] Gleason score � 7 on rebiopsy, rising PSA, increase in volume by biopsy parameters
24 21
Ercole et al. [18] Increase in tumor volume, Gleason score progression, urinary symptoms, change of DRE, patient preference
48 7.8
Hardie et al. [14] Rising PSA, clinical judgment 42 14Klotz et al. [15] PSADT � 2 years Gleason score
� 8 Update 2001: PSADT � 3 years Gleason score � 7 (4 � 3)
64 34
Patel et al. [6] Gleason score increase, PSAV � 0.75 ng/ml per year, increase DRE/TRUS detected lesion, increase biopsy volume
44 35
Roemeling et al. [19] PSADT 40 29Soloway et al. [7] Gleason score increase, PSA and
PSADT increase, stage progression, increase biopsy volume, patient preference
45.3 (mean) � 1
Van As et al. [8] PSAV � 1 ng/ml per year Gleason score � 4 � 3 or � 50% cancer per core
22 20
PSA, prostate-specifi c antigen; PSADT, PSA doubling time; PSAV, PSA velocity; DRE, digital rectal examination; TRUS, transrectal ultrasound.
with prostate cancer, the PCA3 assay could aid in the decision of whether active therapy is needed or active surveillance is appropriate. In a study by Tosoian et al. in patients with low risk prostate cancer who were carefully selected for active surveil-lance PCA3 score was not signifi cantly associated with progressive disease in the short term [26]. While there was a trend toward higher PCA3 scores in patients with high grade disease on surveillance biopsy, signifi cant overlap between the groups prevented the identifi cation of a threshold PCA3 score for clinical use. Therefore, the true value of the test in the setting of AS remains unclear at this point.
Conclusions
AS is a new strategy that aims to individualise therapy by selecting only those patients with signifi cant can-cers for curative therapy. Patients with favourable tumour characteristics are closely monitored using serum PSA concentrations and repeat prostate biop-sies [27]. The choice between radical treatment and continued observation is based on evidence of disease progression, defi ned in terms of the PSADT and ‘ upgrading ’ at repeat biopsy. AS provides an excellent
opportunity for studies to identify molecular markers of prostate cancer behaviour and of assessment therapeutic agents.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Dall ’ Era MA, Cooperberg MR, Chan JM, Davies BJ, Albert-[1] sen PC, Klotz LH, et al. Active surveillance for early-stage prostate cancer: Review of the current literature. Cancer 2008;112:1650 – 9. Johansson JE, Adami HO, Andersson SO, Bergstr ö m R, [2] Krusemo UB, Kraaz W. Natural history of localised prostatic cancer. A population-based study in 223 untreated patients. Lancet 1989;1(8642):799 – 803. Bill-Axelson A, Holmberg L, Ruutu M, H ä ggman M, Andersson [3] SO, Bratell S. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med 2005;352:1977 – 84. Epstein JI, Walsh PC, Carmichael M, Brendler CB. Patho-[4] logic and clinical fi ndings to predict tumor extent of nonpal-pable (stage T1c) prostate cancer. JAMA 1994;271:368 – 74. D ’ Amico AV, Cote K, Loffredo M, Renshaw AA, Schultz D. [5] Determinants of prostate cancer-specifi c survival after radia-tion therapy for patients with clinically localized prostate cancer. J Clin Oncol 2002;20:4567 – 73.

60 R. Montironi et al.
Patel MI, DeConcini DT, Lopez-Corona E, Ohori M, [6] Wheeler T, Scardino PT. An analysis of men with clinically localized prostate cancer who deferred defi nitive therapy. J Urol 2004;171:1520 – 4. Soloway MS, Soloway CT, Williams S, Ayyathurai R, [7] Kava B, Manoharan M. Active surveillance; a reasonable management alternative for patients with prostate cancer: The Miami experience. BJU Int 2008;101:165 – 9. Van As NJ, Norman AR, Thomas K, Khoo VS, Thompson A, [8] Huddart RA, et al. Predicting the probability of deferred radical treatment for localised prostate cancer managed by active surveillance. Eur Urol 2008;54:1297 – 305. Van den Bergh RCN, Roemeling S, Roobol MJ, Roobol W, [9] Schroder FH, Bangma CH. Prospective validation of active surveillance in prostate cancer: The PRIAS study. Eur Urol 2007;52:1560 – 3. Ploussard G, Xylinas E, Salomon L, Allory Y, Vordos D, [10] Hoznek A, et al. The role of biopsy core number in selecting prostate cancer patients for active surveillance. Eur Urol 2009;56:891 – 8. Carter HB, Walsh PC, Landis P, Epstein JI. Expectant man-[11] agement of nonpalpable prostate cancer with curative intent: Preliminary results. J Urol 2002;167:1231 – 4. Carter HB, Kettermann A, Warlick C, Metter EJ, Landis P, [12] Walsh PC, et al. Expectant management of prostate cancer with curative intent: An update of the Johns Hopkins experi-ence. J Urol 2007;178:2359 – 64. Dall ’ Era MA, Konety BR, Cowan JE, Shinohara K, Stauf F, [13] Cooperberg MR, et al. Active surveillance for the manage-ment of prostate cancer in a contemporary cohort. Cancer 2008;112:2664 – 70. Hardie C, Parker C, Norman A, Eeles R, Horwich A, Hud-[14] dart R, et al. Early outcomes of active surveillance for local-ized prostate cancer. BJU Int 2005;95:956 – 60. Klotz L. Active surveillance for prostate cancer: For whom? [15] J Clin Oncol 2005;23:8165 – 9. Klotz L. Active surveillance with selective delayed interven-[16] tion is the way to manage “ good-risk ” prostate cancer. Nat Clin Pract Urol 2005;2:136 – 42. Klotz L. Active surveillance with selective delayed inter-[17] vention for favorable risk prostate cancer. Urol Oncol 2006;24:46 – 50.
Ercole B, Marietti SR, Fine J, Albertsen PC. Outcomes [18] following active surveillance of men with localized prostate cancer diagnosed in the prostate specifi c antigen era. J Urol 2008;180:1336 – 9. Roemeling S, Roobol MJ, de Vries SH, Wolters T, [19] Gosselaar C, van Leenders GJ, et al. Active surveillance for prostate cancers detected in three subsequent rounds of a screening trial: Characteristics, PSA doubling times, and out-come. Eur Urol 2007;51:1244 – 51. Feinberg AP, Vogelstein B. Hypomethylation distinguishes [20] genes of some human cancers from their normal counter-parts. Nature 1983;301:89 – 92. Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, [21] Bova GS, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci USA 1994;91:11733 – 7. Bastian PJ, Palapattu GS, Lin X, Yegnasubramanian S, Man-[22] gold LA, Trock B, et al. Preoperative serum DNA GSTP1 CpG island hypermethylation and the risk of early prostate-specifi c antigen recurrence following radical prostatectomy. Clin Cancer Res 2005;11:4037 – 43. Demichelis F, Fall K, Perner S, Andr é n O, Schmidt F, [23] Setlur SR, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007;26:4596 – 9. Haese A, de la Taille A, van Poppel H, Marberger M, [24] Stenzl A, Mulders PF, et al. Clinical utility of the PCA3 urine assay in European men scheduled for repeat biopsy. Eur Urol 2008;54:1081 – 8. Nakanishi H, Groskopf J, Fritsche HA, Bhadkamkar V, Blase [25] A, Kumar SV, et al. PCA3 molecular urine assay correlates with prostate cancer tumor volume: Implication in selecting candidates for active surveillance. J Urol 2008;179:1804 – 9. Tosoian JJ, Loeb S, Kettermann A, Landis P, Elliot DJ, [26] Epstein JI, et al. Accuracy of PCA3 measurement in predict-ing short-term biopsy progression in an active surveillance program. J Urol 2010;183:534 – 8. Bastian PJ, Carter BH, Bjartell A, Seitz M, Stanislaus P, [27] Montorsi F, et al. Insignifi cant prostate cancer and active surveillance: From defi nition to clinical implications. Eur Urol 2009;55:1321 – 30.

Correspondence: Anders Bjartell, Department of Urology, Sk å ne University Hospital, SE 205 02 Malm ö , Sweden. Tel: � 46 40 332685 (direct), 336398 (secretary). Fax: � 46 40 337049. E-mail: [email protected]
(Received 25 August 2010 ; accepted 18 November 2010 )
REVIEW ARTICLE
Tumor markers in prostate cancer I: Blood-based markers
SHAHROKH F. SHARIAT 1 , AXEL SEMJONOW 2 , HANS LILJA 3 , CAROLINE SAVAGE 4 , ANDREW J. VICKERS 4 & ANDERS BJARTELL 5
1 Department of Urology and Division of Medical Oncology, Weill Cornell Medical College, New York-Presbyterian Hospital, New York, NY, USA, 2 Department of Urology, Prostate Center, University Hospital Muenster, Muenster, Germany, 3 Department of Clinical Laboratories, Surgery (Urology Services), and Medicine (Genito-Urinary Oncology Service), Memorial Sloan-Kettering Cancer Center New York, NY, USA, 4 Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, New York, USA and 5 Department of Urology Malm ö -Lund, Sk å ne University Hospital, Lund University, Sweden
Abstract The introduction of total prostate specifi c antigen (total PSA) testing in blood has revolutionized the detection and management of men with prostate cancer (PCa). The objective of this review was to discuss the challenges of PCa biomar-ker research, defi nition of the type of PCa biomarkers, the statistical considerations for biomarker discovery and validation, and to review the literature regarding total PSA velocity and novel blood-based biomarkers. Methods . An English-language literature review of the Medline database (1990 to August 2010) of published data on blood-based biomarkers and PCa was undertaken. Results . The inherent biological variability of total PSA levels affects the interpretation of any single result. Men who will eventually develop PCa have increased total PSA levels years or decades before the cancer is diagnosed. Total PSA velocity improves predictiveness of total PSA only marginally, limiting its value for PCa screening and prognostication. The combination of PSA molecular forms and other biomarkers improve PCa detection substantially. Several novel blood-based biomarkers such as human glandular kallikrein 2 (hK2), urokinase plasminogen activator (uPA) and its receptor (uPAR), transforming growth factor-beta 1 (TGF- β 1); interleukin-6 (IL-6) and its receptor (IL-6R) may help PCa diag-nosis, staging, prognostication, and monitoring. Panels of biomarkers that capture the biologic potential of PCa are in the process of being validated for PCa prognostication. Conclusions . PSA is a strong prognostic marker for long-term risk of clinically relevant cancer. However, there is a need for novel biomarkers that aid clinical decision making about biopsy and initial treatment. There is no doubt that progress will continue based on the integrated collaboration of researchers, clinicians and biomedical fi rms.
In Western societies, prostate cancer (PCa) is the most common solid malignancy and the second lead-ing cause of cancer death in men [1]. The wide avail-ability of total prostate-specifi c antigen (PSA, formal name human kallikrein 3, hK3) revolutionized PCa screening and ushered in the PSA era resulting in a decrease of PCa metastasis and death. However, the ubiquitous application of PSA screening has also led to over-detection and overtreatment. The lifetime risk of PCa diagnosis is estimated at ∼ 18%, whereas that for death from PCa is ∼ 3%. In addition, PSA is neither cancer specifi c nor a surrogate for the bio-logic behavior of PCa. An elevated PSA level can refl ect the presence of cancer but can also be caused by benign prostatic hyperplasia (BPH), infection,
and/or chronic infl ammation. Virtually all prostate epithelial cells, whether normal, hyperplastic or can-cerous, synthesize PSA. Therefore, there has been a concerted effort to discover and validate novel PCa biomarkers. In this review article, we fi rst discuss the challenge of PCa biomarker research, types of PCa biomarkers, and the statistical considerations for bio-marker discovery and validation. Then, we discuss the limitations of measuring total PSA and its deriv-atives such as total PSA velocity (total PSAV) and different molecular forms (i.e. free PSA, BPSA, pro-PSA, and intact PSA). Moreover, we briefl y dis-cuss several promising novel blood-based biomarkers for PCa diagnosis, staging, prognostication, and monitoring, i.e. human glandular kallikrein 2 (hK2),
Acta Oncologica, 2011; 50(Suppl 1): 61–75
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.542174

62 S. F. Shariat et al.
urokinase plasminogen activator (uPA) and its recep-tor (uPAR), transforming growth factor-beta 1 (TGF- β 1); interleukin-6 (IL-6) and its receptor (IL-6R).
Biomarker challenge in prostate cancer
A PubMed Search on “ prostate cancer ” AND ( “ biomarker ” OR “ marker ” ) in English language yielded 3016 hits (accessed 8/8/2010). The number of articles published on PCa biomarkers has increased steadily over the years. Despite this plethora of bio-markers reported to be “ promising ” , only one bio-marker, (i.e. total PSA in blood) is routinely used by urologists. Why are PCa biomarkers not living up to their promise? For one, there are remarkable analytical and regulatory barriers to the application of biomark-ers in PCa care [2,3]. These include but are not limited to the status of intellectual property protection, availability of standard reference materials for the assay, complexity of assay format, implementation of quality control to assure reproducibility and accuracy, suffi cient market testing size to assess methods of commercialization, lack of clear guidelines for good manufacturing/laboratory practice and quality control requirements for all phases of biomarker development, cost and effort required to accumulate clinical data under appropriately designed and Institutional Review Board-approved prospective trials, and the interval required for resolution of patent issues, assay standard-ization, validation, testing, and regulatory approval.
Besides analytical and regulatory barriers, the lack of PCa biomarker use in daily clinical practice is also a result of poor application of statistics and study design. There has been a lack of effective and effi cient strategies to determine which biomarker candidates justify the great investment of time and money required for assay development, optimization and demonstra-tion of analytical robustness. There are now guidelines intended to ensure that biomarker studies conform to some basic standards of design and reporting [2,3]. For a new biomarker to be clinically useful, it has to answer a clinically relevant question and provide infor-mation that is not available in a more simple and cost-effective way. Any new biomarker needs to provide a benefi t over these standard criteria or at least improve their accuracy. Before a biomarker assay can be imple-mented in the community setting, it needs to address four concepts: “ better, easier, faster and cheaper ” [2].
Conceptually, the development of new biomarkers should be a process that is similar to therapeutic drug evaluation. In 2002, the National Cancer Institute ’ s Early Detection Research Network developed a fi ve phase approach to systematic discovery and validation of biomarkers (Figure 1) [2 – 6]. This schema is not only an intellectual process but also provides a clear scale by which researchers, patients, and investors can evaluate
the status of the biomarker in the development process. The expected failure rate of biomarkers in development can be expected to be similar to the one of drugs. A large concerted effort is required to advance the fi eld of PCa biomarker through systematic discovery, veri fi cation, and validation – each step coupled with adequate statistical analysis.
Refi ning the defi nition of prostate cancer biomarker
According to the National Institute of Health (NIH) in the USA, a biomarker is a characteristic that is objec-tively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharma-ceutical responses to a therapeutic intervention [7]. Cancer biomarkers are either produced by the tumor or by the body in response to the tumor. Six different types of biomarker can be differentiated in PCa:
1) Detection/screening: this biomarker is used for evaluating patients with either risk fac-tors for or symptoms of PCa.
2) Diagnostic: this biomarker can help classical histopathological characteristics in assessing presence or absence of cancer.
3) Prognostic: this biomarker is used to predict the outcome of patients based on different risk of recurrence or progression thereby allowing individualized management.
4) Predictive: this biomarker is used to predict whether the treatment (drug or other ther-apy) will be effective, and/or to monitor the effectiveness of the treatment. It can help identify the best treatment modality.
5) Therapeutic target: this biomarker can help identify the patients who will benefi t from a particular treatment regimen. It identifi es the molecular targets of novel therapies and is affected by therapy. As of now there is no such blood-based biomarker in clinical use for prostate PCa.
6) Surrogate endpoint: this biomarker is used to substitute for a clinical endpoint and/or to meas-ure clinical benefi t, harm or lack of benefi t or harm. Surrogates could replace traditional end-points, such as mortality due to disease or the recurrence or relapse of disease. Biomarkers can reduce time factors and costs for Phase I and II clinical trials by replacing clinical endpoints.
For the purposes of the current review, we will focus on blood-based biomarkers that are the end result of a bioassay, for processing biological material from humans, expressed quantitatively or categorically. Tissue-based and urine-based PCa biomarkers are discussed in separate reports.

Blood-based biomarkers for prostate cancer 63
Statistical considerations for biomarker discovery and validation
An issue that has received less attention is the degree to which research on biomarkers has made suffi cient use of clinically relevant statistics, such as the assess-ment of predictive accuracy, decision analysis, and/or experimental methodology. Most biomarkers do not provide suffi cient information to be used inde-pendent of other information. The optimal use of biomarkers lies in incorporating it in a model that also includes standard clinical data [3,8 – 12]. To determine the value of a new biomarker, it is not suffi cient to show that it is signifi cantly related to the outcome, statistically signifi cant in a multivariable model including the standard clinical and pathologic factors, or more signifi cant than the standard clinical and pathologic factors. A variable that is statistically signifi cant in a multivariable model might not improve the model ’ s predictive accuracy. P-value and odds/hazard ratio do not meaningfully describe a biomark-ers ’ ability to classify patients. For a biomarker to be potentially clinically useful, it is necessary to show that adding the biomarker to an existing model based on the most important clinical and pathologic factors improves the predictive accuracy (discrimination and calibration) of the model [3,11,13 – 16].
One major issue with model development is the need for appropriate validation. There are two general types of validation: internal validation on original dataset and external validation on indepen-dent dataset [2,3]. External validation on a different data set allows for evaluation of the generalizability of the risk prediction tool to wider populations than originally reported. Finally, methods that
incor porate clinical consequences such as decision curve analysis are crucial to the evaluation of biomar kers. Several methods are available including decision curve analysis which combines simplicity with effi cient computations [17 – 20].
Total PSA and its limitations
Neoplastic cells produce somewhat lower and varying tissue levels of PSA compared to benign epithelial cells although both conditions cause total PSA ele-vation in the blood [3]. Therefore, it has been sug-gested that total PSA should be considered as a marker of BPH-related prostate volume, growth, and outcome rather than a reliable marker of PCa [3]. Moreover, some aggressive PCa ’ s do not produce PSA.
PSA levels are inherently variable thereby affect-ing the interpretation of any single result [21]. Varia-tion in total PSA includes both analytical (i.e. pre-analytical sample handling, laboratory process-ing, assay per formance, and standardization) and biological variation (i.e. metabolism, renal elimina-tion, medication, physical and sexual activity, size and integrity of the prostate). Oscillations up to 20 – 30% in the total PSA range 0.1 – 20 ng/ml may be due to biologic variation [22,23]. Furthermore, the use of different detection assays may be another important cause of variation. Differences in assay standardization can give an artifactually high or low estimate of total PSA and total PSAV [24 – 26]. Assays are not interchangeable and caution should be exer-cised when comparing results from different com-mercial total PSA assays. Patients and physicians should be aware of which assay was used each time
Phase Goals/aims Experimentation Sample details
PreclinicalTesting
Exploratory; nominateand rank candidatebiomarker profiles
Preclinical studyfor hypothesis generation
Possible bias: small size andconvenience sampling
0 Develop an assay with clinicallyreproducible results
Reproducibility and robustness ofassay; No assessment of benefit
I Test on small sample todetermine benefit
Marker optimization, establishprediction rules, determine cut-offs
Sample population assaydeveloped from candidatebiomarker profile
II Determine operatingcharacteristics & internalvalidation
Retrospective design be the targetpopulation
Sample population should bethe target population
III External validation Retrospective or prospective,Generalizability, Impact onclinical decision-making
Multi-institutional, largestudy
IV Assess whether biomarkerreduces the burden of disease
Post-approval reporting andtesting for other disease processesor disease stages
Figure 1. Modifi cation of the structured phase-approach to the systematic discovery, evaluation, and validation of biomarkers [2,3].

64 S. F. Shariat et al.
a total PSA measurement is performed, and an effort should be made to use the same assay at the next visit. In addition, studies of total PSA kinetics over time using different assays should be interpreted with caution. Of note, there is an expected 20% lower value when the World Health Organization standard is adopted and laboratories should be obliged to mention the name of the PSA assay used on the lab report as well as stating the assay specifi c reference range and the type of master-calibration (i.e. WHO standardized or traditional calibration) [27 – 29].
The effect of previous BPH treatment on total PSA remains mostly unpredictable. For example, the effect of commonly used 5- α -reductase inhibitors on the pre-dictive value of total PSA kinetics for tumor progres-sion is uncertain. Because 5- α -reductase inhibitors are known to decrease the PSA level with ∼ 50% and mostly suppress the benign components of PSA secre-tion, they may enhance the utility of total PSA [30]. In addition, by shrinking the prostate gland, fi nasteride has been suggested to increase the likelihood of detect-ing a small cancer on needle biopsy [30].
This large normal variability of total PSA requires larger changes between two consecutive measure-ments to distinguish pathological changes from changes resulting from analytical and biological vari-ations. Nixon et al. calculated the coeffi cient of vari-ation over two weeks and demonstrated that a change between two total PSA measurements of approxi-mately 25% indicated a signifi cant change [31,32]. Bunting et al. reported a critical difference, defi ned as the minimum percent change between two con-secutive measurements that suggests a signifi cant change beyond the normal variation, close to 60% over a time period of one year [33]. Bruun et al. recently assessed the long-term variability of the dif-ferent forms of PSA at several different total PSA levels in a randomly selected population of asymp-tomatic and apparently healthy men whose total PSA levels were � 2.0 ng/mL at the end of the 8-year observation period [34]. They found that the total intra-individual variation of total PSA was much less than that reported by Bunting et al. [33] and some-what higher than the intra-individual variation for either free PSA or percent free PSA. This suggests that free PSA concentration in blood may vary less than complexed PSA concentration, which is the major contributor to total PSA. One explanation is that free PSA and complexed PSA may have different elimination pathways, and hence different elimina-tion rates [35 – 38].
Optimal total PSA cut-off values
No single total PSA cut-off separates men at high risk for PCa from men at low risk, nor men affected
with high-grade disease from those with low-grade disease. At a total PSA cut-off of � 4 ng/mL, a sig-nifi cant number of PCas remain undetected [39 – 42]. In addition, intervention at lower total PSA levels has been proposed to improve patient outcomes [43,44]. Catalona et al. found that 22% of men with a normal digital rectal examination (DRE) and a serum total PSA level between 2.6 and 4.0 ng/ml have PCa, and 81% of them have organ-confi ned disease [45]. Data from the Prostate Cancer Prevention Trial (PCPT) revealed that as many as 15% of men with normal DRE and a serum total PSA less than 4.0 ng/ml have PCa [39]. Among men with total PSA levels � 0.5, 0.6 – 1.0, 1.1 – 2.0, 2.1 – 3.0, and 3.1 – 4.0 ng/ml, PCa was detected in 6.6%, 10.1%, 17.0%, 23.9%, and 26.9%, respectively. Moreover, approximately 25% of these men had a tumor with Gleason score of 7 or higher. These and other investigators demon-strated that increasing levels of total PSA are associ-ated with increasing probability of PCa risk within the 0 – 4.0 ng/ml interval [39,41,46]. There is no total PSA threshold at age 62 – 91 below which PCa can be ruled out with high specifi city [39]. No single total PSA cut-off separates men with “ signifi cant ” (high grade, high volume) cancer from those with low-grade, possibly insignifi cant cancer. Similar to PCa presence, high-grade cancer can be found in men with low total PSA levels.
On the other hand, as of now, there is no evidence that lowering the total PSA threshold below 4 ng/ml improves the long-term survival in men with PCa. Lowering the total PSA threshold combined with decreasing the age of total PSA screening may be benefi cial for men who are at an increased risk for PCa (i.e. strong family history of PCa and/or African-American race). However, consideration must be given to the possibility that lowering the total PSA threshold could result in unnecessary biopsies and an increased detection of insignifi cant cancers. Finally, determination of the optimal, institution-specifi c, and management-guiding threshold involves not only clinical and epidemiologic features but should also consider the social and psychological implications of prostate biopsy and possible PCa detection.
The diffi culty in selecting a cut-off to defi ne what constitutes an abnormal total PSA suggests that total PSA is most useful as a continuous variable, provid-ing a spectrum of prostate cancer risk. Therefore, we prefer to include serum total PSA levels in an overall estimate of the risk of cancer, inform the patient of his particular risk, then make a shared decision about a biopsy [39 – 41,47 – 51]. Nam et al., for example, developed a model that predicts an individual’s risk for PCa in a cohort of 3108 men who underwent a prostate biopsy for the fi rst time [51]. This model comprises factors that can be easily determined at

Blood-based biomarkers for prostate cancer 65
the time of screening such as age, ethnicity, family history of PCa, the presence of urinary symptoms, total PSA, percent free PSA, and DRE. Addition of all these risk factors improved the predictive accu-racy of a base model from 0.62 to 0.74. The main advantage of this and other predictive tools [47] is that clinicians can assess PCa risk on an individual basis and make management decisions. However, despite the reasonable accuracy, similar to all predic-tive tools, the exact probability cut-off for undergo-ing or foregoing a biopsy is left with the patient and his treating physician and should be individualized.
Long-term prediction of the future risk of prostate cancer using total PSA
Several studies have suggested that total PSA levels are associated with the risk of PCa years, or even decades, before its diagnosis. The fi rst long-term pre-diction study, which reported that total PSA levels � 2.5 ng/ml predicted diagnosis of PCa over the sub-sequent decade was limited by the small number of cancer cases (n � 44) and by the degradation of total PSA in archived serum samples [52]. In a prospec-tive study involving a large number of cases, the lead time between total PSA levels �4 ng/ml and the sub-sequent clinical diagnosis of PCa was estimated at 5.5 years [53]. Similarly, Fang et al . studied the risk of PCa diagnosis in a cohort of 549 men following a baseline total PSA measurement at age 40 – 60 while providing a median follow-up of − 13 years [54]. They concluded a total PSA value above the age-adjusted median carried a relative risk of subsequent cancer diagnosis of − 3.6.
Two larger studies extended prediction models to lower total PSA ranges and longer follow-up inter-vals. Loeb et al . examined 1178 men in their 40s who had risk factors for Pca [55]. The risk of subsequent PCa diagnosis was 14.6-fold higher for men with a baseline total PSA level between 0.7 and 2.5 ng/ml compared to men with total PSA � 0.7 ng/ml. Lilja et al . assessed PCa risk among 21 277 men younger than 50 years when they attended the Malm ö Preventive Medicine study (MPM), a cardiovascular risk assessment study [56]. The investigators mea-sured total PSA levels in archived plasma obtained from 462 participants diagnosed with PCa within a median of 18 years from start of the study and from 1 222 matched controls. Total PSA level at age 44 – 50 was very strongly associated with the likelihood of developing PCa up to 25 years later. The odds ratio for a PCa diagnosis at a total PSA value of 0.51 – 1.0 ng/ml was 2.51 compared to total PSA � 0.50 ng/ml, which roughly corresponded to the population aver-age. The odds ratio increased to 7.02 for a total PSA of 1.0 – 1.5 ng/ml, and further up to 19.01 for a total
PSA of 2.01 – 3.0 ng/ml compared to a total PSA � 0.50 ng/ml. In a follow-up study, the authors have further shown that total PSA level at age 44 – 50 pre-dicts the likelihood of developing advanced PCa, defi ned as either locally advanced (clinical T3 or higher) or metastatic disease at the time of diagnosis [57]. In another analysis of the MPM-study cohort, the prognostic accuracy of PSA (both total PSA and complexed PSA, described below) decreased with age [58]. The authors hypothesized that these fi nd-ings result from a greater prevalence of BPH (and therefore of non-cancer-related total PSA increase) among older men.
An analysis of the same cohort demonstrated that PSA at 60 is an extremely strong predictor of the risk of prostate cancer metastasis (AUC 0.86) and death (AUC 0.90) by age 85. Almost all deaths (90%) occurred in men in the top quartile of PSA levels ( � 2 ng/ml); men with PSA below the median ( � 1 ng/ml), had an extremely low risk of clinically relevant prostate cancer by age of 85 (0.5% risk of metastasis, 0.2% risk of death from prostate cancer). This suggests that at least half of men can be exempted from prostate cancer screening at age 60, with early detection efforts focusing on a sub-group of men at elevated risk [59].
In summary, these studies indicate that men who will eventually develop PCa have increased total PSA levels years or decades before the cancer is diag-nosed. These total PSA levels may refl ect the long duration of prostate carcinogenesis or could refl ect a causal role of total PSA in PCa development and/or progression. A total PSA measurement before age 50 could help risk-stratify men for frequency and/or type of later PCa screening; a PSA at 60 could deter-mine which men need to continue with screening.
Prostate-specifi c antigen derivatives
Enhancing the diagnostic accuracy of total PSA, particularly specifi city, is critical, since higher speci-fi city would reduce the number of biopsies performed in men not affected by PCa. Several different strate-gies have been investigated, including the use of age-specifi c total PSA cut-offs, total PSA density, total PSA density of the transition zone, total PSA velocity (total PSAV), and the measurement of vari-ous molecular forms of PSA [47,60 – 62]. In this section, we will focus on total PSAV and the measurement of various molecular forms of PSA.
Total PSA velocity
Total PSAV refers to the serial evaluation of serum total PSA concentration over time [63,64]. Different methods of calculating total PSAV are available

66 S. F. Shariat et al.
who were likely to have a low incidence of BPH, Ulmert et al. found no benefi t to calculate total PSAV or the velocity of any other PSA form over total PSA for long-term PCa prediction [71]. Of note, the pre-dictive value of total PSAV alone was 71.2%, while the predictive value of a single total PSA was higher (77.1%) and the combined model including both total PSAV and total PSA did not alter the predictive accuracy. The observed lack of additional predictive value for total PSAV indicates that total PSA levels do not increase sharply before PCa diagnosis but rise gradually and slowly over many years, also in those men who later present with advanced cancer.
Several studies have shown that that a high pre-treatment total PSAV is strongly associated with a poor disease-specifi c survival following diagnosis and could help identify men with low total PSA val-ues who are at increased risk of harboring a poten-tially lethal tumor [72 – 75]. Carter et al. found a strong association between survival and higher total PSAV as early as 10 – 15 years before diagnosis in the Baltimore Longitudinal Study of Aging project [75]. Based on these fi ndings, they proposed that a total PSAV threshold of 0.35 ng/ml per year be used in screening men with low total PSA levels to increase the detection of potentially lethal tumors still in the window of curability. These data have prompted debate as to whether this would suffi ce as evidence to warrant the National Comprehensive Cancer Net-work to recommend a prostate biopsy if the total PSAV is greater than 0.5 ng/ml per year [76]. How-ever, in analysis of the PCPT data, Vickers and col-leagues showed that biopsying men with low PSA but elevated PSAV, led to a large increase in unnecessary biopsies without detecting an important number of clinically signifi cant cancers [77].
D ’ Amico et al. reported that men with a pre-operative total PSAV greater than 2.0 ng/ml per year had a 9.8-fold increased relative risk of death from PCa than men with a lower total PSAV [72]. This analysis is compromised by a low number of events; accordingly, it is impossible to tell whether PSAV adds predictive value to standard predictors such as stage, grade and absolute level of PSA. In a more recent study, these investigators reported that total PSAV was also signifi cantly associated with the risk of cancer-specifi c mortality following external beam radiation therapy [73]. Conversely, using data from 267 Scandinavian men with localized PCa and baseline total PSA levels � 50 ng/ml, Fall and col-leagues found that, although prognostically rele-vant, baseline total PSA levels and relative total PSAV in the fi rst two years following diagnosis were not able to predict accurately which patients would have a lethal PCa outcome [78]. Several other stud-ies have found that PSAV does not aid prediction
(e.g. based on the fi rst and the last measured values only or on a regression line through all available measurements, based on normal or logarithmic val-ues), but only small differences in predictive value have been found among these derivatives. Connolly et al. found that using all available total PSA mea-surements in a linear regression analysis should be the method of choice for calculating total PSAV [65]. When using the fi rst and last measurements only, these should at least be separated by a suffi -ciently long time period.
Carter et al. showed that patients with BPH dem-onstrated a linear increase in total PSA levels over time, whereas patients with PCa had an initial linear increase with a subsequent exponential rise that occurred approximately fi ve years before cancer detection [63]. In men with an initial total PSA level between 4 and 10 ng/ml, a total PSAV cut-off value of 0.75 ng/ml per year provided a sensitivity and specifi city for PCa of 79% and � 90%, respectively. If the initial total PSA concentration was less than 4 ng/ml, the specifi city of total PSAV remained � 90%, but the sensitivity dropped to 11%. These results were questioned using relatively short total PSA intervals of one and two years [66]. Subse-quently, Carter et al. showed that total PSAV values are only useful if a minimum of three consecutive measurements are taken over a two year period [67]. While the specifi city of total PSAV is high, its sensi-tivity is too low to advise against prostate biopsy in a patient with an elevated total PSA level who is oth-erwise healthy and a good candidate for curative therapy. Other limitations of total PSAV include imprecision due to biological and analytical intra-individual variability (see section entitled “ Total PSA and its limitations ” ) and total PSA stability.
Prospective screening studies have reported that total PSAV does not appear to add diagnostic value for PCa detection beyond that of a single total PSA level. In an analysis of PCPT data, Thompson et al. found that when total PSAV was used alone, it was an independent predictor of PCa presence and aggressiveness [40]. However, when total PSAV was adjusted for the effect of total PSA and other standard variables, it lost independent predictive value. Similarly, the fi rst two screening rounds of the Rotterdam section of the ERSPC found that total PSAV did not improve accuracy when combined with total PSA in the prospective setting [68,69]. A recent analysis from the Prostate, Lung, Colon, and Ovarian (PLCO) cancer screening trial showed that although total PSAV was an independent predictor of high-grade disease, addition of total PSAV to total PSA only slightly increased its performance for prediction of high-grade tumors [70]. Using a large population-based cohort of men in early middle age

Blood-based biomarkers for prostate cancer 67
in men treated by radical prostatectomy [79,80] or conservatively [81].
It may be that the observation period necessary for obtaining a valid calculation of total PSAV that is not disturbed by considerable short-term fl uctua-tions is be too long, or that number of total PSA measurements is too high for use in clinical practice. In addition, total PSAV may not correlate with early tumor progression, but could be a mere indicator of aggressive disease for which the window of curability has already closed. Furthermore, a quickly rising total PSA is more common in men with a high start-ing total PSA level [82]. This proportion of men is expected to be much smaller in a screened cohort than in a clinically diagnosed cohort. PSAV is a prac-tical parameter after treatment, when PSA is a sensi-tive measure of cancer; its value in men with a prostate, in whom rises in PSA may be cause by benign disease, remains to be established.
PSA molecular forms
Improvements in measuring PSA isoforms have allowed the measurement of free PSA and its ratio to total PSA [83 – 85]. Other forms include com-plexed PSA, which is a measure of how much PSA in serum is bound to α 2-macroglobulin, α 1-protease inhibitor, or α 1-antichymotrypsin. Currently there is no assay commercially available which specifi cally measures the complex of α 2-macroglobulin with PSA.
The FDA has approved the use of percent free PSA testing [i.e. (fPSA/tPSA) x 100] as an adjunct to total PSA in men with a serum total PSA concentra-tion between 4 and 10 ng/ml. A higher percent free PSA value indicates a lower probability of fi nding PCa on biopsy and raises the likelihood that the elevation in total PSA is due to the presence of BPH [86,87]. In a multicenter, prospective trial, Catalona et al. reported that when a percent free PSA of � 25% is used for triggering a sextant prostate biopsy, it yielded a 95% sensitivity for PCa detection and increased the specifi city by 20% over PSA alone [86]. The area under the curve for percent free PSA was signifi cantly higher than that for total PSA (AUC � 0.72 versus AUC � 0.53). However, in response to the realiza-tion that sextant biopsies misclassify up to one third of patients who have PCa as without cancer, a more recent evaluation of the utility of percent free PSA in patients undergoing extended 10- or 12-core biopsy has suggested a lower diagnostic effi ciency of percent free PSA [88]. While most investigators agree that percent free PSA can improve the diagnostic performance of total PSA between 4 and 10 ng/ml, the most appropriate percent free PSA cut-off value remains debatable. Catalona et al. determined that with a percent free PSA cut-off of less or equal to
27%, they were able to obtain a sensitivity of 90% and avoid 18% of unnecessary biopsies in men over the age of 50 with a total PSA of 2.6 to 4.0 ng/ml [45]. In addition, 83% of these cancers were clini-cally signifi cant. Finally, data on the utility of percent free PSA for the prediction of pathologic grade and stage of PCa is inconclusive.
Data on the usefulness of free PSA to predict clin-ical outcomes is inconclusive. Graefen and coworkers failed to detect an independent association of preop-erative free PSA with biochemical failure in 581 unscreened patients who underwent radical prostate-ctomy for clinically localized CaP [89]. In contrast, Shariat and colleagues found that lower preoperative serum free PSA is an independent predictor of advanced pathologic features, biochemical progres-sion, and patterns of aggressive disease progression in 402 consecutive men treated with radical prostatec-tomy for clinically localized CaP who had total PSA levels less than 10 ng/ml [84].
There are three distinct cleavage isoforms of free PSA in the serum: pro-PSA, BPH-associated PSA (BPSA), and intact free PSA [90]. The precursor of PSA is a 261 amino acid pre-pro-protein. Subse-quent processing by human glandular kallikrein 2 (hK2) and other proteases produces the active 237 amino acid mature PSA [90]. Studies have shown that higher levels of pro-PSA are associated with PCa. In men with PSA levels between 6.0 – 24.0 ng/ml, the [−2]proPSA fraction was found to be signifi cantly higher in men with PCa [90,91]. Moreover, authors demonstrated the utility of the pro-PSA to free PSA ratio for screening patients with PSA levels between 2.5 – 4.0 ng/ml and between 4.0 – 10.0 ng/ml [92]. Elevated pro-PSA to free PSA ratios have also been associated with aggressive pathological features and decreased biochemical disease free survival after radical pros-tatectomy [93,94]. A new automated tool using the [-2]proPSA assay with a percent free PSA based arti-fi cial neural network was capable of detecting PCa and more aggressive disease with higher accuracy than total PSA or percent free PSA alone [95]. In a recent prospective cohort of men enrolled into active surveillance for PCa, serum and tissue levels of pro-PSA at diagnosis were associated with need for sub-sequent treatment [96]. The authors hypothesized that the increase in the ratio of serum pro-PSA to percent free PSA might be driven by increased pro-PSA production from “premalignant” cells.
Molecular forms of PSA may differ in their in-vitro stability properties and information about the pre-analytical conditions is therefore essential for proper clinical interpretation. For proper measure-ment of [-2]proPSA, blood samples should be cen-trifuged within three hours of blood draw. Serum may be stored at room temperature or refrigerated

68 S. F. Shariat et al.
( � 4 ° C) for a maximum of 48 hours and should be frozen if stored for a longer period. Two freeze-thaw cycles have no effect on [-2]proPSA stability [97].
BPH-associated PSA (BPSA) is formed by the internal cleavage of free PSA between Lys182 and Ser183. BPSA is expressed in nodular hyperplasia limited to the transitional zone of men with BPH. BPSA can be detected in semen, blood and prostate, and its levels correlate with transitional zone volume and obstructive voiding symptoms [90,98]. BPSA seems to be a promising marker of BPH since it has been shown a direct association between its secretion and the volume of the transition zone [98]. As such, BPSA is a better predictor of prostate enlargement than total and free PSA [99]. In addition, BPSA is not affected by age and is signifi cantly higher in the presence of BPH symptoms. Adjusting the level of free PSA for BPSA resulted in 13 – 17% improvement in specifi city compared to free PSA alone while maintaining a sensitivity of 90 – 95% [100].
Combination of PSA molecular forms for improved cancer detection
It is most unlikely that a single biomarker will have the single decision as to a diagnosis and/or a prog-nosis of a particular pathology. The future of cancer profi ling relies on the combination of a panel of com-plimentary biomarkers that can give accurate molec-ular staging and indicate the likelihood of aggressive behavior [10,11,101,102].
The group of Vickers and Lilja has developed a statistical model that predicts prostate biopsy out-comes based on age, DRE and a panel of four kal-likrein markers – total PSA, free PSA, intact PSA and hK2. Using data from the randomized prostate can-cer screening trial in G ö teborg, Sweden [one centre of the European Randomized Study of Prostate Can-cer screening (ERSPC)], they estimated that for every 1000 previously unscreened men with elevated total PSA, use of the model to determine biopsy would reduce biopsy rates by 573, while missing only a small number of cancers (31 of 152 low-grade can-cers and 3 of 40 high-grade cancers) [15]. These fi ndings were subsequently replicated in an indepen-dent cohort (reduction in biopsy by 513 per 1000 men with elevated total PSA, missing 54 of 177 low-grade cancer and 12 of 100 high-grade cancers) [103]. These fi ndings have also been verifi ed in men who recently have undergone previous screening, with resultant improvements in predictive accuracy [104,105]. Recently, Gupta et al. demonstrated that the panel of four kallikrein markers can predict the outcome of prostate biopsy in men who had previ-ously undergone prostate biopsy during previous screening [106]. This model in addition to age and
DRE substantially improved the predictive accuracy of a base model (comprising of total PSA, age and DRE), for both low- and high-grade cancers.
Emerging blood-based biomarkers for both prostate cancer detection and prognostication
Human Kallikrein 2 (hK2)
Human kallikrein related peptidase 2 is a secreted serine protease from the same gene family as PSA [107]. They share an 80% sequence homology and are both primarily expressed in the prostate gland [107]. Despite these structural similarities, hK2 and total PSA differ in their enzymatic activity. The levels of hK2 in prostate tissue, plasma, semen, and serum are less than 2% that of total PSA, although hK2 mRNA transcript expression represents half that of total PSA. Similar to total PSA, serum hK2 is present in two forms in the blood: one bound to various protease inhibitors, and the other (preponderant) free in the circulation. Several studies have shown that, when used in conjunction with free and total PSA, serum hK2 could improve the discrimination of men with PCa from men without cancer [108 – 110]. It has also been suggested that hK2 could pre-dict poor differentiation, extra-capsular extension and biochemical recurrence in patients treated with radical prostatectomy [111 – 113]. However, this fi nd-ing has not been validated by other authors [114]. The usefulness of hK2 for the preoperative staging of localized PCa therefore remains controversial. As mentioned above (see “ Combination of PSA molec-ular forms for improved cancer detection ” section), the addition of hK2 to three other kallikreins (total, free, and intact PSA) improved the prediction of prostate biopsy results in men with elevated total PSA (increase of predictive accuracy from 68 – 72% to ∼ 83%) [15]. Considering the risk of PCa at 20%, the number of biopsies would have reduced by half, missing only 3 of 40 high-grade tumors [15].
Emerging blood-based biomarkers for prostate cancer prognostication
Urokinase Plasminogen Activation (uPA)
The urokinase plasminogen activation (uPA) axis represents a potential target for PCa markers by being involved in various phases of tumor development and progression though degradation of the extra-cellular matrix. The serum protease uPA may play a role in cancer progression by binding to the uPA receptor (uPAR) and consequently converting plasminogen to plasmin, which activates proteases related to the degradation of extracellular matrix proteins [115].

Blood-based biomarkers for prostate cancer 69
Immunohistochemical staining of radical prostatec-tomy specimens revealed that overexpression of both uPA and its inhibitor (PAI-1) were associated with aggressive PCa recurrence [116]. In patients with a total PSA level above 2 ng/ml, soluble uPAR and free PSA measured in serum before prostate biopsy improved the regression model accuracy for predic-tion of PCa [117]. Steuber et al. recently showed that uPAR fragments were signifi cant predictors of PCa on biopsy specimens of patients with an elevated PSA [117].
Both uPA and uPAR might also have a prog-nostic value. Elevated circulating levels of uPA and uPAR have been linked to PCa stage and bone metastases [116,118 – 120]. In a study of 429 patients treated with radical prostatectomy, preoperative plasma uPA was a strong predictor of biochemical recurrence after surgery. Both preoperative uPA and uPAR were associated with features of aggressive bio-chemical recurrence such as development of distant metastasis suggesting an association with occult met-astatic disease at time of local therapy. Moreover, elevation of plasma uPA and uPAR levels in PCa patients seemed to be partly caused by local release from the prostate. Larger multi-institutional studies are under way to validate the potential role of uPA and uPAR as markers of metastatic PCa.
Transforming Growth Factor-Beta 1 (TGF- β 1) and Interleukin-6 (IL-6)
TGF- β 1 is a growth factor involved in the regulation of several cellular mechanisms including proliferation, immune response, differentiation and angiogenesis [121]. TGF- β 1 has been shown to promote cell pro-gression in PCa models and its local expression has been associated with higher tumor grade, tumor invasion and metastasis in PCa patients [122,123]. Several studies have shown that increased levels of circulating TGF- β 1 were associated with cancer progression, occult and documented metastasis and biochemical progression in PCa patients [123 – 125].
IL-6 is a cytokine with variable effects on immune and hematopoietic mechanisms. In vitro and in vivo studies have shown that both IL-6 and its receptors (IL-6R) were expressed in PCa [126,127]. Several authors reported that elevated serum levels of IL-6 and IL-6R were associated with metastatic and hor-mone refractory disease, and suggested that IL-6 could predict progression and survival of PCa patients [128,129].
Based on these fi ndings, Kattan and associates developed and internally validated a prognostic model that incorporates plasma TGF- β 1 and IL-6R into a standard nomogram for prediction of biochemical
recurrence following radical prostatectomy [14]. This combination of serum markers and classical clinical parameters improved the predictive accuracy by a statistically and prognostically substantial margin (increase in predictive accuracy from 75 to 84%). However, before a biomarker can become useful in daily clinical management, it needs to be externally validated in an independent cohort of patients (Figure 1) [2,3]. Therefore, in a multi-institutional dataset of 423 patients treated with radical prostatectomy, Sha-riat et al. confi rmed that plasma levels of TGF-beta1 and IL6-SR considerably enhance the accuracy of the standard preoperative nomogram for the prediction of biochemical recurrence (accuracy of clinical features plus biomarkers 87.9% versus 71.1% for clinical fea-tures alone; p � 0.001). Such prog nostic models refi ne our ability to identify patients at a high risk of bio-chemical recurrence after radical pro statectomy who may benefi t from inclusion into perioperative clinical trials and/other intensifi ed follow-up protocols.
Endoglin
Endoglin, or CD 105, is a transmembrane glycopro-tein that is typically expressed by human vascular endothelial cells. Functionally, it is a cell-surface co-receptor for transforming growth factor β 1 (TGF- β 1) and β 3 (TGF- β 3) [130] that modulates cellular responses to TGF- β in the early steps of endothelial cell proliferation. Its critical role in angiogenesis has prompted investigators to evaluate the role of Endo-glin in cancer progression and metastasis. In PCa, Endoglin is preferentially found on new, immature blood vessels and immunohistochemical analysis sup-ports an association between Endoglin expression and disease progression [131]. Urine levels of Endoglin may distinguish patients with PCa and may help in the staging of the disease [132]. In addition, pre-oper-ative plasma Endoglin were found to be associated with metastasis to regional lymph nodes [133], estab-lished features of biologically aggressive PCa such as higher pathologic Gleason sum, and biochemical recurrence following radical prostatectomy [134]. Use of pre-operative plasma Endoglin could help decide whether and how extensively to perform a lymph-adenectomy as well as preoperative identifi cation of patients at risk for disease progression. This would help select patients for neo-adjuvant and/or adjuvant therapy or enrollment into clinical trials. Moreover, Endoglin may be valuable as a surrogate biomarker for occult metastatic disease in patients with presumed organ-confi ned disease. Further investigation is needed to validate Endoglin as a useful biomarker in men with PCa and to elucidate the mechanistic role of this biomarker in the progression of PCa.

70 S. F. Shariat et al.
Combination of blood-based biomarkers for prostate cancer prognostication
A biomarker may refl ect disruption of a biochemical pathway by a particular mechanism. Given the complexity of the molecular abnormalities associated with PCa, it is improbable that a single marker can accurately segregate tumors of similar clinico-pathologic phenotypes into distinct prognostic cate-gories. Therefore, combinations of independent, yet complementary markers, may provide a more accu-rate prediction of outcome compared to a single marker [62].
Shariat et al. found that the addition of a panel comprising pre-operative plasma levels of TGF- β 1, soluble IL-6R, IL-6, Endoglin, vascular endothelial growth factor (VEGF), and vascular cell adhesion molecule-1 (VCAM-1 or CD 106) [120,121,123, 133 – 137] impro ved the predictive accuracy the Kattan pre-operative nomogram [9] by 15.0% (i.e. 71.6 to 86.6%) [11,102]. This increase substantially exceeds accuracy gains obtained from the consider-ation of detailed pathologic descriptors of prostate cancer at radical prostatectomy. Svatek et al. con-fi rmed the strong predictive value of pre -operative levels of the candidate biomarkers after adjusting for the effect of post-operative features [101]. Addition of pre -operative levels of the candidate biomarkers improved the accuracy of the base model (i.e. total PSA, surgical margin status, extracapsular extension, seminal vesicles invasion, lymph node involvement, and pathologic Gleason sum) for prediction of bio-chemical recurrence by a statistically and prognosti-cally signifi cant margin (79 to 86%, p � 0.001). Predictive tools integrating biomarker levels could constitute the new standard for counseling patients regarding their risk of recurrence following curative therapy and for designing clinical trials to test neo-adjuvant and/or adjuvant treatment strategies in high-risk patients. However, while prediction of biochemical recurrence is important, prediction of response to therapy as well as metastasis and survival is more important for the management of PCa patients [138].
Blood-based biomarkers for monitoring of prostate cancer treatment
Total PSA
Treatment monitoring is the most accepted clinical application for total PSA. Total PSA is used for monitoring response to local treatments such as radical prostatectomy, various methods of radiation therapy, and other local therapies including cryo-surgery, as well as systemic treatments such as androgen deprivation therapy and chemotherapy. Post-treatment total PSA levels can provide invaluable
information about the effectiveness of the therapy given and the existence of residual cancer in men treated with local therapy with curative intent. In such patients, rising total PSA levels can signal cancer activity well before any clinical signs of recurrence appear. This lead-time can be further increased by months and even years.
In patients treated with systemic therapy, post-therapy total PSA changes are a seemingly perfect outcome measure because they are easily assessable, quantitative, reproducible, and inexpensive; this is true regardless of whether this outcome measure is applied to evaluate drugs in the clinical trial setting or in clinical practice. Critical to the successful appli-cation of total PSA measurement as an endpoint are the therapeutic objectives of the trial and the mech-anisms of action of the treatment administered. Cri-teria were proposed to screen for treatment effects in prostate cancer clinical trials on the basis of the hypothesis that total PSA declines refl ect signifi cant cell kill in response to agents that cause reduction in overall tumor burden. With the recognition that short-term declines in total PSA levels might simply refl ect the effect of the drug on the marker, and not on cell growth or survival, it is recommended that the declines be documented on more than one occa-sion and, equally importantly, over time. Indeed, dif-ferent total PSA-based outcomes would be required for different classes of drug. For example, drugs that are not anticipated to kill cells would not be expected to produce declines in total PSA. Similarly, ‘ differen-tiating effect ’ might produce an initial rise in total PSA that might be an indication that the drug is actually working. A single set of outcomes would not only be inapplicable to agents that act via diverse mechanisms, but could also be misleading.
Unfortunately, the signifi cance of post-therapy changes in total PSA have been misinterpreted as indi-cators of tumor response, and/or as a measure of clin-ical benefi t. An additional misconception is that the demonstration of a particular degree of decline in total PSA, in a proportion of patients, is an indication that the treatment prolongs life, and as such, should be used as an endpoint for accelerated drug approval.
The use of a post-therapy decline in total PSA as an outcome measure has been justifi ed in part by sta-tistical analyses exploring the relationship between the defi ned outcome measure and survival [139]. Most of these reports include multivariate techniques, and in some, the resultant models were validated on indepen-dent data sets. These early reports did not assess ran-domized comparative trials showing a survival benefi t, a necessary but not suffi cient condition in which to explore measures of surrogacy [139,140].
There is no clear demonstration that a post-therapy total PSA change can account for all of the

Blood-based biomarkers for prostate cancer 71
treatment effects seen is not yet available. Large-scale prospective studies incorporating different post-therapy total PSA change defi nitions, as well as other potential biomarkers, are ongoing. At this point, one can conclude that a cytotoxic drug that does not pro-duce any total PSA declines is unlikely to be effective, but that the corollary is not always true. In addition, it is important to recognize that there is a range of clinical benefi ts to patients that can improve the qual-ity, and more than likely, the duration of survival, which are independent of total PSA levels. This includes differentiating agents that might produce a rise in total PSA before a decline is observed, or act through immunization strategies where the effect on total PSA levels might be delayed or not occur at all, and angiogenesis and growth factor inhibitors. It is also important to note that there remains signifi cant variation in the methodologies and assays used by different laboratories, which makes it diffi cult to com-pare results between groups. All of these factors must be considered carefully to ensure that a drug is not discontinued prematurely on the basis of a total PSA rise that is not relevant to the drug under study.
Conclusions
The introduction of total PSA in clinical practice has resulted in early detection and reduced mortality from PCa [141]. However, PCa screening remains controversial, because of the risk of overdiagnosis reduced mortality and overtreatment and the inability to detect a signifi cant proportion of dangerous tumors. A large concerted effort has been made to improve and/or monitoring the activity of PCa and to guide molecular targeted therapy and/or assess therapeutic response. An integrated approach with blood-based measurement of different molecular forms of PSA in combination with genetic and urine biomarkers hold the promise of improving screening for and diagnosis of PCa. Panels of blood-based biomarkers will allow a fi ngerprinting of the tumors biologic behavior resul-ting in individualized therapy and monitoring. In addition, the emergence of new therapeutic approaches for PCa cannot fl ourish without a set of markers to serve as prognosticators, predictors, therapeutic tar-gets, and/or surrogate end points.
Declaration of interest: Dr. Shariat holds patents for soluble FAS and Biomarker Nomograms. Dr. Lilja holds patents for free PSA, intact PSA, and hK2 assays.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer [1] statistics, 2009. CA Cancer J Clin 2009;59:225 – 49.
Bensalah K, Montorsi F, Shariat SF. Challenges of cancer [2] biomarker profi ling. Eur Urol 2007;52:1601 – 9. Shariat SF, Canto EI, Kattan MW, Slawin KM. Beyond pros-[3] tate-specifi c antigen: New serologic biomarkers for improved diagnosis and management of prostate cancer. Rev Urol 2004;6:58 – 72. Bensalah K, Lotan Y, Karam JA, Shariat SF. New circulating [4] biomarkers for prostate cancer. Prostate Cancer Prostatic Dis 2008;11:112 – 20. Verma M, Srivastava S. New cancer biomarkers deriving [5] from NCI early detection research. Recent Results Cancer Res 2003;163:72 – 84; Discussion 264 – 6. Winget MD, Baron JA, Spitz MR, Brenner DE, Warzel [6] D, Kincaid H, et al. Development of common data ele-ments: The experience of and recommendations from the early detection research network. Int J Med Inform 2003; 70:41 – 8. Ilyin SE, Belkowski SM, Plata-Salaman CR. Biomarker dis-[7] covery and validation: Technologies and integrative approaches. Trends Biotechnol 2004;22:411 – 6. Shariat SF, Karakiewicz PI. Perspectives on prostate cancer [8] biomarkers. Eur Urol 2008;54:8 – 10. Shariat SF, Karakiewicz PI, Roehrborn CG, Kattan MW. An [9] updated catalog of prostate cancer predictive tools. Cancer 2008;113:3075 – 99. Shariat SF, Karam JA, Margulis V, Karakiewicz PI. New blood-[10] based biomarkers for the diagnosis, staging and prognosis of prostate cancer. BJU Int 2008;101:675 – 83. Shariat SF, Karam JA, Walz J, Roehrborn CG, Montorsi F, [11] Margulis V, et al. Improved prediction of disease relapse after radical prostatectomy through a panel of preoperative blood-based biomarkers. Clin Cancer Res 2008;14:3785 – 91. Shariat SF, Park S, Trinh QD, Roehrborn CG, Slawin KM, [12] Karakiewicz PI. Plasminogen activation inhibitor-1 improves the predictive accuracy of prostate cancer nomograms. J Urol 2007;178(4 Pt 1):1229 – 36; Discussion 36 – 7. Kattan MW. Judging new markers by their ability to improve [13] predictive accuracy. J Natl Cancer Inst 2003;95:634 – 5. Kattan MW, Shariat SF, Andrews B, Zhu K, Canto E, [14] Matsumoto K, et al. The addition of interleukin-6 soluble receptor and transforming growth factor beta1 improves a preoperative nomogram for predicting biochemical progres-sion in patients with clinically localized prostate cancer. J Clin Oncol 2003;21:3573 – 9. Vickers AJ, Cronin AM, Aus G, Pihl CG, Becker C, Pettersson [15] K, et al. A panel of kallikrein markers can reduce unnecessary biopsy for prostate cancer: Data from the European Rand-omized Study of Prostate Cancer Screening in Goteborg, Sweden. BMC Med 2008;6:19. Vickers AJ, Jang K, Sargent D, Lilja H, Kattan MW. System-[16] atic review of statistical methods used in molecular marker studies in cancer. Cancer 2008;112:1862 – 8. Vickers AJ, Elkin EB. Decision curve analysis: A novel [17] method for evaluating prediction models. Med Decis Making 2006;26:565 – 74. Vickers AJ, Cronin AM, Elkin EB, Gonen M. Extensions to [18] decision curve analysis, a novel method for evaluating diag-nostic tests, prediction models and molecular markers. BMC Med Inform Decis Mak 2008;8:53. Vickers AJ. Decision analysis for the evaluation of diagnostic [19] tests, prediction models and molecular markers. Am Stat 2008;62:314 – 20. Elkin EB, Vickers AJ, Kattan MW. Primer: Using decision [20] analysis to improve clinical decision making in urology. Nat Clin Pract Urol 2006;3:439 – 48. Ankerst DP, Miyamoto R, Nair PV, Pollock BH, Thompson IM, [21] Parekh DJ. Yearly prostate specifi c antigen and digital rectal

72 S. F. Shariat et al.
examination fl uctuations in a screened population. J Urol 2009;181:2071 – 5; Discussion 6. Soletormos G, Semjonow A, Sibley PE, Lamerz R, Petersen PH, [22] Albrecht W, et al. Biological variation of total prostate-specifi c antigen: A survey of published estimates and conse-quences for clinical practice. Clin Chem 2005;51:1342 – 51. Roehrborn CG, Pickens GJ, Carmody T, 3rd. Variability of [23] repeated serum prostate-specifi c antigen (PSA) measure-ments within less than 90 days in a well-defi ned patient population. Urology 1996;47:59 – 66. Link RE, Shariat SF, Nguyen CV, Farr A, Weinberg AD, [24] Morton RA, et al. Variation in prostate specifi c antigen results from 2 different assay platforms: Clinical impact on 2304 patients undergoing prostate cancer screening. J Urol 2004; 171(6 Pt 1):2234 – 8. Sotelo RJ, Mora KE, Perez LH, Novoa J, Carmona O, [25] De Andrade R, et al. Assay standardization bias: Different prostate cancer detection rates and clinical outcomes result-ing from different assays for free and total prostate-specifi c antigen. Urology 2007;69:1143 – 6. Stephan C, Klaas M, Muller C, Schnorr D, Loening SA, [26] Jung K. Interchangeability of measurements of total and free prostate-specifi c antigen in serum with 5 frequently used assay combinations: An update. Clin Chem 2006;52:59 – 64. Slev PR, La’ulu SL, Roberts WL. Intermethod differences in [27] results for total PSA, free PSA, and percentage of free PSA. Am J Clin Pathol 2008;129:952 – 8. Stephan C, Bangma C, Vignati G, Bartsch G, Lein M, [28] Jung K, et al. 20 – 25% lower concentrations of total and free prostate-specifi c antigen (PSA) after calibration of PSA assays to the WHO reference materials – analysis of 1098 patients in four centers. Int J Biol Markers 2009;24:65 – 9. Semjonow A, Brandt B, Oberpenning F, Roth S, Hertle L. [29] Discordance of assay methods creates pitfalls for the inter-pretation of prostate-specifi c antigen values. Prostate Suppl 1996;7:3 – 16. Thompson IM, Chi C, Ankerst DP, Goodman PJ, [30] Tangen CM, Lippman SM, et al. Effect of fi nasteride on the sensitivity of PSA for detecting prostate cancer. J Natl Cancer Inst 2006;98:1128 – 33. Nixon RG, Wener MH, Smith KM, Parson RE, Blase AB, [31] Brawer MK. Day to day changes in free and total PSA: Sig-nifi cance of biological variation. Prostate Cancer Prostatic Dis 1997;1:90 – 6. Nixon RG, Wener MH, Smith KM, Parson RE, Strobel SA, [32] Brawer MK. Biological variation of prostate specifi c antigen levels in serum: An evaluation of day-to-day physiological fl uctuations in a well-defi ned cohort of 24 patients. J Urol 1997;157:2183 – 90. Bunting PS, DeBoer G, Choo R, Danjoux C, Klotz L, [33] Fleshner N. Intraindividual variation of PSA, free PSA and complexed PSA in a cohort of patients with prostate cancer managed with watchful observation. Clin Biochem 2002; 35:471 – 5. Bruun L, Becker C, Hugosson J, Lilja H, Christensson A. [34] Assessment of intra-individual variation in prostate-specifi c antigen levels in a biennial randomized prostate cancer screening program in Sweden. Prostate 2005;65:216 – 21. Birkenmeier G, Struck F, Gebhardt R. Clearance mechanism [35] of prostate specifi c antigen and its complexes with alpha2-macroglobulin and alpha1-antichymotrypsin. J Urol 1999; 162(3 Pt 1):897 – 901. Bjork T, Ljungberg B, Piironen T, Abrahamsson PA, [36] Pettersson K, Cockett AT, et al. Rapid exponential elimina-tion of free prostate-specifi c antigen contrasts the slow, capacity-limited elimination of PSA complexed to alpha 1-antichymotrypsin from serum. Urology 1998;51:57 – 62.
Lilja H, Haese A, Bjork T, Friedrich MG, Piironen T, [37] Pettersson K, et al. Signifi cance and metabolism of com-plexed and noncomplexed prostate specifi c antigen forms, and human glandular kallikrein 2 in clinically localized pros-tate cancer before and after radical prostatectomy. J Urol 1999;162:2029 – 34; Discussion 34 – 5. Djavan B, Shariat S, Ghawidel K, Guven-Marberger K, [38] Remzi M, Kovarik J, et al. Impact of chronic dialysis on serum PSA, free PSA, and free/total PSA ratio: Is prostate cancer detection compromised in patients receiving long-term dialysis?. Urology 1999;53:1169 – 74. Thompson IM, Pauler DK, Goodman PJ, Tangen CM, [39] Lucia MS, Parnes HL, et al. Prevalence of prostate cancer among men with a prostate-specifi c antigen level � or � 4.0 ng per milliliter. N Engl J Med 2004;350:2239 – 46. Thompson IM, Ankerst DP, Chi C, Goodman PJ, [40] Tangen CM, Lucia MS, et al. Assessing prostate cancer risk: Results from the Prostate Cancer Prevention Trial. J Natl Cancer Inst 2006;98:529 – 34. Thompson IM, Ankerst DP, Chi C, Lucia MS, Goodman [41] PJ, Crowley JJ, et al. Operating characteristics of prostate-specifi c antigen in men with an initial PSA level of 3.0 ng/ml or lower. JAMA 2005;294:66 – 70. Thompson IM, Pauler Ankerst D, Chi C, Goodman PJ, [42] Tangen CM, Lippman SM, et al. Prediction of prostate cancer for patients receiving fi nasteride: Results from the Prostate Cancer Prevention Trial. J Clin Oncol 2007;25: 3076 – 81. Berger AP, Volgger H, Rogatsch H, Strohmeyer D, Steiner H, [43] Klocker H, et al. Screening with low PSA cutoff values results in low rates of positive surgical margins in radical prostatec-tomy specimens. Prostate 2002;53:241 – 5. Catalona WJ, Ramos CG, Carvalhal GF, Yan Y. Lowering [44] PSA cutoffs to enhance detection of curable prostate cancer. Urology 2000;55:791 – 5. Catalona WJ, Smith DS, Ornstein DK. Prostate cancer [45] detection in men with serum PSA concentrations of 2.6 to 4.0 ng/mL and benign prostate examination. Enhancement of specifi city with free PSA measurements. JAMA 1997; 277:1452 – 5. Efstathiou JA, Chen MH, Catalona WJ, McLeod DG, [46] Carroll PR, Moul JW, et al. Prostate-specifi c antigen-based serial screening may decrease prostate cancer-specifi c mortality. Urology 2006;68:342 – 7. Shariat SF, Karakiewicz PI, Margulis V, Kattan MW. [47] Inventory of prostate cancer predictive tools. Curr Opin Urol 2008;18:279 – 96. Kattan MW. When and how to use informatics tools in [48] caring for urologic patients. Nat Clin Pract Urol 2005; 2:183 – 90. Kattan MW, Eastham JA, Wheeler TM, Maru N, Scardino [49] PT, Erbersdobler A, et al. Counseling men with prostate cancer: A nomogram for predicting the presence of small, moderately differentiated, confi ned tumors. J Urol 2003;170: 1792 – 7. Steyerberg EW, Roobol MJ, Kattan MW, van der Kwast TH, [50] de Koning HJ, Schroder FH. Prediction of indolent prostate cancer: Validation and updating of a prognostic nomogram. J Urol 2007;177:107 – 12; Discussion 12. Nam RK, Toi A, Klotz LH, Trachtenberg J, Jewett MA, [51] Appu S, et al. Assessing individual risk for prostate cancer. J Clin Oncol 2007;25:3582 – 8. Stenman UH, Hakama M, Knekt P, Aromaa A, Teppo L, [52] Leinonen J. Serum concentrations of prostate specifi c antigen and its complex with alpha 1-antichymotrypsin before diagnosis of prostate cancer. Lancet 1994;344(8937):1594 – 8.

Blood-based biomarkers for prostate cancer 73
Gann PH, Hennekens CH, Stampfer MJ. A prospective [53] evaluation of plasma prostate-specifi c antigen for detec-tion of prostatic cancer [see comments]. JAMA 1995;273:289 – 94. Fang J, Metter EJ, Landis P, Chan DW, Morrell CH, [54] Carter HB. Low levels of prostate-specifi c antigen predict long-term risk of prostate cancer: Results from the Baltimore Longitudinal Study of Aging. Urology 2001;58:411 – 6. Loeb S, Roehl KA, Antenor JA, Catalona WJ, Suarez BK, [55] Nadler RB. Baseline prostate-specifi c antigen compared with median prostate-specifi c antigen for age group as predictor of prostate cancer risk in men younger than 60 years old. Urology 2006;67:316 – 20. Lilja H, Ulmert D, Bjork T, Becker C, Serio AM, Nilsson JA, [56] et al. Long-term prediction of prostate cancer up to 25 years before diagnosis of prostate cancer using prostate kallikreins measured at age 44 to 50 years. J Clin Oncol 2007;25:431 – 6. Ulmert D, Cronin AM, Bjork T, O’Brien MF, Scardino PT, [57] Eastham JA, et al. Prostate-specifi c antigen at or before age 50 as a predictor of advanced prostate cancer diagnosed up to 25 years later: A case-control study. BMC Med 2008;6:6. Vickers AJ, Ulmert D, Serio AM, Bjork T, Scardino PT, [58] Eastham JA, et al. The predictive value of prostate cancer biomarkers depends on age and time to diagnosis: Towards a biologically-based screening strategy. Int J Cancer 2007; 121:2212 – 7. Vickers AJ, Cronin AM, Bjork T, Manjer J, Nilsson PM, [59] Dahlin A, et al. Prostate specifi c antigen concentration at age 60 and death or metastasis from prostate cancer: Case-control study. BMJ 2010;341:c4521. Lilja H, Ulmert D, Vickers AJ. Prostate-specifi c antigen and [60] prostate cancer: Prediction, detection and monitoring. Nat Rev Cancer 2008;8:268 – 78. Schroder FH, Carter HB, Wolters T, van den Bergh RC, [61] Gosselaar C, Bangma CH, et al. Early detection of prostate cancer in 2007. Part 1: PSA and PSA kinetics. Eur Urol 2008;53:468 – 77. Shariat SF, Karam JA, Roehrborn CG. Blood biomarkers [62] for prostate cancer detection and prognosis. Future Oncol 2007;3:449 – 61. Carter HB, Morrell CH, Pearson JD, Brant LJ, Plato CC, [63] Metter EJ, et al. Estimation of prostatic growth using serial prostate-specifi c antigen measurements in men with and without prostate disease. Cancer Res 1992;52:3323 – 8. Carter HB, Pearson JD, Metter EJ, Brant LJ, Chan DW, [64] Andres R, et al. Longitudinal evaluation of prostate-specifi c antigen levels in men with and without prostate disease [see comments]. J Am Med Assoc 1992;267:2215 – 20. Connolly D, Black A, Murray LJ, Napolitano G, Gavin A, [65] Keane PF. Methods of calculating prostate-specifi c antigen velocity. Eur Urol 2007;52:1044 – 50. Smith DS, Catalona WJ. Rate of change in serum prostate [66] specifi c antigen levels as a method for prostate cancer detec-tion. J Urol 1994;152:1163 – 7. Carter HB, Pearson JD, Waclawiw Z, Metter EJ, Chan DW, [67] Guess HA, et al. Prostate-specifi c antigen variability in men without prostate cancer: Effect of sampling interval on pros-tate-specifi c antigen velocity. Urology 1995;45:591 – 6. Roobol MJ, Kranse R, de Koning HJ, Schroder FH. [68] Prostate-specifi c antigen velocity at low prostate-specifi c antigen levels as screening tool for prostate cancer: Results of second screening round of ERSPC (ROTTERDAM). Urology 2004;63:309 – 13; Discussion 13 – 5. Schroder FH, Roobol MJ, van der Kwast TH, Kranse R, [69] Bangma CH. Does PSA velocity predict prostate cancer
in pre-screened populations? Eur Urol 2006;49:460 – 5; Discussion 5. Pinsky PF, Andriole G, Crawford ED, Chia D, Kramer BS, [70] Grubb R, et al. Prostate-specifi c antigen velocity and pro state cancer gleason grade and stage. Cancer 2007;109: 1689 – 95. Ulmert D, Serio AM, O’Brien MF, Becker C, Eastham JA, [71] Scardino PT, et al. Long-term prediction of prostate cancer: Prostate-specifi c antigen (PSA) velocity is predictive but does not improve the predictive accuracy of a single PSA measure-ment 15 years or more before cancer diagnosis in a large, representative, unscreened population. J Clin Oncol 2008;26: 835 – 41. D’Amico AV, Chen MH, Roehl KA, Catalona WJ. Preopera-[72] tive PSA velocity and the risk of death from prostate cancer after radical prostatectomy. N Engl J Med 2004;351:125 – 35. D’Amico AV, Renshaw AA, Sussman B, Chen MH. Pretreat-[73] ment PSA velocity and risk of death from prostate cancer following external beam radiation therapy. JAMA 2005;294: 440 – 7. Patel DA, Presti JC, Jr., McNeal JE, Gill H, Brooks JD, [74] King CR. Preoperative PSA velocity is an independent prog-nostic factor for relapse after radical prostatectomy. J Clin Oncol 2005;23:6157 – 62. Carter HB, Ferrucci L, Kettermann A, Landis P, Wright EJ, [75] Epstein JI, et al. Detection of life-threatening prostate cancer with prostate-specifi c antigen velocity during a window of curability. J Natl Cancer Inst 2006;98:1521 – 7. National Comprehensive Cancer Network [Internet]. [76] National Comprehensive; 2004. Available from: http://www.nccn.org/professionals/physician_gls/PDF/prostate_detection.pdf. Vickers A, Tangen C, Till C, Lilja H, Thompson I. An empir-[77] ical evaluation of the national cancer center network guide-lines on prostate specifi c antigen velocity in prostate cancer detection. J Urol 2010;183:e771. Fall K, Garmo H, Andren O, Bill-Axelson A, Adolfsson J, [78] Adami HO, et al. Prostate-specifi c antigen levels as a predic-tor of lethal prostate cancer. J Natl Cancer Inst 2007;99:526 – 32. Stephenson AJ, Kattan MW, Eastham JA, Bianco FJ, Jr., [79] Yossepowitch O, Vickers AJ, et al. Prostate cancer-specifi c mortality after radical prostatectomy for patients treated in the prostate-specifi c antigen era. J Clin Oncol 2009;27: 4300 – 5. O’Brien MF, Cronin AM, Fearn PA, Smith B, Stasi J, [80] Guillonneau B, et al. Pretreatment prostate-specifi c antigen (PSA) velocity and doubling time are associated with out-come but neither improves prediction of outcome beyond pretreatment PSA alone in patients treated with radical prostatectomy. J Clin Oncol 2009;27:3591 – 7. Frank O’Brien M, Cronin AM, Fearn PA, Savage CJ, [81] Smith B, Stasi J, et al. Evaluation of prediagnostic prostate-specifi c antigen dynamics as predictors of death from pros-tate cancer in patients treated conservatively. Int J Cancer Epub 2010 Jul 23. Yu X, Loeb S, Roehl KA, Han M, Catalona WJ. The asso-[82] ciation between total prostate specifi c antigen concentration and prostate specifi c antigen velocity. J Urol 2007;177:1298 – 302; Discussion 301 – 2. Lilja H. Signifi cance of different molecular forms of serum [83] PSA. The free, noncomplexed form of PSA versus that complexed to alpha 1-antichymotrypsin. [Review]. Urol Clin North Am 1993;20:681 – 6. Shariat SF, Abdel-Aziz KF, Roehrborn CG, Lotan Y. Pre-[84] operative percent free PSA predicts clinical outcomes in patients treated with radical prostatectomy with total PSA levels below 10 ng/ml. Eur Urol 2006;49:293 – 302.

74 S. F. Shariat et al.
Catalona WJ, Southwick PC, Slawin KM, Partin AW, [85] Brawer MK, Flanigan RC, et al. Comparison of percent free PSA, PSA density, and age-specifi c PSA cutoffs for prostate cancer detection and staging. Urology 2000;56:255 – 60. Catalona WJ, Partin AW, Slawin KM, Brawer MK, Flanigan RC, [86] Patel A, et al. Use of the percentage of free prostate-specifi c antigen to enhance differentiation of prostate cancer from benign prostatic disease: A prospective multicenter clinical trial. JAMA 1998;279:1542 – 7. Woodrum DL, Brawer MK, Partin AW, Catalona WJ, [87] Southwick PC. Interpretation of free prostate specifi c antigen clinical research studies for the detection of prostate cancer. J Urol 1998;159:5 – 12. Canto EI, Singh H, Shariat SF, Kadmon D, Miles BJ, [88] Wheeler TM, et al. Effects of systematic 12-core biopsy on the performance of percent free prostate specifi c antigen for prostate cancer detection. J Urol 2004;172:900 – 4. Graefen M, Karakiewicz PI, Cagiannos I, Hammerer PG, [89] Haese A, Palisaar J, et al. Percent free prostate specifi c anti-gen is not an independent predictor of organ confi nement or prostate specifi c antigen recurrence in unscreened patients with localized prostate cancer treated with radical prostatec-tomy. J Urol 2002;167:1306 – 9. Mikolajczyk SD, Marks LS, Partin AW, Rittenhouse HG. [90] Free prostate-specifi c antigen in serum is becoming more complex. Urology 2002;59:797 – 802. Mikolajczyk SD, Marker KM, Millar LS, Kumar A, [91] Saedi MS, Payne JK, et al. A truncated precursor form of prostate-specifi c antigen is a more specifi c serum marker of prostate cancer. Cancer Res 2001;61:6958 – 63. Sokoll LJ, Chan DW, Mikolajczyk SD, Rittenhouse HG, [92] Evans CL, Linton HJ, et al. Proenzyme psa for the early detection of prostate cancer in the 2.5-4.0 ng/ml total psa range: Preliminary analysis. Urology 2003;61:274 – 6. Catalona WJ, Bartsch G, Rittenhouse HG, Evans CL, Linton [93] HJ, Horninger W, et al. Serum pro-prostate specifi c antigen preferentially detects aggressive prostate cancers in men with 2 to 4 ng/ml prostate specifi c antigen. J Urol 2004; 171(6 Pt 1):2239 – 44. Catalona WJ, Bartsch G, Rittenhouse HG, Evans CL, [94] Linton HJ, Amirkhan A, et al. Serum pro prostate specifi c antigen improves cancer detection compared to free and complexed prostate specifi c antigen in men with prostate specifi c antigen 2 to 4 ng/ml. J Urol 2003;170(6 Pt 1):2181 – 5. Stephan C, Kahrs AM, Cammann H, Lein M, Schrader M, [95] Deger S, et al. A [-2]proPSA-based artifi cial neural network signifi cantly improves differentiation between prostate cancer and benign prostatic diseases. Prostate 2009;69:198 – 207. Makarov DV, Isharwal S, Sokoll LJ, Landis P, Marlow C, [96] Epstein JI, et al. Pro-prostate-specifi c antigen measurements in serum and tissue are associated with treatment necessity among men enrolled in expectant management for prostate cancer. Clin Cancer Res 2009;15:7316 – 21. Semjonow A, Kopke T, Eltze E, Pepping-Schefers B, Burgel [97] H, Darte C. Pre-analytical in-vitro stability of [−2]proPSA in blood and serum. Clin Biochem 2010;43:926 – 8. Mikolajczyk SD, Millar LS, Wang TJ, Rittenhouse HG, [98] Wolfert RL, Marks LS, et al. “BPSA,” a specifi c molecular form of free prostate-specifi c antigen, is found predominantly in the transition zone of patients with nodular benign prostatic hyper-plasia. Urology 2000;55:41 – 5. Canto EI, Singh H, Shariat SF, Lamb DJ, Mikolajczyk SD, [99] Linton HJ, et al. Serum BPSA outperforms both total PSA and free PSA as a predictor of prostatic enlargement in men without prostate cancer. Urology 2004;63:905 – 10; Dis-cussion 10 – 1.
Stephan C, Cammann H, Deger S, Schrader M, Meyer HA, [100] Miller K, et al. Benign prostatic hyperplasia-associated free prostate-specifi c antigen improves detection of prostate cancer in an artifi cial neural network. Urology 2009;74:873 – 7.
Svatek RS, Jeldres C, Karakiewicz PI, Suardi N, Walz J, [101] Roehrborn CG, et al. Pre-treatment biomarker levels improve the accuracy of post-prostatectomy nomogram for predic-tion of biochemical recurrence. Prostate 2009;69:886 – 94.
Shariat SF, Karakiewicz PI, Ashfaq R, Lerner SP, Palapattu [102] GS, Cote RJ, et al. Multiple biomarkers improve prediction of bladder cancer recurrence and mortality in patients undergoing cystectomy. Cancer 2008;112:315 – 25.
Vickers A, Cronin A, Roobol M, Savage C, Peltola M, [103] Pettersson K, et al. Reducing unnecessary biopsy during prostate cancer screening using a four-kallikrein panel: An independent replication. J Clin Oncol 2010;28:2493 – 8.
Vickers AJ, Cronin AM, Aus G, Pihl CG, Becker C, [104] Pettersson K, et al. Impact of recent screening on predicting the outcome of prostate cancer biopsy in men with elevated prostate-specifi c antigen: Data from the European Rand-omized Study of Prostate Cancer Screening in Gothenburg, Sweden. Cancer 2010;116:2612 – 20.
Vickers AJ, Cronin AM, Roobol MJ, Savage CJ, Peltola M, [105] Pettersson K, et al. A four-kallikrein panel predicts prostate cancer in men with recent screening: data from the Euro-pean Randomized Study of Screening for Prostate Cancer, Rotterdam. Clin Cancer Res 2010;16:3232 – 9.
Gupta A, Roobol MJ, Savage CJ, Peltola M, Pettersson K, [106] Scardino PT, et al. A four-kallikrein panel for the prediction of repeat prostate biopsy: Data from the European Rand-omized Study of Prostate Cancer Screening in Rotterdam, Netherlands. Br J Cancer Epub 2010 Jul 27.
Yousef GM, Diamandis EP. The new human tissue kal-[107] likrein gene family: Structure, function, and association to disease. Endocr Rev 2001;22:184 – 204.
Nam RK, Diamandis EP, Toi A, Trachtenberg J, Magklara [108] A, Scorilas A, et al. Serum human glandular kallikrein-2 protease levels predict the presence of prostate cancer among men with elevated prostate-specifi c antigen. J Clin Oncol 2000;18:1036 – 42.
Becker C, Piironen T, Pettersson K, Hugosson J, Lilja H. [109] Clinical value of human glandular kallikrein 2 and free and total prostate-specifi c antigen in serum from a population of men with prostate-specifi c antigen levels 3.0 ng/mL or greater. Urology 2000;55:694 – 9.
Becker C, Piironen T, Pettersson K, Bjork T, Wojno KJ, [110] Oesterling JE, et al. Discrimination of men with prostate cancer from those with benign disease by measurements of human glandular kallikrein 2 (HK2) in serum. J Urol 2000; 163:311 – 6.
Haese A, Graefen M, Steuber T, Becker C, Pettersson K, [111] Piironen T, et al. Human glandular kallikrein 2 levels in serum for discrimination of pathologically organ-confi ned from locally-advanced prostate cancer in total PSA-levels below 10 ng/ml. Prostate 2001;49:101 – 9.
Kwiatkowski MK, Recker F, Piironen T, Pettersson K, [112] Otto T, Wernli M, et al. In prostatism patients the ratio of human glandular kallikrein to free PSA improves the discrimination between prostate cancer and benign hyper-plasia within the diagnostic “gray zone” of total PSA 4 to 10 ng/mL. Urology 1998;52:360 – 5.
Recker F, Kwiatkowski MK, Piironen T, Pettersson K, [113] Lummen G, Wernli M, et al. The importance of human glandular kallikrein and its correlation with different pros-tate specifi c antigen serum forms in the detection of pros-tate carcinoma. Cancer 1998;83:2540 – 7.

Blood-based biomarkers for prostate cancer 75
Kurek R, Nunez G, Tselis N, Konrad L, Martin T, [114] Roeddiger S, et al. Prognostic value of combined “triple”-reverse transcription-PCR analysis for prostate-specifi c antigen, human kallikrein 2, and prostate-specifi c mem-brane antigen mRNA in peripheral blood and lymph nodes of prostate cancer patients. Clin Cancer Res 2004;10:5808 – 14.
Duffy MJ. Urokinase-type plasminogen activator: A potent [115] marker of metastatic potential in human cancers. Biochem Soc Trans 2002;30:207 – 10.
Gupta A, Lotan Y, Ashfaq R, Roehrborn CG, Raj GV, [116] Aragaki CC, et al. Predictive value of the differential expres-sion of the urokinase plasminogen activation axis in radical prostatectomy patients. Eur Urol 2009;55:1124 – 33.
Steuber T, Vickers A, Haese A, Kattan MW, Eastham JA, [117] Scardino PT, et al. Free PSA isoforms and intact and cleaved forms of urokinase plasminogen activator receptor in serum improve selection of patients for prostate cancer biopsy. Int J Cancer 2007;120:1499 – 504.
Hienert G, Kirchheimer JC, Pfl uger H, Binder BR. [118] Urokinase-type plasminogen activator as a marker for the formation of distant metastases in prostatic carcinomas. J Urol 1988;140:1466 – 9.
Miyake H, Hara I, Yamanaka K, Gohji K, Arakawa S, [119] Kamidono S. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate 1999;39:123 – 9.
Shariat SF, Roehrborn CG, McConnell JD, Park S, [120] Alam N, Wheeler TM, et al. Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progres-sion, and metastasis. J Clin Oncol 2007;25:349 – 55.
Shariat SF, Shalev M, Menesses-Diaz A, Kim IY, [121] Kattan MW, Wheeler TM, et al. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol 2001;19:2856 – 64.
Truong LD, Kadmon D, McCune BK, Flanders KC, [122] Scardino PT, Thompson TC. Association of transforming growth factor-beta 1 with prostate cancer: An immunohis-tochemical study. Hum Pathol 1993;24:4 – 9.
Shariat SF, Kattan MW, Traxel E, Andrews B, Zhu K, [123] Wheeler TM, et al. Association of pre- and postoperative plasma levels of transforming growth factor beta(1) and interleukin 6 and its soluble receptor with prostate cancer progression. Clin Cancer Res 2004;10:1992 – 9.
Shariat SF, Kim JH, Andrews B, Kattan MW, Wheeler [124] TM, Kim IY, et al. Preoperative plasma levels of trans-forming growth factor beta(1) strongly predict clinical out-come in patients with bladder carcinoma. Cancer 2001;92: 2985 – 92.
Ivanovic V, Melman A, Davis-Joseph B, Valcic M, Geliebter [125] J. Elevated plasma levels of TGF-beta 1 in patients with invasive prostate cancer. Nat Med 1995;1:282 – 4.
Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, [126] Klocker H, et al. Interleukin-6 regulates prostate-specifi c protein expression in prostate carcinoma cells by activa-tion of the androgen receptor. Cancer Res 1998;58:4640 – 5.
Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine [127] growth factor in human prostate cancer. Am J Pathol 2001; 159:2159 – 65.
Michalaki V, Syrigos K, Charles P, Waxman J. Serum levels [128] of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate can-cer. Br J Cancer 2004;90:2312 – 6.
Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi [129] T, Asakura H, et al. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res 2000;6:2702 – 6.
Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, [130] Massague J, et al. Endoglin is a component of the trans-forming growth factor-beta receptor system in human endothelial cells. J Biol Chem 1992;267:19027 – 30.
Wikstrom P, Lissbrant IF, Stattin P, Egevad L, Bergh A. [131] Endoglin (CD105) is expressed on immature blood vessels and is a marker for survival in prostate cancer. Prostate 2002;51:268 – 75.
Fujita K, Ewing CM, Chan DY, Mangold LA, Partin AW, [132] Isaacs WB, et al. Endoglin (CD105) as a urinary and serum marker of prostate cancer. Int J Cancer 2009;124:664 – 9.
Karam JA, Svatek RS, Karakiewicz PI, Gallina A, [133] Roehrborn CG, Slawin KM, et al. Use of preoperative plasma endoglin for prediction of lymph node metastasis in patients with clinically localized prostate cancer. Clin Cancer Res 2008;14:1418 – 22.
Svatek RS, Karam JA, Roehrborn CG, Karakiewicz PI, [134] Slawin KM, Shariat SF. Preoperative plasma endoglin levels predict biochemical progression after radical prostatectomy. Clin Cancer Res 2008;14:3362 – 6.
Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, [135] Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology 2001;58:1008 – 15.
Shariat SF, Anwuri VA, Lamb DJ, Shah NV, Wheeler TM, [136] Slawin KM. Association of preoperative plasma levels of vascular endothelial growth factor and soluble vascular cell adhesion molecule-1 with lymph node status and biochem-ical progression after radical prostatectomy. J Clin Oncol 2004;22:1655 – 63.
Shariat SF, Walz J, Roehrborn CG, Zlotta AR, Perrotte P, [137] Suardi N, et al. External validation of a biomarker-based preoperative nomogram predicts biochemical recur rence after radical prostatectomy. J Clin Oncol 2008;26:1526 – 31.
Shariat SF, Kattan MW, Erdamar S, Nguyen C, Scardino [138] PT, Spencer DM, et al. Detection of clinically signifi cant, occult prostate cancer metastases in lymph nodes using a splice variant-specifi c rt-PCR assay for human glandular kallikrein. J Clin Oncol 2003;21:1223 – 31.
Gignac GA, Morris MJ, Heller G, Schwartz LH, Scher HI. [139] Assessing outcomes in prostate cancer clinical trials: A twenty-fi rst century tower of Babel. Cancer 2008;113:966 – 74.
Fleming MT, Morris MJ, Heller G, Scher HI. Post-therapy [140] changes in PSA as an outcome measure in prostate cancer clinical trials. Nat Clin Pract Oncol 2006;3:658 – 67.
Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto [141] S, Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009;360: 1320 – 8.

Correspondence: Anders Bjartell, Department of Urology, Sk å ne University Hospital, 205 02 Malm ö , Sweden. E-mail: [email protected]
(Received 23 September 2010 ; accepted 8 October 2010 )
REVIEW ARTICLE
Tumour markers in prostate cancer II: Diagnostic and prognostic cellular biomarkers
ANDERS BJARTELL 1 , RODOLFO MONTIRONI 2 , DANIEL M. BERNEY 3 & LARS EGEVAD 4
1Department of Urology, Sk å ne University Hospital Malm ö , Sweden, 2 Institute of Pathological Anatomy and Histopathology, School of Medicine, Polytechnic University of the Marche Region, Ancona, Italy, 3 Department of Molecular Oncology and Imaging, Institute of Cancer, Queen Mary University of London, London, UK and 4 Department of Pathology, Karolinska University Hospital, Stockholm, Sweden
Abstract The main goal of prostate cancer tissue biomarkers is to improve diagnostic and prognostic accuracy. A particularly impor-tant question is whether the cancer needs immediate treatment or if treatment can be deferred. It is highly unlikely that a single biomarker that provides comprehensive prognostic information about a newly diagnosed prostate cancer will be forth-coming. Despite extensive research efforts, very few biomarkers of prostate cancer have been successfully implemented into clinical practice today. This can be partly explained by a lack of standardised methods for performance and interpretation of immunohistochemistry, but also by poor study design with insuffi cient biomaterial or inappropriate statistical analysis. Also appropriate cohorts to test prostate cancer biomarkers do not exist. It must be kept in mind that unsuccessful integra-tion of new biomarkers in nomograms can also be explained by the good performance of the clinical and pathological base model with serum PSA as the only independent biomarker. A new biomarker must be powerful enough to improve this prediction model and not merely replace. Material and methods . In this report, we focus on diagnostic and prognostic cellular biomarkers in prostate cancer, recent advances and future aspects by reviewing currently available literature. Results. Similar to other malignancies, the proliferation marker Ki-67 seems to be a prognostic tissue biomarker and a strong candidate for integration in prediction models. Circulating tumour cells are promising markers of response to treatments in patients with metastatic disease. Conclusion . Important technical advances together with histological techniques of antibody or probes conjugated with different fl uorophores will certainly improve standardisation and make immunohistochemical biomarker research more reliable and precise in the future. Cellular biomarker studies are also expected to change in the future towards a com-plexed individualised profi ling of human tumours with integrative analysis using different technologies, genome-wide scanning and expression profi ling.
A biomarker can be defi ned as a molecular test to provide additional information over current clinical data. There is a need for biomarkers in prostate can-cer for several reasons:
1. to improve cancer detection and staging; 2. to identify subclasses of prostate cancer; 3. to predict outcome after treatment; 4. to select patients for different treatment options.
The introduction of the well established bio-marker prostate-specifi c antigen (PSA) testing has impacted the detection rate of prostate cancer and is responsible for down-staging at diagnosis, with the vast majority (approximately 80%) of newly diagnosed tumours being localised to the prostate. Gleason score
and clinical stage at the time of diagnosis are impor-tant factors to predict prognosis and outcome after therapy but additional accurate and reliable biomark-ers are warranted. An increasing proportion of tumours fall into the category of clinical stage T1c, low Gleason score (6 or less) and PSA of less than 10 ng/ml and may be considered as indolent cancers. Despite extensive research efforts, very few biomark-ers of prostate cancer have been successfully imple-mented and used in clinical practice today. In fact, the only prostate cancer biomarker routinely used in prediction models is PSA in blood.
The search for diagnostic or prognostic tissue biomarkers in prostate cancer was predominantly based on immunohistochemistry and a large number
Acta Oncologica, 2011; 50(Suppl 1): 76–84
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.531284

Prostate cancer tissue biomarkers 77
of tumour markers with prognostic information were proposed. However, a vast majority of these are not used in clinical practice, probably due to lack of stan-dardised methods to perform and interpret immuno-histochemistry, but also due to inadequate study design with insuffi cient biomaterial and inappropriate end points (e.g., biochemical recurrence instead of death) or misleading statistical analysis. The infl uence of tumour heterogeneity is yet another unsolved prob-lem, especially when only a small part of the tumour volume is available for examination; i.e., in the case of prostate biopsies. Biomarker studies after radical treat-ment often lack suffi cient end point data and only those from radical prostatectomy have plentiful tissue. Diagnostic tissue specimens are all that is available for those treated by radiotherapy, hormone manipu-lation or active surveillance. Subsequently, many reports on promising prognostic tissue biomarkers were never successfully reproduced and validation studies are still missing for most prostate cancer tissue biomarkers.
In 2005, the Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics encour-aged transparent and complete reporting of prognostic tumour marker research by publishing the REMARK document “ Reporting recommendations for tumor MARKer prognostic studies ” , which led to a general reduction in the number of studies describing new immunohis-tochemical markers [1]. More recently, a systematic and comprehensive review recently focused on the use of classical and novel biomarkers as prognostic risk factors for localised prostate cancer and highlighted the poor quality and heterogeneity of studies, which render much of the results inconclusive [2].
In the era of ” omics ” (genomics, proteomics, meta-bolomics, pharmacogenomics, etc.), tissue-based bio-marker research has evolved dramatically. Recent advances in high-throughput technology, microarrays and bioinformatics have opened the door to gain much more information from an individual tumour and to develop multiplex tests to obtain a complete charac-terisation and to predict the behaviour of prostate cancer.
In this report, we focus on a selection of established and promising tissue biomarkers in prostate cancer, recent advances and future aspects. Areas of research based on novel and complexed technology like proteo-mics and genomics will not be covered in this report.
Diagnostic prostate cancer markers
The diagnosis of prostate cancer in histopathological specimens is mainly based on a combination of archi-tectural and cellular atypia. In the vast majority of cases a confi dent diagnosis can be made based on mor-phology alone. However, in some cases the morphologi-cal fi ndings are insuffi cient for a conclusive diagnosis,
either because the atypia is too mild or the atypical focus too small. In a review of the literature, atypia suspicious for cancer was found to have been reported in an average of 5% of needle biopsy series [3]. The diagnostic accuracy can be improved in some of these cases by immunohistochemistry using one or several biomarkers, either on consecutive sections or using a staining cocktail.
High molecular weight cytokeratin
Invasive prostate cancer almost invariably lacks basal cell layer. The cytokeratin profile of basal cells of prostatic glandular epithelium differs from that of luminal cells. The basal cell layer can be labelled with antibodies against high molecular weight cytokeratin or cytokeratin 5. In rare cases (0.3%) there is an aberrant labelling of cancer cells by high molecular weight cytokeratin but the band-like basal cell distri-bution is not seen [4]. More problematic is that benign lesions such as partial atrophy may be negative for high molecular weight cytokeratin [5]. Thus, it is impor-tant that results with this biomarker is interpreted with great caution and carefully correlated with con-ventional morphology.
p63
An alternative to high molecular weight cytokeratin as basal cell marker is p63, a homologue to p53. This biomarker can be used either alone or in a cocktail with high molecular weight cytokeratin [6]. Similar to high molecular weight cytokeratin false negative stains of benign glands occur and correlation with morphol-ogy is of paramount importance. Aberrant diffuse expression of p63 in prostate carcinoma has been described [7]. The sensitivity of p63 and high molec-ular weight cytokeratin is comparable [8].
AMACR
Alfa-methylacyl-CoA-racemase (AMACR) is expressed in cancer and usually negative in benign glands [9]. Staining for AMACR in atypical cases with negative basal cell markers can convert the diagnosis from atypical to cancer in 50% of needle biopsy cases reviewed by an expert uropathologist. The sensitivity varies between 80 – 100% in small atypical foci on nee-dle biopsy [10]. However, variants of prostate cancer that are more diffi cult to diagnose are unfortunately less often positive: 62 – 68% of foamy gland prostate cancers and 70 – 77% of pseudohyperplastic prostate cancers [11]. AMACR is also positive in PIN and occasionally in benign lesions such as atrophy and adenosis, hence, similar to basal cell markers, the results have to be interpreted with caution and carefully com-pared with hematoxylin and eosin stained sections.

78 A. Bjartell et al.
Prognostic tissue biomarkers
Tissue biomarkers may be of value to predict the effect of certain treatments but also as a general prognostic tool. The natural history of prostate cancer has shown that the molecular determinants may change with time by clonal selection of tumour cell populations towards a more aggressive phenotype. However, we also know that some prostate cancers show an aggres-sive behaviour already at an early stage and there is an urgent need to identify tumours that need imme-diate treatment.
Kallikreins
The human tissue kallikrein KLK gene locus consists of 15 genes on chromosome 19q13-4 [12]. The best known of these genes is kallikrein 3 (KLK3), also known as prostate-specifi c antigen (PSA), that is a widely used clinical tumour marker in serum for detection and monitoring of prostate cancer progression. PSA is produced in luminal cells of the benign epithelium and in most tumour cells. Thus it has a prostate-specifi c but not a tumour specifi c expression, which explains the low specifi city of serum PSA measurement to detect small tumours in patients with benign prostatic hyperplasia (BPH). Human kallikrein type 2 (hK2) is similar to PSA in structure and is overexpressed in tumour cells [13]. In an immunohistochemical study hK2 was expressed in every cancer, and the expres-sion incrementally increased from benign epithelium to primary cancer and lymph node metastases [14]. Lintula et al. demonstrated that the ratio of hK2/PSA mRNA increased with grade of tumour [15]. Using a large tissue microarray (TMA) containing samples of 3 261 prostatectomy specimens Erbersdobler and co-workers showed that the loss of PSA expression in tissue samples of prostate cancer is associated with adverse pathologic features and clinical outcome but is not an independent prognostic factor for PSA recurrence after prostatectomy [16]. Immunohis-tochemical studies on other kallikrein family mem-bers have also been suggested to be of prognostic value in prostate cancer patients but validation studies are still lacking.
Prostate-specifi c membrane antigen (PSMA)
The prostate specifi c membrane antigen (PSMA) is a cell surface membrane protein. It is an attractive antigen for antibody-based diagnostic and therapeu-tic intervention in prostate cancer, since it is highly restricted to the prostate and overexpressed in all tum-our stages [17]. In a TMA study, high PSMA expres-sion was independently associated with PSA recurrence in a high-risk cohort and thus might provide insight into the additional use of adjuvant therapy in this
group of patients [18]. Again, validation on other cohorts is required and further evaluation of this marker is needed to determine whether or not it has clinical utility for prostate cancer detection or treat-ment monitoring.
Ki-67
The Ki-67 protein is well known and widely used to assess the tumour proliferation rate and numerous studies have shown Ki-67 to be a prognostic marker in prostate cancer patients treated by radical pros-tatectomy, radiotherapy or androgen deprivation ther-apy (ADT). In a cohort of 808 patients diagnosed by TURP treated conservatively, Ki-67 expression was assessed immunohistochemically in two laboratories, by two different scoring methods and the results com-pared with cancer-specifi c and overall survival [19]. Using both methods, Ki-67 provided additional prog-nostic information beyond that available from Glea-son score, PSA and tumour extent. Although it was confi rmed to be the most promising biomarker, con-fi rmatory studies need to occur in TRUS biopsy mate-rial in order for prospective studies to implement use into routine practice and prediction models.
Androgen receptor
Expression of kallikreins and several other gene products in the benign and malignant prostate gland are dependent on the androgen receptor (AR) func-tion. Studies in animals and humans have demon-strated that AR plays a key role in prostate cancer progression with an overexpression in castration-resistant prostate cancer (CRPC) [20]. However, expression of AR may be of prognostic value also in hormone-na ï ve prostate cancer. Using a total of 640 cases in a TMA setting, Li et al. correlated levels of AR expression with some well-established clinical and pathologic para meters in patients treated with radical prostatectomy [21]. High levels of AR status correlated with proliferation (high Ki-67 index) and a high level of AR expression was an independent predictor of decreased biochemical recurrence-free survival. The prognostic role of AR in prostate cancer was recently confi rmed in a study from an indepen-dent group who described that increased nuclear AR expression in either the diagnostic biopsy and/or radical prostatectomy specimen, from patients with advanced disease, was associated with a reduced time to prostate cancer-specifi c mortality [22]. Simple immunohistochemical analysis of AR may not be suf-fi cient to evaluate AR function, because tumour cells may harbour differences in the AR genotype with variation in the length of CAG repeat, mutations and increased copy number.

Prostate cancer tissue biomarkers 79
Gene fusions
The bioinformatic tool Cancer Outlier Profi le Analysis (COPA) was developed to analyse DNA microarray data for outlier genes and used when they presented the fi rst evidence of current rearrangements in com-mon epithelial tumours as demonstrated by fl uores-cent in situ hybridisation (FISH) [23]. Gene fusions involving members of the erythroblast transforma-tion-specifi c (ETS) family of transcription factors were reported to occur in prostate cancer and with the gene fusion of the 5 ́ -untranslated region of the androgen-regulated gene TMPRSS2 and the ETS family gene ERG as the predominant fi nding (approximately 90%). Several studies have confi rmed the presence of TMPRSS2 – ERG fusions in 36 – 78% of prostate cancers from PSA screened surgical cohorts. Confl ict-ing results have been reported regarding the prog-nostic signifi cance of prostate cancers harbouring TMPRSS2-ETS gene fusions. Duplication of the fusion TMPRSS2 to ERG sequences was shown to identify fatal human prostate cancer in a cohort on 445 men conservatively treated for prostate cancer [24] but in a recent TMA study of 521 cases of clin-ically localised prostate cancer, Fine and co-workers found that TMPRSS2 – ERG fusion was associated with low Gleason scores and not with high-grade morphological features [25]. Taken together, is seems like the TMPRSS2 – ERG fusion gene is an early event related to development of prostate cancer rather than a marker for progressive disease. Duplication of the gene may be merely an indicator of overall genetic instability, aneuploidy and hence, poor prognosis. Several strategies for therapeutically targeting ETS fusions have been identifi ed and are currently being pursued.
PTEN
The phosphatidylinositol 3-kinase (PI3K) is a frequently activated signal transduction pathway in prostate can-cer cells [26] and the most common mechanism of PI3K activation is deletion of the gene encoding the phosphatase and tensin homologue (PTEN) protein. PTEN deletion has been reported to be more fre-quent in metastatic than in organ-confi ned prostate cancer [27]. Decreased expression of PTEN and consequent activation of the PI3K pathway members in prostate cancer tissue samples has been correlated to higher Gleason grade, advanced stage, and devel-opment of androgen resistance [28,29]. These dis-coveries have already been shown to have clinical impact and several phase 1 and 2 clinical trials with specifi c agents targeting key activated proteins in the PI3K pathway are ongoing in prostate cancer patients [30]. A recent fi nding that may have impact on future studies is that PTEN deletion, cooperates with aberrant
ERG expression to promote prostate cancer progres-sion [31]. Use of molecular biomarkers related to the PI3K pathway, may facilitate future selection of patients for these novel therapeutic regimens.
p53
The tumour suppressor gene p53 is mutated in half of human malignancies and has been intensively studied in numerous cancer models. Many of these studies have suggested that p53 may be of prognostic value in prostate cancer after different treatments. In a more recent study on patients treated conserva-tively for prostate cancer, p53 remained prognosti-cally signifi cant in a multivariate model though the risk ratios were less strong compared to Ki-67 [32].
SPINK1/TATI
Tumour-associated trypsin inhibitor (TATI), which is alternatively called pancreatic secretory trypsin inhi-bitor (PSTI) or serine protease inhibitor Kazaltype 1 (SPINK1), is expressed in various normal and malig-nant tissues, and is known as a prognostic tumour marker as reviewed by Paju and Stenman [33]. TATI was fi rst shown to be overexpressed in high-grade prostate cancer [34] and later, outlier expression of SPINK1, the gene coding for the TATI protein was identifi ed exclusively in a subset of ETS rearrange-ment-negative cancers (approximately 10% of total cases) [35]. A series of in vitro and in vivo experi-ments revealed SPINK1/ TATI to be associated with prostate cancer aggressiveness and with invasive growth in a prostate cancer cell line (22RV1) with outlier expression. It was thus shown that SPINK1 outlier expression defi nes an aggressive molecular subtype of prostate cancer (approximately 10% of cases) not attributable to known gene fusion events. Further support of the potential role of SPINK1 expression as a prognostic biomarker was recently demonstrated by Leinonen et al. who studied a cohort of men with endocrine-treated prostate cancer [36]. Additional work is needed to understand the tumour biology behind the role of SPINK1 /TATI as a tissue biomarker in pro-state cancer.
MSMB and EZH2
MSMB (also known as prostate specifi c protein of 94 amino acids (PSP94)) is expressed in benign and malignant prostatic epithelium and is along with pros-tate specifi c antigen (PSA) and prostate acidic phos-phatase (PAP) one of the three most abundant proteins in human seminal plasma [37]. Additionally, MSMB , located at chromosome 10q11.2, has recently been reported as an important candidate gene for prostate

80 A. Bjartell et al.
cancer susceptibility [38,39]. Several groups have employed a variety of approaches in both tissue and serum samples to demonstrate decreasing levels of MSMB in prostate cancer compared to normal con-trols, prompting the suggestion that MSMB may be a promising biomarker for prostate cancer. In a large TMA study of almost 1 000 patients with localised prostate cancer, MSMB was found to be an indepen-dent predictor of recurrence after radical prostatec-tomy, however, addition of MSMB did not importantly improve the performance of existing predictive mod-els [40]. Molecular mechanisms behind the decreased expression of MSMB in prostate cancer is not yet fully clarifi ed. In advanced, castration-resistant pros-tate cancer decrease expression of MSMB was found to correlate with an increased expression of the Poly-comb gene (PcG) protein EZH2 (enhancer of zeste homolog 2), which represses transcription via trim-ethylation of histone H3 on Lys27 (H3K27) [41]. EZH2 has also been attributed the role of a useful tissue biomarker in prostate cancer [42]. Furthermore, EZH2 has also been suggested to silence expression of E-cadherin by trimethylation of H3 lysine 27, providing a functional link between dysregulation of EZH2 and repression of E-cadherin during cancer progression.
Epigenetic silencing of genes by EZH2 and acti-vation of members of the PcG group may be common oncogenic events in pathogenesis of metastatic solid tumours and provide justifi cation for development of small molecule inhibitors of the PcG chromatin silenc-ing pathway as a novel therapeutic modality for treat-ment of metastatic prostate cancer [43].
Heat shock proteins
Heat shock proteins (HSPs) are molecular chaper-ones, protecting cells against stress-related injury [44]. Although HSPs are important for the function of nor-mal cells, cancer cells frequently overexpress HSPs in response to stress and may thereby gain a survival benefi t. It has been demonstrated by proteomics that prostate cancer overexpress HSP60 and HSP70 [45]. In a validation study using immunohistochemistry, HSP60 was an independent predictor of biochemical recurrence after radical prostatectomy in multivariate analysis including extraprostatic extension, margin sta-tus, seminal vesicle invasion and Gleason score, while HSP27 correlated with outcome in univariate analy-sis [46]. Interestingly, HSPs are potential therapeutic targets in prostate cancer patients. Inhibition of the function of HSPs may decrease tumour cell survival by blocking the antiapoptotic activity of HSPs [47]. HSP90 is suggested to play a key role in prostate cancer through interaction with AR and recently designed mito-chondrial Hsp90 chaperones are potentially attractive therapeutic targets in advanced prostate cancer [48].
DNA methylation
Epigenetic events that can affect gene expression without altering the actual sequence of DNA include phenomena such as DNA methylation, chromatin remo deling, histone modifi cation and RNA interfer-ence [49]. Many gene promoters are associated with GC rich regions of the DNA known as CpG islands. Abnormal methylation of CpG islands located within gene promoters is associated with decreased transcri-ptional activity and it occurs in many types of cancers. Abnormal methylation of genes such as those involved with control of cellular growth or detoxifi cation is believed to have a critical role in early stages of PCa progression. Already in 1994, glutathione S-transferase P (GSTP1) hypermethylation was introduced as a central part of prostate carcinogenesis [50]. GSTP1 is a member of a large family of glutathione transferases that function to protect cells from oxidative insult thus, the biological rationale for selecting this marker is its role in preventing damage to cells by neutralising free radicals. GSTP1 has been extensively studied in pros-tate cancer, and its reduced expression, due predom-inantly to promotor hypermethylation, is the most com mon epigenetic alteration associated with this dis-ease. Several studies have shown a high sensitivity for this marker to detect the presence of both PIN and prostate cancer, an ability to distinguish these from BPH, and a prevalence of methylation in the range of 60 – 80% in prostate cancer. Strengths of GSTP1 as a clinical marker are the ability to quantitate the methylation status of the GSTP1 gene in biopsy/prostatectomy tissues and in cells derived from serum, urine, and seminal plasma. Recent studies using quan-titative real-time methylation sensitive PCR demon-strate that epigenetically modifi ed genes are candidate markers for early detection and post-treatment mon-itoring of prostate cancer, however it is more likely that urine and serum will be used instead of tissue samples. This role of hypermethylated genes as prom-ising biomarkers was recently reviewed by Ahmed who emphasised that in addition to clinical validation, assays for methylated genes must be robust, simple, sensitive, specifi c, and made available at affordable costs [51].
HER2
Overexpression, or gene amplifi cation of the human epidermal growth factor receptor 2 (HER2) is evident in 20 – 25% of breast cancers and correlated with dis-ease outcome. Validated methods for determination of HER2-status by immunohistochemistry and/or FISH are used to identify patients with breast cancer who are eligible for treatment with trastuzumab, an HER2-targeted monoclonal antibody that inhibits the proliferation of tumour cells and induces tumour

Prostate cancer tissue biomarkers 81
1. Can the marker be measured accurately and repro ducibly? The value of each biomarker must be tested in large annotated series of patients with reliable independent validation sets. The assay must be robust and reproducible.
2. Does the marker provide additional information to that already available to the clinician? An important step in evaluating the value of a new marker is to compare the predictive accuracy of a model including only standard clinical variables with that of a model including standard clinical var-iables plus the new marker [57]. The statistical analyses must be robust and demonstrate a sig-nifi cantly improvement in receiver operation characteristics (ROC) analyses or Concordance Index (CI) when the new biomarkers is added to the base model.
3. Is the marker associated with outcome in the sort of patients to whom the marker would be applied to in clinical practice? Before we use a marker in clinical practice, we need to know whether it will improve clinical outcome. In other words, we need evidence that measuring the marker would change the decision a doctor would have made in the absence of the marker, and that this changed decision will benefi t the patient. If an assay failed to give reproducible results, it would clearly be of little clinical value and would not be worthy of subsequent research.
Conclusions and future aspects
It is unlikely that a single biomarker will provide all information we need to tell how aggressive a newly diagnosed prostate cancer is. No immunochemical, or genetic marker is currently used to differentiate between aggressive and non-aggressive prostate cancers.
Recent discoveries in genetics, proteomics and bio in formatics using new high-throughput arrays indicate that new multiplex tests will soon be launched and some even commercially available, however, the above mentioned criteria for evalua-tion of new biomarkers must always be strictly fol-lowed. Tissue biomarker research will change in the future as illustrated in a recent report by Taylor and co-workers who used different new technologies for integrative genomic profi ling of human prostate cancer and identifi ed new candidate biomarkers in prostate cancer [58]. Technologies of genome-wide scanning such as gene expression profiling, com-parative genomic hybridisation (CGH), and single nucleotide polymorphism (SNP) arrays are now also applicable to nucleic acids extracted from archival tissues on large series of prostate cancer patients.
cell death through multiple mechanisms. HER2 has also been linked to the clinical progression of CRPC. In a recent meta-analysis, Serpa Neto and collabora-tors [52] investigated the prognostic impact of HER2 over expression in patients with prostate cancer and its correlation with other pathological and clinical variables. Several databases were searched for studies of HER2 protein expression in primary prostate can-cer tissue. The overall relative risk of death in those with HER2 over expression in the primary tumour was 1.63 (95% CI 1.47 – 1.82, p � 0.0001) and the authors found a consistent association of HER2 over-expression with death and recurrence. The clinical usefulness of HER2-status in prostate cancer patients still has to be proven.
NKX3.1 and MYC
Losses of 8p and gains of 8q are two of the most common chromosome aberrations in prostate cancer [53]. Gene products related to these loci are candidates as new important biomarkers in prostate cancer. NKX3.1 (8p21) is an androgen-regulated transcrip-tion factor that regulates the proliferation rate of pros-tate luminal epithelial cells and functions as a tumour suppressor gene [54]. Experimental work from dif-ferent groups shave demonstrated the importance of NKX3.1 in development of prostate cancer, but stud-ies on its role as a tissue biomarker have generated confl icting results.
The well-known oncogene MYC resides on the short arm of chromosome 8. MYC is a well-known regulator of proliferation and biologic activity in prostate cancer cells, and its amplifi cation is associ-ated with the presence of PIN and poor clinical outcome of prostate cancer [55]. Similar to NKX3.1, additional studies are needed to understand how MYC can be clinically used as a tissue biomarker in prostate cancer.
Validation and implementation of tissue biomarkers
Examples of statistical models to predict outcome in prostate cancer patients are the clinical nomograms (www.nomograms.org) [56]. These models incorpo-rate known prognostic variables, such as PSA, Gleason grade, extracapsular extension, lymph node involve-ment, and surgical margins to estimate the probabil-ity of disease recurrence following prostatectomy and other treatments. Unsuccessful integration of new biomarkers in nomograms can be explained by the already high performance of the clinical and patho-logical base models with strong independent biomark-ers. Several questions must be satisfactorily met before a new biomarker can be implemented:

82 A. Bjartell et al.
Development of TMA for large-scale analysis and automated image analysis systems for more precise quantitation and a direct transformation of scoring data to biostatistical analysis are now progressively replacing the subjective, semiquantitative manual scor-ing previously performed by pathologist. These impor-tant technical advances together with histological techniques of antibody or probes conjugated with nanoparticles (quantum dots) and other fl uorescent conjugates will certainly improve the standardisation and make immunohistochemical biomarker research more reliable and precise in the future.
Another challenge is to identify a specifi c tissue biomarker for prostate cancer stem cells. Today it seems like a combination of different markers is needed to identify a cancer stem cell population. Uglokov et al. [59] performed immunohistochemical analysis of pro-posed stem cell markers (CD44, CD133, Oct4, SOX2 and EZH2) in benign and malignant prostatic tissues and suggested that suggest that combined expression of embryonic stem cell markers EZH2 and SOX2 might identify potential cancer stem cells as a minor ( � 10%) subgroup in CD44 � prostatic adenocarci-noma. A new and different approach is aldehyde-dehydrogenase (ALDH) – based sorting of human prostate cells by fl ow cytometry (ALDEFLUOR assay) which is used to simultaneously select putative pros-tate cancer stem cells and metastasis-initiating cells [60]. Analysis of ALDH activity of clinical prostate cancer samples may thus become useful for the strat-ifi cation of prostate cancer patients at risk of develop-ing metastatic disease.
An important question that remains to be answered is the cellular origin of human prostate cancer. Using a combination of immunohistochemical and biochem-ical studies in vitro and in vivo, an American group recently provided evidence that prostate tumours orig-inate from the basal cell layer and not from the lumi-nal cell compartment as earlier believed [61]. Their discoveries may impact future strategies in search for new prostate cancer biomarkers.
Another complex but highly interesting area of research with great expectations in the near future relates to microRNA and other forms of non-coding RNAs which hypothetically can act as important regulators of other biomarkers, but can also function as biomark-ers themselves and as therapeutical targets [62].
Circulating tumour cells (CTC)
The shedding of tumour cells into the circulation is a necessary condition for metastatic spread. Recently, the enumeration of circulating tumour cells (CTC) using the CellSearch ™ system which has been cleared by the US Food and Drug Administration as a prog-nostic clinical biomarker that can be used to monitor
the effectiveness of therapy in patients with meta-static prostate cancer [63]. Although changes in CTC counts over time are predictive of survival, it is the molecular profi ling of these cells that offers insight into the biological status of the tumour. Effective molec-ular profi ling of CTC is complicated by the relatively low cell numbers involved and by dilution from non-tumour material because the current enrichment procedures cannot eliminate all contaminating leu-kocytes. Further, as patients with advanced disease may shed tumour cells from multiple sites, the CTC detected may have diverse characteristics, further diluting potential molecular biomarkers and compli-cating their predictive utility. In a study of 77 patients with progressive castration-resistant disease, Scher and his group have shown that CTC numbers can be monitored in a routine clinical laboratory setting and that cells confi rmed by immunohistochemistry to have features of prostate cancer can be sampled for genetic profi ling by FISH [64]. FISH could be done on CTC (success rate � 87%) supporting its use in the routine management of progressive castra-tion-resistant prostate cancer [65]. The CTC tech-nology is currently evaluated and validated in clinical trials as a predictor and surrogate end point of treat-ment response.
In conclusion, the search for new cellular bio-markers in prostate cancer has resulted in a prolif-eration of tumour marker studies. In order for markers to be successfully incorporated into clinical practice, we need evidence that measuring the marker would change the decision a doctor would have made in the absence of the marker, and that this changed decision will benefi t the patient.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, [1] Clark GM. Statistics subcommittee of the NCI-EORTC working groups on cancer diagnostics. Reporting recommen-dations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst 2005;97:1180 – 4. Sutcliffe P, Hummel S, Simpson E, Young T, Rees A, [2] Wilkinson A, et al. Use of classical and novel biomarkers as prognostic risk factors for localised prostate cancer: A sys-tematic review. Health Technol Assess 2009:13:1 – 219. Epstein JI, Herawi M. Prostate needle biopsies containing pro-[3] static intraepithelial neoplasia or atypical foci suspicious for car-cinoma: Implications for patient care. J Urol 2006;175:820 – 34. Ali TZ, Epstein JI. False positive labeling of prostate cancer [4] with high molecular weight cytokeratin: p63 a more specifi c immunomarker for basal cells. Am J Surg Pathol 2008;32:1890 – 5.

Prostate cancer tissue biomarkers 83
Wang W, Sun X, Epstein JI. Partial atrophy on prostate nee-[5] dle biopsy cores: A morphologic and immunohistochemical study. Am J Surg Pathol 2008;32:851 – 7. Zhou M, Shah R, Shen R, Rubin MA. Basal cell cocktail [6] (34betaE12 � p63) improves the detection of prostate basal cells. Am J Surg Pathol 2003;27:365 – 71. Osunkoya AO, Hansel DE, Sun X, Netto GJ, Epstein JI. [7] Aberrant diffuse expression of p63 in adenocarcinoma of the prostate on needle biopsy and radical prostatectomy: Report of 21 cases. Am J Surg Pathol 2008;32:461 – 7. Wu HH, Lapkus O, Corbin M. Comparison of 34[beta]E12 [8] and P63 in 100 consecutive prostate carcinoma diagnosed by needle biopsies. Appl Immunohistochem Mol Morphol 2004;12:285 – 9. Zhou M, Aydin H, Kanane H, Epstein J. How often does [9] alpha-methylacyl-CoA-racemase contribute to resolving an atypical diagnosis on prostate needle biopsy beyond that pro-vided by basal cell markers? Am J Surg Pathol 2004;28:239 – 43. Magi-Galluzzi C, Luo J, Isaacs WB, Hicks JL, de Marzo AM, [10] Epstein JI. Alpha-methylacyl-CoA racemase: A variably sen-sitive immunohistochemical marker for the diagnosis of small prostate cancer foci on needle biopsy. Am J Surg Pathol 2003;27:1128 – 33. Zhou M, Jiang Z, Epstein JI. Expression and diagnostic util-[11] ity of alpha-methylacyl-CoA-racemase (P504S) in foamy gland and pseudohyperplastic prostate cancer. Am J Surg Pathol 2003;27:772 – 8. Harvey TJ, Hooper JD, Myers SA, Stephenson SA, Ashworth LK, [12] Clements JA. Tissue-specifi c expression patterns and fi ne mapping of the human kallikrein (KLK) locus on proximal 19q13.4. J Biol Chem 2000;275:37397 – 406. Darson MF, Pacelli A, Roche P, Rittenhouse HG, Wolfert RL, [13] Young CY, et al. Human glandular kallikrein 2 (hK2) expres-sion in prostatic intraepithelial neoplasia and adenocarcinoma: A novel prostate cancer marker. Urology 1997;49:857 – 62. Darson MF, Pacelli A, Roche P, Rittenhouse HG, Wolfert RL, [14] Saeid MS, et al. Human glandular kallikrein 2 expression in prostate adenocarcinoma and lymph node metastases. Urol-ogy 1999;53:939 – 44. Lintula S, Stenman J, Bjartell A, Nordling S, Stenman UH. [15] Relative concentrations of hK2/PSA mRNA in benign and malignant prostatic tissue. Prostate 2005;63:324 – 9. Erbersdobler A, Isbarn H, Steiner I, Schlomm T, Chun F, [16] Mirlacher M, et al. Predictive value of prostate-specifi c anti-gen expression in prostate cancer: A tissue microarray study. Urology 2009;4:1169 – 73. Bostwick DG, Pacelli A, Blute M, Roche P, Murphy GP. [17] Prostate specifi c membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: A study of 184 cases. Cancer 1998;82:2256 – 61. Perner S, Hofer MD, Kim R, Shah RB, Li H, M ö ller P, et al. [18] Prostate-specifi c membrane antigen expression as a predictor of prostate cancer progression. Hum Pathol 2007;38:696 – 701. Berney DM, Gopalan A, Kudahetti S, Fisher G, Ambroisine L, [19] Foster CS, et al. Ki-67 and outcome in clinically localised prostate cancer: Analysis of conservatively treated prostate cancer patients from the Trans-Atlantic Prostate Group study. Br J Cancer 2009;100:888 – 93. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, [20] et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33 – 9. Li R, Wheeler T, Dai H, Frolov A, Thompson T, Ayala G. [21] High level of androgen receptor is associated with aggressive clinicopathologic features and decreased biochemical recur-rence-free survival in prostate: Cancer patients treated with radical prostatectomy. Am J Surg Pathol 2004;28:928 – 34.
Donovan MJ, Osman I, Khan FM, Vengrenyuk Y, Capodieci P, [22] Koscuiszka M, et al. Androgen receptor expression is associated with prostate cancer-specifi c survival in castrate patients with metastatic disease. BJU Int 2010;105:462 – 7. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, [23] Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310(5748):644 – 8. Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, Flohr P, [24] et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifi es fatal human prostate cancer. Oncogene 2008;27:253 – 63. Fine SW, Gopalan A, Leversha MA, Al-Ahmadie HA, [25] Tickoo SK, Zhou Q, et al. TMPRSS2-ERG gene fusion is associated with low Gleason scores and not with high-grade morphological features. Mod Pathol Epub 2010 Jun 18. Lee JT Jr, Steelman LS, McCubrey JA. Phosphatidylinositol [26] 30-kinase activation leads to multidrug resistance protein-1 expression and subsequent chemoresistance in advanced prostate cancer cells. Cancer Res 2004;64:8397 – 404. Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, [27] Soares FA, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer 2007;97:678 – 85. McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, [28] Sellers WR. Loss of PTEN expression in paraffi n-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res 1999;59:4291 – 6. Bertram J, Peacock JW, Fazli L, Mui AL, Chung SW, Cox ME, [29] et al. Loss of PTEN is associated with progression to andro-gen independence. Prostate 2006;66:895 – 902. Morgan TM, Koreckij TD, Corey E. Targeted therapy for [30] advanced prostate cancer: Inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets 2009;9:237 – 49. Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, [31] et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 2009;41:619 – 24. Kudahetti S, Fisher G, Ambroisine L, Foster C, Reuter V, [32] Eastham J, et al. p53 immunochemistry is an independent prognostic marker for outcome in conservatively treated prostate cancer. BJU Int 2009;104:20 – 4. Paju A, Stenman UH. Biochemistry and clinical role of [33] trypsinogens and pancreatic secretory trypsin inhibitor. Crit Rev Clin Lab Sci 2006;43:103 – 42. Paju A, Hotakainen K, Cao Y, Laurila T, Gadaleanu V, [34] Hemminki A, et al. Increased expression of tumor-associated trypsin inhibitor, TATI, in prostate cancer and in androgen-independent 22Rv1 cells. Eur Urol 2007;52:1670 – 9. Tomlins SA, Rhodes DR, Yu J, Varambally S, Mehra R, [35] Perner S, et al. The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell 2008;13:519 – 28. Leinonen KA, Tolonen TT, Bracken H, Stenman UH, [36] Tammela TL, Saram ä ki OR, et al. Association of SPINK1 expression and TMPRSS2:ERG fusion with prognosis in endocrine-treated prostate cancer. Clin Cancer Res 2010;16:2845 – 51. Lilja H, Abrahamsson P-A. Three predominant proteins secreted [37] by the human prostate gland. Prostate 1988;12:29 – 38. Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, [38] et al. Multiple loci identifi ed in a genome-wide association study of prostate cancer. Nat Genet 2008;40:310 – 5. Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, [39] Jugurnauth SK, et al. Multiple newly identifi ed loci associated with prostate cancer susceptibility. Nat Genet 2008;40:316 – 21. Bjartell AS, Al-Ahmadie H, Serio AM, Eastham JA, Eggener SE, [40] Fine SW, et al. Association of cysteine-rich secretory protein

84 A. Bjartell et al.
3 and beta-microseminoprotein with outcome after radical prostatectomy. Clin Cancer Res 2007;13:4130 – 8. Beke L, Nuytten M, Van Eynde A, Beullens M, Bollen M. [41] The gene encoding the prostatic tumor suppressor PSP94 is a target for repression by the Polycomb group protein EZH2. Oncogene 2007;26:4590 – 5. Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R, et al. [42] A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res 2007;67:10657 – 63. Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, [43] et al. Repression of E-cadherin by the polycomb group pro-tein EZH2 in cancer. Oncogene 2008;27:7274 – 84. Fuller KJ, Issels RD, Slosman DO, Guillet JG, Soussi T, [44] Polla BS. Cancer and the heat shock response. Eur J Cancer 1994;30A:1884 – 91. Lexander H, Palmberg C, Auer G, Hellstrom M, Franzen B, [45] Jornvall H, et al. Proteomic analysis of protein expression in prostate cancer. Anal Quant Cytol Histol 2005;27:263 – 72. Glaessgen A, Jonmarker S, Lindberg A, Nilsson B, Lewensohn R, [46] Ekman P, et al. Heat shock proteins 27, 60 and 70 as prognos-tic markers of prostate cancer. APMIS 2008;116:888 – 95. Westerheide SD, Morimoto RI. Heat shock response modu-[47] lators as therapeutic tools for diseases of protein conforma-tion. J Biol Chem 2005;280:33097 – 100. Leav I, Plescia J, Goel HL, Li J, Jiang Z, Cohen RJ, et al. [48] Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am J Pathol 2010;176:393 – 401. Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N [49] Engl J Med 2003;349:366 – 81. Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, [50] Bova GS, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci USA 1994;91:11733 – 7. Ahmed H. Promoter methylation in prostate cancer and its [51] application for the early detection of prostate cancer using serum and urine samples. Biomark Cancer 2010;2:17 – 33. Serpa Neto A, Tobias-Machado M, Wroclawski ML, Affonso [52] Fonseca FL, Teixeira GK, Dal Moro Amarante R, et al. HER-2/neu expression in prostate adenocarcinoma: A sys-tematic review and meta-analysis. J Urol Epub 2010 Jul 19. Ribeiro FR, Henrique R, Hektoen M, Berg M, Jer ó nimo C, [53] Teixeira MR, et al. Comparison of chromosomal and array-based
comparative genomic hybridization for the detection of genomic imbalances in primary prostate carcinomas. Mol Cancer 2006; 5:33. Ouyang X, DeWeese TL, Nelson WG, Abate-Shen C. Nkx3.1 [54] promotes increased oxidative damage in prostate carcinogen-esis. Cancer Res 2005;65:6773 – 9. Sato K, Qian J, Slezak JM, Lieber MM, Bostwick DG, [55] Bergstralh EJ, et al. Clinical signifi cance of alterations of chromosome 8 in highgrade, advanced, nonmetastatic prostate carcinoma. J Natl Cancer Inst 1999;91:1574 – 80. Shariat SF, Karakiewicz PI, Margulis V, Kattan MW. Inven-[56] tory of prostate cancer predictive tools. Curr Opin Urol 2008;18:279 – 96. Kattan MW. Judging new markers by their ability to improve [57] predictive accuracy. J Natl Cancer Inst 2003;95:634. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, [58] Carver BS, et al. Integrative genomic profi ling of human prostate cancer. Cancer Cell 2010;18:11 – 22. Ugolkov AV, Eisengart LJ, Luan C, Yang XJ. Expression [59] analysis of putative stem cell markers in human benign and malignant prostate. Prostate Epub 2010 Jun 25. van den Hoogen C, van der Horst G, Cheung H, Buijs JT, [60] Lippitt JM, Guzm á n-Ram í rez N, et al. High aldehyde dehy-drogenase activity identifi es tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res 2010;70:5163 – 73. Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. [61] Identifi cation of a cell of origin for human prostate cancer. Science 2010;329(5991):568 – 71. Gandellini P, Folini M, Zaffaroni N. Emerging role of micro-[62] RNAs in prostate cancer: Implications for personalized med-icine. Discov Med 2010;9:212 – 8. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, [63] Tissing H, et al. Circulating tumor cells predict survival ben-efi t from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 2008;14:6302 – 9. Shaffer DR, Leversha MA, Danila DC, Lin O, Gonzalez-[64] Espinoza R, Gu B, et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin Cancer Res 2007;13:2023 – 9. Leversha MA, Han J, Asgari Z, Danila DC, Lin O, Gonzalez-[65] Espinoza R, et al. Fluorescence in situ hybridization analysis of circulating tumor cells in metastatic prostate cancer. Clin Cancer Res 2009;15:2091 – 7.

Correspondence: Monique J. Roobol, Erasmus MC, University Medical Centre, Department of Urology, P.O. Box 2040, 3000 CA Rotterdam, The Netherlands. E-mail: [email protected]
(Received 15 August 2010 ; accepted 6 September 2010 )
REVIEW ARTICLE
Tumour markers in prostate cancer III: Biomarkers in urine
MONIQUE J. ROOBOL 1 , ALEXANDER HAESE 2 & ANDERS BJARTELL 3
1 Erasmus MC, University Medical Centre, Department of Urology, Rotterdam, The Netherlands, 2Martini Clinic Prostate Cancer Center University Clinic Hamburg Eppendorf, Germany and 3 Department of Urology, Sk å ne University Hospital Malm ö , Sweden
Abstract The serum PSA test still is the most important biomarker for the detection and follow-up of prostate cancer. PSA-based screening can reduce disease specifi c mortality but coinciding unnecessary testing and overdiagnosis warrant further research for more specifi c biomarkers. Numerous studies of both serum and urine-based prostate cancer biomarker can-didates have been presented the last ten years. However, biomarkers for identifying the most aggressive subsets of this malignancy are still missing. Being non-invasive, urine-based tests might be suitable for both clinical and (mass) screening purposes, but also for prediction and to gain prognostic information. Protein-based, DNA-based and RNA-based urine biomarkers have been developed and tested. Protein markers in urine . Data on protein-based urine biomarkers (i.e. Annexin A3, matrix metalloproteinases and the urinary:serum PSA ratio) show up to now contradictory results and further studies are warranted to be able to assess their clinical value in which the cost aspect should not be overlooked. DNA markers in urine . Studies on DNA-based urine biomarkers focus on hypermethylation of gene panels with GSTP1 hypermethylation being the most promising individual marker. Larger prospective clinical studies of single markers and gene panels are however needed to validate their clinical utility. RNA markers in urine . RNA-based urine biomarkers are by far the most developed. The PCA3 test, the TMPRSS2 – ERG fusion gene, transcript expression levels of GOLPH2 , SPINK1 and their combination have been subject of many studies showing encouraging results. Conclusion . Up to now urine-based biomarkers represent a promising alternative or addition to serum-based biomarkers. Prospective studies in a multivariate setting, including larger sample sizes and avoiding attribution bias caused by preselection on the basis of serum PSA are however required.
Acta Oncologica, 2011; 50(Suppl 1): 85–89
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.524935
The serum PSA test still is the most important bio-marker for the detection and follow-up of prostate cancer. The test is well tolerated, quick, cheap and stan-dardised. Physicians are familiar with test results and can easily translate those into a certain risk level for hav-ing the disease or into the risk of tumour progression.
Recently it has been shown that screening with the use of the serum PSA test can reduce disease specifi c mortality [1 – 3]. This very important gain comes how-ever with considerable costs such as 70 – 80% of poten-tially unnecessary prostate biopsies, depending on the PSA cut-off level used [1]. Next to this PSA-based screening leads to overdiagnosis; i.e. detection of “ non-life threatening ” disease, often resulting in overtreatment [4].
New biomarkers for the detection and staging of prostate cancer are therefore an absolute must, and numerous studies of both serum and urine-based
prostate cancer biomarker candidates have been pre-sented the last decade. However, biomarkers for iden-tifying the most aggressive subsets of this malignancy are still missing. Being non-invasive, urine-based tests might be suitable for both clinical and (mass) screening purposes, but also for prediction and to gain prognostic information. Urine-based tests can roughly be divided into three groups; protein-based, DNA-based and RNA-based markers. This report will cover last years development in this area and highlight the most promising urinary markers in prostate cancer.
Protein markers in urine
Initially most research focused on the serum-urinary PSA ratio with confl icting results as compared to total serum PSA alone. In a prospective multicentre trial Irani et al. investigated the clinical relevance of the

86 M. J. Roobol et al.
urinary:serum PSA ratio in enhancing the specifi city of serum PSA in the detection of prostate cancer [5]. In patients with a PSA level of 4 – 10 ng/ml, receiver operating characteristic curves (ROC) showed that the urinary:serum PSA ratio had a larger AUC (0.63) than did total PSA (0.55) or free-to-total PSA ratio (0.60). In a later prospective study, urinary:serum PSA ratio was signifi cantly different, between patients with prostate cancer and those with benign prostatic hyperplasia, with ROC analysis showing a diagnostic cut-off for urinary PSA of � 150 ng/ml, with a sen-sitivity of 92.5% [6]. Promising results from these two studies were contradicted by Pannek et al. who failed to show any improvement in prostate cancer detection or staging in a cohort of 110 men [7].
Annexin A3 (ANXA3) is a calcium-binding pro-tein with decreased production in prostate cancer cells. Quantifi cation by ANXA3 using Western blots of urine samples showed signifi cantly lower values in prostate cancer patients as compared with BPH patients, resulting in an improved sensitivity at high specifi cities compared with total PSA [8]. The com-bination of PSA and urinary ANXA3 showed an AUC of 0.81 in the overall cohort.
Several studies have demonstrated a role of matrix metalloproteinases (MMPs) in growth, invasion and metastatic spread in prostate cancer and other malig-nancies [9]. In a recent study of 103 prostate cancer cases compared with 45 healthy controls, Roy et al. showed that MMP9 in urine was an independent pre-dictor of prostate cancer on multivariate analysis and that prostate cancer could be detected with a speci-fi city of 82% and a sensitivity of 74% with regard to the presence of any MMP in urine [10].
As concluded by Ploussard and de la Taille [11], more recently described urinary protein markers like delta-catenin [12], the heptatocyte growth factor (c-met) [13] and thymosin β 15 [14] have been evaluated in different pilot studies but none of these proposed new prostate cancer markers have yet been validated in independent studies.
The availability of proteomic platforms allowing the analysis of hundreds of peptides simultaneously protein-based urinary marker research has evolved enormously during the last decade. However the complexity of peptide analysis in urine samples was illustrated by Adachi et al. [15] who demonstrated the occurrence of more than 1 500 proteins in the normal urine proteome. A comprehensive review on proteo-mics and prostate cancer was recently published by Goo and Goodlet [16] and this area of research will not be further described in this report.
Metabolomics is a new approach to identify and separate metabolites using technology similar to pro-teomics. These small molecules are often the fi nal prod-ucts of biochemical activity in molecular pathways
and metabolomics could potentially provide oppor-tunity to develop diagnostic and prognostic evalua-tion to stratify patients to choose the best kind of treatment. Using a combination of high-throughput liquid and gas chromatography-based mass spectro-metry, Sreekumar and colleagues identifi ed 1 126 metabolites across 262 clinical samples (plasma, tis-sue and urine) and proposed sarcosine, an N -methyl derivative of the amino acid glycine, to be an impor-tant biomarker for prostate cancer progression and metastasis [17]. Other groups, using somewhat differ-ent methodology, have failed to reproduce these fi nd-ings [18] and the value of sarcosine as an important biomarker in prostate cancer is still under debate.
Taken together, contradictory results are reported on protein-based urinary markers and further studies are warranted to be able to assess their clinical value in which the cost aspect should not be overlooked.
DNA markers in urine
Hypermethylation at various gene loci has been asso-ciated with most malignancies, and several DNA methylation markers have been investigated in pros-tate cancer. The loss of glutathione- S -transferase P ( GSTP1 ) expression as a result of promoter hyperm-ethylation is the most common molecular alteration reported in prostate cancer [19]. Initial studies reported promising results with high sensitive and moderate specifi city in assays detecting GSTP1 hypermethyla-tion in urine samples, whereas subsequent studies have shown confl icting results in terms of predictive accu-racy as recently reviewed by Ploussard and de la Taille [11]. Later reports have mainly focused on hyperm-ethylation of gene panels again showing contradictory results [20 – 22]. A multiplexed, quantitative methy-lation-specifi c PCR assay consisting of the three dif-ferent methylation markers, GSTP1, RARB and APC was recently tested in a prospective multicentre study of post digital rectal examination (DRE) urine sam-ples from 178 patients with prostate cancer and 159 controls [23]. The predictive accuracy (area under the curve, AUC) of the assay for detecting prostate can-cer was 0.72, but this was only a marginal gain in predictive capability with respect to biopsy outcome as compared to total PSA and DRE alone. Results this far have suggested that gene methylation might serve as a useful marker in prostate cancer, and that GSTP1 hypermethylation is the most promising individual marker. Larger prospective clinical studies of single markers and gene panels are needed to validate their clinical utility.
RNA markers in urine
RNA-based urinary tests are by far the most devel-oped. The well known differential display clone 3 or

Prostate cancer urine biomarkers 87
PCA3 test is already commercially available under the trade name Progensa ® PCA3 (Gen-Probe, San Diego, Ca) [24].
The PCA3 test has been subject of many studies virtually all showing superiority of the PCA3 score to the serum PSA level in predicting biopsy outcome when comparing ROC curves. Especially the high specifi cities in the range of 80 to 90% are impressive and can be helpful in avoiding unnecessary biopsies. Looking into more detail at test performance char-acteristics and the study cohort used is however war-ranted. Sensitivity and specifi city of the PCA3 test in men with PSA levels in the so-called grey zone (4 – 10 ng/ml), representing those men that actually would benefi t from an additional test are not that convincing. In a comprehensive review of Vlaeminck-Guillem et al. the test performance in the PSA grey zone is summarised [25]. The specifi city when applying dif-ferent, study dependent, PCA3 cut-off values is indeed impressive ranging from 71 to 93%. However sensitivities are in the range of 53 to 84% [24,26 – 28] and question its clinical value. Data for men with previous negative biopsies on the basis of an elevated PSA level and/or abnormal DRE result show a simi-lar picture. Again specifi cities are high and repeating of unnecessary biopsies will without doubt be avoided with the use of the PCA3 test. However with corre-sponding sensitivities ranging from 47 to 75% one can question whether this is desirable, especially in these men [26 – 30].
Even more important to realise when interpreting these data is the fact that studies of new diagnostic markers are subject to attribution or assignment bias. Usually a more or less arbitrarily chosen cut-off value is used as a “ gold standard ” to determine the indica-tion for the decisive test, a prostatic biopsy, and the assumption is made that no cancers are present below that cut-off value. This assumption has been proved wrong by fi ndings in the control arm of the Prostate Cancer Prevention Trial (PCPT), where more than 5 000 men were biopsied independent of their PSA status. As an example: a PSA cut-off value of 4.0 ng/ml, a commonly used biopsy threshold, missed about 75% of all biopsy-detectable prostate cancer [31]. Most reported studies on PCA3 are in men previously screened with PSA and selected on the basis of an elevated PSA level. When calculating sensi-tivity and specifi city all biopsy detectable prostate can-cer cases present below the PSA cut-off are thus ignored. Resulting sensitivity and specifi city percentages are actually relative sensitivity and specifi city values.
Recently a side study within the ERSPC Rotterdam evaluated the value of PCA3 as a fi rst line screening test. The study design was chosen as such that more than 80% of men had a biopsy indication. Prostate biopsy was indicated if the PSA level was � 3.0 ng/ml
and/or the PCA3 score was � 10. With doing so the attribution bias was avoided as much as reasonably possible [32]. Based on ROC analyses of 721 men all biopsied, PCA3 performed marginally better than total PSA in predicting biopsy outcome. AUCs of PSA and PCA3 were 0.58 and 0.64 (p � 0.143) respectively. A sensitivity of 85% coincided with a PSA cut-off level of 1.0 ng/ml and a PCA3 score cut-off of 20. Specifi cities were similar and relatively low; 27% and 28% respectively. Again high specifi ci-ties coincided with low sensitivities in both PSA and PCA3. For example a specifi city of 75 – 80%, reached with a PSA cut-off of 4.0 ng/ml or a PCA3 score cut-off of 60 rendered sensitivities of 24% and 39% respectively. These data indicate that the PCA3 test, although to a somewhat lesser extent suffers from sim-ilar weaknesses as the serum PSA test. There is no cut-off value at which sensitivity and specifi city achieve a reasonable balance.
The relationship between the PCA3 score and para-meters of cancer aggressiveness has also been studied and differ by outcome. Some studies [33] report a positive relationship between PCA3 scores and param-eters of more serious disease, while other studies could not fi nd such a relationship [34].
Another RNA-based urinary biomarker comes from gene fusions which occur in approximately 50% of the prostate cancer cases. The most common fusion in prostate cancer is between the strong androgen-regulated TMPRSS2 gene transcriptional promoter and the oncogene ERG. This fusion results in an androgen-regulated TMPRSS2 – ERG fusion gene. In 108 men with prostate cancer biopsied on the basis of an elevated PSA level ( � 3.0 ng/ml) Hessels et al. analysed the fusion transcripts in urinary sediments. Again sensitivity was low (37%) with a very high speci-fi city of 93%. No signifi cant relationship was found between the presence of the fusion transcripts and Gleason score in prostate biopsies [35]. This was confi -r med by Rice et al. who detected ERG mRNA in urine samples from 237 men [36].
Combining the PCA3 test and the TMPRSS2-ERG fusion test seems a way to improve diagnostic accuracy. A study done in 105 men showed that the PCA3 alone had an AUC of 0.65, while the combina-tion of PCA3 and TMPRSS2-ERG increased the AUC to 0.77. Adding the PSA level to this multivariate approach resulted in an AUC of 0.80 [37].
If a multiplex analysis might improve the sensitiv-ity of urine-based tests, without sacrifi cing specifi city was evaluated by Laxman et al. [38] who performed quantitative PCR after whole transcriptome amplifi -cation in urine samples from 257 patients, including 152 men with prostate cancer and 105 with negative biopsy results. Different mRNA biomarkers were eval-uated based on bioinformatic analysis and in univariate

88 M. J. Roobol et al.
and multivariate analyses, transcript expression levels of GOLPH2 , SPINK1 , PCA3 , and TMPRSS2 – ETS fusion signifi cant predictors of prostate cancer, with an AUC signifi cantly greater than that of the PCA3 score alone (0.758 versus 0.662). SPINK1 expres-sion in urine samples was higher in TMPRSS2 – ERG -negative than in TMPRSS2 – ERG -positive samples, suggesting the mutual exclusivity of SPINK1 expres-sion and ETS fusions similar to tissue biomarker stud-ies [39]. Validation studies are to be presented.
Conclusion
Urinary biomarkers for prostate cancer are subject of ongoing research and represent a promising alter-native or addition to serum-based biomarkers. Pro-spective studies including larger sample sizes and avoiding attribution bias caused by preselection on the basis of serum PSA are however required. Look-ing at test performances combinations of different, both serum and urinary-based markers in a multi-plex setting will most likely help in resolving the cur-rent problems in the (early) detection and staging of prostate cancer.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Schr ö der FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, [1] Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009;360:1320 – 8. Roobol MJ, Kerkhof M, Schr ö der FH, Cuzick J, Sasieni P, [2] Hakama M, et al. Prostate cancer mortality reduction by prostate-specifi c antigen-based screening adjusted for nonat-tendance and contamination in the European Randomised Study of Screening for Prostate Cancer (ERSPC). Eur Urol 2009;56:584 – 91. Carlsson S, Aus G, Bergdahl S, Khatami A, Lodding P, Pihl CG, [3] et al. Mortality results from the G ö teborg randomised pop-ulation-based prostate-cancer screening trial. Lancet Oncol 2010; 11:725 – 32 . Heijnsdijk EA, der Kinderen A, Wever EM, Draisma G, [4] Roobol MJ, de Koning HJ. Overdetection, overtreatment and costs in prostate-specifi c antigen screening for prostate can-cer. Br J Cancer 2009;101:1833 – 8. Irani J, Salomon L, Souli é M, Zlotta A, de la Taille A, Dor é B, [5] et al. Urinary/serum prostate-specifi c antigen ratio: Compari-son with free/total serum prostate-specifi c antigen ratio in improving prostate cancer detection. Urology 2005;65:533 – 7. Bolduc S, Lacombe L, Naud A, Gr é goire M, Fradet Y, [6] Tremblay RR. Urinary PSA: A potential useful marker when serum PSA is between 2.5 ng/mL and 10 ng/mL. Can Urol Assoc J 2007;1:377 – 81. Pannek J, Rittenhouse HG, Evans CL, Finlay JA, Bruzek DJ, [7] Cox JL, et al. Molecular forms of prostate-specifi c antigen and human kallikrein 2 (hK2) in urine are not clinically use-ful for early detection and staging of prostate cancer. Urology 1997;50:715 – 21.
[8] Schostak M, Schwall GP, Poznanovic S, Groebe K, Muller M, Messinger D, et al. Annexin A3 in urine: A highly specifi c noninvasive marker for prostate cancer early detection. J Urol 2009;181:343 – 53. Egeblad M, Werb Z. New functions for the matrix metallo-[9] proteinases in cancer progression. Nat Rev Cancer 2002;2:161 – 74. Roy R, Louis G, Loughlin KR, Wiederschain D, Kilroy SM, [10] Lamb CC, et al. Tumor-specifi c urinary matrix metallopro-teinase fi ngerprinting: Identifi cation of high molecular weight urinary matrix metalloproteinase species. Clin Cancer Res 2008;14:6610 – 7. Ploussard G, de la Taille A. Urine biomarkers in prostate [11] cancer. Nat Rev Urol 2010;7:101 – 9. Lu Q, Zhang J, Allison R, Gay H, Yang WX, Bhowmick NA, [12] et al. Identification of extracellular deltacatenin accumu-lation for prostate cancer detection. Prostate 2009;69:411 – 8. Russo AL, Jedlicka K, Wernick M, McNally D, Kirk M, [13] Sproull M, et al. Urine analysis and protein networking iden-tify met as a marker of metastatic prostate cancer. Clin Can-cer Res 2009;15:4292 – 8. Hutchinson LM, Chang EL, Becker CM, Ushiyama N, [14] Behonick D, Shih MC, et al. Development of a sensitive and specifi c enzyme-linked immunosorbent assay for thymosin beta15, a urinary biomarker of human prostate cancer. Clin Biochem 2005;38:558 – 71. Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human [15] urinary proteome contains more than 1500 proteins, includ-ing a large proportion of membrane proteins. Genome Biol 2006;7:R80. Goo YA, Goodlett DR. Advances in proteomic prostate can-[16] cer biomarker discovery. J Prot 2010 E-pub doi:10.1016/j.jprot.2010.04.002. Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, [17] Yu J, et al. Metabolomic profi les delineate potential role for sarcosine in prostate cancer progression. Nature 2009;457:910 – 4. Jentzmik F, Stephan C, Miller K, Schrader M, Erbersdobler A, [18] Kristiansen G, et al. Sarcosine in urine after digital rectal exami nation fails as a marker in prostate cancer detection and identifi cation of aggressive tumours. Eur Urol 2010;58:12 – 8. Harden SV, Sanderson H, Goodman SN, Partin AA, Walsh PC, [19] Epstein JI, et al. Quantitative GSTP1 methylation and the detection of prostate adenocarcinoma in sextant biopsies. J Natl Cancer Inst 2003;95:1634 – 7. Hoque MO, Topaloglu O, Begum S, Henrique R, Rosenbaum E, [20] Van Criekinge W, et al. Quantitative methylation specifi c polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol 2005;23:6569 – 75. Vener T, Derecho C, Baden J, Wang H, Rajpurohit Y, Skelton J, [21] et al. Development of a multiplexed urine assay for prostate cancer diagnosis. Clin Chem 2008;54:874 – 82. Payne SR, Serth J, Schostak M, Kamradt J, Strauss A, Thelen P, [22] et al. DNA methylation biomarkers of prostate cancer: Con-fi rmation of candidates and evidence urine is the most sensi-tive body fl uid for non-invasive detection. Prostate 2009;69:1257 – 69. Baden J, Green G, Painter J, Curtin K, Markiewicz J, Jones J, [23] et al. Multicenter evaluation of an investigational prostate cancer methylation assay. J Urol 2009;182:1186 – 93. Groskopf J, Aubin SM, Deras IL, Blase A, Bodrug S, Clark C, [24] et al. APTIMA PCA3 molecular urine test: Development of a method to aid in the diagnosis of prostate cancer. Clin Chem 2006;52:1089 – 95.

Prostate cancer urine biomarkers 89
Vlaeminck-Guillem V, Ruffi on A, Andr é J, Devonec M, [25] Paparel P. Urinary prostate cancer 3 test: Toward the age of reason? Urology 2010;75:447 – 53. Tinzl M, Marberger M, Horvath S, Chypre C. DD3PCA3 [26] RNA analysis in urine — a new perspective for detecting pros-tate cancer. Eur Urol 2004;46:182 – 6. Fradet Y, Saad F, Aprikian A, Dessureault J, Elhilali M, [27] Trudel C, et al. uPM3, a new molecular urine test for the detection of prostate cancer. Urology 2004;64:311 – 5. Deras IL, Aubin SM, Blase A, Day JR, Koo S, Partin AW, [28] et al. PCA3: A molecular urine assay for predicting prostate biopsy outcome. J Urol 2008;179:1587 – 92. Haese A, de la Taille A, van Poppel H, Marberger M, Stenzl A, [29] Mulders PF, et al. Clinical utility of the PCA3 urine assay in European men scheduled for repeat biopsy. Eur Urol 2008;54:1081 – 8. Marks LS, Fradet Y, Deras IL, Blase A, Mathis J, Aubin SM, [30] et al. PCA3 molecular urine assay for prostate cancer in men undergoing repeat biopsy. Urology 2007;69:532 – 5. Thompson IM, Ankerst DP, Chi C, Lucia MS, Goodman PJ, [31] Crowley JJ, et al. Operating characteristics of prostate-specifi c antigen in men with an initial PSA level of 3.0 ng/ml or lower. JAMA 2005;294:66 – 70. Roobol MJ, Schroder FH, van Leeuwen P, Wolters T, van den [32] Bergh RCN, van Leenders GJ, et al. Performance of the Prostate Cancer Antigen3 (PCA3) gene and prostate-specifi c antigen in prescreened men: Exploring the value of PCA3 for a fi rst-line diagnostic test. Eur Urol 2010 Jul 9 [Epub ahead of print] .
Hessels D, van Gils MP, van Hooij O, Jannink SA, Witjes JA, [33] Verhaegh GW, et al. Predicitve value of PCA3 in urinary sediments in determining clinico-pathological characteristics of prostate cancer. Prostate 2010;70:10 – 6. Whitman EJ, Groskopf J, Ali A, Chen Y, Blase A, Furusato B, [34] et al. PCA3 score before radical prostatectomy predicts ext-racapsular extension and tumor volume. J Urol 2008;180:1975 – 8, discussion 1978 – 9. Hessels D, Smit FP, Verhaegh GW, Witjes JA, Cornel EB, [35] Schalken JA. Detection of TMPRSS2-ERG fusion tran-scripts and prostate cancer antigen 3 in urinary sediments may improve diagnosis of prostate cancer. Clin Cancer Res 2007;13:5103 – 8. Rice KR, Chen Y, Ali A, Whitman EJ, Blase A, Ibrahim M, [36] et al. Evaluation of the ETS-related gene mRNA in urine for the detection of prostate cancer. Clin Cancer Res 2010;16:1572 – 6. Aubin SM, Miick S, Williamsen S, Hodge P, Meinke J, Blase A, [37] et al. Improved prediction of prostate biopsy outcome using PCA3, TMPRSS2:ERG gene fusion and serum PSA. J Urol Suppl 2008;179:725. Laxman B, Morris DS, Yu J, Siddiqui J, Cao J, Mehra R, [38] et al. A fi rst-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res 2008;68:645 – 9. Tomlins SA, Rhodes DR, Yu J, Varambally S, Mehra R, [39] Perner S, et al. The role of SPINK1 in ETS rearrange-ment-negative prostate cancers. Cancer Cell 2008;13:519 – 28.

Introduction: Therapy with curative intent
STEN NILSSON 1, GABRIELLA COHN-CEDERMARK1 & PETER WIKLUND 2
1 Department of Oncology-Pathology, Radiumhemmet, Karolinska University Hospital, Stockholm, Sweden and 2 Department of Urology, Karolinska University Hospital, Stockholm, Sweden
Prostate cancer is a disease that is curable when it is still localized to the prostate gland. The rapid devel-opments in early detection as well as surgical- and radio therapeutic modalities have led to an increasing number of patients undergoing curative treatment. Still, there are no scientifi cally unobjectionable ran-domized studies that have compared surgery with radiotherapy. Comparisons between these two treat-ment options are therefore solely based on outcome data from treatment series in which patients have been categorized with respect to commonly recog-nized risk factors such as T stage, Gleason score and prostate specifi c antigen (PSA). Guidelines in most countries state that the two treatment modalities have comparable outcomes with regard to cure and therefore the choice of treatment should be in the hands of patient. In the UK, patient recruitment to the ProtecT (Prostate testing for cancer and Treat-ment) trial was recently completed [1]. This study is expected to provide important information about the three treatment modalities compared: active monitoring, radiotherapy and radical prostatectomy, respectively, in low-risk prostate cancer. Before the results from this trial are obtained, there is an obvious responsibility for treating clinicians to initi-ate, and participate in, additional prospective ran-domized trials evaluating the effi cacy of different therapeutic interventions in patients with localized prostate cancer.
Adjuvant radiotherapy after radical prostatec-tomy has shown excellent results in terms of signifi -cantly reducing the risk of biochemical progression [2], improving biochemical progression-free survival and local control [3] as well as overall survival [4]. One still unanswered question is whether this type of adjuvant therapy can be replaced by “early salvage” radiation therapy, i.e. at the fi rst sign of PSA relapse. Randomized trials are ongoing on this theme and will hopefully provide clarity on this in the next few years: RAVES (Radiotherapy adjuvant versus early salvage) (NCT00860652), RADICALS (Radiotherapy and
androgen deprivation in combination after local surgery) [5] and GETUG-17 (Adjuvant versus salvage radiotherapy) [6].
Another important therapeutic comparison that has to be investigated in curative treatment of patients with localized prostate cancer is radical prostatec-tomy plus adjuvant radiotherapy versus radical radio-therapy. Prospective randomized trials in this area are expected to start in the near future.
The value of neoadjuvant, concomitant and adjuvant endocrine therapy in conjunction with radiotherapy has been investigated in several ran-domized trials. The results from these have been updated in a systematic review and meta-analysis [7]. However, one important question still unanswered is whether dose-escalated radiotherapy can be fully off-set or exceed the effect of combined hormonal- and radiation therapy. We look forward to the initiative also to such studies in the near future.
Trials evaluating outcome of different surgical procedures such as open radical prostatectomy versus robotic assisted laparoscopic prostatectomy are important. One trial currently ongoing on this theme is the Swedish Prospective LAPPRO trial [8]. This trial aims to compare the two surgical tech-niques in aspects of short- and long-term functional and oncological outcome, cost effectiveness and quality of life, supplying new knowledge to support future decisions in treatment strategies for prostate cancer.
The rapid development in radiation therapy tech-niques [9,10] and trials regarding different dose-escalation- and dose-fractionation regimens are ongoing and are expected to shed more light to their place in future curative recommendations in the different prostate cancer risk groups [11].
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
Acta Oncologica, 2011; 50(Suppl 1): 90–91
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.576548

Introduction 91
References
[1] Lane JA, Hamdy FC, Martin RM, Turner EL, Neal DE, Donovan JL. Latest results from the UK trials evaluating prostate cancer screening and treatment: The CAP and ProtecT studies. Eur J Cancer 2010;46:3095 – 101. Wiegel T, Bottke D, Steiner U, Siegmann A, Golz R, [2] Storkel S, et al. Phase III postoperative adjuvant radiotherapy after radical prostatectomy compared with radical prostate-ctomy alone in pT3 prostate cancer with postoperative unde-tectable prostate-specifi c antigen: ARO 96-02/AUO AP 09/95. J Clin Oncol 2009;27:2924 – 30. Bolla M, van Poppel H, Collette L, van Cangh P, [3] Vekemans K, Da Pozzo L, et al. Postoperative radiotherapy after radical prostatectomy: A randomised controlled trial (EORTC trial 22911). Lancet 2005;366(9485):572 – 8. Thompson IM, Tangen CM, Paradelo J, Lucia MS, Miller G, [4] Troyer D, et al. Adjuvant radiotherapy for pathological T3N0M0 prostate cancer signifi cantly reduces risk of metas-tases and improves survival: Long-term followup of a rand-omized clinical trial. J Urol 2009;181:956 – 62. Parker C, Sydes MR, Catton C, Kynaston H, Logue J, [5] Murphy C, et al. Radiotherapy and androgen depriva-tion in combination after local surgery (RADICALS): A new Medical Research Council/National Cancer Institute of
Canada phase III trial of adjuvant treatment after radical prostatectomy. BJU Int 2007;99:1376 – 9. Richaud P, Sargos P, Henriques de Figueiredo B, [6] Latorzeff I, Mongiat-Artus P, Houede N, et al. [Postoperative radiotherapy of prostate cancer]. Cancer Radiother 2010;14: 500 – 3. Shelley MD, Kumar S, Wilt T, Staffurth J, Coles B, [7] Mason MD. A systematic review and meta-analysis of ran-domised trials of neo-adjuvant hormone therapy for localised and locally advanced prostate carcinoma. Cancer Treat Rev 2009;35:9 – 17. Thorsteinsdottir T, Stranne J, Carlsson S, Anderberg B, [8] Bjorholt I, Damber JE, et al. LAPPRO: A prospective mul-ticentre comparative study of robot-assisted laparoscopic and retropubic radical prostatectomy for prostate cancer. Scand J Urol Nephrol 2011;45:102 – 12. Lennernas B, Levitt SH, Nilsson S. Four and fi ve [9] dimensional radiotherapy with reference to prostate cancer – defi nitions, state of the art and future directions. Acta Oncol 2010. Lennernas B, Nilsson S, Levitt SH. Hypofractionation for [10] radiation therapy of prostate cancer – radiobiological and radiophysical considerations. Acta Oncol 2010. Harmenberg U, Hamdy FC, Widmark A, Nilsson S. Curative [11] radiation therapy in prostate cancer. Acta Oncol 2011.

Correspondence: Jonas Hugosson, Sahlgrenska University Hospital, Department of Urology Bruna Str å ket 11 B, SE-41345 G ö teborg, Sweden. Tel: � 46 31 3429017. Fax: � 46 31 415648. E-mail: [email protected]
(Received 26 August 2010 ; accepted 26 October 2010 )
REVIEW ARTICLE
Radical retropubic prostatectomy: A review of outcomes and side-effects
JONAS HUGOSSON , JOHAN STRANNE & SIGRID V. CARLSSON
Institute of Clinical Sciences, Department of Urology, Sahlgrenska Academy at University of Gothenburg, Sweden
Abstract Background. Radical prostatectomy (RP) is worldwide probably the most common procedure to treat localized prostate cancer (PC). Due to a more widespread use of Prostate-Specifi c Antigen (PSA) testing, patients operated today are often younger and have organ confi ned disease justifying a more preservative surgery. At the same time, surgical technique has improved resulting in lower risk of permanent side-effects. This paper aims to give an overview of results from modern surgery regarding cancer control and side-effects. A brief overview of the history is given. Material and Methods. A literature research identifi ed recently published papers focusing on outcome and side-effects after RP. Results. One large randomized study (SPCG-4) compared RP and watchful waiting (WW). The study showed that RP was superior to WW in preventing local progression (RR � 0.36), distant metastasis (RR � 0.65) and death from PC (RR � 0.65). Observational studies also show a better outcome for men treated with RP compared to WW. Peri-operative mortality after RP is low in most material around 0.1%. The risk of stricture of the vesico-urethral anastomosis has decreased with improved technique from his-torically 10 – 20% to a low incidence of around 2 – 9% today. Also the risk of incontinence has declined with improved technique. However, while the rates of severe incontinence is usually very low, as many as 30% still report light incontinence after long-term follow-up. Erectile dysfunction (ED) is still a frequent side-effect after RP. This risk is dependent on age, pre-operative sexual function, surgical technique and other risk factors for ED such as smoking, diabetes, etc. In selected sub-groups the risk of ED is low. Inguinal hernia is a more recently described complication after open retropubic RP with a post-operative incidence of 15 – 20% within three years of surgery. Conclusion. RP is an effective method to achieve cancer control in selected patients. With modern technique it is a safe procedure with a low risk of permanent side-effects except for ED.
Prostate cancer (PC) is a major public health prob-lem. It is one of the most commonly diagnosed can-cers and one of the most common causes of cancer-related death in developed countries [1,2]. The estimated incidence of PC in the USA was nearly 192 280 new cases in 2009 [1] and 345 900 new cases in Europe in 2006 [3]. From being a rare disease in the 19 th century, the incidence has increased rapidly during the past century, especially during the last two decades. A man ’ s lifetime risk of a PC diagnosis has more than doubled from 8% in the early 1980s to almost 18% today [4]. The discov-ery of the Prostate-Specifi c Antigen (PSA) test in the 1970s [5 – 7] is likely to explain most of this increase, refl ected in that the greatest increase in incidence constitutes of non-palpable tumors. Screening with digital rectal exam and/or the PSA test offers detec-tion of early stage, clinically localized and potentially curable disease. These tumors can be managed in
different ways, however, radical prostatectomy (RP) is today considered gold standard for treating local-ized PC [8,9]. The frequency of curative treatment for early-stage disease with surgery as primary treat-ment has increased and evolved enormously since the late 1980s [10 – 14]. Though providing an effec-tive means of treating localized disease [14], feared side-effects of RP are urinary incontinence and impotency. The surgeon ’ s technique put priorities on cancer control, continence and potency [15]. The main indication of RP is patients with low- and inter-mediate- risk localized PC (cT1a-T2b and Gleason score 2 – 7 and PSA � 20) and a life expectancy � 10 years [9]. However, treating high grade as well as locally advanced tumors have also been reported with favorable outcome [16].
The aim of the present paper was to give an over-view of results from modern surgery regarding can-cer control and permanent side-effects. A brief
Acta Oncologica, 2011; 50(Suppl 1): 92–97
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.535848

Radical Retropubic Prostatectomy 93
overview of the history is given. The study did not intend to compare different ways of performing RP or RP with other treatment modalities.
Material and methods
A literature research through April 5, 2011 identi-fi ed recently published papers focusing on outcome and side-effects after RP. Medical electronic data-bases were used (MEDLINE as well as the Cochrane Library to identify systematic reviews). Information was also derived from guidelines. Keywords for the literature search included: radical prostatec-tomy, surgical technique, mortality, side-effects, impotence, incontinence and inguinal hernia. Data extraction and quality assessment were made by the authors.
Results
History
Billroth performed the fi rst perineal prostatectomy for palliation of obstructive PC in the 1860s [17] followed by Young in the beginning of the 20 th cen-tury [18]. In the early 1930s the procedure was replaced by transurethral prostatic resection for this indication [19]. The fi rst retropubic prostatectomy was performed by Millin in 1945 [20]. However, many of the tumors were detected late. The discovery of the PSA changed this and prostatectomy could be regarded as a means of treating early disease. In the early 1980s, Walsh described in detail the anatomy of the prostate, the dorsal venous complex and the neuro-vascular bundles [21] and he also performed the fi rst nerve-sparing radical retropubic prostatec-tomy (RRP) [22]. These efforts led to improvements in postoperative continence and potency as well as reducing peroperative mortality [22]. Peroperative blood loss is aimed at being minimized and the known postoperative complications may be reduced to low levels in expert hands [23]. Modifi cations and improvements of surgery, anesthesia and pre- and postoperative care have been made throughout the years, with a decrease in complication rates [24]. RP can be performed either as open retropubic sur-gery, laparoscopic surgery, robotic-assisted laparo-scopic surgery as well as perineal surgery. Evidence is lacking which technique is superior as regards oncological and functional results as well as cost-effectiveness [9]. Observational studies have com-pared different modalities [25], but no randomized controlled trial exists to date. A large Swedish study (LAPPRO) is currently ongoing comparing open retropubic prostatectomy with robotic-assisted laparoscopic prostatectomy [26].
Outcome
The three primary outcomes of RP, cancer control, continence and potency, are achieved to a high extent today. RP is associated with excellent cancer control in men with organ confi ned disease up to 10 years of follow-up [27]. Prostate cancer-specifi c survival in localized disease has been reported to be 86 – 99.6% at 10 years [9,28 – 30], 82 – 90% at 15 years [31,32] and 76% at 30 years [33].
The benefi ts of RP as compared to expectant management have been reported in one large pro-spective randomized trial (SPCG-4) judged to be of good quality [34]. This Scandinavian landmark study, reported by Holmberg et al., randomized (in 1989 – 1999) 695 men with clinical stage T1-2 tumors to either watchful waiting or RP. During a median of 10.8 years of follow-up, RP signifi cantly reduced the risk of local progression (RR � 0.36), the risks of metastasized disease (RR � 0.65) and disease-specifi c mortality (RR � 0.65, 95% CI 0.45 – 0.94; p � 0.03) [14]. There was a statistically signifi cant dif-ference in overall mortality rates for men � 65 years randomly assigned to RP as compared to watchful waiting (RR 0.59, 95% CI 0.41 – 0.85, p � 0.004) [14]. However, the number needed to treat (NNT) to “ cure ” a single case of PC with RP was calculated to 10 – 19. This rather high NNT could be explained by a high risk of over-treatment (treating too many indolent cancers) or treating too advanced cancers where local treatment no longer is effective. The lat-ter explanation is supported by the fact that in the SPCG-4 trial, the majority of prostate cancers were palpable tumors. However, there is no doubt that also in the SPCG-4-trial there is a high rate of over-treatment emphasizing the demand for a very low rate of permanent side-effects to justify RP in these men. There are three major clinical trials (PIVOT, ProtecT and START) ongoing that investigate the outcomes of RP vs. expectant management for PSA-detected tumors.
Observational studies
In a cohort study of men aged 55 – 74 years with non-metastasized disease diagnosed during 1971 – 1984, RP was compared with external beam therapy and obser-vation. Kaplan – Meier estimates for 10-year disease-specifi c and overall survival were in favor of RP (86% for disease-specifi c survival, 95% CI 84 – 88% and 69% for overall survival, 95% CI 67 – 71%) [29].
In another observational cohort study (including 44 630 men aged 65 to 80 years) of men with low and intermediate risk localized PC, the adjusted haz-ard ratio for overall mortality comparing RP with observation was 0.50 (95% CI 0.47 – 0.53) [35].

94 J. Hugosson et al.
The natural history of today ’ s prostate cancers that are often PSA-detected is not completely under-stood. Stattin et al. reported 10-year outcomes of men in the National Prostate Cancer Register of Sweden Follow-up Study that included men aged � 70 years at diagnosis, local tumor stage T1-2, Nx or N0, Mx or M0, and PSA � 20 ng/ml. For men with low-risk PC, the cumulative 10-year PC-specifi c mortal-ity was 2.4% (95% CI 1.2 – 4.1%) among men on conservative management and 0.4% (95% CI 0.13 – 0.97%) among men who underwent RP [30].
However, non-randomized studies have limita-tions and should be interpreted with caution because of the risk of selection bias and comparison between treatment modalities is beyond the scope of this review.
Side-effects
Short-term side-effects. THIRTY-DAY MORTALITY . We per-formed a nationwide population-based record-link-age study in which the 30-day mortality after RP was low, 0.11 – 0.13% [36], a fi nding consistent with pre-vious studies based on modern series [31,37 – 43]. Co-morbidity [44] and increasing age are factors associated with higher risk of perioperative death [41,45 – 46]. Hospital volume and surgeon volume may also affect the outcome [37,44]. As indicated by our study group [36] and others, ischemic heart disease and pulmonary embolism still seems to constitute the majority of causes of deaths following RP [47 – 50].
Long-term side-effects. All treatments for PC at differ-ent stages have side-effects. The most well-known and bothersome postoperative complications after RRP are urinary incontinence, impotence and stricture of the vesico-urethral anastomosis [51,52]. Large improvements in surgical technique have been made during the past three decades and the risk of side-effects has fallen and together with the earlier detec-tion of PC this results in RP today being a procedure with much lower morbidity [28]. However, while at the same time earlier detection provides a higher chance of cure and treatment with less side-effects, it also implies a higher risk for over-treatment and, in the case of permanent side-effects that still can follow, a long-time suffering for the individual. Due to inconsistency in reporting the outcomes in the literature, the incidence of post prostatectomy uri-nary incontinence ranges from 0% to 87% [53 – 57] and postoperative potency rates vary from 11% to 87% [53]. It has been shown that sexual function can continue to improve even beyond two years postop-eratively [58,59]. At 52 months after RP, 88% have
been reported to suffer from erectile dysfunction and 31% from urinary leakage [60].
However, it should be noted that already before surgery, the prevalence of erectile dysfunction is high in population-based studies. Long et al. have reported that 64% suffered from erectile dysfunction overall pre-operatively; 43% of patients younger than 65 and 84% of patients over 65 [61]. Salonia et al. showed that only 43% of 234 men with localized PC candi-dates for bilateral nerve-sparing RP had a normal erectile function pre-operatively [62].
Risk factors for postoperative impotence include non-nerve sparing surgery, the surgeon ’ s ability to master the technique, age of the patient, baseline sex-ual function, and risk factors for erectile dysfunction including diabetes, hypertension and smoking [63].
The average incidence of severe incontinence is, in most studies, not greater than 5% [55,64], how-ever, severe urinary leakage has a huge negative impact on a patient ’ s quality of life.
The risk of stricture of the vesico-urethral anas-tomosis has decreased with improved technique from historically 10 – 20% [65] to a low incidence at around 2 – 9% today [9].
Quality of Life. Studies have shown that men reported similar (or indicated even better) quality of life after RP compared to men assigned to watchful waiting [52,66]. Better mental health scores and health-related-quality of life scores for men having undergone RP has also been reported compared with men having undergone radiotherapy [66] as well as compared with a control group of men without cancer [67].
Inguinal hernia. Inguinal hernia (IH) as a postopera-tive complication to RP is less well known and was fi rst reported by Regan et al. in 1996 [68]. Later reports indicate an incidence of IH of 12% to 21% within two to three years after RRP [69 – 74]. The background annual incidence of inguinal hernia is approximately 0.5% [75]. These results have recently been confi rmed from the SPCG-4 study, a prospec-tive randomized study between RRP and watchful waiting with a follow-up of over 12 years [14], where 9.3% developed IH after RRP within 48 months as compared to 2.4% in a group of non-operated men with PC and 0.9% in a population control group [76]. Today IH can defi nitely be considered a complication to RRP.
The lower mid-line incision per se has been suggested to be causative [69 – 74]. In a study from 2008, Koie et al. reported a postoperative IH inci-dence of only 2.9% in a group of 272 patients where RRP was performed through a so called “ mini-laparotomy ” incision of only 6 cm [77]. Matsubara et al. also reported an IH incidence of 1.8% after

Radical Retropubic Prostatectomy 95
radical-perineal prostatectomy where the whole procedure is performed through a perineal incision and consequently there is no abdominal incision at all [78]. In a recent study of 946 patients after robot-assisted laparoscopic radical prostatectomy (RALP) 5.8% developed an IH within 48 months as com-pared to 12.2% after RRP. There was no statistical difference in IH incidence after RALP as compared to a population control. Thus, the length, and pos-sibly the placing, of the abdominal incision seems to affect the development of postoperative IH [76].
Concluding remarks
It is 150 years since the fi rst RP was performed. Enormous improvements in surgical technique have been achieved especially during the last 20 years. It is nowadays a safe procedure with excellent cancer control in men with localized disease and few side-effects apart from ED. The main challenge in the future is to further decrease the risk of sexual dysfunction and to avoid over-treatment.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer [1] statistics, 2009. CA Cancer J Clin 2009;59:225 – 49. Jemal A, Ward E, Hao Y, Thun M. Trends in the leading [2] causes of death in the United States, 1970 – 2002. JAMA 2005;294:1255 – 9. Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, [3] Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 2007;18:581 – 92. Ankerst DP. Prostate cancer screening. 2nd ed. New York: [4] Humana Press; 2009. Wang MC, Valenzuela LA, Murphy GP, Chu TM. Purifi ca-[5] tion of a human prostate specifi c antigen. Invest Urol 1979;17:159 – 63. Hara M, Koyanagi Y, Inoue T, Fukuyama T. [Some physico-[6] chemical characteristics of “-seminoprotein”, an antigenic component specifi c for human seminal plasma. Forensic immunological study of body fl uids and secretion. VII]. Nihon Hoigaku Zasshi 1971;25:322 – 4. Li TS, Beling CG. Isolation and characterization of two spe-[7] cifi c antigens of human seminal plasma. Fertil Steril 1973;24:134 – 44. Pirtskhalaishvili G, Hrebinko RL, Nelson JB. The treatment [8] of prostate cancer: An overview of current options. Cancer Pract 2001;9:295 – 306. Heidenreich A, Bolla M, Joniau S, Mason MD, Matveev V, [9] Mottet N, et al. EAU guidelines on prostate cancer. Arnhem: European Association of Urology; 2010 [updated 2010; cited]. Available from: http://www.uroweb.org/guidelines/online-guidelines/. Chu KC, Tarone RE, Freeman HP. Trends in prostate cancer [10] mortality among black men and white men in the United States. Cancer 2003;97:1507 – 16.
Hankey BF, Feuer EJ, Clegg LX, Hayes RB, Legler JM, [11] Prorok PC, et al. Cancer surveillance series: Interpreting trends in prostate cancer – part I: Evidence of the effects of screening in recent prostate cancer incidence, mortality, and survival rates. J Natl Cancer Inst 1999;91:1017 – 24. Mettlin CJ, Murphy GP. Why is the prostate cancer death [12] rate declining in the United States? Cancer 1998;82:249 – 51. Tarone RE, Chu KC, Brawley OW. Implications of stage-[13] specifi c survival rates in assessing recent declines in prostate cancer mortality rates. Epidemiology 2000;11:167 – 70. Bill-Axelson A, Holmberg L, Filen F, Ruutu M, Garmo H, [14] Busch C, et al. Radical prostatectomy versus watchful wait-ing in localized prostate cancer: The Scandinavian Prostate Cancer Group-4 Randomized Trial. J Natl Cancer Inst 2008;100:1144 – 54. Walsh P. Campbell ’ s urology. 8th ed. Walsh PC [15] , Retik AB, Vaughan ED, Wein AJ, editors. Philadelphia: Saunders; 2002. Xylinas E, Dache A, Roupret M. Is radical prostatectomy a [16] viable therapeutic option in clinically locally advanced (cT3) prostate cancer? BJU Int 2010;106:1596 – 600. Billroth T. Carcinoma der prostata. Chir Erfarungen, Zurich, [17] 1860 – 67. Archiv Fur Klinische Chirurgie 1869;Bd X(548). Young HH. The early diagnosis and radical cure of carci-[18] noma of the prostate: Being a study of 40 cases and presen-tation of a radical operation which was carried out in four cases. Bull Johns Hopkins Hosp 1905;XVI(175):315 – 21. Lytton B. Prostate cancer: A brief history and the discovery [19] of hormonal ablation treatment. J Urol 2001;165(6 Pt 1):1859 – 62. Millin T. Retropubic prostatectomy: A new extravesical [20] technique report on 20 cases. Lancet 1945;167:693 – 6. Walsh PC, Donker PJA. Impotence following radical prosta-[21] tectomy: Insight into etiology and prevention. J Urol 1982;128:492 – 7. Walsh PC, Lepor H, Eggleston JC. Radical prostatectomy [22] with preservation of sexual function: Anatomical and patho-logical considerations. Prostate 1983;4:473 – 85. Walsh PCA. Anatomic radical prostatectomy: Evolution of [23] the surgical technique. J Urol 1998;160(6 Pt 2):2418 – 24. Optenberg SA, Wojcik BE, Thompson IM. Morbidity and [24] mortality following radical prostatectomy: A national analysis of Civilian Health and Medical Program of the Uniformed Services benefi ciaries. J Urol 1995;153:1870 – 2. Hu JC, Gu X, Lipsitz SR, Barry MJ, D’Amico AV, Weinberg [25] AC, et al. Comparative effectiveness of minimally invasive vs open radical prostatectomy. JAMA 2009;302:1557 – 64. Thorsteinsdottir T, Stranne J, Carlsson S, Anderberg B, [26] Bjorholt I, Damber JE, et al. LAPPRO: A prospective mul-ticentre comparative study of robot-assisted laparoscopic and retropubic radical prostatectomy for prostate cancer. Scand J Urol Nephrol 2011;45:102 – 12. Walsh PC, Partin AW, Epstein JI. Cancer control and quality [27] of life following anatomical radical retropubic prostatectomy: Results at 10 years. J Urol 1994;152(5 Pt 2):1831 – 6. Galvin DJ, Eastham JA. Critical appraisal of outcomes [28] following open radical prostatectomy. Curr Opin Urol 2009;19:297 – 302. Barry MJ, Albertsen PC, Bagshaw MA, Blute ML, Cox R, [29] Middleton RG, et al. Outcomes for men with clinically nonmetastatic prostate carcinoma managed with radical prostactectomy, external beam radiotherapy, or expectant management: A retrospective analysis. Cancer 2001;91:2302 – 14. Stattin P, Holmberg E, Johansson JE, Holmberg L, [30] Adolfsson J, Hugosson J. Outcomes in localized prostate

96 J. Hugosson et al.
cancer: National Prostate Cancer Register of Sweden follow-up study. J Natl Cancer Inst 2010;102:950 – 8. PMCID: 2897875. Zincke H, Oesterling JE, Blute ML, Bergstralh EJ, Myers RP, [31] Barrett DM. Long-term (15 years) results after radical prostatectomy for clinically localized (stage T2c or lower) prostate cancer. J Urol 1994;152(5 Pt 2):1850 – 7. Han M, Partin AW, Pound CR, Epstein JI, Walsh PC. [32] Long-term biochemical disease-free and cancer-specifi c sur-vival following anatomic radical retropubic prostatectomy. The 15-year Johns Hopkins experience. Urol Clin North Am 2001;28:555 – 65. Lewinshtein D, Teng B, Okura T, Sparks D, Valencia A, [33] Gibbons R, et al. Prostate cancer-specifi c survival thirty years after radical prostatectomy. J Urol 2010;183(4 Suppl abstract 585):e229 – 30. Hegarty J, Beirne PV, Walsh E, Comber H, Fitzgerald T, [34] Wallace Kazer M. Radical prostatectomy versus watchful waiting for prostate cancer. Cochrane Database Syst Rev 2010(11):CD006590. Wong YN, Mitra N, Hudes G, Localio R, Schwartz JS, [35] Wan F, et al. Survival associated with treatment vs observa-tion of localized prostate cancer in elderly men. JAMA 2006;296:2683 – 93. Carlsson S, Adolfsson J, Bratt O, Johansson JE, Ahlstrand C, [36] Holmberg E, et al. Nationwide population-based study on 30-day mortality after radical prostatectomy in Sweden. Scand J Urol Nephrol 2009;43:350 – 6. Judge A, Evans S, Gunnell DJ, Albertsen PC, Verne J, [37] Martin RM. Patient outcomes and length of hospital stay after radical prostatectomy for prostate cancer: Analysis of hospital episodes statistics for England. BJU Int 2007;100:1040 – 9. Dillioglugil O, Leibman BD, Leibman NS, Kattan MW, [38] Rosas AL, Scardino PT. Risk factors for complications and morbidity after radical retropubic prostatectomy. J Urol 1997;157:1760 – 7. Bianco FJ, Jr., Riedel ER, Begg CB, Kattan MW, Scardino [39] PT. Variations among high volume surgeons in the rate of complications after radical prostatectomy: Further evidence that technique matters. J Urol 2005;173:2099 – 103. Bubolz T, Wasson JH, Lu-Yao G, Barry MJ. Treatments for [40] prostate cancer in older men: 1984 – 1997. Urology 2001;58:977 – 82. Lu-Yao GL, Albertsen P, Warren J, Yao SL. Effect of age and [41] surgical approach on complications and short-term mortality after radical prostatectomy – a population-based study. Urology 1999;54:301 – 7. Karakiewicz PI, Bazinet M, Aprikian AG, Tanguay S, Elhilali [42] MM. Thirty-day mortality rates and cumulative survival after radical retropubic prostatectomy. Urology 1998;52:1041 – 6. Arai Y, Egawa S, Tobisu K, Sagiyama K, Sumiyoshi Y, Hashine [43] K, et al. Radical retropubic prostatectomy: Time trends, mor-bidity and mortality in Japan. BJU Int 2000;85:287 – 94. Begg CB, Riedel ER, Bach PB, Kattan MW, Schrag D, [44] Warren JL, et al. Variations in morbidity after radical prosta-tectomy. N Engl J Med 2002;346:1138 – 44. Alibhai SM, Leach M, Tomlinson G, Krahn MD, Fleshner [45] N, Holowaty E, et al. 30-day mortality and major complica-tions after radical prostatectomy: Infl uence of age and comorbidity. J Natl Cancer Inst 2005;97:1525 – 32. Lu-Yao GL, McLerran D, Wasson J, Wennberg JE. An assess-[46] ment of radical prostatectomy. Time trends, geographic variation, and outcomes. The Prostate Patient Outcomes Research Team. JAMA 1993;269:2633 – 6. Alibhai SM, Leach M, Tomlinson G. Examining the location [47] and cause of death within 30 days of radical prostatectomy. BJU Int 2005;95:541 – 4.
Klevecka V, Burmester L, Musch M, Roggenbuck U, Kroepfl [48] D. Intraoperative and early postoperative complications of radical retropubic prostatectomy. Urol Int 2007;79:217 – 25. Noldus J, Michl U, Graefen M, Haese A, Hammerer P, [49] Huland H. Patient-reported sexual function after nerve-sparing radical retropubic prostatectomy. Eur Urol 2002;42:118 – 24. Pedersen KV, Herder A. Radical retropubic prostatectomy [50] for localised prostatic carcinoma: A clinical and pathological study of 201 cases. Scand J Urol Nephrol 1993;27:219 – 24. Besarani D, Amoroso P, Kirby R. Bladder neck contracture [51] after radical retropubic prostatectomy. BJU Int 2004;94:1245 – 7. Steineck G, Helgesen F, Adolfsson J, Dickman PW, [52] Johansson JE, Norlen BJ, et al. Quality of life after radical prostatectomy or watchful waiting. N Engl J Med 2002;347:790 – 6. Alivizatos G, Skolarikos A. Incontinence and erectile dys-[53] function following radical prostatectomy: A review. Sci World J 2005;5:747 – 58. Foote J, Yun S, Leach GE. Postprostatectomy incontinence. [54] Pathophysiology, evaluation, and management. Urol Clin North Am 1991;18:229 – 41. Krane RJ. Urinary incontinence after treatment for localized [55] prostate cancer. Mol Urol 2000;4:279 – 86; discussion 87. Nandipati KC, Raina R, Agarwal A, Zippe CD. Erectile dys-[56] function following radical retropubic prostatectomy: Epide-miology, pathophysiology and pharmacological management. Drugs Aging 2006;23:101 – 17. Grise P, Thurman S. Urinary incontinence following treat-[57] ment of localized prostate cancer. Cancer Control 2001;8:532 – 9. Litwin MS, Melmed GY, Nakazon T. Life after radical pros-[58] tatectomy: A longitudinal study. J Urol 2001;166:587 – 92. Glickman L, Godoy G, Lepor H. Changes in continence and [59] erectile function between 2 and 4 years after radical prosta-tectomy. J Urol 2009;181:731 – 5. Korfage IJ, Essink-Bot ML, Borsboom GJ, Madalinska JB, [60] Kirkels WJ, Habbema JD, et al. Five-year follow-up of health-related quality of life after primary treatment of localized prostate cancer. Int J Cancer 2005;116:291 – 6. Long JA, Lebret T, Saporta F, Herve JM, Lugagne PM, [61] Poulain JE, et al. [Evaluation of sexuality and erectile func-tion of candidates for radical prostatectomy]. Prog Urol 2006;16:450 – 6. Salonia A, Zanni G, Gallina A, Sacca A, Sangalli M, [62] Naspro R, et al. Baseline potency in candidates for bilateral nerve-sparing radical retropubic prostatectomy. Eur Urol 2006;50:360 – 5. Lepor HA. Radical prostatectomy: Status and opportunities [63] for improving outcomes. Cancer Invest 2004;22:435 – 44. Murphy GP, Mettlin C, Menck H, Winchester DP, Davidson [64] AM. National patterns of prostate cancer treatment by radical prostatectomy: Results of a survey by the American College of Surgeons Commission on Cancer. J Urol 1994;152(5 Pt 2):1817 – 9. Tomschi W, Suster G, Holtl W. Bladder neck strictures after [65] radical retropubic prostatectomy: Still an unsolved problem. Br J Urol 1998;81:823 – 6. Litwin MS, Lubeck DP, Spitalny GM, Henning JM, Carroll [66] PR. Mental health in men treated for early stage prostate carcinoma: A posttreatment, longitudinal quality of life anal-ysis from the Cancer of the Prostate Strategic Urologic Research Endeavor. Cancer 2002;95:54 – 60. Mols F, van de Poll-Franse LV, Vingerhoets AJ, Hendrikx A, [67] Aaronson NK, Houterman S, et al. Long-term quality of life among Dutch prostate cancer survivors: Results of a popu-lation-based study. Cancer 2006;107:2186 – 96.

Radical Retropubic Prostatectomy 97
Sun M, Lughezzani G, Alasker A, Isbarn H, Jeldres C, Shar-[74] iat SF, et al. Comparative study of inguinal hernia repair after radical prostatectomy, prostate biopsy, transurethral resec-tion of the prostate or pelvic lymph node dissection. J Urol 2010;183:970 – 5. Stranne J, Hugosson J, Iversen P, Morris T, Lodding P. [75] Inguinal hernia in stage M0 prostate cancer: A comparison of incidence in men treated with and without radical retro-pubic prostatectomy – an analysis of 1105 patients. Urology 2005;65:847 – 51. Stranne J, Johansson E, Nilsson A, Bill-Axelson A, Carlsson [76] S, Holmberg L, et al. Inguinal hernia after radical prostate-ctomy for prostate cancer: Results from a randomized setting and a nonrandomized setting. Eur Urol 2010;58:719 – 26. Koie T, Yoneyama T, Kamimura N, Imai A, Okamoto A, [77] Ohyama C. Frequency of postoperative inguinal hernia after endoscope-assisted mini-laparotomy and conventional retro-pubic radical prostatectomies. Int J Urol 2008;15:226 – 9. Matsubara A, Yoneda T, Nakamoto T, Maruyama S, Koda S, [78] Goto K, et al. Inguinal hernia after radical perineal prosta-tectomy: Comparison with the retropubic approach. Urology 2007;70:1152 – 6.
Regan TC, Mordkin RM, Constantinople NL, Spence IJ, [68] Dejter SW, Jr. Incidence of inguinal hernias following radical retropubic prostatectomy. Urology 1996;47:536 – 7. Lodding P, Bergdahl C, Nyberg M, Pileblad E, Stranne J, [69] Hugosson J. Inguinal hernia after radical retropubic prosta-tectomy for prostate cancer: A study of incidence and risk factors in comparison to no operation and lymphadenec-tomy. J Urol 2001;166:964 – 7. Stranne J. Inguinal hernia after urologic surgery in males [70] with special reference to radical retropubic prostatectomy – A clinical, epidemiological and methodological study [Doctoral thesis]. G ö teborg: G ö teborg University; 2006. Twu CM, Ou YC, Yang CR, Cheng CL, Ho HC. Predicting [71] risk factors for inguinal hernia after radical retropubic pros-tatectomy. Urology 2005;66:814 – 8. Abe T, Shinohara N, Harabayashi T, Sazawa A, Suzuki S, [72] Kawarada Y, et al. Postoperative inguinal hernia after radi-cal prostatectomy for prostate cancer. Urology 2007;69:326 – 9. Ichioka K, Kohei N, Yoshimura K, Arai Y, Terai A. Impact of [73] retraction of vas deferens in postradical prostatectomy inguinal hernia. Urology 2007;70:511 – 4.

Correspondence: Sten Nilsson, Department of Oncology, Karolinska Institutet, Stockholm, SE-17176, Sweden. E-mail: [email protected]
(Received 2 March 2011; accepted 23 March 2011)
ORIGINAL ARTICLE
Curative radiation therapy in prostate cancer
ULRIKA HARMENBERG 1 , FREDDIE C. HAMDY 2 , ANDERS WIDMARK 3 , BO LENNERN Ä S 4 & STEN NILSSON 1
1 Department of Oncology/Pathology, Karolinska University Hospital and Institutet, Stockholm, Sweden, 2 Nuffi eld Department of Surgical Sciences, University of Oxford, Oxford, UK, 3 Department of Radiation Sciences, Oncology, Ume å University, Ume å , Sweden and 4 Department of Oncology, Sahlgrenska Hospital and Academy, University of Gothenburg, Gothenburg, Sweden
AbstractRadiotherapy has experienced an extremely rapid development in recent years. Important improvements such as the intro-duction of multileaf collimators and computed tomography (CT)-based treatment planning software have enabled three dimensional conformal external beam radiation therapy (3DCRT). The development of treatment planning systems and technology for brachytherapy has been very rapid as well. Development of accelerators with integrated on-board imaging equipment and technology , for example image-guided radiation therapy (IGRT) has further improved the precision with reduced margins to adjacent normal tissues. This has , in turn , led to the possibility to administer even higher doses to the prostate than previously. Although radiotherapy and radical prostatectomy have been used for the last decades as curative treatment modalities , still there are no randomized trials published comparing these two options. Outcome data show that the two treatment modalities are highly comparable when used for low- and intermediate-risk prostate cancer.
Keywords: Curative radiation therapy – the rationale for dose escalation – choice of radiation boost techniques
Several randomized trials have shown that a dose-response relationship exists for curative radiotherapy of prostate cancer. A comprehensive summary of these are found in a recent meta-analysis of Viani et al. [1]. Most outcome data are reported with respect to the D ’ Amico risk classifi cation [2]. In patients with inter-mediate-and high-risk prostate cancer doses in the range of 70 – 72 Gy are generally not suffi cient to achieve ablation of the tumor [3 – 7]. These tumors require radiation doses in the range of 78 Gy and above [1]. There also appears to be a dose-response relationship even in low-risk prostate cancer, i.e. these tumors also need dose escalation [1].
Dose escalation is currently being used with dif-ferent approaches at different centers. The majority of these use external radiation therapy alone based on modern technologies such as intensity modulated radiation therapy (IMRT) and IGRT, enabling radi-ation doses in the range of 78 – 81 Gy, or even higher, with conventional fractionation combined with selected radiation boost regimens [1].
Other centers use a combination of 3DCRT, IMRT or IGRT in combination with high dose-rate (HDR) brachytherapy, the latter technique used in the context of radiation boost [8 – 12]. Such com-bined therapy achieves doses � 116 Gy [8]. Two randomized studies have compared the effect of external beam radiotherapy alone with external beam radiotherapy plus HDR brachytherapy boost [13,14]. Early data from one of these studies have been pub-lished [14]. These show the benefi t of combination external beam radiotherapy plus HDR brachyther-apy. However, the external radiation therapy alone utilized a lower biological dose compared with the combination therapy concept. This hampers the com-parison. A systematic review with meta-regression analysis has recently been carried out in which obser-vational studies on external beam radiation therapy (EBRT), seed implantation brachytherapy, and HDR brachytherapy were included [15]. The selection for EBRT included only studies with cohorts treated with at least 75 Gy. The conclusions drawn from this
Acta Oncologica, 2011; 50(Suppl 1): 98–103
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.576115

Curative radiation therapy in prostate cancer 99
analysis are that the combination of external beam radiotherapy and HDR brachytherapy results in a superior biochemical control as well as overall sur-vival [15]. However, there is still a need for random-ized controlled trials comparing the outcome of radiation therapies with different dose fractionation schedules, radiation boost techniques and total doses to the prostate and organs at risk.
Neoadjuvant, concomittant and adjuvant endocrine therapy
The value of combination treatment – radiotherapy and endocrine therapy – has been studied in multiple randomized trials [16,17]. These have in various degrees shown the benefi t of this concept with respect to local control, distant metastasis-free survival, disease-free survival, cancer specifi c mortality and overall survival. However, it is important to take some factors in consideration. The majority of trials have exclusively included patients with high-risk disease, i.e. patients with locally advanced and/or poorly differenti-ated cancer and/or high risk of lymph node metastatic disease. Radiotherapy was generally given at doses that, with today’s knowledge, are not suffi cient to achieve cure. Radiation was in most studies given with whole pelvic fi elds to 50 Gy and a subsequent radiation boost to the prostate [16]. One of the major arguments against the combination concept (radiotherapy com-bined with endocrine treatment) is that the radiother-apy used de facto needed androgen deprivation therapy to compensate, in part, for the inadequate radiation doses that could be achieved with the radiation therapy techniques used at that time. Some observational stud-ies support the assumption that hormonal neoadjuvant and adjuvant treatment can be excluded from the curative treatment provided that adequate radiation therapy is given [18]. The concept of neoadjuvant, con-comittant and adjuvant endocrine treatment will most certainly be challenged by the concept of curative treatment with high-dose radiotherapy as monother-apy (dose-escalated 3DCRT or IMRT/IGRT) or with combination therapy (3DCRT/IMRT/IGRT plus HDR brachytherapy boost) to the primary tumor.
Adjuvant and early salvage radiotherapy
The value of adjuvant radiotherapy after radical prostatectomy has been investigated in three large randomized trials [19 – 21]. Two of these have shown overall survival benefi t in the order of 10%, while the third [21] is not yet mature for overall survival evaluation. All studies showed, in comparison to no postoperative radiotherapy, statistically signifi cant differences in disease-free survival, metastasis-free survival, local progression and cancer-specifi c
mortality at advantage for radiotherapy. The question whether the postoperative treatment is best served in a strictly adjuvant setting or if this treatment could also well be given as early salvage radiotherapy has to be answered. Randomized studies on this concept are under way. The majority of centers have not yet adopted adjuvant radiotherapy into their clinical rou-tine practice. The main reason for this is that the results from the adjuvant trials have not been obtained until quite recently. At least half of the patients expe-riencing recurrence after radical prostatectomy (about 30%) should be considered for early salvage radiotherapy. Still there are no conclusive data on optimal fractionation schedule and the total dose needed in this setting. Questions also remain regard-ing the effi cacy of salvage radiotherapy in patients who never reach undetectable prostate-specifi c anti-gen (PSA) after surgery and/or have already reached a PSA level over 1 ng/ml before salvage radiation therapy and/or with tumor invasion of seminal vesi-cles at surgery. Pending the results from prospective trials the nomogram by Stephenson and coworkers is useful when discussing the benefi t and possible side-effects of salvage radiotherapy for patients experiencing recurrent disease [22].
Treatment of locally advanced disease – the SPCG7/SFUO3 trial
The treatment of choice in patients with locally advanced prostate cancer has over the years been endocrine therapy, either as castration therapy or treatment with antiandrogen as monotherapy. This paradigm was challenged in the Scandinavian SPCG7/SFUO3 randomized phase III trial comparing endo-crine therapy with and without local radiotherapy, followed by castration on progression [23]. The trial recruited 875 patients with locally advanced prostate cancer (T3; 78%; PSA � 70; N0; M0) between Feb-ruary 1996 and December 2002. The patients were randomly assigned to endocrine treatment alone (three months of total androgen blockade followed by continuous endocrine treatment using fl utamide), or to the same endocrine treatment combined with cura-tive radiotherapy. The primary endpoint was prostate-cancer-specifi c survival, and analysis was by intention to treat. After a median follow-up of 7.6 years, 79 men in the endocrine alone group and 37 men in the endocrine plus radiotherapy group had died of pros-tate cancer. The cumulative incidence at 10 years for prostate-cancer-specifi c mortality was 23.9% in the endocrine alone group and 11.9% in the endocrine plus radiotherapy group (difference 12.0%, 95% CI 4.9 – 19.1%), for a relative risk of 0.44 (0.30 – 0.66). At 10 years, the cumulative incidence for overall mortal-ity was 39.4% in the endocrine alone group and

100 U. Harmenberg et al.
29.6% in the endocrine plus radiotherapy group (difference 9.8%, 0.8 – 18.8%), for a relative risk of 0.68 (0.52 – 0.89). Cumulative incidence at 10 years for PSA recurrence was substantially higher in men in the endocrine-alone group (74.7% vs . 25.9%, p � 0.0001; HR 0.16; 0.12 – 0.20). After fi ve years, urinary, rectal, and sexual problems were slightly more frequent in the endocrine plus radiotherapy group. The addition of local radiotherapy to endo-crine treatment, thus, halved the 10-year prostate-cancer-specifi c mortality, and substantially decreased overall mortality in patients with locally advanced or high-risk local prostate cancer.
This is the only trial that has, so far, shown an overall survival benefi t in patients with localized prostate cancer and, in light of these data, endocrine treatment plus radiotherapy should be implemented as the new evidence based (level 1) standard treat-ment in patients with locally advanced disease [23].
Unanswered questions and developmental areas within radiation therapy of prostate cancer
As mentioned above, the early radiotherapy trials included treatment with whole-pelvic fi elds with a subsequent radiation boost to the prostate [16]. However, with the introduction of conformal radia-tion therapy techniques during the 1990s and that only patients with low risk for metastatic disease or patients with N0 disease were accepted for curative treatment, whole-pelvic radiotherapy was abandoned in most centers. With the knowledge of the short-comings of surgical lymph node staging [24] there is now a re-awakening of adjuvant radiation treatment of regional lymph nodes, especially with the advent of conformal techniques such as IMRT and intensity modulated arc therapy (IMAT) [25] and in patients with high risk ( � 15%) of having lymph node meta-static disease [26 – 29]. With these techniques it is now possible to administer radiotherapy to the regional lymph nodes in a more conformal manner, minimizing unwanted radiation dose to organs at risk such as bladder and bowel.
Another area that has attracted much of atten-tion recently is radiobiology of prostate cancer and radiation dose fractionation. Ample data exist to show that this tumor differs from several others by a low level α / β value, speaking in favor of hypofrac-tionation [8,30]. Several studies have now been designed on this concept. Arcangeli et al. have recently published data from the fi rst randomized trial [31]. This is not yet mature for evaluation of treatment effect – other than freedom from PSA recurrence – but the data show that the toxicity of the hypofractionation schedule used is acceptable
[31]. A Swedish-Danish randomized hypofraction-ation trial has recruited over 400 patients with inter-mediate-risk prostate cancer patients comparing EBRT 2 Gy per fraction to 78 Gy with 6.1 Gy in 7 fractions to 42.1 Gy (Widmark, personal communi-cation). It is important to note that the hypofraction-ation concept is based on the assumption that prostate cancer is often a slowly proliferating malig-nancy. However, some tumors also harbor poorly differentiated cancer cells which may not be suitable for hypofractionation [32,33]. The outcome of ongo-ing and future trials will, hopefully, clarify which patients benefi t from hypofractionation.
Radiotherapy versus radical prostatectomy
Prostate cancer is different from many other malig-nancies in that it is one of the few cancers in which radiotherapy is a major curative treatment option pri-mary treatment. Radical prostatectomy is the other curative treatment modality and patients are therefore asked to participate in the decision-making between these two options. Paradoxically, there are still no comparative, randomized studies on the concept radiotherapy versus surgery as primary treatment in prostate cancer – a situation which not seldom con-tributes to a frustrating situation for the patient as well for his partner when choosing therapy. Active monitoring is a valid option for patients with low-risk (T1c, Gleason score � 3 � 3 � 6 and PSA � 10) prostate cancer. The fi rst study on this theme is the ProtecT (Prostate testing for cancer and Treatment) trial undertaken in the UK, in which men with clini-cally localized prostate cancer were randomized to radiotherapy, radical prostatectomy or active monitor-ing [34]. This study has recruited extremely well and has now been closed for inclusion according to pro-tocol. Over 1500 participants agreed to randomiza-tion (63% of those eligible) with annual follow-up at over 90%. The fi rst results are expected in 2015 and overall survival data will be obtained in due course.
In patients with intermediate- and high-risk pros-tate cancer the main options, radiotherapy and sur-gery, remain. One Swedish randomized trial (neoadjuvant endocrine therapy plus radical prostate-ctomy versus neoadjuvant endocrine therapy plus external beam radiotherapy with HDR brachytherapy boost) included 89 patients with HRQoL as main end-point. The data will be published in 2011. To our knowledge, no other prospective randomized trials comparing these modalities are being undertaken.
Discussion
There are very good reasons to expect that the role of radiation therapy will increase in coming years,

Curative radiation therapy in prostate cancer 101
warranting further randomized studies in this area. Healthcare authorities have an important task in sup-porting and commissioning such trials to provide suffi cient evidence to guide practice and improve patient outcomes. Strong growth is expected in the areas of dose escalation, both with external beam radiation therapy as monotherapy and combination treatment with external therapy and brachytherapy. In all dose escalation protocols, the need for mini-mizing the margins of surrounding normal tissue is imperative. The need for improved positioning is becoming increasingly important. The trend toward higher fractions per dose, hypofractionation, will remain, highlighting the need for adequate visualiza-tion of the prostate and its positioning with tech-niques such as IGRT. The need for IMRT will increase for treatment of regional lymph nodes in patients with high-risk disease.
The trend towards higher radiation doses to the prostate and the balance between effi cacy and toxic-ity will place increasing demands on technical devel-opments, their adequate use and understanding of their limitations.
Data from the early 1990s have clearly shown that radiotherapy doses of 70 Gy and below are in the majority of cases insuffi cient for eradication of the cancer [3 – 7,35], and four randomized trials compar-ing 64 – 70 Gy with 74 – 80 Gy have shown improved outcome with dose escalation [36 – 39]. Several stud-ies have thereafter shown a correlation between residual cancer and the risk of local progression, metastatic disease and increased cancer-specifi c mortality [3,40 – 42]. This has also recently been shown in a biopsy study from the above mentioned SPCG-7 trial in which residual cancer was verifi ed in 22% of patients after more than three years and that this presence was associated with the above negative factors including increased cancer-specifi c mortality [43]. Notable is that the residual cancer was poorly differentiated (Gleason sum 8) in all patients with recurrent disease [43]. Data from our own experience have shown that residual cancer is associated with poor overall survival (Ljung et al., unpublished).
The diffi culties of implementing prospective ran-domized studies in radiation therapy as well as sur-gery are obvious, especially for the fact that at least 8 – 10 years of follow-up is needed to detect signifi cant differences in overall survival. The diffi culties in com-paring outcome data from non-randomized studies, especially from different centers, are even more obvi-ous, not only because of patient selection but also because different defi nitions have been used over the years to defi ne biochemical free survival. The diffi cul-ties are compounded by the fact that PSA levels not only refl ect residual disease within the prostate but
also metastatic progression, particularly in patients with intermediate- and high-risk prostate cancer. An alternative endpoint would be to utilize transrectal ultrasound (TRUS)-guided biopsy mapping of the prostate two to three years after completion of radio-therapy, especially when comparing the outcome of different doses and dose fractionation schedules. Analysis of residual cancer is, by defi nition, a surro-gate endpoint, but may have the potential of being more accurate than serum-PSA in assessing local radicality of a given radiation treatment.
The importance of randomized controlled trials comparing modern radiation therapy and radical prostatectomy cannot be stressed enough. The Pro-tecT trial in the UK will provide important informa-tion on the outcome and potential side-effects of these two modalities in comparison with active mon-itoring. However, it will take many years before mature data on overall survival become available. In the meantime it is important to continue the prospective evaluation of new radiation therapies and surgical treatment concepts in large and well conducted randomized controlled trials.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Viani GA, Stefano EJ, Afonso SL. Higher-than-conventional [1] radiation doses in localized prostate cancer treatment: A meta-analysis of randomized, controlled trials. Int J Radiat Oncol Biol Phys 2009;74:1405 – 18. D’Amico AV, Whittington R, Malkowicz SB, Weinstein M, [2] Tomaszewski JE, Schultz D, et al. Predicting prostate specifi c antigen outcome preoperatively in the prostate specifi c anti-gen era. J Urol 2001;166:2185 – 8. Crook JM, Malone S, Perry G, Eapen L, Owen J, Robertson [3] S, et al. Twenty-four-month postradiation prostate biopsies are strongly predictive of 7-year disease-free survival: Results from a Canadian randomized trial. Cancer 2009;115:673 – 9. Ljung G, Egevad L, Norberg M, Holmberg L, Nilsson S, [4] Busch C. Expression of p21 and mutant p53 gene products in residual prostatic tumor cells after radical radiotherapy. Prostate 1997;32:99 – 105. Ljung G, Norberg M, Hansson H, de la Torre M, Egevad L, [5] Holmberg L, et al. Transrectal ultrasonically-guided core biopsies in the assessment of local cure of prostatic cancer after radical external beam radiotherapy. Acta Oncol 1995; 34:945 – 52. Ljung G, Norberg M, Holmberg L, Busch C, Nilsson S. [6] Characterization of residual tumor cells following radical radiation therapy for prostatic adenocarcinoma; immunohis-tochemical expression of prostate-specifi c antigen, prostatic acid phosphatase, and cytokeratin 8. Prostate 1997;31:91 – 7. Almroth A, Ljung G, Eklund T, Nordgren H, Kalafatidis D, [7] Ringqvist I, et al. Value of sextant biopsies in the assessment of local cure following external beam radiotherapy of prostatic adenocarcinoma. Scand J Urol Nephrol 1998;32:111 – 5.

102 U. Harmenberg et al.
Fowler JF. The radiobiology of prostate cancer including [8] new aspects of fractionated radiotherapy. Acta Oncol 2005; 44:265 – 76. Galalae RM, Kovacs G, Schultze J, Loch T, Rzehak P, Wil-[9] helm R, et al. Long-term outcome after elective irradiation of the pelvic lymphatics and local dose escalation using high-dose-rate brachytherapy for locally advanced prostate cancer. Int J Radiat Oncol Biol Phys 2002;52:81 – 90. Vargas CE, Martinez AA, Boike TP, Spencer W, Goldstein [10] N, Gustafson GS, et al. High-dose irradiation for prostate cancer via a high-dose-rate brachytherapy boost: Results of a phase I to II study. Int J Radiat Oncol Biol Phys 2006; 66:416 – 23. Deger S, Boehmer D, Roigas J, Schink T, Wernecke KD, [11] Wiegel T, et al. High dose rate (HDR) brachytherapy with conformal radiation therapy for localized prostate cancer. Eur Urol 2005;47:441 – 8. Kalkner KM, Wahlgren T, Ryberg M, Cohn-Cedermark G, [12] Castellanos E, Zimmerman R, et al. Clinical outcome in patients with prostate cancer treated with external beam radiotherapy and high dose-rate iridium 192 brachytherapy boost: A 6-year follow-up. Acta Oncol 2007;46:909 – 17. Guix B, Bartrina JM, Henriquez I. Dose escalation by com-[13] bined treatment 3D-conformal radiotherapy plus HDR brach-ytherapy as treatment for intermediate- or high-risk cancer: Early toxicity and biochemical outcome of a prospective ran-domized trial. Radiother Oncol 2007;83:S39 [abstract 81]. Hoskin PJ, Motohashi K, Bownes P, Bryant L, Ostler P. High [14] dose rate brachytherapy in combination with external beam radiotherapy in the radical treatment of prostate cancer: Ini-tial results of a randomised phase three trial. Radiother Oncol 2007;84:114 – 20. Pieters BR, de Back DZ, Koning CC, Zwinderman AH. [15] Comparison of three radiotherapy modalities on biochemical control and overall survival for the treatment of prostate can-cer: A systematic review. Radiother Oncol 2009; 93:168 – 73. Gottschalk AR, Roach M, 3rd. The use of hormonal therapy [16] with radiotherapy for prostate cancer: Analysis of prospective randomised trials. Br J Cancer 2004;90:950 – 4. Shelley MD, Kumar S, Wilt T, Staffurth J, Coles B, Mason [17] MD. A systematic review and meta-analysis of randomised trials of neo-adjuvant hormone therapy for localised and locally advanced prostate carcinoma. Cancer Treat Rev 2009;35:9 – 17. Galalae RM, Martinez A, Nuernberg N, Edmundson G, [18] Gustafson G, Gonzalez J, et al. Hypofractionated conformal HDR brachytherapy in hormone naive men with localized prostate cancer. Is escalation to very high biologically equiv-alent dose benefi cial in all prognostic risk groups? Strahlen-ther Onkol 2006;182:135 – 41. Thompson IM, Tangen CM, Paradelo J, Lucia MS, Miller [19] G, Troyer D, et al. Adjuvant radiotherapy for pathological T3N0M0 prostate cancer signifi cantly reduces risk of metas-tases and improves survival: Long-term followup of a rand-omized clinical trial. J Urol 2009;181:956 – 62. Bolla M, van Poppel H, Collette L, van Cangh P, Vekemans [20] K, Da Pozzo L, et al. Postoperative radiotherapy after radical prostatectomy: A randomised controlled trial (EORTC trial 22911). Lancet 2005;366(9485):572 – 8. Wiegel T, Bottke D, Steiner U, Siegmann A, Golz R, Storkel [21] S, et al. Phase III postoperative adjuvant radiotherapy after radical prostatectomy compared with radical prostatectomy alone in pT3 prostate cancer with postoperative undetectable prostate-specifi c antigen: ARO 96-02/AUO AP 09/95. J Clin Oncol 2009;27:2924 – 30. Stephenson AJ, Scardino PT, Kattan MW, Pisansky TM, [22] Slawin KM, Klein EA, et al. Predicting the outcome of
salvage radiation therapy for recurrent prostate cancer after radical prostatectomy. J Clin Oncol 2007;25:2035 – 41. Widmark A, Klepp O, Solberg A, Damber JE, Angelsen A, [23] Fransson P, et al. Endocrine treatment, with or without radiotherapy, in locally advanced prostate cancer (SPCG-7/SFUO-3): An open randomised phase III trial. Lancet 2009;373(9660):301 – 8. Ordon M, Nam RK. Lymph node assessment and lym-[24] phadenectomy in prostate cancer. J Surg Oncol 2009;99:215 – 24. Yu CX, Tang G. Intensity-modulated arc therapy: Principles, [25] technologies and clinical implementation. Phys Med Biol 2011;56:R31 – 54. Nguyen PL, D’Amico AV. Targeting pelvic lymph nodes in [26] men with intermediate- and high-risk prostate cancer despite two negative randomized trials. J Clin Oncol 2008;26:2055 – 6; author reply 2056 – 7. Roach M, 3rd. Targeting pelvic lymph nodes in men with [27] intermediate- and high-risk prostate cancer, and confusion about the results of the randomized trials. J Clin Oncol 2008;26:3816 – 7; author reply 3817 – 8. Wang D, Lawton C. Pelvic lymph node irradiation for pros-[28] tate cancer: Who, why, and when? Semin Radiat Oncol 2008;18:35 – 40. Kim BS, Lashkari A, Vongtama R, Lee SP, Parker RG. Effect [29] of pelvic lymph node irradiation in salvage therapy for patients with prostate cancer with a biochemical relapse fol-lowing radical prostatectomy. Clin Prostate Cancer 2004;3:93 – 7. Williams SG, Taylor JM, Liu N, Tra Y, Duchesne GM, [30] Kestin LL, et al. Use of individual fraction size data from 3756 patients to directly determine the alpha/beta ratio of prostate cancer. Int J Radiat Oncol Biol Phys 2007;68: 24 – 33. Arcangeli G, Saracino B, Gomellini S, Petrongari MG, [31] Arcangeli S, Sentinelli S, et al. A prospective phase III ran-domized trial of hypofractionation versus conventional frac-tionation in patients with high-risk prostate cancer. Int J Radiat Oncol Biol Phys 2010;78:11 – 8. Lennernas B, Nilsson S, Levitt SH. Hypofractionation for [32] radiotherapy of prostate cancer using a low alpha/beta ratio – possible reasons for concerns? An example of fi ve-dimen-sional radiotherapy. Acta Oncol 2011. Miles EF, Lee WR. Hypofractionation for prostate cancer: A [33] critical review. Semin Radiat Oncol 2008;18:41 – 7. Lane JA, Hamdy FC, Martin RM, Turner EL, Neal DE, [34] Donovan JL. Latest results from the UK trials evaluating prostate cancer screening and treatment: The CAP and Pro-tecT studies. Eur J Cancer 2010;46:3095 – 101. Ljung G, Egevad L, Norberg M, de la Torre M, Holmberg [35] L, Busch C, et al. Assessment of proliferation indicators in residual prostatic adenocarcinoma cells after radical external beam radiotherapy. Prostate 1996;29:303 – 10. Pollack A, Zagars GK, Starkschall G, Antolak JA, Lee JJ, [36] Huang E, et al. Prostate cancer radiation dose response: Results of the M. D. Anderson phase III randomized trial. Int J Radiat Oncol Biol Phys 2002;53:1097 – 105. Zietman AL, DeSilvio ML, Slater JD, Rossi CJ, Jr., Miller [37] DW, Adams JA, et al. Comparison of conventional-dose vs high-dose conformal radiation therapy in clinically localized adenocarcinoma of the prostate: A randomized controlled trial. JAMA 2005;294:1233 – 9. Dearnaley DP, Sydes MR, Graham JD, Aird EG, Bottomley [38] D, Cowan RA, et al. Escalated-dose versus standard-dose conformal radiotherapy in prostate cancer: First results from the MRC RT01 randomised controlled trial. Lancet Oncol 2007;8:475 – 87.

Curative radiation therapy in prostate cancer 103
related mortality after external beam radio therapy for pros-tate cancer. J Urol 2008;179:1368 – 73; discussion 1373. Coquard R. Re: Infl uence of local tumor control on distant [42] metastases and cancer related mortality after external beam radiotherapy for prostate cancer: Zelefsky MJ, Reuter VE, Fuks Z, Scardino P, Shippy A. J Urol 2008;179:1368 – 73. J Urol 2008;180:2258; author reply 2258. Solberg A, Haugen OA, Viset T, Bergh A, Tasdemir I, Ahlgren [43] G, et al. Residual prostate cancer in patients treated with endocrine therapy with or without radical radiotherapy: A side study of the SPCG-7 randomized trial. Int J Radiat Oncol Biol Phys.
Peeters ST, Heemsbergen WD, Koper PC, van Putten WL, [39] Slot A, Dielwart MF, et al. Dose-response in radiotherapy for localized prostate cancer: Results of the Dutch multi-center randomized phase III trial comparing 68 Gy of radio-therapy with 78 Gy. J Clin Oncol 2006;24:1990 – 6. Vance W, Tucker SL, de Crevoisier R, Kuban DA, [40] Cheung MR. The predictive value of 2-year posttreatment biopsy after prostate cancer radiotherapy for eventual bio-chemical outcome. Int J Radiat Oncol Biol Phys 2007;67: 828 – 33. Zelefsky MJ, Reuter VE, Fuks Z, Scardino P, Shippy A. Infl u-[41] ence of local tumor control on distant metastases and cancer

Correspondence: Bo Lennern ä s, Department of Oncology, University of Gothenburg, Sahlgrenska University Hospital, s-413 45 Sweden. E-mail: [email protected]
(Received 14 August 2010 ; accepted 28 September 2010 )
ORIGINAL ARTICLE
Four and fi ve dimensional radiotherapy with reference to prostate cancer – defi nitions, state of the art and further directions – an overview
BO LENNERN Ä S 1 , ENRIQUE CASTELLANOS 2 , STEN NILSSON 2 & SEYMOUR LEVITT 2
1 Department of Oncology, Sahlgrenska Hospital and Academy, University of Gothenburg, Gothenburg, Sweden and 2 Department of Oncology/Pathology, Karolinska University Hospital and Institutet, Stockholm, Sweden
Abstract Radiotherapy (RT) always requires a compromise between tumor control and normal tissue side-effects. Technical innova-tion in radiation therapy (RT), such as three dimensional RT, is now established. Concerning prostate cancer (PC), it is reasonable to assume that RT of PC will increase in the future. The combination of small margins, a movable target (pros-tate), few fractions and high doses will probably demand dynamically positioning systems and in real time. This is called four dimensional radiotherapy (4DRT). Moreover, biological factors must be included in new treatments such as hypo-fractionation schedules. This new era is called fi ve dimensional radiotherapy, 5DRT. In this paper we discuss new concepts in RT in respect to PC.
Radiotherapy (RT) always demands a compromise between tumor control and side-effects. Undesir-able side-effects are the result of dose delivered to non-tumor tissue or healthy organs, and can be reduced if the irradiated volume is minimized. However, due to different errors in the radiotherapy chain, and tumor movements, dose to healthy tissue/organs cannot be totally avoided. Radiotherapy will therefore always be associated with some degree of side-effects [1].
Radiotherapy is and will be an important modality in the treatment of PC. There are several randomized studies showing a clear survival benefi t of RT in the treatment of PC and RT shall be considered alone or adjuvant to surgery and hormone therapy [2 – 4].
The goal of developments in radiation therapy is twofold: (1) improve tumor control and/ or (2) decrease side-effects. Suit 2002 [5] has listed up 20 major contributions or steps in the development of radiotherapy from the development of the gantry to computer tomography based dose planning and three dimensional radiotherapy (3DRT).
3DRT became available in the beginning of the 1990s. The Nordic collaboration in the CART program (computer aid in radiotherapy) was an
important contribution to this development, see Figure 1 [6]. In these systems the anatomy of the target could be more complex and the margin sur-rounding the tumor viewed in three dimensions. Compensation for dose absorption in surrounding organs/ tissue (such as the pelvis) became better. Possibly, 3DRT is one of the greatest single improve-ments in radiation therapy.
Even if 3DRT can account for a more complex anatomy, it is still diffi cult in clinical practice to mod-ulate the beam in the axial direction. Wedges can compensate for some inhomogeneous contours, but “ banana-shaped ” or concave targets remain diffi cult to treat with appropriate dose distributions.
The introduction of Intensity modulated radio-therapy (IMRT), or later developments VMAT (volu-metric modulated arc therapy) or Rapid Arc, made it possible to treat banana-shaped targets (lymph node volumes, prostate with seminal vesicles). Proton beams have used since the 1940s, but the accessibility has been low. In recent years, however, the number of facilities increased, and thus possibilities to treat more patients. Because of limitations of space, and the complexity of these techniques, the interested reader is referred to books such as “ The Technical
Acta Oncologica, 2011; 50(Suppl 1): 104–110
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.530003

Radiotherapy in 4D and 5D for PC – an overview 105
Basis of Radiation Therapy, 4 th ed. ” . Springer – Verlag, 2006.
It has always been important to be aware of and control movements of the target and patient. In the use of IMRT and protons this is even more impor-tant. Displacement errors during IMRT treatments can signifi cantly change the outcome of treatment. Considering that the treatment time for IMRT is two to three times longer then that for conventional 3DRT and the steep dose gradient in protons, the need for improved and continuous patient/ target positioning is obvious.
With better positioning of the target and smaller margins this problem can possibly be reduced. Posi-tioning problems associated with 3DRT and IMRT has led to the development of Image Guided Radio-therapy (IGRT). This development has accelerated during recent years and IGRT has initiated a new era in radiation therapy, namely “ four-dimensional radi-ation therapy ” (4DRT).
One major disadvantage of 3DRT is that all treatments are based on the anatomy as defi ned by the treatment planning CT prior to the actual treatment planning. In an optimal 4DRT system positioning information during irradiation is feed-back to the treatment planning system for online corrections. In the clinic this is very complicated
and maybe unnecessary to compensate for small movements.
In the case of prostate cancer, and most other tumors, the development of the treatment protocol involves fi ve critical steps [1,7]. The fi rst step involves the diagnostic procedures including determination of the spread of the tumor (e.g., prostate only or lymph node involvement). Next, what is the best treatment for each patient. Third, if radiation therapy is selected, the target(s) must be accurately defi ned. Fourth, the treatment plan must be optimized (e.g., 3DRT, protons or IMRT) and, fi nally, the treatment must compensate for inter/intra-treatment variations in position of the patient and the prostate (QA, quality assurance).
In this article we will discuss the last three steps from a 4DRT point of view and discuss necessary developments and improvements of radiation ther-apy in general and radiation therapy of prostate can-cer in particular for the next dimension in radiation therapy – namely 4DRT.
We will also discuss the concepts of biological modeling of the physical dose distribution in 4DRT, and this will be referred to as “ fi ve dimensional radio-therapy ” , 5DRT. This is the fi rst time the concept of radiobiology will be integrated with the physical dose, and this is possible because of the exceptional control of the dose distribution using 4DRT.
Figure 1. The Nordic CART (Computer Aid in RadioTherapy) programme (with permission from Hans Dahlin). The drawing summarises 3DRT as we know it today.

106 B. Lennern ä s et al.
Material and methods
The 4DRT and certainly the 5DRT era is in its for-mative stage In April 2005 there were approximately 20 references found on on 4D radiotherapy (RT) or four-dimensional RT (depending on search items) in “ Pubmed ” . On the Internet there were approximately 5 000 references and this refl ects the fact that there were multiple 4D projects globally. In 2010 there are approximately 18 500 Internet hits and approxi-mately 400 hits on Pubmed, although some are not fully appreciable.
There are also numerous publications and proj-ects concerning radiotherapy and movement in the 3DRT milieu and aspects from selected articles will be discussed. Several of these articles could be clas-sifi ed as a 4DRT-publication, but the awareness of the technique has been minimal (that is, the key-words on certain articles do not necessarily include 4D, even though the article itself contains 4D infor-mation). This makes it possible that we might miss important contributions in our discussion.
Results & discussion
There is no clear defi nition of 4DRT. 4DRT can be considered as the logical progression forward from 3DRT. 3DRT treatment is often referred to as any treatment using a 3D based dose-planning system and computer tomography (CT). Since 3DRT is a static situation, 4DRT should be considered to be any treatment which dynamically compensates for target or patient movements during radiotherapy. On the other hand 5DRT is a stand alone concept merging physical and technical aspects of RT with radiobiology. It is of course possible to perform 3DRT and include biological information, but it is impossible to have a defi nition including all combi-nations. We e suggest that the most advanced tech-nology is used as part of the defi nition (5DRT � 4DRT � 3DRT). It is also important to distinguish the six geometric dimensions (X,Y,Z and rotation) from the different levels (dimensions) of radiother-apy. Although they are linked they describe different aspects of RT.
The defi nition of 4DRT does not include treat-ments were motion has been taken in account by merely adding a margin around the tumor. With this defi nition in mind, many of current standard treat-ments cannot be considered as being true 4DRT although they have the technical possibilities to do online corrections.
The fi rst articles in which the term 4DRT is found is in the late 1990s [8]. There are also authors with a slightly different approach, namely that of including time in a biological model, but today
most authors defi ne the motion of the target and its effect on the physical dose distribution as the fourth dimension. This is in concordance with the 4D imaging literature [9]. We suggest that biologi-cal considerations of the physical dose distribution should be defi ned as the fi fth dimension if this is applied to the treatment directly (that is not just during follow-up), and we will briefl y discuss the concept.
Most radiotherapy publications refer to 4DRT in relation to moving targets due to respiratory move-ment (lung, liver and breast) and prostate cancer is only briefl y discussed [9]. The development follows the evolution of the computer tomography [10,11]. However, the concerns about positioning and mov-ing targets is as old as that of radiotherapy. It is also notable that most of the problems discussed in the literature today, were outlined by the Nordic CART project in the 1970s (Figure 1). This relates to the fact that the development of modern radiotherapy is closely linked to the development of computers and imaging devices – 4DRT could hardly be done using laser beams and skin marks.
Two publications worth mention are the entire issue of Seminars in Radiation Oncology [11] deal-ing with moving targets and 4DRT and a recent review by Bucci et al. [12]. These articles present state of the art procedures compensating for moving targets during radiotherapy.
For the clinically oriented reader, literature about moving targets and techniques for dealing with the problem is sometimes confusing for two basic rea-sons. First, techniques like 3DRT, IMRT, IGRT and 4DRT are used as if they were independent tech-niques. In clinical practice 3DRT and 4DRT are the level of treatment precision required, and IMRT, IGRT and are the techniques used to achieve that precision. Second, frequently, positioning only refers to the fi nal step in the treatment chain, but in reality, the 4DRT concept includes all steps from dose plan-ning to the last moment of the treatment.
4D computer tomography (4DCT) and treatment planning
Motion of the target during the dose planning CT can change the shape of the target signifi cantly (Seminars), and this is summarized in Figure 2. Two targets (A and B) are scanned with fi ve CT-slices (1-5). Below are the two resulting 3D images based on the scans. Due to different motion pattern two totally different shapes of the target will be stored in the treatment planning system (A1-5 and B1-5). If we use a high precision RT technique with small margins, this error alone can drastically effect the treatment and it is important to monitor target and

Radiotherapy in 4D and 5D for PC – an overview 107
patient position throughout the whole radiotherapy chain [13].
If we assume that the movement is � /− 2cm (a most common margin) and we divide it in steps of 5 mm, we have nine separate locations of the struc-tures. If the treatment plan has three portals the number of combinations in this very simple example is 9 � 9 � 9 � 729. It is not possible to produce different dose plans for all these combinations. This emphasizes the fact that in clinical practice it is not and it will not be possible to perform ultimate 4DRT, because the number of combinations is to great and probably are not clinically relevant
Four dimensional imaging devices are today well established and as conventional CTs were a major step forward for RT, 4DCTs will be important for RT in the future. This also stresses the importance of a nomenclature and standards for 4DRT in order to stimulate technical development [10 – 12].
An interesting new fi eld of avoiding dose to OAR is the introductions of spacers separating the OAR from the target area, thus reducing the dose to the OAR [13]. These must of course be used on the CT and can possible also reduce the motion of the prostate.
Prostate motion
The prostate can move relative to the beam as a part of a patient movement or internally. With laser posi-tioning, displacement errors of several centimeters can be noted [14]. Figure 3 refers to a 20 minute recording of prostate movements, using an electro-magnetic positioning implant (Raypilot, Micropos Medicial). The maximum error in this example is approximately 14 mm. Usually, prostate movements are minimal (millimeters), but large errors can occur and this is important with fewer fractions and/or long treatment times.
Electronic portal imaging (EPI), cone computer tomography (cone CT) and IGRT
EPI is probably the most commonly used motion detecting device in clinical practice and is the basis of IGRT. Contrast in an EPI image can be enhanced by using markers [15]. Markers in the target-volume (tumor) will also increase the resolution, since EPI does not visualize soft tissue. EPIs are easy to use and give an x-ray verifi cation of the treatment. It is possible to monitor the motion continuously, but this requires manual inspection (although automatic sys-tems have been developed). The quality of EPI in high voltage therapy is sometimes very poor for all portals. EPIs cannot detect movements axial to the beam, but this is seldom a problem for a treatment fi eld. An additional disadvantage of the EPI is that personnel must leave the room during positioning of the patient.
Cone CT was fi rst discussed in isotope applica-tions in the 1980 – 1990s. Today it is used for RT and the principle is to use the whole detecting area of the EPI (not just a small pencil beam such as that in a standard CT). One can use the treatment beam, but it is also possible to improve the image quality by using standard x-ray tubes mounted on the treat-ment gantry (e.g., Electa Synergy). Cone CTs are closely linked to IMRT and the development of VMAT and Rapid Arc but do not solve the problem shown in Figure 3. It is possible to visualize the body, bone and target, but it is not possible in clin-ical practice toperform 4DRT. The largest disadvan-tage with cone CT is that it cannot detect motion during treatment.
Respiration and gating
Most of the work on 4DRT is on motion due to respiration (lung, liver and breast). In this fi eld the problem of Figure 2 is obvious. If one assume that a certain fi lling of the lung is proportional to the posi-tion of the target, it is possible to monitor the respi-ration and use the fl ow of air as a surrogate for target
Figure 2. A schematic view of the result of movements during the dose planning CT. The target A and B is due to movements displayed as A1-5 and B1-5 respectively due to movements. For details, see text.

108 B. Lennern ä s et al.
position. Once the position is known you can treat in two principle different ways, see Figure 3. Either you follow the tumor and treat continuously or you can treat only in well defi ned periods of the respira-tion cycle. The latter will be more time consuming, but is probably easier to plan and perform. However, it is important to notice that the accelerator must have the possibility to start and stop irradiating with small time intervals.
IMRT, VMAT, Rapid Arc and 4DRT
As mentioned above, IMRT made it possible to treat banana-shaped targets such as lymph node volumes and prostate with seminal vesicles. One problem with IMRT is the larger volume of healthy tissue receiving low dose irradiation. Hall and Wuu (2003) have esti-mated that this increased volume of normal tissue irradiation, will almost double the incidence of sec-ond malignancies [17]. VMAT and Rapid Arc can be considered as IMRT with an infi nite number of por-tals. The low-dose volume using VMAT and Rapid Arc seems to lower and treatment time is shorter. Possible advantages in different tumors are currently being investigated [16].
The major step in dose escalation and side-effect reduction is to reduce the Planning Target Volume (PTV), and IMRT, Rapid Arc and VMAT makes it possible to have a uniformly small margin around any shaped target. On the other hand – small margins stress the importance of continuous positioning
monitoring and management of the deviation of the position.
Real time radiotherapy
In order to perform real time corrections in clinical practice, the positioning device must determine the position of the target without human interference. This can be done by using continuous diagnostic x-rays with auto-detection of the target or marker, optical systems (only surface), implanted isotopes, or by using electromagnetic positioning devices (EMP). These techniques have their advantages and disad-vantages, but EMP is the only technique which can determine a target position in the body, such as the prostate, without adding more ionizing radiation to the patient.
There are two EMP systems available, Calypso Medical and Micropos Medical [18]. Figure 4 describes one EMP system. A transmitter is implanted near the target and an external antenna (in the treat-ment table) tracks the signal and thus the position of the target can be determined at sub-millimeter levels. EMP is non-ionizing and the system can therefore be used with personnel in the room.
In the treatment of prostate cancer, the combina-tion of small margins, a moving target, few fractions and high doses requires very high precision, and probably demands more than one positioning sys-tem. At a minimum, one of these systems should be real time and continuously monitoring.
Figure 3. In-vivo prostate movements during 20 minutes. The maximum deviation in 3D is 14 millimeters.

Radiotherapy in 4D and 5D for PC – an overview 109
The aim of 5DRT is to, in the clinic, to integrate biological factors and the physical dose distribution over time (fractionation). Five dimensional radio-therapy, is also essential for understanding side-effects of radiotherapy. For PC better understanding of the nature of PC in individual patients will also improve the selection of patients for curative treat-ments or watchful waiting [21].
For PC, 5DRT is important due to the biological nature of PC and all the possible radiation treat-ments with different fractionation and doses avail-able. However, if 4DRT has recently been established the era of 5DRT has, possibly, just begun. For exam-ple, PC is considered to be a slowly proliferating cancer during treatment [22]. However, recently Thames et al. showed a 6% decrease in biochemical control if the treatment time was extended by one week [23].
During the last decade IMRT in a 3DRT milieu has become very common. One of the goals for QUANTEC (Quantitative Analyses of Normal Tissue Effects in the Clinic) is to summarize 3D dose volume data into constraint parameters to improve IMRT dose planning [24]. However, as pointed out by QUANTEC, data collected from 3DRT is not optimal for the judgment of individual dose plans, since NTCP (Normal Tissue Complica-tion Probability) data extracted from 3DRT are population-based estimates based on a single plan-ning CT with no account for anatomic variation (compared to 4DRT) [25].
The QUANTEC group outlines several other problems which can relate to 5DRT, such as drug interactions, comorbidity and individual radiosen-sitivity. Methodological problems with side-effect sensoring and determination of the delivered dose to the OAR or target in fractionated RT is also high-lighted [24].
The group identifi ed research priorities refl ecting the shortcomings of 3DRT data and summarizes that clinical judgment should not be replaced by NTCP or other constraint parameters for the individual patient [27]. It is also notable that the parametres are extracted from 3DRT 1.8 – 2Gy/fraction and other fractions schedules such as hypofractionation, should be adjusted to conventional fractionation [26]. How-ever, it is not self evident that the Linear Quadratic (LQ) formula which contains no volume parameter is optimal for this conversion for OAR with a non-homogeneous dose distribution.
Conclusion
Radiotherapy is an important modality in the treat-ment of PC. It can be used either alone with or with-out dose escalation, or in combination with surgery
Biological models and 5DRT
The era of Radiobiology started at Radiumhem-met, Karolinska Hospital, by Magnus Strandquist which combined time and fractionation in the “ Stradquist diagram ” [19]. Today the most com-mon calculation is the Linear Quadratic (LQ) and BED (Biological Effective Dose) formula intro-duced by John Fowler [20] using the alfa/beta value. One disadvantage of this particular formula is that it is not suited for heterogeneous dose distributions. Lennern ä s et al. have further developed the LQ/BED formula to incorporate heterogeneous dose distributions, making the it suitable for 4DRT. This formula is called the Dose Volume Inhomogenity – BED (DVIC-BED).
Five dimensional radiotherapy (5DRT) is defi ned as any biological parameter (i.e., sensitivity to RT, drug interactions or r é sistance to RT) that can alter the outcome of radiotherapy. The 5DRT concept is impor-tant to consider in high dose treatments of PC such as high dose rate brachytherapy or hypofractionation.
Figure 4A. The treatment table “ listen ” to the signal from the non-permanent implant and the prostate is localised with sub-millimetre accuracy. The positioning is done continuously with adding more ionisation to the patient (with permission from Micropos Medical, Gothenburg, Sweden). B An implant in the target volume (to the left).

110 B. Lennern ä s et al.
or hormone therapy. It is reasonable to assume that RT of PC will increase in the future. The develop-ment of techniques for 4DRT are important tech-niques for optimizing RT for PC and to improve constraints for IMRT. Furthermore, the combination of small margins, a moving target, few fractions and high doses will probably require more than one posi-tioning system and at least one of these should be real time. The development and research in 5DRT should be accelerated in order to improve risk assess-ment for an individual patient. It is important to evaluate new technologies in respect to excising guidelines, but it is equally important it is that new technologies are investigated and also easier used in the clinic if found to be of advantageous for individual patient [28].
Declaration of interest: Bl Lennernäs, Sten Nils-son and Seymour Levitt are founders of Micropos Medical.
References
Adolfsson J. Screening for prostate cancer – will we ever [1] know? Acta Oncol 2010;49:275 – 7. Widmark A, Klepp O, Solberg A, Damber JE, Angelsen A, [2] Fransson P, et al. Endocrine treatment, with or without radiotherapy, in locally advanced prostate cancer (SPCG-7/SFUO-3): An open randomised phase III trial. Lancet 2009;373(9660):301 – 8. Bolla M, van Poppel H, Collette L, van Cangh P, Vekemans [3] K, Da Pozzo L, et al. Postoperative radiotherapy after radical prostatectomy: A randomised controlled trial (EORTC trial 22911). Lancet 2005;366(9485):572 – 8. Gatta G, Zigon G, Buemi A, Coebergh JW, Colonna M, [4] Contiero P, et al. Prostate cancer treatment in Europe at the end of 1990s. Acta Oncol 2009;48:867 – 73. Suit H. The Gray Lecture 2001: Coming technical advances [5] in radiation oncology. Int J Radiat Oncol Biol Phys 2002;53:798 – 809. Landberg T. Progress in radiotherapy. Acta Oncol 1995;34:[6] 1023 – 9. Ikushima H. Radiation therapy: State of the art and the [7] future. J Med Invest. 2010;57:1–11. Gottlieb KL, Hansen CR, Hansen O, Westberg J, Brink C. [8] Investigation of respiration induced intra- and inter-fractional tumour motion using a standard Cone Beam CT. Acta Oncol 2010;49:1192 – 8. van Dam IE, van S ö rnsen de Koste JR, Hanna GG, Muirhead [9] R, Slotman BJ, Senan S. Improving target delineation on 4-dimensional CT scans in stage I NSCLC using a deform-able registration tool. Radiother Oncol 2010;96:67 – 72. Levitt SH, Perez CA, Hui S, Purdy JA. Evolution of compu-[10] terized radiotherapy in radiation oncology: Potential prob-lems and solutions. Int J Radiat Oncol Biol Phys 2008;70: 978 – 86. Keall P. 4-dimensional computed tomography imaging and [11] treatment planning. Semin Radiat Oncol 2004;14:81 – 90. Bucci MK, Bevan A, Roach M 3rd. Advances in radiation [12] therapy: Conventional to 3D, to IMRT, to 4D, and beyond. CA Cancer J Clin 2005;55:117 – 34.
Isacsson U, Nilsson K, Asplund S, Morhed E, Montelius A, [13] Turesson I. A method to separate the rectum from the pros-tate during proton beam radiotherapy of prostate cancer patients. Acta Oncol 2010;49:500 – 5. Lennern ä s B, Letocha H, Rikner G, Magnusson A, [14] Nilsson S. Field displacement during external radiother-apy in prostatic adenocarcinoma treated with radioactive 198Au implants and external irradiation. Acta Oncol 1995;34:959 – 64. Poulsen PR, Cho B, Sawant A, Ruan D, Keall PJ. Dynamic [15] MLC tracking of moving targets with a single kV imager for 3D conformal and IMRT treatments. Acta Oncol 2010;49:1092 – 100. Johansen S, Cozzi L, Olsen DR. A planning comparison of [16] dose patterns in organs at risk and predicted risk for radiation induced malignancy in the contralateral breast following radiation therapy of primary breast using conventional, IMRT and volumetric modulated arc treatment techniques. Acta Oncol 2009;48:495 – 503. Hall EJ, Wuu CS. Radiation-induced second cancers: The [17] impact of 3D-CRT and IMRT. Int J Radiat Oncol Biol Phys 2003;56:83 – 8. Kindblom J, Ekelund-Olvenmark AM, Syren H, Iustin R, [18] Braide K, Frank-Lissbrant I, et al. High precision trans-ponder localization using a novel electromagnetic positioning system in patients with localized prostate cancer. Radiother Oncol 2009;90:307 – 11. Strandqvist M. Studien iiber die kumulative Wirkung der Ront-[19] genstrahlen bei Fraktionierung. Erfahrungen aus dem Radi-umhemmet an 280 Haut- und Lippenkarzinomen. German. Acta Radiol 1944;(Suppl 55):1 – 300. Fowler JF. The radiobiology of prostate cancer including new [20] aspects of fractionated radiotherapy. Acta Oncol 2005;44:265 – 76. Adami HO. The prostate cancer pseudo-epidemic. Acta [21] Oncol. 2010;49:298 – 304. Gao M, Mayr NA, Huang Z, Zhang H, Wang JZ. When [22] tumor repopulation starts? The onset time of prostate cancer during radiation therapy. Acta Oncol 2010. Thames HD, Kuban D, Levy LB, Horwitz EM, Kupelian P, [23] Martinez A, et al. The role of overall treatment time in the outcome of radiotherapy of prostate cancer: An analysis of biochemical failure in 4839 men treated between 1987 and 1995. Radiother Oncol 2010;96:6 – 12. Epub 2010 Apr 17. Bentzen SM, Constine LS, Deasy JO, Eisbruch A, Jackson A, [24] Marks LB, et al. Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC): An introduction to the scientifi c issues. Int J Radiat Oncol Biol Phys 2010;76(3 Suppl):S3 – 9. Marks LB, Yorke ED, Jackson A, Ten Haken RK, Constine [25] LS, Eisbruch A, et al. Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys 2010;76(3 Suppl):S10 – 9. Michalski JM, Gay H, Jackson A, Tucker SL, Deasy JO. Radi-[26] ation dose-volume effects in radiation-induced rectal injury. Int J Radiat Oncol Biol Phys 2010;76(3 Suppl):S123 – 9. Liu M, Moiseenko V, Agranovich A, Karvat A, Kwan W, Saleh [27] ZH, et al. Normal Tissue Complication Probability (NTCP) modeling of late rectal bleeding following external beam radiotherapy for prostate cancer: A Test of the QUANTEC-recommended NTCP model. Acta Oncol 2010;49:1040 – 4. Stensvold A, Dahl AA, Foss å SD, Axcrona K, Lilleby W, [28] Brennhovd B, et al. Clinicians ’ use of guidelines as illustrated by curative treatment of prostate cancer at a comprehensive cancer center. Acta Oncol 2010;50:408–14.

Correspondence: Bo Lennern ä s, Department of Oncology, University of Gothenburg, Sahlgrenska University Hospital, s-413 45 Sweden. E-mail: [email protected]
(Received 14 September 2010 ; accepted 8 February 2011 )
ORIGINAL ARTICLE
Hypofractionation for radiotherapy of prostate cancer using a low alfa/beta ratio – possible reasons for concerns? An example of fi ve dimensional radiotherapy
BO LENNERN Ä S 1 , STEN NILSSON 2 & SEYMOUR LEVITT 2
1Department of Oncology, Sahlgrenska Hospital and Academy, University of Gothenburg, Gothenburg, Sweden and 2Department of Oncology/Pathology, Karolinska University Hospital and Institutet, Stockholm, Sweden
Abstract It is very attractive, due to the assumed low alfa/beta ratio of prostate cancer (PC), to construct new treatment schedules for prostate cancer using only a few large fractions of radiation (hypofractionation). This will widen the therapeutic window since the ratio for PC might be lower than that of the organs at risk (OAR). PC is an extremely variable disease and often contains both highly and poorly differentiated cells. It is reasonable to assume that different cells have different patterns of radiosensitivity, i.e. alfa/beta ratios and proliferation. In this study we will simulate the effect on the outcome of the treat-ment with different fractionations and different ratios. Material and methods. In this simulation we use an extension of the Linear Quadratic (LQ)/Biological Effective Dose (BED) formula called the dose volume inhomogenity corrected BED (DVIC-BED). In the formula the tumour volume is divided in 50 subvolumes (step of 2%) and it is possible to calculate the relative effect of the treatment with different ratios (1.5, 4 and 6.5) in different subvolumes. Results . The simulations demonstrate that only a small portion (5 – 10%) of cells with a higher ratio will dramatically change the effect of the treat-ment. Increasing the total dose can compensate this, but this will on the other hand increase the dose to the OAR and also the risk for severe side effects. Conclusion . These simulations highlight possible reasons for concerns about the use of hypof-ractionation for pathologically heterogeneous tumours, such as prostate cancer, and also demonstrate the need for testing new treatment schedules using both high and low ratios.
Prostate cancer (PC) is one of the most common types of cancer in western countries [1]. Modern research and development in surgery and radiation treatment techniques have contributed to signifi -cantly improved outcomes for patients who are diag-nosed with PC. In recent years hypofractionated curative radiotherapy of PC has been discussed in order to reduce costs, side effects and possibly improve local control by dose-escalation [2].
In the beginning of the 1990s it was shown that the alfa/beta for PC cell-lines might be lower than that of other tumour cell-lines [3 – 5]. During the past decade evidence for low alfa/beta ratio derived from clinical data has been published, and several retro-spective studies have shown that the ratio could be as low as 1.5 Gy [6,7].
It thus became very attractive to construct new treatment schedules using a few large fractions of
radiation since one gains more cell kill effect per dose if the alfa/beta is low. If the ratio for the tumour is lower than that of the surrounding late reacting tissue, one will widen the therapeutic window.
However, PC is common an extremely variable disease [8]. PC in each patient is also heterogeneous and very often PC shows both highly and poorly dif-ferentiated cells in the same biopsy. Recently the hypothesis that PC is a slowly proliferating tumour has been questioned and this could alter the estima-tion of the alfa/beta ratio.
The assumption of a low alfa/beta ratio implies that certain patients will benefi t from hypofraction-ation. However, it is also reasonable to assume that the variable degree of differentiation implies different ratios in some areas of the tumour. Thus the ratio in some parts of the tumour is similar to that of other common cancers.
Acta Oncologica, 2011; 50(Suppl 1): 111–115
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2011.562536

112 B. Lennern ä s et al.
In this study we present a simulation of external beam hypofractionation with different degrees of tumour differentiation in different volumes (more radioresistant clones varying from 1% to 100%) of the PC. We will demonstrate that using a low ratio when converting a conventional treatment may be problematic since there is a risk of underdosage if only a small part of the tumour volume contains more poorly differentiated cells.
Material and methods
Calculation model
DVIC-BED is an extension of the LQ model, and was initially developed for non-uniform dose dis-tributions with non-standard fractions such as in high dose brachytherapy [9,10]. However, it can easily be modified for both different doses and different radiobiological factors such as the alfa/beta ratio.
The principle of DVIC-BED is to calculate the biological effect in several sub-volumes (with differ-ent a/b ratios or dose) of the target and then to add these sub-effects and express the sum as a dose as if the target had received this uniform dose. The DVIC-BED model is a combination of the linear-quadratic (LQ)-formula and the principle of the critical vol-ume (tissue-rescuing unit) model [10] and the Bio-logically Effective Dose concept (BED). The formula has been described previously [10].
If the number of clonogenic cells is c (c t at a specifi c time), the number of fractions is n, fraction dose d, total dose D and survival fraction SF and E for the effect after several fractions, the following expression for tumour control probability (TCP) can be derived:
TCP � e − c t (1)
c t � c 0 x SF (2)
SF � e(- α D− β dD) (3)
E � −ln(e(− α D− β dD)) (4)
If one assumes that the clonogenic cells are homogeneously distributed in the tumour from the beginning, one can divide the tumour into several sub-volumes (v). The probability for cure will then depend on the number of clonogenic cells in each volume according to:
TCP � e-(( c0 /v x e-E1) � ..... � ( c0 /v x e-Ev)) � (4) � e-(c0/v x (e-E1 � ..... � e-Ev))
To compare different simulated treatments the Biologically Effective Dose is calculated:
BED � E/ α � D(1 � d/( α / β )) (5)
In order to calculate the BED in a target with an inhomogeneous dose distribution it is possible to combine Equation 4 and 5 to the expression:
BED � −(ln(e–E1 � e–E2 � ...e–Evn/vn))/ α (6)
The advantage of using this formula is that it is based on radiobiologically related formulas such as the LQ/ BED and that different fractions and treat-ments can be summated independent of the homo-geneity of the dose distribution or radiobiological parameters.
Alfa/beta ratio
The α / β ratio and α -values have been published by Ekl ö v (4.14, α � 0.12) for 60Co and DeWeese (3.7 – 10.9, ⟨ � 0.064 – 0.115) among others for both high and low dose rate irradiation [3 – 5]. Although these values were determined in vitro PC can be assumed to have both a low α value and a low α / β ratio [3 – 7]. Recently, the ratio has been a subject for discussion by Brenner, Hall and Fowler and the ratio proposed was as low as 1.5. In the simulations dif-ferent combinations of the alfa, beta ratio are used.
Simulation and presentation
In this study we will alter fractionation and radio-biology in eight simulations, see Table I. The fi rst (1) simulation is 3− � 54Gy with the same dose and ratio in all subvolumes; second (2) a 3− � 54Gy with 2 Gy underdose in 1 – 100% of the tumour volume treat-ment; (3) a 3− � 54Gy treatment with change of ratio from 1.5 to 4 in 1 to 100% (step; 2%) of the volume; and (4) a change of ratio from 4 to 6.5. In simulation 1 – 4 the ratios have the same alfa (0.12) and for com-parison a calculation using a different alfa (0.036) for the ratio 1.5 and 4 are presented. In a separate simulation for comparison, the factors are the same as simulation (2) but with the fractionation 6.4− � 32Gy, simulation (6), and the use of a different alfa for different ratios (0.12 for ratio 1.5 and 0.036 for ratio 4).
The simulation was performed on an Apple Com-puter, iMac, using Chipmunk BASIC (http://www.nicholson.com/rhn/basic/). Graphic presentation was performed using EXCEL 2004 for Mac, Microsoft, USA. In simulation 6 and 7 the alfa value differed in the two ratios used. This means that the conversion to DVIC-BED (Equation 6) in absolute terms is only valid on the left side of the Figure 2. However, it is the relative effect which is important in this presentation.

Low alfa/beta ration – reasons for concerns? 113
Results
All simulations are summarised in Table I and the effects are presented in Figures 1 and 2. It is impor-tant to remember that different ratios will give dif-ferent BED ’ s (or DVIC-BED). In Figures 1 and 2 it is the relative effect for each curve (slope of the curve) which is important – not the absolute value.
Simulation one and two were performed for con-trol of the method. Simulation one is the standard treatment with no underdosage and shift in ratio. Simulation two shows the effect of an underdose in 1 – 100% of the volume. With 100% underdosage, the treatment is 36 Gy/2 rather than 54 Gy/3, and this will of course have a signifi cant effect on the treatment.
In simulation 3 – 7 the drastic effect of using a low ratio for treatment conversion can be seen. To the left in the fi gure all subvolumes have a low ratio (1.5 or 4 with a different alfa). To the right it has shifted to a higher value (with the same alfa). It can be seen that if a treatment is constructed based on a low ratio, it will rapidly lose in total effect if only a small portion (less then 5 – 10%) of the volume has a higher ratios.
The effect is as drastic as treating a subvolume of the tumour with an inadequately low total dose (sim-ulation 2). On the other hand, if one uses a ratio of 4, the treatment will be much less sensitive to small por-tions of tumour cells with higher ratios. The last sim-ulations (8) in Figure 2 show that the effect is present even with larger fractions (6.4 Gy x 5) being used.
Discussion and conclusion
PC is an extremely variable disease. Prostate cancer in one patient is often heterogeneous and usually demonstrates both well and poorly differentiated cancer areas in the same biopsy. It is reasonable to assume that different cells have a different radiobiol-ogy including the alfa/beta ratio as compared to most common cancers.
In these simulations, using the DVIC-BED method, we have shown that although a low alfa/beta is an attractive concept it might be dangerous if the tumour volume contains only small proportions of cells with higher ratios. The effect could be very dras-tic and this is due to the mathematical division of fractionation and the alfa/beta ratio in the LQ for-mula. When the ratio is low compared to the numeric number of the fraction size, the relative effect of radiotherapy will increase very steeply. The effect is comparable to under dosage of the target volume (simulation 2). This effect is apparent in all combina-tions of variables and it is important to remember
Table I. The eight simulations described in the paper with different ratio, fractions and dose. The standard treatment is 54/3 (simulation 1). Simulation 2 is same as 1 but with a increasing underdosage of 0 – 100% of the volume. Simulations 3 to 7 is 54/3 with different shift in alfa/beta ratio. Simulation 8 is fi ve fraction treatment to examine the effect in a more extreme hypofraction schedule.
Simulation Fraction Total doseShift in
ratio Alfa
1 3 54 No 0.122 3 to 2 54 or 36 No 0.123 3 54 1.5− � 4 0.124 3 54 4− � 6.5 0.125 3 54 4− � 6.5 0.0366 3 54 1.5− � 4 0.127 3 54 1.5− � 4 0.0368 6.4 32 1.5− � 4 0.12
Simulations treatment 1-5
0
20
40
60
80
100
120
140
160
180
1 6 11 16 21 26 31 36 41 46 51Subvolume
DV
IC
-B
ED
Standard 3->54GyUnderdose 2 Gy
Shift ratio 1.5->4
Shift ratio 4->6.5
Shift 4-6.5 (other alfa)
Figure 1. The result of the eight simulations described in text and Table I (the volume is divided in 50 subvolumes of 2%). The relative effect of the radiation expressed as DVIC-BED is decreasing if the ratio is altered in different subvolumes of the tumour. In can be seen that only a small portion of a higher ratio or underdosage (simulation 2) will have a drastic effect on the relative effect of the treatment (DVIC-BED). From simulation 4–5 it can be seen that the effect is much less pronounced if the ratio is higher then the size of the fraction.

114 B. Lennern ä s et al.
that this study will highlight the relative effect related to the presence of even small number of poorly dif-ferentiated cells with a possibly lower alfa/beta ratio. By using a proper (higher) total dose the relative sub-optimal effect of large fractions of irradiation on poorly differentiated cells will be diminished. This will, on the other hand, increase the dose given in high dose per fraction to the OAR.
PC has a lower alfa/beta ratio compared to other tumours and therefore fractionation may not be the optimal factor to alter compared to other tumours. As mentioned above, by using a proper total dose the relative suboptimal effect on poorly differentiated cells will be diminished.
In order to have some built in safety margin in the hypofractionation schedule, one must prescribe a proper dose for poorly differentiated cells and thus one must utilise physical and technical factors as well to protect OAR, namely; the physical factors of the beam (photons, electrons, protons) or technical factors (number of fi elds, intensity modulation, position).
It has been hypothesised since 1990 that PC is non-proliferating during RT [11]. However, recently Thames et al. showed a 6% decrease in biochemical control if the treatment time was extended one week [12]. These examples emphasises the impor-tance of radiobiology as a dimension (e.g. fi ve dimension radiotherapy, 5DRT) that must be con-sidered along with all other technical and physical factors in the RT of PC [13] and in hypofraction-ation in particular.
Recently Arcangeli et al. showed a 12% improved 3-year freedom from biochemical failure (FFBF) using hypofractionation in high risk cancers [14]. At a glance this could contradict the presented simula-tions – but it actually does not. Assuming 100 cells and 90% with a ratio of 1.5, these cells will be destroyed more effectively and reduce the PSA by 90% using hypofractionation. The problem relates to the remaining 10%. These cells might be poorly dif-ferentiated; more dangerous, and have a higher ratio. Therefore they might not be eradicated. Moreover, it is well known that poorly differentiated PC cells produce less PSA [15]. The use of biochemical con-trol can thus be an illusion of a more effective treat-ment with respect to survival (or local control) of the patient in 10 – 20 years. In the paper of Arcangeli it is also shown that despite the more effective reduction in PSA, the local recurrence rate was four times more common in the hypofractionated arm.
It is important to note that we are not claiming that hypofractionation is not appropriate for PC. However, by using an alfa/beta lower than the numeric value of the fraction dose it might be inap-propriate for the poorly differentiated cells, and this will infl uence local control and survival. Although the PSA may decrease, this decrease may not refl ect the actual effect on all cells in the tumour and a number of the poorly differentiated cells may not be eradicated.
Our recommendation is that, until the radiobiol-ogy of PC is fully understood in each patient, not to use a lower alpha/beta ratio for PC below the ratio of organs at risk (OAR e.g. rectum and bladder). One should perform “ sensitivity analysis ” such as this simulation (with different ratios and total doses) prior to using a new fractionation schedule in the clinic. One should also consider compensating for the lesser effect on poorly differentiated cells by delivering a higher total dose. However, this will also increase the dose to the OAR and this is the dilemma in tumours with low ratios – the lower ratio will diminish the fractionation as a method for decrea sing side effects leaving the physical factors of the beam (photons, electrons, protons) or technical factors (number of fi elds, intensity modulation, position) as the only methods to improve the treatment.
0
100
200
300
400
500
600
1 6 11 16 21 26 31 36 41 46 51Subvolume
DV
IC
-B
ED
Simulation 6-8
1.5>4 alfa=0.12
1.5>4 alfa=0.036
1.5>4 dose=32Gy/6.4
Figure 2. The result of the eight simulations described in text and Table I (the volume is divided in 50 subvolumes of 2%). The relative effect of the radiation expressed as DVIC-BED is decreasing if the ratio is altered in different subvolumes of the tumour. In can be seen that only a small portion of a higher ratio or underdosage (simulation 2) will have a drastic effect on the relative effect of the treatment (DVIC-BED). From simulation 4–5 it can be seen that the effect is much less pronounced if the ratio is higher then the size of the fraction.

Low alfa/beta ration – reasons for concerns? 115
Declaration of interest: There are no confl icts of interest to be declared.
References
Adami HO. The prostate cancer pseudo-epidemic. Acta [1] Oncol 2010;49:298 – 304. Budiharto T, Haustermans K, Kovacs G. External beam radi-[2] otherapy for prostate cancer. J Endourol 2010;24:781 – 9. Ekl ö v S, Essand M, Carlsson J, Nilsson S. Radiation sensiti-[3] zation by Estramustine. Studies on cultured human prostatic cancer cells. Prostate 1992;21:287 – 95. DeWeese T, Dillehay LE, Shao Y, Williams JR. Low dose [4] sensitization of human prostate cancer cells. IJROBP 1994; 30(Suppl):319. Lennern ä s B, Nilsson S. Calculated effects of displacement [5] errors in external beam radiotherapy of prostatic adenocar-cinoma. Acta Oncol 1999;38:203 – 8. Fowler JF. Biological factors infl uencing optimum fractiona-[6] tion in radiation therapy. Acta Oncol 2001;40:712 – 7. Brenner DJ, Hall EJ. Fractionation and protraction for radio-[7] therapy of prostate carcinoma. IJROBP 1999; 15: 1095 – 101. Albertsen PC. WHO International Consultation on Prostate [8] Cancer: A summary. Acta Oncol 2010. Fowler JF. The linear-quadratic formula and progress in frac-[9] tionated radiotherapy. Br J Radiol 1989;62:679 – 94.
Lennern ä s B, Albertsson P, Edgren M, Nilsson S. Calculated [10] and simulated effects of heterogeneous dose distributions in radiotherapy using the dose volume inhomogeneity corrected biological equivalent dose formula with special reference to prostate cancer. Oncol Rep 2007;18:1299 – 303. Gao M, Mayr NA, Huang Z, Zhang H, Wang JZ. When [11] tumour repopulation starts? The onset time of prostate cancer during radiation therapy. Acta Oncol 2010;49: 1269 – 75. Thames HD, Kuban D, Levy LB, Horwitz EM, Kupelian P, [12] Martinez A, et al. The role of overall treatment time in the outcome of radiotherapy of prostate cancer: An analysis of biochemical failure in 4839 men treated between 1987 and 1995. Radiother Oncol 2010;96:6 – 12. Lennernas B, Levitt S, Nilsson S. Four and fi ve dimensional [13] radiotherapy with reference to prostate cancer – defi nitions, state of the art and further directions. Acta Oncol (in press). Arcangeli G, Saracino B, Gomellini S, Petrongari MG, [14] Arcangeli S, Sentinelli S, et al. A prospective phase III ran-domized trial of hypofractionation versus conventional frac-tionation in patients with high-risk prostate cancer. IJROBP 2010;78:11 – 8. Aihara M, Lebovitz RM, Wheeler TM, Kinner BM, Ohori [15] M, Scardino PT. Prostate specifi c antigen and Gleason scale: An immunohistochemical study of prostate cancer. J Urology
1994;151:1558 – 64.
Supplementary material available online
Appendix can be found at http//:www.informahealth-care.com/doi/abs/10.3109/0284186X.2011.562536

Correspondence: Urology Unit, Helsingborg Hospital, SE-251 87 Helsingborg, Sweden. E-mail: [email protected]
(Received 12 September 2010 ; accepted 17 September 2010 )
EDITORIAL
Natural history of prostate cancer, chemopreventionand active surveillance
OLA BRATT & MARTIN C. SCHUMACHER
Urology Unit, Helsingborg Hospital, SE-251 87 Helsingborg, Sweden and Department of Urology, Karolinska University Hospital, Solna, SE-171 76 Stockholm, Sweden
Prostate cancer is a disease with an extreme span of clinical presentations and consequences. Locally advanced and metastatic disease is a major health concern and cause of death in industrialised coun-tries. However, only a minority of the initial, micro-scopic foci of cancer cells in prostatic glands progress to symptomatic and potentially lethal dis-ease. Most elderly men harbour such indolent can-cer foci and die from other causes unaware of their existence. The risk of progression from microscopic prostate cancer to clinical illness is infl uenced by environmental and life-style factors, such as diet, interacting with hereditary genetic factors, but the exact biological events and interactions are diffi cult to unravel [1].
The natural history of prostate cancer
Most often, the natural history of prostate cancer is initially that of a preclinical tumour slowly progressing over decades. Most such preclinical cancer foci do never cause symptoms and remain undetected throughout men ’ s lifetime. The preclini-cal but potentially detectable phase of prostate can-cer is usually very long, compared to other malignant tumours. Many asymptomatic men have palpable prostatic tumours for many years and even more men harbour small cancer foci detectable with sys-tematic prostate biopsies. Whether the more rapidly progressing, poorly differentiated prostate cancers are derived from pre-existing, well differentiated “ latent ” prostate cancers or develop de novo with a much shorter pre-clinical phase, is still unknown.
When a prostatic tumour has grown to such size that it causes symptomatic infravesical obstruction or
invaded adjacent organs producing other local symp-toms, cure is commonly not possible. Locally advanced, symptomatic prostate cancer has at the time of diagnosis usually seeded metastases to the regional lymph nodes or distally to the bone marrow or, less commonly, to other organs. The tumour differentia-tion is highly associated with the progression rate of localised and locally advanced prostate cancer.
There are few studies on the natural history of untreated symptomatic prostate cancer, but the median survival time was considered short before the introduction of endocrine therapy in the 1940s [2]. Untreated, metastatic prostate cancer was a severely disabling disease, usually leading to death within a year or two.
Recent and current research on the natural history of prostate cancer
Research on the natural history of prostate cancer has mainly focused on asymptomatic, localised tumours. Information on prognostic factors is of pro-found importance when deciding the treatment strat-egy for a man with recently diagnosed localised prostate cancer. Since the introduction of PSA test-ing, the detection of small, well differentiated pros-tate cancers has increased dramatically [3,4]. Most patients with such low-risk tumours do not need any therapy at all. However, no defi nition of “ clinically insignifi cant ” prostate cancer in living men (as opposed to autopsy studies) can predict which can-cers will be clinically important and which will not. Since the last WHO consultation on prostate cancer in 2004, active surveillance with selective delayed treatment has evolved to be the method of choice for
Acta Oncologica, 2011; 50(Suppl 1): 116–119
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.527369

Cancer: screening, prevention and therapy 117
reducing overtreatment of low-risk prostate cancer. The critical issues which patients should receive treatment and when were outlined by Peter Albertsen at the WHO International Consultation on Prostate Cancer in Stockholm, 8 – 10 September 2010, and are summarised in this issue of Acta Oncologica [5].
Active surveillance for low-risk prostate cancer
There is no doubt that the adverse effects would vastly overshadow the benefi t if all men diagnosed with low-risk tumours were treated with curative intent. Unfortunately, as long as we continue to biopsy palpably benign prostates, we will continue to detect such clinically insignifi cant prostate can-cers in huge numbers. It seems unlikely that research will in the near future lead to methods for accurate prediction of which localised prostate cancers will develop to life-threatening disease and which will not. Thus, the concept of active surveillance with repeated assessments of the tumour and the patient is here to stay. Active surveillance is proven effective for reducing overtreatment, but knowledge is scarce about the risk of missing the window of curability and of issues related to quality of life. We are thus obliged to do further research on which patients should be recommended active surveillance, how it should be performed, and when radical therapy should be initiated – if ever. Some specifi c areas of uncertainty are listed below:
1. Which criteria defi ne the tumours best managed with active surveillance? The optimal tumour cri-teria vary with the age and co-morbidity of the patient. The current criteria are based on Gleason score, tumour extent and PSA values, but they are largely arbitrary. Most urologists and oncologists agree that patients with a life expectancy of more than ten years should be recommended immediate treatment if there is a substantial amount of cancer with Gleason pattern 4 or 5 in the prostate biopsies. How-ever, Gleason grading is observer-dependent and the defi nitions of the Gleason grades vary over time. Also, the prognostic signifi cance of the extent of cancer in the biopsies is not well studied. Sampling of the prostate with transrec-tal biopsies commonly underestimates the Gleason score and the tumour volume com-pared to prostatectomy specimens [6,7]. More aggressive tumours in the anterior aspect of the prostate may be missed with routine biopsies [8]. The optimal number of biopsies and the location of these for accurate sampling are still to be defi ned. The value of the new variants of
magnetic resonance imaging for assessing the local tumour in potential candidates for active surveillance is unclear, but recent results are promising. Likewise, the total PSA value, the PSA density and the ratio of free to total PSA are all of importance, but optimal cut-off values for recommending active surveillance are unknown. Hopefully, new biomarkers that add prognostic information will appear on the scene within the next few years.
2. What parameters should be followed during active surveillance? The monitoring of the cancer must herald progression before the disease becomes incurable. Most likely, the chance for cure is decreased substantially when tumour progres-sion is obvious with digital rectal examination or transrectal ultrasound. We know little of the risks for tumour dedifferentiation over time or for that poorly differentiated areas of cancer are not sampled at the initial biopsies. Repeat biop-sies are incorporated in most follow-up sched-ules for active surveillance, but how they should be performed and how the results should be interpreted is not studied systematically. Increas-ing PSA is the most common reason for initiat-ing deferred treatment [9]. One problem with PSA as a marker of tumour progression is that poorly differentiated tumours produce less PSA than slowly growing, well-differentiated tumours. Another problem is that many patients with low-risk localised prostate cancer also have benign prostatic hyperplasia, which may con-tribute to most of the PSA measured in blood serum. A small, but comparatively rapidly pro-gressing cancer in a large gland may therefore not give rise to a short PSA doubling time before metastases occur. Furthermore, PSA may fl uctuate for various reasons which may lead to unnecessary intervention or anxiety. The PCPT and REDUCE studies indicate that 5-alpha-reductase inhibitors may be used to sta-bilise the PSA derived from the benign hyper-plasia and enhance the utility of PSA to detect progressive cancer [10,11], but their role during active surveillance remains to be defi ned.
3. What is the optimal assessment interval during active surveillance? For the majority of patients tumour progression is very slow and biannual or even annual assessment is probably adequate. The crucial issue is, however, how short the intervals must be to detect more rapid progres-sion during the “ window of curability ” for the small minority of patient that harbour lethal tumours. Our knowledge on the biology of dis-ease progression from localised prostate cancer to metastatic diseases is still poor.

118 O. Bratt & M. C. Schumacher
4. Can the risk of disease progression be reduced? Sev-eral pharmacological and non-pharmacological interventions, such as physical activity and die-tary modifi cation, are theoretically interesting, but randomised studies are necessary for their evaluation. The potential role of 5-alpha-reductase inhibitors is discussed later in this issue by Roger Rittmaster [12].
5. How does active surveillance affect quality of life? What is the psychological impact of having an untreated cancer, of slowly rising or fl uctuating PSA values, and of the uncertainty of what the next scheduled visit will lead to? Is delayed treatment associated with more side-effects than immediate curative treatment? Delayed treatment may decrease the chance for nerve-sparing surgery and include adjuvant therapy because of a more advanced tumour stage. How are patients affected when deferred treat-ment with curative intent turns out to be initi-ated too late, at a time when the disease has already spread?
Chemoprevention
Large, randomised studies of chemoprevention with vitamins and trace elements have reported negative results and one study with an anti-infl ammatory drug was terminated prematurely because of toxicity [12]. It may be questioned if it is the right way for-ward to initiate further studies on chemoprevention for prostate cancer targeting men in the general pop-ulation. Selecting high-risk populations by the family history of prostate cancer, genetic testing or other biomarkers may be more effi cient.
Primary chemoprevention of prostate cancer must start decades before the age at which symptomatic disease becomes common in the male population. Even secondary chemoprevention, e.g. for patients on active surveillance for low-risk tumours, must most likely include prolonged treatment before positive effects are clinically apparent. It is therefore essential that studies on chemoprevention do not focus on “ prostate health ” only. For the average middle-aged man there are many more threats to future health than prostate cancer, of which cardiovascular diseases are the most important. Smoking men with a low-risk prostate cancer will probably increase their chance of longevity more by stopping smoking than by subject-ing themselves to a radical prostatectomy. Any inter-vention that reduce prostate cancer risks but have negative effects on cardiovascular morbidity or mor-tality may cause more harm than good. It is therefore of major interest that cholesterol-synthesis inhibiting statins, widely used to prevent cardiovascular disease, also may reduce the risk of prostate cancer [13]. Ran-
domised studies of statins with prostate cancer as the primary endpoint are still lacking, though.
As Roger Rittmaster points out in his review later in this issue of Acta Oncologica, the term chemopre-vention implies that a disease is being prevented [12]. For a disease like prostate cancer with its abundant preclinical form, preventing disease progression to clinical disease may be as important as preventing cancer initiation. The 5-alpha-reductase inhibitors fi nasteride and dutasteride reduced the detection of prostate cancer in two large, randomised trials [14,15]. Their effect on the poorly differentiated can-cers with the highest potential of becoming life-threatening is, however, at best small. On the other hand, reducing the detection and progression of low-risk tumours may still be benefi cial to the patients, since a cancer diagnosis per se and the side-effects of (unnecessary) radical treatment may reduce their quality of life. Which patient groups that could be recommended 5-alpha-reductase inhibitors for such risk reduction was discussed at the WHO meeting, but no consensus was reached.
Conclusions
The natural history of small prostate cancers is usu-ally very protracted and most of them will never become clinically important. There is no need to diagnose cancers that will never lead to symptoms. Unfortunately, prostate cancer is commonly incur-able when it has progressed to a stage that is obvi-ously clinically signifi cant. At present and most likely for decades to come, early detection of asymptomatic tumours followed by curative treatment is and will be the most effective measure for reducing morbidity and mortality of prostate cancer. Research on prog-nostic factors and strategies that reduce overtreat-ment of clinically insignifi cant cancers is therefore essential. Pharmacological risk-reduction and active surveillance with selective delayed intervention for low-risk tumours are two very important research fi elds. An expert session was devoted to these topics at the WHO International Consultation on Prostate Cancer in Stockholm, 8 – 10 September 2010. Sum-maries from this session are published in this issue of Acta Oncologica [5,12].
Declaration of interest: Ola Bratt is a member of an advisory board for and has received lecture fees from GlaxoSmithKline, the manufacturer of dutasteride.
References
Damber JE, Aus G. Prostate cancer. Lancet 2008;371:[1] 1710 – 21.

Cancer: screening, prevention and therapy 119
Whitmore WF, Jr. Hormone therapy in prostatic cancer. Am [2] J Med 1956;21:697 – 713. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer [3] statistics, 2009. CA Cancer J Clin 2009;59:225 – 49. Bratt O, Berglund A, Adolfsson J, Johansson JE, [4] Tornblom M, Stattin P. Prostate cancer diagnosed after prostate-specifi c antigen testing of men without clinical signs of the disease: A population-based study from the National Prostate Cancer Register of Sweden. Scand J Urol Nephrol 2010. [Epub 2010-07-14]. Albertsen PC. When is active surveillance the appropriate [5] treatment for prostate cancer? Acta Oncol 2010 (this issue). Conti SL, Dall’era M, Fradet V, Cowan JE, Simko J, [6] Carroll PR. Pathological outcomes of candidates for active surveillance of prostate cancer. J Urol 2009;181:1628 – 33; Discussion 33 – 4. Ploussard G, Salomon L, Xylinas E, Allory Y, Vordos D, [7] Hoznek A, et al. Pathological fi ndings and prostate specifi c antigen outcomes after radical prostatectomy in men eligible for active surveillance—does the risk of misclassifi cation vary according to biopsy criteria? J Urol 2010;183:539 – 44. Duffi eld AS, Lee TK, Miyamoto H, Carter HB, Epstein JI. [8] Radical prostatectomy fi ndings in patients in whom active surveillance of prostate cancer fails. J Urol 2009;182:2274 – 8.
Klotz L, Zhang L, Lam A, Nam R, Mamedov A, Loblaw A. [9] Clinical results of long-term follow-up of a large, active surveillance cohort with localized prostate cancer. J Clin Oncol 2010;28:126 – 31. Thompson IM, Pauler Ankerst D, Chi C, Goodman PJ, Tan-[10] gen CM, Lippman SM, et al. Prediction of prostate cancer for patients receiving fi nasteride: Results from the Prostate Cancer Prevention Trial. J Clin Oncol 2007;25: 3076 – 81. Andriole G, Bostwick D, Brawley O, Gomella L, Marberger [11] M, Montorsi F, et al. The effect of dutasteride on the utility of PSA for the diagnosis of high-grade and clinically relevant prostate cancer in men with a previous negative biopsy: Results from the REDUCE study. J Urol 2011 (in press). Rittmaster RS. Chemoprevention of prostate cancer. Acta [12] Oncol 2010 (this issue). Breau RH, Karnes RJ, Jacobson DJ, McGree ME, Jacobsen [13] SJ, Nehra A, et al. The association between statin use and the diagnosis of prostate cancer in a population based cohort. J Urol 2010;184:494 – 9. Andriole GL, Bostwick DG, Brawley OW, Gomella LG, [14] Marberger M, Montorsi F, et al. Effect of dutasteride on the risk of prostate cancer. N Engl J Med 2010;362:1192 – 202. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller [15] GJ, Ford LG, et al. The infl uence of fi nasteride on the develop-ment of prostate cancer. N Engl J Med 2003;349: 215 – 24.

Correspondence: Peter C. Albertsen, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030, USA. E-mail: [email protected]
(Received 28 July 2010 ; accepted 5 August 2010 )
REVIEW ARTICLE
When is active surveillance the appropriate treatment for prostate cancer?
PETER C. ALBERTSEN
University of Connecticut Health Center, Farmington, USA
Abstract Background . The incidence of prostate cancer has increased dramatically worldwide during the past few decades in part because of increased testing for prostate specifi c antigen (PSA). The aggressive use of this screening tool has resulted in the identifi cation of many localized prostate cancers a majority of which are relatively low volume, low grade tumors. Older autopsy studies have documented that incidental prostate cancer is quite common especially in older men. The fi nasteride chemoprevention trial confi rmed these fi ndings. Many prostate cancers are not destined to progress to clinically signifi cant tumors. Several case series have documented the natural history of clinically detected prostate cancer. The progression of disease identifi ed by PSA testing is less certain. These studies uniformly show that many men with low grade tumors can survive for over two decades in the absence of treatment. Furthermore, randomized clinical trials have shown only a mod-est ten year survival advantage for those men undergoing either surgery or radiation. Results . As a consequence, men with low risk of disease progression may wish to consider active surveillance as a treatment option. To date, several case series have documented that men following an active surveillance protocol that includes regular PSA testing and periodic re-biopsy have an excellent outcome. The majority of these men have not demonstrated evidence of progression during the fi rst decade of follow-up and among those that have the majority have undergone either surgery or radiation without compro-mise of their long-term outcome. Unfortunately, until better biomarkers become available, the outcome of any individual patient defi es accurate prediction. Conclusion . Men with newly diagnosed prostate cancer must weigh the risk of disease progression against the potential effi cacy and safety of treatment when making a decision whether to consider active sur-veillance as an appropriate treatment.
When is active surveillance the appropriate treatment for prostate cancer?
During the past three decades, the diagnosis of pros-tate cancer has increased substantially worldwide. The rise in incidence rates has been most dramatic in countries that have aggressively embraced testing for prostate specifi c antigen (PSA). In USA, for example, the annual age-adjusted prostate cancer incidence rates have almost doubled from 1980 to 2010 [1]. In 2009, over 192 000 men were told that they had prostate cancer. In the UK where PSA test-ing is much less widespread the incidence trends parallel those of USA, but at signifi cantly lower numbers. Prostate cancer is diagnosed at a rate that is 2.5 times higher in USA when compared to the UK [2]. The higher incidence rates have occurred primarily among younger men where the rate ratio
between USA and the UK is even higher. Among men age 45 – 54 years the ratio is 8.2; for men age 55 – 64 years the ratio is 6.7.
These numbers contrast dramatically with the number of prostate cancer deaths recorded world-wide. In 1991 over 33 000 men died from prostate cancer in USA [1]. Similar prostate cancer mor-tality rates were recorded in the UK. Since then there has been a steady decline in prostate cancer mortality in both countries, however the decline in USA has been signifi cantly greater [2]. This is espe-cially true for those men aged 75 years or over. Between 1994 and 2004 the age-adjusted prostate cancer mortality rate declined in USA by 4.17%, a rate four times higher when compared to the UK. In 2009, over 27 000 men died from prostate cancer in USA.
Acta Oncologica, 2011; 50(Suppl 1): 120–126
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.526634

Active surveillance for prostate cancer 121
Which prostate cancers are clinically signifi cant?
The dramatic differences between prostate cancer incidence rates and mortality rates have led many researchers and clinicians to question whether all newly diagnosed prostate cancers pose a clinical threat. For prostate cancer, the ratio of incidence to mortality is now nearly 8:1. This compares with a ratio of only 1.3:1 for lung cancer and 2.1:1 for col-orectal cancer. There is no universally accepted defi nition of clinically signifi cant or insignifi cant dis-ease, but cancer volume, clinical stage and tumor grade at diagnosis have consistently predicted long-term clinical outcome. As a consequence of wide-spread PSA testing, there has been a major stage shift towards localized disease. Many men are now diagnosed with low volume, low grade disease often presenting with only one or two cores positive con-taining Gleason 6 disease. Epstein et al. suggested four criteria to defi ne clinically insignifi cant disease: tumor volume � 0.5 ml, PSA density � 0.15, no Gleason pattern 4 or 5 disease and the presence of less than 3 mm of tissue in a single needle core [3]. Although not prospectively validated, most clini-cians believe that only tumors greater than 0.5 cm 3 are clinically signifi cant.
Although well documented by older autopsy series, several recent publications have demonstrated that prostate cancer is a fairly common fi nding espe-cially among older men. Researchers designing the fi nasteride chemoprevention trial estimated the prev-alence of prostate cancer among men age 55 – 70 years to be 6% and powered the trial to detect a 25% reduction in prostate cancer [4]. After seven years of follow-up, prostate cancer was detected in 24% of men in the control group and 18% of men in the treatment group. Most of the cancers identifi ed by this study were Gleason 6 tumors and were present in men with serum PSA levels below 4.0 ng/ml.
All prostate cancers will progress, but many prostate cancers progress at remarkably slow rates. As a consequence, competing medical hazards often play a more dominant role in patients ’ esti-mates of their long-term prognosis. Some men have aggressive disease that may benefi t from early detec-tion and intervention, but many others harbor can-cers that grow slowly and never progress to clinical signifi cance.
Several key studies have helped shape our under-standing of the natural history of prostate cancer progression. Between 1989 and 2004, Johansson and colleagues published a series of four articles that documented the outcomes of untreated prostate cancer in a population based cohort of patients diagnosed with prostate cancer in Sweden [5]. No
screening for prostate cancer took place during the period when this study population of 648 consecu-tive cases was assembled. Initially the authors found relatively low fi ve and ten year mortality rates among men with clinically localized disease and challenged the use of aggressive initial treatment for all patients with low grade early stage prostate cancer. Long-term follow-up of the study cohort, however, suggested a rising mortality rate from prostate cancer for those men surviving 15 – 20 years following diagnosis.
In 1994 Chodak et al. published a report des-cribing the results of conservative management of clinically localized prostate cancer [6]. Unlike the Johannson report, this study consisted of a pooled analysis of 828 case records from six non-random-ized studies published during the decade preceding the report. Patients with poorly differentiated can-cers had a signifi cantly lower cancer-specifi c survival rate (34%) when compared with men who had well or moderately differentiated cancers (87%). In addi-tion, men with poorly differentiated tumors were much more likely to develop metastases when com-pared to men who were diagnosed with well differ-entiated disease.
In 1998 and 2005, Albertsen et al. reported long-term outcomes of a competing risk analysis of 767 men diagnosed between 1971 and 1984 who were managed expectantly for clinically localized prostate cancer [7]. The results of this study are presented in Figure 1. Few men (4 – 7%) with Gleason 2 to 4 tumors identifi ed by prostate biopsy had progres-sion leading to death from prostate cancer within 20 years of diagnosis. Men with Gleason 5 and 6 tumors identifi ed by prostate biopsy experienced a somewhat higher risk of death from prostate cancer when managed expectantly (6 – 11% and 18 – 30%, respectively). Men with Gleason scores 7 and 8 – 10 tumors identifi ed by prostate biopsy experienced a very high rate of death from prostate cancer regard-less of their age at diagnosis (42 – 70% and 60 – 87%, respectively). Very few of these men of any age sur-vived more than 15 years.
Unfortunately, these studies do not refl ect the impact of widespread PSA testing. Since the intro-duction of PSA screening, more than one million additional men have been diagnosed with prostate cancer in USA. Prior to PSA testing it was rare to diagnose prostate cancer before age 55 years. Since then there has been a dramatic increase in prostate cancer incidence among men in their late 50s and 60s and it is not uncommon to diagnose prostate cancer in men in their late 40s and early 50s. Com-pared to 1986, the relative incidence of prostate can-cer is 1.91 times greater among men aged 60 – 69 years, 3.64 times greater among men aged 50 – 59 years and 7.23 times greater among men younger

122 P. C. Albertsen
than 50 years [8]. Using computer models of inci-dence and mortality, Draisma et al. estimate that PSA testing has advanced the date of diagnosis by approximately 12.3 years for men age 55 years and by 6.0 years for men age 75 years [9].
Recently Grace Lu-Yao et al. explored the ten year outcomes of men over age 65 years with newly diag-nosed localized prostate cancer [10]. They assembled a cohort of over 14 500 men and found that after a median follow-up of seven years most men were either alive or had died of causes other than prostate
cancer (Figure 2). Ten-year prostate cancer mortality was 5.9% (95% CI, 3.6 – 8.2) and 6.2% (95% CI, 4.3 – 8.1) for men aged 66 – 69 years and 70 – 74 years, respectively, diagnosed with moderately differentiated disease. These results were much more favorable than the 15 – 24% mortality rates reported in the studies cited earlier. Similar improvements were observed in poorly differentiated disease. Ten-year cancer-specifi c mortality for T1c (screen detected) and T2 (palpable) disease was 9.8% and. 12.3%, respectively, for mod-erately differentiated cancer (adjusted HR � 0.63,
Figure 1. Survival and cumulative mortality from prostate cancer and other causes up to 20 years after diagnosis, stratifi ed by age at diagnosis and Gleason score.

Active surveillance for prostate cancer 123
Age at DiagnosisPanel A
80+75-7970-7466-69Moderately Differentiated, Stage T1ab
0
20
40
60
80
100
100
80
60
40
20
0
Moderately Differentiated, Stage T1c
0
20
40
60
80
100
100
80
60
40
20
0
Moderately Differentiated, Stage T2
0 2 4 6 8 100
20
40
60
80
100
100
80
60
40
20
0
Aliv
e, %
Deceased, %
Years Following Diagnosis
0 2 4 6 8 10 0 2 4 6 8 10 0 2 4 6 8 10
Age at Diagnosis
80+ 75-7970-7466-69Poorly Differentiated, Stage T1ab
0
20
40
60
80
100
100
80
60
40
20
0
Poorly Differentiated, Stage T1c
0
20
40
60
80
100
100
80
60
40
20
0
Poorly Differentiated, Stage T2
0 2 4 6 8 100
20
40
60
80
100
100
80
60
40
20
0
Aliv
e, %
Deceased, %
Years Following Diagnosis
0 2 4 6 8 10 0 2 4 6 8 10 0 2 4 6 8 10
Panel B
Figure 2. Competing risk of death by age at diagnosis, cancer stage, and grade. Panel A: Moderately-differentiated (Gleason 5 – 7) cancer. Panel B: Poorly-differentiated (Gleason 8 – 10) cancer.

124 P. C. Albertsen
p � 0.001), and 22.2% and 25.5%, respectively, for poorly differentiated cancer (adjusted HR � 0.74, p � 0.001). The use of chemotherapy (1.6%) and surgical or radiological intervention for spinal cord compression (0.9%) was uncommon. They con-cluded that contemporary men over age 65 years with screen-detected prostate cancer had survival out-comes signifi cantly better than those in the pre-PSA era, and had a low-risk of developing cancer related complications that require palliative surgery, radia-tion, or chemotherapy.
Historically, tumor grade has been the most pow-erful predictor of clinically signifi cant disease. When Gleason originally developed his scoring system based on lower power evaluation of glandular archi-tecture, men were usually diagnosed following a tran-surethral resection, an open prostatectomy to treat obstructive urinary symptoms or a needle biopsy of a prostate nodule. Up until 2000 most pathologists used all fi ve patterns described by Gleason, but since then many have become increasingly hesitant to grade any malignant glands lower than pattern 3. During the past decade there has been a steady infl a-tion of Gleason scores such that all low grade tumors previously recorded as Gleason score 2 – 5 are now classifi ed as Gleason score 6 and many Gleason score 6 tumors are now classifi ed as Gleason score 7. Changes in the application of the Gleason scoring system have become so widespread that clinical out-comes are signifi cantly improved if historical classi-fi cations are replace by contemporary classifi cations [11]. It is unusual for men with contemporary Glea-son 6 tumors to have clinically signifi cant progres-sion of their disease.
Active surveillance as a treatment alternative
The rationale for selecting active surveillance over immediate surgical intervention or radiation therapy refl ects the changing understanding of the risk posed by screen detected disease. Prior to the advent of PSA testing, most men presented with clinically advanced disease that often required palliative inter-vention. Men presenting with localized, low grade cancers frequently had slow progression of their dis-ease and often succumbed to competing hazards [5,7]. The recent publication of results from two large randomized trials on prostate cancer screening have shown that in contemporary practice prostate cancer deaths are infrequent during the fi rst ten years following screening and that as many as 48 men must be identifi ed and managed to prevent one prostate cancer death [12,13].
Interest in active surveillance also refl ects a greater understanding of the relative impact of intervention. Aggressive treatments make sense only if they are
effective. Unfortunately, data supporting the effi cacy of surgery and radiation are limited to two random-ized trials both accruing men identifi ed with prostate cancer on the basis of clinical disease rather than as a consequence of PSA testing. The Scandinavian tri-als of both surgery and radiation have shown that some men with localized prostate cancer live longer as a consequence of treatment, but the impact on prostate cancer mortality is modest [14,15]. At ten years, 19 men in the surgery trial and ten men in the radiation trial required treatment to prevent one prostate cancer death. Most patients participating in these trials were diagnosed on the basis of clinical fi ndings and therefore had greater tumor volume when compared to contemporary screen detected patients. As a consequence the relative impact of treatment on contemporary patients is likely to be much less because the threat posed by clinical pro-gression is much less.
These considerations have led clinicians at several academic medical centers to propose criteria that identify men who have a very low risk of disease pro-gression and who may therefore want to consider a treatment strategy that defers immediate interven-tion in favor of a strategy that monitors disease pro-gression and initiates treatment only when a tumor shows signs of becoming clinically signifi cant. They have relied on concepts originally developed by Epstein to propose the following criteria: a) men who present with prostate biopsies that demonstrate pros-tate cancer in two cores or less, b) neither core has more than 50% involvement with disease and c) tumor histology contains no Gleason pattern 4 or 5. Active surveillance is most appropriate for men over 70 years old who have a life expectancy of 15 to 20 years or less, but can also be followed by any man who is willing to defer immediate treatment because he believes the risks associated with more aggressive treatments are not justifi ed by the potential benefi t. Active surveillance does not imply that no treatment will ever be necessary. Should additional clinical information suggest that the risk of disease progres-sion has increased, more aggressive treatments are still available.
Several researchers have published outcomes from large observational cohorts of men who have selected active surveillance as a treatment alterna-tive. Protocols differ slightly among institutions but usually restrict entry to men meeting the criteria outlined above. Most researchers following men on an active surveillance protocol recommend that repeat biopsies and serum PSA values be checked regularly. Protocols differ in the timing of these repeat studies. Some researchers state that a repeat biopsy should be performed within one or two months of the original positive biopsy. Others

Active surveillance for prostate cancer 125
Active surveillance, however, carries several risks. In the Klotz cohort 30% of patients have been offered more aggressive treatment because they were reclassi-fi ed as having higher risk disease. Of these men, half have evidence of biochemical failure on the basis of a rising PSA. Many other patients fi nd active sur-veillance disquieting. As many as one quarter of men enrolled in active surveillance programs have aban-doned these programs after two years in favor of more aggressive treatment on the basis of psychological stress alone. These men have no clinical evidence of disease progression, but feel that the psychological stress of worrying about disease progression outweighs the risk of developing a complication associated with more aggressive treatment. In the SPCG-4 trial Johansson et al. reported that patients randomized to watchful waiting showed more anxiety, depression and a lower overall sense of well being when compared to those men who underwent surgery [24].
Summary
The introduction of PSA testing has dramatically increased the incidence of localized prostate cancer. Many of these newly diagnosed cancers are low vol-ume and low grade and pose minimal threat of pro-gression over ten years. Active surveillance is an experimental approach to managing these cases, especially for those men over age 65 years who have a life expectancy of 10 – 15 years. While one random-ized trial has been initiated, the data supporting this approach are limited to observational cohort studies with follow-up periods of ten years or less. While not confi rmed in Albertsen et al. ’ s observational cohort, Johannson et al. documented an increase in cancer specifi c mortality among men identifi ed with palpa-ble disease and followed beyond 15 years [5,7].
The criteria used to select and monitor men on active surveillance have not been validated. Neither PSA values nor prostate biopsies provide defi nitive information concerning disease progression. PSA values are known to vary widely. In a retrospective analysis of an unscreened population of 972 men with a median age of 62 years, Eastham et al. demonstrated substantial year to year variation in PSA levels [25]. Furthermore, it is unclear whether changes in Gleason score on follow-up biopsies refl ect disease progression or simply refl ect sampling variation. Until better biomarkers for indolent dis-ease are identifi ed, men considering active surveil-lance should carefully weigh the potential of disease progression that persists for 20 years or longer no matter how small or well differentiated the disease appears initially against the potential of complica-tions associated with treatment that often occur shortly after the treatment has been completed.
suggest that a repeat biopsy should be performed at one year, while still others suggest that repeat biop-sies can be deferred for as long as two years follow-ing the initial biopsy. Disagreement also surrounds the frequency of PSA testing. Some clinicians sam-ple PSA every three months while others check them less frequently.
Klotz et al. recently published an update of the clinical outcomes of 450 men enrolled in an active surveillance program in Toronto [16]. After a median follow-up of 6.8 years (range 1 to 13 years) overall survival was 78.6%. The ten year prostate cancer actuarial survival was 97.2%. Most of the patients enrolled in this protocol had a low risk of disease progression. Seventeen percent of patients had Gleason 3 � 4 � 7 disease. All of the men in the latter group were older than age 70. Patients were ini-tially followed every three months with a serum PSA level and a repeat biopsy was performed six to 12 months after the initial diagnostic biopsy and every three to four years thereafter until age 80. If the PSA doubling time was less than three years, the Gleason score increased on a repeat biopsy or there was evidence of clinical progression, patients were reclassifi ed as having higher risk disease and were offered more aggressive therapy. After ten years of follow-up the probability of death from a competing medical condition was 18.6 times more common than prostate cancer among the patients enrolled in the study.
Six other research groups have also published their active surveillance series [17 – 22]. A total of 2500 patients have been enrolled and more than 200 have been followed for over ten years. All of the series rely on PSA kinetics and information obtained at repeat biopsy to identify those patients who are at a higher potential risk of disease progression. To date, the disease specifi c survival associated with active surveillance protocols is 99.7%.
In addition to data from case series, data from a large historical prospective cohort study also support the concept of active surveillance. Shappley et al. recently examined the consequences of deferred treatment as initial management by reviewing the outcomes of 3331 men diagnosed from 1986 to 2007 who were enrolled in the Health Professionals Follow-up Study [23]. Of these men, 342 (10.3%) initially selected no active treatment. Of these, 174 (51%) remain untreated after a mean follow-up of 7.7 years. The remainder were treated an average of 3.9 years after diagnosis. Older men and men with low risk cancer at diagnosis were more likely to defer treatment. Prostate cancer mortality did not differ among men selecting active treatment when com-pared to those who elected to defer treatment at the time of diagnosis.

126 P. C. Albertsen
Declaration of interest: The authors report no confl icts of interest. The authors alone are responsible for the content and writing of the paper.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer [1] statistics, 2009. CA Cancer J Clin 2009;59:225 – 49. Collin SM, Martin RM, Metcalfe C, Gunnell D, Albertsen [2] PC, Neal D, et al. Prostate-cancer mortality in the USA and UK in 1975-2004: An ecological study. Lancet Oncol 2008;9:445 – 52. Epstein JI, Walsh PC, Carmichael M, Brendler CB. Patho-[3] logical and clinical fi ndings to predict tumor extent of nonpalpable (stage T1c) prostate cancer. JAMA 1994;271:368 – 74. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, [4] Miller GJ, Ford LG, et al. The infl uence of fi nasteride on the development of prostate cancer. N Engl J Med 2003;349:215 – 24. Johansson JE, Andren O, Andersson SO, Dickman PW, [5] Holmberg L, Magnuson A, et al. Natural history of early, localized prostate cancer. JAMA 2004;291:2713 – 9. Chodak GW, Thisted RA, Gerber GS, Johansson JE, [6] Adolfsson J, Jones GW, et al. Results of conservative manage-ment of clinically localized prostate cancer. N Engl J Med 1994;330:242 – 8. Albertsen PC, Hanley JA, Fine J. 20 year outcomes follow-[7] ing conservative management of clinically localized prostate cancer. JAMA 2005;293:2095 – 101. Welch AG, Albertsen PC. Prostate cancer diagnosis and treat-[8] ment after the introduction of prostate specifi c antigen screening: 1986 – 2005. J Natl Cancer Inst 2009;101:1325 – 9. Draisma G, Boer R, Otto SJ, van der Cruijsen IW, Damhuis [9] RA, Schroeder FH, et al. Lead times and over detection due to prostate-specifi c antigen screening: Estimates from the European Randomized Study of Screening for Prostate Cancer: J Natl Cancer Inst 2003;95:868 – 78. Lu-Yao GL, Albertsen PC, Moore DF, Shih W, Lin Y, DiPaola [10] RS, et al. Outcomes of localized prostate cancer following conservative management. JAMA 2009;302:1202 – 9. Albertsen PC, Hanley JA, Barrows GH, Penson DF, [11] Kowalczyk PDH, Sanders MM, et al. Prostate cancer and the Will Rogers phenomenon. J Natl Cancer Inst 2005;97:1248 – 53. Andriole GL, Crawford ED, Grubb RL, Buys SS, Chia D, [12] Church TR, et al. PLCO Project Team. Mortality results from a randomized prostate cancer screening trial. N Engl J Med 2009;360:1310 – 9. Schroeder FH, Hugosson J, Roobol MJ, Tammela TL, [13] Ciatto S, Nelen V, et al. ERSPC Investigators. Screening and prostate cancer mortality in a randomized European study. N Eng J Med 2009;360:1320 – 8.
Bill-Axelson A, Holmberg L, Filen F, Ruutu M, Garmo H, [14] Busch C, et al. Scandinavian Study Group Number 4. Rad-ical prostatectomy versus watchful waiting in localized pros-tate cancer: The Scandinavian prostate cancer group-4 randomized trial. J Natl Cancer Inst 2008;100:1144 – 54. Widmark A, Klepp O, Damber JE, Angelsen A, Fransson P, [15] Lund JA, et al. Endocrine treatment, with or without radio-therapy, in locally advanced prostate cancer (SPCG-7/SFUO-3): An open randomized phase III trial. Lancet 2009;373:301 – 8. Klotz L, Zhang L, Lam A, Nam R, Mamedov A, Loblaw A. [16] Clinical results of long-term follow-up of a large, active sur-veillance cohort with localized prostate cancer. J Clin Oncol 2010;28:126 – 31. van AS NJ, Norman AR, Thomas K, Khoo VS, Thompson A, [17] Huddart RA, et al. Predicting the probability of deferred radical treatment for localized prostate cancer managed by active surveillance. Eur Urol 2008;54:1297 – 305. Carter HB, Ketterman A, Warlick C, Metter EJ, Landis P, [18] Walsh PC, et al. Expectant management of prostate cancer with curative intent: An update of the Johns Hopkins experi-ence. J Urol 2007;178:2359 – 64. Soloway MS, Soloway CT, Williams S, Ayyathuria R, Kava B, [19] Manoharan M. Active surveillance: A reasonable manage-ment alternative for patients with prostate cancer – The Miami experience. BJU Int 2008;101:165 – 9. van den Bergh RC, Roemling S, Roobol MJ, Aus G, [20] Hugosson J, Rannikko AS, et al. Outcomes of men with screen detected prostate cancer eligible for active surveil-lance who were managed expectantly. Eur Urol 2009;55:1 – 8. Khatami A, Aus G, Damber JE, Lilja H, Lodding P, Hugosson [21] J. PSA doubling time predicts the outcome after active sur-veillance in screening-detected prostate cancer: Results from the European randomized study of screening for prostate cancer, Sweden section. Int J Cancer 2007;120:170 – 4. Dall ’ Era MA, Konety BR, Cowan JE, Shinohara K, Stauf F, [22] Cooperberg MR, et al. Active surveillance for the manage-ment of prostate cancer in a contemporary cohort. Cancer 2008;112:2664 – 70. Shappley WV, Kenfi eld SA, Kasperzyk JL, QuiW, Stampfer [23] MJ, Sanda MG, et al. Prospective study of determinants and outcomes of deferred treatment or watchful waiting among men with prostate cancer in a nationwide cohort. J Clin Oncol 2009;27:4980 – 5. Johansson E, Bill-Axelson A, Holmberg L, Onelov E, Johans-[24] son JE, Steineck G, Scandinavian Prostate Cancer Group Study No. 4. Time, symptom burden, androgen deprivation, and self-assessed quality of life after radical prostatectomy or watchful waiting: The Randomized Scandinavian Prostate Cancer Group Study Number 4 (SPCG-4) clinical trial. Eur Urol 2009;55:422 – 30. Eastham JA, Riedel E, Scardino PT, Shike M, Fleisher M, [25] Schatzkin A, et al: Polyp Prevention Trial Study Group. Vari-ation of serum prostate-specifi c antigen levels: An evaluation of year-to-year fl uctuations. JAMA 2003;289:2695 – 700.

Correspondence: Roger S. Rittmaster, 5 Moore Drive, Research Triangle Park, North Carolina 27709, USA. E-mail: [email protected]
(Received 13 September 2010 ; accepted 17 September 2010 )
REVIEW ARTICLE
Chemoprevention of prostate cancer
ROGER S. RITTMASTER
Oncology Clinical Development, GlaxoSmithKline, Inc., North Carolina, USA
Abstract Over the past two decades, many more men are diagnosed with prostate cancer then die of the disease. This increase in diagnosis has led to aggressive treatment of indolent disease in many individuals and has been the impetus for fi nding a means of reducing the risk of prostate cancer. In the past decade, there have been eight large trials of prostate cancer risk reduction using dietary supplements, 5 α -reductase inhibitors, or anti-estrogens. The only two trials which have demonstrated effi cacy are those involving 5 α -reductase inhibitors: the PCPT (fi nasteride) and REDUCE (dutasteride). This review exam-ines prostate cancer risk reduction, with emphasis on conclusions that can be drawn from these two landmark studies.
Epidemiology
Prostate cancer is a heterogeneous disease. Whereas most prostate cancers behave indolently and are undiagnosed during life, it is still the second most common cause of cancer death in men. Autopsy studies have indicated that as many as 30% of men in their 30s will have microfoci of prostate cancer with the prevalence increasing by about 10% with each decade of life [1]. With the advent of wide-spread PSA testing, especially in North America, the incidence of prostate cancer diagnosis has greatly increased [2]. At present approximately one in six men will be diagnosed with prostate cancer in regions where PSA testing is widespread, and 3.5% of men will die of the disease. Because up to 90% of men diagnosed with prostate cancer are treated with aggressive therapy (radical prostatectomy, radiation, or androgen ablation) [3], cancers that are consid-ered pathologically insignifi cant are still clinically signifi cant, once they are diagnosed. Even if PSA screening is conclusively shown to decrease the death rate from prostate cancer, it is unlikely that treating low volume, low-grade prostate cancer will contribute to this reduction. Therefore, reducing the overdiagnosis and overtreatment of low-grade prostate cancer is a key benefi t of prostate cancer chemoprevention.
Defi nition of chemoprevention
The term “ chemoprevention ” implies that a disease is being prevented. With respect to prostate cancer, “ risk reduction ” is a more appropriate terminology. Nevertheless, because of its widespread use, “ chemo-prevention ” will continue to be used throughout this manuscript. Primary chemoprevention refers to reducing the risk of cancer development. Secondary chemoprevention involves reducing the risk of progression of cancer that is already present. Because many older men have foci of undiagnosed prostate cancer, chemoprevention involves both primary and secondary cancer risk reduction. This distinction is important, because any successful prostate cancer chemopreventative agent will also need to be a treatment of localized prostate cancer.
Targets for prostate cancer chemoprevention
The two principal targets for prostate cancer chemoprevention have been infl ammation and hor-monal stimulation of the prostate [4,5]. Infl amma-tion has been associated with the development of lung cancer in smokers, hepatic cancer in chronic hepatitis and bowel cancer in infl ammatory bowel disease. In the prostate, inflammation is associ-ated with prostate cancer precursor lesions such as
Acta Oncologica, 2011; 50(Suppl 1): 127–136
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.527367

128 R. S. Rittmaster
proliferative infl ammatory atrophy (PIA) and is believed to increase genetic instability leading to prostate carcinogenesis. Studies involving rofecoxib (a cyclooxygenase-2 inhibitor) and the antioxidants, selenium and Vitamin E, have all been designed to test whether decreasing infl ammation in the prostate would reduce the risk of prostate cancer.
Other trials have involved agents designed to reduce hormonal stimulation to the prostate. Androgens are known to stimulate both benign and malignant prostate growth. Neither benign prostatic hyperplasia nor pros-tate cancer has been described in eunuchs. However, abolishing androgen production and blocking the androgen receptor are unrealistic methods of chemo-prevention, because of the side-effects of hypogonad-ism. On the other hand, testosterone, the major circulating androgen, must be converted to dihydrotes-tosterone by the 5 α -reductase enzymes in order to be active in the prostate. Both fi nasteride and dutasteride, 5 α -reductase inhibitors, target this pathway and have been the subject of successful chemopreventative trials described below.
The role of estrogens in human prostate cancer is not well defi ned. In some rodent models, exogenous estrogens can synergize with androgens to promote prostate cancer [6,7]. In humans the role of estrogens in prostate carcinogenesis is unclear. Clinical trials of estrogens and antiestrogens are diffi cult to interpret, because of the indirect effect of these agents on tes-ticular and adrenal androgen production.
Prostate cancer risk reduction studies
Nearly all of the major prostate cancer risk reduction studies started in the last ten years are now complete
(Table I). The only successful ones have involved 5 α -reductase inhibitors: PCPT (fi nasteride) and REDUCE (dutasteride). The unsuccessful trials will be briefl y reviewed in the order of their completion, followed by a more detailed discussion of the PCPT and REDUCE trials.
ViP trial (rofecoxib) (http://www.clinicalstudyresults.org/documents/company-study_783_0.pdf)
Based on an association of COX-2 overexpression with increased angiogenesis and decreased apoptosis in the prostate, the VIP trial planned to enroll 15 000 men at increased risk of prostate cancer based on an elevated PSA (2.5 – 10 ng/ml), in order to test whether the COX-2 inhibitor rofecoxib could reduce the risk of prostate cancer. The trial was terminated after 4 741 men were enrolled, because rofecoxib was withdrawn from the market due to an excess of isch-emic cardiac events in another study. Therefore, the effi cacy of this approach to prostate cancer risk reduction was never tested.
SELECT trial (Selenium and Vitamin E Cancer Prevention Trial)
Selenium and Vitamin E had been associated with a reduction in prostate cancer in post-hoc analyses of two large studies [8,9]. Prostate cancer was not a pre-defi ned endpoint in either study. In SELECT, 35 000 men over age 50 were randomized to receive 200 μ g/d selenium, 400 IU/day Vitamin E, both or neither. Among the four groups, 1 758 cancers were diagnosed. After a median follow-up of 5.5 years, the trial was terminated for lack of effi cacy [10].
Table I. Prostate cancer prevention studies (in order of completion).
Agent (Trial Name) Population Studied Number of Subjects Date Concluded Results Sponsor
Finasteride (PCPT) Age � 55PSA � 3.0
18,882 02/2003 24.8% decrease in cancers
Southwest Oncology Group
Rofecoxib (ViP Trial) Age 50 – 75PSA 2.5 – 10
15,000 planned 09/2004 Trial cancelled (drug toxicity)
Merck
Vitamin E and C (Physicians Health Study II)
Age � 50 14 641 08/2007 No benefi t from either agent
NIHWyeth
SeleniumVitamin E (SELECT) Age � 50PSA � 4.0
35 533 10/2008 No benefi t from either agent
Southwest Oncology Group
Dutasteride (REDUCE) Age � 50PSA 2.5 – 10
8 231 01/2009 23% decrease in cancers
GlaxoSmithKline
Soy, Selenium, Vitamin E HGPIN 325 2010 No Benefi t NCI CanadaSelenium Age � 40
PSA � 10HGPIN
∼ 435 05/2010 No signifi cant benefi t
U.S. National Cancer Institute
Toremifi ne Age � 30PSA � 10HGPIN
1 590 05/2010 No signifi cant benefi t
GTx

Chemoprevention of prostate cancer 129
Physicians ’ Health Study II (Vitamin E and C)
In this study, 14 641 male physicians over age 49 were randomized to receive 400 IU Vitamin E, 500 mg Vitamin C or placebo for up to ten (median 7.6) years. There were 1 008 prostate cancers diagnosed on for-cause biopsies, with no difference among the groups [11].
Prostate Cancer Prevention Study for Men with High-grade Prostatic Intraepithelial Neoplasia (toremifene vs placebo; ClinTrials.Gov identifi er: NCT00106691)
This trial randomized 1 590 men with high-grade PIN and no cancer on biopsy to 20 mg toremifene, a selective estrogen receptor modulator, or placebo daily for three years [12]. Repeat biopsies were done after one, two and three years. In a press release on May 24, 2010, the sponsor GTx announced that toremifene reduced the incidence of prostate cancer by a non-signfi cant 10.2% (p � 0.385).
L-Selenium Based Chemoprevention of Prostate Cancer Among Men with High-grade Prostatic Intraepithelial Neoplasia (ClinTrials.Gov identifi er: NCT00030901)
This trial recruited approximately 465 men aged 40 – 80 with high-grade PIN on biopsy, who were treated with 200 μ g selenium or placebo for three years. End-of-study biopsies were performed on any man not diagnosed with prostate cancer on for-cause biopsy during the study. This study showed similar incidences of prostate cancer in the two study arms, as reported during a plenary session at the 2010 Annual Meeting of the American Urological Association.
5 α - reductase inhibitors for prostate cancer chemoprevention
Rationale . Testosterone is the principal circulating androgen, but it must be converted to dihydrotestos-terone (DHT) in the prostate to stimulate prostate growth. DHT is intrinsically about twice as potent as testosterone, and it binds more tightly to the androgen receptor [13,14]. There are two enzymes responsible for conversion of testosterone to DHT, Type 1 and 2 5 α -reductase [15]. Type 2 5 α -reductase predominates in benign prostate tissue, and men born with Type 2 5 α -reductase defi ciency have small prostate glands and have never been reported to develop prostate cancer [16]. Type 1 5 α -reductase is upregulated in prostate cancer, especially in high-grade or advanced disease [17,18]. Both fi nasteride, a selective type 2 5 α -reductase inhibitor (5ARI), and dutasteride, a dual 5ARI, have proven effective in the treatment of benign prostatic hyperplasia by
reducing androgen stimulation to the prostate [19]. Based on the knowledge that prostate cancers have not been reported in eunuchs or men with 5 α -reductase defi ciency, fi nasteride and dutasteride have both been considered logical candidates for prostate cancer chemoprevention.
Finasteride and the Prostate Cancer Prevention Trial (PCPT)
The PCPT was funded by the U.S. National Cancer Institute and coordinated by the Southwest Oncol-ogy Group. It was initiated in 1993 at over 200 sites in USA and concluded in 2003. The key inclusion criteria were men � 55 years old, PSA � 3.0 ng/ml, a normal digital rectal examination (DRE), and no suspicion of prostate cancer [20]. There were no baseline biopsies.
Men were randomized to 5 mg fi nasteride/day or placebo for seven years. Annual PSA measurements and digital rectal examinations (DREs) were done and a prostate biopsy ( � 6 cores) was recommended for a PSA � 4.0 ng/ml or a suspicious DRE. PSA values were doubled in the fi nasteride arm in Years 1 – 3, and multiplied by 2.3 after Year 3 to compensate for the mean decrease in serum PSA with fi nasteride in an attempt to equalize for-cause biopsies in the two groups. An end-of-study prostate biopsy was rec-ommended at Year 7 for any man not diagnosed with prostate cancer during the study. For-cause biopsies were defi ned as biopsies that occurred in any man with a PSA � 4.0 ng/ml or an abnormal DRE, whether they were done as an interim or end-of-study biopsy.
Ultimately, 18 882 men were randomized, and 9 060 men (48%) were included in the fi nal analysis (Table II) [21]. The analysis included all men who had an interim biopsy for prostate cancer or who had an end-of-study biopsy (modifi ed crude rate). For-cause biopsies were done in 39% of the participants, and 52% of the cancers were diagnosed on for-cause biopsies. There were 15% fewer for-cause biopsies in the fi nasteride group. Finasteride demonstrated a 24.8% reduction in the primary endpoint, the preva-lence of prostate cancer during the seven-year period (18.4% vs. 24.4% of participants, p � 0.001). Look-ing only at for-cause biopsies, there were 10% fewer cancers in the fi nasteride group (26.5% vs. 29.5%; p � NS). The reduction in overall cancer incidence was entirely due to a reduction in low-grade cancers (Gleason score � 6), and there was an increase in moderate to high-grade cancers: 280 (6.4%) in the fi nasteride group and 237 (5.1%) in the placebo group (p � 0.005). The increase in Gleason 7-10 cancers was almost entirely due to their increased detection in for-cause biopsies. In the end of study

130 R. S. Rittmaster
biopsies, there were 92 and 89 Gleason 7-10 cancers in the fi nasteride and placebo groups, respectively.
These results indicate some limitations of the PCPT trial. Less than 50% of randomized men were included in the fi nal analysis, and over half of the prostate cancers were detected on for-cause biopsies. There were fewer for-cause and end-of-study biop-sies in the fi nasteride arm, suggesting that fi nasteride affected the decision to be biopsied. Subsequent analyses have demonstrated that fi nasteride improved the sensitivity of both PSA and DRE to detect pros-tate cancer, including high-grade cancers [22,23]. Furthermore, if high-grade cancers were present at radical prostatectomy, they were more likely to be detected in the fi nasteride arm than the placebo arm [24]. Also, fi nasteride decreases prostate volume (24% lower in the fi nasteride arm at the time of biopsy), improving the detection of both low and high-grade cancers [25,26].
Based on the increased utility of PSA for prostate cancer detection and prostate volume reduction, one would predict that fi nasteride would increase cancer detection rates, especially on for-cause biopsies. Four separate post hoc analyses have now been conducted to attempt to account for these factors in determin-ing the true effect of fi nasteride on overall and high-grade cancer [27 – 30]. The results have ranged from a 0.88 odds ratio for high-grade cancer with fi nas-teride when prostate volume changes were included to a 27% relative risk reduction when the results from subsequent radical prostatectomies were used instead of prostate biopsies. Because these analyses were retrospective in nature and involved many assumptions, they must be considered hypothesis generating rather than providing defi nitive answers. Although these analyses suggest that fi nasteride did not cause a net increase in high-grade cancers, they also suggest that it was less effective in reducing the risk of high-grade cancer, compared to low-grade cancer.
In summary, in the PCPT fi nasteride causes a highly signifi cant reduction in overall and low-grade prostate cancers. Although induction of high-grade cancer cannot be excluded, the literature suggests
instead that fi nasteride likely led to an earlier detec-tion of high-grade cancer, which was less extensive than in the placebo group [24]. Some authors have challenged the effi cacy of fi nasteride in the PCPT by focusing on the non-signifi cant 10% reduction in prostate cancer in for-cause biopsies only [31]. To do so ignores the factors discussed above. The PCPT was never designed or powered to assess the effect of fi nasteride in a situation where cancers are only detected on for-cause biopsies.
Dutasteride and the Reduction by DUtasteride of prostate Cancer Events (REDUCE) trial
The initial interest in dutasteride for prostate cancer risk reduction came from two observations.
The fi rst was the fi nding that the ratio of type 1 to type 2 5 α -reductase was increased in prostate cancer, suggesting that inhibition of both isoen-zymes might be important [18]. The recent obser-vation that both isoenzymes are increased in localized high-grade prostate cancer compared to low-grade cancer or benign tissue emphasizes the potential relevance of dual 5 α -reductase inhibition [32]. The second observation came from Phase 3 dutasteride trials for benign prostatic hyperplasia, in which the incidence of prostate cancer was 51% less in the dutasteride arm compared to placebo (27 vs 55 cancers) [33]. Although this was a post-hoc observation of adverse event data, such a decrease had not been seen in similarly designed trials with fi nasteride [34,35].
The REDUCE trial was designed in 2002, and the fi rst patient was enrolled in early 2003 [36]. It was a multinational, randomized, placebo-controlled trial designed to test the ability of dutasteride to reduce the risk of biopsy-detectable prostate cancer in men at high risk of being diagnosed with the dis-ease. The key entry criteria were men aged 50 – 75, PSA 2.5 – 10.0 ng/ml, prostate volume � 80 ml, and a single, negative prostate biopsy of 6 – 12 cores done independent of the study and taken within six months prior to study enrollment. After a one-month placebo run-in to assess symptoms of benign prostatic hyper-plasia and prostatitis, 8 122 men were randomized to dutasteride or placebo for four years and took at least one dose of study drug [37]. Repeat, study-mandated prostate biopsies were taken after two and four years; for-cause biopsies could be done at any time. For-cause biopsies during Months 19 – 24 and 43 – 48 replaced the Year 2 or 4 study-mandated biopsies, and hence did not increase a subject ’ s chance of being diagnosed with prostate cancer. Protocol-independent prostate biopsies were those for-cause biopsies done during Months 1 – 18 and Months 25 – 42. Key differences between the REDUCE and PCPT trials are shown
Table II. Summary of subjects, biopsies and cancers in the PCPT.
Total Finasteride Placebo
Subjects randomized 18 882 9 423 9 549Included in analysis 9 060 (48%) 4 368 (46%) 4 692 (50%)For-cause biopsies
(% of men biopsied)
3 573 (39%) 1 639 (38%) 1 934 (41%)
Total cancers 1 950 803 1 147Cancers diagnosed
for cause1 006 (52%) 435 (54%) 571 (50%)

Chemoprevention of prostate cancer 131
in Table III. Because the trials had different study designs and involved different patient populations, they should be viewed as providing complementary results.
Overall, prostate cancer was diagnosed in 858 men in the placebo group (25.1%) and 659 men in the dutasteride group (19.9%) with a relative risk reduction of 23% (p � 0.0001) (Table IV) [37]. Gleason 7-10 cancers were diagnosed in 220 men in the dutasteride group (6.7%) and 233 men in the placebo group (6.8%) (p � 0.81). In the subset of Gleason 8-10 cancers, there were 29 cancers in the dutasteride group and 19 cancers in the placebo group (p � 0.15). During Years 1 – 2 there were 17 and 18 Gleason 8-10 cancers in the dutasteride and placebo groups, respectively. However, during Years 3 – 4 there were 12 Gleason 8-10 cancers in the dutasteride group and only one in the placebo group (of 2 343 biopsies). Although induction of high-grade cancer cannot be excluded, one possible explanation for the paucity of Gleason 8-10 cancers in the placebo arm in Years 3 – 4 was the fact that 141 more Gleason 5-7 cancers were diagnosed in the placebo arm during Years 1 – 2. Because men with cancer were removed from treatment, there was no opportunity for those cancers diagnosed during Years 1 – 2 to be reclassifi ed or upgraded dur-ing Years 3 – 4. Support for this hypothesis comes from an active surveillance study in which 105 men with Gleason 4-7 cancers entered into an active surveillance study [38]. Eight (8%) were upgraded to Gleason 8 cancers on re-biopsy a median of 22 months later. A similar rate of upgrading of lower grade cancers diagnosed during Years 1 – 2 in the REDUCE trial could explain the “ missing ” Gleason 8-10 cancers in the placebo group during Years 3 – 4. Futhermore, in the CombAT study [39], a 4 800-patient, 4-year BPH study comparing dutasteride and tamsulosin monotherapies with the combina-tion of the two, in which all biopsies were done for cause, there was no evidence of an increase in high-grade cancers in the two dutasteride arms compared to the tamsulosin monotherapy arm (Roehrborn CR, Nickel JC, Andriole GL, Gagnier RP, Black L,
Wilson TH, Rittmaster RS. Dutasteride improves the outcomes of benign prostatic hyperplasia when eval-uated for prostate cancer risk reduction: a secondary analysis of the REduction by DUtasteride of prostate Cancer Events (REDUCE) trial., In Press, European Urology, 2010).
Other benefi ts of 5 α -reductase inhibitors in men at risk of prostate cancer
Regardless of the etiology of the increase in high-grade cancers in the PCPT and REDUCE, it is important that such cancers be rapidly diagnosed when they occur. Both fi nasteride and dutasteride have been shown to enhance the utility of PSA for the diagnosis of high-grade prostate cancer [22,40]. Both medications increase the area under the receiver operator characteristics (ROC) curve for PSA detec-tion of prostate cancer. In the REDUCE trial, any rise in PSA after six months of dutasteride treatment characterized a group of men with an increased like-lihood of cancer overall, high-grade cancer and pathologically signifi cant cancer (modifi ed Epstein criteria), compared with men whose PSA was stable or continued to decrease [40]. This relationship was markedly attenuated in the placebo arm. The com-bination of a reduction in the diagnosis of low-grade cancer and enhanced detection of high-grade cancer should lead to a reduction in the overdiagnosis and overtreatment of indolent prostate cancers.
Because most men over age 50 harbor foci of pros-tate cancer, one could argue that the main effect of 5ARIs is to reduce the risk that biopsy-undetectable cancer will grow to biopsy-detectable cancer. 5ARIs also reduce the incidence of high-grade PIN, consid-ered to be a marker of increased risk of prostate can-cer on a subsequent biopsy. In the PCPT fi nasteride reduced the incidence of high-grade PIN without cancer by 15% [41]; in REDUCE dutasteride reduced the incidence of high-grade PIN without cancer by 39% [37]. This data suggest that 5ARIs reduce the stimulus to prostate cancer formation and will decrease the need for follow-up biopsies.
Whether or not 5ARIs have a benefi cial effect on some high-grade cancers, clearly the greatest effect is the reduction in low-grade cancers. Although some authors have called such cancers “ insignifi cant ” , up to 90% of prostate cancers undergo some form of aggressive treatment [3]. For example, in the REDUCE trial, there were 32% fewer treatment interventions for prostate cancer in the dutasteride arm [42].
Men with an elevated PSA are at increased risk not only for prostate cancer, but also for BPH and its complications. In the fi nasteride arm of PCPT, urinary retention occurred 33% less often, there were
Table III. Key differences between the PCPT and REDUCE study designs.
PCPT REDUCE
Drug Finasteride DutasterideDuration (years) 7 4Age range � 55 50 – 75Entry serum PSA (ng/ml) � 3.0 2.5 – 10.0Baseline biopsies No Yes (6 – 12 cores)Study-mandated biopsy timing Year 7 Years 2 and 4Study-mandated biopsy cores � 6 (6 cores
in ∼ 80%)10

132 R. S. Rittmaster
Table IV. Summary of subjects, biopsies and cancers in REDUCE.
Total Dutasteride Placebo
Subjects randomized 8 122 4 049 4 073Subjects Biopsied 6 729 (83%) 3 305 (82%) 3 424 (84%)Number of biopsies 12 024 5 956 6 068Protocol-independent biopsies (percent of biopsies) 810 (6.7%) 344 (5.8%) 466 (7.7%)Total cancers (% of men biopsied) 1 517 (22.5%) 659 (19.9%) 858 (25.1%)Cancers diagnosed on protocol-independent biopsies 98 (6.5%) 41 (6.2%) 57 (6.6%)
47% fewer transurethral prostate resections, and 29% fewer urinary tract infections, compared to the placebo arm [21]. In the dutasteride arm of REDUCE acute urinary retention was reduced by 77%, BPH-related surgery by 73%, and urinary tract infections by 41% [37]. Because men in REDUCE had higher baseline PSA levels than those in PCPT, they were at a higher risk of BPH complications, and hence these results are not directly comparable between the two studies.
Side-effects of 5ARIs
Dutasteride and fi nasteride have similar side-effect profi les, the most common drug-related adverse events being related to sexual function. In a one-year comparative trial, new instances of impotence were noted in 9% of the fi nasteride group and 8% of the dutasteride group; new instances of decreased libido were noted in 6% of the fi nasteride group and 5% of the dutasteride group [43]. In the PCPT, erectile dysfunction occurred in 67% of the fi nasteride group and 61% of the placebo group. Decreased libido occurred in 65% of the fi nasteride group and 60% of the placebo group [21]. Age had a much greater effect on sexual dysfunction in the PCPT than did fi nasteride use, with sexual function deteriorating in both groups over the seven years of the trial [44]. In the four-year REDUCE study, new instances of decreased libido occurred in 5.1% of the dutasteride group and 2.9% of the placebo group [37]. New instances of erectile dysfunction occurred in 9.0% of the dutasteride group and 5.7% of the placebo group. Four point three percent of the dutasteride group and 2.0% of the placebo group dropped out due to drug-related side-effects. Gynecomastia occurred in 4.5% of the fi nasteride arm of the PCPT over seven years and 1.9% of the dutasteride arm of REDUCE over four years, with placebo rates being about half those of the 5ARIs. If decreased libido or erectile dysfunction is going to occur due to 5ARIs, it usually happens within the fi rst six to 12 months of treatment, with rates in the active and placebo groups being similar thereafter [45]. On the other hand, new instances of gynecomastia with 5ARIs occur at a low, but steady rate in excess of placebo.
There have been no life-threatening or serious side-effects proven to be related to either fi nasteride or dutasteride. Both can occasionally be associated with allergic-type skin reactions. In REDUCE, there was an excess of events in the composite term “ heart failure ” in the dutasteride vs. the placebo group (30 vs. 16; 0.7% vs. 0.4%); such an increase has not been seen with dutasteride in other studies.
Balancing the benefi ts and risks of 5ARIs for prostate cancer risk reduction – Who should be treated?
Neither dutasteride nor fi nasteride has yet to be approved in the EU or USA for reducing the risk of prostate cancer. The PCPT and REDUCE trials indicate that both drugs are effective in reducing biopsy-detectable prostate cancer, although the pop-ulations in which each was tested were different. Although the rates of prostate cancer in the placebo groups of the PCPT and REDUCE were similar, the likelihood of a prostate cancer diagnosis in the REDUCE population is much greater outside of a clinical trial. Men with serum PSAs less than 3.0 ng/ml are biopsied infrequently, and biopsying such men yields a low rate of detection of potentially lethal cancers [46]. On the other hand, men with a negative biopsy and elevated PSA are men that are followed closely, often have a rising PSA, and are likely to be re-biopsied. In REDUCE, 72% of men in the placebo group had a rising PSA after Month 6 [40]. Hence, this is a population at high risk of a prostate biopsy and resultant prostate cancer diagnosis and most likely to benefi t from prostate cancer risk reduction.
How about men with a high PSA but below the biopsy threshold? Schroder et al. have shown in the ERSPC prostate cancer screening study that men with a baseline PSA � 1.5 ng/ml were 3.6 and 7.1 times more likely to be diagnosed with high-grade and low-grade cancer, respectively, than men with a baseline PSA � 1.5 ng/ml [47]. Treatment with a 5ARI in this population will likely reduce the number of men in whom a biopsy is indicated and will reduce the overdiagnosis and overtreatment of low-grade prostate cancers. Such men would already be candi-dates for 5ARI treatment if they have symptomatic BPH, and prostate cancer risk reduction would be

Chemoprevention of prostate cancer 133
an added benefi t. Fewer men would undergo aggres-sive treatment for prostate cancer, reducing the fre-quency of radical prostatectomy, radiation therapy and androgen ablation.
5ARIs not only decrease the incidence of low-grade cancer, but enhance the detection of high-grade cancer. It is likely that 5ARIs remove some of the background noise in PSA levels, by suppressing PSA production from benign prostate tissue and indolent prostate cancers. In both the PCPT and REDUCE trials, men whose PSA rose while taking a 5ARI were more likely to have a clinically signifi -cant prostate cancer [40,48]. One could hypothesize that dutasteride and fi nasteride may serve as bioas-says for clinically signifi cant prostate cancer (cancers whose growth is no longer being controlled by the 5ARI] with PSA being the readout. On the other hand, the percent decrease in PSA with dutasteride during the fi rst six months of REDUCE did not pre-dict overall or high-grade cancer detection during the study. To monitor PSA in men taking a 5ARI, reason-able guidance is to wait six months to establish a new PSA baseline. Confi rmed PSA rises from a new base-line should prompt consideration for a prostate biopsy. Continued PSA monitoring is essential in any man taking a 5ARI, if the benefi ts of increased PSA utility are to be realized.
While it cannot be ruled out, it is unlikely that either dutasteride or fi nasteride induces the growth of high-grade cancer. In the PCPT, where PSA drove many biopsies, there was an increase in detection of Gleason 7-10 cancers, but only in the for-cause biop-sies. In REDUCE, where PSA-driven biopsies were uncommon, there was no overall increase in Gleason 7-10 cancer detection. These results are consistent with the increased utility of PSA for detecting high-grade cancer. Both agents reduce prostate volume, making cancers easier to detect. When prostate vol-ume at the time of biopsy was included in a logistic regression in the PCPT, the odds ratio for Gleason 7-10 cancers in the fi nasteride arm was 0.88 [27]. When this same adjustment was done in REDUCE, the odds ratio for Gleason 7-10 cancers in the dutas-teride arm was 0.62 [49]. Nevertheless, it is clear that both 5ARIs are more effective at reducing the risk of low-grade cancer than high-grade cancer.
One concern is that use of 5-ARIs will cause more high-grade cancers to be missed by preventing PSA increases in such men [50]. There is little evi-dence to support this hypothesis. In REDUCE, if only men with a rising PSA are biopsied, there will be about twice as many men with Gleason 7-10 can-cers who would not be biopsied in the dutasteride arm, compared to the placebo arm. However, nor-mally a rise of 0.35 – 0.75 ng/ml/year is required for rebiopsy according to most guidelines in men not
taking a 5ARI. In REDUCE, if one required a 1 ng/ml rise in PSA from Month 6 to the fi nal PSA before biopsy in the placebo arm, the number of Gleason 7-10 cancers that would not be diagnosed would be similar in both arms [40]. In the dutasteride arm these are presumably cancers whose growth is being controlled, preventing a rise in PSA.
Neither the PCPT or REDUCE were of suffi -cient duration to assess whether 5ARIs reduce deaths from prostate cancer, because of the prolonged natu-ral history of biopsy-detected prostate cancer. How-ever, aggressive prostate cancers eventually escape even total androgen ablation, and there is no reason to suspect that such cancers would be controlled by 5ARIs. On the other hand, there is also no reason to suspect that cancers that progress during 5ARI ther-apy would not respond to more aggressive androgen ablation. Even advanced prostate cancers often respond to more complete androgen ablation after GnRH agonists alone have failed.
In addition to the benefi cial effects of 5ARIs in reducing the risk of prostate cancer and high-grade PIN, both fi nasteride and dutasteride are effective treatments for men with symptomatic benign pros-tatic hyperplasia (BPH). They not only improve uri-nary symptoms related to an enlarged prostate, they reduce the risk of acute urinary retention and the need for BPH-related surgery.
Although only 10 – 15% of men taking 5ARIs are likely to experience new or worsening sexual adverse events, the high prevalence of sexual dysfunction in elderly men makes this issue more challenging. It is one thing to tell a man with an elevated PSA and a negative biopsy that his risk of needing another biopsy, and potentially being diagnosed with cancer, will be reduced by taking a 5ARI. It is a more diffi cult prop-osition to expect an asymptomatic man with a mildly elevated PSA to take a medication to reduce the risk of a disease he would prefer not thinking about.
In summary, both fi nasteride and dutasteride have been shown to reduce the risk of biopsy-detectable prostate cancer. The concern over induction of high-grade cancers and lack of regulatory approval have prevented their widespread use for this indication. Dutasteride is now being submitted for approval worldwide for prostate cancer risk reduction. In the near future we will learn if it meets the strict benefi t:risk balance required for a cancer risk reduction indication.
Future directions
There have been many epidemiological or retrospec-tive studies suggesting that different dietary factors, supplements or medications may reduce the risk of prostate cancer [51]. For example, there has been

134 R. S. Rittmaster
increased emphasis on statins as a potential class of drugs for prostate cancer risk reduction. However, the data on primary prevention of prostate cancer with statins has been less convincing than their effects on reducing the risk of prostate cancer progression or advanced prostate cancer [52,53]. Complicating the interpretation of these studies is evidence that statins suppress PSA. It is impossible to be certain whether this represents a primary effect on prostate cancer or a primary effect on PSA leading to an ascertainment bias in identifying disease occurrence or progression. The stakes are high, because appro-priately designed and powered studies on prostate cancer risk reduction are expensive and take many years to achieve defi nitive results.
If 5 α -reductase inhibitors are going to be widely accepted for prostate cancer risk reduction, they will need the approval of regulatory agencies. Continued research is also needed on the issue of high-grade cancers. With respect to dutasteride, the 3-year REDEEM study, comparing dutasteride to placebo in 302 men with low volume, Gleason 6 cancers being followed by active surveillance [54], will pro-vide data on the rate of upgrading to higher Gleason scores, as well as the utility of dutasteride in an active surveillance setting. This results of this study are scheduled to be published in early 2011. There still remains many unanswered questions regarding 5 α -reductase inhibitors, such as whether they can be safely used in men who have never had a biopsy in order to better select men for biopsy, the ideal age at which such therapy should be initiated for prostate cancer risk reduction, and whether they should be continued indefi nitely or stopped after a man reaches a certain age.
Prostate cancer can now be counted among the few cancers whose risk can be lowered through dietary or medicinal means. The prospects are excellent for improved management of this disease, especially with respect to the overdiagnosis and overtreatment of indolent prostate cancers that are unlikely to cause death or disability in the absence of treatment.
Declaration of interest statement . Dr. Rittmaster is an employee of GlaxoSmithKline, Inc. Glaxo-SmithKline is the manufacturer of dutasteride. Dutas-teride and fi nasteride are approved for the treatment of benign prostatic hyperplasia. Neither medication is approved for prostate cancer risk reduction.
References
Sakr WA, Grignon DJ, Crissman JD, Heilbrun LK, [1] Cassin BJ, Pontes JJ, et al. High-grade prostatic intraepithe-lial neoplasia (HGPIN) and prostatic adenocarcinoma
between the ages of 20 – 69: An autopsy study of 249 cases. In Vivo 1994;8:439 – 43. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics CA Cancer [2] J Clin 2010 Jul 7. Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia D, [3] Church TR, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009;360:1310 – 9. Rittmaster RS, Fleshner NE, Thompson IM. Pharmacologi-[4] cal approaches to reducing the risk of prostate cancer. Eur Urol 2009;55:1064 – 73. Nelson WG. Prostate cancer prevention. Curr Opin Urol [5] 2007;17:157 – 67. Prins GS, Korach KS. The role of estrogens and estrogen [6] receptors in normal prostate growth and disease. Steroids 2008;73:233 – 44. Ricke WA, Wang Y, Cunha GR. Steroid hormones and car-[7] cinogenesis of the prostate: The role of estrogens. Differen-tiation 2007;75:871 – 82. Prostate disease trial to study 3,000 MTOPS. Clinical [8] Operations Dept News Release. 1996. The Alpha-Tocopherol BCCPSG. The effect of vitamin E [9] and beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Caro-tene Cancer Prevention Study Group. N Engl J Med 1994;330:1029 – 35. Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thomp-[10] son IM, Ford LG, et al. Effect of Selenium and Vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2008 Dec 9. Gaziano JM, Glynn RJ, Christen WG, Kurth T, Belanger C, [11] MacFadyen J, et al. Vitamins E and C in the prevention of prostate and total cancer in men: The Physicians ’ Health Study II randomized controlled trial. JAMA 2009;301:52 – 62. Marshall JR, Sakr W, Wood D, Berry D, Tangen C, Parker F, [12] et al. Design and progress of a trial of selenium to prevent prostate cancer among men with high-grade prostatic intraepithelial neoplasia. Cancer Epidemiol Biomarkers Prev 2006;15:1479 – 84. Grino PB, Griffi n JE, Wilson JD. Testosterone at high con-[13] centrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 1990;126:1165 – 72. Wright AS, Thomas LN, Douglas RC, Lazier CB, Rittmaster [14] RS. Relative potency of testosterone and dihydrotestosterone in preventing atrophy and apoptosis in the prostate of the castrated rat. J Clin Invest 1996;98:2558 – 63. Wright AS, Douglas RC, Thomas LN, Lazier CB, Rittmaster [15] RS. Androgen-induced regrowth in the castrated rat ventral prostate: Role of 5alpha-reductase. Endocrinology 1999;140:4509 – 15. Imperato-McGinley J, Gautier T, Zirinsky K, Hom T, [16] Palomo O, Stein E, et al. Prostate visualization studies in males homozygous and heterozygous for 5 alpha-reductase defi ciency. J Clin Endocrinol Metab 1992;75:1022 – 6. Luo J, Dunn TA, Ewing CM, Walsh PC, Isaacs WB. Decreased [17] gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: Implications for fi nasteride therapy of pros-tate carcinoma. Prostate 2003;57:134 – 9. Thomas LN, Douglas RC, Lazier CB, Too CK, Rittmaster RS, [18] Tindall DJ. Type 1 and type 2 5alpha-reductase expression in the development and progression of prostate cancer. Eur Urol 2008;53:244 – 52. Gravas S, Oelke M. Current status of 5alpha-reductase [19] inhibitors in the management of lower urinary tract symp-toms and BPH. World J Urol 2010;28:9 – 15.

Chemoprevention of prostate cancer 135
Feigl P, Blumenstein B, Thompson I, Crowley J, Wolf M, [20] Kramer BS, et al. Design of the Prostate Cancer Prevention Trial (PCPT). Control Clin Trials 1995;16:150 – 63. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, [21] Miller GJ, Ford LG, et al. The infl uence of fi nasteride on the development of prostate cancer. N Engl J Med 2003;349:215 – 24. Thompson IM, Chi C, Ankerst DP, Goodman PJ, Tangen [22] CM, Lippman SM, et al. Effect of fi nasteride on the sensitiv-ity of PSA for detecting prostate cancer. J Natl Cancer Inst 2006;98:1128 – 33. Thompson IM, Tangen CM, Goodman PJ, Lucia MS, Parnes [23] HL, Lippman SM, et al. Finasteride improves the sensitivity of digital rectal examination for prostate cancer detection. J Urol 2007;177:1749 – 52. Lucia MS, Epstein JI, Goodman PJ, Darke AK, Reuter VE, [24] Civantos F, et al. Finasteride and high-grade prostate cancer in the Prostate Cancer Prevention Trial. J Natl Cancer Inst 2007;99:1375 – 83. Basillote JB, Armenakas NA, Hochberg DA, Fracchia JA. [25] Infl uence of prostate volume in the detection of prostate can-cer. Urology 2003;61:167 – 71. Serfl ing R, Shulman M, Thompson GL, Xiao Z, Benaim E, [26] Roehrborn CG, et al. Quantifying the impact of prostate volumes, number of biopsy cores and 5alpha-reductase inhibitor therapy on the probability of prostate cancer detection using mathematical modeling. J Urol 2007;177:2352 – 6. Cohen YC, Liu KS, Heyden NL, Carides AD, Anderson [27] KM, Daifotis AG, et al. Detection bias due to the effect of fi nasteride on prostate volume: A modeling approach for analysis of the Prostate Cancer Prevention Trial. J Natl Cancer Inst 2007;99:1366 – 74. Kaplan SA, Roehrborn CG, Meehan AG, Liu KS, Carides [28] AD, Binkowitz BS, et al. PCPT: Evidence that fi nasteride reduces risk of most frequently detected intermediate- and high-grade (Gleason score 6 and 7) cancer. Urology 2009;73:935 – 9. Pinsky P, Parnes H, Ford L. Estimating rates of true high-[29] grade disease in the Prostate Cancer Prevention Trial. Cancer Prevent Res 2008;1:182 – 6. Redman MW, Tangen CM, Goodman PJ, Lucia MS, [30] Coltman CA, Jr., Thompson IM. Finasteride does not increase the risk of high-grade prostate cancer: A bias-adjusted mode-ling approach. Cancer Prevent Res 2008;1:182 – 6. Walsh PC. Re: The Prostate Cancer Prevention Trial: Design, [31] biases and interpretation of study results. Goodman PJ, Thompson IM, Jr., Tangen CM, Crowley JJ, Ford LG, Colt-man CA, Jr., J Urol 2006;175:2234 – 42. J Urol 2006;176:2744; author reply. Thomas LN, Douglas RC, Lazier CB, Gupta R, Norman [32] RW, Murphy PR, et al. Levels of 5alpha-reductase type 1 and type 2 are increased in localized high-grade compared to low-grade prostate cancer. J Urol 2008;179:147 – 51. Andriole GL, Roehrborn C, Schulman C, Slawin KM, [33] Somerville M, Rittmaster RS. Effect of dutasteride on the detection of prostate cancer in men with benign prostatic hyperplasia. Urology 2004;64:537 – 41. Andriole G, Bautista M, Crawford D, Kusec J, McConnell [34] JD, Lucia S, et al. Prostate cancer (CAP) detection in the medical therapy of prostatic symptoms (MTOPS) trial. J Urol 2003;169:120 Abstract. Andriole GL, Guess HA, Epstein JI, Wise H, Kadmon D, [35] Crawford ED, et al. Treatment with fi nasteride preserves use-fulness of prostate-specifi c antigen in the detection of prostate cancer: Results of a randomized, double-blind, placebo-con-
trolled clinical trial. PLESS Study Group. Proscar long-term effi cacy and safety study. Urology 1998;52:195 – 201. Andriole G, Bostwick D, Brawley O, Gomella L, Marberger [36] M, Tindall D, et al. Chemoprevention of prostate cancer in men at high risk: Rationale and design of the reduction by dutasteride of prostate cancer events (REDUCE) trial. J Urol 2004;172:1314 – 7. Andriole GL, Bostwick DG, Brawley OW, Gomella LG, [37] Marberger M, Montorsi F, et al. Effect of dutasteride on the risk of prostate cancer. N Engl J Med 2010;362:1192 – 202. Choo R, Danjoux C, Morton G, Szumacher E, Sugar L, [38] Gardner S, et al. How much does Gleason grade of follow-up biopsy differ from that of initial biopsy in untreated, Gleason score 4-7, clinically localized prostate cancer? Prostate 2007;67:1614 – 20. Roehrborn CG, Siami P, Barkin J, Damiao R, Major-Walker [39] K, Nandy I, et al. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol 2010;57:123 – 31. Andriole G, Bostwick D, Brawley OW, Gomella L, [40] Marberger M, Montorsi F, et al. The effect of dutasteride on the utility of PSA for the diagnosis of high-grade and clinically relevant prostate cancer in men with a previous negative biopsy: Results from the REDUCE study. J Urol 2011 (in press). Thompson IM, Lucia MS, Redman MW, Darke A, La Rosa [41] FG, Parnes HL, et al. Finasteride decreases the risk of prostatic intraepithelial neoplasia. J Urol 2007;178:107 – 9; Discussion 10. Rittmaster R, Montorsi F, Marberger M, Roehrborn C, [42] Andriole G, Somerville M, et al. Reduction in surgical and non-surgical interventions for prostate cancer with dutas-teride treatment in the REduction by DUtasteride of prostate Cancer Events (REDUCE) Study (abstract). Eur Soc Med Oncol 2010 Milan. Nickel JC. Comparison of clinical trials with fi nasteride and [43] dutasteride. Rev Urol 2004;6(Suppl 9):S31 – S9. Moinpour CM, Darke AK, Donaldson GW, Thompson IM, [44] Jr., Langley C, Ankerst DP, et al. Longitudinal analysis of sexual function reported by men in the Prostate Cancer Pre-vention Trial. J Natl Cancer Inst 2007;99:1025 – 35. Roehrborn CG, Lukkarinen O, Mark S, Siami P, Ramsdell [45] J, Zinner N. Long-term sustained improvement in symptoms of benign prostatic hyperplasia with the dual 5alpha-reduct-ase inhibitor dutasteride: Results of 4-year studies. BJU Int 2005;96:572 – 7. Schroder FH, Bangma CH, Roobol MJ. Is it necessary to [46] detect all prostate cancers in men with serum PSA levels � 3.0 ng/ml? A comparison of biopsy results of PCPT and outcome-related information from ERSPC. Eur Urol 2008;53:901 – 8. Schroder FH, Roobol MJ, Andriole GL, Fleshner N. Defi n-[47] ing increased future risk for prostate cancer: Evidence from a population based screening cohort. J Urol 2009;181:69 – 74; Discussion. Thompson IM, Pauler Ankerst D, Chi C, Goodman PJ, [48] Tangen CM, Lippman SM, et al. Prediction of prostate cancer for patients receiving fi nasteride: Results from the Prostate Cancer Prevention Trial. J Clin Oncol 2007;25:3076 – 81. Andriole G, Rittmaster RS. Dutasteride and prostate cancer [49] [letter to the editor]. N Engl J Med 2010;363:794 – 5. Walsh PC. Effect of dutasteride on the risk of prostate cancer [50] [editorial]. J Urol. 2010;194:174 – 5. Syed DN, Khan N, Afaq F, Mukhtar H. Chemoprevention [51] of prostate cancer through dietary agents: Progress and

136 R. S. Rittmaster
hormone therapy for prostate cancer. J Clin Oncol 2010;28:2651 – 2. Fleshner N, Gomella LG, Cookson MS, Finelli A, Evans A, [54] Taneja SS, et al. Delay in the progression of low-risk prostate cancer: Rationale and design of the Reduction by Dutasteride of Clinical Progression Events in Expectant Management (REDEEM) trial. Contemp Clin Trials 2007;28:763 – 9.
promise. Cancer Epidemiol Biomarkers Prev 2007;16:2193 – 203. Platz EA. Epidemiologic musing on statin drugs in the pre-[52] vention of advanced prostate cancer. Cancer Epidemiol Biomarkers Prev 2007;16:2175 – 80. D’Amico AV. Statin use and the risk of prostate-specifi c [53] antigen recurrence after radiation therapy with or without

Correspondence: Anders Ull é n, Department of Oncology-Pathology, Karolinska Institutet, Karolinska University Hospital Solna, Radiumhemmet, 17176 Stockholm, Sweden. E-mail: [email protected]
(Received 11 August 2010 ; accepted 23 September 2010 )
EDITORIAL
Management of advanced prostate cancer – new drugs
ANDERS ULL É N 1 , CARL-HENRIK SHAH 1 & ZIYA KIRKALI 2
1 Department of Oncology-Pathology Karolinska Institutet, Stockholm, Sweden and 2 Department of Urology, Dokuz Eylul University School of Medicine, Izmir, Turkey
Androgen deprivation therapy by chemical or surgical castration remains the cornerstone in the manage-ment of advanced disease. Although initially effective, the effect of currently available androgen deprivation therapies is transient and most patients develop pro-gressive disease despite low levels of testosterone. It is generally believed that tumour progression is asso-ciated with continued signalling via the androgen receptor pathway through mechanisms such as andro-gen receptor mutation, androgen receptor amplifi ca-tion, ligand-independent androgen receptor activation or enhanced local production of androgens. The rec-ognition of the antiandrogen withdrawal response, which demonstrates the continuous importance of the androgen receptor signalling pathway in castrate resistant prostate cancer, has stimulated the develop-ment of new exciting androgen targeting therapies. MDV3100, which is a nonsteroidal compound and RD162, are examples of two new orally available interesting agents that target the androgen receptor with higher affi nity than bicalutamide [1]. MDV3100 has shown promising phase I/II activity [2] and is currently evaluated in a placebo-controlled ran-domised phase III trial in patients progressive on docetaxel therapy. EPI-001 represents from a mech-anistic point-of-view, a conceptually new and inter-esting compound. It binds to the N-terminal domain of the androgen receptor and has a new mode of action as it targets the transactivation of the andro-gen receptor, regardless of the presence of androgen [3,4]. Other efforts are made to further improve the blockage of testosterone production. Abiraterone acetate is an interesting, potent small molecule that irreversibly inhibits cytochrome p17 which catalyzes two key reactions in androgen biosynthesis [5]. This compound has been evaluated in a phase III trial in
castrate-resistant patients previously treated with docetaxel (results are pending) and a second phase III trial is ongoing in patients who have not received prior ketoconazole or chemotherapy.
Since the data from the TAX 327 [6] and SWOG 9916 [7] trials were presented in 2004, every-three-week docetaxel has become a standard treatment for patients with castrate resistant prostate cancer. Indeed, these were the fi rst trials demonstrating signifi cant and principally important overall survival benefi t using cytotoxic therapy for this patient category. Although signifi cant, the survival benefi ts achieved by docetaxel-based therapy are rather small, improving the overall median survival compared to mitoxan-trone with approximately three months, to less than 20 months [6,7]. Clearly, more effective regimens continue to be needed.
The last years, efforts have been made to improve the effects of docetaxel by either adding an agent to docetaxel/prednisone, develop more effective front line therapy or to fi nd an effective second line ther-apy. Recently cabazitaxel, a novel taxane with a favourable low affi nity to PGP (multidrug resis-tance P-glycoprotein) in combination with predni-sone was approved by the FDA following a phase III trial demonstrating an overall survival benefi t of 2.4 months as compared to mitoxantrone, in patients previously treated with docetaxel-containing che-motherapy [8]. If approved also by EMA, cabazi-taxel is the reasonable second line option and furthermore the comparator for subsequent second line trials. The epothilone analogues are examples of other chemotherapeutics with promising phase I/II data under current evaluation [9,10].
Our understanding of the complex molecular pathogenesis of prostate cancer has continued to
Acta Oncologica, 2011; 50(Suppl 1): 137–140
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.529458

138 A. Ull é n et al.
expand and several novel drugs that target specifi c molecules in pathways involved in cell signalling, proliferation, apoptosis, angiogenesis, and immune modulation are also under intensive investigation. Examples of candidate progression pathways include epidermal growth factor (EGFR) signalling, vascular endothelial growth factor (VEGF) signalling path-ways, phosphatidylinositol 3-kinase (PI3K) / Akt mammalian target of rapamycin (mTOR) pathway as well as the insulin-like growth factor pathway.
Multiple tyrosine kinase inhibitors with a typical antiangiogenic profi le, such as sunitinib and sorafenib are evaluated in prostate cancer but data are yet limited. So far, their effi cacy appears to be modest or the toxicity signifi cant [11 – 15]. Of note are the reported discordant radiological and PSA evalua-tions, indicating that PSA might not be valuable as a marker for response for these compounds and furthermore that the antitumoural effects caused by these substances in prostate cancer are not com-pletely understood [11 – 15]. The anti-VEGF anti-body bevacizumab has been evaluated in several phase II studies with promising results when given in combination with docetaxel regimens. An awaited large phase III frontline trial, CALGB 90401, was recently reported evaluating the addition of bevaci-zumab to standard docetaxel regimens. Unfortu-nately, this trial failed to meet the primary endpoint of demonstrating an overall survival benefi t follow-ing the addition of bevacizumab [16] and the place for this compound in the treatment arsenal remains unclear.
Src and src-family kinases represent interesting target molecules since they are involved in multiple signalling pathways central to prostate cancer devel-opment and in the pathogenesis of bone metastases. For example is the value of adding the src-inhibitor dasatinib, which has also activity against bcr/abl, to standard docetaxel regimen, under evaluation in an ongoing phase III trial [17]. Other examples of novel targeted agents under investigation in advanced pros-tate cancer include the mTOR inhibitors everolimus and temsirolimus, various inhibitors of IGF-1R and custirsen (OGX-011), an innovative antisense oligo-nucleotide directed against the cytoprotective chap-erone, clusterin [18].
Histone deacetylase inhibitors such as panobinos-tat (LBH589) are currently evaluated and represent principally interesting approaches to target tumour-induced epigenetic aberrations believed to be critical for androgen receptor mediated signalling [19].
In addition to targeted therapies, the slowly-growing nature and high expression of tumour-associated antigens of prostate cancer have stimulated the development of several immunotherapeutic approa ches. Just some months ago, FDA approved
the fi rst therapeutic vaccine ever, sipuleucel-T, for asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer [20,21]. Sipuleucel-T consists of autologous dendritic cells derived from a patient ’ s own peripheral blood mononuclear cells. The dendritic cells are collected through leukaphere-sis and exposed to a recombinant fusion protein com-posed of the tumour associated antigen PAP (prostatic acid antigen) linked to granulocyte-macrophage col-ony stimulating factor [20,21]. In the pivotal phase III trial, patients receiving sipuleucel-T had a median overall survival of 25.8 months compared to 21.7 for the patients receiving placebo, without any substan-tial side-effects related to the vaccine [21]. Other examples of vaccine approaches under evaluation are vector-based strategies (Prostvac) [22] or whole tumour cell vaccines (GVAX), the latter however with recently reported negative phase III trials [23,24]. Ipilipumab, an anti-CTLA4 monoclonal antibody, represents a principally different immunotherapeutic approach under evaluation since treatment with ipilipumab is supposed to enhance T-cell activation. Phase I and II data have indicated activity [25,26] and phase III studies are under way.
Several agents targeting the bone with different mechanisms of action are under intensive investiga-tion. Denosumab, a fully human monoclonal anti-body that specifi cally bind to the ligand of RANK has recently been approved by the EMA for the treatment of bone loss associated with hormone ablation in men with prostate cancer at increased risk of fractures [27]. Furthermore, denosumab recently was reported superiority over zoledronic acid in delaying or pre-venting SREs (skeletal related events) in patients with bone metastases from castrate resistant prostate can-cer [28]. Radium-223 is a new promising alpha-emitting bone-seeking radiopharmaceutical which has proven well tolerated with demonstrated effects on bone-ALP concentrations and survival parameters in a randomised phase II study [29]. A subsequent phase III study is running and in late planning phase is a trial evaluating combined treatment with doc-etaxel. Finally, a number of endothelin A receptor (ETa) targeted agents are under evaluation, which rely on the importance of this receptor in the devel-opment of prostate cancer progression and formation of bone metastases. Atrasentan and ZD4054 are agents, proven to selectively inhibit ETa, with encour-aging phase II data that are under extensive evalua-tion in several large, randomised phase III trials.
Considering the large number of compounds under evaluation, the next decade appears to be an exciting time for prostate cancer research and management of advanced disease. The emergence of therapies thatrely on improved molecular understanding of bio-logical processes behind prostate cancer progression

Advanced prostate cancer - new drugs 139
and therapeutic resistance is promising and stimulate to further research efforts.
How to best combine and evaluate signifi cantly different treatments modalities such as radionu-clides, vaccines, hormonal-, cytotoxic-, targeted- and other immunomodulatory treatments in terms of sequence, dosages, etc are examples of impor-tant issues that remain unclear and obviously will require signifi cant work and multidisciplinary col-laboration. Another important challenge is to strive for biomarker driven trials with the aim to clarify and defi ne if certain populations, based on their tumour biological and/or clinical characteristics, are particularly sensitive to any of these treatment alternatives. By use of such strategies, we can better tailor and optimise the treatment of the individual patient while improving the cost/benefi t for the soci-ety as a whole.
References
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. [1] Development of a second-generation antiandrogen for treat-ment of advanced prostate cancer. Science 2009;324(5928):787 – 90. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME,[2] Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1-2 study. Lan-cet 2010;375(9724):1437 – 46. Skinner M. Prostate cancer: A new tARget. Nat Rev Cancer [3] 2010;10:534 – 5. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung [4] JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010;17:535 – 46. Attard G, Reid AH, A’Hern R, Parker C, Babu Oommen N, [5] Folkerd E, et al. Selective inhibition of CYP17 with abirater-one acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol 2009;27:3742 – 8. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi [6] KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502 – 12. Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones [7] JAM, Taplin ME, et al. Docetaxel and estramustine com-pared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004;351:1513 – 20. de Bono JS, Ozguroglu M, Hansen S, Machiels JH, Shen L. [8] Cabazitaxel or mitoxantrone with prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) previously treated with docetaxel: Final results of a multina-tional phase III trial (TROPIC). J Clin Oncol 2010;28:15s(Suppl; abstract 4508). Galsky MD, Small EJ, Oh WK, Chen I, Smith DC, Colevas [9] AC, et al. Multi-institutional randomized phase II trial of the epothilone B analog ixabepilone (BMS-247550) with or without estramustine phosphate in patients with progressive castrate metastatic prostate cancer. J Clin Oncol 2005;23:1439 – 46. Rosenberg JE, Ryan CJ, Weinberg VK, Smith DC, Hussain M, [10] Beer TM, et al. Phase I study of ixabepilone, mitoxantrone, and prednisone in patients with metastatic castration-resistant
prostate cancer previously treated with docetaxel-based ther-apy: A study of the department of defence prostate cancer clinical trials consortium. J Clin Oncol 2009;27:2772 – 8. Aragon-Ching JB, Jain L, Gulley JL, Arlen PM, Wright JJ, [11] Steinberg SM, et al. Final analysis of a phase II trial using sorafenib for metastatic castration-resistant prostate cancer. BJU Int 2009;103:1636 – 40. Chi KN, Ellard SL, Hotte SJ, Czaykowski P, Moore M, [12] Ruether JD, et al. A phase II study of sorafenib in patients with chemo-naive castration-resistant prostate cancer. Ann Oncol 2008;19:746 – 51. Dahut WL, Scripture C, Posadas E, Jain L, Gulley JL, Arlen [13] PM, et al. A phase II clinical trial of sorafenib in androgen-independent prostate cancer. Clin Cancer Res 2008;14:209 – 14. Dror Michaelson M, Regan MM, Oh WK, Kaufman DS, [14] Olivier K, Michaelson SZ, et al. Phase II study of sunitinib in men with advanced prostate cancer. Ann Oncol 2009;20:913 – 20. Sonpavde G, Periman PO, Bernold D, Weckstein D, Fleming [15] MT, Galsky MD, et al. Sunitinib malate for metastatic cas-tration-resistant prostate cancer following docetaxel-based chemotherapy. Ann Oncol 2010;21:319 – 24. Kelly WK, Halabi S, Carducci DJ, George DJ, Mahoney JF, [16] Stadler WM, et al. A randomized, double-blind, placebo-controlled phase III trial comparing docetaxel, prednisone and placebo with docetaxel, prednisone and bevacizumab in men with metastatic castration-resistant prostate cancer (mCRPC): Survival results of CALBG 90401. J Clin Oncol 2010;28:18s(Suppl. abstract LBA4511). National Institutes of Health. A randomized double-blind [17] phase III trial comparing docetaxel combined with dasatinib compared to docetaxel combined with placebo in castrate-resistant prostate cancer. NCT00744497. Zoubeidi A, Chi K, Gleave M. Targeting the cytoprotective [18] chaperone, clusterin, for treatment of advanced cancer. Clin Cancer Res 2010;16:1088 – 93. Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, [19] et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res 2009;69:958 – 66. Higano CS, Small EJ, Schellhammer P, Yasothan U, [20] Gubernick S, Kirkpatrick P, et al. Sipuleucel-T. Nat Rev Drug Discov 2010;9:513 – 4. Patel PH, Kockler DR. Sipuleucel-T: A vaccine for meta-[21] static, asymptomatic, androgen-independent prostate cancer. Ann Pharmacother 2008;42:91 – 8. Madan RA, Arlen PM, Mohebtash M, Hodge JW, [22] Gulley JL. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin Investig Drugs 2009;18:1001 – 11. National Institutes of Health. Docetaxel in combination with [23] GVAX ® immunotherapy versus docetaxel and prednisone in prostate cancer patients (VITAL-2). NCT00133224. National Institutes of Health. GVAX [24] ® Vaccine for Prostate Cancer vs Docetaxel and Prednisone in patients with meta-static hormone refractory prostate cancer (VITAL-1). NCT00089856. Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, [25] Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res 2007;13:1810 – 5. Slovin SF BT, Higano CS. Initial phase II experience of [26] ipilimumab (IPI) alone and in combination with radiother-apy (XRT) in patients with metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol 2009;27:15s (abstr 5138).

140 A. Ull é n et al.
Smith MR, Egerdie B, Hernandez Toriz N, Feldman R, [27] Tammela TL, Saad F. Denosumab in men receiving andro-gen-deprivation therapy for prostate cancer. N Engl J Med 2009;361:745 – 55. Fizazi K, Carducci M, Smith M. A randomized phase III trial [28] of denosumab versus zoledronic acid in patients with bone
metastases from castrate-resistant prostate cancer. J Clin Oncol 2010;28:18s (Suppl. abstract LBA 4507). Nilsson S, Franzen L, Parker C, Tyrrell C, Blom R, Tennvall J, [29] et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: A randomised, multicentre, placebo-controlled phase II study. Lancet Oncol 2007;8:587 – 94.

Correspondence: Ian F. Tannock, Princess Margaret Hospital, Suite 5-208, 610 University Avenue, Toronto, ON M5G 2M9, Canada. E-mail: [email protected]
(Received 10 August 2010 ; accepted 6 September 2010 )
REVIEW ARTICLE
Broadening horizons in medical management of prostate cancer
SAROJ NIRAULA & IAN F. TANNOCK
Division of Hematology and Medical Oncology, Princess Margaret Hospital and University of Toronto, Canada
Abstract Hormonal therapy . Testosterone suppression achieved either medically or surgically is the standard initial treatment for men with advanced prostate cancer. Most men respond but the disease progresses after a median of 1 – 2 years. Clinical trials suggest that intermittent androgen deprivation therapy (ADT) provides equal or longer time to castration-independence than continuous ADT, and is preferred, especially since there are subtle long-term toxicities associated with ADT. Further hormonal manipulations (including addition and withdrawal of peripheral antiandrogens, steroid synthesis inhibitors such as ketoconazole, and estrogens) can be transiently effective in selected patients with castration-resistant prostate cancer (CRPC). Androgen-dependent signalling pathways remain active in most men with CRPC and are associated with muta-tion, changes in expression or modulation of the androgen receptor (AR); abiraterone acetate and MDV3100 are promis-ing drugs being evaluated in clinical trials that may lead to further hormonal response. Chemotherapy . Eventually men who progress rapidly, are symptomatic, and/or develop metastasis to visceral organs require chemotherapy. Three-weekly docetaxel with prednisone has been shown to improve survival and relieve symptoms but eventually men develop progres-sive disease or become intolerant to docetaxel. Multiple trials are evaluating new drugs (mainly molecular targeted agents) either given fi rst line with docetaxel chemotherapy, or to men who have progressive disease after receiving docetaxel. Cabazitaxel was shown recently to improve survival as compared to mitoxantrone when used second line and has been approved by the United States Food and Drug Administration (FDA). Conclusion . Despite major advances, treatment of men with advanced CRPC remains a challenge both for the seeker and giver of care.
Dr. Charles Huggins was awarded the 1966 Nobel prize in physiology and medicine for his discovery in the 1940s that hormones (specifi cally estrogens that led to decreased testosterone) could control the spread of prostate cancer. Orchiectomy became a standard treatment (since estrogens were associ-ated with cardiac toxicity) and in the 1980s Leu-tinising Hormone Releasing Hormone (LHRH) agonists were introduced as a medical alternative. Medical or surgical castration remains the standard initial treatment for men with advanced prostate cancer. Despite clinical and biochemical response in � 80% of patients, mean duration of response to initial androgen deprivation therapy (ADT) is around 18 to 24 months, but is highly variable depending on the biological behaviour of the tumour. Some men with castration-resistant pros-tate cancer (CRPC) may respond transiently to further hormonal manipulations but eventually become resistant. Current standards for managing
these patients and evolving treatment strategies are described below.
Hormonal treatment
Two variants of initial ADT have been investigated in multiple randomised trials: combined androgen blockade (CAB) and intermittent hormonal therapy. Studies have shown that initial CAB (orchiectomy or LHRH agonist with a peripheral anti-androgen) adds cost and toxicity without meaningful benefi t as compared to monotherapy or surgery alone [1] and this strategy should not be used. In contrast, emerg-ing data support the use of intermittent ADT, which is based on preclinical observations that intermittent suppression of testosterone delays the time to castra-tion independence in animal models [2]. Several tri-als have confi rmed that intermittent ADT, in which therapy is interrupted when the serum PSA falls to a low value and is reintroduced when it rises above
Acta Oncologica, 2011; 50(Suppl 1): 141–147
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.524936

142 S. Niraula & I. F. Tannock
a certain level, provides equal or superior duration of disease control compared with continuous ADT [3]. Intermittent ADT can now be regarded as stan-dard treatment for men with a good initial response to treatment, especially as there are evolving data showing subtle long-term toxicities associated with ADT, including bone loss, metabolic syndrome and cardiac events [3 – 5] (Table I).
Awareness of the chronic toxicities associated with ADT that are summarised in Table I should lead to caution in introducing such therapy, especially in men with predisposing factors, such as a history of diabetes or cardiovascular events. While men with metastatic disease who are symptomatic or have a rapidly rising PSA should be treated, there is no evi-dence to support initiation of ADT in men with slowly rising PSA after initial local therapy, or those with slowly progressive asymptomatic disease. All men on ADT should be evaluated for bone density, with routine co-administration of calcium and vita-min D, and selective use of bisphosphonates in those with continuing bone loss [4].
Some men whose disease is progressing after pri-mary ADT may achieve further transient response by adding a peripheral anti-androgen. About 20% of those that respond to addition of an anti-androgen may respond to its withdrawal, because the classical agents (fl utamide, bicalutamide and nilutamide) can initially inhibit but subsequently stimulate the andro-gen receptor (AR) [6]. Further responses to hormonal manipulations can be obtained in some men by treat-ment with agents that inhibit synthesis of adrenal androgens (e.g. ketoconazole used with hydrocorti-sone), estrogens, glucocorticoids, and by switching from one peripheral anti-androgen to another.
Androgen-dependent signalling in men with CRPC
Despite the high rate of initial response, men eventu-ally develop resistance to the above forms of hor-monal therapy via mechanisms that include persistent signalling through the AR, mutation or changes in the expression or modulation of the AR, and activity of alternate cellular pathways that stimulate proliferation
of prostate cancer cells [7]. Even in the castrate state, substantial levels of androgens may be produced within prostate tumour tissue and can stimulate the AR [8]. Around one quarter of prostate cancers in men with CRPC have been reported to carry a point mutation, especially Thr-Ala877 [9], resulting in acti-vation by commonly used hormonal agents such as peripheral anti-androgens and estrogens. The AR-dependent signalling pathways have also been shown to cross talk with other growth signalling pathways in prostate cancer cells including those dependent on epidermal growth factors, vascular endothelial growth factor, fi broblast growth factors and transforming growth factor, thereby leading to mechanisms of cell survival despite the castrate state [7]. Two novel agents that target AR-associated signalling in men who are resistant to current forms of hormonal manipulation, abiraterone acetate and MDV 3100, are being evaluated in Phase III clinical trials.
Abiraterone acetate . Abiraterone acetate (hereafter ‘ abi-raterone ’ ) is an orally active compound that functions by irreversibly inhibiting enzymatic activity of 17-alpha mono-oxygenase (17 alpha-hydrolase/C17,20 lyase complex), a member of the cytochrome p450 family. This enzyme catalyses the 17-alpha hydroxyla-tion of steroids, required in two key steps for the con-version of cholesterol to androgens. Hence, abiraterone suppresses androgen synthesis in the testis, the adre-nal glands and in other sites, including prostatic tissue. Abiraterone is reported to be well tolerated with easily managed side effects such as hypertension and kypokalemia, which can be reduced by co-administra-tion of low-dose prednisone. In a multinational Phase II trial [10], which recruited 58 men with CRPC after docetaxel chemotherapy, abiraterone was associated with PSA response (i.e. 50% or greater maximal decline) in 43% of men including 30% of those pre-treated with ketoconazole and 55% who were keto-conazole-na ï ve. Another post-chemotherapy Phase II trial reported PSA response in 24 (51%) of 47 patients. In both these trials treatment with abiraterone was associated with reduction in circulating tumour cell (CTC) counts – a marker reported to be predictive of survival of men with metastatic CRPC [11]. High baseline serum levels of dehydroepiandrostenedione (DHEA), DHEA-sulfate (DHEA-S) and estradiol have been recognised as markers predicting biochem-ical response and time to progression after abiraterone [12]. Large multinational Phase III studies have com-pleted recruitment including a double-blind placebo-controlled trial of abiraterone and prednisone versus prednisone alone in men with CRPC who have pro-gressed after receiving docetaxel, and a related trial for chemotherapy-na ï ve men with CRPC who were min-imally symptomatic.
Table I. Toxicities associated with ADT reported in clinical trials.
Decrease in bone mineral density, leading to increased risk of fracture
Loss of muscle mass Increase in body fat, especially abdominal subcutaneous fat Gynecomastia Impotence Hot fl ashes and symptoms of male menopause Anaemia Increased risk of diabetes Increased risk of coronary artery disease Increased risk of sudden cardiac death

New drugs for prostate cancer 143
MDV3100 . MDV3100 is an orally available potent antagonist of the AR that inhibits nuclear transloca-tion, co-activator peptide recruitment and DNA binding, and lacks agonist activity [13]. A Phase I/II study in 140 men with progressive CRPC showed an impressive PSA response rate in 57% (37/65) of chemotherapy-na ï ve men and 45% (22/49) in men who had received chemotherapy [14]. The authors also reported that 92% of patients who had CTC counts � 5 (favourable) at baseline maintained favourable CTC counts post-treatment and 53% (21/40) of those with � 5 CTC (unfavourable) con-verted to favourable CTC counts post-treatment, indicating a promising role of this agent in treatment of prostate cancer. A Phase III placebo-controlled trial with 2:1 randomisation in favour of MDV3100 in the post-chemotherapy setting is underway with overall survival as the primary endpoint.
Chemotherapy
Mitoxantrone, used with low-dose prednisone, was the fi rst chemotherapy drug approved for treatment of men with CRPC, on the basis of improvement in symptoms (especially pain) in randomised trials compared to prednisone alone [15,16]. Mitoxan-trone has the advantage of low toxicity, provided the total dose is limited to prevent cardiac toxicity, and is well tolerated even by very old men. There were no trends to improved survival in the mitox-antrone trials, although they were too small to detect them. Subsequently the TAX-327 trial demon-strated that docetaxel, given every three weeks at 75 mg/m 2 with prednisone 5 mg bid prolonged overall survival of men with CRPC by about three months, as compared to mitoxantrone and predni-sone, and improved the quality of life of symptom-atic men [17]. Surprisingly weekly docetaxel was not as effective, and not less toxic, in the 3-arm TAX327 trial. The SWOG 99-16 trial [18] com-pared docetaxel and estramustine with mitoxantrone and prednisone: it also showed a small benefi t for the docetaxel arm, but together these trials suggest that estramustine adds toxicity without benefi t, and there is no basis for the continued use of this drug. Only about half of men with CRPC respond to docetaxel chemotherapy, and all of them eventu-ally discontinue this treatment because of toxicity or disease progression.
There are multiple causes of resistance to tax-anes including cellular causes related to drug uptake and retention in cells, and changes to its binding to and action to stabilise β -tubulin. The drug may also have limited distribution within solid tumour tissue. Preclinical evidence has suggested that tax-anes exert their antineoplastic activity in prostate
cancer partly by blocking AR signalling [19], so that overexpression, amplifi cation and mutation of the AR may also play a role in the development of a chemo-resistant state [20].
New types of chemotherapy, and molecular tar-geted agents are being (or have been) evaluated with the goal of either enhancing the effectiveness of doc-etaxel when used in combination, or of providing further benefi t when used in men whose disease has progressed during or after treatment with docetaxel. Some of these agents are described below.
Second-line chemotherapy
Retreatment with docetaxel . For men with CRPC who demonstrate clinical and/or biochemical response to docetaxel as fi rst-line chemotherapy, followed by a period off-treatment, re-introducing docetaxel is a reasonable option if the treatment was toler-ated well.
Mitoxantrone . About 15% of men with CRPC respond to mitoxantrone after progressing on docetaxel [21], and given the excellent tolerance of this agent, it is a reasonable choice of second-line chemotherapy in symptomatic men with acceptable cardiac function.
Satraplatin . Satraplatin is a third generation oral platinum compound which had shown activity in a Phase II trial. Consequently, a large (n � 950) randomised Phase III clinical trial compared satra-platin to placebo, each with low-dose prednisone in men with CRPC after fi rst line chemotherapy (only about half of the participants had received docetaxel) [22]. Risk of disease progression was reduced by satraplatin (Hazard Ratio, HR � 0.67; p � 0.001) with signifi cant reduction in time to pain progression but there was no difference in overall survival. The FDA and European Medicines Agency (EMEA) did not approve this drug for use in men with prostate cancer.
Cabazitaxel . Cabazitaxel is a semi-synthetic deriva-tive of the natural taxoid 10-deacetylbaccatin III and like docetaxel acts by binding and stabilising tubulin, resulting in the inhibition of microtubule depolymerisation and cell division, cell cycle arrest in the G2/M phase, and the inhibition of tumour cell proliferation. It differs from other taxanes in that it is a poor substrate for the membrane-associ-ated, multidrug resistance, P-glycoprotein effl ux pump and crosses the blood-brain barrier. A recent randomised controlled trial compared cabazitaxel (25 mg/m 2 ) with mitoxantrone (12 mg/m 2 ), each given with prednisone (10 mg/day) as second line

144 S. Niraula & I. F. Tannock
chemotherapy in 755 men with CRPC who had received docetaxel [23]. There was a statistically signifi cant difference in overall survival favouring cabazitaxel (HR � 0.70, p � 0.0001) with an increase in median survival of about 2.5 months. This led to approval of cabazitaxel by the FDA and the drug will probably also be approved by EMEA. However, this drug may exacerbate peripheral neuropathy, a common residual side effect in men post-docetaxel – so that careful selection of patients will be crucial. Also, there was a ∼ 5% rate of toxic death in the cabazitaxel arm of the randomised trial that might have been reduced by administering a lower dose. A full article describing this trial is awaited, and should address how long after treat-ment with docetaxel the patients were enrolled; as described above, many patients who respond initially to docetaxel and have a break from treatment may benefi t from re-treatment with docetaxel.
Ixabepilone and patupilone . Ixabepilone and patupil-one are water soluble epothilones with a related mechanism of action to taxanes. They exhibit their anti-neoplastic activity primarily by inhibition of microtubule formation. A randomised Phase II trial comparing ixabepilone and mitoxantrone with pred-nisone in docetaxel pre-treated men demonstrated PSA response rates of 17% and 20% respectively and overall survival of 9.8 months in the entire cohort [24]. Given this disappointing result, it seems unlikely that ixabepilone will be studied further in men with CRPC.
A Phase II study of patupilone in 45 patients (55% had previous taxane therapy) was well-tolerated but only 13% of patients had a PSA response and the median overall survival was 13.4 months [25]. A multicentre Canadian study evaluating patupilone in CRPC after fi rst line docetaxel is planned.
Molecular targeted agents
Multiple targeted agents have been evaluated in Phase II trials at various stages of disease in men with CRPC, and some of them have been (or are being) evaluated in Phase III randomised trials to determine whether they augment the activity of docetaxel. These trials are described briefl y below. However, as yet no molecular targeted agent (other than those acting via hormonal mechanisms) has shown suffi cient activity to warrant approval for treatment of men with prostate cancer.
Inhibitors of angiogenesis . Formation of blood vessels is a requirement for growth of solid tumours including prostate cancer, and agents targeting angiogenesis
are being evaluated. Bevacizumab, a monoclonal antibody blocking Vascular Endothelial Growth Factor-A (VEGF-A) has been approved for use with chemotherapy in people with advanced colorectal and lung cancer, on the basis of small improvements in survival. A recent randomised controlled trial evaluated bevacizumab with docetaxel and predni-sone for men with CRPC but did not show a sig-nifi cant survival difference as compared to docetaxel and prednisone alone [26].
A large trial comparing docetaxel and prednisone with or without afl ibercept, a potent inhibitor of VEGF-A, VEGF-B and placental growth factor, has completed accrual. Other inhibitors of angiogenesis such as sunitinib, and thalidomide and its analogues (e.g. lenolidamide), have shown activity in Phase II clinical trials in men with CRPC [27,28]. Ran-domised Phase III clinical trials evaluating sunitinib in post-chemotherapy patients and lenalidomide in combination with docetaxel are underway. One prob-lem with this approach might be the presence of redundant pathways that may stimulate angiogenesis, such that inhibitors of only one pathway may have limited effects.
Inhibitors of Endothelin-A . Endothelin-A antago-nists are reported to exhibit antitumour properties mainly by inhibition of cell proliferation, induction of apoptosis, decrease in osteoclastic bone resorp-tion and inhibition of VEGF. However a ran-domised trial evaluating atresantan, an orally available inhibitor of endothelin-A did not meet its endpoint of time to disease progression in men with CRPC [29]. ZD4054, another Endothelin-A antagonist, is being evaluated in a randomised Phase III clinical trial in non-metastatic CRPC with dual primary end - points of progression-free survival and overall survival.
Apoptotic pathways . Some agents that are known to stimulate apoptosis have been evaluated in combi-nation with chemotherapy for men with CRPC. Custirsen (OGX-011), an antisense oligonucleotide targeting clusterin gave encouraging results in a Phase II clinical trial in combination with docetaxel [30] leading to an ongoing Phase III clinical trial. Gataparsen sodium, an antisense oligonucleotide targeting survivin, and AT-101, an oral, pan-Bcl-2 inhibitor are being evaluated in Phase II clinical trials in combination with docetaxel and prednisone.
Inhibitors of other pathways . Agents targeting cell-survival pathways that have cross-talk with AR-dependent pathways have been evaluated in early phase clinical trials but the results are generally

New drugs for prostate cancer 145
disappointing. Studies of inhibitors of epidermal growth factor receptors (EGFR1 and 2; e.g. erlotinib, gefi tinib, trastuzumab and pertuzumab) alone or in combination with docetaxel have not been encour-aging. Similarly, the anti-interleukin-6 monoclonal antibody siltuximab (CNTO 328) led to increased mortality when combined with mitoxantrone as compared with mitoxantrone alone and the trial was prematurely terminated [31]. Despite discouraging results using targeted monoclonal antibodies, others including cetuximab, cixutumumab and fi gitu-mumab are being evaluated in Phase II clinical trials. A study of TKI258, an inhibitor of fi broblast growth factor (FGF) is recruiting men with CRPC focused at evaluating markers of FGF signalling in bone marrow and plasma.
Additional strategies
Immunomodulation . Men diagnosed with prostate cancer are generally old and may have multiple co-morbidities leading to poor tolerance of cytotoxic chemotherapy. Immuno-modulation might be a preferable treatment strategy for these men. Two Phase III clinical trials evaluated administration of a vaccine (GVAX) consisting of two allogeneic pros-tate cancer cell lines (PC3 and LNCaP) ,which were genetically modifi ed through adenoviral trans-fer to secrete granulocyte macrophage – colony-stim-ulating factor and lethally irradiated. Two randomised trials comparing GVAX to docetaxel/prednisone (VITAL1) and GVAX � docetaxel/prednisone to docetaxel/prednisone (VITAL2) failed to meet their primary end-point of an increase in survival and were terminated prematurely [32,33]. In a novel but complex approach, dendritic cells were taken from patients, transported to a centre where they were exposed to prostatic acid phosphatase (PAP), and transported back to be re-infused into the patient. A recent Phase III randomised clinical trial evaluat-ing this approach (known as Sipuleucel-T) in men with metastatic CRPC demonstrated a signifi cant survival advantage over placebo (median survival 25.8 versus 21.7 months; hazard ratio 0.78; p � 0.03) [34]. This led to approval of this vaccine by the FDA (not yet by EMEA). There may however be challenges in bringing such agents into clinical practice because of the cost and logistics associated with the production process.
Bisphosphonates and Denosomab . Bone metastasis in CRPC is associated with RANKL-mediated osteo-clast activation [35]. The potent bisphosphonate, zoledronic acid has demonstrated an effect to delay the time to skeletal related events (SREs: defi ned by time to pathologic fracture of the bones, need
for surgical/radiotherapeutic intervention or spinal cord compression) in men with CRPC and bone metastasis [36]. More recently, a fully human mono-clonal antibody against RANKL, denosumab was compared with zoledronate in a Phase III ran-domised controlled clinical trial with 1901 patients. There was delayed time to fi rst SRE in favour of denosumab [37], but overall survival and time to cancer progression were similar and more frequent adverse events such as osteonecrosis of the jaw and hypocalcemia were seen in the denosumab arm. It seems unlikely that this (undoubtedly expensive) agent will provide substantial benefi t compared to standard bisphosphonates.
Analogues of vitamin D . After a Phase II trial (ASCENT) [38] with favourable PSA-response and survival with high-dose calcitriol (DN-101) as com-pared to placebo in combination with docetaxel, a Phase III clinical trial (ASCENT-2) was designed. However interim analysis after randomisation of 953 patients revealed inferior survival in men receiving DN-101 with docetaxel than those receiving doc-etaxel alone [39]. Median overall survival was 16.8 months for DN-101 subjects and 19.9 months for controls (HR � 1.33; p � 0.019) and hence the trial was terminated prematurely.
Radioisotopes . The bone-seeking radioisotopes, 89 Strontium and 153 Samarium are useful for pallia-tion of pain in men with CRPC and widespread bone metastases, although they may suppress bone mar-row function and increase the toxicity of subsequent chemotherapy. 89 Strontium was reported to show substantial survival benefi t (27.7 months versus 16.6 months, p � 0.001) when given as consolidation treatment post-chemotherapy in a Phase II study of men with CPRC and bone metastases [40], and is being evaluated in a randomised Phase III clinical trial. Similarly, the combination of 153 Samarium with docetaxel in a Phase II study for men with CRPC and bone metastases was well tolerated, and showed long-lasting pain control and favourable survival [41]. Encouraging results were also reported with 223 Radium in a randomised Phase II study [42]; a randomised Phase III trial of radium-223 versus pla-cebo in men with symptomatic bone metastates from prostate cancer is underway.
In summary, drug development aimed at improv-ing survival and its quality for men with advanced prostate cancer is a very active area of preclinical and clinical research that includes strategies involving hormonal agents, classical chemotherapy, molecular targeted agents, bone seeking radio-isotopes and immune modulation.

146 S. Niraula & I. F. Tannock
Kantoff PW, Halabi S, Conaway M, Picus J, Kirshner J, [16] Hars V, et al. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: Results of the cancer and leukemia group B 9182 study. J Clin Oncol 1999;17:2506 – 13. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, [17] Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502 – 12. Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr., Jones [18] JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004;351:1513 – 20. Gan L, Chen S, Wang Y, Watahiki A, Bohrer L, Sun Z, et al. [19] Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res 2009;69:8386 – 94. Seruga B, Ocana A, Tannock IF. Drug resistance in meta-[20] static castration-resistant prostate cancer. Nat Rev Clin Oncol 2010 Sept 21 (Epub ahead of print). Berthold DR, Pond GR, de Wit R, Eisenberger M, Tannock [21] IF. Survival and PSA response of patients in the TAX 327 study who crossed over to receive docetaxel after mitox-antrone or vice versa. Ann Oncol 2008;19:1749 – 53. Sternberg CN, Petrylak DP, Sartor O, Witjes JA, Demkow T, [22] Ferrero JM, et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: The SPARC trial. J Clin Oncol 2009;27:5431 – 8. de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels [23] JH, Shen L, et al. Cabazitaxel or mitoxantrone with pred-nisone in patients with metastatic castration-resistant pros-tate cancer (mCRPC) previously treated with docetaxel: Final results of a multinational phase III trial (TROPIC). J Clin Oncol (Meeting Abstracts) 2010;28:4508. Rosenberg JE, Weinberg VK, Kelly WK, Michaelson D, [24] Hussain MH, Wilding G, et al. Activity of second-line chem-otherapy in docetaxel-refractory hormone-refractory prostate cancer patients: Randomized phase 2 study of ixabepilone or mitoxantrone and prednisone. Cancer 2007;110:556 – 63. Hussain A, DiPaola RS, Baron AD, Higano CS, [25] Tchekmedyian NS, Johri AR. Phase II trial of weekly patu-pilone in patients with castration-resistant prostate cancer. Ann Oncol 2009;20:492 – 7. Kelly WK, Halabi S, Carducci MA, George DJ, Mahoney JF, [26] Stadler WM, et al. A randomized double-blind placebo con-trolled phase III trial comparing docetaxel, prednisone and placebo with docetaxel, prednisone and bevacizumab in men with metastatic castration-resistant prostate cancer: Survival results of CALGB 90401 . J Clin Oncol (Meeting Abstracts) 2010;28:LBA4511. Sonpavde G, Periman PO, Bernold D, Weckstein D, Fleming [27] MT, Galsky MD, et al. Sunitinib malate for metastatic cas-tration-resistant prostate cancer following docetaxel-based chemotherapy. Ann Oncol 2010;21:319 – 24. Dahut WL, Gulley JL, Arlen PM, Liu Y, Fedenko KM, [28] Steinberg SM, et al. Randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer. J Clin Oncol 2004;22:2532 – 9. Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep [29] DJ, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer 2008;113:2478 – 87. Chi KN, Hotte SJ, Yu EY, Tu D, Eigl BJ, Tannock IF, [30] et al. A randomized Phase 2 study of docetaxel and pred-nisone with or without OGX-011 in patients with metastatic
Declaration of interest: Dr Tannock has advised several companies about prostate cancer trials and has also chaired such trials. He has received contri-butions to his resarch fund but does not accept personal remuneration from companies.
References
Prostate Cancer Trialists ’ Collaborative Group. Maximum [1] androgen blockade in advanced prostate cancer: An overview of the randomised trials. Lancet 2000;355:1491 – 8. Sato N, Gleave ME, Bruchovsky N, Rennie PS, Goldenberg [2] SL, Lange PH, et al. Intermittent androgen suppression delays progression to androgen-independent regulation of prostate-specifi c antigen gene in the LNCaP prostate tumour model. J Steroid Biochem Mol Biol 1996;58:139 – 46. Seruga B, Tannock IF. The changing face of hormonal [3] therapy for prostate cancer. Ann Oncol 2008;19(Suppl 7):vii79 – 85. Smith MR, McGovern FJ, Zietman AL, Fallon MA, Hayden [4] DL, Schoenfeld DA, et al. Pamidronate to prevent bone loss during androgen-deprivation therapy for prostate cancer. N Engl J Med 2001;345:948 – 55. Keating NL, O’Malley AJ, Smith MR. Diabetes and cardio-[5] vascular disease during androgen deprivation therapy for prostate cancer. J Clin Oncol 2006;24:4448 – 56. Miyamoto H, Rahman MM, Chang C. Molecular basis for [6] the antiandrogen withdrawal syndrome. J Cell Biochem 2004;91:3 – 12. Zhu ML, Kyprianou N. Androgen receptor and growth fac-[7] tor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer 2008;15:841 – 9. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn [8] TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resist-ant tumor growth. Cancer Res 2008;68:4447 – 54. Gaddipati JP, McLeod DG, Heidenberg HB, Sesterhenn IA, [9] Finger MJ, Moul JW, et al. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res 1994;54:2861 – 4. Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade [10] SR, Smith MR, et al. Phase II multicenter study of abirater-one acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol 2010;28:1496 – 501. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller [11] MC, Tissing H, et al. Circulating tumor cells predict survival benefi t from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 2008;14:6302 – 9. Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, [12] Folkerd E, et al. Selective inhibition of CYP17 with abirater-one acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol 2009;27:3742 – 8. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. [13] Development of a second-generation antiandrogen for treat-ment of advanced prostate cancer. Science 2009;324:787 – 90. Scher HI, Beer TM, Higano CS, Taplin M, Efstathiou E, [14] Anand A, et al. Antitumour activity of MDV3100 in castra-tion-resistant prostate cancer : A phase 1-2 study. Lancet 2010;375:1437 – 46. Tannock IF, Osoba D, Stockler MR, Ernst DS, [15] Neville AJ, Moore MJ, et al. Chemotherapy with mitox-antrone plus prednisone or prednisone alone for sympto-matic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J Clin Oncol 1996;14:1756 – 64.

New drugs for prostate cancer 147
castration resistant prostate cancer. J Clin Oncol 2010 (in press). de Bono JS, Fizazi K, Flechon A, Heidenreich A, Voog E, [31] Davis NB, et al. Randomized phase II study of CNTO 328 (3), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone (M) versus M alone in metastatic castration-resistant prostate cancer (CRPC). ASCO Genitourinary Cancer Symposium 2010 (abstract 86). Small E, Demkow T, Gerritsen WR, Rolland F, Hoskin P, [32] Smith DC, et al. A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel versus docetaxel plus prednisone in symptomatic, castration-resistant prostate cancer (CRPC). ASCO Genitourinary Cancers Symposium 2009 (Abstract 7). Higano C, Saad F, Somer B, Curti B, Petrylak D, Drake G, [33] et al. A phase III trial of GVAX immunotherapy for prostate cancer in combination with docetaxel versus docetaxel plus prednisone in symptomatic, castration-resistant prostate cancer (CRPC). ASCO Genitourinary Cancer Symposium 2010 (LBA150). Kantoff PW, Higano CS, Shore ND, Berger ER, Small [34] EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411 – 22. Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, [35] Singh RK, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell 2005;7:485 – 96. Saad F, Gleason DM, Murray R, Tchekmedyian S, Venner P, [36] Lacombe L, et al. Long-term effi cacy of zoledronic acid for the prevention of skeletal complications in patients with
metastatic hormone-refractory prostate cancer. J Natl Cancer Inst 2004;96:879 – 82. Fizazi K, Carducci MA, Smith MR, Dami ã o R, Brown JE, [37] Karsh L, et al. A randomized phase III trial of denosumab versus zoledronic acid in patients with bone metastases from castration-resistant prostate cancer. J Clin Oncol (Meeting Abstracts) 2010 28: LBA4507. Beer TM, Ryan CW, Venner PM, Petrylak DP, Chatta GS, [38] Ruether JD, et al. Double-blinded randomized study of high-dose calcitriol plus docetaxel compared with placebo plus doce-taxel in androgen-independent prostate cancer: A report from the ASCENT Investigators. J Clin Oncol 2007;25:669 – 74. Scher HI, Chi KN, de Wit R, Berry WR, Albers P, Henick B, [39] et al. Docetaxel (D) plus high-dose calcitriol versus D plus prednisone (P) for patients (Pts) with progressive castration-resistant prostate cancer (CRPC): Results from the phase III ASCENT2 trial. J Clin Oncol (Meeting Abstracts) 2010;28:4509. Tu SM, Millikan RE, Mengistu B, Delpassand ES, Amato RJ, [40] Pagliaro LC, et al. Bone-targeted therapy for advanced androgen-independent carcinoma of the prostate: A ran-domised phase II trial. Lancet 2001;357:336 – 41. Fizazi K, Beuzeboc P, Lumbroso J, Haddad V, Massard C, [41] Gross-Goupil M, et al. Phase II trial of consolidation docetaxel and samarium-153 in patients with bone metas-tases from castration-resistant prostate cancer. J Clin Oncol 2009;27:2429 – 35. Nilsson S, Franz é n L, Parker C, Tyrrell C, Blom R, Tennvall J, [42] et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: A randomised, multicentre, placebo-controlled phase II study. Lancet Oncol 2007;8:587 – 94.

Correspondence: Louis J. Denis, Europa Uomo, Oncology Centre Antwerp, Lange Gasthuisstraat 35-37, 2000 Antwerp, Belgium. Tel: � 32 33389150. Fax: � 32 33389152. E-mail: [email protected]
(Received 10 August 2010 ; accepted 28 September 2010 )
ORIGINAL ARTICLE
Prostate cancer from the horizon of the patient
LOUIS J. DENIS 1 , MONIQUE ROOBOL 2 & BRIGITTE DOURCY-BELLE-ROSE 1
1 Oncology Center, Antwerp, Belgium and 2 Department of Urology, Erasmus MC, Rotterdam, The Netherlands
Abstract The democratization of civil society and the development of modern medicine changed the sacrosanct doctor-patient relationship to a doctor-partner dialogue. Values and respect were lost in the process where common courtesy and empathy in trust were replaced by patient rights. Launch of Europa Uomo . Europa Uomo, the European prostate cancer coalition, represents 22 national, autonomous patient support groups. Its aim includes increasing the awareness of prostate diseases, support individualized treatment as a balance between optimal medical treatment and personalized care delivered by a multiprofessional team. We expect our information/education from dedicated professional societies while in return we share care for properly informed members as well as a fast, unbiased and cheap distribution of information/innovation across the European continent. The role of a patient group . Our advocacy role is focused on quality of life, tailored treatment, knowledge of risk factors, support for research and last but not least active partnerships. We believe that we can play a modest but basic role in common actions to overcome inequalities in treatment and care in Europe. Our responsibilities range from defi ning patient obligations to facilitating translational research and saving scarce health resources. The horizon of the patient . Our hope is to plead for a treatment policy on the man fi rst and then on his cancer and to improve treatment outcomes by multiprofessional collaboration and the development of expert Prostate Units . Future expectations . A transparent, open communication line between the multiprofessional team and the patient is mandatory. The existing uncertainties should be discussed with common sense but always leave a factor of hope in survival or quality of life.
In the ascent of mankind disease and its conse-quences was a calamity for the individual and the population. This resulted in a deep respect towards the healers of the times as it was accepted that the Gods used disease to punish populations and indi-viduals alike for their committed sins. The patient could only hope for divine support to survive and recover sometimes despite the treatments received.
We all honor the great Hippocrates of Kos as the father of modern, human health care especially for his golden rule “ do not harm ” . Still each one of us knows that we do have to harm the patient with can-cer to restore his health and end up with a new post treatment quality of life. For most of the history of civilization the patient had to submit to the Latin roots of the word. He had to have patience and endure his misery.
The actual reality is not so much different as we blame the environment as the major cause of cancer and reproach the lifestyle of the patient in smoking, alcohol consumption, lack of exercise resulting in
obesity as causes of cancer. Major changes as secu-larization and democratization coupled to the devel-opment of the information technology and relative, widespread wealth in our Western societies created a social health care where the major income of most health care professionals comes out of the collected taxes from the population [1].
The net result is a development towards the man-tra of the French population: “ Libert é , egalit é , fra-ternit é ” . We are still recovering from the 1968 wave where titles and expertise were brought down to the lowest possible denominator. Our elected politicians contributed so much to the process that they receive in population polls the lowest fi gures of trust while fortunately physicians and fi refi ghters still enjoy the trust of the great majority of the population.
It is clear that this trust is based on the merits of the previous generation and that this generation has to earn their own credit in trust.
All these changes opened the chance for the patients to claim rights in the organization and practice in
Acta Oncologica, 2011; 50(Suppl 1): 148–154
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.528446

Prostate cancer: A patient ’ s view 149
social health care. It is of course true that they are at the bottom of the hierarchy of established stakehold-ers in health care and that the overall atmosphere of doctor – patient relation and communication could and still can be improved (Table I) [2].
This was effectively achieved in the USA with women ’ s groups as the National Women ’ s Health Network exercising pressure in civil society to defend their viewpoints on a number of public health issues quickly followed by many.
The launch of Europa Uomo
The Europa Donna association was launched in 1995 to improve breast cancer management in Europe and they lobbied successfully into the European Parlia-ment. As this movement was supported by the Euro-pean School of Oncology (ESO) it was predictable that a patient coalition against prostate cancer was to follow. In effect at the end of 2002 the concept of Europa Uomo was discussed in the Italian govern-ment at the time that the prime minister Berlusconi was diagnosed and treated for prostate cancer. After a number of preparatory meetings, Europa Uomo
was formally established in 2004 in Milan as its legal site with the support of ESO and the Oncologic Center Antwerp (OCA). The original confederation formed with 12 autonomous national organizations expanded rapidly to 23 groups representing their respective countries in 2010. It represents and sup-ports patient groups focused on prostate diseases in general and prostate cancer in particular. The aims include increasing the awareness of prostate dis-eases, the support of individualized treatment based on optimal medical treatment and personalized care as well as patients ’ advocacy as a priority focused on quality of life, based on solidarity and mutual respect.
These goals are clearly expressed in our 2004 Manifesto (Table II). The ten objectives speak for themselves where one should note the emphasis on quality of life as well as the promotion of prostate cancer research [3].
Our trust in optimal medical treatment which we consider the responsibility of the treating multipro-fessional group brought us the sympathy and genuine support of the professional groups. Our updated and evidence based information/education standards are exclusively from top of the bill professional support. For Europe we have received fi rst quality instructions from our multiprofessional scientifi c committee and the executive board of the European Association of Urology (EAU). Please note that in the European health care organization based on national health care systems our national membership groups have their own scientifi c committee to implement the updated European guidelines adapted to their cul-ture and standards.
On the other hand we want to be involved in patient-centered care that we like to share with a broad sup-port on all aspects of care including psycho-social,
Table I. Health care stakeholders.
Health authorities
Insurance agencies PublicPrivate
Professionals ClinicalResearch
Industry PharmaTechnology
Cancer leaguesConsumersPatients
Table II. Manifesto Europa Uomo.
1. To fi nd ways and means to promote quality of life for prostate cancer patients and their families;
2. To promote the dissemination and exchange of evidence-based as well as factual and up-to-date information on prostate cancer;
3. To promote prostate awareness and appropriate diagnosis and prognosis;
4. To emphasize the need for appropriate early detection; 5. To campaign for provision of and access to optimum
treatment; 6. To ensure quality, supportive care throughout and after
treatment; 7. To promote multiprofessional quality care and appropriate
medical infrastructure; 8. To acknowledge good clinical practice and promote its
development; 9. To ensure that all men fully understand any proposed
treatment options, including entry into clinical trials and their right to a second opinion;
10. To promote the advancement of prostate cancer research.
Table III. Europa Uomo ’ s view on cancer management.
1. Optimal medical treatment Evidence/conscious based Multiprofessional2. Patient-centered care Shared care with broad support Holistic/reciprocal respect
Table IV. Proactive prostate cancer call out.
- Governments to be aware of prostate diseases- Governments to support research biomarkers- Remember the risk factors of prostate cancer- Tailored treatment to the individual patient through
appropriate use of PSA test- Partnership building to reduce burden of disease, identify
common actions and overcome inequalities in medical treatment and holistic care

150 L. J. Denis et al.
emotional, spiritual and fi nancial problems related to disease (Table III).
This policy worked so well that we organized a proactive prostate cancer call out in 2009 with our European Association of Urology (EAU) and ESO partners [4]. The call out is presented in Table IV. This call proved to be an instant hit and was imme-diately endorsed by the main oncological and patient organizations (Figure 1).
This European network contains all the exper-tise represented by surgeons, radiation and medical oncologists, nursing, technicians and all health and social personnel needed for its ultimate goal to pro-vide optimal treatment and holistic patient care
Partnerships Europa Uomo
EPPOSI OECIESOP ECP
EORTC GU EAUNECCO – ESMO – ESSO – ESTRO – EONS
ECPC Eurocan+PlusEuropa Donna PROCABIO
PRO-NESTWWPCC
EAU - EUomo - ESO
Europa Uomo 2010Europa Uomo (EUomo) European Association of Urology (EAU) European School of Oncology (ESO) European CanCer Organization (ECCO) European Society for Medical Oncology (ESMO) European Society of Surgical Oncology (ESSO) European Society for Therapeutic Radiology and Oncology (ESTRO) European Oncology Nursing Society (EONS) Genito-Urinary group of the European Organization for Research and Treatment of Cancer (EORTC GU)European Society of Oncology Pharmacy (ESOP) European Platform for Patients’ Organizations, Science and Industry (EPPOSI)European Association of Urology Nurses (EAUN) European Cancer Prevention organization (ECP) Organisation of European Cancer Institutes (OECI) European Cancer Patient Coalition (ECPC) Europa Donna WorldWide Prostate Cancer Coalition (WWPCC) Eurocan+Plus European initiative to develop tailored treatment of PROstate CAncer by BIOmarkers (PROCABIO) Prostate Research Organizations-Network of Early Stage Training (PRO-NEST)
Figure 1. Partnerships Europa Uomo.
The C taboo word
Tsunami information(professionals, media, friends)
The medical labyrinth
Evidence Based, Guidelines,Nomograms
Loss of personality
OutcomesStatistics
PANIC
Europa Uomo 2010
Figure 2. The C taboo word. The word cancer is a taboo word for patients in the diagnostic process causing a vision of death by cancer and acute panic in many instances.

Prostate cancer: A patient ’ s view 151
covering psychological, emotional and social needs of the patient.
The decision of the European Commission to invite patient support groups to participate in the European Partnership Action Against Cancer (EPAAC) program is a milestone in the integration of the patient as a partner in cancer clinical research in Europe.
The role of a patient group
And yet despite of all this hard work and dedication by many we still face the sad problem that a newly diagnosed unprepared cancer patients faces the spell of the great C taboo word that his life is at stake and his quality of life gone for the rest of his remaining days (Figure 2) [5].
We call it the “ lost ” patient syndrome. A state of mind that stays for a number of days or weeks despite concerted efforts of the general practitioner, the nursing or data manager. A well known moment in the disease history where the patient has an endless list of questions and the doctor has never time to answer them. Here starts the supporting role of the patient support groups united in a shared mission, to mobilize the solidarity of the survivors to provide correct, updated, clear and validated information in a stepwise manner on the medical pathway from diagnosis to beyond primary treatment [6]. Ideally public awareness on the history of the disease has been available for the general public before the diag-nosis. Further education, a sympathetic ear and understanding psychological and emotional distress can be improved by the shared experience of being treated for cancer [7].
The pitfalls next to the need for the “ perfect ” infor-mation/education are the number of experts to be encountered in a less than smooth circuit, the infl exible guidelines or nomograms representing cohorts but
never individuals and unfortunately the incomparable, uncontrollable outcomes in the world of medical prac-tice. Here we hope to see the development of reference expert centers in the near future with breast cancer as a good example.
A special attention should be given to the loss of personality in the hospital environment when one becomes a room number or worse a disease number. In our modern society where good manners and eti-quette seem to be fading for the egocentric approach patients resent that they are improperly informed, the lack of time from the doctor for their needs, the exclusion of choice in their own treatment and, yes a lack of respect from the doctor. Typical examples are a number of phone calls during the medical ana-mnesis, a decision chat with a naked or undressing patient and ridicule any alternative treatment that the patient has chosen (Table V).
It is less of a problem in private practice but it should be avoided as a general rule in any hospital or hospice. An atmosphere of relaxed respect and proper communication solves many misunderstand-ings in the daily routine where an informed, prepared patient makes the chores of the nursing and medical staff much easier.
Let it be known that Europa Uomo in this era of legal patient rights and charters equivocally sees a number of recommendations/obligations of patients and their relatives and friends to the multiprofes-sional treating team. These obligations involve respecting the standing rules in the different areas
Table V. Typical patient complaints.
Improper, incorrect informationThe doctor has no timeNo choice in their own treatmentLack of respect
Table VI. Role of prostate cancer patient support groups.
1. Involve in all aspects of holistic care;2. Establish fast and cheap networks of reliable, updated
information/education;3. Support optimal medical treatment for all as well as research
be it translational or basic;4. Sharing best practice and aim for expert prostate centers;5. Save health resources by avoiding duplication or unreliable
diagnostic procedures.
Table VII. Partnership with patient groups.
1. Strength in numbers (votes) can be infl uential in political (Health Authorities) policies;
2. A plus factor in negotiations with government bodies (Landmark 2010);
3. Enables an independent fl ow/exchange of medical information and best practice (Watch the source);
4. Partnerships reinforce recognition.
Figure 3. A ying and yang balance is advocated in information, and treatment of cancer.

152 L. J. Denis et al.
of treatment, understanding the communication fail-ures that can happen in busy centers, see the staff as trained individuals and above all inform them on any kind of other treatment or advice that one receives.
The fact that many cancer diseases especially breast and prostate become long time chronic dis-eases by the disease or by the treatment should encourage both doctors and patients to engage in a mutual congenial partnership [8].
As a conclusion one sees the constructive role of the patient support organizations as effective to reach holistic care, organize cheap and effi cient grass roots information, support basic and clinical research and by being partner save money for unnecessary exam-inations in follow-up, chronic care (Table VI) [9]. These efforts and outcome results in improving health care can be easily multiplied by expanding the network. Basic requirements are shared interests, similar objectives and a good transparency of the positive but different goals of the participating part-ners including all possible stakeholders. Each asso-ciation or project has its own broad or targeted agenda. The last thing one needs is an hidden agenda as a successful partnership is based on win-win situ-ations where the expertise and/or the means of the different partners are utilized to reach a common goal (Table VII).
The horizon of the patient
Coming back to the individual prostate cancer patient still his horizon is the treating urologist. It is under-standable as the track record of the urological specialty involves the mortality reduction of benign prostatic
hyperplasia (BPH) to zero following an orderly tran-sition from surgery to medical treatment of lower urinary tract symptoms (LUTS) and still looking for less invasive treatments.
The track record of managing prostate cancer has been less spectacular and has been uneven in the balance of facts and remaining open questions. In a way we watched the importance of two Noble prizes to reward progress in the clinical treatment of pros-tate cancer. The fi rst one bestowed on Charley Hug-gins 25 years after his fundamental studies on the physiology of the prostate resulting in the surgical castration as primary treatment of prostate cancer. The second one bestowed on Andrew Schally for his isolation and analysis of the natural LHRH decapep-tide leading to a tsunami of medical castration.
All patients with prostate cancer have been treated or threatened with some endocrine manipulation and most have enjoyed the palliative power of primary endocrine treatment in a far advanced, metastatic prostate cancer. It is only in the last decades and after the outcomes of randomized, clinical trials that it became clear that managing prostate cancer involves more than endocrine treatment [10].
As simple observers of clinical progress and trans-lational/basic research in cancer we are sometimes surprised by the professional groups ignoring clean facts and unanswered questions alike in proposing clinical decisions to their patients. We prefer to bal-ance facts and uncertainties according to old wisdom before we try to come to a shared decision on impor-tant crossroads in the treated history of dealing with one ’ s prostate cancer (Figure 3).
A patient diagnosed with prostate cancer goes through different phases of the disease (Table VIII).
Table VIII. Phases in the treated history of prostate cancer.
1. Suspicion: Serendipity according to age Risk factors Positive marker(s) (PSA)2. Confi rmation: Biopsies3. No diagnosis without prognosis4. Primary treatment: Overdiagnosis (AS) Underdiagnosis5. Secondary treatment (WW)6. Castration Resistant Prostate Cancer (CRPC)
Table IX. Active surveillance vs. watchful waiting.
Active Surveillance Watchful Waiting
Fit patient Co-morbidity/ageLow risk cancer High risk cancerPSA dynamics defi ne treatment
( � biopsies)Symptoms defi ne treatment
Option: cure Option: palliation
Table X. In a nutshell.
1. Prostate cancer is a heterogeneous disease with a long, natural history.
2. This chronicity is specifi c for the disease but includes treatment related illnesses.
3. Age is the most important risk factor increasing the burden in an ageing society.
Table XI. Incontinentince/erectile dysfunction by treatment or age.
Treated vs. Normgroep
Incontinence urine 23 – 48% vs. 4%
Incontinence bowel 5 – 14% vs. 2%
Erectile Dysfunction 40 – 74% vs. 18%
Thesis F. Mols, 2007

Prostate cancer: A patient ’ s view 153
patient. It would start in choosing treatment, now based on nomograms and guidelines [12,13], with objective, reliable treatment results and side-effects. The side-effects of curative treatment are sometimes underesti-mated for the individual patient. The fi gures related to primary treatment are important (Table XI).
Treat the available diagnostic uncertainties with common sense to keep the confi dence of the patient. These uncertainties include the PSA numbers, the biopsies, the nomograms, the Gleason score and the imaging procedures. Improvements are possible and expected in all domains. For screening, prevention and primary treatment results we wait for the ongo-ing clinical trials (Table XII) [14].
Last we have seen improvement in treatment results of castration resistant prostate cancer (CRPC). Here the patients expect access to these clinical trials or new treatments. If one looks for a bottom line we should advise to treat the man and his co-morbidities fi rst and then his cancer (Table XIII).
All these factors are condensed in Table XIV where we express hope that the existing gap between the available knowledge and the practiced reality may close in the near or distant future.
Declaration of interest: The authors report no confl icts of interest. The authors alone are respon-sible for the content and writing of the paper.
References
Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of can-[1] cer incidence and mortality in Europe in 2008. Eur J Cancer 2010;46:765 – 81. One step closer to patients ’ rights. HSCNEWS International [2] 2009;49. Denis L. Europa Uomo. BJU Int 2007;99:1329 – 34. [3] Van daele H. Prostaatkanker 2009. Wat zijn de feiten? PROS-[4] TAATinfo 2009;8:4 – 5. Denis L. Cancer Prostatique localis é d é butant: Surveillance [5] active ou traitement imm é diat? Andrologic 2009;5:123 – 6. Dourcy-Belle-Rose B. Prostaatproblemen. Gids voor man-[6] nen en vrouwen. 4th ed. OCA; Antwerp. 2010. van Muilekom HAM, Spil JA. Handboek Prostaatcarcinoom. [7] Elsevier Gezondheidszorg; Amsterdam. 2006.
First of course the serendipity of the cancer and an increased PSA number. Second confi rmation of the suspicion and then going for a reliable prognosis. Once established it is time to discuss primary treat-ment among the available procedures including active surveillance.
The next phase, many times disappointing, is the outcome result of the primary treatment and its side-effects. Again another shared decision in secondary treatment is important and the outcome of this treat-ment and its side-effects.
Last of course is the recognition that our prostate cancer is progressive and ultimately lethal. Here the treatments look more invasive as they don ’ t carry the promise of cure. The choice should include watchful waiting. The latter is very different from active sur-veillance as here there is no chance to return to pri-mary, curable treatment (Table IX) [11].
By this time the need for a multiprofessional team is so obvious that the urge to establish expert prostate units in our health system becomes more attractive to the patient and professional alike.
Despite the label of incurable cancer many people can and do enjoy good quality lives. It is usually in the last year of their lives that prostate cancer suffer from the disease progress expressed in back pain, bone fractures, anemia, fatigue and lower urinary tract obstruction.
Prostate cancer is a chronic, heterogeneous dis-ease with high incidences in the seventh and eighth decade of life with a specifi c mortality in the ninth decade. The low mortality, meaning death by pros-tate cancer, is relative (2 – 4%) as the number of patients is so high and most patients still die by their concomitant lethal diseases (Table X).
Future expectations
What do patients and their respective groups expect in future, optimal treatment?
First and above all a good communication line between the different members of the multiprofessional team with a transparent, open information to the
Table XIII. A plea from Europa Uomo.
Focus on the man fi rst and then on his cancer.Improve outcome by multiprofessional action and expert
Prostate Units.
Table XIV. Priorities to improve management of cancer.
Knowledge Reality
Prevention Treatment
Rich Poor
Collaboration Olympic stand
Transparent Pandora’s box
Table XII. Existing uncertainties in the management of prostate cancer.
1. PSA fi gures, biopsies, Gleason grading2. Imaging TRUS, MRI3. Improvements in surgery, radiation4. The biology of man and his cancer(s)

154 L. J. Denis et al.
Valdagni R, Scardino PT, Denis L, Kattan MW. Predictive [12] modeling in prostate cancer: A conference summary. Eur Urol 2009;55:300 – 2. European Association of Urology. Pocket guidelines. 2010 [13] edition. EAU 2010. Available from: https://www.uroweb.org/publications/eau-guidelines/ Roobol MJ, Kerkhof M, Schroder FH, et al. Prostate cancer [14] mortality reduction by prostate-specifi c antigen-based screening adjusted for nonattendance and contamination in the European Randomised Study of Screening for Prostate Cancer (ERSPC). Eur Urol 2009;56:584 – 91.
Bangma CH. Het prostaat pensioen plan. Elsevier Gezond-[8] heidszorg; Rotterdam. 2008. Kirkali Z. There has to be a way. Eur Urol 2007;52:1558 – 9. [9] Tannock IF, Osoba D, Stockler MR, et al. Chemotherapy with [10] mitoxantrone plus prednisone or prednisone alone for sympto-matic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J Clin Oncol 1996;14:1756 – 64. Bangma CH, van den Bergh RCN, Denis L, Roobol MJ. [11] Understanding active surveillance. A new treatment option for PSA positive low risk prostate cancer. Central Eur J Urol 2009;62:70 – 3.

Correspondence: Peter C. Albertsen, Department of Urology, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030, USA. E-mail: [email protected]
(Received 10 September 2010; accepted 18 November 2010)
COMMENTARY
WHO International Consultation on Prostate Cancer: A summary
PETER C. ALBERTSEN
Division of Urology, University of Connecticut Health Center, Farmington, USA
During the past three decades, prostate cancer has assumed an increasingly prominent role in health care policy and delivery in many western countries. Prostate cancer is now the most commonly diag-nosed non-skin tumor and the second leading cause of death after lung cancer. The dramatic rise in the incidence of this disease has been directly linked to the dissemination of testing for prostate specifi c anti-gen (PSA) and is highest in regions where annual PSA testing has become commonplace.
The introduction of PSA testing has resulted in several signifi cant changes concerning how scientists, clinicians and public health offi cials deal with pros-tate cancer. PSA testing has impacted how we iden-tify prostate cancer, the type of cancers we diagnose, and how we stage and manage this disease. This manuscript summarizes the papers presented at the WHO International Consultation on Prostate Can-cer concerning the advances in screening, prevention and therapy for prostate cancer and highlights the future challenges that lie ahead.
A) Early detection and screening
In 2009 two publications appeared in the New England Journal of Medicine that advanced our understanding of the impact of PSA testing on the early detection of prostate cancer. On fi rst glance, the conclusions from these studies appeared to be con-tradictory. The Prostate, Lung, Colorectal and Ovar-ian (PLCO) Cancer Screening Trial demonstrated no benefi t from PSA testing, whereas the European Randomized Study of Screening for Prostate Cancer (ERSPC) trial suggested that PSA testing reduced prostate cancer mortality by 20% but with the caveat that at least 48 men required treatment to prevent one prostate cancer death.
Closer inspection of the study designs of these two trials reveals that the PLCO trial evaluated the impact of intensive annual PSA screening against modest PSA testing, whereas the ERSPC trial evalu-ated the impact of PSA screening versus no screen-ing. The ERSPC trial tested whether PSA testing is effi cacious, while the PLCO trial explored whether PSA testing is effective from a population perspec-tive. When considering broad public health policies, this distinction helps guide appropriate clinical appli-cation. PSA screening must be effi cacious if it is to be effective, but it may be not effective from a public health perspective even when it is effi cacious for an individual patient.
In the PLCO trial, men were recruited to study centers and randomly assigned to receive either annual PSA tests for six years and digital rectal examinations for four years or usual care. If a study participant was found to have an abnormal PSA test or digital rectal examination, study researchers referred him to his primary care physician or other health care provider for further evaluation, a possible prostate biopsy, and treatment if the biopsy specimen was positive for cancer cells. In the ERSPC trial, potential study participants were identifi ed from population registries and invited to participate in the study. Men who were found to have an abnormal PSA level were offered a transrectal ultrasound and prostate biopsy. Men with positive biopsies were pro-vided defi nitive treatment.
Both of these studies have been the subject of numerous analyses and editorials identifying many technical problems inherent to both studies. Both tri-als, however, have identifi ed the major public health issue of over diagnosis of prostate cancer as a result of repeated PSA testing. In the ERSPC study 42 cancers had to be identifi ed and managed in order to prevent
Acta Oncologica, 2011; 50(Suppl 1): 155–160
ISSN 0284-186X print/ISSN 1651-226X online © 2011 Informa HealthcareDOI: 10.3109/0284186X.2010.542175

156 P. C. Albertsen
one prostate cancer death. Many men who would not otherwise have been diagnosed with prostate cancer are undergoing biopsy and treatment. Without more information concerning the impact of PSA testing on health care quality of life and the economic costs asso-ciated with widespread PSA screening and treatment, it is currently inappropriate to adopt population based PSA testing as public policy.
One promising method to manage the problem of over-diagnosis of prostate cancer is genetic testing. Available markers currently explain approximately 25% of the heritability of this disease and researchers anticipate that within the next two years, the number of identifi ed markers will increase from 35 to over 100 and will have the potential of explaining almost 50% of the heritable factors associated with prostate cancer. Potential clinical applications of these genetic tests include: a) defi ning a sub-population of healthy men who have an excessive risk of developing clini-cally signifi cant prostate cancer, and b) integrating the information provided by genetic factors to assess an individual patient ’ s risk of developing clinically signifi cant disease. Unfortunately, there is no data presently available that can be used to differentiate men with clinically signifi cant disease from those who have clinically insignifi cant disease.
B) Assessing the tumor
The past decade has witnessed an explosion in the number of tools that can be used to assess prostate cancer. Traditional analysis of histology remains the gold standard for diagnosing prostate cancer and assessing the aggressiveness of this disease. The Gleason scoring system has been adopted world wide, but the system is not static. Consensus confer-ences conducted within the last few years have con-tinued to modify how the Gleason system is applied. Gleason patterns 1 and 2 are rarely used by contem-porary pathologists. Pathologists have also modifi ed the defi nition of Gleason pattern 3. Increasingly men who harbor cribriform glandular patterns are classi-fi ed as having Gleason pattern 4 disease. Glandular differentiation has become a key factor for many pathologists. Glandular differentiation is preserved in specimens classifi ed as Gleason pattern 3 � 3 dis-ease. When glandular differentiation is only partially preserved the specimen is usually classifi ed as Gleason pattern 3 � 4 or 4 � 3. When glandular dif-ferentiation is absent the specimen is classifi ed as having Gleason patterns 4 and 5.
Other factors that continue to demonstrate impor-tant prognostic signifi cance include the number of positive cores at biopsy and the extent of tumor in each core. Perineural invasion is often reported, but it is unclear whether this factor provides independent
prognostic information. Important changes in staging include the classifi cation of microscopic bladder neck invasion and the intraprostatic seminal vesicle inva-sion as T3a disease. The diameter of the largest node is also clinically relevant. Sub-classifi cations of T2 disease provide no valuable information.
Prostate specifi c antigen remains the most impor-tant and widely used biomarker to assess the presence and potential extent of prostate cancer. Unfortunately PSA, like all biomarkers, has signifi cant problems with false negative and false positive values. No single biomarker is capable of distinguishing clinically sig-nifi cant disease from normal pathology or indolent disease. In the future panels of biomarkers will be combined and entered into prediction models. Dur-ing the past few years several nomograms and risk calculators have been developed.
Researchers have explored the performance of PSA using multiple approaches including PSA kine-tics. The inherent analytical and biological variability of total PSA levels affects the interpretation of any single result. Men who will eventually develop pros-tate cancer have increased total PSA levels years or decades before the cancer is diagnosed. PSA velocity marginally improves the specifi city of total PSA, but has limited use for screening or prognostication. The combination of PSA molecular forms and other biomarkers promises to improve the detection of prostate cancer. Examples include the blood markers PHI (�2proPSA, %PSA, tPSA), a four kallikrein panel (tPSA, %PSA, intact PSA and hK2), tissue markers (uPA axis, TGF-bets 1, IL-6R, Endoglin, Ki-67) and urine markers (PCA3).
Panels of biomarkers that capture the biological potential of prostate cancer are in the process of being validated. For advanced prostate cancer, cir-culating tumor cells appear to offer the greatest promise for predicting and monitoring the response to therapy. Unfortunately, at the present time no single biomarker is clinically useful as a predictor of prostate cancer progression.
Imaging techniques have also undergone signifi -cant advances during the past decade. Magnetic resonance imaging in particular shows promise in the diagnosis and clinical assessment of tumor grade and local extent. Prostate cancer is more diffi cult to detect and localize in the central gland as compared with the peripheral zone. MR offers the potential to improve our ability to identify and biopsy these lesions when compared to transrectal ultrasound (TRUS) and biopsy. Multiparametric MR imaging combines T2 weighted imaging, diffusion weighted imaging and dynamic contrast enhanced imaging. The typical prostate cancer lesion has low signal intensity on T2 weighted imaging, a low diffusion coeffi cient value, a high choline and creatine/citrate

WHO Consultation Summary 157
ratio, high contrast agent permeability and rapid washout. Multiparametric imaging is needed because not all prostate cancer lesions exhibit all of these fea-tures. Aggressive cancers appear to demonstrate a high diffusion coeffi cient and a high choline and creatine/citrate ratio.
MR offers the potential to improve prostate biop-sies. Lesions that are often missed on TRUS can be imaged on MR. An experienced radiologist can per-form an MR directed biopsy within 30 minutes. An endorectal coil is needed for prostate cancer staging if only a 1.5T magnet is available. If a 3T magnet is available, an endorectal coil is only required to identify minimal capsular penetration. MR imaging can be used for conventional nodal staging using standard criteria for size and shape. Novel contrast agents are still considered experimental and require FDA and EMEA approval. MR offers the potential of outper-forming bone scintigraphy when evaluating the axial skeleton for bone metastases. Dynamic contrast-enhanced MR imaging may play an important role in detecting tumor recurrence after surgery or radiation.
Positron Emission Tomography (PET) combined with computerized tomography also shows promise as a technique to measure the extent of prostate can-cer. Many new tracers are available. FDHT targets the androgen receptor and therefore can be used to either guide prostate biopsies to assess the presence of metastatic disease or to assess a patient ’ s response to various chemotherapeutic agents.
Technitium-99 based bone scans have been tra-ditionally used to assess patients who are likely to harbor bone metastases. Recent data suggest that technetium bone scans can also be used as a second-ary end point in clinical trials. Patients who develop more than two new lesions on bone scan when com-pared to their base line scan have clinical evidence of clinical progression.
C) Therapy with curative intent
Men with moderate or high grade prostate cancers (Gleason score � 7) face a substantial risk of disease progression when compared to men with low grade disease. This is especially true for those men who present with clinically palpable disease (stage T2 and T3). Two randomized trials conducted in Scandina-via revealed a survival benefi t for men who received either surgery or radiation. The SPCG-4 trial ran-domized patients between immediate surgical inter-vention and deferred endocrine treatment. A total of 695 men were enrolled during the period 1989 – 1999. A review of the study cohort reveals that 76% of the participants had T2 disease and only 11% of the par-ticipants had disease detected on the basis of PSA testing. The distribution of Gleason scores was as
follows: 48% Gleason 5 – 6, 23% Gleason 7, and 5% Gleason 8 – 10.
After a median follow-up of 10.8 years, 39% of the men had died. There was a small absolute differ-ence in overall survival of 7% favoring men undergo-ing radical prostatectomy, but the difference was not statistically different. After 12 years the overall mor-tality rate was 33% among men undergoing surgery and 40% among men receiving deferred androgen deprivation therapy. Interestingly the survival advan-tage only favored those men who were age 65 or less at the time of surgery. Men older than this had sim-ilar outcomes in each of the two arms of the study.
The SPCG-7/SFUO-3 trial randomized men to either immediate androgen deprivation alone or androgen deprivation plus radiation therapy. A total of 875 patients were enrolled from 1996 to 2002. As with the SPCG-4 trial, most of the men enrolled had more advanced disease and presented because of clinical fi ndings rather than on the basis of PSA screening. A review of the study cohort shows that 98% of the patients had T2 disease or higher with the majority presenting with T3 disease. Unfortu-nately, tumors were not graded by Gleason score, but 85% of the patients had WHO grade II disease or higher and 23% had seminal vesicle invasion. A review of PSA values at entry showed that only 24% of patients had a PSA lower than 10.0 ng/ml. These patients had considerably more advanced disease when compared to the typical patient presenting with newly diagnosed localized prostate cancer in the United States during the past decade.
After a median follow-up of 7.6 years, 79 men in the androgen deprivation alone group and 37 men in the radiation plus androgen deprivation group had died of prostate cancer. The ten year overall mortal-ity was 39.4% in the androgen deprivation group and 29.6% in the radiation plus androgen deprivation group. Ten year prostate cancer-specifi c mortality was 23.9% in the androgen deprivation alone group compared with 11.9% in the radiation therapy plus androgen deprivation group.
The fi ndings in both of these trials are remarkably similar. Men with clinically advanced, localized pros-tate cancer have a survival advantage after receiving surgery or radiation therapy. Despite their relatively advanced disease, a majority of the men enrolled died from a cause unrelated to prostate cancer within ten years of diagnosis. Among those men who ultimately died from prostate cancer, radiation or surgery low-ered this probability by about 50%. Twelve years after surgery and ten years after radiation 12.5% of men undergoing surgery and 11.9% of men receiving radi-ation died from their disease. For those men in the control arms 17.9% and 23.9% respectively died of prostate cancer after 12 years and 10 years.

158 P. C. Albertsen
Very few of the prostate cancers treated in the Scandinavian trials were diagnosed on the basis of PSA testing and therefore it is diffi cult to generalize these fi ndings to a contemporary population of men in the United States. At minimum these fi ndings refl ect 15 or 20 year outcomes if the lead time intro-duced by PSA testing is added. Surgery and radiation therapy, however, come at a price. The side effects of surgery and radiation therapy are well known and often include problems with urinary function, bowel function and especially sexual function.
In 2010 several problems remain. Low risk pros-tate cancer is poorly stratifi ed and suffers from under or overtreatment. Active surveillance protocols are inadequate. Intermediate risk disease for men with a ten year life expectancy can be treated equally well with either radiation therapy or surgery after careful counseling. Men with high risk disease are often undertreated with monotherapy and have poor out-comes. Surgery is often underutilized for these patients who often require combination therapies for optimal outcomes. Randomized clinical trials are urgently needed to refi ne active monitoring proto-cols, to defi ne low risk disease and to evaluate mini-mally invasive therapies.
D) Natural history of prostate cancer and the role of active surveillance
During the past two decades there has been a dra-matic increase in the incidence of prostate cancer in those countries that have adopted screening for pros-tate specifi c antigen (PSA). Prostate cancer is now diagnosed at a rate that is 2.5 times higher in the USA where PSA testing is commonplace when com-pared to the UK where PSA testing is much less frequent. The dramatic differences between prostate cancer incidence rates and mortality rates have led many researchers and clinicians to recognize that prostate cancer is increasingly being over-diagnosed in many countries. While all prostate cancers are likely to progress, many cancers progress at remark-ably slow rates and therefore are not destined to become clinically signifi cant.
Several key studies have shaped our understand-ing of the natural history of disease progression. Between 1989 and 2004, Johansson and colleagues published a series of four articles that documented the outcomes of untreated prostate cancer. Albertsen et al. also reported on the long-term outcomes of a competing risk analysis of men diagnosed with local-ized disease between 1971 and 1984. Together these studies have shown that men with low grade disease (Gleason score � 6) can frequently survive up to 20 years without evidence of disease progression. Conversely men with high risk disease (Gleason
score � 7) often die of this disease within ten years of diagnosis.
Unfortunately these studies do not refl ect the impact of widespread PSA testing that has advanced the date of diagnosis by as much as ten years. As a consequence, men diagnosed with low volume, low grade T1c disease are at very low risk of disease pro-gression. For these men intervention with either sur-gery or radiation frequently carries higher risks than surveillance. Interest in active surveillance of prostate cancer refl ects this greater understanding of the rela-tive impact of intervention. These considerations have led clinicians at several academic medical centers to propose criteria that identify men who have a low risk of disease progression. They have relied on concepts originally developed by Epstein that were based on pathological analysis that correlated biopsy fi ndings with those found following radical prostatectomy. The core criteria include: a) men who present with pros-tate cancer in two cores or less ( � 25% of the total biopsy specimen), b) neither core has more than 50% involvement with disease, and c) tumor histology that contains no Gleason patterns 4 or 5.
These concepts have yet to be validated by clini-cal trials. To date, support for this approach comes from several case series. Klotz et al. have published the longest follow-up of over 450 men participating in an active surveillance program. After a median follow-up of seven years overall survival is 78.6% and prostate cancer specifi c survival is 97.2%. Death from competing risks has been 16 times greater than the number of deaths from prostate cancer. Unfor-tunately, active surveillance carries risks. In the Klotz cohort, over 30% of patients have undergone more aggressive treatment because their disease has been reclassifi ed as higher risk. Of these men, half have evidence of biochemical failure on the basis of a ris-ing PSA. Whether these patients will ultimately die from prostate cancer remains to be determined.
Most likely, the concept of repeated assessments of the tumor over time is here to stay. We know that active surveillance leads to reduced overtreatment, but the risk of missing the window of curability is unknown, as is the optimal parameters for assessing the tumor. Issues related to quality of life must also be evaluated thoroughly.
E) Chemoprevention of prostate cancer
The rising incidence of prostate cancer and the rec-ognition that many cancers are not clinically signifi -cant has caused many clinicians to pursue the concept of chemoprevention as a way to prevent overdiagno-sis and overtreatment of this disease. The term “ chemoprevention ” implies that prostate cancer can be prevented. While this is a noble goal, a more

WHO Consultation Summary 159
accurate description of current efforts is “ risk reduc-tion ” . Primary chemoprevention refers to the reduc-tion of the risk of prostate cancer development while secondary chemoprevention refers to the reduction of the risk of prostate cancer progression.
The two principal targets of prostate cancer chemoprevention have been infl ammation and hor-monal stimulation of the prostate. Infl ammation has been associated with the development of lung cancer in smokers, hepatic cancer in chronic hepatitis and bowel cancer in infl ammatory bowel disease. In pros-tate cancer, infl ammation is associated with prostate cancer precursor lesions such as proliferative infl am-matory atrophy and is believed to increase genetic instability leading to prostate carcinogenesis. Unfor-tunately, several studies such as the SELECT trial have failed to show that anti infl ammatory agents such as the antioxidants selenium and vitamin E can reduce prostate cancer mortality.
To date, interest in chemoprevention of prostate cancer has centered on the fi ve alpha reductase inhib-itors. Two trials, the fi nasteride chemo prevention trial (PCPT) and the reduction by Dutasteride of prostate cancer events (REDUCE) trial, have both demon-strated that the incidence of prostate cancer can be lowered in men who take these medications. These two trials, however, were fundamentally different. The PCPT trial tested whether prostate cancer incidence could be lowered among men who had normal PSA levels at entry. The REDUCE trial tested whether prostate cancer incidence could be lowered in the sub-set of men who were previously identifi ed as having elevated PSA levels and therefore were at higher risk of being diagnosed with prostate cancer. The PCPT trial followed men for an average of seven years; the REDUCE trial followed men for an average of four years.
Both trials demonstrated that prostate cancer incidence was lower in men taking 5 alpha reductase inhibitors. The incidence of prostate cancers of all Gleason grades was lowered in both studies, but the primary impact was in the number of low grade can-cers. Controversy surrounds both of these trials. The incidence of prostate cancer was four times higher in both arms of the PCPT trial than originally anticipated. Therefore it is unclear whether 5 alpha reductase inhibitors actually prevent prostate cancer or simply decrease the probability that a man will undergo prostate biopsy. Furthermore, these trials have raised a concern that 5 alpha reductase inhibi-tors may induce high grade lesions. Follow-up in both of these trials have been insuffi cient to deter-mine whether these agents ultimately lead to lower prostate cancer mortality. Information from the REDEEM study should provide new data concern-ing the role of 5 alpha reductase inhibitors in
men choosing active surveillance as a treatment alternative.
F) Management of advanced disease, new drugs
Treatment of advanced prostate cancer has posed challenges for both clinicians and research scientists. Many drugs have been tested, but few have shown much effi cacy in altering the outcome of this disease. Docetaxel, approved in 2004, was the fi rst agent that showed evidence of a survival advantage. Since then researchers have explored many drugs targeting dif-ferent biological mechanisms. Currently androgen deprivation therapy by either surgical or chemical castration remains the cornerstone for the manage-ment of advanced prostate cancer. Unfortunately, the effect of this approach is transient. Many patients developed androgen independent disease that pro-gresses despite low levels of testosterone.
Since data from the TAX 327 and the SWOG 9916 trials were presented in 2004, docetaxel admin-istered every three weeks has become the standard treatment for men with androgen resistant disease. Unfortunately, treatment has improved survival only modestly with an average median survival increase of less than 20 months. Recently cabazitaxel, a novel taxane with a favorable low affi nity to multidrug resistant P-glycoprotein, in combination with pred-nisone has been approved by the United States Federal Drug Administration. A phase III trial dem-onstrated an overall survival benefi t of 2.4 months when compared to mitoxantrone in patients previ-ously treated with docetaxel alone.
The recognition that the withdrawal of anti-androgens can lead to a clinical response has dem-onstrated the continued importance of the androgen receptor signaling pathway in castrate resistant pros-tate cancer. Several new drugs have been developed to exploit biological mechanisms of androgen recep-tor mutation, androgen receptor amplifi cation, ligand-dependent androgen receptor activation or enhanced local production of androgens. MDV3100, a non-steroidal compound, and RD162 are examples of two new agents that target the androgen receptor with higher affi nity than bicalutamide. EPI-001 is another new compound that binds to the N-terminal domain of the androgen receptor and targets the trans-activation of the androgen receptor regardless of the presence of androgens. Abiraterone acetate is another interesting compound. This agent inhibits cytochrome P17 which catalyzes two key reactions in androgen biosythesis.
Our understanding of the complex molecular pathogenesis of prostate cancer has led researchers to explore several novel drugs that target specifi c

160 P. C. Albertsen
trials that will target specifi c subgroups of patients with varying biological characteristics.
G) Prostate cancer from the patient ’ s perspective
The past two decades have witnessed a changing role for patients in the diagnosis and management of pros-tate cancer. Patients are demanding a more active role in decision making and no longer rely exclusively on the treating physician ’ s advice. These changes are part cultural, but are also a consequence of changing tech-nology. Internet search engines enable patients to access enormous amounts of information with mini-mal effort. The physician is now frequently challenged to keep up with medical literature so he can interpret fi ndings accurately for his patients.
Prostate cancer patients are demanding more information concerning their personal prognosis and whether a specifi c intervention is likely to prolong their life. Patients are increasingly aware that all treatments carry side effects and are demanding more information concerning the frequency and severity of treatment mishaps. These demands have become quite personal. Patients now seek informa-tion on surgeons ’ individual skills and the frequency of complications. This information is often not avail-able, but the adoption of electronic medical record keeping will greatly facilitate this process in the future.
Patients have become more sophisticated con-sumers. They recognize the potential trade off between the risks of a disease and the risks of inter-vention. They are demanding more quantitative information to help them decide what treatments are best for them. They recognize that many treatments may fail and demand information on treatment alter-natives and salvage therapies. They recognize that increased longevity may or may not be the primary goal. A high quality of life is more often the preferred outcome.
Historically the role of a physician has been one of a trusted advisor who can help guide a patient by helping them understand how their disease will unfold in the future. Some physicians have become blinded by their own technology and now appear as salesmen touting their wares. The more sophisticated patients will seek the advisor and shun the salesmen. Clinical trials are protected by an ethical code that states that investigators must provide suffi cient information to patients to help them make an informed decision. Many patients assume that this also occurs in clinical practice. Patient advocacy groups are working to ensure that physicians perform at this higher standard. If physicians do not, they risk losing the professional respect and privilege that took centuries to build.
molecular pathways such as: epidermal growth fac-tor (EGFR) signaling, vascular endothelial growth factor (VEGF) signaling pathways, phosphati-dylinositol 3-kinase (PI3K)/Akt mammalian target of rapamycin (mTOR) pathway as well as the insulin-like growth factor pathway. Multiple tyrosine kinase inhibitors with a typical anti-angiogenic pro-fi le such as sunitinib and sorafenib have also been evaluated. Unfortunately data concerning long-term outcomes are limited. The anti-VEGF antibody bev-acizumab has also been evaluated but the results from a recent phase III trial, CALGB 90401 have been disappointing.
Src and src-family kinases that are involved in multiple signaling pathways central to the develop-ment of prostate cancer and the pathogenesis of bone metastases are currently being tested in phase III tri-als. Other agents under investigation include the mTOR inhibitors everolimus and temsirolimus, vari-ous inhibitors of IGF-1R and custirsen (OGX-011), an innovative antisense oligonucleotide directed against the cytoprotective chaperone, clusterin. Tumor-induced epigenetic aberrations believed to be critical for androgen receptor mediated signaling have been targeted with histone deacetylase inhibitors such as vorinsostat and panobinostat (LHB589).
The slow progression and high expression of tumor associated antigens have stimulated the devel-opment of several immutherapeutic approaches. The FDA recently approved the fi rst therapeutic vaccine, sipuleucel-T, for asymptomatic or minimally symp-tomatic metastatic androgen resistant prostate can-cer. Sipuleucel-T consists of autologous dendritic cells derived from a patient ’ s own peripheral mono-nuclear cells. A recent phase III trial reported a sur-vival advantage of four months among men receiving this agent versus those receiving placebo. Other examples of vaccine approaches under evaluation include vector based strategies (Prostvac) or whole tumor cell vaccines (GVAX). Ipilipumab, an anti-CTLA4 monoclonal antibody, represents another immunotherapeutic approach under evaluation.
Several agents targeting bone metastases include denosumab, a fully human monoclonal antibody that specifi cally binds to the ligand of RANK. This agent was recently reported to be superior to zolendronic acid in delaying or preventing bone fractures. Radium-223 is a new alpha-emitting bone-seeking radiopharmaceutical which shows promise against bone metastases. A number of endothelin A receptor targeted agents such as Atrasentan and ZD4054 are also under evaluation.
Considering the large number of compounds under evaluation the next decade shows promise for the treatment of advanced, androgen resistant pros-tate cancer. New biomarkers should drive clinical
Copyright © 2022 FDOKUMEN




















