
Weyl spin-orbit-coupling-induced interactions in uniform and trapped atomic quantum fluids
Upload
khangminh22Category
view
3download
0
Quantum Memory in Atomic
Ensembles
Joshua Nunn
St. John’s College, Oxford
Submitted for the degree of Doctor of PhilosophyHilary Term 2008
Supervised byProf. Ian A. Walmsley
Clarendon LaboratoryUniversity of Oxford
United Kingdom

For glory. I mean Gloria.

Abstract
This thesis is a predominantly theoretical study of light storage in atomic en-
sembles. The efficiency of ensemble quantum memories is analyzed and optimized
using the techniques of linear algebra. Analytic expressions describing the memory
interaction in both EIT and Raman regimes are derived, and numerical methods
provide solutions where the analytic expressions break down. A three dimensional
numerical model of off-axis retrieval is presented. Multimode storage is considered,
and the EIT, Raman, CRIB and AFC protocols are analyzed. It is shown that
inhomogeneous broadening improves the multimode capacity of a memory. Raman
storage in a diamond crystal is shown to be feasible. Finally, experimental progress
toward implementing a Raman quantum memory in cesium vapour is described.

Acknowledgements
I have been very lucky to work with a fantastic set of people. I owe a debt ofgratitude to my supervisor Ian Walmsley, whose implacable good humour alwaystransmutes frustration into comedy, and who has overseen my experimental failureswith only mild panic. Much of the theory was conceived in the course of meetingswith Karl Surmacz, who has been Dr. Surmacz for a year already. I fried my firstlaser in the lab with Felix Waldermann, and his patience and subtle sense of humourare missed — he is also qualified and long-gone! My current postdocs Virginia Lorenzand Ben Sussman have been a continual source of exciting discussion, and NorthAmerican optimism. We’re gonna make it work guys! And no misunderstandingcan survive a keen frisking at the hands of Klaus Reim, who is currently completinghis D.Phil — and mine — within our group. KC Lee has made the transitionfrom theorist to experimentalist, without a blip in his coffee intake, and he remainsan inspiration. The help and encouragement of Dieter Jaksch, and more recentlyChristoph Simon, are greatly appreciated. Thanks must also be due to my officeneighbours: Pete Mosley, who introduced me to the concept of a progress chart, andwith it a reification of inadequacy, and Dave Crosby, who go-karts better than hesings.
The rest of the ultrafast group divides cleanly into those who drink tea and thosewho do not. A great big thank you to the tea drinkers: you understand that a teabreak is more than a bathroom break with a drink. It lies at the heart of whatit means to prevaricate. Adam Wyatt knows this. He is a tea soldier. As for thetea-less philistines (you know who you are), I have nothing to say to you. (dramaticpause). Nothing.
Thanks to my Oxford massif, Andy Scott and Tom Rowlands-Rees, who know agood lunch when they see one. And in that vein, thanks to Matthijs Branderhorst,who along with Ben, introduced me to the burrito. There is a growing ultrafastdiaspora — good people in far-off places — and of these I should like to big-upDaryl Achilles and Jeff Lundeen, who are awethome even when in errr-rr.
I could not have made it this far without the support of Sonia: there can be fewless attractive prospects than the unshaven maniac that is a D.Phil student writingup. Thank you for keeping me sane!
And lastly my parents. Thanks mum and thanks dad! Next up: driving license.

Contents
1 Introduction 1
1.1 Classical Computation . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.2 Quantum Computation . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.1 Qubits . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.2 Noise . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.2.3 No cloning . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1.2.4 Universality . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.3 Quantum Memory . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.4 Linear Optics Quantum Computing . . . . . . . . . . . . . . . . . . 11
1.5 Quantum Communication . . . . . . . . . . . . . . . . . . . . . . . . 13
1.6 Quantum Repeaters . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
1.6.1 The Ekert protocol . . . . . . . . . . . . . . . . . . . . . . . . 21
1.6.2 Entanglement Swapping . . . . . . . . . . . . . . . . . . . . . 22
1.6.3 Entanglement Purification . . . . . . . . . . . . . . . . . . . . 24
1.6.4 The DLCZ protocol and number state entanglement . . . . . 24

CONTENTS v
1.7 Modified DLCZ with Quantum Memories . . . . . . . . . . . . . . . 31
2 Quantum Memory: Approaches 35
2.1 Cavity QED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
2.2 Free space coupling . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
2.3 Ensembles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
2.3.1 EIT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
2.3.2 Raman . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
2.3.3 CRIB . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
2.3.4 AFC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
2.4 Continuous Variables . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
3 Optimization 67
3.1 The Singular Value Decomposition . . . . . . . . . . . . . . . . . . . 70
3.1.1 Unitary invariance . . . . . . . . . . . . . . . . . . . . . . . . 75
3.1.2 Connection with Eigenvalues . . . . . . . . . . . . . . . . . . 75
3.1.3 Hermitian SVD . . . . . . . . . . . . . . . . . . . . . . . . . . 76
3.1.4 Persymmetry . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
3.2 Norm maximization . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
3.3 Continuous maps . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
3.3.1 Normally and Anti-normally ordered kernels. . . . . . . . . . 81
3.3.2 Memory Optimization. . . . . . . . . . . . . . . . . . . . . . . 81
3.3.3 Unitary invariance . . . . . . . . . . . . . . . . . . . . . . . . 82

CONTENTS vi
3.4 Optimizing storage followed by retrieval . . . . . . . . . . . . . . . . 85
3.5 A Simple Example . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
4 Equations of motion 92
4.1 Interaction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
4.2 Electric Field . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
4.3 Dipole Operator . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
4.3.1 Parity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
4.4 Hamiltonian . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
4.5 Linear approximation (1) . . . . . . . . . . . . . . . . . . . . . . . . 102
4.6 Rotating Wave Approximation . . . . . . . . . . . . . . . . . . . . . 103
4.7 Unwanted Coupling . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
4.8 Linear Approximation (2) . . . . . . . . . . . . . . . . . . . . . . . . 106
4.9 Propagation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
4.10 Paraxial and SVE approximations . . . . . . . . . . . . . . . . . . . 110
4.11 Continuum Approximation . . . . . . . . . . . . . . . . . . . . . . . 112
4.12 Spontaneous Emission and Decoherence . . . . . . . . . . . . . . . . 116
5 Raman & EIT Storage 120
5.1 One Dimensional Approximation . . . . . . . . . . . . . . . . . . . . 120
5.2 Solution in k-space . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
5.2.1 Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . 123
5.2.2 Transformed Equations . . . . . . . . . . . . . . . . . . . . . 124

CONTENTS vii
5.2.3 Optimal efficiency . . . . . . . . . . . . . . . . . . . . . . . . 125
5.2.4 Solution in Wavelength Space . . . . . . . . . . . . . . . . . . 129
5.2.5 Including the Control . . . . . . . . . . . . . . . . . . . . . . 134
5.2.6 An Exact Solution: The Rosen-Zener case . . . . . . . . . . . 137
5.2.7 Adiabatic Limit . . . . . . . . . . . . . . . . . . . . . . . . . . 145
5.2.8 Reaching the optimal efficiency . . . . . . . . . . . . . . . . . 152
5.2.9 Adiabatic Approximation . . . . . . . . . . . . . . . . . . . . 155
5.3 Raman Storage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
5.3.1 Validity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
5.3.2 Matter Biased Limit . . . . . . . . . . . . . . . . . . . . . . . 167
5.3.3 Transmitted Modes. . . . . . . . . . . . . . . . . . . . . . . . 168
5.4 Numerical Solution . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182
5.4.1 Dispersion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 187
5.5 Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 188
6 Retrieval 190
6.1 Collinear Retrieval . . . . . . . . . . . . . . . . . . . . . . . . . . . . 190
6.1.1 Forward Retrieval . . . . . . . . . . . . . . . . . . . . . . . . 191
6.2 Backward Retrieval . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
6.3 Phasematched Retrieval . . . . . . . . . . . . . . . . . . . . . . . . . 207
6.3.1 Dispersion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
6.3.2 Scheme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 211
6.4 Full Propagation Model . . . . . . . . . . . . . . . . . . . . . . . . . 213

CONTENTS viii
6.4.1 Diffraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215
6.4.2 Control Field . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
6.4.3 Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . 218
6.4.4 Read out . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 220
6.4.5 Efficiency . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 222
6.5 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223
6.6 Angular Multiplexing . . . . . . . . . . . . . . . . . . . . . . . . . . . 228
6.6.1 Optimizing the carrier frequencies . . . . . . . . . . . . . . . 229
6.6.2 Capacity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 231
7 Multimode Storage 234
7.1 Multimode Capacity from the SVD . . . . . . . . . . . . . . . . . . . 235
7.1.1 Schmidt Number . . . . . . . . . . . . . . . . . . . . . . . . . 237
7.1.2 Threshold multimode capacity . . . . . . . . . . . . . . . . . 239
7.2 Multimode scaling for EIT and Raman memories . . . . . . . . . . . 241
7.2.1 A spectral perspective . . . . . . . . . . . . . . . . . . . . . . 242
7.3 CRIB . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 245
7.3.1 lCRIB . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 246
7.3.2 Simplified Kernel . . . . . . . . . . . . . . . . . . . . . . . . . 249
7.3.3 tCRIB . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 254
7.4 Broadened Raman . . . . . . . . . . . . . . . . . . . . . . . . . . . . 262
7.5 AFC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 268

CONTENTS ix
8 Optimizing the Control 276
8.1 Adiabatic shaping . . . . . . . . . . . . . . . . . . . . . . . . . . . . 277
8.2 Non-adiabatic shaping . . . . . . . . . . . . . . . . . . . . . . . . . . 279
9 Diamond 286
9.1 Diamond Scheme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
9.2 Quantization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 288
9.3 Acoustic and Optical Phonons . . . . . . . . . . . . . . . . . . . . . 290
9.3.1 Decay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 291
9.3.2 Energy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 293
9.4 Raman interaction . . . . . . . . . . . . . . . . . . . . . . . . . . . . 294
9.4.1 Excitons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 294
9.4.2 Deformation Potential . . . . . . . . . . . . . . . . . . . . . . 296
9.5 Propagation in Diamond . . . . . . . . . . . . . . . . . . . . . . . . . 299
9.5.1 Hamiltonian . . . . . . . . . . . . . . . . . . . . . . . . . . . . 300
9.5.2 Electron-radiation interaction . . . . . . . . . . . . . . . . . . 301
9.5.3 Electron-lattice interaction . . . . . . . . . . . . . . . . . . . 307
9.5.4 Crystal energy . . . . . . . . . . . . . . . . . . . . . . . . . . 309
9.6 Heisenberg equations . . . . . . . . . . . . . . . . . . . . . . . . . . . 310
9.6.1 Adiabatic perturbative solution . . . . . . . . . . . . . . . . . 311
9.7 Signal propagation . . . . . . . . . . . . . . . . . . . . . . . . . . . . 314
9.8 Coupling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 318
9.9 Selection Rules . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 321

CONTENTS x
9.10 Noise . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 322
10 Experiments 323
10.1 Systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 323
10.2 Thallium . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 325
10.3 Cesium . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 326
10.4 Cell . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 328
10.4.1 Temperature control . . . . . . . . . . . . . . . . . . . . . . . 329
10.4.2 Magnetic shielding . . . . . . . . . . . . . . . . . . . . . . . . 329
10.5 Buffer gas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 331
10.6 Control pulse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 333
10.6.1 Pulse duration . . . . . . . . . . . . . . . . . . . . . . . . . . 333
10.6.2 Tuning . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 336
10.6.3 Shaping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337
10.7 Pulse picker . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337
10.8 Stokes scattering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 338
10.9 Coupling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 342
10.9.1 Optical depth . . . . . . . . . . . . . . . . . . . . . . . . . . . 342
10.9.2 Rabi frequency . . . . . . . . . . . . . . . . . . . . . . . . . . 344
10.9.3 Raman memory coupling . . . . . . . . . . . . . . . . . . . . 346
10.9.4 Focussing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 347
10.10Line shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 349
10.11Effective depth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 351

CONTENTS xi
10.12Optical pumping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 353
10.12.1 Pumping efficiency . . . . . . . . . . . . . . . . . . . . . . . . 355
10.13Filtering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 356
10.13.1 Polarization filtering . . . . . . . . . . . . . . . . . . . . . . . 358
10.13.2 Lyot filter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358
10.13.3 Etalons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 359
10.13.4 Spectrometer . . . . . . . . . . . . . . . . . . . . . . . . . . . 362
10.13.5 Spatial filtering . . . . . . . . . . . . . . . . . . . . . . . . . . 362
10.14Signal pulse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364
10.15Planned experiment . . . . . . . . . . . . . . . . . . . . . . . . . . . 366
11 Summary 369
11.1 Future work . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 372
A Linear algebra 374
A.1 Vectors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 375
A.1.1 Adjoint vectors . . . . . . . . . . . . . . . . . . . . . . . . . . 377
A.1.2 Inner product . . . . . . . . . . . . . . . . . . . . . . . . . . . 378
A.1.3 Norm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 379
A.1.4 Bases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 380
A.2 Matrices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 381
A.2.1 Outer product . . . . . . . . . . . . . . . . . . . . . . . . . . 384
A.2.2 Tensor product . . . . . . . . . . . . . . . . . . . . . . . . . . 385

CONTENTS xii
A.3 Eigenvalues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 388
A.3.1 Commutators . . . . . . . . . . . . . . . . . . . . . . . . . . . 389
A.4 Types of matrices . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 391
A.4.1 The identity matrix . . . . . . . . . . . . . . . . . . . . . . . 391
A.4.2 Inverse matrix . . . . . . . . . . . . . . . . . . . . . . . . . . 392
A.4.3 Hermitian matrices . . . . . . . . . . . . . . . . . . . . . . . . 393
A.4.4 Diagonal matrices . . . . . . . . . . . . . . . . . . . . . . . . 394
A.4.5 Unitary matrices . . . . . . . . . . . . . . . . . . . . . . . . . 396
B Quantum mechanics 399
B.1 Postulates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 400
B.1.1 State vector . . . . . . . . . . . . . . . . . . . . . . . . . . . . 400
B.1.2 Observables . . . . . . . . . . . . . . . . . . . . . . . . . . . . 400
B.1.3 Measurements . . . . . . . . . . . . . . . . . . . . . . . . . . 400
B.1.4 Dynamics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 401
B.2 The Heisenberg Picture . . . . . . . . . . . . . . . . . . . . . . . . . 403
B.2.1 The Heisenberg interaction picture . . . . . . . . . . . . . . . 405
C Quantum optics 407
C.1 Modes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 407
C.2 Quantum states of light . . . . . . . . . . . . . . . . . . . . . . . . . 410
C.2.1 Fock states . . . . . . . . . . . . . . . . . . . . . . . . . . . . 410
C.2.2 Creation and Annihilation operators . . . . . . . . . . . . . . 411

CONTENTS xiii
C.3 The electric field . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 414
C.4 Matter-Light Interaction . . . . . . . . . . . . . . . . . . . . . . . . . 415
C.4.1 The A.p Interaction . . . . . . . . . . . . . . . . . . . . . . . 415
C.4.2 The E.d Interaction . . . . . . . . . . . . . . . . . . . . . . . 418
C.5 Dissipation and Fluctuation . . . . . . . . . . . . . . . . . . . . . . . 422
D Sundry Analytical Techniques 427
D.1 Contour Integration . . . . . . . . . . . . . . . . . . . . . . . . . . . 427
D.1.1 Cauchy’s Integral Formula . . . . . . . . . . . . . . . . . . . . 429
D.1.2 Typical example . . . . . . . . . . . . . . . . . . . . . . . . . 430
D.2 The Dirac Delta Function . . . . . . . . . . . . . . . . . . . . . . . . 432
D.3 Fourier Transforms . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434
D.3.1 Bilateral Transform . . . . . . . . . . . . . . . . . . . . . . . 434
D.3.2 Unitarity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434
D.3.3 Inverse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 435
D.3.4 Shift . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 435
D.3.5 Convolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . 436
D.3.6 Transform of a Derivative . . . . . . . . . . . . . . . . . . . . 436
D.4 Unilateral Transform . . . . . . . . . . . . . . . . . . . . . . . . . . . 437
D.4.1 Shift . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 438
D.4.2 Convolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . 438
D.4.3 Transform of a Derivative . . . . . . . . . . . . . . . . . . . . 439
D.4.4 Laplace Transform . . . . . . . . . . . . . . . . . . . . . . . . 440

CONTENTS xiv
D.5 Bessel Functions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 440
D.5.1 Orthogonality . . . . . . . . . . . . . . . . . . . . . . . . . . . 441
D.5.2 Memory Propagator . . . . . . . . . . . . . . . . . . . . . . . 442
D.5.3 Optimal Eigenvalue Kernel . . . . . . . . . . . . . . . . . . . 445
E Numerics 447
E.1 Spectral Collocation . . . . . . . . . . . . . . . . . . . . . . . . . . . 449
E.1.1 Polynomial Differentiation Matrices . . . . . . . . . . . . . . 452
E.1.2 Chebyshev points . . . . . . . . . . . . . . . . . . . . . . . . . 453
E.2 Time-stepping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 455
E.3 Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . 457
E.4 Constructing the Solutions . . . . . . . . . . . . . . . . . . . . . . . . 459
E.5 Numerical Construction of a Green’s Function . . . . . . . . . . . . . 462
E.6 Spectral Methods for Two Dimensions . . . . . . . . . . . . . . . . . 465
F Atomic Vapours 471
F.1 Vapour pressure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 471
F.2 Oscillator strengths . . . . . . . . . . . . . . . . . . . . . . . . . . . . 474
F.3 Line broadening . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 476
F.3.1 Doppler broadening . . . . . . . . . . . . . . . . . . . . . . . 476
F.3.2 Pressure broadening . . . . . . . . . . . . . . . . . . . . . . . 477
F.3.3 Power broadening . . . . . . . . . . . . . . . . . . . . . . . . 480
F.4 Raman polarization . . . . . . . . . . . . . . . . . . . . . . . . . . . 481

List of Figures
1.1 The state space of a qubit . . . . . . . . . . . . . . . . . . . . . . . . 5
1.2 The BB84 protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
1.3 Entanglement swapping . . . . . . . . . . . . . . . . . . . . . . . . . 23
1.4 Single-rail entanglement swapping . . . . . . . . . . . . . . . . . . . 26
1.5 QKD with single-rail entanglement . . . . . . . . . . . . . . . . . . . 27
1.6 Λ-level structure of atoms for DLCZ . . . . . . . . . . . . . . . . . . 29
1.7 Generation of number state entanglement in DLCZ . . . . . . . . . . 31
1.8 Modification to DLCZ with photon sources and quantum memories . 33
2.1 The simplest quantum memory . . . . . . . . . . . . . . . . . . . . . 36
2.2 Adding a dark state . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
2.3 Cavity QED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
2.4 Confocal coupling in free space . . . . . . . . . . . . . . . . . . . . . 40
2.5 Atomic ensemble memory . . . . . . . . . . . . . . . . . . . . . . . . 41
2.6 EIT. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
2.7 Stopping light with EIT. . . . . . . . . . . . . . . . . . . . . . . . . . 45

LIST OF FIGURES xvi
2.8 Raman storage. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
2.9 CRIB storage. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
2.10 tCRIB vs. lCRIB. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
2.11 AFC storage. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
2.12 Wigner distributions. . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
2.13 Atomic quadratures. . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
2.14 QND memory. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
2.15 Level scheme for a QND memory. . . . . . . . . . . . . . . . . . . . . 66
3.1 Storage map. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
3.2 Linear transformation . . . . . . . . . . . . . . . . . . . . . . . . . . 72
3.3 Persymmetry. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
4.1 The Λ-system again. . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
4.2 Time-ordering. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
4.3 Useful and nuisance couplings. . . . . . . . . . . . . . . . . . . . . . 105
5.1 Quantum memory boundary conditions. . . . . . . . . . . . . . . . . 124
5.2 Bessel zeros. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
5.3 Optimal storage efficiency. . . . . . . . . . . . . . . . . . . . . . . . . 133
5.4 The Rosen-Zener model. . . . . . . . . . . . . . . . . . . . . . . . . . 140
5.5 Raman efficiency. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
5.6 Raman storage as a beamsplitter. . . . . . . . . . . . . . . . . . . . . 181
5.7 Modified DLCZ protocol with partial storage. . . . . . . . . . . . . . 182

LIST OF FIGURES xvii
5.8 Comparison of predictions for the optimal input modes in the adia-
batic limit. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
5.9 Comparison of predictions for the optimal input modes outside the
adiabatic limit. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
5.10 Broadband Raman storage. . . . . . . . . . . . . . . . . . . . . . . . 187
5.11 Broadband EIT storage. . . . . . . . . . . . . . . . . . . . . . . . . . 188
6.1 Forward retrieval. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
6.2 Phasematching considerations for backward retrieval. . . . . . . . . . 200
6.3 Backward Retrieval. . . . . . . . . . . . . . . . . . . . . . . . . . . . 206
6.4 Non-collinear phasematching. . . . . . . . . . . . . . . . . . . . . . . 209
6.5 Efficient, phasematched memory for positive and negative phase mis-
matches. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 209
6.6 Focussed beams. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
6.7 Effectiveness of our phasematching scheme. . . . . . . . . . . . . . . 224
6.8 Comparing phasematched and collinear efficiencies. . . . . . . . . . . 225
6.9 Angular multiplexing. . . . . . . . . . . . . . . . . . . . . . . . . . . 230
6.10 Minimum momentum mismatch. . . . . . . . . . . . . . . . . . . . . 232
7.1 Bright overlapping modes are distinct. . . . . . . . . . . . . . . . . . 236
7.2 Visualizing the multimode capacity. . . . . . . . . . . . . . . . . . . 237
7.3 The appearance of a multimode Green’s function. . . . . . . . . . . . 239
7.4 Multimode scaling for Raman and EIT memories. . . . . . . . . . . . 244

LIST OF FIGURES xviii
7.5 Scaling of Schmidt number with broadening. . . . . . . . . . . . . . 252
7.6 Comparison of the predictions of the kernels (7.23) and (7.18). . . . 253
7.7 Understanding the linear multimode scaling of lCRIB. . . . . . . . . 254
7.8 Multimode scaling for CRIB memories. . . . . . . . . . . . . . . . . . 261
7.9 The multimode scaling of a broadened Raman protocol. . . . . . . . 268
7.10 The multimode scaling of the AFC memory protocol. . . . . . . . . . 275
8.1 Adiabatic control shaping. . . . . . . . . . . . . . . . . . . . . . . . . 284
8.2 Non-adiabatic control shaping. . . . . . . . . . . . . . . . . . . . . . 285
9.1 The crystal structure of diamond. . . . . . . . . . . . . . . . . . . . . 287
9.2 Phonon aliasing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 290
9.3 Phonon dispersion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 292
9.4 Band structure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 295
9.5 An exciton. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 296
9.6 The Raman interaction in diamond. . . . . . . . . . . . . . . . . . . 298
10.1 Observing Stokes scattering as a first step. . . . . . . . . . . . . . . . 325
10.2 Thallium atomic structure. . . . . . . . . . . . . . . . . . . . . . . . 326
10.3 Cesium atomic structure. . . . . . . . . . . . . . . . . . . . . . . . . 327
10.4 First order autocorrelation. . . . . . . . . . . . . . . . . . . . . . . . 334
10.5 Second order interferometric autocorrelation. . . . . . . . . . . . . . 336
10.6 Stokes scattering efficiency. . . . . . . . . . . . . . . . . . . . . . . . 342
10.7 Cesium optical depth. . . . . . . . . . . . . . . . . . . . . . . . . . . 345

LIST OF FIGURES xix
10.8 Cesium D2 absorption spectrum. . . . . . . . . . . . . . . . . . . . . 350
10.9 Absorption linewidth. . . . . . . . . . . . . . . . . . . . . . . . . . . 352
10.10Equal populations destroy quantum memory. . . . . . . . . . . . . . 354
10.11Optical pumping. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 355
10.12Verifying efficient optical pumping. . . . . . . . . . . . . . . . . . . . 357
10.13Lyot filter. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 360
10.14Stokes filtering. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 361
10.15Backward Stokes scattering. . . . . . . . . . . . . . . . . . . . . . . . 364
10.16A possible design for demonstration of a cesium quantum memory. . 368
A.1 A vector. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 376
A.2 The inner product of two vectors. . . . . . . . . . . . . . . . . . . . . 380
A.3 A matrix acting on a vector. . . . . . . . . . . . . . . . . . . . . . . . 383
A.4 Eigenvectors and eigenvalues. . . . . . . . . . . . . . . . . . . . . . . 389
A.5 Non-commuting operations. . . . . . . . . . . . . . . . . . . . . . . . 390
A.6 A unitary transformation. . . . . . . . . . . . . . . . . . . . . . . . . 396
C.1 Symmetrized photons. . . . . . . . . . . . . . . . . . . . . . . . . . . 413
D.1 Contour integrals. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 429
D.2 Upper closure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 432
D.3 Integration limits. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 439
D.4 Lower closure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 444
E.1 The method of lines. . . . . . . . . . . . . . . . . . . . . . . . . . . . 449

LIST OF FIGURES xx
E.2 Periodic extension. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 451
E.3 Chebyshev Points. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 455
E.4 Example solutions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 462
E.5 A numerically constructed Green’s function. . . . . . . . . . . . . . . 464
E.6 Spectral methods in two dimensions. . . . . . . . . . . . . . . . . . . 470
F.1 Vapour pressure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 473
F.2 The Doppler shift. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 477
F.3 Collisions in a vapour. . . . . . . . . . . . . . . . . . . . . . . . . . . 478
F.4 Polarization selection rules. . . . . . . . . . . . . . . . . . . . . . . . 483
F.5 Alternative scattering pathways. . . . . . . . . . . . . . . . . . . . . 486

Chapter 1
Introduction
The prospect of building a quantum computer, with speed and power far outstripping
the best possible classical computers, has motivated an enormous and sustained
research effort over the last two decades. In this thesis we explore a number of
candidates for the ‘memory’ that would be required by such a device. As we will
see, building a quantum memory is considerably harder than fabricating the RAM
chips used by modern computers. For instance, it is not possible to copy quantum
information, nor can quantum information be digitized. These facts make quantum
storage particularly vulnerable to noise, and loss — problems for which solutions
must be found before quantum computation can mature into a viable technology.
The bulk of this thesis is concerned with optimization of the efficiency and storage
capacity of a quantum memory. We focus on optical memories, in which a pulse of
light is ‘stopped’ for a controllable period, before being re-released.
The structure of the thesis is as follows. In this chapter we introduce the concepts

2
of quantum computing and quantum communication, and we discuss the context
and motivation for the present work on quantum memories. In Chapter 2 we survey
the various approaches to quantum memory, and we describe the principles behind
the memory protocols analyzed later. Chapter 3 introduces the mathematical basis
for our approach to analyzing and optimizing ensemble memories — the Green’s
function and its singular value decomposition. In Chapter 4 we derive the equations
of motion describing the quantum memory interaction in an ensemble of Λ-type
atoms. In Chapter 5 we apply the techniques of Chapter 3 to this interaction. Several
new results are derived, and connections are made with previous work. Chapter 6
is concerned with retrieval of the stored excitations from a Λ-ensemble. It is shown
that both forward and backward retrieval are problematic. A numerical model
is presented that confirms the efficacy of an off-axis geometry, which solves these
problems. In Chapter 7 we move on to consider multimode storage. Our formalism
provides a natural way to calculate the multimode capacity of a memory, and we
study the multimode scaling of all the memory protocols introduced in Chapter 2.
Chapter 8 describes how to optimize a Λ-type memory by shaping the ‘control pulse’.
In Chapter 9 we study the Raman interaction in a diamond crystal: we show that a
diamond Raman quantum memory is feasible. Finally in Chapter 10 we review our
attempts to implement a Raman quantum memory in the laboratory, using cesium
vapour.
But let us begin at the beginning.

1.1 Classical Computation 3
1.1 Classical Computation
Classical computers are conventional computers, like the one I am using to typeset
this document. Their importance as enablers of technological progress, as well as
their utility as a technology in their own right, attest to the fantastic potential of
classical computation. They are typified by the use of bit1 strings — sequences of
1’s and 0’s — to encode information. Information is processed by application of
binary logic to the bits. That is, Boolean operations such as OR, AND or not-AND
(NAND). This last operation is a universal gate, because any logic operation can be
constructed using only NAND gates. Such a gate can be implemented electronically
using a pair of transistors, millions of which can be combined on a single silicon
chip. The rest is history.
Computers have progressed in leaps and bounds over the last fifty years. In 1965
computers were developing so fast that Gordon Moore, a founder of the industrial
giant Intel, proposed a ‘law’, stipulating that the number of transistors comprising
a processor would double every year [2]. Incredibly, this exponential improvement in
computing power has persisted for over 40 years. But improvement by miniaturiza-
tion cannot continue indefinitely. The reason for this is that the physics of electronic
components undergoes a qualitative change at small scales: classical physics becomes
quantum physics. In his 1983 lectures on computation [3], Richard Feynman consid-1‘Bit’ first appeared in Claude Shannon’s 1948 paper on the theory of communication as a
contraction of ‘binary digit’ [1]; the name is apposite, since one bit is the smallest ‘piece’ or ‘chunk’of information there can be: one bit of information is one bit of information. Shannon attributesthe term to John Tukey, a creator of the digital Fourier transform, who is also credited with coiningthe word ‘software’.

1.2 Quantum Computation 4
ers the fate of classical computation as shrinking dimensions bring quantum effects
into play. Two years later David Deutsch published the first explicitly quantum
algorithm [4], demonstrating how quantum physics actually permits more powerful
computation than classical physics allows. A quantum computer, capable of har-
nessing this greater power, must process quantum information, encoded not with
ordinary bits, but with quantum bits.
1.2 Quantum Computation
1.2.1 Qubits
A quantum bit — a qubit2 — is an object with two mutually exclusive states, 0
and 1, say. The only difference with a classical bit is that the object is described
by quantum mechanics. Accordingly, we label the two states by the kets |0〉 and |1〉
(see Appendix B). These kets are to be thought of as vectors in a two dimensional
space: the state space of the qubit (see Figure 1.1). The classical property of mutual
exclusivity is manifested in the quantum formalism by requiring that |0〉 and |1〉 are
perpendicular to one another in the state space. In general, the state of the qubit
can be any vector, of length 1, in the state space. Since both the kets |0〉 and |1〉
have length 1, and since they point in perpendicular directions, an arbitrary qubit
state |ψ〉 can always be written as a linear combination of them,
|ψ〉 = α|0〉+ β|1〉, (1.1)2The term ‘qubit’ first appears in a paper by Benjamin Schumacher in 1995 [5]; he credits its
invention to a conversation with William Wootters.

1.2 Quantum Computation 5
where α and β are two numbers which must satisfy the normalization condition
|α|2 + |β|2 = 1. States like (1.1), which are a combination of the two mutually
exclusive states |0〉 and |1〉, are called superposition states, or just superpositions.
What does it mean to say that a qubit is in a superposition between its two mutually
exclusive states? Somehow it is both 0 and 1 at the same time. Physically, this is
like saying that a switch is both ‘open’ and ‘closed’, or that a lamp is both ‘on’ and
also ‘off’. Already, for the simplest possible system, without any real dynamics —
no interactions, nothing happening — we see that the basic structure of quantum
mechanics does not sit well with our intuition. Despite these interpretational diffi-sta
te space
Figure 1.1 A visual representation of the state space of a qubit
culties, superposition is central to the success of quantum mechanics. Atomic and
molecular physics, nuclear and particle physics, optics and electronics all make use of
superpositions to successfully explain processes and interactions. From the point of
view of computation, the existence of states like (1.1) provides a clue to the greater
capabilities of a quantum computer. Each qubit has two ‘parts’, the |0〉 part and the

1.2 Quantum Computation 6
|1〉 part; logical operations on qubits act on both parts together, and the output of
a calculation also has these two parts. So there’s some sense in which a qubit plays
the role of two classical bits, stuck ‘on top of eachother’. David Deutsch coined the
term quantum parallelism for this property — he considers it to be the strongest
evidence for the existence of parallel universes. The B-movie-esque connotations
of this ‘many-worlds’ view make it generally unpopular among physicists, but the
appeal of quantum computing remains, independently of how it is understood.
1.2.2 Noise
A potential difficulty associated with quantum computing is also apparent from
(1.1): the numbers α and β can be varied continuously (subject to the normaliza-
tion constraint). So the number of possible states |ψ〉 of a qubit is infinite! This also
hints at their greater information carrying capacity, but it means that they must
be carefully protected from the influence of noise. Classical bits have exactly two
states; if noise introduces some distortions, it is usually possible to correct these
simply by comparing the distorted bit to an ideal one. Only very large fluctuations
can make a 0 look like a 1, so the discrete structure of classical bits makes them
very robust. By contrast, a perturbed qubit state is also a valid qubit state. In
this respect, the difference between bits and qubits can be likened to the difference
between digital and analogue musical recordings: The quality of music reproduced
by a CD does not degrade gradually with time, whereas old cassettes sound progres-
sively worse as distortions creep into the waveform imprinted on the tape. In fact,

1.2 Quantum Computation 7
it is possible to correct errors by constructing codes involving bunches of qubits.
The invention of these codes in 1996 by Calderbank, Shor and Steane [6,7] was a
major milestone in demonstrating the practical viability of quantum computation.
Nonetheless these error correcting schemes currently require that noise is suppressed
below thresholds of a few percent, which makes techniques for isolating qubits from
noise a technological sine qua non.
1.2.3 No cloning
Another problematic aspect of quantum information is that it cannot be copied.
The proof of this fact is known as the no-cloning theorem [8]. Suppose that we have
a device which can copy a qubit. If we give it a qubit in state |ψ〉, and also a ‘blank’
qubit in some standard initial state |blank〉, this machine spits out our original qubit,
plus a clone, both in the state |ψ〉. In Dirac notation, using kets, the action of our
qubit photocopier is written as
U |blank〉|ψ〉 = |ψ〉|ψ〉. (1.2)
Here U is the unitary transformation implemented by our machine. Unitary trans-
formations are those which preserve the lengths of the kets upon which they act.
Since all physical states have length 1, and any process must produce physical states
from physical states, it follows that all processes are described by length-preserving
— unitary — transformations (see §B.1.4 in Appendix B). Had we fed our machine

1.2 Quantum Computation 8
a different state, for example |φ〉, we would have
U |blank〉|φ〉 = |φ〉|φ〉. (1.3)
The length of a ket |ϕ〉 is defined by taking the scalar product 〈ϕ|ϕ〉 of |ϕ〉 with
itself (see §A.1.2 in Appendix A). To prove the impossibility of cloning, we take the
scalar product of the first relation (1.2) with the second, (1.3).
〈ψ|〈blank|U †U |blank〉|φ〉 = 〈ψ|〈ψ||φ〉|φ〉. (1.4)
The U acting on |blank〉 on the left hand side does not change its length, which is
just equal to 1, so the result simplifies to
〈ψ|φ〉 = 〈ψ|φ〉2. (1.5)
Clearly, this expression does not hold for arbitrary choices of |ψ〉 and |φ〉, and there-
fore cloning an arbitrary qubit is impossible. In fact, (1.5) is only true when 〈ψ|φ〉 is
either 1 or 0. The first case corresponds to |ψ〉 = |φ〉, which says that it is possible to
build a machine that can make copies of one particular, pre-determined state. The
second case occurs only when |ψ〉 and |φ〉 are perpendicular, as is the case for |0〉 and
|1〉. This says that it is possible to clone mutually exclusive states. Indeed, this is
precisely what classical computers are doing when they copy digitized information.
An immediate consequence of the no-cloning theorem is that a quantum memory

1.2 Quantum Computation 9
must work in a qualitatively different way to a classical computer memory. To
store quantum information, that information must be transferred to the memory,
rather than simply copied to it. It is never possible to ‘save a back-up’, as we
routinely do with classical computers. To build a quantum memory, we must find
an interaction between information carrier and storage medium which ‘swaps’ their
quantum states, so that the storage medium ends up with all the information, with
nothing left behind. In this thesis we will examine various ways of accomplishing this
optically, by considering collections of atoms which essentially ‘swallow’ a photon
in a controlled way, completely transferring the quantum state of an optical field to
that of the atoms.
1.2.4 Universality
Any classical computation is possible provided that one is able to apply NAND gates
to pairs of bits. What is required to perform arbitrary quantum computations?
This question is not trivial, but the answer is fortuitously simple [9]. Any quantum
computation can be performed, provided that one is able to arbitrarily control the
state of any qubit (single-qubit rotations), and provided that one can make pairs of
qubits interact with one another (two-qubit gates). It is generally sufficient to have
only a single type of interaction, so long as the final state of both interacting qubits
depends in some way on the initial state of both qubits. Such a gate is known as an
entangling gate, and they are notoriously difficult to implement.

1.3 Quantum Memory 10
1.3 Quantum Memory
In the light of the preceding discussion, a quantum memory can be understood as a
physical system that is well protected from noise, and that can be made to interact
with information carriers so that their quantum state is transferred into, or out of,
the memory. Note how we have distinguished the system comprising the memory
from the information carriers. In many cases, this distinction is artificial. For in-
stance, in ion trap quantum computing [10], the hyperfine states of calcium ions are
used as qubits. These ions are isolated from their noisy environment by trapping
them with oscillating electric fields; the quantum states of the qubits therefore re-
main un-distorted for long periods (on the order of seconds), and so there is no need
to transfer these states into a separate memory. But there are other proposals for
quantum computing that make explicit use of quantum memories. An example is
the use of nitrogen-vacancy centres in diamond for quantum computing [11,12]. Here
a single electron from a nitrogen atom, lodged in a diamond crystal and surrounded
by carbon atoms, is used as a qubit. The electron qubit can be controlled with laser
pulses to perform computations, but this very sensitivity to light makes it suscepti-
ble to damage from noise. Therefore a scheme was devised to transfer the quantum
state of the electron to that of a nearby carbon nucleus. The carbon nucleus is
de-coupled from the optical field, and it can be used to store quantum information
for many minutes.
A common theme among such computation schemes is an antagonism between
controllability and noise-resilience. That is, systems which are easily manipulated

1.4 Linear Optics Quantum Computing 11
and controlled with external fields are susceptible to noise from those same fields,
while well-isolated systems that are not badly affected by noise are generally hard
to access and control in order to perform computations. This trade-off leads to a
natural division of labour between systems that are easily manipulated, but short-
lived, and systems that are not easily controlled, but long-lived. Many quantum
computing architectures put both types of system to use, the former as quantum
processor, the latter as quantum memory.
1.4 Linear Optics Quantum Computing
Since James Clerk Maxwell wrote down the equations of electromagnetism in 1873,
physics has undergone profound upheavals at least twice, with the development of
both Relativity and Quantum Mechanics in the early twentieth century. Maxwell’s
equations have weathered these storms with astonishing fortitude, being both rel-
ativistically covariant and directly applicable in quantum field theory. They are
probably the oldest correct equations in physics. Implicit within them is a descrip-
tion of the photon, the quantum of the electromagnetic field. Photons come with
one of two polarizations, and superposition states of these polarizations are readily
prepared in the lab. In addition, they are themselves discrete entities, and it is pos-
sible to generate superpositions of different numbers of photons. Photons therefore
embody the archetypal qubit, and for this reason Maxwell’s equations remain as
central to the emerging discipline of quantum information processing as they were
to the pioneers of telegraphy and radio.

1.4 Linear Optics Quantum Computing 12
Photons occupy a frustrating territory on the balance sheet of usefulness for
quantum computation. They are ideal qubits, and arbitrary manipulation of their
polarization and number states can be accomplished with simple waveplates, beam-
splitters and phase-shifters. That is, single-qubit rotations are ‘cheap’. Unfortu-
nately, entangling gates between photons are much more difficult to realise. This
is unsurprising, since such a gate requires that two photons be made to interact
with one another, and it is well known that light does not generally interact with
light: torch beams do not ‘bounce off’ each other; rather they pass through each
other unaffected. In 2001 Emanuel Knill, Raymond Laflamme and Gerard Mil-
burn showed how to overcome these difficulties by careful use of measurements [13],
making universal quantum computation possible with only ‘linear optics’. Further
developments [14,15] have cemented linear optics quantum computing (LOQC) as an
important paradigm for the future of quantum computation. However the two-qubit
gates proposed are generally non-deterministic. As the number of gates required in
a computational step increases, the probability that all gates are implemented suc-
cessfully decreases exponentially, so that large computations must be repeated many
times for yielding reliable answers. This problem of scalability can be mitigated if
the photons output from successful gates can be stored until all the required gates
succeed. But photons generally have a short lifetime because they travel at the
speed of light: if they are confined in a laboratory they must be trapped by mirrors
(in a cavity) or by a waveguide (optical fibre), and absorption or scattering losses
are inevitable on time scales of milliseconds or greater [16]. Therefore the ability to

1.5 Quantum Communication 13
transfer the quantum state of a photon into a quantum memory would be a boon to
LOQC.
Another possibility for quantum computing with photons is to implement two-
qubit gates inside a quantum memory. Single-qubit operations are easily performed
on the photons directly; when interactions are needed, the photons are transferred
to atomic excitations which can be manipulated with external fields to accomplish
the entangling gates [17–19].
Applications such as these constitute the most ambitious motivation for the study
of optical quantum memories. In the next section we will see that quantum mem-
ories are also required in extending the range of so-called quantum communication
protocols, which provide guaranteed security from eavesdroppers.
1.5 Quantum Communication
Although practical quantum computing remains beyond the reach of current tech-
nology, another application of quantum mechanics has already made the leap into
the commercial sector. Quantum Key Distribution (QKD) is a technique which al-
lows two communicating parties to be absolutely certain, in so far as the laws of
physics are known to be correct, that their messages have not been intercepted [20].
It is possible to purchase QKD systems from two companies: MagiQ based in New
York, and ID Quantique in Geneva; many other businesses are incumbent, and the
market for such guaranteed-secure communication is estimated at around a billion
dollars annually. The idea behind QKD is simple: if Alice sends a message to Bob

1.5 Quantum Communication 14
in which she has substituted each letter for a different one, in a completely ran-
dom way, neither Bob, nor anyone else, can decode the message, unless Alice tells
them how she did the substitution. This information is known as the key, and only
someone in possession of the full key has access to the contents of Alice’s message.
Encrypting messages in this way is the oldest and simplest method of encryption. It
is absolutely and completely secure, provided that only the intended recipient has
access to the key. Once the key has been used, it should not be used again, since
with repeated use an eavesdropper, conventionally called Eve, might start to see
patterns in the encrypted messages and begin to guess the substitution rule. For
this reason this encryption protocol is known as the one time pad. For each message
sent, a new, completely random key must be used by both Alice and Bob. How does
Alice send the keys to Bob? If these are encrypted, she will need to send another
key beforehand, and our perfect security is swallowed by an infinite regression. If
she sends the keys unencrypted, can she be sure that Eve has not intercepted them?
If she has, then Eve has access to all of Alice’s subsequent messages, and there’s no
way for Alice or Bob to know their code has been cracked until the paparazzi arrive.
These issues are eliminated by public key cryptography. Here, Bob tells Alice
the substitution rule she should use for her message. The encryption is done in
such a way that Alice’s message cannot be decoded using this rule, so it doesn’t
matter if Eve discovers it. Alice then sends her coded message to Bob, who knows
how to decrypt the message. An implementation of this idea using the mathemat-
ics of large prime numbers was developed in 1978 by Ron Rivest, Adi Shamir and

1.5 Quantum Communication 15
Leonard Adleman [21]. The RSA cryptosystem is the industry standard for secure
communication over the internet; much of modern finance relies on its security. But
unlike the one time pad, no-one has proved that it is secure. The RSA algorithm
relies on the empirical fact that it is computationally very demanding to find the
prime factors of a large number. That is, if two large prime numbers p and q are
multiplied together to give their product n, it is not practically possible to find p
and q, given knowledge of n alone. The best algorithm, the number field sieve, can
find the factors of n in roughly eN1/3 log
2/32 N computational steps [22], where N is
the number of bits needed to represent n. This exponential scaling means that it
is easy to make the calculation intractably long by making n just a little larger.
But this is not the whole story. In 1994 Peter Shor showed how a quantum com-
puter could be used to perform this factorization much faster [23]. Shor’s algorithm
requires just N2(log2N) log2(log2N) steps, an exponential improvement over the
best conventional methods. A quantum computer that can implement this algo-
rithm efficiently does not yet exist, although proof-of-principle experiments using
LOQC have been performed [24–26]. But it is now known that the RSA cryptosystem
is living on borrowed time: if a practical quantum computer is ever made, modern
secure communications will be spectacularly compromised.
Enter QKD. QKD makes use of quantum mechanics to distribute the keys re-
quired for a one time pad protocol in a secure way, avoiding the infinite regress
arising from a classical protocol, and obviating the need to rely on the fatally flawed
RSA system. The goal is to provide both Alice and Bob with an identical string

1.5 Quantum Communication 16
of completely random bits, which they can use as keys to encrypt and decrypt a
message. The most widely known protocol used to do this is known as BB84, after
the 1984 paper by Bennett and Brassard [27]. Alice sends photons, one at a time, to
Bob. Alice can choose the polarization of each photon to point in one of four possi-
ble directions: horizontal, vertical, anti-diagonal or diagonal (|H〉, |V 〉, |A〉 or |D〉;
see Figure 1.2). These directions form a pair of perpendicular polarizations, with a
quarter-turn between them. Each of these pairs is known as a basis. The unrotated
basis contains the |H〉 and |V 〉 polarizations, and is known as the rectilinear basis.
The rotated basis contains the |A〉 and |D〉 polarizations, and is referred to as the
45-degree basis. Bob can measure the polarization of the photons he receives from
Alice, but to do so he has to line up his detector with either the rectilinear or the
45-degree basis — he has to choose. The measurement gives one of two possible
results, either 0 or 1, but these results mean nothing unless the photon polariza-
tion belonged to the basis that Bob chose to measure. For example, a |D〉 photon
polarized in the 45-degree basis will give a completely random result, either 0 or 1
with equal probability, if Bob aligns his detector with the rectilinear basis. So if he
gets the basis wrong, his measurement results are useless. If he chooses correctly
however, and the photon is polarized in the same basis that he measures in, then the
0 or 1 results tell him to which of the two possible directions in that basis the photon
polarization belonged. So for instance if Bob aligns his detector with the 45-degree
basis, the |D〉 photon will always give a 1 result. An |A〉 photon would give a 0 result
for this measurement, while either |H〉 or |V 〉 photons would give random results.

1.5 Quantum Communication 17
This strange property of photons, that measurements give useful or uncertain results
depending on the measurement basis, is a manifestation of Heisenberg’s uncertainty
principle [28]. It is uniquely quantum mechanical.
rectilinear
45-degree
Figure 1.2 BB84 protocol. Alice sends photons to Bob with polar-izations chosen randomly from the four possible directions |H〉/|V 〉and |A〉/|D〉, represented here as qubit states. To measure the polar-ization, Bob (or Eve) must choose a basis, rectilinear or 45-degree,for their measurement. Only photons polarized in this basis will yielduseful information.
To proceed with QKD, Alice generates two completely random, unrelated, bit
strings. For each photon, she uses the first bit string to decide in which basis to
polarize her photon. For instance, 0 could signify rectilinear and a 1 would mean 45-
degree. Then she uses the second bit string to decide which of the two perpendicular
polarizations in that basis to use. When Bob receives Alice’s photons, he records
the results of his measurements, and the basis he used for each measurement. Alice
then sends Bob her first bit string. This tells him the bases each photon belonged
to. He knows that his results are useless every time he chose the wrong basis for
his measurement. So he discards these results. The remaining results tell him the

1.5 Quantum Communication 18
correct polarization of each photon. That is, Bob’s remaining results now tally
exactly with Alice’s second bit string. Alice and Bob now share a cryptographic
key they can use for a one-time pad. But what about Eve? Well, Eve may have
intercepted Alice’s first bit string, but this only contains information about which
results to discard, it tells Eve nothing about what those measurement results were,
so this does not help her in cracking Alice and Bobs’ code. She could also have
intercepted the photons that Alice sent, and tried to measure their polarizations.
But, just like Bob, she has to guess at which basis to measure in. She gets a result,
but she has no idea whether the basis she chose is correct. She has to send photons
on to Bob, otherwise he will receive nothing and get suspicious. But Eve does not
know what polarization to give her photons, because she doesn’t know whether her
measurements are reliable or not. Suppose she just decides to send photons polarized
according to her measurement results in the basis she chose to measure in. Bob
receives these photo ns and measures them, none the wiser. But after Alice sends
her first bit string, and Bob discards his unreliable measurements, Bob’s remaining
results may not tally perfectly with Alice’s anymore. This is because sometimes Eve
will have chosen a different basis to Alice, obtained useless measurement results, and
sent photons to Bob with the wrong polarization. So Bob and Alice can compare
their keys, or a small part of them, and if they do not match up, they know that Eve
has been tampering with their photons. There is no way for Eve to listen in without
Alice and Bob discovering her presence. In quantum mechanics, measurements affect
the system being measured, and the BB84 protocol exploits this fact to guarantee

1.6 Quantum Repeaters 19
secure communication.
Quantum memories are not needed in the above protocol, provided Alice’s pho-
tons survive to reach Bob. As mentioned in Section 1.4 in the context of LOQC,
photons generally do not survive for longer than around 1 ms in an optical fibre, so
Bob should not be further than around 200 miles from Alice, otherwise her photons
will be scattered or absorbed before they reach him. In order to extend the distance
over which QKD is possible, some kind of amplifier is needed, which can give the
photons a ‘boost’, while maintaining their quantum state — that is, their polar-
ization. But the photons are qubits. They cannot simply be copied; we know this
from the no-cloning theorem (see Section 1.2.3). What is required is a modification
to the protocol described above, and a device known as a quantum repeater. Such
a device requires a quantum memory. If quantum memories can be made efficient,
with storage times on the order of 1 s, intercontinental quantum communication be-
comes possible. In the next section we introduce the quantum repeater, and discuss
the usefulness of quantum memories in this context.
1.6 Quantum Repeaters
A quantum repeater is a device designed to extend entanglement. Entanglement is
a purely quantum mechanical property that can be used as a resource to perform
QKD. In this section we will introduce entanglement, examine how it degrades over
long distances, and how quantum repeaters ameliorate this degradation.
Entanglement is a property of composite quantum systems. As an example,

1.6 Quantum Repeaters 20
consider two qubits. Classically, a system composed of two parts could be described
by the states of each part. In quantum mechanics this is not always true. Just as
a qubit can exist in a superposition of different states, so a system comprising two
qubits can exist in a superposition of different combined states. Such states arise
from a blend of correlation and indeterminism. To see this, suppose that we have a
machine that produces two photons, each with the same polarization. Now suppose
that the direction of this polarization is not fixed. It might polarize both photons
horizontally, we’ll label this polarization |0〉, or vertically, |1〉. We have no way of
knowing which of these two polarizations the machine uses, we only know that both
photons will have the same polarization. Such a state cannot be described by talking
about each photon in turn, as is clear from the language we used to describe the set
up. Using subscripts to denote the two photons, the state is written as
|ψ〉 =1√2
(|0〉1|0〉2 + |1〉1|1〉2) . (1.6)
The factor of 1/√
2 appears simply to fix the length of the state vector |ψ〉 to 1. This
state is an entangled state, because there is no way to write it as a product of states
of the first photon with states of the second. It expresses the two properties of our
machine: first, that the two photons always have the same polarization, and second,
that it is not certain which of the two polarizations will be produced. In fact, because
the two possible states |0〉 and |1〉 are mutually exclusive, (1.6) is a maximally
entangled state, sometimes known as a Bell state. Bell states represent much of

1.6 Quantum Repeaters 21
what is counter-intuitive about quantum mechanics. Their name derives from John
Bell’s famous 1964 paper [29] in which he proves that these states are incompatible
with local realism. A ‘local’ world is one in which no effect can propagate faster
than the speed of light; a ‘real’ world is one in which all properties can be assigned
definite values at all times. That modern physics describes states which do not admit
a local realistic interpretation is intriguing and controversial. Below we will see that
these states are also a resource for quantum communication. If their use becomes
widespread, we will be in the awkward position of deriving practical benefits from
a technology based on a philosophical conundrum!
1.6.1 The Ekert protocol
In 1991 Artur Ekert proposed a modification of the BB84 QKD protocol based on
the use of Bell states like (1.6) [30]. In this protocol, our machine for generating
entangled photon pairs is used. One photon from each pair is sent to Alice, the
other to Bob. Now Alice and Bob both have polarization detectors; they each have
to choose a basis to measure their photons in. Sometimes they will choose the same
basis as eachother, sometimes they will choose different bases. When they choose
differently, their results are meaningless, but when they choose the same basis, their
results are perfectly correlated. This is obvious for the rectilinear basis by inspection
of the form of (1.6). A little algebra shows that the same perfect correlations also
hold if both Alice and Bob measure in the 45-degree basis. The QKD is accomplished
in the same way as for the BB84 protocol: Alice tells Bob the measurement bases

1.6 Quantum Repeaters 22
she used; Bob discards the results of all measurements where his basis differed from
Alice’s. Alice and Bob then compare part of the remaining results to check that
they are correlated, as they should be. Poor correlations signify the presence of an
eavesdropper.
The importance of this modified protocol is that entanglement is a transferrable
resource. Below we will see how entanglement can be swapped between photons to
extend the range of quantum communication.
1.6.2 Entanglement Swapping
Entanglement swapping allows one to entangle two photons that have never encoun-
tered eachother. The situation is sketched in Figure 1.3. Two sources each emit
a pair of entangled photons in the state (1.6). One photon from each pair is sent
into a polarizing beam splitter, which transmits horizontally polarized photons, and
reflects vertically polarized photons. Behind the beamsplitter are a pair of photon
detectors. The beamsplitter has the effect of limiting the information we can learn
about the photons from the detectors. For instance, if both photon detectors D1
and D2 fire together, it could be that photons (2) and (3) were both vertically po-
larized, or that they were both horizontally polarized. That is, a ‘coincidence count’
from D1 and D2 only tells us that photons (2) and (3) had the same polarization;
it reveals nothing about what that polarization was. But we know from the state
(1.6) that photon (1) has the same polarization as photon (2), and similarly that
photon (4) has the same polarization as photon (3). So if photons (2) and (3) have

1.6 Quantum Repeaters 23
the same polarization, so do photons (1) and (4). Their polarization is unknown,
but correlated. Therefore, after a coincidence count, the two remaining photons,
(1) and (4), are in a Bell state. The entanglement between photons (1)-(2) and
(3)-(4) has been swapped to photons (1)-(4). This procedure was first demonstrated
experimentally by Jian Wei-Pan et al. in 1998 [31], and is now an essential tool for
LOQC.
&
1 2 3 4
PBS
D1 D2
S1 S2
Figure 1.3 Entanglement swapping. Two independent sources, S1and S2, emit pairs (1)-(2) and (3)-(4) of polarization entangled pho-tons. Photons (2) and (3) are directed into a polarizing beam splitter(PBS). When both detectors D1 and D2 fire behind the PBS, photons(1) and (4), which have never met, become entangled.
It’s clear from the above arguments that entanglement swapping is not much
more than a re-assignment of our knowledge regarding correlations, in the light of
a measurement carefully designed to reveal only partial information. Nonetheless,
a real resource — entanglement — has been extended over a larger distance by this
procedure. And QKD can now be performed using photons (1) and (4).

1.6 Quantum Repeaters 24
1.6.3 Entanglement Purification
So far we have shown how entanglement can be extended over large distances by
swapping perfect Bell states, each distributed over shorter distances. In practice,
however, propagating even over short distances can distort the polarizations of the
photons. Small distortions do not completely destroy the entanglement; rather there
is a smooth degradation in the usefulness of the photons for QKD as the distortions
become worse. Nevertheless, with each entanglement swap, these deleterious effects
are compounded, so that the entanglement vanishes after only a few swapping op-
erations. However, it is possible to transform several poorly entangled photon pairs
into one photon pair with near-perfect entanglement using an ‘entanglement pu-
rification protocol’. Several of these exist [32–36], generally they involve mixing and
measuring photons in a way that strengthens the correlations between the remaining
photons. Such procedures allow entanglement to be ‘topped up’, at the expense of
having to use more photons. QKD across distances much larger than those over
which distortions affect photons can then be implemented. Below we will introduce
a paradigm for constructing a quantum repeater which does not explicitly make
use of polarization entanglement, but which combines entanglement swapping and
entanglement purification into a single step.
1.6.4 The DLCZ protocol and number state entanglement
As mentioned previously, photons have several different degrees of freedom that can
be used to encode qubits. We have mostly focussed on polarization qubits for QKD,

1.6 Quantum Repeaters 25
but in the following protocol qubits are encoded in the number of photons occupying
a given optical mode, so-called single-rail encoding. Consider a machine similar to
the Bell state sources discussed above, which emits one photon either to the left, or
to the right. Using subscripts L and R for these directions, the state produced by
this machine is
|ψ〉 =1√2
(|0〉L|1〉R + |1〉L|0〉R) . (1.7)
Note that the states |0〉 and |1〉 now refer to the number of photons, rather than the
photon polarization as before. This state is also a maximally entangled state. Like
(1.6) it is also a Bell state. It expresses the correlation that a photon in one mode
always signifies the absence of a photon in the other mode. And it expresses the
indeterminacy, built into our device, that there is no way of knowing whether the
emitted photon will be found in the left or the right mode. How does entanglement
swapping work on such a state? A slightly different set-up is used (see Figure 1.4).
The action of the measurement is particularly clear in this example: the beamsplitter
(BS) mixes the two modes (2) and (3), so that a detection at D1 or D2 tells us only
that one of those modes contained a photon, but not which one. A bit of epistemic
book-keeping reveals the entanglement swap: if (2) contained a photon but (3) did
not, that means (1) had no photon while (4) carries a photon. Similarly if (2) was
empty but (3) contained a photon, (1) must carry a photon while (4) does not.
There is no way to distinguish these possibilities, and therefore a single detection
behind the BS puts the modes (1) and (4) into the entangled state (1.7).
For single-rail encoding, the most damaging effect of propagation is the pos-

1.6 Quantum Repeaters 26
|
1 2 3 4
BS
D1 D2
S1 S2
Figure 1.4 Single-rail entanglement swapping. Two independentsources, S1 and S2, emit single photons into modes (1)-(2) and (3)-(4)in the state (1.7). Modes (2) and (3) are mixed on a beam splitter(BS). When one of the detectors D1 or D2 (but not both) fires behindthe BS, modes (1) and (4) become entangled.
sibility of photon loss, through absorption or scattering. This has the effect of
introducing a third term into the state (1.7) of the form |0〉L|0〉R, corresponding to
no photons in either mode (they’ve all been lost!). With the addition of this term,
the quality of the entanglement is reduced. But this ‘vacuum’ component can never
cause any detection events. Therefore if either of the detectors D1 or D2 fire, sig-
naling a successful entanglement swap, the vacuum component is removed, since the
detection of a photon renders it counterfactual. For this type of state, entanglement
swapping also accomplishes a degree of entanglement purification.
The ability to perform both swapping and purification using such a simple mea-
surement makes this type of encoding attractive as a means of distributing en-
tanglement over large distances for quantum communication. However, it is not
immediately obvious how to generalize the Ekert QKD protocol to states encoded in
this way. For example, to perform a measurement in the analogue of the 45-degree
basis, one would require detectors that are sensitive to superpositions of photon

1.6 Quantum Repeaters 27
number states. These issues are avoided by combining two states of the form (1.7);
the measurement scheme is shown below in Figure 1.5. Two entangled states of the
1 2
3 4
BS
D2
D4D3
D1
BS
&
&
3
Figure 1.5 QKD with single-rail entanglement. Two entangledstates are distributed between Alice — detectors D1 and D3 on theleft — and Bob — detectors D2 and D4 on the right. The photonson each side are mixed on a beamsplitter (BS). A ‘polarization mea-surement’ is made when a single detector fires on each side. Both D1and D2 firing is a ‘0’. D3 and D4 firing is a ‘1’. Adjusting the phasesφA,B allows to select the measurement bases.
form (1.7), are distributed between Alice and Bob. Alice, on the left hand side,
has two detectors, D1 and D3. Bob, on the right, has detectors D2 and D4. If
Alice only records measurements when one of her detectors fires, she knows that one
photon came to her, while the other went to Bob. Similarly a single detection at
Bob’s side tells him that Alice received the other photon. The phases φA and φB
are independently chosen by Alice and Bob from the set 0, π/2. These phases,
in combination with the beamsplitters, allow Alice and Bob to control the basis in
which their detectors measure, in direct analogy with the rectilinear and 45-degree
bases of the Ekert protocol. When they choose the same phases, their measurements

1.6 Quantum Repeaters 28
should be correlated, with D3 and D4 firing together for a ‘1’ result, and D1 and D2
firing together for a ‘0’ result. Their is no correlation if they choose different phases.
Alice and Bob publicly announce their basis choices, and then compare some of their
results to check for the presence of Eve.
The above discussion shows how number state entanglement can be used for
quantum communication over long distances. A specific proposal for implement-
ing this protocol using atomic ensembles to generate the entangled states was first
made in 2001 by Lu-Ming Duan, Michael Lukin, Ignacio Cirac and Peter Zoller [37].
The DLCZ protocol is exciting because it is firmly grounded in feasible technol-
ogy. Several improvements have since been suggested [38–42], which make the scheme
more robust to phase instability, photon loss, detector noise and inefficiency. Below
we briefly introduce the principle behind the original protocol, since this will serve
as a useful introduction to the uses of atomic ensembles in quantum information
processing technologies.
We consider two clouds of atoms L and R, each with internal energy levels ar-
ranged as depicted in Figure 1.6. This type of Λ-structure is ubiquitous in quantum
information protocols. We will encounter it many times in our survey of quantum
memory protocols. It also arises in all but the simplest quantum systems: not just
atoms, but crystals, quantum dots and molecules, as we will see. The atomic ensem-
bles L and R will together play the role of the entangled photon source introduced
earlier. Here’s how it works. Both ensembles are pumped simultaneously by a laser
pulse. The pump pulse is tuned out of resonance with the atomic state |2〉, so that

1.6 Quantum Repeaters 29
Generation Readout
(a) (b)
Figure 1.6 Λ-level structure of atoms used in the DLCZ protocol.(a): A pump pulse excites an atom out of the ground state |1〉, whichdecays down to a long-lived metastable state |3〉, emitting a ‘Stokesphoton’. (b): A readout pulse brings the excited atom back to theground state, which emits an ‘anti-Stokes photon’ in the process.
most of the time nothing happens. But provided that the number of atoms in each
cloud is large enough, there is a small probability that the pump pulse will cause a
two-photon Raman transition in one of the atoms (see §2.3.2 in Chapter 2), exciting
it to a long-lived metastable state |3〉, as shown in Figure 1.6 (a). Such an excita-
tion is always accompanied by the emission of a Stokes photon. The probability of
two atoms being excited this way is negligibly small. Therefore at most one Stokes
photon is emitted from the ensembles. Due to the extended, pencil-like geometry
of the ensembles, any Stokes photons tend to be emitted in the forward direction,
so that they can be captured and directed as required [43]. Detectors D1 and D2
are placed behind a beamsplitter in front of the ensembles, as shown in Figure 1.7.
The beamsplitter mixes the optical modes from the two ensembles, so that if one
of the detectors fires, we know that a Stokes photon was emitted from one of the
ensembles, but we don’t know which. After a detection then, the state of the atomic

1.6 Quantum Repeaters 30
ensembles is of precisely the form (1.7), where |1〉 and |0〉 now refers to the presence
or absence of an excited atom in an ensemble.
We have now generated the number state entanglement required for the repeater
protocol. A difference with the preceding discussion is that the entanglement is
between the atomic ensembles, rather than optical modes. But this is an advan-
tage, because the atomic excitations are stationary, and can last for a long time,
while photons are generally short lived, as mentioned earlier. Since all the steps of
the protocol involve waiting for particular detectors to fire, it is essential that the
entanglement can be preserved until all the required steps are completed. When
the entanglement is ‘needed’, for instance to perform an entanglement swapping
operation, the atomic excitations can be converted back into photons. This is done
by applying a ‘readout’ pulse to the ensembles, which returns any excited atoms to
their ground states via an anti-Stokes transition, as shown in Figure 1.6 (b). The
anti-Stokes modes inherit the entanglement from the ensembles. If the anti-Stokes
mode from ensemble L is sent off in one direction (left), and the anti-Stokes mode
from the R ensemble is sent in the other direction (right), we now have a device
that can emit entangled states on-demand. The sources S1 and S2 appearing in
Figure 1.4 should now be thought of as each comprised of a pair of entangled atomic
ensembles, waiting to be ‘read-out’ when desired.
So far we have seen how atomic ensembles show promise for quantum communi-
cation protocols, but we have not encountered a specific need for quantum optical
memories. To conclude this section, we describe how the DLCZ protocol can be

1.7 Modified DLCZ with Quantum Memories 31
BS
D1
D2
L
R
Figure 1.7 Generation of number state entanglement using atomicensembles in the DLCZ quantum repeater protocol. A click in oneof the detectors D1 or D2 tells us that one atom, in either of theensembles L or R, is excited, but because of the beamsplitter (BS),we don’t know to which ensemble this atom belongs. Therefore theensembles are left in the entangled state (1.7).
improved by using memories in combination with single photon sources [41].
1.7 Modified DLCZ with Quantum Memories
Recall that we claimed the probability for two atoms in the ensembles L and R to be
excited by the pump pulses was so small as to be insignificant. If two atoms could
be excited, it would mean that sometimes one Stokes photon from L and one from
R is emitted simultaneously. Suppose that one of these photons is lost somehow.
Perhaps it is absorbed in an optical fibre. Then only one of the detectors D1 or
D2 behind the beamsplitter will fire, but both ensembles are in fact excited. To
account for this possibility, we should add a term to the state (1.7) of the form
|1〉L|1〉R — that is, the ensembles are no longer in a maximally entangled state. Of
course the probability of a double excitation can always be made arbitrarily small
by making the pump pulses weaker. But this also reduces the probability of even

1.7 Modified DLCZ with Quantum Memories 32
a single excitation, so that it is necessary to wait a long time before one of the
detectors fires. If the waiting time becomes too long, the entanglement stored in the
ensembles starts to degrade as the atoms drift and collide, so this limits the distance
over which entanglement can be distributed.
This issue can be resolved if the entanglement is generated using a single-photon
source and a beamsplitter. The modified set up is shown in Figure 1.8. The atomic
ensembles L and R are now used as quantum memories, which we label QML and
QMR. We will explore the details of atomic ensemble quantum memories in Chapters
4–8. For now we only need to know that a photon entering a quantum memory is
trapped until it is needed. We have assumed that we have access to single photon
sources (SL and SR) that each emit one and only one photon, on-demand. Sources
like this are actually rather difficult to make, but research into this technology has
advanced greatly over the last few years [44–47], and including such sources into the
design is no less realistic than the inclusion of quantum memories. To generate
number-state entanglement, we start by triggering both sources SL and SR. Each
emits a photon, which then encounters a beamsplitter (BS). At the beamsplitters
the photons can either be reflected into a quantum memory, or transmitted, in which
case they are brought together on a final BS placed in front of detectors D1 and
D2. In the case that just one of the detectors fires, we know that only one photon
was transmitted at the first BS; the other photon must have been reflected, in which
case it is now stored in a quantum memory. But the final BS prevents us knowing
which quantum memory contains the photon. We therefore have a superposition

1.7 Modified DLCZ with Quantum Memories 33
state, with either QML excited and QMR empty, or vice versa. This is the desired
entangled state (1.7). What if both photons are transmitted, but one of them is
BS
BS
BS
D1
D2
QML
QMR
SL
SR
Figure 1.8 Modification to the DLCZ protocol using single photonsources (SL and SR) and quantum memories (QML and QMR). Thenumber state entanglement is generated by beamsplitters.
somehow lost? In that case only one detector fires, but neither quantum memory
is excited. We therefore have to add a vacuum component of the form |0〉L|0〉R
to the state (1.7). This certainly degrades the entanglement in the same way as
the |1〉L|1〉R component did for the corresponding error in the DLCZ protocol. But
when we perform entanglement swapping to extend the entanglement, we always
wait until the appropriate detector fires (see Section 1.6.2), and this purifies the
entanglement, removing the vacuum term (see Section 1.6.3). Therefore vacuum
errors do not damage the protocol, and we are able to run it faster and further than
the original DLCZ scheme permits.
Hopefully the possible uses for quantum optical memories is now clear. Presum-

1.7 Modified DLCZ with Quantum Memories 34
ably future proposals will develop further applications for them. In the next chapter,
we will review the various techniques used for quantum storage.

Chapter 2
Quantum Memory: Approaches
The aim of a quantum optical memory is to convert a flying qubit — an incident
photon — into a stationary qubit — an atomic excitation. This conversion should
be reversible, so that the photon may be re-emitted some time later, at the behest of
the user. In this chapter we review a number of approaches for achieving this kind
of quantum storage. More detailed calculations are deferred until the next chapter.
The simplest system one could imagine for storing a photon would consist of
a single atom, coupled to a single optical mode via an electric dipole transition
(see Figure 2.1). An incident photon, resonant with the transition, is absorbed,
promoting the atom from its ground state |1〉 to its excited state |2〉. The photon
is now ‘trapped’ as a stationary excitation of the atom, and the quantum storage
is complete. However, quantum mechanics is always invariant under time-reversal.
That is to say, the electric dipole interaction (see §C.4 in Appendix C), which allows
the photon to be absorbed by the atom, also causes the atom to re-emit the photon,

36
00
0.5
1
time/
Excita
tio
n p
rob
ab
ility
(a) (b) (c)
Figure 2.1 The simplest quantum memory. (a): A single atomis coupled to a single optical mode. (b): An incident photon is ab-sorbed, exciting the atom. (c): Unfortunately, time reversal symme-try requires that the photon is immediately re-emitted.
almost immediately afterwards. In fact, these two processes compete continuously,
so that over time the population of the excited state oscillates back and forth as
the atom absorbs and re-emits the photon; see Figure 2.1 (c). This behaviour is
known as Rabi flopping, after Isidor Isaac Rabi, who first used the phenomenon
in the context of nuclear magnetic resonance [48]. The Rabi frequency Ω of these
oscillations is proportional to the strength of the coupling between the atom and
the electromagnetic field.
The above scheme needs some modification if it is to store a photon in a controlled
way. One solution is to introduce a third state |3〉 — a dark state — that is not
coupled to the photon mode we want to store. We should have some control field
we can apply that transfers the atomic state from |2〉 to |3〉 once the storage is
complete. In this way the Rabi oscillations are ‘frozen’, and the photon remains
trapped as an excitation of the dark state for as long as is desired. Provided the
atom is well isolated, the dark state will persist for as long as is needed. To retrieve

37
the photon, the control field is applied again, transferring the atomic state from
|3〉 back to |2〉. The Rabi flopping of Figure 2.1 (c) continues and the photon is
re-emitted. The scheme is shown in Figure 2.2. This typifies the approach taken in
(a) (b) (c)
input
control
storage retrieval
Figure 2.2 Adding a dark state. (a): We address the atom withan auxiliary control. (b): After the input photon is absorbed, thecontrol transfers the atom to |3〉. (c): When the photon is needed,the control is re-applied.
many quantum optical memories, although the details of the protocols differ widely.
While single atoms have been used, ensembles of many atoms, each with the Λ-type
structure of Figure 2.2, are also commonly employed. The atom(s) can be enclosed
in an optical cavity, trapped at the centre of a confocal microscope, or addressed
by collimated beams. The optical fields may be resonant, or off-resonant with the
atomic transitions; the shape and timing of the control fields can vary, and indeed
non-optical controls, such as magnetic or electric fields, may be used. We will briefly
review this menagerie of memory protocols in the following sections.

2.1 Cavity QED 38
2.1 Cavity QED
In the forerunning discussion we presumed that a single atom could be coupled to a
single optical mode. This is a highly unnatural state of affairs, since the electromag-
netic field pervades all of space, so that atoms are generally surrounded by a bath of
electromagnetic field modes. The most dramatic effect of this ‘reservoir’ of modes
is that any atomic excitations coupled to the field tend to leak away rather quickly:
an emitted photon is very unlikely to couple back to the atom, so the atomic popu-
lation does not exhibit Rabi flopping; rather it decays exponentially. This is known
as spontaneous emission — the stronger the coupling between an atomic state and
the field, the shorter the lifetime of that state. An associated consequence is that
an incident photon is very unlikely to couple to the atom. Therefore a single atom
in free space cannot be used for quantum storage.
In order to recover strong coupling between an atom and an optical mode, it
is necessary to suppress the interaction with all unwanted field modes. This can
be done interferometrically, but introducing a highly reflective cavity around the
atom (see Figure 2.3). Any fields inside the cavity are reflected back on themselves
by the cavity mirrors. Only those fields with a wavelength equal to a half integer
multiple of the cavity length add constructively when folded back on themselves; all
other wavelengths interfere destructively. The optical modes supported by the cavity
are therefore spaced regularly in frequency. If the resonant frequency of the atomic
transition |1〉 ↔ |2〉 is close to one of these supported modes, the atomic coupling will
be confined to this single mode. All other cavity modes being too far from resonance

2.2 Free space coupling 39
to contribute significantly. If the volume of the cavity is sufficiently small, and the
Figure 2.3 Cavity QED. An atom is confined in a high-finesse op-tical cavity, which supports only a discrete set of optical frequencies.Only one optical mode, resonant with the atomic transition, couplesto the atom.
cavity mirrors sufficiently reflective, it is possible to bring an atom into the so-called
strong-coupling regime [49–53], where the light matter interaction behaves broadly as
described in Figures 2.1 and 2.2. This so-called Cavity QED approach to light-matter
interfaces has been widely applied. Both trapped and moving beams of atoms are
used [16,45,54,55], as well as quantum dots [56–59] and molecules [60]. However, cavities
are rather difficult to fabricate; approaches which do away with this requirement
would be easier to scale up.
2.2 Free space coupling
Another possibility is to focus an incident photon onto an atom in a way that
maximizes the probability of it being absorbed. The cavity is replaced with a pair of
microscope objectives, as shown in Figure 2.4. The rationale behind this approach
is that good coupling should be possible if the spatial shape of an incoming photon
matches the spatial pattern of an outgoing spontaneously emitted photon. This
time-reversal argument suggests that an incident photon should be converted into a

2.3 Ensembles 40
collapsing dipole pattern. The dipole pattern of an atom is very far from a narrow
collimated beam — it is nearly isotropic, covering almost 4π steradians. Therefore
the lenses should be as wide as possible: this in itself represents a technical challenge.
Only preliminary experiments have been done [61], but theoretical work [62,63] suggests
that efficient coupling can be achieved if the numerical aperture of the objective
lenses is made large enough.
Figure 2.4 Confocal coupling in free space. An atom is trappedat the focus of a pair of microscope objectives. If the solid anglesubtended by an incident photon is sufficiently large, the couplingefficiency is predicted to approach unity.
2.3 Ensembles
In addition to the practical difficulties of building a high-finesse cavity, or indeed
a high numerical aperture confocal microscope, there are technological hurdles as-
sociated with trapping and placing single atoms. Another possibility for quantum
storage is to use ensembles of atoms, as shown in Figure 2.5. An incident pho-
ton may have a small probability with interacting with any given atom, but as the
number of atoms in the ensemble increases, the probability that the photon fails
to interact with all the atoms decreases exponentially. Therefore it is possible to
asymptotically approach unit interaction probability simply by adding more atoms.

2.3 Ensembles 41
This approach is the main focus of this thesis. As we will see, introducing many
atoms makes it possible to store more than one photon, or more than one optical
mode. Complicated optical arrangements and cavities are not required, and a wide
range of possible storage media — from atomic vapours to Bose-Einstein conden-
sates, quantum dots, crystals and fibres — are well-suited for protocols of this kind.
We are primarily concerned with schemes based on the absorption, and subsequent
Figure 2.5 Atomic ensemble memory. If an incident photon en-counters enough atoms, it is almost certain to be absorbed.
re-emission, of a freely propagating photon by an atomic ensemble. These schemes
divide broadly into four protocols, all closely related, which we will now introduce.
2.3.1 EIT
EIT stands for Electromagnetically Induced Transparency. It was first observed
by Boller et al. in 1991 [64]. In EIT, a strong laser — the control — is shone
into an atomic ensemble with a Λ-structure, as shown below in Figure 2.6 (a).
Ordinarily, a weak probe beam would be absorbed by the atoms, but the interaction
with the control laser causes the ensemble to become transparent to the probe.
This can be understood by considering the dressed states of the atom, under the
influence of the control [65,66]. The control field couples the states |2〉 and |3〉, so the
Hamiltonian for the electric dipole interaction of an atom with the control is of the

2.3 Ensembles 42
(a) (b) (c)
Detuning (arb. units)
Su
sce
ptib
ility
(a
rb. u
nits)
0
Figure 2.6 EIT. (a): a weak probe beam propagates through anensemble of Λ-type atoms, while a strong control field couples theexcited and metastable states. (b): the control mixes states |2〉 and|3〉 to produce an Autler-Townes doublet. (c): The imaginary part ofthe probe susceptibility (solid line) exhibits a double resonance, witha transparency window at the atomic |1〉 ↔ |2〉 transition frequency.The real part (dotted line) changes quickly within this window, caus-ing marked dispersion.
form H = Ω|2〉〈3| + h.c., where Ω is the Rabi frequency of the control laser on the
|2〉 ↔ |3〉 transition. This can be simply re-written as H = Ω (|+〉〈+| − |−〉〈−|),
where the dressed states are defined by |±〉 = (|2〉 ± |3〉) /√
2. These dressed states
are simply rotated versions of the natural atomic states |2〉 and |3〉. In fact, although
we have changed our notation, the relationship between these states is the same as
between the 45-degree and rectilinear bases discussed in the context of QKD in
chapter 1. The Hamiltonian is diagonal in the dressed basis, and we can read off
the energies of the dressed states as ±Ω. That is, the combination of the control
with the atom produces a system with a double-resonance — known as an Autler-
Townes doublet [67] — with a splitting between the dressed states set by Ω. This is
a manifestation of the dynamic Stark effect: the control brings the energy of state
|3〉 up to that of |2〉, and the dipole interaction induces an anti-crossing.
From the above arguments, we might already expect the probe absorption spec-

2.3 Ensembles 43
trum to divide into two peaks, with a transparency window in between. Figure 2.6
(c) shows the results of a steady-state calculation of the linear susceptibility for the
probe field as a function of its detuning from the |2〉 ↔ |3〉 resonance. The probe
absorption is proportional to the imaginary part, which shows the expected doublet
structure. But the depth of the transparency window cannot be explained simply
by superposing two identical resonances. As is clear from the plot, the absorption
actually vanishes completely in the centre of the transparency window. This total
transparency is due to quantum interference: the contributions to the susceptibility
from the two dressed states are of opposite sign, because one is shifted above, and
the other below the original resonance. There is therefore an exact cancellation at
this resonance, and the susceptibility is identically zero at this point.
In addition to propagating without absorption, the probe beam also propagates
extremely slowly. More precisely, the group velocity of the probe is reduced by
the strong dispersion within the transparency window. The refractive index n of
the atomic ensemble is given by n =√
1 + Re(χ), where χ is the probe suscep-
tibility. Inspection of part (c) of Figure 2.6 shows that the real part of χ varies
extremely rapidly across the transparency window; therefore the refractive index is
also changing quickly in this spectral region. To see why this slows the group veloc-
ity, consider a pulsed probe. A pulse is composed of a range of frequencies, covering
a spectral bandwidth δω, all interfering constructively, so that the phase variation
over the pulse bandwidth is roughly zero. Each frequency component of the pulse
accumulates an optical phase kδz − ωδt over spatial and temporal increments δz,

2.3 Ensembles 44
δt, where k = nω/c is the wavevector of each component. The trajectory of the
pulse is the locus of those points at which all the components of the pulse remain
in phase, so that δkδz = δωδt, with δk the range of wavevectors spanned by the
pulse. The group velocity — the velocity of the pulse — is therefore defined by
vg = dz/dt = dω/dk = c/(n+ωn′), where n′ = dn/dω. The steep increase in Re(χ)
across the transparency window makes n′ large, and therefore vg is small. Group
velocities as low as 17 ms−1 have been demonstrated in the laboratory [68].
The use of EIT as a method for storing light is an elegant application of these
effects. It was first described by Misha Lukin and Michael Fleischhauer in 2000 [69],
and has since been demonstrated experimentally many times [70–74]. The protocol
works as follows. An atomic ensemble of Λ-type atoms is illuminated by a control
field, preparing a transparency window. A probe pulse — to be stored — is now
directed into the ensemble, tuned to the centre of the transparency window. As
described above, it propagates slowly, but without loss. Even if the spatial extent of
the pulse is much longer than the ensemble initially, the sudden slowing of the pulse
as it enters the ensemble causes it to bunch up, so that it fits within the ensemble
as it propagates (see Figure 2.7). As soon as the entire pulse is inside the ensemble,
the control beam is attenuated. That is, the Rabi frequency Ω is reduced, so that
the splitting of the Autler-Townes doublet decreases. The transparency window gets
smaller, and the variation in Re(χ) becomes steeper, so the group velocity of the
probe falls. This process continues until the control is switched off entirely, at which
point the transparency window completely collapses; the dispersion diverges, and

2.3 Ensembles 45
the group velocity vanishes: the probe has been brought to a complete stop! In
fact, the quantum state of the optical field has been transferred to the atoms, and
the state can be stored for as long as the coherence of the atoms survives without
distortions. If the control field is switched back on, the probe field is re-accelerated,
and emerges — hopefully — unchanged, as if the ensemble had not been present.
Figure 2.7 Stopping light with EIT. A long probe pulse bunchesup as it enters the EIT medium, due to the slow group velocity atthe centre of the transparency window.
The arguments just given provide a useful physical picture for the mechanism
behind light-stopping by EIT. In isolation they don’t provide a satisfactory expla-
nation for why the probe is not simply absorbed as the transparency window is
made narrower, nor for precisely how the probe light is transferred to an atomic
excitation. A more complete discussion can be found in the paper by Lukin and
Fleischhauer [69]. Nonetheless we can still draw some conclusions about the circum-
stances under which this procedure will work. The probe spectrum should not be
wider than the transparency window, otherwise it will be partially absorbed, so the
control must be sufficiently intense when the probe enters the ensemble. The control
cannot be turned off too quickly, since the dressed states producing the transparency

2.3 Ensembles 46
window are only a good approximation in the steady, or nearly steady state. The
control intensity must be reduced adiabatically ; we’ll examine this condition more
closely in Chapter 5 (see §5.2.9). Finally, the control should be turned off before
the probe escapes from the ensemble, so the initial group velocity should be suf-
ficiently slow — this means the ensemble density should not be too low. These
considerations tell us that the bandwidth of probe pulses that can be stored via
EIT, and the efficiency of the storage, is limited by the density of the ensemble, and
the available control intensity. We now introduce a related protocol, with the aim
of circumventing some of these limitations. As more detailed calculations show, this
attempt is only partially successful, but the flexibility of the new protocol makes it
an attractive alternative.
2.3.2 Raman
In 1928 Chandrasekhara Venkata Raman was the first to observe the weak inelastic
scattering of light from the internal excitations of vapours and liquids. No lasers
were available, and he used a focussed beam of sunlight to generate the required
intensity [75]. His work earned him the Nobel Prize in 1930; the eponymous Ra-
man scattering is now used routinely in spectroscopy, industrial sensing and indeed
quantum optics.
Raman scattering can be understood rather simply in the context of an atomic
Λ-system. It is represented schematically in Figure 1.6 (a) from Chapter 1 — the
interaction used in the DLCZ repeater protocol is precisely Raman scattering. It

2.3 Ensembles 47
is a two-photon process — that is, second-order in the electric dipole interaction
— in which a pump photon (green arrow in the diagram) is absorbed, and at the
same time a Stokes photon (blue wavy arrow) is emitted. Energy conservation is
satisfied if the frequency difference between the pump and Stokes photons is equal
to the frequency splitting between the states |1〉 and |3〉 in the atoms. The optical
fields need not be tuned into resonance with the |1〉 ↔ |3〉 transition: the likelihood
of Raman scattering decreases as the fields are tuned further away from resonance,
but given a sufficiently intense pump field, and sufficiently many atoms, the Raman
interaction can be made rather strong (see §§10.8, 10.9 in Chapter 10).
In a Raman quantum memory, the Raman interaction is turned on its head: a
signal photon is absorbed, and a strong control field stimulates the emission of a
photon into the control beam (see Figure 2.8). The signal and control fields are
tuned into two-photon resonance, that is, the difference in their frequencies is equal
to the frequency splitting between |1〉 and |3〉.
The process is conceptually very similar to that outlined at the beginning of
this chapter (c.f. Figure 2.2). But in a Raman memory, no atoms are ever excited
into the state |2〉. This is because the fields are tuned out of resonance with this
state, with a common detuning ∆. Instead, the control field creates a virtual state
— represented by the dotted lines in Figure 2.8 — and an atom is excited into
this virtual state by the signal photon. The control field then transfers the atom
into the state |3〉 for storage. Of course, all possible time-orderings for this process
contribute to the interaction, but from this perspective it is clear that no storage is

2.3 Ensembles 48
(a)
Storage Retrieval
(b)
Figure 2.8 Raman storage. Both fields are off-resonant, with adetuning ∆ from the excited state. (a): A signal photon is absorbed,and at the same time a photon is emitted into the control beam. Thistwo-photon Raman transition promotes one atom in the ensemblefrom |1〉 to |3〉. (b): The strong control is applied again, and theinverse Raman process returns the excited atom to its ground state|1〉, re-emitting the signal photon.
possible without the presence of the control field. Hence the name.
We might expect the physics of Raman storage to relate very closely to that of
EIT storage, and indeed they are confusingly similar. To connect Raman storage
with our previous discussion of EIT, we can consider the dressed states of the atoms
in a Raman memory, under the influence of the control. In fact, we have already
done so. The virtual state into which the signal photon is absorbed is precisely the
dressed state produced from the atomic state |3〉 when the control is present. Just
as in the case of the |−〉 state of EIT, the virtual state is really a combination of
the states |2〉 and |3〉. The difference is that it contains a very small amount of
the excited state |2〉, because of the large detuning ∆. The other dressed state, the
equivalent of |+〉 for the Raman memory, is not shown in Figure 2.8, since it is so
close to the bare atomic state |2〉 as to be indiscernible. Again, the large detuning
means that this state is made up almost entirely of |2〉, with almost no contribution

2.3 Ensembles 49
from |3〉. We can therefore frame the difference between EIT and Raman storage in
the following way. In an EIT memory, the photon to be stored is tuned between the
dressed states; it is brought to a halt by turning the control field off. In a Raman
memory, the signal photon is tuned into resonance with one of the dressed states,
which is made up almost entirely of the storage state |3〉. Once it has been absorbed,
the control field is turned off, and the state becomes ‘dark’ — decoupled from the
electromagnetic field.
One motivation for studying Raman storage is the possibility of broadband stor-
age. That is, the storage of temporally short photons, which necessarily comprise a
large range of frequencies. EIT storage requires that the spectral bandwidth of the
input photon should fit within the transparency window. Raman storage does not
rely on a transparency effect, so this limitation does not pertain. In addition, the
spectral width of the virtual state into which the signal photon is absorbed is set by
the spectral width of the control field. We might therefore expect that broadband
photons can be stored with a broadband control. We should ensure that the excited
state |2〉 is never populated, since this state will eventually decay and our photon
will be lost. This means that the detuning ∆ must greatly exceed the bandwidth of
the signal photon. Nonetheless, given sufficiently many atoms, the large detuning
need not weaken the interaction, and efficient broadband storage should be possible.
In fact, as discussed in §5.2.9 of Chapter 5, the Raman memory protocol is ideally
suited to broadband storage, although its advantages over the EIT protocol in this
respect are not entirely clear-cut.

2.3 Ensembles 50
A Raman memory was first proposed by Kozhekin et al. in 2000 [76]. They did
not explicitly address the problem of storing a single photon; rather they considered
the transfer of quadrature squeezing — a phenomenon we’ll describe in the next
section on continuous variables memories — from light to atoms. The theoretical
treatment of a Raman memory for photon storage was published in 2007 [77], and
forms part of this thesis. Raman storage is yet to be implemented experimentally,
but we are currently attempting to demonstrate the protocol in cesium vapour;
details are given in Chapter 10.
2.3.3 CRIB
CRIB stands for Controlled Reversible Inhomogeneous Broadening. In this protocol,
a spatially varying electric or magnetic field is applied to the ensemble. This shifts
the resonant frequency of the |1〉 ↔ |2〉 transition in the atoms, with a frequency
shift proportional to the strength of the field. Therefore, depending on their posi-
tion, some atoms experience a large shift; others a smaller shift, or a negative shift.
The net effect is to produce an inhomogeneous broadening of the atomic resonance.
That is, each atom has a narrow resonance, but the ensemble as a whole absorbs
light over a broad range of frequencies, as determined by the applied field (see Figure
2.9). Storage is accomplished via the general procedure sketched in Figure 2.2 (b).
A signal photon is tuned into resonance with the broadened |1〉 ↔ |2〉 transition,
and is completely absorbed. An important feature of the scheme is that broadband
photons can be stored, because the ensemble resonance covers a wider spectral range

2.3 Ensembles 51
than the unbroadened transition. In particular, photons with a temporal duration
much shorter than the spontaneous emission lifetime of the state |2〉 can be stored.
Therefore the absorption process is finished long before the atoms have had time to
decay, and losses due to spontaneous emission are minimal. Note that so far in the
protocol we have not applied any optical control fields, so there are no dressed states
— the signal photon has simply been resonantly absorbed. When the absorption is
complete, a single atom in the ensemble has been excited into state |2〉. The broad-
ening field is now switched off, so that the atoms recover their natural resonance
frequencies. Then a control field — a laser pulse tuned to the |2〉 ↔ |3〉 transition
— transfers the excited atom to the storage state |3〉.
Storage has now been completed, but the stored excitation is rather mixed up.
To understand why, it is easiest to drop our consideration of photons for a moment,
and consider the memory as it would be described classically. The atoms can be
thought of as a collection of dipoles (separated charges) that are set in motion by an
impinging field — the signal field. Due to the applied inhomogeneous broadening,
each dipole is oscillating at a slightly different frequency. Therefore, although they
all begin oscillating together, driven by the signal pulse, they soon drift out of phase
with one another. When the control pulse is applied, the dipoles are essentially
frozen, since their motion is transferred to the dark state |3〉. In being stored, the
signal pulse has had its phase scrambled. That is, information about the time-of-
arrival of the signal pulse has been lost. The crucial step in retrieving the signal is to
recover this information, by reversing the dephasing process. This is the vital ‘R’ in

2.3 Ensembles 52
CRIB. Because the inhomogeneous broadening is man-made, we are able to switch
it around, by changing the polarity of the external field. When the broadening
is flipped in this way, every atom that was shifted to a higher frequency is now
shifted to a correspondingly lower frequency, and vice versa. This provides us with
a way to completely reverse the dynamics of the memory. Retrieval works like this:
the control pulse is sent into the ensemble. This unfreezes the stored excitation,
transferring the atoms from |3〉 to |2〉. The atomic dipoles are still out of phase, but
then the inhomogeneous broadening is re-applied, this time with the reverse polarity;
see Figure 2.9 (b). The atoms that were red-shifted at storage are now blue-shifted,
and those previously blue-shifted are now red-shifted. The atomic dipoles therefore
eventually re-phase. The collective oscillation of the entire ensemble, in phase, then
acts as a source for the electric field, and the signal pulse is re-emitted.
(a)
Storage Retrieval
(b)
Figure 2.9 CRIB storage. (a): A signal photon is absorbed res-onantly by the broadened ensemble, exciting an atom to state |2〉.Then a strong control pulse transfers the excited atom to the stor-age state. (b): To retrieve the stored photon, the control is appliedagain, and the inhomogeneous broadening is switched on, this timewith reversed polarity. The optical dipoles eventually re-phase, andthe signal photon is re-emitted.
CRIB storage was first proposed by Nilsson and Kroll [78], following analyses of

2.3 Ensembles 53
a generalized photon echo by Moiseev [79–81]; the acronym appeared in a proposal
by Kraus et al. [82]. The protocol has been implemented experimentally by several
groups, all using rare-earth ions doped into solids [83–85].
The reasons for choosing these materials as storage media are manifold. First,
use of atoms in the solid state eliminates limitations to the storage time arising from
atomic collisions, or from atoms drifting out of the interaction region (see §10.5 in
Chapter 10). Second, the rare-earth elements all share a rather peculiar feature in
their electronic structure, namely that the radius of the 4f shell is smaller than the
radii of the (filled) 5s and 5p shells [86]. The optically active electrons in the 4f shell
are therefore shielded by the 5s and 5p shells. These filled shells are spherically
symmetric, and can be thought of as metallic spheres that isolate the f electrons
from external fields, much as would a Faraday cage. Therefore, even when doped
into a solid, rare-earths maintain much of their electronic structure. Perturbations
due to the surrounding ‘host’ material may induce frequency shifts, but noise and
fluctuations, which might reduce the possible storage time, are effectively eliminated.
Optical transitions between different states within the f shell are generally forbidden,
since all these states have the same parity (see §4.3.1 in Chapter 4). This means that
spontaneous emission from these states is greatly suppressed, making them ideal
for use in a quantum memory. Spontaneous lifetimes of several hours have been
measured [87], and photon storage times of up to 30 s are realistic [88]. Of course, if
no transitions are allowed at all, no incident photons can ever be absorbed. But
one effect of the host is to alter the atomic potential so that the f shell acquires an

2.3 Ensembles 54
admixture of d orbital states, making electric dipole transitions possible [89].
Finally, the 4f 4I15/2 ↔ 4I13/2 transition in Erbium has a wavelength of 1.5 µm,
which matches the wavelength at which optical fibres used for telecommunications
have minimal absorption. Particular attention has therefore been paid to the pos-
sibilities for building a solid state quantum memory based on Erbium ions, since it
would integrate extremely well with existing telecoms systems.
We can distinguish two categories of CRIB, based on the direction of variation
of the external field with respect to the propagation direction of the signal (see
Figure 2.10). If these directions are perpendicular, so that the atomic frequencies
are broadened across the ensemble, we call this tCRIB (transverse CRIB). If they
are parallel, with the atoms broadened along the ensemble, we call this lCRIB
(longitudinal CRIB). The latter of these is sometimes referred to as GEM (gradient
echo memory [84,90]). As we will see in Chapter 7, the performance of these two
schemes for photon storage is essentially the same.
(b)
(a)
Figure 2.10 tCRIB vs. lCRIB. (a): In tCRIB, an external fieldbroadens the ensemble resonance in a direction transverse to thepropagation direction of the light to be stored. (b) In lCRIB, thebroadening field is applied parallel to the propagation direction.

2.3 Ensembles 55
2.3.4 AFC
The atomic frequency comb memory protocol was proposed recently [91] by Afzelius
et al.. Their research group in Geneva have focussed on the implementation of CRIB,
and AFC takes a number of cues from their experience in this connection. In the
AFC protocol, we suppose that it is possible to prepare an atomic ensemble with
a large number of absorption lines equally spaced in frequency (see Figure 2.11).
This atomic frequency comb plays a similar role to the inhomogeneous broadening
in CRIB. It increases the bandwidth of the absorptive resonance, so that a tempo-
rally short signal is efficiently absorbed. As with CRIB, this means that the entire
absorption process can be completed long before the excited state |2〉 has any time
to decay. Again, for long-term storage, the excitation is mapped to the dark state
|3〉. The ‘trick’ in the design of the AFC protocol becomes clear at retrieval. Recall
that, after transferring the stored excitation back to |2〉, the atomic dipoles must be
brought back into phase with one another before the signal can be re-emitted. In
CRIB this is done by reversing the atomic detunings, and this would certainly work
for AFC. But even if the atomic resonances are not altered in any way, the AFC
memory still re-emits the signal! The reason is that the atomic frequency comb has
a discrete spectral structure. Therefore, the optical polarization undergoes periodic
revivals — re-phasings — at a rate given by the frequency of the beat note associ-
ated with the comb frequencies. If the frequency separation between adjacent comb
teeth is ∆, the time between re-phasings is roughly 1/∆.
Having introduced the principle behind AFC storage, a number of comments

2.3 Ensembles 56
(a)
Storage Retrieval
(b)
Figure 2.11 AFC storage. (a): A broadband signal photon, witha bandwidth covering many comb teeth, is absorbed by the atomicfrequency comb. The excitation is then transferred to state |3〉 bya control pulse. (b) To retrieve the signal, the control is re-appliedto return the excitation to the frequency comb. The atomic dipolesre-phase, due to the discrete structure of the comb, and the signal isre-emitted.
are in order. First, it is not obvious that an ensemble with a comb-like resonance
will smoothly absorb the signal photon. One might expect that only those parts of
the signal spectrum overlapping with the comb teeth would be absorbed, with the
intervening frequencies simply transmitted and lost. In fact, the absorption between
the comb teeth never vanishes completely. Provided the ensemble contains enough
atoms, the combined absorption over the whole ensemble is enough to store all the
frequencies in the signal.
Second, it is not obvious how the control field can transfer all the atoms in the
frequency comb from state |2〉 to state |3〉 and back again. This must be done
in AFC so as to prevent the periodic revivals in polarization from re-emitting the
signal too early. A sufficiently bright and short control pulse will accomplish the
state transfer — the control pulse bandwidth should span the full spectral width of
the comb. So-called coherent control can be used to shape the control pulse so as to

2.3 Ensembles 57
maximize the transfer efficiency [92–98].
Third, it is not obvious how to prepare an atomic frequency comb. The proce-
dure suggested by Afzelius et al. is based around an implementation with rare-earths
doped into solids. In these materials, natural variations in the position within the
host occupied by the rare-earths causes the resonant frequencies to be shifted ran-
domly over a broad spectral range. This natural inhomogeneous broadening is not
useful for CRIB, since it cannot be ‘reversed’. Therefore in the CRIB protocol, it is
necessary to remove all the atoms except those with the desired resonant frequency,
before applying the external field to artificially broaden the resonances of only these
atoms. To ‘remove’ undesired atoms, an optical pump is employed [99] (see §10.12 in
Chapter 10). This is a series of laser pulses with frequencies tuned so as to transfer
unwanted atoms from the ground state |1〉 into a new state |4〉 (not shown in any
diagrams so far), where they are ‘shelved’ for the duration of the memory protocol.
The shelf state can be chosen to have an extremely long lifetime (several hours, as
mentioned above in Section 2.3.3); this is why we can consider the atoms as having
been simply removed from the ensemble. To prepare the frequency comb for the
AFC protocol, a similar optical pumping procedure is used: all atoms with resonant
frequencies in between the comb teeth are shelved. A significant practical advantage
of AFC over CRIB is now clear. CRIB requires that we pump out all but a single
narrow resonance within the ensemble: we ‘throw away’ a lot of atoms. In AFC,
we pump out all but N narrow resonances, where N is the number of comb teeth,
which may be quite large. Therefore we throw away fewer atoms, and this allows for

2.4 Continuous Variables 58
a much stronger absorption — a much more efficient memory — using an ensemble
with the same doping concentration as a less efficient CRIB protocol. In Chapter
7 we will show that AFC is also well-suited to the parallel storage of multiple sig-
nal fields, making it attractive for use in quantum repeaters. AFC storage has not
yet been demonstrated in its entirety, but proof-of-principle experiments have been
performed by de Riedmatten et al. [100].
2.4 Continuous Variables
So far, our discussion of quantum memories has focussed on the storage of single
photons. For the purposes of QKD and computation discussed in Chapter 1, the
quantum information encoded into these photons is of a discrete nature. Either the
number of photons, or their polarization, can be used to represent the two basis
states of a qubit. This is not the only paradigm for quantum information process-
ing, however. Photons possess other degrees of freedom that are not discrete. The
position of a photon, or its momentum, for example. These quantities don’t lend
themselves to representation in terms of qubit states. But it is still possible to use
such continous variables to encode information (as is done in classical analogue com-
puting). And the specifically quantum features of qubits that confer their increased
computational power — superposition; entanglement — all carry over to continuous
variables. Therefore quantum computing protocols [101,102], and indeed QKD pro-
tocols [103–106], that take advantage of the continuous degrees of freedom possessed
by photons, have all been developed. In general their relationship to the equivalent

2.4 Continuous Variables 59
qubit-based protocols is similar to that between analogue and digital classical com-
putation. On the one hand, the continuous versions are robust to noise, in the sense
that information is not completely erased in the presence of distortions. On the
other hand, below a certain threshold, digital algorithms can be made essentially
impervious to noise, producing ‘perfect’ outputs, whereas analogue computations
are always subject to fluctuations — they never work perfectly. Anyone who has
ever witnessed the catastrophic failure of ‘crisp and clear’ digital television with a
poor signal will appreciate the fuzzy watchability of analogue television in the same
conditions.
In this section we briefly introduce the concepts of continuous variables, so as
to understand a class of memory based on continuous variables storage. The most
commonly used variables are the field quadratures. These are defined in terms of the
electric field E associated with an optical mode,
E = x cos(ωt) + p sin(ωt), (2.1)
where ω is the optical carrier frequency, and t is the time. The coefficients x and
p for the amplitude of the field are known as the in-phase and out-of-phase field
quadratures, respectively [107]. By convention, the same symbols as would normally
be associated with position and momentum are used, and this is motivated by their
formal similarity. Like the position and momentum of a classical pendulum, the
quadratures are continuous variables that describe the oscillation of the field. Even

2.4 Continuous Variables 60
more strikingly, in quantum mechanics, x and p are complementary in the same
way as are the position and momentum of a harmonic oscillator. That is to say,
a measurement of one quadrature ‘disturbs’ the other, so that it is impossible to
precisely measure both simultaneously. In appropriately scaled units, the Heisenberg
uncertainty principle applies [103]:
∆x∆p ≥ 1, (2.2)
where ∆x, ∆p are the precisions with which the quadratures are simultaneously
known. Just as in classical physics, the quantum state of an optical field can be
completely described in terms of x and p. Due to the uncertainty principle (2.2),
the state is not a single point in (x,p)-space, or phase space as it is more often called,
but rather a kind of ‘blob’, whose spread represents the uncertainties ∆x and ∆p.
This blob is known as the Wigner distribution [108], and it is the representation of
choice for states parameterized by continuous variables. The most ‘normal’ state
of an optical field — the coherent state — is a monochromatic beam, like that
produced by a laser. It is completely classical, in the sense that the formalism
of quantum mechanics is not required to describe it. Classical electromagnetism
is sufficient. Its Wigner distribution is a Gaussian ‘hill’, with equal uncertainties
in x and p (see Figure 2.12 (a)). Clearly this is not the only type of distribution
compatible with 2.2. Figure 2.12 (b) shows an example of a squeezed state, with
a small uncertainty in x. The price for this increased precision in x is that the

2.4 Continuous Variables 61
uncertainty in p grows, but states like this can be extremely useful for reducing
the noise on measurements associated with just one of the quadratures. Squeezed
states of light would appear to be rather exotic, but they arise naturally in any
process that converts one frequency into another. Such non-linear processes only
require that the potential in which optically active electrons move is not exactly
quadratic1 in the electrons’ displacement. This happens to some extent in nearly all
materials, and the technology for generating efficient squeezing is now rather well
developed [109–112].
(b)
(a)
Figure 2.12 Wigner distributions in quadrature phase space fortwo different states of an optical mode. (a): A classical coherentstate, with equal uncertainties in x and p. (b) A squeezed state, with∆x halved, but ∆p doubled. The lengths of the dotted lines give thebrightness of the states; their angles give their relative phases.
Research into squeezed light is being actively pursued as a means to improve the
sensitivity of gravitational wave detectors [113,114], and to toughen the noise tolerance
of communication systems [115]. As might be expected from the ubiquity of uncer-1If the potential is not quadratic (i.e. anharmonic), the ‘restoring force’ on the electrons is
non-linear.

2.4 Continuous Variables 62
tainty relations like (2.2) in quantum mechanics, the phenomenon of squeezing is
not confined to light, and their are metrological benefits that accrue if atoms can be
squeezed [116]. Increased sensitivity to magnetic fields [117], enhanced spectroscopic
precision [118], and better atomic clocks [119–121], as well as quantum computation and
communication, are all enabled by the ability to manipulate non-classical states of
light and matter.
One of the most successful demonstrations of quantum memory has grown out
of research in this area. The mechanism by which storage is accomplished is most
transparent when couched in the language of continuous variables. The memory
works by transferring the quadratures of a light beam into an ensemble of atoms,
where the quadratures X, P associated with the atoms are their ‘coronal’ angular
momenta Jx = X, Jy = P (see Figure 2.13). The storage interaction involves two
steps. In the first, a control and signal beam — both tuned far off-resonance —
are directed through an atomic ensemble. A strong magnetic field is applied to the
ensemble, which aligns the atomic spins, so that all the atoms are initially in state
|1〉 (see Figure 2.15). The control is polarized parallel to the field, so that it does not
induce a turning moment. On the other hand, the signal is polarized perpendicular to
the field. Two-photon Raman transitions, involving both the control and the signal,
can therefore change the z-component of the collective atomic angular momentum,
transferring atoms from |1〉 to |3〉 or vice versa. In terms of the atomic quadratures,

2.4 Continuous Variables 63
the passage of the optical fields through the ensemble induces the transformation
X → X = X + p, (2.3)
where the tilde denotes the quadrature value at the end of the interaction. At the
same time, the x quadrature of the light is also shunted — a manifestation of the
Faraday effect,
x→ x = x+ P. (2.4)
The other quadratures p, P of both the signal and the atoms are unchanged. For this
reason the interaction is known as a quantum non-demolition (QND) measurement,
since information about the p quadrature of the signal is transferred to the atoms,
without altering it.
In the second step of the storage protocol, the x quadrature of the signal is mea-
sured using balanced homodyne detection [107]. As shown in Figure 2.14, a polarizing
beamsplitter, aligned in the 45-degree basis, mixes the control and signal beams.
The difference in the intensities detected at the two output ports of the beamsplit-
ter is directly proportional to x. This measurement result is then used to determine
the strength of a radio frequency pulse that applies a controlled torque to the atoms,
producing a shift
P → P = P − x = P − (x+ P ) = −x. (2.5)
The two maps (2.3) and (2.5) taken together almost constitute an ideal memory.

2.4 Continuous Variables 64
Figure 2.13 Atomic quadratures. In the presence of a strong mag-netic field, the atomic spins in an ensemble align with the z-axis. Thequadratures X and P for the atoms are then given by small displace-ments of their angular momenta in the coronal plane, normal to thez-axis.
Apart from an unimportant minus sign in (2.5), the only difficulty is the presence
of the initial atomic quadrature X in (2.3). The average value of X can be made to
vanish, but the finite spread of the initial atomic Wigner distribution still introduces
unwanted fluctuations. This spread can be reduced by squeezing the Wigner distri-
bution of the atoms, so that ∆X → 0, before attempting the memory, and schemes
for doing this are in development [116].
The above type of continuous variables memory, based on a QND interaction
followed by measurement and feedback, was first implemented by Julsgaard et al.
at the Niels Bohr intitute in Copenhagen [122] using an ensemble of cesium atoms.
The same research group, led by Eugene Polzik, has since refined and extended
the technology, and have demonstrated quantum teleportation of light onto atoms,
deterministic entanglement generation, efficient spin squeezing of atomic ensembles

2.4 Continuous Variables 65
Figure 2.14 QND memory. A magnetic field aligns the atomicspins in an ensemble. A strong control, polarized parallel, and aweak signal, polarized perpendicular to the spins, are sent throughthe atoms. The control cannot rotate the atomic spins, but the signaldoes. A homodyne measurement extracts the resulting x quadratureof the signal, and a radio frequency pulse, generated by the coilsshown, applies a final rotation.
and many other continuous variables protocols. An excellent review of their progress
in this area can be found in the recent review article by Hammerer et al [123].
The conceptual shift between the above description in terms of continuous vari-
ables, and the rather intuitive picture of ‘reversible absorption’ we have used for all
the other memory protocols, makes comparisons difficult. Certainly the optimiza-
tion of this protocol does not fit into the general scheme we apply to the optimization
of the other memory protocols in this thesis. The scheme has enjoyed considerable
success, despite its technical complexity, probably due in large part to the experience
and prowess of those working at the Niels Bohr institute. Nonetheless, it does not
perform as well as the other protocols as a component of a DLCZ-type quantum
repeater. This is because, as described in Chapter 1, the entanglement purification

2.4 Continuous Variables 66
Figure 2.15 Level scheme for a QND memory. The atoms beginwith their spins aligned with the external magnetic field, in state|1〉. State |3〉 cannot be reached by interaction with the control alone(green arrows), since it is π-polarized — parallel to the B-field —and it cannot induce a spin flip (see Figure F.4 in Appendix F).Raman transitions involving both the control and signal (blue wavyarrows) can transfer atoms to state |3〉. The intermediate excitedstates are collectively labelled |2〉. Both types of Raman interactionare involved: Stokes scattering, as in the DLCZ protocol (see Figure1.6 in Chapter 1), and the anti-Stokes scattering used in the Ramanmemory protocol (see Figure 2.8).
in these repeaters is effective against photon loss, but not against photon addition,
and the Raman transitions shown in Figure 2.15 can produce extra photons that
contaminate the number state entanglement. Other applications for which photon
loss is particularly damaging would benefit from a continuous variables memory,
since loss can be essentially eliminated by increasing the power of the RF pulses
applied in the feedback step, at the expense of distorting the stored state. In any
case, we will not discuss continuous variables memory further.
In the next chapter we introduce the optimization scheme relevant for the absorption-
based memories we have discussed.

Chapter 3
Optimization
Here we introduce the optimization scheme that we will apply to the Raman, EIT,
CRIB and AFC ensemble memory protocols. In all these protocols, an input field,
the signal field, is transferred to a stationary excitation inside an atomic ensemble.
The aim of the optimization is to maximize the efficiency of this transfer, which
amounts to maximizing the amount of stored excitation, given a fixed input. Fortu-
nately, a technique borrowed from the mathematical toolbox of linear algebra makes
this optimization extremely easy to perform.
Suppose that the signal field is a pulse, with a time-dependent amplitude A(τ),
where τ is the time. The action of the memory is to absorb this input pulse, and
convert the incident energy into some kind of long-lasting excitation within the
ensemble (see Figure 3.1). We’ll denote the amplitude of this excitation by B(z),
where z is the position along the ensemble. Here the z dependence allows for the
possibility that the excitation may be distributed over the length of the ensemble

68
in a non-uniform way. This kind of collective, de-localized excitation is typically
referred to as a spin wave, since in many cases the atomic states involved are states
of different spin angular momentum (as in the QND memory discussed at the end
of Chapter 2). We’ll use this term indiscriminately, to describe any distributed
excitation relevant to quantum storage, regardless of the nature of the quantum
numbers associated with the atomic states. In the next chapter, we’ll define the spin
wave more precisely. For the purposes of optimization, it is sufficient to understand
the spin wave as the stationary counterpart of the propagating signal field. That
is, the storage process is a mapping A→ B, and the retrieval process is the inverse
map B → A.
Figure 3.1 Storage map. An incident signal field A(τ) is mappedto a stationary spin wave B(z) within an atomic ensemble.
If the signal field initially contains NA = N photons, we would ideally like the
spin wave at the end of the storage process to contain the same number of excited
atoms NB = N , so that all the input light has been stored. In practice, some of
the input light will pass through the ensemble and be lost, and some of the excited
atoms will decay back down to their ground state, or drift out of the interaction
region. The efficiency η of the storage interaction is simply the ratio of the number

69
of excited atoms to the number of input photons, η = NB/NA, where the number of
quanta, either material of optical, is found by integrating the squared norm of the
relevant amplitude,
NA =∫ ∞−∞|A(τ)|2 dτ, NB =
∫ L
0|B(z)|2 dz. (3.1)
Here L denotes the length of the ensemble. We note that in this type of memory
there is no process that can produce extra excitations. At least in principle, there
is no source of background noise. The failure mode of the memory is photon loss:
photons directed into the memory are not stored, and so are not recovered in the
retrieval process. This differs from the QND memory mentioned in the previous
chapter, which need not suffer from photon loss, but which may introduce noise at
retrieval. Therefore the performance of this type of memory is optimal when the
efficiency is maximized. No other figure of merit, involving the suppression of atomic
fluctuations for instance, is relevant.
A key requirement of a quantum memory is linearity. That is, suppose that we
store a signal field A = αA1 + βA2 built from two contributions A1 and A2. This is
a superposition state, like (1.1) in Chapter 1, and to preserve its encoded quantum
information, the coefficients α, β should not be altered by the storage process. The
resulting spin wave should be of the form B = αB1 +βB2, where B1, B2 are the spin
waves generated by storage of A1, A2 only. This property, required to faithfully store
superpositions, restricts the storage map A → B to be a linear map (and similarly

3.1 The Singular Value Decomposition 70
for the retrieval process). Fortunately, as we will see, all the memory protocols we
will analyze are indeed linear. In general, then, we can always write the storage map
in the following way
B(z) =∫ ∞−∞
K(z, τ)A(τ) dτ. (3.2)
The integral kernel K is known as the Green’s function, or the propagator for the
storage interaction. It contains all the information about how the memory behaves.
In the next chapter we will show how to derive expressions for K in some cases.
Generally, it is possible to construct the form of K numerically, as we will show in
Chapter 5 (see §5.4). For the moment, we suppose that we are able to write the
memory interaction in the form (3.2). It is clear that the efficiency of the memory
depends on achieving a good ‘match’ between K and the shape, in time, of A. For
instance, no storage is possible if K ∼ 0 during the arrival time of the signal. K
and A should ‘overlap’. Optimizing a quantum memory involves finding the shape
of A that maximizes this overlap, so that all of A ends up in B. In the next section
we introduce the singular value decomposition, a valuable analytical tool that can
be used to find this optimal shape, and more besides.
3.1 The Singular Value Decomposition
The SVD is most commonly encountered in the context of matrices. It is one of
the most useful results in linear algebra, and it finds applications from face recog-
nition [124] to weather prediction [125]. Under the name Schmidt decomposition it is

3.1 The Singular Value Decomposition 71
of critical importance in quantum information theory as a tool for the analysis of
bi-partite entanglement [126]. It seems to have been discovered independently several
times in the 19th century [127–129]. Erhard Schmidt applied the decomposition to
integral operators (an example of which is (3.2)) in 1907 [130], and Bateman coined
the term ‘singular values’ in 1908 [131]. The proof of the decomposition for arbitrary
matrices was given in 1936 by Eckart and Young [132].
At the heart of the SVD is a geometric interpretation for the action of a linear
operator. A linear operator, or linear map, takes some vector a as input, and spits
out another vector b as output. Representing the linear operator as a matrix M , we
have
b = Ma. (3.3)
Looking at Figure 3.2 (a), it is clear that such a transformation is equivalent to the
following procedure. (i) rotate a until it lies along one of the coordinate axes (ii)
re-scale the axes so that the length of the rotated version of a matches the length of
b (iii) rotate this re-scaled vector until it sits on top of b. This suggests that it should
be possible to decompose an arbitrary linear transformation by combining rotations
and coordinate re-scaling. The SVD is nothing more than this representation of a
general linear map as a rotation, a re-scaling, and a final rotation. An example is
shown in Figure 3.2 (b), where the action of M is shown on the set a of all vectors
with a certain length. The tips of these vectors trace out the surface of a sphere.
The effect of M is to ‘squish’ this sphere into an ellipsoid. This has to be done in
such a way that the black dot on the sphere ends up as the red dot on the ellipsoid.

3.1 The Singular Value Decomposition 72
(a) (b)
Figure 3.2 Linear transformation. (a): a vector a is mapped ontoa vector b by M , which may be seen as implementing a rotation ofa onto the x-axis, followed by a coordinate re-scaling to increase thelength of a until it matches the length of b, followed by a rotationonto b. (b): The action of M on the set of all initial vectors a(the grey sphere) produces a set of vectors b (the red ellipsoid). Asan example, the red dot is the image of the black dot under underM . The final ellipsoid can be generated from the initial sphere by:rotating the sphere, re-scaling the axes and then rotating again.
To produce the ellipsoid from the sphere, it is necessary to first rotate the sphere
until the black dot is placed appropriately. Then the x, y and z axes are re-scaled
to deform the rotated sphere into the ellipsoid ‘shape’. Finally, a second rotation
puts the ellipsoid in the correct orientation, with the black dot sitting on top of the
red dot.
The above discussion is limited to the intuitive case of a real vector in three
dimensions being mapped to another real vector in three dimensions. In fact, the
SVD exists for all matrices M — all linear transformations. That is, M could be a
complex rectangular matrix of any size, that maps a complex n-dimensional vector
to a complex m-dimensional vector. In general, M can always be written in the
form
M = UDV †. (3.4)

3.1 The Singular Value Decomposition 73
Here U and V are both unitary matrices, and D is a real, positive, diagonal matrix.
The properties of these types of matrices are reviewed in Appendix A. In terms
of the discussion above, V † represents a rotation into a new coordinate system,
D represents a re-scaling of this new coordinate system, and U represents a final
coordinate rotation. The elements of D, lying along its diagonal, are the factors
by which the coordinates are re-scaled in the second step. They are known as the
singular values of M , and they contain a great deal of useful information about the
transformation represented by M . Geometrically, the singular values correspond
to the lengths of the semi-axes of the ellipsoid in Figure 3.2 (b). In this thesis,
singular values are generally denoted by the symbol λ, the same as eigenvalues. It
should always be clear from the context what is meant. By convention, the singular
values, which are all positive, real numbers, are ordered in descending magnitude,
so that D11 = λ1 is the largest singular value, D22 = λ2 is smaller, and so on. It is
sometimes useful to visualize the structure of the matrices in the SVD, and so for
reference we provide the following tableau,
M =
|u1〉
. . . |um〉
︸ ︷︷ ︸
U
λ1
. . .
λm
︸ ︷︷ ︸
D
〈v1|
...
〈vm|
︸ ︷︷ ︸
V †
. (3.5)

3.1 The Singular Value Decomposition 74
Performing the matrix multiplications explicitly, we obtain
M =∑j
λjMj , (3.6)
where each of the matrices Mj = |uj〉〈vj | is an outer product of the jth columns
of U and V (see Section A.2.1 in Appendix A). This last representation provides
a natural way to interpret the action of M , as a sum of independent mappings
from the column-space of V to the column-space of U . To understand why this is
useful, recall that the sets of column vectors |vj〉 and |uj〉 of U and V are both
orthonormal bases, because U and V are unitary (see Section A.4.5 in Appendix
A). Therefore any vector |a〉 to which M is applied can be written in the coordinate
system defined by the |vj〉, and the result |b〉 can always be written in terms of
the |uj〉,
a = |a〉 = a1|v1〉+a2|v2〉+. . .+an|vn〉, and b = |b〉 = b1|u1〉+b2|u2〉+. . .+bm|um〉.
(3.7)
Applying (3.6) to |a〉, and making use of the orthonormality of the u’s and v’s,
〈ui|uj〉 = 〈vi|vj〉 = δij , we find that
b1 = λ1a1, b2 = λ2a2, etc . . . . (3.8)
So M can be viewed as a set of mappings between two special coordinate systems,
each with a different ‘fidelity’, given by the singular values. M may seem like

3.1 The Singular Value Decomposition 75
a complicated transformation, but as long as we use the |vj〉 to define our input
coordinate system, and the |uj〉 to define our output coordinate system, the action
of M is always extremely simple. It just maps each coordinate from the input onto
the corresponding output coordinate, re-scaled by the corresponding singular value.
3.1.1 Unitary invariance
If the input coordinate system is rotated, before performing the SVD, this cannot
change the singular values, but only the input basis that we should use. The same
is true if the output coordinate system is rotated. More generally, consider applying
some unitary transformation W to M . The SVD of this compound operator is then
WM = WUDV † = UDV †, (3.9)
where U = WU is a unitary matrix, since both W and U are unitary. The output
basis vectors are modified by W , but the singular values — the elements of D —
are unchanged. By the same token, the product MW also has the same singular
values as M , but V must be replaced by W †V . Sometimes it is only possible to find
rotated versions of a matrix M , in which case this property of the SVD is useful.
3.1.2 Connection with Eigenvalues
The SVD is connected with the eigenvalues of the ‘square’ of M . More precisely, con-
sider the normally and antinormally ordered productsKN = M †M andKA = MM †.
These two products are both Hermitian, since K†N = KN and K†A = KA. Therefore,

3.1 The Singular Value Decomposition 76
they both have real eigenvalues, and their eigenvectors each form orthonormal bases
(see Section A.4.3 in Appendix A for a derivation of this fact). And inserting the
decomposition (3.4), we see that
KN = V D2V †, and KA = UD2U †, (3.10)
where we have used the relations U †U = V †V = I, which must hold for unitary
matrices. Therefore, the eigenvalues of KN and KA are both given by the squares
of the singular values.
3.1.3 Hermitian SVD
Note also that if M is a Hermitian matrix, with M = M †, we must have that U = V .
That is,
M = UDU †, (3.11)
which is precisely the spectral decomposition of M , with the eigenvalues of M lying
along the diagonal of D, and the eigenvectors of M making up the columns of U . So
the SVD and the spectral decomposition of M are identical for Hermitian matrices.
3.1.4 Persymmetry
Another case, which we will encounter in our treatment of Raman storage, is that
of persymmetry. Suppose that M is a real, square matrix, with n = m. Then M is
persymmetric if it is symmetric under reflection in its anti-diagonal — the diagonal

3.1 The Singular Value Decomposition 77
running from bottom left to top right, as shown in Figure 3.3. M = MP, where
(MP)ij = Mm−j+1m−i+1. This is rather an unusual type of symmetry, and it is
Figure 3.3 Persymmetry. The upper left and lower right portionsof a real persymmetric matrix are mirror images of eachother underreflection in the anti-diagonal, represented as a grey stripe.
not often discussed in textbooks. But the action of a persymmetric matrix can be
viewed as very similar to that of a real Hermitian matrix, the only difference being
that the result is ‘flipped around’. To see the implications of persymmetry for the
SVD, let us write M = XH, where H is a real Hermitian matrix (i.e. symmetric
under reflection in its main diagonal), and where X is a ‘flip’ matrix, with ones
along its anti-diagonal, and zeros everywhere else (left blank for clarity below),
X =
1
1
. . .
1
. (3.12)
The action of X when multiplying a matrix is to flip it around a horizontal axis, so
that its last row becomes its first, and vice versa. For every persymmetric matrix M ,
there must always be some Hermitian matrix H such that M = XH. The property

3.2 Norm maximization 78
of persymmetry can also be easily written in terms of X. A little mental acrobatics
will verify thatMP = XM †X, so that persymmetry requiresM = XM †X. Inserting
the spectral decomposition of H, we obtain
M = XM †X = X(XH)†X = XH†X†X = XHX2 = XUDU † = UDU †, (3.13)
where we used the Hermiticity of H and X, along with the fact that X2 = I (two
horizontal flips cancel each other out), and where U = XU is a unitary matrix whose
columns have been flipped round. Therefore the SVD of a persymmetric matrix is
such that the columns of U , the basis for the output coordinate system, are flipped
versions of the columns of V , the input basis. This result will be of use to us in
Chapter 5.
3.2 Norm maximization
Suppose that we would like to know how to choose a in order to maximize the norm
(the length) of b. Since M is a linear transformation we can always increase the
norm of b just by increasing the norm of a: if we double the length of a, the length
of b also doubles. But this is not interesting. Clearly the direction of a matters;
some directions will result in a larger norm for b. If the norm of a is fixed, how
should we choose its direction? This question is easily answered if we are able to
compute the SVD of M . Without losing generality, let ||a|| = a = 1. That is,

3.3 Continuous maps 79
|a1|2 + |a2|2 + . . .+ |an|2=1. Using (3.8), the norm of b is then given by
b2 = |b1|2 + |b2|2 + . . .+ |bm|2
= λ21|a1|2 + λ2
2|a2|2 + . . . λ2n|an|2. (3.14)
But by definition, λ1 is the largest of the singular values, so that
b2 < λ21(|a1|2 + |a2|2 + . . .+ |an|2)
= λ21, (3.15)
That is, the largest possible norm of b is b = λ1. This maximum norm is obtained
by choosing a1 = 1, with all other components vanishing. So the ‘optimal’ vector
a, with regard to maximizing b, is a = v1 (or |a〉 = |v1〉). From the perspective
of Figure 3.2 (b), this amounts to choosing a so that the resulting b lies along the
largest semi-axis of the ellipsoid generated by M .
3.3 Continuous maps
The preceding discussion of matrices can be extended, without essential modifica-
tion, to the continuous map (3.2) describing the storage interaction in a quantum
memory. The Green’s function K(z, τ) has the same basic structure as a matrix,
except that it has two continuous arguments, instead of two discrete indices. And
it is also amenable to the SVD. The expression (3.6) for a matrix becomes, in the

3.3 Continuous maps 80
continuous case
K(z, τ) =∑j
λjψj(z)φ∗j (τ). (3.16)
As before, the λ’s are the singular values. The functions ψj and φj are the continuous
analogues of the input and output basis vectors |uj〉 and |vj〉. We will refer to them
as modes; φ1 is the first input mode, ψ2 is the second output mode, and so on.
Alternatively, since the signal field and spin wave both appear in (3.2), it may be
physically more meaningful to talk of the φ’s as the signal modes, and the ψ’s as
spin wave modes. The inner product between two vectors a, b takes the form of an
overlap integral, for continuous functions a(x), b(x),
a†b = 〈a|b〉 =∑i
a∗i bi −→∫a∗(x)b(x) dx. (3.17)
The orthonormality conditions 〈ui|uj〉 = 〈vi|vj〉 = δij for basis vectors are therefore
replaced by the relations
∫ L
0ψ∗i (z)ψj(z) dz =
∫ ∞−∞
φ∗i (τ)φj(τ) dτ = δij . (3.18)
These conditions tend to produce sets of ‘wiggly’ functions, so that the product of
two different modes alternates between positive and negative values, and integrates
to zero. Sets of oscillating modes, satisfying orthonormality conditions like (3.18),
are common in harmonic analysis and acoustics; they are also the bread and butter
of much of quantum physics — quantum memories are no exception. In general, the

3.3 Continuous maps 81
first modes, associated with the largest singular value λ1, are slowly varying. They
represent the basic ‘shape’ of the Green’s function, with broad brushstrokes. Higher
modes tend to oscillate faster, and they represent smaller corrections, at increasingly
fine levels of detail.
3.3.1 Normally and Anti-normally ordered kernels.
The continuous analogues of the matrices KN and KA in (3.10) are found by inte-
grating the product of two K kernels over one of their arguments,
KN (τ, τ ′) =∫ L
0K∗(z, τ)K(z, τ ′) dz,
KA(z, z′) =∫ ∞−∞
K(z, τ)K∗(z′, τ) dτ. (3.19)
These two kernels share the same eigenvalues; they satisfy the eigenvalue equations
∫ ∞−∞
KN (τ, τ ′)φj(τ ′) dτ ′ = λjφj(τ ′),∫ L
0KA(z, z′)ψj(z′) dz′ = λjψj(z′). (3.20)
Sometimes these kernels are more convenient to work with than the kernel K.
3.3.2 Memory Optimization.
Now it is clear how to optimize the performance of a quantum memory. We want
the largest efficiency η = NB/NA. But from the expressions in (3.1), we see that
NB is just the continuous analogue of b2, the squared norm of the output spin wave.

3.3 Continuous maps 82
Suppose that we fix NA = 1, just as we fixed a2 = 1 in Section 3.2. How should
we choose the signal field A(τ) to maximize η? The answer carries over directly
from our considerations of matrices. We should choose A(τ) = φ1(τ). With this
choice, the resulting spin wave is B(z) = λ1ψ1(z), and the optimal storage efficiency
is η = λ21.
In practice, the SVD of the Green’s function is almost always computed numer-
ically, and to do this, the continuous function K is discretized on a fine grid: it is
converted into a matrix. Therefore our treatment of matrices applies directly when
calculating the singular values, and the mode functions, for a quantum memory.
Note that, since we must of course have η ≤ 1, any physical Green’s function will
have λ1 ≤ 1, and this condition is a useful check that K has been sampled with a
sufficiently fine grid.
3.3.3 Unitary invariance
When deriving a form for the Green’s function K, it will sometimes be easier to
work in terms of a transformed coordinate system. Suppose that we introduce a
new variable y = y(z), where y(z) is some monotonic single-valued (i.e. invertible)
function. The Green’s function, expressed in terms of y, has the same singular values
as in the original coordinates, provided that we make the transformation unitary by
including a Jacobian factor:
K(y, τ) = K [z(y), τ ]× 1√J(y)
, (3.21)

3.3 Continuous maps 83
where z(y) is the inverse transformation, relating z to y, and where J(y) = ∂zy is
the Jacobian, relating the line elements dy and dz. Including the factor involving J
ensures that the norm of K is preserved in the new coordinate system,
∫|K(y, τ)|2 dy =
∫|K [z(y), τ ] |2 × ∂z
∂ydy
=∫|K(z, τ)|2 dz.
The output modes transform accordingly,
ψj(y) =ψj [z(y)]√
J(y). (3.22)
An identical procedure is used when transforming the time coordinate.
Instead of re-parameterizing the kernel, we may wish to transform to a frequency
representation, instead of a temporal one. That is, we might be able to work out an
expression for the Fourier transformed Green’s function K
K(z, ω) =1√2π
∫K(z, τ)eiωτ dτ. (3.23)
Inserting the SVD expansion (3.16) into (3.23), we obtain
K(z, ω) =∑j
λjψj(z)φ∗j (ω), (3.24)
where the modes φj are Fourier transforms of the φj . These Fourier transformed

3.3 Continuous maps 84
modes also form an orthonormal set,
∫φ∗i (ω)φj(ω) dω =
12π
∫ [∫φ∗i (τ)e−iωτ dτ
∫φj(τ ′)eiωτ ′ dτ ′
]dω
=∫ ∫
δ(τ − τ ′)φ∗i (τ)φj(τ ′) dτ ′dτ
= δij , (3.25)
where in the penultimate step we used the plane-wave expansion of the Dirac delta
function (see §D.2 in Appendix D),
δ(x) =1
2π
∫e±ixy dy. (3.26)
The expression (3.24) is therefore precisely the SVD of the transformed kernel K.
The singular values of K are the same as those of K. In fact the temporal Fourier
transform we applied is just an example of a unitary transformation; unitary because,
by Parseval’s theorem, it is norm-preserving. That is to say,
∫|f(τ)|2 dτ =
∫|f(ω)|2 dω, (3.27)
for any function f and its Fourier transform f . As described in Section 3.1.1, the
singular values of a mapping are never altered by a unitary transformation.
Other useful possibilities include Fourier transforming over the spatial variable,

3.4 Optimizing storage followed by retrieval 85
to find the Green’s function in so-called k-space,
K(k, τ) =1√2π
∫K(z, τ)eikz dz. (3.28)
Again the singular values are the same as those for K, but the output modes are
k-space versions of the ψj .
Generally the integration limits in these Fourier transforms would be [−∞,∞],
but when boundary conditions are needed, it will sometimes be useful instead to
implement a unilateral transform, where the integration runs from 0 to ∞. The
utility of these techniques will become clear when we examine the memory inter-
action more closely in the next chapter. A brief review of both the bilateral and
unilateral Fourier transforms can be found in Appendix D.
3.4 Optimizing storage followed by retrieval
Suppose that we are not interested in the efficiency of storage alone, but rather
the combined efficiency of storage into, followed by retrieval from the memory. In
many situations it is this combined efficiency that is most experimentally relevant.
The same techniques as outlined above are directly applicable. The entire memory
interaction can be characterized as a map between the input and the output signal
fields,
Aout(τ) =∫ ∞−∞
Ktotal(τ, τ ′)Ain(τ ′) dτ ′. (3.29)

3.5 A Simple Example 86
The total efficiency of the memory is given by the ratio of the norms of Aout and
Ain. The input mode that maximizes this efficiency is therefore found from the
SVD of the Green’s function Ktotal, and the resulting optimal efficiency is given by
ηcombined = λ21, where λ1 is the largest singular value of Ktotal.
If we neglect any decoherence of the spin wave (as we do throughout this thesis),
the kernel Ktotal can be constructed from the kernels describing the storage and
retrieval processes individually. Under certain circumstances — when the retrieval
process is precisely the time-reverse of the storage process [133] — the combined
kernel Ktotal is equal to the normally ordered kernel KN defined in (3.19), and
ηcombined = η2. This is the optimal situation, and in general a mismatch between the
storage and retrieval processes reduces the memory efficiency so that ηcombined < η2.
These issues are explored in more detail in Chapter 6.
3.5 A Simple Example
Before embarking on a detailed derivation of the equations of motion for an ensemble
quantum memory, we run through a simple example that contains many of the
features that emerge from a more rigorous analysis.
We consider a classical optical signal pulse propagating through an ensemble of
identical atoms. We use a classical Lorentz model for the atoms [134], in which the
electric field of the light pulse ‘pulls’ on an electron in each atom, while a harmonic
restoring force ‘pulls back’, keeping the electrons bound around their equilibrium
positions. Let the average displacement at time τ , away from equilibrium, of an

3.5 A Simple Example 87
electron located at position z, be given by x(z, τ). The average is taken over all
the atoms with the same position coordinate z, all of which behave identically. The
restoring force on each electron is given by Frestore = −kx, for some constant k.
This is a good approximation for any kind of restoring force, provided the typical
displacement x is small enough. The force on an electron due to the electric field
E of the signal pulse is Flight = eE, where e is the electronic charge. The classical
equation of motion for x is then given by Newton’s second law, Ftotal = ma, where
m is the electronic mass and a = ∂2τx is the acceleration of the electron. Putting
this together yields the equation
(m∂2
τ + k)x = eE. (3.30)
We can write the signal field as E = Aeiωsτ , where ωs is the optical carrier frequency
of the signal, and where A is a slow modulation describing the temporal profile of
the pulse. Irradiating the atoms will produce a response with a similar temporal
structure, so we write x = Beiωsτ , where B is some slowly varying envelope. Sub-
stituting this into the equation of motion, and neglecting the term ∂2τB, gives the
equation
∂τB = −iαA, (3.31)
where α = e2mωs
. Here for simplicity we have assumed that the signal field frequency
is tuned perfectly into resonance with the atoms, so that ωs =√k/m. This equation
describes the response of the atoms to the field; to complete the picture, we would

3.5 A Simple Example 88
like to understand how the field responds to the atoms. As a first step, we apply a
(bilateral) Fourier transform from τ −→ ω, so that (3.31) becomes
B =α
ωA, (3.32)
where the tildes denote the transformed variables. The electronic displacement B
acts as a source for the signal field. More precisely, each oscillating electron generates
an electric field E(r) = ae/4πε0c2r in proportion to its acceleration1. Here ε0 as
usual denotes the permittivity of free space; r is the distance from the electron.
Summing the contributions from all the atoms in a thin slice of the ensemble with
thickness δz, we find the total field generated by the dipoles is
Etot = −iδzωsen
4πε0cx, (3.33)
where n is the atomic number density [137]. If we consider the propagation of the
signal through a thin slice of the ensemble of thickness δz, we therefore have
A(z + δz, ω) = A(z, ω)− iδzωsen
4πε0cB(z, ω)
=(
1− iβδz
ω
)A(z, ω), (3.34)
1This can be derived rather neatly from relativistic equivalence. The electrostatic potentialenergy V = e2/4πε0r of two electrons separated by r produces a relativistic mass increase M = V/c2
of the pair of electrons. The extra weight 12gM of one of the electrons, in a gravitational field g,
must have its origin in a vertical force eE exerted by the electric field E of the other electron. Theequivalence principle demands that we cannot tell if the field g is swapped for an acceleration a.Putting this all together, we find the field due to an accelerating charge is E ∼ ae/4πε0c2r [135,136].

3.5 A Simple Example 89
where β = e2n8πε0mc
. Taking the limit as δz −→ 0, we derive Beer’s law of exponential
absorption for the signal field, with absorption coefficient β,
A(z, ω) = limδz→0
(1− i
βδz
ω
)z/δzAin(ω)
= e−iβz/ωAin(ω), (3.35)
where Ain(ω) = A(z = 0, ω) is the initial spectrum of the signal field, at the start of
the ensemble [138]. Substituting this into (3.32) gives an expression for the average
electronic displacement in the Fourier domain,
B(ω, z) = αe−iβz/ω
ω× Ain(ω). (3.36)
Using the result (D.40) from §D.5 in Appendix D, along with the convolution theo-
rem (D.21), we can take the inverse Fourier transform of this to obtain the map
Bout(z) = −iα∫ T
0J0
[2√βz(T − τ)
]Ain(τ) dτ ′, (3.37)
where Bout(z) = B(τ = T, z) is the electronic displacement at time τ = T , with
T some time chosen to define the end of the interaction, after the signal pulse has
passed through the ensemble. Here J0 is a zero’th order ordinary Bessel function
of the first kind — we will encounter this function, through similar inverse Fourier
transforms, frequently in Chapter 5. We have arranged for the notation in (3.37) to
appear suggestive of the storage map described at the start of this Chapter. If we

3.5 A Simple Example 90
identify the electronic displacement Bout as the amplitude of a spin wave excited by
the signal field, then the storage kernel K in (3.2) can be identified with the Bessel
function appearing in the integrand of (3.37). A numerical SVD of the Green’s
function
K(z, τ) = −iαJ0
[2√βz(T − τ)
](3.38)
would reveal the temporal shape of Ain that maximizes the degree of atomic exci-
tation. We have not taken care to normalize the signal field A, or the electronic
displacement B, and so (3.1) is not quite true, using the current definitions. But
up to some constant normalization, the optimal efficiency of a quantum memory
described by the above model would be provided by squaring the largest singular
value of (3.38).
Our purpose in the above exposition has not been to describe a real quantum
memory in any quantitative detail. But this simple example contains many of the
ingredients we will encounter in the following Chapters. We have used a one di-
mensional propagation model, combined with some atomic physics, to obtain two
equations of motion — the first describing the atomic response to the field, and the
second describing the influence on the field due to the induced atomic polarization.
The solution was found using a Fourier transform, and the result took the form of
the storage map (3.2), with the kernel given by a Bessel function. This story applies
equally to the fuller treatment given shortly. Finally, note that we have made no
use of the formalism of quantum mechanics so far. Of course, a correct description
of the atoms at least requires that we quantize the electronic energies. But the

3.5 A Simple Example 91
propagation is simply Maxwellian electrodynamics. It may help to keep in mind
that, although we will treat the signal field quantum mechanically for the sake of
completeness, the quantum memories we consider behave essentially classically. Or,
we might say that their efficiencies may be derived classically, since efficiencies do
not depend on correlation functions of the optical fields, and it is only in the photon
statistics revealed by these correlation functions that non-classicality is manifest.

Chapter 4
Equations of motion
In this chapter we derive the dynamical equations describing the interaction of light
with the atoms of an ensemble quantum memory. We focus on EIT and Raman stor-
age, which can be treated together; inhomogeneously broadened memories, such as
CRIB and AFC, are covered in Chapter 7. We borrow techniques from the treatment
of Stokes scattering by Mostowski et al. [139], and some notation from the later treat-
ment of quantum memories by Gorshkov et al. [140]. The derivation divides broadly
into three parts. First, we write down the Hamiltonian describing the interaction
of a single atom with the signal and control fields, and we obtain Heisenberg equa-
tions for the atomic evolution. Next, we introduce Maxwell’s equations, describing
the coupling of the signal field to the macroscopic atomic polarization as it propa-
gates through the ensemble. Finally, we add up the contributions from many atoms
to form the macroscopic variables describing the atomic polarization and the spin
wave. Having derived the dynamical equations in a convenient form, we investigate

4.1 Interaction 93
various methods of solution, in order to optimize the performance of the memory,
in the next chapter.
4.1 Interaction
We consider the propagation of a weak signal field through an ensemble of Λ-type
atoms, in the presence of a bright control field, tuned into two-photon resonance
with the signal, as depicted in Figure (4.1).
Figure 4.1 The signal (blue) and control (green) fields involved ina Λ-type ensemble quantum memory.
As shown in Section C.4 in Appendix C, the interaction of a light beam with an
atom is well described by the electric dipole Hamiltonian HED = −E.d, where E
is the electric field associated with the light at the atomic position, and where d is
the dipole moment associated with an optically active electron in the atom.

4.2 Electric Field 94
4.2 Electric Field
The electric field is composed of two parts, a bright classical control field, and a
weak signal field that we wish to store, both propagating along the z-axis,
E = Ec +Es. (4.1)
The control field is sufficiently intense that it is not affected by its interaction with
the atoms of the memory, so that we do not need to treat it as a dynamical variable
in the Hamiltonian. We therefore represent it as a classical field,
Ec(t, z,ρ) = vcEc(t, z,ρ)eiωc(t−z/c) + c.c., (4.2)
where vc is the control polarization vector1, ωc is the central frequency of the control
field, and Ec(t, z,ρ) is the slowly varying envelope of the control, describing the
spatio-temporal profile of the control pulse. Here ρ = xx + yy is a transverse
position vector, as shown in Figure 4.1.
The signal field is much weaker than the control, and in general it may be in
a non-classical state (for example, a Fock state — see Appendix C). Therefore we
treat the signal field quantum mechanically. The signal field, when well-collimated,1Note that if the polarization is not linear (circular, for instance), the polarization vector is
complex, satisfying v∗.v = v†v = 1.

4.2 Electric Field 95
can be written in the form
Es(z,ρ) = ivs∫g(ω)a(ω,ρ)e−iωz/c dω + h.c., (4.3)
where vs is the signal polarization vector, g(ω) =√
~ω/4πε0c is the mode amplitude,
and a(ω,ρ) is an annihilation operator for a signal photon with frequency ω, and
transverse position ρ, which satisfies the equal-time commutation relation
[a(ω,ρ), a†(ω′,ρ′)] = δ(ω − ω′)δ(ρ− ρ′). (4.4)
The form of (4.3) is very similar to the expression (C.10) in Appendix C, the only
difference being the inclusion of the transverse position ρ, which allows us to treat
diffraction (this is covered in Chapter 6). In the next chapter we will drop the
transverse coordinate and work with a one dimensional propagation model. Note
that all these operators have no time-dependence in the Schrodinger picture (see
Appendix B). In the Heisenberg picture, commutation with the optical free-field
Hamiltonian (see (4.14) below) gives the annihilation operators the simple time-
dependence
a(ω,ρ, t) = a(ω,ρ)eiωt. (4.5)
It will be useful to factorize the signal field into a carrier wave, and a slowly vary-
ing envelope, as we did with the control field in (4.2). To do this, we make use of
the approximation that the bandwidth of the signal field will be very small in com-

4.2 Electric Field 96
parison to its central frequency ωs. Therefore, only terms with frequencies rather
close to ωs will be important in the integral in (4.3). Since the dependence of the
mode amplitude g(ω) on frequency is quite weak (only ‘square-root’), we make the
replacement g(ω) −→ g(ωs). We are then able to perform the frequency integral
explicitly, to obtain
Es(t, z,ρ) = ivsgsA(t, z,ρ)eiωs(t−z/c) + h.c., (4.6)
where gs =√
2πg(ωs), and where we have defined the slowly varying time-domain
annihilation operator A according to the relation
A(t, z,ρ) = e−iωs(t−z/c) × 1√2π
∫a(ω,ρ, t)e−iωz/c dω. (4.7)
That it retains the property of a photon annihilation operator, albeit for spatio-
temporal, rather than spectral modes, can be seen from its commutator with its
Hermitian adjoint. Inserting (4.5) into (4.7), we find
[A(t, z,ρ), A†(t′, z′,ρ′)] = δ(t− t′ − (z − z′)/c)δ(ρ− ρ′). (4.8)
Aside from the leading phase factor, which removes the rapid time-dependence due
to the carrier frequency ωs, the action of A(t, z,ρ) in free space can be understood
as the annihilation of a signal photon in a spatio-temporal mode centred at position
ρ and retarded time τ = t− z/c.

4.3 Dipole Operator 97
4.3 Dipole Operator
Having developed a convenient notation for the signal and control fields, we now
consider the atomic variables. The Coulomb interaction between the atomic nu-
cleus and the electrons is of course rather complex in general, but the formalism
of quantum mechanics comes to the rescue. The energy levels labelled |1〉, |2〉, |3〉
are eigenstates of the atomic Hamiltonian, and therefore they form an orthonormal
basis for the Hilbert space of quantum states of the atom (see Appendices B and C).
That is, any state of the atom can be described in terms of these states. And any
operator acting on the atomic states can be expressed using the coordinate system
defined by these states. In particular, the electric dipole operator d can be written
in the following way,
d =∑j,k
djkσjk, (4.9)
where the coefficients djk = 〈j|d|k〉 are the matrix elements of the dipole opera-
tor, the σjk = |j〉〈k| are ‘flip operators’ (sometimes known as transition projection
operators), and where the double summation runs over the three atomic states.
To preempt possible confusion, we should clarify that d is a three-dimensional
vector in space, whose elements are quantum mechanical operators acting on the
three-dimensional Hilbert space of the Λ-level atom. The Dirac notation (|1〉, |2〉,
etc...) refers to vectors and/or operators in/on this Hilbert space, while the bold
font notation (d, E, v, etc...) refers to vectors in ordinary space (whether their
elements are numbers, or operators).

4.3 Dipole Operator 98
4.3.1 Parity
So far, we have not used any properties of the dipole operator specifically — (4.9) is
an identity that holds for any atomic operator. But now we can use the parity of the
dipole operator to remove some terms from the sum in (4.9). Parity refers to the way
a quantity transforms under the operation of inversion, when all spatial coordinates
are reflected in the origin. That is, r −→ −r, where r is any position vector. As
discussed in Appendix C, the dipole operator is simply given by d = −er, where now
r is the position, with respect to the atomic centre-of-mass, of the optically active
electron. Therefore under inversion, we have d −→ −d. The atomic dipole operator
has negative parity. This means that all the diagonal dipole matrix elements must
vanish, djj = 0. To see this, we can express the matrix element djj in terms of the
wavefunction ψj of the state |j〉,
djj = 〈j|d|j〉
=∫ψ∗j (r)dψj(r) d3r. (4.10)
The integral runs over all space, but we can divide it into a pair of integrals: the
first over half of all space, with positive coordinates r (+), and the second over the
remaining half of space, with negative coordinates −r (−). Since the dipole operator

4.3 Dipole Operator 99
changes sign under inversion, the second integral exactly cancels with the first,
djj =∫
+|ψj(r)|2d d3r +
∫−|ψj(r)|2d d3r
=∫
+|ψj(r)|2(d− d) d3r
= 0. (4.11)
Here we used the fact that |ψj |2 has positive parity (i.e. is unaffected by inversion).
This must be true since |ψj |2 describes the electronic charge density associated with
the state |j〉, and this must be spherically symmetric: there is no interaction to
break the spherical symmetry of the bare atom.
The electric dipole interaction is therefore completely off-diagonal, meaning that
it only couples different states together, never the same state to itself. Furthermore,
we require that the state |3〉 is long-lived, in order that it can serve as an effective
storage state. Therefore we neglect any dipole coupling between the states |1〉 and
|3〉, so that no direct transitions between these states are mediated by the dipole
operator: our goal, instead, is to implement an indirect transition, mediated by the
control field. In the light of these arguments, we arrive at the somewhat pared-down
expression
d = d12σ12 + d23σ23 + h.c.. (4.12)

4.4 Hamiltonian 100
4.4 Hamiltonian
We are now in a position to write down the Hamiltonian for the atom-light system,
H = HA +HL +HED. (4.13)
Here HL is the free-field energy of the light field,
HL =∫ω
∫Aa†(ω,ρ)a(ω,ρ) d2ρdω, (4.14)
where the transverse integral runs over the transverse area A of the signal field.
Here we have neglected the zero-point energy, and also the fixed energy associated
with the control field. HA is the Hamiltonian for the bare atom. The atomic states
|1〉, |2〉, |3〉 are by definition eigenstates of HA, and therefore when written in terms
of these states, HA is purely diagonal (See Section A.4.3 in Appendix A),
HA =∑j
ωjσjj , (4.15)
where ωj is the resonant frequency of state |j〉.
We can now use the Heisenberg equation (see B.10 in Appendix B) to find the
time evolution of the atomic flip operators. Since these operators always commute
with the optical free-field Hamiltonian HL, we drop this from the Hamiltonian (it

4.4 Hamiltonian 101
has no effect on the atoms), and we work with the equation
∂tσjk = i[σjk, HA +HED]. (4.16)
The flip operators satisfy the following multiplicative identity,
σijσkl = σilδjk, (4.17)
and under Hermitian conjugation we have σ†jk = σkj . Using these relations we obtain
five independent atomic equations; two for the atomic populations,
∂tσ11 = −iE. (d12σ12 − h.c.) ,
∂tσ33 = iE. (d23σ23 − h.c.) , (4.18)
and three for the atomic coherences,
∂tσ12 = iω21σ12 − iE. [d∗12 (σ11 − σ22) + d23σ13] ,
∂tσ13 = iω31σ13 − iE. [d∗23σ12 − d∗12σ23] ,
∂tσ23 = iω32σ23 − iE. [d∗23 (σ22 − σ33)− d12σ13] , (4.19)
where we have defined ωjk = ωj − ωk as the frequency difference between the states
|j〉 and |k〉. Note that∑
j σjj = I, the identity, so that the sum of the populations
commutes with the Hamiltonian, and therefore has no time-dependence. This simply

4.5 Linear approximation (1) 102
expresses the fact that the atom remains in one of the states |1〉, |2〉, |3〉, at all times.
Therefore ∂tσ22 = −∂t(σ11 + σ33).
The coupled equations (4.18) and (4.19) constitute a rather complex system,
and it is not possible to extract an analytic solution, in general. Fortunately, the
equations simplify considerably in the linear regime.
4.5 Linear approximation (1)
Provided that we store a small number of photons in the quantum memory, such
that most of the atoms remain in their ground states, with only very few atoms
excited, we can ignore the dynamics of the atomic populations. We replace the
operators σjj by their expectation values on the atomic ground state,
σ11 −→ 1, σ22 −→ 0, σ33 −→ 0. (4.20)
This leaves us with just the three equations for the coherences,
∂tσ12 = iω21σ12 − iE. [d∗12 + d23σ13] ,
∂tσ13 = iω31σ13 − iE. [d∗23σ12 − d∗12σ23] ,
∂tσ23 = −iω23σ23 + iE.d12σ13. (4.21)

4.6 Rotating Wave Approximation 103
4.6 Rotating Wave Approximation
The leading terms in each of the equations in (4.21), of the form iωkjσjk, simply de-
scribe rapid oscillations. The electric field E is also oscillating rapidly; the combined
dynamics of the coherences will therefore contain components oscillating at both the
sum and difference frequencies of these oscillations. Physically, these contributions
give rise to different time-orderings of the two-photon Raman transition between
states |1〉 and |3〉, as shown in Figure 4.2. When the detuning is small compared to
optical frequencies, the sum frequencies are many orders of magnitude higher than
the difference frequencies, and so the sum frequencies average out to zero: they can
be neglected. This is the content of the rotating wave approximation (RWA).
rotating
counter-rotating
Figure 4.2 Time-ordering. The rotating wave approximation ne-glects counter-rotating terms, which correspond to strongly sup-pressed time-orderings for the Raman process.
To implement the RWA, we define rotating coherences by the ansatz
σjk = σjkeiωjkτ , (4.22)

4.6 Rotating Wave Approximation 104
where τ = t− z/c is the retarded time (see the text following (4.8)). Including the
dependence on z here will be useful when we consider propagation. Inserting (4.22)
into (4.21) yields the equations
∂tσ12 = −iE.[d∗12e
−iω21τ + d23σ13e−iω23τ
],
∂tσ13 = −iE.[d∗23σ12e
iω23τ − d∗12σ23e−iω21τ
],
∂tσ23 = iE.d12σ13eiω21τ . (4.23)
Inserting the expressions (4.2) and (4.6) for Ec and Es into E, and multiplying out
the resulting expressions, we find terms multiplying rapidly varying exponentials,
like e−i(ω21+ωs)τ , for instance, and also terms multiplying slowly varying exponen-
tials, like e−i(ω21−ωs)τ = e−i∆τ . Neglecting the fast oscillating terms, we obtain the
equations
∂tσ12 = −id∗12.[vcEce
−i∆+τ + ivsgsAe−i∆τ]− id23.
[vcEce
−i∆τ + ivsgsAe−i∆−τ]σ13,
∂tσ13 = −id∗23.[v∗cE
∗c e
i∆τ − iv∗sgsA†ei∆−τ
]σ12 + id∗12.
[vcEce
−i∆+τ + ivsgsAe−i∆τ]σ23,
∂tσ23 = id12.[v∗cE
∗c e
i∆+τ − iv∗sgsA†ei∆τ
]σ13, (4.24)
where we have defined ∆+ = ω21 − ωc and ∆− = ω23 − ωs (see Figure 4.3).

4.7 Unwanted Coupling 105
(a) (b) (c)
Figure 4.3 Useful and nuisance couplings. (a) The desired quan-tum memory coupling. (b) The control field couples to the groundstate, initiating spontaneous Stokes scattering. (c) The signal fieldcouples to the storage state: it is very weak, and there is no significantpopulation in this state, so the effect of this coupling is negligible.
4.7 Unwanted Coupling
We have already succeeded in dramatically simplifying the dynamical equations, but
it is still not obvious how the behaviour described by this system of equations allows
for the implementation of a quantum memory. Other physical processes obscure
the useful features of the system. For instance, the term involving d∗12.vc represents
the coupling of the control field to the |1〉 ↔ |2〉 transition. If this term is strong,
the control field can initiate spontaneous Stokes scattering, as shown in Figure 4.3
(b). Aside from complicating the equations, this process can be problematic, since it
generates excited atoms in the state |3〉 that are not correlated with the signal field.
When we attempt to retrieve the signal field, some of these uncorrelated atoms may
contribute a noisy background emission. In practice, it is possible to distinguish
the signal from the noise using phasematched retrieval, which we discuss in §6.3.2
in Chapter 6. In any case, a description of the memory interaction requires that we
can eliminate this unwanted coupling. As can be seen from Figure 4.3, the Stokes
scattering process is detuned further from resonance, with detuning ∆+ > ∆. If

4.8 Linear Approximation (2) 106
∆+ is sufficiently large (this requires that the splitting ω31 is large compared to ∆),
the term representing Stokes scattering will oscillate quickly enough to be neglected.
Alternatively, it may be possible to choose the polarization of the control such that
the product d12.vc vanishes due to a selection rule, although strict selection rules
usually require the application of an external magnetic field. Polarization selection
rules are discussed in the context of cesium in §F.4 of Appendix F. Regardless of
the justification, in the following analysis we set d12.vc = 0. By the same token, we
set d23.vs = 0, thereby disregarding any coupling of the signal field to the |2〉 ↔ |3〉
transition, as depicted in Figure 4.3 (c). The equations of motion are now given by
∂tσ12 = d∗12.vsgsAe−i∆τ − id23.vcEce
−i∆τ σ13,
∂tσ13 = −id∗23.v∗cE∗c e
i∆τ σ12 − d∗12.vsgsAe−i∆τ σ23,
∂tσ23 = d12.v∗sgsA
†ei∆τ σ13. (4.25)
4.8 Linear Approximation (2)
Now that we have arrived at the more transparent set of equations (4.25), we can
identify some terms in these equations that are ‘small’, in the sense that ‘weakly
excited’ operators are involved. To be more concrete, we analyse the size of each
term perurbatively. We attach a parameter ε to each of the coherences σjk, and also
to the signal field A. This parameter just labels these quantities as ‘small’; in the
case of the coherences, they are initially vanishing, and in the case of the signal field,

4.8 Linear Approximation (2) 107
only a very few signal photons are sent into the memory. The equations become
ε∂tσ12 = d∗12.vsgsεAe−i∆τ − id23.vcEce
−i∆τ εσ13,
ε∂tσ13 = −id∗23.v∗cE∗c e
i∆τ εσ12 − d∗12.vsgsε2Ae−i∆τ σ23,
ε∂tσ23 = d12.v∗sgsε
2A†ei∆τ σ13. (4.26)
There are two terms proportional to ε2. They correspond to ‘second-order’ pertur-
bative corrections to the dynamics, and they can be neglected. Dropping the label
ε, the linearized equations of motion for the atomic coherences are
∂tσ12 = d∗12.vsgsAe−i∆τ − id23.vcEce
−i∆τ σ13,
∂tσ13 = −id∗23.v∗cE∗c e
i∆τ σ12. (4.27)
These equations contain the important physics of the quantum memory interaction.
They describe how the coherence σ12 is directly excited by the signal field A, and
how this excitation is then coupled to the coherence σ13 through the control field
Ec. Macroscopically, when many identical atoms are involved, we can identify σ12
with the atomic polarization, and σ13 with the spin-wave. Before considering the
collective dynamics of the ensemble as a whole, we first consider the propagation of
the signal field.

4.9 Propagation 108
4.9 Propagation
Heisenberg’s equations describe evolution in time, but it is not obvious how to treat
the spatial propagation of the signal field in this formalism. In fact, it is possible to
do this, but a more transparent derivation utilizes Maxwell’s equations. The atomic
ensemble behaves as a dielectric medium in the presence of the signal field, for which
the appropriate formulation is as follows,
∇.D = ρfree, ∇.B = 0,
∇×E = −∂tB, ∇×H = Jfree + ∂tD. (4.28)
Here D is the displacement field, H is the magnetic field, E is the electric field and
B is the magnetic induction. The distinction between B and H will not be very
important for us, since all the materials we are concerned with are non-magnetic.
We simply take B = µ0H, where µ0 is the permeability of free space, and refer to
B as the magnetic field. The quantities ρfree and Jfree are the free charge density
and current, respectively. The designation ‘free’ refers to the fact that they are not
induced by the fields: they are charges and currents that are not associated with
any material dielectric properties. In any case we only deal with materials in which
there are no free charges or currents, and therefore we set ρfree = Jfree = 0. A wave
equation for the electric field is found by differentiating the equation for H, and

4.9 Propagation 109
substituting in the equation for E,
∇× ∂tH = ∂2tD,
⇒ −∇× (∇×E) = µ0∂2tD. (4.29)
The double curl derivative on the left can be simplified using the vector calculus
identity
∇× (∇×E) = ∇ (∇.E)−∇2E. (4.30)
The signal field is a transverse propagating wave, which is divergence free2. This
is evident from the definition of the Coulomb gauge (see Appendix C), in which
∇.A = 0, with E = ∂tA, where A is the magnetic vector potential. We therefore
drop the first term in (4.30), to obtain
∇2E = µ0∂2tD. (4.31)
The displacement field is formed from the sum of the electric field, and the material
polarization P ,
D = ε0E + P , (4.32)
where P is the polarization density, defined as the dipole moment per unit volume.
Substituting (4.32) into (4.31), and using the relation ε0µ0 = 1/c2, we arrive at the2These arguments apply to a homogeneous and isotropic dielectric containing no source
charges [141]

4.10 Paraxial and SVE approximations 110
wave equation [∇2 − 1
c2∂2t
]E = µ0∂
2tP . (4.33)
This equation relates the propagation of the optical fields to the atomic polarization
in the memory. Since the control field is so intense, it is not significantly affected by
its interaction with the ensemble, and so we do not consider its propagation further.
For the signal field, we use the equation
[∇2 − 1
c2∂2t
]Es = µ0∂
2tPs, (4.34)
where Ps is the component of the atomic polarization which acts as a source for the
signal field. That is, Ps is the component of the polarization oscillating at the signal
carrier frequency ωs. Further simplification is accomplished by making use of the
fact that the signal and control fields are collimated beams.
4.10 Paraxial and SVE approximations
The slowly varying envelope approximation (SVE), and the paraxial approximation,
are both implicit in the decomposition (4.6). We assume that the amplitude A (its
status as an operator is not important for this discussion) is a smooth, slowly varying
function of time and space. The exponential factor then represents the optical carrier
wave, oscillating in time with frequency ωs, and oscillatiing along the z-axis with
wavevector ks = ωs/c.
The paraxial approximation allows us to treat the signal field as a beam traveling

4.10 Paraxial and SVE approximations 111
along the z-axis, with negligible divergence. This approximation is satisfied as long
as the transverse spatial profile of A is much larger than the signal wavelength
λs = 2π/ks.
The SVE approximation allows us to treat the propagation of the signal field
purely in terms of the envelope of the signal pulse, represented by the temporal
shape of A, without having to explicitly model the very fast time-dependence of the
carrier wave, which oscillates much faster. To make this approximation successfully,
we should have that the temporal duration of the signal field is much longer than
the optical period 2π/ωs.
To see how these approximations simplify the theoretical description, we insert
the signal field (4.6) into the wave equation (4.34). We consider only the positive
frequency component of the signal field, since only this component is coupled to
the atoms through the system (4.27). We also define the slowly varying atomic
polarization Ps by factorizing out the signal frequency,
Ps = Pseiωsτ . (4.35)
The resulting wave equation is
[∇2 − 1
c2∂2t
] [ivsgsAeiωs(t−z/c)
]= µ0∂
2t
[Pse
iωs(t−z/c)]. (4.36)
We take the scalar product of both sides with the polarization vector v∗s (this is the
same as taking the inner product with v†s, see Appendix A), and apply the chain

4.11 Continuum Approximation 112
rule for the derivatives to obtain
∇2⊥ +
(∂2z −
1c2∂2t
)− 2i
ωsc
(∂z +
1c∂t
)−[(ωs
c
)2− 1c2ω2s
]A = −i
µ0
gsv∗s .(∂2t + 2iωs∂t − ω2
s
)Ps,
(4.37)
where we have divided out the exponential factor eiωsτ . The transverse Laplacian
∇2⊥ = ∂2
x+∂2y describes diffraction of the signal field as it propagates. Note that the
term in square brackets on the left hand side vanishes. Concerning the remaining
terms, we observe that according to the SVE approximation
∣∣∣∣ks(∂z +1c∂t
)A
∣∣∣∣ ∣∣∣∣(∂2z −
1c2∂2t
)A
∣∣∣∣ . (4.38)
We therefore drop the smaller term. For the same reason, we drop all but the last
term on the right hand side, and we arrive at the equation
(i
2ks∇2⊥ + ∂z +
1c∂t
)A = − µ0ω
2s
2gsksv∗s .Ps. (4.39)
This equation describes the coupling of the atoms to the signal field amplitude.
It now remains for us to connect the atomic evolution equations (4.27) with the
macroscopic polarization Ps.
4.11 Continuum Approximation
We have in mind an ensemble of sufficient density that the atoms form an effective
continuum. Consider a small region within the ensemble at position r = (z,ρ),

4.11 Continuum Approximation 113
with volume δV . We’ll call this a voxel. To make the continuum approximation,
we should have many atoms in each voxel; nδV 1, where n is the number den-
sity of atoms in the ensemble. Each voxel should be ‘pancake-shaped’, so that its
thickness δz along the z-axis satisfies δz λs, while its transverse area δA satis-
fies δA λ2s. The condition on δz ensures that the longitudinal optical phase ksz
is roughly constant throughout the voxel. The condition on δA ensures that the
typical interatomic separation is much larger than the signal wavelength λs, so that
dipole-dipole interactions between the atoms can be neglected, and we can treat the
atoms as isolated from one another. The macroscopic polarization at position r is
then found by adding up the dipole moments in the voxel located at r,
P =1δV
∑β(r)
dβ, (4.40)
where the index β runs over all the atoms in the voxel at position r. Using the
expression (4.12) for each atom, and recalling that the coherence σ23 is negligible
(see Section 4.8), we have
P =1δV
∑β(r)
(d12σ
β12 + h.c.
). (4.41)
We now define macroscopic variables involving sums over the coherences σjk, in order
to ‘tie up’ the system of equations (4.27) with the propagation equation (4.39). For

4.11 Continuum Approximation 114
the macroscopic polarization, we define the operator
P =1√nδV
∑β(r)
σβ12ei∆τ . (4.42)
Note that P is not simply the magnitude of the vector P . They are closely related
(see (4.47) below), but some book-keeping is required to ensure that we keep track of
all the relevant constants. In the same vein, for the spin wave we define the operator
B =1√nδV
∑β(r)
σβ13. (4.43)
These definitions are motivated by analogy with the slowly varying photon annihi-
lation operator A. For instance, the equal-time commutator of B with its Hermitian
adjoint is given by
[B(z,ρ, t), B†(z′,ρ′, t)
]=
1(δV )2n
∑β(r)
∑γ(r′)
[σβ13, σ
γ31
]
=
1
(δV )2n× nδV (σ11 − σ33) if r and r′ label the same voxel,
0 otherwise.
(4.44)
Using the linear approximation (4.20), we find, in the continuum limit δV −→ 0,
[B(z,ρ, t), B†(z′,ρ′, t)
]= δ(z − z′)δ(ρ− ρ′). (4.45)

4.11 Continuum Approximation 115
Identical arguments yield the commutator
[P (z,ρ, t), P †(z′,ρ′, t)
]= δ(z − z′)δ(ρ− ρ′), (4.46)
for the polarization. Therefore both the operators P and B satisfy bosonic commuta-
tion relations. We interpret them as annihilation operators for an atomic excitation
at position r. That is, P (z,ρ) annihilates a distributed excitation of the excited
state |2〉 at position (z,ρ). And B(z,ρ) annihilates a distributed excitation of the
storage state |3〉.
Substituting the definition (4.42) into (4.41), and taking the positive frequency
component (i.e. the component oscillating at a frequency +ωs), we find
Ps =√nd12P, (4.47)
for the slowly varying macroscopic polarization. We can now write down equations
governing both the propagation of the signal field, and the atomic dynamics. These
equations are,
(i
2ks∇2⊥ + ∂z +
1c∂t
)A = −κ∗P,
∂tP = i∆P + κA− iΩB,
∂tB = −iΩ∗P, (4.48)

4.12 Spontaneous Emission and Decoherence 116
where we have defined the control field Rabi frequency
Ω =d23.vc
~Ec, (4.49)
and the coupling constant
κ =d∗12.vs
~×√ngs =
d∗12.vs~
√~ωsn2ε0c
. (4.50)
The equations (4.48) describe the propagation and diffraction of a weak signal field
through an ensemble of ideal atomic Λ-systems, prepared initially in their ground
states. We have not yet included any description of decay processes, such as spon-
taneous emission from the excited state, or collisional de-phasing of the spin wave.
These two processes are expected to happen on very different time-scales, but they
can be treated in exactly the same way, which we now introduce.
4.12 Spontaneous Emission and Decoherence
In Section (C.5) in Appendix (C), we discuss the description of Markovian decay
in the Heisenberg picture using Langevin equations. We used a model in which a
bosonic system was coupled to a large reservoir, also composed of bosons. In the
present discussion, all the operators we consider — A, B and P — are bosonic in
character (see Eqs. (4.8), (4.45) and (4.46) for their commutators). In the case of
spontaneous emission, these operators are coupled to the electromagnetic field, which
is a large reservoir of bosons, and so the model of Appendix C is applicable. Using

4.12 Spontaneous Emission and Decoherence 117
this model, spontaneous emission at the rate γ is described by incorporating an ap-
propriate decay term into the dynamical equation for P , along with a Langevin noise
operator FP , which introduces fluctuations that preserve the bosonic commutation
relations of P . The noise operator is delta-correlated, meaning that no correlations
exist between its values at different times. And since all the atoms in the ensemble
are subject to independent fluctuations, no correlations exist between the noise at
different positions. These properties are summarized by the relations
〈F †P (t, z,ρ)FP (t′, z′,ρ′)〉 = 2γnP × δ(t− t′)δ(z − z′)δ(ρ− ρ′),
〈FP (t, z,ρ)F †P (t′, z′,ρ′)〉 = 2γ(nP + 1)× δ(t− t′)δ(z − z′)δ(ρ− ρ′), (4.51)
where the expectation value is taken on the initial state of both the ensemble and
the reservoir (i.e. the electromagnetic field to which the atoms are coupled). Here
the number nP is the initial number of atoms thermally excited into state |2〉, on
average. When dealing with optical transitions, we typically have nP = 0, of course.
Similarly, to treat decoherence of the spin wave B at a rate γB, we add a decay
term and a noise operator FB, which satisfies identical relations to (4.51), again with
nB = 0. The dynamical equations including these dissipative processes are then
(i
2ks∇2⊥ + ∂z +
1c∂t
)A = −κ∗P,
∂tP = −γP + i∆P + κA− iΩB + FP ,
∂tB = −γBB − iΩ∗P + FB. (4.52)

4.12 Spontaneous Emission and Decoherence 118
Note that these decay rates are for the atomic coherences. The atomic populations
decay at twice these rates. For instance, the number of spin wave excitations is
given by
NB =∫ L
0
∫AB†(z,ρ)B(z,ρ) d2ρdz. (4.53)
In the absence of any optical fields (with Ω = 0), we find that
∂t〈NB〉 = −2γB〈NB〉, (4.54)
where the expectation value is taken on an arbitrary state. Any terms involving the
noise operator FB vanish when taking the expectation value because its fluctuations
average to zero. The relation (4.54) shows that the number of spin wave excitations
decays at the rate 2γB. The same argument applied to P shows that the number
of excited atoms, in the state |2〉, decays at the rate 2γ, so that the spontaneous
lifetime of the state |2〉 is 1/2γ.
Now that we have introduced losses and decoherence with a degree of formal
rigour, we see that in fact the noise operators FB, FP may be neglected when
optimizing the performance of a quantum memory. The reason is that the efficiency
of a quantum memory depends only on the ratio of stored to input excitations.
That is, only the number operators for the signal field and spin wave are involved.
These number operators involve only normally ordered products, of the form A†A
or B†B, and therefore only normally ordered products of the F operators enter into
the efficiency. Since we have n ∼ 0, any products of the form F †F vanish when an

4.12 Spontaneous Emission and Decoherence 119
expectation value is taken, as is clear from (4.51). Therefore the Langevin operators
can be safely dropped from the system of equations (4.52). Of course the decay
terms are important!

Chapter 5
Raman & EIT Storage
Here we use the equations of motion derived in the last chapter to study the opti-
mization of the EIT and Raman quantum memory protocols.
5.1 One Dimensional Approximation
The analysis is greatly simplified if we use a one dimensional model, so that we
only consider propagation along the z-axis. This can always be made a good ap-
proximation by using laser-beams with low divergence. The effects of diffraction
are considered in Chapter 6. In the following, we will average over the transverse
coordinate ρ, to produce a one dimensional propagation model. We re-define the

5.1 One Dimensional Approximation 121
variables A, P and B by integrating over the transverse area A of the signal field,
A(t, z,ρ) −→ A(t, z) =1√A
∫AA(t, z,ρ) d2ρ,
P (t, z,ρ) −→ P (t, z) =1√A
∫AP (t, z,ρ) d2ρ,
B(t, z,ρ) −→ B(t, z) =1√A
∫AB(t, z,ρ) d2ρ. (5.1)
Having averaged the variables in this way, we now drop the transverse Laplacian
∇2⊥ from the propagation equation for A in (4.52). Further simplifications follow.
For instance, in the absence of any transverse structure, the control field envelope
Ec, propagating undisturbed at the speed of light along the z-axis, can be written
as a function of the retarded time τ = t − z/c only. We therefore make a change
of variables from (t, z) to (τ, z), which enables us to write the control field Rabi
frequency as Ω = Ω(τ). Furthermore, the mixed derivative in the propagation
equation for A is simplified, since
∂z
)t+
1c∂t
)z
= ∂z
)τ, ∂t
)z
= ∂τ
)z, (5.2)
where the subscripted parentheses indicate the variables held constant. In this new
coordinate system, the one dimensional equations of motion for a Λ-type quantum

5.1 One Dimensional Approximation 122
memory are therefore given by
∂zA(z, τ) = −κ∗P (z, τ),
∂τP (z, τ) = −ΓP (z, τ) + κA(z, τ)− iΩ(τ)B(z, τ),
∂τB(z, τ) = −iΩ∗(τ)P (z, τ), (5.3)
where Γ = γ−i∆ is the complex detuning. Note that we have dropped the decay term
associated with the decoherence of the spin wave B: by assumption this is negligible
on the time-scale of the storage process, which is what we seek to optimize. We have
also dropped the Langevin noise operator FP associated with spontaneous decay of
the polarization P , since this noise does not affect the efficiency (see the end of
Section 4.12 in the previous chapter). The equations (5.3) are mercifully rather
simple. Certainly they are easier on the eye than any of their previous incarnations
in Chapter 4! The elimination of the Langevin noise operators means that these
equations are now entirely classical in nature: we can treat (5.3) as a system of
coupled partial differential equations in three complex-valued functions A, P and
B. Since the equations are linear, the solutions will be linear, and we need not
worry about issues involving commutators or operator ordering. Our aim is to
find an expression for the Green’s function K(z, τ), relating the input signal field
Ain(τ) = A(z = 0, τ) to the final spin wave Bout(z) = B(z, τ −→ ∞). In the
absence of decoherence of the spin wave, the limit τ −→∞ is simply a mathematical
shorthand for “the end of the storage interaction”, when the control and signal fields

5.2 Solution in k-space 123
have fallen away to zero. As described in (3.2) in Chapter 3, taking the SVD of K
will tell us how to optimize the memory efficiency. We now attempt a solution of
the system (5.3).
5.2 Solution in k-space
5.2.1 Boundary Conditions
We must solve three first order partial differential equations in three functions, and
therefore there must be three boundary conditions. For the storage process, we
begin with no excitations of the atomic polarization, and no spin wave excitations,
so the boundary conditions for the functions P and B are simply
Pin(z) = P (z, τ −→ −∞) = 0, Bin(z) = B(z, τ −→ −∞) = 0. (5.4)
As mentioned above, the boundary condition for the signal field is set by the initial
temporal profile of the signal envelope A, as it impinges on the front face of the
ensemble at z = 0,
Ain(τ) = A(z = 0, τ). (5.5)
Our analysis will tell us the shape for Ain that maximizes the memory efficiency.
These boundary conditions are represented by the tableau in Figure 5.1.

5.2 Solution in k-space 124
Figure 5.1 Quantum memory boundary conditions. Example solu-tions for the functions A, P and B are shown in each panel, with the zcoordinate running from top to bottom and the τ coordinate runningfrom left to right. The red lines indicate the boundary conditionsthat must be specified to generate the solutions.
5.2.2 Transformed Equations
To proceed with solving the equations of motion, it will be useful to reduce them to
a system of coupled ordinary differential equations. This can be done by applying
a unilateral Fourier transform over the z coordinate (see Appendix D). We define
Fourier transformed variables accordingly,
A(k, τ) =1√2π
∫ ∞0
A(z, τ)eikz dz,
P (k, τ) =1√2π
∫ ∞0
P (z, τ)eikz dz,
B(k, τ) =1√2π
∫ ∞0
B(z, τ)eikz dz.
(5.6)

5.2 Solution in k-space 125
Using the result (D.27) for the transform of the spatial derivative ∂z, we obtain
−ikA− 1√2πAin = −κ∗P ,
∂τ P = −ΓP + κA− iΩB,
∂τ B = −iΩ∗P . (5.7)
We remark that the independence of Ω from z is critical to the usefulness of this
transformation. The spatial propagation has now been reduced to an algebraic
equation, which we can solve for A.
5.2.3 Optimal efficiency
Even given unlimited energy for the control pulse, the storage efficiency is limited
by spontaneous emission from the excited state |2〉. The storage into the dark state
|3〉, which is not affected by spontaneous emission, is always mediated via coupling
to |2〉. Even with perfect transfer between states |2〉 and |3〉, we can never store
more efficiently into |3〉 than we can couple to |2〉. Therefore the storage efficiency
is bounded by the efficiency with which we can transfer population into |2〉. To
evaluate this upper bound, we simply neglect the spin wave, and solve the equation
for P with Ω = 0. Including the solution for A, we obtain
∂τ P = −(
Γ + i|κ|2
k
)P +
iκ√2πk
Ain. (5.8)

5.2 Solution in k-space 126
This equation can be integrated directly to give
P (k, τ) = Pin(k)e−(Γ+i|κ|2/k)τ +iκ√2πk
∫ τ
−∞e−(Γ+i|κ|2/k)(τ−τ ′)Ain(τ ′) dτ ′. (5.9)
Since we are concerned with storage, we can set the initial polarization to zero.
We next assume that we are somehow able to transfer all the excitations from P
to the spin wave B, with no loss, at some time τ = T , which marks the end of
the storage interaction. We can then make the substitution Pout → Bout, and the
optimal storage process is described by the map
Bout(k) =∫ ∞−∞
K(k, T − τ)Ain(τ) dτ, (5.10)
where the k-space storage kernel is given by
K(k, τ) =iκ√2πk
e−(Γ+i|κ|2/k)τ . (5.11)
Note that for times τ > T , we set K = 0, so that no storage takes place after τ = T .
Now, some comments are warranted. First, the optimal storage efficiency does not
depend on the detuning ∆. To see this, note that e−Γτ = e−γτ × ei∆τ . The latter
factor, involving the detuning, represents a pure phase rotation. We could absorb it
into the definition of Ain without altering its norm. Therefore we can drop it from
the kernel — it has no effect on its singular values, and no effect on the optimal
efficiency. Second, the optimal efficiency only depends on the resonant optical depth,

5.2 Solution in k-space 127
defined by
d =|κ|2Lγ
. (5.12)
To see this, we normalize the time and space coordinates, along with the spin wave
and signal field amplitudes, to make them dimensionless. The spontaneous decay
rate γ and the ensemble length L provide natural time and distance scales for this
normalization. We denote the normalized variables by an overbar,
τ = γτ, k = kL,
A =A√γ, B =
B√L. (5.13)
This notation is rather clumsy, but it serves to clarify the re-scaling. With these
definitions, the storage map becomes
Bout
(k)
=∫ ∞−∞
K(k, T − τ
)Ain (τ) dτ , (5.14)
where the kernel has been converted into the dimensionless form
K(k, τ)
=i√d
k√
2πe−(1+id/k)τ . (5.15)
Here we have assumed for simplicity that κ is real, (i.e. κ = κ∗), and we have
dropped the detuning for the reason mentioned above. It is now clear that the
optical depth d is the only parameter associated with the interaction that plays any
role in determining the efficiency of the storage process. This was first shown by

5.2 Solution in k-space 128
Gorshkov et al [133]. Their explanation is that, regardless of the memory protocol
used, the branching ratio between loss — spontaneous emission — and storage —
absorption — is fixed by the optical depth. We have derived the result by arguing
that we cannot do better than is possible through direct, linear absorption into the
state |2〉. Clearly detuning from resonance cannot improve matters, and so from
this perspective it is unsurprising that the best possible efficiency is only limited
by the resonant coupling, parameterized by d. Below we examine the quantitative
behaviour of the optimal efficiency in more detail.
The optimal efficiency is given by the square of the largest singular value of K.
Note that it makes no difference whether the argument τ or the ‘flipped’ argument
T − τ is used; this just flips the input modes without affecting the singular values.
Unfortunately, K has a singularity at k = 0, and this means we cannot approxi-
mate it as a finite matrix, in order to compute the singular values numerically. To
proceed further, we need to transform to a different coordinate system to remove
the singularity. One possibility is to apply an inverse Fourier transform, and this
works well. Before doing this, however, we first introduce another way to analyse
the kernel (5.15), which gives a degree of insight into its structure. In what follows,
we will drop the overbar notation for the normalized parameters, as a concession to
legibility.

5.2 Solution in k-space 129
5.2.4 Solution in Wavelength Space
Recall that the singular values of K are unaffected by a unitary transformation.
Consider the coordinate transformation k −→ λ = 2π/k, from k-space to ‘wave-
length space’. The kernel (5.15) is no longer singular in this coordinate system.
To guarantee unitarity, the transformed kernel must include a Jacobian factor of
1/√∂kλ = −i
√2π/λ (see §3.3.3 in Chapter 3). We obtain the result
K(λ, τ) =
√d
2πe−(1+idλ/2π)τ . (5.16)
We now form the anti-normally ordered product KA, as described in (3.19) in Chap-
ter 3, which takes the form
KA(λ, λ′) =1
2πi× 1λ− 4π
d i− λ′. (5.17)
The eigenvalues of KA give the singular values of K, so we should try to solve the
following eigenvalue problem,
12πi
∫ ∞−∞
ψj(λ′)λ− 4π
d i− λ′dλ′ = ηjψj(λ). (5.18)
The integrand has a singularity at λ′ = λ− 4πd i. We consider extending the integral
into the complex plane, and integrating along a semicircle-shaped contour, closed
in the lower half of the complex plane (as depicted in Figure D.4 in Appendix D),
so that the contour encloses the singularity. We assume that the mode functions

5.2 Solution in k-space 130
ψj fall away to zero along the curved portion of the contour, in the limit that its
radius is made infinitely large. Under this assumption, the only contribution to the
integral comes from the straight portion of the contour, along the real line, which
is precisely the integral in (5.18). We can now use Cauchy’s integral theorem (see
§D.1.1 in Appendix D) to evaluate the left hand side of (5.18),
ψj(λ− 4π
d i)
= ηjψj(λ). (5.19)
By inspection, a possible form for the modefunctions is
ψj(λ) ∝ e±iαjλ. (5.20)
That is, plane waves in λ-space, each with some eigenfrequency αj . We should choose
the minus sign in (5.20), so that the modefunctions are exponentially damped in the
lower half of the complex plane, as we assumed above. The storage efficiencies ηj
are then given by
ηj = e−4παj/d. (5.21)
This form is reassuring, since the ηj −→ 1 in the limit d −→∞, which makes sense.
However, it is not obvious that there is any constraint on the eigenfrequencies αj .
We need a ‘quantization condition’ on the modes. One possibility is to transform
back into ordinary z-space, and look for a physically reasonable condition. First, we

5.2 Solution in k-space 131
transform back in to k-space. Remembering to include the Jacobian factor, we get
ψj(k) ∝ 1k× e−2πiαj/k. (5.22)
The proportionality symbol reflects the fact that the modes should be properly
normalized; at the moment we are simply concerned with their functional form.
Transforming back into z-space requires taking the inverse Fourier transform of
(5.22). The method is described in §D.5.2 of Appendix D. The result is
ψj(z) ∝ J0
(2√
2παjz), (5.23)
where J0 is a zero’th order ordinary Bessel function of the first kind. In the limit of
large d, we expect that the signal field is completely absorbed, so that there is no
signal left at the end of the ensemble. We are working in normalized coordinates, so
the end of the ensemble is located at z = 1. With no remaining signal field, no spin
wave excitations can be excited, so we should expect ψj(1) ∼ 0. This is satisfied if
we choose αj = .23, 1.21, 2.98, 5.53, etc..., as shown in Figure 5.2. This choice of
quantization is consistent with the orthogonality requirement on the modes,
∫ 1
0ψi(z)ψ∗j (z) dz = 0, if i 6= j, (5.24)
which follows from the orthogonality condition (D.34) of the Bessel functions (see
§D.5 in Appendix D).

5.2 Solution in k-space 132
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5
0
1
Figure 5.2 The first three zeros of the function J0(2√
2πx). Thecondition that the spin wave should vanish at z = 1 picks out thesezeros as the eigenfrequencies αj .
The optimal storage efficiency is then given by
η1 = e−4πα1/d ≈ 1− 2.9/d, (5.25)
where the approximation holds in the limit of large d.
The above quantization procedure used to derive the optimal efficiency (5.25)
was rather ad hoc, and we should check it against a numerical SVD. We therefore
return to the k-space kernel (5.15). As noted previously the singularity at k = 0
makes this form of the kernel inconvenient. Fortunately it is easy to take the inverse
Fourier transform from k-space back into ordinary space (again using the method
described in §D.5.2 of Appendix D). The result is
K (z, τ) =√de−τJ0
(2√dτz). (5.26)
We could perform a numerical SVD on this kernel directly. The numerical problem

5.2 Solution in k-space 133
converges better, however, if we form the anti-normally ordered product kernel KA,
which is given by
KA
(z, z′
)=d
2e−d(z+z′)/2I0
(d√zz′). (5.27)
Here I0 is a zero’th order modified Bessel function of the first kind (see §D.5.3 in
Appendix D for a clue as to how to perform the required integral). The optimal
efficiency η1 is the largest eigenvalue of this kernel. In Figure 5.3, we plot the analytic
prediction (5.25) alongside the numerical result, over a range of optical depths. The
analytic formula is an excellent approximation for optical depths larger than ∼ 50.
This scaling of the optimal storage efficiency was first noted by Gorshkov et al. [133].
They also provided an elegant proof of the optimality of the kernel (5.27), that does
not rely on the heuristic assertion that ‘we cannot map more efficiently to B than
we can to P ’. Nonetheless, the result is the same.
0 400 800 1200 1600 200010
−3
10−2
10−1
100
101
Figure 5.3 Optimal storage efficiency. The plot shows the differ-ence between the optimal efficiency η1 and unity, on a logarithmicscale, as a function of the optical depth d. The analytic formula1− η1 ≈ 2.9/d derived above (green), is in excellent agreement withthe numerical result (blue), found by diagonalizing the kernel (5.27).
Having identified the best possible storage efficiency, we now continue with our

5.2 Solution in k-space 134
analysis of the equations of motion, including the control field. This analysis will
reveal how the temporal profile of the optimal input mode depends on the control
pulse, and in what regimes it is possible to effectively shape the optimal input mode
by shaping the control.
5.2.5 Including the Control
We return to the system (5.7). Solving the first equation for A, and substituting
the result into the second equation for P , yields a pair of coupled linear differential
equations in time only. For each spatial frequency k, we need to solve for the
temporal dynamics of the system (P , B). To do this, we define a vector |ψ〉 whose
two elements are the functions P and B,
|ψ(τ)〉 = P (τ)| ↑〉+ B(τ)| ↓〉, (5.28)
where the basis kets | ↑〉 and | ↓〉 are given by
| ↑〉 =
1
0
, and | ↓〉 =
0
1
. (5.29)
We have suppressed the dependence on the spatial frequency k, which is no longer
a dynamical variable, but which of course must not be forgotten! The equation of
motion for |ψ〉 is found to be
∂τ |ψ〉 = −iM |ψ〉+ |α0〉, (5.30)

5.2 Solution in k-space 135
where the time-dependent matrix M is given by
M(τ) =
|κ|2k − iΓ Ω(τ)
Ω∗(τ) 0
, (5.31)
and where the time-dependent vector |α0〉 includes the signal field boundary cond-
tion,
|α0(τ)〉 = iκ√2πk
Ain(τ)| ↑〉. (5.32)
Using the normalized variables introduced in the previous section, in which all
lengths are scaled by L and all frequencies by γ, we simply replace κ with√d. The
structure of (5.30) is very similar to Schrodinger’s equation (B.6) for a two-level
system, except that the evolution is not unitary, because M is not quite Hermi-
tian, and because of the ‘driving term’ |α0〉. Nonetheless, techniques applied to the
solution of Schrodinger’s equation remain useful. First, we write down the formal
solution. Suppose that we are able to construct a propagation matrix V (τ) such
that ∂τV = iVM . We then have that
∂τ (V |ψ〉) = (∂τV ) |ψ〉+ V ∂τ |ψ〉
= iVM |ψ〉+ V (−iM |ψ〉+ |α0〉)
= V |α0〉. (5.33)

5.2 Solution in k-space 136
Integrating this gives
|ψ(τ)〉 = V −1(τ)Vin|ψin〉+ V −1(τ)∫ τ
−∞V (τ ′)|α0(τ ′)〉 dτ ′, (5.34)
where |ψin〉 contains the boundary conditions for P and B, and where Vin is the
propagation matrix at the start of the interaction. Clearly we must have Vin = I,
the identity operator. To find the spin wave at the end of the storage process, for
which |ψin〉 = 0, we take the limit τ −→∞ to obtain
Bout = 〈↓ |V −1out
∫ ∞−∞
V (τ)|α0(τ)〉 dτ, (5.35)
where Vout = V (τ −→∞). The k-space storage kernel is then given by
K(k, τ) =ik
√d
2π〈↓ |V −1
out (k)V (k, τ)| ↑〉, (5.36)
where we have now included the dependence of the V matrices on k explicitly, lest
we forget it. If we can find an expression for V , we can construct the storage kernel,
and so find the optimal input mode by means of its SVD. Here we comment that if
the matrix M(τ) were replaced by an ordinary function, we would simply have
V (τ) = exp[i∫ τ
−∞M(τ ′) dτ ′
]. (5.37)
And in fact this is still true wheneverM is a diagonal matrix, since then [M(τ),M(τ ′)] =
0 (diagonal matrices always commute). Alternatively, if M is constant in time, it

5.2 Solution in k-space 137
can be pulled out of the integral in (5.37), and there is no issue with commutation
at different times. However, in general, when M is non-diagonal and time-varying,
as we have whenever Ω is not simply a constant, the solution (5.37) is not correct1.
This is an example of what is sometimes known as the great matrix tragedy : the sim-
ple fact that eAeB 6= eA+B when [A,B] 6= 0 is responsible for most of the difficulty
arising in quantum mechanical calculations!
5.2.6 An Exact Solution: The Rosen-Zener case
It is possible to find an exact solution for V , in the particular case that the shape
of the control field envelope is given by a hyperbolic secant,
Ω(τ) =Ω0
Tcsech
(τ
Tc
), (5.38)
where Tc sets the duration of the control pulse, and Ω0 is a dimensionless constant
that sets the strength of the pulse (see Figure 5.4 (a)). The method of solution
is due to Rosen and Zener [142–144]. We include details of the derivation here for
completeness, but in §5.4 below we introduce a numerical approach that is faster,
more accurate and more general. The analytic solutions presented here do provide
useful points of comparison, of course.
We first transform to the interaction picture, to remove the rapid oscillations
generated by the diagonal elements of M . To do this, we define the diagonal and1Sometimes the formal solution is written like this, but in general it is understood that the
exponential must be time ordered

5.2 Solution in k-space 138
off-diagonal matrices
M0 =
2βTc
0
0 0
, MX =
0 Ω
Ω∗ 0
, (5.39)
where for later convenience we have defined 2β/Tc = d/k− iΓ. Clearly we have that
M = M0 + MX . The interaction picture evolution operator VI is then defined by
VI = V V −10 , where V0 satisfies ∂τV0 = iV0M0. We can write out V0 explicitly as
V0(τ) = eiM0τ since M0 is a constant. The equation of motion for VI is found to be
∂τVI = (∂τV )V −10 + V
(∂τV
−10
)= (iVIV0M)V −1
0 + VIV0
(−iM0V
−10
)= iVIMI , (5.40)
where MI = V0MXV−1
0 is the operator that generates time evolution in the interac-
tion picture, given by
MI =
0 Ωe2iβτ/Tc
Ω∗e−2iβτ/Tc 0
. (5.41)
As described by Pechukas and Light [143], the matrix elements of VI each satisfy a
second order differential equation, which we find by differentiating the equation of

5.2 Solution in k-space 139
motion,
∂τ (∂τVI) = i (∂τVI)MI + iVI (∂τMI)
= −VIM2I + iVI (∂τMI) . (5.42)
The square of MI is simply given by M2I = |Ω|2I, where I is the identity matrix. We
can express the derivative of MI as ∂τMI = MIG, where G is the diagonal matrix
G =
∂τΩ∗
Ω∗ − 2i βTc 0
0 ∂τΩΩ + 2i βTc
. (5.43)
Inserting this into (5.42) gives
∂2τVI − (∂τVI)G+ |Ω|2VI = 0. (5.44)
The boundary conditions are
VI∣∣τ→−∞ = I, ∂τVI
∣∣τ→−∞ = iMI
∣∣τ→−∞. (5.45)
The equation (5.44) is solved by making a temporal coordinate transformation.
We define the normalized integrated Rabi frequency by
ω(τ) =1W
∫ τ
−∞|Ω(τ ′)|2 dτ ′, (5.46)

5.2 Solution in k-space 140
where W is a normalization related to the total energy in the control pulse,
W =∫ ∞−∞|Ω(τ)|2 dτ. (5.47)
The coordinate ω runs from 0 to 1, as τ runs from −∞ to ∞ (see Figure 5.4 (b)). ω
can be thought of as the time coordinate marked out by a clock that is powered by
the control field. Using the control field profile (5.38), we can evaluate the integral
in (5.46) explicitly, to get
ω(τ) = 12tanh
( τT
)+ 1
2 . (5.48)
0-6 -4 -2 0 2 4 6
1
-6 -4 -2 0 2 4 6
(a) (b)
Figure 5.4 The Rosen-Zener model. The control field, shown in(a) with Ω0 = Tc = 1, takes the form of hyperbolic secant. (b): theintegrated Rabi frequency ω marks out time at a rate given by |Ω|2.
Under the transformation τ −→ ω, the temporal derivative ∂τ transforms as

5.2 Solution in k-space 141
follows,
∂τ = (∂τω) ∂ω
=1
2Tcsech2
(τ
Tc
)∂ω
=1
2Tc
(ΩTcΩ0
)2
∂ω, (5.49)
where we used the control shape (5.38). The second derivative is then given by
∂2τ =
(∂2τω)∂ω + (∂τω)2 ∂2
ω
=1T 2c
[(1− 2ω)
(ΩTcΩ0
)2
∂ω +14
(ΩTcΩ0
)4
∂2ω
]. (5.50)
Putting all this together, and using
(ΩTcΩ0
)2
= 4ω(1− ω), (5.51)
we find the equation
ω(1− ω)∂2ωVI + (∂ωVI)
[12
+ iβZ − ω]
+ Ω20VI = 0, (5.52)
where Z is the Pauli matrix
Z =
1 0
0 −1
. (5.53)

5.2 Solution in k-space 142
The boundary conditions, in terms of the new variable ω, are given by
VI∣∣ω→0
= I, ∂ωVI∣∣ω→0
= iΩ0
0 ω−θ−
ω−θ+ 0
, (5.54)
where θ± = 12 ± iβ. The equation (5.52) is known as a hypergeometric differential
equation. The solutions are known as hypergeometric functions, denoted2 by the
symbol F , and parameterized by the coefficients appearing in the equation. The
hypergeometric functions are special functions that can be evaluated using a mathe-
matics application such as Matlab or Mathematica. Matching the general solutions
to the boundary conditions (5.54), the solution for VI is given by
VI(ω) =
F (Ω0,−Ω0, θ+, ω) iΩ0θ+ωθ+F (θ+ + Ω0, θ+ − Ω0, 1 + θ+, ω)
iΩ0θ−ωθ−F (θ− + Ω0, θ− − Ω0, 1 + θ−, ω) F (Ω0,−Ω0, θ−, ω)
.
(5.55)
The properties of these special functions can be used to show that at the end of the
storage process we have
VI∣∣ω→1
=
Γ2(θ+)
Γ(θ++Ω0)Γ(θ+−Ω0)i sin(πΩ0)cosh(πβ)
i sin(πΩ0)cosh(πβ)
Γ2(θ−)Γ(θ−+Ω0)Γ(θ−−Ω0)
, (5.56)
where Γ(x) is the Euler Gamma function. We substitute these matrices into (5.36) to
obtain an expression for the storage kernel in terms of k and ω. Some manipulations2Sometimes the symbol 2F1 is used, and the designation Gauss hypergeometric function then
distinguishes this from the generalized hypergeometric functions pFq.

5.2 Solution in k-space 143
reveal that the determinant of the matrix (5.56) is 1, which simplifies forming the
inverse V −1out . The exponential factor ei2βτ/Tc that enters when transforming from VI
back to V = VIV0 translates into the factor
ei2βτ/Tc =(
ω
1− ω
)iβ
. (5.57)
After some algebra, we arrive at the result
K(k, ω) =i√d
k√
2π
(ω
1− ω
)iβ
× iQ(k, ω)√2ω(1− ω)/Tc
, (5.58)
where we’ve defined the function Q in the following way
Q(k, ω) =Γ2(θ+)
Γ(θ+ + Ω0)Γ(θ+ − Ω0)Ω0
θ−ωθ−F (θ− + Ω0, θ− − Ω0, 1 + θ−, ω)
− sin(πΩ0)cosh(πβ)
F (Ω0,−Ω0, θ+, ω). (5.59)
It helps to keep in mind that θ± is a function of k through β. Note we have included
the Jacobian factor√
2ω(1− ω)/Tc =√∂τω in the denominator of (5.58), to make
the transformation from τ to ω a unitary one (see §3.3.3 in Chapter 3). We would
now like to extract the optimal input mode and its associated optimal efficiency by
applying the SVD. However, just as in the case of (5.15), the kernel in (5.58) has a
singularity at k = 0. As before, we remove this by transforming form k-space into
λ-space, where λ = 2π/k is the wavelength of the spin wave excitation. Including

5.2 Solution in k-space 144
the Jacobian factor, the expression for K in terms of λ and ω is,
K(λ, ω) =i
2π
√Tcd
2ω−θ−(1− ω)−θ+Q(λ, ω), (5.60)
where Q is given, as before, by (5.59), the only difference being that in λ-space, the
parameter β takes the form 2β/Tc = dλ2π − iΓ. There is a minor pathology associated
with the points ω = 0 and ω = 1, which blow up, but in practice this is easily
addressed by introducing a small regularization ε that shifts the singularities into
the complex plane,
ω → ω + iε. (5.61)
After these steps, we have a non-singular kernel that is amenable to a numerical SVD.
From this we obtain the singular values λj, and a set of input modes φj(ω). The
optimal storage efficiency is already given by η1 = λ21. To find the temporal mode
of the signal field that is stored with this optimal efficiency, we need to transform
the mode φ1(ω) back into the temporal domain. Including the Jacobian factor, we
have
φ1(τ) =
2Tcω(τ) [1− ω(τ)]
1/2
φ1 [ω(τ)]
=sech
(τT
)√
2Tcφ1
[12tanh
(τ
Tc
)+ 1
2
]. (5.62)
The analytic solution for K in (5.60) provides a check on the numerical optimiza-
tions we present in §5.4 (see Figures 5.8 and 5.9). In fact, evaluating the function

5.2 Solution in k-space 145
F (a, b, c, ω) with complex a, b or c can be time-consuming, since these values are
generated by analytic continuation of F into the complex plane. This procedure
is not always accurate, and so the direct numerical optimizations presented at the
end of this chapter are both faster and more reliable. Finally, the analytic solution
(5.60) is, of course, only valid for the particular control (5.38). It would be more
convenient if we could derive an expression that holds for a range of control field
profiles. We now show how to construct an approximation to V that holds in the
adiabatic limit, which is essentially the limit of a slowly varying control.
5.2.7 Adiabatic Limit
The idea behind adiabatic evolution is to adjust Ω sufficiently slowly that at each
moment we can neglect the time dependence of M , and treat the problem as if
it were time-stationary. In this limit, the state |ψ〉 remains in an instantaneous
eigenstate of M at all times. As M changes, the eigenstates of M slowly evolve,
and we arrange for the populated eigenstate at the end of the storage interaction
to overlap with the | ↓〉 state; in this way excitations are transferred into the spin
wave. To see how this works, we re-cast the equation of motion for V in terms of
the adiabatic basis, which is the basis formed by the instantaneous eigenstates of M .
Suppose M has the following eigenvalue decomposition (see §A.4.4 in Appendix A),
M = RDR−1. (5.63)

5.2 Solution in k-space 146
We define the operator Vad = V R, and differentiate it to obtain its equation of
motion,
∂τVad = (∂τV )R+ V ∂τR
= iVMR+ V ∂τR
= iV RDR−1R+ V R(R−1∂τR
)= iVadMad, (5.64)
where Mad = D− iR−1∂τR generates the time evolution in the adiabatic basis. The
content of the adiabatic approximation is to neglect the term R−1∂τR in Mad, so
that Mad is a purely diagonal matrix. This allows us to solve the equation of motion
for Vad, using the result (5.37). That is,
Vad(τ) = exp[i∫ τ
−∞Mad(τ ′) dτ ′
]. (5.65)
Armed with this solution, we can construct the propagation matrix V = VadR−1, and
therefore the storage kernel (5.36). We now implement this programme explicitly,
after which the conditions under which the adiabatic approximation is justifiable
will become clearer.
Here we remark that corrections to the adiabatic approximation can be gen-
erated, in the current formalism, by making use of the Magnus expansion [143,145],
or Salzman’s expansion [146], which provide approximations to the propagator V

5.2 Solution in k-space 147
when Mad is non-diagonal and time-dependent. These corrections quickly become
unwieldy however, and so we do not present them here: the numerical approach
presented in §5.4 obviates the need for them.
To find the adiabatic kernel, we start by finding the instantaneous eigenvalues
of M(τ), by solving the equation |M − λI| = 0, where the vertical bars denote the
determinant (see §A.4.2 in Appendix A). The resulting eigenvalues are
λ± = b±√b2 + |Ω|2, (5.66)
where we have defined 2b = d/k − iΓ (thus replacing the notation 2β/Tc defined in
the previous section). Solving the equation M |±〉 = λ±|±〉 for the elements of the
eigenvectors |±〉, we find
|+〉 ∝
λ+
Ω∗
, |−〉 ∝
λ−
Ω∗
. (5.67)
The diagonalizing transformation R is the matrix with |+〉 as its first column and |−〉
as its second. But there is some freedom as to the normalization of the vectors |±〉.
This is fixed by requiring that limτ→−∞R = I, the identity. This just codifies our
knowledge that Ω = 0 at the start of the storage interaction, so that M is initially
diagonal, which means that no transformation need be applied to diagonalize it at

5.2 Solution in k-space 148
τ −→ −∞. A suitable form for R, that satisfies this boundary condition, is
R =
1 λ−Ω∗
Ω∗
λ+1
. (5.68)
Note that limτ→−∞ λ− = 0. The inverse transformation is then given by
R−1 =1
1− λ−λ+
1 −λ−Ω∗
−Ω∗
λ+1
. (5.69)
We have Mad = diag(λ+, λ−), so that Vad is given by
Vad(τ) =
exp[i∫ τ−∞ λ+(τ ′) dτ ′
]0
0 exp[i∫ τ−∞ λ−(τ ′) dτ ′
] . (5.70)
Combining these results together and substituting them into (5.36), we find there is
only a single non-vanishing term contributing to the storage kernel,
K(k, τ) = − ik
√d
2π×
Ω∗(τ) exp[−i∫∞τ λ−(τ ′) dτ ′
]λ+(τ)− λ−(τ)
. (5.71)
We can further simplify this result if we make the assumption that
|b| |Ω| (5.72)
at all times. This does not have anything to do with the rate at which we change
the control field, but it is usually considered as part of the adiabatic approximation,

5.2 Solution in k-space 149
as discussed in §5.2.9 below. With this approximation, we can write
λ+ ≈ 2b+|Ω|2
2b, and λ− ≈ −
|Ω|2
2b. (5.73)
Inserting these expressions into (5.71), and making the replacement 2b = d/k − iΓ,
we find, after a little algebra,
K(k, τ) =1√2π×√d
ΓΩ∗(τ) exp
[− 1
Γ
∫ ∞τ|Ω(τ ′)|2 dτ ′
]× 1k + i dΓ
exp
[idΓ2
∫∞τ |Ω(τ ′)|2 dτ ′
k + i dΓ
].
(5.74)
The first exponential factor represents the accumulation of phase due to the dynamic
Stark effect, in which the strong control field ‘dresses’ the atoms and causes a time
dependent shift in the |2〉 ↔ |3〉 transition frequency. The second exponential factor
represents the ‘meat’ of the interaction: the response of the atoms to the incident
signal field. The kernel (5.74) is already amenable to a numerical SVD. However, to
make a connection with previous work, we take the inverse Fourier transform. We
use the method detailed in §D.5.2 in Appendix D, and apply the shift theorem, to
get the adiabatic z-space kernel
K(z, τ) = −i
√d
ΓΩ∗(τ) exp
− 1
Γ
[∫ ∞τ|Ω(τ ′)|2 dτ ′ + dz
]×J0
(2i
√d
Γ
√∫ ∞τ|Ω(τ ′)|2 dτ ′z
).
(5.75)
Note the additional contribution dz to the exponential factor. This contribution
represents the change in refractive index experienced by the signal field as it prop-
agates through the ensemble. On resonance (with Γ = 1 in normalized units), this

5.2 Solution in k-space 150
term describes exponential attenuation i.e. absorption, with an absorption coeffi-
cient of d. This explains why the quantity d is known as the optical depth: it directly
quantifies the optical thickness of the ensemble on resonance.
A numerical SVD could also be applied to the kernel (5.75) to obtain the optimal
temporal input mode φ1(τ), and its associated optimal efficiency η1 = λ21. This
adiabatic solution appears in the work of Gorshkov et al. [133], although the method
of derivation differs slightly. They provided a method to find the optimal input
mode, in the limit of large control field energy, but they did not use the SVD: The
SVD is a more direct method, and it does not require the assumption of large control
energy. We will discuss the differences between the SVD method and the method of
Gorshkov et al. shortly. First, we make a coordinate transformation that removes
the explicit dependence of K on the shape of the control field. As in the Rosen-Zener
solution above, we transform from τ to ω, where ω is the normalized integrated Rabi
frequency, defined by (5.46). We do not assume a particular shape for the control:
its profile can be arbitrary in the present case (within the limits of the adiabatic
approximation, to be discussed below). By removing the dependence on the control
field with this coordinate transformation, we only need to perform the SVD once, in
the transformed coordinate system, in order to obtain the optimal input mode for
any control field shape. To make the coordinate transformation unitary, we include
a Jacobian factor of√W/Ω∗(τ) in the transformed kernel (see §3.3.3). This rather
conveniently cancels with the factor of Ω∗(τ) in (5.75), so the transformed kernel

5.2 Solution in k-space 151
can be written as
K(z, ω) = −i
√dW
Γe−(W−Wω+dz)/ΓJ0
(2i√d
Γ
√(W −Wω)z
), (5.76)
A final, cosmetic simplification is achieved by flipping the ω coordinate, ω −→
1 − ω. This has no effect on the singular values of the kernel, but simply flips the
input modes around. The optimal mode for adiabatic storage in a Λ-type quantum
memory, with an arbitrary control field profile, is now found by taking the SVD of
K(z, ω) = −i
√dW
Γe−(Wω+dz)/ΓJ0
(2i√dWΓ
√ωz). (5.77)
We note that both ω and z run from 0 to 1. The optimal temporal input mode, is
then found from the mode φ1(ω) by the relation
φ1(τ) =Ω(τ)√Wφ1 [1− ω(τ)] . (5.78)
The factor of Ω/√W arises from the Jacobian relating the coordinates ω and τ . We
now have a simple prescription for finding the optimal input mode for a quantum
memory. Given a fixed value for W , which essentially quantifies the total energy in
the control pulse, and given values for the detuning and the optical depth, we form
the kernel (5.77) and take the SVD. Then, for any arbitrary shape of the control,
we can construct the optimal input mode φ1, using the transformation (5.78) above.
Some examples of the optimal input modes predicted using this approach can be

5.2 Solution in k-space 152
found in Figures 5.8, 5.9, 5.10 and 5.11 in §5.4 at the end of this chapter.
In general, the storage efficiency depends on the optical depth, the detuning
(through Γ), and the control pulse energy, through W , since the kernel (5.77) de-
pends on all of these quantities. We also know however, from the discussion in
§5.2.3, that the best possible storage efficiency only depends on the optical depth.
Below we connect these two results together.
5.2.8 Reaching the optimal efficiency
In §5.2.3 we derived an expression for the optimal storage efficiency possible in a
Λ-type ensemble memory. In fact, it is possible to reach this optimal efficiency in the
adiabatic limit. As was first shown by Gorshkov et al. [133], the anti-normally ordered
kernel formed from the adiabatic storage kernel (5.77) is equal to the optimal kernel
(5.27), in the limit of large control pulse energy. To see this, we substitute (5.77) into
the expression (3.19) for the anti-normally ordered kernel, and perform the integral
over ω. In the limit W −→∞, we can evaluate the integral analytically. After some
leg work — see §D.5.3 in Appendix D — we find that the result is exactly (5.27).
That is,
limW→∞
∫ 1
0K(z, ω)K∗(z′, ω) dω =
∫ ∞0
K(z, ω)K∗(z′, ω) dω =d
2e−d(z+z′)/2I0(d
√zz′).
(5.79)
This shows that it is possible to saturate the upper bound on the storage efficiency,
even in the adiabatic limit. The adiabatic limit is not only useful because we can

5.2 Solution in k-space 153
construct the optimal input mode explicitly. It is also useful because it is possible
to shape the input mode by shaping the control. From the form of (5.78), it is
clear that, by an appropriate choice of Ω(τ), we can choose the shape of φ1(τ). In
particular, we can choose the control so that φ1(τ) matches the temporal profile of
some ‘given’ input field. This is of considerable practical importance, since it may
be experimentally much easier to shape the bright control field, than to shape the
weak input field (this is discussed in Chapter 8). The combination of these two facts
— that adiabatic storage can be optimal, and also that it enables one to shape the
input mode — makes the adiabatic limit an important regime for the operation of
a quantum memory.
Under what circumstances is the equality in (5.79) achieved? The limit of W −→
∞ is not really required. Examining the form of (5.77), it is clear that we just
need to make W large enough that the exponential factor e−Wω/Γ, evaluated at the
limit ω = 1, is sufficiently small that extending the integral further would make no
difference. We should certainly have that W |Γ| then. In addition, we should
ensure that any contribution from the Bessel function is negligible at ω = 1. Using
the approximation J0(ix) = I0(x) ∼ ex/√x for x 1, we see that we should also
have that W d. To summarize, adiabatic storage is optimal if the control pulse
is sufficiently energetic that the conditions
W max (|∆|, d) (5.80)

5.2 Solution in k-space 154
are satisfied. Recall that ∆ is the common detuning of the signal and control fields
from resonance. We can connect W with the total energy Ec in the control pulse.
The total energy is
Ec = A∫ ∞−∞
Ic(τ) dτ, (5.81)
where Ic = 2ε0c|Ec|2 is the cycle-averaged control pulse intensity. From the definition
of the Rabi frequency (4.49), we then find
Ec =2ε0cA∣∣∣d23.vc
~
∣∣∣2γW. (5.82)
Here the presence of the factor of γ indicates that we have converted back into
ordinary units (rather than normalized units). From the definition of W (5.47), we
find it has the dimensions of frequency, and so in ordinary units it is accompanied
by the factor γ. Let us define the number of photons in the control pulse by Nc =
Ec/~ωc. We also define Na as the number of atoms in the ensemble addressed by
the optical fields, Na = nLA. Using (4.50) and (5.12), we can express the optical
depth as
d =∣∣∣∣d∗12.vs
~
∣∣∣∣2 ~ωs2ε0cAγ
×Na. (5.83)
With these definitions, we see that the condition W d in (5.80) amounts, essen-
tially, to the condition Nc Na (we have used ωs ≈ ωc and |d∗12.vs| ≈ |d23.vc|).
We might therefore describe this condition as describing a ‘light-biased’ interaction,
where the number of control photons dominates over the number of atoms; this is

5.2 Solution in k-space 155
why it is the latter quantity that limits the efficiency.
In the work of Gorshkov et al. [133], a method is presented for optimizing the
adiabatic storage efficiency in this light-biased limit. The method works by combin-
ing the optimal anti-normally ordered kernel (5.27), whose eigenfunctions are the
optimal spin waves, with the adiabatic storage kernel (5.75), which connects the
control field profile to the signal field profile. Combining these two kernels together
is possible only when Nc Na, and also W ∆. In this limit their method works
extremely well. One advantage of the SVD method however, is that we can apply
it directly to the kernel (5.77), without making this approximation, and we can
therefore find the optimal input modes for arbitrary values of W .
5.2.9 Adiabatic Approximation
We have employed several approximations in the name of adiabaticity. We now
examine the physical content of these approximations. The first assumption we
made, in the text following (5.64), was to neglect the term R−1∂τR in the adiabatic
generator Mad. Clearly this term vanishes if the control field is held constant, and
so the size of this term is set by the rate of variation of the control field. To make
the adiabatic approximation, we must therefore limit the bandwidth of the control
pulse. To find this limit, we introduce the second approximation we made in (5.72),
namely that
|b| |Ω|. (5.84)

5.2 Solution in k-space 156
With this approximation, we can write the diagonalizing transformations R, R−1 in
the form
R =
1 − Ω2b
Ω∗
2b 1
; R−1 =
1 Ω2b
−Ω∗
2b 1
. (5.85)
For adiabatic evolution, we should have that ||R−1∂τR|| ||D||, where D =
diag(2b + |Ω|2/2b,−|Ω|2/2b). Here the double bars represent the Frobenius norm,
which is found by adding in quadrature the magnitudes of all the elements in a
matrix. We neglect terms of order |Ω/b|, which are small by assumption. We then
arrive at the condition ∣∣∣∣∂τΩ2b
∣∣∣∣2 |Ω|2. (5.86)
Recall that b = 12(d/k − iΓ) depends on the wavevector k, and that Ω varies with
time, so we should be careful to satisfy this condition for all the values of these two
parameters that play a role in the storage process.
We can distinguish two regimes. First, the EIT regime (see §2.3.1 in Chapter
2), in which the optical fields are tuned into resonance with the excited state. In
this case, ∆ = 0 and so Γ = 1 (in normalized units). Since d 1 for reasonably
efficient storage, the contribution to b from the complex detuning Γ is small, and we
have |b| ∼ d/2k. Therefore the adiabatic conditions (5.84) and (5.86) vary strongly
with k, and we must take some care to identify the range of wavevectors that are
important for the storage process.
The second regime is the Raman regime (see §2.3.2 in Chapter 2), in which the
detuning is large compared to the excited state linewidth; ∆ 1 in normalized

5.2 Solution in k-space 157
units. In this regime |b| ∼ 12(d/k−∆). In this case the large contribution to b from
the detuning makes the adiabatic conditions less dependent on k.
What range of wavevectors are important in the storage process? One answer is
provided by inspection of the k-space kernel (5.74). Using the expansion (k+i dΓ)−1 =
−iΓd + Γ2
d2 k + iΓ3
d3 k2 + . . ., we can re-write the k-space map in the form
K(k, ω) ≈ − i√2π
√W
d× ei
Wd ωk × e−
ΓWd2 ωk2
, (5.87)
where we have introduced the integrated Rabi frequency ω, and ‘flipped’ the kernel,
as we did in deriving (5.77). This expression is a good approximation if |d/Γ| is
large, which is not guaranteed in the Raman regime, but which is generally true in
the EIT regime. The final exponential factor describes a Gaussian profile in k-space,
with a characteristic width given by
δk = δk(ω) =d√Wω
, (5.88)
in the EIT regime with Γ = 1. In this case the most restrictive form of the adiabatic
condition (5.84) can be written as (dropping an unimportant factor of two)
Ωmax d
δk(ωmax), (5.89)
where Ωmax = max(Ω) is the peak Rabi frequency of the control, and where ωmax is
the value of the integrated Rabi frequency when this peak occurs. For a symmetric

5.2 Solution in k-space 158
control pulse, we would have ωmax = 12 ; in general ωmax will be some fraction that
for our purposes we may approximate as ∼ 1. Substituting (5.88) into (5.89), we
find that this condition in fact restricts the bandwidth δc of the control pulse:
δc 1. (5.90)
Here we made the approximation W ∼ Ω2max/δc. The condition (5.90), in ordinary
units, is δc γ. That is, the bandwidth of the control should not exceed the natural
linewidth of the |2〉 ↔ |3〉 transition in the ensemble, to achieve adiabatic storage on
resonance. This suggests that EIT is a memory protocol best suited for the storage of
narrowband fields. However, the analysis by Gorshkov et al. [133] reaches a different
conclusion: that the adiabatic restriction on the bandwidth of the control is δc dγ
(in ordinary units). This much less stringent condition is derived by evaluating the
adiabatic condition (5.86) using b ∼ d/max(δk) with max(δk) ∼ 1. Identifying the
maximum of the quantity |∂τΩ/Ω| with the bandwidth δc, one arrives at their result.
One justification for using δk ∼ 1 is that for optimal storage, we only need to access
the optimal spin wave mode; higher modes are irrelevant. Since the optimal mode
is that mode which is most slowly varying in space, its width in k-space is limited
to a relatively small region, and the adiabatic approximation need only be satisfied
within this range. It is difficult to argue rigorously about these approximations, but
as we will see, numerics reveal that the reality lies somewhere between these two
cases: the adiabatic approximation breaks down rather quickly in the EIT regime

5.3 Raman Storage 159
as the bandwidth approaches the natural linewidth, although it is true that this is
mitigated somewhat by increasing the optical depth.
In the Raman regime, the analysis is simpler. The adiabatic condition is simply
that
Ωmax |∆|, (5.91)
independent of k. We might comment that there may be some particular value of k
such that there is a cancellation, and b becomes small, but the effect of this isolated
point is generally negligible. The limitation on the control bandwidth comes from
(5.86), which yields the condition
δc |∆|. (5.92)
Therefore adiabatic evolution is guaranteed in the Raman case whenever the detun-
ing ∆ is the dominant frequency involved in the interaction, with both the Rabi
frequency and the bandwidth of the control field small by comparison.
5.3 Raman Storage
So far we have studied the properties of storage in a Λ-type ensemble for arbitrary
values of the detuning. We now specialize to the case of large detunings, ∆ γ
(or ∆ 1 in normalized units). In this Raman regime the storage kernel simpli-
fies further, and an interesting connection between the input and spin wave modes
emerges. The following treatment forms the basis of our Rapid Communication on

5.3 Raman Storage 160
Raman storage [77].
Taking the limit ∆ 1, we can write Γ ≈ −i∆, and the storage kernel (5.77)
becomes
K(z, ω) = C × e−i(Wω+dz)/∆ × J0(2C√ωz), (5.93)
where we have defined the Raman memory coupling by
C =
√Wd
|Γ|≈√Wd
∆. (5.94)
The exponential factor in (5.93) represents only phase rotations applied to the ω
and z coordinates. We can absorb these phases into the signal field and spin wave,
and therefore we can drop them from the kernel (we will be careful to ‘put them
back’ when we write down the optimal modes). We can now write down a very
simple recipe for constructing the optimal input mode in the Raman regime. First,
we form the kernel
K(z, ω) = CJ0(2C√ωz). (5.95)
This depends only on the memory coupling C, therefore this parameter uniquely
determines the efficiency of the memory in the Raman limit [77]. We note that
the kernel K is real, and symmetric under the exchange of ω and z. That is, K
is Hermitian (see §A.4.3 in Appendix A). Therefore, its SVD is the same as its
spectral decomposition (see §3.1.3 in Chapter 3). The singular values of K are also
its eigenvalues, and the input modes φj, as functions of ω, have the same form as

5.3 Raman Storage 161
the spin wave modes ψj, as functions of z. Their phases differ though, because of
the phase rotations we ‘absorbed’. To be precise, let us define the functions ϕj as
the eigenfunctions of the kernel (5.95). That is to say, ϕj satisfies
∫ 1
0CJ0(2C
√xy)ϕj(x) dx = λjϕj(y). (5.96)
The optimal input mode for the signal field, including the correct phase rotation
and transforming back from ω to τ , is given by
φ1(τ) =1√W
Ω(τ)× exp
iW [1− ω(τ)]∆
× ϕ1[1− ω(τ)]. (5.97)
The optimal output mode for the spin wave, to which this optimal input mode is
mapped by the storage process, is given by
ψ1(z) = e−idz/∆ϕ1(z). (5.98)
The optimal storage efficiency is given by the square of the largest eigenvalue in
(5.96); η1 = λ21. Figure 5.5 shows the variation of this optimal efficiency with C.
In taking the Raman limit, we have neglected spontaneous emission. This means
that the predicted efficiency can approach unity, as long as C is large enough, even
if the optical depth is low. That is, a smaller d can be ‘compensated’ by a larger
W — a more energetic control. Of course the optimal efficiency derived in §5.2.3
remains correct, even in the Raman limit: the best achievable efficiency is always

5.3 Raman Storage 162
limited, through spontaneous emission, by the optical depth. If the efficiency pre-
dicted by (5.96) is larger than the upper limit (5.25), then we have reached a regime
where spontaneous emission dominates over other losses. However, generally a Ra-
man memory requires a large optical depth to operate efficiently, as we discuss
below. Therefore the dominant loss mechanism in a Raman memory is not sponta-
neous emission, but insufficient coupling. That is, the large detuning from resonance
makes the interaction weak, so that the biggest problem in getting a Raman memory
to work is to make the coupling strong enough that the signal field is completely
absorbed. The utility of the kernel (5.95), is that it provides a simple way to analyze
the Raman limit before spontaneous emission becomes a limitation. Examples of
the optimal modes predicted by the Raman kernel are shown in Figures 5.8 and
5.10 in §5.4 at the end of this chapter. What is notable about the behaviour of the
0 0.5 1 1.5 2 2.5 3 3.5 410
−5
10−4
10−3
10−2
10−1
100
0 0.5 1 1.5 2 2.5 3 3.5 40
0.2
0.4
0.6
0.8
1
(a) (b)
Figure 5.5 Raman efficiency. (a) the optimal storage efficiencyη1 = λ2
1 predicted by the kernel (5.95) in the Raman limit ∆ 1,versus the memory coupling C =
√Wd/|Γ|. The efficiency ‘saturates’
at around C ∼ 2. (b) A logarithmic plot of the difference 1 − η1between the predicted efficiency and unity.
Raman efficiency is that it rises steeply for small values of C, before ‘saturating’

5.3 Raman Storage 163
at C ∼ 2. Physically, this saturation point coincides with the stimulated scattering
regime. For ordinary Stokes scattering, the scattering process becomes efficient as
the coupling is increased beyond this point (see Figure 10.6 in Chapter 10). In the
case of a Raman quantum memory, the transmission of the signal field through the
ensemble drops sharply, and the efficiency of the memory becomes limited by spon-
taneous emission, rather than insufficient coupling. Therefore in designing a Raman
memory, one need only ensure that C & 2, in order that the scheme is viable (see
§9.8 in Chapter 9 and §10.9 in Chapter 10).
5.3.1 Validity
As mentioned above, in deriving the Raman kernel we neglected spontaneous emis-
sion. We did this tacitly when we dropped the real part of Γ in the exponential
factor appearing in the storage kernel. This is valid so long as neither W nor d is
too large. To see this, we define the balance R according to the relation
R =
√W
d. (5.99)
Note that the balance has no relation to the matrix R introduced in §5.2.7 above.
Using the arguments employed in §5.2.8, we see that the balance really expresses
the extent to which the interaction is dominated by light, or matter:
R2 ∼ Nc
Na. (5.100)

5.3 Raman Storage 164
That is, the case R 1 corresponds to a light-biased interaction — as described ear-
lier, this is the limit in which the adiabatic kernel saturates the upper bound on the
storage efficiency — and the case R 1 describes a matter-biased interaction, with
a weak control, but a large/dense ensemble. Using the balance, and the definition
C =√Wd/|Γ|, we can re-express the adiabatic storage kernel (5.77) as follows,
K(z, ω) = Ce−iθ × e−iC cos θ(Rω+z/R) × eC sin θ(Rω+z/R) × J0(2Ce−iθ√ωz), (5.101)
where we have defined the phase angle θ such that tan θ = 1/∆; the complex detun-
ing being given by
Γ = −i|Γ|eiθ. (5.102)
The Raman limit is the limit of large detuning, which corresponds to the limit θ 1.
The exponential factor involving cos θ in (5.101) corresponds to the phase rotations
we dealt with previously. On the other hand, the exponential factor involving sin θ
comes from the real part of Γ, and represents spontaneous emission. It cannot
be removed by a unitary transformation of the modes, and it reduces the storage
efficiency. This is the term we neglect in the Raman limit. For this approximation to
hold for both the optical and spin wave modes together, both terms in the exponent
should be small. This is true provided we satisfy the conditions
C sin θR 1, and C sin θ × 1R 1. (5.103)

5.3 Raman Storage 165
Using sin θ ≈ tan θ, setting C ∼ 1 (as we should have for a reasonably efficient
memory), and using (5.100), we find that the Raman kernel is a good approximation
whenever
∆ γ, andγ2
∆2 Nc
Na ∆2
γ2, (5.104)
using ordinary units. That is to say, the interaction should be roughly ‘balanced’,
with broadly equal contributions to the coupling originating from atoms and light.
The larger the detuning, the more ‘leeway’ there is to bias the interaction one way
or the other.
Suppose that we set R ∼ 1, so that d ∼W . For reasonable efficiency, we should
have C ∼ 1, or thereabouts. Squaring this, and using R ∼ 1 then, we find d ∼ ∆. In
a Raman memory, ∆ 1, and therefore d 1. That is, a Raman memory described
by (5.95) generally requires a large optical depth. This explains why spontaneous
emission is not as important a limiting factor as is the issue of sufficiently strong
coupling.
Finally, we comment that since ∆ ∼ d, the adiabatic condition on the bandwidth
of the control field can be written as δc d, or in ordinary units δc dγ. This
is the same as the condition derived by Gorshkov et al. for the limitation on the
control bandwidth for resonant EIT storage. It is therefore arguable that a Raman
memory does not allow for more broadband storage than an EIT memory does.
But the adiabatic condition in the Raman case is rather more robust that it is in
the EIT case, since it does not depend on identifying an ‘important’ region in k-
space. Numerics show that the adiabatic storage kernel is a better approximation

5.3 Raman Storage 166
in the Raman case, for more broadband control pulses, than it is in the EIT case.
Broadband storage provided the motivation for studying the Raman memory, but
its advantages in this respect are not clear cut.
Other considerations that may favour detuning from resonance include the possi-
bility of dealing with a more complex excited state manifold. Suppose that, instead
of a single excited state |2〉, there are a host of states, perhaps resulting from spin-
orbit or hyperfine splitting (see, for example, Figure 10.3 in Chapter 10). Tuning
into resonance with one of these states could make the dynamics rather complicated.
There may be some direct absorption of the signal field into the nearby states, fol-
lowed by spontaneous emission and loss. Detuning away from all of the states puts
the contribution from each state on an equal footing, so that the dynamics can be
treated just as we did the simple three level system, where we need only swap the
coupling C to a single state for an equivalent coupling that includes the scattering
amplitudes for all the states (see §F.4 in Appendix F). And by detuning we eliminate
the possibility of absorption losses. In addition, a Raman memory is tunable, since
if we tune further from resonance, we can maintain strong coupling by increasing
the control pulse energy. The Raman memory is also affected less adversely by inho-
mogeneous broadening of the excited state than an EIT memory might be. Again,
this is because the coupling to the ensemble is not dominated by resonance with a
single frequency.

5.3 Raman Storage 167
5.3.2 Matter Biased Limit
We have already shown how the anti-normally ordered product formed from the
adiabatic storage kernel tends to the optimal kernel (5.27) in the limit of large
control pulse energy. This is the light-biased limit, with R 1. To reach this
limit, the balance R should exceed the upper limit in (5.104), so we should have
R > ∆ (in normalized units). Now, there is a degree of symmetry to the structure
of (5.101): The kernel is unchanged when we swap z and ω, if at the same time we
send R −→ 1/R. It therefore follows that in the matter-biased limit R 1 — that
is Na > ∆Nc/γ in ordinary units — the normally ordered kernel tends to a limit
defined not by the optical depth, but by the control pulse energy:
limd→∞
∫ 1
0K∗(z, ω)K(z, ω′) dz =
W
2e−W (ω+ω′)/2I0(W
√ωω′). (5.105)
The kernel on the right hand side has precisely the same form as (5.27), except that
the efficiency is limited by W rather than by d. Therefore, if we are limited by the
energy of the control, the optimal efficiency achievable is given by η1 = 1− 2.9/W .
Of course, extremely high energy lasers are readily available, whereas the size of the
ensemble is generally not easily varied. Nonetheless, in cases where the ensemble
may be damaged by a high energy laser pulse — as may be the case for a solid state
memory — it may be that the control energy becomes a limitation.

5.3 Raman Storage 168
5.3.3 Transmitted Modes.
In this section we describe a connection between the modes that are stored in the
memory, and the modes that are transmitted through it when the efficiency is not
perfect. This connection holds in the Raman limit, and it is clearest when we
form the equations of motion for the signal field and the spin wave in the adiabatic
approximation. This is the way the adiabatic approximation is most commonly
introduced, and so it is informative to run through the procedure. We start with
the equations of motion (5.3), which we reproduce below in normalized units,
∂zA(z, τ) = −√dP (z, τ),
∂τP (z, τ) = −ΓP (z, τ) +√dA(z, τ)− iΩ(τ)B(z, τ),
∂τB(z, τ) = −iΩ∗(τ)P (z, τ). (5.106)
The adiabatic approximation is made by setting ∂τP = 0 on the left hand side of
the second equation. This is reasonable when the natural dynamics of the optical
polarization P are overwhelmed by the motion driven by the signal and control fields
— known as adiabatic following. In this situation, 1/|Γ| is the shortest timescale
in the problem, so that when far-detuned we must have (δc,Ωmax) |∆|, precisely
the adiabatic conditions (5.91), (5.92) derived above. We can then solve the sec-
ond equation for P algebraically, and substitute the result into the first and third

5.3 Raman Storage 169
equations. The result is
(∂z +
d
Γ
)A = i
Ω√d
ΓB,(
∂τ +|Ω|2
Γ
)B = −i
Ω∗√d
ΓA. (5.107)
We then switch coordinates from (τ, z) to (ω, z), and we define new variables α and
β for the signal field and spin wave as follows
α(z, ω)e−(Wω+dz)/Γ =√WA(z, τ)Ω(τ)
,
β(z, ω)e−(Wω+dz)/Γ = B(z, τ). (5.108)
These new variables incorporate the Jacobian factor associated with the coordinate
transformation, and also the phase rotations associated with the dynamic Stark shift
and the ensemble refractive index. In general, the transformation linking A and B to
α and β is not quite unitary, because Γ is not strictly imaginary, so the exponential
factors on the left hand side of (5.108) make the norms of A, B different to the
norms of α, β — they are not pure phase rotations. However, taking the Raman
limit ∆ 1, we find Γ ≈ −i∆, and in this case, the transformation is unitary. This
will be important shortly. For now, observe that the equations of motion simplify
greatly when they are cast in terms of the transformed variables α, β. We have
removed the control field, and also the homogeneous terms on the left hand side, so

5.3 Raman Storage 170
that we obtain the system
∂zα = −Cβ,
∂ωβ = Cα. (5.109)
Note the symmetry of this system of equations. If we swap α for β, and β for −α,
and then swap z and ω, the system of equations is unchanged. As we will see below,
this symmetry simplifies the form of the solution, so that there are only two different
Green’s functions, instead of a potential four.
We solve these equations by applying a unilateral Fourier transform. Having
eliminated the control field, we can choose either to apply the transform over the ω
coordinate or the z coordinate. We will apply the transform over the z coordinate,
as we have done previously, and solve the equations in k-space. Using tildes to
denote the transformed variables, we have
−ikα− α0√2π
= −Cβ,
∂ωβ = Cα, (5.110)
where α0 = α(z = 0, ω) represents the boundary condition for the signal field.
Solving the first equation for α yields
α = −iC
kβ +
i√2πk
α0. (5.111)

5.3 Raman Storage 171
Substituting this result into the second equation, and integrating, we obtain the
solution for β,
β(k, ω) = e−iC2ω/kβ0 + iC√2πk
∫ ω
0e−iC2(ω−ω′)/kα0(ω′) dω′, (5.112)
where β0 is the Fourier transform of the spin wave boundary condition β0 = β(z, ω =
0). Finally, substituting this back into (5.111) gives the solution for α,
α(k, ω) = −iC
ke−iC2ω/kβ0 +
i√2πk
α0(ω) +C2
√2πk2
∫ ω
0e−iC2(ω−ω′)/kα0(ω′) dω′.
(5.113)
These k-space solutions are singular at k = 0, but fortunately it is possible to take
the inverse Fourier transform analytically. We use the results described in §D.5 in
Appendix D, along with the convolution theorem for the unilateral Fourier transform
(see §D.4.2 in Appendix D), to obtain the rather formidable-looking solution
α(z, ω) = α0(ω)− C∫ ω
0
√z
ω − ω′J1
[2C√z(ω − ω′)
]α0(ω′) dω′
−C∫ z
0J0
[2C√
(z − z′)ω]β0(z′) dz′,
β(z, ω) = β0(z)− C∫ z
0
√ω
z − z′J1
[2C√
(z − z′)ω]β0(z′) dz′
+C∫ ω
0J0
[2C√z(ω − ω′)
]α0(ω′) dω′. (5.114)
We re-write this in terms of Green’s functions, or propagators, in order to bring out

5.3 Raman Storage 172
its structure.
αout(ω) =∫ 1
0L(ω, ω′)αin(ω′) dω′ −
∫ 1
0K(ω, z)βin(z) dz,
βout(z) =∫ 1
0L(z, z′)βin(z′) dz′ +
∫ 1
0K(z, ω)αin(ω) dω, (5.115)
where αout(ω) = α(z = 1, ω) is the transmitted signal field, and βout = β(z, ω = 1)
is the spin wave at the end of the interaction. We have introduced the Green’s
functions L and K, defined as follows,
L(x, y) = δ(x− y)− CΘ(x− y)× 1√x− y
J1(2C√x− y),
K(x, y) = CJ0
[2C√x(1− y)
]. (5.116)
Note that only two distinct Green’s functions are required, because of the symmetry
between α and β in the adiabatic equations of motion (5.109). The Green’s function
K(x, 1−y) is precisely the adiabatic Raman storage kernel (5.95), and this is why we
have used the same notation. The kernel L describes the relation of the transmitted
signal field to the input field, or equivalently it relates the final to the initial spin
wave. We are considering storage, so that βin = 0, but note that the same solutions
can be used to describe retrieval from the memory; retrieval is dicussed in Chapter
6. The Heaviside step function Θ(x) in L makes the kernel causal, so that the
transmitted signal field is never influenced by future values of the input field.
We now show that there is a connection between the SVD of K, which tells
us about the optimal input mode and its associated efficiency, and the SVD of L,

5.3 Raman Storage 173
which tells us about the transmitted fields. Note that it is only correct to associate
the singular values of the Green’s function K with storage efficiencies when the
transformation connecting A, B to α, β is unitary, and this is only true in the
Raman limit ∆ 1. Therefore the following analysis applies only in this limit, and
we will assume that we are tuned far from resonance in the remainder of this section.
To see the connection between the SVDs of K and L, it will help us to use matrix
notation, since it is much more compact. As discussed in Chapter 3, the solutions
5.115 may be considered as the infinite-dimensional limit of the following matrix
equations,
|αout〉 = L|αin〉 −K|βin〉,
|βout〉 = L|βin〉+K|αin〉. (5.117)
To be more precise, we define the vector |αout〉 as a discretized version of the con-
tinuous function αout,
|αout〉 =
αout(0)
αout( 1N−1)
αout( 2N−1)
...
αout(1)
, (5.118)
where N is the number of discretization points. The other vectors are defined

5.3 Raman Storage 174
similarly. The matrices K and L are given in terms of the continuous kernels by
Kjk = K
(j − 1N − 1
,k − 1N − 1
)× 1N − 1
, Ljk = L
(j − 1N − 1
,k − 1N − 1
)× 1N − 1
.
(5.119)
The continuous equations are recovered by taking the limit N −→∞, when matrix
multiplication is replaced by integration, and the factors 1/(N − 1) become the
increments dz, dω. It is useful to be able to swap between the continuous and
discretized representations, in order to use both the machinery of calculus and linear
algebra in their ‘natural habitats’.
The relationship between the SVDs of the matrices K and L is fixed by the
equations of motion (5.109), which are unitary. To see this, observe that these
equations imply the identity
∂z(α†α) + ∂ω(β†β) = 0. (5.120)
We have used the † notation for Hermitian conjugation, rather than the ∗ notation
for complex conjugation, since this equation holds whether or not we treat α, β as
ordinary functions, or as quantum mechanical annihilation operators3. Integrating
this identity over all z and ω gives the condition
Nα,out +Nβ,out = Nα,in +Nβ,in, (5.121)
3Strictly, the Langevin noise operators introduced in §4.12 of Chapter 4 contribute unless anexpectation value is taken, but in the Raman limit we neglect spontaneous emission, and theseoperators along with it.

5.3 Raman Storage 175
where Nα,out, Nα,in are the numbers of transmitted and incident signal photons,
respectively, and where Nβ,out, Nβ,in are the numbers of final and initial spin wave
excitations:
Nα,out =∫ 1
0α†out(ω)αout(ω) dω, Nα,in =
∫ 1
0α†in(ω)αin(ω) dω,
Nβ,out =∫ 1
0β†out(z)βout(z) dz, Nβ,in =
∫ 1
0β†in(z)βin(z) dz. (5.122)
The condition (5.121) fixes the combined transformation of signal field and spin
wave, implemented by the memory interaction, as unitary, meaning that the total
number of ‘particles’ is conserved. This unitarity holds in the Raman limit approx-
imately, since we have neglected spontaneous emission, which process would scatter
particles into modes other than the signal field or the spin wave, thus violating the
conservation law (5.121). As discussed in §5.3.1, this approximation is generally a
good one for a Raman memory, since large optical depths are required, making spon-
taneous emission losses negligible. The conservation condition (5.121) has the same
form as that for a beamsplitter, which mixes a pair of input modes without loss, to
produce a pair of output modes. Indeed, this is a helpful perspective from which to
view the action of the Raman memory (see Figure 5.6 below). The difference with
a conventional beamsplitter, as we will see, is that the Raman interaction couples
multiple modes together in a pairwise fashion, with each pair of modes ‘seeing’ a
different reflectivity. We derive this fact by combining the solutions for α and β into

5.3 Raman Storage 176
a single transformation,
|xout〉 = U |xin〉, (5.123)
where the vectors |xout〉 and |xin〉 are defined by
|xout〉 =
|αout〉
|βout〉
, |xin〉 =
|αin〉
|βin〉
, (5.124)
and where the matrix U is given by
U =
L −K
K L
. (5.125)
In this notation, the conservation condition (5.121) is written as
〈xout|xout〉 = 〈xin|xin〉. (5.126)
Substituting the transformation (5.123) into the above condition, we find that U †U =
I, where I is the identity matrix. Using this result, we multiply (5.123) by U † from

5.3 Raman Storage 177
the left, to obtain the inverse transformation
|xin〉 = U †|xout〉. (5.127)
Substituting this into the conservation condition, we then find UU † = I. So we see
that U is indeed a unitary transformation (see §A.4.5 in Appendix A). To see the
implications of this for the matrices K and L, we substitute the form (5.125) for U
into the conditions U †U = UU † = I, and perform the matrix multiplications. This
yields the conditions
L†L+K†K = LL† +KK† = I. (5.128)
In terms of the normally and antinormally ordered products, these conditions are
written as
LN +KN = I; LA +KA = I. (5.129)
Therefore we must have that LN and KN commute:
[LN ,KN ] = [LN , I − LN ] = [LN , I]− [LN , LN ] = 0. (5.130)
And similarly for the antinormally ordered products, [LA,KA] = 0. As discussed
in §3.1.2 of Chapter 3, the eigenvectors of KA are the output modes of K, and
the eigenvectors of KN are its input modes. That is, if we write the SVD of K as
K = UKDKV†K , then KA = UKD
2KU†K , and KN = VKD
2KV
†K . The fact that KA
commutes with LA implies that the eigenvectors of LA are the same as those of KA

5.3 Raman Storage 178
(see §A.3.1 in A). So we can write LA as
LA = UKD2LU†K , (5.131)
where D2L is a diagonal matrix of positive eigenvalues. The same argument applies
for the normally ordered products, so that we have
LN = VKD2LV†K . (5.132)
Note that the eigenvalues of LN are necessarily the same as those of LA. To sum-
marize, the unitarity of the memory interaction constrains the matrices K and L
such that their SVDs are built from a common set of input and output modes:
L = UKDLV†K K = UKDKV
†K . (5.133)
And substituting these expressions into the conditions 5.129, we find the following
relationship between the singular values,
D2L +D2
K = I. (5.134)
Our analysis is nearly complete. Let us review what we have discovered. A general
transformation from |xin〉 to |xout〉 could involve four Green’s functions — that is,
there could have been four matrices to deal with, one for each of the elements of
U in (5.125). But the symmetry of the equations of motion (5.109) allows us to

5.3 Raman Storage 179
express the transformation in terms of just two Green’s functions, L and K. Each
of these has an SVD, so there are potentially four sets of orthogonal modes to deal
with, associated with the input and output modes of the two Green’s functions L
and K. The further condition of unitarity on U , however, allows us to reduce the
number of orthogonal sets from four down to two, since L and K must share the
same input modes, and also the same output modes, as one another. We now show
that in fact the output modes are just flipped versions of the input modes, so that
the entire interaction can be fully described using just a single set of orthogonal
modes. The final step is accomplished by noticing that the matrices L and K are
persymmetric. That is, they are symmetric under reflection in their anti-diagonal
(see §3.1.4 in Chapter 3). This can be seen by examining the functional forms
(5.116) of the Green’s functions. For example, the value of L(x, y) only depends on
the difference x − y. Therefore the contours of L all lie parallel to the line y = x,
which corresponds to the main diagonal of the matrix L. L is accordingly unchanged
by a reflection in the main anti-diagonal, that is to say, L is persymmetric. That
K is also persymmetric follows from the fact that K(x, 1 − y) is Hermitian. As
shown in §3.1.4 in Chapter 3, the input and output modes associated with the SVD
of a persymmetric matrix are simply ‘flipped’ versions of one another. Putting this
last result together with our previous analysis, we can express the Green’s functions

5.3 Raman Storage 180
entirely in terms of a single set of modefunctions ϕj,
K(x, y) =∑j
ϕj(x)λjϕj(1− y),
L(x, y) =∑j
ϕj(x)µjϕj(1− y), (5.135)
where the singular values add in quadrature to unity,
µ2j + λ2
j = 1. (5.136)
The modes ϕj can be found from a numerical SVD of the kernel K. Or, equivalently,
they are given by the eigenvalue equation (5.96) introduced previously in §5.3.
The procedure used to connect the SVDs of K and L through the unitarity of
U is known as the Bloch-Messiah Reduction [147–149]. The resulting decomposition
makes the assertion that Raman storage may be understood by analogy with a
beamsplitter into a rigorous correspondence. If we define aj , (bj) as the annihilation
operator for a photon (spin wave excitation) in the jth input mode ϕj , and if Aj
(Bj) annihilates a photon (spin wave excitation) in the jth output mode, then for
each mode, the memory interaction can be written as
Aj = µjaj − λjbj ,
Bj = µjbj + λjaj . (5.137)
These relations are precisely those arising in the quantum mechanical description of

5.3 Raman Storage 181
an optical beamsplitter, coupling input modes aj , bj , to output modes Aj , Bj , with
reflectivity R = ηj = λ2j (see Figure 5.6). Optimal storage corresponds to the case
R = 1, so that the incident signal field is entirely ‘reflected’ into a spin wave mode.
Figure 5.6 Raman storage as a beamsplitter. Optical and spinwave modes are mixed pairwise by the storage interaction, as lightbeams are on a beamsplitter. The ideal quantum memory, with unitstorage efficiency, would see the beamsplitter replaced by a perfectmirror.
A possible use of a Raman quantum memory with non-unit storage efficiency
is as part of the modified DLCZ quantum repeater protocol described in §1.7 in
Chapter 1. Instead of using a 50 : 50 beamsplitter in combination with an ideal
quantum memory to generate number state entanglement, a single Raman quantum
memory with η1 = 50% can be used, as shown in Figure 5.7. The quality of the
entanglement generated relies on good overlap of the transmitted field modes on
the final beamsplitter, so the preceding theoretical characterization of the temporal
structure of these modes simplifies the analysis of this type of protocol.

5.4 Numerical Solution 182
BS
D1
D2
QML
QMR
SL
SR
Figure 5.7 Modified DLCZ protocol with partial storage. Whena single detector fires behind the final beamsplitter, number stateentanglement is generated between the memories. The protocol isexplained in §1.7 in Chapter 1. The only difference is that here,a single memory with 50% efficiency replaces the combination of abeamsplitter and an ideal memory.
5.4 Numerical Solution
So far we have succeeded in deriving a form for the storage kernel in the adiabatic
limit, or for arbitrary bandwidths in the case of a hyperbolic secant control. The
general problem of finding the storage kernel for both arbitrary bandwidths and
arbitrary control pulse profiles has not been solved analytically. But it is possible
to construct the kernel numerically. This can be done by integrating the system
of coupled equations (5.106) multiple times, each time with a different boundary
condition. Provided the boundary conditions form a set of orthogonal functions, the
Green’s function can be reconstructed. The easiest set of boundary conditions to
implement is the set of ‘impulses’ — delta functions. To see why this works, recall

5.4 Numerical Solution 183
the definition of the storage kernel,
Bout(z) =∫ ∞−∞
K(z, τ)Ain(τ) dτ, (5.138)
where we used normalized units for z. Now, if we insert a delta function δ(τ − τj)
as the signal field boundary condition Ain(τ), where τj is some particular time slot,
the resulting spin wave Bout,j is
Bout,j(z) = K(z, τj). (5.139)
We can therefore reconstruct an approximation to the entire Green’s function by
numerically solving for Bout,j repeatedly, with the times τj chosen from a finite
grid. The grid should range over a sufficient range of times that all of the Green’s
function is sampled, and we should make the grid sufficiently fine that no features
of the Green’s function are missed. So long as these requirements can be met while
keeping the computation reasonably fast, this is a convenient way to find the storage
kernel without requiring the adiabatic approximation, and without imposing any
restriction on the temporal profile of the control field. This method was previously
used to reconstruct the adiabatic Green’s functions describing stimulated Stokes
scattering in a dispersive ensemble by Wasilewski and Raymer [148]. In this thesis
we solve the equations of motion numerically using Chebyshev spectral collocation
for the spatial derivatives, and a second order Runge-Kutta (RK2) method for the
time stepping. The method of solution is explained in detail in Appendix E.

5.4 Numerical Solution 184
In the figures below we compare the optimal input modes predicted by the various
methods presented in this chapter. In Figures 5.8 and 5.9 we plot, side by side, the
optimal input modes found from numerically constructed Green’s functions, the
Rosen-Zener kernel (5.60), the adiabatic kernel (5.77) and the Raman kernel (5.95).
In Figure 5.8 the adiabatic approximation is well satisfied; the Raman approxi-
mation only poorly so, and therefore there is good agreement between all predictions,
save for the Raman prediction, which deviates slightly. But note that the adiabatic
and Raman kernels do slightly overestimate the phase due to the dynamic Stark-
shift.
−5 0 50
0.5
1Rosen Zener
−5 0 5
Numerical
−5 0 5
Adiabatic
−5 0 5
Raman
0
2
4
6
Inte
nsity
(arb
. u
nits)
Ph
ase
Time
Figure 5.8 Comparison of predictions for the optimal input modesin the adiabatic limit. Here we used a hyperbolic secant control, givenby (5.38) with Tc = 1 and Ω0 = Ωmax = 3, an optical depth of d = 300and a detuning ∆ = 15, in normalized units. The control intensityprofile |Ω(τ)|2 is indicated by the dotted lines, scaled for clarity. Theblue lines show the predicted optimal intensity profiles |Ain(τ)|2 =|φ1(τ)|2, and the red lines show the variation of the temporal phaseof the mode φ1 in radians, referred to the axes on the right handside. The storage efficiency is ∼ 90% in all cases, and the predictionsare in good agreement generally. But both the adiabatic and Ramankernels overestimate the phase shift due to the dynamic Stark shift.And the detuning is not quite large enough to render the Ramankernel correct.
In Figure 5.9 the fields are tuned into resonance, the optical depth is reduced,

5.4 Numerical Solution 185
and the control intensity is increased, so that the adiabatic approximation is no
longer satisfied. There is rough agreement between the numerical and Rosen-Zener
predictions, which do not rely on the adiabatic approximation, although there is
a slight discrepancy that we attribute to the accumulation of numerical errors in
the evaluation of the Hypergeometric functions in the Rosen-Zener kernel (5.60).
The optimal modes predicted by these two methods exhibit oscillations, as can be
seen from the phase jumps indicating sign changes. These oscillations are missing
from the predictions of the adiabatic kernel; the prediction of the Raman kernel is
catastrophically wrong, as we would expect on resonance.
−5 0 50
0.5
1Rosen Zener
−5 0 5
Numerical
Time
−5 0 5
Adiabatic
−5 0 5
Raman
0
2
4
6
Inte
nsity
(arb
. u
nits)
Ph
ase
Figure 5.9 Comparison of predictions for the optimal input modesoutside the adiabatic limit. Again, the control takes the form of ahyperbolic secant, this time with Tc = 1 and Ω0 = Ωmax = 5. Wereduce the optical depth down to d = 10, and we tune into resonance,putting ∆ = 0. The numerical and Rosen-Zener predictions roughlycoincide, and both the predictions exhibit oscillations characteristic ofnon-adiabatic ‘ringing’. Numerical errors in the Rosen-Zener kernelamplify the size of these oscillations slightly. The adiabatic kerneldoes not correctly reproduce the oscillations, while the Raman kernelis totally inappropriate for modelling this resonant interaction. Theoptimized storage efficiency predicted by the numerics is around 82%.
The numerically constructed Green’s functions yield the most reliable predic-
tions. In addition, they take less time to calculate then the Rosen-Zener predictions

5.4 Numerical Solution 186
— the Matlab code for the numerical method runs in ∼ 40 s on a 3 GHz machine;
the Rosen-Zener kernel takes several hours to construct, simply because the hyperge-
ometric functions are so difficult to evaluate efficiently. Also, the numerical method
is more flexible, since arbitrary control profiles can be used. In general, then, the
direct numerical approach is the method of choice for optimizing a Λ-type quantum
memory.
In the adiabatic and Raman limits, however, it is faster to use the analytic
kernels derived using these approximations. And of course, the optimization can
then be trivially generalized to arbitrary control pulse shapes using the transforma-
tions (5.78) and (5.97). In Figures 5.10 and 5.11 below we compare the numerical
predictions with those of the adiabatic and Raman kernels in the resonant and
Raman limits, using much more broadband control pulses. As expected, there is
good agreement between all three predictions in the Raman limit. In the resonant
case, describing EIT storage, the Raman kernel of course fails completely, but the
adiabatic kernel remains reasonably reliable. That said, its agreement with the nu-
merical prediction is not as good as in the Raman limit, which is symptomatic of
the fragility of the adiabatic approximation on resonance.
When far into the Raman limit, the numerical, adiabatic and Raman methods
all predict a significant temporal phase variation due to the dynamic Stark shift.
Whereas in the EIT case, the optimal input mode has a flat phase. Depending on
the flexibility of the technology used to shape the optical pulses before storage, it
may be easier to implement EIT storage for this reason. On the other hand, Raman

5.4 Numerical Solution 187
−0.2 0 0.20
0.5
1Numerical
−0.2 0 0.2
Raman
0
2
4
6
−0.2 0 0.2
Adiabatic
Time
Inte
nsity
(arb
. u
nits)
Ph
ase
Figure 5.10 Broadband Raman storage. Here the control is aGaussian pulse Ω(τ) = Ωmaxe
−(τ/Tc)2, where Tc = 0.1 and W =
302.5, so that Ωmax = (2/π)1/4√W/Tc = 49.1. The optical depth is
d = 300, and the detuning is ∆ = 150. As usual all these quantitiesare in normalized units. These parameters give a Raman memorycoupling of C = 2.0 and a balanced interaction with R = 1.0. Notethat the control pulse duration is roughly one tenth of the sponta-neous emission lifetime of the excited state |2〉. Nonetheless, due tothe large detuning, the adiabatic and Raman approximations are wellsatisfied, and the agreement between the numerical and analytic pre-dictions is clear. The optimized storage efficiency is predicted by allthe methods shown to be ∼ 98%.
storage allows for more freedom in the carrier frequencies of the signal and control
fields.
5.4.1 Dispersion
All of the optimal modes shown in the figures above are slightly delayed in time
with respect to the control pulse. This is due to the characteristic dispersion associ-
ated with an absorption process, which produces a superluminal group velocity. Of
course, there is no question of violating causality, it is simply that as the trailing
edge of the signal field is absorbed, the ‘centre of mass’ of the signal pulse advances,
giving the appearance of superluminal propagation. The efficiency of the memory

5.5 Summary 188
−0.2 0 0.20
0.5
1Numerical
−0.2 0 0.2
Adiabatic
−0.2 0 0.2
Raman
Time
Inte
nsity
(arb
. u
nits)
Ph
ase
0
2
4
6
Figure 5.11 Broadband EIT storage. All parameters are the sameas above in Figure 5.10, except that the storage is performed onresonance, with ∆ = 0. The Raman kernel fails utterly, but theadiabatic prediction fairs better, comparing well with the numericalprediction. That the agreement is poorer on resonance is a signaturethat the adiabatic approximation is less robust on resonance. Theoptimized storage efficiency predicted by the numerical method is∼ 99%.
is maximized by ‘pre-compensating’ for this effect, and this explains the time shift
common to the optimal input modes. In the case of resonant storage, this view con-
flicts with our characterization of EIT in §2.3.1 of Chapter 1 as working by slowing
the group velocity of the signal. Perhaps a better perspective is therefore that on
resonance the control must precede the signal in order to ‘prepare’ the transparency
window.
5.5 Summary
We have covered rather a lot of material in this chapter. Here we review the main
results.
1. The analysis of the storage process is greatly simplified if we use a one di-

5.5 Summary 189
mensional propagation model. All the results listed below make use of this
model.
2. The best possible storage efficiency is limited by the optical depth, being given
by η = 1− 2.9/d.
3. The method of Rosen and Zener yields an analytic expression for the storage
kernel for a hyperbolic secant control. It is, however, difficult to evaluate this
efficiently and accurately (at least using Matlab).
4. In the adiabatic approximation, an analytic expression for the storage kernel
can be derived that holds for all control pulse profiles. This is quick to evaluate.
5. When far detuned, the expression further simplifies, yielding the Raman kernel,
which only depends on the Raman memory coupling C. The Raman memory
can be decomposed as a set of beamsplitter transformations between light and
matter, using just a single set of modefunctions.
6. If none of the above methods are appropriate, the storage kernel can be directly
constructed by repeated numerical integration of the equations of motion for
the memory. This method yields the correct optimal input mode for arbitrary
control profiles and detunings, for all values of the optical depth.
In the next chapter, we consider retrieval from a Λ-type memory.

Chapter 6
Retrieval
So far we have considered the optimization of storage in a Λ-type quantum mem-
ory. In some circumstances this optimization is sufficient to maximize the combined
efficiency of storage into, followed by retrieval from the memory. Specifically, this is
true when the retrieval process is the time reverse of the storage process [133]. When
this is not the case, the optimization of this combined efficiency is a distinct prob-
lem. In this Chapter, we discuss various strategies for retrieval, and we present the
results of a numerical analysis of their effectiveness.
6.1 Collinear Retrieval
In order to convert the stored excitation back into a propagating optical signal, we
send a second control pulse — a read out pulse — into the ensemble. Since the
control mediates coupling between the signal field and the spin wave, the rationale
is that the spin wave excitations will transfer back to the optical field, emerging

6.1 Collinear Retrieval 191
as a collimated pulse traveling in a well-defined direction. We have met with some
success in analyzing the storage process using a one dimensional propagation model,
and so it is natural to consider read out from this perspective. We will see later
that certain advantages accrue if a small angle is introduced between the signal
and control fields. For the moment, suppose that we have stored a signal photon
collinearly, as described in the previous chapter. Confining ourselves to the same
one dimensional model, there are two possible read out geometries: forward, and
backward retrieval.
6.1.1 Forward Retrieval
We can describe forward retrieval using the same equations of motion as we used
for storage — (5.3), or (5.106) in normalized units. For the retrieval process, the
signal field boundary condition is set to zero, and the spin wave boundary condition
is set by the coherence generated in the medium during the storage process. Let us
denote quantities associated with the retrieval process with a superscript r. In this
notation the boundary conditions at read out are
Arin(τ r) = 0, Br
in(z) = Bout(z). (6.1)
The second condition assumes that there is no loss of coherence during the time that
the excitations are stored. This kind of loss is homogeneous in space, and so it would
appear only as a constant factor reducing the amplitude of Brin. The optimization
of the memory is therefore unaffected by the neglect of decoherence.

6.1 Collinear Retrieval 192
The first step in analyzing the efficiency of the retrieval process is to examine
the form of the map from the stored excitations to the retrieved signal field,
Arout(τ
r) =∫ 1
0Kr(τ r, z)Br
in(z) dz. (6.2)
In fact, in the adiabatic limit, the retrieval kernel Kr is identical in form to the
storage kernel K, when expressed in terms of the integrated Rabi frequency ωr,
rather than the time τ r. This is guaranteed by the symmetrical form of the equations
of motion in the adiabatic limit, which is discussed in §5.3.3 in Chapter 5. For
completeness, we verify this by explicit calculation. In the notation of §5.2.5 in
Chapter 5, the retrieved signal field is expressed in k-space as
Ar(k, τ r) = −i
√d√
2πk〈↑ |V r−1(k, τ r)V r
in(k)| ↓〉Brin(k). (6.3)
Using V rin = I, along with the adiabatic approximations (5.70) and (5.73), we obtain
Ar(k, τ r) = − 1√2π
√d
ΓrΩr(τ r) exp
[− 1
Γr
∫ τ r
−∞|Ωr(τ ′)|2 dτ ′
]× 1k + i dΓr
exp
[id
Γr2
∫ τ r
−∞ |Ωr(τ ′)|2 dτ ′
k + i dΓr
]Br
in(k). (6.4)
Here Ωr is the Rabi frequency describing the temporal profile of the read out control
pulse, which may differ from that of the storage pulse, and Γr = γ − i∆r allows for
the possibility that the detuning is changed for the readout process. We transform
the time coordinate from τ r to ωr, and take the inverse Fourier transform from k

6.1 Collinear Retrieval 193
to z-space. Using the result (D.40), along with the shift and convolution theorems
(D.18), (D.26) in Appendix D, we find that the retrieval kernel is given by
Kr(τ r, z) =Ωr∗(τ r)√W r
×Kr [ωr(τ r), z] , (6.5)
where
Kr(ωr, z) = i
√dW r
Γre−[W rωr+d(1−z)]/Γr
J0
[2i√dW r
Γr
√ωr(1− z)
]. (6.6)
This kernel has precisely the same form as (5.76), except that ω has been switched
for z, and z has been switched for ωr. Now we can write down the adiabatic map
describing the combined processes of storage followed by forward retrieval,
Arout(τ
r) =∫ ∞−∞
Ktotal(τ r, τ)Ain(τ) dτ. (6.7)
The kernel Ktotal describes the entire memory interaction (see §3.4 in Chapter 3),
and is given by
Ktotal(τ r, τ) =Ωr∗(τ r)Ω(τ)√
W rW×Ktotal [ωr(τ r), ω(τ)] , (6.8)
where
Ktotal (ωr, ω) =∫ 1
0Kr(ωr, z)K(z, ω)dz. (6.9)
Here the kernel K is the adiabatic storage kernel given in (5.76) in Chapter 5, and
Kr, given in (6.6), is related to K by the symmetry connecting the storage and

6.1 Collinear Retrieval 194
retrieval kernels just derived above.
The input mode that optimizes the total memory efficiency is found from the
SVD of Ktotal in (6.9). The kernel looks superficially similar in form to the normally
ordered kernel KN formed from the storage kernel K. But Ktotal 6= KN because
Kr 6= K∗. If these two kernels were equal to one another, then the optimal input
mode found from the SVD of Ktotal would be equal to the mode found from the
SVD of K; this follows from the properties of KN (see §3.3.1 in Chapter 3). Since
these two kernels are not equal to each other, the optimization of storage followed
by forward retrieval is different to the optimization of storage alone.
Note that the optimal input mode does not depend on the shape Ωr of the read
out control; it only depends on the total energy in the read out pulse, parameterized
by W r. But changing the shape of Ωr changes the temporal profile of the signal field
retrieved from the memory. This is useful: one can optimize the memory efficiency
once, for a fixed energy W r, and then changing the shape of Ωr allows one to produce
an output signal pulse of any shape, within the limits of the adiabatic approximation.
Novikova et al. have already demonstrated optimal storage, followed by shaped
retrieval, experimentally, on resonance [74,150,151], using the theory of Gorshkov et
al. [133], which applies in the light-biased limit (see the end of §5.2.8 in Chapter 5).
In Figure 6.1, we show an example of how the optimal input mode for storage,
followed by retrieval, differs from the optimal storage mode. We used the adiabatic
kernel (5.76) to model the storage interaction, and we evaluated the expression (6.9)
for Ktotal numerically. This is easily done by discretizing the coordinates so that K

6.1 Collinear Retrieval 195
becomes a matrix. We use the same control pulse and detuning for both storage
and readout, Ω = Ωr and ∆ = ∆r, and then Ktotal is given by the product of
two copies the matrix K. Taking the SVD of the result yields a radically different
optimal input mode to that found from the SVD of K alone. The difference can be
understood by considering the shape, in space, of the spin wave Bout(z) produced by
the storage interaction. When optimizing storage alone, this is given by the optimal
output mode ψ1(z) found from the SVD of the storage kernel K. The shape of this
mode generally takes the form of a decaying exponential, as shown in part (b) of
Figure 6.1. This shape is consistent with Beer’s law absorption: as the signal pulse
propagates through the ensemble it is increasingly likely to be absorbed, so that at
the exit face, at z = 1, there is very little probability for the signal pulse to excite
an atom. Therefore the spin wave decays in magnitude steadily from the input to
the exit face. But forward retrieval from a spin wave of this shape is problematic:
if the amplitude of the spin wave is concentrated close to the entrance face of the
ensemble, a retrieved excitation, that has been converted into a signal photon, must
propagate a large distance through the ensemble before reaching the exit face. There
is therefore a high probability that the retrieved photon will be re-absorbed, so that
it never emerges from the ensemble, and this greatly reduces the efficiency of the
read out process. For forward retrieval, it is much better that the bulk of the spin
wave is concentrated towards the exit face of the ensemble, so that re-absorption
losses are minimized. In fact, the optimal spin wave mode for retrieval is precisely
the space-reverse of the mode generated by the storage process; this can be derived

6.1 Collinear Retrieval 196
from the symmetry relating the storage and retrieval kernels. It is the fact that the
optimal shape of the spin wave for retrieval is improperly matched to the spin wave
mode generated by optimal storage that complicates the combined optimization of
storage followed by retrieval. A compromise between these two shapes must be
found. Such a compromise is shown in part (d) of Figure 6.1: the amplitude of
the spin wave at z = 1 is much larger than for the spin wave that results from only
optimizing storage. And there is a marked suppression of the spin wave amplitude at
z = 0, since even though the storage interaction naturally excites atoms close to the
entrance face, it is extremely deleterious to the retrieval efficiency to concentrate the
stored excitations there. If we could run the storage process backwards, so that the
retrieval process was precisely the time-reverse of the storage process, we should be
able to achieve a retrieval efficiency equal to the storage efficiency, ηretrieve = ηstorage.
It should then be possible to achieve a combined efficiency of storage followed be
retrieval of ηcombined = ηstorage × ηretrieval = η2storage. However, even after optimizing
the combined efficiency, it still falls far short of this optimum, ηcombined η2storage.
In the example shown in Figure 6.1, ηcombined = 28%, whereas η2storage = 80%. The
tension between Beer’s law during storage and re-absorption during retrieval makes
forward retrieval generally inefficient. In the next section we consider backward
retrieval, which performs better.

6.2 Backward Retrieval 197
0
0.5
1Spin waveInput mode
(a) (b)
(d)(c)In
ten
sity
(arb
. u
nits)
Inte
nsity
(arb
. u
nits)
Pha
se
Pha
se
0
2
4
6
−2 0 20
0.5
1
0 0.5 10
2
4
6
Figure 6.1 Forward retrieval. We consider collinear storage, fol-lowed by forward retrieval, using a Gaussian control pulse Ω(τ) =Ωmaxe
−τ2, with W = 9, so that Ωmax = (2/π)1/4
√W = 2.68. The
optical depth is d = 300, and the detuning is ∆ = 15, in normalizedunits. (a) shows the input mode that optimizes just the storage ef-ficiency, and (c) shows the input mode that optimizes the combinedefficiency of storage followed by retrieval. The shape of the controlintensity profile is indicated by the black dotted lines. The formeroptimization gives a storage efficiency of ∼ 90%, whereas the latteroptimization gives a combined efficiency of only 28%. In parts (b) and(d) we show the ‘intensity’ |Bout(z)|2 and spatial phase of the spinwave generated in the ensemble after the storage process is complete,for the storage and combined optimizations, respectively. The com-bined optimization produces a spin wave that reduces re-absorptionlosses by shifting the ‘centre of mass’ of the spin wave away from theentrance face at z = 0.
6.2 Backward Retrieval
The arguments given in the previous section suggest that retrieving the signal field
backwards should be much more efficient than forward retrieval. If the storage in-
teraction produces a spin wave with its amplitude concentrated toward the entrance
face of the ensemble, re-absorption losses are minimized if retrieved signal photons
propagate backwards to re-emerge from the entrance face. This is indeed the case,
but issues of momentum conservation arise if the energy splitting between the ground

6.2 Backward Retrieval 198
and storage states |1〉 and |3〉 is non-zero. To see why, consider the diagram shown
in part (a) of Figure 6.2. Momentum conservation requires that the wavevectors
ks, kc and κ associated with the signal, control and spin wave, respectively, sum
to zero. When this is the case, the storage interaction is said to be phasematched,
since the spatial phases accumulated by the optical fields as they propagate through
the ensemble are ‘matched’ to the spatial phase of the spin wave. This means that
the slowly varying envelopes of these fields are strongly coupled to the spin wave,
as described by the equations of motion for the quantum memory (see (5.106) in
Chapter 5, for instance). As we will see below, when momentum is not conserved,
the interaction is not phasematched, and destructive interference greatly reduces the
strength of the coupling, and with it the memory efficiency. Fortunately, the storage
process is always phasematched, because there is no fixed dynamical relationship be-
tween the spatial phase of the spin wave and its energy. This is because the atoms
comprising the spin wave do not interact with another, so that there is no coupling
at all between the spatial shape of the spin wave and the frequency splitting between
the ground and storage states. In the storage process, a signal photon is absorbed,
and a control photon is emitted. The spin wave therefore acquires a wavevector that
‘takes up the slack’, given by the difference of the signal and control wavevectors,
κ = ks − kc =ωs − ωc
cz, (6.10)

6.2 Backward Retrieval 199
where z is a unit vector pointing along the positive z axis. In the last equality we
used the optical dispersion relation for a plane wave,
k = ω/c, (6.11)
where k is the magnitude of the wavevector and ω is the angular frequency of
the wave (not the integrated Rabi frequency!). This relation fixes the momentum
associated with the spin wave as given by the difference between the signal and
control field frequencies, which is in turn fixed by the energy splitting that separates
the ground and storage states, in order to satisfy two-photon resonance. Even though
there is no intrinsic connection between the spatial phase of the spin wave and its
energy, the kinematics of the scattering process that generates the spin wave does, in
fact, tie these two quantities together. The efficiency of the retrieval process depends
critically on whether it is possible to phasematch the retrieval interaction. When we
attempt to retrieve the spin wave excitations by sending the readout control pulse
in the backward direction, momentum conservation requires that the wavevector of
the retrieved signal field is formed from the sum of the spin wave and the read out
control wavevectors,
krs = κ+ kr
c. (6.12)
Substituting (6.10) into (6.12), we obtain
krs − kr
c = ks − kc. (6.13)

6.2 Backward Retrieval 200
For collinear storage, we have ks = ksz and kc = kcz. For backward retrieval, we
have krs = −kr
sz and krc = −kr
cz. Inserting these expressions into (6.13), and using
the optical dispersion relation (6.11) along with the two-photon resonance condition,
we get to the condition
ωc − ωsc
=ωs − ωc
c. (6.14)
As is clear from part (b) of Figure 6.2, this condition can only be satisfied when
ωs = ωc, and κ = 0, which requires that the ground and storage states are degenerate
in energy. Experimentally, it is important to be able to distinguish the weak signal
field from the bright control pulse, and the ability to spectrally filter these two fields
should not be surrendered lightly. Therefore it is very useful to consider storage in
memories where ωs 6= ωc, and then the issue of momentum conservation becomes
important when considering backward retrieval.
(a) (b)
Figure 6.2 Phasematching considerations for backward retrieval.(a) The momenta of the control field and spin wave must sum tothat of the signal field, from which they are ‘scattered’. The storageprocess is automatically phasematched, since the magnitude of κ isinitially a free parameter, that is determined by ks and kc duringstorage. (b) The spin wave momentum is pointing in the ‘wrongdirection’, when backward retrieval is attempted: it is not possible tosimultaneously satisfy the two-photon resonance condition kr
s − krc =
(ωs − ωc)/c, and the phasematching condition krs = κ+ kr
c.
In order to optimize the combined efficiency of storage, followed by backward

6.2 Backward Retrieval 201
retrieval, we need to find an expression for Ktotal that describes the entire memory
interaction in this case. The SVD of this kernel will then provide us with the optimal
input mode. The equations of motion for the retrieval process have the same form
as those describing storage, but they must describe propagation in the backward
direction, with the z coordinate is reversed. The kernel Ktotal is simply constructed
using the adiabatic solutions for storage and retrieval separately, provided we are
careful about how the boundary conditions are ‘stitched together’. Let us denote the
flipped z coordinate for the retrieval process by zr, so that zr = L − z, in ordinary
units, where z is the coordinate describing propagation during storage. The atomic
coherence at the start of the retrieval process is identical — as always assuming no
decoherence — to that generated by the storage process. We can write this as
σr13,in(zr) = σ13,out(z), (6.15)
where the σ13(x) denotes the Raman coherence associated with an atom located at
the longitudinal position x. The spin wave B, defined in (4.43), makes use of the
slowly varying operators σ13, introduced in (4.22) (see Chapter 4), which incorporate
an exponential factor eiω13τ . Recall that τ is in fact the retarded time, τ = t− z/c,
so there is a spatial phase built into the spin wave; this represents the momentum
imparted to the spin wave by the optical fields that create it during the storage

6.2 Backward Retrieval 202
process. Equating the boundary conditions, as in (6.15), gives the relation
Brin(zr) = Bout(z)× e−iω13τout × eiω13τ r
in ,
= Bout(z)× e−iω13(trin−tout) × e−iω13(z−zr)/c,
= Bout(L− zr)e2iω13zr/c. (6.16)
Here we used the definition τ r = tr − zr/c = tr − L/c + z/c, and in the last line
we dropped some unimportant constant phase factors. The spatial phase factor in
(6.16) represents the phase mismatch shown in Figure 6.2 (b). Note that it vanishes
if ω13 = 0. The phase mismatch causes oscillations of the spin wave in space that
can dramatically reduce the retrieval efficiency. This can be seen by considering the
form of the retrieval map
Arout(τ
r) =∫ 1
0Kr(τ, zr)Br
in(zr) dzr
=∫ 1
0Kr(τ r, zr)eiδkzr
Bout(1− zr) dzr. (6.17)
Here we have switched to normalized units, and defined the dimensionless phase
mismatch
δk =2ω13L
c. (6.18)
Note that δk is a negative quantity if the storage state |3〉 is more energetic than the
ground state |1〉. It is clear that, regardless of the form of the retrieval kernel Kr, or
of the spin wave Bout, the integral can be made to vanish if δk is made large enough.

6.2 Backward Retrieval 203
This explains why a phase mismatch can be very detrimental to the efficiency of
backward retrieval. More precisely, if kmax represents the width in k-space of the
integrand Kr × Bout, the value of the integral becomes strongly suppressed when
δk kmax. It is therefore possible to mitigate the effect of the phase mismatch,
to some extent, by localizing the spin wave strongly in space, so that kmax becomes
large. Below, we show explicitly how to optimize storage, followed by backward
retrieval in the presence of a phase mismatch; the result essentially generates a more
strongly localized spin wave, for exactly this reason.
To perform the optimization, we construct the kernel Ktotal using the solution
(6.4) for the retrieval process, making the replacement z −→ zr. The result is
Arout(τ
r) =∫ ∞−∞
Ktotal(τ r, τ)Ain(τ) dτ, (6.19)
with
Ktotal(τ r, τ) =Ωr∗(τ r)Ω(τ)√
W rW×Ktotal [ωr(τ r), ω(τ)] , (6.20)
and where
Ktotal (ωr, ω) =∫ 1
0Kr(ωr, zr)K(1− zr, ω)eiδkzr
dzr. (6.21)
Suppose that the readout control pulse is identical in shape and frequency to the
storage control pulse, with Ωr = Ω and ∆r = ∆. Suppose also that the storage state
is degenerate with the ground state, so δk = 0. Then consider the kernel Ktotal in

6.2 Backward Retrieval 204
(6.21) with its output time argument flipped around,
Ktotal(1− ωr, ω) =∫ 1
0Kr(1− ωr, zr)K(1− zr, ω) dzr
=∫ 1
0K(1− zr, ωr)K(1− zr, ω) dzr, (6.22)
The second line follows from the symmetry Kr(1−x, y) = K(1−y, x) that connects
the retrieval kernel (6.6) to the storage kernel (5.76). This shows that Ktotal is,
in the case under consideration, very similar in structure to the normally ordered
kernel KN that would be formed from K(1 − zr, ω). The only difference is that
the first instance of K in (6.22) is replaced by its complex conjugate in KN . If K
is real, then this complex conjugation is not important, and Ktotal = KN , which
means that the optimal input modes derived from the SVD of Ktotal are equal to
the optimal input modes derived from the SVD of K alone. This observation shows
that the optimal input mode for storage alone is close to the optimal input mode
for storage followed by backward retrieval, if identical storage and readout control
pulses are used, and if there is no phase mismatch. The two optimizations are the
same whenever K is real, and more generally whenever ψ1(z), the output spin wave
mode generated by optimal storage, is real.
In Figure 6.3 below we show an example of how the optimal input mode for
storage alone can differ from the optimal mode for storage followed by backward
retrieval. Without a phase mismatch (parts (c) and (d)), the difference between the
two optimizations is rather small, though non-negligible, for the example shown.

6.2 Backward Retrieval 205
When a significant phase mismatch is introduced (parts (e) and (f)) the optimiza-
tions differ more markedly. It is a general feature of the optimization of storage
with backward retrieval, that as |δk| is increased, the shape of the optimal input
mode approaches that of the control pulse. The reason is as follows. Recall that
the dispersion experienced by the signal field as it propagates causes its group ve-
locity to differ from that of the control (see the end of §5.4 in Chapter 5). If the
signal initially overlaps with the control, then the coupling between these two fields
is initially high, but as the signal walks off from the control, the coupling decays
away. The bulk of the storage interaction therefore occurs near the entrance face
of the ensemble at z = 0. As is clear from part (f) of Figure 6.3, the spin wave
generated by the optimal input mode is indeed concentrated more closely towards
the entrance face. This makes the spin wave more strongly localized, which reduces
the ‘wash-out’ caused by the phase mismatch, as discussed above, while at the same
time reducing re-absorption losses.
A discussion of similar optimizations can be found in the work of Gorshkov
et al. [133]. In this work time-reversal arguments are employed to show that for
optimized storage with backward retrieval, ηcombined = η2storage if ψ1(z) is real. This
condition is always satisfied in the light-biased limit (see the end of §5.2.8 in Chapter
5), when the anti-normally ordered kernel KA becomes real, independent of whether
or not the adiabatic approximation is satisfied (see (5.27 in Chapter 5)). As shown
above in the adiabatic limit, when ψ1 is not real, implementing ‘true’ time reversal
requires that the phase of the spin wave is conjugated. Without a practical method

6.2 Backward Retrieval 206
0246
0
0.5
1
0
5
10
0
0.5
1
0246
0246
!2 0 20
0.5
1
02468
0 0.5 10246
Spin waveInput mode
Inte
nsity
(arb
. unit
s)In
tens
ity(a
rb. u
nits)
Inte
nsity
(arb
. unit
s)Phase
PhasePhase
(a) (b)
(d)(c)
(f)(e)
Figure 6.3 Backward Retrieval. We consider collinear storage fol-lowed by backward retrieval, using a Gaussian control pulse Ω(τ) =Ωmaxe
−(τ/Tc)2, with Tc = 0.1 and W = 100, so that Ωmax =
(2/π)1/4√W/Tc = 28.25. The optical depth is d = 300, and the
detuning is ∆ = 150, in normalized units. Parts (a) and (b) showthe optimal input mode for storage alone, alongside the spin wavemode ψ1(z) generated in the medium. We use the adiabatic formfor the storage kernel; comparison with the prediction derived fromthe numerically constructed kernel verifies that the adiabatic approx-imation is well satisfied. The optimal efficiency for storage alone isηstorage ∼ 96%. In parts (c) and (d) we show the optimal input modefor storage followed by backward retrieval, and the generated spinwave, with degenerate ground and storage states, δk = 0. The opti-mized total efficiency is ∼ 88%, which is about 5% less than η2
storage.The most notable difference between these two optimizations is theappearance of a ‘wiggle’ in the phase of the input mode for the com-bined optimization. But this occurs while the signal intensity is ratherlow, so it is actually less important than the more subtle re-shapingthat occurs. In parts (e) and (f), we show the optimal input modefor storage and backward retrieval, along with the generated spinwave, with a phase mismatch δk = −5. Note that this optimizationresults in a spin wave with its ‘centre of mass’ concentrated moreclosely towards the entrance face of the ensemble at z = 0 than theother optimizations. This reduces the effective length of the ensemble— the length over which there is significant excitation — which di-minishes the effect of the phase mismatch. The optimized combinedefficiency of storage and retrieval is around 75% in this case.

6.3 Phasematched Retrieval 207
for doing this, ηcombined < η2storage, and the optimal mode for storage alone differs
from the mode that optimizes the combination of storage and backward retrieval.
In this and the previous section we have shown that retrieving the stored excita-
tions from a quantum memory efficiently is a non-trivial problem. Forward retrieval
is plagued by re-absorption losses — a modematching issue. Backward retrieval is
beset by momentum conservation problems — a phasematching issue. In the next
section we present a solution to both of these problems, which requires a departure
from the one dimensional treatment we have worked with thus far.
6.3 Phasematched Retrieval
The best memory efficiency possible is achieved by optimizing collinear storage,
followed by backward retrieval, with δk = 0. But to spectrally filter the signal field
from the control, we should have δk 6= 0. By introducing a small angle between the
signal and control beams, both at storage and at read out, we can maintain proper
phasematching, even when δk 1, which allows for efficient retrieval. This idea
was proposed by Karl Surmacz, and the following treatment is adapted from our
paper [152]; the numerical simulations were performed by me. The principle of the
scheme is easily understood by considering the phasematching diagrams in Figure
6.4. Here we assume for simplicity that all the beams used, both at storage and at
read out, are confined to a common plane, so that we need only consider two space
dimensions. The spin wave momentum is determined by the signal and control field
wavevectors according to (6.10). If we fix the detuning, so that ∆r = ∆, then

6.3 Phasematched Retrieval 208
the magnitudes of ks and kc are unchanged at read out, and then the condition
(6.12) uniquely defines the direction of krs that is phasematched. By symmetry, the
angle θ between the signal and control beams is the same at read in as at read out.
Provided we adhere to the geometry shown in Figure 6.4 (a), the retrieval process
remains correctly phasematched for all choices of θ. To maximize the efficiency of
the memory, we should reduce θ as far as is possible, so that
1. we approach a collinear geometry, which maximizes the overlap of the signal
and control pulses, and
2. the spin wave generated by the storage process overlaps well with the optimal
spin wave mode for retrieval.
An heuristic choice of θ that satisfies these desiderata is that shown in part (b)
of Figure 6.4, with ks cos θ = kc. The signal and control wavevectors are close to
parallel, but they are arranged so that the spin wave momentum κ is orthogonal
to kc. This way, when the control field direction is reversed for backward retrieval,
no phase mismatch is introduced and the signal field emerges at an angle θ with
respect to the read out control pulse, as shown in part (a) of Figure 6.5. This choice
for θ assumes that the storage state is more energetic that the ground state, so
that δk is negative, and ks > kc. We also consider the possibility that the storage
state lies energetically below the ground state. In many systems the ground state is
prepared artificially by optical pumping (see §10.12 in Chapter 10), and it is then
quite feasible to select the ground state so that ω1 > ω3. In this case, δk > 0 and

6.3 Phasematched Retrieval 209
ks < kc. We then choose θ so that kc cos θ = ks; the geometry is shown in part (b)
of Figure 6.5.
(a) (b)
Figure 6.4 Non-collinear phasematching. (a) the general geometryrequired to phasematch storage and retrieval, when the signal beammakes an angle θ with the control. (b) a geometry that closely ap-proaches the collinear one, while preserving correct phasematching.In the next section we include dispersion, which modifies the lengthof ks and kr
s.
Storage
(a)
(b)
Retrieval
Figure 6.5 Efficient, phasematched memory for positive and neg-ative phase mismatches. We show the beam directions of the con-trol (green) and signal (blue) fields at storage and retrieval, with (a)δk < 0, and (b) δk > 0. Off-resonance, the angles used depend onboth δk and on the material dispersion (see §6.3.1 below).

6.3 Phasematched Retrieval 210
6.3.1 Dispersion
So far we have used the dispersion relation (6.11), which applies to light propagation
in vacuo. The phase mismatch δk quantifies the spatial phase imparted to the
spin wave with collinear storage calculated using only this vacuum dispersion. But
even in the case of degenerate ground and storage states, the spin wave is not
always entirely real, as discussed in the previous section (see part (b) of Figure 6.3).
This spatial phase arises dynamically, and has its origin in the material dispersion
experienced by the signal field as it propagates through the ensemble. The control
field is unaffected, since it couples states |2〉 and |3〉, both of which are unpopulated.
But the signal field couples the populated ground state |1〉 to state |2〉, and therefore
it propagates subject to an augmented refractive index. This effect can be described
by incorporating a dispersive term into our definition for the magnitude of the signal
field wavevector,
ks −→ kd = ks − kdisp, (6.23)
where, working in ordinary units, we define
kdisp =|κ|2∆|Γ|2
=dγ∆
(γ2 + ∆2)L. (6.24)
This definition is motivated by inspection of the adiabatic storage kernel (5.77)
derived in Chapter 5, which includes an exponential factor with an exponent whose
imaginary part contains the spatial phase kdispz. The form of kdisp is consistent with
the refractive index associated with an absorptive resonance at ∆ = 0. Note that on

6.3 Phasematched Retrieval 211
resonance, kdisp vanishes, and the signal field wavevector assumes its vacuum value.
At large detunings, kdisp ∝ 1/∆, so it is generally small in the Raman limit, but not
negligible.
Incorporating the dispersive phase into the signal wavevector allows us to use
the phasematching scheme outlined in the previous section to compensate both for
non-degeneracy of the ground and storage states and for material dispersion in the
ensemble. By eliminating even this latter dynamical phase, we essentially render
the spin wave real, so that the efficiency of backward retrieval (albeit at a small
angle to the z-axis) is equal to the storage efficiency, which is optimal. And with all
spatial phases removed in this way, the optimization of storage alone is the same as
the optimization of storage with backward retrieval, and so it is only necessary to
perform the former, simpler optimization.
6.3.2 Scheme
Including material dispersion, then, the phasematching scheme is summarized by
the following choice for θ,
θ = cos−1 r ; r = maxkdkc,kckd
, (6.25)
where kc = ωc/c, and kd is given by (6.23). This formula holds for arbitrary values
of δk, and for all detunings.
Provided that θ is sufficiently small, we expect that many of the results pertaining
to the optimization of collinear storage remain approximately valid. Our general

6.3 Phasematched Retrieval 212
strategy is to use the one dimensional theory of Chapter 5 — in particular the
adiabatic storage kernel (5.77) — to find the optimal temporal input mode, and
then implement the above phasematching scheme by introducing the small angle θ
between the signal and control fields.
Note that this scheme is experimentally appealing for three reasons. First, it
allows for storage with non-degenerate ground and storage states, so the signal and
control fields can be spectrally filtered. Second, introducing an angle between the
signal and control beams makes it possible to spatially filter the signal from the
control, with a pinhole or an optical fibre tip, for example. Third, the effect of
unwanted Stokes scattering can be eliminated. This is because any spin wave exci-
tations produced by Stokes scattering — the process shown in part (b) of Figure 4.3
in Chapter 4 — will not have the same momentum as those produced by absorption
of the signal field. At read out, we choose the direction of the control pulse so that
retrieval of the stored signal field is correctly phasematched. This same control pulse
could potentially drive anti-Stokes scattering, converting the unwanted excitations
into noise photons with the same frequency as the retrieved signal. But this process
will generally not be correctly phasematched, and is therefore greatly suppressed.
All these features of the scheme constitute a compelling case for the utility of a
non-collinear phasematched memory protocol.
Here we should remark that this type of phasematching scheme produces a spin
wave with a larger momentum than arises from collinear storage, since |κ| ≥ |δk|. If
the atoms in the ensemble are free to move — for instance if the storage medium is

6.4 Full Propagation Model 213
a warm atomic vapour — then any large spatial frequencies in the spin wave may be
washed out as atoms diffuse over the phase fronts of the spin wave. Therefore our
phasematching scheme may be more susceptible to decoherence, if D > λκ = 2π/|κ|,
where D is the distance over which the atoms diffuse during the storage time (see
§10.5 in Chapter 10). This problem does not affect solid state memories, which are
appealing for this and many other reasons.
To investigate the efficacy of our phasematching scheme, we should consider the
effect of walk-off between the signal and control fields as they propagate, since when
θ 6= 0 their paths inevitably diverge. In the next section we present the results of
numerical simulations that account both for walk-off and diffraction. These vindicate
the scheme (6.25), and they show, as might be expected, that the beams should be
loosely focussed, so as to maximize the overlap of the pulses.
6.4 Full Propagation Model
We model the propagation of the signal and control fields in three dimensions: two
spatial dimensions, and time. The signal field propagates along the z-axis, and the
control field is launched at an angle θ to the z-axis, in the (x, z)-plane, intersecting
the signal beam in the centre of the ensemble. We consider that both fields are
Gaussian beams — defined below — focussed at the centre of the ensemble, as
depicted in Figure 6.6. The equations of motion for the interaction are given in (4.52)
at the end of Chapter 4. For numerical convenience, we work in normalized units that
render all quantities dimensionless, and as close as possible to ∼ 1. We summarize

6.4 Full Propagation Model 214
the normalizations we employ below; they differ slightly from the normalized units
used in the preceeding analytic treatments.
1. The z coordinate is measured in units of L, and the magnitudes ks, kc, are
measured in units of 1/L. We define the z coordinate so that it runs from −12
to 12 , so that the centre of the ensemble lies at z = 0. This definition is useful
for parameterizing the control field.
2. The x coordinate is measured in units ws, where ws is the transverse beam-
waist of the signal field. That is, at z = 0, the transverse signal field amplitude
is a Gaussian e−(x/ws)2in ordinary units, and by definition e−x
2in normalized
units. To capture all relevant dynamics, x runs from −3 to 3.
3. The time coordinate τ , which as usual is the retarded time τ = t − z/c, is
measured in units of Tc, the duration of the control field. This is a departure
from the time normalization used previously, in which times were measured
in units of 1/γ. With this new normalization, the control field has roughly
unit duration, as does the signal field (since the optimal signal pulse is gen-
erally comparable in duration to the control). All relevant dynamics are then
captured by considering times from τ = −3 up to τ = 3, or thereabouts.

6.4 Full Propagation Model 215
The equations of motion, written using these normalized units, are given by
(iα2
2ks∂2x + ∂z
)A = −
√dγP,
∂τP = −ΓP +√dγA− iΩB,
∂τB = −iΩ∗P. (6.26)
As in all of our previous analyses, we have dropped the Langevin noise operators
associated with spontaneous emission and decoherence, since they have no effect
on the efficiency of the memory. The normalization factor α = L/ws is the aspect
ratio of the ensemble, which appears to correctly account for the difference in units
used for the x and z coordinates. Note that the amplitudes A, P and B are not
the averaged quantities defined in (5.1) at the start of Chapter 5. They are closer
to those quantities given in (4.7), (4.42) and (4.43) in Chapter 4, which depend
on the transverse spatial coordinates ρ, as well as τ and z. In fact, since we have
dropped any dependence on the y-coordinate, we have implicitly averaged over this
dimension. But this has no effect on the equations.
6.4.1 Diffraction
The transverse derivative ∂2x in (6.26) describes the diffraction of the signal field
as it propagates. Its importance in the dynamics is set by the ratio of α2 to ks.
Although both of these are generally large quantities, their ratio can be small, or
large, depending on how tightly the signal is focussed: if ws is small, diffraction

6.4 Full Propagation Model 216
Figure 6.6 Focussed beams. The signal (blue) and control (green)beams are cylindrically symmetric Gaussian beams focussed at thecentre of the ensemble, with a small angle between them.
can become significant. In fact, the signal field Rayleigh range, which quantifies the
length of the region over which the focussed signal beam is well-collimated, is given
in our normalized units by zs = ks/(2α2). As expected, the contribution from the
diffractive term becomes significant when the Rayleigh range falls below 1, which is
the length of the ensemble.
6.4.2 Control Field
Walk-off between the signal and control field appears through the spatial depen-
dence of the control field Ω. To construct the correct expression, consider first the
representation of a pulse with a Gaussian transverse profile, and also a Gaussian

6.4 Full Propagation Model 217
temporal profile, propagating along the z-axis,
Ω(x, τ, z) =Ωmax
W (z/zc)exp
[i
R(z/zc)− 1W 2(z/zc)
](x
wc
)2
− i tan−1
(z
zc
)×e−τ2
.
(6.27)
Here zc = kcw2c/(2α
2) is the Rayleigh range of the control field, with wc the beam-
waist of the control, measured in units of ws. The structure of (6.27) is well known
in optics, since the Gaussian transverse profile arises naturally as the lowest order
mode supported by the confocal cavity in a laser. When a laser beam of this form
is directed through a lens, it retains its Gaussian profile, but its width narrows as it
approaches the focal plane, and then widens out aftwerwards. The functions W and
R parameterize the beam size and the radius of curvature of the field phase fronts,
and are given by
W (z) =√
1 + z2,
R(z) = z
(1 +
1z2
). (6.28)
The focus lies at z = 0, at which point R −→ ∞ and W = 1. The term involving
tan−1 is known as the Gouy phase; its effect on the memory efficiency is negligible,
but we include it for completeness. Direct substitution shows that (6.27) indeed
satisfies the paraxial wave equation
(iw2c
4zc∂2x + ∂z
)Ω = 0. (6.29)

6.4 Full Propagation Model 218
When the control propagates at an angle θ to the z-axis, the control amplitude is de-
scribed by a similar expression to (6.27), except that we rotate the (x, z) coordinates
by the angle θ. We should be careful to apply this rotation with t held constant,
not τ , and we must take account of the different units used for z and x. The correct
transformation is found by making the replacements
x −→ x′ = cos(θ)x+ α sin(θ)z,
z −→ z′ = cos(θ)z − sin(θ)α
x
and τ −→ τ ′ = τ +z − z′
c. (6.30)
The factors of α ensure that the units of x and z are interconverted as they should
be, and the modification to τ represents the change in the apparent velocity of the
control pulse in the reference frame of the signal field.
6.4.3 Boundary Conditions
The boundary conditions must be specified in much the same way as in the previ-
ous one dimensional analyses. One additional feature is the dependence on the x
coordinate. The boundary conditions associated with this new degree of freedom
are simply that all the quantities A, P and B vanish as |x| −→ ∞. In practice, this
is achieved by fixing the value of the signal amplitude A to zero at x = ±3 (i.e.
at the edges of the region covered by the numerics). We use Chebyshev spectral
collocation to treat the spatial derivatives when solving the system (6.26), and we

6.4 Full Propagation Model 219
build this boundary condition directly into the differentiation matrices; the method
is explained in Appendix E. Fixing A at these boundaries also fixes P and B, since
the interaction is local: there can be no atomic excitation where there is no input
light. To model the storage process, we launch the signal field along the z-axis with
a Gaussian transverse profile at the entrance face of the ensemble,
Ain(x, τ) = A(z = −1
2 , x, τ)
= exp
[i
R(−1
2/zs) − 1
W 2(−1
2/zs)]x2
φ1(τ),
(6.31)
where φ1(τ) is the optimal input mode for storage alone, found using the one dimen-
sional theory of the previous Chapter — we use the adiabatic kernel (5.77), so that φ1
is given by (5.78). As described above, the phasematching scheme should eliminate
the spatial phase of the spin wave, so that the optimal mode for collinear storage
alone should be close to optimal for phasematched storage and retrieval. Note that
the absolute magnitude of the signal field is irrelevant, because the memory interac-
tion is linear. This boundary condition is also incorporated into the differentiation
matrices we use to solve the dynamics numerically.
For the storage process, both the spin wave B and the polarization P are initially
zero,
Bin(x, z) = B(τ −→ −∞, x, z) = 0, Pin(x, z) = P (τ −→ −∞, x, z) = 0. (6.32)
We use an RK2 method for the time-stepping, and these boundary conditions are
trivially implemented by zeroing the vectors representing B and P at the collocation

6.4 Full Propagation Model 220
points on the first iteration. See Appendix E for a description of the RK2 method,
and how it is used in combination with spectral collocation to arrive at a solution.
6.4.4 Read out
To model the read out process, we solve the equations of motion once more, this time
with no incident signal field, and a pre-existing Raman coherence determined by the
storage process. We are only able to model the build up of the signal field along a
specific direction, the z-axis, and so we must re-orient our coordinate system so that
the z-axis points along the direction in which the retrieval process is phasematched.
This is the direction of krs in Figure (6.4). The angle through which we rotate our
coordinate system is therefore the angle between ks and krs in Figure (6.4), which is
given by
θ′ = −2 sin−1
kc sin(θ)√k2d + k2
c − 2kckd cos(θ)
=
π if kc > kd
2θ − π if kd > kc
. (6.33)
The first result holds for general values of θ, while the second result applies to the
case where θ is chosen according to the scheme (6.25).
At read out, the signal field is initially zero,
Arin(xr, τ r) = 0, (6.34)

6.4 Full Propagation Model 221
as is the polarization,
P rin(xr, zr) = 0. (6.35)
Here xr, zr are the rotated coordinates describing the retrieval process,
xr = cos(θ′)x+ α sin(θ′)z,
zr = cos(θ′)z − sin(θ′)α
x. (6.36)
As in (6.30), the factors of α appear to interconvert the units of x and z.
The initial spin wave at read out is set by the spin wave generated by the storage
process, but we must be careful to include any spatial phase factors arising from
the above coordinate transformation. When the signal and control fields are not
collinear, the definition of the slowly varying coherence σ13 (see (4.22) in Chapter
4) must be modified accordingly,
σ13(r) = σ13(r)eiω13t+i(ks−kc).r. (6.37)
Here the argument r indicates that the coherence refers to an atom at that position
in the ensemble. The boundary condition for B is found by equating the coherences
at the end of storage and the start of read out,
σr13,in(rr) = σ13,out(r). (6.38)
The spin wave amplitudes are built from the slowly varying coherences. Dropping

6.4 Full Propagation Model 222
some unimportant temporal phase factors, we therefore obtain
Brin(rr) = Bout(r)ei[(kr
s−krc).r
r−(ks−kc).r]. (6.39)
Writing this boundary condition out explicitly in terms of the retrieval coordinate
system gives the relation
Brin(xr, zr) = Bout(xrc′ − s′αzr, zrc′ + s′xr/α)× (6.40)
exp
i[(ks − kcc)(1− c′)− kcss′
]zr + i
[(ks − kcc)s′ − kcs(1 + c′)
]xr/α
,
where c and s denote cos(θ) and sin(θ), respectively, and c′, s′ are equal to cos(θ′)
and sin(θ′). Note that in the absence of dispersion, with ks = kd, the phase factor
identically vanishes; it is removed by the phasematching scheme. When dispersion is
significant, the slowly varying envelope Bout itself contains a spatial phase variation.
The phasematching scheme is then designed so that the exponential factor in the
second line of (6.40) cancels this spatial phase, rendering Br as smooth as possible,
for efficient retrieval.
6.4.5 Efficiency
The efficiency of the memory is calculated in two stages. First, we simulate storage
and evaluate the storage efficiency, which is given by
ηstorage =
∫∞−∞
∫ 1/2−1/2 |Bout(x, z)|2 dz dx∫∞
−∞∫∞−∞ |Ain(x, τ)|2 dτ dx
. (6.41)

6.5 Results 223
We then simulate retrieval, and we calculate the retrieval efficiency,
ηretrieval =
∫∞−∞
∫∞−∞ |A
rout(x
r, τ r)|2 dτ r dxr∫∞−∞
∫ 1/2−1/2 |B
rin(xr, zr)|2 dzr dxr
. (6.42)
The total memory efficiency is then given by ηcombined = ηstorage × ηretrieval.
6.5 Results
In Figures 6.7 and 6.8, we present the results of simulations performed to examine the
effectiveness of our phasematching scheme. We simulated both resonant EIT storage,
and also off-resonant Raman storage. Each simulation was run twice, once with a
tightly focussed control field, with wc = 1, and once with a loosely focussed control,
with wc = 2. In the latter case the energy in the control pulse was quadrupled, so
that the intensity of the control was the same in both cases.
Figure 6.7 shows the angle θ at which the combined efficiency of storage and
retrieval is largest, over a range of values of the phase mismatch δk. The numerical
solutions generally bear out the analytic prediction (6.25) of our phasematching
scheme. But the agreement with our prediction is much better when the control is
loosely focussed. This is to be expected, since transverse walk-off is less important
when the control is wider than the signal, and since a wider control diffracts less, so
that its intensity is more homogeneous over the ensemble. For EIT, collinear storage
with θ = 0 is only optimal when δk = 0, since there is no dispersion on resonance.
But for the Raman memory, collinear storage is optimal when δk ∼ −2, when the

6.5 Results 224
−4 −3 −2 −1 0 1 2 3 40
2
4
6
8x 10
−3
θ (
rad
ian
s)
−4 −3 −2 −1 0 1 2 3 40
2
4
6
8x 10
−3
θ (
radia
ns)
(a)
(b)
EIT
Raman
Figure 6.7 Effectiveness of our phasematching scheme. We plotthe angle θ at which the combined efficiency of storage followed byretrieval is optimized. The blue solid line shows the prediction of ourphasematching scheme (6.25). The black filled circles correspond totight control focussing, with wc = 1; the red open diamonds corre-spond to loose control focussing with wc = 2. We plot the results fora typical EIT protocol in (a): we set γ = 1, d = 30 and ∆ = 0, inunits normalized as described in §6.4 above. In (b) we present equiv-alent results for a Raman protocol d = 300, γ = 0.1 and ∆ = 15.The bandwidth of the control is 10 times larger, with respect to thenatural atomic linewidth γ, in this latter protocol. To make up forthe increased detuning in the Raman protocol, the optical depth isalso much larger. In both protocols, the control field amplitude wasgiven by Ωmax = 5.
storage state lies significantly above the ground state in energy. This is because the
material dispersion alters the signal field wavevector, kd 6= ks, so that even with
degenerate ground and storage states, the spin wave acquires a spatial phase.
In Figure 6.8 we plot the optimal memory efficiencies obtained from the numer-
ical simulations alongside the analytic predictions for the best collinear efficiencies,
again over a range of values of δk. The efficiencies obtained using the phasematching
scheme (6.25) are generally very close to the best efficiencies achieved in the sim-

6.5 Results 225
−4 −2 0 2 4
effic
ien
cy
0
0.2
0.4
0.6
0.8
1
−4 −2 0 2 40
0.2
0.4
0.6
0.8
1
−4 −2 0 2 4
effic
ien
cy
0
0.2
0.4
0.6
0.8
1
−4 −2 0 2 40
0.2
0.4
0.6
0.8
1
analytic
collinear backwards
collinear forwards
phasematched
)b()a(
(c) (d)
Narrow EIT Wide EIT
Narrow Raman Wide Raman
Figure 6.8 Comparing phasematched and collinear efficiencies.Plots (a) and (b) contain the results for the EIT protocol, with tightand loose focussing, respectively. Plots (c) and (d) show equivalentresults for the Raman protocol. The parameters used are the sameas those used in produciing Figure 6.7. The solid lines represent theefficiencies obtained if the phasematching scheme (6.25) is used. Theblack filled circles are the best efficiencies achievable. The green dot-ted lines show the optimal efficiencies predicted for collinear storagewith backward retrieval, calculated using the one dimensional adia-batic kernel (6.21). The dashed red lines show the efficiency achievedby collinear storage with forward retrieval, where the input signalpulses were shaped using the adiabatic kernel (6.9), where this im-proved the efficiency.
ulations. These efficiencies are greatest when θ = 0, since at these points walk-off
between the signal and control is eliminated. As δk is increased, so does the angle θ
required for optimal efficiency, and walk-off therefore reduces the memory efficiency.
But the efficiency falls only slowly with increasing δk. With loose focussing it is
possible to exceed the optimal collinear efficiency when δk is large enough, using
either EIT or Raman storage. This demonstrates what we sought to show with our
simulations, that even including the effects of diffraction and walk-off, non-collinear
storage and retrieval, with proper phasematching, is preferable over collinear stor-

6.5 Results 226
age, with either forward or backward retrieval. The lower efficiencies observed for
tight control focussing confirms that diffraction and walk-off can be detrimental, but
provided sufficient laser energy is available, it is possible to reduce their effects by
simply widening the control beam waist.
The red dashed lines in Figure 6.8 are the result of simulating collinear storage
with forward retrieval, using the full numerical propagation model. For the EIT
protocol, we used the one dimensional adiabatic kernel (6.9) to predict the optimal
input profiles, and these input pulse shapes performed well — better than any other
pulse profiles we tried. This at least confirms the utility of the one dimensional
theory in this context. However, for the Raman protocol, this optimization did not
produce the best pulse profiles. In fact, optimal pulse profiles for backward retrieval
worked better, even though the excitation was retrieved in the forward direction.
So these input profiles were used to produce the red dashed lines in parts (c) and
(d). The reason for the failure of the forward retrieval optimization is that the
Raman protocol is more sensitive to the control intensity than is EIT. The control
intensity is highest in the centre of the ensemble, where it is focussed; its intensity
falls off towards the exit face of the ensemble. But as noted in §6.1.1, optimizing for
forward retrieval tends to shift the bulk of the spin wave towards the exit face. When
diffraction is introduced in the full model, this optimization is no longer beneficial,
since it shifts more of the spin wave to where the control intensity is lower. This
explains why the pulses optimized for backward retrieval performed better in the
simulations of forward retrieval for the Raman protocol. If the control beam waist

6.5 Results 227
is increased above 2.4, diffraction effects become small enough that the optimization
for forward retrieval does indeed produce better results than the backward retrieval
optimization, for both the EIT and Raman protocols.
Comparison of Figures 6.7 and 6.8 reveals that the efficiency falls off sharply in
the region kd > kc, to the left of the point at which θ = 0. That is, the EIT efficiency
falls sharply for δk < 0, and the Raman efficiency falls quickly as δk is reduced below
around −2. The efficiency remains high even for large angles when kd < kc. This
is easily explained by considering the geometry in part (b) of Figure 6.4. When
kd < kc, the retrieved signal field propagates exactly backwards with respect to the
direction of the input signal beam, since θ′ = π (see (6.33)). Therefore the region
over which the retrieved signal experiences gain overlaps precisely with the region
in which the spin wave is deposited during the storage process. We could say that
the ‘retrieval overlap’ is high, and the efficiency is correspondingly large. On the
other hand, when kc < kd, θ′ 6= π, and the retrieved signal field propagates at
an angle to the direction defined by the input signal field. The retrieval overlap
suffers as θ is increased, and this explains the sharp drop in memory efficiency in
this regime: although the memory is correctly phasematched, the atomic excitations
are not distributed favourably for efficient retrieval.
The foregoing discussion suggests that, as a rule, positive phase mismatches are
preferable to negative ones, although dispersion complicates things slightly. That is
to say, for efficient phasematched retrieval, it is generally better to arrange for the
ground state |1〉 to have a higher energy than the storage state |3〉. Although this

6.6 Angular Multiplexing 228
may be rather non-standard, when the ground state manifold is prepared by optical
pumping, it is not problematic.
We have succeeded in showing that proper phasematching can indeed boost the
efficiency of retrieval from a quantum memory. In the next section we describe how
the angular selectivity of phasematching offers the possibility of storing multiple
modes in a single ensemble.
6.6 Angular Multiplexing
There is a sense in which an ensemble memory represents a degree of ‘overkill’ if
just a single photon is to be stored. The number of atoms is many times larger
than the number of input photons, so there is an enormous redundancy built into
the memory. A more efficient use of resources would aim to store multiple input
pulses; in this way a single physical memory allows for highly parallel information
processing. Several schemes that make use of the idea of multimode memories have
already been proposed [17,38,153,154], in connection with both quantum computing
and quantum repeaters. A general difficulty with multimode storage in an ensemble
is the possibility that the control field used for storing one signal pulse, may also
retrieve another signal pulse that was previously stored. Phasematching provides a
way to suppress this kind of unwanted coupling.
We consider storing multiple pulses in a single ensemble by changing the angle θ
between the signal and control for each input pulse. After a pulse has been stored,
momentum conservation picks out a single, unique direction, in which the retrieval

6.6 Angular Multiplexing 229
process is phasematched, as shown in part (a) of Figure 6.4. As long as the control
field used to store a subsequent pulse propagates in a different direction to this, the
previously stored pulse will not be retrieved. By successively scanning the angle
of either the signal or control field, it is possible to store many pulses in a single
ensemble. And it is also possible to selectively retrieve just one of the stored pulses,
by aligning a retrieval control pulse with the appropriate direction for the chosen
mode.
The directional selectivity afforded by phasematching is demonstrated using our
numerical model in part (a) of Figure 6.9. Here we simulated EIT storage, with θ
chosen using the phasematching scheme (6.25). Upon retrieval, however, we rotated
away from the optimal angle by θd. In the case δk = 0, we have θ = 0 and the spin
wave momentum vanishes, so that all retrieval directions are phasematched. The
efficiency is therefore only limited by walk-off, so it remains high over a large range
of deviation angles θd. But when we set δk = 1, phasematching selects a unique
retrieval direction, and the efficiency falls off quickly as θd is increased. This shows
how it is possible to ‘switch off’ retrieval from a particular spin wave by tuning the
angle of the control.
6.6.1 Optimizing the carrier frequencies
Angular multiplexing requires that we implement storage with a range of angles θ
between the signal and control fields. Now, for a given pair of frequencies ωc, ωs for
the control and signal fields, there is only one angle that satisfies the phasematching
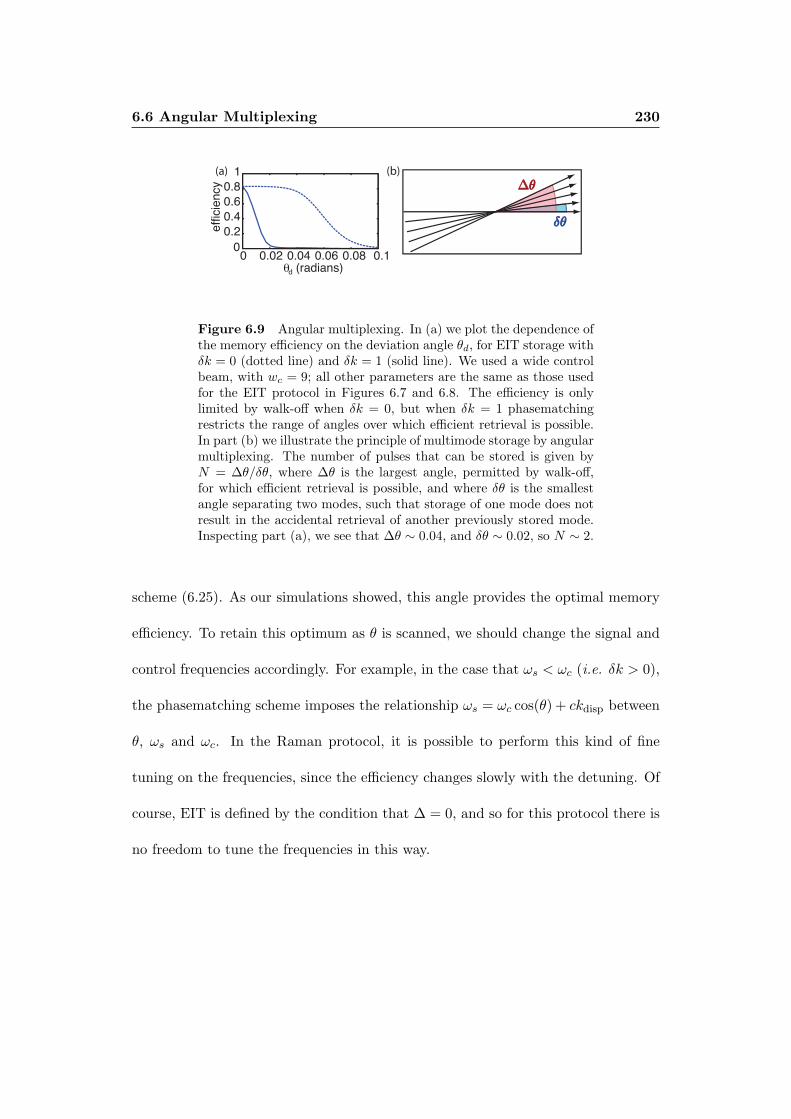
6.6 Angular Multiplexing 230
(b)
0 0.02 0.04 0.06 0.08 0.10
0.2
0.4
0.6
0.8
1
θ (radians)
effic
ien
cy
(a)
d
Figure 6.9 Angular multiplexing. In (a) we plot the dependence ofthe memory efficiency on the deviation angle θd, for EIT storage withδk = 0 (dotted line) and δk = 1 (solid line). We used a wide controlbeam, with wc = 9; all other parameters are the same as those usedfor the EIT protocol in Figures 6.7 and 6.8. The efficiency is onlylimited by walk-off when δk = 0, but when δk = 1 phasematchingrestricts the range of angles over which efficient retrieval is possible.In part (b) we illustrate the principle of multimode storage by angularmultiplexing. The number of pulses that can be stored is given byN = ∆θ/δθ, where ∆θ is the largest angle, permitted by walk-off,for which efficient retrieval is possible, and where δθ is the smallestangle separating two modes, such that storage of one mode does notresult in the accidental retrieval of another previously stored mode.Inspecting part (a), we see that ∆θ ∼ 0.04, and δθ ∼ 0.02, so N ∼ 2.
scheme (6.25). As our simulations showed, this angle provides the optimal memory
efficiency. To retain this optimum as θ is scanned, we should change the signal and
control frequencies accordingly. For example, in the case that ωs < ωc (i.e. δk > 0),
the phasematching scheme imposes the relationship ωs = ωc cos(θ) + ckdisp between
θ, ωs and ωc. In the Raman protocol, it is possible to perform this kind of fine
tuning on the frequencies, since the efficiency changes slowly with the detuning. Of
course, EIT is defined by the condition that ∆ = 0, and so for this protocol there is
no freedom to tune the frequencies in this way.

6.6 Angular Multiplexing 231
6.6.2 Capacity
How many modes can be stored in such a multiplexed memory? One constraint is
that it must be possible to resolve one retrieved signal field from another. Therefore
the minimum angle δθ by which the direction of phasematched retrieval must change
between two modes must be greater than the angular divergence of the retrieved
signal field. This angular divergence is set by Fraunhofer diffraction, which yields
the condition δθ & λs/ws — the angle between consecutive modes must exceed the
ratio of the wavelength and beam waist of the signal field.
Equally important is the requirement that there is no ‘cross-talk’ between neigh-
bouring modes — that δθ is large enough that retrieval of an undesired mode is
well-suppressed by its momentum mismatch. An estimate of the magnitude of the
momentum mismatch is given by δθks (see Figure 6.10). This momentum mismatch
is directed approximately perpendicular to the z-axis, so we can approximate the
phase accumulated by the mismatch as δθksws (working in ordinary units). For good
suppression of unwanted retrieval, this phase should exceed 2π, roughly, and from
this we again derive the condition δθ & λs/ws. Therefore the multimode capacity of
a multiplexed memory is bounded by the number of diffraction limited modes that
can be addressed. For the example in Figure 6.9 (a) this condition implies δθ ∼ 0.01,
which appears to be broadly correct.
The number of modes that can be stored is given by N = ∆θ/δθ, where ∆θ is the
largest angle at which efficient retrieval is possible, as limited by walk-off between
the signal and control fields. We approximate ∆θ as the angle subtended by the

6.6 Angular Multiplexing 232
Figure 6.10 Minimum momentum mismatch. Two signal fieldswith momenta k1 and ks differ in their directions by a small angleδθ. When retrieval for one field is phasematched, retrieval for theother is not, with the momentum difference shown in red, whichpoints essentially across the ensemble. It’s magnitude is roughly δθks(ignoring any dispersion that may slightly alter the lengths of k1, ksaway from ks). The total phase accumulated through propagationacross the ensemble, of width ws, is then roughly δθksws. Whenthis phase is larger than 2π, phasematching is sufficiently selective toeffectively suppress any accidental retrieval of stored excitations.
control waist across the length of the cell, ∆θ ∼ wc/L (in ordinary units). The
multimode capacity is then given by N ∼ wcws/λsL. This is essentially equal to
the geometric mean of the Fresnel numbers Fs, Fc associated with the signal and
control fields,
N ∼√FsFc, where Fs,c =
w2s,c
Lλs,c, (6.43)
where we used the fact that the control wavelength λc is generally rather close to
the signal wavelength. The Fresnel number of a beam quantifies the number of
diffraction limited modes it supports, so this result has the interpretation that the
number of modes that may be stored in a multiplexed memory is given by the average
(in the sense of the geometric mean) number of modes supported by the optical fields
used. For the example used in Figure 6.9 (a), the formula (6.43) predicts N ∼ 3,
and this agrees reasonably well with what might be estimated ‘by eye’ (see caption
of Figure 6.10).

6.6 Angular Multiplexing 233
A high capacity multiplexed memory requires wide beams, with Fs,c 1. In
addition, our numerics showed that wc should be at least 2 (better 3) times larger
than the signal beam waist. The feasibility of this kind of memory depends on the
availability of energetic lasers that can maintain a high intensity, even when very
loosely focussed. But laser technology has advanced sufficiently that this is certainly
possible for beams with a width on the order of centimeters, giving multimode
capacities on the order of 100.
In the next chapter we continue to investigate multimode storage, both in the
Raman and EIT protocols covered so far, and in a number of alternative memory
protocols.

Chapter 7
Multimode Storage
At the end of the last Chapter we described a way to store multiple pulses in a single
ensemble memory. We considered scanning the propagation direction of either the
control or the input signal beams: each independently stored pulse represented a
separate spatial mode, characterized by the direction of its wavevector. However, in
many practical situations, it is desirable to fix the alignment of all components in an
optical system. It may therefore be more useful to consider using a different degree
of freedom to define a basis of modes. As discussed at the start of Appendix C, we
might also consider polarization or spectral modes. And in fact since polarization is
only two dimensional, any such consideration of multimode storage must focus on
spectral modes. Equivalently, we might talk about temporal modes, since time and
frequency are Fourier conjugates; they are two interchangeable representations of
the same underlying space. This space can be thought of as the space of all possible
input profiles for the signal field. The method used in the previous Chapters to

7.1 Multimode Capacity from the SVD 235
optimize the storage efficiency of a quantum memory is immediately useful in this
connection. The SVD of the storage kernel provides a ‘natural’ basis for the space
of signal profiles: the input modes φk. In this Chapter we apply the SVD to the
study of multimode storage: we show how to optimize and quantify the multimode
capacity of the Raman, EIT, CRIB and AFC memory protocols. The following
treatment expands upon the account published in Physical Review Letters [155].
7.1 Multimode Capacity from the SVD
Suppose that we would like to store a ‘message’ in a quantum memory. We must
select an alphabet in which to write the message. If we encode the message in an
optical signal, each letter in the alphabet is represented by a mode of the optical
field. The multimode capacity of the quantum memory is the size of the largest
alphabet we can use for our message compatible with efficient storage. In general,
this capacity will depend on the particular alphabet chosen: some modes of the
optical field cannot be stored at all. To take an absurd example, we certainly could
not encode any letters as X-rays! The multimode capacity also depends on the
brightness of the signals used to encode the message. For instance, when two similar
modes are bright, we can distinguish them and encode two letters, whereas when
they are dim we can encode only a single letter (see Figure 7.1).
In a quantum communication protocol, security requires that we encode each
letter using just a single photon (see §1.5 in Chapter 1). This means that there can
be no overlap whatsoever between the modes comprising the alphabet. That is, we

7.1 Multimode Capacity from the SVD 236
Bright
A B
Dim
Figure 7.1 Bright overlapping modes are distinct. Two “letters”A and B are encoded with similar optical pulses. When they arebright, the letters can be distinguished; not so when their intensityis reduced.
must find an orthonormal basis of modes. The multimode capacity of a quantum
memory in this context is therefore the number of orthonormal modes that can
be stored. It remains true that this capacity depends on the choice of basis. For
example, it may not be the same for time bin modes as it is for frequency modes
(see §C.1 in Appendix C). What, then, is the optimal basis? Which encoding
maximizes the capacity? It is the basis formed by the input modes φk determined
from the SVD of the storage kernel K. That is, for any number N , storage of the
first N input modes φ1, . . . , φN is more efficient than the storage of any other set
of N orthonormal modes. Each input mode φk is stored with efficiency ηk = λ2k. In
the optimal situation of phasematched backward retrieval without dispersion, the
retrieval efficiency is also equal to ηk (see the discussion towards the end of §6.2 in
6). The total memory efficiency for the kth mode is then η2k = λ4
k. The multimode
capacity is given by the width of the ‘distribution’ of efficiencies (see Figure 7.2).
Below we consider two ways of quantifying this width.

7.1 Multimode Capacity from the SVD 237
Figure 7.2 Visualizing the multimode capacity. The total memoryefficiency — the efficiency of storage followed by retrieval — for thekth mode is η2
k = λ4k. These efficiencies define a decaying distribu-
tion, and the multimode capacity is related to the width, or ‘variance’of this distribution. The Schmidt number quantifies this width, buttakes no account of the absolute efficiencies. If we introduce a thresh-old efficiency ηthreshold, we can define an operational measure N forthe multimode capacity by considering the number of modes withmemory efficiency greater than the threshold.
7.1.1 Schmidt Number
The Schmidt number S is defined as follows [156,157],
S =1∑∞
k=1 η4k
, where η2k =
η2k∑∞
j=1 η2k
. (7.1)
The η2k are defined so that they sum to unity, turning them into effective probabili-
ties. In fact, if we consider that these probabilities are the eigenvalues of a quantum
mechanical density matrix, describing a statistically mixed state, then S is the in-
verse of the purity of this state [158]. From the definition (7.1) it’s clear that S = 1
if there is just one non-zero singular value. In general, if there are N equal singular
values, with all others vanishing, then S = N , so the Schmidt number does indeed
count the number of modes that may be stored with equal efficiency. When the
singular values are not equal, but instead decay in magnitude smoothly as the index

7.1 Multimode Capacity from the SVD 238
k increases, S still provides a sensible measure of the number of coupled modes. The
normalization of the η’s makes S independent of the overall efficiency of the memory:
it measures how many modes are involved in the memory interaction with compa-
rable strength, independent of what that strength is. This makes S an independent
figure of merit for characterizing the performance of a quantum memory. One might
choose to optimize the efficiency (i.e. the optimal efficiency) of a quantum memory,
or the Schmidt number, or both. In most situations, however, it is more useful to
quantify the ability of a memory to store multiple modes efficiently, so that a metric
that combines these two measures is desirable. We introduce such a metric in the
next section. But before abandoning the Schmidt number entirely, we observe that
it is possible to develop an intuition for the value of S — at least whether or not
it is large — by looking at the form of the Green’s function describing the storage
interaction (see Figure 7.3). The reason is that the contribution of a single mode to
the Green’s function necessarily takes the form of a product of two functions; each
with contours orthogonal to the other’s: one function represents the input mode;
the other the corresponding output mode. Now, the Schmidt number is independent
of the basis of modes we consider, because the singular values are. We are therefore
always free to choose a basis of modes comprised of localized pulses. Each mode
is orthogonal to the others if the pulses do not overlap. The product of two such
modes produces a contribution to the Green’s function in the form of a rectangular
‘blob’, with sides parallel to the coordinate axes, and rounded edges if the pulses
have smooth edges. Counting the number of such blobs required to make up the

7.1 Multimode Capacity from the SVD 239
Green’s function provides an estimate of the number of modes required to construct
it. Any feature of the Green’s function that traces out a curve is ‘multimode’, as is
any feature that runs at an angle to the coordinate axes. The Schmidt number is
sometimes useful because it is amenable to this kind of dead reckoning, as we will
see in §7.3.2 below.
(a) (b)Single mode Multimode
Figure 7.3 The appearance of a multimode Green’s function. In-spection of the contours of the Green’s function describing a memoryinteraction offers insight into the degree to which it is multimode, asquantified by the Schmidt number S. By reconstructing the approx-imate shape of these contours using non-overlapping rectangles, onecan estimate the number of modes of which it is composed. In (a)we show an archetypal single mode Green’s function, which looks likean isolated ‘hill’; a single elliptical contour is drawn. The contour isroughly approximated by just a single rectangle, indicating that onlyone mode contributes to its structure. In (b) we illustrate how curvedor angled contours admit a decomposition involving more modes.
7.1.2 Threshold multimode capacity
To be more quantitative, we introduce a threshold efficiency ηthreshold that allows
us to delineate those modes that are stored and retrieved with acceptable efficiency
from those that are not. We then define the multimode capacity as the largest
number of modes N for which the mean memory efficiency, averaged over all N

7.1 Multimode Capacity from the SVD 240
modes, exceeds ηthreshold. The average memory efficiency of the first k input modes
is given by
Λk =
∑kj=1 η
2j
k. (7.2)
Then the multimode capacity can be written as
N =∞∑k=1
βk, where βk =
1 if Λk > ηthreshold
0 otherwise
. (7.3)
A more conservative estimate could replace the average efficiency (7.2) with the
minimum efficiency η2k. The results presented below are not qualitatively altered by
making this alternative definition, but as N becomes large, this definition represents
an increasingly pessimistic estimate of the performance of a memory for storing
random ‘words’ drawn from the alphabet defined by the φk. The capacity defined
in (7.3) is a convenient measure, with a clear interpretation.
In the next section, we investigate the scaling of N for various memory protocols.
We set ηthreshold = 70%; this value makes the form of the scaling apparent for
parameters that are tractable numerically, and indeed experimentally. We return
to the one dimensional model used in Chapter 5, since we are able to write down
explicit expressions for the storage kernels in this case. The simulations presented in
the previous Chapter confirm that the one dimensional approximation works well,
provided that the control field is more loosely focussed than the signal field (see §6.5
in Chapter 7).

7.2 Multimode scaling for EIT and Raman memories 241
7.2 Multimode scaling for EIT and Raman memories
In Chapter 5 we showed that the best possible storage efficiency for a Λ-type quan-
tum memory is found from the SVD of the anti-normally ordered kernel (5.27) [133].
We repeat it here for convenience,
KA
(z, z′
)=d
2e−d(z+z′)/2I0
(d√zz′). (7.4)
Here we are using normalized units for the z coordinates. The kernel (7.4) is valid
for both EIT and Raman protocols, in the light-biased limit where the control pulse
is sufficiently energetic (see §5.2.8 in Chapter 5). The eigenvalues of this optimal
kernel are equal to the largest possible values of the storage efficiencies ηk = λ2k,
so we can calculate the multimode capacity N by diagonalizing KA. This can be
done numerically without difficulty, and we present the results of such a numerical
diagonalization in part (a) of Figure 7.4 below. In part (b) we plot the capacity
predicted by the Raman kernel (5.95) introduced in §5.3 of Chapter 5. In both cases
the capacity rises only slowly with the optical depth d. To explain these results, we
briefly return to the treatment given in §5.2.3 in Chapter 5. There we derived an
approximate expression (5.21) for the storage efficiencies, valid in the limit d 1:
ηk = e−4παk/d, (7.5)

7.2 Multimode scaling for EIT and Raman memories 242
where αk is the kth zero of the function J0(2√
2πx). The zeros of J0 are distributed
approximately linearly along the real line (see §15.4 in Chapter XV in G.N. Watson’s
famous treatise [159]). Therefore the αk increase, roughly, with the square of the index
k. Setting αk ∼ qk2 for some constant q, we find that the index of the mode whose
total efficiency η2k falls below ηthreshold is given approximately by
k ∼√ηthreshold
8πq×√d. (7.6)
The multimode capacity N should scale in the same way, so that we expect
N ∼√d, (7.7)
for large optical depths, regardless of the threshold ηthreshold. Inspection of Figure
7.4, which was generated by setting ηthreshold = 70%, indeed reveals a scaling of
N ≈√d/3.
7.2.1 A spectral perspective
The kernel (7.4) is derived by considering the optimal efficiency for absorption into
the excited state |2〉, and the square root scaling of N with d can be understood by
considering the bandwidth of the absorption line associated with an ensemble of two-
level atoms. If each atom has a Lorentzian lineshape with natural linewidth 2γ, the
absorption profile of the entire ensemble is given by exponentiating the single-atom

7.2 Multimode scaling for EIT and Raman memories 243
profile,
F (ω) = exp[− 2dγ2
γ2 + ω2
]. (7.8)
The full width at half maximum (FWHM) of F is given by
∆ω = 2
√2dγ2
ln 2− γ2 ≈ 2γ
√2dln 2
, (7.9)
where the approximation holds for d 1. An estimate of the multimode capacity is
provided by counting the number of spectral modes with FWHM 2γ that ‘fit’ inside
this absorption profile, which procedure yields
N ∼ ∆ω2γ∼√d. (7.10)
We can therefore identify the origin of the square root scaling of N with d as the
scaling of the bandwidth of the absorption of an ensemble with its optical thickness.
This scaling is rather poor: in order to store two modes in such a memory, we must
quadruple the optical depth required for storing a single mode. If this optical depth
were divided up into four separate ensemble memories, we could store four modes
— one mode in each memory — and so the multimode scaling for EIT and Raman
memories is decidedly sub-optimal. The reason is that when the optical depth is
increased, new atoms are added ‘behind’ the old: they absorb at the same frequencies
as the other atoms, and so they improve the absorption at these frequencies, but
they do not provide much coupling at other frequencies — other modes. In the next

7.2 Multimode scaling for EIT and Raman memories 244
section we will show that adding an inhomogeneous broadening to the ensemble
can improve the multimode scaling, by redistributing optical depth over a range of
frequencies. This turns the square root scaling with d into a linear one, consistent
with what one might expect to achieve from operating separate memories in parallel.
And this is essentially the way such broadened protocols work, albeit within a single
physical ensemble.
0 20 40 60 80 1000 100 200 300 400 5000
2
4
6
8
Optimal kernel Raman kernel(a) (b)
Figure 7.4 Multimode scaling for Raman and EIT memories. (a):the multimode capacity found by numerical diagonalization of theoptimal kernel KA in (7.4). This represents the best possible scalingfor both EIT and Raman memories; the capacity scales only with thesquare root of the optical depth. (b): we also show the multimodecapacity found by diagonalizing the Raman kernel (5.95), which isparameterized by the Raman memory coupling C (see §5.3 in Chapter5). This kernel is valid in the far off-resonant limit — though if thecontrol pulse intensity is increased sufficiently, the optimal kernelKA should be used instead. We plot the Raman capacity against C2,since this quantity is proportional to d. The same square root scalingwith ensemble density is evident. Equivalently, if d is held constant,the Raman capacity scales with the square root of the control pulseenergy. We used ηthreshold = 70% in both plots.

7.3 CRIB 245
7.3 CRIB
The CRIB memory protocol is introduced in §2.3.3 in Chapter 2. Storage is achieved
by direct absorption into the excited state |2〉, which is artificially broadened by
application of an external field. Once storage is complete, the excitation is ‘shelved’
by application of a short, bright control pulse, which transfers the excitation to
the storage state |3〉. To implement retrieval, another control pulse transfers the
excitation back to |2〉, and the inhomogeneous broadening is ‘flipped’, so that the
atomic dipoles re-phase and the signal field is re-emitted.
The same considerations regarding retrieval discussed in the previous Chapter
apply to this protocol, just as they do to Raman and EIT memories: retrieval in
the forward direction is inefficient due to re-absorption losses [160], while backward
retrieval is vulnerable to phasematching problems. However, in this protocol, the
control pulse is applied after the signal has been absorbed, so it is possible to
distinguish the two fields temporally. Spectral and spatial filtering is therefore less
important, and so it is feasible to use an ensemble where the states |1〉 and |3〉 are
degenerate. This allows for efficient collinear storage, followed by phasematched
backward retrieval. Since there is no dispersion on resonance, the stored excitation
has no spatial phase, and the retrieval efficiency is equal to the storage efficiency,
with ηcombined = η2storage. In the following, we restrict our attention to this situation,
which is optimal.

7.3 CRIB 246
7.3.1 lCRIB
We first consider lCRIB, in which the broadening is applied longitudinally [84,161,162].
The resonant frequency of the |1〉 ↔ |2〉 transition varies linearly along the z-axis.
Since the control field is only applied after the signal field has been resonantly
absorbed, the equations of motion for lCRIB are given by the system (5.106), with
Ω = 0, and with the spatial variation of the detuning included.
∂zA(z, τ) = −√dP (z, τ),
∂τP (z, τ) = −Γ(z)P (z, τ) +√dA(z, τ), (7.11)
where Γ(z) = 1 − i∆(z), with ∆(z) = ∆0
(z − 1
2
). Here we have returned to the
normalized units of Chapter 5, with all frequencies measured in units of γ, and with
z running from 0, at the entrance face of the ensemble, up to 1, at the exit face. The
width of the applied spectral broadening is ∆0. As usual, we solve these equations
by applying a unilateral Fourier transform over the z-coordinate. Using the formula
(D.27) from Appendix D, we obtain the transformed system
−ikA− 1√2πAin = −
√dP ,
∂τ P = −(1−∆0∂k + i∆0
2
)P +
√dA, (7.12)

7.3 CRIB 247
whereAin is the temporal profile of the incident signal field. Solving the first equation
for A and substituting the result into the second equation yields
(∂τ −∆0∂k) P = −(1 + i∆0
2 + i dk)P + i
√d√
2πkAin. (7.13)
Now if we temporarily define the composite variable s = k + ∆0τ , we can replace
the derivatives on the left hand side with a single time derivative,
∂τ P = −f(s, τ)P + i
√d√
2π(s−∆0τ)Ain, (7.14)
where the partial derivative ∂τ is taken with s held constant, and where we have
defined the function
f(s, τ) = 1 + i∆02 + i d
s−∆0τ. (7.15)
Integrating (7.14) gives the solution
Pout(s) = Pin(s)e−R T−∞ f(s,τ) dτ + i
√d√
2π
∫ T
−∞Ain(τ)e−
R Tτ f(s,τ ′) dτ ′ dτ, (7.16)
where Pout(s) = P (s, T ) is the atomic excitation at the moment τ = T that the
short control pulse is applied, marking the end of the storage interaction. We do not
explicitly model the atomic dynamics induced by the control; we simply assume that
it is sufficiently intense to transfer — effectively instantaneously — all the atomic
excitations into the storage state |3〉, so that Pout −→ Bout. We set Pin = 0, since no
atoms are excited at the start of the storage process. We then perform the integral

7.3 CRIB 248
in the exponent of the integrand in the second term, and convert back from s to k,
to arrive at the following expression for the lCRIB storage map in k-space,
Bout(k) =∫ ∞−∞
K(k, T − τ)Ain(τ) dτ, (7.17)
with the kernel K defined by
K(k, τ) = i
√d√
2πe−(1+i∆0/2)τ × kid/∆0(k + ∆0τ)−id/∆0−1 (7.18)
for τ ≥ 0, with K = 0 when τ < 0. The multimode scaling of lCRIB is determined
by the singular values of this kernel. Unfortunatey we cannot extract these singular
values directly because there is a singularity at k = −∆0τ . A number of alternatives
are open to us. First, we can return to the equations of motion, and construct the
Green’s function in (z, τ)-space directly by numerical integration (see §5.4 in Chapter
5 and §E.5 in Appendix E). This kernel is not singular, and is therefore amenable to
a numerical SVD. We’ll refer to this as simply the ‘numerical method’. Second, we
can remove the singularity in (7.18) by applying a small regularization to k. That is,
we replace k by k− iε, where ε is some small real number. This shifts the singularity
off the real axis, making the kernel well-behaved. But it also changes the singular
values, since it is not a unitary transformation. We fix this with the following
procedure. We apply a numerical inverse Fourier transform from k-space back to z-
space. This can be performed very efficiently with a fast Fourier transform (FFT), an
implementation of which is standard in Matlab. The Fourier transform is unitary, so

7.3 CRIB 249
it does not affect the singular values. Next, we multiply the result by the exponential
factor e−εz. By the shift theorem (see §D.3.4 in Appendix D), this compensates for
the regularization, so that the result is equal to the Fourier transform of (7.18)
without the regularization applied. Because the Fourier transform is unitary, we
can extract the singular values of (7.18) by taking a numerical SVD of the kernel
generated by this procedure. We will refer to this as the ‘Fourier method’.
We use both of these methods below. But to gain some insight into the form of
the multimode scaling of lCRIB, we now introduce a third approach. The approach is
only valid for very large broadenings, but it clarifies the scaling behaviour exhibited
by the numerical techniques just described.
7.3.2 Simplified Kernel
We perform a series of unitary transformations that simplify the form of the kernel
(7.18), while leaving the singular values unchanged. The first of these transforma-
tions is to trop the exponential factor iei∆0τ/2, since it is just a phase rotation. Next
we drop the factor kid/∆0 . This is, again, a pure phase rotation, which is well defined
as long as ∆0 6= 0 and k 6= 0. The second of these conditions arises because there is a
logarithmic singularity in the phase of kid/∆0 at k = 0. The effect of this singularity
is small when d/∆0 is small, so in the following we assume a large broadening, with
∆0 d. The resulting kernel is
K(k, τ) =
√d√
2πe−τ (k + ∆0τ)−id/∆0−1. (7.19)

7.3 CRIB 250
We now take the inverse Fourier transform, from k-space back to z-space. I have to
confess to not knowing how to perform this transform, but Mathematica provides
an answer! Combining this with the shift theorem gives
K(z, τ) = α (d/∆0)√de−τzid/∆0ei∆0zτ , (7.20)
where α is given by
α(x) = − 1πe−πx/2sinh(πx)Γ(ix), (7.21)
with Γ denoting the Euler Gamma function (not the complex detuning!). Finally,
we note that the factor zid/∆0 is another pure phase rotation, well-defined if ∆0 6= 0
and z 6= 0. We can drop it without affecting the singular values, again in the limit
∆0 d, which gives the simple kernel,
K(z, τ) = α(d/∆0)√de(i∆0z−1)τ . (7.22)
Now we form the anti-normally ordered kernel KA, by integrating the product of
two of these kernels from τ = 0 to τ =∞ (see §3.3.1 in Chapter 3),
KA(z, z′) =d
∆0|α(d/∆0)|2 × 1
2/∆0 − i(z − z′). (7.23)
This kernel has the simple structure we have been seeking. It takes the form of a
Lorentzian peak centred on the line z = z′, with a width set by ∆0, and a height
determined by the ratio d/∆0. We can draw two conclusions from the form of this

7.3 CRIB 251
kernel.
First, in the limit of large broadening ∆0 d, the Schmidt number becomes
independent of the optical depth, being determined only by ∆0. This follows from
the fact that the functional form of (7.23) does not depend on d, which only affects its
overall magnitude. The Schmidt number does not depend on this overall magnitude,
because of the normalization of the η’s in (7.1), and so S is independent of d.
The contours of (7.23) form a strip along the diagonal line z = z′. Application
of the estimation technique described in Figure 7.3 suggests that the multimode
capacity is proportional to the ratio of the length to the width of the strip, so that
S ∝ (2/∆0)−1 ∼ ∆0 (see Figure 7.5). Numerics confirm this to be the case: the
Schmidt number of an lCRIB memory rises linearly with the applied broadening
(see Figure 7.6).
The second conclusion we can draw from the structure of (7.23) is that the
threshold multimode capacity N rises linearly with the optical depth d — a signifi-
cant improvement over the square root scaling derived for EIT and Raman memories
in §7.2. To see why, consider a situation where the optimal memory efficiency η21
exceeds the threshold efficiency ηthreshold by a reasonable margin, so that N ≈ S
(see Figure 7.7). If we double the applied broadening, the Schmidt number doubles.
But the ratio d/∆0 is then halved, so that the overall efficiency falls below ηthreshold,
and N ≈ 0. In fact, the function α(x) ≈ 1 for x 1, so that halving d/∆0 ap-
proximately halves the ηk, and divides the total memory efficiencies η2k by four. To
bring the overall efficiency back to its previous value, above the threshold, we must

7.3 CRIB 252
(a) (b)Many modes Few modes
0 0.5 10
0.5
1
0 0.5 10
0.5
1
Figure 7.5 Scaling of Schmidt number with broadening. In (a)and (b) we plot |KA| for large and small broadenings of ∆0 = 100and ∆0 = 10, respectively, with d = 30 in both cases. Below theseplots we illustrate the mode-counting procedure described in Figure7.3, which provides a way to understand why the Schmidt numberincreases linearly with the applied broadening (see Figure 7.6).
double the optical depth. The multimode capacity N is then doubled, because we
have both doubled S and maintained the correspondence N ≈ S by keeping the
overall efficiency above ηthreshold. This argument is quite general: we can increase
the number of modes contributing to the storage interaction by increasing the ap-
plied broadening, but the coupling is ‘shared’ between all these modes, so we must
increase the optical depth at the same time in order to maintain efficient storage
over all the modes. If our aim is to maximize N , there is an optimal value for the
ratio d/∆0, which depends on ηthreshold. If the ratio is too large, we can afford to
increase the broadening and introduce more modes without bringing the efficiency
below the threshold. Converseley, if the ratio is too small, we should sacrifice some

7.3 CRIB 253
0 100 200 3000
0.5
1
(a) (b)
0 100 200 3000
10
20
30
Figure 7.6 Comparison of the predictions of the kernels (7.23) and(7.18). (a): the optimal total memory efficiency η2
1 predicted by thesimplified kernel (7.23) (green dotted line) is plotted alongside theefficiency predicted by the kernel (7.18), using the ‘Fourier method’(blue solid line), as a function of the applied broadening. The opticaldepth is set at d = 30; the simplified kernel compares well with theFourier method for ∆0 & 3d. (b): the Schmidt number predicted bythe two kernels. The agreement between the two, even at the bound-aries of the regime of validity of the simplified kernel, is excellent.The linear scaling of S with ∆0 expected from the form of (7.23) isclear.
modes and reduce the broadening to boost the efficiency above the threshold. This
analysis shows that the multimode capacity N of an lCRIB memory scales linearly
with d, provided that the broadening ∆0 is increased linearly with d at the same
time.
The preceding discussion applies in the limit ∆0 d. We confirm the persistence
of linear multimode scaling outside this regime using the direct numerical method
described earlier, in which we construct the Green’s function by integrating the
equations of motion. The result is plotted in Figure 7.8 at the end of §7.3.3 below,
alongside the results for EIT and Raman shown earlier, and the results for tCRIB,
which we now turn to.

7.3 CRIB 254
(a) (b) (c)
Figure 7.7 Understanding the linear multimode scaling of lCRIB.In the limit of large broadening, the Schmidt number of the lCRIBstorage interaction depends linearly on the width of the applied spec-tral broadening, but the overall storage efficiency is determined bythe ratio d/∆0. To double the multimode capacity N , given a fixedthreshold efficiency ηthreshold, we need to double the Schmidt number,while keeping the overall efficiency the same. The process is shownin parts (a) to (c). We begin with η2
1 > ηthreshold, so that S ≈ N . In(b) we double the spectral broadening, which doubles S, but reducesthe efficiency (in fact, by roughly a quarter). To return to the sameefficiency as in (a), in part (c) we double the optical depth. Thishas no effect on S, but it returns the ratio d/∆0 to the same valueas in part (a). By doubling the optical depth, we have suceeded indoubling the multimode capacity. These arguments explain why thethreshold capacity N depends linearly on d for lCRIB.
7.3.3 tCRIB
In a tCRIB memory, the direction of the broadening is perpendicular to the z-axis.
That is, the resonant frequency of the atoms varies across the ensemble (see part
(a) of Figure 2.10 in Chapter 2). The theoretical treatment requires that we divide
the ensemble into frequency classes, where all the atoms in one frequency class have
the same detuning from the signal field carrier frequency. Thorough treatments are
given by Gorshkov et al. [163], and also by Sangouard et al. [160], and we adapt their
techniques to our purpose, which is to derive the Green’s function for the tCRIB
memory interaction.
Suppose that the applied broadening produces an inhomogeneous line with spec-

7.3 CRIB 255
tral absorption profile p(∆). This means that a proportion p(∆)d∆ of the the atoms
have their resonant frequencies shifted by ∆ away from their nominal frequency. The
profile is normalized, so that ∫p(∆) d∆ = 1. (7.24)
The total optical depth d is divided amongst all the frequency classes, so that the
optical depth contributed by atoms detuned by ∆ is d(∆)d∆ = dp(∆)d∆. The
equations of motion for the storage process are found by a straightforward general-
ization of the system (7.11) to the case of multiple frequency classes (and with no
variation of ∆ with z in this case, of course),
∂zA = −∫ √
d(∆)P (∆) d∆,
∂τP (∆) = −Γ(∆)P (∆) +√d(∆)A. (7.25)
Here we have emphasized the functional dependence of the complex detuning Γ(∆) =
1− i∆, and we have defined P (∆) as the slowly varying polarization associated with
the frequency class of atoms detuned by ∆. We quickly encounter difficulties if we
attempt to solve this system of equations with our usual trick of Fourier transforming
over z. Instead, we get to the solution by applying a unilateral Fourier transform
over time, from τ to ω. Note that here ω is the frequency conjugate to the retarded
time τ ; it certainly has no relation to the integrated Rabi frequency used in Chapter

7.3 CRIB 256
5! The transformed equations are given by
∂zA = −∫ √
d(∆)P (∆) d∆,
−iωP (∆)− Pin(∆)√2π
= −Γ(∆)P (∆) +√d(∆)A. (7.26)
Solving the second equation, and substituting the result into the first yields the
following equation for the signal field,
[∂z + df(ω)] A(ω, z) = −√
d
2π
∫ √p(∆)Pin(z; ∆)Γ(∆)− iω
d∆, (7.27)
where we have defined the lineshape function
f(ω) =∫
p(∆)1− i(∆ + ω)
d∆, (7.28)
which is essentially the convolution of the Lorentzian spectral response of the atoms
with the inhomogeneous profile p(∆). Recall that in our normalized units the natural
atomic linewidth is defined to be equal to 1. We can immediately solve for the signal
field,
A(ω, z) = e−df(ω)zAin(ω)−√
d
2π
∫ √p(∆)
∫ z
0e−df(ω)(z−z′) Pin(z′; ∆)
Γ(∆)− iωdz′ d∆.
(7.29)
Here Ain is the spectrum of the incident signal field. Now that we are in possession of
a solution for the signal, it is a matter of algebra to construct the Green’s function.

7.3 CRIB 257
But it is not enough to find the storage map alone. We can write down an expression
for the atomic excitation at the end of the interaction, but since it is distributed over
all frequency classes, it is not clear what we should optimize for efficient storage.
There is no single spin wave whose norm represents the storage efficiency. Therefore,
we construct the kernel Ktotal that relates the input signal to the retrieved signal
field (see §3.4 in Chapter 3). The modes found from the SVD of this kernel have a
clear interpretation as those modes that are eventually retrieved from the memory.
To find this kernel, we first solve for the polarization Pout at the end of the storage
process, setting Pin = 0. This requires taking an inverse Fourier transform from ω
back to τ = T , where T marks the end of the storage interaction,
Pout(z,∆) =1√2π
∫ ∞−∞
e−iωT P (ω, z; ∆) dω, (7.30)
where, using the second line of (7.26),
P (ω, z; ∆) =
√d(∆)A(ω, z)Γ(∆)− iω
=
√d(∆)e−df(ω)z
Γ(∆)− iω× Ain(ω), (7.31)
where in the second line we used (7.29). This completes the description of the storage
process. At time τ = T , the control pulse shelves all the excited atoms. To describe
retrieval of the excitations, we again use (7.29). As usual we use a superscript r to
identify quantities associated with the retrieval process. There is no input field for
retrieval, so Arin = 0. The retrieved signal field, at the exit face of the ensemble with

7.3 CRIB 258
zr = 1, is given by
Arout(ω
r) = −√
d
2π
∫ √pr(∆r)
∫ 1
0e−df
r(ωr)(1−zr) Prin(∆r, zr)
Γ(∆r)− iωrdzr d∆r. (7.32)
We ‘stitch’ together the storage and retrieval interactions by making the identifica-
tion
P rin(∆r, zr) = Pout(∆, z). (7.33)
This says that the atomic polarization is the same at the end of the storage process
as it is at the start of the retrieval process. But in between storage and retrieval, the
inhomogeneous profile is flipped, so that red-detuned atoms become blue-detuned
and vice-versa. This is crucial for reversing the atomic dynamics so that they re-emit
the signal field. We model this flipping of the detunings by setting
∆ = −∆r, (7.34)
so that frequency classes with positive and negative detunings swap places. For
backward retrieval with no phase mismatch — the optimal situation — we also
swap the z-coordinate:
z = 1− zr. (7.35)
Note that in this case the frequency ωr is conjugate to the retarded time τ r, which is
the time coordinate in a frame moving at the speed of light backwards with respect to
the initial z-axis used for the storage process. We now combine the relations (7.35)

7.3 CRIB 259
and (7.34) with (7.33), and substitute this into (7.32), using (7.30) and (7.31). The
result is
Arout(ω
r) =∫ ∞−∞
Ktotal(ωr, ω)Ain(ω) dω, (7.36)
where the Green’s function for the memory interaction is given by
Ktotal(ωr, ω) = −e−iωT
∫ √pr(∆r)p(−∆r)
[Γ(∆r)− iωr][Γ(−∆r)− iω]d∆r× d
2π
∫ 1
0e−d(1−zr)[f r(ωr)+f(ω)] dzr.
(7.37)
Now we assume that the inhomogeneous profile is the same for both the storage
and retrieval processes, and symmetric about the unbroadened resonance. then
pr(∆r) = p(∆r) = p(−∆r), and the lineshape is also unchanged, f r = f . Performing
the integrals over ∆r and zr, we find
Ktotal(ωr, ω) =1
2πe−d[f(ωr)+f(ω)] − 1
2 + i(ωr + ω), (7.38)
where we have dropped the exponential factor of e−iωT , since it represents only
an unimportant phase rotation. Here we note that a nearly identical treatment
was first given by Gorshkov et al. [163]. The crucial difference for us is that we
used a unilateral Fourier transform, rather than a Laplace transform, to solve the
equations. This means that the transformed amplitudes Ain and Arout have a natural
interpretation as the spectra of the input and retrieved signal fields. The norm of
the signal spectrum is the same as the norm of the temporal profile, by Parseval’s
theorem (or, alternatively, by energy conservation), and so the SVD of the kernel

7.3 CRIB 260
Ktotal in (7.38) tells us about the storage efficiency of the memory, and indeed, its
multimode capacity.
Now that we have found an explicit form for the Green’s function describing
tCRIB, the multimode capacity can be found by taking its SVD: the singular values
of Ktotal are equal to the ηk. In Figure 7.8 we plot the resulting prediction for the
multimode scaling of tCRIB. We used a rectangular broadening profile with total
width ∆0,
p(∆) =
1/∆0 if |∆| ≤ ∆0/2,
0 otherwise.
(7.39)
And we optimized N with respect to the width ∆0, using a threshold ηthreshold =
70%. It is clear that the scaling of N with d is linear. Furthermore, it is the same
as the scaling of lCRIB, whose multimode capacity is also plotted. This suggests
that the multimode capacity of both CRIB protocols is identical. The scaling of
unbroadened EIT and Raman protocols, as derived from (7.4), is shown for com-
parison. Clearly CRIB dramatically outperforms equivalent unbroadened protocols:
given the same optical depth — the same total number of atoms — more modes of
the optical field can be stored by adding a controlled broadening. We found that for
both tCRIB and lCRIB, the multimode capacity scaled roughly as N ∼ d/25, and
the optimal width for the spectral broadening scaled as ∆opt0 ∼ 9d/5. This confirms
the validity of the arguments given at the end of §7.3.2 for lCRIB.
Just as the poor multimode scaling of unbroadened protocols can be explained
by considering the absorption bandwidth of a homogeneous ensemble (see §7.2.1), so

7.3 CRIB 261
we can understand the improved scaling of CRIB by considering the inhomogeneous
linewidth. Consider the storage of a spectral mode with bandwidth 2γ. An optical
depth of order 10 is required to efficiently absorb the incident light. To store N
such spectral modes ‘side by side’ in frequency, we should have a total optical depth
d ∼ 10N , spread over a spectral width ∆0 ∼ 2γN . That is, the multimode capacity
scales linearly with d, and so does the broadening ∆0, which is precisely what we have
found to be the case for CRIB. And it is clear why there is an optimal broadening:
if ∆0 is too large, there is insufficient optical depth over the bandwidth of a mode
for efficient absorption.
In the next section, we consider a modification to the Raman protocol which
takes advantage of spectral broadening to improve its multimode scaling.
0 100 200 300 400 5000
10
20
Figure 7.8 Multimode scaling for CRIB memories. The multi-mode capacity N is shown as a function of the total optical depth dfor tCRIB (green dashed line), lCRIB (red dotted line) and, for com-parison, unbroadened EIT and Raman protocols (solid blue line).For the tCRIB calculation, a numerical SVD was applied directlyto the kernel (7.38). For lCRIB, we constructed the storage kernelby solving the equations of motion (7.11) numerically — we foundthis to be the most reliable method, and of course no approximationsare required. Nonetheless some numerical error is apparent at largebroadenings, since the numerical problem becomes increasingly stiffas ∆0 increases. For both the CRIB protocols, we optimized N over∆0, and as expected, the optimal broadening width was found toscale linearly with d. We set ηthreshold = 70% for all calculations.

7.4 Broadened Raman 262
7.4 Broadened Raman
Is it possible to improve the multimode capacity of the EIT and Raman protocols by
incorporating a spectral broadening? Here we consider a simple modification to the
Raman protocol, in which a longitudinal broadening is applied to the storage state.
That is, the energy of the state |3〉 varies linearly along the z-axis, covering a range
of frequencies ∆0. The treatment is very similar to that of lCRIB given earlier,
and we will see that it is indeed possible to recover the linear multimode scaling
characteristic of CRIB. As usual, we start with the equations of motion describing
one dimensional propagation. Adapting the system (5.106) to the present scenario,
with an added broadening, we have
∂zA(z, τ) = −√dP (z, τ),
∂τP (z, τ) = −ΓP (z, τ) +√dA(z, τ)− iΩ(τ)B(z, τ),
∂τB(z, τ) = i∆0
(z − 1
2
)B(z, τ)− iΩ∗(τ)P (z, τ). (7.40)
The first term on the right hand side of the Heisenberg equation for B just describes
a position-dependent energy shift of the storage state. We make no attempt to solve
these equations exactly. Rather, we proceed directly to study the adiabatic limit,
in which any driven dynamics are much slower than the timescale 1/∆ set by the
detuning. In this limit, we can eliminate the polarization P , by setting the left hand
side of the second equation to zero (see §5.3.3 in Chapter 5, and also the papers by
Gorshkov et al. [133]). Solving the resulting algebraic equation for P , and substituting

7.4 Broadened Raman 263
the result into the other two equations yields the system
[∂z +
d
Γ
]A(z, τ) = i
Ω(τ)√d
ΓB(z, τ),[
∂τ +|Ω(τ)|2
Γ− i∆0
(z − 1
2
)]B(z, τ) = −i
Ω∗(τ)√d
ΓA(z, τ). (7.41)
As we did for lCRIB, we now apply a unilateral Fourier transform over z. The
adiabatic equations of motion in k-space are then
A(k, τ) =Γ√2πAin(τ) + iΩ(τ)
√dB(k, τ)
d− ikΓ,[
∂τ −∆0∂k + i∆0
2+|Ω(τ)|2
Γ
]B(k, τ) = −i
Ω∗(τ)√d
ΓA(k, τ). (7.42)
Again we encounter the combined derivatives ∂τ − ∆0∂k, which we deal with by
transforming from the coordinates (k, τ) to (s, τ), where s = k+∆0τ . The derivatives
can then be replaced with ∂τ , where now s is held constant. Substituting the first
equation of (7.42) into the second, and integrating, we arrive at the solution
Bout(s) = Bin(s)e−R∞−∞ f(s,τ) dτ−i
√d
2π
∫ ∞−∞
Ω∗(τ)d− iΓ(s−∆0τ)
e−R∞τ f(s,τ ′) dτ ′Ain(τ) dτ,
(7.43)
where the function f is given by
f(s, τ) = i∆0
2+|Ω(τ)|2
Γ
[1− d
d− iΓ(s−∆0τ)
]. (7.44)

7.4 Broadened Raman 264
To model the storage process, we set Bin = 0. At the end of the storage interaction,
at time τout −→∞, the coordinate s is given by s = k+∆0τout. That is, k is related
to s by a constant offset. This does not affect the norm of Bout, so it does not affect
the efficiency of the memory. In the following we therefore make the replacement
s −→ k, since k has the clearer physical meaning: it is the spatial frequency of the
spin wave. We also drop the first term i∆0/2 from f , since it represents only a phase
rotation, which also does not affect the memory efficiency. The storage map for a
broadened Raman memory can then be written as
Bout(k) =∫ ∞−∞
K(k, τ)Ain(τ) dτ, (7.45)
where the k-space storage kernel is given by
K(k, τ) = −i
√d
2πΩ∗(τ)g(k, τ)e−
1Γ
R∞τ |Ω(τ ′)|2[1−dg(k,τ ′)] dτ ′ , (7.46)
with g defined by
g(k, τ) =1
d− iΓ(k −∆0τ). (7.47)
Note that the broadening introduces a timescale into the dynamics that is not set
by the control field, so we cannot conveniently write this kernel in terms of the
integrated Rabi frequency used in Chapter 5 (see (5.46)). But if we set ∆0 = 0,
(7.46) does indeed reduce to the standard adiabatic storage kernel (5.74) for a Λ-type
memory.

7.4 Broadened Raman 265
We can extract the singular values from (7.46) with a numerical SVD. This can be
a little problematic, since for large detunings the kernel becomes nearly singular. But
it is easy to resolve this issue by introducing a regularization, Fourier transforming,
and then compensating — this ‘Fourier method’ is described at the end of §7.3.1
above, where we applied it to the k-space storage kernel for lCRIB, which is also
singular. In Figure 7.9 we show how the square root scaling of the unbroadened
protocol is transformed into linear scaling upon application of a broadening. To
see how this scaling arises from the structure of the storage kernel, we repeat the
arguments used for lCRIB which allow us to simplify the kernel. We consider the
special case of control field with a rectangular profile, since this allows us to perform
the integral in the exponent of K,
Ω(τ) =
Ωmax for 0 ≤ τ ≤ T ,
0 otherwise.
(7.48)
The kernel evaluates to
K(k, τ) = −i
√d
2πΩmaxg(k, τ)e−Ω2
maxT/Γ
[g(k, τ)g(k, T )
]idΩ2
maxΓ2∆0
. (7.49)
In the Raman limit ∆ 1, we can write Γ ≈ −i∆, to obtain
K(k, τ) = −iC∆√2πT
e−iΩ2maxT/∆ × [g(k, T )]i
C2
∆0T × [g(k, τ)]1−i C2
∆0T , (7.50)

7.4 Broadened Raman 266
where C is the Raman coupling (this is defined in (5.94) in §5.3 of Chapter 5). In
this far-detuned limit, the exponential factor is a pure phase rotation that we are
free to remove, and so is the factor involving g(k, T ), since g is purely real and it is
here raised to a purely imaginary power. As in our treatment of lCRIB, there is a
logarithmic singularity at g(k, T ) = 0, but its effect becomes negligible in the limit
∆0 C2/T of large broadening. Dropping these terms, and some other spurious
phase factors, we find
K(k, τ) =C√2πT
(d
∆− k −∆0τ
)i C2
∆0T−1
. (7.51)
Taking the inverse Fourier transform, with the help of Mathematica, and the shift
theorem, and dropping a further phase factor, we find the z-space kernel
K(z, τ) = α(C2/∆0T
)√C2/Te−i∆0zτ , (7.52)
where the function α is defined in (7.21) in §7.3.2 above. This kernel is nearly
identical in form to (7.22). Instead of being damped in time by the factor e−τ due
to the spontaneous lifetime of the excited state, the kernel is instead truncated at
τ = T , when the control field is switched off. But the functional form is the same:
an exponential parameterized only by the spectral broadening ∆0. The magnitude
of the kernel is also the same as for (7.22), except the optical depth d has been
swapped for the quantity C2/T . By analogy with the facts we know for lCRIB, we
therefore conclude the following. First, in the limit of large broadening, the Schmidt

7.4 Broadened Raman 267
number of the Raman memory depends only on ∆0. Second, the multimode capacity
N , given some threshold efficiency, scales linearly with C2/T , provided that as this
quantity is increased, the applied broadening ∆0 is also increased proportionately.
Note finally that C2 ∝ d, so the multimode capacity scales linearly with the optical
depth, as it does for CRIB.
These assertions are confirmed by the results of a numerical SVD performed on
the kernel (7.46) using the ‘Fourier method’ described above. We used a Gaussian
control pulse, but the shape of the control makes no difference to the multimode
scaling in the adiabatic limit. This is to be expected, since in this regime the dy-
namics adiabatically follow the control, and so its temporal profile only affects the
shapes of the input modes, not the efficiency of the memory. For the parameters
shown, we found N ∼ d/300, with an optimal broadening width of ∆0 ∼ d/77.
We should remark that the multimode capacity is much smaller than that found
for CRIB, or even than is predicted by the optimal kernel KA in (7.4). This is
because the memory operates far off resonance, so that a much higher optical depth
is required to achieve a strong interaction. Of course the other advantages of Ra-
man storage — for instance, broadband capability, tunability, and insensitivity to
unwanted inhomogeneous broadening — are retained in the present scheme. Since
investigating this protocol, a demonstration has been implemented by Hetet et al.
in Canberra [164], although only a single optical mode was stored and retrieved: this
is quite difficult enough to start with! An analysis of this protocol was also recently
conducted by Moiseev and Tittel [165].

7.5 AFC 268
0 1000 2000 30000
5
10
Figure 7.9 The multimode scaling of a broadened Raman proto-col. The blue solid line shows the multimode capacity calculatedfrom the kernel (7.46) using the ‘Fourier method’, optimized over thewidth ∆0 of the applied broadening. The green dashed line showsthe square-root scaling obtained if ∆0 is set equal to zero. We used aGaussian control pulse, with Ω(τ) =
√10de−(τ/0.1)2 , and a detuning
of ∆ =√
90d. These parameters maintain the adiabatic conditionthat ∆ Ωmax, so that we remain in the Raman limit as the cou-pling is increased. We used a threshold efficiency of 70% for bothcalculations.
The last memory protocol we consider is the AFC memory protocol proposed by
Afzelius et al. in Geneva [91], which is introduced in §2.3.4 of Chapter 2.
7.5 AFC
In the AFC protocol, an ensemble with a naturally broad inhomogeneous absorption
line is prepared by optical pumping, producing an atomic frequency comb. That is,
atoms are removed (or pumped into a ‘shelf’ state) that have resonant frequencies
lying between the ‘teeth’ of a spectral comb, so that the ensemble only absorbs light
at the evenly spaced frequencies of the comb teeth. The great advantage of this
approach, is that the broad spectral bandwidth covered by the ensemble absorp-
tion arises naturally. Adding more teeth to the comb to increase the absorption

7.5 AFC 269
bandwidth only requires that fewer atoms are removed, the density or size of the
ensemble need not be increased. This should be contrasted with CRIB, in which
broadening the absorption bandwidth requires an increase in the total optical depth,
if the same level of absorption is to be maintained. As a result, the multimode ca-
pacity of AFC does not depend on the density of the ensemble, making it by far the
most ‘multimode’ protocol yet proposed.
We model AFC using precisely the same approach as we used for tCRIB. We
assume that we have succeeded in preparing an ensemble so that it has an inho-
mogeneous absorption profile that takes the form of a series of M equally spaced
resonances, covering a total spectral width ∆0, each with optical depth d,
p(∆) =M∑j=1
δ(∆− δj), where δj = ∆0
[j − 1M − 1
− 12
]. (7.53)
Note that we have defined p so that∫p(∆) d∆ = M . The total optical depth associ-
ated with the entire frequency comb is then dtotal = Md. We reserve the designation
d for the optical depth associated with a single tooth of the frequency comb, since
this is set by the density of the ensemble. This definition allows direct comparison
of AFC with the other protocols studied in this Chapter, where d quantifies the
physical resources — density, length — required to build the memory.
The lineshape function for AFC is a sum of Lorentzian lines,
f(ω) =M∑j=1
11− i(δj + ω)
(7.54)

7.5 AFC 270
(again, recall that the ‘1’ in the denominator represents the natural atomic linewidth
in our normalized units). We construct the Green’s function for the AFC memory
in the same way as we did for tCRIB. The only difference is that the inhomogeneous
profile is not flipped around for retrieval: the discrete structure of the comb means
that the phase of the atomic dipoles undergoes periodic revivals, without requiring
any external meddling. We still consider phasematched retrieval in the backward
direction, however, since this is the optimal situation. The equivalent expression to
(7.37) for AFC is therefore
Ktotal(ωr, ω) = −e−iωT
∫p(∆)
[Γ(∆)− iωr][Γ(∆)− iω]d∆× d
2π
∫ 1
0e−d(1−zr)[f(ωr)+f(ω)] dzr,
(7.55)
where we used ∆r = ∆, pr = p and f r = f . Performing the integrals, and dropping
the unnecessary phase factor, we find the Green’s function for AFC to be
Ktotal(ωr, ω) =1
2πf(ωr)− f(ω)
ωr − ω× e−d[f(ωr)+f(ω)] − 1
f(ωr) + f(ω). (7.56)
The first quotient on the right hand side exhibits a removable singularity at ωr = ω,
but this can be dealt with using L’Hopital’s rule:
Ktotal(ω, ω) =1
2πf ′(ω)
e−2df(ω) − 12f(ω)
, where f ′(ω) =M∑j=1
iω[1− i(δj + ω)]2
.
(7.57)
In Figure 7.10 we show the multimode scaling for AFC derived from the SVD of this
kernel. In part (a) we show the multimode capacityN as function of the tooth optical

7.5 AFC 271
depth d, for various numbers of comb teeth. For each number of teeth M , the square
root scaling characteristic of an unbroadened memory is apparent. But it is possible
to increase the multimode capacity arbitrarily, just by adding more teeth to the
comb. In principle, the multimode capacity of this memory protocol is infinite! Of
course, there is a limitation in practice. First, the number of modes stored can never
exceed the number of atoms in the ensemble. A more important restriction however
comes from the spectral width of the initial inhomogeneous line. To achieve efficient
retrieval, the teeth comprising the comb must be ‘well-separated’, so that they do
not overlap in frequency. If they overlap, the re-phasing of the atomic dipoles will
not be complete, and there will be only partial retrieval of the signal field. Because
the lineshape function f is a sum of Lorentzians, which are functions that do not
have compact support, a rather large frequency separation between the comb teeth
is desirable. Therefore, as more teeth are added to the comb, the total width ∆0 of
the comb must be increased, in order to accomodate the increased number of teeth
with the same separation between them. Eventually, the width of the comb will
approach the width of the initial inhomogeneous absorption profile from which the
comb was prepared. More teeth cannot then be added without compromising the
efficiency of the retrieval process. We found that the memory efficiency begins to
suffer seriously if the finesse F falls below around 30, with F defined by
F =∆0
2(M − 1). (7.58)

7.5 AFC 272
The finesse is just the ratio of the tooth separation δj+1− δj and the natural atomic
linewidth 2 (the FWHM in normalized units). For our threshold of ηthreshold = 70%,
a finesse of greater than 100 was required.
In part (b) of Figure 7.10 we show the multimode scaling of AFC as a function
of the total optical depth dtotal, alongside the scaling for CRIB and unbroadened
protocols. For these protocols, dtotal = d; the plots are simply reproduced from
Figure 7.8. This plot shows that, as we might expect, the multimode capacity of
AFC does indeed scale linearly with dtotal. That is, N remains proportional to the
total number of atoms available for the protocol. The advantage of AFC lies in the
use of a ‘natural resource’ of atoms, so that increasing the number of atoms used in
the protocol does not require a higher ensemble density. Note however, that even
as a function of dtotal, the AFC protocol still outperforms CRIB, with a scaling of
N ∼ 2d/25, and an optimal comb width of ∆opt0 ∼ 5d.
This concludes our investigation of multimode storage. The techniques used are
quite general, and are readily applicable to new quantum memory protocols as they
are invented. Experimental implementations of multimode storage will likely by
challenging, but the technical advantages for applications of memory to quantum
communication makes the research a worthwhile endeavour.
Practically, it is hard to imagine how one might encode or detect photons in
the optimal input modes φk(τ). These modes have non-trivial shapes, which do
not necessarily overlap well with the ‘natural’ modes of photon detectors. Gener-
ally these detectors, be they avalanche photodiodes (APDs), photomultiplier tubes

7.5 AFC 273
(PMTs) or superconducting bolometers (SSPDs), are engineered to have a broad,
flat spectral response. This means they couple to a temporal mode that looks like
a short, sharp, spike. The natural modes for single photon detection are therefore
time-bin modes, where information is encoded in the time of arrival of a photon.
These time-bin modes are not usually the same as the φk, so time-bin encoding is
sub-optimal, and the multimode capacity predicted by the SVD cannot be reached.
But the discrepancy between the optimal capacity N and the achieved capacity
depends on the overlap between the subspace of signal profiles spanned by the time-
bin modes with the subspace spanned by the first N optimal modes. As N becomes
large, this overlap grows, and the capacity for time-bin modes rapidly approaches
the optimal capacity. Without attempting a detailed calculation, the plausibility of
this claim can be appreciated by comparing parts (a) and (b) in Figure 7.5 in §7.3.2.
Here, we estimated the Schmidt number by decomposing the Green’s function us-
ing non-ovelapping rectangular pulses as spatial modes. If we consider a Green’s
function defined in the temporal domain, such pulses are precisely time-bin modes.
And it is clear that in part (a), the multimode Green’s function is more faithfully
reconstructed than its few-mode counterpart in part (b). This is quite general. As
a Green’s function becomes multimode, it admits an increasingly fine-grained de-
composition in terms of time-bin modes. This shows that the time-bin basis quickly
becomes ‘just as good’ as the φk. Therefore the multimode capacity for time-bin
modes fast approaches that calculated from the SVD. A similar argument applies
if a frequency-bin encoding is used, or, for that matter, if any other orthonormal

7.5 AFC 274
basis is chosen for the signal alphabet. The multimode capacities calculated in this
chapter can accordingly be characterized as a tight upper bound on the performance
of the protocol concerned.
In the next chapter, we wrap up our analysis of quantum memory by addressing
the optimization of storage when the signal field is given, and cannot be ‘shaped’.

7.5 AFC 275
0 100 200 300 400 5000
50
100
150
2
8
14
20
0 100 200 300 400 500
0
10
20
30
40
(a) (b)
Figure 7.10 The multimode scaling of the AFC memory protocol.(a): We show the multimode capacity of AFC for various numbers ofcomb teeth — indicated by the numbers in the plot — as a functionof the optical depth d associated with a single comb tooth. For eachnumber of comb teeth M , the capacity scales with the square root ofd, just like an unbroadened protocol. But adding more teeth allowsto increase N arbitrarily, so the multimode capacity is not limitedby the optical depth. (b): Here we show the multimode capacityof AFC (green dashed line) as a function of the total optical depthdtotal = Md associated with the entire frequency comb. This showsthat N scales linearly with the total number of atoms involved inthe protocol, just as it does for the CRIB protocols. For comparison,the capacities of tCRIB, lCRIB and EIT/Raman protocols are alsoshown (red dotted line, black dot-dashed line and blue solid line,respectively). For these latter protocols dtotal = d, so these capacitiesare just taken from Figure 7.8. It is interesting that the capacity ofAFC is larger than that of CRIB, even when evaluated as a functionof dtotal. Of course, d is the more relevant physical resource for AFC,since this sets the ensemble density. We optimized the capacitiesplotted for AFC in both (a) and (b) over the spectral width ∆0 of thefrequency comb. The threshold efficiency used was ηthreshold = 70%,as usual.

Chapter 8
Optimizing the Control
The SVD has proved an extremely useful tool for the analysis of quantum storage.
Given a set of parameters, including the ensemble geometry and density, as well
as the detuning and the control pulse profile, it is possible to construct a Green’s
function that contains all the information we might want to know about the memory
interaction. In particular, taking its SVD provides us with the optimal input mode,
so that we can achieve efficient storage by shaping the signal field. But suppose that
we are simply given a signal field, and asked to store it. This is probably the more
likely situation in practical applications, where we have control over our memory,
but not over the source that generates the signal field to be stored. In this case we
are not able to shape the signal so that it matches the optimal mode φ1. How do we
achieve efficient storage in this situation? We must try to deform the optimal mode
φ1 into the shape of our signal! This can be done by shaping the control field in
order to sculpt the Green’s function; in this chapter we will explore how the correct

8.1 Adiabatic shaping 277
shape for the control may be found.
8.1 Adiabatic shaping
The adiabatic limit is precisely the limit in which the natural atomic response is
sufficiently fast that the atoms can ‘follow’ the driving fields. In this limit, the atomic
response function is completely determined by the control field profile. Therefore
the adiabatic limit is the regime we should work in, if our aim is to be able to affect
the Green’s function by shaping the control. Fortunately, another consequence of
adiabatic following is that the Green’s function may be completely parameterized by
the integrated Rabi frequency ω, defined in (5.46) in §5.2.6 of Chapter 5. Later in
Chapter 5 we derived an analytic solution for the adiabatic storage kernel of a Λ-type
ensemble memory (5.77). It is extremely convenient that the optimal input mode
found from the SVD of this kernel applies universally for all control profiles (within
the adiabatic limit, of course). Given a control profile, it is easy to find the temporal
shape of the optimal input mode, simply by converting back from the ω to τ . The
conversion is given by (5.78). The optimization problem in this case is then simple.
We must adjust the control field until the shape of the optimal temporal mode is the
same as the shape of the signal field we intend to store. Changing the shape of the
control pulse has no effect on the singular values, since the storage kernel depends
only the control pulse energy through W . Therefore, when the shaping is complete,
the same storage efficiency is achieved, as would have been if we had shaped the
signal field and left the control fixed.

8.1 Adiabatic shaping 278
In Figure 8.1 we show some examples of this kind of optimization. The control
field is parameterized by a vector Ω of 2N real numbers, giving the real and imagi-
nary parts of Ω(τ) at a set τj of N discrete points in time. We choose the points τj
to lie on a Chebyshev grid, since this makes polynomial interpolation of the control
field to other times numerically stable (see §E.1.2 in Appendix E). Starting with
the initial guess Ω(τj) = Ain(τj), we optimize Ω using a simplex search algorithm —
‘fminsearch’ in Matlab — to minimize the norm of the difference Ain(τ) − φ1(τ).
At each iteration the optimal mode φ1(τ) is determined from Ω by interpolating to
find the envelope Ω(τ), and then using (5.78). Convergence does not take longer
than 1 minute on a 3 GHz machine. After each optimization, we use the optimized
control profile along with the numerical method described in §5.4 of Chapter 5 to
construct the storage kernel without the adiabatic approximation. The resulting op-
timal mode is then shown for comparison with the signal profile to be stored. This
provides a direct way to examine the accuracy of the optimizations. The method
used relies on the validity of the adiabatic kernel (5.77), and it is discernible that the
optimization performs better in the Raman limit than for EIT, since the adiabatic
approximation is less robust on resonance (this is discussed in §5.2.9 of Chapter 5).
As with so much of this thesis, an excellent account of closely related work can
be found in the papers of Gorshkov et al. [133], who studied this problem in the light-
biased limit of large control pulse energy, where the optimal kernel (5.27) is valid (see
§5.2.8 in Chapter 5). They did not use the SVD, or the integrated Rabi frequency
ω, so their approach is a little more convoluted, but nonetheless it is accurate and

8.2 Non-adiabatic shaping 279
ingenious.
8.2 Non-adiabatic shaping
The opposite to the adiabatic limit is the limit of a very short, broadband control
pulse. If the pulse is sufficiently short, there is no time for the coherence induced
on the |2〉 ↔ |3〉 transition to couple to the optical polarization P connecting states
|1〉 and |2〉. In this limit, the shape of the control field makes no difference to the
form of Green’s function describing absorption of the signal. Instead, the control
simply induces Rabi oscillations between |2〉 and |3〉 that transfer population from
the excited state to the storage state. The efficiency of this population transfer
is given by sin2(θ/2), where θ = 2∫
Ω(τ) dτ is the pulse area (note the difference
between this dimensionless quantity and the integrated Rabi frequency ω, which is
relevant in the adiabatic limit). Perfect transfer is achieved by an infinitely short
π-pulse: this kind of instantaneous map is assumed in the CRIB and AFC protocols
treated in the last Chapter, and in the derivation of the optimal kernel (5.27) in
Chapter 5. If we use such a control pulse, the optimal signal input mode is fixed
by the homogeneous and inhomogeneous lineshape of the ensemble. If the signal
field mode does not coincide with this optimal mode, there is nothing we can do to
optimize the storage efficiency.
But there is a large middle-ground between the adiabatic regime and the extreme
case of a π-pulse control. In this middle-ground, the analytic solution for the storage
kernel (5.77) is of limited use, but the shape of the control field still has an effect, at

8.2 Non-adiabatic shaping 280
least to some degree, on the form of the storage Green’s function. Since the analytic
solution breaks down, we must resort to numerical solutions of the equations of
motion to optimize the control. The method we use is the most direct method one
could imagine. Given a vector Ω parameterizing the control, we construct the full
control profile Ω(τ) by interpolation, and then we integrate the equations of motion
numerically, and we extract the storage efficiency η. We then use a simplex search
algorithm to optimize the elements of Ω so as to maximize η — using fminsearch
in Matlab, we minimize −η. This approach works well, but relies on the ability to
repeatedly solve the equations of motion quickly and accurately. Fortunately, this
is possible using the method described in Appendix E, where we use Chebyshev
spectral collocation for the spatial propagation, and RK2 for the time-stepping.
To improve the convergence of the optimization, it helps to start with an initial
guess that is already close to optimal. We can make use of the SVD in this con-
nection. We start with an initial guess Ω, determined by setting Ω(τ) = Ain(τ).
This is motivated by the fact that φ1(τ) = Ω(τ) in the adiabatic limit, when the
coupling is small (i.e. when d or C is small), so it is a reasonable opening gambit.
Next we use the numerical method described in §5.2.9 of Chapter 5 to construct the
resulting Green’s function, and we take its SVD to extract the optimal input mode
φ1(τ). In general, since the coupling is not small, φ1(τ) 6= Ain(τ) — this is why the
optimization is necessary. By fitting a cubic spline interpolant to φ1, and also to
Ain, we are able to build a sequence of n functions φ(k)1 that represent a smooth
deformation of this optimal mode into Ain. That is, φ(1)1 = φ1, and φ(n)
1 = Ain, with

8.2 Non-adiabatic shaping 281
the intervening functions lying ‘between’ these two. This is easily done in Matlab by
building the φ(k)1 using splines with coefficients found by interpolating between the
coefficients for φ1 and Ain. Now, Ω already describes the optimal control for storing
the mode φ(1)1 , by construction. We now use the optimization method described
in the previous paragraph to optimize Ω for storing the mode φ(2)1 . The hope is
that this is sufficiently ‘close’ to φ(1)1 that the optimization will converge quickly.
We then repeat the optimization, this time using the newly optimized vector Ω as
our initial guess, and using the mode φ(3)1 as the target input mode. The pattern
is now clear. We iterate these steps, each time switching the target input mode
from φ(k)1 to φ(k+1)
1 , and using the previous result for Ω as an initial guess. As we
procede, the target modes approach Ain, and Ω approaches the vector describing
the optimal control for storing Ain. The rationale behind this approach is that the
control remains near-optimal at all times. We begin with an optimal control for the
‘wrong’ target, and by degrees we deform this target into Ain, all the time ‘bringing
along’ the control. We found that this method is helpful in the most non-adiabatic
situations, where the initial guess Ω(τ) = Ain(τ) is particularly poor. When the
dynamics are more adiabatic, optimizing for Ain directly in a single step is often
sufficient.
In Figure 8.2 we show some examples of this type of optimization. Clearly the
numerical optimization works well where the analytic optimization described in the
previous section fails. The method is not particularly time consuming on a modern
computer, although it is of course slower than the analytic optimization. It is quite

8.2 Non-adiabatic shaping 282
general though, and it provides an easy way to optimize the storage efficiency for a
given input, and also to check the final proximity of the optimal input mode to the
desired shape.
Gorshkov et al. have also studied the optimization of the control field outside the
adiabatic limit, in the fourth of their series of papers on the subject [166]. They use
an interesting approach involving gradient ascent, in which they derive an explicit
formula for the incremental change in the control profile that will improve the storage
efficiency. Numerical solution of the equations of motion provides them with this
incremental change, and by iterating they are able to generate the optimal control
for storage. This is a robust and efficient optimization that produces very similar
results to the method we describe here. They comment that they are not able to
verify the optimality of their results, and this is true for our optimizations too. But
in both cases, the SVD of the numerically constructed Green’s function does make it
possible to check the resemblance between the desired input mode, and the optimal
mode resulting from the control. When these two modes match up, as they do in
the cases shown in Figure 8.2, it is clear that the optimization has converged. When
these modes do not match, of course, it is not obvious whether the result is a global
or simply a local optimum.
This completes our theoretical treatment of optimal storage and retrieval from
ensemble memories. In the next Chapter we present a derivation of the equations
of motion for the memory interaction in diamond, since this is one of the media we
have used in our experiments, and the theory of Chapter 4 is not directly applicable.

8.2 Non-adiabatic shaping 283
Finally, in Chapter 10 we review the status of our experiments, before concluding
the thesis in Chapter 11.

8.2 Non-adiabatic shaping 284
0
0.5
1Broadband Raman
Inte
nsi
ty (
arb
. un
its)
Inte
nsi
ty (
arb
. un
its)
0
2
4
6
8
Broadband EIT
Ph
ase
Ph
ase
0
2
4
6
−2 0 20
0.5
1Narrowband EIT
0
2
4
6
8
−2 0 2
−0.2 0 0.2 −0.2 0 0.2
Non-adiabatic
0
2
4
6
(a) (b)
(d)(c)
Figure 8.1 Adiabatic control shaping. Given an input signal pro-file, as well as the available control pulse energy W , optical depth dand detuning ∆ we construct the adiabatic kernel (5.77), and extractthe optimal input mode, as a function of the integrated Rabi fre-quency ω. We then optimize the control profile Ω(τ) until the tempo-ral profile of the optimal mode matches the given signal. In parts (a)to (d), the signal is assumed to be a Gaussian, Ain(τ) = e−[(τ−τs)/Ts]2 .Its intensity profile |Ain(τ)|2 is shown as a dashed black line. Thetiming of the signal pulse τs is arbitrary, and in each case we chooseit so as to aid the convergence of the optimization. The temporalintensity profile |Ω(τ)|2 of the optimized control is represented bythe blue solid line, with its temporal phase shown by the red solidline (referred to the axes on the right-hand side). We used N = 21Chebyshev points to parameterize the control in all cases. The greendotted lines show the temporal intensity profiles of the optimal inputmode determined from the optimized control using numerical integra-tion to construct the Green’s function. These should coincide withthe signal mode, if the optimization has succeeded. Part (a) showsthe result for an optimization with W = 2.5, ∆ = 150 and d = 300,and a signal duration of Ts = 0.1, all in normalized units. This de-scribes off-resonant Raman storage. The adiabatic approximation iswell satisfied, and the optimization performs well. In part (b) weshow the result for the same optimization when the detuning is setto zero, which describes broadband EIT. Here the adiabatic approx-imation is not well satisfied, and the optimization performs ratherpoorly. In part (c) we optimize the control for a narrowband signalwith Ts = 1 (note the difference in time scale on the horizontal axis).The optimization performs much better. Finally in part (d) we re-duce the optical depth to d = 10, and we increase the control energyto W = 49. The detuning is set to 0, and the signal duration remainsTs = 1. Despite the narrow signal bandwidth, the adiabatic approx-imation that worked well for part (c) is now ‘broken’, because of thelarge Rabi frequency, and the optimization fails entirely. The storageefficiencies achieved in parts (a) to (d) were 94%, 74%, 94% and 54%,respectively. For comparison, the efficiencies that would have beenachieved if the signal field were exactly equal to φ1(τ) were 97%, 99%,96% and 82%.

8.2 Non-adiabatic shaping 285
0
5
10
0
2
4
6
0
5
10
0
2
4
6
0
0.5
1In
ten
sity
(a
rb. u
nit
s)In
ten
sity
(a
rb. u
nit
s)
0
0.5
1
Ph
ase
Ph
ase
Broadband Raman Broadband EIT
−2 0 2
Narrowband EIT
−2 0 2
−0.2 0 0.2 −0.2 0 0.2
Non-adiabatic
(a) (b)
(d)(c)
Figure 8.2 Non-adiabatic control shaping. Parts (a) to (d) showthe results of the direct numerical optimization described above,where n = 10 steps were used in deforming the initial target Ain(τ) =φ
(1)1 (τ) into the final target Ain(τ) = e−[(τ−τs)/Ts]2 . The optimiza-
tions each ran in around 2 minutes on a 3 GHz machine. The pa-rameters are identical to those used in parts (a) to (d) of Figure 8.1.The numerical optimization copes well with non-adiabatic dynam-ics, and in all cases comparison of the target signal mode with theoptimized input mode shows that the optimizations have met withsome success. In part (b), the broad bandwidth of the signal makesthe adiabatic approximation poor, and it is noticeable that the opti-mized control profile features a small oscillation which is not presentin the control in part (c). The adiabatic approximation fails entirelyin part (d), and here the control involves a large oscillation, with itsenergy distributed into two ‘pulses’. These oscillations are typicalof non-adiabatic shaping, since the ‘ringing’ of the atomic dynam-ics must be compensated by the control to produce a smooth inputmode. The storage efficiencies achieved in parts (a) to (d) were 97%,99.5%, 97% and 81%, respectively. For comparison, the efficienciesthat would have been achieved if the signal field were exactly equalto φ1(τ) were 97%, 99.6%, 97% and 82%.

Chapter 9
Diamond
In this chapter we build a theoretical description of quantum memory in diamond.
The end result is a set of equations with precisely the same form as (5.107) describing
Raman storage in a vapour, so that our analysis of optimal storage and retrieval
applies in diamond just as it does to atomic vapours (see §5.3.3 in Chapter 5). But
some justification of this claim is required, and so we present a derivation below.
9.1 Diamond Scheme
Diamond is a singular material, both physically and aesthetically. It is the hardest
and most transparent mineral, with the highest thermal conductivity of any material,
and a very large refractive index (around 2.4). These properties arise in part from
its extremely simple structure. It is comprised entirely of carbon atoms; each is
connected to four others by strong covalent bonds. The bonds are all equivalent,
and this symmetry produces a tetrahedral arrangement of atoms that is exceedingly

9.1 Diamond Scheme 287
robust. The diamond structure can be visualized by convolving a basis of two carbon
atoms with a face-centred cubic bravais lattice [167], as shown in Figure 9.1.
Figure 9.1 The crystal structure of diamond. The diamond latticeis FCC (face-centred cubic); here we show just a single unit cell,outlined for clarity with thin ‘rods’. Each basis is shown as a pairof atoms connected by a thick ‘tube’, one atom is coloured red; theother grey. The grey atom of each basis is located at the sites of theFCC lattice. Each atom is connected by four covalent bonds (notshown) to its neighbours, forming a tetrahedral pattern.
If the basis is deformed, the bonds produce a large restoring force, and so in
response to an impulse the atoms of the basis can undergo harmonic oscillations
relative to one another. The frequency of these ‘internal’ vibrations is rather high,
because the interatomic bonds are very ‘stiff’. It is these high frequency vibrational
modes we seek to excite when implementing a quantum memory in diamond.
Eventually any relative motion within a basis couples to collective motion of
the basis with respect to its neighbouring bases, and the energy is dissipated as

9.2 Quantization 288
waves of motion of the lattice sites with respect to one another: sound waves. This
process limits the lifetime of a diamond quantum memory, and in fact the lifetime
— on the order of picoseconds — is much too short to make diamond a useful
medium for quantum storage. Nonetheless, it is possible to store very broadband
pulses in diamond, because the oscillation frequency is so high, and a solid-state,
room-temperature, broadband quantum memory is interesting in its own right.
9.2 Quantization
Just as the electromagnetic field can be quantized, revealing photons as the con-
stituents of light, so the harmonic oscillations of a crystal can be quantized. The
quanta of crystal vibrations are known as phonons. Our aim in the present context
is to describe the coherent mapping of a single photon to a single phonon in the
diamond crystal.
The crystal vibrations may be quantized by imposing periodic boundary con-
ditions on the atomic displacements within a cubic crystal of side length L. To
illustrate this procedure we consider a one dimensional wave b(z) = eikz describing
the atomic displacement at position z. Periodic boundary conditions require that
b(0) = b(L), so that we must have kL = 2πm for some integer m. Therefore the
momenta associated with crystal vibrations are quantized, with k = 2πm/L. The
quanta are phonons.
The smallest non-zero wavevector allowed is found by setting m = 1, whence we
obtain the mode separation δk = 2π/L. There is also an upper limit to the allowed

9.2 Quantization 289
wavevectors. The wavelength of any vibration cannot be meaningfully defined if it
falls below 2a, where a is the lattice constant — the distance separating neighbouring
atoms. The wave is only ‘sampled’ at the atomic positions, and the spatial frequency
of the waves cannot exceed the sampling rate. The maximum wavevector, set by
this coarseness of the crystal structure, is ∆k = π/a. As shown in Figure 9.2 below,
any wave with a larger wavevector k is physically indistinguishable from a wave with
momentum k−2∆k, and so we only consider phonons with wavevectors lying within
the range [−∆k,∆k]. This region in k-space is known as the first Brillouin zone,
or just the Brillouin zone. That all physically distinct phonon modes are contained
within the Brillouin zone can be confirmed by counting them: the number of modes
in the Brillouin zone is 2∆k/δk = L/a = N , where N is the number of atoms in
the crystal. So the number of phonons is equal to the number of atoms. This must
be true in one dimension, since each atom has one vibrational degree of freedom.
The generalization to three dimensional vibrations turns the Brillouin zone into a
three dimensional volume in k-space, often with a non-trivial shape. But this is not
important for us. As in most of the rest of this thesis, we will use a one dimensional
model to describe the quantum memory.
The lattice constant in diamond is around 3.6 A, so the Brillouin zone boundary
lies at ∆k ∼ 1010 m−1. By comparison, the wavevector associated with visible light
at around 500 nm is about 107 m−1. Any momentum imparted to the crystal by
interaction with optical fields is therefore very small, on the scale set by the crystal
lattice. The excitations produced by a quantum memory may safely be considered to

9.3 Acoustic and Optical Phonons 290
lie at the zone centre, with δk ∼ 0. This, of course, greatly simplifies our theoretical
description.
dis
pla
cem
en
t
Figure 9.2 Phonon aliasing. A crystal vibration with a wavelengthsmaller than 2a is physically equivalent to one with a longer wave-length. In the example shown, the blue solid line represents the profileof a short wavelength vibration, and the dashed blue line shows theprofile of the equivalent longer wavelength vibration. Note that theatomic positions, indicated by the red circles, are identical for bothwaves. The black circles show the equilibrium positions of the atoms,equally spaced by the lattice constant a.
9.3 Acoustic and Optical Phonons
Phonons come in two varieties, as we have already hinted. Acoustic phonons are
the quanta of sound waves in a crystal. They represent compression and rarefac-
tion within the crystal lattice, and the energy associated with this lattice distortion
clearly vanishes as its wavelength becomes large, since in the limit of infinite wave-
length there is no distortion, and therefore no restoring force. As mentioned above,
optical wavelengths are already much larger than a unit cell, so the energies of op-
tically accessible acoustic phonons are very small. A typical dispersion relation for
acoustic phonons is shown in part (a) of Figure 9.3. The energy of acoustic phonons
rises linearly with their wavevector k for k ∆k. Scattering from low-energy zone-

9.3 Acoustic and Optical Phonons 291
centre acoustic phonons is conventionally known as Brillouin scattering, but it is not
of interest to us here. We will focus instead on Raman scattering, which in crystals
refers to the scattering of light from so-called optical phonons. These represent the
second variety of phonon; they are the quanta associated with the high-frequency
internal vibrations of the crystal bases. The bases oscillate essentially independently
of their neighbours, provided the wavelength of the oscillation is not so small that
their neighbours are ‘pulling’ on them. In the limit of infinite wavelength, the op-
tical phonon energy does not vanish, but is set by the frequency associated with
the normal modes describing the natural internal oscillations of the basis. A typi-
cal dispersion relation for optical phonons is shown in part (a) of Figure 9.3. The
non-vanishing energy of optical phonons at the zone-centre distinguishes them from
acoustic phonons. The Raman interaction in diamond couples an incident signal
field to these zone-centre optical phonons, and it is these phonons that play the
role of the storage state in a diamond quantum memory. Broadly speaking, these
phonons are like the metastable state |3〉 used for storage in the atomic systems
discussed previously.
9.3.1 Decay
In many crystals, the basis is composed of unlike atoms or ions, and so the optical
phonons are associated with an electric dipole moment. This means they couple
strongly to the electromagnetic field — that is why they are given the designation
‘optical’. However in diamond, which is homopolar, with both atoms in its basis

9.3 Acoustic and Optical Phonons 292
Optical branch
Acoustic branch
(a) (b)
Figure 9.3 Phonon dispersion. (a): typical dispersion curves foracoustic and optical phonons. The former have negligible energy nearthe zone-centre, which is the region to which we have access withoptical fields. The latter have large energies, and a flat dispersionrelation close to the zone-centre. It is the zone-centre optical phononsthat we use to store an incident photon. (b): The decay of opticalphonons is dominated by the Klemens channel, in which anharmoniccoupling allows a zone-centre optical phonon (black dot) to decayinto a pair of acoustic phonons with large, opposite momenta (reddots). We only show the dispersion in the first Brillouin zone. Thedotted lines on the left and right hand sides indicate the boundariesof this zone, which are identified, meaning that any we could wrapthe plots around a cylinder and stitch these two lines together: theyrepresent physically equivalent momenta.
identical, there is no separation of charge associated with internal oscillations. The
optical phonons in diamond are therefore not, in fact, optically active. That is, they
do not directly radiate or absorb electromagnetic radiation. This is advantageous for
quantum memory, since the optical phonons in diamond are accordingly longer-lived
than in many other materials. As touched upon above, the dominant decoherence
process for these phonons is the decay into acoustic phonons via anharmonic cou-
plings: the covalent bonds do not behave like perfect springs, and their deviation
from Hooke’s law allows the optical and acoustic phonons to exchange energy. To
conserve momentum the acoustic phonons are produced in pairs with approximately
opposite momenta, as illustrated in part (b) of Figure 9.3. This process is known

9.3 Acoustic and Optical Phonons 293
as the Klemens channel [168,169]. The anharmonicities that give rise to the Klemens
channel are largely geometrical in origin, and the lifetime of optical phonons is only
weakly affected by temperature [170]. The purity and quality of the crystal also con-
tribute, but all diamonds have an optical phonon lifetime1 τp . 10 ps.
9.3.2 Energy
There are three optical phonons in diamond, corresponding to basis oscillations in
three orthogonal directions. The three phonons are degenerate at the zone centre,
because diamond is symmetric with respect to the interchange of these three direc-
tions. With a one dimensional interaction we will excite just a single phonon mode;
the degeneracy of the modes means we need not worry about which mode this is.
The optical phonon energy at zone centre is Ep = 0.17 eV, which corresponds to a
wavenumber of νp = 1332 cm−1, or an angular frequency of ωp = 2.5 × 1014 s−1.
This would correspond to an infra-red wavelength of λp = 7.5 µm. The large phonon
energy in diamond is advantageous for the following reasons:
First, the energy scale kBT associated with room temperature (T ∼ 300 K) is
around 1/40 eV, which is much smaller than the phonon energy. Therefore there
are very few thermally excited optical phonons at room temperature: using the
Boltzmann formula pthermal = e−Ep/kBT we predict a population of around 1.7×10−3
thermal phonons per mode. Therefore demonstrating quantum memory at room1These phonon lifetimes were studied in our research group by Felix Waldermann, and later
by K.C. Lee and Ben Sussman, using a technique they named TCUPS [171]. In these experiments,a pair of delayed pump pulses directed through a diamond crystal produces a corresponding pairof Stokes pulses. The phase coherence, as measured from the visibility of spectral interference,between the two Stokes pulses directly measures the coherence of the optical phonons.

9.4 Raman interaction 294
temperature in diamond is feasible.
Second, the bandwidth of the stored signal field cannot exceed the phonon fre-
quency, since ωp sets the frequency difference between the signal and control fields,
and these should not overlap spectrally. Since the phonon frequency is so large, the
signal bandwidth can be large, meaning that a short pulse can be stored. Taking
τs ∼ 1/ωp as a rough estimate of the shortest signal pulse duration that can be
stored, we find τs ∼ 1000τp. If the ratio of the shortest storable pulse duration
to the maximum storage time τs/τp is taken as a figure of merit for a memory, a
diamond quantum memory is actually rather impressive!
9.4 Raman interaction
9.4.1 Excitons
The Raman interaction in diamond involves an intermediate state, just as it does in
the atomic case considered in earlier chapters. The optical fields are detuned from
resonance with this state, but the interaction nonetheless requires strong coupling
to this state to mediate the storage of the signal field. The relevant intermediate
state in diamond is an exciton. To understand what this is, recall that the electronic
orbitals in an extended crystal arrange themselves into disjoint bands. Electronic
band structure arises from Bragg scattering of electrons from the periodic potential
associated with the regular lattice of atomic nuclei in the crystal. As the De Broglie
wavelength of an electron approaches 2a, the reflected and transmitted components

9.4 Raman interaction 295
of the electronic wavefunctions interfere destructively, leading to the appearance of
forbidden energy bands, containing no allowed electronic states, at the edges of the
Brillouin zone. This is illustrated in Figure 9.4.
Free(a) Periodic(b) Bands(c)
Figure 9.4 Band structure. (a): the dispersion relation of a freeelectron is parabolic, since its kinetic energy E = p2/2m is quadraticin k. (b): electrons in a periodic lattice must have a periodic dis-persion relation. To a first approximation, this is found by simplyadding ‘copies’ of the free electron dispersion relation at intervals of2∆k = 2π/a in k-space. (c): scattering of the electrons from the pe-riodic potential produced by the atomic cores results in anti-crossingsin the dispersion relations, leaving energy gaps, shown shaded in gray.Here we have removed any electronic states lying outside the firstBrillouin zone (red shaded area), since they are not physically mean-ingful.
An exciton is produced when an electron in the lower energy band, the valence
band, is promoted into the upper energy band, the conduction band, leaving behind
a hole. The hole in the valence band simply represents the absence of an electron,
but it is convenient to think of it as a particle in its own right, with positive charge,
negative energy and negative mass. An exciton is the combined system of an electron
in the conduction band and a hole in the valence band (see Figure 9.5). In fact, since
these two particles have opposite charge, they attract one another, and it is possible
for them to form a bound system, similar to a hydrogen atom, or the positronium

9.4 Raman interaction 296
of particle physics. The binding energy is rather small, however, and here we will
treat the electron and hole as if they were free particles2.
Conduction band
Valence band
Figure 9.5 An exciton. A photon (not shown) promotes an elec-tron (blue dot) from the valence band into the conduction band,leaving behind a positively charged hole (red dot). Note that thecurvature of the valence band for the hole has been reversed, sincethe hole has negative energy. In this picture, the electron and holehave approximately equal and opposite momenta, so that the totalmomentum of the exciton is small, consistent with the small momen-tum of the photon.
9.4.2 Deformation Potential
An incident photon can produce an exciton if it has sufficient energy to breach
the band gap. This process provides the coupling between the optical fields and
the diamond. To excite an optical phonon, there should be some coupling between
excitons and phonons. In polar crystals, there are direct couplings between the
dipole fields of excitons and phonons, in the form of the Frohlich and piezoelectric
interactions [173–176]. In diamond, these long-range electric couplings are absent, but2A precise characterization of the Raman cross-section in diamond does require an account of
bound excitons [172], but our aim is simply to study the feasibility of using this interaction forquantum storage, so we sacrifice rigour for simplicity

9.4 Raman interaction 297
there remains a short range coupling known as the deformation potential. The origin
of this interaction can be understood as follows. The diamond structure takes the
form of two interpenetrating FCC lattices, offset from one another. A zone centre
optical phonon — with infinite wavelength — may then be interpreted as the rigid
displacement of one sub-lattice with respect to the other. At any instant, the crystal
structure is accordingly deformed, much as if it were subject to an external strain,
and the electronic band structure, which depends on electron scattering from the
crystal potential, is altered. Therefore electronic energy levels are coupled to crystal
vibrations. More specifically, excitons are coupled to optical phonons. Optical
phonons are not energetic enough to create or destroy an exciton outright, but
an exciton with some momentum k can scatter from the deformation potential to
produce an exciton with a momentum k′, and an optical phonon with momentum κ.
Phasematching of this process over the length of the crystal ensures that momentum
is conserved, with κ = k − k′.
We now have to hand all the ingredients necessary to unpick the Raman inter-
action in diamond, which is shown in Figure 9.6,
1. A signal photon produces a virtual exciton — ‘virtual’ because the energy
of the signal photon is smaller than the band gap. This is analogous to the
virtual, or dressed state to which the signal couples in atomic systems, when
it is detuned from the excited state |2〉.
2. The virtual exciton scatters from the deformation potential to produce an
exciton with a different momentum, and also a zone-centre optical phonon.

9.4 Raman interaction 298
3. The remaining virtual exciton recombines — the electron decays back into the
valence band, filling the hole — and emits a control photon.
From this description, it’s clear that the Raman interaction in diamond is third
order, rather than second order as it is in the atomic systems considered in earlier
Chapters. In principle this makes it weaker than in atomic systems, but the ex-
tremely high density of electrons in the solid state more than makes up for the extra
perturbative order, and the Raman cross section in diamond is, in fact, extremely
large.
(a) (b)
Figure 9.6 The Raman interaction in diamond. (a): An energylevel diagram for the Raman quantum memory interaction. The rel-evant states are written in the form |n,m〉, where n is the numberof excitons involved, and m is the number of optical phonons. A sig-nal photon (blue wavy arrow) produces a virtual exciton (indicatedby the dotted line, detuned from the real exciton state). The defor-mation potential interaction then produces another virtual exciton,and an optical phonon (orange arrow). Finally, the virtual excitonrecombines, emitting a control photon (green arrow) and leaving asingle optical phonon behind. (b): A Feynman diagram for the sameprocess. Here the dotted lines indicate the world lines of the virtualelectron and hole comprising the intermediate exciton. The opticalphonon is indicated by the orange spiral.

9.5 Propagation in Diamond 299
9.5 Propagation in Diamond
It is advantageous to describe both the optical fields and the excitations of the
diamond crystal in the Heisenberg picture, in order to treat propagation. This
was done for the atomic case in Chapter 4 by studying the dynamics of the flip
operators σjk describing the atomic evolution. The analysis was greatly simplified
by considering just three discrete atomic states. In diamond however the electronic
states form a quasi-continuum in each energy band. It is therefore not immediately
obvious how the approach of Chapter 4 carries over to the present case.
In addition to this issue, there is the problem of how to describe the local dynam-
ics of the crystal excitations. To examine the spatial distribution of these excitations,
we would like to obtain equations for the phonon or exciton amplitudes at some po-
sition z within the crystal. In the atomic case the interaction was entirely local,
since each atom scattered light at a point, and independently of all other atoms.
The local dynamics was therefore determined by the Hamiltonian of a single atom,
and propagation was treated by summing these local contributions. The situation
in diamond is conceptually different. First, phonons are global excitations of the
crystal. Second, the electrons in a crystal are not localized around the atomic cores;
rather they form a quasi-free Fermi gas distributed over the volume of the crystal.
The above problems are essentially cosmetic, as we’ll see below. Our general
strategy is as follows. To work in the Heisenberg picture, we write the crystal

9.5 Propagation in Diamond 300
Hamiltonian H in second-quantized form,
H =∑α,β
Hαβ|α〉〈β|, (9.1)
where in the sum, both α and β run over a complete set of states, and where the
Hαβ = 〈α|H|β〉 are the matrix elements of the Hamiltonian connecting these states.
The dynamics are then determined from the Heisenberg equations of motion for the
flip operators |α〉〈β|. This approach broadly mirrors that used in Chapter 4 (see
§4.3).
To extract the spatial variation of the crystal excitations we seek to express the
crystal Hamiltonian in the form
H =1L
∫ L
0H(z) dz, (9.2)
where H is an effective local Hamiltonian. It turns out that this representation of
the Hamiltonian emerges naturally from the periodic structure of the crystal lattice.
9.5.1 Hamiltonian
The Hamiltonian for a diamond quantum memory is comprised of three parts,
H = HER +HEL +H0. (9.3)

9.5 Propagation in Diamond 301
The first two contributions represent the interaction of the electrons in the diamond
with the radiation field and with the lattice respectively. The last part accounts for
the energy of the excitations in the diamond. We neglect the Hamiltonian HL of
the free radiation field, as we did in §4.4 in Chapter 4, since it plays no part in the
equations of motion.
9.5.2 Electron-radiation interaction
The Hamiltonian HER is simply the A.p interaction introduced in §C.4 in Appendix
C. This form of the light-matter interaction is more appropriate than theE.d electric
dipole Hamiltonian, because the electrons in diamond are not localized around the
atomic cores. Instead, they are spread over the entire volume of the crystal, in so
called Bloch waves3.
Signal and control fields We divide the vector potential into two parts; the
weak signal field and the strong classical control,
A = As +Ac. (9.4)
The control field is written as
Ac(t, z) = vcAc(t, z)eiωc(t−nz/c) + c.c., (9.5)3The vector potential is not actually a physical field, and strictly we should apply the PZW
transformation (C.20) to the Bloch states, in order that our treatment is gauge invariant [177,178].However, this transformation simply shifts the electron momentum (c.f. (C.23)), and has no effecton the transition matrix elements. In any case, in the Heisenberg picture we are free to choose agauge such that A(t = 0) = 0, whence the PZW transformation becomes trivial [177].

9.5 Propagation in Diamond 302
where we have included the factor n = 2.417 in the exponent, which is the refractive
index of diamond. We treat the signal field quantum mechanically, so it is written
in second-quantized notation as
As(z) = vs
∫g(ω)ω
a(ω)e−iωnz/c dω + h.c., (9.6)
where we have used the one dimensional formula (C.10) along with (C.13) from
§C.4 in Appendix C. The mode amplitude g(ω) =√
~ω/4πε0nAc includes the
refractive index. Just as we did in (4.6) in Chapter 4, we anticipate the compact
spectral support of the signal pulse about its carrier frequency ωs by pulling the
mode amplitude g(ω)/ω out of the integral and defining a slowly varying envelope
operator A,
As(t, z) =vsgsωs
A(t, z)eiωs(t−nz/c) + h.c.. (9.7)
Here gs =√
2πg(ωs). We have also introduced the time dependence of the operators
arising in the Heisenberg picture (see Appendix B).
Electron wavefunctions It is sufficient to consider the interaction of the optical
fields with just a single active electron. This is because the complicated many-body
physics governing the behaviour of all the electrons in the crystal can be swept under
the rug of Fermi-Dirac statistics: the Pauli-exclusion principle prevents more than
one electron from occupying each state, and since all valence band states in the
crystal are initially occupied, any electron-electron scattering in this band is ‘frozen

9.5 Propagation in Diamond 303
out’ because no electron can change its state without occupying a previously filled
orbital. The upshot of this is that we may consider p to be the momentum operator
for a single electron.
The wavefunction ψk,n(r) of an electron with wavevector k in the nth energy band
is given by the product of a spatial phase factor with a periodic Bloch function,
ψk,n(r) = eikzuk,n(r). (9.8)
The Bloch functions have the same translational symmetry as the crystal lattice,
uk,n(z + a) = uk,n(z), which is a consequence of Floquet’s theorem. Note that such
states are not exactly eigenstates of the momentum operator; this is why the A.p
interaction can induce electronic transitions.
Matrix elements We are interested in the matrix element 〈α|HER|β〉 connecting
two quantum states. Neglect for the moment the state of the signal mode. This ma-
trix element is then given by the spatial overlap of the electronic orbitals describing
the initial and final states, with the operator A.p inserted between the two orbitals:
〈α|HER|β〉 = − e
m
∫crystal
ψ∗α(r)A(z).pψβ(r) d3r. (9.9)
Here the indices α, β are standing in for the wavevectors and band indices of the
initial and final orbitals. Now, the coordinate representation of the momentum
operator is p = −i∇. Applying this to ψβ(r), and using (9.8), we can write the

9.5 Propagation in Diamond 304
matrix element as
〈α|HER|β〉 = − e
m
∫crystal
ei(kβ−kα)zA(z). [u∗α(r)(p+ kβ)uβ(r)] d3r. (9.10)
Since all the momenta are very close to the zone-centre, the spatial variation of
the exponential factors in the integrand is very slow, and this is also true for any
variation of the optical field A(z) (which contains only slowly varying envelopes
or similarly long-wavelength exponential factors). On the other hand, any rapid
spatial variation of the Bloch functions uα,β is periodic, repeated in every unit cell.
We therefore factorize the integral into two parts, as follows,
〈α|HER|β〉 = − e
m
∑j
ei(kβ−kα)zjA(zj).[∫
unit cellu∗α(r)(p+ kβ)uβ(r) d3r
], (9.11)
where zj is the position of the jth unit cell. We define the matrix element pαβ as N
times the overlap integral inside the square brackets, where N = AL/a3 is the total
number of unit cells in the crystal. Taking the continuum limit for the sum over the
zj , we can write
〈α|HER|β〉 = − e
mLpαβ.
∫ L
0A(z)ei(kβ−kα)z dz. (9.12)
We have now succeeded in separating out the local from the bulk dynamics. It only
remains for us to introduce the flip operators |α〉〈β| in a convenient form.

9.5 Propagation in Diamond 305
Excitons The momentum operator has negative parity, just as the atomic dipole
operator djk does (see §4.3.1 of Chapter 4). Therefore pαα = 0, so there is no
coupling of any state to itself. As mentioned above, if all electrons are in the valence
band, an electron has ‘nowhere to go’, since all the valence band states are occupied.
The only possibility is to promote an electron into the empty conduction band,
creating an exciton. This process, and its time-reverse — exciton recombination
— are the only important scattering processes involved in HER. Once an exciton
has been created, it is of course possible for either the conduction band electron or
one of the valence electrons to undergo scattering (this latter process is the same as
scattering of the hole) within their respective bands via the A.p interaction. But
the energies involved are much smaller than the photon energies in the signal or
control fields, so these processes do not conserve energy, and may be neglected. The
Hamiltonian can therefore be written as
HER(t) = − e
mL
∫ L
0
A(t, z).∑ν,k
pνk,0s†νke
ikz + h.c.
dz, (9.13)
where the subscript 0 on p denotes the crystal ground state, and where s†νk creates an
exciton with momentum k and energy ων . In the notation of (9.12), k = kβ−kα. The
Hermitian conjugate component destroys an exciton, representing recombination.
The energy of an exciton does not depend on its wavevector k, because the electron
and hole comprising the exciton might have large but opposite momenta, giving
a small total momentum, but a large total energy. Therefore the exciton state

9.5 Propagation in Diamond 306
is independently parameterized by the two quantities ν and k. Excitons, being
composed of pairs of fermions (a hole is a quasiparticle obeying fermionic statistics),
are bosons. The annihilation operators sνk therefore satisfy the same commutation
relation as photon mode annihilation operators (see Appendix C),
[sνk, s
†µk′
]= δν,µδk,k′ . (9.14)
A merciful simplification is achieved by neglecting any dependence of the matrix
elements pνk,0 on ν or k, since the dependence of the Bloch functions describing the
electron and hole on wavevector is rather weak. We thus write pνk,0 = ip, where p
is the constant magnitude of the matrix element, and where the factor of i appears
because the momentum operator is purely imaginary. With these simplifications,
the Hamiltonian can now be written in the form of (9.2), with the effective local
Hamiltonian given by
HER(t, z) = − iemp.A(t, z)
∑ν,k
s†νkeikz − h.c.
. (9.15)
As a final step, we can perform the sum over momenta in (9.15) explicitly. We define
the local exciton operator
Sν(z) =1√L
∑k
sνke−ikz, (9.16)

9.5 Propagation in Diamond 307
which satisfies the commutation relation
[Sν(z), S†µ(z′)
]= δν,µδ(z − z′). (9.17)
The local Hamiltonian then takes the form
HER(t, z) = − ie√L
mp.A(t, z)
[∑ν
S†ν(z)− h.c.
]. (9.18)
9.5.3 Electron-lattice interaction
The Hamiltonian HEL for the electron lattice interaction is just given by
HEL = V |with phonon − V |no phonon, (9.19)
where V is the potential experienced by an electron, generated by all the atomic
cores and other electrons. Let the displacement between the sub-lattices of diamond
caused by a zone-centre optical phonon by given by u. If this displacement is small,
a Taylor expansion to first order gives
HEL =∂V
∂u.u. (9.20)
When we quantize the lattice vibrations, the leading factor becomes the deformation
potential matrix elements, or just the ‘deformation potentials’ [174,179,180], and the
second factor becomes the operator for the optical phonon amplitude. For vibrations

9.5 Propagation in Diamond 308
along the direction vp with wavevector κ the phonon amplitude operator can be
written [180,181]
uκ = gκvpeiκz(b†κ + b−κ). (9.21)
Here b†κ creates an optical phonon with momentum κ. Phonons are bosons, so that
we have (see (C.8) in Appendix C)
[bκ, b†κ′ ] = δκ,κ′ . (9.22)
The phonon mode amplitude gκ, which has the dimensions of length (it is the lattice
displacement due to a single phonon), is given by
gκ =√
~NMωκ
, (9.23)
where M is the mass of a carbon atom and ωκ is the phonon frequency.
For reasons discussed in the previous section, the only states where electronic
scattering can occur are the exciton states, with an electron in the conduction band
and hole in the valence band. The action of the deformation potential is therefore to
destroy an exciton with energy and momentum (ν, k), and to produce a new exciton
with modified parameters (ν ′, k′). Summing over all possibilities gives the expression
HEL =∑
κ,µ,ν,k,k′
∫ L
0
1aLvp.Dνµkk′κs
†µk′sνke
i(k′−k)z × gκeiκz(b†κ + b−κ)
dz. (9.24)
The factor of 1/a appears to give the deformation potentials D the dimensions of

9.5 Propagation in Diamond 309
energy. The integral over space, along with the factor of 1/L, arises in precisely
the same way as it did in (9.12) above. A dramatic simplification of (9.24) is pos-
sible, since the deformation potentials D depend only very weakly on the phonon
and exciton momenta so close to the zone centre. By the same token, the phonon
frequency ωκ = ωp is independent of κ close to the zone centre (see Figure 9.3).
Dropping these dependencies, we can perform the summations over k, k′ and κ to
obtain the following expression for the effective local Hamiltonian,
HEL(z) =L3/2g
a
∑µ,ν
DµνS†µ(z)Sν(z)
[B†(z) +B(z)
], (9.25)
where Dµν = Dνµ = vp.Dµν is the real magnitude of the deformation potential
connecting excitons with energies ωµ and ων , and where we have defined the local
phonon operator
B(z) =1√L
∑κ
bκe−iκz, (9.26)
which has the commutator
[B(z), B†(z′)
]= δ(z − z′). (9.27)
9.5.4 Crystal energy
The energy H0 of the excited crystal is simply found by counting the number of
excitons and phonons. Using the number operators for these particles (see (C.9) in

9.6 Heisenberg equations 310
Appendix C), we find
H0 =∑ν,k
ωνs†νksνk +
∑κ
ωκb†κbκ. (9.28)
Or, in terms of the operators S and B, the local energy takes the form
H0
L=∑ν
ωνS†ν(z)Sν(z) + ωpB
†(z)B(z). (9.29)
9.6 Heisenberg equations
Now that we have constructed the Hamiltonians describing the Raman interaction,
we can write down the Heisenberg equations governing time evolution of the opera-
tors Sν and B. The dynamics of A are derived in the next section using Maxwell’s
equations, as was done for the atomic case in Chapter 4.
Commutation of Sν and B with H, using the relations (9.17) and (9.26), yields
the equations
∂tSν = iωνSν +e
m√Lp.A+ i
√Lg
a
∑µ
DµνSµ(B† +B),
∂tB = iωpB + iD√Lg
a
∑µ,ν
DµνS†µSν . (9.30)

9.6 Heisenberg equations 311
9.6.1 Adiabatic perturbative solution
Let us define local operators Sν and B in a rotating frame, so that
Sν = Sνe−iων(t−nz/c),
B = Be−iωp(t−nz/c). (9.31)
For notational convenience, let us also define
B(t, z) = B†(t, z) +B(t, z)
= B†(t, z)e−iωp(t−nz/c) + B(t, z)eiωp(t−nz/c). (9.32)
The equations of motion for the slowly varying operators Sν and B are then given
by
∂tSν =e
m√Lp.Ae−iων(t−nz/c) + i
√Lg
a
∑µ
Dµν Sµeiωµν(t−nz/c)B,
∂tB = i√Lg
ae−iωp(t−nz/c)
∑µ,ν
Dµν S†µSνe
−iωµν(t−nz/c), (9.33)
where ωµν = ωµ − ων . The spatial phase factors are included for convenience when
considering the exponentials within A.
Our aim is to obtain an equation for B, the local phonon amplitude, in terms
of the signal and control fields. We achieve this by eliminating the intermediate
excitons Sν adiabatically. The procedure is related to that used in §5.3.3 in Chapter

9.6 Heisenberg equations 312
5. We start by formally integrating the equation for Sν in (9.33),
Sν(t) =e
m√Lp.
∫ t
0A(t′)e−iων(t′−nz/c) dt′
+i√Lg
a
∑µ
Dµν
∫ t
0Sµ(t′)B(t′)eiωµν(t′−nz/c) dt′. (9.34)
We have set S(t = 0) = 0, since there are no excitons in the crystal initially.
Unfortunately (9.34) does not provide a direct solution for Sν , because it is coupled
to all the other excitons through the summation on the right hand side. We settle
instead for a perturbative solution. Substituting the first term on the right hand
side of (9.34) into the second term yields the approximate result
Sν(t) =e
m√Lp.
∫ t
0A(t′)e−iων(t′−nz/c) dt′ (9.35)
+i√Lg
a
∑µ
Dµν
∫ t
0
[e
m√Lp.
∫ t′
0A(t′′)e−iωµ(t′′−nz/c) dt′′
]B(t′)eiωµν(t′−nz/c) dt′,
This solution is correct to first order in the deformation potential. To perform the
integrals in (9.35), we use the fact that the time variation of the exponential factors
is much faster than the temporal dynamics of the optical fields, and also faster than
the dynamics of the crystal excitations produced by these fields. This adiabatic
approximation requires that the detunings ∆ν = ων − ωs of the excitons from the
signal frequency are all much larger than the bandwidths of the signal or control
fields. In diamond, the bandgap is in the ultraviolet, so this condition is always very
well satisfied if optical frequencies are used. We proceed by pulling all slowly varying

9.6 Heisenberg equations 313
amplitudes out of the integrals and integrating only the exponentials. The resulting
expression for Sν contains 12 terms. Inserting this into the equation (9.33) for B
provides us with the dynamical description we have been looking for, although now
there are 144 terms! Fortunately, of the first order terms in the product S†µSν , only
very few contribute significantly to the memory dynamics: most terms are oscillating
at high frequencies, so that they average to zero. After some legwork, we obtain
∂tB = iKΩ∗A, (9.36)
where we have defined the control field Rabi frequency
Ω(t, z) =evc.pAc(t, z)
m~, (9.37)
and where the coupling constant K, with the dimensions of (length)−1/2×(time)1/2,
is given by
K =g
~a× ep.vsgs
~ωsm× 1√
L
∑µν
[Dµν
(ωµ + ωc)(ων + ωs)+
Dµν
(ωµ − ωc)(ων − ωs)
]. (9.38)
Let us suppose that we are sufficiently close to resonance that ων + ωs ων − ωs.
This need not be the case, because the bandgap in diamond is very large, but it
is a convenient simplification to neglect the ‘counter-rotating’ terms with summed
frequencies in their denominators (c.f. §4.6 in Chapter 4). The coupling now takes

9.7 Signal propagation 314
the approximate form
K =g
~a× ep.vsgs
~ωsm× 1√
L
∑µ,ν
Dµν
(∆µ + ωp)∆ν, (9.39)
where we have used the Raman resonance condition ωs = ωc + ωp. The double
appearance of the detuning in the denominator is characteristic of third-order scat-
tering. Equation (9.36) is very similar in form to the equation for the spin wave
(5.107) derived in Chapter 5. Thus encouraged, we proceed in the next section to
derive the equation describing the propagation of the signal field.
9.7 Signal propagation
The electric field of the signal is the solution to Maxwell’s wave equation, with the
polarization in the diamond acting as a driving term (see (4.34) in Chapter 4). The
polarization is the dipole moment per unit volume. That is, er =∫P dV , where
r is the one-electron position operator. We can relate the matrix elements of r to
those of the momentum operator p, as follows (see (F.6) in §F.2 of Appendix F),
pαβ = −imωαβrαβ, (9.40)

9.7 Signal propagation 315
where ωαβ is the frequency splitting between the energy eigenstates |α〉, |β〉. Using
this relation, a second-quantized form for the polarization operator can be found:
P =ei
mAL∑ν,k
(pνk,0ων
s†νkeikz −
p0,νk
ωνsνke
−ikz
)= − ep
mA√L
∑ν
1ων
(S†νe
−iων(t−nz/c) + h.c.). (9.41)
Making the SVE approximation (see (4.39) in Chapter 4), we find the propagation
equation (∂z +
n
c∂t
)A = − µ0ω
2s
2gsksv∗s .Ps, (9.42)
where Ps is the component of the polarization oscillating at the signal frequency ωs.
Substituting the solution (9.35) into (9.41), performing the integrals with the help
of the adiabatic approximation, and retaining only those terms with the appropriate
time dependence, we arrive at the final equation for the signal,
(∂z +
n
c∂t + iχ
)A = iK∗ΩB, (9.43)
where K is very similar to K,
K =g
~a× ep.vsgs
~ωsm× 1√
L
∑µ,ν
ωsDµν
ων(∆µ + ωp)∆ν, (9.44)

9.7 Signal propagation 316
and where χ represents a spatial phase picked up by the signal due to the crystal
dispersion,
χ =∣∣∣∣evs.pgs~mωs
∣∣∣∣2 × 1L
∑ν
ωsων∆ν
. (9.45)
The discrepancy between K and K is probably spurious, and may arise from the
secular approximation we use in eliminating terms oscillating at the ‘wrong’ frequen-
cies [177,178]. In any case, the difference between the two expressions is small close
to resonance, and never more than an order of magnitude. In what follows, we set
K −→ K.
The coupling constants are admittedly rather dense combinations of various ma-
terial and optical parameters. In the next section we will try to extract a prediction
for the coupling strength of a diamond quantum memory. But for the moment,
what is important is the form of the equations (9.36) and (9.43). They are es-
sentially identical to the Raman memory equations of §5.3.3 in Chapter 5. We
can make the similarity explicit with a number of coordinate transformations and
re-normalizations. First, we introduce the retarded time τ = t−nz/c (note the pres-
ence of the refractive index n). When the equations are written in terms of τ and
z, the derivative ∂z + nc ∂t becomes simply ∂z, while the time derivative ∂t becomes
∂τ . Next, we remove the dispersive factor χ with a phase rotation, by making the
transformation
A −→ A = Aeiχz,
B −→ B = Beiχz. (9.46)

9.7 Signal propagation 317
To remove the dependence on the control field profile, we introduce the normalized
integrated Rabi frequency ω = ω(τ) — defined in (5.46) in Chapter 5 — and the
dimensionless transformed variables
α(z, ω) = i√WA(z, τ)Ω(τ)
, β(z, ω) =√LB(z, τ). (9.47)
Here we have also re-scaled the longitudinal coordinate by L so that z runs from
0 up to 1. As a final simplification, we replace K with K (as mentioned above),
and we assume that K is real. The equations of motion for the diamond quantum
memory then take the form
∂zα = −Cβ, ∂ωβ = Cα, (9.48)
where the dimensionless Raman memory coupling is
C =√LWK. (9.49)
This tells us that a diamond memory will behave in precisely the same way as a
Raman memory based on an atomic vapour (see (5.109) in §5.3.3 of Chapter 5). We
know that efficient storage is possible if the Raman memory coupling C is of the
order of unity (we should have C & 2). And all of the results pertaining to optimal
shaping carry over. So given the coupling C, we can directly find the optimal input
mode φ1 for the signal, using (5.97).

9.8 Coupling 318
9.8 Coupling
Here we show that efficient storage is possible in a small sample of diamond, so that
diamond quantum memory need not be a luxury enjoyed only by the super rich.
To estimate the memory coupling C, we must evaluate the summations in (9.39).
To do this we first write Dµν = Dδµ,ν , which holds approximately, since the wave-
functions of distinct excitons overlap poorly [172]. Next we assume that both con-
duction and valence bands are parabolic around the zone centre (as shown in Figure
9.5), so that we may write the energy of an exciton as [182]
ων −→ ω(k) = ω0 +~k2
2m?, (9.50)
where ~k is the momentum of the electron relative to the hole, and where m? is
an effective reduced mass for the exciton that describes the local band curvature.
Parameterizing the exciton energies in this way, the summations in K may be re-
written approximately as an integral over a sphere in k-space,
∑µ,ν
Dµν
(∆µ + ωp)∆ν−→
∑ν
D
(∆ν + ωp)∆ν(9.51)
≈ DAL
(2π)3
∫ kmax
0
4πk2
(ω0 − ωs + ωp + ~k2/2m?) (ω0 − ωs + ~k2/2m?)dk,
where kmax is an arbitrary, suitably large cut-off. The integral can be performed

9.8 Coupling 319
with a partial fraction expansion and a trigonometric substitution,
∫ κ
0
k2
(a+ bk2)(c+ bk2)dk =
b−3/2
a− c
[∫ √bκ0
a
a+ x2dx−
∫ √bκ0
c
c+ x2dx
](9.52)
=b−3/2
(a− c)
[√a tan−1
(√b/aκ
)−√c tan−1
(√b/cκ
)].
Choosing kmax sufficiently large, we get to the result [182]
∑µν
Dµν
(∆µ + ωp)∆ν≈ ALD
4πωp
(2m?
~
)3/2 (√∆ + ωp −
√∆), (9.53)
where ∆ = ω0 − ωs is the detuning of the signal field from the conduction band
minimum.
We can express W in terms of the energy Ec in the control pulse as follows,
W =∫ ∞−∞|Ω(τ)|2 dτ =
2πα|vc.p|2
~m2ω2cAn
× Ec, (9.54)
where here α = e2/4πε0~c = 1/137 is the fine structure constant.
We estimate the momentum matrix elements as vc.p ≈ vs.p = ~ × 2π/a. This
is justified by noting that the band gap at the zone centre is produced by Bragg
scattering that mixes electrons with k = 0 and k = 2∆k = 2π/a, and it is this latter
component that is responsible for interband transitions.
Other parameters are as follows. The bandgap4 in diamond is ~ω0 = 13 eV, and4This energy corresponds to the direct gap in diamond (at the zone centre, or ‘Γ-point’). The
conduction band minima occur elsewhere in the Brillouin zone (~ω0 ∼ 5 eV at the ‘L-point’), buttransitions to these states are mediated by phonons. They are therefore suppressed somewhat.Although they may not be insignificant, we neglect these processes here.

9.8 Coupling 320
the deformation potential has a value of around D ≈ 7~ω0 ≈ 90 eV. As mentioned
before, the phonons have wavenumber νp = 1332 cm−1. The lattice constant in
diamond is a = 3.6 A. The refractive index is n = 2.417, and the mass of a carbon
atom is M = 12 a.m.u. (indeed, by definition). For simplicity we assume a reduced
exciton mass m? = m. We then consider a crystal of length L = 1 mm, illuminated
by beams with waists 100 µm in diameter. If the control pulse is taken from a
modelocked pulse train with an 80 MHz repetition rate and an average power of
10 mW, it has an energy of 0.13 nJ. Using a signal field with central wavelength
λs = 800 nm, we estimate a memory coupling of
C = 2.4. (9.55)
The optimal storage efficiency is therefore around 99.9% (see Figure 5.5 in §5.3 of
Chapter 5). This demonstrates that efficient storage in a small sample of diamond
is extremely feasible. Of course, the estimate we have made is very crude, but
since neither the laser energy nor the sample size required are close to any practical
limitations, a downward revision of C by a factor as large as 100 could still be
accommodated. In fact (9.55) is something of an underestimate, since we have
neglected the counter-rotating terms in K. When the signal is so far detuned from
the conduction band edge, these terms still contribute significantly.

9.9 Selection Rules 321
9.9 Selection Rules
The large Stokes shift (i.e. the large phonon energy) in diamond makes it easy
to distinguish the signal and control fields spectrally. For instance, if the signal
field wavelength is 800 nm, the control wavelength is around 894 nm. In addition,
however, it turns out that the crystal symmetry requires the signal and control fields
to be orthogonally polarized. The Raman interaction that couples the input and
output fields is constrained to be proportional to an irreducible representation of
the symmetry group associated with the optical phonons. The zone-centre phonons
are always described by a subgroup of the crystal point group — the group of
reflections and rotations that leaves the crystal unchanged. In the case of diamond,
the crystal point group is the cubic group m3m (sometimes written Oh), and the
optical phonons transform as the subgroup Γ+5 (sometimes written T2g). The optical
fields are 3 dimensional vectors, and so the Raman interaction must be proportional
to the 3× 3 irreducible representation of the group Γ+5 , which takes the form [180]
Γ+5 (x) =
1
1
, Γ+5 (y) =
1
1
, Γ+5 (z) =
1
1
,
where zero elements have been left blank for clarity, and we have assumed that the
z-axis is aligned with the [001] direction (that is, parallel to a vertical edge of the
cubic unit cell; see Figure 9.1). The Raman interaction requires that vc = Γ+5 (z)vs,
from which it is clear that vc should be perpendicular to vs. This polarization

9.10 Noise 322
selection rule adds to the experimental attractiveness of a diamond memory.
9.10 Noise
The foregoing analysis of storage in diamond implicitly ignored the possibility of
unwanted couplings. For instance, the intense control pulse can stimulate strong
Stokes scattering, creating optical phonons and Stokes photons in pairs. This is the
same problem as that shown for the atomic case in part (b) of Figure 4.3 in §4.7 of
Chapter 4. Here it is exacerbated because the detuning from resonance is typically
so large that both processes — storage of the signal and Stokes scattering — occur
with roughly equal probabilities. In §6.3.2 of Chapter 6, a solution to this problem
is described that involves introducing a small angle between the signal and control
beams, and this solution certainly carries over to the diamond memory. Another
interesting possibility is that of modifying the optical dispersion so that the unwanted
Stokes light cannot propagate. It would be possible to do this by building a Bragg
grating — alternating layers of diamond and air — with a spatial frequency equal
to that of the unwanted Stokes light. Interference within the Bragg structure would
then suppress any Stokes generation. But we won’t consider this further; a proof-
of-principle demonstration of broadband storage in room temperature diamond is
challenging enough, without engaging in micro-fabrication [183].
In the next chapter, we review the experimental progress made in our group
towards the goal of demonstrating a Raman quantum memory.

Chapter 10
Experiments
Although most of this thesis is theoretical, I had initially intended to build a working
quantum memory. I have not succeeded as yet, but the ‘Memories’ subgroup is
continuing its efforts in this direction. In this chapter we discuss some of the ongoing
experimental work; its goals and future prospects.
10.1 Systems
The experimental programme divides into three projects.
1. Diamond,
2. Quantum dots,
3. Atomic vapour.
My experimental research has been focussed on the last of these; the theoretical
analysis in the preceeding chapter constitutes my contribution to the first. We will

10.1 Systems 324
not describe the quantum dot project here; suffice it to say that quantum dots may
be thought of as artificial atoms, so that a sample containing many dots behaves
like an atomic ensemble, in which Raman storage may be implemented.
Light storage in atomic vapour is becoming standard in quantum optics. In al-
most all cases, resonant EIT is used, and narrowband diode lasers provide the signal
and control fields [74,150,151]. Our research group has some considerable experience
with ultrafast lasers, however, and it was decided that a broadband Raman memory
would be interesting. Atomic vapour is an ideal system for demonstrating such a
memory; indeed this is the system that is considered when deriving the memory
equations in Chapter 4.
A common feature of all the experiments is the requirement of strong Raman
coupling between a laser field and a material sample. The easiest way to verify the
existence of strong coupling is to observe strong Stokes scattering. This is therefore
the first step in all our experiments — the general strategy is shown schematically
in Figure 10.1. Unfortunately, we have not yet been able to achieve this first pre-
requisite in the atomic vapour experiments. This is rather depressing, but we are
persevering, since there are many improvements to be made. In the rest of this chap-
ter, we will introduce the atomic species used in our experiments with atomic vapour.
We then discuss the theory of Stokes scattering, and we estimate the strength of the
coupling we expect to achieve. Finally, we describe the experimental techniques we
have developed in our attempts to see strong Stokes scattering. We finish with a
discussion of the planned realization of a Raman quantum memory.

10.2 Thallium 325
Vapour cellRaman pumpStokes
Filter
Laser
Figure 10.1 Observing Stokes scattering as a first step. StrongRaman coupling between a bright laser and the atomic ensemble isrequired for a Raman quantum memory. The ability to produce,and detect, strong, stimulated Stokes scattering is a sine qua non forimplementing the memory.
10.2 Thallium
The first incarnation of the atomic vapour experiment used thallium (Tl) as the
storage medium. Thallium is an extremely toxic poor metal, with a history of use
in rat poison, and homicide generally. However the atomic structure of thallium
exhibits a well-defined Λ-system, with a large Stokes shift (see Figure 10.2). For
this reason, attempts were made to build a Raman quantum memory with thallium
vapour, provided by means of a heated glass cell containing solid thallium. After
around a year of unsuccessful attempts to observe strong Raman scattering from
thallium vapour, it was realized that the vapour pressure of thallium is too low for
a strong Raman interaction to be engineered (see the discussion in §10.9.1, and part
(a) of Figure F.1 in Appendix F).

10.3 Cesium 326
1/ 27S
1/ 26P
378 nm
1283 nm
F=0
F=1
F=0
F=1
3/ 26P
F=1
F=2
Figure 10.2 Thallium atomic structure. The three levels of the Λ-system are discernible, marked by the thickened line segments. Thehyperfine structure (indicated by the fine branches) is ignored, beingnegligible in the ground P -wave manifolds. The large Stokes splittingbetween the J = 1/2, 3/2 states in the P -state manifold makes it idealfor broadband storage. Unfortunately Thallium has a low vapourpressure, making an efficient thallium quantum memory impractical.
10.3 Cesium
To increase the Raman coupling, an atomic species with a much higher vapour
pressure was required. Our current experiment uses cesium1 (Cs), which is many
orders of magnitude denser than thallium at room temperature (see part (b) of
Figure F.1 in Appendix F). Cesium is a soft, gold-coloured alkali metal that is,
thankfully, non-toxic, although it reacts explosively on contact with water, even the
water vapour in air! Our cesium is sealed in an evacuated glass cell, along with a
small amount of neon (10Ne), which acts as a buffer gas (see §§10.4 and 10.5 below).
There is only one stable isotope of cesium, namely 133Cs. Needless to say we refer
only to this isotope in what follows; our sample is naturally isotopically pure. The1Cesium is the American spelling; the British spelling Caesium retains some of the latinate
flavour if its etymology. The word derives from the latin for ‘sky’, because the 7P ↔ 6S1/2 doubletlines are a brilliant blue colour. However American spell-checkers and journal styles have worn medown, and now the British spelling seems odd.

10.3 Cesium 327
Λ-system is implemented in the so-called cesium D2 line at 852 nm. This is the
second of the strong ‘doublet lines’ (the D1 line is at 894 nm) characteristic of alkali
metals — the same doublet in Sodium illuminates the night-time activities of most
of this planet. The relevant atomic structure is shown in Figure 10.3.
9.2 GHz
852 nm
Figure 10.3 Cesium atomic structure. The F = 3 and F = 4hyperfine levels in the 6S1/2 ground state — the famous ‘clock states’— provide the ground and metastable states for the Λ-type quantummemory. The 6P3/2 manifold collectively provides the excited state.
The upper 6P3/2 state is split by the hyperfine interaction: The nuclear spin
of I = 7/2 combines with the total electronic spin of J = 3/2 to produce four
hyperfine levels with total angular momentum quantum numbers F = 2, 3, 4 and 5.
The interaction is weak however, and the splitting between each state is around 200
MHz. For the purposes of the memory, we therefore treat the excited state manifold
as a single state, which plays the role of |2〉 in the Λ-system.
The hyperfine interaction is much stronger in the 6S1/2 electronic ground state.
The reason is that the ground state is an S-wave, meaning that it has no orbital
angular momentum. It therefore has no azimuthal phase, and so it remains well-

10.4 Cell 328
defined at the origin without vanishing — a wavefunction with such a phase is multi-
valued at the origin unless it is zero, so higher orbitals must disappear at the origin.
In the ground state, then, the electron penetrates into the cesium nucleus. There is
therefore a strong magnetic dipole coupling between the nuclear and electronic spins,
known as the Fermi contact interaction, which acts to separate the two hyperfine
states with F = 3 and F = 4 by an enormous 9.2 GHz. These two states are
sometimes known as the clock states because coherent oscillations between them are
used in cesium atomic clocks. In fact the hyperfine splitting is now defined to be
exactly 9, 192, 631, 770 Hz, with the duration of the second being a derived quantity.
The clock states form the two lower states in the quantum memory Λ-system.
10.4 Cell
We contain the cesium vapour in a glass cell (the TT-CS-75-V-Q-CW from Triad
Technology in Colorado, USA). The cell is 10 cm in length, and is made of glass that
is resistant to heating up to temperatures of 500 C. The cell windows are 25 mm
across, and are made of optically polished quartz, to which an anti-reflection coating
has been applied which reduces reflection losses for light at the D2 wavelength of
852 nm down to around 0.1%. The cell is evacuated, and then a small sample of
solid cesium is introduced. Finally, the cell is backfilled up to a pressure of 20 torr
(∼ 2700 Pa) with neon gas. The reason for introducing this buffer gas is explained in
§10.5. The cell is then hermetically sealed, by pinching shut the glass tube through
which the cell contents are delivered.

10.4 Cell 329
10.4.1 Temperature control
The cell is wrapped in heating tape: a thin weave of high resistance wires surrounded
by thermal insulation. A thermocouple connected to an electronic temperature
controller allows one to set and maintain the cell temperature as desired. The glass
protuberance remaining after the cell is sealed provides a convenient ‘cold finger’
— an unheated region protruding from the cell where the cesium preferentially
condenses. As long as the cell windows remain hotter than this cold finger, cesium
does not collect on the cell windows, and the cell remains transparent to our laser
beams.
10.4.2 Magnetic shielding
The hyperfine levels in cesium are not pure quantum states. A hyperfine state with
total angular momentum quantum number F is (2F+1)-degenerate, being comprised
of Zeeman sublevels with quantum numbers m = −F,−F + 1, . . . , F − 1, F . These
quantum numbers represent the projection of the atomic angular momentum along
some axis, known as the quantization axis (see §F.4 in Appendix F). In the presence
of a magnetic field, the atomic angular momenta precess around the direction of the
field, and it is natural to define the quantization axis as aligned with this direction.
With this definition, the quantum numbers m remain good quantum numbers, but
the degeneracy of the Zeeman sublevels is lifted — this is known as the Zeeman
shift. Optical transitions between the Zeeman sublevels occur subject to selection
rules, which determine whether or not a transition conserves angular momentum,

10.4 Cell 330
based on the polarization of the incident light. With a judicious choice of polarized
lasers, one can prepare the ensemble in just one Zeeman sublevel. Such a ‘spin
polarized’ ensemble is used by Julsgaard et al. when implementing their continuous-
variables memory in cesium [122] (see §2.4 of Chapter 2). This type of ensemble
state is exquisitely sensitive to external magnetic fields, and it is common to build
a magnetic shield of so-called µ-metal (an alloy with a high magnetic permeability
that deflects magnetic field lines) around the vapour cell.
In our system, the spectral bandwidth of the laser pulses comprising the signal
and control fields is much larger than any feasible Zeeman shift one could produce
(see §10.6). Therefore the Zeeman sublevels cannot be resolved in the quantum
memory interaction, and so we neglect the Zeeman substructure of the hyperfine
states. There is no need to spin-polarize the ensemble, and no need for magnetic
shielding.
In §F.4 of Appendix F, we show that orthogonal circular polarizations are not
coupled by the Raman interaction. This is a further reason why it is not useful to
polarize the ensemble: it might have been possible to use the Zeeman selection rules
to our advantage (as is explained in the Appendix), but this result obviates this
possibility.
An unpolarized ensemble is in a mixed quantum state. It can be thought of as
several independent sub-ensembles, each with a different spin polarization. Each sub-
ensemble interacts coherently with the optical fields, however, and the theoretical
description of the memory interaction given in Chapter 4, 5 remains valid for each

10.5 Buffer gas 331
sub-ensemble.
The lack of any magnetic shielding means that there may be stray magnetic
fields that introduce a distribution of frequencies into the evolution of the Raman
coherence through the Zeeman shift. This may cause the spin wave to dephase, so
that the coherence is lost. However, as long as the magnetic fields remain constant
over the memory lifetime, the spin wave will re-phase, because the number of Zeeman
components is finite (this periodic re-phasing of a discrete set of oscillators is the
principle behind retrieval from the AFC quantum memory [91]; see §2.3.4 in Chapter
2). The periodic beating between the Zeeman sublevels restricts the times at which
efficient retrieval is possible to those times where the spin wave is ‘in phase’, but the
efficiency of the memory is not adversely affected.
The above discussion justifies our decision not to build a magnetic shield for our
cesium cell, and indeed not to attempt to polarize the vapour. However, since we
have not yet been able to observe strong Stokes scattering, we cannot be sure that
a magnetic shield would not help. We are investigating the construction of such a
shield.
10.5 Buffer gas
The 20 torr of neon buffer gas is added in order to extend the time that the cesium
atoms spend in the interaction region. The cesium atoms are deflected by collisions
with the neon atoms, so that their motion is diffusive rather than ballistic. The
mean time between collisions is given by 1/γp, as can be verified by computing

10.5 Buffer gas 332
〈τ〉 =∫ τ
0 τps(τ) dτ using the ‘survival’ distribution (F.15) in Appendix F. If we
follow the trajectory of a cesium atom, it will trace out a random walk with an
average step length l = vth/γp, where vth =√
2kBT/M is the average thermal
velocity of the atom. After N such steps, the root-mean-square displacement of the
atom from its starting point is D =√Nl. The beam diameter is roughly
√A, so an
atom escapes the beam after a time tescape, where
D(tescape) ∼√A,
⇒ tescape ∼ AMγp2kBT
. (10.1)
A typical diffusion time for a beam with diameter 100 µm is around 10 µs at room
temperature, which compares with around 0.5 µs in the absence of a buffer gas.
During the course of this Brownian motion, the atom undergoes around 105 col-
lisions. As described in Appendix F, these collisions randomize the phase of the
optical polarization, causing pressure broadening. But the hyperfine coherence —
the spin wave — is unaffected. The ground state hyperfine levels represent different
orientations of the cesium nuclear spin, but neon is spinless, so there is no magnetic
dipole interaction between the atomic nuclei in a collision. The Raman coherence is
therefore maintained, despite the frequent collisions of cesium atoms with the buffer
gas. The natural lifetime of the hyperfine coherence is several thousand years, so
the memory lifetime is set by tescape.
As well as slowing the escape of the cesium atoms, collisions with the buffer gas

10.6 Control pulse 333
change their velocities, so that the cesium atoms diffuse spectrally : as the cesium
atoms’ velocities change, so do their Doppler shifted resonant frequencies. Before
an atom leaves the interaction region, its resonance will ‘wander’ across the entire
Doppler profile (see §10.10 below). This means that the atoms can be optically
pumped (see §10.12 below) using a narrowband laser tuned into resonance with
stationary atoms, since all atoms will eventually drift into resonance with the pump
laser. Without a buffer gas, it is necessary to dither the pump laser frequency in
order to address all the atoms, which is inconvenient (although certainly possible).
10.6 Control pulse
The control pulse, or alternatively the Raman pump light, is sourced from a bespoke
Ti:Sapphire laser oscillator: a Spectra-Physics Tsunami. The oscillator is actively
modelocked using an acousto-optic modulator installed in the laser cavity, and the
laser produces a pulse train with a repetition rate of 80 MHz, which is set by the
cavity round-trip time.
10.6.1 Pulse duration
The bandwidth of the control pulse cannot exceed the 9.2 GHz Stokes splitting be-
tween the two lower hyperfine states, if a quantum memory is to be implemented.
However the gain bandwidth of Ti:Sapphire is very wide (several hundred nanome-
ters!), so some care was taken to limit the bandwidth of the laser. A Gires-Tournois
Interferometer (GTI) is installed inside the laser cavity. This is essentially a Fabry-

10.6 Control pulse 334
Perot etalon, with the rear of the two plates having a reflectivity of 100%. All
incident energy is therefore reflected, but the spectral phase inside the cavity under-
goes periodic jumps, with the free spectral range determined by the plate separation.
Any pulse in the laser cavity with a bandwidth spanning one of these phase jumps
experience catastrophic dispersion, which greatly reduces the efficiency with which
the pulse — being heavily distorted — can extract energy from the Ti:Sapphire
crystal. The bandwidth of the laser is therefore limited to bandwidths smaller than
the free spectral range of the GTI. Use of a GTI with an unusually large plate sep-
aration produces a modelocked pulse train with a spectral bandwidth or around 1.5
GHz (∼ 3.6 pm at 852 nm).
Beamsplitter Retroreflector
Photodiode
Mirror
Mirror
150 200 250 3000
500
1000
x (mm)
x
inte
nsity (
arb
. units)
Figure 10.4 First order autocorrelation. The set up is essentiallywhat is known as a Fourier transform spectrometer. An incidentpulse is split into two components, and one is delayed with respect tothe other. The arrangement with the retroreflector and facing mirrormakes the movable arm insensitive to misalignment as the retroflectoris translated over a large distance.
To characterize the pulse train, measurements of the first and second order cor-
relation functions of the output light were made. A Michelson interferometer was

10.6 Control pulse 335
built, so that interference of the optical field with a delayed copy of itself could
be observed. The first order field autocorrelation, shown in Figure 10.4, produces
a fringe pattern whose envelope is equal to the Fourier transform of the spectral
intensity I(ω) of the laser output (see (F.16) in Appendix F for an example of this
relationship). The spectral intensity is found to be approximately Gaussian, with
a FWHM bandwidth of 1.5 GHz, as mentioned above. This is consistent with a
Fourier-transform-limited pulse duration of 300 ps, but it may be that distortions
to the spectral phase of the pulse train ‘smear out’ the pulses, producing longer du-
rations. To investigate this possibility, we performed a second order interferometric
autocorrelation. The experimental set up is shown in Figure 10.5. Two delayed
copies of an incident pulse are focussed into a non-linear crystal — a small piece of
β-Barium Borate (BBO). Blue light scattered in the forward direction results from
the frequency upconversion of one pulse with its delayed counterpart. The envelope
of the resulting fringe pattern can be related to the pulse duration. Although the
shape of the autocorrelation envelope is not uniquely related to the pulse envelope
(it is not possible to ‘invert’ the autocorrelation to retrieve the pulse profile), it is
possible to infer the presence of spectral phase distortions by inspection of the lower
portion of the interferogram. In the presence of ‘chirp’ (a drift in the carrier fre-
quency of the pulse as a function of time), interference between the leading edge of
one pulse, and the trailing edge of the other, is suppressed, since the carrier frequen-
cies are no longer commensurate. This causes a distinctive narrowing of the lower
portion of the interferogram that is not present in our measurement. On the basis of

10.6 Control pulse 336
this result, we conclude that the pulses produced by our laser source are close to be-
ing transform-limited, with approximately Gaussian spectral and temporal profiles.
The FWHM duration of each pulse is around 300 ps, and the spectral bandwidth is
approximately 1.5 GHz. On the one hand, this is much narrower than the Stokes
splitting, as required. On the other hand, the pulses are much more broadband than
have been used to date in quantum memory experiments (pulse durations of 100’s
of microseconds are the shortest that are employed for EIT [74]).
BBO
Lens
150 200 250 3000
200
400
600
x (mm)
inte
nsity (
arb
. u
nits)
Beamsplitter Retroreflector
Photodiode
Mirror
Mirror
x
Filter
Figure 10.5 Second order interferometric autocorrelation. A non-linear crystal is inserted, and the frequency upconversion of one pulsewith its delayed counterpart is detected. The inset shows the mea-sured envelope of the interferogram. Again, the red lines are Gaus-sian fits. These data, along with the data in Figure 10.4, are the onlyexperimental data in this thesis that were taken by me!
10.6.2 Tuning
The plate separation of the GTI can be temperature tuned using a knob on top of the
laser, and this also provides a convenient way to smoothly tune the laser frequency
over small frequency shifts, of order 10 or 20 GHz (it appears that the spectral phase

10.7 Pulse picker 337
introduced by the GTI introduces differential gain over its free spectral range, which
allows small changes to the FSR to sweep the laser frequency). Larger frequency
shifts can be ‘dialled in’ using a birefringent filter (this is actually an intra-cavity
Lyot filter — see §10.13.2 below).
10.6.3 Shaping
Although a great deal of the theoretical work in this thesis deals with the optimiza-
tion of quantum storage by appropriate shaping of the control or signal pulse profiles,
it is not feasible to shape the pulses used in this experiment. They are too short
to be shaped electronically, and two narrowband to be shaped spectrally! However,
since we are still struggling to demonstrate storage in the first place, we are content
to walk before attempting a steeplechase. The excellent work of Novikova et al. [74]
on resonant shaped storage is a vindication of the theory of Gorshkov et al. [133], and
by extension the theory in this thesis, since they are so closely related (at least at
optical depths less than 50 or so [151]).
10.7 Pulse picker
The time between consecutive pulses in the output of our laser is 12.5 ns. This
is much shorter than the lifetime of the Raman coherence (see §10.5 above), so if
the pulse train from the laser is sent ‘as is’ into the cell, the ensemble does not
recover after each Raman interaction. If one’s aim is to generate strong Stokes
scattering, this may be beneficial. The Stokes process can be stimulated both the

10.8 Stokes scattering 338
presence of Stokes photons, and by the presence of spin wave excitations, so the
Raman coherence produced by a previous pulse may stimulate the Raman interaction
in subsequent pulses. However it is not possible to investigate the efficiency of a
quantum memory if different realizations of the memory interaction are coupled. To
demonstrate the quantum memory, it is necessary to reduce the repetition rate of the
laser so that each implementation of the memory is independent of all others. This
is done with a ‘pulse picker’, which is a fast optical switch based on the electro-optic
effect, known as a Pockels cell. The device is synchronized with the laser output,
and the optical switch is set to transmit every 80, 000th pulse, reducing the laser
repetition rate to 1 kHz. The ensemble now has 1 ms to ‘reset’ between pulses,
which is plenty of time.
In the next section we introduce the theory of Stokes scattering.
10.8 Stokes scattering
The requirements for observing strong Stokes scattering are very similar to the
requirements of a Raman quantum memory. A medium must be prepared in the
ground state of a Λ-system. There should be strong Raman coupling between an
incident laser pulse and the excitations of the medium. And it should be possible
to observe scattering at the Stokes-shifted wavelength by filtering out the strong
Raman pump.
A description of Stokes scattering runs along very similar lines to that of a Raman
quantum memory: the signal and control fields simply trade places, becoming pump

10.8 Stokes scattering 339
and Stokes fields (see part (a) of Figure 1.6, or part (b) of Figure 4.3). However,
instead of absorption, the Stokes field experiences gain, with energy being transferred
from the pump field into the Stokes field, while at the same time excitations are
generated in the Raman medium.
We are interested in transient Stokes scattering, which refers to the regime in
which the duration of the Raman pump pulse is much shorter than the lifetime of
the excitations in the medium. This is consistent with the requirement that the
same medium should be useful as a quantum memory, which requires that the spin
wave excitations far outlive the optical pulses.
The equations describing transient Stokes scattering in the adiabatic limit can
be written in the following simple form,
∂zα = Cβ†, ∂ωβ = Cα†. (10.2)
Here all the notation has the same meaning as in (5.109) from §5.3.3 in Chapter 5,
except that α is a dimensionless annihilation operator for the Stokes mode. Recall
that ω is the integrated Rabi frequency (defined in (5.46) in §5.2.6 of Chapter 5),
where now the relevant Rabi frequency is that of the Raman pump pulse. The cou-
pling constant C is the same as the Raman memory coupling, so the efficiency of
both Stokes scattering and quantum memory are characterized by the same number.
The equations (10.2) describe a squeezing, or Bogoliubov transformation as opposed
to a beamsplitter interaction: optical and material excitations are produced in cor-

10.8 Stokes scattering 340
related pairs. This interaction is the basis of the DLCZ quantum repeater protocol,
since the Stokes mode becomes entangled with the ensemble (see §1.6.4 in Chapter
1). The equations can be solved in precisely the same manner as the system (5.109).
The solution for the Stokes field is
αout(ω) = α0(ω) +∫ ω
0
√C2
ω − ω′I1
[2C√ω − ω′
]α0(ω′) dω′ (10.3)
+C∫ 1
0I0
[2C√
(1− z)ω]β0(z) dz.
Here the I’s denote modified Bessel functions, which describe exponential gain. We
are interested in the intensity of spontaneously initiated Stokes scattering, when
there are initially no spin wave excitations and no photons in the Stokes mode.
The average number of Stokes photons produced is given by the normally ordered
product
〈Nout〉 =∫ 1
0〈α†out(ω)αout(ω)〉 dω. (10.4)
On substitution of (10.3) into (10.4), a number of terms vanish. The cross terms
involving the products 〈α†0(ω)β†0(z)〉 and 〈β0(z)α0(ω)〉 are both zero, since in the
first case the inner product between singly-excited and vacuum states is taken, and
in the second case the vacuum state is annihilated. The same is true of the term
involving 〈α†0(ω)α0(ω)〉. The only non-vanishing term involves the anti-normally
ordered combination 〈β0(z)β†0(z)〉. To evaluate this term, we observe that β is a
bosonic annihilation operator which satisfies the same commutation relation as B

10.8 Stokes scattering 341
(see (4.45) in Chapter 4, or (9.27) in Chapter 9),
[β(z), β†(z′)
]= δ(z − z′). (10.5)
We therefore have that 〈β0(z)β†0(z′)〉 = 〈δ(z−z′)+β†0(z′)β0(z)〉 = δ(z−z′). Inserting
this result into the expression for 〈Nout〉, we obtain2
〈Nout〉 = 2C2[I2
0 (2C)− I21 (2C)
]− CI0(2C)I1(2C). (10.6)
Figure 10.6 shows the average number of scattered Stokes photons as a function of
the coupling C. C = 1 corresponds to 〈Nout〉 ∼ 1, and this may be thought of as
marking the onset of the stimulated scattering regime, when previously scattered
Stokes photons stimulate further scattering, so that the scattering efficiency begins
to grow exponentially with C. For efficient Raman storage, a quantum memory
requires C ∼ 1 also, so the possibility of producing stimulated Raman scattering is
a necessary condition for implementing a Raman quantum memory in any system.
In the next section we explain how to calculate a prediction for the coupling
constant C, when an atomic vapour is used as the memory medium (a calculation
of C for the case of a diamond quantum memory was undertaken at the end of the
previous Chapter).2This result appears as equation (38) in the seminal paper on Stokes scattering by Michael
Raymer and Jan Mostowski [139], who specialized to the case of a square pump pulse.

10.9 Coupling 342
0 2 4 6 810
−5
100
105
1010
Figure 10.6 Stokes scattering efficiency. As the coupling C in-creases, the number of Stokes photons scattered rises, growing expo-nentially for C & 1. The model neglects depletion of the energy inthe pump pulse: of course the number of Stokes photons scatteredcannot exceed the number of photons in the pump pulse, or indeedthe number of atoms in the ensemble.
10.9 Coupling
Throughout this thesis we have made reference to the quantities d, Ω, C etc...
Fortunately these quantities are not difficult to calculate in the context of an atomic
vapour.
10.9.1 Optical depth
Combining the definition (5.12) in §5.2.3 of Chapter 5 with (4.50) in §4.11 of Chapter
4, the optical depth can be written as
d =|d∗12.vs|2ωsnL
2γε0~c. (10.7)
The spontaneous emission rate 2γ can be expressed in terms of the dipole moment,
using Fermi’s golden rule to derive the stimulated emission rate, and then Einstein’s

10.9 Coupling 343
relations to connect this to the spontaneous rate [107]. The result is
2γ =ω3
21|d12|2
3πε0~c3. (10.8)
Here we have neglected any factors arising from degeneracy of the states involved.
The factor of 1/3 represents an average over all spatial direcitons. Substituting
(10.8) into (10.7), and assuming that the signal polarization vs is aligned with the
dipole moment d∗12, gives the result
d =3
4π× nλ2L
∼ nλ2L, (10.9)
where λ = 2πc/ω21 is the wavelength associated with the |1〉 ↔ |2〉 transition, and
where we have made the approximation ωs ≈ ω21 (any detuning is much smaller than
an optical frequency). (10.9) is consistent with the notion that the scattering cross-
section of an atomic transition is roughly λ2. The approximation in the second line of
(10.9) is not generally accurate, but it is extremely useful as an ‘order of magnitude’
estimate for the optical depth. The number density n can be found from the vapour
pressure, given the temperature of the atomic ensemble (see §F.1 in Appendix F).
For an optical transition with λ ∼ 1 µm and a typical ensemble length of L ∼ 1 cm,
we have d ∼ n[m−3]× 10−14. As a rule of thumb, this allows one to easily estimate
the atomic number density required for an efficient memory (with d & 100).
In general, a more accurate value for the optical depth should be calculated by

10.9 Coupling 344
using empirical values for the rate γ (recall that 2γ is the spontaneous emission rate)
and for the dipole moment d12. Sometimes data tables list the values of oscillator
strengths associated with atomic transitions. These are dimensionless numbers that
quantify the dominance of a transition over other possibilities within the atom. The
connection between the oscillator strength and the dipole moment for a transition
is derived in §F.2 of Appendix F. The optical depth can be expressed in terms of
the oscillator strength f12 for the |1〉 ↔ |2〉 transition as follows,
d =(π~αm
)× f12nL
γ, (10.10)
where m is the electron mass and α = 1/137 is the fine structure constant. Figure
10.7 below shows the variation of the optical depth of our cesium cell, with a length
of 10 cm, as a function of temperature.
10.9.2 Rabi frequency
The Rabi frequency is given by
Ω =d23.vcEc
~, (10.11)
where Ec is the electric field amplitude of the control associated with its positive
frequency component. The instantaneous intensity of the control field is given by
Ic =12ε0c× |2Ec|2. (10.12)

10.9 Coupling 345
300 350 400 450 50010
2
104
106
108
Temperature, K
Opt
ical
dep
th
Figure 10.7 Cesium optical depth. This is the optical depth as-sociated with the complete 6P3/2 manifold. The optical depth is thesame whether we consider transitions from the F = 3 ground statelevel, or the F = 4 level. The dipole moment for the cesium D2
line is around 10−29 Cm, according to the data provided by DanielSteck [184] (this figure has been divided by a factor of 3, because weconsider linearly polarized incident fields). The length of our cell is10 cm. The number density is found from the vapour pressure curvein Figure F.1 in Appendix F.
An estimate of the energy in the control pulse is then given by Ec = IcATc. Neglect-
ing any complex phase in Ec, we can express the peak Rabi frequency of the control
in terms of this energy,
Ωmax ≈d23.vc
~×
√Ec
2ε0cATc. (10.13)
This can, of course, be expressed in terms of the oscillator strength for the |2〉 ↔ |3〉
transition using the conversion formula given in Appendix F. If the control pulse
is taken from a pulse train — for example the output of a modelocked laser — we
can write Ec = P/R, where P is the average power in the beam, and where R is the
pulse repetition rate. The pulse duration Tc needs to be measured (see §10.6 above).

10.9 Coupling 346
10.9.3 Raman memory coupling
The Raman memory coupling is defined in ordinary units as C =√dγW/∆. An
approximate expression, assuming a top-hat shape for the control pulse, is
C2 ≈ Tcdγ ×(
Ωmax
∆
)2
. (10.14)
If (10.13) is used in this expression, the factors of Tc cancel, and the assumption of a
top-hat profile can be dropped. Since adiabatic evolution requires that ∆ Ωmax,
the above form of the Raman coupling makes it easy to see why an efficient, adiabatic
Raman memory should have Tcdγ 1 [133]. That is to say, the bandwidth δc ∼ 1/Tc
of the pulses stored (the optimal signal bandwidth is close to the control bandwidth)
should be small compared with dγ.
When estimating the magnitude of the Raman coupling, the following form can
be useful3,
C2 ≈(
~mA∆
)2
× (πα)2 × (f12f23)×NaNc, (10.15)
where Na is the number of atoms in the ensemble that are illuminated by the optical
fields, and where Nc is the number of photons in the control pulse. Putting f12 ≈
f23 ≈ 1 provides an upper limit to the Raman coupling that is achievable with a
given number of atoms and control photons.
As an example, an optical depth of d ∼ 3.8×104 is predicted for our cesium cell,
of length 10 cm, when heated to 90 C. Setting the control field beam waist to 3303This form of the coupling appears in our Rapid Communication on Raman storage [77], but
there is a typo! The form given here is correct.

10.9 Coupling 347
µm gives a Rayleigh range (see §6.4 of Chapter 6) of 5 cm, so that the beam remains
collimated over the full length of the cell. Setting the average power from our laser
oscillator to 600 mW gives a pulse energy of 7.5 nJ. One such pulse, focussed as
just described and detuned by ∆ = 20 GHz from the D2 line is sufficient to produce
a Raman memory coupling of C ∼ 2.4, corresponding to a storage efficiency of
∼ 99.9%. The memory balance is then R ∼ 0.5, so the memory is well within
the Raman regime (i.e. the interaction is balanced; see §5.3.1 in Chapter 5). The
detuning is around 50 times larger than the Doppler linewidth (see §10.10 below),
so the inhomogeneous broadening may be neglected. Adiabaticity is guaranteed
because the detuning is by far the largest frequency involved in the interaction,
∆/γ ∼ 104, ∆Tc ∼ 40, ∆/Ωmax ∼ 6.
10.9.4 Focussing
Substitution of (10.10) and (10.13) into (10.14) reveals that the Raman coupling
depends on the geometry of the ensemble in the following way,
C2 ∝ L
A. (10.16)
This is because the optical depth only depends on the length, and the squared Rabi
frequency depends only on the inverse of the beam area. The ensemble is addressed
by collimated laser beams, which in the ideal case (when the laser is working well)
have a Gaussian transverse profile. The Rayleigh range zR of a focussed Gaussian

10.9 Coupling 348
beam of wavelength λ is related to its cross-sectional area at the focus by diffraction,
zR =Aλ. (10.17)
The beam remains well collimated over a region of length zR either side of the focus,
but after this is quickly diverges, and the intensity drops rapidly. Therefore we may
consider that the length of the ensemble over which the memory interaction is rea-
sonably strong is limited by the Rayleigh range, L ∼ zR. Making this identification
in (10.16), and using the relation (10.17), we find
C2 ∝ 1λ. (10.18)
That is, the geometrical dependence of C drops out. The Raman coupling is inde-
pendent of how the beams are focussed. Loosely focussed beams are better described
by the one-dimensional theory, and indeed numerical simulations show that a loosely
focussed control beam improves the memory efficiency (see 6.5 of Chapter 6). But
if the beams are too loosely focussed, their Rayleigh range will extend beyond the
cell length, and the coupling will be limited by the dimensions of the cell. Therefore
the optimal situation obtains when the Rayleigh range of the beams is matched to
the cell length, zR = L/2.

10.10 Line shape 349
10.10 Line shape
Figure 10.8 shows an absorption spectrum for the D2 line, measured using a laser
with a spectral bandwidth of 1.5 GHz, with the cell heated to 70 C. A number
of factors contribute to the shape of the D2 absorption lines in a warm cesium
vapour. The absorption linewidth of each transition in the D2 line is around 250
MHz at room temperature for a single atom. But the large optical depth widens
the absorption lines of the ensemble (see §7.2.1 in Chapter 7, and §10.11 below).
The dominant line broadening mechanism is Doppler broadening, with pressure
broadening contributing at the level of ∼ 10 MHz. These mechanisms are explained
in §F.3 of Appendix F.
The Doppler width is calculated using (F.12) by setting M = 133 a.m.u. — the
mass of a cesium atom — and ω0 = 2πc/λ0, with λ0 = 852 nm the cesium D2 line
wavelength.
The pressure-broadened linewidth is calculated from (F.18), but the parameters
used are those of the buffer gas. The cesium cell contains neon, which was back-filled
up to a nominal pressure of pbuffer = 20 torr. The buffer gas reduces the mean free
path of the cesium atoms, so that they stay in the interaction region — the volume
illuminated by the light beams — for a longer time than they otherwise would (see
§10.5 below). The number density of neon is much greater than the number density
of cesium, at all reasonable temperatures, so a cesium atom is much more likely
to collide with a neon atom than with another cesium atom. The relevant number
density in estimating the pressure-broadened linewidth is therefore n = pbuffer/kBT0,

10.10 Line shape 350
−10 0 10 200
200
400
600
800
1000
Detuning, GHz
Sig
nal,
mV
Absorption spectrumNo cell
Figure 10.8 Cesium D2 absorption spectrum. This plot appearsthanks to Klaus Reim, a D.Phil student currently dividing his timebetween the cesium and quantum dot projects. A weak laser isscanned across the cesium D2 line at 852 nm. The blue line showsthe signal from a photodiode placed after the cesium cell. The reddotted line indicates the signal detected if the cell is removed. Thetwo dips correspond to the two ground state hyperfine levels: on theleft is the F = 3 state; the F = 4 state is on the right. The 9.2 GHzsplitting between these states is evident. The hyperfine structure ofthe upper 6P3/2 state is not resolved. The laser used has a Gaussianspectrum with a FWHM bandwidth of ∼ 1.5 GHz (see §10.6), andthis contributes to the wide absorption linewidth. Doppler broad-ening, along with the ‘smearing’ effect of the hyperfine splitting inthe upper state, and the large optical depth (d ∼ 104 at 70 C), ac-counts for the remainder of the linewidth, with pressure broadeningcontributing negligibly.
where T0 ∼ 300 K is the temperature at which the cell was filled. The cell is sealed,
so the number density of the buffer gas is fixed. The relevant collision velocity is
the thermal velocity of Neon atoms, calculated using M = 10 a.m.u.. A reasonable
figure for the collision cross-section σ is found by choosing datom = (300 + 50)/2 pm,
which represents an average of the atomic diameters of a cesium and neon atom.
Figure 10.9 shows the variation in the Doppler and pressure-broadened linewidths
with temperature for our cesium cell. The temperature dependence is very weak,

10.11 Effective depth 351
since both linewidths scale linearly with the thermal velocity of the atoms in the
vapour cell, which in turn scales with√T . It’s clear that Doppler broadening dom-
inates over pressure broadening. The linewidth is similar to the hyperfine splitting
in the 6P3/2 excited state manifold, so these hyperfine states are not resolved in our
sample. Fortunately it is not important, either for optical pumping (see §10.12 be-
low), or for the quantum memory interaction, to distinguish the hyperfine structure
of the excited state. Provided that Raman storage is implemented with a detuning
that is large compared to both the Doppler linewidth and the hyperfine splitting,
a theoretical description that ignores these complications remains appropriate. In
fact, Gorshkov et al. have shown that even resonant storage is unaffected by Doppler
broadening provided the ensemble is sufficiently optically thick [163]. However, char-
acterizing the optical depth of the ensemble is not entirely straightforward, in the
presence of line-broadening.
10.11 Effective depth
If a weak probe beam is tuned into resonance with one of the D2 line transitions,
the measured attenuation is much less than would be predicted on the basis of the
optical depth estimated using (10.9). The reason for this is the redistribution of
optical depth over a wide spectral range, because of Doppler broadening. Suppose
one measures the transmitted power Pout, and the input power Pin. The effective
optical depth is defined by
Pout = Pin × e−2deff . (10.19)

10.11 Effective depth 352
300 350 400 450 50010
6
107
108
109
Temperature, K
Lin
ew
idth
, Hz
Pressure
Doppler
Figure 10.9 Absorption linewidth. The pressure (green) andDoppler (blue) linewidths γ/2π for our cesium sample, calculatedusing the formulae (F.12) and (F.18) derived in Appendix F. Thered dotted line shows the half-width at half-maximum of the result-ing Voigt profile, which is the line profile resulting from the convo-lution of the pressure-broadened Lorentzian and Doppler-broadenedGaussian lineshapes. Doppler broadening dominates however, andthe lineshape is essentially Gaussian.
The spectrum of the transmitted beam can be found from (7.29) in Chapter 7,
setting z = 1 and Pin = 0. In the presence of pressure-broadening, the polarization
decay rate is modified,
γ −→ γ′ = γ + γp/2, (10.20)
and the pressure broadened optical depth dp = d×γ/γ′ should be used. The effective
optical depth is then given by
deff =12
ln ∫
Iin(ω) dω∫Iin(ω)e−2dp<[f(ω)] dω
, (10.21)
where Iin(ω) is the spectral intensity profile of the probe beam, and where the
lineshape function f(ω) is defined in (7.28) in Chapter 7, the only difference being

10.12 Optical pumping 353
that all frequencies are normalized by γ′ instead of γ.
Using a beam with an average power of 1 µW, taken from the output of a
modelocked Ti:Sapphire oscillator with a spectral bandwidth of 1.5 GHz (see §10.6),
we measure deff ∼ 3 at a cell temperature of 90 C. If we invert the formula (10.21),
using γ = 16.4 MHz (the spontaneous lifetime of the cesium D2 line is τ = 1/2γ = 30
ns), γ′ ≈ 70 MHz and γd ≈ 260 MHz, we infer a ‘real’ optical depth of d ≈ 3,500.
This is consistent with the prediction d = 3,700, found from (10.7) using the number
density plotted in part (b) of Figure F.1.
10.12 Optical pumping
Even though the hyperfine splitting is extremely large, it represents a very small en-
ergy gap at room temperature, with Esplitting ≈ kBT/680. Therefore the populations
of both the clock states are equal. From the perspective of quantum storage, this
means there is a large thermal background of incoherent material excitations that
would swamp any stored signal. But the situation is worse than this: the memory
interaction is totally destroyed if the populations of the two lower states are equal.
As signal photons are absorbed, they are also produced by Stokes scattering from
the thermal population in the storage state. The gain from thermally seeded Stokes
scattering exactly balances the quantum memory absorption, and the memory is
rendered useless. This effect is demonstrated in Figure 10.10.
For an efficient quantum memory, therefore, one of the lower two states must be
emptied. This is done in the laboratory by by optical pumping.
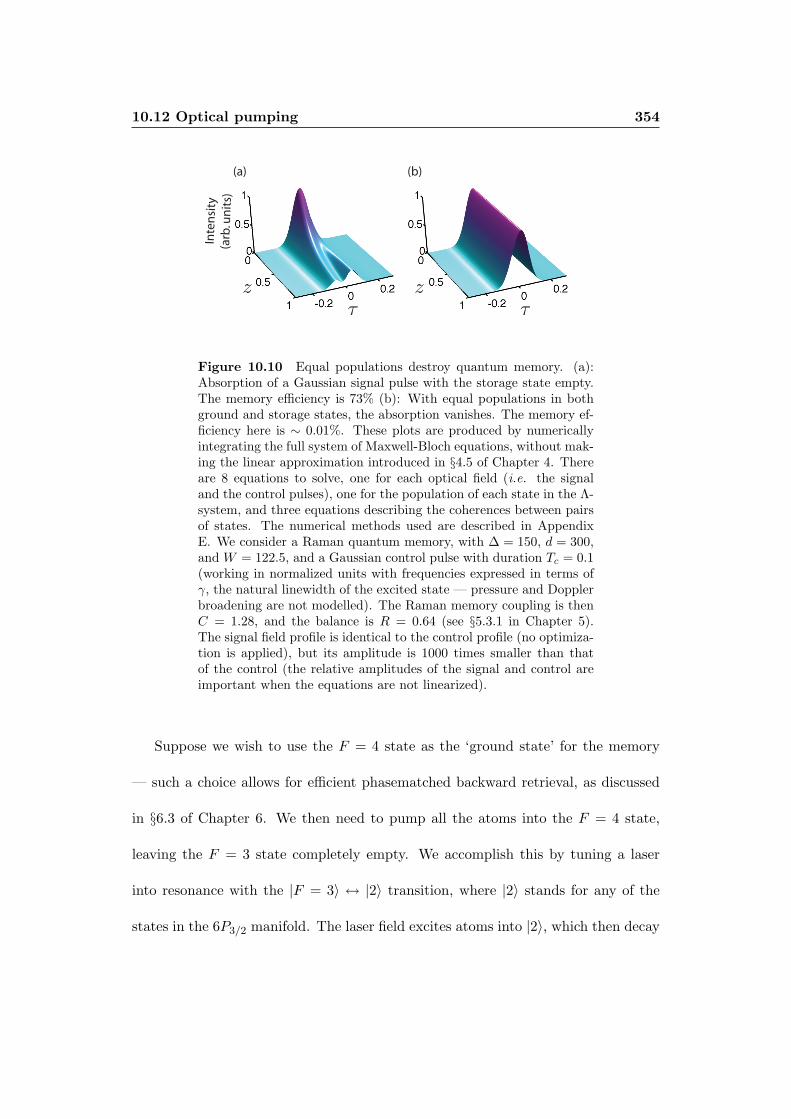
10.12 Optical pumping 354
Inte
nsi
ty(a
rb. u
nit
s)
(a) (b)
Figure 10.10 Equal populations destroy quantum memory. (a):Absorption of a Gaussian signal pulse with the storage state empty.The memory efficiency is 73% (b): With equal populations in bothground and storage states, the absorption vanishes. The memory ef-ficiency here is ∼ 0.01%. These plots are produced by numericallyintegrating the full system of Maxwell-Bloch equations, without mak-ing the linear approximation introduced in §4.5 of Chapter 4. Thereare 8 equations to solve, one for each optical field (i.e. the signaland the control pulses), one for the population of each state in the Λ-system, and three equations describing the coherences between pairsof states. The numerical methods used are described in AppendixE. We consider a Raman quantum memory, with ∆ = 150, d = 300,and W = 122.5, and a Gaussian control pulse with duration Tc = 0.1(working in normalized units with frequencies expressed in terms ofγ, the natural linewidth of the excited state — pressure and Dopplerbroadening are not modelled). The Raman memory coupling is thenC = 1.28, and the balance is R = 0.64 (see §5.3.1 in Chapter 5).The signal field profile is identical to the control profile (no optimiza-tion is applied), but its amplitude is 1000 times smaller than thatof the control (the relative amplitudes of the signal and control areimportant when the equations are not linearized).
Suppose we wish to use the F = 4 state as the ‘ground state’ for the memory
— such a choice allows for efficient phasematched backward retrieval, as discussed
in §6.3 of Chapter 6. We then need to pump all the atoms into the F = 4 state,
leaving the F = 3 state completely empty. We accomplish this by tuning a laser
into resonance with the |F = 3〉 ↔ |2〉 transition, where |2〉 stands for any of the
states in the 6P3/2 manifold. The laser field excites atoms into |2〉, which then decay

10.12 Optical pumping 355
by spontaneous emission back into both the F = 3 and F = 4 ground states (the
branching ratio is roughly equal). The laser field then re-excites the atoms, and
the process repeats (see Figure 10.11). Consider the effect of consecutive cycles of
excitation and decay. With each cycle, only around 50% of the initial population
of the F = 3 state ends up back in F = 3. The rest ends up in the F = 4 state.
Since there is no laser field exciting atoms out of this state, population builds up in
F = 4, while the F = 3 state is eventually emptied entirely. This is the principle
behind optical pumping.
Figure 10.11 Optical pumping. A CW diode laser is tuned intoresonance with the |F = 3〉 ↔ |2〉 transition. Spontaneous emissionredistributes the excited population more-or-less equally between thetwo ground hyperfine levels. After several cycles, population buildsup in the un-pumped F = 4 state, while the F = 3 state is emptied.
10.12.1 Pumping efficiency
To characterize the optical pumping efficiency a simple measurement was made (this
was done by Virginia Lorenz and Klaus Reim), based on an experiment performed
by Jean-Louis Picque in 1974 [185]. The set-up is illustrated in Figure 10.12. We use
an external cavity diode laser to provide the pumping light. This is a single-mode

10.13 Filtering 356
diode laser with a diffraction grating placed in front of it in the Littrow orientation:
the first order of diffraction is directed back into the laser. By fine-tuning the
grating angle, it is possible to select those frequencies that experience the greatest
optical gain (other frequencies are misaligned by the grating and are lost). The
diode laser frequency can therefore by stabilized and tuned using the grating. The
specular reflection from the grating provides the laser output, which is a continuous
wave (CW) beam with a linewidth of around 100 MHz. Using the measurement
scheme shown in Figure 10.12, we detect the fluorescence signal from a weak probe
beam. The suppression of this signal with the pump beam set at 30 mW over the
signal with the pump blocked indicates that we achieve a pumping efficiency of
around 95%, with the cell temperature set at T = 50 C. At higher temperatures,
the optical pumping efficiency drops as the energy in the pump is absorbed by the
larger number of atoms. We are currently installing an ECDL with an output power
of 100 mW, which should improve our pumping efficiency for higher temperatures.
10.13 Filtering
The greatest experimental challenge posed by the implementation of a Raman quan-
tum memory is the ability to filter out the very weak — even single photon — signal
field from the strong classical control pulse. The problem is particularly difficult in
cesium, because the Stokes shift of 9.2 GHz corresponds to a very small spectral
shift. At 852 nm — the D2 resonance wavelength — the signal and control frequen-
cies differ by around 20 pm. The required filter contrast is of order 108 or better,

10.13 Filtering 357
Laser diode
Lock-in
Pump
Probe
Cs cell
Heaters
Lens
Photodiode
Chopper
Grating
Beamsplitter
Figure 10.12 Verifying efficient optical pumping. The beam froman external cavity diode laser (ECDL) tuned into resonance withthe |F = 3〉 ↔ |2〉 transition is split into two parts at an asymmetricbeamsplitter (just a microscope slide). 96% of the diode beam is usedto pump the cesium atoms in the heated cell. 4% of the beam is sentthrough an optical chopper, which applies a 1 kHz modulation, beforebeing directed through the cell at a small angle to the pump beam.Fluorescence is collected by a lens and focussed onto a photodiode. Alock-in amplifier is used to isolate the component of the fluorescencedue to the modulated probe beam. This signal quantifies the opticalpumping efficiency. The inset shows the reduction in the fluorescencesignal as the pump power is increased. A pumping efficiency of 95%is achieved. These data appear thanks to Virginia Lorenz and KlausReim.
so that no single filter provides sufficient rejection of the pump: several filters must
be used in tandem. In this section we give details of the various filtering techniques
we have at our disposal.

10.13 Filtering 358
10.13.1 Polarization filtering
The Raman interaction couples orthogonal linear polarizations: a horizontally po-
larized control pulse will store a vertically polarized signal pulse, and vice versa,
provided the detuning is sufficiently large. And any Stokes scattering induced by a
linearly polarized pump pulse is polarized orthogonally to the pump. These facts are
derived in §F.4 in Appendix F. Therefore the pump can be effectively rejected after
the cell using a polarizer aligned orthogonally to the pump polarization. We use
Glan-Laser polarizers from Foctek, which use total internal reflection at the bound-
ary of two calcite crystals with perpendicular optic axes to preferentially transmit
one linear polarization. The extinction ratio of these polarizers is of order 10−6.
However, stress-induced birefringence of the cell windows can distort the pump po-
larization, causing leakage of the pump light through the polarizer. The effective
polarization extinction is therefore of order 10−4.
10.13.2 Lyot filter
A Lyot filter is a polarization-based spectral filter, popular among astronomers be-
cause of its wide working numerical aperture. In general a Lyot filter consists of
a number of concatenated filter stages, with each stage improving the finesse of
the filter (that is, reducing the spectral width of the pass-band, while increasing
the spectral width of the region over which frequencies are blocked). We built the
simplest possible type of Lyot filter, which involves a single stage. The filter then
has a sinusoidal transmission as a function of frequency. Figure 10.13 shows the

10.13 Filtering 359
structure of the filter. It is essentially a Mach-Zender interferometer for polariza-
tion: an incident pulse is split into fast and slow polarization components inside a
birefringent retarder. The polarizations are then combined on a polarizer, where
they interfere to produce spectral fringes. The free spectral range (i.e. the period
of the fringes in frequency) depends inversely on the length and birefringence of the
material used for the retarder. In order to be useful as a spectral filter for Stokes
scattering on the cesium D2 line, a free spectral range of ∆f = 2 × 9.2 GHz is
required, so that transmission of the Stokes light is accompanied by rejection of the
Raman pump light (see Figure 10.14). Calcite has the largest birefringence avail-
able, with |no − ne| = 0.17, but 10 cm of calcite are still required to produce the
required free spectral range! We use three pieces of calcite, each around 3 cm long,
mounted in series, along with a pair of Foctek Glan-laser polarizers (see previous
section). The filter contrast is limited to 99%, however, because of phase distortions
arising from the surface roughness of the calcite faces. The Lyot filter can be tuned
in frequency over one free spectral range by tilting one of the calcite crystals slightly,
which alters the optical path length.
10.13.3 Etalons
Since the Lyot filter does not have sufficient contrast for filtering the Stokes light, we
ordered some custom Fabry-Perot etalons. These are fixed air-gap etalons — pairs
of optically flat glass plates that form a planar cavity with a discrete transmission
spectrum. The free spectral range of the etalons depends on the separation L of

10.13 Filtering 360
optic axis
P1
B1
B2
P2
Retarder
Fringes(a) (b)
Figure 10.13 Lyot filter. (a): The filter consists of a piece ofbirefringent material of length L — a retarder — placed betweenpolarizers P1 and P2. The optic axis of the retarder is aligned at45 to P1, so that an incident pulse of wavelength λ (purple) is splitinto ordinary and extraordinary polarization components (red andblue pulses). One component is delayed with respect to the other,because the refractive indices no, ne associated with each componentare different. The phase retardance is given by φ = 2π|no − ne|L/λ.When the components recombine at P2, they interfere, producingsinusoidal spectral fringes, with a free spectral range ∆f = c/L|no −ne|. (b): Analogy with a Mach-Zender interferometer, in which twodelayed pulses are mixed on a beamsplitter, producing fringes.
the plates as ∆f = c/2L, since 2L is the distance an optical wave must traverse
to make a round-trip of the cavity. Requiring a free spectral range of 18.4 GHz,
as in the case of the Lyot filter, sets the etalon plate separation to be 8.2 mm. As
mentioned in §10.6 below, the laser bandwidth is around 1.5 GHz, so the width of
the pass band should not be smaller than this, in order to transmit the full Stokes
spectrum (which follows the Raman pump spectrum). The maximum finesse is then
F = 18.4/1.5 ∼ 12. The finesse of a Fabry-Perot etalon is fixed by the reflectivity
R of the plates comprising the cavity, according to the formula
F =π√R
1−R. (10.22)

10.13 Filtering 361
9.2 GHz
Stokes
Pump
Figure 10.14 Stokes filtering. A Lyot filter with a free spectralrange of 18.4 GHz can be used to suppress the Raman pump, whiletransmitting the weaker Stokes field. The finesse F = ∆f/δf , whereδf is the FWHM of the pass band, is only 2, so this filter is far fromideal.
The above considerations therefore fix the plate reflectivity to be 78%. This limits
the out-of-band extinction to around 95%, so one of these etalons is not quite as
good a filter as the Lyot filter. However, they are considerably more convenient,
being very easy to align, and much smaller! Six custom etalons with these specifi-
cations, and a large clear aperture of 15 mm, were ordered from CVI Melles Griot.
Initially it was intended to concatenate several etalons together, in series, so as to
combine their extinction. However, when the etalons are placed one after another,
their behaviour becomes complicated by interference effects arising from reflections
between the etalons. This is mitigated somewhat because the etalons we use have
anti-reflection coatings on their outside faces, which are also deliberately wedged.
Nonetheless it can be problematic to use the etalons consecutively.

10.13 Filtering 362
10.13.4 Spectrometer
Most recently, a grating spectrometer was built to filter the Stokes light (this was
done by Virginia Lorenz and Klaus Reim). The resolution required is R = λ/δλ >
852/0.02 ≈ 106. The resolution of a grating spectrometer is limited by the number
of lines ruled on the grating, R ≈ N . The spacing between consecutive lines cannot
fall below half the optical wavelength, or the first order of diffraction does not exist.
For light at λ = 852 nm, this limits the maximum groove frequency to less than
2300 lines mm−1. Using this groove frequency, a grating roughly 40 cm across is
required. Such a large grating is not available, but a spectrometer with a grating
around 6 cm across has been built, using a large off-axis parabolic mirror to focus
the diffracted light onto a photon-counting CCD camera. Although the resolution
of this spectrometer is sub-optimal, the ability to visualize the spectrum of the light
downstream from the cesium cell makes it an extremely useful apparatus.
All the above techniques have been used in an attempt to observe strong Stokes
scattering from the cesium cell. So far we have not been successful, but our combined
filter contrast is still improving.
10.13.5 Spatial filtering
The utility of spatial filtering for a quantum memory is discussed in §6.3.2 of Chapter
6. In a quantum memory, where both signal and control beams are directed by the
experimenter, it is possible to introduce a small angle between the beams so that
the strong control pulse can be blocked after the cell. When looking for stimulated

10.13 Filtering 363
Stokes scattering however, it is necessary to look along the axis of the Raman pump,
since the Raman gain is restricted to this direction. Therefore it is not generally
possible to spatially filter Stokes scattering, although attempting to detect scattered
light at very small angles from the pump direction may be sensible. A pinhole, or
even an optical fibre — fibre in-coupling is very spatially selective — can be used;
we are investigating these possibilities.
It is possible, however, to look for Stokes scattering in the backward direction.
The duration of our pulses is sufficiently long that they extend, in space, over the
length of the cell: cTc ∼ L. This means that a Stokes photon emitted near the exit
face of the cell in the backward direction, while illuminated by the leading edge of
the pump pulse, can propagate backwards, nearly all the way to the entrance face
of the cell, while all the time the cell remains illuminated by the pump, so that the
photon experiences Raman gain all along the cell’s length (see part (a) of Figure
10.15). The experimental set-up shown in part (b) of Figure 10.15 has been used
to look for this type of Stokes scatter. Strong stimulated fluorescence has been
observed, and the signal is extremely clear, because there is no need to filter out
the control pulse: it is propagating in the opposite direction! However the search
is still on for Stokes scattering, the signature being that the Stokes signal should
tune in frequency as the pump frequency is tuned, rather than remaining at the D2
resonance, as it does currently.
Similar arguments would suggest that a Raman quantum memory in which the
control and signal pulses are counter-propagating might be efficient, as long as the

10.14 Signal pulse 364
(a)
(b)
Figure 10.15 Backward Stokes scattering. (a): If the spatial ex-tent of the Raman pump pulse is comparable to the cell length,a backward scattered Stokes photon experiences Raman gain as itpropagates backward. (b): A polarizing beamsplitter can be usedto deliver the control pulse, and separate the orthogonally polarizedstimulated Stokes light that is scattered backwards. This essentiallyeliminates the background from the Raman pump. We are currentlylooking for Stokes light using this method.
pulses are sufficiently long. The propagation theory of Chapters 4 and 5 is not ap-
plicable in this situation, but the numerical model presented in Chapter 6 can be
used to show that the memory efficiency indeed remains high in this case. Such
an arrangement removes the demanding filtering requirements, and is therefore an
appealing possibility. A classical optical memory based on off-resonant Brillouin
scattering using counter-propagating pulses in an optical fibre has in fact been im-
plemented [186], so there is some precedent for this approach.
10.14 Signal pulse
Before implementing a single-photon quantum memory, a proof-of-principle demon-
stration using a weak coherent pulse for the signal field is planned. A single-photon

10.14 Signal pulse 365
source for the signal field is not, therefore, of immediate concern. However it remains
challenging to generate the signal field. Currently we have only one laser oscillator.
It is therefore necessary to generate the signal pulse from the control. The idea is to
sample a small portion of the control field, perhaps using a beamsplitter, and then
to apply a frequency modulation to shift the carrier frequency of the sampled pulse
by 9.2 GHz, so that this frequency shifted pulse can act as the signal.
Raman modulator One way to achieve this frequency modulation is to use the
cesium atoms themselves, by inducing strong Stokes scattering of a control pulse.
The Stokes sideband can then be sent into a second cesium cell along with the re-
maining control, where storage can be implemented. However, the Stokes scattering
process is somewhat aleatoric: large fluctuations in the intensity of the Stokes light
may make it difficult to assess the reliability of the memory.
EOM A second possibility is to use an Electro-Optic Modulator (EOM) to apply
the frequency shear. This is a device containing an electro-optically active crystal,
whose refractive index can be altered by the application of an external voltage.
Subjecting the crystal to a sinusoidally varying potential with frequency Ω will
imprint the waveform onto the phase of an optical wave passing through the crystal,
E(t) −→ E(t)× eiφ sin(Ωt), (10.23)

10.15 Planned experiment 366
where φ quantifies the amplitude of the phase modulation. Fourier transforming
(10.23) reveals the presence of sidebands, separated by the modulation frequency,
E(ω) =∞∑
k=−∞sgn(k)|k|J|k| (φ) E(ω + kΩ), (10.24)
where the sideband amplitudes are given by Bessel functions. With the choice
φ ∼ 0.2, the energy in the first sideband represents around 2% of the total trans-
mitted energy, while all higher order sidebands may be neglected. Use of such a
modulator allows one to reproducibly generate a weak signal pulse with the correct
frequency shift from the control pulse. A modulator with the ability to operate in
the microwave X-band at 9.2 GHz has been ordered from New Focus.
10.15 Planned experiment
The current experimental set-up is in a state of flux, we are continually improving
our filtering contrast and optical pumping efficiency, in the hope of detecting the
strong stimulated Stokes signal that indicates there is sufficient Raman coupling to
implement a Raman memory. Plans for the final implementation of the memory are
tentative, being contingent on the eventual success of these early stages. However in
the spirit of optimism we present in Figure 10.16 below a schematic of the possible
layout of an experimental demonstration of a cesium Raman memory. The optical
pumping beams are not shown, for clarity, although of course efficient optical pump-
ing is critical. The off-axis geometry for phasematched retrieval described in Chapter

10.15 Planned experiment 367
6 is used, and we assume that the atoms have been prepared by optical pumping in
the upper F = 4 state for this purpose. The angle between the control and signal
beams is around 2, and we assume that the control is more loosely focussed than
the signal (the signal focus can be tightened by expanding the signal beam before it
enters the confocal system, but we have not shown the beam expander). Since we
seek only to demonstrate the feasibility of the memory, a long memory lifetime is
not important, and so the delay between the storage and retrieval control pulses is
adjusted by a mechanical delay stage. It is only feasible to move such a stage over a
few feet, which corresponds to just a few nanoseconds of variability in the memory
storage time, but this is sufficient for our purposes. The quarter wave plate in the
control beam following the cell rotates the control polarization through 90 (since
the control beam traverses it twice, the combined effect being that of a half-wave
plate). The rotated control then retrieves the signal field into the orthogonal polar-
ization mode to the polarization mode of the incident signal field. This allows the
retrieved signal to be re-directed to a detector using a polarizing beamsplitter. The
efficiency of the memory can be quantified by comparing the energy in the retrieved
signal field to that of the incident signal. The incident signal pulse is generated from
the control using an EOM (see §10.14 above), and an etalon removes the fundamen-
tal (the unmodulated light transmitted through the EOM). A Pockels cell is used
to reduce the repetition rate of the Ti:Sapphire laser, as described in §10.7.
We look forward to overcoming our present difficulties and assembling the above
apparatus, or a variation thereupon, in the near future.

10.15 Planned experiment 368
EOM
Ti:Sapphire
Pulse
Picker Block L1 L2
QWP
Delay stage
E1
E2
Cs cell
Retrieved signal
Figure 10.16 A possible design for demonstration of a cesiumquantum memory. The pulse train from a Ti:Sapphire oscillatorpasses through a pulse picker to reduce the pulse repetition rate.Consider a single pulse. A beamsplitter redirects a portion of thepulse into an EOM, and the first sideband, shifted by 9.2 GHz is iso-lated from the output using a Fabry-Perot etalon E1 (see §10.13.3).This is the input signal field. The remainder of the initial pulse is usedas the control field. It is directed through the cesium cell at a smallangle to the signal pulse using a confocal arrangement (lenses L1 andL2): the signal is (hopefully) stored in the cell. The transmitted con-trol field is sent through a variable delay line, and its polarization isrotated through 90, before being sent back through the cell. Thestored signal field is retrieved with the orthogonal polarization to theincident signal, and is sent by a polarizing beamsplitter through anetalon E2 (to remove any residual control) to a detector. Note thatwe have not shown the optical pumping beams, which are critical.
This concludes the thesis. In the next chapter we summarize the results of the
present research.

Chapter 11
Summary
This thesis has been concerned with the problem of storing light. I can recall feeling
some puzzlement, lying in bed on a school night, at how completely my room dark-
ened when the light was switched off. Why did you have to keep pouring more and
more light into a room? Well, light is an ephemeral beast. But the possibility of
a material that remembers its illumination — not with the feeble pallor of glow-in-
the-dark paint, but with the unmistakable vigour of a laser pulse — is remarkable. I
am not the first to study such media, and the current research was undertaken in the
aftermath of the successes of light stopping by EIT. The main contributions of this
thesis are theoretical: a fairly general framework for the analysis and optimization
of light storage has been developed. The framework provides a unified description
of both EIT and Raman storage, and generalizes to tCRIB, lCRIB, AFC and broad-
ened Raman protocols. Further applications of the formalism are expected. The
use of the SVD has been crucial to the success of the theoretical programme. Many

370
of the results in the thesis are simple adaptations of well-known facts from linear-
algebra to the particular case under study. Another important ingredient of the
thesis is numerical simulation. The propagation of optical fields through an atomic
ensemble is always described by a set of coupled linear partial differential equations,
and these are particularly easy to solve on a modern computer.
The ‘take-home’ results are as follows.
1. Any quantum memory is a linear system, with storage and retrieval interac-
tions described by Green’s functions, which are essentially large matrices.
2. The SVD of the Green’s functions provides a complete characterization of the
memory. It allows one to immediately identify the optimal input mode.
3. The singular values of the Green’s function are invariant under unitary trans-
formations. This fact can be very useful in analyzing the memory interaction.
One can work in the Fourier domain, or indeed in an entirely unfamiliar coor-
dinate system.
4. The efficiency of a memory is limited by its optical depth [133].
5. Explicit expressions for the Green’s functions describing storage in a Λ-type
atomic ensemble are provided. The Rosen-Zener kernel holds whenever the
control has a hyperbolic secant temporal profile. The adiabatic kernel holds for
all detunings and control pulse shapes that satisfy the adiabatic approximation.
The Raman kernel holds for adiabatic interactions that are far-detuned and
‘balanced’.

371
6. In the adiabatic limit, a coordinate transformation links the results for different
control profiles. Therefore the SVD only needs to be computed once for a
given control pulse energy. Changes to the control profile simple change the
coordinate transformation.
7. A Raman memory may be characterized as a multimode beamsplitter inter-
action between optical and material modes. A single set of modes describes
both the transmitted fields and the stored excitations. A single number C
characterizes the efficiency of a Raman memory.
8. The Green’s function can always be constructed numerically, so that the op-
timal input modes and memory efficiencies can be found at any detuning,
regardless of whether or not the interaction is adiabatic.
9. Retrieval of the stored excitations can be problematic. Forward retrieval suf-
fers from re-absorption losses. Backward retrieval is not phasematched if the
ground and storage states are non-degenerate. Numerical simulations ver-
ify that an off-axis geometry allows for efficient backward retrieval with non-
degenerate states. Loose focussing of the control field is desirable.
10. The multimode capacity of a quantum memory can be evaluated by consid-
ering the SVD of the Green’s function. The multimode scaling of EIT, Ra-
man, tCRIB, lCRIB, AFC, and a broadened Raman protocol is studied. Un-
broadened ensembles have poor multimode scaling. Adding an inhomogeneous
broadening improves the scaling. The AFC protocol has the best multimode

11.1 Future work 372
scaling of all the protocols studied.
11. If one is not able to shape the signal pulse, optimal storage can still be achieved
by instead shaping the control. But to solve the optimization problem, the
equations of motion for the memory must be solved numerically. This can
be done rather quickly, however. A simple optimization algorithm works well
for finding the optimal control profiles. The SVD allows one to verify the
optimality of the numerical solutions. This optimality suffers as the interaction
becomes less adiabatic.
12. A Raman memory in bulk diamond, based on the excitation of optical phonons,
is feasible. It is shown that the equations of motion describing the Raman
interaction in diamond have precisely the same form as the Raman equations
describing storage in an atomic vapour. The form of the coupling constant C
is derived.
13. Attempts to implement Raman storage in cesium vapour have been made, but
it is proving difficult even to generate and detect Stokes scattering. I am not
a good experimentalist!
11.1 Future work
There is a great deal of experimental work to do. It may be that there is a good
reason why our attempts to build a Raman memory have been unsuccessful: we
should either make the memory work, or find this reason, in the coming year.

11.1 Future work 373
An intriguing theoretical challenge is how to make use of the stored excitations
once they are in place. Is it possible to perform computationally interesting oper-
ations on the spin wave in an atomic ensemble? Can operations be designed that
allow different stored modes in a multimode memory to interact? Work on this front
has begun in the literature [17,187,188], but this is likely to be a rich seam.
If you have survived this far, I am very grateful for your attention! I hope that
some of the results in this thesis are useful to other workers in the field, even if only
as a warning of what not to try.

Appendix A
Linear algebra
Physics is generally concerned with change: the evolution of a system over time, or
the response of a system to an external agent. The easiest, and most uninspiring
situation to analyze, is when there is no change and no response. Linear algebra
is concerned with the much more interesting situation arising when the response
depends linearly on some parameter. On the one hand, this is almost always an
approximation that only holds for small changes in a parameter, and small responses.
So linear algebra rarely provides an exact description. On the other hand, the
linear approximation can be successfully applied to nearly every physical system!
Linear algebra is therefore useful in almost every branch of physics, as well as in
mathematics and science generally. In our case, the linear response of a quantum
memory is certainly an approximation, valid when the signal field does not contain
too many photons.
Here we summarize various concepts that are needed to properly understand the

A.1 Vectors 375
singular value decomposition as it pertains to the optimization of quantum storage.
There is a significant overlap with the formalism of quantum mechanics, so we
will also take this opportunity to review some aspects of that formalism. A clear
and comprehensive introduction can be found in Nielsen and Chuang’s quantum
information bible [158].
A.1 Vectors
A vector is essentially a list of numbers. It also helps to keep in mind the image of
a vector as an arrow (see Figure A.1). This analogy cannot always be made with
rigour, but it provides a convenient visualization. The numbers comprising a vector
are the components of the arrow along the coordinate axes. When writing down
these components, implicit reference is therefore always made to some coordinate
system. It’s clear that we could rotate the coordinate axes — altering the vector’s
components — without changing the arrow in Figure (A.1), and in this sense, a
vector transcends its components. Nonetheless, it will be useful to write down the
vector components for concreteness.
We will use two equivalent sets of notation for a vector labelled ‘v’. Either v, or
|v〉. The first symbol, in bold face, is in general use. The second — a ket — is an
example of ‘Dirac notation’, used only in the context of quantum mechanics. Dirac
notation is at times very convenient, and it will help to be able to use these two
types of notation interchangeably.
The number of components of a vector is called the dimension of the vector. If

A.1 Vectors 376
Figure A.1 A vector. On the left is a representation in ‘componentform’. On the right the same mathematical object is drawn as anarrow. The direction and length of the arrow are determined by itscomponents α, β. Implicitly, a coordinate system (thinner arrows) isused to define the components.
there are n components, the vector is said to be n-dimensional. We will adopt the
convention that v is a column vector;
v = |v〉 =
v1
v2
...
vn
. (A.1)
Vectors can be added together, provided they have the same dimension,
v +w = |v〉+ |w〉 =
v1 + w1
v2 + w2
...
vn + wn
. (A.2)

A.1 Vectors 377
And a vector can be multiplied by a number, say α, like this
αv = α|v〉 =
αv1
αv2
...
αvn
. (A.3)
Using these operations, vectors can be combined together to make new vectors, and
it is convenient to think of all these possible vectors as inhabiting a ‘space’, known
as an n-dimensional vector space. Our own universe, with three spatial dimensions,
can be thought of as a 3-space.
A.1.1 Adjoint vectors
The components of a vector do not have to be real numbers. In quantum mechanics,
and many other applications, they are generally complex numbers. A useful concept
in this case is the Hermitian adjoint v† of a vector v. This is simply another vector,
this time a row vector, with each component equal to the complex conjugate of the
corresponding component of v,
v† = 〈v| =(v∗1 v∗2 . . . v∗n
). (A.4)
The symbol 〈v| is known, rather unfortunately, as a ‘bra’, for reasons that will
become clear. These row vectors (bras) can be added or multiplied by numbers in
the same way as column vectors (kets), and so they form their own vector space,

A.1 Vectors 378
sometimes known as the adjoint space.
Every vector v has a corresponding Hermitian adjoint v†; every ket has its
corresponding bra. And Hermitian conjugation is involutive: The Hermitian adjoint
of v† is v again.
A.1.2 Inner product
Vectors can be multiplied together in ways as various as mathematicians are inven-
tive. The inner product — sometimes scalar product — is defined as the sum of the
component-wise products of a bra and a ket with the same dimension,
v†w = 〈v|w〉 = v∗1w1 + v∗2w2 + . . .+ v∗nwn. (A.5)
This type of product between two vectors is not another vector; it’s just a number.
For some reason it’s rather satisfying to take an inner product, and Paul Dirac’s
notation anticipates something of this satisfaction. When a bra 〈v| encounters a ket
|w〉 they merge to become a ‘braket’ 〈v|w〉, and so fulfill their destiny.
It’s quite common to speak of taking the inner product of two kets, |v〉 and
|w〉. In this case it is understood that one of the kets has to be replaced by its
corresponding bra before using (A.5). Note that 〈v|w〉 = 〈w|v〉∗, so one should be
consistent about which of the two kets is replaced.
A complex vector space with an inner product defined as in (A.5) is known as
a Hilbert space. Quantum mechanics is a theory about vectors in Hilbert space; as
such it is extremely simple. It is the reconciliation of this mathematical structure

A.1 Vectors 379
with what we know about the real world that makes the theory so difficult to pin
down.
A.1.3 Norm
Having defined the inner product, we can now define the norm of a vector. This is
defined as the square root of the inner product of a vector with itself,
v = ||v|| = || |v〉 || =√v†v =
√〈v|v〉. (A.6)
This is a positive, real quantity that grows with the size of the components of v. In
fact, substituting in the definition (A.5) and applying Pythagoras’ theorem shows
that the norm is simply the length of the arrow representing the vector v. Some
further geometrical manipulations reveal that the inner product is related to the
angle θ between the arrows representing two vectors, as follows (see Figure A.7),
v†w = 〈v|w〉 = vw cos θ. (A.7)
An immediate consequence of this is that the inner product 〈v|w〉 vanishes when
θ = π/2, that is, when v is perpendicular to w. Often, the word orthogonal is
used instead of perpendicular, especially in the case that the vectors involved are
complex, or when they have dimension greater than 3, since then the notion of an
angle is less transparent.

A.1 Vectors 380
Figure A.2 The inner product of two vectors.
A.1.4 Bases
We have already mentioned in passing the concept of a coordinate system, with
respect to which the components of a vector are defined. We drew the coordinate
axes in Figure A.1 as black arrows. The axes themselves are therefore described by
a pair of vectors, one pointing along the x-axis; the other along the y-axis. Let’s
call them |x〉 and |y〉. If we fix the length (the norm) of these vectors as 1, then we
can write any vector v directly in terms of |x〉 and |y〉,
|v〉 =
vx
vy
= vx|x〉+ vy|y〉. (A.8)
The set of two vectors |x〉, |y〉 is a basis from which we can construct any other
2-dimensional vector. In fact, it’s clear that any two vectors, as long as they point
in different directions, can serve as a basis. The nice feature of the set |x〉, |y〉 is
that these two vectors are orthogonal to each other, 〈x|y〉 = 0. This is particularly
convenient, because the components of |v〉 can be found directly by taking inner
products, vx = 〈x|v〉, vy = 〈y|v〉. A basis of this kind is almost always preferable to
non-orthogonal bases. Such a basis, with mutually orthogonal basis vectors of unit

A.2 Matrices 381
norm, is called an orthonormal basis. When we speak of a coordinate system, or
coordinate axes, we are implicitly making reference to an orthonormal basis.
A.2 Matrices
A matrix is essentially an array of numbers, laid out on a rectangular grid, as follows:
M =
M11 M12 . . . M1n
M21 M22 . . . M2n
......
. . ....
Mm1 Mm2 . . . Mmn
(A.9)
The numbers Mij comprising a matrix are known as its elements. The dimension
of a matrix is specified by two numbers, the number of rows, and the number of
columns in the matrix. In the example (A.9) M has dimension m× n. Just as with
vectors, matrices are greater than the sum of their parts: the actual values of the
elements of a matrix are not important. To visualize a matrix, one should imagine
a process, in which a vector is transformed into another vector (see Figure A.3).
For this reason, matrices are sometimes referred to as maps, since they map one
vector onto another. The term operator is also used, since a matrix can be viewed
as an operation — rotation, or reflection, say — applied to a vector. The way this
operation is performed mathematically is via matrix multiplication, written like this,
w = Mv, or |w〉 = M |v〉. (A.10)

A.2 Matrices 382
This multiplication is evaluated by combining the elements of M and the components
of v to form w, in the following way. Define a set of column vectors
mj = |mj〉 =
M1j
M2j
...
Mmj
, (A.11)
so that each column of the matrix M is given by one of these vectors,
M =
m1
m2
. . .
mn
. (A.12)
The vector w is then given by a weighted sum of the mj , with coefficients equal to
the components of v,
w = v1m1 + v2m2 + . . .+ vnmn. (A.13)
From (A.13) it is clear that the number of columns of M must be the same as the
dimension of v for this multiplication to be possible. Thus the number of columns
of M sets the dimension of the vectors upon which M can act. Similarly the number
of rows of M sets the dimension of the vectors mj , and therefore the dimension of
the output vector w. So an m×n matrix is an operator that acts on n dimensional

A.2 Matrices 383
vectors to produce m dimensional ones.
Figure A.3 A matrix acting on a vector. Here M maps the initialvector v (black) onto the final vector w (red), via a rotation and a‘dilation’ (length increase). The values of the matrix elements Mij
depend on the components of v and w, which in turn depend on thedirection of the coordinate axes (thin arrows). Rotating the coordi-nate axes would change the Mij , but the transformation representedby M would be the same. In this sense, a matrix is more fundamentalthan its elements.
Matrices with the same dimensions can be added together; M + N is just the
matrix whose elements are given by the sum of the corresponding elements of M
and N . And of course they can be multiplied by numbers. αM is a matrix whose
elements are αMij . Incidentally, these properties mean that the space of all matrices
is actually also a vector space. But this will not be important for us.
Two matrices can be multiplied together to produce a new matrix. In the product
MN = Q, each of the column vectors qj of Q are formed from the corresponding
column vector nj of N , by combining the column vectors mj of M in a weighted
sum like (A.13), with coefficients given by the components of nj . So M acts on each
column of N to produce the columns of Q.
To multiply a row vector (or a bra) by a matrix, we simply treat the row vector

A.2 Matrices 384
as a 1× n matrix, and apply the above rule. We therefore obtain
〈v|M =(〈v|m1〉 〈v|m2〉 . . . 〈v|mn〉
). (A.14)
Matrices also have Hermitian adjoints. The Hermitian adjoint M † of M is given
by swapping the rows and columns of M , and taking the complex conjugate of all
its elements,
M † =
M∗11 M∗21 . . . M∗m1
M∗12 M∗22 . . . M∗m2
......
. . ....
M1n∗ M∗2n . . . M∗mn
. (A.15)
Using this definition, it’s easy to check that Q† = N †M † (note the reversed order of
M and N), and that (M |v〉)† = 〈v|M †. These facts are useful when manipulating
expressions involving several matrices.
A.2.1 Outer product
The outer product of two vectors |v〉 and |w〉 is a matrix, written as
|v〉〈w| = vw† =
v1w∗1 v1w
∗2 · · · v1w
∗m
v2w∗1 v2w
∗2 · · · v2w
∗m
......
. . ....
vnw∗1 vnw
∗2 · · · vnw
∗m
. (A.16)

A.2 Matrices 385
Each column of this matrix is just |v〉, multiplied by the corresponding element of
〈w|, so that its structure can be visualized as that of a row vector 〈w| with column
vectors |v〉 ‘hanging’ from it. The Dirac notation is very satisfying in this context,
since the result of applying the operator |v〉〈w| to a third vector |x〉 is written like
this,
|v〉〈w||x〉 = |v〉〈w|x〉 = 〈w|x〉|v〉. (A.17)
That is, the matrix product of |v〉〈w| with |x〉 is just the same as the inner product
of 〈w| and |x〉, multiplied by the vector |v〉. If |x〉 = |w〉, the result is w2|v〉.
As |x〉 deviates away from |w〉, the inner product 〈w|x〉 gets smaller and smaller,
until it vanishes, when |x〉 is orthogonal to |w〉. A natural interpretation for the
operator (A.16) is therefore as a kind of ‘switch’ that maps an input from |w〉 to |v〉.
Operators of this kind are sometimes known as flip operators, or transition operators,
in quantum mechanics. Breaking down larger operators into flip operators can often
provide valuable insights.
A.2.2 Tensor product
A further generalization of the outer product is the tensor product. The tensor
product is used to combine vector spaces together to produce a new, larger space.
Suppose we have an n-dimensional vector space V, and also an m-dimensional space
W. The tensor product V ⊗W of these two spaces would be the space of all vectors
with dimension nm. Vectors v and w from the smaller spaces can be combined
together via the tensor product to produce a vector v⊗w inhabiting the larger space.

A.2 Matrices 386
And similarly matrices M and N acting on the smaller spaces can be combined
together to produce an operator M ⊗N , that acts on vectors in the tensor product
space V ⊗W. The result of applying the combined operator to the combined vector
is the same as the result of applying the operators to the vectors separately, and
then taking the tensor product:
(M ⊗N)(v ⊗w) = (Mv)⊗ (Nw). (A.18)
A common example arising in quantum mechanics is the tensor product of a pair
of 2-dimensional vectors, representing the state of a pair of qubits (a pair of elec-
tron spins perhaps). Suppose one qubit is in the state labelled |0〉, and the other
is in the state |1〉. The combined state |ψ〉 of both is found by taking the ten-
sor product of these two vectors, |ψ〉 = |0〉 ⊗ |1〉. Sometimes the more compact
notation |01〉 is employed, where the meaning should be clear from the context.
But other 4-dimensional vectors, which cannot be represented as tensor products
of 2-dimensional vectors, can exist in the 4-dimensional tensor product space. For
instance, the vector |ψ〉 = (|01〉 + |10〉)/√
2 is a valid state in quantum mechan-
ics (see (1.7) in Section 1.6.4 of Chapter 1). It cannot be written in the form
|state 1〉 ⊗ |state 2〉, but it is a 4-dimensional vector, produced by adding together
two vectors that can be written in this form. Vectors of this kind, that exist in the
tensor product space, but cannot be written as a tensor product of vectors from
the component spaces, are known as non-separable. In quantum mechanics, they

A.2 Matrices 387
represent states that are entangled.
The tensor product of two matrices M (with dimension m × n) and N (with
dimension p×q) is found by ‘attaching’ a copy of M to each element of N , as shown
below,
M ⊗N =
M11
N11 · · · N1q
.... . .
...
Np1 · · · Npq
· · · M1n
N11 · · · N1q
.... . .
...
Np1 · · · Npq
...
. . ....
Mm1
N11 · · · N1q
.... . .
...
Np1 · · · Npq
· · · Mmn
N11 · · · N1q
.... . .
...
Np1 · · · Npq
.
(A.19)
The procedure for vectors is identical; the vectors are just treated as matrices with a
single column (or row, in the case of bras). A bit of head scratching will verify that
this definition, when combined with standard matrix multiplication (A.13), satisfies
the requirement (A.18). An important property of the tensor product is as follows.
If |i〉 is an orthonormal basis for one space, and |j〉 is an orthonormal basis for
a second space, the set of tensor product vectors |i〉 ⊗ |j〉 is an orthonormal basis
for their tensor product space.

A.3 Eigenvalues 388
A.3 Eigenvalues
Consider a matrix R representing a reflection about the x-axis, as shown in Figure
A.4. A vector |1〉 lying along the x-axis is not changed by the action of this matrix.
That is, it is its own reflection. So we have R|1〉 = |1〉. A second vector |2〉 lying
along the y-axis is flipped around by R. Its reflection points in the opposite direction
to itself, so R|2〉 = −|2〉. Other vectors are altered in more complicated ways when
they are reflected, so that the vector resulting from the application of R is not related
to the original vector by a simple numerical factor (1 or −1 in the two cases above).
The vectors |1〉 and |2〉 are examples of vectors for which the action of R is the same
as multiplication by a number. These ‘special’ vectors are known as eigenvectors of
R. In general, any matrix M has a set of eigenvectors |i〉, such that
M |i〉 = λi|i〉. (A.20)
Here the number λi is the eigenvalue corresponding to the eigenvector |i〉. For the
example given above, we had λ1 = 1 and λ2 = −1. The eigenvectors and eigenvalues
contain all the information required to reconstruct the transformation implemented
by M ; they represent the essence of a transformation, and as such they are of
paramount importance in linear analysis, and central to quantum mechanics.

A.3 Eigenvalues 389
Figure A.4 Eigenvectors and eigenvalues. The matrix R representsreflection in the x-axis (horizontal axis). The eigenvectors of R arethose vectors pointing along, or perpendicular to the x-axis, sincethe application of R to one of these vectors produces the same vectoragain, multiplied by some number.
A.3.1 Commutators
In general, matrix multiplication is not commutative. That is, MN 6= NM ; the order
in which matrices are multiplied is important. This makes sense when matrices are
viewed as representing transformations of vectors (see Figure A.5).
Often it is useful to examine the commutator of two matrices, defined by
[M,N ] = MN −NM. (A.21)
If M and N were just numbers, their commutator would always vanish, but for
matrices often it does not. In quantum mechanics, the commutator of different
physical quantities may be non-zero, and this non-vanishing of the commutator
can be viewed as the source of a great many of the strange features of quantum
mechanics.

A.3 Eigenvalues 390
1
2
3
1
23
Figure A.5 Non-commuting operations. Here M represents a re-flection around the y-axis, while N is an anti-clockwise rotationthrough 90 degrees. The red arrows are numbered in order, with1 the initial vector, 2 the result after the application of one of thetransformations, and 3 the result after both transformations havebeen applied. On the left, M is applied first, and then N . On theright, N is applied first, followed by M . The results are differentbecause the matrices M and N do not commute. The notation canbe counter-intuitive: the product MN represents the application ofN first, with M applied afterwards.
If the two matrices N , M do commute, then they have common eigenvectors. To
see this, suppose that |u〉 is an eigenvector of N , with eigenvalue λ. If we take the
product MN |u〉 (that is, we apply N to |u〉 first, and then M), the result is simply
λM |u〉. On the other hand, if [M,N ] = 0, we can swap the order of M and N , to
get NM |u〉. That is,
N(M |u〉) = λ(M |u〉). (A.22)
Therefore the vector M |u〉 is also an eigenvector of N , with the same eigenvalue
λ. M |u〉 must be parallel to |u〉, so that M |u〉 = µ|u〉. That is, |u〉 is also an
eigenvector of M , with some new eigenvalue µ. This fact is intimately connected
with the epistemology of quantum mechanics.

A.4 Types of matrices 391
A.4 Types of matrices
There are some types of matrix that are particularly important, both for the calcu-
lations in this thesis, and for quantum mechanics generally.
A.4.1 The identity matrix
The identity matrix, often denoted by I, is the matrix equivalent of the number 1.
It is the matrix that results in no change when it is multiplied by another matrix —
it represents the operation ‘doing nothing’. That is, IM = MI = M . And of course
the identity does not change a vector either, I|v〉 = |v〉, 〈v|I = 〈v|. The identity
matrix is a square matrix (i.e. dimension m×m), with ones along its main diagonal,
and zeros everywhere else (the zero elements are left blank below to avoid clutter),
I =
1
1
. . .
1
. (A.23)
Sometimes care should be taken to ensure that the correct dimension m of I is used,
so that the multiplication is possible. Usually this is quite clear from the context,
but the symbol Im can be used when the size of I needs to be specified.

A.4 Types of matrices 392
A.4.2 Inverse matrix
The inverse M−1 of a matrix M is the matrix that ‘undoes’ the action of M . It is
the matrix equivalent of a reciprocal. The inverse satisfies the relations M−1M =
MM−1 = I. It is clear that taking the inverse of a matrix is also involutive, since the
inverse of an inverse is just the original matrix, (M−1)−1 = M . If M is rectangular,
with m < n, then M describes a map from a larger space into a smaller space,
so that some information is inevitably lost, in the sense that there are different
vectors in the input space that are mapped to the same vector in the output space.
Therefore M cannot have an inverse — it is impossible to ‘undo’ this type of map. It
is generally the case that only square matrices, with m = n, have a matrix inverse.
It is possible to define a pseudo-inverse, that represents the closest approximation
of a true inverse, for any matrix (even rectangular ones), but we will not make use
of the pseudo-inverse [189–192]. Calculating the inverse of a matrix can be rather
involved, and although an algorithm for inverting 3 × 3 dimensional matrices is
taught to students in school, matrix inversion is rarely performed explicitly. Lloyd N.
Trefethen is a prominent numerical analyst who teaches a course on computational
linear algebra at Oxford University. His reaction to a suggestion that students should
consult W. H. Press’s famous book on numerical techniques was
The only way to annoy a numerical analyst more than by inverting
a matrix, is to use Numerical Recipes.

A.4 Types of matrices 393
The formula for the inverse of a 2× 2 matrix M is simple however,
M−1 =
a b
c d
−1
=1
ad− bc
d −b
−c a
. (A.24)
The quantity ad−bc is known as the determinant of the matrix — sometimes denoted
by vertical bars, |M | — since it determines whether or not the inverse of M exists:
if |M | = 0, the formula for the inverse ‘blows up’. In this case, the matrix does not
have in inverse, which implies that there exists some vector |v〉 such that M |v〉 = 0.
Clearly it is not possible to invert this expression. This provides a convenient way to
find the eigenvalues of a matrix. If we want to find |u〉 such that M |u〉 = λ|u〉, then
we must have that (M − λI)|u〉 = 0, and therefore we require that |M − λI| = 0.
A.4.3 Hermitian matrices
A Hermitian matrix is equal to its Hermitian adjoint, H = H†. It is the matrix
equivalent of a real number, and in fact its eigenvalues are all real numbers. To see
this, consider the quantity k = 〈i|H|j〉. On the one hand, using the definition of H,
along with (A.20), we have
k = (H†|i〉)†|j〉 = (H|i〉)†|j〉 = (λi|i〉)†|j〉 = λ∗i 〈i|j〉.
On the other hand, we have
k = 〈i|(H|j〉) = λj〈i|j〉.

A.4 Types of matrices 394
Taking the difference of these, we get k − k = (λ∗i − λj)〈i|j〉 = 0. If we set i = j,
we must have that λ∗j − λj = 0, since 〈j|j〉 > 0 is the square of the norm of |j〉.
Therefore λj = λ∗j , that is, the eigenvalues of H are real numbers. At the same
time, if we set i 6= j, we must have that 〈i|j〉 = 0, which means that different
eigenvectors of H are all orthogonal to one another. Note that we are free to scale
the eigenvectors |i〉 so that they have length 1. If we do this, the set of eigenvectors
|i〉 of a Hermitian matrix is an orthonormal basis. The eigenvectors define a
‘natural’ coordinate system for the space of vectors upon which H acts. And it’s
very practical to work with this coordinate system, since the effect of H on each
basis vector reduces to multiplication by the corresponding eigenvalue,
H|v〉 = H (v1|1〉+ v2|2〉+ . . .+ vn|n〉) = v1λ1|1〉+ v2λ2|2〉+ . . .+ vnλn|n〉. (A.25)
A.4.4 Diagonal matrices
A diagonal matrix D is a matrix with zeros everywhere except along its main diag-
onal,
D =
D11
D22
. . .
Dmm
. (A.26)
Clearly diagonal matrices must always be square, with n = m. The identity matrix
is a diagonal matrix with Djj = 1. Diagonal matrices are very easy to work with.

A.4 Types of matrices 395
For example, the square of a diagonal matrix D2 = DD is another diagonal matrix
with its elements equal to D2jj . The inverse D−1 of a diagonal matrix is just another
diagonal matrix with all its elements equal to 1/Djj . Any two diagonal matrices
commute with one another, [D1, D2] = 0, and the eigenvalues of a diagonal matrix
are just equal to its elements, λj = Djj , with its eigenvectors being the basis vectors
of the coordinate system with respect to which the matrix elements are defined.
This last property is important. Diagonal matrices are wonderfully simple to
manipulate, but surely it is very unlikely that any interesting matrices are diagonal.
The point is that all matrices are diagonal matrices (or more correctly, most square
matrices), as long as you write them down with reference to the correct coordinate
system! This coordinate system is the one defined by the eigenvectors of the matrix,
and when written down using this basis, the elements of the matrix are just its
eigenvalues.
A brief inspection of (A.25) reveals that in fact, when written down with reference
to the coordinate system defined by the eigenvectors |i〉, H is actually diagonal, with
elements H11 = λ1, H22 = λ2, etc...
For this reason, the process of finding the eigenvalues and eigenvectors of a matrix
is sometimes referred to as diagonalization, since this calculation is simply what is
required to convert a matrix into a diagonal one. The eigenvalue decomposition can
be written as
M = WDW−1, (A.27)
where D is a diagonal matrix containing the eigenvalues of M , and where the eigen-

A.4 Types of matrices 396
vectors of M comprise the columns of the matrix W . Very efficient algorithms exist
for finding this decomposition; the results in this thesis rely heavily on the speed
and precision of the LAPACK routines implemented in MATLAB.
A.4.5 Unitary matrices
A unitary matrix U is a matrix whose inverse is equal to its Hermitian adjoint,
U−1 = U †. A unitary matrix represents a rotation in space, so that |w〉 = U |v〉 is a
vector pointing in a different direction to |v〉, but with the same norm — the same
length. To see why, consider the norm of |w〉, w2 = 〈w|w〉 = 〈v|U †U |v〉. But since
U † = U−1, we have that U †U = I, so w2 = 〈v|I|v〉 = v2. That is, unitary matrices
preserve the norm of vectors upon which they act.
Figure A.6 A unitary transformation. U represents a rotationfrom an initial (black) into a new (red) coordinate system. Thecolumns of U are unit vectors comprising an orthonormal basis forthe new coordinate system.
A rotation can be thought of as a transformation from one orthonormal coordi-
nate system to another, as shown in Figure A.6. Associated with this new coordinate
system is an orthonormal basis |i〉, and these vectors are actually the columns of
U , ui = |ui〉 = |i〉. To see this, consider the product K = U †U . From the definition

A.4 Types of matrices 397
of the Hermitian adjoint, the rows of U † are the bras 〈ui|,
U † =
〈u1|
...
〈um|
. (A.28)
Applying the matrix multiplication described in (A.13), we find that each element
of the product matrix K is given by an inner product, Kij = 〈ui|uj〉. But since U
is unitary, K = I, the identity, so that we must have 〈ui|uj〉 = δij , where δij is the
kronecker delta symbol (δij = 1 if i = j, and 0 otherwise). Therefore the column
vectors |ui〉 form an orthonormal basis. If U is applied to a vector |v〉 pointing
along the x-axis, with components v1 = 1, vj 6=1 = 0, the result is |u1〉. In the same
way, each coordinate axis is mapped by U to a new axis |ui〉, and so U represents a
rotation into a new orthonormal coordinate system defined by its columns.
Incidentally, the above arguments serve to demonstrate that the inner product
〈u|v〉 of two vectors |u〉 and |v〉 is always independent of the coordinate system used
for writing out the components of |u〉 and |v〉. Changing the coordinate system
is done by applying a rotation |u〉 → U |u〉, |v〉 → U |v〉, and the inner product is
then 〈u|U †U |v〉 = 〈u|v〉. Changing coordinates makes no difference. This is to be
expected of course, since (A.7) makes no reference to any coordinates.
It is worth noting that U † is unitary, if U is. Therefore the columns of U † also
form an orthonormal basis, and so the rows of U form an orthonormal basis.
Note also that the product of two unitary matrices U , V is also unitary: UV (UV )† =

A.4 Types of matrices 398
UV V †U † = UIU † = I. Two rotations composed together can always be thought of
as a single rotation.
Unitary matrices play a central role in quantum mechanics, and we will encounter
them in the optimization of quantum memories.

Appendix B
Quantum mechanics
In this Appendix we give a brief review of the structure of quantum mechanics. This
is intended as a pedagogical precursor to Appendix C, on quantum optics. We will
make use of the concepts developed in Appendix A.
Quantum Mechanics was developed in the early twentieth century, primarily as a
theory of atomic physics. In the days before Google, interdisciplinary communication
was more difficult, and in fact Werner Heisenberg re-invented matrices in order to
formulate his version of quantum theory [193]. The incarnation we present here uses
the notation introduced by Paul Dirac [194], and we follow broadly the excellent
account given by Nielsen and Chuang [158].

B.1 Postulates 400
B.1 Postulates
B.1.1 State vector
In quantum mechanics, the state of a system is described by a ket |ψ〉. The simplest
vector is a 2-dimensional one, and this describes the simplest type of quantum system
— a qubit. More complicated systems are described by higher dimensional vectors.
B.1.2 Observables
Quantities, like energy, momentum or position — any observable that might be mea-
sured — are represented by matrices that act on the state vector. These matrices are
always Hermitian, and this guarantees that their eigenvalues are real numbers (see
Section A.4.3 in Appendix A). In addition, the eigenvectors of Hermitian matrices
form an orthonormal basis: they define a coordinate system.
B.1.3 Measurements
Quantum mechanics provides the following recipe for making predictions about mea-
surements. The observable being measured is assigned to a Hermitian operator H.
Making this assignment correctly is left up to the skill and imagination of the physi-
cist. This operator is diagonalized, yielding its eigenvalues λi and eigenvectors
|i〉. The eigenvalues are real numbers, and each one represents a possible nu-
merical outcome of the measurement: the number you might see on an oscilloscope
screen, for example. Each eigenvalue λi is associated with an eigenvector |i〉, and
these eigenvectors define a coordinate system. The state vector of the system |ψ〉 is

B.1 Postulates 401
written with reference to these coordinates, known as the measurement basis,
|ψ〉 = ψ1|1〉+ ψ2|2〉+ . . .+ ψm|m〉. (B.1)
The probability pi that the measurement yields the result λi is then given by |ψi|2,
the squared magnitude of the ith component of |ψ〉 in the measurement basis. A
more compact way to write this is
pi = |〈ψ|i〉|2. (B.2)
This is known as the Born rule, after Max Born who proposed it in 1926 [195]. Imme-
diately after the measurement has been completed, the state of the system changes,
essentially instantaneously, according to the measurement result, |ψ〉 → |i〉. This is
known as the collapse postulate.
The average value of the measurement result is often useful. This is sometimes
called the expectation value of the quantity H, since it is the number one would
expect when repeating the measurement many times. The expectation value is
given by 〈H〉 =∑
i piλi, and a bit of thought shows that this is equal to 〈ψ|H|ψ〉.
The fact that 〈H〉 = 〈ψ|H|ψ〉 is another convenience of Dirac notation.
B.1.4 Dynamics
It must always be the case that the probabilities pi sum to unity,∑
i pi = 1. This just
codifies the assertion that we must always get some result from a measurement, even

B.1 Postulates 402
if the result is ‘no signal’. Using the Born rule, this means that∑
i |ψi|2 = 〈ψ|ψ〉 = 1.
That is, the norm of a state vector in quantum mechanics is always exactly equal
to 1. The norm can never be altered by any dynamical process, which immediately
fixes all dynamics in quantum theory to be unitary. In other words, given some
initial state |ψ0〉, and a final state |ψ〉, we must have
|ψ〉 = U |ψ0〉, (B.3)
where U is a unitary operator that advances the system from the initial to the final
state. Differentiating (B.3) with respect to the time t, we obtain the equation of
motion
∂t|ψ〉 = U |ψ0〉
= UU †|ψ〉, (B.4)
where the overdot indicates the time derivative of U . Now, the requirement that
the norm of |ψ〉 does not change can be expressed by the condition ∂t(〈ψ|ψ〉) = 0.
Substituting in (B.3) gives
〈ψ0|U †U + U †U |ψ0〉 = 0, (B.5)
from which we derive the condition that the operator (UU †) is skew-Hermitian,
meaning that it changes sign under Hermitian conjugation. Any skew-Hermitian

B.2 The Heisenberg Picture 403
operator can be represented as the product of the imaginary unit i with a Hermitian
operator H, and making this replacement in (B.4) gives us the Schrodinger equation
∂t|ψ〉 = iH|ψ〉. (B.6)
The operator H is known as the Hamiltonian. Schrodinger’s great insight was to
identify H as the operator associated with the energy of the system. In (B.6) it is the
energy, represented by H, that sets the rate of change of the state vector. Systems
with high energy evolve quickly, with fast oscillations, while low energy systems are
more sluggish.
B.2 The Heisenberg Picture
The above discussion was based on the so-called Schrodinger picture, in which the
quantum state |ψ〉 evolves in time. It is possible to formulate quantum mechanics
differently, and sometimes it is easier to solve a problem by using this different
formulation. The results are identical, regardless of how the calculations are done. In
Heisenberg’s formulation, the quantum state |ψ0〉 of a system at some initial time is
fixed. It does not change with time. Instead, the operators acting on the state vector
evolve in time. As an example, consider a Hermitian operator A associated with
some quantity that we might want to measure. Here we use the symbol A instead
of H; we reserve the symbol H for the Hamiltonian from now on. In the Heisenberg
picture the operator A depends on the time at which we make the measurement.

B.2 The Heisenberg Picture 404
In order for this formulation to work, we must have that the expectation value
〈A〉 predicted by either formalism is the same. Denoting the fixed operator in the
Schrodinger picture with a subscript S, we must have
〈ψ0|A|ψ0〉 = 〈ψ|AS|ψ〉,
⇒ 〈ψ0|A|ψ0〉 = 〈ψ0|U †ASU |ψ0〉,
∴ A = U †ASU. (B.7)
That is, the time evolution of an operator in the Heisenberg picture is found by sand-
wiching the Schrodinger operator between two copies of U , the same operator that
generates the time evolution of the state in the Schrodinger picture. Differentiating
(B.7) with respect to time, we find
∂tA = U †ASU + U †ASU
= U †UA+AU †U . (B.8)
Note also that [U †, U ] = 0, since
U †U = U †UUU † = U †UUU † = UU †, (B.9)
where we used the fact that [U, U ] = 0 (a little thought shows that an operator must
always commute with its derivative; see Section A.3.1 in Appendix A). Therefore
(B.8) can be re-written in terms of the Hamiltonian, to produce the Heisenberg

B.2 The Heisenberg Picture 405
equation
∂tA = i[A,H]. (B.10)
This is the fundamental equation of motion in the Heisenberg picture; it plays the
same role as the Schrodinger equation does in the Schrodinger picture — generating
time evolution.
B.2.1 The Heisenberg interaction picture
Often we are interested in analysing the behaviour of a system when it is subjected
to a weak external field. Of specific relevance in this thesis is the case of an atom
illuminated by a laser: the internal electric fields generated by the charges within
the atom are much stronger than the electric fields within the laser beam, so the
laser acts as a weak external perturbation, on top of the much stronger interactions
binding the atom together. In such cases, it is convenient to separate out the strong
and weak contributions to the energy of a system. Suppose that we can divide
the Hamiltonian into two parts, H = H0 + Hint, where H0 dominates, and Hint
represents a comparatively small interaction. The large contribution H0 will make
the operator A change very quickly (as can be seen from the form of (B.10), where a
large energy produces rapid oscillations in time). This rapid oscillation can obscure
any interesting effects arising from the interaction Hamiltonian Hint. To extract
these interesting effects, we define a slowly varying operator A in the following way,
A = U0AU†0 . (B.11)

B.2 The Heisenberg Picture 406
Here U0 is the time evolution operator associated with the Hamiltonian H0. That
is, U0 satisfies U0U†0 = iH0. Differentiating (B.11) with respect to time, and using
the Heisenberg equation (B.10), we find
∂tA = U0AU†0 + U0(∂tA)U †0 + U0AU
†0 ,
= iH0A+ U0 (i[A,H0 +Hint])U†0 − iAH0,
= −i[A,H0] + i[A,H0] + i(U0AHintU
†0 − U0HintAU
†0
). (B.12)
Conveniently, the first two terms cancel. The last term can be re-written in a
compact form, if we define a modified Hamiltonian H = U0HintU†0 , whence we
obtain the interaction picture equation of motion
∂tA = i[A, H]. (B.13)

Appendix C
Quantum optics
Quantum optics is the study of the quantum features of light. The theory requires a
treatment of ensembles of identical photons, which are easily created and destroyed
in their interaction with atoms. Therefore the techniques of quantum field theory
must be employed, in order to deal with the creation and destruction of identical
particles. In this Appendix we briefly review the quantum mechanical description
of the electric field associated with a propagating light beam, before describing the
form of the interaction between light and matter.
C.1 Modes
Classically, light is a transverse electromagnetic wave. Apart from its amplitude,
it has three degrees of freedom that must be specified to uniquely determine its
properties. These are (i) its polarization, (ii) its frequency and (iii) its propagation
direction.

C.1 Modes 408
The polarization is the direction along which the electric field oscillates; it is
a vector in a plane perpendicular to the propagation direction. It is easy to see
that the space of polarizations is simply a 2-dimensional vector space. In fact,
due to the possibility of phase delays between different polarization directions, it is
actually a complex vector space — a Hilbert space (see Section A.1.2 in Appendix
A). Nonetheless it is a 2-dimensional vector space.
The same is true for the other degrees of freedom. That is, the space of fre-
quencies is a vector space. It is a space with an uncountably infinite number of
dimensions, since the different possible frequencies are infinitely closely spaced, but
it is no different in character to the space of polarizations. And similarly for the
propagation direction: there are an infinite number of infinitely closely spaced prop-
agation directions, and the set of all of these forms a vector space.
Already, in talking of these vector spaces, we have made implicit reference to a
basis for each of them. We talk of two perpendicular directions for polarization. Or
different directions of propagation. These are labels that we use to keep track of
dimensions in a vector space, and they are intuitive and natural. But any basis is as
good as any other. For example, instead of talking about different frequencies, we
could talk about different arrival times. Or we could think of left and right circular
polarizations as the polarization basis. It is useful in quantum optics to be flexible
about the basis we use to describe an optical field. A common concept is therefore
that of the optical mode.
A mode is a member of an orthonormal basis for one of the vector spaces associ-

C.1 Modes 409
ated with a light field. So, horizontal polarization is a polarization mode, since it is
one of a pair of perpendicular polarizations that form a basis for the space of possi-
ble light polarizations. The other, orthogonal mode, is vertical polarization. And a
single frequency ω labels a spectral mode. It is orthogonal to another frequency ω′,
because two plane-waves with these frequencies have a vanishing inner product,
∫eiωτe−iω′τ dτ = 0, (C.1)
when ω 6= ω′. Equivalently, we could label different temporal modes t and t′. These
are orthogonal because two delta-functions with these timings also have a vanishing
inner product, ∫δ(τ − t)δ(τ − t′) dτ = 0, (C.2)
when t 6= t′.
An optical mode is a member of a basis for the full space of all possible optical
fields. This space of all possible fields is just the tensor product of the vector spaces
associated with each degree of freedom. And a basis for the full space is found
by taking the tensor product of the bases used for each degree of freedom (see
Section A.2.2 in Appendix A). That is, an optical mode is the tensor product of a
polarization mode, a spectral mode and a spatial mode.
Once a basis of modes is settled upon, it is possible to introduce the concept of
a photon. A photon is an excitation of an optical mode. Sometimes it is useful to
remember that photons are only defined with respect to a basis of modes. Although

C.2 Quantum states of light 410
photons are often contrasted with waves as an embodiment of the particulate nature
of light, they do not have to be localized, like tiny bullets. The ‘shape’ of a photon
is the shape of the mode of which it is an excitation.
C.2 Quantum states of light
C.2.1 Fock states
Suppose we consider a plane wave optical mode. That is, a mode with a linear po-
larization (horizontal, say), a single frequency ω, and a single propagation direction
k, where k is the wavevector of the mode. An excitation of this mode has a fixed
energy, given by the Planck formula E = ~ω, so a single photon in this mode is an
eigenstate of the Hamiltonian for the field. Similarly, if we excite two photons in
this mode, we have a state with twice the energy, E = 2~ω. This is also an energy
eigenstate, but with a different eigenvalue. It follows that these two states must
be orthogonal. And by extension, each photon number state is orthogonal to every
other photon number state. If we use the notation |n〉 to denote the state with n
photons, we must have
〈n|m〉 = δnm. (C.3)
Changing the basis of optical modes from plane waves to some other basis cannot
change this orthogonality, since the inner product is invariant under unitary trans-
formations. Therefore (C.3) holds generally, for different photon number states of
an arbitrary optical mode.

C.2 Quantum states of light 411
The orthonormal basis of photon number states |n〉, associated with excitations
of some given optical mode, is known as the Fock basis for that mode. The photon
number states are sometimes known as Fock states, and the space for which they
form a basis is accordingly Fock space. The Fock space represents the final degree of
freedom associated with an electromagnetic quantum state: the amplitude. That is
to say, the more photons in a mode, the more intense the field. Thus the quantum
state of an electromagnetic field is fully specified by the tensor product of 4 vector
spaces: the polarization, spectral and spatial modes (collectively specifying an opti-
cal mode), and finally the Fock space (specifying the photon number: the energy in
the field; its brightness).
C.2.2 Creation and Annihilation operators
A marked difference between optical fields and material systems is the imperma-
nence of photons. Generally the atoms and electrons in a quantum memory are
considered to be indestructible. They are not created or destroyed by their interac-
tions. But photons can be absorbed and re-emitted. So we must describe processes
that change one Fock state into another — processes that change the number of
photons excited into a given mode. This description is accomplished by introducing
a creation operator a†, that adds a single photon to an optical mode. The simi-
larity of the symbol ‘†’ for Hermitian conjugation to a ‘+’ sign serves as a useful
mnemonic for this operator’s function. The effect of applying a† to an empty optical
mode |0〉, containing no photons, is to produce the state |1〉, with a single photon.

C.2 Quantum states of light 412
Further applications of a† add extra photons, with contributions from all possible
permutations of arranging these photons (see Figure C.1). These contributions must
be included, since photons are bosons, meaning that their state must be unchanged
by swapping any pair of photons. The Fock states created by the action of a† are
not correctly normalized, so that a numerical factor, accounting for the number of
permutations, must be included,
(a†)n|0〉 =√n!|n〉. (C.4)
Another way to write this is
a†|n〉 =√n+ 1|n+ 1〉. (C.5)
Taking the norm of (C.5), we have
〈n|aa†|n〉 = (n+ 1)〈n+ 1|n+ 1〉 = n+ 1,
⇒ aa†|n〉 = (n+ 1)|n〉. (C.6)
That is, the Hermitian conjugate a = (a†)† is an annihilation operator that removes
a photon. And from (C.6) we see that
a|n〉 =√n|n− 1〉. (C.7)

C.2 Quantum states of light 413
Note that a ‘kills’ the empty vacuum state, a|0〉 = 0, which is fortunate, since there
cannot be fewer than zero photons in a mode! It is often useful, when manipulating
Figure C.1 Symmetrized photons. n applications of the photoncreation operator a† to the vacuum state |0〉 produces a symmetrizedn photon state, with contributions from all n! permutations of the nphotons. Swapping any two photons leaves the state unchanged, asrequired by Bose statistics.
expression involving these operators, to be able to reverse their ordering. This is
done using their commutator which, applying (C.5) and (C.7), is given by
〈n|[a, a†]|m〉 = 0, 〈n|[a, a†]|n〉 = 1,
⇒ [a, a†] = 1. (C.8)
The commutator (C.8) expresses what is known as the canonical commutation rela-
tion; commutators of this form are common to creation and annihilation operators
for all bosonic fields.
Another useful operator is the number operator N = a†a, a Hermitian operator
that satisfies the eigenvalue equation
N |n〉 = n|n〉, (C.9)
so that N counts the number of photons excited into a particular mode.

C.3 The electric field 414
C.3 The electric field
Electric fields are associated with separated charges, while magnetic fields are asso-
ciated with moving charges. Electrons move rather slowly in most ordinary forms
of matter, and accordingly their interaction with light is dominated by its electric
component. In this thesis, we treat light fields as if they were purely electric waves,
an approximation that is very well satisfied provided that light intensities are not
sufficient to produce a relativistic electron plasma.
The electric field associated with a beam of light, as might be generated by a
laser, can be expressed in terms of the annihilation operators a(ω) associated with
plane waves propagating along the beam [107],
E(z) = iv∫g(ω)a(ω)e−iωz/c dω + h.c., (C.10)
where z is the longitudinal position along the beam, v is a unit polarization vec-
tor in the plane perpendicular to the beam and g(ω) =√
~ω/4πε0Ac is the mode
amplitude. Here A is the cross-sectional area of the beam, ε0 is the permittivity
of free space and c is the speed of light. Note that in principle the electric field is
an observable quantity, that we could measure (although at optical frequencies it is
not generally possible to directly measure the electric field, at radio frequencies it
certainly is feasible). And so E is a Hermitian operator, as expected.
The annihilation operators a(ω), labelled by the frequency ω of the mode upon

C.4 Matter-Light Interaction 415
which they act, satisfy the commutation relation
[a(ω), a†(ω′)] = δ(ω − ω′). (C.11)
This expresses the fact that operators for different frequency modes do not ‘see’
eachother, so they commute, while when ω = ω′, the canonical relation (C.8) is
satisfied. The delta function is the appropriate generalization for the case when the
modes are labelled by a continuous parameter, such as ω.
C.4 Matter-Light Interaction
Generally light interacts with matter through electrons. In most quantum memory
protocols these are the optically active outer electrons bound to some atoms. We
will also consider scattering in a diamond crystal, and here the electrons are more
appropriately described as free, or quasi -free particles. The Hamiltonian describing
the interactions are slightly different in these two cases; here we briefly review their
origin, and the relationship between them.
C.4.1 The A.p Interaction
The interaction of an electron with the electromagnetic field is found by incorpo-
rating the appropriate electromagnetic term, associated with so-called U(1) gauge
symmetry, into the Lagrangian density. The effect of this term is to modify the

C.4 Matter-Light Interaction 416
momentum p of the electron,
p −→ p− eA, (C.12)
where e is the electronic charge, and where A is the magnetic vector potential.
The potential A is not actually an observable field. The electric and magnetic
fields are related to its derivatives, but the absolute value of A is arbitrary to some
extent. Different choices for the functional form ofA— known as different gauges —
produce different Hamiltonians, with differing degrees of calculational convenience;
the physical predictions of the theory are unchanged of course. A standard choice of
gauge in quantum optics is the Coulomb gauge, which requires that A is divergence
free, ∇.A = 0. With this choice, the physical electric and magnetic fields are given,
respectively, by
E = −∂tA, B = ∇×A. (C.13)
Using (C.13) we can express the potentialA in the form (C.10), with the replacement
ig(ω) −→ g(ω)/ω.
The Hamiltonian for an electron, with mass m, in an electromagnetic field is
found by substituting the ‘canonical momentum’, given by (C.12), into the Hamil-
tonian for a ‘bare’ electron,
H =(p− eA)2
2m+A2ε0
∫ [E2 + c2B2
]dz. (C.14)
This is known as the minimal coupling Hamiltonian. The first term is the kinetic

C.4 Matter-Light Interaction 417
energy of the electron, with the transformation (C.12) included. The second term,
in square brackets, represents the ‘free field’ energy: this is the energy of the electro-
magnetic fields, in the absence of the electron. The integral extends over all space,
or at least, over the entire region occupied by the fields. The contribution from the
magnetic field B is very small, but the contribution from the electric field E is more
significant. Inserting (C.10) for E shows that the free field energy takes the form∫(N + 1
2)~ω dω. The term involving the number operator N simply expresses the
Planck formula E = ~ω, so that the energy in the field increases with the number of
photons excited. The term proportional to 12 is known as the zero-point energy: the
energy of the vacuum. It is rather unfortunate that this energy is infinite (since it is
integrated over all frequencies), but it is possible to work around these technicalities
with some mathematical sleight-of-hand, known as renormalization [196]. In any case
we will not be concerned with the zero point energy.
Multiplying out the first term in (C.14), we obtain a term of the form p2/2m,
which just describes the ‘bare’ kinetic energy of the electron, without the field.
There is a term A2/2m, which describes the field acting back on itself — this type
of non-linear back action is generally negligibly small. And there is a term of the
form −eA.p/m. This describes the coupling of electronic momenta to the vector
potential. In situations where electrons are spread over an extended region, such as
in a crystal, this interaction dominates the atom-light coupling.

C.4 Matter-Light Interaction 418
C.4.2 The E.d Interaction
When electrons are well-localized, such as when bound into atoms, a more con-
venient form of the interaction Hamiltonian can be derived. This is accomplished
formally by means of a unitary transformation due to Power, Zienau and Woolley
(PZW) [197,198]. In general it is desirable to eliminate explicit reference to the vector
potentialA in the Hamiltonian, since then the equations are manifestly gauge invari-
ant — it is quite clear that there can be no-dependence on the choice of gauge. The
PZW transformation removes A from the Hamiltonian, and introduces interactions
between the physical field E and the moments of the atomic charge distribution. To
see how this is done, we will need two results. The first is the equal-time commutator
of E and A, the amplitudes of the electric field and the vector potential,
[A(z), E(z′)] = −i~e
Aε0δ(z − z′), (C.15)
This is easily derived from (C.10) and (C.13) using (C.11). It is well known that in
quantum mechanics momentum and position generally satisfy the relation [x, p] = i~,
and indeed the form of (C.15) when z = z′ reflects the fact that in the Coulomb
gauge the field E is actually the ‘momentum’ that is conjugate to the ‘coordinate’
A in the electromagnetic Hamiltonian.
The second result we need is that
eCDe−C = D + [C,D], (C.16)

C.4 Matter-Light Interaction 419
whenever [C,D] is just a number (i.e. not another operator). Here the exponential
of an operator is defined according to the series
eC =∞∑n=1
Cn
n!. (C.17)
The result (C.16) is straightforward to derive. Consider the product CnD. Using
the commutator, we can ‘pull’ the operator D through C, in the following way,
CnD = Cn−1(CD) = Cn−1(DC + [C,D]) = (Cn−1D)C + [C,D]Cn−1. (C.18)
Repeating this procedure recursively, we obtain
CnD = DCn + n[C,D]Cn−1. (C.19)
Re-writing the left hand side of (C.16) using the series (C.17), and applying (C.19),
we arrive at (C.16).
With these preliminaries, we can introduce the PZW transformation. Suppose
that the action of the light is to make an optically active electron oscillate; it re-
mains bound to an atom, but it is ‘wiggled’ by the field. This is certainly what we
expect would happen classically. The atomic polarization, distinct from the opti-
cal polarization, is a useful concept in this situation. It is the ‘dipole moment per
unit volume’, where the dipole moment is the product of the electronic charge and
displacement. Suppose that the electron, with charge −e, is displaced a distance x

C.4 Matter-Light Interaction 420
along the polarization direction v of the incident light field. The dipole moment is
d = −exv, and the atomic polarization is P (z) = dδ(z)/A, where the delta function
describes a single dipole placed at the position z = 0. To express the interaction en-
ergy associated with the atomic polarization, we introduce a unitary transformation
of the Hamiltonian,
H → UHU †, with U = exp[
iA~
∫P (z′).A(z′) dz′
]. (C.20)
This transformation simply changes the coordinate system with respect to which the
quantum states |ψ〉 of the atom-light system are defined. Essentially it is nothing
more than a cosmetic change, but it has a marked effect on the form of the Hamilto-
nian. Applying the transformation to the free-field part of (C.14), we have UE2U †
= (UEU †).(UEU †), with
UEU † = vUEU † = v
E(z) +
iA~
∫P (z′)[A(z′), E(z)] dz′
= E(z)− 1
ε0P (z). (C.21)
That is, the PZW transformation adds a component proportional to the electron dis-
placement into the electric field. Using (C.21), the free-field Hamiltonian, neglecting
the small contribution from the magnetic field B, becomes
A2ε0
∫E2 dz −→ A
2ε0
∫ [E − 1
ε0P
]2
dz
=A2ε0
∫ [E2 +
1ε20P 2
]dz′ − d.E(z = 0). (C.22)

C.4 Matter-Light Interaction 421
The term proportional to E2 represents the ‘bare’ free-field energy, with no electron
present. The P 2 term represents an unimportant ‘self-interaction’ of the electron.
But the last term, proportional to E.d, represents the interaction of the physical
electric field, at the position of the electron, with the electronic dipole moment. It
is known as the electric dipole interaction Hamiltonian, and it serves as the basis
for the analysis of all the atomic quantum memory protocols in this thesis.
Finally, we note that the electron momentum acquires a component proportional
to the vector potential under the PZW transformation,
p −→ p+iA~
∫A(z′)[P (z′), p] dz′
= p+ eA(z = 0), (C.23)
where this time the commutator of the electronic momentum and position was used,
[x, px] = i~. In the approximation that the wavelength of the light is much longer
than the spatial extent of the atom — a limit valid for all interactions at optical fre-
quencies — we can set A ≈ A(z = 0), and then the vector potential A is completely
eliminated from the Hamiltonian.
Thus when electrons are tightly bound into atoms, the only significant interaction
with optical fields occurs through the electric dipole interaction.

C.5 Dissipation and Fluctuation 422
C.5 Dissipation and Fluctuation
In this section we address the issue of loss in quantum systems. Specifically, we seek
a theoretical description of the decoherence in a quantum memory: the constituent
atoms may emit photons into random directions, or collide with one another, and
these processes partially destroy the quantum information stored in the memory. In
Chapter 4 we use the Heisenberg picture to describe the propagation of light through
a quantum memory, and so we should account for losses using the Heisenberg picture.
In the following we use a simple model to show how the equations of motion for a
quantum system are modified by the presence of losses. Fortunately it is well known
that the results are not significantly altered by refining the model.
Our model consists of a single bosonic mode, our system, coupled to a bath of
other bosons — a reservoir. We use bosons because their commutation relations
are simple, but this model applies rather well to an ensemble quantum memory.
As shown in §4.11 in Chapter (4), both the atomic polarization and the spin wave
appearing in the equations of motion of an ensemble memory have approximately
bosonic commutation relations. In fact, in diamond, the optical phonons that con-
stitute the spin wave really are bosons (see Chapter 9). The reservoir of modes to
which the system is coupled could be the electromagnetic field, and indeed this is
an excellent description of spontaneous emission losses affecting the atomic polar-
ization.
The Hamiltonian contains the free field energy of the bath, and also terms that
represent processes in which a photon in the system is destroyed, while a photon in

C.5 Dissipation and Fluctuation 423
the reservoir is created, or vice versa. Working in the Heisenberg interaction picture,
so that the free-field energy of the system (and zero point energy of the reservoir)
is removed, we have
H =∫ωb†(ω)b(ω) dω + κ
∫ [a†b(ω) + b†(ω)a
]dω, (C.24)
where b(ω) destroys a photon with frequency ω in the reservoir, and where a destroys
a photon in the system. The equal-time commutators of these operators are given
by
[a, a†] = 1, and [b(ω), b†(ω′)] = δ(ω − ω′). (C.25)
Using the Heisenberg equation (B.13), we derive the following equations of motion
for the annihilation operators,
∂ta = −iκ∫b(ω) dω, (C.26)
∂tb(ω) = −iωb(ω)− iκa. (C.27)
Integrating (C.27) and substituting the result into (C.26), we obtain
∂ta(t) = −iκ∫
e−iωtb0(ω)− iκ∫ t
0e−iω(t−t′)a(t′) dt′
dω, (C.28)
where the operators b0(ω) = b(ω, t = 0) represent the initial state of the reservoir.
Performing the frequency integral in the second term produces a delta-function (see

C.5 Dissipation and Fluctuation 424
(D.9) in Appendix D), which selects out the time t = t′, and then (C.28) becomes
∂ta(t) = −γa(t) + F (t), (C.29)
where
F (t) = −iκ∫b0(ω)e−iωt dω (C.30)
is known as a Langevin noise operator, and where γ = πκ2 is an exponential de-
cay rate. Equations of this form were first used to describe the classical Brownian
motion of colloidal particles buffeted by the molecules of a warm fluid. There, the
term involving F represents the fluctuating force arising from collisions with the
randomly moving molecules, and a similar interpretation is helpful in the present
case. The operator F mixes a component of ‘white noise’ into the dynamics of a,
which otherwise would simply decay exponentially with a rate γ. That the noise is
white, with a flat power spectrum, can be seen from examining its temporal corre-
lation functions. Suppose that initially the reservoir contains some small thermal
excitations, so that there are n photons, on average, in a mode with frequency ω.
Then we have
〈b†0(ω)b0(ω′)〉 = nδ(ω − ω′). (C.31)

C.5 Dissipation and Fluctuation 425
The normally ordered correlation function of the noise operator is then given by
〈F †(t)F (t′)〉 = κ2
∫ ∫nδ(ω − ω′)ei(ωt−ω′t′) dω dω′,
= 2πκ2n× 12π
∫eiω(t−t′) dω (C.32)
= 2γnδ(t− t′), (C.33)
and the anti-normally ordered correlation function is similarly given by
〈F (t)F †(t′)〉 = 2γ(n+ 1)δ(t− t′), (C.34)
where we used the commutator in (C.25). The noise is therefore delta-correlated,
meaning that it is completely random from moment to moment. There is no cor-
relation with earlier times, and the noise changes ‘infinitely quickly’; a signature of
unlimited-bandwidth white noise. The infinite bandwidth is a consequence of the
fact that the reservoir coupling κ was assumed to be frequency independent. This
assumption is known as a Markov approximation, since it means that the dynamics
of a does not explicitly depend on its past values. As is clear from the form of
(C.29), the Markov approximation gives rise to exponential decay of a: the ubiquity
of exponential decay in quantum systems serves to confirm the robustness of this
approximation as a model for a wide variety of dissipative processes.
The noise F is just sufficient to preserve the expectation value of the equal-time
commutator in (C.25), so that a remains a bona fide bosonic operator. This can be

C.5 Dissipation and Fluctuation 426
seen by inserting the formal solution to (C.29),
a(t) = a(0)e−γt +∫ t
0e−γ(t−t′)F (t′) dt′, (C.35)
into the commutator (C.25),
〈[a(t), a†(t)]〉 = 〈[a(0), a(0)†]〉e−2γt +∫ t
0
∫ t
0e−γ(2t−t′−t′′)〈[F (t′), F †(t′′)]〉dt′ dt′′
= e−2γt
[1 + 2γ
∫ t
0e2γt′ dt′
]= 1. (C.36)
This close connection between fluctuations, represented by F , and damping, rep-
resented by γ, is a manifestation of the fluctuation-dissipation theorem, discovered
first by Einstein.
Finally, we note that the solution (C.35) allows us to solve for the time evolution
of the number operator for the system, giving the result
〈N〉 = 〈a†(t)a(t)〉 = e−2γt〈a†(0)a(0)〉+(1− e−2γt
)n. (C.37)
This shows that in the infinite future, with t −→ ∞, the system relaxes into ther-
mal equilibrium with the reservoir, with N −→ n. And this thermal equilibrium
condition can often be taken as the initial state of the system, if we want to analyze
processes which drive it out of equilibrium.

Appendix D
Sundry Analytical Techniques
In this Appendix we define the Dirac delta function and the unilateral and bilateral
Fourier transforms used in this thesis. We discuss some of their properties, and
finally we demonstrate some results relating to the Fourier transform of certain
Bessel functions. First though, we introduce a useful technique that expedites these
calculations.
D.1 Contour Integration
Contour integration is a powerful method for evaluating integrals that would oth-
erwise be difficult, or impossible, to perform. To see how it works, we consider the
integral
Ix =∫ b
af(x) dx (D.1)

D.1 Contour Integration 428
of some smooth function f(x). The anti-derivative F (x) of f is the function that
satisfies ∂xF = f , and (D.1) can be simply expressed in terms of F as Ix = F (b)−
F (a). That is, the integral of f between two points is given by the change in
‘height’ of its anti-derivative between these same two points (see Figure D.1 (a)).
Now suppose that we introduce the possibility of complex arguments for f , so that
we consider all the values of f(z), where z = x+ iy is an arbitrary complex number.
We can now think of f as a two dimensional surface, lying above the (x, y)-plane:
as we vary the real and imaginary coordinates x and y, the value of f traces out a
characteristic landscape. We could define an integral
Iz =∫ B
Af(z) dz (D.2)
from some initial point A = xA + iyA to a final point B = xB + iyB. But now that
we are working in a two dimensional plane, the endpoints aren’t enough to specify
the integral completely. We need to know the path that we should take to get from
A to B. On the other hand, the anti-derivative F (z) also describes some kind of
surface in the (x, y)-plane, as shown in part (b) of Figure D.1, and the integral is
again given by Iz = F (B) − F (A). So in fact, the integral does not depend on the
path taken, it only depends on its endpoints. The proof of this fact is known as the
Cauchy integral theorem. Suppose that we integrate along a closed loop, so that the
endpoint B is equal to the starting point A. Then F (B) = F (A) and the integral
vanishes. This is interesting, since we could build the first integral (D.1) into the
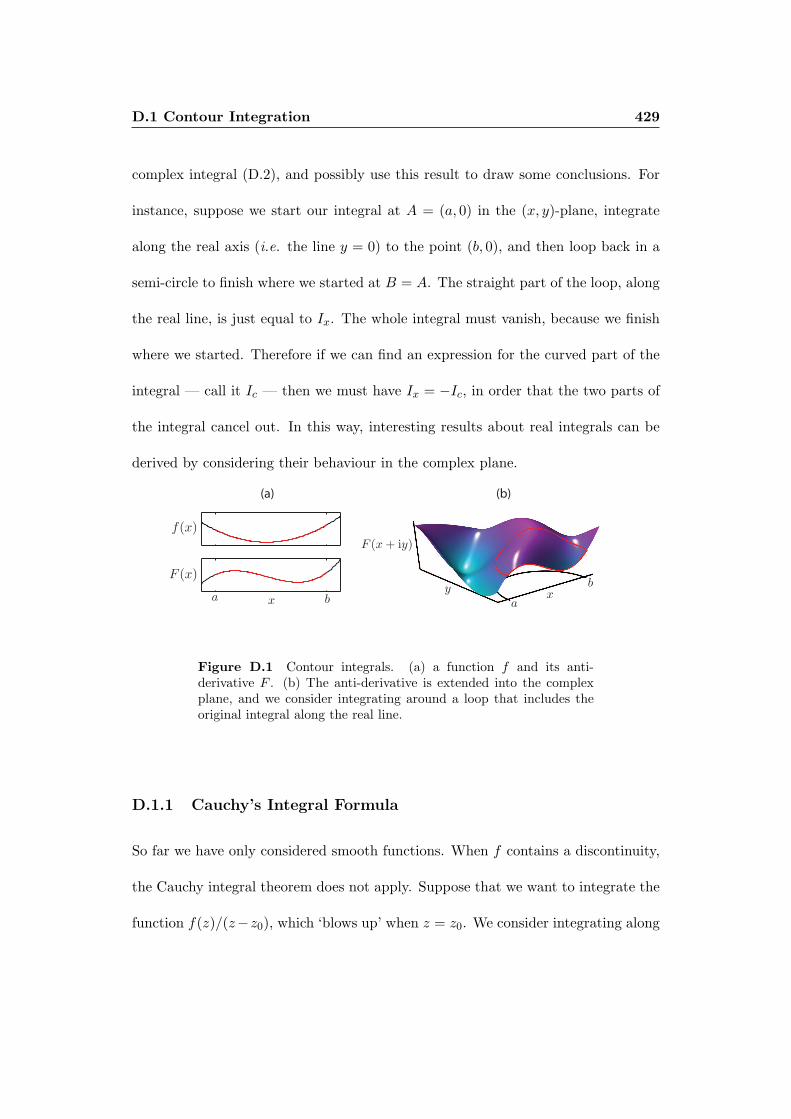
D.1 Contour Integration 429
complex integral (D.2), and possibly use this result to draw some conclusions. For
instance, suppose we start our integral at A = (a, 0) in the (x, y)-plane, integrate
along the real axis (i.e. the line y = 0) to the point (b, 0), and then loop back in a
semi-circle to finish where we started at B = A. The straight part of the loop, along
the real line, is just equal to Ix. The whole integral must vanish, because we finish
where we started. Therefore if we can find an expression for the curved part of the
integral — call it Ic — then we must have Ix = −Ic, in order that the two parts of
the integral cancel out. In this way, interesting results about real integrals can be
derived by considering their behaviour in the complex plane.
(a) (b)
Figure D.1 Contour integrals. (a) a function f and its anti-derivative F . (b) The anti-derivative is extended into the complexplane, and we consider integrating around a loop that includes theoriginal integral along the real line.
D.1.1 Cauchy’s Integral Formula
So far we have only considered smooth functions. When f contains a discontinuity,
the Cauchy integral theorem does not apply. Suppose that we want to integrate the
function f(z)/(z−z0), which ‘blows up’ when z = z0. We consider integrating along

D.1 Contour Integration 430
a circular loop around the singularity at z0. We can parameterize the integral using
polar coordinates z = z0 + reiθ, with dz = ireiθdθ. We are free to make the radius
r as small as we like, since as above the details of the path taken don’t matter; it
only matters that we encircle the singularity. We find
∮f(z)z − z0
dz = limr→0
∫ 2π
0
f(z0 + reiθ)reiθ
× ireiθ dθ = 2πi× f(z0), (D.3)
where the symbol∮
indicates integration around a loop. The singularity is like a
witness to our passage around the loop, so that we do not quite get back to where
we started on completing our roundtrip. The contribution 2πif(z0) to the integral is
known as the residue associated with the singularity at z = z0. In general, wherever
a function blows up, it leaves a residue that contributes to an integral that encloses it.
Differentiating the formula (D.3) with respect to z0 tells us how to handle ‘stronger’
singularities of the form 1/(z − z0)n,
∮f(z)
(z − z0)ndz = 2πi×
∂n−1z0 f(z0)(n− 1)!
. (D.4)
D.1.2 Typical example
We now give an example of how to use these formulas to evaluate an integral. The
method is typical. Consider the integral
Ix =∫ ∞−∞
eix
xdx. (D.5)

D.1 Contour Integration 431
This is equivalent to the contour integral
Iz = limR→∞
∮eiz
zdz, (D.6)
where the integration path is a closed semicircle with radius R, such that the straight
portion runs along the real line, as shown in Figure D.2. To see that Iz = Ix, note
that the function eiz is damped with a positive imaginary argument, ei(x+iy) =
eixe−y. Therefore any contribution to Iz from the curved portion of the path, all of
which has y > 0, vanishes in the limit R −→ ∞. Note that if the integrand were
conjugated, so that it contained the exponential e−ix, we would have to choose to
close the contour in the lower half of the plane, in order that the curved part of the
integral should vanish.
Now the integral Iz is not zero, because the integrand has a singularity at z = 0.
It’s a little inconvenient that this lies exactly on the integration path. We deal with
this by moving the singularity inside the integration path by a small amount ε, in
order to apply (D.3); we then take the limit ε −→ 0 when we are done:
Ix = Iz = limε→0
limR→∞
∮eiz
z − iεdz
= limε→0
2πi× e−ε (D.7)
= 2πi. (D.8)
This technique for performing integrals is indispensable in Fourier analysis, as dis-
cussed below. And of course it is quite elegant!

D.2 The Dirac Delta Function 432
Figure D.2 Upper closure. The integration path is comprised of astraight portion running along the real axis, and a semicircle in theupper half of the complex plane. In the limit of infinite radius R, thestraight portion becomes equal to the real integral Ix, and the curvedportion vanishes.
D.2 The Dirac Delta Function
We use the Dirac delta function δ(x) a great deal in this thesis. Where it is not
introduced ‘by hand’, prompted by physical arguments, it is most often encountered
in the form of an integral over plane waves,
δ(x) =1
2π
∫ ∞−∞
eikx dk. (D.9)
This integral is not really well defined, but its properties become apparent from the
following limit,
δ(x) = limT→∞
12π
∫ T
−Teikx dk
= limT→∞
T sinc(xT )π
, (D.10)
where sinc(θ) = sin(θ)/θ. For each value of T , this describes a narrow peak of width
∼ 1/T and height T , centred at x = 0. As T →∞, the peak becomes infinitely tall

D.2 The Dirac Delta Function 433
and narrow. But its integral remains finite. To see this, consider the integral
Ix =∫ ∞−∞
sinc(x) dx =−i2
∫ ∞−∞
eix
xdx−
∫ ∞−∞
e−ix
xdx. (D.11)
The first integral on the right hand side is given by (D.8). Applying the same
technique to the second integral, we must use an integration path that is closed
in the lower half of the complex plane, as explained in §D.1.2. When we shift the
singularity using z → z − iε, it moves into the upper half of the plane, so the
contour used for the second integral does not enclose the singularity, and therefore
the integral vanishes. Therefore we find that
Ix =−i22πi− 0 = π. (D.12)
Integrating the delta function then yields
∫ ∞−∞
δ(x) dx = limT→∞
1π
∫ ∞−∞
T sinc(xT ) dx = 1. (D.13)
The Dirac delta function is an ideal ‘spike’, with unit area, and so it has the extremely
useful property that ∫ ∞−∞
f(x)δ(x− x0) dx = f(x0). (D.14)

D.3 Fourier Transforms 434
D.3 Fourier Transforms
D.3.1 Bilateral Transform
The most familiar Fourier transform is the bilateral transform f(k) of a function
f(z),
f(k) = Fz f(z) (k) =1√2π
∫ ∞−∞
f(z)eikz dz. (D.15)
The name ‘bilateral’ refers to the lower limit of integration, which is −∞ in this
case, so that the whole function f(z), including its values for negative z, is involved
in forming the Fourier transform f .
D.3.2 Unitarity
The factor of 1/√
2π makes the transform unitary, meaning that the norm of the
the transform f is the same as the norm of f ,
∫ ∞−∞|f(k)|2 dk =
12π
∫ ∞−∞
∫ ∞−∞
∫ ∞−∞
f(z)f∗(z′)eik(z−z′) dk dz dz′
=∫ ∞−∞
∫ ∞−∞
δ(z − z′)eik(z−z′)f(z)f∗(z′) dz dz′
=∫ ∞−∞|f(z)|2 dz, (D.16)
where the plane wave expansion (D.9), and the property (D.14) were used.

D.3 Fourier Transforms 435
D.3.3 Inverse
The inverse Fourier transform takes essentially the same form, except that the inte-
gral kernel is conjugated,
f(z) = F−1k
f(k)
(z) =
1√2π
∫ ∞−∞
f(k)e−ikz dk. (D.17)
This is easily demonstrated by substituting (D.15) into (D.17) and using (D.9).
D.3.4 Shift
From the definition (D.15) we can derive a simple formula for the Fourier transform
of a shifted function f(z + a), where a is some constant,
Fz f(z + a) (k) =1√2π
∫ ∞−∞
f(z + a)eikz dz
=1√2π
∫ ∞−∞
f(x)eik(x−a) dx
= e−ikaf(k), (D.18)
where we changed integration variables from z to x = z+ a in the second line. This
shows that a shift in the coordinate z corresponds to a phase rotation in the Fourier
domain. Identical arguments show that for the inverse transform
F−1k
f(k + a)
(z) = eizaf(z). (D.19)

D.3 Fourier Transforms 436
D.3.5 Convolution
The bilateral convolution of two functions f and g is defined by the integral
f ∗ g(z) =1√2π
∫ ∞−∞
f(x)g(z − x) dx =1√2π
∫ ∞−∞
f(z − x)g(x) dx. (D.20)
The Fourier transform of a convolution is given by the product of the Fourier trans-
forms of the convolved functions. That is,
Fz f ∗ g(z) (k) =1
2π
∫ ∞−∞
∫ ∞−∞
f(x)g(z − x)eikz dz dx
=1
2π
∫ ∞−∞
∫ ∞−∞
f(x)eikxg(y)eiky dxdy
= f(k)g(k), (D.21)
where we used a change of variables y = z−x with dy = dz. This shows also that the
inverse Fourier transform of a product of two functions is given by the convolution
of their individual inverse transforms.
D.3.6 Transform of a Derivative
The nth derivative ∂nz f(z) of a function can be expressed using the inverse (D.17) as
∂nz f(z) =1√2π
∫ ∞−∞
f(k)∂nz e−ikz dk
= F−1k
(−ik)nf(k)
(z), (D.22)

D.4 Unilateral Transform 437
and therefore we must have that
Fz ∂nz f(z) (k) = (−ik)nf(k). (D.23)
That is, the Fourier transform converts differentiation into ordinary multiplication;
clearly this is a boon when tackling differential equations.
D.4 Unilateral Transform
The unilateral Fourier transform is identical to its bilateral counterpart, except that
it involves an integral over positive coordinates only,
f(k) = Fz f(z) (k) =1√2π
∫ ∞0
f(z)eikz dz. (D.24)
We have used the same notation to denote both types of transform: we will be
careful to distinguish between them when the difference is important. This type of
transform is appropriate for functions f(z) which vanish, or are undefined, when
z < 0. As an example, the function f might describe the temporal response to
an interaction at ‘time’ z = 0, in which case causality requires that f vanishes for
z < 0. For these types of functions, the unitarity property (D.16) still holds, and
the inverse transform is given precisely by (D.17). That is, the inverse is bilateral.

D.4 Unilateral Transform 438
D.4.1 Shift
The shift theorem (D.18) is not directly applicable to the unilateral transform, be-
cause of the semi-infinite integral domain. But since the inverse transform is bilat-
eral, the shift theorem (D.19) does apply to the inverse transform.
D.4.2 Convolution
The unilateral convolution, or causal convolution of two functions f and g is defined
similarly to the bilateral convolution (D.20), except that the integral is limited by
causal consistency,
f ∗ g(z) =1√2π
∫ z
0f(x)g(z − x) dx =
1√2π
∫ z
0f(z − x)g(x) dx. (D.25)
To calculate the transform of this convolution, we need to take care with the limits
of integration, as shown in Figure D.3. We then obtain
Fz f ∗ g(z) (k) =1
2π
∫ ∞0
∫ z
0f(x)g(z − x)eikx dxdz
=1
2π
∫ ∞0
∫ ∞x
f(x)g(z − x)eikz dzdx
=1
2π
∫ ∞0
∫ ∞0
f(x)eikxg(y)eiky dydx
= f(k)g(k). (D.26)
Just as for the bilateral case above, the unilateral Fourier transform of a causal
convolution is given by the product of the transforms of the convolved functions.

D.4 Unilateral Transform 439
(a) (b)
Figure D.3 Integration limits. (a) Schematic of the integral∫∞0
∫ z0
dxdz. (b) Schematic of the same integral with the order of in-tegration reversed,
∫∞0
∫∞x
dz dx. Each arrow represents an instanceof the inner integral at a fixed value of the outer variable.
D.4.3 Transform of a Derivative
The real utility of the unilateral transform is in the treatment of derivatives. Con-
sider the unilateral Fourier transform of ∂zf . Integrating parts,
1√2π
∫ ∞0
eikz∂zf(z) dz =1√2π
[eikzf(z)
]∞0− 1√
2π
∫ ∞0
f(z)ikeikz dz
= −ikf(k)− 1√2πf(0). (D.27)
This is the same result as (D.23) for the bilateral transform, except for the presence
of the boundary condition f(0). Note that we assumed f(z −→ ∞) = 0: all the
functions we will consider satisfy this property, referred to alternately as boundedness
or integrability. Physically, this simply says that the effects of any interaction fall
away to nothing as we move away from their source.

D.5 Bessel Functions 440
D.4.4 Laplace Transform
The unilateral Fourier transform occupies a middle ground between the bilateral
Fourier transform on the one hand, and the well-known Laplace transform on the
other. The Laplace transform Lz f(z) (s) is generated from the unilateral Fourier
transform by making the replacement ik → s. The Laplace transform is more com-
mon since even unbounded functions generally possess a Laplace transform, due
to the exponential damping provided by the integral kernel. However this damp-
ing destroys the unitarity of the transform. For this reason, the unilateral Fourier
transform better suits our purposes in this thesis, since the efficiency of a quantum
memory is unchanged by working with this type of transform.
D.5 Bessel Functions
In this section we define the ordinary and modified Bessel functions that represent
the propagators for quantum memories operated adiabatically (see Chapter 5). The
nth order ordinary Bessel function of the first kind — denoted by Jn — is defined
by the infinite series
Jn(2z) =∞∑m=0
(−1)mz2m+n
m!(m+ n)!, (D.28)
where n is a non-negative integer. The corresponding modified Bessel funtion In is
defined identically, except that the factor of (−1)m is missing from each term of the
sum. Conversion between ordinary and modified Bessel functions is reminiscent of

D.5 Bessel Functions 441
the relation between trigonometric and hyperbolic ratios,
Jn(iz) = inIn(z). (D.29)
Indeed the J functions have the appearance of decaying cosines, while the I functions
all grow exponentially with increasing z. Just as sin(x) and cos(x) take the values
0 and 1, respectively, at the point x = 0, so it is also true that J0(0) = I0(0) = 1,
while Jn>0(0) = In>0(0) = 0.
D.5.1 Orthogonality
As can be easily shown using the series (D.28), the Bessel functions Jn(z) satisfy
the differential equation
z∂z (z∂z) Jn = (n2 − z2)Jn. (D.30)
Consider the ith zero ai of Jn, which satisfies Jn(ai) = 0. Defining s = z/ai and
X(s) = Jn(ais), we can re-write (D.30) in the ‘normalized’ form
s∂s (s∂s)X = (n2 − a2i s
2)X, (D.31)
with X(1) = 0. We can repeat this procedure for another zero of Jn, say aj , giving
s∂s (s∂s)Y = (n2 − a2js
2)Y, (D.32)

D.5 Bessel Functions 442
where now s = z/aj and Y (s) = Jn(ajs), so that Y (1) = 0. We now multiply (D.32)
by X and (D.31) by Y, subtract the two resulting equations and divide through by
s, to obtain
(a2i − a2
j )sXY = ∂s [Xs∂sY − Y s∂sX] .
Integrating this from s = 0 to s = 1, and using the fact that X(1) = Y (1) = 0, the
right hand side vanishes. Dividing through by a2i − a2
j and converting back into the
Jn notation, we find the orthogonality condition
∫ 1
0sJn(ais)Jn(ajs) ds = 0, when i 6= j. (D.33)
Alternatively, writing z = s2, we have
∫ 1
0Jn(ai
√z)Jn(aj
√z) dz = 0, when i 6= j. (D.34)
D.5.2 Memory Propagator
In Chapter 5 we are faced with taking the inverse Fourier transform of the function
f(k) =e−ia/k
kn+1, (D.35)
where a is some constant and n = 0, 1, 2.... This is done with the help of con-
tour integration, as described at the beginning of this Appendix. Using the series

D.5 Bessel Functions 443
expansion of the exponential, we have
f(z) =1√2π
∫ ∞−∞
1kn+1
[ ∞∑m=0
1m!
(−iak
)m]e−ikz dk. (D.36)
We must therefore perform the integral
Ik =∫ ∞−∞
e−ikz
kn+m+1dk. (D.37)
We can equate this to the complex integral
Iz = limε→0
limR→∞
∮e−ikz
(k + iε)n+m+1dk, (D.38)
where the integration contour is closed in the lower half of the complex plane, as
shown in Figure D.4, and where the regularization ε shifts the singularity into the
interior of the contour. Using Cauchy’s integral formula (D.4), we find
Iz = limε→0
−2πi×
[∂n+mk e−ikz
(n+m)!
]k=−iε
= −2πi× (−iz)n+m
(n+m)!. (D.39)
Substituting this result into the series (D.36), and comparing with (D.28), we obtain
the result
f(z) = (−i)n+1√
2πΘ(z)(za
)n/2Jn(2√az). (D.40)

D.5 Bessel Functions 444
The function Θ(z) is known as the Heaviside step function, defined so that Θ(z) = 0
when z < 0, and Θ(z) = 1 for z > 0. We include it in the above expression to
represent its causal nature. That f(z) = 0 for negative z can be seen by considering
the integration contour used to evaluate the Fourier transform. When z becomes
negative, the integration contour must be closed in the upper half of the complex
plane, so that it no longer encloses the singularity, which therefore no longer leaves
a residue, and the integral vanishes.
Figure D.4 Lower closure. Here the integrand is damped in thelower half of the complex plane, so we close the integration contourin this region.
We will also need the inverse transform of (D.35) when n = −1,
f(z) = F−1k
e−ia/k
(z). (D.41)
Again, inserting the series expansion for the exponential, we have
f(z) =1√2π
∞∑m=0
(−ia)m
m!
∫ ∞−∞
e−ikz
kmdk. (D.42)
The first term, with m = 0, involves an integral with no singularity. In fact, it is an

D.5 Bessel Functions 445
integral over plane waves — a delta function. We separate off this first term, and
re-index the remaining terms,
f(z) =1√2π
∫ ∞−∞
e−ikz dk +1√2π
∞∑m=0
(−ia)m+1
(m+ 1)!
∫ ∞−∞
e−ikz
km+1dk. (D.43)
Then we perform the singular integrals using contour integration, as before, to obtain
f(z) =√
2πδ(z)− ia√2π
∞∑m=0
(−ia)m(−iz)m
(m+ 1)!m!Θ(z)
=√
2πδ(z)−Θ(z)
√a
zJ1
(2√az)
. (D.44)
We make use of this result in Chapter 5, where it relates transmitted and incident
signal fields in a Raman quantum memory.
D.5.3 Optimal Eigenvalue Kernel
In Chapter (5) we claim that it is possible to derive a certain kernel KA from a
storage kernel K by direct integration — the result is used in the work of Gorshkov
et al. The Fourier transforms presented above provide one way to confirm it. The
two results we need are as follows. First, we need the unilateral Fourier transform
of an integral over the product of two storage kernels:
A = Fz,z′∫ ∞
0e−ax × J0(2
√bxz)e−uz × J0(2
√cxz′)e−vz
′dx
(k, k′). (D.45)

D.5 Bessel Functions 446
Using the result (D.40) along with the shift theorem (D.19) we obtain
A =1
2π
∫ ∞0
e−ax × e−ibx/(k+iu)
k + iu× eicx/(k′+iv)
k′ + ivdx
=1
2π× 1akk′ + i(va+ c)k + i(ua+ b)k′ − uva− bv − cu
. (D.46)
The second result we need is the unilateral Fourier transform of an anti-normally
ordered kernel:
B = Fz,z′e−α(z+z′)J0(2
√βzz′)
(k, k′). (D.47)
Transforming over z first, and then z′, we get
B =i√2πFz′e−αz
′ × e−iβz′/(k+iα)
k + iα
(k′)
=i
2π
∫ ∞0
e−[α+iβ/(k+iα)−ik′]z′
k + iαdz′ (D.48)
= − 12π× 1kk′ + iα(k + k′)− β − α2
. (D.49)
Comparing these two results, and grinding through some tedious algebra, it is not
hard to demonstrate the claimed equality.

Appendix E
Numerics
In this Appendix we review the numerical methods used in this thesis. Our aim
is to solve a system of coupled linear partial differential equations (PDEs) in space
and time. For pedagogical purposes, we will consider the simple example presented
in §3.5 in Chapter 3: A resonant signal pulse with amplitude A propagates through
an ensemble of classical atoms, whose response to the optical field is described by
the average displacement B of the atomic electrons from their equilibrium positions.
The equations of motion are given by
∂τB = −iαA, and ∂zA = −iβ
αB. (E.1)
(The second equation follows from the first line of (3.34)). Since we are only con-
cerned with the method of numerical solution, we set α = β = 1, and we normalize
the τ and z coordinates so that they are dimensionless, with z running from 0 to 1,

448
and τ running from 0 to 10. The boundary conditions are Ain = A(z = 0, τ) and
Bin = B(z, τ = 0): the profile of the incident signal pulse, and the initial atomic
excitation, respectively. Generally Ain will take the form of a pulse, reminiscent of a
Gaussian, and Bin = 0, if the atoms are prepared in their ground states. To find the
solution numerically, we discretize the space and time coordinates on a finite grid,
z −→ zj , τ −→ τk, (E.2)
and we represent the continuous functions A(z, τ), B(z, τ) as matrices A, B whose
elements approximate those functions sampled at the grid points,
Ajk = A(zj , τk), Bjk = B(zj , τk). (E.3)
We solve the system (E.1) via the method of lines [199]. The technique is so named
because we integrate the spatial derivative ∂z ‘in one go’, whereas we integrate the
temporal derivative ∂τ incrementally, stepping forwards in time iteratively. The so-
lution, starting as a series of points in space at τ = 0, evolves gradually forwards
in time, and each of the points in space traces out a line, over time, that describes
the solution at that position in space. It is a feature of the implementation of the
technique that the temporal discretization is much finer than the spatial one, and
so the numerics are conducive to this mental picture, which is illustrated schemat-
ically in Figure E.1. The spatial derivative is performed using a spectral method,
which uses the value of the function at all the spatial points in order to compute

E.1 Spectral Collocation 449
Figure E.1 The method of lines. The spatial coordinate is dis-cretized on a coarse grid. The solution at each spatial point tracesout a line as the temporal integration proceeds stepwise on a finegrid.
the derivative. For smooth functions, this is extremely accurate, even when very
few spatial points are used. Before describing the method used for the temporal
derivative, we introduce this spectral method more fully.
E.1 Spectral Collocation
An excellent introduction to this field of numerical analysis is provided in Nick
Trefethen’s book Spectral Methods in Matlab [200] — which is hosted on the internet.
Other treatments include the books by Gottlieb and Orzag [201], and by Boyd [202].
The easiest spectral method to understand is that involving a Fourier transform. As
shown in §D.3.6 in Appendix D, Fourier transformation of a function f from z to
k converts differentiation by z into multiplication by −ik. It is also clear that the
operation of taking a Fourier transform, or indeed its inverse, is a linear one. Suppose
that we discretize the coordinates z and k, so that the function f is approximated by

E.1 Spectral Collocation 450
a vector f , with components fj = f(zj). In this context the points zj at which f(z)
is sampled are known as collocation points. The linearity of the Fourier transform
then allows us to represent it as a matrix acting on f . The composition of the
operations of taking the Fourier transform, multiplying by −ik, and then taking the
inverse Fourier transform, can therefore also be represented as a single matrix, D.
An approximation fz to the derivative of f at the collocation points is then given
by
fz = Df . (E.4)
The matrix D is dense, meaning that there are generally very few elements of D
that are zero, so all of the elements of f are involved in determining fz. As a result,
spectral methods can be very accurate, even if very few collocation points are used.
This accuracy comes at the price of requiring a dense matrix multiplication, which is
computationally more expensive than a more ‘local’ method — such as the method
we employ for the temporal integration (see below). As it happens there is a more
efficient way to implement a discrete Fourier transform (DFT), known as the fast
Fourier transform (FFT), which was developed by Cooley and Tukey in 1965 [203].
The FFT does not use explicit matrix multiplication, and it is generally employed
for Fourier spectral methods. We do not use a Fourier spectral method in this
thesis, however. One reason for this is that a DFT works by fitting a finite number
of complex exponentials — which are periodic — to the vector f . Since the basis
functions are periodic, the vector f must be periodic. Of course any function f(z)
defined on a finite domain [0, 1] can be made periodic by ‘gluing’ copies of the

E.1 Spectral Collocation 451
function onto either end of the domain, as shown in Figure E.2. But if f(0) 6= f(1),
the resulting ‘extended’ function will contain discontinuous jumps, where the copies
were glued together. These discontinuities represent a very rapid change in f , so
that the spectrum f(k) contains very high spatial frequencies. Given that f , and
the approximated Fourier transform f , are comprised of only a finite number of
collocation points, the accuracy of the DFT is therefore severely compromised by
these discontinuities. Fourier spectral methods are not well suited to dealing with
functions that are not ‘really’ periodic, for this reason. Since we are modeling the
spatial distribution of atomic excitations, which need have no intrinsic periodicity,
it behooves us to use a method that is not subject to this limitation. Such a method
is provided by polynomial differentiation matrices.
(a)
(b)
Figure E.2 Periodic extension. (a): the function f satisfies f(0) =f(1), so that its periodic extension is smooth. The Fourier spectralderivative of this function is accurate. (b): now f(0) 6= f(1), and theperiodic extension of f contains sharp discontinuities, which erodethe accuracy of a Fourier spectral derivative.

E.1 Spectral Collocation 452
E.1.1 Polynomial Differentiation Matrices
To generalize spectral differentiation to the case of non-periodic functions, we should
use non-periodic basis functions. Instead of fitting the vector f with complex expo-
nentials, as in the Fourier case, we fit f with an algebraic polynomial p(z). We can
then easily differentiate this to get p′(z), and the vector approximating the deriva-
tive of f is found by evaluating p′ at the collocation points. The basis functions
from which we construct p are specified by requiring that p evaluates to f at the
collocation points. We write p in terms of a set of basis functions pj, and the
elements of f , as
p(z) =∑k
fkpk(z). (E.5)
Fixing p(zj) = fj imposes the condition
pk(zj) = δjk. (E.6)
The interpolant p(z) is a polynomial of degree N − 1, if there are N collocation
points, and therefore the basis functions pk are also polynomials of degree N − 1.
The condition (E.6) means that each of the N collocation points, except for zk, are
roots of pk(z), and therefore
pk(z) =1ak
∏i 6=k
(z − zi), with ak =∏i 6=k
(zk − zi). (E.7)

E.1 Spectral Collocation 453
Here ak is just the normalization required so that pk(zk) = 1. Differentiating the
relation (E.5), and setting z = zj gives
p′(zj) =∑k
p′k(zj)fk. (E.8)
Identifying the elements of fz with p′(zj), and using the definition (A.13) in Ap-
pendix A for matrix multiplication, we see the elements of the differentiation matrix
D are given by
Djk = p′k(zj). (E.9)
Taking the logarithm of (E.7) and differentiating, we find
p′k(z) = pk(z)∑i 6=k
1z − zk
. (E.10)
The elements of D are then given by the general formulae
Djk =
∑
i 6=j1
zj−zi if j = k,
ajak(zj−zk) otherwise.
(E.11)
E.1.2 Chebyshev points
We are now able to construct a differentiation matrix that works on an arbitrary
set of collocation points zj. A grid of equally spaced points is the natural choice,
but this is in fact disastrously unstable: the interpolant p develops pathological
oscillations near the edges of the domain, close to z = 0 and z = 1. This is known

E.1 Spectral Collocation 454
as the Runge phenomenon, and it is dealt with by using a set of collocation points
that cluster together near the domain edges. To develop an intuition for why this
might be, observe that the magnitude of one of the basis functions can be written
in the form
|pk(z)| =eV (z)
|ak|, where V (z) =
∑i 6=k
ln |z − zi|. (E.12)
Notice that V (z) has the same functional form as the electrostatic potential due to
infinite lines of charge intersecting the z-axis at the collocation points zi. There-
fore the size of the basis functions is exponentially sensitive to the potential energy
associated with the ‘charge distribution’ describing the collocation points. The in-
terpolant p is well-behaved when this energy is constant across the domain [0, 1].
If we allowed the collocation points to move along the z-axis according to the mu-
tual repulsion between them represented by V , they would arrange themselves in
a minimum-energy configuration which renders the potential flat, and the inter-
polant would be stable. In this configuration, the points are clustered together at
the domain boundaries, and this explains why choosing a set of collocation points
according to this configuration avoids the Runge phenomenon.
A stable set of collocation points is provided by the Chebyshev points, which are
the projections onto the domain of equally spaced points along a semicircle joining
the domain boundaries (see Figure E.3):
zj = 12
1− cos
[π(j−1)N−1
]. (E.13)

E.2 Time-stepping 455
By substituting the points (E.13) into the formula (E.11), the appropriate Cheby-
collocation points
(a) (b)
Figure E.3 Chebyshev Points. (a) Chebyshev collocation pointsare clustered towards the boundary of the domain. They are thedownward projections of equally spaced points along a semicircle ofradius 1
2 centred at z = 12 . (b) The method of lines with Chebyshev
spectral collocation.
shev differentiation matrix can be calculated. We use Matlab for all of our numer-
ical computations, and the differentiation matrices are conveniently generated by
a simple script called cheb.m, which is available online, and can be found in Nick
Trefethen’s book [200], along with many detailed examples of its application.
Having introduced the method we use for the spatial derivatives, we now discuss
the temporal derivatives, before describing how these two techniques are combined
to solve the system (E.1).
E.2 Time-stepping
Suppose that we would like to solve the differential equation
f ′(τ) = g [f(τ), τ ] , with f(0) = f0, (E.14)

E.2 Time-stepping 456
where the prime now denotes differentiation with respect to τ . We assume that
the boundary condition f0 and the function g are known. Note that g may depend
on f . For instance, in our example, B′ = −iA, but A depends on B. Now, the
simplest numerical approach is to make a finite difference approximation to the
time derivative. In terms of the discretized functions and coordinates, we have
fk+1 − fkδτ
= g (fk, τk)
⇒ fk+1 = fk + δτg (fk, τk) , (E.15)
where δτ = τk+1 − τk is the time step — assumed to be independent of k. The
second line of (E.15) is a recursion relation, relating the future value of f to the
present values of f and g. Starting with the boundary condition at τ = 0, we
can use this relation to step forward in time, gradually building up the solution
for f . This method is known as a first order Euler method, since errors of order
δτ accumulate in the numerical solution. In our numerics, we use a second-order
Runge-Kutta (RK2) method, which is only slightly more complicated, but accurate
up to errors of order δτ2. The Runge-Kutta method reduces the errors by using
values of the known function g at intermediate points, between τk and τk+1. The
recursion formula therefore requires that we discretize our functions on two grids: a
primary grid, with times τk, which we denote with the same notation as used above,
and a secondary, intermediate grid, with times τk = τk + 23δτ . The second order

E.3 Boundary Conditions 457
Runge-Kutta method we employ for time stepping is then written as
fk+1 = fk +δτ
4[g (fk, τk) + 3g
(fk, τk
)], (E.16)
where the value of f on the secondary grid is approximated with the first order Euler
formula (E.15), so that
fk = fk + 23δτg (fk, τk) . (E.17)
E.3 Boundary Conditions
It is clear how to implement the boundary condition Bin on B using the above
time-stepping algorithm. This boundary condition simply tells us the initial values
to use, on the first time step. However it is not so clear, from our discussion of
spectral methods, how to implement the boundary condition Ain on A, which holds
at z = 0 at all times. This is done by incorporating the boundary condition into
the dynamical equation for A. First consider the discretized version of the equation
∂zA = −iB, which is
DA = −iB, (E.18)
where D is a Chebyshev differentiation martrix, which acts on each column of A to
produce the corresponding column of −iB. There is no mention of the boundary
condition so far. The discretized form of the boundary condition on A can be written

E.3 Boundary Conditions 458
as
1× [A11 A12 A13 · · · A1N ] = [Ain(τ1) Ain(τ2) Ain(τ3) · · · Ain(τN )] . (E.19)
That is, 1 multiplied by the first row of A — which corresponds to the values of A
at z = z1 = 0 — is equal to the discretized boundary condition. This equation can
be ‘built in’ to the dynamical equation (E.18) simply by replacing the first row of
D with the first row of the identity matrix I, and by replacing the first row of −iB
with the discretized boundary condition. The equation becomes
DbA = −iBb, (E.20)
where the superscript b — for ‘boundary’ — indicates the modifications
[Db
11 Db12 D
b13 · · · Db
1N
]= [1 0 0 · · · 0] , and[
Bb11 B
b12 B
b13 · · · Bb
1N
]= i× [Ain(τ1) Ain(τ2) Ain(τ3) · · · Ain(τN )] .(E.21)
All the other elements of Db and Bb are the same as those of D and B.
Now that we have fixed the boundary condition for A, we can solve for A in
terms of B. Formally, we can invert the modified differentiation matrix Db to get
A = −iDb−1Bb. (E.22)

E.4 Constructing the Solutions 459
Of course, we do not know all the values of B until we have performed the time-
stepping, and this requires knowledge of A. In order to get to the solutions for A and
B, we must build up B column by column, using the time-stepping, and solving for
each column of A in turn using the above procedure. We are now ready to describe
how this works.
E.4 Constructing the Solutions
Let us denote the columns of A by ak, and similarly the columns of B by bk:
A =
a1
a2
. . .
aM
, B =
b1
b2
. . .
bM
,
(E.23)
where M is the number of temporal discretization points. The algorithm proceeds
as follows. First, we use the boundary condition Bin to set the values of b1,
B11
B21
...
BN1
=
Bin(z1)
Bin(z2)
...
Bin(zN )
. (E.24)
Then, we solve for a1 using the formula
ak = −iDb−1bbk. (E.25)

E.4 Constructing the Solutions 460
Here bbk is the kth column of Bb. That is to say, bb
k = bk, except that its first element
is replaced by the signal field boundary condition, (bbk)1 = iAin(τk). Now that we
have both b1 and a1, we implement the first stage of the RK2 iteration, which is to
approximate the first time step on the intermediate grid, b1, using (E.17),
bk = bk − i23δτak. (E.26)
Before we can implement the second part of RK2, we must approximate a1. This is
done by using (E.25) again, this time replacing bbk with bb
k. That is,
ak = −iDb−1bbk. (E.27)
This time, the modified vector contains the signal boundary condition evaluated
at τ = τk. That is,(b
bk
)1
= iAin
(τk + 2
3δτ). Finally, the second part of RK2 is
implemented, which provides us with b2. The formula is
bk+1 = bk − iδτ
4(ak + 3ak) . (E.28)
We have now succeeded in constructing b2 from our knowledge of b1, and along the
way we have found a1. Iterating this procedure, we can construct b3 and a2, and
then b4 and a3, and so on. In this way we proceed forward in time until we reach
the end of our time domain, at τ = τM = 10. This completes the numerical solution.
Here we comment that a dramatic increase in computational speed is achieved

E.4 Constructing the Solutions 461
if Gaussian elimination is used instead of matrix inversion in (E.25) and (E.27).
Gaussian elimination is an efficient method for solving the matrix equation Ax =
y to obtain x, without explicitly calculating the inverse A−1. In Matlab, this is
implemented by the ‘backslash’ operator. For example, the Matlab code for (E.25)
might look like
A(:,k)=-i*inv(Db)*Bb(:,k),
where the ‘inv’ function calls a matrix inversion routine. A much more efficient
way to perform the same calculation would be coded as follows,
A(:,k)=-i*( Db\Bb(:,k) ).
This latter method becomes crucial to the feasibility of our chosen spectral
method when modeling complicated dynamics.
In order to produce an informative plot, we can use polynomial interpolation in
order to replace the coarse Chebyshev grid of spatial collocation points with a finer,
equally spaced grid. This utilizes the full accuracy of the spectral method, since the
values at the collocation points are implicitly derived from the global interpolant p(z)
at each time step. In practice, it is often faster, and quite acceptably accurate, to
use a piecewise spline interpolant, which glues together multiple cubic polynomials
in a continuous way, to join the collocation points. Routines for implementing this
are standard in Matlab. In Figure E.4 we plot example solutions for A and B. Even
using only 5 spatial collocation points, the solutions are accurate to ∼ 1%.

E.5 Numerical Construction of a Green’s Function 462
Figure E.4 Example solutions. We plot the the squared moduli|A|2 and |B|2 of the solutions for A and B, found using the methodof lines, as a function of z and τ . We used N = 5 spatial collocationpoints, on a Chebyshev grid, and M = 20 equally spaced time steps.The Matlab code runs in 0.02 seconds on a 3 GHz machine. Thesignal boundary condition is a Gaussian pulse, Ain(τ) = e−(τ−2.5)2 ,and we assume there are no atomic excitations initially: Bin(z) = 0.The black lines indicate the time evolution of the spatial collocationpoints. The smooth surfaces are generated by spline interpolation inbetween these points. The red lines indicate the boundary conditions.The interpolated solutions are accurate to ∼ 1%, even with only 5collocation points - this rapid convergence is a remarkable feature ofspectral collocation.
The behaviour is as might be expected: the signal field is absorbed as it propa-
gates through the ensemble, with the transmitted field having undergone significant
temporal distortion due to the spectral hole burnt by the atomic absorption line.
The atomic excitation grows in time, assuming a roughly exponential shape in space,
at the end of the interaction, consistent with Beer’s law.
E.5 Numerical Construction of a Green’s Function
Now that we are able to solve the system of coupled PDEs describing a quantum
memory, for given boundary conditions, we can find the Green’s function — the
storage kernel — for the interaction. As described in §5.4 in Chapter 5, this is done

E.5 Numerical Construction of a Green’s Function 463
by solving with delta function boundary conditions for the signal field, successively
varying the timing of the delta function to construct an approximation to the kernel.
As usual for storage, we set Bin(z) = 0. Of course, it is not possible to implement
a true delta function numerically. Instead, the delta function Ain(τ) = δ(τ − τk) is
represented as
[A11 A12 · · · A1k · · · A1N ] = [0 0 · · · 1 · · · 0] . (E.29)
That is, along the line z = 0, the signal field is identically vanishing at all times,
except at τ = τk, where it takes the value 1. Since the equations are linear, the
absolute magnitude of Ain is not important, it only matters that the incident signal
field is non-zero at just the single time step τ = τk. Due to the causal nature of the
interaction, it is not necessary to numerically integrate from τ = 0 up to τ = τk.
Since the signal field is zero in this region, and since there is no atomic excitation,
the dynamics are trivial in this region. Therefore we only integrate from τ = τk up
to τ = τM .
Let bkM denote the vector representing the atomic excitation at the end of the
interaction at τ = τM , produced by a delta function incident signal at τ = τk. The
numerical approximation to the Green’s function is the matrix whose kth column is

E.5 Numerical Construction of a Green’s Function 464
bkM ,
K =
b1M
b2M
. . .
bMM
. (E.30)
Constructing the Green’s function is a more demanding computation than simply
solving an instance of the equations with some particular boundary condition. First,
the system of PDEs must be solved multiple times, and second, the sharp nature of
the delta function boundary condition generally requires a smaller time step than
is required for a smooth boundary condition. Below, in Figure E.5, we plot the
numerically constructed Green’s function for our example system, alongside the
analytic result, expressed as a Bessel function, which is derived in (3.38) in §3.5
of Chapter 3. With a sufficiently fine grid, the numerical and analytic results are
indistinguishable.
numerical analytic
Figure E.5 A numerically constructed Green’s function. We usedN = 30 spatial collocation points, and M = 4000 time steps. Thecomputation takes around 10 minutes on a 3 GHz machine; the agree-ment with the analytic result is excellent.

E.6 Spectral Methods for Two Dimensions 465
E.6 Spectral Methods for Two Dimensions
In Chapter 6 we describe simulations carried out in three dimensions — two space
dimensions, and time. Needless to say, these simulations are much more time con-
suming than those involving only one spatial dimension. The method we use is
essentially identical to that described in §E.4 above, except that now we use spec-
tral collocation to deal with both spatial dimensions: we still use RK2 for the time
stepping. In this section, we explain how to extend the spectral method to two
dimensions. For concreteness, we will consider solving the simple example system
(E.1) using only spectral collocation. The method is rather memory-intensive, and
it quickly becomes unwieldy as the problem becomes complex, but it has the appeal
of being very direct: we encode all of the dynamics into a single matrix equation,
from which we extract the solutions in a single step. To see how this works, consider
the following representation of the dynamical equations (E.1),
∂z i
i ∂τ
A
B
= 0. (E.31)
If we could invert the matrix of derivatives, we might solve for A and B. But
this matrix has no unique inverse: boundary conditions are needed. We proceed by
discretizing the coordinates, and approximating (E.31) numerically using Chebyshev
differentiation matrices to represent the differential operators. We then incorporate
the boundary conditions into the resulting matrix equation, much as we did in §E.3
previously. Finally, we extract the solutions for A and B using Matlab’s backslash

E.6 Spectral Methods for Two Dimensions 466
operator.
In order to represent (E.31) numerically, we must convert A and B into column
vectors. First, we discretize both the z and τ coordinates on a Chebyshev grid. We
use N spatial collocation points and M temporal collocation points. The functions
A and B are then represented as N ×M matrices, whose columns describe spatial
variation, and whose rows describe evolution in time. Let us denote the columns of
A and B by ak and bk, as before (see (E.23)). We vectorize the matrices A and
B by concatenating adjacent columns into a single column vector, of total length
NM ,
A = vec(A) =
a1
a2
...
aM
, B = vec(B) =
b1
b2
...
bM
. (E.32)
We now generate the differentiation matrices that will simulate the action of ∂z and
∂τ on A and B. Suppose that Dz is the differentiation matrix designed for the
spatial Chebyshev grid defined on [0, 1] (Dz is therefore exactly the same as the
matrix D discussed in §E.4). This acts on a column ak of A to produce the required
derivative. The matrix Dz, which acts on A as a partial space derivative, is therefore

E.6 Spectral Methods for Two Dimensions 467
given by
Dz =
Dz
Dz
. . .
Dz
= I ⊗Dz, (E.33)
where on the right hand side we have used the tensor product notation (see §A.2.2 in
Appendix A). This expresses the fact that Dz acts on the spatial part of the vector
A (within each column of A), whereas the temporal part (across columns of A) is
unaffected. In a similar vein, we assemble the temporal partial derivative ∂τ by first
generating a Chebyshev differentiation matrix Dτ for the temporal Chebyshev grid.
Since the τ coordinate runs from 0 up to 10, the collocation points are given by
τk = 10× 12
1− cos
[π(k−1)M−1
]. (E.34)
Because of the factor of 10 appearing here, we have Dτ = 110D, where D is an
M ×M Chebyshev differentiation matrix generated for the domain [0, 1], like the
one used for the spatial derivative previously. The operator Dτ representing a partial
temporal derivative on B is then given by
Dτ = Dτ ⊗ I. (E.35)
This operator acts only on the rows B, leaving its columns unaffected. We are now

E.6 Spectral Methods for Two Dimensions 468
able to write down our numerical approximation to (E.31), which is
Dz i
i Dτ
A
B
= 0. (E.36)
We can express this compactly as
LX = 0, (E.37)
in an obvious notation. In order to solve for X — i.e. for A and B — we must
insert the boundary conditions for the signal field and atomic excitations. As in
§E.3 above, the general approach is to incorporate the equation
1× ( X at the boundary ) = ( appropriate boundary condition ) . (E.38)
For instance, to fix the signal field boundary condition Ain(τ), we identify those
elements of X which describe A at z = 0. This is the first row of the matrix A,
corresponding to the elements
1, N + 1, 2N + 1, . . . , (M − 1)N + 1, (E.39)
of X. The left hand side of (E.38) is then implemented in two steps. First, we set
the rows of L with row indices given by (E.39) to zero. Second, we set the diagonal
elements of L, with both indices given by (E.39), equal to one. The resulting modified

E.6 Spectral Methods for Two Dimensions 469
matrix has the property that it maps the signal field at the boundary z = 0 to itself.
The right hand side of (E.38) is realized by introducing a vector C into the right
hand side of (E.37), with all elements zero, except for those with indices given by
(E.39), which are set equal to the discretized boundary condition,
C1 = Ain(τ1), (E.40)
CN+1 = Ain(τ2), (E.41)
... (E.42)
C(M−1)N+1 = Ain(τM ). (E.43)
The same procedure is used to implement the boundary condition Bin for the atomic
excitations. This time the boundary is the first column of B, which is indices 1 to
N of B, and therefore indices NM + 1 to NM +N of X. Finally, we are left with
a modified equation
LbX = C, (E.44)
where the superscript b distinguishes the modified matrix, and where C contains all
the relevant boundary conditions. The formal solution is then
X = Lb−1C. (E.45)
As mentioned previously, this is calculated much more efficiently if Gaussian elim-
ination, rather than explicit matrix inversion, is used. In Matlab, we invoke the

E.6 Spectral Methods for Two Dimensions 470
backslash operator. This completes the numerics. We are now in possession of
approximations to both A and B at all spatial and temporal collocation points.
Polynomial interpolation, or piecewise spline interpolation, provides us with an ac-
curate representation of the solution for arbitrary z and τ . In Figure E.6 we plot
the solutions for A and B found using this method.
Figure E.6 Spectral methods in two dimensions. We use N = 5spatial collocation points, andM = 15 temporal collocation points, tosolve the example system (E.1), with the same boundary conditionsas shown previously in Figure E.4. The code runs in 0.01 seconds ona 3 GHz machine, and the interpolated solutions shown are identicalto those generated using spectral collocation and RK2 time stepping.

Appendix F
Atomic Vapours
In this final Appendix we cover some results pertaining to atomic vapours, and in
particular to our ongoing experiments with cesium vapour. We review the behaviour
of vapour pressure with temperature, the concept of oscillator strength and its rela-
tion to the dipole moment associated with a transition, and the various mechanisms
of line broadening. We finish with an analysis of the polarization of Stokes scattering
in cesium.
F.1 Vapour pressure
No solid is infinitely sticky. Although the constituents may be bound tightly, there
remains a finite probability that an atom will escape. Every solid is therefore ac-
companied by a diffuse cloud of free atoms, the pressure of which is known as its
vapour pressure. It is this diffuse vapour that is used for the storage medium in our
experiments (and many others). The density of the vapour determines its optical

F.1 Vapour pressure 472
depth, and with it the achievable storage efficiency (see Chapter 5). To estimate
this density, we treat the atoms as a classical statistical ensemble. The probability
that an atom occupies a state with energy E is given by the Boltzmann distribution,
p(E ) ∼ e−E /kBT . (F.1)
The energy E required for an atom to escape depends on the potentials within the
solid, and is largely independent of temperature. The ratio between the probabilities
for escape at two temperatures T0, T , is therefore
p
p0= e− EkB
(1/T−1/T0). (F.2)
This is, in fact, a good approximation for the vapour pressure above a solid, if p is
now taken to be the pressure. The relation can be derived much more rigourously
from the Clausius-Clapeyron equation [204]. Generally the reference state temper-
ature T0 is taken to be the boiling point of the material, since then the vapour
pressure p0 must be equal to the ambient pressure at this temperature. The energy
E is commonly given as the molar enthalpy of sublimation ∆H. Boltzmann’s con-
stant kB is then switched for the molar gas constant R = 8.31 JK−1mole−1. Part
(a) of Figure F.1 shows the vapour pressure of thallium — the storage medium used
in our preliminary experiments — using the values ∆H = 182 kJmole−1, T0 = 1746
K and p0 = 5.8 × 105 Pa. Application of the ideal gas law p = nkBT allows us to
convert the vapour pressure into the atomic number density, which is also shown in

F.1 Vapour pressure 473
the plot.
Calculation of the vapour pressure of cesium, which is shown in part (b), is
complicated by the fact that cesium melts at 28 C. The qualitative behaviour is
unchanged, but we use an empirical formula taken from the excellent document by
Daniel Steck [184], which is available online.
300 350 400 450 50010
−30
10−20
10−10
10
1610
510
010
510
1010
1510
1810
2010
2210
0
300 350 400 450 500 300 350 400 450 50010
−4
10−2
100
102
300 350 400 450 500
Pressure (Pa)
Number density (m
-3)
(a) (b)Thallium Cesium
Temperature (K) Temperature (K)
Figure F.1 The vapour pressure of (a) thallium, and (b) cesium.The black curves show the vapour pressure in Pascals (left axis) asa function of temperature, measured in Kelvin. The red curves showthe corresponding number density (right axis), calculated using theideal gas law.
As described in §10.9.1 of Chapter 10, a rough estimate of the optical depth
of an ensemble is given by d = nλ2L ∼ n × 10−14, assuming optical wavelengths
and a vapour cell a few centimeters long. From this it is clear that thallium has
insufficient density for efficient storage at reasonable temperatures. Raising the
temperature further starts to introduce a thermal background into the signal field
from blackbody radiation! On the other hand, cesium has an optical depth of order
100, even at room temperature.

F.2 Oscillator strengths 474
F.2 Oscillator strengths
The oscillator strength fjk is a dimensionless measure of the dominance of an atomic
transition |j〉 ↔ |k〉, compared to all other possible transitions. The oscillator
strengths for all transitions from the state |j〉 sum to unity,
∑k
fjk = 1, ∀j. (F.3)
The relation between the fjk and the dipole matrix elements djk = 〈j|er|k〉 may be
derived solely from this, and one other condition, which is that
fjk ∝ |djk|2. (F.4)
This simply relates the f ’s to the transition probabilities. The following derivation is
due to Charles Thiel at Montana State University. To determine the proportionality
constant, we insert (F.4) into (F.3).
∑k
fjk = 1 =∑k
αjk |〈j|er|k〉|2
=∑k
e2αjk ×12
[〈j|r|k〉〈k|r|j〉+ 〈j|r|k〉〈k|r|j〉] . (F.5)
The development in the second line seems a little obtuse, but we now make use of
the following relation between the matrix elements of the position and momentum

F.2 Oscillator strengths 475
operators,
pjk = 〈j|m∂tr|k〉
= im〈j|[r,H]|k〉
= −imωjkrjk, (F.6)
where m is the electron mass, and where ωjk is the frequency splitting between
the states |j〉, |k〉, which are taken to be energy eigenstates of the Hamiltonian H.
Substitution of this relation into (F.5) yields
1 =∑k
ie2αjk2mωjk
[〈j|r|k〉〈k|p|j〉 − 〈j|p|k〉〈k|r|j〉]
=−ie2α
2m〈j| [r, p] |j〉
=e2~α2m
, (F.7)
where in the penultimate line we used the decomposition of the identity I =∑
j |j〉〈j|,
and where we somewhat heuristically set αjk = αωjk. In the final line we made use
of the canonical commutation relation [r, p] = i~. The resulting expression for the
oscillator strength is
fjk =2mωjk
~e2|djk|2. (F.8)
The inverse relation is useful for calculating the dipole moment from the oscillator
strengths listed in data tables.

F.3 Line broadening 476
F.3 Line broadening
There are three processes that increase the absorption linewidth in our cesium
vapour: Doppler broadening, pressure broadening and power broadening. An ex-
cellent treatment of these effects can be found in The Quantum Theory of Light by
Loudon [107]. Here we briefly review the physics of line broadening.
F.3.1 Doppler broadening
Doppler broadening refers to the variation in the resonance frequencies of atoms
moving with different velocities. Consider an atom emitting light of wavelength λ0
while moving at a velocity v (see Figure F.2). Over the course of an optical period
T0, the atom moves so that the wavelength appears compressed, λ = λ0−vT0. Using
the relations λ0 = 2πc/ω0, λ = 2πc/ω and T0 = 2π/ω0, we derive the Doppler shift
δω = −ω0v
c, (F.9)
where δω = ω − ω0 is the shift in angular frequency caused by the motion of the
atom. The spectral intensity is then given by the distribution of atomic velocities
in the vapour. An atom with velocity v and mass M has a kinetic energy
E = 12Mv2. (F.10)

F.3 Line broadening 477
Using the Boltzmann distribution (F.1), and substituting for v using (F.9) gives the
spectrum
I(δω) ∝ e−(δω/γd)2
, (F.11)
where the Doppler linewidth is given by
γd =
√2kBT
M× ω0
c. (F.12)
Figure F.2 The Doppler shift. An atom moving with velocityv ‘catches up’ with the light it emits, so that the wave appearssquashed.
F.3.2 Pressure broadening
Collisions between atoms in a vapour cause disruptions to the wavetrain of light
emitted by them. In particular, the phase of the light is randomized by each collision.
To understand the effect on the spectrum of the light we should consider the statistics
of collisions.
An atom with cross sectional area σ travelling at velocity v sweeps out a volume

F.3 Line broadening 478
Figure F.3 Collisions in a vapour. An atom with cross section σtravelling with velocity v sweeps out a volume vσdτ in a short timedτ . Any other atom within this volume gets hit!
σvdτ in a short time dτ (see Figure F.3). The probability of a collision is just the
probability of finding another atom within this volume, which is given by n×σvdτ ,
where n is the number density of atoms in the vapour. Now consider ps(τ), the
probability that an atom ‘survives’ without colliding for a time τ . We do not know
this probability yet, but we can say the following,
ps(τ + dτ) = ps(τ)× (1− nσvdτ) . (F.13)
That is, the probability of surviving a further time dτ after τ is given by the proba-
bility of surviving for a time τ , and then not colliding during dτ . Taylor expanding
(F.13) gives
ps(τ) + ∂τps(τ)dτ = ps(τ)− nσvps(τ)dτ,
⇒ ∂τps(τ) = −nσvps(τ). (F.14)

F.3 Line broadening 479
The survival probability is therefore given by an exponential distribution,
ps(τ) = γpe−γpτ , (F.15)
where γp = nσv, and where the preceeding factor of γp ensures that the distribution
is correctly normalized.
We are now in a position to derive the form of a collision-broadened spectrum.
The spectral intensity profile of the emitted light is given by
I(ω) =∣∣∣E(ω)
∣∣∣2= Fτ
1√2π
∫E∗(t)E(t+ τ) dt
(ω), (F.16)
where E(ω) and E(t) = E0eiω0t+φ(t) are the spectral and temporal electric field
amplitudes of the light, and where in the second line we have used the convolution
theorem (see §D.3.5 in Appendix D). The convolution inside the curly braces is the
first order correlation function of E(t). In the absence of a collision, there is no
change to E, and this correlation function is constant. After a collision, the phase
φ is randomized, and when this is averaged over all atoms, the correlation function
vanishes. The average correlation function is thus proportional to the probability
that there is no collision in the interval [0, τ ]. Factorizing out the carrier frequency

F.3 Line broadening 480
ω0 using the shift theorem (see §D.3.4 in Appendix D), the spectrum is given by
I(δω) =|E0|2√
2π×Fτ
1−
∫ τ
0ps(τ ′)dτ ′
(δω)
=|E0|2√
2π×Fτ
e−γp|τ |
(δω)
=|E0|2
2π× γp
γ2p + δω2
, (F.17)
where in the second line the modulus sign indicates that the correlation function is
symmetric in time. (F.17) describes a Lorentzian lineshape with a width γp. Al-
though this spectrum was derived by considering emission, the absorption spectrum
is identical, since absorption is simply the time reverse of emission. An estimate of
the collision-broadened linewidth is given by
γp ≈ n(πd2atom)×
√2kBT
M. (F.18)
The number density n can be obtained from the vapour pressure as described in the
start of this Appendix. The term in brackets approximates the collision cross-section
σ as the area of a circle with radius given by the atomic width datom. The last factor
is found by setting the kinetic energy (F.10) equal to the thermal energy kBT and
solving for the average atomic velocity.
F.3.3 Power broadening
In the presence of an intense laser field, an atomic absorption line broadens. One
way to understand this effect is to consider that the presence of light at the atomic

F.4 Raman polarization 481
resonance frequency stimulates emission from the excited state, and this reduces
the lifetime of the state, which introduces a concomitant broadening. Alternatively,
consider that the atomic dynamics are governed by Hamiltonian evolution. In the
loosest possible sense, the resulting behaviour can be characterized as a mixing of all
the frequencies in the Hamiltonian. Thus if there exists an optical frequency ω and
a Rabi frequency Ω, there will be driven oscillations at frequencies of ω ± Ω. Such
oscillations may be resonant with the atomic transition at ω0, even when neither
frequency is separately. Therefore absorption is possible at large detunings, provided
the laser is sufficiently intense. The atomic transition is rendered broader by the laser
field. In fact, the power-broadened linewidth is indeed given by γpb = Ω, as a more
detailed derivation from the damped optical Bloch equations shows [107]. This effect
is essentially the same as the dynamic Stark effect that generates the instantaneous
frequency shift in (5.74) in Chapter 5, and the Autler-Townes splitting discussed in
§2.3.1 in Chapter 2.
F.4 Raman polarization
In Chapter 10 we describe the preliminary steps taken toward building a Raman
quantum memory in cesium vapour. Here we explain how we arrive at the conclusion
that the Stokes light scattered from a linearly polarized pump will be polarized
orthogonally to the pump, when far detuned.
We consider Raman scattering from the 6S1/2 ↔ 6P3/2 D2 line in cesium, at 852
nm. There are two hyperfine levels in the ground state, with F = 3 and F = 4.

F.4 Raman polarization 482
Atoms prepared in one of these levels are transferred by the Raman interaction to
the other level, with the transfer mediated by one of the excited states in the 6P3/2
manifold. In the process, a photon from the Raman pump is absorbed, and a Stokes
photon is emitted1. Each photon carries only one quantum of angular momentum,
so the intermediate state cannot differ in its angular momentum quantum number
by more than 1 from the initial and final states. Of the four hyperfine levels —
F = 2, 3, 4, 5 — in the excited state manifold, only the central two with F = 3, 4 are
compatible with this requirement, so we consider only these two intermediate levels.
Within each hyperfine level, there are 2F + 1 Zeeman sublevels, with magnetic
quantum numbers m = −F,−F+1, . . . , F−1, F . We specify one of these states with
the notation |F,m〉. The magnetic quantum numbers correspond to the component
of the atomic angular momentum along some axis, known as the quantization axis.
We must choose the direction for this axis before proceeding further. If a magnetic
field is applied to the atoms, the only sensible choice is to align the quantization
axis with the field. But in the absence of a magnetic field, the choice is arbitrary,
and we may choose the quantization direction in whichever way is convenient for
our calculations. As mentioned above, a single photon carries one unit of angular
momentum, and so it can change the magnetic quantum number by at most ±1, or
indeed not at all, if the spin of the photon has no component along the quantization1Strictly the term ‘Stokes’ refers to Raman scattered photons with less energy than the pump
photons. When they have more energy than the pump, they are commonly termed anti-Stokesphotons. In the present case, Stokes scattering only occurs if the atoms are prepared in the lower ofthe two ground states, with F = 3. The distinction between Stokes and anti-Stokes is not importantfor us, however. For simplicity, we always refer to the light emitted in Raman scattering as Stokeslight, regardless of whether or not its frequency is lower than that of the Raman pump.

F.4 Raman polarization 483
axis. Figure F.4 shows how the direction and polarization of a photon with respect to
the quantization axis is connected with the changes in m induced by its absorption.
quantization axis
Figure F.4 Polarization selection rules. Right and left circularpolarizations propagating along the quantization axis change m by+1 and −1; they are known as σ+ and σ− polarizations, respectively.It is intuitive that as the electric field follows a corkscrew trajectory, itapplies a torque to the atoms about the quantization axis, increasingor decreasing m appropriately. π polarized light does not change m.It is polarized linearly along the quantization axis and propagatesat right angles to it, so it cannot induce a turning moment. Theorthogonal linear polarization can, however, change m by either +1or −1.
We choose the quantization axis so that the linearly polarized Raman pump
is π-polarized. With this choice, absorption of a pump photon cannot change the
magnetic quantum number m. Given the initial state |Fi,mi〉 of an atom, this
narrows down the number of possible intermediate states to two: either |Fint =
3,mint = mi〉 or |Fint = 4,mint = mi〉 (the only exception is if mi = 0 initially. Then
F must change, so there is only one possible intermediate state, with Fint 6= Fi). The
atom now decays from the intermediate excited state into the ‘final’ ground hyperfine
level with Ff 6= Fi. There is no restriction on the polarization of the emitted Stokes
photon, so there are three possibilities for this latter part of the interaction. The

F.4 Raman polarization 484
possible final states are |Ff ,mf = mi〉 and |Ff ,mf = mi±1〉. As shown in Figure F.5,
each final state |Ff ,mf〉 can be reached from the given initial state |Fi,mi〉 by two
alternative ‘paths’. The first via the excited state with Fm = 3, and the second with
Fm = 4. When two different paths connect the same starting and ending points,
there is the possibility of interference. The present case is archetypical: π-polarized
Stokes emission is forbidden because of destructive interference between the two
interaction pathways. To see this, we explicitly evaluate the quantum mechanical
amplitude Aππ for π-polarized Stokes emission given a π-polarized pump, given the
initial state |Fi,mi〉,
Aππ(Fi,mi) =∑
Fm=3,4
d(J = 1/2, Fi,mi → J = 3/2, Fm,mm = mi)× (F.19)
d(J = 3/2, Fm,mm = mi → J = 1/2, Ff 6= Fi,mf = mi).
Here d denotes a dipole matrix element connecting the states with angular momen-
tum quantum numbers indicated by its arguments. Recall that J = 3/2 describes
the intermediate excited states. The selection rules set by fixing both photon polar-
izations to π keep the magnetic quantum number equal to its initial value throughout
the interaction. We have factorized out any dependence of the dipole matrix ele-
ments on the radial (or principle) quantum number. That this is always possible
is a consequence of the Wigner-Eckhart theorem. There exists a sophisticated and
confusing mathematical apparatus for dealing with the resulting ‘reduced’ matrix

F.4 Raman polarization 485
elements. In standard notation, we can write [184]
d(J, F,m→ J ′, F ′,m′) = (−1)F+J+I+1√
(2F + 1)(2J + 1)
J J ′ 1
F ′ F I
C1FF ′
(m′−m)mm′ ,
(F.20)
where the curly braces denote a Wigner 6j symbol, and where Cj1j2Jm1m2Mis a Clebsch-
Gordan coefficient. Here I = 7/2 is the cesium nuclear spin. The 1’s appearing
in various places represent the angular momentum of the photon involved in the
transition. Angular momentum conservation requires that the lower indices of the
Clebsch-Gordan coefficient obey the sum rule m1 +m2 = M . The symbols in (F.20)
can be evaluated by standard routines in Mathematica. Matlab routines have also
been written, and are available freely on the web. Performing the sum in (F.19)
using (F.20), we find that Aππ is identically zero for all choices of the initial state
(i.e. either Fi = 3 or 4, and any value of mi). No doubt there is a deep group-
theoretical reason for this, but I do not know what it is! Nonetheless the calculation
shows that Stokes scattered photons emerge with the orthogonal linear polarization
to that of the pump. A very fortunate outcome for the experimenter who wishes to
filter the weak Raman signal out from the bright pump.
In evaluating the scattering amplitude, we have given equal weight to both path-
ways. This implicitly assumes that the optical fields are sufficiently far detuned from
the excited state manifold that the small energy splitting between the intermediate
hyperfine levels — around 200 MHz — makes a negligible difference to the Raman
coupling. If instead we tune into resonance with one of the levels, we can neglect

F.4 Raman polarization 486
-3 -2 -1 0 1 2 3 -3 -2 -1 0 1 2 3
-3 -2 -1 0 1 2 3
-4 4
-3 -2 -1 0 1 2 3-4 4
Figure F.5 Alternative scattering pathways. Each initial state iscoupled by the Raman interaction to a final state via two possibleinteraction pathways; the first involves an intermediate excited statewith Fm = 3 (black-blue), the second with Fm = 4 (black-red). Thefigure illustrates this with the example of an atom initially preparedin the state Fi = 4,mi = 1. Absorption of a π-polarized Raman pumpphoton couples this state to the intermediate states |Fm = 3,mm = 1〉and |Fm = 4,mm = 1〉. Finally emission of a Stokes photon reducesthe magnetic quantum number by one: both paths end with the finalstate |Ff = 3,mf = 0〉.
scattering involving the other level, and there is no longer any interference. Isolating
just a single term from the sum in (F.19), we calculate a Stokes π-polarization of
40% on resonance with the Fm = 3 state, averaged over all possible initial states.
This averaging assumes an unpolarized ensemble, with atoms populating all Zeeman
substates in the initial hyperfine level equally. If we tune into resonance with the
Fm = 4 state, on average only 26% of the Stokes light is π-polarized. These calcu-
lations show that even on resonance, there remains a significant proportion of the
Stokes scattering that is polarized orthogonally to the pump, so that polarization
filtering is always feasible.
Note that these conclusions hold equally well for the polarizations of the the
signal and control fields in a cesium quantum memory, which has at its heart the very

F.4 Raman polarization 487
same Raman interaction. Therefore a vertically polarized signal field can be stored
by a horizontally polarized control pulse, when both are tuned far from resonance.
If we choose a circularly polarized Raman pump — this time aligning the quanti-
zation axis with the pump propagation direction for convenience — we find that, far
from resonance, all of the Stokes light is emitted into the same circular polarization
as the pump. That is, right-circular scatters into right-circular, and left-circular into
left-circular. This is rather unfortunate, because it precludes the use of what might
otherwise have been a rather ingenious ‘trick’2: Suppose that the atomic ensemble
is prepared in the the state |Fi = 4,mi = 4〉. Then a σ+-polarized photon can
only couple to a state with mm = 5, which lies within the Fm = 5 hyperfine level
in the excited state. This level cannot participate in the Raman interaction (see
the discussion at the start of this section), and so a σ+-polarized photon cannot
act as a Raman pump. If the ensemble is used as a quantum memory, then a σ+-
polarized control pulse cannot cause spontaneous Stokes scattering. This eliminates
the unwanted ‘noise’ process mentioned in §4.6 of Chapter 4 (see part (b) of Figure
4.3). Unfortunately, the signal photon must be σ−-polarized, and then the memory
coupling vanishes because quantum interference between the two possible scatter-
ing pathways destroys any coupling between orthogonal circular polarizations. This
explains why we make no attempt to spin-polarize our cesium ensemble: it is more
trouble than it is worth!
2This was suggested by Jean Dalibard when he came to Oxford to give a colloquium.

Bibliography
[1] CE Shannon. A mathematical theory of communication. ACM SIGMOBILEMobile Computing and Communications Review, 5(1):3–55, 2001.
[2] G.E. Moore. Cramming more components on to integrated circuits. ElectronicsJournal, 38(8):114–117, 1965.
[3] R.P. Feynman. Feynman Lectures on Computation. Da Capo Press, 1996.
[4] D. Deutsch. Quantum theory, the church-turing principle and the universalquantum computer. Proceedings of the Royal Society of London. Series A,Mathematical and Physical Sciences (1934-1990), 400(1818):97–117, 1985.
[5] Benjamin Schumacher. Quantum coding. Phys. Rev. A, 51(4):2738–2747, Apr1995.
[6] A. M. Steane. Error correcting codes in quantum theory. Phys. Rev. Lett.,77(5):793–797, Jul 1996.
[7] A. R. Calderbank and Peter W. Shor. Good quantum error-correcting codesexist. Phys. Rev. A, 54(2):1098–1105, Aug 1996.
[8] W.K. Wootters and W.H. Zurek. A single quantum cannot be cloned. Nature,299(5886):802–803, 1982.
[9] M.A. Nielsen and I.L. Chuang. Quantum Computation and Quantum Infor-mation. Cambridge University Press, 2000.
[10] A. Steane. The ion trap quantum information processor. Applied Physics B:Lasers and Optics, 64(6):623–643, 1997.
[11] M. V. Gurudev Dutt, L. Childress, L. Jiang, E. Togan, J. Maze, F. Jelezko,A. S. Zibrov, P. R. Hemmer, and M. D. Lukin. Quantum register based on indi-vidual electronic and nuclear spin qubits in diamond. Science, 316(5829):1312–1316, 2007.
[12] L. Childress, MV Gurudev Dutt, JM Taylor, AS Zibrov, F. Jelezko,J. Wrachtrup, PR Hemmer, and MD Lukin. Coherent Dynamics of CoupledElectron and Nuclear Spin Qubits in Diamond, 2006.

BIBLIOGRAPHY 489
[13] E. Knill, R. Laflamme, and G. J. Milburn. A scheme for efficient quantumcomputation with linear optics. Nature, 409(6816):46–52, 2001.
[14] N. Yoran and B. Reznik. Deterministic linear optics quantum computationwith single photon qubits. Phys. Rev. Lett., 91(3):037903, Jul 2003.
[15] Michael A. Nielsen. Optical quantum computation using cluster states. Phys-ical Review Letters, 93(4):040503, 2004.
[16] J. M. Raimond, M. Brune, and S. Haroche. Manipulating quantum entangle-ment with atoms and photons in a cavity. Rev. Mod. Phys., 73(3):565–582,Aug 2001.
[17] Karl Tordrup, Antonio Negretti, and Klaus Molmer. Holographic quantumcomputing. Physical Review Letters, 101(4):040501, 2008.
[18] C.A. Muschik, I. de Vega, D. Porras, and J.I. Cirac. Quantum processingphotonic states in optical lattices. Arxiv preprint quant-ph/0611093, 2006.
[19] S. D. Barrett, P. P. Rohde, and T. M. Stace. Scalable quantum computingwith atomic ensembles, 2008.
[20] N. Gisin, G. Ribordy, W. Tittel, and H. Zbinden. Quantum cryptography.Reviews of Modern Physics, 74(1):145–195, 2002.
[21] RL Rivest, A. Shamir, and L. Adleman. A method for obtaining digital signa-tures and public-key cryptosystems. Communications of the ACM, 21(2):120–126, 1978.
[22] M.A. Nielsen and I.L. Chuang. Quantum Computation and Quantum Infor-mation. Cambridge University Press, 2000.
[23] P.W. Shor. Algorithms for quantum computation: Discrete logarithms andfactoring. Proceedings of the 35th Annual Symposium on Foundations of Com-puter Science, 124, 1994.
[24] C.Y. Lu, D.E. Browne, T. Yang, and J.W. Pan. Demonstration ofShor’s quantum factoring algorithm using photonic qubits. Arxiv preprintarXiv:0705.1684, 2007.
[25] BP Lanyon, TJ Weinhold, NK Langford, M. Barbieri, DFV James,A. Gilchrist, and AG White. Experimental Demonstration of a CompiledVersion of Shor’s Algorithm with Quantum Entanglement. Physical ReviewLetters, 99(25):250505, 2007.
[26] L.M.K. Vandersypen, M. Steffen, G. Breyta, C.S. Yannoni, R. Cleve, and I.L.Chuang. Experimental Realization of an Order-Finding Algorithm with anNMR Quantum Computer. Physical Review Letters, 85(25):5452–5455, 2000.

BIBLIOGRAPHY 490
[27] C.H. Bennett, G. Brassard, et al. Quantum cryptography: Public key dis-tribution and coin tossing. Proceedings of IEEE International Conference onComputers, Systems, and Signal Processing, 175, 1984.
[28] W. Heisenberg. The Physical Principles of the Quantum Theory. CourierDover Publications, 1949.
[29] JS Bell. On the Einstein-Podolsky-Rosen paradox. Physics (US) Discontinuedwith Vol. 4, no. 1, 1968, 1, 1964.
[30] Artur K. Ekert. Quantum cryptography based on bell’s theorem. Phys. Rev.Lett., 67(6):661–663, Aug 1991.
[31] Jian-Wei Pan, Dik Bouwmeester, Harald Weinfurter, and Anton Zeilinger. Ex-perimental entanglement swapping: Entangling photons that never interacted.Phys. Rev. Lett., 80(18):3891–3894, May 1998.
[32] D. Deutsch, A. Ekert, R. Jozsa, C. Macchiavello, S. Popescu, and A. Sanpera.Quantum Privacy Amplification and the Security of Quantum Cryptographyover Noisy Channels. Physical Review Letters, 77(13):2818–2821, 1996.
[33] W. Dur, H.-J. Briegel, J. I. Cirac, and P. Zoller. Quantum repeaters based onentanglement purification. Phys. Rev. A, 59(1):169–181, Jan 1999.
[34] Charles H. Bennett, Herbert J. Bernstein, Sandu Popescu, and Benjamin Schu-macher. Concentrating partial entanglement by local operations. Phys. Rev.A, 53(4):2046–2052, Apr 1996.
[35] Jian-Wei Pan, Christoph Simon, Caslav Brukner, and Anton Zeilinger. En-tanglement purification for quantum communication. Nature, 410(6832):1067–1070, 2001.
[36] Christoph Simon and Jian-Wei Pan. Polarization entanglement purificationusing spatial entanglement. Phys. Rev. Lett., 89(25):257901, Dec 2002.
[37] L.M. Duan, MD Lukin, JI Cirac, and P. Zoller. Long-distance quantum com-munication with atomic ensembles and linear optics. Nature, 414:413–418,2001.
[38] Nicolas Sangouard, Christoph Simon, Bo Zhao, Yu-Ao Chen, Hugues de Ried-matten, Jian-Wei Pan, and Nicolas Gisin. Robust and efficient quantum re-peaters with atomic ensembles and linear optics. Physical Review A (Atomic,Molecular, and Optical Physics), 77(6):062301, 2008.
[39] L. Jiang, J. M. Taylor, and M. D. Lukin. Fast and robust approach to long-distance quantum communication with atomic ensembles. Physical Review A(Atomic, Molecular, and Optical Physics), 76(1):012301, 2007.

BIBLIOGRAPHY 491
[40] Zeng-Bing Chen, Bo Zhao, Yu-Ao Chen, Jorg Schmiedmayer, and Jian-WeiPan. Fault-tolerant quantum repeater with atomic ensembles and linear optics.Physical Review A (Atomic, Molecular, and Optical Physics), 76(2):022329,2007.
[41] Nicolas Sangouard, Christoph Simon, Jiri Minar, Hugo Zbinden, Huguesde Riedmatten, and Nicolas Gisin. Long-distance entanglement distributionwith single-photon sources. Physical Review A (Atomic, Molecular, and Opti-cal Physics), 76(5):050301, 2007.
[42] Bo Zhao, Zeng-Bing Chen, Yu-Ao Chen, Jorg Schmiedmayer, and Jian-WeiPan. Robust creation of entanglement between remote memory qubits. Phys-ical Review Letters, 98(24):240502, 2007.
[43] L.M. Duan, JI Cirac, and P. Zoller. Three-dimensional theory for inter-action between atomic ensembles and free-space light. Physical Review A,66(2):23818, 2002.
[44] Peter J. Mosley, Jeff S. Lundeen, Brian J. Smith, Piotr Wasylczyk, Alfred B.U’Ren, Christine Silberhorn, and Ian A. Walmsley. Heralded generation ofultrafast single photons in pure quantum states. Physical Review Letters,100(13):133601, 2008.
[45] Axel Kuhn, Markus Hennrich, and Gerhard Rempe. Deterministic single-photon source for distributed quantum networking. Phys. Rev. Lett.,89(6):067901, Jul 2002.
[46] Christian Kurtsiefer, Sonja Mayer, Patrick Zarda, and Harald Weinfurter. Sta-ble solid-state source of single photons. Phys. Rev. Lett., 85(2):290–293, Jul2000.
[47] A. Beveratos, S. Kuhn, R. Brouri, T. Gacoin, J. P. Poizat, and P. Grang-ier. Room temperature stable single-photon source. The European PhysicalJournal D - Atomic, Molecular, Optical and Plasma Physics, 18(2):191–196,2002.
[48] I. I. Rabi, J. R. Zacharias, S. Millman, and P. Kusch. A new method ofmeasuring nuclear magnetic moment. Phys. Rev., 53(4):318, Feb 1938.
[49] H. Mabuchi and A. C. Doherty. Cavity quantum electrodynamics: Coherencein context. Science, 298(5597):1372–1377, 2002.
[50] P. Rabl, D. DeMille, J. M. Doyle, M. D. Lukin, R. J. Schoelkopf, and P. Zoller.Hybrid quantum processors: Molecular ensembles as quantum memory forsolid state circuits. Physical Review Letters, 97(3):033003, 2006.
[51] J. I. Cirac, P. Zoller, H. J. Kimble, and H. Mabuchi. Quantum state transferand entanglement distribution among distant nodes in a quantum network.Phys. Rev. Lett., 78(16):3221–3224, Apr 1997.

BIBLIOGRAPHY 492
[52] M. Hennrich, T. Legero, A. Kuhn, and G. Rempe. Vacuum-stimulated ramanscattering based on adiabatic passage in a high-finesse optical cavity. Phys.Rev. Lett., 85(23):4872–4875, Dec 2000.
[53] EM Purcell. Spontaneous emission probabilities at radio frequencies. Phys.Rev, 69(681):166, 1946.
[54] AD Boozer, A. Boca, R. Miller, TE Northup, and HJ Kimble. Reversible StateTransfer between Light and a Single Trapped Atom. Physical Review Letters,98(19):193601, 2007.
[55] J. Bochmann, M. Muecke, G. Langfahl-Klabes, C. Erbel, B. Weber, H. P.Specht, D. L. Moehring, and G. Rempe. Fast Excitation and Photon Emissionof a Single-Atom-Cavity System. Phys. Rev. Lett., 67(13):1727–1730, Sep 2008.
[56] J. P. Reithmaier, G. Sek, A. Loffler, C. Hofmann, S. Kuhn, S. Reitzenstein,L. V. Keldysh, V. D. Kulakovskii, T. L. Reinecke, and A. Forchel. Strongcoupling in a single quantum dot-semiconductor microcavity system. Nature,432(7014):197–200, 2004.
[57] A. Kiraz, P. Michler, C. Becher, B. Gayral, A. Imamoglu, L. Zhang, E. Hu,WV Schoenfeld, and PM Petroff. Cavity-quantum electrodynamics using asingle InAs quantum dot in a microdisk structure. Applied Physics Letters,78:3932, 2001.
[58] A. ImamogZlu, DD Awschalom, G. Burkard, DP DiVincenzo, D. Loss,M. Sherwin, and A. Small. Quantum Information Processing Using Quan-tum Dot Spins and Cavity QED. Physical Review Letters, 83(20):4204–4207,1999.
[59] R.J. Young, S.J. Dewhurst, R.M. Stevenson, P. Atkinson, A.J. Bennett, M.B.Ward, K. Cooper, D.A. Ritchie, and A.J. Shields. Single electron-spin memorywith a semiconductor quantum dot. Arxiv preprint arXiv:0706.2143, 2007.
[60] A. Andre, D. Demille, JM Doyle, MD Lukin, SE Maxwell, P. Rabl,RJ Schoelkopf, and P. Zoller. A coherent all-electrical interface between po-lar molecules and mesoscopic superconducting resonators. Nature Physics,2(9):636–642, 2006.
[61] M.K. Tey, S.A. Aljunid, F. Huber, B. Chng, Z. Chen, G. Maslennikov, andC. Kurtsiefer. Interfacing light and single atoms with a lens. Arxiv preprintarXiv:0804.4861, 2008.
[62] M.K. Tey, Z. Chen, S.A. Aljunid, B. Chng, F. Huber, G. Maslennikov, andC. Kurtsiefer. Strong interaction between light and a single trapped atomwithout a cavity. Arxiv preprint arXiv:0802.3005, 2008.

BIBLIOGRAPHY 493
[63] M. Sondermann, R. Maiwald, H. Konermann, N. Lindlein, U. Peschel, andG. Leuchs. Design of a mode converter for efficient light-atom coupling in freespace. Applied Physics B: Lasers and Optics, 89(4):489–492, 2007.
[64] K.-J. Boller, A. Imamolu, and S. E. Harris. Observation of electromagneticallyinduced transparency. Phys. Rev. Lett., 66(20):2593–2596, May 1991.
[65] Michael Fleischhauer, Atac Imamoglu, and Jonathan P. Marangos. Electro-magnetically induced transparency: Optics in coherent media. Reviews ofModern Physics, 77(2):633, 2005.
[66] J. H. Eberly, M. L. Pons, and H. R. Haq. Dressed-field pulses in an absorbingmedium. Phys. Rev. Lett., 72(1):56–59, Jan 1994.
[67] S. H. Autler and C. H. Townes. Stark effect in rapidly varying fields. Phys.Rev., 100(2):703–722, Oct 1955.
[68] Lene Vestergaard Hau, S. E. Harris, Zachary Dutton, and Cyrus H. Behroozi.Light speed reduction to 17 metres per second in an ultracold atomic gas.Nature, 397(6720):594–598, 1999.
[69] M. Fleischhauer and M. D. Lukin. Dark-state polaritons in electromagneticallyinduced transparency. Phys. Rev. Lett., 84(22):5094–5097, May 2000.
[70] Chien Liu, Zachary Dutton, Cyrus H. Behroozi, and Lene Vestergaard Hau.Observation of coherent optical information storage in an atomic medium usinghalted light pulses. Nature, 409(6819):490–493, 2001.
[71] K. S. Choi, H. Deng, J. Laurat, and H. J. Kimble. Mapping photonic en-tanglement into and out of a quantum memory. Nature, 452:67–71, March2008.
[72] Julien Laurat, Chin wen Chou, Hui Deng, Kyung Soo Choi, Daniel Felinto,Hugues de Riedmatten, and H J Kimble. Towards experimental entanglementconnection with atomic ensembles in the single excitation regime. New Journalof Physics, 9(6):207, 2007.
[73] T. Chaneliere, D. N. Matsukevich, S. D. Jenkins, S.-Y. Lan, R. Zhao, T. A. B.Kennedy, and A. Kuzmich. Quantum interference of electromagnetic fieldsfrom remote quantum memories. Physical Review Letters, 98(11):113602, 2007.
[74] Irina Novikova, Nathaniel B. Phillips, and Alexey V. Gorshkov. Optimal lightstorage with full pulse shape control, 2008.
[75] CV Raman and KS Krishnan. A new type of secondary radiation. Nature,121(3048):501, 1928.
[76] A. E. Kozhekin, K. Mølmer, and E. Polzik. Quantum memory for light. Phys.Rev. A, 62:033809, 2000.

BIBLIOGRAPHY 494
[77] J. Nunn, I. A. Walmsley, M. G. Raymer, K. Surmacz, F. C. Waldermann,Z. Wang, and D. Jaksch. Mapping broadband single-photon wave packetsinto an atomic memory. Physical Review A (Atomic, Molecular, and OpticalPhysics), 75(1):011401, 2007.
[78] Mattias Nilsson and Stefan Krll. Solid state quantum memory using completeabsorption and re-emission of photons by tailored and externally controlledinhomogeneous absorption profiles. Optics Communications, 247(4-6):393–403, 2005.
[79] S. A. Moiseev and S. Kroll. Complete reconstruction of the quantum state of asingle-photon wave packet absorbed by a doppler-broadened transition. Phys.Rev. Lett., 87(17):173601, Oct 2001.
[80] SA Moiseev, VF Tarasov, and BS Ham. Quantum memory photon echo-liketechniques in solids. Journal of Optics B: Quantum and Semiclassical Optics,5(4):S497–S502, 2003.
[81] S. A. Moiseev and B. S. Ham. Photon-echo quantum memory with efficientmultipulse readings. Phys. Rev. A, 70(6):063809, Dec 2004.
[82] B. Kraus, W. Tittel, N. Gisin, M. Nilsson, S. Kroll, and J. I. Cirac. Quan-tum memory for nonstationary light fields based on controlled reversible inho-mogeneous broadening. Physical Review A (Atomic, Molecular, and OpticalPhysics), 73(2):020302, 2006.
[83] AL Alexander, JJ Longdell, MJ Sellars, and NB Manson. Photon EchoesProduced by Switching Electric Fields. Physical Review Letters, 96(4):43602,2006.
[84] G. Hetet, JJ Longdell, AL Alexander, PK Lam, and MJ Sellars. Electro-OpticQuantum Memory for Light Using Two-Level Atoms. Physical Review Letters,100(2):23601, 2008.
[85] M. U. Staudt, S. R. Hastings-Simon, M. Nilsson, M. Afzelius, V. Scarani,R. Ricken, H. Suche, W. Sohler, W. Tittel, and N. Gisin. Fidelity of anoptical memory based on stimulated photon echoes. Physical Review Letters,98(11):113601, 2007.
[86] E. Desurvire, C.R. Giles, and J.L. Zyskind. Erbium-doped fiber amplifier,June 25 1991. US Patent 5,027,079.
[87] Nicklas Ohlsson, R. Krishna Mohan, and Stefan Krll. Quantum computerhardware based on rare-earth-ion-doped inorganic crystals. Optics Communi-cations, 201(1-3):71–77, 2002.
[88] E. Fraval, M. J. Sellars, and J. J. Longdell. Dynamic decoherence control ofa solid-state nuclear-quadrupole qubit. Physical Review Letters, 95(3):030506,2005.

BIBLIOGRAPHY 495
[89] B. R. Judd. Optical absorption intensities of rare-earth ions. Phys. Rev.,127(3):750–761, Aug 1962.
[90] G. Hetet, JJ Longdell, MJ Sellars, PK Lam, and BC Buchler. Bandwidth andDynamics of the Gradient Echo Memory. Arxiv preprint arXiv:0801.3860,2008.
[91] Mikael Afzelius, Christoph Simon, Hugues de Riedmatten, and Nicolas Gisin.Multi-Mode Quantum Memory based on Atomic Frequency Combs, 2008.
[92] Luıs E. E. de Araujo, Ian A. Walmsley, and C. R. Stroud. Analytic solutionfor strong-field quantum control of atomic wave packets. Phys. Rev. Lett.,81(5):955–958, Aug 1998.
[93] L. E. E. de Araujo and I. A. Walmsley. Quantum control of molecularwavepackets: An approximate analytic solution for the strong-response regime.Journal of Physical Chemistry A, 103(49):10409–10416, 1999.
[94] Luıs E. E. de Araujo and Ian A. Walmsley. Quantum control of rydberg wavepackets in the strong-response regime. Phys. Rev. A, 63(2):023401, Jan 2001.
[95] L.E.E. de Araujo and I.A. Walmsley. Analytic solution for quantum control ofatomic and molecular wavepackets. JOURNAL OF OPTICS B QUANTUMAND SEMICLASSICAL OPTICS, 5(1):27–42, 2003.
[96] J. Oreg, G. Hazak, and JH Eberly. Multilevel inversion schemes in and beyondthe adiabatic limit. Physical Review A, 32(5):2776–2783, 1985.
[97] S. Guerin, S. Thomas, and H. R. Jauslin. Optimization of population transferby adiabatic passage. Phys. Rev. A, 65(2):023409, Jan 2002.
[98] B. W. Shore, K. Bergmann, A. Kuhn, S. Schiemann, J. Oreg, and J. H. Eberly.Laser-induced population transfer in multistate systems: A comparative study.Phys. Rev. A, 45(7):5297–5300, Apr 1992.
[99] B. Lauritzen, S. R. Hastings-Simon, H. de Riedmatten, M. Afzelius, andN. Gisin. State Preparation by Optical Pumping in Erbium Doped Solids us-ing Stimulated Emission and Spin Mixing. Messenger of Mathematics, 19:1–5,2008.
[100] Hugues de Riedmatten, Mikael Afzelius, Matthias U. Staudt, Christoph Simon,and Nicolas Gisin. A solid-state light-matter interface at the single-photonlevel. Nature, 456(7223):773–777, 12 2008.
[101] S.L. Braunstein and P. van Loock. Quantum information with continuousvariables. Reviews of Modern Physics, 77(2):513–577, 2005.
[102] Samuel L. Braunstein and Peter van Loock. Quantum information with con-tinuous variables. Reviews of Modern Physics, 77(2):513, 2005.

BIBLIOGRAPHY 496
[103] T. C. Ralph. Continuous variable quantum cryptography. Phys. Rev. A,61(1):010303, Dec 1999.
[104] C. Silberhorn, TC Ralph, N. Lutkenhaus, and G. Leuchs. Continuous Vari-able Quantum Cryptography: Beating the 3 dB Loss Limit. Physical ReviewLetters, 89(16):167901, 2002.
[105] Stefano Pirandola, Stefano Mancini, Seth Lloyd, and Samuel L. Braunstein.Continuous-variable quantum cryptography using two-way quantum commu-nication. Nat Phys, advanced online publication:–, 2008.
[106] Lijian Zhang, Christine Silberhorn, and Ian A. Walmsley. Secure quantumkey distribution using continuous variables of single photons. Physical ReviewLetters, 100(11):110504, 2008.
[107] Rodney Loudon. The Quantum Theory of Light. Oxford University Press,2004.
[108] E. Wigner. On the quantum correction for thermodynamic equilibrium. Phys.Rev., 40(5):749–759, Jun 1932.
[109] T. Hirano, K. Kotani, T. Ishibashi, S. Okude, and T. Kuwamoto. 3 dB squeez-ing by single-pass parametric amplification in a periodically poled KTiOPO4crystal. Optics Letters, 30(13):1722–1724, 2005.
[110] RJ Senior, GN Milford, J. Janousek, AE Dunlop, K. Wagner, H.A. Bachor,TC Ralph, EH Huntington, and CC Harb. Observation of a comb of opticalsqueezing over many gigahertz of bandwidth. Optics Express, 15(9):5310–5317,2007.
[111] Henning Vahlbruch, Moritz Mehmet, Simon Chelkowski, Boris Hage, Alexan-der Franzen, Nico Lastzka, Stefan Gossler, Karsten Danzmann, and RomanSchnabel. Observation of squeezed light with 10-db quantum-noise reduction.Physical Review Letters, 100(3):033602, 2008.
[112] Y. Eto, T. Tajima, Y. Zhang, and T. Hirano. Observation of quadraturesqueezing in a?ˆ(2) nonlinear waveguide using a temporally shaped local os-cillator pulse. Optics Express, 16(14):10650–16657, 2008.
[113] Alex Abramovici, William E. Althouse, Ronald W. P. Drever, Yekta Gursel,Seiji Kawamura, Frederick J. Raab, David Shoemaker, Lisa Sievers, Robert E.Spero, Kip S. Thorne, Rochus E. Vogt, Rainer Weiss, Stanley E. Whitcomb,and Michael E. Zucker. Ligo: The laser interferometer gravitational-waveobservatory. Science, 256(5055):325–333, 1992.
[114] Carlton M. Caves. Quantum-mechanical noise in an interferometer. Phys.Rev. D, 23(8):1693–1708, Apr 1981.

BIBLIOGRAPHY 497
[115] H. Yuen and J. Shapiro. Optical communication with two-photon coherentstates–part i: Quantum-state propagation and quantum-noise. InformationTheory, IEEE Transactions on, 24(6):657–668, Nov 1978.
[116] T. Fernholz, H. Krauter, K. Jensen, J. F. Sherson, A. S. Sorensen, and E. S.Polzik. Spin squeezing of atomic ensembles via nuclear-electronic spin entan-glement. Physical Review Letters, 101(7):073601, 2008.
[117] Vivi Petersen, Lars Bojer Madsen, and Klaus Molmer. Magnetometry withentangled atomic samples. Physical Review A (Atomic, Molecular, and OpticalPhysics), 71(1):012312, 2005.
[118] D. Leibfried, M. D. Barrett, T. Schaetz, J. Britton, J. Chiaverini, W. M.Itano, J. D. Jost, C. Langer, and D. J. Wineland. Toward heisenberg-limitedspectroscopy with multiparticle entangled states. Science, 304(5676):1476–1478, 2004.
[119] C. F. Roos, M. Chwalla, K. Kim, M. Riebe, and R. Blatt. /‘designer atoms/’for quantum metrology. Nature, 443(7109):316–319, 2006.
[120] P. J. Windpassinger, D. Oblak, P. G. Petrov, M. Kubasik, M. Saffman,C. L. Garrido Alzar, J. Appel, J. H. Muller, N. Kjaergaard, and E. S. Polzik.Nondestructive probing of rabi oscillations on the cesium clock transition nearthe standard quantum limit. Physical Review Letters, 100(10):103601, 2008.
[121] Masahiro Kitagawa and Masahito Ueda. Nonlinear-interferometric generationof number-phase-correlated fermion states. Phys. Rev. Lett., 67(14):1852–1854,Sep 1991.
[122] Brian Julsgaard, Jacob Sherson, J. Ignacio Cirac, Jaromir Fiurasek, and Eu-gene S. Polzik. Experimental demonstration of quantum memory for light.Nature, 432(7016):482–486, 2004.
[123] K. Hammerer, A.S. Sorensen, and E.S. Polzik. Quantum interface betweenlight and atomic ensembles. Not yet published, 2008.
[124] Jun Liu, Songcan Chen, and Xiaoyang Tan. Fractional order singular valuedecomposition representation for face recognition. Pattern Recogn., 41(1):378–395, 2008.
[125] T. N. Palmer, R. Buizza, F. Molteni, Y.-Q. Chen, and S. Corti. Singular vec-tors and the predictability of weather and climate. Philosophical Transactions:Physical Sciences and Engineering, 348(1688):459–475, 1994.
[126] Guifre Vidal. Entanglement of pure states for a single copy. Phys. Rev. Lett.,83(5):1046–1049, Aug 1999.
[127] E. Beltrami. Sulle funzioni bilineari. Giornale di Matematiche ad Uso degliStudenti Delle Universita, 11:98–106, 1873.

BIBLIOGRAPHY 498
[128] C. Jordan. Sur la reduction des formes bilineaires. Comptes Rendus del’Academie des Sciences, Paris, 78:614–617, 1874.
[129] JJ Sylvester. A new proof that a general quadric may be reduced to its canon-ical form (that is, a linear function of squares) by means of a real orthogonalsubstitution. Messenger of Mathematics, 19:1–5, 1889.
[130] E. Schmidt. Zur Theorie Der Linearen Und Nichtlinearen Integralgleichungen.I Teil: Entwicklung WillkuLicher Funktion Nach Systemen Vorgeschriebener.Mathenatische Annalen, 63:433–476, 1907.
[131] H. Bateman. A formula for the solving function of a certain integral equation ofthe second kind. Transactions of the Cambridge Philosophical Society, 20:179–187, 1908.
[132] C. Eckart and G. Young. The approximation of one matrix by another one oflower rank. Psychometrika, 1:211–218, 1936.
[133] Alexey V. Gorshkov, Axel Andre, Mikhail D. Lukin, and Anders S. Sorensen.Photon storage in lambda-type optically dense atomic media. ii. free-spacemodel. Physical Review A, 76:033805, 2007.
[134] J.M. Supplee. Optical Bloch equations: Informal motivation without theSchrodinger equation. American Journal of Physics, 68:180, 2000.
[135] T.H. Boyer. Electrostatic potential energy leading to a gravitational masschange for a system of two point charges. American Journal of Physics,47(2):129–31, 1979.
[136] B.R. Holstein and A.R. Swift. Elementary derivation of the radiation fieldfrom an accelerated charge. American Journal of Physics, 49:346, 1981.
[137] R.P. Feynman, R.B. Leighton, M. Sands, and EM Hafner. The FeynmanLectures on Physics; Vol. I. American Journal of Physics, 33:750, 1965.
[138] A. Kalachev. Quantum storage on subradiant states in an extended atomicensemble. Physical Review A, 76(4):43812, 2007.
[139] M. G. Raymer and J. Mostowski. Stimulated Raman scattering: Unifiedtreatment of spontaneous initiation and spatial propagation. Phys. Rev. A,24(4):1980–1993, 1981.
[140] Alexey V. Gorshkov, Axel Andre, Mikhail D. Lukin, and Anders S. Sorensen.Photon storage in lambda-type optically dense atomic media. ii. free-spacemodel. Physical Review A (Atomic, Molecular, and Optical Physics),76(3):033805, 2007.
[141] R.W. Boyd. Nonlinear Optics. Academic Press, 2003.

BIBLIOGRAPHY 499
[142] N. Rosen and C. Zener. Double Stern-Gerlach Experiment and Related Colli-sion Phenomena. Physical Review, 40(4):502–507, 1932.
[143] P. Pechukas and J.C. Light. On the Exponential Form of Time-DisplacementOperators in Quantum Mechanics. The Journal of Chemical Physics, 44:3897,2004.
[144] RT Robiscoe. Extension of the Rosen-Zener solution to the two-level problem.Physical Review A, 17(1):247–260, 1978.
[145] S. Klarsfeld and JA Oteo. Magnus approximation in the adiabatic picture.Physical Review A, 45(5):3329–3332, 1992.
[146] WR Salzman. An alternative to the magnus expansion in time-dependentperturbation theory. The Journal of Chemical Physics, 82:822, 1985.
[147] Samuel L. Braunstein. Squeezing as an irreducible resource. Physical ReviewA (Atomic, Molecular, and Optical Physics), 71(5):055801, 2005.
[148] W. Wasilewski and MG Raymer. Pairwise entanglement and readout ofatomic-ensemble and optical wave-packet modes in traveling-wave Raman in-teractions. Physical Review A, 73(6):63816, 2006.
[149] W. Wasilewski, AI Lvovsky, K. Banaszek, and C. Radzewicz. Pulsedsqueezed light: Simultaneous squeezing of multiple modes. Physical ReviewA, 73(6):63819, 2006.
[150] Irina Novikova, Alexey V. Gorshkov, David F. Phillips, Anders S. Sorensen,Mikhail D. Lukin, and Ronald L. Walsworth. Optimal control of light pulsestorage and retrieval. Physical Review Letters, 98(24):243602, 2007.
[151] Nathaniel B. Phillips, Alexey V. Gorshkov, and Irina Novikova. Optimal lightstorage in atomic vapor, 2008.
[152] K. Surmacz, J. Nunn, K. Reim, K. C. Lee, V. O. Lorenz, B. Sussman,I. A. Walmsley, and D. Jaksch. Efficient spatially resolved multimode quan-tum memory. Physical Review A (Atomic, Molecular, and Optical Physics),78(3):033806, 2008.
[153] C. Simon, H. de Riedmatten, M. Afzelius, N. Sangouard, H. Zbinden, andN. Gisin. Quantum repeaters with photon pair sources and multimode mem-ories. Phys. Rev. Lett., 98:190503, 2007.
[154] O. A. Collins, S. D. Jenkins, A. Kuzmich, and T. A. B. Kennedy. Multiplexedmemory-insensitive quantum repeaters. Physical Review Letters, 98(6):060502,2007.

BIBLIOGRAPHY 500
[155] J. Nunn, K. Reim, K. C. Lee, V. O. Lorenz, B. J. Sussman, I. A. Walmsley,and D. Jaksch. Multimode memories in atomic ensembles. Physical ReviewLetters, 101(26):260502, 2008.
[156] J. H. Eberly. Schmidt Analysis of Pure-State Entanglement. ArXiv QuantumPhysics e-prints, August 2005.
[157] R. Grobe, K. Rzazewski, and JH Eberly. Measure of electron-electron correla-tion in atomic physics. Journal of Physics B: Atomic, Molecular, and OpticalPhysics, 27(16):L503–L508, 1994.
[158] M. A. Nielsen and I. L. Chuang. Quantum Computation and Quantum Infor-mation. Cambridge University Press, 2004.
[159] G.N. Watson. A Treatise on the Theory of Bessel Functions. CambridgeUniversity Press, 1995.
[160] Nicolas Sangouard, Christoph Simon, Mikael Afzelius, and Nicolas Gisin.Analysis of a quantum memory for photons based on controlled reversibleinhomogeneous broadening. Physical Review A, 75:032327, 2007.
[161] JJ Longdell, G. Hetet, MJ Sellars, and PK Lam. Analytic treatment ofCRIB Quantum Memories for Light using Two-level Atoms. Arxiv preprintarXiv:0807.0067, 2008.
[162] G. Hetet, J. J. Longdell, M. J. Sellars, P. K. Lam, and B. C. Buchler. Multi-modal properties and dynamics of gradient echo quantum memory. PhysicalReview Letters, 101(20):203601, 2008.
[163] Alexey V. Gorshkov, Axel Andre, Mikhail D. Lukin, and Anders S. Sorensen.Photon storage in lambda-type optically dense atomic media. iii. effects ofinhomogeneous broadening. Physical Review A, 76:033806, 2007.
[164] G. Hetet, M. Hosseini, B. M. Sparkes, D. Oblak, P. K. Lam, and B. C. Buchler.Photon echoes generated by reversing magnetic field gradients in a rubidiumvapor. Opt. Lett., 33(20):2323–2325, 2008.
[165] S.A. Moiseev and W. Tittel. Raman-Echo Quantum Memory. 2008.
[166] Alexey V. Gorshkov, Tommaso Calarco, Mikhail D. Lukin, and Anders S.Sorensen. Photon storage in lambda-type optically dense atomic media. iv.optimal control using gradient ascent. Physical Review A (Atomic, Molecular,and Optical Physics), 77(4):043806, 2008.
[167] C. Kittel and P. McEuen. Introduction to solid state physics. Wiley New York,1986.
[168] PG Klemens. Anharmonic Decay of Optical Phonons. Physical Review,148(2):845–848, 1966.

BIBLIOGRAPHY 501
[169] A. Debernardi, S. Baroni, and E. Molinari. Anharmonic Phonon Lifetimesin Semiconductors from Density-Functional Perturbation Theory. PhysicalReview Letters, 75(9):1819–1822, 1995.
[170] M.S. Liu, L.A. Bursill, S. Prawer, and R. Beserman. Temperature depen-dence of the first-order Raman phonon line of diamond. Physical Review B,61(5):3391–3395, 2000.
[171] FC Waldermann, B.J. Sussman, J. Nunn, VO Lorenz, KC Lee, K. Surmacz,KH Lee, D. Jaksch, IA Walmsley, P. Spizziri, et al. Measuring phonon dephas-ing with ultrafast pulses using Raman spectral interference. Physical ReviewB, 78(15), 2008.
[172] A. Cantarero, C. Trallero-Giner, and M. Cardona. Excitons in one-phononresonant raman scattering: Deformation-potential interaction. Phys. Rev. B,39(12):8388–8397, Apr 1989.
[173] W. Potz and P. Vogl. Theory of optical-phonon deformation potentials intetrahedral semiconductors. Physical Review B, 24(4):2025–2037, 1981.
[174] GD Whitfield. Theory of Electron-Phonon Interactions. Physical Review Let-ters, 2(5):204–205, 1959.
[175] H. Frohlich. Interaction of electrons with lattice vibrations. Proceedings ofthe Royal Society of London. Series A, Mathematical and Physical Sciences(1934-1990), 215(1122):291–298, 12 1952.
[176] Kun Huang. On the interaction between the radiation field and ionic crys-tals. Proceedings of the Royal Society of London. Series A, Mathematical andPhysical Sciences (1934-1990), 208(1094):352–365, 09 1951.
[177] K. Rzazewski and RW Boyd. Equivalence of interaction Hamiltonians in theelectric dipole approximation. Journal of Modern Optics, 51(8):1137–1147,2004.
[178] M. O. Scully and M. S. Zubairy. Quantum Optics. Cambridge UniversityPress, 1997.
[179] R. P. Feynman. Forces in molecules. Phys. Rev., 56(4):340–343, Aug 1939.
[180] W. Hayes, R. Loudon, and J.F. Scott. Scattering of Light by Crystals. Amer-ican Journal of Physics, 47:571, 1979.
[181] R. Loudon. Theory of the First-Order Raman Effect in Crystals. Proceedingsof the Royal Society of London. Series A, Mathematical and Physical Sciences(1934-1990), 275(1361):218–232, 1963.
[182] R. Loudon. Theory of Stimulated Raman Scattering from Lattice Vibrations.Proceedings of the Physical Society, 82:393–400, 1963.

BIBLIOGRAPHY 502
[183] A.D. Greentree, P. Olivero, M. Draganski, E. Trajkov, J.R. Rabeau, P. Re-ichart, B.C. Gibson, S. Rubanov, S.T. Huntington, D.N. Jamieson, et al. Crit-ical components for diamond-based quantum coherent devices. JOURNAL OFPHYSICS CONDENSED MATTER, 18(21):825, 2006.
[184] Daniel A. Steck. Cesium D line data. 2008.
[185] J.L. Picque. Hyperfine optical pumping of cesium with a CW GaAs laser.Quantum Electronics, IEEE Journal of, 10(12):892–897, 1974.
[186] Zhaoming Zhu, Daniel J. Gauthier, and Robert W. Boyd. Stored Light in anOptical Fiber via Stimulated Brillouin Scattering. Science, 318(5857):1748–1750, 2007.
[187] Ashok K. Mohapatra, Mark G. Bason, Bjorn Butscher, Kevin J. Weatherill,and Charles S. Adams. A giant electro-optic effect using polarizable darkstates. Nat Phys, 4(11):890–894, 11 2008.
[188] D. E. Chang, V. Gritsev, G. Morigi, V. Vuletic, M. D. Lukin, and E. A.Demler. Crystallization of strongly interacting photons in a nonlinear opticalfibre. Nat Phys, 4(11):884–889, 11 2008.
[189] G. Golub and W. Kahan. Calculating the singular values and pseudo-inverseof a matrix. Milestones in Matrix Computation: The Selected Works of GeneH. Golub with Commentaries, 2007.
[190] GW Stewart. On Scaled Projections and Pseudo-inverses. University of Mary-land, 1988.
[191] A.E. Albert. Regression and the Moore-Penrose pseudoinverse. AcademicPress, New York, 1972.
[192] TNE Greville. Some Applications of the Pseudoinverse of a Matrix. SIAMReview, 2:15, 1960.
[193] M. Born and P. Jordan. On quantum mechanics. Z. Phys, 34(11-12):858–888,1925.
[194] P.A.M. Dirac. The Principles of Quantum Mechanics. Oxford UniversityPress, 1958.
[195] M. Born. Quantenmechanik der Stoßvorgange. Zeitschrift fur Physik AHadrons and Nuclei, 38(11):803–827, 1926.
[196] D.J. Amit and V. Martin-Mayor. Field theory, the renormalization group, andcritical phenomena. World Scientific Singapore, 1984.
[197] RG Woolley. Gauge invariant wave mechanics and the Power-Zienau-Woolleytransformation. Journ. Phys. A, 13:2795, 1980.

BIBLIOGRAPHY 503
[198] M. Babiker and R. Loudon. Derivation of the power-zienau-woolley hamil-tonian in quantum electrodynamics by gauge transformation. Proceedings ofthe Royal Society of London. Series A, Mathematical and Physical Sciences,385(1789):439–460, 1983.
[199] S.C. Reddy and L.N. Trefethen. Stability of the method of lines. NumerischeMathematik, 62(1):235–267, 1992.
[200] L.N. Trefethen. Spectral Methods in MATLAB. Society for Industrial Mathe-matics, Philadelphia, 2000.
[201] D. Gottlieb and S.A. Orszag. Numerical Analysis of Spectral Methods: Theoryand Applications. Society for Industrial Mathematics, 1977.
[202] J.P. Boyd. Chebyshev and Fourier Spectral Methods. Courier Dover Publica-tions, 2001.
[203] J.W. Cooley and J.W. Tukey. An algorithm for the machine computation ofcomplex Fourier series. Mathematics of Computation, 19(90):297–301, 1965.
[204] RG Gareev, FA Arslanov, and GG Telyashev. THEORETICAL PRINCI-PLES. Chemistry and Technology of Fuels and Oils, 37(4), 2001.
Copyright © 2022 FDOKUMEN




















