
Qualitative and comparative proteomic analysis ofXanthomonas campestris pv.campestris 17
12
RESEARCH ARTICLE Qualitative and comparative proteomic analysis of Xanthomonas campestris pv. campestris 17 Wei-Jen Chung 1 * , Hung-Yu Shu 1 * , Chi-Yu Lu 2 , Chiu-Yi Wu 1 ,Yi-Hsiung Tseng 3, 4 , Shih-Feng Tsai 1, 5, 6 and Chao-Hsiung Lin 2, 6, 7 1 Sequencing Core, Genome ResearchCenter, National Yang-Ming University, Taipei, Taiwan 2 Proteome ResearchCenter, National Yang-Ming University, Taipei, Taiwan 3 Institute of Molecular Biology, National Chung Hsing University, Taichung, Taiwan 4 Center for Research and Development, Chungtai Institute of Health Sciences and Technology, Taichung, Taiwan 5 Division of Molecular and Genomic Medicine, National Health Research Institutes, Taipei, Taiwan 6 Department of Life Sciences and Institute of Genome Sciences, National Yang-Ming University, Taipei, Taiwan 7 Department of Education and Research,Taipei City Hospital, Taipei, Taiwan The bacterium Xanthomonas campestris pathovar campestris (XCC) 17 is a local isolate that causes crucifer black rot disease in Taiwan. In this study, its proteome was separated using 2-DE and the well-resolved proteins were excised, trypsin digested, and analyzed by MS. Over 400 protein spots were analyzed and 281 proteins were identified by searching the MS or MS/MS spectra against the proteome database of the closely related XCC ATCC 33913. Functional categorization of the identified proteins matched 141 (50%) proteins to 81 metabolic pathways in the Kyoto Encyclo- pedia of Genes and Genomes (KEGG) database. In addition, we performed a comparative pro- teome analysis of the pathogenic strain 17 and an avirulent strain 11A to reveal the virulence- related proteins. We detected 22 up-regulated proteins in strain 17 including the degrading enzymes EngXCA, HtrA, and PepA, which had been shown to have a role in pathogenesis in other bacteria, and an anti-host defense protein, Ohr. Thus, further functional studies of these up-regulated proteins with respect to their roles in XCC pathogenicity are suggested. Received: August 24, 2006 Revised: February 16, 2007 Accepted: March 7, 2007 Keyword: Plant pathogen / Xanthomonas campestris pv. campestris Proteomics 2007, 7, 2047–2058 2047 1 Introduction Xanthomonas campestris is a Gram-negative phytopathogen consisting of more than 141 pathovars based on their specific host range. Under conditions like high humidity and tem- perature, one of the pathovars, X. campestris pv. campestris (XCC), can infect crucifer family members such as cabbage, cauliflower, radish and Arabidopsis thaliana, resulting in black rot and serious agricultural loss. Black rot disease turns leaves yellow and black with a V-shaped lesion along the margin of leaf and finally gives rise to necrosis [1]. Despite its pathogenicity, XCC is used to produce xanthan gum, which is widely applied in the food industry, agriculture and petro- leum production. To date, the genome of two XCC strains, ATCC 33913 (5.08 Mb) and 8004 (5.15 Mb) have been completely sequenced [2, 3]. These genomes contain more than 4100 predicted coding sequences (CDS). Sequence comparison Correspondence: Dr. Chao-Hsiung Lin, Department of Life Sciences and Institute of Genome Sciences, National Yang-Ming University, 155Li-Nong St., Sec. 2, Taipei 11221, Taiwan E-mail: [email protected] Fax: 1886-2-2820-2449 Abbreviations: CDS, coding sequences; COG, clusters of ortholo- gous groups; KEGG, Kyoto Encyclopedia of Genes and Ge- nomes; XCC, Xanthomonas campestris pv. campestris * These authors contributed equally to this work. DOI 10.1002/pmic.200600647 © 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Transcript of Qualitative and comparative proteomic analysis ofXanthomonas campestris pv.campestris 17

RESEARCH ARTICLE
Qualitative and comparative proteomic analysis of
Xanthomonas campestris pv. campestris 17
Wei-Jen Chung1*, Hung-Yu Shu1*, Chi-Yu Lu2, Chiu-Yi Wu1,Yi-Hsiung Tseng3, 4,Shih-Feng Tsai1, 5, 6 and Chao-Hsiung Lin2, 6, 7
1 Sequencing Core, Genome Research Center, National Yang-Ming University, Taipei, Taiwan2 Proteome Research Center, National Yang-Ming University, Taipei, Taiwan3 Institute of Molecular Biology, National Chung Hsing University, Taichung, Taiwan4 Center for Research and Development, Chungtai Institute of Health Sciences and Technology, Taichung, Taiwan5 Division of Molecular and Genomic Medicine, National Health Research Institutes, Taipei, Taiwan6 Department of Life Sciences and Institute of Genome Sciences, National Yang-Ming University, Taipei, Taiwan7 Department of Education and Research, Taipei City Hospital, Taipei, Taiwan
The bacterium Xanthomonas campestris pathovar campestris (XCC) 17 is a local isolate that causescrucifer black rot disease in Taiwan. In this study, its proteome was separated using 2-DE and thewell-resolved proteins were excised, trypsin digested, and analyzed by MS. Over 400 protein spotswere analyzed and 281 proteins were identified by searching the MS or MS/MS spectra againstthe proteome database of the closely related XCC ATCC 33913. Functional categorization of theidentified proteins matched 141 (50%) proteins to 81 metabolic pathways in the Kyoto Encyclo-pedia of Genes and Genomes (KEGG) database. In addition, we performed a comparative pro-teome analysis of the pathogenic strain 17 and an avirulent strain 11A to reveal the virulence-related proteins. We detected 22 up-regulated proteins in strain 17 including the degradingenzymes EngXCA, HtrA, and PepA, which had been shown to have a role in pathogenesis inother bacteria, and an anti-host defense protein, Ohr. Thus, further functional studies of theseup-regulated proteins with respect to their roles in XCC pathogenicity are suggested.
Received: August 24, 2006Revised: February 16, 2007
Accepted: March 7, 2007
Keyword:
Plant pathogen / Xanthomonas campestris pv. campestris
Proteomics 2007, 7, 2047–2058 2047
1 Introduction
Xanthomonas campestris is a Gram-negative phytopathogenconsisting of more than 141 pathovars based on their specifichost range. Under conditions like high humidity and tem-
perature, one of the pathovars, X. campestris pv. campestris(XCC), can infect crucifer family members such as cabbage,cauliflower, radish and Arabidopsis thaliana, resulting inblack rot and serious agricultural loss. Black rot disease turnsleaves yellow and black with a V-shaped lesion along themargin of leaf and finally gives rise to necrosis [1]. Despite itspathogenicity, XCC is used to produce xanthan gum, whichis widely applied in the food industry, agriculture and petro-leum production.
To date, the genome of two XCC strains, ATCC 33913(5.08 Mb) and 8004 (5.15 Mb) have been completelysequenced [2, 3]. These genomes contain more than 4100predicted coding sequences (CDS). Sequence comparison
Correspondence: Dr. Chao-Hsiung Lin, Department of LifeSciences and Institute of Genome Sciences, National Yang-MingUniversity, 155 Li-Nong St., Sec. 2, Taipei 11221, TaiwanE-mail: [email protected]: 1886-2-2820-2449
Abbreviations: CDS, coding sequences; COG, clusters of ortholo-gous groups; KEGG, Kyoto Encyclopedia of Genes and Ge-nomes; XCC, Xanthomonas campestris pv. campestris * These authors contributed equally to this work.
DOI 10.1002/pmic.200600647
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2048 W.-J. Chung et al. Proteomics 2007, 7, 2047–2058
revealed that these two XCC strains are highly conservedover a large proportion of their gene contents; and up to 80%of the CDS are identical at nucleic acid level. Nevertheless,significant chromosome rearrangements were found be-tween the strains when the genome sequences were alignedand compared [3].
XCC has been an important model organism for study-ing host-pathogen interactions. The invasion process of XCCrelies on the secretion of cellulase, peptidase, and otherdegrading enzymes [4, 5]. In a recent analysis of the XCCB100 extracellular proteome, 13 degrading enzymes wereidentified [6]. Most of these degrading enzymes possesssecretion signals, implying they are secreted by the type IIsecretion pathway. Moreover, various virulence-related genesof XCC 8004 were found using a Tn5-based mutagenesisscreening system, including several cell-signaling proteinsand metabolic enzymes [3].
In this study, we analyzed the proteomes of XCC 17, alocal pathogenic strain [7]. Among the 281 proteins identifiedin the soluble proteome, 141 could be assigned to 81 meta-bolic pathways within the Kyoto Encyclopedia of Genes andGenomes (KEGG) database [8]. In addition, in order to iden-tify virulence-related genes, we also compared the cellularproteomes of XCC strain 17 and an avirulent mutant strain11A [9]. A total of 22 proteins, including three degradingenzymes showing a higher level of expression in strain 17were identified. Taken together, our data provides a usefulprotein list for the general understanding of the organism aswell as a list of putative pathogenicity-related proteins ofXCC that may aid further investigation of the invasion pro-cesses.
2 Materials and methods
2.1 Bacterial strains and culture conditions
A fully pathogenic XCC strain 17 and a spontaneous aviru-lent mutant, strain 11A, were used in this study. Starter cul-tures were prepared by inoculating a single colony in 5 mLTYG (1% tryptone, 0.6% yeast extract, 0.5% glucose, and1 mM MgSO4) and grown overnight at 307C with shakingand then 1 mL of the culture was transferred into 50 mL offresh TYG for a further 16 h of incubation to early stationaryphase.
2.2 Protein preparation from cell lysate
Bacterial cells were transferred to a centrifuge tube andsedimented at 50006g for 15 min. The cells in the pelletwere resuspended and washed with 8 mL PBS and subse-quently centrifuged at 60006g for 15 min at 47C. Theresulting pellet was repeatedly washed with 5 mL water, andthen lysed in 5 mL lysis buffer containing one complete miniprotease inhibitor tablet (Roche) in 10 mL 40 mM Tris-HCl(pH 7.2) and 4% Triton X-100 followed by sonication for
4 min (cycles of 10 s on and 10 s off). To avoid the co-purifi-cation of xanthan gum from the cell wall, which mightinterfere with the protein purification and 2-DE perfor-mance, the proteins were then purified by chloroform/methanol precipitation [10].
2.3 2-DE
Approximately 600 mg of each protein sample (450 mL) con-taining 0.1 M DTT and 0.5% v/v IPG buffer pH 3–10(Amersham Biosciences) were applied to a 24-cm linearpH 3–10 Immobiline DryStrips (Amersham Biosciences) forin-gel rehydration. The first-dimension IEF was carried outon an IPGphor™ (Amersham Biosciences) system using thefollowing program: 0 V63.5 h; 50 V63.5 h; 200 V61 h;500 V61 h; and 1000 V61 h. The gradient was raised from1000 to 8000 V within 30 min and the IEF was terminatedafter 70 000 Vh. Before separating on SDS-PAGE, thefocused IPG strips were placed in an equilibrium buffer(50 mM Tris-HCl, pH 8.8, 6 M Urea, 30% v/v glycerol, and2% w/v SDS) containing 1% w/v DTT for 15 min and soakedin the same buffer containing 2.5% w/v iodoacetamide for15 min. The strips were then placed on a 12% T acrylami-de:bis-acrylamide (37:1) SDS-PAGE gel and immobilizedwith agarose sealing solution (0.5% w/v agarose, 25 mMTris, 192 mM glycine, 0.1% w/v SDS, and a trace of bromo-phenol blue). The second-dimension SDS-PAGE was per-formed at 25 mA per gel until the bromophenol blue dyefront reached the bottom of the gel. The gels were fixed infixing buffer (45% v/v methanol and 10% v/v acetic acid) for30 min and stained in CBB solution (ammonium sulfate17% w/v, phosphoric acid 3% v/v, methanol 34% v/v, andCBB G-250 0.1% w/v) overnight. Finally, the gels weredestained in water to clear the background and scanned on aflatbed scanner. The resulting images were analyzed byPDQuest v7.2 software (BioRad, Hercules, CA, USA), whichmeasured the MW, pI and relative intensities of all proteinspots.
2.4 In-gel digestion
Protein spots were automatically excised on a Spot Cutter(BioRad) according to the manufacturer’s instructions. Eachgel piece (1.2 mm in diameter) was placed into a well of a 96-well plate. For destaining, the gel pieces were washed threetimes with 100 mL of 25 mM NH4HCO3/50% (v/v) ACN for15 min. The solution was then removed and 100 mL of 100%ACN was added to dehydrate the gel pieces. Next, the desic-cated gel pellets were rehydrated with 1.6 mL of 25 mMNH4HCO3 containing 20 ng/mL trypsin (Promega) at 47C for40 min. An extra 2 mL of 25 mM NH4HCO3 was then addedand the reaction was incubated at 557C for 1 h and vacuumdried. Finally, the peptides were released from the gel piecesby sonication in 7 mL of 1% formic acid for 15 min and ana-lyzed by MS.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 2047–2058 Microbiology 2049
2.5 MS
MS analysis by MALDI-TOF-MS was performed on anUltraflex MALDI-TOF-MS (Brucker, Bremen, Germany).The tryptic peptide mixture was mixed with equal volume of2 mg/mL of CHCA in a water/ACN/TFA (50:50:0.1 v/v/v)solution and deposited on an AnchorChip™ 600/384(Brucker). The peptide mixtures of angiotensin II (MH1
1046.5418), angiotensin I (MH1 = 1296.6848), substance P(MH1 = 1347.7354), bombesin (MH1 = 1619.8223), ACTH(1-17, MH1 = 2093.0862), and ACTH (18-39, MH1 =2465.1983) were used as standards for external calibration.The samples were analyzed by MALDI-TOF at a detectionrange from 800 to 3500 m/z. The batch process for the peaklist analysis was carried out using flexAnalysis 2.0 (Brucker),in which the SNAP algorithm was used for peak detection,and the criterion for the S/N was set to 4.
For those samples that could not be identified by MALDI-TOF, additional MS/MS analyses were carried out using a Q-TOF-2 (Micromass, Manchester, UK). The tryptic peptideswere separated on a reversed-phase C18 capillary column[20-mm id (90-mm od)6length] and directed to the electro-spray source of mass spectrometer. The MS was operated inpositive ion mode with a source temperature of 807C and acone voltage of 45 V. A voltage of 3.2 kV was applied to thesource capillary. The TOF analyzer was set in the V-mode.The MS/MS spectra were obtained in the data-dependentacquisition mode whereby the four most abundant doubly-or triply-charged ions were selected for CID. A dynamicexclusion was implemented so the analyzed parent ion wasexcluded if it re-occurred within 90 s. Collision energies wereset to 10 and 30 V for, respectively, the MS and MS/MS scans.Mass spectra were processed using the MassLynx 4.0 soft-ware (Micromass) and the protein identities were analyzedusing the MS/MS peak lists exported from MassLynx.
The protein identification for PMF and MS/MS data wascarried out by in-house MASCOT Server 2.0 software (MatrixScience, London, UK) using the XCC ATCC 33913 proteomesequences from NCBI. The search parameters were: digestedenzyme: trypsin; missed cleavage site: one; variable mod-ification: carbamidomethylation (cysteine) and oxidation(methionine, histidine, and tryptophan); peptide mass toler-ance: less than 300 ppm; and mass values: MH1 and mono-isotopic. Only proteins with a MOWSE score above the sig-nificant level were considered as reliable identifications.
2.6 Functional categorization and pathway
reconstruction of the identified proteins
All of the XCC ATCC 33913 proteins were classified accord-ing to their cellular function using a Perl script and theCOGNITOR tool in Cluster of Orthologous Groups (COG)database. During the computational process, the criterionwas set to 3 clades. Well-annotated ORF represented the ORFthat could be classified into COG categories, except categoryR and S. Poor-annotated ORF represented the ORF belong-
ing to a COG category R, S or those that could not be classi-fied into a COG category. The metabolic pathway recon-struction was conducted based on the XCC ATCC 33913reference pathways in the KEGG database. Operon predic-tion was carried out using FGENESB from Softberry, whichpredicts genes and operons as well as functional RNA(http://www.softberry.com/berry.phtml?topic=gfindb).
3 Results
3.1 Identification of soluble proteins from XCC strain
17
The XCC strain 17 bacteria were harvested at early stationaryphase. To obtain the highest possible resolution, 600 mg ofXCC protein extracts was separated by 2-DE using a linearpH 3–10 IPG strip (24 cm) and SDS-PAGE. Approximately800 spots could be detected on the gel (Fig. 1). Among these,474 well-separated spots were excised, trypsin-digested, andsubjected to MALDI-TOF-MS analysis. The PMF data wereanalyzed by MASCOTsearch against an in-house XCC ATCC33913 protein database [2]. To raise the confidence level oftrue-positive identifications, a parallel search was carried outagainst the NCBI nr database. When the MALDI-TOF spec-trum of any sample failed to provide a confident proteinidentification, the peptide sample was sequenced by ESI-Q-TOF-MS/MS. A total of 381 spots were successfully identi-fied, namely, 243 by PMF and 138 by peptide sequencing.The identified proteins correspond to 281 named genes ofXCC ATCC 33913 and one hypothetical protein of X. oryzaepv. oryzae KACC10331 (XOO1805) as described in Support-ing Information Tables S1, S2 and S3.
3.2 Functional classification of the identified proteins
The 281 XCC protein sequences were assigned by COG-NITOR program [11] for their corresponding COG families.In summary, 35 of the proteins in “Energy production andconversion”, 31 in “Amino acid transport and metabolism”,26 in “Post-translational modification, protein turnoverchaperones”, 25 in “Translation, ribosomal structure andbiogenesis”, and the remaining proteins dispersed across 15remaining categories were identified (Fig. 2). Notably, thecategories of “transcription” and “DNA replication, recombi-nation and repair” have the relative low percentage of pre-dicted proteins identified, while there are higher identifica-tion ratios in “Nucleotide transport and metabolism” (15/64,23.4%), “Lipid metabolism” (19/106, 17.9%), “Post-transla-tional modification, chaperones” (26/147, 17.7%), and“Energy production and conversion” (35/198, 17.7%).
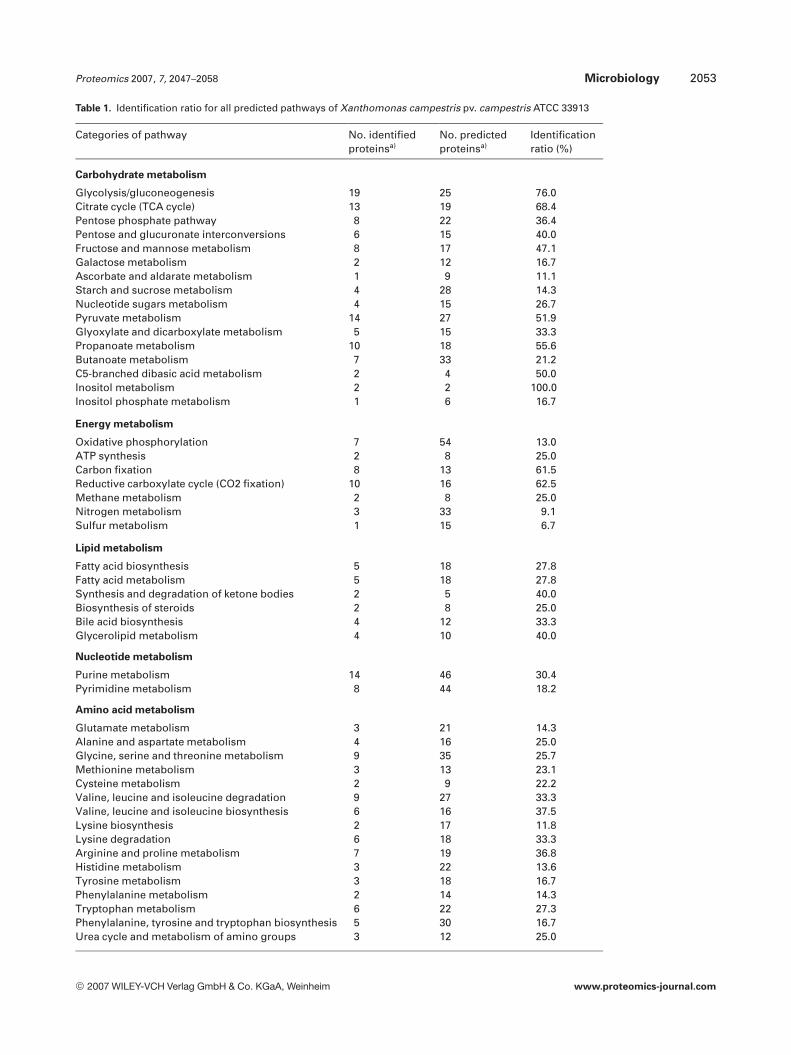
Based on the KEGG database, the identified proteinswere used to reconstruct the metabolic network of XCC 17and the results are summarized in Table 1. Briefly, 141 pro-teins could be assigned to 81 metabolic pathways. A detailed
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2050 W.-J. Chung et al. Proteomics 2007, 7, 2047–2058
description of these pathways is shown in Fig. S1 of theSupporting Information, with all the identified proteinsindicated. Our results showed that high percentages of pro-teins involved in citrate cycle (13/19, 68.4%), carbon fixation(9/14, 63.4%), reductive carboxylate cycle (10/16, 62.5%), andglycolysis/gluconeogenesis (20/35, 57.1%) were identified inthis study. Notably, more than half of predicted proteins inthe category of translation factors were identified, includingTufA, FusA, TufB, Frr, Tsf, eIF-2B, YeiP, and EF-P.
3.3 Differential study of cellular proteins associated
with pathogenicity
Previous studies showed that the pathogenicity and xanthanproduction of XCC were regulated by diffusible signal factor(DSF), which positively regulates the synthesis of extracellularenzymes expressed in mediums to high levels during the lateexponential and stationary phase [12–14]. XCC 11A is a spon-taneous avirulent mutant of the virulent strain 17 [9]. In thisstudy, we carried out a preliminary comparison of the proteinexpression profiles of these two strains grown to early sta-
tionary phase to detect the virulence-associated proteins(Fig. 3). Among all the increased proteins in the virulent strain17, 56 most intense spots were excised for mass analysis and22 proteins were identified (Table 2). Among these, GroESandXCC1535 expressed more than 15-fold higher in strain 17 thanin strain 11A. Two degrading enzymes, endoglucanase(EngXCA) and high-temperature-requirement A (HtrA) pro-tease, known to be pathogenesis-related, were also identified[3]. In addition, the hydroperoxide-resistance protein (Ohr)that plays an essential role in the resistance to organic hydro-peroxides from plant host [15] was also observed to be over-expressed in XCC strain 17. Interestingly, several differentiallyexpressed proteins (TufA, Asd, EngXCA, PepA, XCC4094,GroES and XCC1535) were also identified in the extracellularproteome of another pathogenic strain, XCC B100 [6].
4 Discussion
The 2-DE of the XCC strain 17 soluble proteome resolvedapproximately 800 protein spots and MS analysis of excised
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 2047–2058 Microbiology 2051
Figure 1. 2-DE gels of Xanthomonas campestris pv. campestris strain 17. (A) Total proteins of X. campestris pv. campestris strain 17 wereseparated according to charge (IEF) and mass (SDS-PAGE). Around 800 visible spots can be detected on a gel and a total of 419 spots werefurther analyzed by MS. The identified protein spots are numbered and summarized in Supporting Information Table S1. (B) A regionwithin the dotted lines of Figure 1A (pH 5.0–6.5 and MW 40–100 kDa) is expanded.
proteins led to the identification of 282 proteins. This repre-sents 6.7% of the predicted proteome. These results comparefavorably with the extracellular proteomics analysis of XCCB100 by Watt et al. [6], who identified 62 extracellular pro-teins, 27 most abundant cytosolic proteins, and 49 peri-plasmic proteins. Among these, 35 extracellular proteins, 24cytosolic proteins and 35 periplasmic proteins were alsoidentified in this study, which provides comparable results.The operon prediction using FGENESB program revealed858 putative operons of XCC ATCC 33913 (http://www.soft-berry.com/berry.phtml?topic=gfindb). In this study, 184identified proteins could be assigned to 143 operons encod-ing 487 genes, indicating that more functionally related pro-teins in these operons remain to be identified (Table 3).
Among these operons, we identified a two-componentsystem containing the xanthan biosynthesis-related pro-teins, XanA (XCC0626) and XanB (XCC0625). Xanthan, anextracellular polysaccharide and important industrial prod-uct, is believed to be a virulence determinant in XCC [1].XanA and XanB are bifunctional enzymes involved in thebiosynthesis of UDP-glucose and GDP-mannose, the sugarnucleotide precursors for xanthan gum biosynthesis [16,17]. However, unlike other exopolysaccharide biosyntheticsystems, the XanA and XanB operons are physically sepa-rated from the other genes in the gum operon, which areinvolved in assembly, polymerization, and secretion of xan-than [2, 3, 18]. It is known that phosphorylated RpfG, aspart of two-component system respond for DSF signal
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2052 W.-J. Chung et al. Proteomics 2007, 7, 2047–2058
Figure 2. Functional analysis ofidentified X. campestris pv.campestris proteins. (A) The 281identified proteins of X. cam-pestris pv. campestris strain 17were categorized according totheir function using COGNITOR.There are 19 COG categories intotal. The relative percentagesof identified proteins in eachcategory are shown. Some ofthe proteins were assigned withmore than one category. (B) Forall the predicted ORFs of X.campestris pv. campestris ATCC33913, their COG categorieswere determined. The ratios ofidentified proteins to predictedgenes in each category werecalculated.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Proteomics 2007, 7, 2047–2058 Microbiology 2053
Table 1. Identification ratio for all predicted pathways of Xanthomonas campestris pv. campestris ATCC 33913
Categories of pathway No. identifiedproteinsa)
No. predictedproteinsa)
Identificationratio (%)
Carbohydrate metabolism
Glycolysis/gluconeogenesis 19 25 76.0Citrate cycle (TCA cycle) 13 19 68.4Pentose phosphate pathway 8 22 36.4Pentose and glucuronate interconversions 6 15 40.0Fructose and mannose metabolism 8 17 47.1Galactose metabolism 2 12 16.7Ascorbate and aldarate metabolism 1 9 11.1Starch and sucrose metabolism 4 28 14.3Nucleotide sugars metabolism 4 15 26.7Pyruvate metabolism 14 27 51.9Glyoxylate and dicarboxylate metabolism 5 15 33.3Propanoate metabolism 10 18 55.6Butanoate metabolism 7 33 21.2C5-branched dibasic acid metabolism 2 4 50.0Inositol metabolism 2 2 100.0Inositol phosphate metabolism 1 6 16.7
Energy metabolism
Oxidative phosphorylation 7 54 13.0ATP synthesis 2 8 25.0Carbon fixation 8 13 61.5Reductive carboxylate cycle (CO2 fixation) 10 16 62.5Methane metabolism 2 8 25.0Nitrogen metabolism 3 33 9.1Sulfur metabolism 1 15 6.7
Lipid metabolism
Fatty acid biosynthesis 5 18 27.8Fatty acid metabolism 5 18 27.8Synthesis and degradation of ketone bodies 2 5 40.0Biosynthesis of steroids 2 8 25.0Bile acid biosynthesis 4 12 33.3Glycerolipid metabolism 4 10 40.0
Nucleotide metabolism
Purine metabolism 14 46 30.4Pyrimidine metabolism 8 44 18.2
Amino acid metabolism
Glutamate metabolism 3 21 14.3Alanine and aspartate metabolism 4 16 25.0Glycine, serine and threonine metabolism 9 35 25.7Methionine metabolism 3 13 23.1Cysteine metabolism 2 9 22.2Valine, leucine and isoleucine degradation 9 27 33.3Valine, leucine and isoleucine biosynthesis 6 16 37.5Lysine biosynthesis 2 17 11.8Lysine degradation 6 18 33.3Arginine and proline metabolism 7 19 36.8Histidine metabolism 3 22 13.6Tyrosine metabolism 3 18 16.7Phenylalanine metabolism 2 14 14.3Tryptophan metabolism 6 22 27.3Phenylalanine, tyrosine and tryptophan biosynthesis 5 30 16.7Urea cycle and metabolism of amino groups 3 12 25.0
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2054 W.-J. Chung et al. Proteomics 2007, 7, 2047–2058
Table 1. Continued
Categories of pathway No. identifiedproteinsa)
No. predictedproteinsa)
Identificationratio (%)
Metabolism of other amino acids
b-Alanine metabolism 4 10 40.0Selenoamino acid metabolism 4 20 20.0Cyanoamino acid metabolism 1 10 10.0Glutathione metabolism 1 13 7.7
Glycan biosynthesis and metabolism
Lipopolysaccharide biosynthesis 1 12 8.3Peptidoglycan biosynthesis 1 20 5.0
Biosynthesis of polyketides and nonribosomal peptides
Polyketide sugar unit biosynthesis 2 5 40.0
Metabolism of cofactors and vitamins
Riboflavin metabolism 1 10 10.0Nicotinate and nicotinamide metabolism 2 12 16.7Pantothenate and CoA biosynthesis 2 12 16.7Folate biosynthesis 1 21 4.8One carbon pool by folate 2 13 15.4Ubiquinone biosynthesis 2 20 10.0
Biosynthesis of secondary metabolites
Limonene and pinene degradation 3 14 21.4Alkaloid biosynthesis I 1 4 25.0Streptomycin biosynthesis 3 8 37.5Novobiocin biosynthesis 1 7 14.3
Xenobiotics biodegradation and metabolism
Caprolactam degradation 2 13 15.4Toluene and xylene degradation 1 3 33.3Gamma-hexachlorocyclohexane degradation 1 4 25.01,2-Dichloroethane degradation 1 2 50.0Styrene degradation 1 5 20.0Nitrobenzene degradation 1 2 50.0Benzoate degradation via CoA ligation 2 19 10.5Benzoate degradation via hydroxylation 1 14 7.11- and 2-Methylnaphthalene degradation 3 12 25.0Metabolism of xenobiotics by cytochrome P450 1 12 8.3
Transcription
RNA polymerase 2 15 13.3
Translation
Ribosome 8 60 13.3Aminoacyl-tRNA synthetases 7 24 29.2
Replication and repair
DNA polymerase 1 13 7.7
Membrane transport
ABC transporters 1 77 1.3
Signal transduction
Two-component system 1 20 5.0
Cell motility
Bacterial chemotaxis 1 51 2.0
a) Assignment of pathways were based on the annotation of Xanthomonas campestris pv. campestris ATCC33913 in KEGG PATHWAY database.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 2047–2058 Microbiology 2055
Figure 3. 2-DE gels of Xanthomonas campestris pv. campestris strain 17 and 11A. Images of silver-stained 2-DE gels from Xanthomonascampestris pv. campestris strain 17 and 11A were compared. Proteins (100 mg) were separated by IEF on nonlinear strips (pH 3–10, 17 cm),and 12% SDS-PAGE at second dimension. Only the identified spots with up-regulated expression in Xanthomonas campestris pv. cam-pestris strain 17 were numbered and listed in Table 2.
Table 2. List of up-regulated proteins of Xanthomonas campestris pv. campestris strain 17 compared to Xanthomonas campestris pv.campestris strain 11A
Spotno.
Foldchange
XCC No. Gene name Annotation Gel MW(kDa)/pI
Calc. MW(kDa)/pI
382 3.97 XCC1286 ScoF Cold shock protein 10.0/4.96 7.3/5.67383 2.44 XCC0880 TufA Elongation factor Tu 26.9/5.03 43.2/5.45384 2.11 XCC2549 Asd Aspartate semialdehyde dehydrogenase 40.3/4.96 36.7/5.11385 2.03 XCC0880 TufA Elongation factor Tu 45.1/4.99 43.2/5.45386 4.97 XCC3521 EngXCA Cellulase 44.5/8.39 52.2/6.71387 1.75 XCC3898 HtrA Protease Do 46.8/8.97 49.8/7.94388 2.99 XCC1985 XCC1985 Hypothetical protein XCC1985 20.8/5.94 23.0/5.82389 1.99 XCC0264 Ohr Organic hydroperoxide resistance protein 11.8/4.93 14.4/5.06390 4.97 XCC3521 EngXCA Cellulase 44.4/7.12 52.2/6.71391 3.68 XCC0793 SurA Peptidyl-prolyl cis-trans isomerase 49.2/4.89 49.9/5.21392 2.26 XCC3335 Pcm L-isoaspartate protein carboxylmethyltransferase 23.8/4.76 24.1/4.79393 1.98 XCC0269 Oxidoreductase Oxidoreductase 26.3/5.67 29.5/5.58394 1.74 XCC0928 Mdh Malate dehydrogenase 33.9/5.65 34.9/5.52395 1.68 XCC1079 Oxidoreductase Oxidoreductase 31.4/4.93 32.0/4.87396 4.16 XCC0874 RplY 50S ribosomal protein L25 26.2/4.60 23.1/5.01397 1.80 XCC0649 PepA Aminopeptidase A/I 49.24.99 51.6/5.11398 2.08 XCC0591 UptA Fumarylacetoacetate hydrolase 33.4/4.82 35.3/4.85399 2.08 XCC1565 RplI 50S ribosomal protein L9 15.1/5.01 15.7/5.20400 3.34 XCC0897 RplW 50S ribosomal protein L23 11.4/4.78 11.0/10.29401 2.05 XCC4094 XCC4094 Hypothetical protein XCC4094 35.7/4.83 32.4/5.11402 2.06 XCC0880 TufA Elongation factor Tu 24.9/5.91 43.2/5.45403 1.67 XCC3314 Ppa Inorganic pyrophosphatase 20.6/4.77 19.7/4.86404 1.60 XCC3683 XCC3683 Conserved hypothetical protein 32.9/4.79 32.4/4.70405 64.56 XCC0522 GroES 10kDa chaperonin 10.7/5.72 10.0/5.78406 15.95 XCC1535 XCC1535 Peptidyl-prolyl cis-trans isomerase 24.9/9.40 25.0/9.40
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2056 W.-J. Chung et al. Proteomics 2007, 7, 2047–2058
Table 3. List of the identified operons in Xanthomonas campes-tris pv. campestris ATCC 33913
Identified proteins No. ofgenes inoperon
List of genes in operon
XCC0002 2 XCC0001–XCC0002XCC0011 4 XCC0008–XCC0011XCC0081 2 XCC0081–XCC0082XCC0093 2 XCC0093–XCC0094XCC0151, XCC0152 2 XCC0151–XCC0152XCC0181 3 XCC0179–XCC0181XCC0191 2 XCC0190–XCC0191XCC0206 3 XCC0206–XCC0208XCC0270 2 XCC0269–XCC0270XCC0307 3 XCC0306–XCC0308XCC0358 2 XCC0358–XCC0359XCC0427, XCC0430 4 XCC0427–XCC0430XCC0523 2 XCC0522–XCC0523XCC0542 2 XCC0542–XCC0543XCC0552, XCC0554 9 XCC0547–XCC0555XCC0581 2 XCC0581–XCC0582XCC0591 2 XCC0591–XCC0592XCC0597, XCC0598 2 XCC0597–XCC0598XCC0605, XCC0606 7 XCC0600–XCC0606XCC0619, XCC0620 2 XCC0619–XCC0620XCC0621, XCC0622 2 XCC0621–XCC0622XCC0625, XCC0626 2 XCC0625–XCC0626XCC0627 2 XCC0627–XCC0628XCC0696 4 XCC0695–XCC0698XCC0764 2 XCC0764–XCC0765XCC0779 3 XCC0778–XCC0780XCC0793 3 XCC0792–XCC0794XCC0830 2 XCC0830–XCC0831XCC0850 2 XCC0849–XCC0850XCC0883, XCC0885,
XCC0887, XCC08888 XCC0882–XCC0889
XCC0892, XCC0893 4 XCC0890–XCC0893XCC0904, XCC0905,
XCC0907, XCC090817 XCC0894–XCC0910
XCC0919 5 XCC0916–XCC0920
transduction, positively regulates the expression of gumoperon, but the role of regulation of xanA and xanB remainsunclear [14].
A comparative genome analysis of XCC ATCC 33913 and8004 revealed that less than 2% genes in each were strainspecific [3]. Similarly, the proteins in 412 of 413 spots identi-fied in this study correspond to 281 proteins from XCCATCC 33913; the exception is spot 18, whose MS/MS spec-trum matched to XOO1805, a hypothetical protein of X. ory-zae pv. oryzae KACC10331 (Fig. 4). A gene organizationanalysis of XOO1805 and its flanking genes in the sameoperon was carried out for X. oryzae pv. oryzae KACC10331 aswell as XCC ATCC 33913 and 17 (the partial XCC 17 genomeis available at http://xcc.life.nthu.edu.tw/). As shown inFig. 4, XOO1803, XOO1804, XOO1806 and XOO1807 are
conserved across these three genomes. In addition,XOO1805 only resembles to Orf3 in XCC 17 with a 91%identity in amino acid sequence. However, Orf3 only shares28% identity with the corresponding XCC2867 in XCCATCC 33913. Thus, the Orf3 in XCC strain 17 is likely astrain-specific protein and needs further study for its biolog-ical role.
A total of 227 predicted ORF from XCC ATCC 33913can be classified into the category of “transcription”. How-ever, only five proteins (NusG, RpoB, RpoA, ColR, PhoP)were identified in this study. Notwithstanding this, thetranslation/transcription processing factors of XCC,including Fus, Tsf, Tuf, RpoA, RpoB, RpoC, RpoD andTypA, were all predicted as highly expressed genes basedon their biased codon usage [19, 20]. Perhaps, the reasonthat RpoB and RpoC were not identified in this study isdue to their large molecular weights (154.89 and155.30 kDa). In addition, the predicted transmembranedomain of FusE may cause its absence in sample prepara-tion of 2-DE. However, the failure to identify many of theseproteins remains puzzling, but perhaps suggests othertranscriptional controls.
Our preliminary COG analysis of the 4181 ORF of XCCATCC 33913 reveals that there are 2499 well-annotated genesand 1682 poorly annotated genes. In this study, 229 identi-fied proteins are among the well-annotated genes and theremaining 53 proteins are among the poorly annotated. Thehigher identification ratio (229/2499) observed in well-anno-tated ORF is probably due to the fact that the poor-annotatedgenes generally have a shorter average protein length andthus have a decreased chance to be identified. By calculation,the average protein length is 390 amino acids for the well-annotated ORF and 272 amino acids for the poorly annotatedORF in XCC ATCC 33913. The result is consistent withobserved molecular weights (MW) on the 2-DE, where theaverage MW of the well-annotated proteins is 52.48 kDa andthat of the poorly annotated proteins is 34.64 kDa. Longerproteins usually generate more tryptic peptides and thus aremore likely to be identified by MS analysis. Therefore, theaverage length of the identified proteins is 429 for well-annotated genes and 306 for the poorly annotated genes. Onthe other hand, the lower identification ratio (53/1682)observed for the poorly annotated ORF may also imply moregene prediction errors [21].
The secretion systems of XCC are known to have a majorfunctional role in pathogenicity including transporting viru-lent proteins into host cells [22]. However, no proteins fromthese secretion systems were identified in this study. Thismay be due to the fact that all components of secretion sys-tems are located at membrane or extracellular and such pro-teins are usually insoluble during sample preparation of2-DE. On the other hand, the type-III secretion system ofXCC may resemble the one in X. campestris pv. vesicatoria,which is only induced by the plant host or XVM2 medium,and is thus not normally expressed under standard growthconditions [23, 24]. Nevertheless, a significant proportion of
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2007, 7, 2047–2058 Microbiology 2057
Figure 4. Identification of astrain-specific gene (Orf3) in X.campestris pv. campestris 17. (a)Gene organizations of X. cam-pestris pv. campestris 33913, 17,and X. oryzae pv. oryzaeKACC10331 were examined. Thepercentages of amino acid iden-tity between each two proteincounterparts were calculated.(b) Sequence alignment ofamino acids 1–250 of Orf3 andXOO1805. Identical amino acidsare boxed and the shadedsequences of Orf3 indicate thetwo major tryptic peptides mat-ched to XOO1805 by MS.
the identified proteins belong to the category “post-transla-tional modification, protein turnover, chaperons” (26/147)(Fig. 2B). Fifteen putative chaperones were predicted fromXCC ATCC 33913 genome and seven of them (GroEL, Tig,CbpA, GrpE, DnaK, DnaJ, HtpG) were identified in XCCstrain 17. Furthermore, GroEL was shown to be stronglydown-regulated in the non-pathogenic XCC strain 11A,while the others were not affected. Previous studies haverevealed that, in some bacteria, many type III-secreted pro-teins require type III secretory chaperones to allow proteinsecretion and stabilization [25]. In this study, identification ofchaperone proteins implies that the secretion pathways areactive in XCC 17 even though components of the secretionmachinery cannot be directly detected in this study.
Similarly, the 12 predicted proteins of the gum operonwere not identified in this study. The genetic functions of thegum operon are well studied, yet the location and enzymaticactivity of these gene products remain uncertain. A recentstudy indicated that GumK protein is a membrane-asso-ciated b-glucuronosyltransferase [26]. This is in agreementwith previous studies that xanthan biosynthesis requiresinnermembrane polyisoprenol phosphate as an acceptor [17,27, 28]. Therefore, during our analysis of XCC 17, proteins ingum operon might be associated with the membrane and arethus not present in the 2-DE.
Previous study has shown that the endoglucanase, pro-tease and polygalacturonate lyase will accumulate to highlevel in late exponential and stationary phase [12], whichimplies XCC will degrade the host and its metabolic path-ways will convert the reactant into energy to adapt the envi-ronment. In order to identify proteins whose abundance maydirectly correlated with XCC pathogenicity, a comparativeproteomic analysis was performed between strain 17 andavirulent strain 11A. In total, we observed 22 proteins thatwere significantly up-regulated in XCC strain 17, includingthree degrading enzymes and six putative secretory proteins.Among these degrading proteins, EngXCA is a major extra-cellular endoglucanase of XCC [29], and had recently been
shown to be involved in pathogenesis by transposon muta-genesis [3]. This suggests that EngXCA is the primary viru-lent cellulase candidate among the nine predicted cellulasegenes in the XCC ATCC 33913 genome [2].
HtrA, known as a protein containing both serine pro-tease and chaperone activities in other bacteria, was alsoobserved to be up-regulated in XCC strain 17 compared tostrain 11A. While the functional switch of HtrA has beendemonstrated to be temperature-modulated, the exact func-tion of this switch in Xanthomonas has yet to be characterized[30]. However, HtrA has been shown to participate in patho-genesis in several animal pathogens, a phenomenon prob-ably associated with its ability to protect cells from environ-mental stress. For example, HtrA contributes to host defensesystem resistance in Salmonella typhimurium and Brucellaabortus [31, 32]. Furthermore, the ability of Streptococcuspneumoniae to cause pneumonia and bacteremia is atten-uated in a HtrA-deficient strain [33]. Thus, it is likely thatabundant basal expression of HtrA prepares XCC to over-come the plant defense mechanism.
The predicted protein, XCC1535, expressed at a level ofmore than 15-fold higher in the virulent strain 17, is a puta-tive peptidyl-prolyl cis/trans isomerase (PPIase). It had beenreported that PPIase accelerates the rate-limiting isomeriza-tion of cis/trans peptidyl-prolyl bonds during protein folding,and possesses a chaperone-like function [34, 35]. PPIase canalso promote the binding of GroEL to its substrate, andGroEL-dependent degradation [36, 37]. At present, the exactfunction of PPIase in XCC has not been verified but theobservation of concurrent overexpression of GroES andXCC1535 in strain 17 suggests that further study is needed toexamine the relationship of these proteins with virulence.
Previous analysis of X. campestris pv. phaseoli had shownthat both alkyl hydroperoxide reductase (AhpC) and organichydroperoxide resistance enzyme (Ohr) have important andnon-redundant roles in the detoxification of host-producedlinoleic hydroperoxide [15]. The Ohr and AhpC were bothidentified in this study, and the former is overexpressed in
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

2058 W.-J. Chung et al. Proteomics 2007, 7, 2047–2058
XCC strain 17. It had recently been found that in order tosurvive and inactivate lipid hydroperoxides from the host,AhpC and Ohr expressed in the bacteria and reduce the lipidhydroperoxides to the corresponding alcohols [15]. Previousevidence had shown that peroxides could efficiently neu-tralize OhrR repression of the OhrR-regulated P1 promoterand thus induce Ohr expression [38]. In this study, the dif-ferential expression of Ohr in XCC strain 17 would seem tobe correlated with its role in neutralizing the host defensesystem.
In summary, the data reported here provides usefulinformation regarding the cellular processes of XCC as wellas the virulence-associated proteins of this group of micro-organisms. The pattern of protein expression will serve as abasis and reference source for future proteome analysis. Atotal of 281 proteins were identified in this study, includingmany metabolic enzymes. These enzymes can be classifiedinto 81 XCC metabolic pathways and probably play impor-tant roles in cell growth. In addition, the identification of theup-regulated proteins in XCC strain 17 versus strain 11Aallows us to further study the pathogenic interaction betweenthe bacteria and their plant host.
We thank Drs. Wailap Victor Ng and Ming-Ji Fann for crit-ical reading of the manuscript and valuable suggestions. Thiswork was supported in part by grants (NSC 91-B-FA05-1-4, 95-2120-M-010-002, and 95-2627-M-007-002) from the NationalScience Council in Taiwan.
5 References
[1] Alvarez, A. M., in: Slusarenko, A. J., Fraser, R. S. S., van Loon,L. C. (Eds.),Mechanisms of resistance to plant diseases,Kluwer Academic Publications, Dordrecht, Netherlands 2000,21–52.
[2] da Silva, A. C., Ferro, J. A., Reinach, F. C., Farah, C. S. et al.,Nature 2002, 417, 459–463.
[3] Qian, W., Jia, Y., Ren, S. X., He, Y. Q. et al., Genome Res. 2005,15, 757–767.
[4] Lindgren, P. B., Annu. Rev. Phytopathol. 1997, 35, 129–152.
[5] Van Sluys, M. A., Monteiro-Vitorello, C. B., Camargo, L. E.,Menck, C. F. et al., Annu. Rev. Phytopathol. 2002, 40, 169–189.
[6] Watt, S. A., Wilke, A., Patschkowski, T., Niehaus, K., Prote-omics 2005, 5, 153–167.
[7] Yang, B. Y., Tseng, Y. H., Bot. Bull. Acad. Sin. 1988, 29, 93–99.
[8] Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F. et al.,Nucleic Acids Res. 2006, 34, D354–D357.
[9] Chen, J. H., Hsieh, Y. Y., Hsiau, S. L., Lo, T. C., Shau, C. C., J.Bacteriol. 1999, 181, 1220–1228.
[10] Wessel, D., Flugge, U. I., Anal. Biochem. 1984, 138, 141–143.
[11] Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R.et al., BMC Bioinformatics 2003, 4, 41.
[12] Barber, C. E., Tang, J. L., Feng, J. X., Pan, M. Q. et al., Mol.Microbiol. 1997, 24, 555–566.
[13] Dow, J. M., Crossman, L., Findlay, K., He, Y. Q. et al., Proc.Natl. Acad. Sci. USA 2003, 100, 10995–11000.
[14] Slater, H., Alvarez-Morales, A., Barber, C. E., Daniels, M. J.,Dow, J. M., Mol. Microbiol. 2000, 38, 986–1003.
[15] Klomsiri, C., Panmanee, W., Dharmsthiti, S., Vattanaviboon,P., Mongkolsuk, S., J. Bacteriol. 2005, 187, 3277–3281.
[16] Koplin, R., Arnold, W., Hotte, B., Simon, R. et al., J. Bacteriol.1992, 174, 191–199.
[17] Ielpi, L., Couso, R., Dankert, M., FEBS Lett. 1981, 130, 253–256.
[18] Becker, A., Katzen, F., Puhler, A., Ielpi, L., Appl. Microbiol.Biotechnol. 1998, 50, 145–152.
[19] Sharp, P. M., Li, W. H., Mol. Biol. Evol. 1987, 4, 222–230.
[20] Fu, Y. J., Yin, L. T., Wang, W., Chai, B. F., Liang, A. H., Bio-technol. Lett. 2005, 27, 1597–1603.
[21] Jaffe, J. D., Stange-Thomann, N., Smith, C., DeCaprio, D. etal., Genome Res.2004, 14, 1447–1461.
[22] Buttner, D., Bonas, U., EMBO J. 2002, 21, 5313–5322.
[23] Schulte, R., Bonas, U.J. Bacteriol. 1992, 174, 815–823.
[24] Wengelnik, K., Van den Ackerveken, G., Bonas, U., Mol. PlantMicrobe Interact. 1996, 9, 704–712.
[25] Hueck, C. J., Microbiol. Mol. Biol. Rev., 1998, 62, 379–433.
[26] Barreras, M., Abdian, P. L., Ielpi, L., Glycobiology 2004, 14,233–241.
[27] Ielpi, L., Couso, R. O., Dankert, M. A., Biochem. Biophys. Res.Commun. 1981, 102, 1400–1408.
[28] Ielpi, L., Couso, R. O., Dankert, M. A.,J. Bacteriol. 1993, 175,2490–2500.
[29] Gough, C. L., Dow, J. M., Keen, J., Henrissat, B., Daniels, M.J., Gene 1990, 89, 53–59.
[30] Spiess, C., Beil, A., Ehrmann, M., Cell 1999, 97, 339–347.
[31] Baumler, A. J., Kusters, J. G., Stojiljkovic, I., Heffron, F.,Infect. Immun. 1994, 62, 1623–1630.
[32] Elzer, P. H., Phillips, R. W., Robertson, G. T., Roop, R. M., 2nd,Infect. Immun. 1996, 64, 4838–4841.
[33] Ibrahim, Y. M., Kerr, A. R., McCluskey, J., Mitchell, T. J.Infect.Immun. 2004, 72, 3584–3591.
[34] Kofron, J. L., Kuzmic, P., Kishore, V., Colon-Bonilla, E., Rich,D. H., Biochemistry 1991, 30, 6127–6134.
[35] Scholz, C., Stoller, G., Zarnt, T., Fischer, G., Schmid, F. X.,EMBO J. 1997, 16, 54–58.
[36] Kandror, O., Busconi, L., Sherman, M., Goldberg, A. L., J.Biol. Chem. 1994, 269, 23575–23582.
[37] Kandror, O., Sherman, M., Rhode, M., Goldberg, A. L., EMBOJ. 1995, 14, 6021–6027.
[38] Panmanee, W., Vattanaviboon, P., Eiamphungporn, W.,Whangsuk, W. et al., Mol. Microbiol. 2002, 45, 1647–1654.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com




















