
Purification of Hematopoietic Stem Cells for Further Biological Study
17
Purification of Hematopoietic Stem Cells for Further Biological Study Terry E. Thomas, Cindy L. Miller, and Connie J. Eaves StemCell Technologies Inc., Vancouver, British Columbia, Canada; and Terry Fox Laboratory, BC Cancer Agency, Vancouver, British Columbia V5Z 1L3, Canada For many years, the hematopoietic system has provided a convenient and fascinating model for studies of the molec- ular processes regulating cell growth and differentiation. However, this system also poses considerable challenges because the most primitive “stem” cells as well as their initial differentiating progeny are normally present in hema- topoietic tissues at extremely low frequencies and no unique, stable phenotype has yet been identified to allow hematopoietic cells with specific stem and progenitor func- tions to be measured directly. Rather, this requires the use of functional assays that detect their developmental prop- erties and take several weeks to complete. Accordingly, many investigations of primitive hematopoietic cell behav- ior and their responses to molecular cues in the environ- ment have relied on the development of cell separation techniques specifically designed for obtaining highly en- riched populations of primitive hematopoietic cells. Key to these procedures is the use of a preenrichment step(s) in which differences in cell density, size, or sensitivity to phar- macological agents or surface phenotype are exploited to first “debulk” the sample. This step can then be followed by a more selective antibody-mediated procedure to generate useful numbers of highly purified cells. Batchwise immuno- adsorption techniques offer many advantages for obtaining enriched populations of hematopoietic progenitors because they avoid the nonspecific toxicity seen with antibody- mediated cell killing and are suitable for rapidly processing large samples. For any cell separation procedure, a balance must be struck between the purity and the recovery of the desired cells because steps to increase cell purity usually reduce yields. Both the negative and the positive selection techniques are useful strategies but negative selection usu- ally requires one less manipulation step and circumvents potential effects incurred by the presence of antibody on the surface of the cell being isolated. Specific details for the use and results obtained with an immunomagnetic negative column selection technique are then presented. © 1999 Academic Press Adult hematopoiesis is normally maintained by the continuous production of large numbers of blood cells from a relatively small number of hematopoi- etic stem cells residing in the bone marrow. Tradi- tionally, hematopoiesis has been viewed as a multi- step process in which uncommitted stem cells, with very extensive proliferative potential, give rise to successive generations of progenitor cells that differ- entiate into multiple myeloid and lymphoid lin- eages. This hierarchical system provides an enticing model for the study of biochemical processes that regulate the behavior of stem cells. This includes their transition from a quiescent to a cycling state (and vice versa), maintenance of their stem cell properties (self-renewal) versus differentiation to lineage committed progenitors, and maintenance of their viability versus initiation of apoptosis. Suspen- sions containing these cells are obtained with rela- tive ease from harvesting samples of bone marrow, blood, cord blood, and various fetal tissues such as the liver. Numerous techniques have been used to divide these suspensions into fractions that are en- riched in their content of progenitor cells based on their differences from mature blood cells in cell size and density, expression of cell surface antigens, dif- ferential dye uptake, and sensitivity to cytotoxic drugs. Several general points regarding the biology of primitive hematopoietic cells are important when METHODS: A Companion to Methods in Enzymology 17, 202–218 (1999) Article ID meth.1998.0731, available online at http://www.idealibrary.com on 202 1046-2023/99 $30.00 Copyright © 1999 by Academic Press All rights of reproduction in any form reserved.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Purification of Hematopoietic Stem Cells for Further Biological Study

Pf
T
SB
cuHbituhtoemimtrtwmfiauaetmlmdrtapt
METHODS: A Companion to Methods in Enzymology 17, 202–218 (1999)
Article ID meth.1998.0731, available online at http://www.idealibrary.com on
2
urification of Hematopoietic Stem Cellsor Further Biological Study
erry E. Thomas, Cindy L. Miller, and Connie J. Eaves
temCell Technologies Inc., Vancouver, British Columbia, Canada; and Terry Fox Laboratory,
C Cancer Agency, Vancouver, British Columbia V5Z 1L3, Canadatn©
tcetsvseemrt(pltstbtdrtafd
For many years, the hematopoietic system has provided aonvenient and fascinating model for studies of the molec-lar processes regulating cell growth and differentiation.owever, this system also poses considerable challengesecause the most primitive “stem” cells as well as their
nitial differentiating progeny are normally present in hema-opoietic tissues at extremely low frequencies and nonique, stable phenotype has yet been identified to allowematopoietic cells with specific stem and progenitor func-ions to be measured directly. Rather, this requires the usef functional assays that detect their developmental prop-rties and take several weeks to complete. Accordingly,any investigations of primitive hematopoietic cell behav-
or and their responses to molecular cues in the environ-ent have relied on the development of cell separation
echniques specifically designed for obtaining highly en-iched populations of primitive hematopoietic cells. Key tohese procedures is the use of a preenrichment step(s) inhich differences in cell density, size, or sensitivity to phar-acological agents or surface phenotype are exploited to
rst “debulk” the sample. This step can then be followed bymore selective antibody-mediated procedure to generate
seful numbers of highly purified cells. Batchwise immuno-dsorption techniques offer many advantages for obtainingnriched populations of hematopoietic progenitors becausehey avoid the nonspecific toxicity seen with antibody-
ediated cell killing and are suitable for rapidly processingarge samples. For any cell separation procedure, a balance
ust be struck between the purity and the recovery of theesired cells because steps to increase cell purity usuallyeduce yields. Both the negative and the positive selectionechniques are useful strategies but negative selection usu-lly requires one less manipulation step and circumventsotential effects incurred by the presence of antibody on
he surface of the cell being isolated. Specific details for p02
he use and results obtained with an immunomagneticegative column selection technique are then presented.1999 Academic Press
Adult hematopoiesis is normally maintained byhe continuous production of large numbers of bloodells from a relatively small number of hematopoi-tic stem cells residing in the bone marrow. Tradi-ionally, hematopoiesis has been viewed as a multi-tep process in which uncommitted stem cells, withery extensive proliferative potential, give rise touccessive generations of progenitor cells that differ-ntiate into multiple myeloid and lymphoid lin-ages. This hierarchical system provides an enticingodel for the study of biochemical processes that
egulate the behavior of stem cells. This includesheir transition from a quiescent to a cycling stateand vice versa), maintenance of their stem cellroperties (self-renewal) versus differentiation toineage committed progenitors, and maintenance ofheir viability versus initiation of apoptosis. Suspen-ions containing these cells are obtained with rela-ive ease from harvesting samples of bone marrow,lood, cord blood, and various fetal tissues such ashe liver. Numerous techniques have been used toivide these suspensions into fractions that are en-iched in their content of progenitor cells based onheir differences from mature blood cells in cell sizend density, expression of cell surface antigens, dif-erential dye uptake, and sensitivity to cytotoxicrugs.Several general points regarding the biology of
rimitive hematopoietic cells are important when
1046-2023/99 $30.00Copyright © 1999 by Academic Press
All rights of reproduction in any form reserved.

csrmfcmttvecen
W
sfiplmopnrft(mi“ipepc
A
I
ptm
ctitommmtltseggpspart(b
qtindacwCl1m
I
pvvouatmdwl
203PURIFICATION OF HEMATOPOIETIC STEM CELLS
onsidering what purification strategy may be mostuitable for a given investigation. First, it must beemembered that a universal set of phenotypic ororphological criteria that are completely selective
or hematopoietic stem cells does not exist. The spe-ific identification and quantitation of primitive he-atopoietic cells must, therefore, rely on procedures
hat detect their unique proliferative and differen-iative properties expressed in either in vivo or initro assay systems. Second, all types of hematopoi-tic progenitor cells are present at very low frequen-ies in hematopoietic tissues, which presents annormous challenge for their isolation in sufficientumbers and purity for further analysis.
HAT IS A HEMATOPOIETIC STEM CELL?
In the early 1960s, Till and McCulloch demon-trated that single mouse bone marrow cells couldorm macroscopic spleen colonies upon injection intorradiated recipients. Some of these colonies containrogeny belonging to cells of all the major myeloidineages as well as other progeny that form new
ultilineage colonies upon transplantation into sec-ndary irradiated recipients (1–3). These findingsrovided the first direct evidence of the presence inormal adult mouse marrow of a population of plu-ipotential hematopoietic cells with some capacityor self-renewal. However, later it was shown thathe majority of the initial colony-forming unit-spleenCFU-S) do not contribute to the long-term engraft-ent obtained when marrow cells are transplanted
nto irradiated mice (4–6). The term hematopoieticstem” cell is therefore now usually reserved to des-gnate those cells that can be shown to sustain theroduction of blood cells of all hematopoietic lin-ages for extended periods of time (months) and areresumably also capable of generating daughterells with similar pluripotent properties.
SSAYS FOR HEMATOPOIETIC STEM CELLS
n Vivo AssaysThe competitive repopulating unit (CRU) assay
rovides a reproducible and specific method for de-ermining the frequency of transplantable lympho-
yeloid cells with long-term in vivo reconstituting wapacity (7). To perform this assay, different doses ofhe cells to be evaluated for their CRU content arenjected into groups of irradiated congenic mice andhe proportions of recipients showing reconstitutionf both their lymphoid and their myeloid compart-ents with donor-derived cells is determined 3–4onths later. Poisson statistics and the method ofaximum likelihood can then be used to calculate
he frequency of CRU in the original test cell popu-ation from the proportion of negative animals de-ected within each group. Commercially availabletrains of mice with distinguishable allelic differ-nces including Ly-5 (CD45), hemoglobin, andlucose-phosphate isomerase allow readily distin-uishable donor and recipient genotypes to be com-letely histocompatible. To enable all recipients tourvive independently of whether they are trans-lanted with any stem cells, either they are given andditional minimal but radioprotective graft of mar-ow cells (of the same genotype as the recipient) orhe recipient is given a sublethal dose of irradiation7, 8). The frequency of CRU in normal adult murineone marrow is approximately 1 per 104 cells.In vivo model systems that can be used for the
uantitation of an analogous type of human hema-opoietic stem cell have also been developed. Thesenvolve the transplantation of human cells into xe-ogeneic hosts, such as fetal sheep (9) and immuno-eficient mice (10). The same principles are thenpplied to calculate human stem cell (CRU) frequen-ies from analysis of groups of recipients injectedith graded doses of test cells. The frequency of suchRU in normal adult bone marrow is 1 per 3 3 106
ight density cells (11, 12) and in cord blood is 1 per06 light density cells or approximately 5 CRU perilliliter of cord blood (12, 13).
n Vitro AssaysAlthough such transplantation-based assays at
resent provide the only definitive methods for in-estigating hematopoietic stem cells, a number of initro assays that are believed to detect related orverlapping cell types have also been of great use innderstanding how early events in hematopoiesisre regulated. One of these is based on the observa-ion first made by Dexter et al. that murine bonearrow cells when placed in culture at high cell
ensities form an adherent layer of stromal cellshich would support the production of CFU-S and
arge numbers of mature granulocytes for many
eeks (14). The subsequent discovery that cortico-
ssttidtpcdscsaiw(atac(tcAlespautLihota
tahcmettCtws
PC
sscoptIaoeieensewpnh
P
frl1oecfasi(saeswfyCpp
204 THOMAS, MILLER, AND EAVES
teroids are important to the rapid establishment ofuch cultures led to the successful extension of thisype of culture to human bone marrow (15). Quan-ification of the hematopoietic cells responsible fornitiating the long-term hematopoiesis obtained un-er such conditions became possible by eliminatinghe requirement of the test cells to establish a com-etent feeder layer (16). The cells thus detected arealled long-term culture-initiating cells (LTC-IC). Toetermine the LTC-IC content of a test cell suspen-ion, the cells are added to cultures in which a semi-onfluent feeder layer of irradiated primary marrowtromal cells or a suitable fibroblast cell line haslready been established. These cocultures are thenncubated for $4 (mouse) or $5 (human) weeks afterhich the myeloid activity of the culture is assessed
4, 16, 17). The most objective, specific, and gener-lly applicable way of detecting myeloid cells inhese cultures involves harvesting the entire culturend assessing its content of myeloid colony-formingells (CFC) using standard in vitro semisolid assayssee below). Some investigators prefer to determinehe presence of these cells by adding the cytokine-ontaining semisolid medium directly into the LTC.lternatively, the presence of hematopoietic cell is-
ands developing within the adherent layer can bevaluated. These are often referred to as “cobble-tone areas” because of their cobblestone-like ap-earance (hence the derivative term cobblestonerea-forming cell) (4). Limiting dilution analysis issed to determine the frequency of LTC-IC in theest cell population. For both murine and humanTC-IC, the duration of the LTC reflects how prim-
tive the detected input cell is (18–22). For both, itas also been possible to devise modifications of theriginal LTC-IC assay to permit the in vitro detec-ion of a subset of LTC-IC that have both myeloidnd lymphoid potential (LTC-ICML) (6, 21, 23, 24).Semisolid cultures containing defined combina-
ions of soluble cytokines support the proliferationnd differentiation of a variety of different types ofematopoietic progenitor cells. These form coloniesontaining varying numbers of cells of one or moreyeloid lineages (erythroid, neutrophil, basophil,
osinophil, monocytic, and megakaryocytic). Al-hough some of these CFC, including those referredo as high proliferative potential CFC (25) and blastFC (26) exhibit limited self-renewal as well as mul-
ilineage differentiation potential, it is not clearhether any CRU are capable of cytokine-
timulated proliferation in semisolid media (27, 28). p
HENOTYPIC CHARACTERISTICS OF STEMELLS
Thus far, it has not been possible to identify apecific and stable stem cell phenotype. However,ystematic functional analyses of hematopoieticells expressing particular cell surface antigens orther markers have led to the identification of rarehenotypes whose isolation can yield suspensionshat are highly enriched for stem cell (CRU or LTC-C) activity. Some of these phenotypes are simplyssociated with the quiescent status of the majorityf stem cells in the normal adult (6, 29–31). How-ver, these markers are clearly not reliable for thesolation of cycling stem cell populations. Other rel-vant markers rely on the fact that many antigensxpressed on the surface of mature blood cells areot normally expressed on their primitive precur-ors or vice versa (13, 23, 32, 33). However, thextent to which these correlations are maintainedhen hematopoiesis is perturbed or differently sup-orted is not yet clear. In fact there are alreadyumerous examples where antigen expression onematopoietic cells can be deregulated (34–36).
rimitive Human CellsA commonly used group of monoclonal antibodies
or isolating primitive human hematopoietic cellseacts with CD34 (37). CD34 is a heavily glycosy-ated transmembrane protein that is expressed on–4% of normal adult human bone marrow cells andn ,0.1% of the cells present in steady-state periph-ral blood. However, large numbers of CD341 cellsan be “mobilized” from the marrow into the bloodollowing the administration of certain cytokinesnd/or myelotoxic therapies (38). In such circum-tances, the CD341 cell content of the blood mayncrease to .1% of the total light density fraction39). Most CD341 cells do not express many of theurface antigens (“lineage markers”) that are char-cteristic of terminally differentiating hematopoi-tic cells. Thus removal of such lin1 cells leaves auspension of predominantly immature cells most ofhich are CD341 (40). The isolation of CD341 cells
rom normal human bone marrow or mobilized bloodields populations that are greatly enriched in theirFC, LTC-IC, and CRU content (12), although bothhenotypically and functionally these CD341 cellopulations are still very heterogeneous. For exam-
le, approximately only 10% have in vitro colony-
fmscladp
ff(hC(CCghmTmb
ICnfia
P
atmsuctnmorCceCeap7(fismfmmmpafnsMct4a(5(g
Fnssr(oCpCC
205PURIFICATION OF HEMATOPOIETIC STEM CELLS
orming potential of which ,10% of these will formultilineage colonies. CD34 is also expressed in
ome nonhematopoietic but ontologically relatedells (37, 41). Although it has been generally be-ieved for many years that all human CFC, LTC-IC,nd CRU express CD34 (9, 13, 42, 43), recent evi-ence indicates that a proportion of human hemato-oietic stem cells may be CD342 (44).Several cell surface markers have proven useful in
urther dividing the CD341 population into moreunctionally homogeneous progenitor populations17, 45, 46). Figure 1 illustrates how this approachas been useful in separating the most primitiveD341CD45RA2CD712 adult human marrow cells
containing all of the LTC-IC) from theD341CD45RA1CD716 granulopoietic CFC and theD341CD45RA2CD711 erythroid CFC. Other anti-ens found to be selectively expressed on primitiveuman hematopoietic progenitors and not on theajority of lineage-restricted human CFC includehy-1 and AC133. Thy-1 is expressed on approxi-ately 25% of CD341 cells in human fetal liver, cord
lood, and bone marrow and on a majority of LTC-
IG. 1. The progenitor potential of different subpopulations oformal human bone marrow CD341 cells. CD341 cells wereorted based on their expression of CD45RA and CD71, and theorted cell populations were assayed for CFC and LTC-IC usingecombinant growth factors and engineered feeders as describedHogge et al., Blood 88: 3765 (1996)). All of the LTC-IC, one-halff the CFU-GM, and one-third of the BFU-E were found in theD341CD45RA2CD712 population. The CD341CD45RA1CD712
opulation contained the other half of the CFU-GM. All of theFU-E and 70% of the BFU-E were found in the
1 2 1
rD34 CD45RA CD71 population.C, whereas a majority of the CFC are in theD341Thy-12 fraction (23, 46). The AC133 monoclo-al antibody binds to a recently identified cell sur-ace antigen present on 20–60% of CD341 cells,ncluding those with long-term in vivo repopulatingctivity, but is not expressed on all CFC (47).
rimitive Murine CellsMonoclonal antibodies that bind to cell surface
ntigens and certain lectins have been widely usedo identify and purify different classes of primitiveurine hematopoietic cells. As in the case of human
tem cells, no single marker has been found that isniquely present on murine hematopoietic stemells. Indeed, given the biological heterogeneity ofhe stem cell compartment, such a marker may wellot exist. Strategies for achieving maximal enrich-ent of CRU have, therefore, had to rely on the use
f combinations of procedures, usually one to firstemove the majority of mature (lin1) cells and someFC and then another to positively select for a stemell-enriched subpopulation. The most commonlymployed lineage markers include B220 (i.e.,D45RA which expressed preferentially on B lin-age cells); CD3, CD4, CD8, and CD5 (all of whichre T cell markers); Mac-1 (CD11b) and F480 (ex-ressed on monocytes and macrophages); GR-1 and-4 (both expressed on granulocytes); and Ter119expressed on erythroid cells). Spangrude et al. (32)rst described the use of Sca-1 (Ly6A/E) to isolate atem cell-enriched subpopulation from adult mousearrow lin2Thy1.1lo cells. This Sca-11lin2Thy1.1lo
raction was also shown to contain the majority ofultilineage CFC and all CFU-S. Use by others ofonoclonal antibodies directed against Thy-1 forurine stem cell purification has been limited, in
art, because this antigen exists in two allelic formsnd is expressed in different mouse strains at dif-erent levels. The Sca-11lin2Thy1.1lo population inormal adult mouse bone marrow can be furtherubdivided based on levels of expression of CD4 andac-1. The majority of the long-term repopulating
ells are Mac-12CD42 whereas those with short-erm repopulating potential are Mac-1loCD4lo/2 (48,9). Many studies have suggested that most, if notll, of the long-term repopulating cells express c-kitCD117) (50, 51), the receptor for Steel Factor (52,3). The extent to which cells retain Hoechst 3334254, 55) and/or rhodamine-123 (56, 57), bind wheaterm agglutinin (34, 58, 59), or express CD38 (mu-
1
ine CRU are CD38 ) (60) has also been used to
dmi
iofmtbasdbastlsctcpb1cm
C
stetTmcmitacfausti
seFpt
“tpsmtroi
C
wtoofecdvprtrdousioagaci
fco(m(
206 THOMAS, MILLER, AND EAVES
iscriminate between CRU and other cells in adultouse marrow that have primitive progenitor activ-
ty.In the past decade, interest has focused increas-
ngly on the biology and properties of stem cells fromther hematopoietic tissues including cord blood andetal liver. Although these studies have revealedany phenotypic and functional similarities be-
ween the stem cells present in such tissues, a num-er of phenotypic and functional differences havelso been identified. For example, in the murineystem, the frequency of CRU (on a per cell basis) inay 14 fetal liver and adult bone marrow is similar,ut fetal CRU cells display a greater proliferativend self-renewal potential in vivo (61, 62). Bothhare a Sca-1, c-kit1lin2 phenotype but, in contrasto their adult counterparts, the majority of fetaliver CRU are Mac-11 and AA4.11 (62–64). Expres-ion of CD34 on primitive murine hematopoieticells has been useful for identifying primitive hema-opoietic progenitors in murine tissues, but its spe-ific utility for fractioning murine hematopoietic cellopulations is still unclear. Interestingly, CRU haveeen detected in both Sca-11lin2CD341 and Sca-1lin2CD342 fractions of adult mouse bone marrowell suspensions (65, 66), but none of the CRU inurine fetal liver appears to express CD34 (8).
ELL SEPARATION METHODS
Cell separation involves the physical removal orelective killing of cells in a mixed suspension. His-orically, these techniques have been developed toxploit differences in cell size or density, sensitivityo pharmacologic agents, or cell surface phenotype.ypically, the cells being enriched or depleted areonitored by assessment of their phenotypic, bio-
hemical, physical, or functional properties. Theore difficult a cell type is to identify, or the rarer it
s, the greater the challenge to achieve its purifica-ion. Hence, even though much has been learnedbout the unique features of hematopoietic stemells, separation efforts to develop simpler andaster procedures continue since many questionsbout stem cell biology and regulation require these of larger numbers of highly enriched cell suspen-ions than have been generally available on a rou-ine basis. The extremely low frequency of stem cells
n particular has fostered the development of multi- ftep procedures. These are usually most useful ifach step exploits a different separation parameter.or example, a density separation is often used as areenrichment step before an antibody-mediatedechnique.
Initial “preenrichment” steps can be viewed asdebulking” procedures and can be very useful forhis purpose even if they are quite crude as highurity can then be reserved for subsequent, moreelective separation methods. When performingultiple separation steps, it is important to consider
he overall cell loss anticipated since the final cellecovery is likely to be compromised in order tobtain cell suspensions of greater purity. (This issues discussed in more detail below.)
ell Separation Based on Size and Density
The rate at which a cell moves through mediumhen a centrifugal force is applied is dependent on
he density of the medium and the size and densityf the cell. Mature hematopoietic cells vary in bothf these parameters. This makes it possible to dif-erentially remove certain mature cell types such asrythrocytes and granulocytes from hematopoieticell suspensions by their centrifugation on a me-ium of a higher density, e.g., Ficoll (d # 1.077 g/cm3
ersus regular medium, d 5 1.000 g/cm3). This ap-roach has proven to be a highly effective way toemove normal mature red cells from samples buthe accompanying enrichment of stem cells achievedelative to other nucleated cells is low and depen-ent on the abundance in the sample of other typesf quite dense cells such as granulocytes. If the gran-locytes constitute 90% of the nucleated cells in aample, their elimination could enrich the remain-ng cells (including the stem cells) up to a maximumf only 10-fold. For adult murine bone marrow, sep-ration of the cells on a discontinuous metrizimideradient in a density range of 1.063–1.077 g/cm3 hasllowed the recovery of most of the CFU-S and stemells and yields a population that is enriched 40-foldn pluripotent CFC content (67).
Counterflow elutriation adds a second separationorce (counterflow) in the opposite direction of theentrifugal force, thus enabling further separationf lymphocytes from monocytes (68, 69). The CFCand, in the mouse, CFU-S) elute primarily with the
onocytes, but the smaller more primitive cellsCRU) are found predominantly in the lymphocyte
raction (5, 70).
S
essmundtscbsgCtmrctttmc
S
ntt4fhfpef(5spce(Cmm
SA
bdahkamcaau
traelopt
iwtncAmtie
aioasjttr“i
207PURIFICATION OF HEMATOPOIETIC STEM CELLS
elective Cell Agglutination/Rosetting
Hydroxyethyl starch and methylcellulose causerythrocytes to form large “rouleaux” which thenediment out of mixed hematopoietic cell suspen-ions faster than the other cell types. This allowsost of the mature red cells to be rapidly removed,sually without significant losses of the originalucleated cell population. However, as with theensity and counterflow procedures just described,his approach also offers little enrichment of thetem cell population relative to the other mononu-lear cells. T cells can be removed from a sample oflood or bone marrow by their agglutination withheep red blood cells. The formation of these ag-lutinates is mediated by an interaction betweenD2 on the T cell surface and a specific ligand on
he sheep erythrocyte (71). The cells are firstixed with sheep red blood cells and then the
osetted and nonrosetted cells are separated byentrifugation over a density gradient (72, 73). Ifhe sample contains a high number of T cells (as ishe case for peripheral blood mononuclear cells),his strategy can achieve a three- to sixfold enrich-ent of stem cells relative to other mononuclear
ells.
elective Cell Killing Based on Exposure toPharmacologic Agents
The slow turnover of hematopoietic stem cells inormal adult bone marrow makes the majority ofhem insensitive to short-term exposure to agentshat kill cycling but not quiescent cells.-Hydroperoxycyclophosphamide and mafos-amide are examples of pharmacologic agents thatave been used to purge cycling malignant cellsrom clinical blood and bone marrow autograftsrior to their reinfusion (74). Such treatments alsoliminate most of the CFC but not the bulk of theully differentiated cells that are not cycling75). Prolonged (days) in vitro exposure to-fluorouracil (5-FU) has also recently been de-cribed as a strategy for obtaining highly enrichedopulations of primitive human hematopoieticells (24). In mice, it has been more convenient toxpose hematopoietic cells to such agents in vivo29, 67, 76) and an increase in the frequency ofRU (on a per cell basis) is seen in murine adultarrow 2 days after a single injection of 150
g/kg of 5-FU (6). bELECTION STRATEGIES BASED ONNTIBODY-MEDIATED TECHNIQUES
The specificity and diversity of monoclonal anti-odies offer enormous possibilities for separatingistinct subsets of cells through the use of differentntibodies in appropriate combinations. Antibodiesave been coupled to toxins to achieve selective cellilling, to fluorochromes to allow fluorescent-ctivated cell sorting (FACS), and to various solidatrices to allow selective cell adherence to flasks,
olumns of beads, and magnetic particles. Binding ofntibodies to cell surface antigens and subsequentctivation of the complement cascade has also beensed to lyse selected cell populations.A great advantage of FACS-based separations is
hat they can generate cell suspensions of high pu-ity (as defined by the presence or absence of thentibody–fluorochrome conjugate(s) used). How-ver, FACS is limited by its inability to process veryarge numbers of cells. Thus, FACS technology hasften been found to be most useful when applied to areenriched lin2 population as a later positive selec-ion procedure.
Positive selection is the term used to describe thesolation of a cell population which will be labeledith a given type (or combination) of antibodies and
hen removed from the remaining unlabeled cells. Inegative selection techniques, it is the unwantedells that are targeted with antibodies and removed.ntibody/complement treatment and the use of im-unotoxins are, by definition, negative selection
echniques, whereas both FACS and most batchwisemmunoadsorption techniques can be adapted forither positive or negative selection applications.Techniques that rely on selective cell killing, such
s with antibody-bound complement or immunotox-ns, are suitable for eliminating defined populationsf cells but may be problematic if used as primarypproaches to the isolation of rare cell types liketem cells. In the latter circumstances, a vast ma-ority of the cells in the cell suspension must beargeted and killed for a useful degree of enrichmento be achieved. Even if this is successful, the finalesult is a small number of viable stem cells in asea” of cell debris and potential toxins. Thus, phys-cal removal of the antibody-labeled cells is often a
etter approach than selective cell killing.
I
fn(eupaosmeb
tum2tptmtstg(flc
s
imt
C
nrp.rnpTmamtm
inw
FeF
p
208 THOMAS, MILLER, AND EAVES
mmunoadsorption TechniquesIn immunoadsorption techniques, the cells to be
ractionated are differentially labeled with monoclo-al antibodies and then bound to a solid matrixflask surface, column of beads, magnetic particles,tc.) to allow removal of the labeled cells from thenlabeled cells. Immunoadsorption techniques haveroven very attractive for both clinical and researchpplications because they exploit the high specificityffered by monoclonal antibodies but avoid the non-pecific toxicity of immunotoxins and complement-ediated lysis. Another advantage is their relative
ase of being scaled up to deal with the large num-ers of cells present in clinical transplants.Immunoadsorption techniques have many varia-
ions. In addition to the numerous types of matricessed, selective binding of the labeled cells to theatrix surface can be either direct or indirect (Fig.
). With direct techniques, the primary antibodyhat binds to an antigen on the cell surface is cou-led directly to the separation matrix. Thus, whenhe cell suspension is placed in contact with theatrix, the cells are bound directly to it. In indirect
echniques, the separation surface is coated with aecondary reagent that then binds the primary an-ibody. Typical secondary reagents are sheep (oroat) anti-murine immunoglobulin (Ig), strepavidinwhich binds biotinylated antibodies), or anti-uorescein isothiocyanate (FITC, which binds FITC-onjugated antibodies).Direct immunoadsorption techniques usually re-
ult in less nonspecific binding of cells to the matrix
IG. 2. Direct and indirect immunoadsorption techniques. (Re-
printed with permission from StemCell Technologies Inc.)n comparison to indirect methods. However, directethods do not have the same flexibility for allowing
he antibody combination used to be altered.
ritical Variables
Purity versus recovery. The objective of an immu-oadsorption technique is to transfer the selectiveeactivity of a particular monoclonal antibody to thehysical separation of whole cells (that have a mass109 times that of antibody molecules). It must be
emembered that cells are not inert or homoge-eous, but highly heterogeneous entities with com-lex and continuously changing surface properties.wo selection processes are at work: the desired cellust be selectively bound to antibodies, and the
ntibodies must be selectively bound to a separationatrix. The goal is to maximize the specific interac-
ions between cells and the separation matrix whileinimizing all nonspecific interactions.Separation conditions typically affect cell–matrix
nteractions in general rather than just specific oronspecific interactions. This manifests itself inhat appears to be a balance between purity and
IG. 3. Immunoadsorption surfaces. The total surface area ofach matrix is equivalent to eight panning flasks. (Reprinted with
ermission from StemCell Technologies Inc.)
rFfic(ieitctrt
mpiCasotsueb
ticotnstfns
nwtprirtrssw
FhitaOa(
FmsbCuIaad
209PURIFICATION OF HEMATOPOIETIC STEM CELLS
ecovery of the desired cells in the final suspension.or example, in positive selection techniques, any
actor that increases cell–matrix interactions willncrease both specific and nonspecific interactions toause an increase in the recovery of the desired cellsmore desired cells are bound) as well as a decreasen the purity (more undesired cells are trapped). Anxample of this would be to slow down the flow raten an immunoadsorption column. In negative selec-ion techniques, increasing both specific and nonspe-ific cell–matrix interactions increases the purity ofhe desired cells (more of the unwanted cells areemoved) but also tends to decrease the recovery ofhe desired cells (more are removed nonspecifically).
Positive selection. The most commonly used im-unoadsorption technique for obtaining enriched
opulations of primitive human hematopoietic cellsnvolves the positive selection of cells that expressD34. The initial cell suspension is incubated withn anti-CD34 antibody and the labeled cells are thenelectively adsorbed onto a surface such as a columnf beads or magnetic particles. The selected cells arehen recovered from the surface in a second step. Ineparating rare cells by positive selection, it is of thetmost importance that the extent of nonspecificntrapment of unwanted cells be kept very low andelow the percentage of “target” (positive) cells in
IG. 4. Schematic drawing of StemSep magnetic labeling ofuman cells. Human cells are cross-linked to magnetic dextran
ron particles using tetrameric antibody complexes comprisingwo murine IgG1 monoclonal antibodies held in a tetramericrray by two rat anti-mouse IgG1 monoclonal antibody molecules.ne murine antibody molecule recognizes the cell surface antigennd the other recognizes the dextran on the magnetic particle.
Reprinted with permission from StemCell Technologies Inc.) fhe original cell suspension (77). The specific bind-ng of the target cells is often considered to be not asritical but will, in fact, be reflected in the final yieldf cells obtained (e.g., 60% of the target cells cap-ured is 60% recovery). Since positive selection tech-iques require cell suspensions that exhibit low non-pecific binding of cells to antibodies and surfaces,hey are frequently difficult to apply to previouslyrozen samples of hematopoietic cells because of theumerous numbers of dead and “sticky” cells suchuspensions typically contain.
Negative selection. In negative selection tech-iques, the nonspecific “stickiness” of such un-anted cells is not a problem and may even aid in
heir removal. Negative selection can, therefore, bearticularly useful for isolating primitive cells fromelatively “poor” starting cell suspensions. The crit-cal issue in this case is the efficiency of labeling andemoving the unwanted cells. In situations wherehe desired cells are rare, small inefficiencies in theemoval of unwanted cells results in a cell suspen-ion of low purity. One major advantage of negativeelection is that the cells of interest are not labeledith antibody. This avoids any concern about poten-
IG. 5. Schematic drawing of StemSep magnetic labeling ofurine cells. Indirect magnetic labeling is used for StemSep
eparation of murine cells. Cells are first labeled with a cocktail ofiotinylated antibodies directed against cell surface markers.ells are then cross-linked to magnetic dextran iron particlessing tetrameric antibody complexes comprosed of two murinegG1 monoclonal antibodies held in a tetrameric array by two ratnti-mouse IgG1 monoclonal antibody molecules. One murinentibody molecule recognizes biotin and the other recognizesextran on the magnetic particle. (Reprinted with permission
rom StemCell Technologies Inc.)
tfaheimtcidahbpcthta
btta
fcW
Ff
F
210 THOMAS, MILLER, AND EAVES
ial effects of the antibody on cell function or theurther characterization of the isolated cells (for ex-mple, by FACS). It also eliminates the step(s) in-erent to positive selection procedures that are nec-ssary to release the attached cells from themmunoadsorption matrix surface. The simplest
ethod for achieving this is to use physical agitationo allow shear forces to break the cell–matrix bondsreated. This must entail a certain amount of phys-cal stress to the cells, the significance of which willepend on the cell type being isolated, its condition,nd what it is to be used for. Enzymatic methodsave also been used to cleave the cell–antibodyond. The most commonly used agent is chymopa-ain, which cleaves proteins from the surface of theells. A more gentle competitive release method (De-achabead) for the positive selection of CD341 cellsas been developed by Dynal (Oslo, Norway). Poten-ial effects of these treatments, as well as priorntigen–antibody interactions on the subsequent
IG. 6. StemSep for human cells. (Reprinted with permission
rom StemCell Technologies Inc.) fehavior of the cells isolated, can also be an impor-ant consideration as at least some of these interac-ions can activate intracellular signaling pathwaysnd ultimately gene expression.
Capacity. A variety of surfaces, ranging in sizerom a panning dish to submicron magnetic parti-les, have been used for immunoadsorption of cells.hatever the surface, it must allow physical sepa-
IG. 7. StemSep for murine cells. (Reprinted with permission
rom StemCell Technologies Inc.)
rstrprpfpgctslilitps
tatmMStt
M
ffltcsarbstfapctbTps
tamitswuagw
S
Btmwcflsyaautdaa
gt
Fb
211PURIFICATION OF HEMATOPOIETIC STEM CELLS
ation of the bound cells from the remaining celluspension. Figure 3 gives a visual comparison ofhe various cell separation devices (particles/beads)equired to offer the same total surface area as eightanning flasks (600 cm2). Flasks offer the lowestatio of surface area to volume and have, not sur-risingly, been replaced with more compact devicesor stem cell enrichment. Columns of beads, heavyarticles, and magnetic particles all offer muchreater surface areas for selective cell binding andan be used for both positive and negative selectionechniques. However, the use of negative stem cellelection approaches to isolate rare cells requiresabeling and removal of the vast majority of the cellsn the initial suspension. This in turn requires aarger immunoadsorption surface than is required tosolate the same cells by positive selection. This lat-er drawback has thus focused most applications ofanning and immunoadsorption columns on positiveelection procedures.The increased surface area and efficient specific
argeting features inherent in particle technologiesre reflected in the range of commercially availableechniques for stem cell enrichment. All make use ofagnetic or dense particle separations (Baxter,iltenyi Biotec, StemCell Technologies Inc., MICRA
cientific Inc., and Coulter Cell Therapeutics) withhe exception of the CellPro CD34 immunoadsorp-ion column.
agnetic Cell SeparationBoth heavy particles and magnetic particles utilize a
orce (gravity or magnetic forces) in addition to simpleow/replacement of the medium to physically removehe unbound cells from the separation matrix and theells that bind to it. This usually results in lower non-pecific entrapment of cells. A magnetic force has andvantage over gravity in that it can be “turned off” oremoved during the cell labeling steps. This facilitatesinding of the cells to the particle surface. The celluspension is then placed in the magnetic field duringhe cell separation procedure. Dense particles must beairly large (2–10 mm) and this restricts the surfacerea available for cell targeting (78–80). Magneticarticles or beads range in size from 0.01 to 10 mm andan be divided into two basic categories depending onhe magnetic field to be used. The larger 1–10 mmeads can be separated with a standard magnet (,0.3esla), dragging any attached cell to the magneticole. Their disadvantages are that they do not stay in
uspension, thus hampering their delivery to cells, and (hat their volume to surface area ratio is not as favor-ble as with smaller particles. Colloidal (,100 nm)agnetic particles have the great advantage of form-
ng a stable solution and also offer a very large surface-o-volume ratio. They can be used in concentrationsufficient to saturate cell labeling and do not interfereith subsequent FACS analyses. The disadvantage ofsing colloidal magnetic particles is that they requirehigh-gradient magnetic field for separation. High-
radient magnetic fields can be produced by placing aire matrix in a direct magnetic field (81, 82).
temSep CELL SEPARATION
StemSep (StemCell Technologies Inc., Vancouver,C, Canada) is a high-yield negative selection sys-
em in which the unwanted cells are labeled withultiple antibodies and colloidal magnetic particles,hich are then bound to a high-gradient magnetic
olumn. The desired cells, collected in the columnowthrough, are not antibody labeled and are thusuitable for further antibody-based functional anal-ses. Extensive enrichment of stem cells using neg-tive selection requires capturing a large numbernd variety of cells. Specific labeling/binding of thenwanted cells for removal using the StemSep sys-em is achieved using unmodified colloidal magneticextran–iron particles and noncovalent bispecificntibody cross-linking reagents called tetramericntibody complexes (83) (Figs. 4 and 5).The small size of the colloidal magnetic particles
reatly facilitates delivery of the magnetic label tohe cells. The use of bispecific tetrameric antibody
IG. 8. Typical FACS histogram results for mobilized peripherallood separated using the StemSep progenitor enrichment cocktail.
Reprinted with permission from StemCell Technologies Inc.)
ccitplbdq
MS
r
C
A
c
a
c
a
O
pb
a
fl
S
r
C
A
c
p
c
c
O
w
a
MFBF
ea
212 THOMAS, MILLER, AND EAVES
omplexes avoids expensive and inefficient covalentoupling of antibodies to magnetic particles, ensur-ng reproducibility and ease of scale-up. The use ofhis cell labeling technique together with large ca-acity columns allows .3 log (99.9%) depletion ofabeled cells to be reproducibly achieved. The com-ination of antibodies used in the depletion cocktailetermines which cells will be removed and conse-uently which cells will be recovered.
ethodstemSep for Human CellsThe following is an abbreviated description of the
ecommended protocol (see also Fig. 6).
ell Preparation:
1. Resuspend cells at 5 3 107/ml.
ntibody Labeling of Cells:
2. Add 100 ml antibody cocktail per milliliter ofells. Mix well.3. Incubate on ice for 30 min or at room temper-
ture for 15 min.4. Add 60 ml of magnetic colloid per milliliter of
ells. Mix well.5. Incubate on ice for 30 min or at room temper-
ture for 15 min.
peration of Column:
6a. Pump feed—assemble column and prime withhosphate-buffered saline (PBS). Check for air bub-les. Place in magnet.6b. Gravity feed—place column in magnet and
ssemble. Prime with PBS. Check for air bubbles.
TAB
Purity and Yield of Human CD341CPrimitive Progenitor
Cell sample nCD341CD382
start fraction (%)
obilized PB 3 0.02 6 0.01rozen CB 1 0.16M 3 0.03 6 0.01rozen BM 6 0.05 6 0.01
a Percentage of CD341CD382 cells in start fractions are typicstimates. Accordingly, percent yield values, which represent the
lso relatively inaccurate.7. Wash sample with 33 column vol of medium.8. Load sample.9. Collect 33 column vol and sample volume as
owthrough.
temSep for Murine CellsThe following is an abbreviated description of the
ecommended protocol (see also Fig. 7).
ell Preparation:
1. Resuspend cells at 5 3 107/ml.2. Add 5% normal rat serum.
ntibody Labeling of Cells:
3. Add 10 ml of antibody cocktail per milliliter ofells. Mix well.4. Incubate at 4 to 8°C for 15 min.5. Wash and resuspend at 5 3 107 nucleated cells
er milliliter in medium.6. Add 100 ml of anti-biotin tetrameric antibody
omplexes per milliliter of cells. Mix well.7. Incubate at 4 to 8°C for 15 min.8. Add 60 ml of magnetic colloid per milliliter of
ells. Mix well.9. Incubate at 4 to 8°C for 15 min.
peration of Column:
10a. Pump feed—assemble column and primeith PBS. Check for air bubbles. Place in magnet.10b. Gravity feed—place column in magnet and
ssemble. Prime with PBS. Check for air bubbles.11. Wash sample with 33 column vol of medium.
1
82 Cells Obtained Using StemSeprichment Procedurea
CD341CD382
enriched fraction (%)Yield (%)
CD341CD382 cells
67 6 6 50 6 578 20
61 6 11 80 6 1034 6 6 90 6 20
too low to detect accurately; therefore, values given are rougho input vs recovered absolute numbers of CD341CD382 cells, are
LE
D3En
allyrati
fl
R
H
spOrstrgpe(fMcdaCptrarst
Adi(coafirpCofspo
Cdttvoatre
CSpC
FscI
CD
F
B
M
B
M
213PURIFICATION OF HEMATOPOIETIC STEM CELLS
12. Load sample.13. Collect 33 column vol and sample volume as
owthrough.
esults
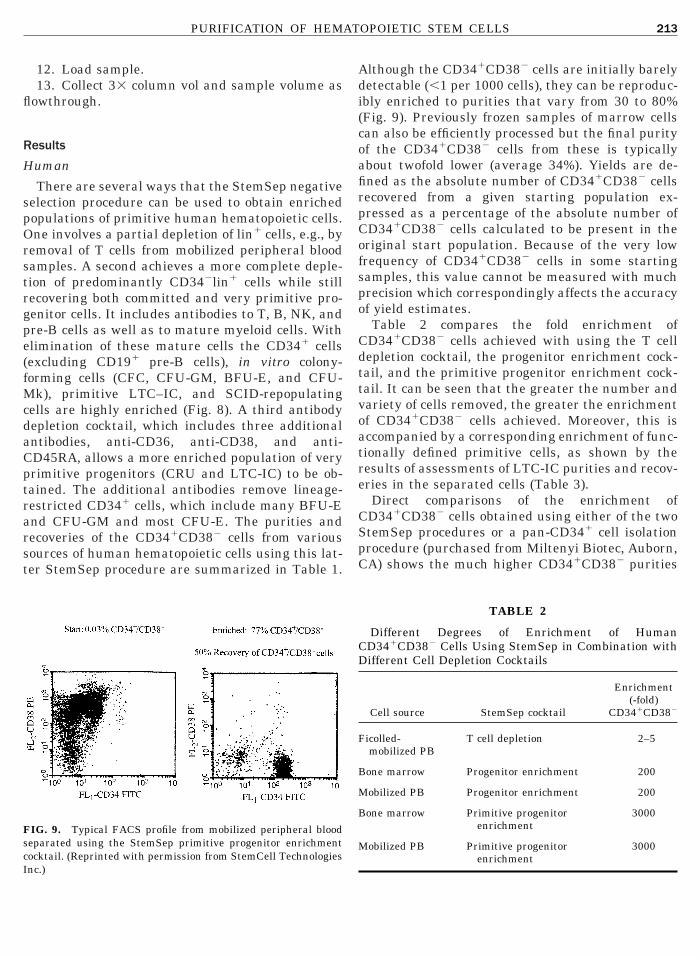
uman
There are several ways that the StemSep negativeelection procedure can be used to obtain enrichedopulations of primitive human hematopoietic cells.ne involves a partial depletion of lin1 cells, e.g., by
emoval of T cells from mobilized peripheral bloodamples. A second achieves a more complete deple-ion of predominantly CD342lin1 cells while stillecovering both committed and very primitive pro-enitor cells. It includes antibodies to T, B, NK, andre-B cells as well as to mature myeloid cells. Withlimination of these mature cells the CD341 cellsexcluding CD191 pre-B cells), in vitro colony-orming cells (CFC, CFU-GM, BFU-E, and CFU-
k), primitive LTC–IC, and SCID-repopulatingells are highly enriched (Fig. 8). A third antibodyepletion cocktail, which includes three additionalntibodies, anti-CD36, anti-CD38, and anti-D45RA, allows a more enriched population of veryrimitive progenitors (CRU and LTC-IC) to be ob-ained. The additional antibodies remove lineage-estricted CD341 cells, which include many BFU-End CFU-GM and most CFU-E. The purities andecoveries of the CD341CD382 cells from variousources of human hematopoietic cells using this lat-er StemSep procedure are summarized in Table 1.
IG. 9. Typical FACS profile from mobilized peripheral bloodeparated using the StemSep primitive progenitor enrichmentocktail. (Reprinted with permission from StemCell Technologies
nc.)lthough the CD341CD382 cells are initially barelyetectable (,1 per 1000 cells), they can be reproduc-bly enriched to purities that vary from 30 to 80%Fig. 9). Previously frozen samples of marrow cellsan also be efficiently processed but the final purityf the CD341CD382 cells from these is typicallybout twofold lower (average 34%). Yields are de-ned as the absolute number of CD341CD382 cellsecovered from a given starting population ex-ressed as a percentage of the absolute number ofD341CD382 cells calculated to be present in theriginal start population. Because of the very lowrequency of CD341CD382 cells in some startingamples, this value cannot be measured with muchrecision which correspondingly affects the accuracyf yield estimates.Table 2 compares the fold enrichment ofD341CD382 cells achieved with using the T cellepletion cocktail, the progenitor enrichment cock-ail, and the primitive progenitor enrichment cock-ail. It can be seen that the greater the number andariety of cells removed, the greater the enrichmentf CD341CD382 cells achieved. Moreover, this isccompanied by a corresponding enrichment of func-ionally defined primitive cells, as shown by theesults of assessments of LTC-IC purities and recov-ries in the separated cells (Table 3).Direct comparisons of the enrichment ofD341CD382 cells obtained using either of the twotemSep procedures or a pan-CD341 cell isolationrocedure (purchased from Miltenyi Biotec, Auborn,A) shows the much higher CD341CD382 purities
TABLE 2
Different Degrees of Enrichment of HumanD341CD382 Cells Using StemSep in Combination withifferent Cell Depletion Cocktails
Cell source StemSep cocktail
Enrichment(-fold)
CD341CD382
icolled-mobilized PB
T cell depletion 2–5
one marrow Progenitor enrichment 200
obilized PB Progenitor enrichment 200
one marrow Primitive progenitorenrichment
3000
obilized PB Primitive progenitorenrichment
3000

op
llmtatnbp
M
po(SpCibtsde
H
tsfist
rcaamlatsoti
f
R
S
w
tcr
a
Cr
M
FB
UP
CFFFFFFF
psC
214 THOMAS, MILLER, AND EAVES
btained when the StemSep primitive progenitorrocedure is used (Table 4).The StemSep strategy can also be adapted to al-
ow the selective enrichment of other cell subpopu-ations by the use of appropriate antibodies for re-
oving unwanted cell types. For example, additiono the standard lin2 cocktail of anti-CD45RA, CD33,nd CD10 antibodies allows a population of ery-hroid progenitors to be selectively enriched. Alter-atively, the addition of anti-CD71 and CD10 anti-odies allows a population of granulopoieticrogenitors to be selectively isolated.
urineEnriched populations of murine hematopoietic
rogenitors can be similarly obtained from samplesf murine hematopoietic tissues by removing cellslin1) that express murine lineage markers. A Stem-ep depletion cocktail designed to enrich murinerogenitors contains antibodies to murine CD5,D45RA, CD11b, GR-1, and TER119. FACS profiles
llustrating the Sca-11lin2 cell enrichment obtainedy subjecting normal adult mouse bone marrow cellso this procedure are shown in Fig. 10. The corre-ponding results for different types of functionallyefined cells contained within these Sca-11lin2 cellnriched populations are summarized in Table 5.
elpful HintsThe following section provides additional informa-
ion for optimizing the use of StemSep magnetic celleparations. The single most significant factor af-ecting separation efficiencies (with any technique)s the quality of the single-cell suspension. Fresh cellamples are usually easier to work with thanhawed cell samples and additional steps may be
TABLE 3
Enrichment and Yield of Long-Term Culture-Initiatingells (LTC-IC) Using StemSep Primitive Progenitor En-ichment Procedure
Type of cellsYield (%)of LTC-IC
Enrichment (-fold)of LTC-IC
obilized PB 160 2,00070 1,000
rozen CB 180 4,000M 110 10,000
110 5,500
iequired to overcome the problems associated withlumping of many thawed cell samples. The use ofppropriate anticoagulant in sufficient amounts isnother important consideration. For example, itay be necessary to first centrifuge the sample at a
ow speed to allow platelets to be removed if there islikelihood of platelet clumps or platelets sticking
o the cells to be processed. In general, the cellshould be kept at all times under conditions thatptimize their viability and minimize their exposureo any conditions (e.g., Ficoll or high-speed vortex-ng) that promote clumping.
To ensure optimal results when using StemSep,ollow these suggestions:
eagents:Store reagents as indicated:
Do not freeze the tetrameric antibody complex.tore at 4°C.
The magnetic colloid may be stored for up to 6eeks at 4°C or at 220°C for up to 1 year.
Repeated freezing and thawing will not affecthe physical or chemical properties of the magneticolloid but it should be frozen in aliquots in order toeduce the risk of contamination.Use buffered salt solutions without Ca21 or Mg21
nd with 2 to 6% fetal bovine serum (FBS).
TABLE 4
Comparison of Human CD341CD382 Cell Enrichmentsing StemSep Primitive or Regular Progenitor Enrichmentrocedure Versus Pan-CD34 Isolation Method
Cell type
% CD341CD382
StemSepprimitiveprogenitor
enrichment
StemSepa
progenitorenrichment
CD34b positiveselection
ord blood 77 ND 9.4resh BM 78 2.7 1.6rozen BM 43 6.2 1.4rozen BM 56 1.8 NDrozen BM 23 4.8 NDrozen BM 34 4.4 NDrozen BM 24 3.2 NDrozen BM 40 4.2 ND
a StemSep lineage depletion to enrich for both committed andrimitive hematopoietic progenitors. The depletion cocktail isimilar to the primitive progenitor cocktail but lacks antibodies toD38, CD36, and CD45RA.b A magnetic positive selection for CD34. Performed as per
nstructions (Miltenyi Biotec).

C
w
p
u
twsn
t
C
e(atN
1
sca
to
C
2
sca
Tcap
s
a
bt
C
c
t
g
i
F
tftwt
fF
FCCLC
FmmS
215PURIFICATION OF HEMATOPOIETIC STEM CELLS
olumn Preparation:
Check all the connections during priming andashing to ensure they do not leak.Prime the column from the bottom up.Use PBS without FBS (or any other protein) to
rime the column.Ensure that there are no air bubbles in the col-
mn.Use PBS with FBS, or Hanks’ balanced salt solu-
ion with FBS to wash the column. Protein in theash solution blocks nonspecific protein binding
ites on the mesh in the column, thus reducing theonspecific binding of cells to the column.Ensure that the column does not run dry at any
ime.
ell Labeling (Human):
Ensure that the RBC content in the sample is notxcessive. If the hematocrit of a sample exceeds 5%nucleated cell concentration at 5 3 107/ml) or therere greater than 20 erythrocytes per nucleated cell,reat as whole blood and lyse the red blood cells inH4Cl prior to labeling.Resuspend cells at 5 3 107/ml (a range of 2–8 3
07 is acceptable).Ensure that the cells are in a single-cell suspen-
ion. If you are running a gravity separation and theells are clumping, filter the cell suspension through200-mm mesh prior to labeling.Incubate the cells using the correct time/
emperature combination. Cells should be incubatedn ice for 30 min or at room temperature for 15 min.
IG. 10. Typical FACS profiles of normal adult mouse bonearrow cells before and after their separation using the StemSepurine progenitor enrichment. (Reprinted with permission from
temCell Technologies Inc.)ell Labeling (Murine):Resuspend the cells at 5 3 107/ml (a range of
–8 3 107 is acceptable).Ensure that the cells are in a single cell suspen-
ion. If you are running a gravity separation and theells are clumping, filter the cell suspension through200-mm mesh prior to labeling.Add normal rat serum to the cells prior to labeling.
he normal rat serum blocks Fc receptors on murineells, thereby preventing the nonspecific binding of ratntibodies. This requires at least 15 min of incubationrior to adding the antibody cocktail.Adhere to recommended incubation times. Exces-
ive delays may affect product performance.Incubate the cells at 4 to 8°C, i.e., in the refriger-
tor, not on ice.Wash the cells before adding the tetrameric anti-
ody complexes. Note: Washing removes excess an-ibodies.
ell Separation:Load the appropriate number of cells into the
olumn—use the correct column size.Collect the cells that run through the column, not
hose that have adhered to the column.During the separation, place the column in the
ap of the magnet and not in the side.Use a 0.6-Tesla magnet.Use the correct pump speed or in the case of grav-
ty feed, the correct gauge blunt-end needle.
actors Affecting PurityPoor antibody binding to unwanted cells reduces
he purity of the recovered cells. This could result,or example, from incubating the cells with the an-ibody and tetrameric antibody complexes at therong temperature and/or time or using antibodies
hat are too dilute.
TABLE 5
Yield and Enrichment of Primitive Hematopoietic Cellsrom Murine Bone Marrow Defined by Phenotypic andunctional End Points
Assay n Enrichment (-fold) Yield (%)
ACS Lin2SCA11 33 14 6 1 20 6 2FC 33 18 6 3 30 6 4FU-S 5 12 6 1 30 6 5TC-IC 5 11 6 5 22 6 5RU 5 12 6 5 35 6 10

p
wia
c
F
t
t
sr
aum
C
sfatmttptsh(emdpa1oSmg
masbwatecst
A
trFCFlr
R
1
1
216 THOMAS, MILLER, AND EAVES
Overloading the column may also decrease cellurity.An excess of red blood cells can reduce cell purity.Cell purity may be decreased if the column is notashed or primed correctly, if air bubbles are lodged
n the column, or if the column is allowed to run dryt any time.In separations of murine cells, delays may de-
rease cell purity.
actors Affecting RecoveryPoor cell recovery may result from underloading
he column.If cells are not in a single-cell suspension (i.e.,
hey are “clumpy”), cell recovery may be reduced.Neglecting to block murine cells with normal rat
erum prior to the addition of antibody can alsoeduce recoveries in murine cell separations.If the column is not washed or primed correctly, if
ir bubbles are lodged in the column, or if the col-mn is allowed to run dry at any time, cell recoveryay be reduced.
ONCLUSIONS
The very heterogeneous nature of hematopoietictem cells makes them difficult to isolate in “pure”orm. In addition they are typically rare elements inny hematopoietic tissue and possess very few dis-inguishing physical characteristics or cell surfacearkers. More than one separation technique is
herefore usually required to obtain suspensionshat are highly enriched in their content of hemato-oietic stem cells. Antibody-mediated FACS separa-ion techniques offer the greatest opportunities forelectivity and enrichment, but FACS alone does notave the capacity for processing very large samples.109 cells). However, FACS can become a very pow-rful tool when combined with a prior antibody-ediated batch separation technique. The lack of
efinitive stem cell markers has limited batchwiseositive selection techniques to pan-CD34 selectionpproaches. These have a potential to provide up to00-fold enrichments in stem cell content. On thether hand, negative selection techniques (e.g.,temSep), which can make use of antibodies toarkers expressed on lineage-restricted CD341 pro-
enitor cells as well as those that allow removal of
ost mature CD342 cells, offer the possibility ofpproximately 1000- to 10,000-fold enrichments intem cell content. All separation techniques or com-inations of techniques must balance gain of purityith decreased recovery. This is currently bestchieved through the use of an initial preenrichmentechnique that offers good yields and a 10- to 50-foldnrichment of the cells of interest. This procedurean then be effectively followed by the use of a moreelective technique targeting several cell surface an-igens to give the desired purity and recovery.
CKNOWLEDGMENTS
This work was supported in part by the National Cancer Insti-ute of Canada (with funds from the Terry Fox Run) and by aesearch grant from StemCell Technologies Inc. C.J.E. is a Terryox Cancer Research Scientist of the National Cancer Institute ofanada. The authors thank Dr. C. Horrocks and Maureenairhurst for assistance in generating the previously unpub-
ished StemSep data and Julie Davidson for manuscript prepa-ation.
EFERENCES
1. Till, J. E., and McCulloch, E. A. (1961) Radiat. Res. 14,213–222.
2. Siminovitch, L., McCulloch, E. A., and Till, J. E. (1963) J. CellComp. Physiol. 62, 327–336.
3. Wu, A. M., Till, J. E., Siminovitch, L., and McCulloch, E. A.(1968) J. Exp. Med. 127, 455–464.
4. Ploemacher, R. E., van der Sluijs, J. P., Voerman, J. S., andBrons, N. H. (1989) Blood 74, 2755–2763.
5. Jones, R. J., Wagner, J. E., Celano, P., Zicha, M. S., andSharkis, S. J. (1990) Nature 347, 188–189.
6. Lemieux, M. E., Rebel, V. I., Lansdorp, P. M., and Eaves, C. J.(1995) Blood 86, 1339–1347.
7. Szilvassy, S. J., Humphries, R. K., Lansdorp, P. M., Eaves,A. C., and Eaves, C. J. (1990) Proc. Natl. Acad. Sci. USA 87,8736–8740.
8. Miller, C. L., and Eaves, C. J. (1997) Proc. Natl. Acad. Sci.USA 94, 13648–13653.
9. Zanjani, E. D., Pallavicini, M. G., Ascensao, J. L., Flake,A. W., Langlois, R. G., Reitsma, M., MacKintosh, F. R.,Stutes, D., Harrison, M. R., and Tavassoli, M. (1994) J. Clin.Invest. 89, 1178–1188.
0. Dick, J. E., Lapidot, T., and Pflumio, F. (1991) Immunol. Rev.124, 25–43.
1. Holyoake, T. L., Nicolini, F., Cashman, J., and Eaves, C. J.
(1997) Blood 90, 159a. [Abstract]
1
1
1
1
1
1
1
1
2
2
2
2
2
22
2
2
2
3
3
3
3
3
3
3
3
3
3
4
4
4
4
4
4
4
4
4
4
5
5
5
5
5
5
5
5
5
5
6
6
217PURIFICATION OF HEMATOPOIETIC STEM CELLS
2. Wang, J. C. Y., Doedens, M., and Dick, J. E. (1997) Blood 89,3919–3924.
3. Conneally, E., Cashman, J., Petzer, A., and Eaves, C. (1997)Proc. Natl. Acad. Sci. USA 94, 9836–9841.
4. Dexter, T. M., Allen, T. D., and Lajtha, L. G. (1977) J. CellPhysiol. 91, 335–344.
5. Gartner, S., and Kaplan, H. S. (1980) Proc. Natl. Acad. Sci.USA 77, 4756–4759.
6. Sutherland, H. J., Lansdorp, P. M., Henkelman, D. H., Eaves,A. C., and Eaves, C. J. (1990) Proc. Natl. Acad. Sci. USA 87,3584–3588.
7. Sutherland, H. J., Eaves, C. J., Eaves, A. C., Dragowska, W.,and Lansdorp, P. M. (1989) Blood 74, 1563–1570.
8. Ploemacher, R. E., van der Sluijs, J. P., van Beurden, C. A. J.,Baert, M. R. M., and Chan, P. L. (1991) Blood 78, 2527–2533.
9. Neben, S., Anklesaria, P., Greenberger, J., and Mauch, P.(1993) Exp. Hematol. 21, 438–443.
0. Verfaillie, C. M., Catanzarro, P. M., and Li, W. (1994) J. Exp.Med. 179, 643–649.
1. Hao, Q.-L., Thiemann, F. T., Smogorzewska, E. M., andCrooks, G. M. (1995) Blood 86, 3745–3753.
2. Conneally, E., Eaves, C. J., and Humphries, R. K. (1998)Blood 91, 3487–3493.
3. Baum, C. M., Weissman, I. L., Tsukamoto, A. S., Buckle,A.-M., and Peault, B. (1992) Proc. Natl. Acad. Sci. USA 89,2804–2808.
4. Berardi, A. C., Wang, A., Levine, J. D., Lopez, P., and Scad-den, D. T. (1995) Science 267, 104–108.
5. Bradley, T. R., and Hodges, G. S. (1979) Blood 54, 1446–1450.6. Nakahata, T., and Ogawa, M. (1982) Proc. Natl. Acad. Sci.
USA 79, 3843.7. Petzer, A. L., Hogge, D. E., Lansdorp, P. M., Reid, D. S., and
Eaves, C. J. (1996) Proc. Natl. Acad. Sci. USA 93, 1470–1474.8. Sitnicka, E., Lin, N., Priestley, G. V., Fox, N., Broudy, V. C.,
Wolf, N. S., and Kaushansky, K. (1996) Blood 87, 4998–5005.9. Hodgson, G. S., and Bradley, T. R. (1979) Nature 281, 381–
382.0. Fleming, W. H., Alpern, E. J., Uchida, N., Ikuta, K., Span-
grude, G. J., and Weissman, I. L. (1993) J. Cell Biol. 122,897–902.
1. Ponchio, L., Conneally, E., and Eaves, C. (1995) Blood 86,3314–3321.
2. Spangrude, G. J., Heimfeld, S., and Weissman, I. L. (1988)Science 241, 58–62.
3. Gan, O. I., Murdoch, B., Larochelle, A., and Dick, J. E. (1997)Blood 90, 641–650.
4. Rebel, V. I., Dragowska, W., Eaves, C. E., Humphries, R. K.,and Lansdorp, P. M. (1994) Blood 83, 128–136.
5. Spangrude, G. J., Brooks, D. M., and Tumas, D. B. (1995)Blood 85, 1006–1016.
6. Zandstra, P. W., Conneally, E., Petzer, A. L., Piret, J. M., andEaves, C. J. (1997) Proc. Natl. Acad. Sci. USA 94, 4698–4703.
7. Civin, C. I., Trischmann, T. M., Fackler, M. J., Bernstein,I. D., Buhring, H.-J., Campos, L., Greaves, M. F., Kamoun,
M., Katz, D. R., Lansdorp, P., Look, A. T., Seed, B., Suther-land, D. R., Tindle, R. W., and Uchanska-Ziegler, B. (1990) inLeukocyte Typing IV (Knapp, W., Ed.) pp. 818–825, OxfordUniversity Press, Oxford.
8. To, L. B., Haylock, D. N., Simmons, P. J., and Juttner, C. A.(1997) Blood 89, 2233–2258.
9. Siena, S., Bregni, M., Brando, B., Ravagnani, F., Bonadonna,G., and Gianni, A. M. (1989) Blood 74, 1905–1914.
0. Thomas, T. E., Fairhurst, M. A., and Lansdorp, P. M. (1997)Blood 90, 347b. [Abstract]
1. Fina, L., Molgaard, H. V., and Robertson, D. (1990) Blood 75,2417–2426.
2. Civin, C. I., Almeida-Porada, G., Lee, M.-J., Olweus, J., Ter-stappen, L. W. M. M., and Zanjani, E. (1996) Blood 88, 4102–4109.
3. Bhatia, M., Wang, J. C. Y., Kapp, U., Bonnett, D., and Dick,J. E. (1997) Proc. Natl. Acad. Sci. USA 94, 5320–5325.
4. Zanjani, E., Almeida-Porada, G., Livingston, A. G., Flake,A. W., and Ogawa, M. (1998) Exp. Hematol. 26, 353–360.
5. Lansdorp, P. M., and Dragowska, W. (1992) J. Exp. Med. 175,1501–1509.
6. Craig, W., Kay, R., Cutler, R. L., and Lansdorp, P. M. (1993)J. Exp. Med. 177, 1331–1142.
7. Yin, A. H., Miraglia, S., Zanjani, E., Almeida-Porada, G.,Ogawa, M., Leary, A. G., Olweus, J., Kearney, J., and Buck,D. W. (1997) Blood 90, 5002–5012.
8. Wineman, J. P., Gilmore, G. L., Gritzmacher, C., Torbett,B. E., and Muller-Sieburg, C. E. (1992) Blood 80, 1717–1724.
9. Morrison, S. J., and Weissman, I. L. (1994) Immunity 1,661–673.
0. Nakauchi, H., Nagayoshi, K., Nishikawa, S.-I., Miura, Y., andSuda, T. (1992) Blood 80, 3044–3050.
1. Orlic, D., Fischer, R., Nishikawa, S.-I., Nienhuis, A. W., andBodine, D. M. (1993) Blood 82, 762–770.
2. Chabot, B., Stephenson, D. A., and Chapman, V. M. (1988)Nature 335, 88–89.
3. Geissler, E. N., Ryan, M. A., and Houseman, D. E. (1988) Cell55, 185–192.
4. Wolf, N. S., Kone, A., Priestly, G. V., and Bartelmez, S. H.(1993) Exp. Hematol. 21, 614–622.
5. Goodell, M. A., Brose, K., Paradis, G., Conner, A. S., andMulligan, R. C. (1996) J. Exp. Med. 183, 1797–1806.
6. Spangrude, G. J., and Johnson, G. R. (1990) Proc. Natl. Acad.Sci. USA 87, 7433–7437.
7. Zijlmans, J. M., Visser, J. W., Kleiverda, K., Kluin, P. M.,Willemze, R., and Fibbe, W. E. (1995) Proc. Natl. Acad. Sci.USA 92, 8901–8905.
8. Visser, J. W., Brauman, J. G., Mulder, A. H., Eliason, J. F.,and de Leeuw, A. M. (1984) J. Exp. Med. 59, 1576–1590.
9. Ploemacher, R. E., van der Loo, J. C. M., von Beurden, C. A.and Baert, M. R. (1993) Leukemia 7, 120–130.
0. Randall, T. D., Lund, F. E., Howard, M. C., and Weissman,I. L. (1996) Blood 87, 4057–4067.
1. Rebel, V. I., Miller, C. L., Eaves, C. J., and Lansdorp, P. M.
(1996) Blood 87, 3500.
6
6
6
6
6
6
6
6
7
7
7
7
7
7
7
7
7
7
8
8
8
8
218 THOMAS, MILLER, AND EAVES
2. Rebel, V. I., Miller, C. L., Thornbury, G. R., Dragowska,W. H., Eaves, C. J., and Lansdorp, P. M. (1996) Exp. Hematol.24, 638–648.
3. Jordan, C. T., McKearn, J. P., and Lemischka, I. R. (1990)Cell 61, 953–963.
4. Morrison, S. J., Hemmati, H. D., Wandycz, A. M., and Weiss-man, I. L. (1995) Proc. Natl. Acad. Sci. USA 92, 10302–10306.
5. Morel, F., Szilvassy, S. J., Travis, M., Chen, B., and Galy, A.(1996) Blood 88, 3774–3784.
6. Osawa, M., Hanada, K., Hamada, H., and Nakauchi, H.(1996) Science 273, 242–245.
7. Shih, J. P., Zeng, H. Q., and Ogawa, M. (1992) Leukemia 6,193–198.
8. Wagner, J. E., Donnenberg, A. D., Noga, S. J., and Cremo,C. A. (1988) Blood 72, 1168–1176.
9. Noga, S. J., Wagner, J. E., and Rowley, S. D. (1990) in BoneMarrow Purging and Processing (Gross, S., Gee, A. P., andWorthington-White, D. A., Eds.), pp. 345–361, Wiley–Liss,New York.
0. Noga, S. J., Seber, A., Davis, J. M., Berenson, R. J., Vogel-sang, G. B., Braine, H. G., Hess, A. D., Marcellus, D., Miller,C. A., Sharkis, S. J., Goodman, S. N., Santos, G. W., andJones, R. J. (1998) J. Hematother. 7, 151–157.
1. Ogasawara, H., Kusui, K., and Takasaki, S. (1995) Immunol.Lett. 48, 35–38.
2. North, J., and Henry, C. (1980) in Selected Methods in CellularImmunology (Mishell, B.B., and Shiigi, S. M., Eds.), Vol. 8.7, pp.
186–208, W. H. Freeman and Company, San Francisco.3. Ownby, D. R., and McCullough, J. (1983) J. Immunol. Meth-ods 56, 281–284.
4. Shpall, E. J., Stemmer, S. M., Johnston, C. F., Hami, L.,Bearman, S. I., Berenson, R., and Jones, R. B. (1992) J. He-matother. 1, 45–54.
5. Rowley, S. D., Zuehlsdorf, M., Braine, H. G., Colvin, O. M.,Davis, J., Jones, R. J., Saral, R., Sensenbrenner, L. L., Yea-ger, A., and Santos, G. W. (1987) Blood 70, 271–275.
6. Lerner, C., and Harrison, D. E. (1990) Exp. Hematol. 18,114–118.
7. Thomas, T. E., and Lansdorp, P. M. (1998) in AutologousStem Cell Transplantation (Carella, A.M., Ed.), pp. 5–13,Harwood Academic Press, London.
8. Shapero, M., Laus, R., and Van Vlasselaer, P. (1995) Blood86, 625a. [Abstract]
9. Zwerner, R. K., Schmittling, R., and Russell, T. (1996) J. Im-munol. Methods 198, 199–202.
0. Kenyon, N. S., Ricordi, C., Gribben, J. G., Nadler, L. M.,Zwerner, R. K., and Russell, T. R. (1998) in Cell SeparationMethods and Applications (Recktenwald, D., and Radbruch,A., Eds.), pp. 103–132, Marcell Dekker, New York.
1. Molday, R. S., and MacKenzie, D. (1982) J. Immunol. Meth-ods 52, 353–367.
2. de Latour, C., Schmitz, G., Maxwell, E., and Kelland, D.(1983) IEEE Trans. Magn. Mag-19, 2127–2129.
3. Lansdorp, P. M., Aalberse, R. C., Bos, R., Schutter, W. G., andVan Bruggen, E. F. J. (1986) Eur. J. Immunol. 16, 679–
683.



















