
Productive infection and subsequent interaction of CD4-gp120 at the cellular membrane is required...
10
Journal of General Virology (1995), 76, 681 690. Printed in Great Britain 681 Productive infection and subsequent interaction of CD4-gpl20 at the cellular membrane is required for HIV-induced apoptosis of CD4 + T cells Jacques Corbeil 1. and Douglas D. Richman 1,2 1 University of California San Diego, Departments of Medicine and Pathology and 2 San Diego Veterans Affairs Medical Center, La Jolla, CA 92093-0679, USA One of the hallmarks of human immunodeficiency virus type 1 (HIV-1) infection is the decline in CD4 + T lymphocytes which precedes the progression from an asymptomatic state to AIDS. Apoptosis (programmed cell death) is one of the mechanisms proposed to mediate this depletion. Infectious and inactivated pre- parations of HIV-1 ~A1 were compared for their potential to induce apoptosis. Analysis with fluorescence- activated cell sorting using the DNA intercalative compound propidium iodide demonstrated that apop- tosis occurred only with infectious HIV-1, implying that cell surface binding and signalling by the virus alone were insufficient to trigger apoptosis. Apoptosis was further confirmed by the presence of characteristic digestion of host cell DNA and morphologically by nuclear condensation observed by transmission electron microscopy. HIV infection of CD4 + T cell lines generated an accumulation of the cells in GJM phase of the cell cycle and cells undergoing apoptosis appeared to originate from the pool of cells in the 61 phase. Inhibitors of HIV replication were used to identify the point in the virus replicative cycle at which apoptosis is induced. The reverse transcriptase inhibitor, ddI, or the HIV protease inhibitor, RO31-8959 (Saquinavir), were added either 2 h before or 6 h after HIV inoculation. Only ddI inhibited HIV-induced apoptosis when added before inoculation; however, neither treatment was effective in preventing HIV-induced apoptosis when applied 6 h after inoculation. These data indicate that apoptosis requires a single round of reverse transcription and the expression of virion proteins, but not the maturation of progeny virions. Two agents which compete with HIV for binding to CD4 + T cells, dextran sulphate and the anti-CD4 MAb Leu3a, were effective at preventing apoptosis when added 6 h after infection, implying that a subsequent gpl20-CD4 interaction at the surface of an infected cell was required to complete the apoptotic process. Introduction The decline in CD4 + T lymphocytes is one of the hallmarks of HIV infection (Levy, 1993; Rosenberg & Fauci, 1991; Stein et al., 1992). The level of virus replication in asymptomatic infection is greater than previously thought (Embretson et al., 1993; Pantaleo et al., 1993; Piatak et al., 1993; Scadden et al., 1992). Because human immunodeficiency virus type 1 (HIV-1) can be toxic to CD4 + T lymphocytes, it is plausible that HIV may account directly for a significant proportion of the CD4 + T cell depletion seen in AIDS. In addition to immune-based mechanisms such as cytotoxic T cell killing (Zarling et al., 1990), natural killer cell-mediated killing of infected cells (Brenner et al., 1989) and antibody-dependent cellular cytotoxicity (Tanneau et al., 1990), HIV-induced apoptosis may also contribute to * Author for correspondence. Fax +1 619 552 7445. e-mail [email protected] this depletion. Several mechanisms have been proposed to explain how HIV-1 infection leads to apoptotic cell death (Ameisen, 1994; Ameisen & Capron, 1991 ; Banda et al., 1992; Gougeon et al., 1993a; Groux et al., 1992; Laurent-Crawford et al., 1991; Meyaard et al., 1992; Terai et al., 1991); however, some uncertainty remains concerning the factors and pathways that trigger this process (Ameisen, 1992; Cohen, 1993; Gougeon et al., 1993b; Gougeon & Montagnier, 1993; Weiss, 1993). A recent report from Laurent-Crawford and collaborators concluded that HIV-induced apoptosis of infected CD4 + T cells is due to gpl20-gp41 heterodimer complex programming death in these lymphocytes. The expres- sion of the env gene in the lymphoblastoid T cell line CEM was sufficient to induce apoptosis (Laurent- Crawford et al., 1993). The study presented here was aimed at elucidating the components necessary to induce apoptosis during HIV- 1 infection in the absence of immune responses by defining the steps in viral replication that contribute to this process. It was found that virions containing 0001-2734 © 1995SGM
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Productive infection and subsequent interaction of CD4-gp120 at the cellular membrane is required...

Journal of General Virology (1995), 76, 681 690. Printed in Great Britain 681
Productive infection and subsequent interaction of CD4-gpl20 at the cellular membrane is required for HIV-induced apoptosis of CD4 + T cells
Jacques Corbeil 1. and Douglas D. Richman 1,2
1 University o f California San Diego, Departments o f Medicine and Pathology and 2 San Diego Veterans Affairs Medical Center, La Jolla, CA 92093-0679, USA
One of the hallmarks of human immunodeficiency virus type 1 (HIV-1) infection is the decline in CD4 + T lymphocytes which precedes the progression from an asymptomatic state to AIDS. Apoptosis (programmed cell death) is one of the mechanisms proposed to mediate this depletion. Infectious and inactivated pre- parations of HIV-1 ~A1 were compared for their potential to induce apoptosis. Analysis with fluorescence- activated cell sorting using the DNA intercalative compound propidium iodide demonstrated that apop- tosis occurred only with infectious HIV-1, implying that cell surface binding and signalling by the virus alone were insufficient to trigger apoptosis. Apoptosis was further confirmed by the presence of characteristic digestion of host cell DNA and morphologically by nuclear condensation observed by transmission electron microscopy. HIV infection of CD4 + T cell lines generated an accumulation of the cells in GJM phase of the cell cycle and cells undergoing apoptosis appeared to
originate from the pool of cells in the 61 phase. Inhibitors of HIV replication were used to identify the point in the virus replicative cycle at which apoptosis is induced. The reverse transcriptase inhibitor, ddI, or the HIV protease inhibitor, RO31-8959 (Saquinavir), were added either 2 h before or 6 h after HIV inoculation. Only ddI inhibited HIV-induced apoptosis when added before inoculation; however, neither treatment was effective in preventing HIV-induced apoptosis when applied 6 h after inoculation. These data indicate that apoptosis requires a single round of reverse transcription and the expression of virion proteins, but not the maturation of progeny virions. Two agents which compete with HIV for binding to CD4 + T cells, dextran sulphate and the anti-CD4 MAb Leu3a, were effective at preventing apoptosis when added 6 h after infection, implying that a subsequent gpl20-CD4 interaction at the surface of an infected cell was required to complete the apoptotic process.
Introduction
The decline in CD4 + T lymphocytes is one of the hallmarks of HIV infection (Levy, 1993; Rosenberg & Fauci, 1991; Stein et al., 1992). The level of virus replication in asymptomatic infection is greater than previously thought (Embretson et al., 1993; Pantaleo et al., 1993; Piatak et al., 1993; Scadden et al., 1992). Because human immunodeficiency virus type 1 (HIV-1) can be toxic to CD4 + T lymphocytes, it is plausible that HIV may account directly for a significant proportion of the CD4 + T cell depletion seen in AIDS. In addition to immune-based mechanisms such as cytotoxic T cell killing (Zarling et al., 1990), natural killer cell-mediated killing of infected cells (Brenner et al., 1989) and antibody-dependent cellular cytotoxicity (Tanneau et al., 1990), HIV-induced apoptosis may also contribute to
* Author for correspondence. Fax +1 619 552 7445. e-mail [email protected]
this depletion. Several mechanisms have been proposed to explain how HIV-1 infection leads to apoptotic cell death (Ameisen, 1994; Ameisen & Capron, 1991 ; Banda et al., 1992; Gougeon et al., 1993a; Groux et al., 1992; Laurent-Crawford et al., 1991; Meyaard et al., 1992; Terai et al., 1991); however, some uncertainty remains concerning the factors and pathways that trigger this process (Ameisen, 1992; Cohen, 1993; Gougeon et al., 1993b; Gougeon & Montagnier, 1993; Weiss, 1993). A recent report from Laurent-Crawford and collaborators concluded that HIV-induced apoptosis of infected CD4 + T cells is due to gpl20-gp41 heterodimer complex programming death in these lymphocytes. The expres- sion of the env gene in the lymphoblastoid T cell line CEM was sufficient to induce apoptosis (Laurent- Crawford et al., 1993).
The study presented here was aimed at elucidating the components necessary to induce apoptosis during HIV- 1 infection in the absence of immune responses by defining the steps in viral replication that contribute to this process. It was found that virions containing
0001-2734 © 1995 SGM

682 J. Corbeil and D. D. Richman
expressed glycoproteins alone were not sufficient to trigger apoptosis. Rather, HIV-induced apoptosis re- quired an initial round of replication followed by engagement of expressed gpl20 with CD4 molecules to complete the process.
Methods Celt culture and virus preparations. The lymphoblastoid CD4 + T cell
line SupT1 was obtained from Dr J. Hoxie and is available from the repository of the NIH AIDS Research and Reference Reagent Program. CD4+-enriched peripheral blood lymphocytes (PBL) were obtained from healthy HIV-seronegative donors after Ficoll-paque separation and depletion of CD8 ÷ T cells and monocytes using a commercially available kit as described by the manufacturer (AIS microcellector; Applied Immune Sciences). All cells were cultured with RPMI 1640 (Gibco-BRL) supplemented with I0 % FBS (v/v) and with 2 mM-glutamine and penicillin G (100 units/ml) and streptomycin (100 gg/ml). Ten U/ml of interleukin (IL-2) were added to the PBL culture 3 days after stimulation with phytohaemagglutinin (PHA) (3 lag/ml). The cells were passaged (1:5) 24 h prior to the experiments to ensure that the cells were in log-phase growth.
A high titre stock of HIV-1LA I (5 X 107 TCIDs0/ml ) was prepared and titrated using the endpoint dilution method of K~rber (1931). The high titre stocks were prepared by inoculating 107 CEM cells in 25 ml of culture medium at an m.o.i, of 0.001 and growing the cells for 6 to l0 days. Ten ml of this culture was then added to 400 ml of uninfected CEM (5 x 105 cells/ml) and grown for 7 to 10 days. The cells were pelleted (300g for 10min) and the supernatant was clarified by centrifugation (800 g for 10 min).
The stock of HIV-ILA ~ was inactivated with 4'-aminomethyl- trioxsalen (10 lag/ml) (Calbiochem-Behring) in combination with long- wavelength UV light (320 to 380 nm) for 12 min to render the viral preparation non-infectious while preserving most of its antigenicity and ability to bind to CD4 + T cells (Hanson, 1992: Redfield et al., 1981; Watson et al., 1990). UV-activated intercalating compunds such as 4/- anainomethyltrioxsalen form covalent adducts with nucleic acids, thereby inactivating cell-free and cell-associated viral infectivity (Redfield et al., 1981 ; Dodd. 1994). After this treatment, the infectious titre was reduced at least one million-fold to less than 10 TCIDs0/ml as assessed with MT-2 cells. To determine the binding ability of the virus preparations, SupT1 cells were inoculated with HIV-ILA I or UV psoralen-treated H1V-1LA ~ at equal amounts (p24 equivalent to an m.o.i, of 1) for 4 h at 37 °C. Virions were removed from the cell surface with a brief treatment (4 min at 37 °C) with a solution of trypsin/EDTA (Gibco BRL). The cells were then washed three times with cold PBS before measuring internalized p24 antigen by using an ELISA as described by the manufacturer (Abbott Laboratories).
HIV-1 infection and utili=ation of HIV inhibitors. The infection protocol was identical for each of the cell types utilized. Briefly, between 5 x 106 and 10 x 106 cells were inoculated with HIV-1LA I at different m.o.i.s in 1 ml of culture medium containing 1 lag/ml of polybrene to facilitate infection. The cells were incubated for 2 h at 37 °C in 5 % CO 2 in air. The cells were then washed once with RPMI 1640 and resuspended at a density of 5 x 105 cells/ml in culture medium. Samples of 1 x 106 to 2 x 10 ~ cells were taken daily to perform the analyses.
The following HIV inhibitors were added to the culture: dextran sulphate (Mbemba et al., 1992; Mitsuya et al., 1988), (M r 6000-8000) (ICN); the reverse transcriptase inhibitor, dideoxyinosine (ddI) (Gao et al., 1993), (Bristol-Myers); the HIV proteinase inhibitor Saquinavir RO31-8959 (chemical name: N-tert-Butyl-decahydro-2-[2(r)-hydroxy- 4-phenyl-3(s)-[[N-2-quinolylcarbonyi-L-asparaginyl] amino] butyl]-
4aS, 8aS-isoquinoline-3(S)-carboxamide methanesulphonate) (Roche Products) (Craig et al., 1991) and the anti-CD4 antibody Leu3a (Becton-Dickinson) (Attanasio et al., 1991).
Quantification of surface CD4 receptor expression by flow cytometry. Uninfected and HIV-infected cells (106) were washed in PBS and resuspended in 200 gl of PBS+2% FBS. The anti-CD4 monoclonal antibody OKT4 (Johnson & Johnson) was added (20 lal) to the cells (except the secondary antibody control) and kept at 4 °C for 30 min for staining. The cells were then washed twice in PBS and resuspended in 200 pl of PBS + 2 % FBS to which 4 pl of goat anti-mouse IgG-FITC (Tago) was added and kept at 4 °C for 30 rain. The cells were then washed in PBS and resuspended in 400 gl of 0.5 % paraformaldehyde in PBS and kept in the dark at 4 °C until analysed by fluorescence- activated cell sorting (FACS).
Detection of apoptosis-associated chrornat#t degradation by flow cytometry. Cells (2 x 106) were washed in PBS and resuspended in 30 % ethanol and kept at 4 °C. The cells were stained with propidium iodide (PI) as previously described (Darzynkiewicz et al., 1992; Telford et al., 1992) with slight modifications. Briefly, the cells were centrifuged and resuspended in PBS containing 0"1 mM-EDTA(Na)z, RNaseA at 50 gg/ml (50 units/mg) and PI (50 lag/ml). Cell cycle analysis was performed using a EPICS V fluorescence-activated cytometer with a cell cycle analysis doublet discrimination protocol. PI was excited using a 488 nm argon laser and detected with a 620-700 nm long-pass filter.
Electron microscopy. The cells (106) were washed twice with PBS and centrifuged at 300 g for 5 min in a microcentrifuge tube. The pellet was treated with a modified Karnovsky's fixative containing 2% paraformaldehyde and 1% glutaraldehyde in 0'1 M-sodium cacodylate buffer pH 7.2, for 1 h at room temperature, washed twice with 0.1 M- cacodylate buffer and fixed with a solution of 1% osmium tetroxide in 0.1 M-cacodylate buffer for 45 min at room temperature. The cells were then rinsed three times with 0-1 M-cacodylate buffer. The cells were stained with a saturated uranyl acetate solution in 50 % ethanol. The pellets were dehydrated with a graded series of ethanol solutions (30-100%). The cell pellets were infiltrated with a propylene oxide/ epon resin (Scipoxy 812 resin: Energy Beam Sciences) mixture overnight. The excess solution was removed and 100 % resin was added and the blocks were polymerized at 65 °C overnight. Thin sections (60-90 nm) were cut using a Reichert-Jung Ultracut E and post-stained in a saturated uranyl acetate solution in ethanol, rinsed in distilled water and stained again with a bismuth subnitrate solution before visualization in a Zeiss EM10 transmission electron microscope.
Results
Cell surface signalling by HIV-1 is insufficient to trigger apoptosis o f target cells
The ability of infectious and non-infectious HIV to induce apoptosis was compared. To prepare non- infectious virus, HIV-1LA ~ was treated with 4'-amino- methyltrioxsalen (10 pg/ml) in combination with long- wavelength UV light (Watson et al., 1990). This rendered a high titre stock (5 × 107 TCIDs0/ml) essentially non- infectious (< 10 TCIDs0/ml). We assessed the ability of untreated and UV-treated HIV-1LA ~ to bind to CD4 + T cells (4h at 37 °C) and quantified the amount of internalized virus. Treated virions were shown to bind specifically to the surface and to penetrate the cells as assessed by p24 ELISA ( 2 5 9 0 120 pg/ml per l0 s cells

50
40
30
20
1 2 3
10
-~ 0 o .,= O
20 tz~ <
HIV-induced apoptosis requires infection 683
(b)
15
10
0 1 2 Time post-infection (days)
Fig. 1. Percentage of HIV-induced apoptotic cells in SupT1 cells and CD4+-enriched PBL. (a) Percentage of apoptotic cells present when SupT1 cells were treated with HIV-ILA ~ at an m.o.i, of 0.5 and the equivalent amount (as assessed by p24 concentration) of long wavelength UV psoralen-treated HIV-I at days 1, 2 or 3 post- inoculation. (b) Percentage of apoptotic cells present when CD4 ÷- enriched PBL were treated with HIV-I~AI at an rn.o.i, of 0-5 and long wavelength UV psoralen-treated HIV-1. Representative experiments are shown. Key: (11), control; ([]), UV psoralen-treated HIV-ILA~; (~5:]), H I V - l LAI"
after removal of surface virions). Infectious virions penetrated with a greater efficiency (two- to threefold more efficient). However, addition of UV psoralen- treated virus at higher m.o.i. (5; to compensate for this discrepancy in binding and internalization) to SupT1 cells did not result in apoptosis being detected even at later time points (day 7; results not shown).
FACS analysis using a cell cycle discrimination protocol and the use of the intercalative compound propidium iodide permitted the assessment of the proportion of cells in a specific stage of the cell cycle and the amount of DNA present in this population. A comparison of the percentage of cells in an apoptotic state for each group demonstrated that apoptosis occurred only with infectious HIV-1, implying that cell surface signalling resulting from viral binding alone was insufficient to trigger apoptosis in both SupT1 (Fig. 1 a)
and CD4+-enriched PBL (Fig. 1 b). Furthermore, all cells excluded the viability dye trypan blue ( > 95 %) implying that the plasma membrane was still intact. This ex- perimental approach also excluded the possibility that non-infectious cell-associated virus induced apoptosis through CD4-gpl20 interaction. Variation in the per- centage of apoptosis observed was noted among experi- ments, more so in the CD4+-enriched T lymphocytes. This variation was probably due to the time elapsed between fixation and the time when flow cytometric analysis was performed (Darzynkiewicz et al., 1992). The divergence for the PBL may also be partly attributed to donor cell variation. Nevertheless the inability of inactivated virus to induce apoptosis was documented in all experiments.
HIV infection induced a drastic reduction of cell surface CD4 receptor expression [Fig. 2a; (i) to (iii)]. For example, a viral inoculum at an m.o.i, of 0-5 required 3 days to induce the complete disappearance of CD4 from the surface of nearly all SupT1 cells. This effect was accompanied by an increase in apoptotic cell death; however, only a minority of cells (19% at day 3 for this same experiment) was demonstrated to be apoptotic (Fig. 2b).
The presence of characteristic digestion of host cell DNA and nuclear condensation are hallmarks of apoptosis, To confirm that the process observed by FACS analysis was indeed apoptosis, morphological examination by electron microscopy and D NA frag- mentation analysis were performed. Fig. 3(a) is a photomicrograph of an uninfected SupT1 cell. The nucleolus appeared as a darker electron-dense structure within the nucleus. Fig, 3(b) is a representative field at low magnification of SupT1 cells 48 h after infection with HIV-1LA~ at an m.o.i, of 0.5. Cells were at various stages of apoptotic death as exemplified by the different degrees of chromatin condensation. This phenomenon could be seen at a higher magnification in Fig. 3(c) where extensive chromatin condensation occurred with invagination of the nucleus. Fig. 3 (d) is a higher magnification of the inset of figure 3 (c) showing that, despite the extensive viral budding and chromatin condensation, the mem- brane was still intact and the mitochondria appeared to retain their normal morphology. Fragmented DNA could be detected within 48 h in HIV-inoculated cell cultures (results not shown).
Exam#tation by FACS analysis of the point of induction of apoptosis in SupT1 cells
The percentage of apoptotic cell death was assessed by FACS analysis in the presence of two HIV-1 inhibitors: ddI, a nucleoside analogue that prevents reverse tran- scription of input viral RNA and RO31-8959, an

684 J. Corbeil and D. D. Richman
(a)
240
160
80
0 240
160
o o
80
0 240
160
80
CD4 expression
(i) 2 nd Ab CD4
(ii)
99 %
30 %
0 0 v,,...._ 0 64 128 192 256 0 64 128 192 256 0 64 128 192 256 Relative fluorescence (log scale) DNA content
Fig. 2. (a) Cell surface expression of the CD4 receptor during infection with HIV- 1LAI Samples were taken at days 1 and 3, The controls (i) were uninfected SupT1 cells stained either with the anti-CD4 antibody OKT4 and FITC-labelled goat anti-mouse antibody or with the FITC-labelled goat anti-mouse antibody only (2 nd Ab). (ii) Represents the amount of cell surface expression of CD4 1 day post- infection where 30 % of the cells still expressed CD4. (iii) Represents SupT1 cells, 3 days post-infection where only 4 % of the cells retained CD4 expression on their surface. The control, uninfected SupT1 cells, at day 3 were similar to day 1 (results not shown). (b) Shows the percentage of HIV-induced apoptotic cells present when SupT 1 cells were infected with HIV-1 ~ai at an m.o.i, of 0-5. Aliquots were taken at days 1, 2 and 3 and the percentage of apoptotic cells assessed by flow cytometry using propidium iodide (see Methods). The percentages of apoptotic cells present are indicated in the top right corner of each panel (labelled apo). Control cells had less than 3 % of ceils in the sub-G 1 region for all three days. An accumulation of cells in the G2/M period of the cell cycle was detected during the course of the infection with a concomitant reduction of cells in G 1.
(b) Control HIV- 1
1200 G 1 5 %
\
Controls 800 [ X ~
Day 1 ' ' Day 1 p.i. G2/M
APO I ~ . . ~ 400 i
0j<<u 1200
6%
HIV-l-infected 800 i Day 1 p,i.
400 IAPO' I A Day 2 p.i.
1200 19 % 4%
HIV- 1 -infected
Day 3 p.i. 800
Day 3 p.i.
I APO 400
inhibitor of HIV aspartic protease that prevents viral maturation after budding and therefore yields non- infectious viral particles. These compounds had no intrinsic effect on cell viability when added alone. The reverse transcriptase inhibitor, ddI at a concentration that completely inhibited infection (20 gM), prevented apoptosis when added 2 h before viral inoculation; however, when added 6 h after inoculation ddI failed to prevent apoptosis (Table 1). At this time into the first round of the viral replicative cycle (which takes 20 to 24h for HIV-1LA~) reverse transcription has been accomplished in a significant proportion of the target cells. The experiments were conducted in the continuous presence of the drugs and therefore only the cells infected
in the first 6 h contribute to the apoptotic process because no viral spread should occur in the presence of the inhibitors.
The HIV protease inhibitor, RO31-8959 (2pM), marginally reduced apoptotic cell death (but not to the extent of ddI) when added either before or after viral inoculation. A decrease in the percentage of apoptotic cell death was noted for HIV-l-inoculated SupT1 cells pretreated for 2 h with the protease inhibitor. This small decline may be attributed to some inhibitory activity in the early phase of the virus life cycle (Nagy et al., 1994). Apoptotic cell death peaked at day 2 or 3 for each of the experiments consistent with the amount of virus utilized and the rapid pace of the apoptotic process. However,

HIV-induced apoptosis requires infection 685
(a)
!
(c)
(b)
IJ,
Fig. 3. Morphological changes associated with HIV-induced apoptosis. Photomicrographs of SupT 1 cells infected with HIV-1LAI at an m.o.i, of 0.5. The cells were processed at 48 h p.i. (a) Uninfected SupT1 cell. Bar marker represents 1 lain. (b) HIV-infected SupT1 cells. Bar marker represents 5 gm. HIV-I is seen budding out of an infected cell (arrowhead). Chromatin condensation and nuclear invagination can be seen in most cells with a very late stage cell at the bottom left corner of the photomicrograph. Apoptosis occurs at the single cell level and is independent of syncytium formation. (c) An HIV-infected SupT 1 cell. Bar marker represents 1 ~tm. HIV- infected SupT1 cell showing budding and characteristic apoptotic features. (d) Inset of boxed region of (c). Bar marker represents 0.1 ~tm. Note the viral budding indicative of viral production and the chromatin condensation and that the mitochondria remain morphologically unaffected at this stage; both are characteristic of apoptosis. This is also an example of active viral synthesis in a cell at an early stage of apoptosis.
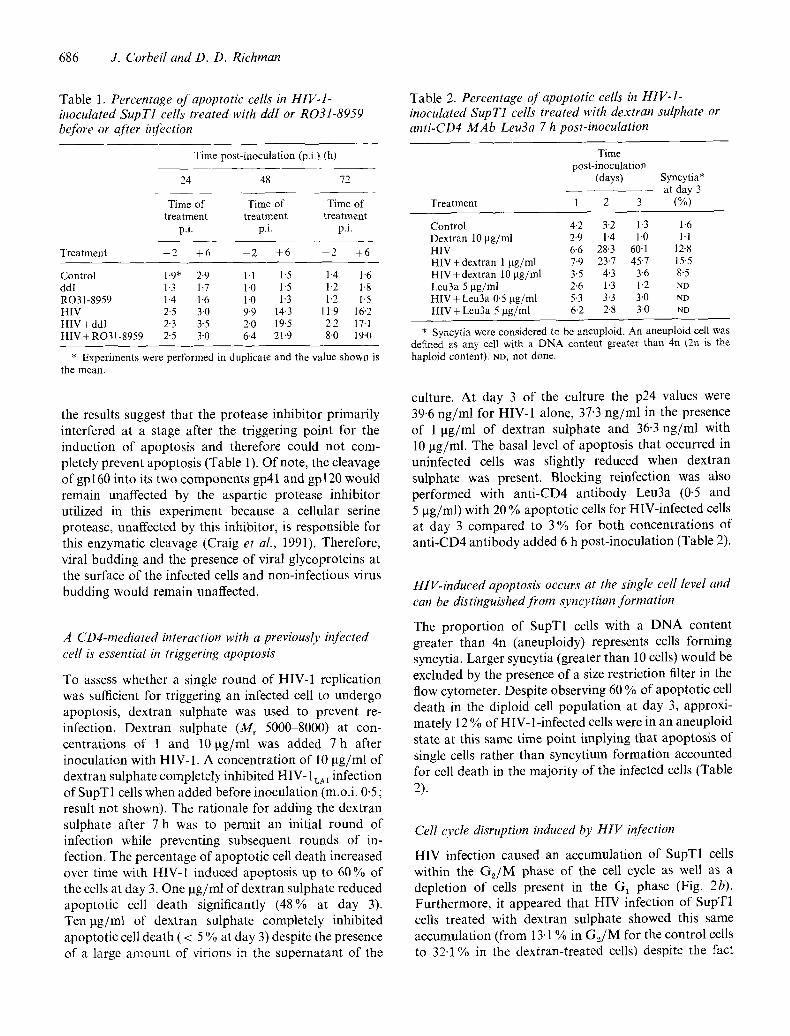
686 J. Corbeil and D. D. Richman
Table 1. Percentage of apoptotic cells in HIV.1- inoculated SupT1 cells treated with ddI or R031-8959 before or after infection
Treatment
Time post-inoculation (p.i.) (h)
24 48 72
Time of Time of Time of treatment treatment treatment
p.i. p.i. p.i.
- 2 +6 - 2 +6 - 2 +6
Control 1.9" 2.9 1-1 l 5 1.4 1-6 ddI 1.3 1.7 1-0 1"5 1.2 1.8 RO31-8959 1.4 1.6 10 1"3 1.2 1'5 HIV 2.5 3,0 9.9 1+3 11.9 16"2 H I V + d d l 2.3 3.5 2.0 19-5 2.2 17,1 HIV+ RO31-8959 2-5 3-0 6-4 21-9 8-0 19,0
* Experiments were performed in duplicate and the value shown is the mean.
the results suggest that the protease inhibitor primarily interfered at a stage after the triggering point for the induction of apoptosis and therefore could not com- pletely prevent apoptosis (Table 1). Of note, the cleavage of gpl60 into its two components gp41 and gpl20 would remain unaffected by the aspartic protease inhibitor utilized in this experiment because a cellular serine protease, unaffected by this inhibitor, is responsible for this enzymatic cleavage (Craig et al., 1991). Therefore, viral budding and the presence of viral glycoproteins at the surface of the infected cells and non-infectious virus budding would remain unaffected.
A CD4-mediated interaction with a previously infected cell is essential in triggering apoptosis
To assess whether a single round of HIV-1 replication was sufficient for triggering an infected cell to undergo apoptosis, dextran sulphate was used to prevent re- infection. Dextran sulphate (M r 5000-8000) at con- centrations of 1 and 10 gg/ml was added 7 h after inoculation with HIV-1. A concentration of 10 gg/ml of dextran sulphate completely inhibited HIV-1 LA~ infection of SupT1 cells when added before inoculation (m.o.i. 0.5; result not shown). The rationale for adding the dextran sulphate after 7 h was to permit an initial round of infection while preventing subsequent rounds of in- fection. The percentage of apoptotic cell death increased over time with HIV-1 induced apoptosis up to 60 % of the cells at day 3. One lag/ml of dextran sulphate reduced apoptotic cell death significantly (48% at day 3). Ten gg/ml of dextran sulphate completely inhibited apoptotic cell death ( < 5 % at day 3) despite the presence of a large amount of virions in the supernatant of the
Table 2. Percentage of apoptotic cells in HIV-1- inoculated SupT1 cells treated with dextran sulphate or anti-CD4 MAb Leu3a 7 h post-inoculation
Time post-inoculation
(days)
Treatment 1 2 3
Syncytia* at day 3
(%)
Control 4.2 3.2 1-3 1-6 Dextran 10 ~tg/ml 2.9 1.4 10 1.1 HIV 6.6 28.3 60-1 12.8 HIV + dextran 1 gg/ml 7.9 23-7 45.7 15.5 HIV+dext ran 10 gg/ml 3.5 4.3 3-6 8.5 Leu3a 5 gg/ml 2.6 1.3 1.2 ND HIV + Leu3a 0.5 gg/ml 5-3 3.3 3.0 ND HIV+Leu3a 5 gg/ml 6.2 2.8 3.0 ND
* Syncytia were considered to be aneuploid. An aneuploid cell was defined as any cell with a DNA content greater than 4n (211 is the haploid content). YD, not done.
culture. At day 3 of the culture the p24 values were 39.6 ng/ml for HIV-1 alone, 37"3 ng/ml in the presence of 1 tJg/ml of dextran sulphate and 36.3 ng/ml with 10 ~tg/ml. The basal level of apoptosis that occurred in uninfected cells was slightly reduced when dextran sulphate was present. Blocking reinfection was also performed with anti-CD4 antibody Leu3a (0-5 and 5 ~tg/ml) with 20 % apoptotic cells for HIV-infected cells at day 3 compared to 3 % for both concentrations of anti-CD4 antibody added 6 h post-inoculation (Table 2).
H1V-induced apoptos& occurs at the single cell level and carl be distingu&hed from syncytium formation
The proportion of SupT1 cells with a DNA content greater than 4n (aneuploidy) represents cells forming syncytia. Larger syncytia (greater than 10 cells) would be excluded by the presence of a size restriction filter in the flow cytometer. Despite observing 60 % of apoptotic cell death in the diploid cell population at day 3, approxi- mately 12 % of HIV-l-infected cells were in an aneuploid state at this same time point implying that apoptosis of single cells rather than syncytium formation accounted for cell death in the majority of the infected cells (Table 2).
Cell o, cle disruption induced by HIV infection
HIV infection caused an accumulation of SupT1 cells within the G J M phase of the cell cycle as well as a depletion of cells present in the G 1 phase (Fig. 2b). Furthermore, it appeared that HIV infection of SupT1 cells treated with dextran sulphate showed this same accumulation (from 13" 1% in G J M for the control cells to 32.1% in the dextran-treated cells) despite the fact

HIV-induced apoptosis requires bTfection 687
1600
1200
800
1600 o
-~ 1200
(a)
400
APO: 3.2 % G2/M: 13-1% GI: 53.6 %
800
G2/M
/ i ^
0 '-""-----'--~ " ~ ' ~ ' ~ ~',,-.--"
(d) APO: 23-7 % G2/M: 24.9 % Gl: 26"9 %
400
(b)
(e)
APO: 1,4 % G2/M: 17-7 % G~: 51.7 %
APO: 4.3 % G2/M: 32.1% GI: 35.2 %
0 0 40 80 120 160 200
. , - - - , - . - ~
200 0 40 80 120 160 DNA content
(c) APO: 28-3 % G2/M: 24.8 % G1:25.7 %
0 40 80 120 160 20
Fig. 4. Flow cytometric profiles and cell cycle analysis of SupT1 cells 2 days p.i. (a) Uninfected cells. (b) Uninfected cells treated with dextran sulphate (10 [ag/ml). (c) HIV-1LA~-infected SupT1 cells (m.o.i. 0.5). (d) HIV-1LAi-infected SupTI cells with dextran sulphate (1 gg/ml). (e) HIV-1Lai-infected SupT1 cells with dextran sulphate (10 gg/ml). A significant accumulation of cells in G2/M was detected in HIV-infected cells with a concomitant reduction of cells present in G r This G J M accumulation was not abrogated by the presence of dextran sulphate (10 gg/ml); however the G1 reduction and the extent of apoptosis were drastically reduced at this concentration of dextran sulphate.
that apoptosis was completely blocked in these cells (4 % only) [compare Fig. 4(c) and (e)]. Interestingly, this prevention of apoptosis by dextran sulphate (10 gg/ml) also increased the proportion of cells in G 1 (from 25"7 % for the HIV-infected cells to 35.2% in the dextran- treated cells) as compared to HIV-infected SupTl cells (Fig. 4c) and HIV-infected SupT1 cells treated with a suboptimal dose of dextran sulphate (1 gg/ml) (Fig. 4d).
Discussion
These studies were initiated to investigate the mechanism of HIV-induced apoptosis. Lymphoid cells are especially prone to apoptosis, a natural phenomenon in the life history of these ceils. Apoptosis in mature T cells has previously been demonstrated to occur by separate ligation of CD4 and the T cell receptor (TCR) for antigen (Newell et al., 1990) and affect activated T cells (Wesselborg et al., 1993). At the onset, it appeared that aberrant cell surface signalling between HIV virions and an uninfected cell was insufficient to trigger the cascade of events resulting in cell death. Inactivated UV psoralen- treated HIV did not induce apoptosis despite being able to bind CD4 and be internalized in a manner similar to
an infectious viral preparation. Interestingly, as de- scribed by Schwartz and collaborators, membrane- bound HIV-1 envelope glycoproteins stably expressed in HeLa cells were capable of inducing apoptosis in a human CD4 + T cell clone (SPB21) and primary blood mononuclear cells possibly through multimerization of CD4 molecules that impairs normal TCR stimulation and presumably this anergic state would result in apoptosis (Schwartz et al., 1994). In this particular system, however, the level of expression of gpl20 may differ from a true infection because the cells express env products under dhfr selection. Moreover, cultured T cell clones are exquisitely susceptible to apoptosis. Laurent- Crawford and collaborators have demonstrated that gpl20-gp41 heterodimer complex may be programming cell death in CD4 + lymphocytes (Laurent-Crawford et al., 1993). The expression of the env gene in the lymphoblastoid T cell line CEM was sufficient to induce apoptosis possibly by generating the multimerization of surface CD4. In the vaccinia virus system used to express their constructs, the presence of this virus, the absence of any mechanisms for CD4 downregulation (in particular Net) and the disruption of intracellular signalling processes may also be triggering factors independently of

688 J. Corbeil and D. D. R i c h m a n
gp120/41 complexes being present. The expression of these same constructs in CD8 + T cells as a control would be reassuring.
In our experimental model, direct infection appeared to be an essential requirement for the induction of apoptosis. This phenomenon had been previously alluded to by a number of investigators (Groux et al., 1992; Laurent-Crawford et al., 1991; Terai et al., 1991). The extent of apoptosis observed was in direct cor- relation with the amount of virus inoculated suggesting that a viral factor must accumulate before initiating the steps leading to apoptosis or that a certain threshold of virus must be reached before inducing the necessary cellular factor(s) required for apoptosis (results not shown). Alternatively, HIV-! may also inhibit essential cellular processes, as suggested by the cell cycle anomalies detected, and drive the celt to undergo apoptosis.
Downregulation of the CD4 receptor, through the expression of the n e f g e n e product (Garcia et al., 1993), trapping by gpl60 (Crise et al., 1990), or vpu induced degradation of CD4 (Willey et al., 1992), is an important feature of HIV infection. This nearly complete down- regulation may be a prerequisite for the persistence of infected cells in the presence of high viral load and implies that CD4 downmodulation may be a mechanism for the virus to prolong its host cell's survival to increase viral propagation. Of note, the propensity of target cells to undergo apoptosis seemed to correlate with the amount of surface CD4 and the generation of chronically HIV-infected cell lines reflect both this resistance to apoptosis and downregulation of CD4.
The use of different HIV inhibitors allowed the selective blockade of different steps in the viral replicative cycle and thus demonstrated that completion of the replicative cycle with expression of viral proteins on the surface of the infected cell was necessary to induce apoptosis. It can be surmised that the machinery for cell death is fully operational at a point past reverse transcription and that HIV-infected ceils are suitably primed to undergo apoptosis (Cohen et al., 1993; Gougeon et aL, 1993a). Only ddI added 2h before inoculation to block infection, prevented apoptosis (p24 < 100 pg/ml). The HIV protease inhibitor RO31- 8959 added either 2 h before or 6 h after (p24 > 40 ng/ml for both conditions) did not completely inhibit apoptosis presumably because it did not prevent the membrane expression of HIV glycoproteins. In addition, dextran sulphate and the anti-CD4 monoclonal antibody Leu3a blocked apoptosis in already infected cells indicating that a subsequent interaction mediated through the CD4 molecule was the triggering event which completed the process. It should be noted that the anti-CD4 antibody Leu3a binds the CD4 receptor at the binding site of gp120 (Sattentau et al., 1986) and paradoxically did not
induce apoptosis although the interaction of membrane- bound gp120 and CD4 must be quantitatively (valence) and qualitatively (affinity) different than that of an antibody. The utilization of novel HIV inhibitors and anti-CD4 antibodies targeted to different sites on the CD4 receptor may further refine this analysis and delineate the critical interaction needed to commit the cell to undergo apoptosis and which cellular processes are affected.
A number of different triggering signals and alternate mechanisms might induce apoptosis of a target cell (McConkey et al., 1990). Death by apoptosis is the endpoint of a large number of biological and signalling events (Allen et al., 1993; McCabe & Orrenius, 1992). The actual intracellular events occurring during HIV infection that trigger apoptosis have yet to be uncovered. Following HIV infection of SupT1 cells, a substantial accumulation of cells in the G J M phase of the cell cycle can be detected (Fig. 2 & 4). This accumulation was present despite completely preventing apoptosis by the addition of dextran sulphate (101ag/ml) 7h post- inoculation. This suggests that the block in G J M is insufficient to trigger apoptosis. Another anomaly of the cell cycle profile of SupT1 ceils upon HIV infection, apart from the appearance of an apoptotic population, was the selective depletion of the cells in the G 1 pool (from 53.6 % in the control to 25'7 % in HIV-infected cells). This depletion was partly prevented by the addition of dextran sulphate (10 ~tg/ml) suggesting that the apoptotic cells possibly originated from the G 1 pool of cells and were therefore unable to pass the G1-S transition checkpoint. We cannot however completely exclude the possibility that G, cells kept cycling and were not renewed due to apoptotic depletion of G~./M cells, implying that the cells may be undergoing apoptosis during the G2/M-G 1 transition. This situation is not unique to HIV infection since a number of virally transformed or tumour cells arrest in G2 (Traganos et al., 1994). A possible mechanism for this accumulation of cell in G J M could be for HIV to induce the over- production of Rb protein. The Rb protein is known to exert its activity at decision points in the G 1 phase of the cell cycle and is interacting with some component(s) of the cell cycle-regulatory machinery during the G 2 phase. It has been reported that overproduction of the Rb protein after the G1/S boundary causes G 2 arrest in a number of cell lines (Karantza et al., 1993).
The mechanism for apoptosis described here is distinct from the one reported by Banda (1992) where cross- linking of bound gpl20 on CD4 + T cells followed by signalling through the TCR by antigen-induced activation-dependent cell death. This cell surface sig- nalling is also a different mechanism to the one described by Groux et al. (1992) where CD4 + T cells isolated from

HIV-infected individuals, despite being mostly HIV- negative, were predisposed to undergo apoptosis and some did so when confronted with another immuno- logical challenge such as superantigens. Our results are in agreement with the apoptosis mechanism presented by Laurent-Crawford and collaborators (Laurent-Crawford et al., 1993) where membrane expression of HIV envelope glycoprotein, or in our case productive viral infection, would trigger apoptosis. However, subsequent CD4 engagement by gpl20 in the context of an infected cell would be required according to our observations. Binding to CD4 by gpl20 of virions alone was not sufficient to induce apoptosis (results not shown).
Our data suggest that HIV-1 penetrates the CD4 + T cell and generates a state perhaps analogous to the pre- apoptotic state found in thymocytes as proposed by Cohen et al. (1993). Upon further engagement of the CD4 molecule this process then proceeds to completion. This requirement for a productive HIV-1 infection may explain the slow decline of CD4 + T cells seen in vivo in the presence of persistently circulating virus (Piatek et al., 1993) since apoptosis is a very rapid mechanism which would quickly deplete the immune system if cell surface signalling events alone were required for triggering apoptosis. Hypothetically, a rate of only one cell death in a thousand per day would parallel the decline observed in most HIV-infected individuals (Adleman & Wofsy, 1993) and this rate could possibly be accounted for by the level of infection of CD4 + T cells found in asymptomatic individuals.
The authors acknowledge Dr Judy Nordberg for the cell cycle analysis by flow cytometry, Ms Abbyann Sisk for preparing samples for transmission electron microscopy, Dr Meredith L. Howell for generating the Southern blot of HIV-infected cells to confirm apoptosis and Dr Richard Kornbluth for critical comments on the manuscript. J.C. is supported by a Fellowship of the Commonwealth of Australia AIDS Research Committee. D. D. R. is supported by grants AI-27670, AI-30457 and AI-29164 from the NIH. This work was supported by the Research Center for AIDS and HIV Infection of the San Diego Veterans Affairs Medical Center and the UCSD Center for AIDS Research (AI-36214).
References ADLEMAN, L. M. & WOFSY, D. (1993). T-cell homeostasis: implications
in HIV infection. Journal of Acquired Immune Deficiency Syndromes 6, 144~152.
ALLEN, P. D., BUSTIN, S. A. & NEWLAND, A. C. (1993). The role of apoptosis (programmed cell death) in haemopoiesis and the immune system. Blood Reviews 7, 63-73.
AMEISEN, J,C. (1992). Programmed cell death and AIDS: from hypothesis to experiment, hnmunology Today 13, 388 391.
AMEISEN, J.C. (1994). Programmed cell death (apoptosis) and cell survival regulation: relevance to AIDS and cancer. AIDS 8, 1197-1213.
A~EISEN, J. C. & CAVRON, A. (1991). Cell dysfunction and depletion in AIDS: the programmed cell death hypothesis. Immunology Today 12, 102-105.
ATTANASIO, R., DILLEY, D., BUCK, D., MA1NO, V. C., LOHMAN, K. L., KANDA, P. & KENNEDY, R. C. (1991). Structural characterization of
HIV-induced apoptosis requires infection 689
a cross-reactive idiotype shared by monoclonal antibodies specific for the human CD4 molecule. Journal of Biological Chemistry 266, 14611-14619.
BANDA, N.K., BERNIER, J., KURAHARA, D.K., KURRLE, R., HAIGWOOD, N., SEKALV, R. P. & FINKEL, T. H. (1992). Crosslinking CD4 by human immunodeficiency virus gpl20 primes T cells for activation-induced apoptosis. Journal of Exper#nental Medicine 176, 1099-1106.
BRENNER, B. G., DASCAL, A., MARGOLESE, R. G. & WAINBERG, M. A. (1989). Natural killer cell function in patients with acquired immunodeficiency syndrome and related diseases. Journal of Leukocyte Biology 46, 75-83,
COHEN, J. (1993). What causes the immune system collapse seen in AIDS? Science 260, 1256.
COHEN, G.M., SuN, S.M.. SNOWDEN, R.T., ORMEROD, M.G. & DINSOALE, D. (1993). Identification of a transitional preapoptotic population of thymocytes. Journal of Immunology 151, 566-574.
CRAIG, J. C., DUNCAN, I. B., HOCKLEY, D., GRIEF, C., ROBERTS, N. A. & MILLS, J.S. (1991). Antiviral properties of RO31-8959, an inhibitor of human immunodeficiency virus (HIV) proteinase. Antiviral Research 16, 295-305.
CRISE, B., BUONOCORE, L. & ROSE, J. K. (1990). CD4 is retained in the endoplasmic reticulum by the human immunodeficiency virus type 1 glycoprotein precursor. Journal of Virology 64, 5585-5593.
DARZYNKIEWICZ, Z., BRUNO, S,, DEL BINO, G., GORCZYCA, W., HOTZ, M. A., LASSOTA, P. & TRAGANOS, F. (1992). Features of apoptotic cells measured by flow cytometry. Cytometry 13, 795-808.
Door), R.Y. (1994). Viral inactivation in platelet concentrates. Transfusion Clinique et Biologique l, 181-186.
EMBRETSON, J., ZUPANCIC, M,, RIBAS, J. L,, BURKE, A., RACZ, P., TENNER-RACZ, K. & HAASE, A. T. (1993). Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 362, 359-362.
GAO, W-Y., SHIRASAKA, T., JOHNS. D. J., BRODER, S. & MITSUYA, H. (1993). Differential phosphorylation of azidothymidine, dideoxy- cytidine, and dideoxyinosine in resting and activated peripheral blood mononuclear cells. Journal of Clinical Investigation 91, 2326-2333.
GARCIA, J. V., ALFANO, J. & MILLER, A. D. (1993). The negative effect of human immunodeficiency virus type 1 Nef on cell surface CD4 expression is not species specific and requires the cytoplasmic domain of CD4. Journal of Virology 67, 1511-1516.
GOUGEON, M.L. & MON'rAGNIER, L. (t993). Apoptosis in AIDS. Science 260, 1269-1270.
GOUGEON, M. L., GARCIA, S., HEENEY, J., TSCHOPP, R., LECOEUR, H., Gta~TARD, D., RAM'E, V., DAUGtrET, C. & MONTAGNIER, L. (1993a). Programmed cell death in AIDS-related HIV and SlY infections. AIDS Research and Human Retroviruses 9, 553-563.
GOUGEON, M. t . , LAURENT-CRAWFORD, m. G., HOVANESSIAN, A. G. & MONTAGNmR, L. (1993b). Direct and indirect mechanisms mediating apoptosis during HIV infection: contribution to in vivo CD4 T cell depletion. Seminars in Immunology 5, 187-194.
GROUX, H., TORPIER, G., MONTE, D., MOUTON, Y., CAPRON, A. & AMEISEN, J.C. (1992). Activation-induced death by apoptosis in CD4+ T cells from human immunodeficiency virus-infected asymptomatic individuals. Journal of Experimental Medicine 175, 331-340.
HANSON, C.V. (1992). Photochemical inactivation of viruses with psoralens: an overview. Blood Cells 18, 7-25.
KARANTZA, V., MAROO, A., FAY, D. & SEDIVY, J.M. (1993). Overproduction of Rb protein after the G1/S boundary causes G~ arrest. Molecular and Cellular Biology 13, 6640-6652.
K);.RBER, G. (1931). Beitrag zur kollektiven behandlung pharmako- logisher reihenversuche. Archiv fiir Experbnentelle Pathologic und Pharmakolog& 162, 480.
LAURENT-CRAWI~ORD, A. G., KRUST, B., MULLER, S., RWI~RE, Y., REY- CUILLE, M.A., BECHET, J. M., MONTAGNIER, L. t~. HOVANESSIAN, A.G. (1991). The cytopathic effect of HIV is associated with apoptosis. Virology 185, 829-839.
LAURENT-CRAWFORD, A. G., KRUST, B., RIVIERE, Y., DESGRANGES, C., MULLER, S., KIENY, M.P., DAUGUET, C. & HOVANESSIAN, A. G. (1993). Membrane expression of HIV envelope glycoproteins triggers

690 J. Corbeil and D. D. Richman
apoptosis in CD4 cells. AIDS Research and Human Retroviruses 9, 761 773.
LEVY, J.A. (1993). Pathogenesis of human immunodeficiency virus infection. Microbiological Reviews 57, 183-289.
MCCAaL M. J. JR & ORReNIUS, S. (1992). Deletion and depletion: the involvement of viruses and environmental factors in T-lymphocyte apoptosis. Laboratory Investigation 66, 403~,06.
McCONKEY, D.J., ORRENIUS, S. & JONDAL, M. (1990). Cellular signalling in programmed cell death (apoptosis). Immunology Today l l , 120-121.
MBEMBA, E., CHAMS, V., GLUCKMAN, J.C., KLATZMAN, D. & GATTEGNO, L. (1992). Molecular interaction between HIV-I major envelope glycoprotein and dextran sulfate. Biochemica et Biophysica Acta 1138, 62-67.
MEYAARD, L., OTTO, S.A., JONKER, R.R., MIJNSTER, M.J., KEET, R. P. & MIEDEMA, F. (1992). Programmed death of T cells in HIV- 1 infection. Science 257. 217-219.
MITSUYA, H., JARRETT, R. F., MATSUKURA, M., DI MARZO VERONESE, F., DEV1CO, A. L., SARNGADHARAN, M.G., JOHNS, D.G., REITZ, M.S. & BRODER, S. (1987). Long-term inhibition of human T- lymphotropic virus type III/lymphadenopathy-associated virus (human immunodeficiency virus) DNA synthesis and RNA ex- pression in T cells protected by 2',Y-dideoxynucleosides in vitro. Proceedings of the National Academy of Sciences, USA 84, 2033~037.
MITSUYA, H., LOONEY, D. J., KUNO, S., UENO, R., WONG-STAAL, F. & BRODER, S. (1988). Dextran sulfate suppression of viruses in the HIV family: inhibition of virion binding to CD4+ cells. Science 240, 646-648.
NAGY, K., YOUNG, M., BABOONIAN, C., MERSON, J., WHITTLE, P. & OROSZLAN, S. (1994). Antiviral activity of human immunodeficiency virus type 1 protease inhibitors in a single cycle of infection: evidence for a role of protease in the early phase. Journal of Virology 68, 757-765.
NEWELL, M. K , HAUGHN, L.J., MAROUN, C.R. & JULIUS, M.H. (1990). Death of mature T cells by separate ligation of CD4 and the T-cell receptor for antigen. Nature 347, 286-289.
PANTALEO, G., GRAZIOSI, C., DEMAREST, J. F., BUTINI, L., MONTRONI, M., Fox, C. H., ORENSTEIN, J. M., KOTLER, D. P. & FAUCI, A. S. (1993). HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362, 355-358.
PIATAK, M., JR, SAAG, M. S., YANG, L. C., CLARK, S. J., KAPPES, J. C., LUK, K. C., HAHN, B. H., SHAW, G. M. t~ LIFSON, J. D. (1993). High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 259, 1749-1754.
REDFIELD, D.C., RICHMAN. n. D., OXMAN, M. N. t~ KRONENBERG, L. H. (1981). Psoralen inactivation of influenza and herpes simplex viruses and of virus-infected cells. Infection and Immunity 32, 1216-1226.
ROSENBERG, Z. F. & FAUCI, A. S. (1991). Immunopathogenesis of HIV infection. FASEB Journal 5, 2382-2390.
SATTENTAU, Q.J., DALGLEISH, A.G., WEISS. R.A. & BEVERLEY, P. C. L. (1986). Epitopes of CD4 antigen and HIV infection. Science 234, ll20~1127.
SCADDEN, D. T., WANG, Z. t~ GROOPMAN, J. E. (1992). Quantitation of plasma human immunodeficiency virus type l RNA by competitive polymerase chain reaction. Journal of Infectious Diseases 165, 1119-1123.
SCHWARTZ, O., ALIZON, M., HEARD, J.M. & DANOS, O. (1994). Impairment of T cell receptor-dependent stimulation in CD4+ lymphocytes after contact with membrane-bound HIV-1 envelope glycoprotein. Virology 198. 360-365.
STEIN, D.S., KORVICK, J.A. & VERMUND, S.H. (1992). CD4+ lymphocyte cell enumeration for prediction of clinical course of human immunodeficiency virus disease. Journal of Infectious Diseases 165, 352 363.
TANNEAU, F., MCCHESNEV, M., LOPEZ, O., SANSONETTI, P., MONTAGNIER, L. &; RIVI~RE, Y. (1990). Primary cytotoxicity against the envelope glycoprotein of human immunodeficiency virus-l: evidence for antibody-dependent cellular cytotoxicity h7 vivo. Journal of lnfectious Diseases 162, 837-843.
TELFORD, W. G., KING, L. E. & FRAKER, P.J. (1992). Comparative evaluation of several DNA binding dyes in the detection of apoptosis-associated chromatin degradation by flow cytometry. Cytometrv 13, 132143.
TERAI, C., KORNBLUTH, R.S., PAUZA, C.D., RICHMAN, D.D. & CARSON, D. A. (1991). Apoptosis as a mechanism of cell death in cultured T lymphoblasts acutely infected with HIV-1. Journal of Clinical Investigation 87, 1710-1715.
TRAGANOS, F., GONG, J., ARDELT, B. • DARZYNKIEWICZ, Z. (1994). Effect of staurosporine on MOLT-4 cell progression through G2 and on cytokinesis. Journal of Cell Physiology 158, 535-544.
WATSON, A. J., KLANIECKI, J. & HANSON, C. V. (1990). Psoralen/UV inactivation of HIV-l-infected cells for use in cytologic and immunologic procedures. AIDS Research and Human Retroviruses 6, 503-513.
WEISS, R.A. (1993). How does HIV cause AIDS? Science 260, 1273-1279.
WESSELBORG, S., JANSSEN, 0. & KABELITZ, D. (1993). Induction of activation-driven death (apoptosis) in activated but not resting peripheral blood T cells. Journal of Immunology 150, 4338~,345.
WILLEY, R. L., MALDARELLI, F., MARTIN, M. A. & STREBEL, K. (1992). Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. Journal of Virology 66, 7193 7200.
ZARLING, J. M.~ LEDBETTER, J. A., SIAS, J., FULTZ, P., EICHBERG, J., GJERSET, G. & MORAN, P. A. (1990). HIV-infected humans, but not chimpanzees, have circulating cytotoxic T lymphocytes that lyse uninfected CD4 + cells. Journal of Immunology 144, 2992 2998.
(Received 24 June 1994, Accepted 14 October 1994)




















