Comparative Study of the Selective Sorption of Organic Dyes ...
Upload
independentCategory
view
7download
0
lable at ScienceDirect
Journal of Environmental Radioactivity 141 (2015) 90e96
Contents lists avai
Journal of Environmental Radioactivity
journal homepage: www.elsevier .com/locate / jenvrad
Plutonium(IV) sorption to montmorillonite in the presence of organicmatter
Mark A. Boggs*, Zurong Dai, Annie B. Kersting, Mavrik ZavarinGlenn T. Seaborg Institute, Physical and Life Sciences, Lawrence Livermore National Laboratory, PO Box 808 L-231, 94550 CA, USA
a r t i c l e i n f o
Article history:Received 10 October 2014Received in revised form11 December 2014Accepted 12 December 2014Available online
Keywords:PlutoniumColloidsHumic acidSiderophoreSorptionClay
* Corresponding author.E-mail address: [email protected] (M.A. Boggs).
http://dx.doi.org/10.1016/j.jenvrad.2014.12.0050265-931X/© 2014 Elsevier Ltd. All rights reserved.
a b s t r a c t
The effect of altering the order of addition in a ternary system of plutonium(IV), organic matter (fulvicacid, humic acid and desferrioxamine B), and montmorillonite was investigated. A decrease in Pu(IV)sorption to montmorillonite in the presence of fulvic and humic acid relative to the binary Pu-montmorillonite system, is attributed to strong organic aqueous complex formation with aqueousPu(IV). No dependence on the order of addition was observed. In contrast, in the system where Pu(IV)was equilibrated with desferrioxamine B (DFOB) prior to addition of montmorillonite, an increase inPu(IV) sorption was observed relative to the binary system. When DFOB was equilibrated with mont-morillonite prior to addition of Pu(IV), Pu(IV) sorption was equivalent to the binary system. X-raydiffraction and transmission electron microscopy revealed that DFOB accumulated in the interlayer ofmontmorillonite. The order of DFOB addition plays an important role in the observed sorption/desorp-tion behavior of Pu. The irreversible nature of DFOB accumulation in the montmorillonite interlayer leadsto an apparent dependence of Pu sorption on the order of addition in the ternary system. This workdemonstrates that the order of addition will be relevant in ternary systems in which at least onecomponent exhibits irreversible sorption behavior.
© 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Decades of nuclear weapons production and testing, disposal ofnuclear material, scheduled and accidental discharges from nuclearpower reactors have all lead to plutonium (Pu) being introducedinto the environment. For instance, approximately 4.8 � 1018 Bq ofPu have been released into the subsurface at the Nevada NationalSecurity Site, Nevada USA (Smith et al., 2003). The large quantitiesof Pu present in the environment combined with its radiologicaland chemical toxicity (Taylor, 1989) and long half-life (239Pu t1/2 ¼ 2.4 � 104) make understanding and predicting the fate of Pu inthe environment important.
Sorption of Pu to montmorillonite is of particular importance asclays are ubiquitous in the environment. In addition, bentonite,composed primarily of montmorillonite, has been proposed as abackfill material for long-term radioactive waste disposal sites inmany countries. While Pu sorption to montmorillonite may lead toimmobilization of Pu, sorption of Pu to inorganic colloidal matter
(Kersting et al., 1999; Novikov et al., 2006) (including montmoril-lonite) as well as natural organic matter (Zhao et al., 2011) has beenlinked to increases in Pu environmental mobility. Thus Pu sorptionto montmorillonite may lead to immobilization and/or mobiliza-tion depending on the nature and conditions of Pu interaction withmontmorillonite.
The behavior of Pu on mineral and organic surfaces is a functionof its oxidation state, where Pu is known to exist primarily in the IVand V oxidation states in natural waters (Choppin, 2003). Previousstudies have observed reduction of Pu(V) to Pu(IV) on the surface ofmontmorillonite (Zavarin et al., 2012), though the rates are farslower than those of iron minerals (Powell et al., 2005, 2004).Sorption of Pu(IV) to montmorillonite has been shown to be linearover 10 orders of magnitude in Pu concentration. (Begg et al., 2013)Under atmospheric low ionic strength conditions, sorption ishighest at pH 4 and decreases as pH increases (Table 1).
Humic substances are large organic molecules derived from thedegradation of organic matter in the environment. Two majorclassifications of humic substances are humic acid (HA) and fulvicacid (FA), which are operationally differentiated by the pH range inwhich they are soluble. FA is soluble at all pH while HA is insolubleat low pH. In addition to solubility, HA has greater carbon content

Table 1Thermodynamic constants for Pu(IV) interaction with organic ligands and mineralsurface used in these studies. All stability constants are expressed at zero ionicstrength unless noted in the table. *PuFA stability constants from personalcommunicationwith Brian Powell ([email protected]) and are representative ofthe species present at pH 4. As there is currently no comprehensive list of Pu(IV) eFA complexation constants, data in this table have been used to predict speciationover the range of pH studied.
pH log(Kd) Source
4 6.2 ± 0.28 Begg et al. 20146 4.92 ± 0.03 Begg et al. 20148 4.48 ± 0.02 Begg et al. 201410 4.18 ± 0.02 Begg et al. 2014
Species logb Source
Pu4þ þ FA$PuFA 0.92 ± 0.02 Powell *Fe3þ þ FA$FeFA 2.4 ± 0.2 Tipping et al., 1998Pu4þ þ HA$PuHA 16.6 ± 0.3 Szabo et al., 2010Pu4þ þ ðHAÞHþ H2O$PuðOHÞ2HAþ 3Hþ 6.8 ± 0.3 Zimmerman et al.,
2014Fe3þ þ HA$FeHA 2.5 ± 0.2 Tipping et al., 1998Pu4þ þ DFOB2� þ 2Hþ$PuH2DFOB
4þ 35.5 ± 0.5 Boukhalfa et al.,2007
Pu4þ þ DFOB2� þ Hþ$PuHDFOB3þ 34.9 ± 0.3 Boukhalfa et al.,2007
Pu4þ þ DFOB2�$PuDFOB2þ 35.8 ± 0.9 Boukhalfa et al.,2007
Pu4þ þ H2Oþ DFOB2�$PuDFOBðOHÞþþHþ 27.33 ± 0.05 Boukhalfa et al.,2007
Fe3þ þ DFOB2�$FeDFOBþ 30.99 Evers et al., 1989(0.1 M KCl)
M.A. Boggs et al. / Journal of Environmental Radioactivity 141 (2015) 90e96 91
and is generally larger than FA. Humic substances play an importantrole in the transport of metals in the environment and studies ofnuclear fallout in Japan and Korea have found that up to 60% of Puwas associated with HA and FA (Fujikawa et al., 1999; Lee and Lee,2000).
As a tool for solubilizing iron necessary for metabolic activity, anumber of bacteria and fungi produce strong chelators calledsiderophores. With few exceptions, fungal siderophores containhydroxamic acid functional groups (Renshaw et al., 2002), givingthem a distinct chemical difference to the carboxylic acid functionalgroups of humic and fulvic acid. Siderophores are known to formsoluble complexes with more than just iron, and increases in Puoxide dissolution have been observed in the presence of naturallyoccurring siderophores (Brainard et al., 1992). One commonlystudied siderophore is desferrioxamine B (DFOB); consisting ofthree hydroxamic acids linked by succinic acid and dia-minopentane chains (Fig. 1). DFOB is critical for iron acquisition in avariety of cell strains (Roberts et al., 2012) and select microbes havebeen shown to produce up to 0.136 mM DFOB in iron deficientenvironments (Kraemer et al., 2002). In metal-free solutions, DFOBexists primarily as a linear chain, though upon complexation withmost metals it forms a cyclical complex (Kiss and Farkas, 1998). The
Fig. 1. Structure of desferrioxam
tendency to form cyclical multidentate complexes results in verystrong complexes, such as those of FeIII and Pu(IV) (Table 1) (Everset al., 1989). DFOB is attributed to decreases in metal sorption tozeolites (Karimzadeh et al., 2013), kaolinite (Neubauer et al., 2000),and iron minerals (Neubauer et al., 2000), while increases insorption have been observed with swelling clays (Neubauer et al.,2000; Siebner-Freibach et al., 2004).
Experiments where the order of addition of metal, ligand, andmineral were examined have been generally limited to HA and FA,of which no consensus in the literature on its importance has beenreached. For instance, no effect was observed with thorium, ananalog for Pu, for the Th(IV)-HA-bentonite system (Song et al.,2013), while up to a 90% difference in Th(IV) sorption wasobserved for Th(IV)-HA-hematite system (Reiller et al., 2005). In thecase of the Pu(IV)-HA-kaolinite system, the order of addition had noeffect (Buda et al., 2008). However, in both the Pu(IV)-HA-kaoliniteand Th(IV)-HA-bentonite systems, an increase in sorption at lowpHin the presence of humic substances was observed and attributed tosurface ternary complexes. Order of addition effects in the Cm(III)-HA-silica system varied with pH and were partially attributed toCm(III) incorporation into the silica (Kar et al., 2011). Sorption ofNi(II) in the Ni(II)-HA/FA-montmorillonite system showed no effectin the order of addition (Xu et al., 2008). Alternatively in the Ni(II)-FA-bohemite (g-AlO(OH)) system order of addition did not affectsurface sorption on the macroscale, but different amounts of thesurface species Ni(II)-FA-AlOOH and FA-Ni(II)eAlOOH wereobserved by Extended X-ray Absorption Fine Structure (EXAFS)spectroscopy (Strathmann and Myneni, 2005). The different Ni(II)-FA-bohemite surface complexes were investigated at 250 mg/Lfulvic acid. At these concentrations, FA is known to form highmolecular weight aggregates, which may not reflect the chemistryof the smaller molecules present at lower concentrations. Nostudies examining the order of addition of DFOB are present in theliterature to date. However, order of addition of arginine, a nitrogencontaining small ligand, had an effect on Ni(II) sorption to a loamysand while the carboxylic acid containing citrate showed no effect(Poulsen and Hansen, 2000).
Thermodynamic stability constants of Pu(IV)-organic com-plexes, and some comparable Fe(III) complexes, used in our studiesare shown in Table 1. Binding constants of Pu(IV)-HA have beendetermined using HA immobilized on silica gel and ultrafiltration,resulting in different apparent binding constants (Szabo et al.,2010; Zimmerman and Powell, 2010). The representation of thethermodynamic data differs by almost ten orders of magnitude.However, when the thermodynamic data are adjusted for the dif-ferences in complex formed (e.g Pu(IV)HA vs. Pu(IV) (OH)3HA) theyare in better agreement. For the sake of simplicity, the HA complexof Pu(IV) is referred to as Pu(IV)HA in this manuscript. The mostrecent complexation data of Zimmerman et al. (2014) were used inour speciation model. In the presence of HA, Pu(V) and Pu(VI) areknown to reduce to Pu(IV) (Andre and Choppin, 2000; Nash et al.,
ine B mesylate (DFOB) salt.

M.A. Boggs et al. / Journal of Environmental Radioactivity 141 (2015) 90e9692
1981). This reduction simplifies speciation calculations as it can beassumed that Pu(IV) is the only oxidation state present in ourternary sorption experiments.
In this paper, we present the results of a study investigating thesorption of Pu(IV) tomontmorillonite in the presence of three typesof organic matter (HA, FA, and DFOB). The order of organic matteraddition was investigated to determine if order of addition mayinfluence Pu(IV) sorption characteristics. In addition, transmissionelectron microscopy (TEM) and x-ray diffraction (XRD) wereemployed to examine whether sorption of organic matter can leadto changes in the morphology and interlayer structure ofmontmorillonite.
2. Methods
Unless otherwise noted all chemicals were of high purity (re-agent grade) and used without any additional purification. A 238Pu(98.9% 238Pu, 0.11% 241Pu, and 0.1% 239Pu by activity) stock wasprepared for all experiments. Impurities were removed by loadingPu onto a Bio Rad AG 1-8X resin, in 8M HNO3, and eluting as Pu(III)with an HI:HCl mixture. The eluent was heated to dryness severaltimes to remove excess HI and subsequently dissolved in 1.0N HCl.The oxidation state of the Pu stock solution was verified by UVeVisand LaF3 precipitation (Kobashi et al., 1988) to be IV (99%). 238Puconcentrations were determine by Liquid Scintillation Counting(Packard Tri-Carb TR2900 LSA) for all experiments.
Preparation of montmorillonite colloids from SWy-1 (SourceClays Repository of the Clay Minerals Society) used in this study isdescribed in Zavarin et al. (2012). Briefly, montmorillonite wastreated with 1 mM HCl and 30 mM H2O2 to remove salts and limitredox active species. The clay was homoionized with 10 mM NaCland dialized in >18 MU water (Milli-Q Gradient System). Afterhomoionization, the clay was suspended in >18 MU water andcentrifuged at 180 g for 5 min to sediment particles >2 mm. Theremaining supernatant was then centrifuged at 2500 g for 6 h tosediment all particles <50 nm. The suspension was dried at 40 �Cand solutions of 10 g/mL montmorillonite were prepared in NaCl/NaHCO3. The surface area and iron content of SWy-1 Na-mont-morillonite prepared under these conditions was previouslydetermined to be 31.5 ± 0.17 m2 and 9.5 ± 0.6 � 10�6 molFe/gclay,respectively (Begg et al., 2013). Considering possible soluble Fe(III)and Fe(III) organic complexes an excess of uncomplexed ligand willbe present under the conditions of our experiments.
Elliot Soil HA standard and Suwannee River FA were purchasedfrom the International Humic Substances Society and used with nofurther purification. Solid HA/FA was dissolved in 0.002 M freshlyprepared NaOH to reach 100 ppm in carbon. Desferrioxamine Bmesylate (SigmaeAldrich) was dissolved in >18 MU water to reach100 ppm C. All solutions were made fresh monthly and stored inamber glass at 3 �C.
Sorption experiments were performed by equilibrating 0.1 g/Lmontmorillonite, 0.8 ppm carbon (as HA, FA, or DFOB) and1 � 10�10 M 238Pu. Freshly prepared NaOH and HCl were used tocontrol pH. Ionic strength was held constant at 10 mM using NaCland NaHCO3 and equilibrated with atmospheric conditions. Sam-ples were shielded from light and equilibrated under gentleagitation at room temperature for the entirety of the experiment.Independent of pH, sorption of Pu(IV) to montmorillonite reachedequilibrium after 1 month (Supplementary Table 1). Ternary sam-ples were initially prepared as binary systems of Pu-ligand orligand-montmorillonite and allowed to equilibrate for 1 month.Either Pu(IV) or montmorillonite was added after the initial binaryequilibration. Samples were then equilibrated as ternary systemsfor another month prior to sampling. Aqueous Pu concentrationswere determined by centrifugation at 9200 g for 2 h to remove
solids (~50 nm cutoff), after which an aliquot of the supernatantwas taken for analysis by liquid scintillation counting (LSC). Sys-tems where Pu(IV) and an organic complexant were equilibratedprior to the addition of montmorillonite will be referred to asPu(IV)-complexant-montmorillonite, while those where theorganic complexant was equilibratedwithmontmorillonite prior tothe addition of Pu(IV) will be referred to as complexant-montmorillonite-Pu(IV) from here forward.
XRD and TEM characterization was performed on samples of5 mg dry Na-montmorillonite dissolved in 1.0 mL of 0.01 M NaCl inthe presence of DFOB. After a 24 h equilibration, aliquots wererinsed three times with >18 MU water and placed on a zero back-ground plate for XRD analysis or on a carbon coated copper grid forTEM analysis and air dried. XRD analysis was performed on aBruker D8 X-ray diffractometer using radiation from a copper targettube (Cu Ka) and operating at 40 kV, 40 mA and a scanning speed of0.5�/min, while TEM analysis was performed on a Philips CM300FEG super-twin microscope operating at 300 kV.
3. Results/discussion
3.1. Binary sorption
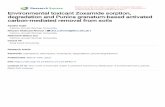
Sorption of Pu(IV) to montmorillonite in the absence of anycomplexing ligand is shown in Fig. 2. Sorption is greatest betweenpH 4 and 6 and decreases under alkaline conditions, which agreeswith published Kd values (Table 1) (Begg et al., 2013). The decreasein sorption at pH greater than 8 may be attributed to Pu(IV) hy-drolysis and the formation of aqueous carbonate complexes. LaF3precipitation method was used to determine that the Pu remainingin solution after equilibrium was in the IV oxidation state at pH 8and 10. There was insufficient Pu in solution at pH 4 and 6 foroxidation state analysis.
3.2. Pu sorption in the presence of humic acid
In the ternary system where HA was added, the sorptionbehavior at pH > 6 was also independent of the order of addition(Fig. 2A). Although the order of addition was not significant, thepresence of HA significantly decreased the total sorption of Pu tomontmorillonite. At pH>6, total Pu(IV) sorption tomontmorillonitedecreased by 20e50% relative to the binary system independent ofthe order of addition. At pH 6 a nearly 50% decrease in Pu(IV)sorption is observed. At pH > 4, a decrease in Pu(IV) sorption tomontmorillonite observed in these experiments results from thegreater stability of the Pu(IV)-HAaqueous complex relative to Pu(IV)surface species. The effect of HA decreases as aqueous carbonatespecies becomemore favorable at pH> 8 (Fig. 3A). Similar decreasesin Pu(IV) sorptionwere also observed in ternary experiments of HA-Pu(IV)-gibbsite at pH 6 (Zimmerman and Powell, 2010). In our ex-periments, the presence of HA strongly affected the behavior of Puresulting in increased aqueous Pu concentrations.
At pH 4, Pu(IV) sorption to montmorillonite was not affected bythe addition of HA. In these experiments, the surface sorption wasnear 100% for the binary system, and as a result we cannot deter-mine if Pu(IV)-HA-montmorillonite ternary surface species played arole in Pu sorption. Precipitation of HA under these low pH condi-tions precludes any discussion of the nature of the sorbed Pucomplex.
3.3. Pu sorption in the presence of fulvic acid
Sorption of Pu(IV) to montmorillonite in the presence of FA isshown in Fig. 2B. Similar to the HA, altering the order of FA additiondid not have a large effect on Pu(IV) sorption to montmorillonite. In
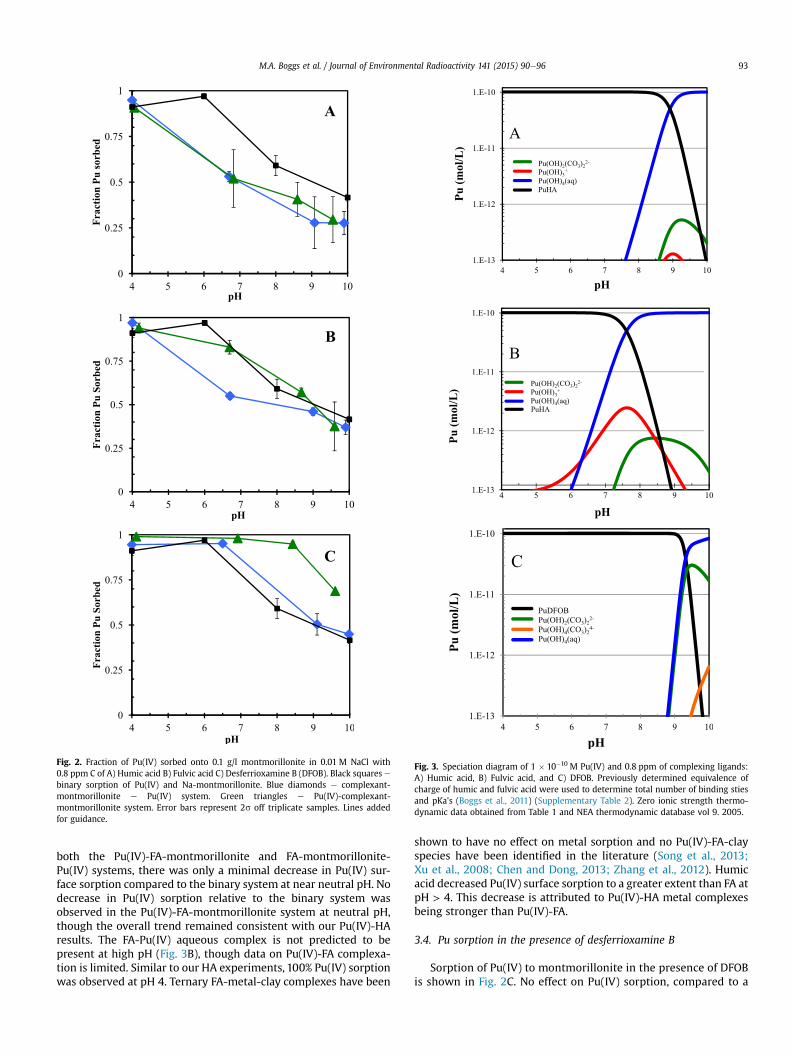
Fig. 2. Fraction of Pu(IV) sorbed onto 0.1 g/l montmorillonite in 0.01 M NaCl with0.8 ppm C of A) Humic acid B) Fulvic acid C) Desferrioxamine B (DFOB). Black squares ebinary sorption of Pu(IV) and Na-montmorillonite. Blue diamonds e complexant-montmorillonite e Pu(IV) system. Green triangles e Pu(IV)-complexant-montmorillonite system. Error bars represent 2s off triplicate samples. Lines addedfor guidance.
Fig. 3. Speciation diagram of 1 � 10�10 M Pu(IV) and 0.8 ppm of complexing ligands:A) Humic acid, B) Fulvic acid, and C) DFOB. Previously determined equivalence ofcharge of humic and fulvic acid were used to determine total number of binding stiesand pKa's (Boggs et al., 2011) (Supplementary Table 2). Zero ionic strength thermo-dynamic data obtained from Table 1 and NEA thermodynamic database vol 9. 2005.
M.A. Boggs et al. / Journal of Environmental Radioactivity 141 (2015) 90e96 93
both the Pu(IV)-FA-montmorillonite and FA-montmorillonite-Pu(IV) systems, there was only a minimal decrease in Pu(IV) sur-face sorption compared to the binary system at near neutral pH. Nodecrease in Pu(IV) sorption relative to the binary system wasobserved in the Pu(IV)-FA-montmorillonite system at neutral pH,though the overall trend remained consistent with our Pu(IV)-HAresults. The FA-Pu(IV) aqueous complex is not predicted to bepresent at high pH (Fig. 3B), though data on Pu(IV)-FA complexa-tion is limited. Similar to our HA experiments, 100% Pu(IV) sorptionwas observed at pH 4. Ternary FA-metal-clay complexes have been
shown to have no effect on metal sorption and no Pu(IV)-FA-clayspecies have been identified in the literature (Song et al., 2013;Xu et al., 2008; Chen and Dong, 2013; Zhang et al., 2012). Humicacid decreased Pu(IV) surface sorption to a greater extent than FA atpH > 4. This decrease is attributed to Pu(IV)-HA metal complexesbeing stronger than Pu(IV)-FA.
3.4. Pu sorption in the presence of desferrioxamine B
Sorption of Pu(IV) to montmorillonite in the presence of DFOBis shown in Fig. 2C. No effect on Pu(IV) sorption, compared to a
M.A. Boggs et al. / Journal of Environmental Radioactivity 141 (2015) 90e9694
binary system, was observed in the DFOB-montmorillonite-Pu(IV)system, indicating that the ligand is not out competing themontmorillonite for metal complexation (e.g. humic and fulvicacid systems). Pu(IV)-DFOB complexes are thermodynamicallystable (Table 1) and predicted to be present over the range of pHstudied (Fig. 3C). However, the Pu(IV)-DFOB-montmorillonitesystem shows an increase in sorption compared to the binarysystem. Unlike the FA and HA systems, the order of additionhas a significant effect on the sorption of Pu(IV) to montmoril-lonite. Thus, different sorption processes are most likely con-trolling these two ternary systems and will be further discussedbelow.
In order to determine how DFOB interacts with montmoril-lonite, montmorillonite equilibrated with DFOB was furtheranalyzed by XRD and TEM. Fig. 4 shows XRD patterns obtainedfrom 3� to 10� (2q). In these diffraction patterns, a dominant peakand a broad shoulder can be identified occurring at low angle in thepatterns of DFOB concentration lower than 1000 ppm (indicated byupward arrowheads). This indicates a broad range on interlayerspacing (supplementary Fig. 1). The dominant peaks are repre-sentative of the basal spacing of the predominant clay, d001, that canbe determined according to Bragg's law:
2dsinq ¼ l (1)
where q is the diffraction angle, d is the spacing of correspondingcrystal plane, and l is the wavelength of X-ray source (for Cu target:l (Ka1,2) ¼ 1.5418 Å). As the concentration of DFOB added to thesystem increased, from 0 to 10 ppm, the d001 peak shifted veryslightly (<0.2� in 2q) to a higher diffraction angle (i.e., the basal
Fig. 4. X-ray diffraction patterns of 5 mg of Na-montmorillonite in 1.0 mL of 0.01 NaClwith 0 ppm DFOB (green), 5 ppm DFOB (mocha), 10 ppm DFOB (blue), 100 ppm DFOB(red), 1000 ppm DFOB (purple) and 10,000 ppm DFOB (yellow). See SupplementaryFigure 1 for details. (For interpretation of the references to color in this figurelegend, the reader is referred to the web version of this article.)
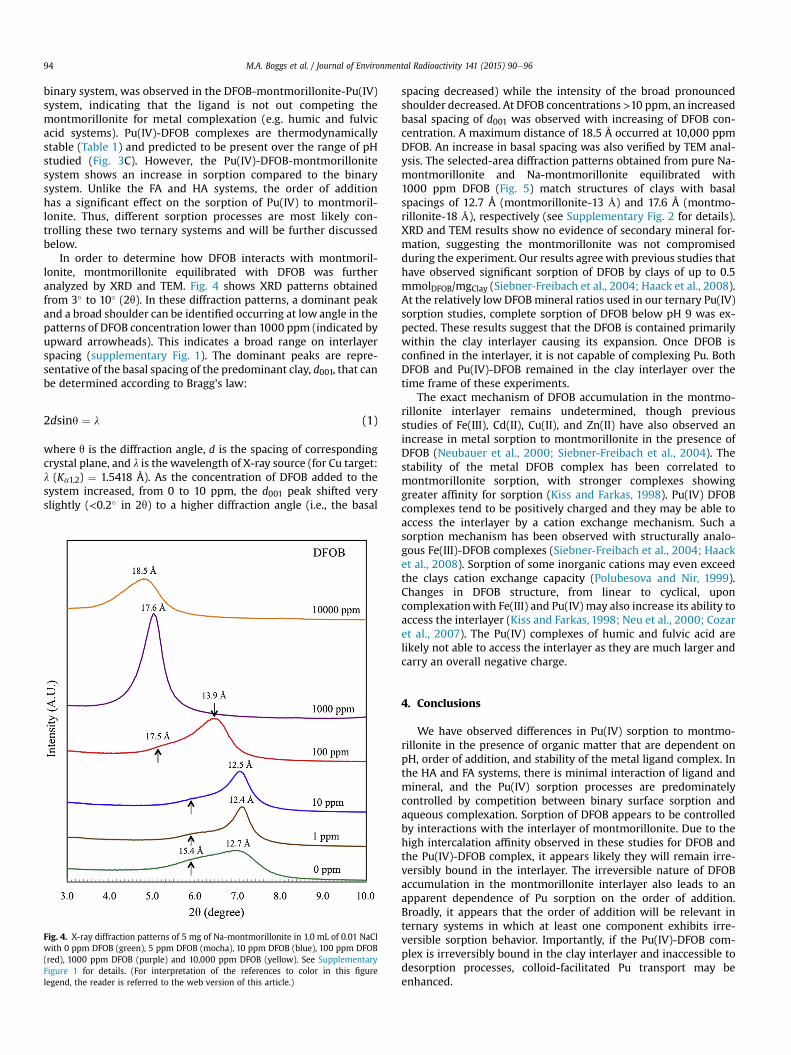
spacing decreased) while the intensity of the broad pronouncedshoulder decreased. At DFOB concentrations >10 ppm, an increasedbasal spacing of d001 was observed with increasing of DFOB con-centration. A maximum distance of 18.5 Å occurred at 10,000 ppmDFOB. An increase in basal spacing was also verified by TEM anal-ysis. The selected-area diffraction patterns obtained from pure Na-montmorillonite and Na-montmorillonite equilibrated with1000 ppm DFOB (Fig. 5) match structures of clays with basalspacings of 12.7 Å (montmorillonite-13 Å) and 17.6 Å (montmo-rillonite-18 Å), respectively (see Supplementary Fig. 2 for details).XRD and TEM results show no evidence of secondary mineral for-mation, suggesting the montmorillonite was not compromisedduring the experiment. Our results agree with previous studies thathave observed significant sorption of DFOB by clays of up to 0.5mmolDFOB/mgClay (Siebner-Freibach et al., 2004; Haack et al., 2008).At the relatively low DFOBmineral ratios used in our ternary Pu(IV)sorption studies, complete sorption of DFOB below pH 9 was ex-pected. These results suggest that the DFOB is contained primarilywithin the clay interlayer causing its expansion. Once DFOB isconfined in the interlayer, it is not capable of complexing Pu. BothDFOB and Pu(IV)-DFOB remained in the clay interlayer over thetime frame of these experiments.
The exact mechanism of DFOB accumulation in the montmo-rillonite interlayer remains undetermined, though previousstudies of Fe(III), Cd(II), Cu(II), and Zn(II) have also observed anincrease in metal sorption to montmorillonite in the presence ofDFOB (Neubauer et al., 2000; Siebner-Freibach et al., 2004). Thestability of the metal DFOB complex has been correlated tomontmorillonite sorption, with stronger complexes showinggreater affinity for sorption (Kiss and Farkas, 1998). Pu(IV) DFOBcomplexes tend to be positively charged and they may be able toaccess the interlayer by a cation exchange mechanism. Such asorption mechanism has been observed with structurally analo-gous Fe(III)-DFOB complexes (Siebner-Freibach et al., 2004; Haacket al., 2008). Sorption of some inorganic cations may even exceedthe clays cation exchange capacity (Polubesova and Nir, 1999).Changes in DFOB structure, from linear to cyclical, uponcomplexationwith Fe(III) and Pu(IV) may also increase its ability toaccess the interlayer (Kiss and Farkas, 1998; Neu et al., 2000; Cozaret al., 2007). The Pu(IV) complexes of humic and fulvic acid arelikely not able to access the interlayer as they are much larger andcarry an overall negative charge.
4. Conclusions
We have observed differences in Pu(IV) sorption to montmo-rillonite in the presence of organic matter that are dependent onpH, order of addition, and stability of the metal ligand complex. Inthe HA and FA systems, there is minimal interaction of ligand andmineral, and the Pu(IV) sorption processes are predominatelycontrolled by competition between binary surface sorption andaqueous complexation. Sorption of DFOB appears to be controlledby interactions with the interlayer of montmorillonite. Due to thehigh intercalation affinity observed in these studies for DFOB andthe Pu(IV)-DFOB complex, it appears likely they will remain irre-versibly bound in the interlayer. The irreversible nature of DFOBaccumulation in the montmorillonite interlayer also leads to anapparent dependence of Pu sorption on the order of addition.Broadly, it appears that the order of addition will be relevant internary systems in which at least one component exhibits irre-versible sorption behavior. Importantly, if the Pu(IV)-DFOB com-plex is irreversibly bound in the clay interlayer and inaccessible todesorption processes, colloid-facilitated Pu transport may beenhanced.
Fig. 5. Bright-field TEM images of pure Na-montmorillonite (A) and Na-montmorillonite equilibrated with 1000 mg/l of desferrioxamine B (B). (C) and (D) are selected-area electrondiffraction patterns corresponding to images of (A) and (B), and match crystal structures of montmollonite-12.7 Å and montmollonite-17.5 Å, respectively (see SupplementaryFigures 2 and 3 for details).
M.A. Boggs et al. / Journal of Environmental Radioactivity 141 (2015) 90e96 95
Acknowledgments
This work was supported by the Subsurface BiogeochemicalResearch Program of the U.S. Department of Energy's Office ofBiological and Environmental Research. Prepared by LLNL underContract DE-AC52-07NA27344. LLNL-JRNL-662319.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jenvrad.2014.12.005
References
Andre, C., Choppin, G.R., 2000. Radiochim. Acta 88, 613e616.Begg, J.D., Zavarin, M., Zhao, P.H., Tumey, S.J., Powell, B., Kersting, A.B., 2013. Environ.
Sci. Technol. 47, 5146e5153.Begg, J.D., Zavarin, M., Kersting, A.B., 2014. Plutonium desorption from mineral
surfaces at environmental concentrations of hydrogen peroxide. Environ. Sci.Technol 48, 6201e6210. http://dx.doi.org/10.1021/es500984w.
Boggs, M.A., Minton, T., Dong, W., Lomasney, S., Islam, M.R., Gu, B., Wall, N.A., 2011.Environ. Sci. Technol. 45, 2718e2724.
Boukhalfa, H., Reilly, S., Neu, M., 2007. Complexation of Pu(IV) with the naturalsiderophore desferrioxamine B and the redox properties of Pu(IV)(siderophore)complexes. Inorganic Chem 46, 1018e1026. http://dx.doi.org/10.1021/ic061544q.
Brainard, J.R., Strietelmeier, B.A., Smith, P.H., Langstonunkefer, P.J., Barr, M.E.,Ryan, R.R., 1992. Radiochim. Acta 58-9, 357e363.
Buda, R.A., Banik, N.L., Kratz, J.V., Trautmann, N., 2008. Radiochim. Acta 96,657e665.
Chen, L., Dong, Y.H., 2013. J. Radioanal. Nucl. Chem. 295, 2117e2123.Choppin, G.R., 2003. Radiochim. Acta 91, 645e649.Cozar, O., Leopold, N., Tomoaia-Cotisel, M., Mocanu, A., Jelic, C., 2007.
J. Optoelectron. Adv. Mater. 9, 3912e3916.Evers, A., Hancock, R.D., Martell, A.E., Motekaitis, R.J., 1989. Inorg. Chem. 28,
2189e2195.Fujikawa, Y., Zheng, J., Cayer, I., Sugahara, M., Takigami, H., Kudo, A., 1999.
J. Radioanal. Nucl. Chem. 240, 69e74.Haack, E.A., Johnston, C.T., Maurice, P.A., 2008. Geochimica Cosmochimica Acta 72,
3381e3397.Kar, A.S., Kumar, S., Tomar, B.S., Manchanda, V.K., 2011. J. Hazard. Mater. 186,
1961e1965.Karimzadeh, L., Nair, S., Merkel, B.J., 2013. Aquat. Geochem. 19, 25e37.Kersting, A.B., Efurd, D.W., Finnegan, D.L., Rokop, D.J., Smith, D.K., Thompson, J.L.,
1999. Nature 397, 56e59.Kiss, T., Farkas, E., 1998. J. Inclusion Phenom. Mol. Recognit. Chem. 32, 385e403.Kobashi, A., Choppin, G.R., Morse, J.W., 1988. Radiochim. Acta 43, 211e215.Kraemer, S.M., Xu, J.D., Raymond, K.N., Sposito, G., 2002. Environ. Sci. Technol. 36,
1287e1291.Lee, M.H., Lee, C.W., 2000. J. Environ. Radioact. 47, 253e262.Nash, K., Fried, S., Friedman, A.M., Sullivan, J.C., 1981. Environ. Sci. Technol. 15,
834e837.Neu, M.P., Matonic, J.H., Ruggiero, C.E., Scott, B.L., 2000. Angew. Chemie-
International Ed. 39, 1442e1444.Neubauer, U., Nowack, B., Furrer, G., Schulin, R., 2000. Environ. Sci. Technol. 34,
2749e2755.Novikov, A.P., Kalmykov, S.N., Utsunomiya, S., Ewing, R.C., Horreard, F., Merkulov, A.,
Clark, S.B., Tkachev, V.V., Myasoedov, B.F., 2006. Science 314, 638e641.Polubesova, T., Nir, S., 1999. Clays Clay Minerals 47, 366e374.Poulsen, I.F., Hansen, H.C.B., 2000. Water Air Soil Pollut. 120, 249e259.Powell, B.A., Fjeld, R.A., Kaplan, D.I., Coates, J.T., Serkiz, S.M., 2004. Environ. Sci.
Technol. 38, 6016e6024.Powell, B.A., Fjeld, R.A., Kaplan, D.I., Coates, J.T., Serkiz, S.M., 2005. Environ. Sci.
Technol. 39, 2107e2114.

M.A. Boggs et al. / Journal of Environmental Radioactivity 141 (2015) 90e9696
Reiller, P., Casanova, F., Moulin, V., 2005. Environ. Sci. Technol. 39, 1641e1648.Renshaw, J.C., Robson, G.D., Trinci, A.P.J., Wiebe, M.G., Livens, F.R., Collison, D.,
Taylor, R.J., 2002. Mycol. Res. 106, 1123e1142.Roberts, A.A., Schultz, A.W., Kersten, R.D., Dorrestein, P.C., Moore, B.S., 2012. Fems
Microbiol. Lett. 335, 95e103.Siebner-Freibach, H., Hadar, Y., Chen, Y., 2004. Soil Sci. Soc. Am. J. 68, 470e480.Smith, D.K., Finnegan, D.L., Bowen, S.M., 2003. J. Environ. Radioact. 67, 35e51.Song, X.P., Wang, Y.J., Cai, J.J., 2013. J. Radioanal. Nucl. Chem. 295, 991e1000.Strathmann, T.J., Myneni, S.C.B., 2005. Environ. Sci. Technol. 39, 4027e4034.Szabo, G., Guczi, J., Reiller, P., Miyajima, T., Bulman, R.A., 2010. Radiochim. Acta 98,
13e18.Taylor, D.M., 1989. Sci. Total Environ. 83, 217e225.
Tipping, E., 1998. Humic ion-binding model VI: An improved description of theinteractions of protons and metal ions with humic substances. AquaticGeochemistry 4, 3e48. http://dx.doi.org/10.1023/a:1009627214459.
Xu, D., Zhou, X., Wang, X.K., 2008. Appl. Clay Sci. 39, 133e141.Zavarin, M., Powell, B.A., Bourbin, M., Zhao, P., Kersting, A.B., 2012. Environ. Sci.
Technol. 46, 2692e2698.Zhang, L.C., Luo, L., Zhang, S.Z., 2012. Colloids Surfaces a-Physicochem. Eng. Aspects
406, 84e90.Zhao, P., Zavarin, M., Leif, R.N., Powell, B.A., Singleton, M.J., Lindvall, R.E.,
Kersting, A.B., 2011. Appl. Geochem. 26, 308e318.Zimmerman, T.N., Powell, B.A., 2010. Geochimica Cosmochimica Acta 74, A1237.Zimmerman, T., Zavarin, M., Powell, B.A., 2014. Radiochim. Acta 102, 629e643.
Copyright © 2022 FDOKUMEN