
EWAT-B - Multi scroll chiller with R-32 refrigerant - Daikin ...
Upload
johnshopkinsCategory
view
0download
0
PLEASE SCROLL DOWN FOR ARTICLE
This article was downloaded by: [JHU John Hopkins University]On: 24 June 2011Access details: Access Details: [subscription number 930492920]Publisher Psychology PressInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Journal of Clinical and Experimental NeuropsychologyPublication details, including instructions for authors and subscription information:http://www.informaworld.com/smpp/title~content=t713657736
Modeling longitudinal change in motor and cognitive processing speed inpresymptomatic Huntington's diseaseDavid Aaron Maroofa; Alden L. Grossb; Jason Brandtabcd
a Department of Psychiatry and Behavioral Sciences, The Johns Hopkins University School ofMedicine, Baltimore, MD, USA b Department of Mental Health, Johns Hopkins Bloomberg School ofPublic Health, Baltimore, MD, USA c Department of Neurology, The Johns Hopkins University Schoolof Medicine, Baltimore, MD, USA d The Copper Ridge Institute, Sykesville, MD
First published on: 01 June 2011
To cite this Article Maroof, David Aaron , Gross, Alden L. and Brandt, Jason(2011) 'Modeling longitudinal change inmotor and cognitive processing speed in presymptomatic Huntington's disease', Journal of Clinical and ExperimentalNeuropsychology,, First published on: 01 June 2011 (iFirst)To link to this Article: DOI: 10.1080/13803395.2011.574606URL: http://dx.doi.org/10.1080/13803395.2011.574606
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article may be used for research, teaching and private study purposes. Any substantial orsystematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply ordistribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae and drug dosesshould be independently verified with primary sources. The publisher shall not be liable for any loss,actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directlyor indirectly in connection with or arising out of the use of this material.

JOURNAL OF CLINICAL AND EXPERIMENTAL NEUROPSYCHOLOGY2011, iFirst, 1–9
Modeling longitudinal change in motor and cognitiveprocessing speed in presymptomatic Huntington’s
disease
David Aaron Maroof1, Alden L. Gross2, and Jason Brandt1,2,3,4
1Department of Psychiatry and Behavioral Sciences, The Johns Hopkins University School of Medicine,Baltimore, MD, USA2Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD,USA3Department of Neurology, The Johns Hopkins University School of Medicine, Baltimore, MD, USA4The Copper Ridge Institute, Sykesville, MD
Persons who have the genetic mutation responsible for Huntington’s disease (HD) have been shown to exhibitlower cognitive test scores years prior to manifest HD. Most studies have examined cognitive performance inpresymptomatic persons by using estimated times to manifest HD based on published algorithms. We followed 19gene-positive persons who were presymptomatic using an objective criterion (i.e., Quantified Neurological Examscore ≤ 10) at baseline for up to 21 years (median = 5 years) with periannual neuropsychological assessments untila diagnosis of manifest HD. Results showed that our tests of information- and psychomotor-processing speedthat place minimal demands on cognition, worsening performance became evident 5–10 years prior to the devel-opment of manifest HD. In conclusion, cognitive decline precedes motor signs in HD and may be an importanttarget in clinical trials and early intervention. Cognitive test scores may also improve the ability to predict dis-ease onset among gene mutation carriers and help families to better plan for potential personal and economichardship.
Keywords: Huntington’s disease; Speed; Longitudinal; Modeling.
Following the discovery of the genetic mutation responsi-ble for Huntington’s disease (HD; Huntington’s DiseaseCollaborative Research Group, 1993), there have beenmany attempts to determine whether people who carrythe mutation but remain asymptomatic can be dis-tinguished from those without the mutation (Aylwardet al., 2000; Larsson, Almkvist, Luszcz, & Wahlin, 2008;Paulsen et al., 2008; Paulsen et al., 2001). Brain mor-phometric (Aylward et al., 2004) and functional (Paulsenet al., 2004) changes have been documented years prior toclinical symptoms sufficiently severe and specific for diag-nosis, as have lower cognitive performance scores (Brandtet al., 2008; Kirkwood et al., 1999; Solomon et al.,2007). Quantifying neuropsychological performance in
This research was supported, in part, by Grant NS16375 from the National Institute on Neurological Disorders and Stroke and aCenter of Excellence Award from the Huntington’s Disease Society of America. The authors thank Barnett Shpritz for assistance withdata collection.
Address correspondence to Jason Brandt, Department of Psychiatry and Behavioral Sciences, The Johns Hopkins University Schoolof Medicine, 600 N. Wolfe Street, Meyer 218, Baltimore, MD 21287–7218, USA. (E-mail: [email protected]).
asymptomatic HD allows us to infer the very earliest cog-nitive manifestations of the HD mutation. Declines incognition may serve as useful predictors of imminent clin-ical onset, as well as markers of disease pathogenesis toserve as surrogate outcomes in clinical trials.
The majority of studies that have investigated cogni-tive decline in presymptomatic HD have used predic-tions of time to “conversion” (development of symp-toms sufficient for diagnosis) in their statistical mod-eling (Duff et al., 2010; Larsson et al., 2008; Paulsenet al., 2008; Solomon et al., 2007). For example, Brandt,Shpritz, Codori, Margolis, and Rosenblatt (2002) exam-ined 203 neurologically normal offspring of HD patientswho were tested for the HD mutation. Comparisons
© 2011 Psychology Press, an imprint of the Taylor & Francis Group, an Informa business
http://www.psypress.com/jcen DOI: 10.1080/13803395.2011.574606
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011

2 MAROOF, GROSS, BRANDT
of neuropsychological functioning between mutation-positive and mutation-negative groups failed to detectsignificant differences. However, when proximity to clini-cal onset (a product of cytosine–adenine–guanine [CAG]repeat length and parental age of disease onset) was esti-mated among the mutation-positive persons, those closerto onset were more severely impaired on 7 of 12 tests. Thiswas not accounted for by the minute differences betweenclose-to-onset and far-from-onset groups in neurologicexam scores. Duff and colleagues (2010) examined theprevalence of mild cognitive impairment (MCI; Petersenet al., 1999) in three at-risk groups of asymptomatic HDgene carriers: those believed to be close to disease onset(<9 years to estimated diagnosis), those 9–15 years toestimated onset, and those far (>15 years) from esti-mated diagnosis. Of all pre-HD persons, approximately40% met the criteria for MCI. A nonamnestic MCI diag-nosis (where performance on tasks other than memory isimpaired) was twice as frequent as an amnestic MCI diag-nosis; impairments on tasks requiring processing speedwere the most common. Persons estimated to be closerto HD diagnosis (estimated from CAG repeat length andcurrent age of the asymptomatic gene-positive partici-pant. Langbehn, Brinkman, Falush, Paulsen, & Hayden,2004) had higher rates of MCI. Taken together, the extantcross-sectional research indicates that proximity to dis-ease onset is a salient predictor of decline in cognitivefunction.
Many fewer studies have examined neuropsychologi-cal functioning longitudinally from preclinical to actualclinical onset of HD. Using data from the multicenterPREDICT-HD study, Paulsen et al. (2001) found thatbaseline cognitive performances on the Symbol DigitModalities Test and Stroop Color–Word Test were con-sistently worse for the at-risk group who converted tomanifest HD after two years than for those who wereat risk but did not convert. In fact, longitudinal changescores showed that performance in those who convertedduring the study showed significant declines on all tests(the above tests plus a measure of verbal fluency). Personswho remained asymptomatic showed improvements onall of these cognitive tests. A major limitation of thisstudy was the relatively short follow-up period (an aver-age of two years). Snowden, Craufurd, Thompson, andNeary (2002) examined cognitive functioning at baselineand follow-up in 15 presymptomatic HD individuals whoprogressed to manifest HD after an average of five years.Memory declined precipitously as disease onset neared.Smaller but still significant declines were observed onpsychomotor tasks. More recently, Brandt et al. (2008)found subtle differences between “early” converters and“late” converters at baseline on a single problem-solvingtask (Wisconsin Card Sorting Test) among mutation car-riers who became diagnosable (“converted”) some yearslater. More specifically, when the participants were strat-ified into early converters (i.e., an average time to con-version of 3.7 years) and late converters (i.e., an averagetime to conversion of 11.8 years), the early convertersachieved fewer sorts, were less efficient (i.e., they requiredmore cards to achieve completed sorts), and made moreperseverative and nonperseverative errors.
The current study examines the rate of change inmotor and cognitive task performance in asymptomaticindividuals who tested positive for the HD mutation aftertheir initial evaluation, who were reassessed periodically(periannually, in many cases), became symptomatic, andwere eventually diagnosed with clinical HD. The lengthof this prospective study (up to 21 years of longitudi-nal follow-up) until clinical symptoms of HD (a medianof 5 years to conversion) is a strength found in fewprevious studies (Aylward et al., 2000; Paulsen et al.,2008; Solomon et al., 2007), as is the statistical modelingprocedure (generalized estimating equation; GEE).
METHOD
Participants and procedure
All participants in the current study underwent presymp-tomatic genetic testing for HD at the BaltimoreHuntington’s Disease Research Center at the JohnsHopkins University School of Medicine. They wereenrolled in a prospective, longitudinal study trackingmotor, psychological (e.g., affective, behavioral, and cog-nitive), and brain morphometric changes (Aylward et al.,2000; Brandt et al., 1989; Campodonico et al., 1998).The parent study from which these data were drawnwas fully reviewed and approved by the Johns HopkinsMedicine Institutional Review Board. All participantsgave informed consent.
At-risk asymptomatic persons were enrolled in thisstudy between February 1986 and May 2010. All par-ticipants were required to have definitive DNA tests forthe huntingtin mutation, neuropsychological examina-tions, and scores ≤10 on the Quantified Neurologic Exam(QNE) at enrollment before genetic testing (Folstein,Jensen, Leigh, & Folstein, 1983). Of 248 persons whomet these criteria, 138 persons tested negative for theHD mutation, while 110 tested positive (i.e., presymp-tomatic). During the study period, 19 developed symp-toms and were diagnosed with disease onset by neuropsy-chiatrists specialized in HD and returned for at leastone follow-up as an affected person. At each visit, theQNE was administered, as was a battery of neuropsy-chological tests (discussed below). Years-to-diagnosis wascalculated as the difference between time of participant’sbaseline (presymptomatic) visit and time of the visit inwhich they were first diagnosed with HD. Characteristicsof the sample, including neurological performance atbaseline and at the time of conversion, are displayed inTable 1.
Neuropsychological tests
At each visit, participants were administered four testsrequiring rapid mental processing and motor respond-ing. These tasks were the Symbol Digit Modalities Test(SDMT; written trial; Smith, 1973), Trail Making Test(TMT; Army Individual Test Battery, 1944; includingthe discrepancy between Part A time and Part B time,
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011
MOTOR AND COGNITIVE SPEED IN PRESYMPTOMATIC HD 3
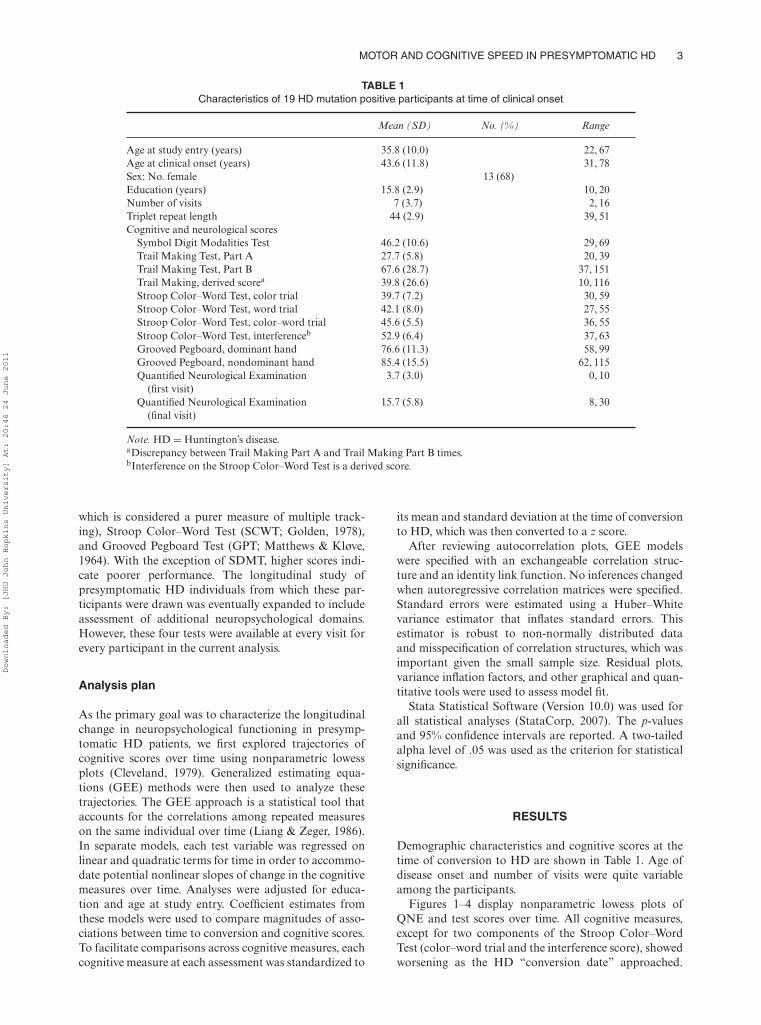
TABLE 1Characteristics of 19 HD mutation positive participants at time of clinical onset
Mean (SD) No. (%) Range
Age at study entry (years) 35.8 (10.0) 22, 67Age at clinical onset (years) 43.6 (11.8) 31, 78Sex: No. female 13 (68)Education (years) 15.8 (2.9) 10, 20Number of visits 7 (3.7) 2, 16Triplet repeat length 44 (2.9) 39, 51Cognitive and neurological scores
Symbol Digit Modalities Test 46.2 (10.6) 29, 69Trail Making Test, Part A 27.7 (5.8) 20, 39Trail Making Test, Part B 67.6 (28.7) 37, 151Trail Making, derived scorea 39.8 (26.6) 10, 116Stroop Color–Word Test, color trial 39.7 (7.2) 30, 59Stroop Color–Word Test, word trial 42.1 (8.0) 27, 55Stroop Color–Word Test, color–word trial 45.6 (5.5) 36, 55Stroop Color–Word Test, interferenceb 52.9 (6.4) 37, 63Grooved Pegboard, dominant hand 76.6 (11.3) 58, 99Grooved Pegboard, nondominant hand 85.4 (15.5) 62, 115Quantified Neurological Examination
(first visit)3.7 (3.0) 0, 10
Quantified Neurological Examination(final visit)
15.7 (5.8) 8, 30
Note. HD = Huntington’s disease.aDiscrepancy between Trail Making Part A and Trail Making Part B times.bInterference on the Stroop Color–Word Test is a derived score.
which is considered a purer measure of multiple track-ing), Stroop Color–Word Test (SCWT; Golden, 1978),and Grooved Pegboard Test (GPT; Matthews & Kløve,1964). With the exception of SDMT, higher scores indi-cate poorer performance. The longitudinal study ofpresymptomatic HD individuals from which these par-ticipants were drawn was eventually expanded to includeassessment of additional neuropsychological domains.However, these four tests were available at every visit forevery participant in the current analysis.
Analysis plan
As the primary goal was to characterize the longitudinalchange in neuropsychological functioning in presymp-tomatic HD patients, we first explored trajectories ofcognitive scores over time using nonparametric lowessplots (Cleveland, 1979). Generalized estimating equa-tions (GEE) methods were then used to analyze thesetrajectories. The GEE approach is a statistical tool thataccounts for the correlations among repeated measureson the same individual over time (Liang & Zeger, 1986).In separate models, each test variable was regressed onlinear and quadratic terms for time in order to accommo-date potential nonlinear slopes of change in the cognitivemeasures over time. Analyses were adjusted for educa-tion and age at study entry. Coefficient estimates fromthese models were used to compare magnitudes of asso-ciations between time to conversion and cognitive scores.To facilitate comparisons across cognitive measures, eachcognitive measure at each assessment was standardized to
its mean and standard deviation at the time of conversionto HD, which was then converted to a z score.
After reviewing autocorrelation plots, GEE modelswere specified with an exchangeable correlation struc-ture and an identity link function. No inferences changedwhen autoregressive correlation matrices were specified.Standard errors were estimated using a Huber–Whitevariance estimator that inflates standard errors. Thisestimator is robust to non-normally distributed dataand misspecification of correlation structures, which wasimportant given the small sample size. Residual plots,variance inflation factors, and other graphical and quan-titative tools were used to assess model fit.
Stata Statistical Software (Version 10.0) was used forall statistical analyses (StataCorp, 2007). The p-valuesand 95% confidence intervals are reported. A two-tailedalpha level of .05 was used as the criterion for statisticalsignificance.
RESULTS
Demographic characteristics and cognitive scores at thetime of conversion to HD are shown in Table 1. Age ofdisease onset and number of visits were quite variableamong the participants.
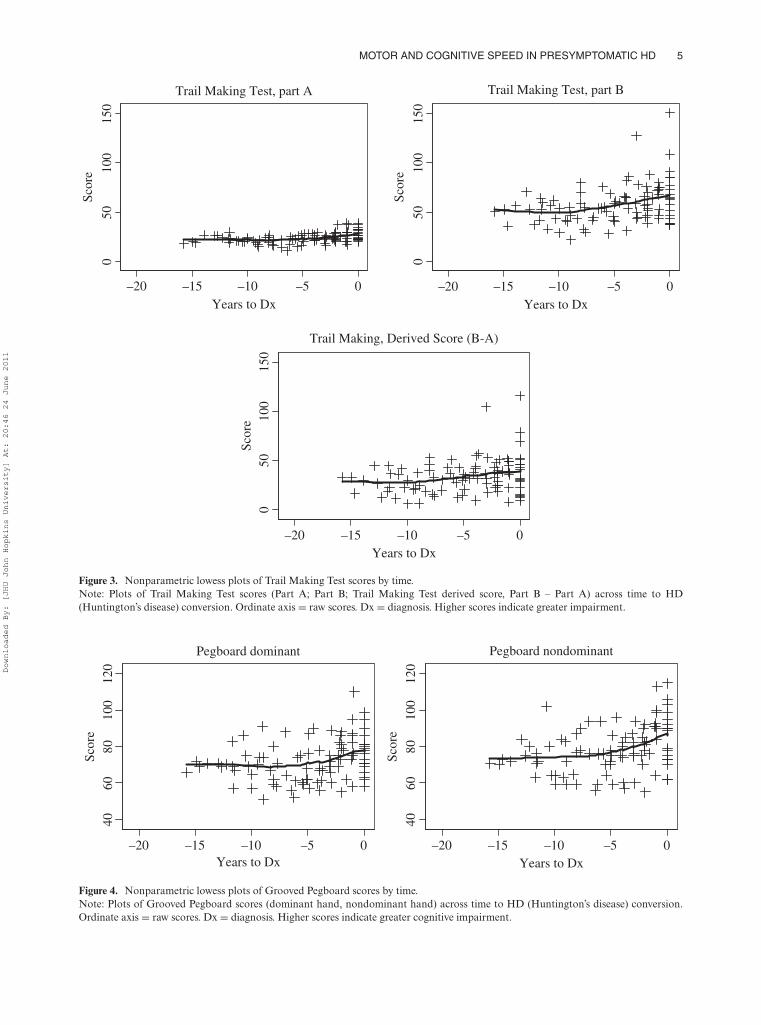
Figures 1–4 display nonparametric lowess plots ofQNE and test scores over time. All cognitive measures,except for two components of the Stroop Color–WordTest (color–word trial and the interference score), showedworsening as the HD “conversion date” approached.
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011

4 MAROOF, GROSS, BRANDT
010
2030
Scor
e
–20 –15 –10 –5 0
Years to Dx
Quantified Neurological Exam
2040
6080
100
Scor
e
–20 –15 –10 –5 0
Years to Dx
Symbol Digit Modalities Test
Figure 1. Nonparametric lowess plots of Quantified Neurological Examination and Symbol Digit Modalities Test scores by time.Note: Plots of Quantified Neurological Examination and Symbol Digit Modalities Test scores across time to HD (Huntington’s disease)conversion. Ordinate axis = raw scores. Dx = diagnosis. Higher scores on the Quantified Neurological Examination and lower scoreson the Symbol Digit Modalities Test scores indicate greater impairment.
3040
5060
70Sc
ore
–20 –15 –10 –5 0
Years to Dx
Stroop color trial30
4050
6070
Scor
e
–20 –15 –10 –5 0Years to Dx
Stroop word trial
3040
5060
70Sc
ore
–20 –15 –10 –5 0Years to Dx
Stroop color–word trial
3040
5060
70
Scor
e
–20 –15 –10 –5 0Years to Dx
Stroop interference score
Figure 2. Nonparametric lowess plots of Stroop Color–Word Test scores by time.Note: Plots of Stroop Color–Word Test scores (color, word, color–word trials, Stroop derived interference score) across time to HD(Huntington’s disease) conversion. Ordinate axis = raw scores. Dx = diagnosis. Lower scores indicate greater impairment.
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011
MOTOR AND COGNITIVE SPEED IN PRESYMPTOMATIC HD 5
050
100
150
Scor
e
–20 –15 –10 –5 0Years to Dx
Trail Making Test, part A
050
100
150
Scor
e
–20 –15 –10 –5 0Years to Dx
Trail Making Test, part B
050
100
150
Scor
e
–20 –15 –10 –5 0Years to Dx
Trail Making, Derived Score (B-A)
Figure 3. Nonparametric lowess plots of Trail Making Test scores by time.Note: Plots of Trail Making Test scores (Part A; Part B; Trail Making Test derived score, Part B – Part A) across time to HD(Huntington’s disease) conversion. Ordinate axis = raw scores. Dx = diagnosis. Higher scores indicate greater impairment.
4060
8010
012
0Sc
ore
–20 –15 –10 –5 0Years to Dx
Pegboard dominant
4060
8010
012
0Sc
ore
–20 –15 –10 –5 0Years to Dx
Pegboard nondominant
Figure 4. Nonparametric lowess plots of Grooved Pegboard scores by time.Note: Plots of Grooved Pegboard scores (dominant hand, nondominant hand) across time to HD (Huntington’s disease) conversion.Ordinate axis = raw scores. Dx = diagnosis. Higher scores indicate greater cognitive impairment.
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011
6 MAROOF, GROSS, BRANDT
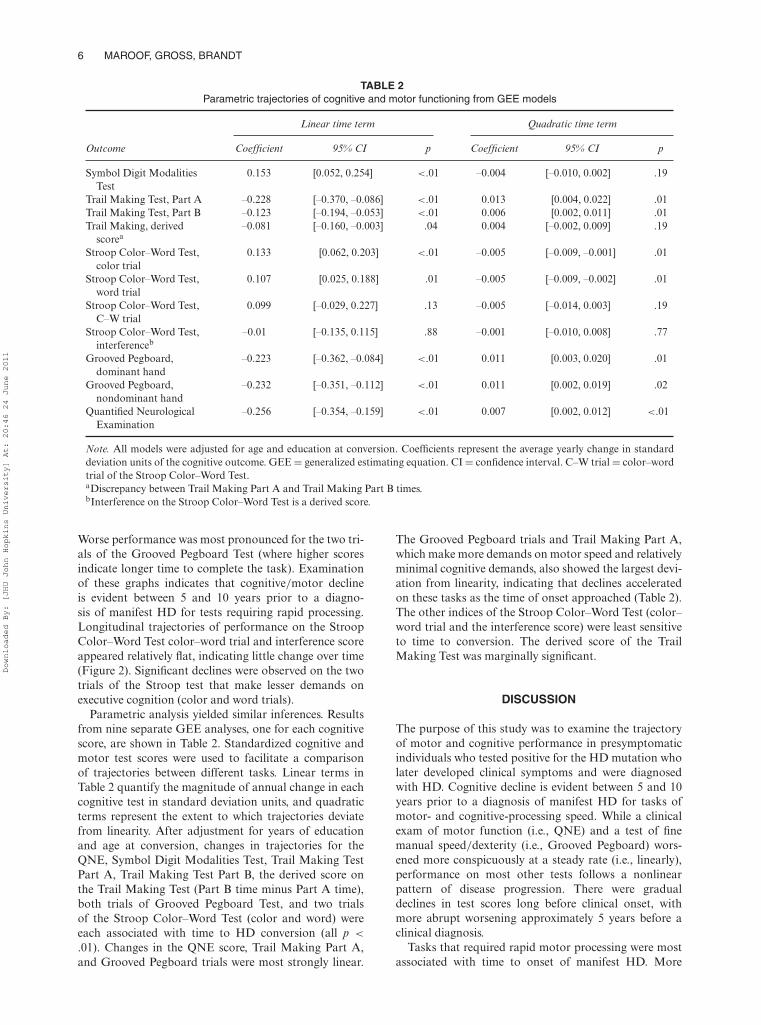
TABLE 2Parametric trajectories of cognitive and motor functioning from GEE models
Linear time term Quadratic time term
Outcome Coefficient 95% CI p Coefficient 95% CI p
Symbol Digit ModalitiesTest
0.153 [0.052, 0.254] <.01 –0.004 [–0.010, 0.002] .19
Trail Making Test, Part A –0.228 [–0.370, –0.086] <.01 0.013 [0.004, 0.022] .01Trail Making Test, Part B –0.123 [–0.194, –0.053] <.01 0.006 [0.002, 0.011] .01Trail Making, derived
scorea–0.081 [–0.160, –0.003] .04 0.004 [–0.002, 0.009] .19
Stroop Color–Word Test,color trial
0.133 [0.062, 0.203] <.01 –0.005 [–0.009, –0.001] .01
Stroop Color–Word Test,word trial
0.107 [0.025, 0.188] .01 –0.005 [–0.009, –0.002] .01
Stroop Color–Word Test,C–W trial
0.099 [–0.029, 0.227] .13 –0.005 [–0.014, 0.003] .19
Stroop Color–Word Test,interferenceb
–0.01 [–0.135, 0.115] .88 –0.001 [–0.010, 0.008] .77
Grooved Pegboard,dominant hand
–0.223 [–0.362, –0.084] <.01 0.011 [0.003, 0.020] .01
Grooved Pegboard,nondominant hand
–0.232 [–0.351, –0.112] <.01 0.011 [0.002, 0.019] .02
Quantified NeurologicalExamination
–0.256 [–0.354, –0.159] <.01 0.007 [0.002, 0.012] <.01
Note. All models were adjusted for age and education at conversion. Coefficients represent the average yearly change in standarddeviation units of the cognitive outcome. GEE = generalized estimating equation. CI = confidence interval. C–W trial = color–wordtrial of the Stroop Color–Word Test.aDiscrepancy between Trail Making Part A and Trail Making Part B times.bInterference on the Stroop Color–Word Test is a derived score.
Worse performance was most pronounced for the two tri-als of the Grooved Pegboard Test (where higher scoresindicate longer time to complete the task). Examinationof these graphs indicates that cognitive/motor declineis evident between 5 and 10 years prior to a diagno-sis of manifest HD for tests requiring rapid processing.Longitudinal trajectories of performance on the StroopColor–Word Test color–word trial and interference scoreappeared relatively flat, indicating little change over time(Figure 2). Significant declines were observed on the twotrials of the Stroop test that make lesser demands onexecutive cognition (color and word trials).
Parametric analysis yielded similar inferences. Resultsfrom nine separate GEE analyses, one for each cognitivescore, are shown in Table 2. Standardized cognitive andmotor test scores were used to facilitate a comparisonof trajectories between different tasks. Linear terms inTable 2 quantify the magnitude of annual change in eachcognitive test in standard deviation units, and quadraticterms represent the extent to which trajectories deviatefrom linearity. After adjustment for years of educationand age at conversion, changes in trajectories for theQNE, Symbol Digit Modalities Test, Trail Making TestPart A, Trail Making Test Part B, the derived score onthe Trail Making Test (Part B time minus Part A time),both trials of Grooved Pegboard Test, and two trialsof the Stroop Color–Word Test (color and word) wereeach associated with time to HD conversion (all p <
.01). Changes in the QNE score, Trail Making Part A,and Grooved Pegboard trials were most strongly linear.
The Grooved Pegboard trials and Trail Making Part A,which make more demands on motor speed and relativelyminimal cognitive demands, also showed the largest devi-ation from linearity, indicating that declines acceleratedon these tasks as the time of onset approached (Table 2).The other indices of the Stroop Color–Word Test (color–word trial and the interference score) were least sensitiveto time to conversion. The derived score of the TrailMaking Test was marginally significant.
DISCUSSION
The purpose of this study was to examine the trajectoryof motor and cognitive performance in presymptomaticindividuals who tested positive for the HD mutation wholater developed clinical symptoms and were diagnosedwith HD. Cognitive decline is evident between 5 and 10years prior to a diagnosis of manifest HD for tasks ofmotor- and cognitive-processing speed. While a clinicalexam of motor function (i.e., QNE) and a test of finemanual speed/dexterity (i.e., Grooved Pegboard) wors-ened more conspicuously at a steady rate (i.e., linearly),performance on most other tests follows a nonlinearpattern of disease progression. There were gradualdeclines in test scores long before clinical onset, withmore abrupt worsening approximately 5 years before aclinical diagnosis.
Tasks that required rapid motor processing were mostassociated with time to onset of manifest HD. More
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011

MOTOR AND COGNITIVE SPEED IN PRESYMPTOMATIC HD 7
cognitively demanding tasks were less sensitive to time ofonset of diagnosable HD. For example, while the word-reading and color-naming trials of the Stroop Color–Word Test showed marked declines closer to onset ofmanifest HD, the interference trial (Color–Word) andthe interference score—the difference between a person’sactual Color–Word score and his or her predicted Color–Word score, (C × W)/(C + W), which is considereda purer measure of interference as it partials out theinfluence of color-naming and word-reading speeds—showed more modest declines as diagnosable HD becameimminent. This is consistent with two recent prospectivestudies of patients with mild to moderate HD (Beglingeret al., 2010; Snowden, Craufurd, Griffiths, Thompson, &Neary, 2001), which showed greater decline on tasks thatplaced minimal demands on higher level cognition, pre-sumably a reflection of primarily subcortical (i.e., striatal)neuropathology (Snowden et al., 2001; Snowden et al.,2002). Snowden and colleagues (2001) initially observedthis phenomenon when they examined cognitive func-tion in symptomatic HD patients longitudinally. Greaterdeclines were detected on less cognitively demandingtasks, leading the authors to conclude that such findingswere in part a reflection of change that occurs in HD forrelatively perfunctory, speed-based tasks. In fact, declinesin psychomotor- and cognitive-processing speed may besome of the earliest indicators of illness onset and are sen-sitive to striatal dysfunction (Aylward et al., 2004; Duffet al., 2010; Harris et al., 1999; Snowden et al., 2002).
Our study revealed that different neuropsychologicalmeasures are differentially sensitive to HD conversion atdifferent points in time relative to HD conversion. Thesefindings highlight a critical limitation of those studies thatuse composite or summary scores from individual neu-ropsychological measures to predict time to an HD diag-nosis (e.g., Duff et al., 2010; Paulsen et al., 2004). Suchcomposite measures, besides making inferences difficultto generalize to settings where not all these measureswere administered to a patient, mask domain-specificassociations.
Although some studies have shown processing speedto be the hallmark of early cognitive decline in HD(Kirkwood et al., 2000; Kirkwood et al., 1999; Siemerset al., 1996), others have shown different patterns ofimpairment. Lawrence et al. (1998) examined cognitivetask performance in 54 at-risk carriers of the HD muta-tion and found that they performed significantly worsethan noncarriers on tasks of cognitive set-shifting andfluency, deficits attributed to inhibitory control mediatedby striatofrontal circuitry. Similarly, Brandt et al. (2008)reported an isolated impairment at baseline on a testof mental flexibility and concept formation in mutationcarriers who were closer to disease onset. The fact thatour study was longitudinal rather than cross-sectionalmay help explain the differences found among previousstudies.
Our longitudinal study could have examined cogni-tive trajectories differently, using piecewise spline termsfor time instead of quadratic terms (e.g., Paulsen et al.,2008). Spline terms to punctuate longitudinal growthwould allow us to examine the statistical significance of
the change in slope of cognitive scores at particular times(e.g., 2 years) prior to manifest HD conversion. However,given our small sample size and the likelihood that dif-ferent individuals probably show accelerated decline atdiffering times relative to a clinical HD diagnosis, pos-sibly depending on age, sex, and other characteristics,we concluded that such a modeling technique was lessappropriate than the use of quadratic terms to charac-terize cognitive trajectories. Nonetheless, the fact that wefound strong associations with a relatively small samplesize speaks to the robustness of our findings. We alsofocused our analyses on nonparametric models because,unlike parametric models, they are not unduly influencedby outliers and nonlinear data (Box & Draper, 1987).We did not control for sex in our analyses because thiswould simply explain between-person variability. And, ofcourse, individuals serve as their own controls in repeatedmeasures analyses.
Limitations of the current study include a small sam-ple size as well as our inability to assess, prospectively,a broader range of neuropsychological functions (e.g.,memory) due to the very limited cognitive test batteryin the early years of this study. For example, a recentstudy (Stout et al., 2011) showed that patients closestto predicted disease onset had poorer test performanceacross multiple cognitive domains. However, mountingresearch suggests that the deficits observed in our sampleare among the earliest (Rowe et al., 2010) and most com-mon cognitive problems encountered in asymptomaticHD mutation carriers. In the future, we hope to addressissues of low statistical power by using a greater numberof subjects and determining not only whether cognitivechanges are apparent beyond the time frame suggestedby this study, but whether they include other domains ofcognition as well.
Additional limitations might include not formally cor-recting for multiple tests. However, the pattern of findingsin this study is more informative with regard to the mainconclusions rather than any particular p-value. A fur-ther limitation of the present study is that because wedid not include a control group of nonconverters, causalinferences about whether the dramatic decline in perfor-mance over time is attributable to the development ofHD or normal aging are limited. Last, practice effects arelikely minimal, since such effects typically present onlyafter initial exposure to a test. Most patients in this studyunderwent several test administrations.
Strengths of our study include the prospective designwith up to 21 years of longitudinal follow-up, actualdiagnosis times based on neuropsychiatric evaluations,and sequential assessments of cognitive task performancefrom preclinical to clinical stages of HD. Despite the rel-atively small sample size, we used an objective measure(QNE), with a relatively stringent cutoff, to ensure thatparticipants in this study were presymptomatic from theoutset.
In conclusion, our findings both of the type andof the temporal course of cognitive decline paralleldocumented abnormalities in both brain morphometry(Aylward et al., 2004) and function (Paulsen et al., 2004)seen in preclinical HD individuals. Our results help to
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011

8 MAROOF, GROSS, BRANDT
further characterize the parameters of disease onset andprogression in HD, as the illness’s cognitive signs oftenprecede choreiform movements and therefore diagnos-able HD. Regression methods that incorporate CAGrepeat length and age to predict time of onset of man-ifest HD have been relatively accurate, though incorpo-rating cognitive test scores may help us better predictdisease onset and perhaps disease severity. The abilityto reliably and sensitively measure cognitive signs veryearly on in the disease process is the first step towardexploring the development and use of neuroprotectiveagents—targeting these early clinical/cognitive markersof illness—that might delay the onset or the progressionof cognitive decline. Increasing the degree of certainty insymptoms sufficiently severe and specific for a diagnosisof HD may also help families to better plan for per-sonal needs (e.g., caretaking) and to anticipate economichardships.
Original manuscript received 18 October 2010Revised manuscript accepted 8 March 2011
First published online day month year
REFERENCES
Army Individual Test Battery: Manual of directions and scor-ing. (1944). Washington, DC: War Department, AdjutantGeneral’s Office.
Aylward, E. H., Codori, A. M., Rosenblatt, A., Sherr, M.,Brandt, J., & Stine, O. C. (2000). Rate of caudate atrophyin presymptomatic and symptomatic stages of Huntington’sdisease. Movement Disorders, 15, 552–560.
Aylward, E. H., Sparks, B. F., Field, K. M., Yallapragada, V.,Shpritz, B. D., Rosenblatt, A., et al. (2004). Onset and rate ofstriatal atrophy in preclinical Huntington disease. Neurology,63, 66–72.
Beglinger, L. J., Duff, K., Allison, J., Theriault, D., O’Rourke, J.J. F., Leserman, A., et al. (2010). Cognitive change in patientswith Huntington disease on the Repeatable Battery for theAssessment of Neuropsychological Status. Journal of Clinicaland Experimental Neuropsychology, 32, 573–578.
Box, G., & Draper, N. (1987). Empirical model-building andresponse surfaces. New York, NY: John Wiley & Sons.
Brandt, J., Inscore, A. B., Ward, J., Shpritz, B., Rosenblatt, A.,Margolis, R. L., et al. (2008). Neuropsychological deficitsin Huntington’s disease gene carriers and correlates ofearly “conversion.” Journal of Neuropsychiatry and ClinicalNeurosciences, 20, 466–472.
Brandt, J., Quaid, K. A., Folstein, S. E., Garber, P., Maestri,N. E., Abbott, M., et al. (1989). Presymptomatic diagno-sis of delayed-onset disease with linked DNA markers: Theexperience in Huntington’s disease. Journal of the AmericanMedical Association, 261, 3108–3114.
Brandt, J., Shpritz, B., Codori, A. M., Margolis, R., &Rosenblatt, A. (2002). Neuropsychological manifestationsof the genetic mutation for Huntington’s disease inpresymptomatic individuals. Journal of the InternationalNeuropsychological Society, 8, 918–924.
Campodonico, J. R., Aylward, E., Codori, A., Young, C.,Krafft, L., Magdalinski, M., et al. (1998). When doesHuntington’s disease begin? Journal of the InternationalNeuropsychological Society, 4, 467–473.
Cleveland, W. S. (1979). Robust locally weighted regression andsmoothing scatterplots. Journal of the American StatisticalAssociation, 74, 829–836.
Duff, K., Paulsen, J., Mills, J., Beglinger, L. J., Moser, D. J.,Smith, M. M., et al. (2010). Mild cognitive impairment inprediagnosed Huntington disease. Neurology, 75, 500–507.
Folstein, S. E., Jensen, B., Leigh, R. L., & Folstein, M. F. (1983).The measurement of abnormal movement: Methods devel-oped for Huntington’s disease. Neurobehavioral Toxicologyand Teratology, 5, 605–609.
Golden, C. (1978). Stroop Color and Word Test Manual.Chicago, IL: Stoelting.
Harris, G. J., Codori, A. M., Lewis, R. F., Schmidt, E., Bedi, A.,& Brandt, J. (1999). Reduced basal ganglia blood flow andvolume in pre-symptomatic, gene tested personas at-risk forHuntington’s disease. Brain, 122, 1667–1678.
Huntington’s Disease Collaborative Research Group. (1993). Anovel gene containing a trinucleotide repeat that is expandedand unstable on Huntington’s disease chromosomes. Cell, 72,971–983.
Kirkwood, S. C., Siemers, E., Bond, C., Conneally, P. M.,Christian, J. C., & Foroud, T. (2000). Confirmation of sub-tle motor changes among presymptomatic carriers of theHuntington disease gene. Archives of Neurology, 57, 1040–1044.
Kirkwood, S. C., Siemers, E., Stout, J. C., Hodes, M.E., Conneally, P. M., Christian, J. C., et al. (1999).Longitudinal cognitive and motor changes among presymp-tomatic Huntington disease gene carriers. Archives ofNeurology, 56, 563–568.
Langbehn, D. R., Brinkman, R. R., Falush, D., Paulsen, J. S.,& Hayden, M. R. (2004). A new model for prediction of theage of onset and penetrance for Huntington’s disease basedon CAG length. Clinical Genetics, 65, 267–277.
Larsson, M. U., Almkvist, O., Luszcz, M. A., & Wahlin, T. R.(2008). Phonemic fluency deficits in asymptomatic gene car-riers for Huntington’s disease. Neuropsychology, 22, 596–605.
Lawrence, A. D., Hodges, J. R., Rosser, A. E., Kershaw,A., Ffrench-Constant, C., Rubinsztein, D. C., et al.(1998). Evidence for specific cognitive deficits in preclinicalHuntington’s disease. Brain, 121, 1329–1341.
Liang, K., & Zeger, S. (1986). Longitudinal data anal-ysis using generalized linear models. Biometrika, 73,13–22.
Matthews, C., & Kløve, H. (1964). Instruction manual forthe Adult Neuropsychology Test Battery. Madison, WI:University of Wisconsin Medical School.
Paulsen, J. S., Langbehn, D. R., Stout, J. C., Aylward, E., Ross,C. A., Nance, M., et al. (2008). Detection of Huntington’sdisease decades before diagnosis: The Predict-HD study.Journal of Neurology, Neurosurgery, & Psychiatry, 79, 874–880.
Paulsen, J. S., Zhao, H., Stout, J. C., Brinkman, R. R.,Guttman, M., Ross, C. A., et al. (2001). Clinical markers ofearly disease in persons near onset of Huntington’s disease.Neurology, 57, 658–662.
Paulsen, J. S., Zimbelman, J. L., Hinton, S. C., Langbehn, D.R., Leveroni, C. L., Benjamin, M. L., et al. (2004). fMRIbiomarker of early neuronal dysfunction in presymptomaticHuntington’s disease. American Journal of Neuroradiology,25, 1715–1721.
Petersen, R. C., Smith, G. E., Waring, S. C., Ivnik, R. J.,Tangalos, E. G., & Kokmen, E. (1999). Mild cognitiveimpairment: Clinical characterization and outcome. Archivesof Neurology, 56, 303–308.
Rowe, K. C., Paulsen, J. S., Langbehn, D. R., Duff, K.,Beglinger, L. J., Wang, C., et al. (2010). Self-paced timingdetects and tracks change in prodromal Huntington disease.Neuropsychology, 24, 435–442.
Siemers, E., Foroud, T., Bill, D. J., Sorbel, J., Norton,J. A., Hodes, M. E., et al. (1996). Motor changes inpresymptomatic Huntington disease gene carriers. Archivesof Neurology, 53, 487–492.
Smith, A. (1973). Symbol Digit Modalities Test manual. LosAngeles, CA: Western Psychological Services.
Snowden, J. S., Craufurd, D., Griffiths, H., Thompson, J., &Neary, D. (2001). Longitudinal evaluation of cognitive dis-order in Huntington’s disease. Journal of the InternationalNeuropsychological Society, 7, 33–44.
Snowden, J. S., Craufurd, D., Thompson, J., & Neary, D. (2002).Psychomotor, executive, and memory function in preclinical
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011

MOTOR AND COGNITIVE SPEED IN PRESYMPTOMATIC HD 9
Huntington’s disease. Journal of Clinical and ExperimentalNeuropsychology, 24, 133–145.
Solomon, A. C., Stout, J. C., Johnson, S. A., Langbehn, D.R., Aylward, E. H., Brandt, J., et al. (2007). Verbal episodicmemory declines prior to diagnosis in Huntington’s disease.Neuropsychologia, 45, 1767–1776.
StataCorp. (2007). Stata Statistical Software: Release 10[Computer software]. College Station, TX: StataCorp LP.
Stout, J. C., Paulsen, J. S., Queller, S., Solomon, A. C.,Whitlock, K. B., & Campbell, C. (2011). Neurocognitivesigns in prodromal Huntington disease. Neuropsychology, 25,1–14.
Downloaded By: [JHU John Hopkins University] At: 20:46 24 June 2011
Copyright © 2022 FDOKUMEN




















