
Piryanka Sasidharan - TSpace - University of Toronto
80
i The Development of Small Manganese(III) Porphyrins as Blood Pool Agents for Magnetic Resonance Angiography and the Analyses of Their Binding to Human Serum Albumin by Piryanka Sasidharan A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Piryanka Sasidharan 2018
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Piryanka Sasidharan - TSpace - University of Toronto

i
The Development of Small Manganese(III) Porphyrins as Blood
Pool Agents for Magnetic Resonance Angiography and the Analyses
of Their Binding to Human Serum Albumin
by
Piryanka Sasidharan
A thesis submitted in conformity with the requirements
for the degree of Master of Science
Department of Chemistry
University of Toronto
© Copyright by Piryanka Sasidharan 2018
2019

ii
The Development of Small Manganese(III) Porphyrins as Blood
Pool Agents for Magnetic Resonance Angiography and the Analyses
of their Binding to Human Serum Albumin Piryanka Sasidharan
Master of Science
Department of Chemistry
University of Toronto
2018
Abstract Magnetic Resonance Angiography (MRA) allows for the visualization of the vascular system
and the diagnosis of various pathologies. A contrast agent (CA) is often used to enhance tissue
contrast. Currently, gadolinium-based CAs (GBCAs) are most widely used but due to the
emerging issues related to Gd-toxicity and suboptimal r1 at high field, there is an increasing need
to replace GBCAs. To overcome these limitations, we have developed water-soluble
Manganese(III) porphyrins (MnPs) as alternatives to conventional GBCAs. In this thesis, the
design, synthesis and characterization of MnTriCPP, a small MnP is reported to show high
relaxivity (21.0 mM-1s-1 at 31.6 MHz and 14.8 mM-1s-1 at 3T) upon non-covalent interaction with
Human Serum Albumin (HSA). Furthermore, to understand the mechanism of HSA binding,
Isothermal Titration Calorimetry (ITC) was chosen to study the binding properties of MnTriCPP
in comparison with previously synthesized MnPs, and all exhibit detectable entropy driven
affinity to HSA.
2019

iii
Acknowledgments I would like to thank my research supervisors, Dr. Xiao-an Zhang and Dr. Kagan Kerman, for
the opportunity to pursue this research project under their supervision and their financial support.
I am incredibly grateful for the assistance and supervision provided to me during my studies as a
Master student. I would like to acknowledge our previous graduate students, Weiran Cheng and
Inga Haedicke whose work has continually provided a foundation for many of the other students
and my own work. I would also like to thank Hanlin Liu, Bhargav Patel and Hassan Seifi Fini
who taught me the practical skills needed for organic syntheses, analytical instrumentation and
provided guidance through my research. Thank you to Dr. Ruby Sullen for being my thesis
reader and providing further discussions. I am very thankful for your guidance on organic
synthetic techniques and analytical chemistry, which have been essential to my undergraduate
and graduate studies. Thanks to the Zhang and Kerman Lab members for the continual support
and time spent together over past three years. Thank you Maryam Abdinejad, Vlad
Constantinescu, Ryan Correa, Haixia Lyu, Guanlin Qiao, Maegan Sweeney, Lida Tan, Keith
Tang, Henry Tieu, Han Su, Shaopei Li, Anthony Singh, Qusai Hassan, Aruna Raja, Hashwin
Ganesh, Dhesmon Lima, Ana Carolina Mendes Hacke, Celia Ferrang and Ari Chow. Thank you
for the stimulating discussions, support and help around the lab. It was a great pleasure to work
with these individuals and the best of luck in future endeavors. Thank you to Tony Adamo, our
TRACES center lab manager at UTSC, for the help and advice involving the use of the analytical
instruments and his effort in maintaining the instruments to be available as needed. I would also
like to thank my family for providing the support and accepting my extended absence during the
duration of my studies.
Thanks to the University of Toronto Scarborough travel grant, for funding my travel to ImNO
2018 conference. This work was supported by the University of Toronto department of
chemistry, the University of Toronto Scarborough, Canada Foundation for Innovation, and
Ontario Research Fund.

iv
Table of Contents Abstract ii.
Acknowledgments iii.
Table of Contents
iv.
List of Tables
vii.
List of Figures
vii.
List of Abbreviations
ix.
List of Appendices
xi.
Preface and Contributions
xiv.
Chapter 1 Introduction
1.2 Angiography
1
1.2.1 Magnetic Resonance Angiography 3
1.2.2 Contrast agent free MRA techniques 4
1.2.3 Blood Pool Agents for Contrast Enhanced MRA 4
1.2.4 MRI Contrast Agents 5
1.2.4.1 MRI BPAs 6

v
1.2.4.2 Gadofosveset and Human Serum Albumin 7
1.2.4.3 Manganese (III) Based Contrast Agents used for Blood Pool Imaging 11
1.2.5 Methods of Analyzing Interactions with HSA 14
1.3 Isothermal Titration Calorimetry 17
1.3.1 Binding Thermodynamics 18
1.3.2 Calorimetry Theory and Operation 20
1.3.3 Data analysis 21
1.3.3.1 Mathematical models 22
1.3.4 Applications of ITC in the Detection of Protein-Protein and Protein-Small
Molecule Interactions
1.4 Objectives of this Thesis
24
26
Chapter 2 Development of Small Manganese (III) Porphyrins as Contrast Agents
2.1 Introduction
27
27
2.2 Results and Discussion
2.2.1 Molecular Design
27
27
2.4.2 Synthesis of MnTriCPP
28

vi
2.2.3 Field Dependent Relaxivity Measurements of MnTriCPP 30
2.3 Conclusions
32
2.4 Materials and Methods
2.4.1 Materials and Instrumentation
32
32
2.4.2 Synthesis 33
Chapter 3 Isothermal Titration Calorimetry Binding Studies
3.1 Introduction
36
36
3.2 Results and Discussion
3.2.1 Isothermal Titration Calorimetry of MnTPPS and HSA
36
36
3.2.2 Isothermal Titration Calorimetry of MnP2 and HSA
37
3.2.3 Isothermal Titration Calorimetry of MnTriCPP and HSA
40
3.3 Conclusions
42
3.4 Materials and Methods
42
Chapter 4 Conclusions and Future Outlooks
43
References
45
Supplementary Information
49

vii
List of Figures and Tables Table 1: Extracellular MR contrast agents suitable for MRA1
Figure 1: Chemical structure of Gadofosveset Vasovist® GdDTPA
Figure 2: Field dependent relaxivity measurement of free floating (black squares) and
HSA bound (red circles) Gadofosveset Vasovist® Gd-DTPA
Figure 3: General schematic of manganese based contrast agents
Figure 4: Chemical structures of (left to right) MnTPPS, MnTCP and MnP2
Figure 5: Field dependent molar relaxivity (two Mn atoms per molecule) of free floating
(black squares) and HSA bound (red circles) MnP2
Figure 6: Left: General schematic of isothermal titration calorimetry instrument. Middle:
Thermogram measuring the heat compensation per injection. Right: Binding curve.
Figure 7: A small MnP, MnTriCPP, as developed based on the chemical structure of tetra-
substituted MnTCP. MnTriCPP is comprised of one paramagnetic centre, three carboxylates
and a single phenyl group to assist in HSA binding.
Figure 8: Field dependent r1 of MnTriCPP (black) and MnTriCPP-HSA (red) at 25°C in pH
7.2 25 mM HEPES. At 3T, MnTriCPP-HSA r1 = 14.8 mM-1s-1.
Figure 9: Isothermal titration calorimetry thermogram and binding curve of 1.984mM MnTPPS
titrated into 150 µM fatty-acid free HSA at 25˙C, 50 mM HEPES buffer, 2 µL per injection and
750 rpm.
Figure 10: Isothermal titration calorimetry thermogram and binding curve of 1.3mM MnP2
titrated into 30µM fatty-acid free HSA at 25˙C, 50 mM HEPES buffer, 2 µL per injection and
750 rpm.

viii
Figure 11: Normalized overlap of injection shown between 550 – 750 minutes of MnTPPS (red)
and MnP2 (blue)
Figure 12: Isothermal titration calorimetry thermogram and binding curve of 4.22 mM
MnTriCPP titrated into 300 µM fatty-acid free HSA at 25˙C,10 mM PBS buffer, 2 µL injections
and 750rpm.

ix
List of Abbreviations CA
CD
CTA
Contrast agent
Circular Dichroism
Computed tomography angiography
DCM Dichloromethane
DIPEA N-N-Diisopropylethylamine
DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
DMF Dimethyl formamide
DSA Digital Subtraction Angiography
DSC Differential scanning calorimetry
ECG Electrocardiogram
EDP Ethyl 2,2-di(1H-pyrrol-2-yl)acetate
ESI Electrospray ionization
FAAS Flame atomic absorption spectroscopy
GBCA Gadolinium-based contrast agents
Gd Gadolinium
Gd-DTPA Gadolinium diethylene triaminepentaacetic acid
HEPES (4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid
HPLC High performance liquid chromatography
IR Infrared spectroscopy
ITC Isothermal titration calorimetry
IV Intravenous
HSA Human serum albumin
Mn Manganese
MnP Manganese porphyrin
MnP2 Manganese (III) porphyrin dimer
MRA Magnetic resonance angiography
MRI Magnetic resonance imaging
MnTriCDP Manganese (III) 20-([1,1'-biphenyl]-4-yl)porphyrin-5,10,15-tricarboxylic acid
MnTriCPP Manganese (III) 20-phenylporphyrin-5,10,15-tricarboxylic acid

x
MnTriEPP Manganese (III) 20-phenylporphyrin-5,10,15-tricarboxylateethylester
MWCO Molecular weight cut off
NMRD Nuclear magnetic resonance dispersion
NSF Nephrogenic systemic fibrosis
PET Positron emission tomography
q Number of coordinated water molecules
r1 Longitudinal relaxivity
RF Radiofrequency
SBM theory Solomon-Bloembergen-Morgan theory
SPECT Single photon emission computed tomography
SPR Surface Plasmon Resonance
T1 Spin-lattice or longitudinal relaxation time
T2 Spin-spin or transverse relaxation time
THF Tetrahydrofuran
TLC Thin layer chromatography
TOF Time of flight
TriEPP Triethyl 20-phenylporphyrin-5,10,15-tricarboxylate
UV-Vis Ultraviolet visible
X-ray/CT X-ray computed tomography
!! Electron relaxation time
!! Life time of coordinated water molecule
!! Rotational correlation time

xi
List of Appendices
Figure S1. 1H-NMR spectrum of DPM-Phe in CDCl3.
Figure S2. 1H-NMR spectrum of EDP in CDCl3.
Figure S3. 1H-NMR spectrum of TriEPP in CDCl3.
Figure S4. UV-vis absorption spectra of MnTriCPP and its synthetic precursors. Dominant
peaks: TriEPP 411nm, MnTriEPP 476nm, MnTriCPP 466nm.
Figure S5. ESI-MS of MnTriEPP. ESI-MS found m/z = 655.2 ([M]+) Calculated for
C35H28MnN4O6+, m/z = 655.14
Figure S6. HPLC trace of MnTriCPP. Area % dominant peak 93.91% purity.
Figure S7. ESI-MS of MnTriCPP. ESI-MS found m/z = 284.1 ([M]2-), calculated for
C29H13MnN4O62-, m/z = 284.01
Figure S8. Isothermal titration calorimetry thermogram and binding curve of 1.3 mM MnTPPS
titrated into 30 µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2 µL per injections and
750 rpm. Left: Trial 2 Right: Trial 3.
Figure S9. Isothermal titration calorimetry thermogram of control experiments of 1.948mM
MnTPPS titrated into 50mM HEPES at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm.
Figure S10. Isothermal titration calorimetry thermogram of control experiments of 50uM
HEPES titrated into 150µM HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm.

xii
Figure S11. Isothermal titration calorimetry thermogram of control experiments 50mM HEPES
titrated into 50 mM HEPES at 25˙C, 2 µL per injections and 750rpm.
Figure S12. Isothermal titration calorimetry thermogram and binding curve of 1.3 mM MnP2
titrated into 30 µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and
750 rpm. Left: Trial 2 Right: Trial 3.
Figure S13. Isothermal titration calorimetry thermogram of control experiments of 1.3 mM
MnP2 titrated into 50 mM HEPES at 25˙C, 50 mM HEPES buffer, 2 µL per injection and
750rpm.
Figure S14. Isothermal titration calorimetry thermogram of control experiments of 50 µM
HEPES titrated into 150 µM HSA at 25˙C, 50mM HEPES buffer, 2 µL per injection and 750
rpm.
Figure S15. Isothermal titration calorimetry thermogram and binding curve of 2 mM MnP2
titrated into 30 µM fatty-acid free HSA at 25˙C, 50 mM HEPES buffer, 2 µL per injection and
750 rpm. Left: Trial 2 Right: Trial 3.
Figure S16: Overlay of 3rd injection of 2 mM MnP2 titrated into 30 µM fatty-acid free HSA at
25˙C, 50 mM HEPES buffer, 2 µL per injection and 750 rpm. Red: 180 second injection
separation Right: 250 second injection separation.
Figure S17: Isothermal titration calorimetry thermogram 9.84mM MnTriCPP titrated into 300
µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2 µL per injections and 750 rpm. Top:
First three injections show substantial baseline noise due to risk posed to instrument the
experiment was run no further. Bellow: Solution contained within sample cell post three
injections show significant aggregation upon MnTriCPP and HSA introduction.

xiii
Figure S18: Isothermal titration calorimetry thermogram 5 mM MnTriCPP titrated into 300 µM
fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2 µL per injection and 750 rpm. First three
injections show substantial baseline noise due to risk posed to instrument the experiment was run
no further.
Figure S19: Isothermal titration calorimetry thermogram 2.5mM MnTriCPP titrated into 300
µM fatty-acid free HSA at 25˙C, 50 mM HEPES buffer, 2 µL per injection and 750 rpm.
Figure S20: Isothermal titration calorimetry thermogram and binding curve of 4.22 mM
MnTriCPP titrated into 300 µM fatty-acid free HSA at 25˙C, 10 mM PBS, 2 µL per injection and
750 rpm. Left: Trial 2 Right: Trial 3.
Figure S21: Isothermal titration calorimetry thermogram of control experiments of 10 mM PBS
titrated into 300 µM HSA at 25˙C, 50 mM HEPES buffer, 2 µL per injection and 750rpm.
Figure S22: Isothermal titration calorimetry thermogram of control experiments of 4.22 mM
MnTriCPP titrated into 10 mM PBS at 25˙C, 50 mM HEPES buffer, 2 µL per injection and 750
rpm.
Figure S23: Isothermal titration calorimetry thermogram of control experiments 10 mM PBS
titrated into 10 mM PBS at 25˙C, 2 µL per injection and 750 rpm.

xiv
Preface and Contributions All studies were conceptualized and executed through the collaborative efforts of PS, KK and
XaZ. The work presented herein has been written by PS and revised by KK and XaZ. The
specific contributions are outlined as follows:
Chapter 2
Liu, H., Sasidharan, P., Zhang, X.A. A Small, Polar and Stable Manganese(III) Porphyrin with
Serum Albumin Affinity as a High Relaxivity MRI Contrast Agent for Angiography at High
Clinical Field (3T)
Unpublished
PS, HL, XaZ designed the experiments. PS performed all synthetic steps and collection of all
spectroscopic data with assistance from HL. PS, HL and XaZ analyzed the data.
Chapter 3
PS, KK, XaZ designed the experiments. All measurements were collected by PS. HL provided
MnP2 and MnTriCPP for analysis. PS, KK and XaZ analyzed the data.

1
Chapter 1 Introduction
1.2 Angiography Angiography is a medical imaging technique used to visualize the vasculature system and allows
for the diagnosis of cardiovascular pathologies and malformations. The resulting image is termed
as an angiogram. A variety of techniques are used within angiography, with the vast majority
making use of X-rays. Wilhelm Röntgen, a German physicist, first discovered X-ray in 1895. 2
Due to it’s the ability of X-rays to penetrate the body deeper than visible light, it permitted
imaging of the internal body. The technique relies on producing an image through interaction of
X-ray photons with electrons. Areas which have a high electron densities, such as hard tissues
like bone, can be clearly visualized with X-ray imaging. The vasculature system has
comparatively less electron density and thus is hardly visible. Since its discovery and inability to
visualize the vasculature system, various attempts were made by use of contrast media or a dye,
particularly the use of halide salts, to image the vasculature system.3 Due to these initial findings,
the use of halide salts proved to be promising for the field of radiographic imaging. Further
developments occurred by the Mayo Clinic team of Osborne in 1923 who had administered
orally and intravenously sodium iodide as treatment of syphilis. Post treatment, the urinary
bladder was seen to become radiopaque in the abdominal radiograph. The technique of
angiography was first developed in the mid-1920s by Professor Antonio Caetano de Abreu Freire
Egas Moniz a Portuguese physician and neurologist at the University of Lisbon. During his
Neurology residency in France, he had trained with renowned doctors in neurological centers
within Paris and Bordeaux. Moniz later performed the first cerebral angiogram in 1927 with a
sodium iodide contrast solution to visualize a pituitary tumour. These images were produced
through use of X-rays deducing the location and size of an intracranial tumour within a patient. 4
Since Moniz’s initial image acquisition, several techniques of angiography have been developed
and used to diagnose a variety of pathologies. Computed tomography angiography (CTA) is one
of such techniques, which make use of a dye (also termed as contrast media or contrast agent)
and X-rays to visualize coronary arteries to produce cross-sectional images of the body. A
catheter is inserted into a blood vessel to supply an X-ray visible dye, which enhances the
visualisation of vascular system. An image is obtained by rotating an X-ray source around an

2
object, with a positioned detector. In order to achieve high levels of X-ray attenuation in
comparison to that which is observed for the biological tissue elements of higher atomic number
are often incorporated into the contrast dye. Historically iodine is the atom of choice for CTA
imaging applications with an atomic number of 53. An incident X-ray is used with energy levels
equal or slightly greater than the binding energy of the electrons of the atom. A large sudden
increase in absorption coefficient is then observed, a series of attenuation profiles of the body is
obtained and the data is transformed into images displaying visible contrast to the surrounding
tissue. 5 CTA produces adequate image quality allowing for stenosis to be ruled out with high
predictive values. Though the X-ray radiation exposure is high due to the nature with which the
data is collected. The data is collected by helical acquisition, which consequently leads to
substantial oversampling of a single sample volume. This translating to the same level within the
patient irradiated with several consecutive rotations of X-ray gantry. Ultimately, this results in
high levels of radiation exposure with reports of doses as high as 30 mSv. 6
Digital Subtraction Angiography (DSA) a second technique is used to image the blood vessels in
bony or dense soft tissue environments with high specificity with reduced background signal
with use of a contrast dye. Before the dye is injected, a background image is often acquired. This
is referred to as the mask image. A dye is delivered through a catheter and X-ray images are
taken of the vessels. To produce the final image, a subtraction of the mask image is carried out.
By subtracting the background signal, DSA provides the advantage of unwanted densities such
as overlying bone or soft tissue to be reduced, thus allowing for the high contrast of blood vessel
and visualization of low concentration dye media. Due to the use of the subtraction technique,
there is increased specificity for the vasculature system is created. Though the methodology still
remains invasive.
A third technique, Pulmonary Angiogram, involves X-ray imaging of blood vessels with a dye to
obtain measurements of the pressure of the blood vessels which carry blood to the lungs and
evaluate blockages or narrowing. Fluoroscopy is often used where continuous X-ray imaging is
carried out. As a consequence, high radiation doses especially in complex interventional
procedures, which require fluoroscopy, is administered for a long period of time.
In addition of X-ray, higher energy γ-ray can also be applied for angiography. Radionuclide
Angiogram alternatively makes use of a radionuclide, which emits γ-ray photon to produce an
image. The technique permits for the evaluation of the heart chambers while in motion with

3
measurements taken while resting and in exercise. Often the radionuclide, usually technetium-
99m (99mTc), is injected into an arm vein tagging the blood cells and allowing monitoring of their
progress through the heart. A gamma camera makes the necessary recordings of the heart wall
and a correlation to the heartbeat can be made by use of electrocardiogram (ECG). Exercise is
performed by the patient and monitored, assisting the physician in assessing the heart's function
during rest and exercise. It is believed that the amount of the radionuclide injected for the
procedure is small enough that there is no need for precautions against radioactive exposure.
Lastly Magnetic Resonance Angiography (MRA) is a truly non-invasive diagnostic procedure
unlike the methods mentioned above. MRA is based on magnetic resonance technology (MRI)
signals, which fundamentally avoid the ionizing radiation, to visualize blood vessels. MRA is
discussed further below.
1.2.1 Magnetic Resonance Angiography MRA is used to visualize the vasculature system and makes use of MRI technology. Due to the
use of non-ionizing radiation this permits for safer means of obtaining anatomical and functional
information. Moreover, MRI has good spatial resolution, high soft tissue contrast and deep tissue
penetration. MRI mainly relies on 1H-NMR signal of water, fat and other biomolecule. With the
vast majority of contributions from water as it is most abundance in vivo. 1H nuclei have a spin
of ½ and will align with or against the direction of an applied magnetic field. The net
magnetization vector then is parallel to the applied magnetic field along the z-axis. For common
practice, usually a 90˚ RF pulse can subsequently be applied resulting in the net magnetization
flipping to the transverse plane (the x-y plane). The RF pulse is then removed and excess energy
is released as the net magnetization vector returns to its original state of thermal equilibrium.
This return is known as relaxation. Relaxation is made up of longitudinal and transverse
relaxation, and the time constants for these two processes are termed as longitudinal relaxation
time (T1) and transverse relaxation time (T2), respectively. T1 is the return of the net
magnetization to equilibrium in longitudinal direction and T2 is return of the net magnetization to
equilibrium in the transverse plane. The variation of MRI signal intensity (contrast) on one hand
is based on variable proton densities, and on the other hand is caused by variable T1 and T2
relaxation times. 7

4
1.2.2 Contrast Agent Free MRA Techniques There are various techniques applied within MRA. One of such is Time of flight (TOF)
angiography, which allows for the deduction of flow related enhancements within the
vasculature system. When TOF angiography is employed, repetitive RF pulses magnetically
saturates stationary tissue. There remains continuous blood flows into the imaging area and due
to having not experienced the pulses a high initial magnetization is seen. This results in a
brighter appearance of blood stream compared to the background tissue. Very often
overestimation of stenosis is seen to occur due to turbulence signal losses. Additionally, when
the pulse applied is in plane of the flow there is signal loss as signal can only be high when the
direction of flow is perpendicular to the RF pulse. 8 The diagnosis of retrograde flow, such as in
subclavian steal syndrome where collaterals around stenotic lesions cause retrograde flow, is not
possible with TOF. This is due to the technique employing a series of “traveling saturation
pulses” as a way to eliminate any signals from veins which are flowing in the opposite direction. 9 Phase contrast angiography, an alternative method of angiography, is applied to obtain
cardiovascular flow measurements and cerebrospinal fluid (CSF) flow studies. Phase contrast
angiography relies on moving spins subjected to bipolar gradients applied along one anatomical
axis leading to changes in phase shifts. This gives sensitivity to the flow in a specific direction,
measurements of phase shifts are obtained and velocity can be deduced.
1.2.3 Blood Pool Agents for Contrast Enhanced MRA CAs can also be referred to as blood pool agents (BPAs) when they are specifically optimized for
MRA. Unlike common CAs, BPAs provide the advantage of limited extravasation from healthy
blood vessels, resulting in prolonged blood circulation time in the body and consequently
prolonged imaging. Moreover, targeting BPA design to the proteins or other large molecules
existing in the vasculature system can assist in increasing the circulation time. The long retention
times provided by BPAs are a desirable property when investigating any vasculature related
diseases within MRA.

5
1.2.4 MRI Contrast Agents The application of CAs shorten the relaxation times of the 1H nuclei within the surrounding bulk
water, in turn enhancing the signal in the vicinity of the CA.7 CAs often contain a paramagnetic
transition metal with unpaired electrons whereby enhanced relaxation is achieved by electron-
nuclear coupling between the 1H nuclei and the unpaired electron(s).7 The properties of the
paramagnetic metal used as well as its surrounding chelate influences the relaxation rate. Tissue
selectivity can also be realized by altering the molecular structure on the chelate.
Conventional CAs employed in vivo shortens the T1 or T2 relaxation times of the surrounding
bulk water molecules and therefore enhancing the signal. Moreover, employing a CA shortens T1
and T2 to varying dependent upon their chemical design and the applied field. Due to this, they
are often classified as a T1 or T2 agents. T1 agents lead to an increase in signal intensity and a
brightening of the image and T2 agents lead to a decrease and darkening of the image. Most often
for clinical MRA, T1 agents tend to be most commonly used for in vivo.
The effectiveness of a CA to enhance T1 relaxation is referred as relaxivity (r1) given in unit of
mM-1s-1 and describes the rate increase in relaxation per millimolar of paramagnetic metal. The
relaxivity of the CA depends upon the direct coordination of the metal ion with water (inner
sphere relaxation), interaction of water in near proximity of the metal due to hydrogen bonding
with nearby functional groups on chelate (second sphere relaxation) and the bulk water
interactions (outer sphere relaxation). Moreover, the Solomon-Bloembergen-Morgan (SBM)
paramagnetic relaxation theory quantitatively describes the molecular parameters related to r1.10
Among them, the prominent parameters are the number of water molecules that are coordinated
to the metal (q), the distance between the water protons and electron spin (r), the molecular
rotational correlation time (!!), life time of the inner sphere water remains coordinated to the
metal (!!) and the electron relaxation time (!!).10 Modification to CA design can subsequently
be made based upon the SBM theory whereby an increase in q, decrease in r or an optimization
of !!, !! and/or !! can increase r1 of a CA.

6
1.2.4.1 MRI BPAs CAs commonly used for MRI are Gd-based contrast agents (GBCAs) which is often introduced
intravenously as a 20 mL injection ranging from 0.5-1 M administered over 10 seconds
following a saline flush.11 The employment of a CA removes the confounding factors that had
played a role in non-contrast enhanced MRA like velocities, flow direction or turbulence.
Moreover, signal intensity is determined principally by the concentration of the Gd(III) within
the vessels. A variety of GBCAs are clinically available spanning two categories: extracellular
fluidic (ECF) agents for first pass MRA and intravascular agents for steady state MRA. The
agents all contain Gd(III) within its core, coordinate to one water molecule and differ in iconicity
and have variable chelates as shown within Table 1. The majority of these ECF CAs for MRA
are non-protein binding and are not tissue specific.
Table 1: Extracellular MR contrast agents suitable for MRA1
Ionic Gadopentetic acid dimeglumine salt
Magnevist®
Gd(DTPA)2-
Gadobenate dimeglumine
MultiHance®
Gd(BOPTA)2-
Non
Ionic Gadodiamide hydrate
Omniscan®
Gd(DTPA-BMA)
Gadoteridol
ProHance®
Gd(HP-DO3A)
Gadobutrol
Gadovist®
Gd(DO3A-butriol)
O
O-
Gd3+
NN
O-O O-O
N
O- OO-
O
ON
Gd3+
O-
O
NN
OO-
O
O-O
O-
O
HN
O
Gd3+N
NN
O
NH
O-O
O-
O
O-
O
OGd3+
NN
O-O
N
O-
ON
O-
O
OHN
Gd3+
O
HO
NN
N
O-
O
O-O
O-O

7
1.2.4.2 Gadofosveset and Human Serum Albumin The use of extracellular CAs for MRA has been an established methodology. Though these
nonspecific extracellular agents can lead to diffusion into the extracellular space and therefore
cleared quickly through renal filtration. This extravasation from the circulation system causes
intravascular signal decrease leading to a shorter available window for MRA image acquisition.
Additionally, these agents generally display lower r1 and so its use requires multiple, high
concentration injections for contrast enhanced MRA. To overcome these limitations, a more
selective BPA can be beneficial. Due to the variety of molecules present within the circulation
system, these macromolecules have been explored extensively as a potential sites of covalent
attachment to Gd-based CAs.12
Lauffer et al. designed a small Gd-based CA, MS-325 (Gadofosveset, GdDTPA, Vasovist®) as
shown within Figure 1, which displays non-covalently binding to human serum albumin (HSA).
In addition, increased relaxivity is seen due to the slower tumbling rate (τR).13 Due to its success,
it was the first intravascular contrast agent approved for MRA in European Union and USA. It is
prepared as a single injection of 244 mg/ml and administered as a dosage of 0.03 mmol/kg.
Figure 1: Chemical structure of MS-325 Gadofosveset Vasovist® GdDTPA
Gadofosveset has been shown to be advantageous due to the high relaxivity provided at 1 tesla
(T) with an r1 of 31.7 mM-1s-1, persistent high intravascular enhancement, high resolution image
in steady state, its use in first pass arterial imaging and does not require following bolus in first
pass as most other contrast agents do. 14 Often when the initial injection of a CA is applied it is
done as a fast, large bolus of contrast medium. The time of contrast material bolus arrival and
organ enhancement is highly correlated to each other and therefore a required piece of
information during imaging. Due to the large variation that is seen in downstream contrast

8
material bolus flow, it often can be challenging to determine a precise scan delay based on when
the contrast medium arrives at the area of interest. Following the bolus represents the time of
contrast material arrival or the contrast material bolus transit. With this information, the scan
delay is individualized for each organ, ultimately optimizing the injection protocol and
parameters for desirable organ specific enhancement.15 There is greater convenience with
Gadofosveset as there is an extended diagnostic window due to the decreased dependency of
enhancement on bolus dynamics.
Gadofosveset is known to reversibly bind to human serum albumin (HSA) as it contains
gadolinium diethylene triaminepentaacetic acid (Gd-DTPA) chelate attached with a hydrophobic
diphenylcyclohexyl (via a phosphate linker) which is believed to be the primary site of binding
with HSA. It has been found to display a dissociation constant (Kd) of 85-90.9 ± 26.7 µM with
HSA as determined through Ultrafiltration studies with a 5 kDa molecular weight cut off filter. 13,16
HSA is a prominent protein in the blood plasma at concentrations of approximately 700 µM,
synthesized in the liver and is known to have a variety of ligand binding capabilities. It is 66.5
kDa in size, contains three prominent binding regions, referred to as Sudlow’s site I, II and III,
and each made of 2 separate helical domains (A and B). Most often bulky heterocyclic anions
bind to Sudlow’s site I, aromatic carboxylates bind Sudlow site II and subdomain IB is known to
bind heme. 17 For intravenous CAs, binding to HSA influences kinetic and pharmacodynamic
properties. Specifically, it will keep the CA with long retention in the blood stream, and reduce
extravasation to non-vascular tissue. Therefore, it improves the selectivity of vascular
enhancement and increase the time window for imaging. Moreover, the binding is seen to
improve efficacy of CAs due to the water proton relaxation time dependency on the tumbling
motion (!!) of metal-chelate complex. 17 The phosphodiester linkage of Gadofosveset limits
hepatic clearance and GBCA is seen to remain within the intravascular space with minimum
extravasation. Moreover, it is primarily excreted renally with 94% excreted in first 72 hours and
the remaining in feces. In individuals with renal impairment, an alteration to the plasma half-life
is seen to occur with an increase of 2-3 fold. 16

9
Though GBCAs have dominated the market for more than 30 years, they do exhibit some
toxicities in patients with renal dysfunction. Often these patients already have persisting kidney
pathologies, which lead to poor clearance of GBCAs. This coupled with the disassociation of
free Gd3+ cation and subsequent accumulation in tissues would then be implicated in the
development of nephrogenic systemic fibrosis (NSF). 18 The precise mechanism of NSF
development is unknown but is postulated that endogenous transition metal cations, such as zinc,
iron and copper ions, induces Gd3+ dissociation from chelate. These free cations competitively
bind to the chelate to replace Gd3+ which is released to and then binds to any available
endogenous ligand such as phosphate, carbonate, hydroxide or other salts. For example,
gadofosveset contains a linear ligand, which is less stable than a cyclic. Due to this, and
gadofosveset’s intrinsically longer vascular retention time there is an increased risk of Gd
release. The free or bound Gd3+ resulting then passes from the intravascular to extravascular
space to later interact with surrounding tissue and also macrophages. The details of each step
needs to be confirmed but this interaction results in a series of inflammatory events. Activation
of macrophages or monocytes and the production of proinflammatory and profibrotic cytokines
and chemokines are seen to occur resulting in the symptoms of NSF development.19
Moreover, Gd3+ deposition also has been observed to occur within the brain and eyes of
individuals with healthy kidneys.18,20 Experimentation conducted by Kanda et al. Used
inductively coupled plasma mass spectroscopy (ICP-MS) to evaluate gadolinium accumulation
in 23 post-mortem brain tissue subjects. Areas, which were specifically examined, were the
dentate nucleus and globus pallidus regions within the brain. Subjects with a previous history of
GBCA administration but no renal impairment were selected in addition to those with no
previous history and no renal impairment. These samples with a history of GBCA use were
found to contain higher concentrations of gadolinium (mean concentration = 0.25 ± 0.44 µg/g
brain tissue) than those who have had no previous history of GBCA administration (mean
concentration = 0.0025 ± 0.005 µg/g brain tissue). Due the body not naturally having
gadolinium, its presence was associated with the GBCA.18 Additionally to this, Hitomi et al. had
unexpectedly found gadolinium leakages to occur in ocular structures in patients who have had
acute strokes. This was further studied in 167 patients who underwent administration of a
GBCA. It was found 67% of patients displayed the leakage in the aqueous chambers of the eyes.
10
Though the mechanism was unknown, it has shown that there is some permeability of
gadolinium across the blood-ocular barrier.20 These findings prove to be impactful, as the use of
GBCAs is seen to effect even individuals with healthy kidneys.
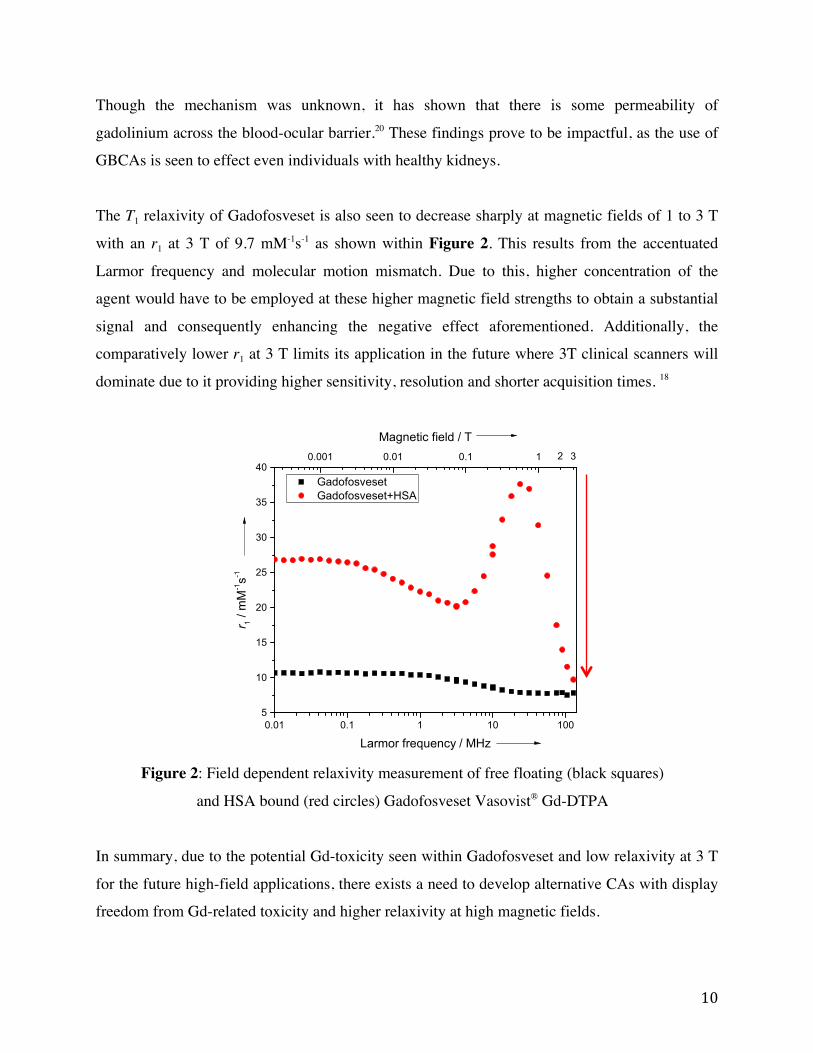
The T1 relaxivity of Gadofosveset is also seen to decrease sharply at magnetic fields of 1 to 3 T
with an r1 at 3 T of 9.7 mM-1s-1 as shown within Figure 2. This results from the accentuated
Larmor frequency and molecular motion mismatch. Due to this, higher concentration of the
agent would have to be employed at these higher magnetic field strengths to obtain a substantial
signal and consequently enhancing the negative effect aforementioned. Additionally, the
comparatively lower r1 at 3 T limits its application in the future where 3T clinical scanners will
dominate due to it providing higher sensitivity, resolution and shorter acquisition times. 18
Figure 2: Field dependent relaxivity measurement of free floating (black squares)
and HSA bound (red circles) Gadofosveset Vasovist® Gd-DTPA
In summary, due to the potential Gd-toxicity seen within Gadofosveset and low relaxivity at 3 T
for the future high-field applications, there exists a need to develop alternative CAs with display
freedom from Gd-related toxicity and higher relaxivity at high magnetic fields.
0.01 0.1 1 10 1005
10
15
20
25
30
35
40
Gadofosveset Gadofosveset+HSA
r 1 /
mM-1s-1
Larmor frequency / MHz
2 30.001 0.01 0.1 1
Magnetic field / T

11
1.2.4.3 Manganese (III) Based Contrast Agents used for Blood Pool
Imaging In order to avoid Gd(III) related toxicity, our research group aims to study different platforms for
safer Gd-free T1 contrast agents. As an alternative, Manganese(III) porphyrins (MnPs) have been
proposed for applications as a CA as shown within Figure 3.21 MnPs display high
thermodynamic and kinetic stability preventing Mn dissociation, low toxicity as manganese is a
naturally occurring micronutrient within the body. MnP displays two water coordination sites as
opposed to the one of GBCA, a rigid backbone by use of a porphyrin chelate, a natural product
with various derivative created by nature, and versatile modifications on the porphyrin backbone. 22 Our group has developed a few synthetic strategies to introduce different functional groups in
the meso-positions (marked as R – R”’ in Figure 3) which tuned the balance of hydrophobicity
and water-solubility.
Figure 3: General schematic of manganese based contrast agents
The potential of MnPs for application as MRI contrast agent was first evaluated by Chen et al. in
1984 with MnTPPS (structure see Figure 4). The r1 (10.4 mM-1s-1) was first reported at a single
magnetic field strength of 0.47 T, higher than regular small Gd-complexes. 23 A later study, led
by Koenig, investigated the magnetic field dependent behaviour relaxivity of MnTPPS using
nuclear magnetic relaxation dispersion (NMRD) method, and reported an increased r1 from 1
MHz onwards to 40 MHz , this unlike typical small GBCA.24 MnTPPS has also been shown to
localize in tumours though the precise mechanism is unknown.25 Many in vivo studies do indicate
MnTPPS to consistently show tumour enhancements but contradictory results has been seen
with regards to its toxicity. Multiple reports indicate no toxic side effects post-IV administration
in animal tumour models.26 Though a LD50 of ~ 0.5 mmol/kg for IV administration was
published by Lyon et al.27 and has been frequently cited by others.28,29 Though this preliminary
N N
N N
R''
R
R'''MnR'

12
study was only reported in an unpublished personal communication article and is likely an area
where more studies are required.
MnTCP was later developed in our group, as shown within Figure 4, based upon the structural
modification of MnTPPS with the replacement of phenylsulfonate groups with carboxylate
groups. As a result, MnTCP is a smaller and more polar manganese porphyrin. This decreasing
the second sphere relaxivity shell and perhaps assisting in inner sphere relaxation. Moreover, an
r1 at 3 T (7.90 mM-1s-1) was seen to be greater than Gadofosveset. It provided stronger signal
enhancements in vivo with efficient elimination of the agent through renal filtration with full
clearance approximately 24 hours post injection. Studies conducted with MnTCP in six tumour
bearing female nude rats suggested it to be a more sensitive alternative to extracellular GBCAs
for tumour imaging. 30
Meanwhile, our research group also developed MnP2, an MnP dimer, based upon the MnTPPS
monomer building block. MnP2 was designed with aims of improving r1 by increase number of
paramagnetic centers consequently increasing number of inner sphere water coordination sites.
Moreover, the design also included optimization of a relaxation process, by an increasing !!,
based on the SBM theory described in section 1.2.4. This has been approved as an effect strategy
for GBCAs to increase r1. By covalently dimerize MnTPPS while maintaining the rigid
structure, slowing rotation (longer !!) was expected to yield an increased r1. Indeed, MnP2 was
shown to have high relaxivity of 20.9 mM-1s-1 per Mn at 1 T and 16.3 per Mn at 3 T. 22 Which
has been found to be higher than that of Gadofosveset and thus more applicable in a clinical
market where 3 T clinical scanners will dominate.
In addition to the increase of r1, the structural modification also led to a drastic difference on the
pharmacokinetic properties of MnP2 in contrast to MnTPPS and MnTCP. A much longer
vascular enhancement retention time was observed with a relatively low dose of 0.05 mmol of
Mn injection in rats. 31 Upon in vivo IV injection significant T1 contrast enhancements were seen
to be present in rats. Specifically liver enhancements persisted with a decrease in contrast across
a 3 day period, after which the agent was metabolized by the liver. No bladder enhancements
were seen across the entire experimental period with confirmation of no MnP2 signal as found

13
when analyzing urine samples as tested through Mn AAS and UV-VIS. The desired property of
blood pool imaging was achieved where long persistent enhancement of the blood vessels and
heart were found. Due to the significantly high r1 that was achieved with MnP2, the findings
suggested a lower dosage was sufficient for contrast enhancement. Additionally, all rats were
found to remain healthy after the in vivo studies suggesting MnP2 displays good
biocompatibility.22 Successful imaging of cardiovascular and lungs was also seen to be present.31
The structural design of MnP2 consisted of two water-soluble MnP units that were joined with a
hydrophobic biphenyl bridge. This biphenyl structures has been previously found to display
strong interactions with HSA.32 To evaluate this optical spectroscopic method such as circular
dichroism (CD) was utilized to quantify this binding. Induced optical acitivity was seen to be
present upon MnP2 binding to HSA this suggesting a specific and tight interaction. A 1:1
stoichiometric ratio was determined due to a plateau in induced ellipticity after addition of 1
equivalence of HSA. A dissociation constant (Kd) of 2 µM was determined through fitting with a
least squares fit based model. Despite the targeted 1:1 ratio of MnP2 and HSA and the high
binding affinity a decreased relaxivity was seen to occur upon HSA binding as shown within
Figure 5 across all magnetic field strengths.
Figure 4: Chemical structures of (left to right) MnTPPS, MnTCP and MnP2
N N
N N
O+Na-O
+Na-O O
O
O-Na+Mn
O
+Na-O
N N
N NMn
SO3
O3S SO3
SO3
MnTPPS MnTCP MnP2

14
Figure 5: Field dependent molar relaxivity (two Mn atoms per molecule) of free
floating (black squares) and HSA bound (red circles) MnP2
Experimentation conducted within the group indicate this decrease in r1 is seen to occur due to
deep hydrophobic pocket binding and therefore blockage of the coordination sites within MnP2.
Additionally, this included binding to a hydrophobic heme-binding pocket. This creates an area
of improvement in the design of MnPs for MRA.
1.2.5 Methods of Analyzing Interactions with HSA The prominence of HSA within the blood plasma at an approximate concentration of 700 µM
makes it an ideal candidate of targeted binding with CA for intravascular imaging. Ideally this
binding should be reversible and the equilibrium will result between free and bound species.
Therefore, there is a need to obtain a complete understanding of the drug-protein interaction
because such information is crucial to influence the pharmacodynamics and pharmacokinetics
(absorption, distribution, metabolism and elimination) properties of the drugs, including imaging
agents and therapeutic agents. This evaluation is often carried out in the earlier stages of drug
development and can assist in drug design and modification.
0.01 0.1 1 10 100
5
10
15
20
25
30
35
40
45
MnP2 MnP2+HSA
r 1 /
mM-1s-1
Larmor frequency / MHz
0.001 0.01 0.1 1
Magnetic field / T2 3

15
Currently, a variety of approaches exist to assess drug-protein binding, these being separative
and non-separative methodologies. Equilibrium dialysis, a separative methodology relying
primarily on molecular size and weight differences, evaluates binding across a two-compartment
semipermeable membrane. The membrane allows molecules of certain size to pass through and
very often this includes a permeable drug molecule and impermeable proteins. The complex of
drug-protein occurs with incubation and later equilibrium. The remaining free drug fraction is
then measured. Equilibrium dialysis poses advantages of studying drug interactions as free
floating with equilibrium maintenance mimicking in vivo circumstances. Though despite this,
equilibration may be long and the equilibrium time should be known. Additionally, volume shifts
due to oncotic pressure occur and this seen often with albumin. Nonspecific adsorption of drug
or protein on dialysis membranes can occur skewing results. 33
Ultrafiltration, a similar methodology to equilibrium dialysis, makes use of a similar method
with decreased analysis time due to pressure applied across the membrane. Similar advantages
and disadvantages, however, are seen. Ultracentrifugation utilizes a mixture of drug and protein
molecules and places the mixture within a centrifugal field resulting in sedimentation. Any free
drug molecules remain within solution due to lower sedimentation coefficients. The free drug
within the supernatant is then quantified. Though this methodology is within solution, the
physical phenomena of centrifugation can change free drug concentrations due to its own
sedimentation, any back diffusion or viscosity effects. 33
Alternatively, size exclusion chromatography relies on separation based upon size or the
hydrodynamic volume. Most often the drug and protein solutions are eluted through a packed
porous column. Free protein and drug-protein complexes elute due to being too large and unable
to penetrate the pores of the packing. In contrast, the free drug molecule elutes later due to its
interaction with the stationary phase. This methodology, though in free solution, can display low
column efficiency and poor protein recovery. 33
Lastly, high performance affinity chromatography relies on the immobilization of protein on a
support and subsequent injection of a drug molecule into the column. Drug molecules, which
display a high affinity, interact with the protein. This technique is advantageous as a small

16
amount of protein is required, the protein can be reused with disassociation treatments, minimal
run-to-run variations, the opportunity for automation, ease in studying racemic mixtures and
lastly the prevalence of albumin drug binding studies. Though it requires the immobilization of
proteins, which can affect protein behaviours such as denaturation, improper orientation
preventing binding or steric hindrances. 33
An alternative to separative methodologies includes non-separative methods. Spectroscopy
studies such as Ultraviolet visible spectroscopy (UV-VIS), fluorescence, infrared spectroscopy
(IR), nuclear magnetic resonance (NMR), or circular dichroism (CD) studies of the drug-protein
binding make use of changes of electronic or spectroscopic energy levels upon binding. These
methodologies provide significantly more advantages as they enrich the information known
about the binding mechanism. Fluorescence allows for the quantification of the binding kinetics
and affinity, identification of binding sites of a drug and calculations can be carried out deducing
the binding distance between a fluorophore on the protein and drug.34 IR permits for the study
secondary structures of protein, and how binding impacts it. NMR is versatile yet technically
challenging method to provide kinetic, thermodynamic and structural information. For example,
it can be used to deduce the groups that are involved in binding process. Finally CD can be used
to deduce the 3D structure of complex, including the drug binding site. 33
Calorimetric techniques are another non-separative methodology. For example, differential
scanning calorimetry (DSC) can be made use of to characterize protein stability and folding. A
reaction cell within the DSC instrument contains mixture of drug and protein molecules
subjected to heated. The technique primarily relies on the transition midpoint of protein ligand
complex, this being the temperature at which 50% protein in native conformation and 50% is
denatured, where by an indirect method is used to find the binding constant. The technique poses
some advantages as it allows for the measure of large binding constant (1015 M) and provides full
thermodynamic picture. Though there is low throughput and large sample consumption. 33
Alternatively Surface Plasmon Resonance (SPR) based assays immobolize one reactant on
surface and monitor the interaction with a second component that flows over the surface. A
change in refractive index is seen as a complex is formed occurs at the sensor surface. A visual
inspection provides information on complex formation (increase resonance), equilibrium

17
(plateau), and later reversible reactions (decrease response). The technique provides kinetic
stability information with a time and concentration dependency to give quantitative information
on stoichiometry, binding constant and kinetic rate constants. SPR is advantageous real time
characterization can be carried out, quantitative information is obtained, small material use and a
wide range of affinity constants can be determined (Kd mmol/L to pmol/L ranges). Though the
immobilization of protein or drug molecule may alter the binding patterns and a high
maintenance cost is associated with instrument upkeep. 33,35 For most GBCA-BPAs, HSA
binding was measured by use of separative methodologies as opposed to non-separative due to
primarily the lack of spectroscopic signal. Isothermal titration calorimetry (ITC) permits for the
study of the thermodynamics associated with complex formation. Due to the technique not
requiring any structural modifications and being a technique completely within solution it is
discussed further and utilized as a means of preliminary binding affinity studies within this
thesis.
1.3 Isothermal Titration Calorimetry Calorimeters are one of the first scientific instruments utilized to study various heat evolving
processes. Calorimetry is often considered to be a universal detector as any chemical reaction or
physical change will involve a change in heat or the enthalpy. The first reported use of
calorimeters was by Black et al. in 1760s where measurements of heat capacity and latent heat of
water were carried out. Later Lavoisier designed ice calorimetry and utilized the instrument to
measure the metabolic heat produced by a guinea pig in measurement chamber. Christensen et
al. first described the use of titration calorimetry in 1966 and it was initially applied to weak
acid-base equilibria and metal ion complexation reactions. These initial systems were limited to
Keq values of less than 104-105 M-1 at the time. 36 Beaudette and Langerman were later the first to
publish calorimetric binding studies of biological system in 1978. 37 Their studies had marked the
beginning of using calorimetry to study biological equilibria and it is now commercialized and
routinely used to characterize thermodynamics of biological binding interactions.
ITC, a type of calorimeter, is used to characterize the thermodynamics of a binding interaction. It
utilizes a constant temperature (isothermal) while the titrant is titrated into a sample cell during
which time the change in heat is measured (calorimetry). Moreover there may be heat taken from

18
surroundings (endothermic) or given up to surroundings (exothermic). The given amount of
enthalpy change for a particular reaction (∆H) is given in kcal/mol or kJ/mol. ITC primarily
makes a measure of this enthalpy change and from this resulting information of stoichiometry of
binding interaction (n), affinity constant (Ka), enthalpy (∆H) and entropy (∆S) is obtained. ITC
provides some advantages as it is done within solution, without any ligand or protein
modifications, with reactants that appear spectroscopically silent and can be done over various
conditions such as temperature, salt content or pH for example. Modern ITC instruments, such as
MicroCal iTC200 or MicroCal VP-ITC, allow determination of binding constants within the
ranges of 104-109 M-1. Though ITC poses difficulties when low affinity systems are being
analyzed and aggregation or precipitation occur. To obtain conclusive data a user must design
the optimum experiment, carry out complete data analysis and adjust fitting parameters. The
specifics of each discussed further below.
1.3.1 Binding Thermodynamics Thermodynamics is a branch of science that correlates heat and temperature and their
relationship to energy and work. Three laws of thermodynamics govern all behaviour. These
laws define fundamental physical quantities such as temperature, energy and entropy and relate
them to thermodynamic systems and thermal equilibriums. Moreover, they describe the
expected behaviour under certain circumstances and forbid certain phenomena. The laws are as
follows:
First law of thermodynamics: Energy can neither be created nor destroyed only
transferred from one form to another
Second law of thermodynamics: The entropy of the universe only increases and
never decreases
Third law of thermodynamics: Entropy of system approaches a constant value as
the temperature approaches absolute zero
These thermodynamic parameters are utilized to describe a certain binding process. Binding
involves a ligand interacting with a specific site on a second molecule. A generalization of

19
binding is shown in equation (1) where upon binding three species of molecules result; these
include free binding site, free ligand and complex.
!"#"$%&' + !"#$%& ↔ !"#$%&' (1)
To understand the thermodynamics of the binding interaction knowledge of Ka, n, ∆H and ∆S are
required. Moreover the driving forces of reactions can supplement this understanding.
Reactions can be considered as spontaneous because they give off energy in form of heat (∆H <
0) or alternatively lead to increase in disorder of system (∆S > 0). Calculations of ∆H and ∆S can
determine driving force of reaction. When one is favourable and the other is not Gibbs free
energy (G) is used to define the thermodynamics of a system. This is given as equation (2) where
T is the temperature in Kelvin, ∆G Gibbs free energy change given in kJ/mol, ∆H is the enthalpy
change and ∆S is the entropy change for complex formation.
∆! = ∆! − !∆! (2)
Equation (2) defines the extent to which the enthalpy and entropy terms act as the driving force
in a reaction and can be utilized to determine if a reaction is spontaneous or nonspontaneous.
When ∆G is negative the reaction is said to be favourable and spontaneous. Moreover, as entropy
is related to temperature this suggests that the entropy term becomes prominent as the
temperature increases.
The relationship between free energy of reaction (∆G) and the standard state free energy (∆G˙)
given by equation (3). ∆Go is standard Gibbs free energy change, R is the universal gas constant
and Q the reaction quotient a specific time.
∆! = ∆!! − !"#$% (3)
When the reaction is at equilibrium Q = Keq and ∆G = 0 equation (3) can be written in the form
of equation (4) wherein Keq is that which is shown in equation (5). Thus permits for the
calculation of the equilibrium constant of any reaction at standard state free energy. Equilibrium
constants furthermore are seen to change with temperature with this relationship. Additionally,
∆G˙ to is dependent on the temperature as shown in equation (6).
∆!! = −!"#$!!" (4)

20
!!" =!"#$%&'
!"#"$%&' [!"#$%&] (5)
∆!! = ∆!! − !∆!! (6)
The following equations are utilized in deducing thermodynamic relationships. ITC provides the
advantage of providing accurate values of K, ∆G, ∆H, -T∆S, and n in a single experiment
provided it is done under optimal experimental conditions.
1.3.2 Calorimetry Theory and Operation
A general schematic shown of ITC operation is shown within figure 6. Where often the ligand at
defined concentration is placed within the syringe and the macromolecule at defined
concentration placed in sample cell. Reference contains the solution within which the
ligand/macromolecule were dissolved in. The results from a single titration are recorded within
a thermogram where each peak represents the thermal effect associated with the injection.
Figure 6: Left: General schematic of isothermal titration calorimetry instrument. Middle:
Thermogram measuring the heat compensation per inject. Right: Binding curve.
A titration mode is used where there are incremental injections of the ligand at predefined time
intervals. The experiment therefore must be designed in a manner where the heat change is
measurable per injection and varies for subsequent injections producing a curved thermogram.
The curvature within the thermogram is a function of the concentration of macromolecule
Syringe
Sample Reference

21
([M]tot) and the equilibrium constant (Keq) such that it satisfies the c parameter as shown in
equation (7).38 An acceptable value of c is considered to be within the range of 10-500.
! = ! ! !"!!! (7)
Provided it is within range, the thermogram is considered to have an acceptable curvature for the
nonlinear regression analysis to obtain accurate thermodynamic parameters. Very often
experiments with large Ka constants (Ka > 108 M-1) must be done with low macromolecule
concentrations and opposite for small Ka values (Ka < 104 M-1). It is of paramount importance
accurate concentrations of the ligand/macromolecule are prepared and the ligand and
macromolecule must be matched with regards to pH, buffer and salt concentrations. If not
matched there may be dilution heats, which overwhelm the heat signal of the binding reaction.
Additionally, it is generally recommended the concentration in the syringe be 15-30 fold larger
than the concentration within the sample cell. Ultimately it is the user who must find the ideal
compromise between concentrations, c value, injection volume and spacing to obtain a well
defined symmetrical sigmoidal binding curve.
The calorimetric measurements are taken by power compensation where the sample cell is
controlled at a constant temperature. A temperature controller and heater are used to keep cell
temperature constant with reference to the reference cell. As titration is carried out chemical
reaction takes place and heat input or use from a chemical reaction is sensed and the power
applied is compensated to maintain this constant temperature. The raw signal in power
compensation is recorded as µcal/sec on the thermogram. The individual peaks are representative
of the time required for the return to a baseline reference temperature. Very often the initial heats
are larger as there is larger excess of unpopulated binding sites. As the titration proceeds less
binding events occurs and three species remain; these include free ligand, free macromolecule
and ligand macromolecule complex
1.3.3 Data analysis Heat effects can arise from a variety of events these include binding interaction, ligand dilution,
macromolecule dilution, heat effects from buffer and mixing heat effects. Due to this, heat of
dilution of macromolecule for example must be accounted for in control experiments. These
include titrations whereby the buffer is titrated into sample cell containing the macromolecule.

22
The same is done for ligand control experiment where by the ligand is titrated into the sample
cell. Finally a buffer into buffer control experiment is done for thoroughness. These individual
dilutions of heat generated from macromolecule and ligand must be measured in separate
experiments and a correction of the most prominent control experiment is done through the
software in accordance to equation (8).
!!"##$!%$& = !!"#$%&"' − !!"#$%"&' !"#$%& − !!"#$%"&' !"#$%!%&'#(&' − !!"#$%"&' !"##$% (8)
Upon conducting this correction, an integration of the thermogram is taken to produce the
binding curve. It is represented as the µcal/sec evolved per injection as a function of
ligand:protein molar ratio. To determine relevant thermodynamic parameters involved in the
binding process a binding model must be assumed. These include the following:
One-set-of-sites: n number of non-interacting thermodynamically indistinguishable sites
Two-set-of-sites: Two independent thermodynamically distinguishable sites
Sequential Binding: n > 1 number of sites displaying some co-operativity
The use of these models is considered to be a curve-fitting process by nonlinear regression fitting
to the corrected titration integration values. The model outputs the best values of K, ∆H, and n
within experimental error determined through an iterative process. The model itself is made up
of a predefined mathematical description of the physical processes taking place in the
calorimeter. The dependent variable therefore is the heat or heat rate and it is defined as a
function of a variety of independent variable.
1.3.3.1 Mathematical models To obtain thermodynamic data by use of the one-set-of-sites model a series of mathematical
equations are utilized. Under this model there exists ! ≥ 1 set of thermodynamically
indistinguishable sites. The following is a discussion of the simplest mathematics associated with
the binding curve fitting process of the one-set-of-sites model. Other models discussed above are
composed of more complex mathematical representations and for this reason they are not delved
any further in this section.

23
A general term referred to as the binding parameter (!) is related to the fractional saturation (!)
and the number of binding sites (!) through the equation (9). The fractional saturation is defined
as the fraction of macromolecules that are saturated with the ligand and can be written further as
shown in equation (10) where [M] is the concentration of the free macromolecule, [X] the
concentration of free ligand and [MX] the concentration of complex. Additionally, the binding
constant Ka can also be written as a function of fractional saturation in the following equation
(11). 39
! = !" (9)
! = !"! + !" = ! ! !!
! + ! ! !!= !! !1+ !! !
(10)
!! =!
(1− !)[!] (11)
The concentration of free ligand is related to the total ligand ! ! and bound ligand ! ! by mass
conservation law as shown within equation (12). Equation (12) can be rewritten to include
equation (9) in form shown within equation (13)
! = ! ! − ! ! (12)
! = ! ! − !" ! ! (13)
When combining equation (11) and (13) the quadratic equation shown in equation (13) is
obtained. Moreover the only root is given as equation (14). 39
!! − ! 1+ 1!!! ! !
+ ! !! ! !
+ ! !! ! !
= 0 (13)
! = 12 1+ 1
!!! ! !+ ! !![!]!
− 1+ 1!!! ! !
+ ! !![!]!
!− 4 ! !! ! !
(14)
The integral of the thermogram is taken and the binding heat after the ith injection is given by
equation (15). Where !! is the cell volume and ∆!! the molar enthalpy change of the ith injection.
A correction is conducted as the volume with the sample cell changes with each progressive
titration as shown in equation (16). 34,40
! = ! ! !!!∆!!!! (15)

24
! ! = ! ! + !!!!!! ! + ! ! − 1
2 − !(! − 1) (16)
The heat of the ith injection is given by equation (17). Where ∆ ! ! is difference in bound ligand
concentration between ith and (i-1)th injection. When utilitizing one-set-of-sites model equation
(17) becomes that which is shown in equation (18) involving the substitution of equation (10). 39
! = !!∆!!∆ ! ! = !!∆!!! ! !(!! − !!!!) (17)
! = !!∆!!! ! !!! ! !
1+ !! ! !− !! ! !!!1+ !! ! !!!
(18)
The experimental data is non-linearly fit using the equations defined by (13) and (18) to form a
!! vs ! !! !
binding curve. The model gives predictive vales for Ka, ∆Ht and n utilizing equations
(13)-(16). The best fit of this nonlinear regression is one that has minimum error square sum as
determined through the Marquardt Method, an algorithm utilized by the software during the
curve fitting process.
1.3.4 Applications of ITC in the Detection of Protein-Protein and
Protein-Small Molecule Interactions The thermodynamics of biomacromolecule to ligand interaction is very important in
understanding the structure function relationship in proteins. ITC experiments involve a titration
of a biomacromolecule solution and a reactant at constant temperature to obtain the exchanged
heat of the reaction. One of the classical applications of ITC studies includes the investigation of
the relationships and the thermodynamics of protein–protein or protein-small molecule
interaction. Briefly within this section, before delving into the theory and operations of ITC, a
discussion of some representative biological systems that have been studied with ITC will be
examined.
Azo dyes are a dominant group of colorant utilized in industrial dye manufacturing industry;
with usage that ranges from 60-70%, Though various genotoxic, mutagenic and carcinogenic
characteristics are associated with these dyes. Experimentation conducted by Patel et al.
examined one of such azo dyes, particularly Congo red (1-naphthalenesulfonic acid, 3,3′-(4,4′-
biphenylenebis (azo)) bis(4-amino-) disodium salt). Congo Red even at low concentrations has

25
been shown to be extremely toxic to the body with a mechanism of binding between Congo Red
and HSA. Upon conducting a series of ITC studies it was found Congo Red displayed an entropy
driven and spontaneous reaction, with an affinity constant (Ka) of 1.12 x 10 6 ± 0.2 L mol-1 and a
stoichiometry of 0.216 ± 0.09 HSA to Congo Red.34 Understanding the binding can allow for one
to determine the protein function during Congo Red’s circulation, a key component in its toxicity
to the body. Additionally, the study shed light into a potential development of protein-based
probes, which can assist in clean up and remediation procedures.
An alternative study by Nisius et al. utilized ITC to study a class G protein-coupled receptor
(GPCRs) the human chemokine receptor CCR5. GPCRs have the capability to bind to many
different types of ligands these being a photon, small molecules or proteins. Moreover, these
binding events often play important developmental roles regulation of host immune responses.
Within literature there is only one study, which reports the use of ITC for studying GPCR
activity. Within this study ITC was used to characterize the characteristics of a recombinant
purified CCR5 receptor and its binding to its native ligand RANTES. The binding was to be
exothermic with a measured apparent binding affinity of ~1 μM. 41 This value being lower than
the nanomolar binding affinities that is commonly observed for the native membrane bound
receptor. This variation in observed binding between purified and membrane-bound proteins are
not uncommon and reflect the influence a membrane can have on protein function. Additionally
it was found the CCR5 receptor, which contains two binding sites, Site I and Site II, does not
exhibit cooperative binding whereby binding at Site I does not induce conformational change for
binding at Site II. The study provides to be a good example of proof-of-principle whereby ITC
can be used as a technique for the study of GPCRs.
Briefly discussed herein, show two varied applications of ITC both seeking out to determine the
specifics of a particular protein-protein binding interaction or a protein-small molecule
interaction. When coupled with alternative techniques a better picture of the binding event can be
determined.

26
1.4 Objectives of this Thesis This thesis will explore the potential for MnPs as an alternative to GBCAs for intravascular
imaging within MRA as there exists a need to replace clinically used GBCAs due to Gd-related
toxicity issues and low relaxivity at high magnetic field strengths. The first part of this thesis will
investigate the development, synthesis and relaxivity of novel small MnPs. The second will
investigate the binding properties of previously and currently synthesized MnPs by use of ITC to
deduce thermodynamic parameters of binding.

27
Chapter 2 Development of Small Manganese(III) Porphyrins as
Contrast Agents
2.1 Introduction As mentioned in section 1.2.2.1 and 1.2.2.2, MnTPPS exhibits an abnormally high T1 relaxivity
and an increase in relaxivity beyond 1 MHz to approximately 40 MHz, this being unlike most
GBCAs. 23 This posed to be a promising discovery as the use of MnPs would avoid the use of
intrinsically toxic Gd within CAs. As a result increase relaxivity at high magnetic field strengths
can decrease the dosage required for in vivo imaging, and increase biocompatibility. Based upon
the MnTPPS tetra-substituted model, MnP2 was developed as a first-generation MnP-based
blood pool agent for MRA imaging. Improvements of MnP2 included the following: multiple
paramagnetic metal centers into one molecule increasing the number of coordinated water
molecules and increasing rotational correlation time (!! ) and improved relaxivity at high
magnetic filed strengths.22 But due to the decreased relaxivity upon HSA introduction, this
created an area of improvement within which second generation of blood pool agents can aim to
target. Moreover, we seek to tune these small MnPs with variable affinity to HSA as this is
influenced the blood retention time and creates a toolbox of CAs to cater to the specific needs of
a single MRA.
2.2 Results and Discussion
2.2.1 Molecular Design We embarked on the synthesis of a small MnP as a second generation Gd-free high relaxivity
MRA BPA (Figure 7). MnTriCPP was developed as a small, polar and tri-substituted molecule
with one unique substitution to both increase r1 and display affinity to HSA. MnTCP, as
mentioned in section 1.2.2.2, is the smallest water soluble MnP that has been shown to display
high r1 with a molecular weight of 600 g/mol.30 MnTriCPP with its three-carboxylate groups
retains the small and polar nature as found through MnTCP and the hydrophilic carboxylate
groups are expected to increase the water-accessibility to the nearby paramagnetic Mn-centre. In
the fourth single hydrophobic phenyl group is expected to assist in specific hydrophobic

28
interactions for HSA binding. HSA is well known for binding hydrophobic organic anion, such
as fat acids. For example, Sudlow’s site II is an area prefers aromatic carboxylates. 17
Figure 7: A small MnP, MnTriCPP, as developed based on the chemical structure of
tetra-substituted MnTCP. MnTriCPP is comprised of one paramagnetic centre,
three carboxylates and a single phenyl group at miso-positions to assist in HSA
binding.
2.2.2 Synthesis of MnTriCPP The step-wise synthesis shown in Scheme 1 was developed as a flexible strategy of synthesizing
MnTriCPP by use of a 2+2 dipyrromethane approach.42 Two dipyrromethene intermediates,
namely DPM-Phe and EDP were generated by the reaction of aldehyde and pyrolle with BF3 •
OEt2, a Lewis acid catalyst. As pyrrole displays the propensity to polymerize, it was first
distilled before carrying out the synthesis. Moreover, the reaction was carried out in the dark and
under anaerobic conditions as this prevents pyrrole polymerization and any additional
unfavourable side reactions. Crude DPM-Phe and EDP were purified by column
chromatography and subsequently characterized by 1H-NMR (Figure S1 and Figure S2).
Generation of TriEPP was also carried out in the dark under anaerobic conditions as a means to
avoid premature oxidation and generation of alternate polypyrroles. A porphyrinogen was first
formed within a one-pot synthetic methodology, followed by the use of DDQ to generate the
conjugated system. A yield of 13% was obtained due to the generation of isomeric side products,
N N
N NMn
SO3
O3S SO3
SO3
MnTPPS
N N
N N
O+Na-O
+Na-O O
O
O-Na+Mn
O
+Na-O
MnTCP MnP2
1st generation blood pool agent for MRA
N
HN
NH
NO
O O
O
OO
N
N
N
NO
O O
O
OO
Mn
TriEPP
MnTriEPP
N
N
N
NO
+Na-O O-Na+
O
O+Na-O
Mn MnTriCPP
MnTriCPP 2
nd generation blood pool agent for MRA

29
which may include porphryin isomers with inverted pyrrole rings or formation of alternate rings
through 2,2’-pyrrole ligation, well-documented in the literature.43 1H-NMR spectrum was used to
confirm the desired structure (Figure S3). The UV-Vis spectrum indicated a λmax of 411nm
(Figure S4), which agrees with a typical Soret band absorption of meso-substituted porphyrins.
Insertion of manganese into the porphyrin core was subsequently carried out on TriEPP, to yield
MnTriEPP. This was done by means of refluxing with MnCl2 • 4H2O with DMF whereby two
pyrollic protons were lost to the non-nucleophilic base, DIPEA, and Mn(II) was reversibly
inserted in the porphyrin resulting in a manganese(II)porphryin. Upon the instantaneous
oxidation by air, the more stable manganese (III) porphryin, MnTriEPP, was generated at a
yield of 99.17%. The reaction was monitored by means of UV-Vis spectroscopy (Figure S4)
where a change in the absorption spectrum upon metal insertion was observed. Consistent with
known Mn(III)porphyrin absorption spectras, the spectrum of the product exhibited three distinct
regions; two intense high-energy Soret bands (!!"# of 476nm and 368 nm) and several weak Q
bands red-shifted from the Soret band. Additionally, ESI-MS indicated an m/z of 655.2 ([M]+);
this was consistent with the calculated m/z ratio for C35H28MnN4O6+ (655.14) (Figure S5). Any
subsequent characterizations by 1H-NMR could not be carried out as the paramagnetic Mn(III)
makes for difficult structural characterization.
MnTriCPP, was generated by base-catalyzed hydrolysis of MnTriEPP. This was done by
means of refluxing with 2 M NaOH aqueous solution with co-solvent system of EtOH and THF
to improve the solubility of starting material at the beginning of the reaction. MnTriCPP was
generated at a yield of 96% and a purity of 93.9%, as deduced by the HPLC trace (Fig. S6). The
UV-Vis spectrum of MnTriCPP (Figure S4) was consistent with metal inserted porphyrins
where a !!"# of 466 nm and 379 nm were obtained. An ESI-MS was obtained wherein an m/z of
284.1 ([M]2-) was found (Figure S7), which was consistent with the calculated m/z ratio for
C29H13MnN4O62- (284.01). A molar coefficient of 66957.5 M-1cm-1 was obtained by use of Mn-
AAS a value with is consistent with typical Mn(III)Ps such as MnTCP (68000 M-1cm-1).
30
Scheme 1: a) NaHCO3, BF3 • OEt2, DCM, RT; b) NaHCO3, DCM, RT, 20h; c) NaHCO3, BF3 •
OEt2, DCM, RT; d) TEBAC, BF3 • OEt2, DCM. DDQ; e) DIPEA, MnCl2 • 4H2O, 12h, DMF; f)
NaOH (2M), 72˚C, 25h, EtOH: THF (1.3:1)
2.2.3 Field Dependent Relaxivity Measurements of MnTriCPP To evaluate the MRI contrast enhancement efficiency at different magnetic fields, nuclear
magnetic relaxation dispersion (NMRD) profiles of MnTriCPP and MnTriCPP with HSA were
acquired (Figure 8), using a SMARTracer™ Bench-top Fast Field Cycling NMR Relaxometer.
Upon reaching clinically relevant fields, free MnTriCPP achieves maximal r1 of 9.9 mM-1s-1 at
0.56 T though this not greater than MnP2 which exhibited an r1 of 20.2 mM-1s-1 at 0.56 T. At 3T,
MnTriCPP displayed an r1 of 8.4 mM-1s-1, though again still lower than that of free MnP2 (16.3
mM-1s-1). Both MnPs do display greater r1 at 0.56 T and 3 T in comparison to Gadofosveset with
values of 7.9 mM-1s-1 and 7.8 mM-1s-1 respectively.
NH
+H
O a
NH HN
+
HO
OO
HN
HN OH
OOHO
OO
b
NH HN
OO
N HN
NH N
OO
O O
O
O
N N
N N
OO
O O
O
OMn
d
eN N
N N
O+Na-O
+Na-O O
O
O-Na+Mn
+
NH
+H
O
OO
TriEPPMnTriEPPMnTriCPP
2
2
f
DPM-Phe
EDP
c
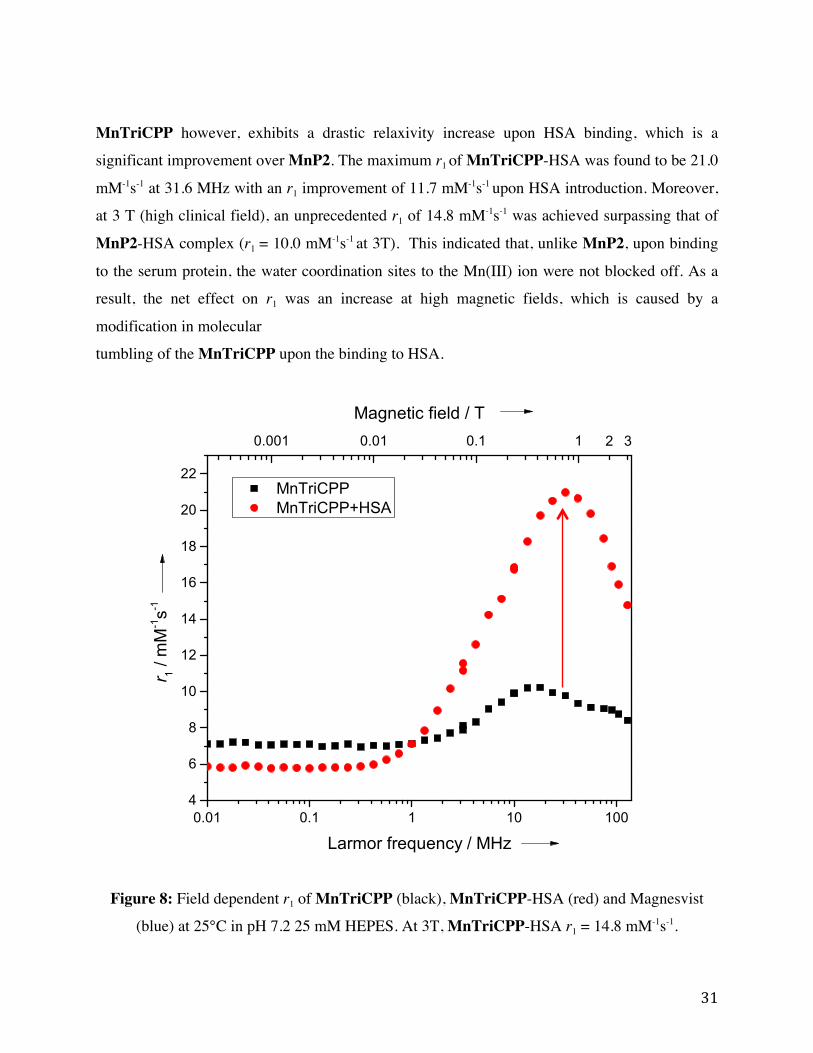
31
MnTriCPP however, exhibits a drastic relaxivity increase upon HSA binding, which is a
significant improvement over MnP2. The maximum r1 of MnTriCPP-HSA was found to be 21.0
mM-1s-1 at 31.6 MHz with an r1 improvement of 11.7 mM-1s-1 upon HSA introduction. Moreover,
at 3 T (high clinical field), an unprecedented r1 of 14.8 mM-1s-1 was achieved surpassing that of
MnP2-HSA complex (r1 = 10.0 mM-1s-1 at 3T). This indicated that, unlike MnP2, upon binding
to the serum protein, the water coordination sites to the Mn(III) ion were not blocked off. As a
result, the net effect on r1 was an increase at high magnetic fields, which is caused by a
modification in molecular
tumbling of the MnTriCPP upon the binding to HSA.
Figure 8: Field dependent r1 of MnTriCPP (black), MnTriCPP-HSA (red) and Magnesvist
(blue) at 25°C in pH 7.2 25 mM HEPES. At 3T, MnTriCPP-HSA r1 = 14.8 mM-1s-1.
0.01 0.1 1 10 1004
6
8
10
12
14
16
18
20
22
MnTriCPP MnTriCPP+HSA
r 1 /
mM-1s-1
Larmor frequency / MHz
0.001 0.01 0.1 1
Magnetic field / T32

32
2.3 Conclusions MnTriCPP has been designed, successfully synthesized, purified and characterized.
Additionally, NMRD profiles indicate that for HSA bound MnTriCPP, an r1 of 14.8 mM-1s-1 was
achieved at 3 T, the highest achieved for any MnP developed within our lab. MnTriCPP
exhibits significant improvement to previously synthesized MnPs and GBCAs, with high
relaxivity enhancement upon HSA binding, and displays potential for future in vivo MRA
applications. Due to this increase in r1 upon HSA binding, an understanding of the binding
properties between the MnP and HSA can give insight into the pharmacodynamic properties of
the interaction and its behaviours in vivo clinical applications.
2.4 Materials and Methods
2.4.1 Materials and Instrumentation All reagents and solvents were of commercial grade and used without further purification unless
otherwise stated. Pyrrole, benzaldehyde, N,N-Diisopropylethylamine (DIPEA), boron trifluoride
diethyl etherate (BF3∙OEt2) and albumin from human serum (HSA) were obtained from Sigma
Aldrich. 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was obtained from AK Scientific
Inc. Triethylbenzylammonium chloride (TEBAC) was obtained from Alfa Aesar. Column
chromatography was set up using Silica Gel 60, 250-400 microns from Desican Inc. Basic
alumina was obtained from Caledon. Cation exchange was done by use of Amberlite® IR120 H
resin. Dialysis was performed by use of Spectra/Por® 3 Dialysis membrane 1K MWCO.
1H NMR spectra were obtained by use of a Bruker NMR (500MHz) spectrometer. Mass spectra
were obtained by use of Agilent 6500 LCMS system electrospray ionization mass spectrometer
(ESI-MS) equipped with an ESI source. The purity of MnTriCPP was determined by use of an
Agilent 1100 high performance liquid chromatography (HPLC) system equipped with a Agilent
1100 series diode array UV-VIS detector and an Eclipse C-19 reverse phase column (4.6 mm x
150 mm x 5 mm). The concentration of MnTriCPP was determined by use of Mn-FAAS
(Thermo iCE 3500 Flame AA Spectrometer). NMR, HPLC, FAAS and ESI-MS spectroscopy
data were obtained using the research facilities provided by the University of Toronto

33
Scarborough TRACES center. UV-VIS measurements were recorded on an Agilent Cary 60 UV-
VIS Spectrophotometer. NMRD profiles were acquired with a SMARTracer™ Bench-top Fast
Field Cycling NMR Relaxometer for magnetic fields below 10 MHz (~0.2 T). The high field
relaxivity from 10–120 MHz (~0.2 T to 3 T) were obtained on a high temperature
superconductor (HTS)-110 NMR system from Scott® Technology Limited. UV-VIS and NMRD
profiles were obtained in Dr. Xiao-an Zhang’s laboratory at the University of Toronto
Scarborough.
2.4.2 Synthesis Synthesis of 2,2’-(phenylmethylene)bis(1H-pyrrole) (DPM-Phe)
DCM (50mL) was combined with NaHCO3 (5.0764g). The resulting suspension
mixture was stirred at 300rpm and deoxygenated with argon for 20 min at RT.
Pyrrole (10mL, distilled) and benzaldehyde (1.048g) was added and the solution
stirred at 300rpm for 30 min at RT, and the reaction flask was covered with
aluminum foil. The solution was filtered and washed with fresh DCM (50mL). To the mixture,
BF3∙OEt2 (16!") was added and stirred at 280rpm for 2 hours at RT. The reaction was stopped
by addition of 5% NaHCO3 (100mL) in a separatory funnel. The organic layer was collected and
dried over Na2SO4. The crude 1 was concentrated on a rotary evaporator. To remove excess
pyrrole, the crude was subjected to vacuum distillation (80oC, 100 mbar, H2O bath). Purification
was conducted by column chromatography (stationary phase: silica; mobile phase: 6.5:3.5
DCM/Hexane initially, then 7.5:2.5 DCM/Hexane and finally 8:2 DCM/Hexane) to afford DPM-
Phe as a yellow powder. 1H-NMR (500MHz, CDCl3): != 7.95 (s, 2H, -NH), 7.27-7.33 (dd, 3H,
meta & para-benzene H), 7.21-7.23 (d, 2H, ortho-benzene H), 6.73 (q, 2H, pyrrole-!H), 6.19 (q,
2H, pyrrole-!H), 5.94(s, 2H, pyrrole-!H) and 5.51 (s, 1H, -CH), as shown in figure S1.
Synthesis of ethyl 2,2-di(1H-pyrrol-2-yl)acetate (EDP)
DCM (50mL) was combined with NaHCO3 (5.2016g). The resulting solution
was stirred at 300rpm and degassed with argon for 20 min at RT. Pyrrole
(10mL, distilled) and ethyl glyoxylate (2mL) was added and the solution
NH HN(1)
NH HN
OO
(2)

34
stirred at 300rpm covered with aluminum foil for 30 min at RT. The solution was filtered and
washed with DCM (50mL). To the mixture, BF3∙OEt2 (6 !") was added and stirred at 280rpm
for 2 hours at RT. The reaction was stopped by addition of 5% NaHCO3 (100mL) in a separatory
funnel. The organic layer was collected and dried over Na2SO4. The crude EDP was
concentrated on a rotary evaporator. Purification was conducted by column chromatography
(stationary phase: silica; mobile phase: 7:3 DCM/Hexane initially, then 9:1 DCM/Hexane) to
afford EDP as a light pink solid (63%). 1H-NMR (500 MHz, CDCl3): != 8.46 (s, 2H, -NH), 6.72
(q, 2H, pyrrole-!H), 6.16 and 6.08 (q, 4H, pyrrole-!Hs), 5.5 (s, 1H, -CH), 4.23 (q, 2H, -CH2)
and 1.31 (t, 3H,-CH3) , as shown in figure S2.
Synthesis of triethyl 20-phenylporphyrin-5,10,15-tricarboxylate (TriEPP)
To filtered DCM (1mL), DPM-Phe (0.1042g) was added. To this
solution, ethyl glyoxylate (178!L) and NaHCO3 (0.1254g) was added.
The resulting solution was stirred at 280rpm covered in aluminum foil
for 20 hours at RT. The reaction was quenched with excess DCM and
the solution was filtered to remove excess NaHCO3. The crude was
concentrated on a rotary evaporator. TEBAC (8.2mg), EDP (0.1089g) and the crude was
dissolved in DCM and added to the mixture with argon purging for 20 min. DCM (225mL) was
added and the solution and stirred at 280rpm for 20 minutes in an ice bath. Subsequently,
BF3∙OEt2 (12 !L) was added drop wise slowly with stirring. DDQ (0.4825g) was added after 3
hours and the solution was left to stir at 280rpm overnight at RT. The resulting black mixture
was filtered through column chromatography (stationary phase: alumina, mobile phase: DCM)
resulting in crude TriEPP. Crude TriEPP was concentrated by a rotary evaporator and purified
by column chromatography (stationary phase: silica, mobile phase: 7.5:2.5 DCM:hexane) to
afford TriEPP as a purple powder (40mg, 13.31%). 1H-NMR (500MHz, CDCl3): != 9.55 (s, 4H,
pyrrole-!H), 9.4-9.41 (d, 2H, pyrrole-!H), 8.97 (d, 2H, pyrrole-!H), 8.19 (d, 2H, meta-benzene
H), 7.79-7.87 (m, 3H, ortho/para benzene H), 5.09-5.16 (overlapping q, 6H, -CH2-), 1.8-1.86
(overlapping t, 9H, -CH3) and 3.07 (s, 2H, -NH). UV spectrums were obtained, which further
confirmed product formation, as shown in figure S3.
N HN
NH N
OO
O O
O
O
(3)

35
Synthesis of Manganese (III) triethyl 20-phenylporphyrin-5,10,15-tricarboxylate
(MnTriEPP)
TriEPP (0.0203g) was dissolved in DMF (6mL). To the solution
DIPEA (35!L) and MnCl2∙4H2O (0.0725g) was added and refluxed in
an oil bath (130oC) with stirring at 180rpm for 2 hours. The solution
was left to stir at 180rpm overnight at RT exposed to air. A rotary
evaporator was then used to concentrate the solution. Crude
MnTriEPP was purified by column chromatography (stationary phase: silica, mobile phase:
9.5:0.5 DCM:methanol) to produce dark-red powder of MnTriEPP (20.19mg, 99.17%). ESI-MS
and UV-Vis spectrums were obtained, which further confirmed product formation.
Synthesis of Manganese(III) 20-phenylporphyrin-5,10,15-tricarboxylic acid (MnTriCPP)
MnTriEPP (20.19mg) was dissolved in 1.31:1 EtOH:THF (9.28mL)
and 2M NaOH (10mL) was added. The solution was refluxed (72oC)
with stirring at 180rpm for 24 hours. The solution was neutralized to
pH 8 using 1M HCl and 1M NaOH. A rotary evaporator was then used
to concentrate the solution. An extraction was subsequently carried out
with ethyl acetate (10 times, 20mL) and the resulting aqueous phase concentrated using a rotary
evaporator. Dialysis was carried out (MWCO 1K). Crude MnTriCPP was passed through a
cation exchange column (stationary phase: Amberlite® IR120 H resin, mobile phase: double
distilled H2O) to remove excess Mn(II) salts, and 15.8mg (96%) of brown powder was collected
as MnTriCPP. ESI-MS, UV-Vis spectrums were obtained , as shown in figure S4 and figure S5.
N N
N N
ONaO
NaO O
O
ONaMnIII
(5)
N N
N N
OO
O O
O
OMnIII
(4)

36
Chapter 3 Isothermal Titration Calorimetry Binding Studies
3.1 Introduction MnTriCPP was shown to display a large increase in r1 upon binding to HSA. To understand this
interaction ITC was utilized to determine the thermodynamic properties of this binding.
Additionally as mentioned in section 1.3, ITC provides advantages over alternative
methodologies as the binding can be studied within solution and without any addition ligand or
protein modifications. In addition to the studies of MnTriCPP, MnTPPS and MnP2 were
examined as reference compounds. Preliminary studies utilized fatty acid free HSA.
3.2 Results and Discussion
3.2.1 Isothermal Titration Calorimetry of MnTPPS and HSA The titration of MnTPPS into fatty acid free HSA is shown within Figure 9. Upon initial
titration of MnTPPS to HSA at a ratio of 0.07 [MnTPPS]:[HSA] detectable binding was seen to
be present. The experiment was carried out in triplicate with additional trials shown in Figure
S8. Moreover, the initial binding of MnTPPS to HSA was seen to be an endothermic process.
Upon the heat of binding subsiding peaks hypothesized to correspond to the control experiments
was seen to be present (as shown within Figure S9-S11).
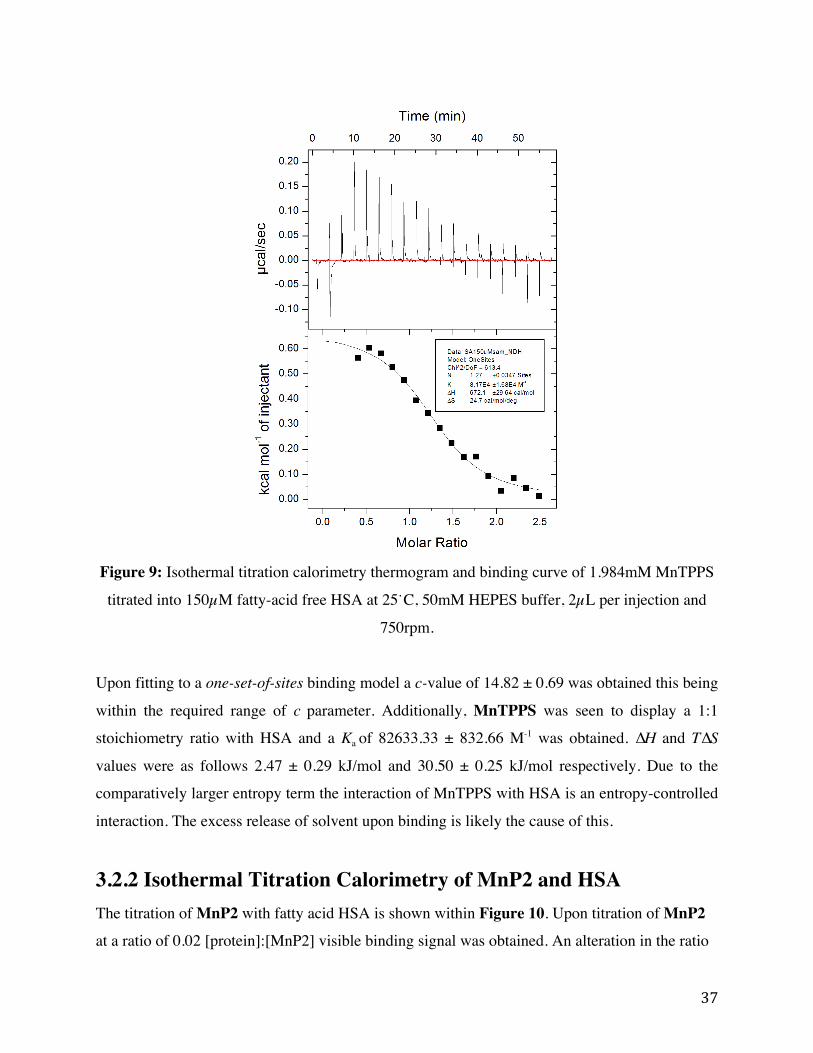
37
Figure 9: Isothermal titration calorimetry thermogram and binding curve of 1.984mM MnTPPS
titrated into 150µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm.
Upon fitting to a one-set-of-sites binding model a c-value of 14.82 ± 0.69 was obtained this being
within the required range of c parameter. Additionally, MnTPPS was seen to display a 1:1
stoichiometry ratio with HSA and a Ka of 82633.33 ± 832.66 M-1 was obtained. ∆H and T∆S
values were as follows 2.47 ± 0.29 kJ/mol and 30.50 ± 0.25 kJ/mol respectively. Due to the
comparatively larger entropy term the interaction of MnTPPS with HSA is an entropy-controlled
interaction. The excess release of solvent upon binding is likely the cause of this.
3.2.2 Isothermal Titration Calorimetry of MnP2 and HSA The titration of MnP2 with fatty acid HSA is shown within Figure 10. Upon titration of MnP2
at a ratio of 0.02 [protein]:[MnP2] visible binding signal was obtained. An alteration in the ratio

38
and concentrations was required to obtain ITC data from which accurate fitting can be carried
out and reasonably reliable thermodynamic parameters can be extracted. A concentration ratio of
0.07 [protein]:[MnP2] was initially tested to mimic the ITC carried out with MnTPPS as shown
above in section 3.4.1, this is shown within Figure S12. Under these conditions a constant signal
upon progressive titrations was not obtained and thus a state of protein saturation can be said to
not have occurred yet. Due to this, optimization processes were carried out yielding an optimized
ratio of 0.02 [protein]:[MnP2] where by constant signal is obtained towards the end of the
titration.
Figure 10: Isothermal titration calorimetry thermogram and binding curve of 1.3mM MnP2
titrated into 30µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm.
Upon each injection an endothermic signal was obtained. Fitting to a one-set-of-sites binding
model a c-value of 19.67 ± 3.45 was obtained this being within the required range of c
parameter. Additionally, MnP2 was seen to display a 4:1 stoichiometry ratio with HSA and a Ka
of 160000.00 ± 21702.53 M-1 was obtained. ∆H and T∆S values were as follows 9.99 ± 0.47
kJ/mol and 39.6 ± 0.50 kJ/mol respectively. Due to the comparatively larger entropy term the

39
interaction of MnP2 with HSA is an entropy-controlled interaction. As mentioned above this is
likely due to solvent release upon binding.
Additionally MnP2 displays a “skirting” effect upon each injection where by continual heat
production is seen to occur and return to the baseline occurred just before the next injection. A
visual representation of this is shown within Figure 11 below. Upon overlaying a single
injection of MnP2 with MnTPPS it is seen that the binding persists for a comparatively longer
period of time across a single injection with MnP2. Upon lengthening the amount of time
between injection (250 seconds) as shown in Figure S16 the skirting remains. Previous
unpublished studies within the research group too indicate MnP2 to display a deep pocket that
presents for an extended period of time.
Figure 11: Normalized overlap of injection shown between 550 – 750 minutes of MnTPPS (red)
and MnP2 (blue)

40
3.2.3 Isothermal Titration Calorimetry of MnTriCPP and HSA Initial experimentation utilized MnTriCPP and HSA at concentrations of 9.84mM and 300µM
within HEPES buffer. Additionally it was observed the solubility of MnTriCPP decreases
beyond 10mM. Upon initial injection of MnTriCPP a clean baseline was not produced wherein
substantial signal was obtained between progressive injections. This is often suggestive of
aggregation or precipitation occurring with the sample cell. Upon drawing up the solution within
the sample cell visible signs of coagulation was seen to occur. This is shown within Figure S17.
In attempts to reduce the coagulation concentrations of MnTriCPP was reduced through to
5mM as shown in Figure S18. Due to the risk posed to the instrument the experiment was run no
further. Similar observations were seen when 2.5mM of MnTriCPP was used though between
injection noise was seen towards the end of the titration. This is shown in Figure S19. Due this
modification to a PBS buffer was done.
Previous unpublished studies within our research group of NMRD titrations of MnTriCPP with
HSA indicated MnTriCPP to display a Ka of 4761.90 M-1 and a 1:1 binding stoichiometry. Due
to this comparatively lower affinity constant to MnP2 and MnTPPS a need to optimize the
concentration parameters of the ITC experiment was required to obtain an acceptable a c-value
and the required sigmoidal curve for fitting. A titration of MnTriCPP was carried out in HSA at
a ratio of 0.07 [protein]:[MnTriCPP] as shown in Figure 12.

41
Figure 12: Isothermal titration calorimetry thermogram and binding curve of 4.22 mM
MnTriCPP titrated into 300 µM fatty-acid free HSA at 25˙C,10 mM PBS buffer, 2 µL injections
and 750rpm.
The thermogram does not appear to show a reduction in the binding signal and peaks
representative of the control experiments (as shown in Figures S21-S23). Though this is not seen
within the thermogram, a visibly sigmoidal curve was obtained when the integration of the heat
peaks was taken. Despite this, binding is seen to occur as a result of ITC signal generation.
Though the reasoning for a lack of visible reduction in the binding signal to the heats of dilution
represented by the control experiments is not known and requires further studies. Regardless
fitting of this experimental binding data was fit to a one-set-of-sites model with a c-value of 7.83
± 2.19. It was found that MnTriCPP displays a 1:1 binding stoichiometry with a Ka of 24766.66
± 2857.15 M-1, higher than that which was found in the studies mentioned above. ∆H and T∆S
values were as follows 1.55 ± 0.12 kJ/mol and 26.5 ± 0.25 kJ/mol respectively. Due to the
comparatively larger entropy term the interaction of MnTriCPP with HSA is an entropy-

42
controlled interaction. As mentioned previously this too is likely due to solvent release upon
binding.
3.3 Conclusions To understand the binding of MnTriCPP with HSA, ITC was chosen as preliminary technique to
study the binding as it is within solution and no modifications to ligand or protein is needed. The
binding properties of two previously synthesized MnPs, MnTPPS and MnP2 (the 1st generation
MnP-Based BPA) were additionally examined as reference compounds. All MnPs were shown to
display detectable affinity to HSA. Moreover, the binding was found to be entropy driven due to
de-solvation upon binding. MnP2 has been found to bind the tightest due to its large hydrophobic
area with possible multiple binding sites.
3.4 Materials and Methods All reagents and solvents were of commercial grade and used without further purification unless
otherwise stated. Albumin from human serum (HSA) was obtained from Sigma Aldrich. HEPES
was obtained from Fisher BioReagents. Potassium phosphate dibasic, potassium phosphate
monobasic and potassium chloride minimum 99.0% was obtained from Sigma-Aldrich. Sodium
chloride Biotechnology Grade was obtained from Bioshop.
ITC was carried on the various MnP samples with the use of fatty acid free HSA placed in the
sample cell and the MnP in the syringe at their respective concentrations. All ITC experiments
were performed on a MicroCal iTC200 (GE Healthcare) instrument within Dr. Kagan Kerman’s
laboratory. Titrations were carried out by 180 seconds spacing between each injection and a
stirring speed of 750 rpm. Experiments were performed at 25°C. Control experiments were
additionally conducted by titrating the following: buffer into HSA, MnP into HSA and buffer
into buffer. The heat evolution was recorded and a subtraction procedure was carried out using
the control experiments. Origin 7.0 (OriginLab) was used to analyze binding isotherms. A
standard Marquardt method was used to evaluate the errors associated with iteration.

43
Chapter 4 Conclusions and Future Outlooks MnTriCPP was designed and successfully synthesized by a 2+2 method to construct the
porphyrin backbone, and was found to exhibit an affinity to HSA. Moreover, upon binding to
HSA r1 of MnTriCPP increases approximately 2 fold to 21.0 mM-1s-1 at 31.6 MHz and 14.8
mM-1s-1 at 3T, significantly higher than those of previously synthesized MnPs and GBCAs. To
understand the binding of MnTriCPP with HSA, Isothermal Titration Calorimetry was chosen,
as a preliminary technique, to study the binding as it is within solution and no modifications to
ligand or protein is needed. The binding properties of two previously synthesized MnPs,
MnTPPS and MnP2 (the 1st generation MnP-Based BPA) were additionally examined as
reference compounds. All MnPs were shown to display detectable affinity to HSA. Moreover,
the binding was found to be entropy driven due to de-solvation upon binding. MnP2, a dimer
with large areas of hydrophobicity, has been found to display a high binding affinity to HSA
with possible multiple binding sites.
The platform of a mono-substituted MnP permitted for a concentrated area of hydrophobicity.
Consequently, this allows for further exploration of structural based design permitting for
targeted affinity to HSA by means of synthetic based structural tuning at this area. Post
preliminary studies of the binding affinity of the various MnPs, it requires further more
comparative studies of the binding allowing for structure based evaluation and its related
applicability as a BPA. Furthermore, in cases of multisite binding, such as with MnP2, a deeper
understanding of the specifics of each binding event is required. This can be done by means of
competitive ligand binding studies with commercial reagents for which the binding site locations
were determined. Additionally, all preliminary studies carried out herein made use of fatty acid
free HSA. As HSA in vivo is primarily fatty acid bound and moreover it has been shown fatty
acids to play a role within binding events sequential introduction of fatty acids and subsequent
binding studies should be carried out to gain a broader understanding of the binding.17
In addition to the aforementioned points of exploration, alternative instrumentation can be
utilized to study the binding affinities of the various MnPs to HSA. Experimentation conducted
by Zhu et al. carried out binding studies of 5,10-di(N- methyl-pyridiniumyl)-15,-20-(4-trimethyl
- phenyl - ammo- niumyl) porphyrin with HSA by SPR. The study utilized a gold film

44
preparation containing carboxyl groups and subsequent HSA immobilization was carried out.44
The MnP of interest was prepared in a series of concentrations as well as different capacity
surfaces of HSA were prepared. The use of capacity surfaces allowed for optimization of
immobilization avoiding mass transport, steric hindrance, crowding and any aggregation that
might result.45 Values of affinity constants can then be determined through fitting software. It is
perhaps suggested, that the next steps with regards to this project would be to utilize SPR
technology and conduct MnP binding studies using a methodology similar to that of Zhu et al.
In summary, MnTriCPP has been designed and synthesized as a second generation Gd-free
BPA. Additionally, it has been shown to display detectable binding to HSA by means of NMRD
and ITC. Due to the high r1 of MnTriCPP upon HSA binding, it displays potential for future
structural tuning as well as in vivo MRA applications.

45
References (1) Knopp, M. V.; Von Tengg-Kobligk, H.; Floemer, F.; Schoenberg, S. O. Contrast Agents
for MRA: Future Directions. J. Magn. Reson. Imaging 1999, 10 (3), 314–316.
(2) Sternbach, G.; Varon, J. Wilhelm Konrad Roentgen: A New King of Rays. Am J Emerg
Med. 1993, 11, 743–745.
(3) Franch, R.; SB, K.; JS, D. Techniques of Cardiac Catheterization Including Coronary
Arteriography, 7th ed.; JW, H., RC, S., CE, R., Eds.; McGraw-Hill: New York, 1990.
(4) Artico, M.; Spoletini, M.; Fumagalli, L.; Biagioni, F.; Ryskalin, L.; Fornai, F.; Salvati, M.;
Frati, A.; Pastore, F. S.; Taurone, S. Egas Moniz: 90 Years (1927–2017) from Cerebral
Angiography. Front. Neuroanat. 2017, 11 (September), 1–6.
(5) Lusic, H.; Grinstaff, M. W. X-Ray Computed Tomography Contrast Agent. Chem. Rev.
2014, 113 (3).
(6) Meyer, T.; Hermann, F.; Hadamitzky, M.; Gerber, T. C.; Mccollough, C.; Achenbach, S.
Estimated Radiation Dose Associated With Cardiac CT Angiography. JAMA 2009, 301
(5), 500–507.
(7) Lauffer, R. B. Paramagnetic Metal Complexes as Water Proton Relaxation Agents for
NMR Imaging: Theory and Design. Chem. Rev. 1987, 87 (5), 901–927.
(8) Saloner, D. The AAPM / RSNA : : Tutorial for Residents. Imaging 1998, 18, 731–744.
(9) Kaufman, J. A.; Mccarter, D.; Geller, S. C.; Arthur, C. The Lower Extremities : Artifacts
and Pitfalls. Am. J. Roentgenol. 1998, 171 (July), 129–135.
(10) Canet, D.; Nancy, I. General Theory of Nuclear Relaxation. Adv. Inorg. Chem. 2005, 57
(05), 3–40.
(11) Helm, A.; Lothar, M.; Tóth, É. The Chemistry of Contrast Agents in Medical Magnetic
Resonance Imaging; John Wiley & Sons, Ltd, 2013.
(12) Villaraza, A.; Bumb, A.; Brechbiel, M. Macromolecules, Dendrimers and Nanomaterials
in Magnetic Resonance Imaging: The Interplay Between Size, Function and
Pharmacokinetics. Chem Rev. 2011, 110 (5), 2921–2959.
(13) Caravan, P.; Cloutier, N. J.; Greenfield, M. T.; McDermid, S. A.; Dunham, S. U.; Bulte, J.
W. M.; Amedio, J. C.; Looby, R. J.; Supkowski, R. M.; Horrocks, W. D. W.; et al. The
Interaction of MS-325 with Human Serum Albumin and Its Effect on Proton Relaxation
Rates. J. Am. Chem. Soc. 2002, 124 (12), 3152–3162.

46
(14) Goyen, M. Gadofosveset-Enhanced Magnetic Resonance Angiography. Vasc. Health Risk
Manag. 2008, 4 (1), 1–9.
(15) Bae, K. T. Intravenous Contrast Medium Administration and Scan Timing at CT:
Considerations and Approaches. Radiology 2010, 256 (1), 32–61.
(16) Huang, M. S.; Sage, A. P.; Manuscript, A.; Thompson, K. L.; Miller, T. J.; Bonventre, J.
V; Goering, P. L.; Szczech, L. a; Barnhart, H. X.; Inrig, J. K.; et al. Biodistribution of
Gadolinium-Based Contrast Agents, Including Gadolinium Deposition. J Magn Reson
Imaging 2010, 30 (6), 1259–1267.
(17) Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.;
Ascenzi, P. The Extraordinary Ligand Binding Properties of Human Serum Albumin.
IUBMB Life 2005, 57 (12), 787–796.
(18) Kanda, T.; Fukusato, T.; Matsuda, M.; Toyoda, K.; Oba, H.; Kotoku, J.; Haruyama, T.;
Kitajima, K.; Furui, S. Gadolinium-Based Contrast Agent Accumulates in the Brain Even
in Subjects without Severe Renal Dysfunction: Evaluation of Autopsy Brain Specimens
with Inductively Coupled Plasma Mass Spectroscopy. Radiology 2015, 276 (1), 228–232.
(19) Nielsen, Y. W.; Thomsen, H. S. Contrast-Enhanced Peripheral MRA: Technique and
Contrast Agents. Acta radiol. 2012, 53 (7), 769–777.
(20) Hitomi, E.; Simpkins, A. .; Luby, M.; Latour, L. .; Leigh, Latour, R. J.; Leigh, R. Blood-
Ocular Barrier Disruption in Patients with Acute Stroke. Neurology 2018, 90 (11), e915–
e923.
(21) Liu, H.; Zhang, X.-A. Manganese-Based Magnetic Resonance Imaging Contrast Agents.
Encycl. Inorg. Bioinorg. Chem. 2018, 1–16.
(22) Cheng, W.; Haedicke, I. E.; Nofiele, J.; Martinez, F.; Beera, K.; Scholl, T. J.; Cheng, H.
M.; Zhang, X. Complementary Strategies for Developing Gd-Free High-Field T1 MRI
Contrast Agents Based on Mn(III) Porphyrins. J. Med. Chem. 2014, 57 (2), 516–520.
(23) Chen, C. wan; Cohen, J. S.; Myers, C. E.; Sohn, M. Paramagnetic Metalloporphyrins as
Potential Contrast Agents in NMR Imaging. FEBS Lett. 1984, 168 (1), 70–74.
(24) Koenig, S. H.; Brown, R. D.; Spiller, M. The Anomalous Relaxivity of Mn3+(TPPS4).
Magn. Reson. Med 1987, 4, 252−260.
(25) Fiel, R. J.; Mark, E.; Button, T.; Gilanp, S.; Musser, D. Mechanism of the Localization OF
Manganese (III) Mesotetra(4-Sulfonatophenyl) Porphine in Mice Bearing L1210 Tumors.

47
Cancer Lett. 1988, 40, 23–32.
(26) Patronas, N. J.; Cohen, J. S.; Knop, R. H.; Dwyer, A. J.; Colcher, D.; Lundy, J.; Mornex,
F.; Hambright, P.; Sohn, M.; \Myers, C. E. Metalloporphyrin Contrast Agents for
Magnetic Resonance Imaging of Human Tumors in Mice. Cancer Treat. Rep. 1986, 70
(3), 391–395.
(27) Lyon, R. C.; Faustino, P. J.; Cohen, J. S.; Katz, A.; Mornex, F.; Colcher, D.; Baglin, C.;
Koenig, S. H.; Hambright, P. Tissue Distribution and Stability of Metalloporphyrin MRI
Contrast Agents. Magn. Reson. Med. 1987, 4 (1), 24–33.
(28) Place, D. A.; Faustino, P. J.; Berghmans, K. K.; van, Z. P. C.; Chesnick, A. S.; Cohen, J.
S. MRI Contrast-Dose Relationship of Manganese(III)Tetra(4-Sulfonatophenyl) Porphyrin
with Human Xenograft Tumors in Nude Mice at 2.0 T. Magn. Reson. Imaging 1992, 10
(6), 919–928.
(29) Els, T.; Bockhorst, K.; Hoehn-berlage, M. Nmr Contrast Enhancement of Brain Tumours:
Comparison of the Blood Brain Barrier Tracer GdDTPA and the Tumour-Selective
Contrast Agent MnTPPS. MAGMA 1993, 1 (3–4), 126–133.
(30) Nofiele, J. T.; Haedicke, I. E.; Zhu, Y. L. K.; Zhang, X. A.; Cheng, H. L. M. Gadolinium-
Free Extracellular MR Contrast Agent for Tumor Imaging. J. Magn. Reson. Imaging
2015, 41 (2), 397–403.
(31) Cheng, W.; Ganesh, T.; Martinez, F.; Lam, J.; Yoon, H.; Macgregor, R. B.; Scholl, T. J.;
Cheng, H.-L. M.; Zhang, X. Binding of a Dimeric Manganese Porphyrin to Serum
Albumin: Towards a Gadolinium-Free Blood-Pool T1 MRI Contrast Agent. J. Biol. Inorg.
Chem. 2014, 19 (2), 229–235.
(32) Jung, K.; Kim, H.; Park, J.; Nam, K. S.; Lee, G. H.; Chang, Y.; Kim, T. Gd Complexes of
DO3A-(Biphenyl-2,2′-Bisamides) Conjugates as MRI Blood-Pool Contrast Agents. ACS
Med. Chem. Lett 2012, 3, 1003–1007.
(33) Vuignier, K.; Schappler, J.; Veuthey, J.-L.; Carrupt, P.-A.; Martel, S. Drug–Protein
Binding: A Critical Review of Analytical Tools. Anal. Bioanal. Chem. 2010, 398 (1), 53–
66.
(34) Patel, B. R.; Kerman, K. Calorimetric and Spectroscopic Detection of the Interaction
between a Diazo Dye and Human Serum Albumin. Analyst 2018, 143 (16), 3890–3899.
(35) Nguyen, H. H.; Park, J.; Kang, S.; Kim, M. Surface Plasmon Resonance: A Versatile

48
Technique for Biosensor Applications. Sensors (Switzerland) 2015, 15 (5), 10481–10510.
(36) Christensen, J. J.; Izatt, R. M.; Hansen, L. D.; Partridge, J. A. Entropy Titration. A
Calorimetric Method for the Determination of ΔG, ΔH, and ΔS from a Single
Thermometric Titration 1a,B. J. Phys. Chem. 1966, 70 (6), 2003–2010.
(37) Langerman, N.; R.L, B. Microcalorimetry for Biological Chemistry : Experimental Design
, Data Analysis , and Interpretation; 1979; Vol. 10.
(38) Chilom, C. G.; Craescu, C. T.; Popescu, A. I. Parameters of Interaction between Proteins
and Their Specific Ligands, Deduced by Isothermal Titration Calorimetry. Rom. Journ.
Phys. 2004, 51 (July 2004), 443–457.
(39) Garcia-Fuentes, L.; Tellez-Sanz, R.; Quesada-Soriano, I.; Baro n, C. Thermodynamics of
Molecular Recognition by Calorimetry. IntechOpen 2011.
(40) Basu, A.; Kumar, G. Natural and Artificial Flavoring Agents and Food Dyes, 1st ed.; A.
Grumezescu and A. M. Holban, Ed.; Academic Press: London, 2018.
(41) L. Nisius, M. Rogowski, L. Vangelista, S. G. Large-Scale Expression and Purification of
the Major HIV-1 Coreceptor CCR5 and Characterization of Its Interac- Tion with
RANTES. Protein Expr. Purif. 2008, 61 (155–162).
(42) Terazono, Y.; North, E. J.; Moore, A. L.; Moore, T. A.; Gust, D. Base-Catalyzed Direct
Conversion of Dipyrromethanes to 1,9-Dicarbinols: A [2 + 2] Approach for Porphyrins.
Org. Lett. 2012, 14 (7), 1776–1779.
(43) The Porphyrin Handbook, 1st ed.; Guilard, R., Kadish, K., Smith, K., Eds.; Academic
Press, 2012.
(44) Zhou, B. O.; Zhang, Z. H. I.; Zhang, Y. U. E.; Li, R. A. N.; Xiao, Q. I.; Liu, Y. I.; Li, Z.
Binding of Cationic Porphyrin to Human Serum Albumin Studied Using Comprehensive
Spectroscopic Methods. J. Pharm. Sci. 2009, 98 (1), 105–113.
(45) Roden, L.; Myszka, D. Global Analysis of a Macromolecular Interaction Measured on
BIAcore. Biochem Biophys Res Commun 1996, 225, 1073–1077.

49
Supplementary Information
Figure S1. 1H-NMR spectrum of DPM-Phe in CDCl3.
NH HNA
B
CD
E
F
GH

50
Figure S2. 1H-NMR spectrum of EDP in CDCl3.
NH HN
OO
A B
C
DE
FG

51
Figure S3. 1H-NMR spectrum of TriEPP in CDCl3.
N HN
NH N
O O
O O
O
O
HH
D
E
EC
B
FG
AA
F
G
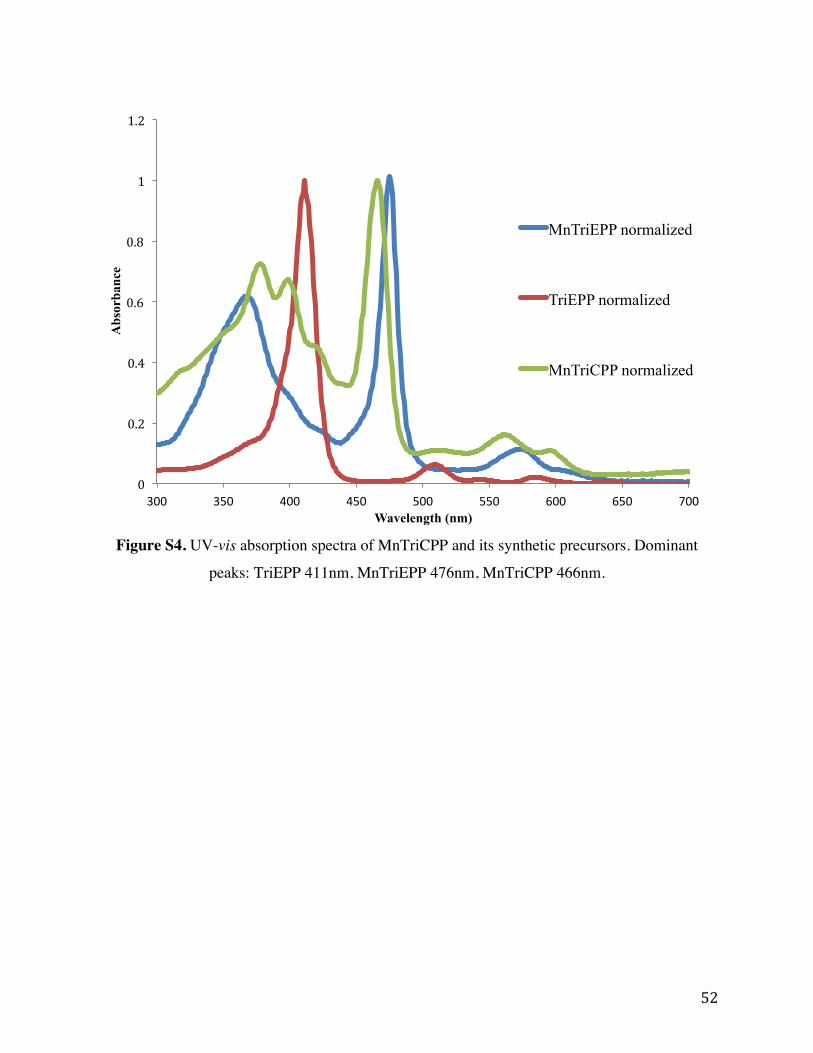
52
Figure S4. UV-vis absorption spectra of MnTriCPP and its synthetic precursors. Dominant
peaks: TriEPP 411nm, MnTriEPP 476nm, MnTriCPP 466nm.
0
0.2
0.4
0.6
0.8
1
1.2
300 350 400 450 500 550 600 650 700
Abs
orba
nce
Wavelength (nm)
MnTriEPP normalized
TriEPP normalized
MnTriCPP normalized

53
Figure S5.ESI-MS of MnTriEPP. ESI-MS found m/z = 655.2 ([M]+) Calculated for
C35H28MnN4O6+, m/z = 655.14
N N
N N
O O
O O
O
OMnIII
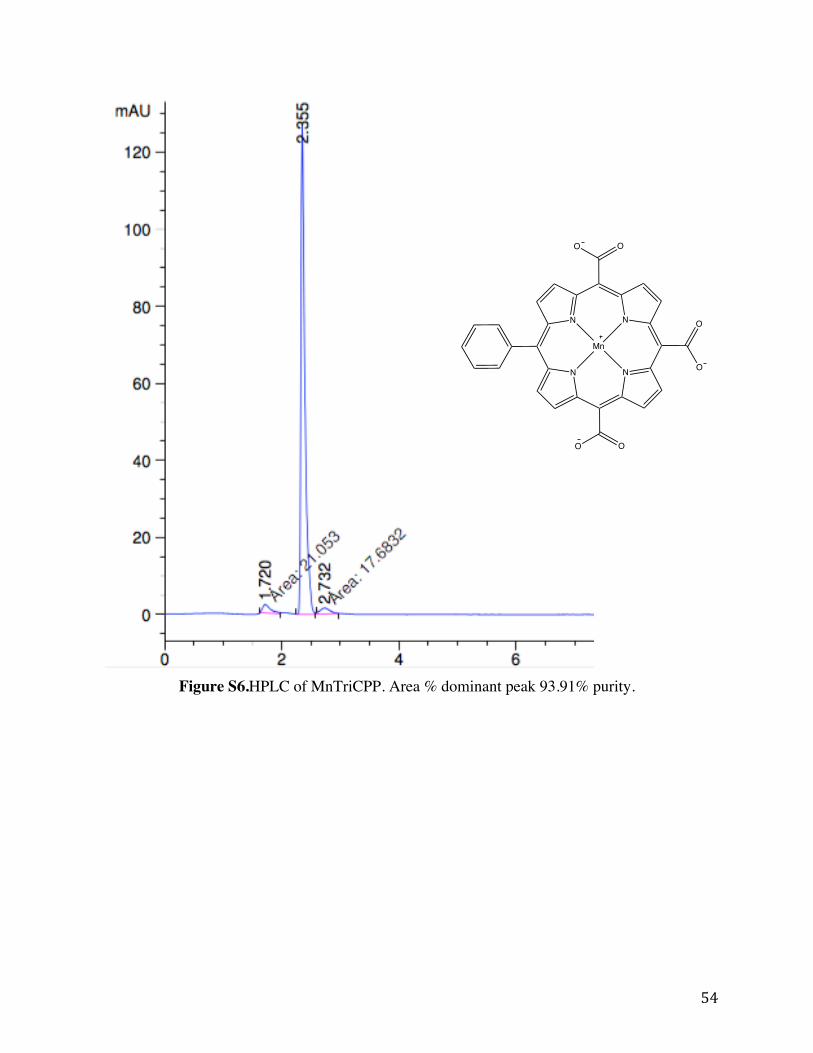
54
Figure S6.HPLC of MnTriCPP. Area % dominant peak 93.91% purity.
N N
N N
OO
O O
O
O
Mn
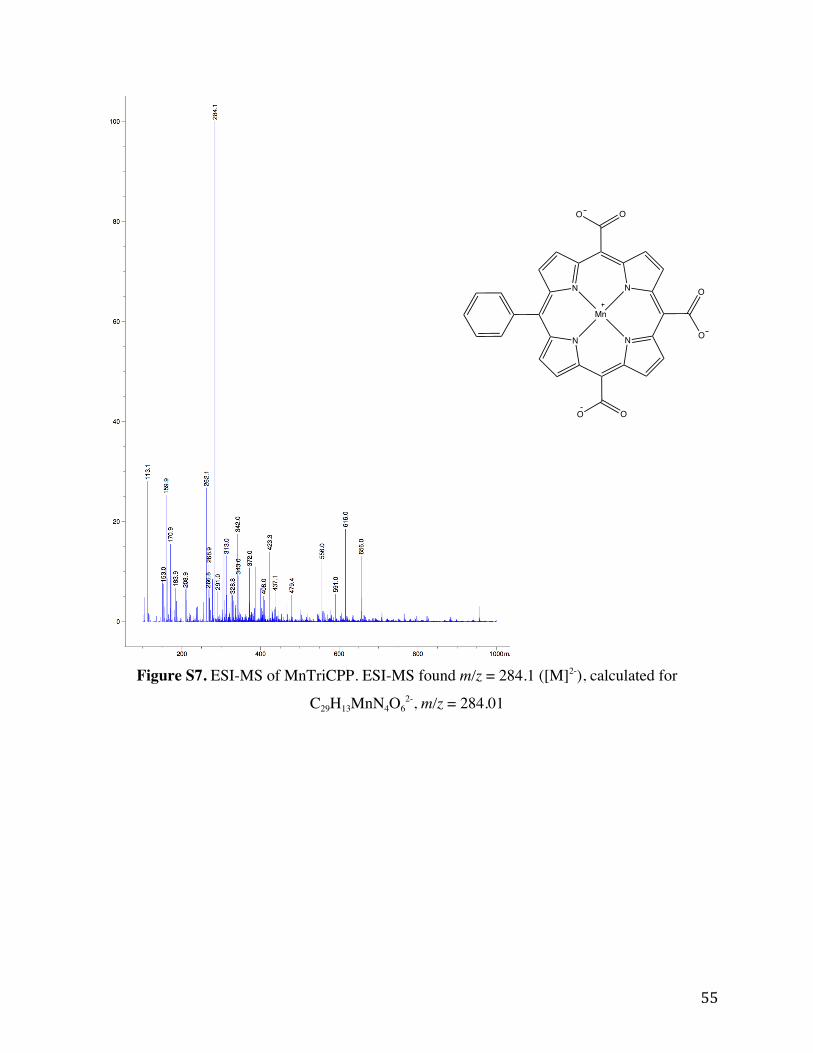
55
Figure S7. ESI-MS of MnTriCPP. ESI-MS found m/z = 284.1 ([M]2-), calculated for
C29H13MnN4O62-, m/z = 284.01
N N
N N
OO
O O
O
O
Mn

56
Figure S8. Isothermal titration calorimetry thermogram and binding curve of 1.3mM MnTPPS
titrated into 30µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm. Left: Trial 2 Right: Trial 3.
57
Figure S9. Isothermal titration calorimetry thermogram of control experiments of 1.948mM
MnTPPS titrated into 50mM HEPES at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm.
Figure S10. Isothermal titration calorimetry thermogram of control experiments of 50uM
HEPES titrated into 150µM HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm.
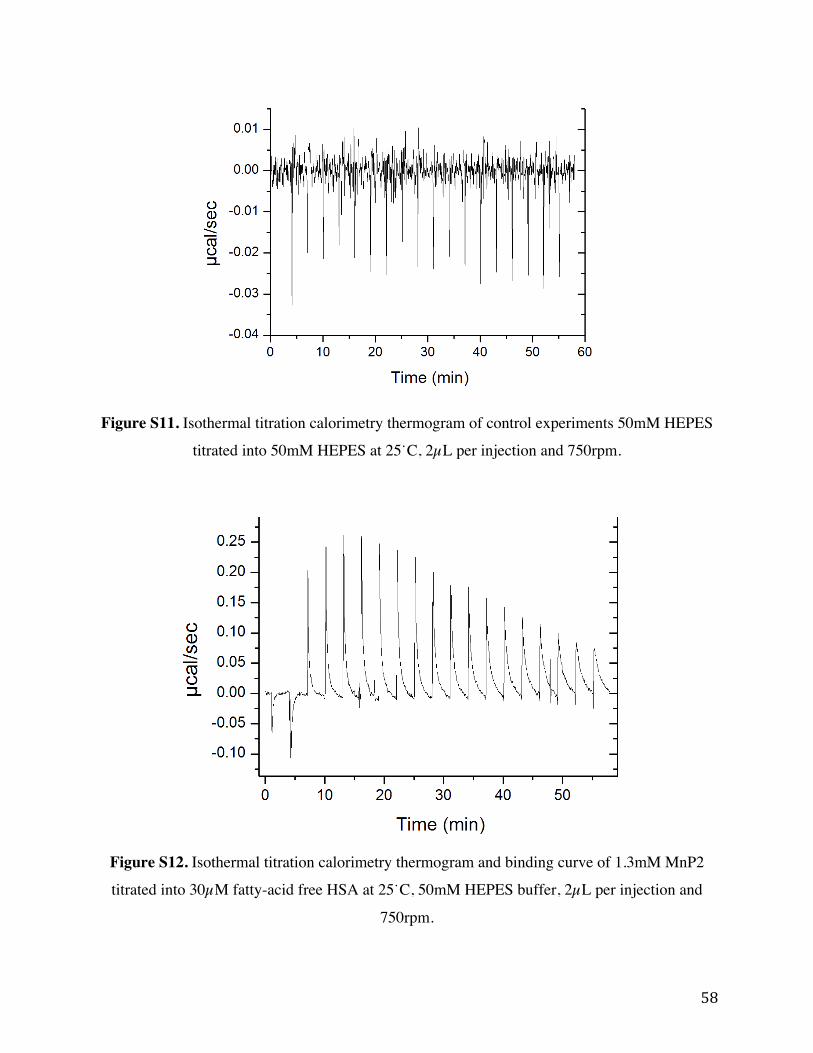
58
Figure S11. Isothermal titration calorimetry thermogram of control experiments 50mM HEPES
titrated into 50mM HEPES at 25˙C, 2µL per injection and 750rpm.
Figure S12. Isothermal titration calorimetry thermogram and binding curve of 1.3mM MnP2
titrated into 30µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm.

59
Figure S13. Isothermal titration calorimetry thermogram and binding curve of 2mM MnP2
titrated into 30µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm. Left: Trial 2 Right: Trial 3.
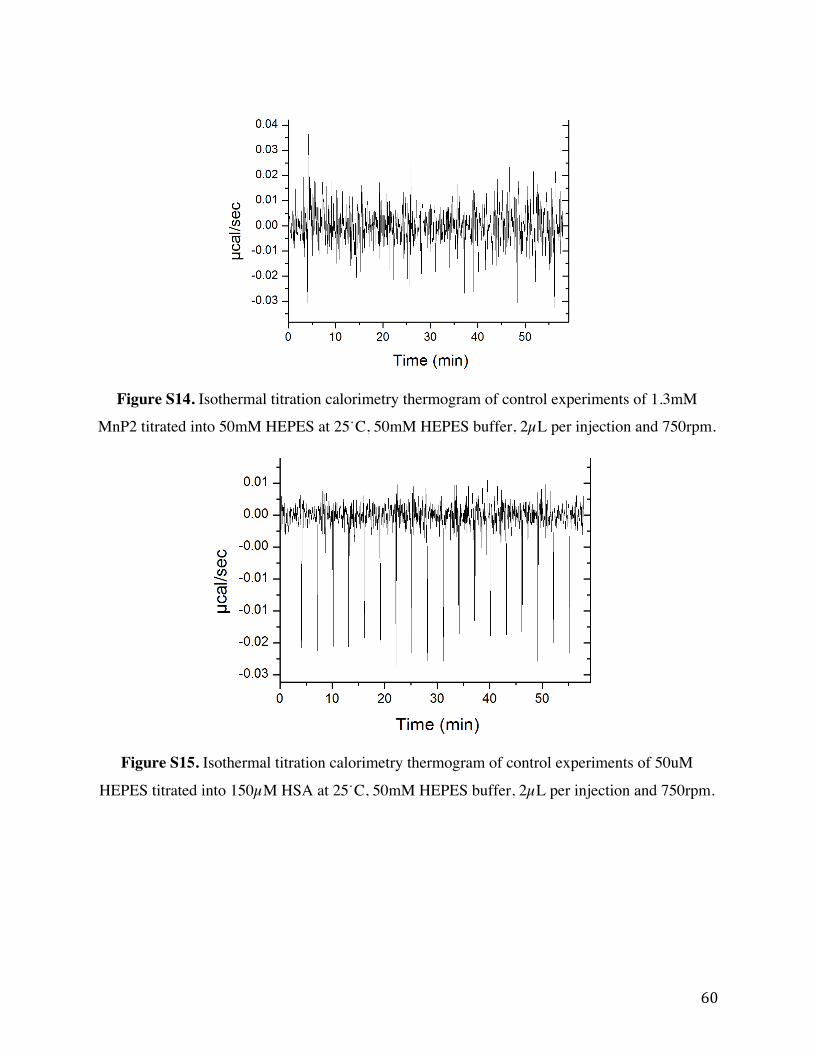
60
Figure S14. Isothermal titration calorimetry thermogram of control experiments of 1.3mM
MnP2 titrated into 50mM HEPES at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm.
Figure S15. Isothermal titration calorimetry thermogram of control experiments of 50uM
HEPES titrated into 150µM HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm.

61
Figure S16: Overlay of 3rd injection of 2mM MnP2 titrated into 30µM fatty-acid free HSA at
25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm. Red: 180 second injection separation
Right: 250 second injection separation.

62
Figure S17: Isothermal titration calorimetry thermogram 9.84mM MnTriCPP titrated into 300
µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm. Top: First
three injections show substantial baseline noise due to risk posed to instrument the experiment
was run no further. Bellow: Solution contained within sample cell post three injections show
significant aggregation upon MnTriCPP and HSA introduction.

63
Figure S18: Isothermal titration calorimetry thermogram 5mM MnTriCPP titrated into 300 µM
fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm. First three
injections show substantial baseline noise due to risk posed to instrument the experiment was run
no further.
Figure S19: Isothermal titration calorimetry thermogram 2.5mM MnTriCPP titrated into 300
µM fatty-acid free HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm.

64
Figure S20: Isothermal titration calorimetry thermogram and binding curve of 4.22mM
MnTriCPP titrated into 300 µM fatty-acid free HSA at 25˙C, 10 mM PBS, 2µL per injection and
750rpm. Left: Trial 2 Right: Trial 3.
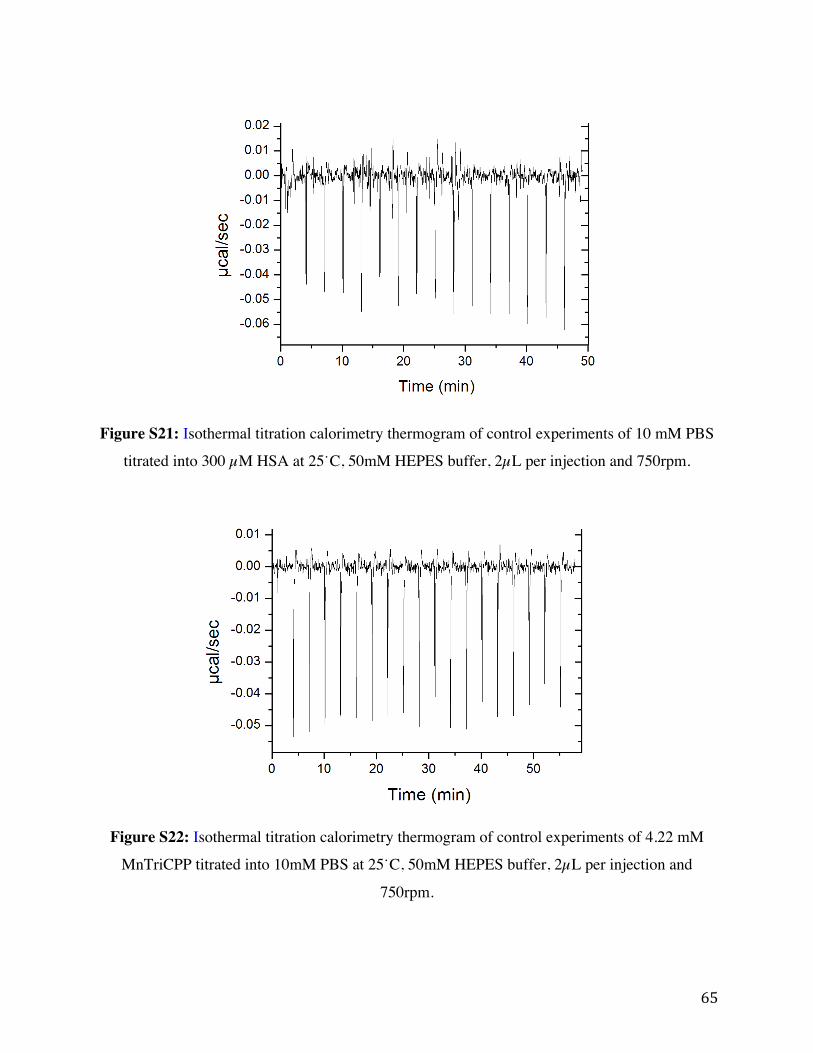
65
Figure S21: Isothermal titration calorimetry thermogram of control experiments of 10 mM PBS
titrated into 300 µM HSA at 25˙C, 50mM HEPES buffer, 2µL per injection and 750rpm.
Figure S22: Isothermal titration calorimetry thermogram of control experiments of 4.22 mM
MnTriCPP titrated into 10mM PBS at 25˙C, 50mM HEPES buffer, 2µL per injection and
750rpm.

66
Figure S23: Isothermal titration calorimetry thermogram of control experiments 10 mM PBS
titrated into 10 mM PBS at 25˙C, 2µL per injection and 750rpm.




















