
PINK1 mutation in Taiwanese early-onset parkinsonism
9
J Neurol (2007) 254:1347–1355 DOI 10.1007/s00415-007-0534-7 ORIGINAL COMMUNICATION Yi-Hsin Weng Yah-Huei Wu Chou Wen-Shiang Wu Kun-Ju Lin Hsiu-Chen Chang Tzu-Chen Yen Rou-Shayn Chen Shiaw-Pyng Wey Chin-Song Lu PINK1 mutation in Taiwanese early-onset parkinsonism Clinical, genetic, and dopamine transporter studies JON 2534 Received: 27 April 2006 Received in revised form: 30 October 2006 Accepted: 17 January 2007 Published online: 15 October 2007 Dr.Y.-H.Weng · Dr. R.-S. Chen · C.-S. Lu, MD () · W. S.Wu, Msc · H.-C. Chang, BS Neuroscience Research Center Section of Movement Disorders Dept. of Neurology Chang Gung Memorial Hospital and Chang Gung University 33305 5 Fu-Shin St. Kweishan, Taoyuan, Taiwan Tel.: +886-3/328-1200 ext -8414 Fax: +886-3/397-1504, +886-3/328-7226 E-Mail: [email protected] Dr. Y.-H. Wu Chou Human Molecular Genetic Laboratory Chang Gung Memorial Hospital Chang Gung University College of Medicine 33305 5 Fu-Shin St. Kweishan, Taoyuan, Taiwan Dr. K.-J. Lin · Dr. T.-C. Yen Dept. of Nuclear Medicine Chang Gung Memorial Hospital Chang Gung University College of Medicine. 33305 5 Fu-Shin St. Kweishan, Taoyuan, Taiwan Dr. S.-P. Wey Dept. of Medical Imaging and Radiological Sciences Chang Gung Memorial Hospital Chang Gung University College of Medicine. 33305 5 Fu-Shin St. Kweishan, Taoyuan, Taiwan Dr.Y.-H.Weng · Dr.Y.-H.Wu Chou · Dr. K.-J. Lin · Dr. T.-C. Yen · Dr. R.-S. Chen · Dr. C.-S. Lu Neuroscience Research Center Chang Gung Memorial Hospital Chang Gung University College of Medicine ■ Abstract The PINK1 gene muta- tion is probably the second most common genetic cause of early- onset Parkinson’s disease (EOPD). The frequency and the characteris- tics of the PINK1 mutation in the Taiwanese population are un- known. This study was designed to investigate the genotype, pheno- type and dopaminergic function of PINK1 in a cohort of EOPD pa- tients. The genetic settings were to detect the PINK1 gene mutations in 138 EOPD patients and in 191 con- trols. Using the 99m Tc-TRODAT-1 (TRODAT) scan, we investigated the differences in the dopamine transporter (DAT) activities be- tween the PINK1 patients, late-on- set Parkinson’s disease (LOPD) pa- tients and healthy controls. Four EOPD patients with 3 genotypic mutations in the PINK1 gene were found: a compound heterozygous mutation (Q239X/R492X) in 2 sis- ters, a novel homozygous mutation (R492X) in a woman, and a novel heterozygous mutation (G193R) in a man. The three PINK1 patients had typical phenotype with juve- nile onset, benign course, and fre- quently with dyskinesias. The TRO- DAT scan showed a rather even and symmetrical reduction of uptake in PINK1 patients, unlike the domi- nant decline in the putamen in the LOPD patients. The annual reduc- tion rate of uptake in the striatum was much slower in PINK1 patients than that in the LOPD patients (1.7 % vs. 4.1 %; p < 0.005). In the patient with a heterozygous muta- tion in the PINK1 gene, the reduc- tion ratio in the striatum, as well as the annual reduction rate, were closer to those in the LOPD group. We conclude that the incidence of carrying PINK1 mutations in the present cohort of Taiwanese EOPD patients was low, accounting for 2/39 (5.1 %) in familial cases, and 2/99 (2 %) in sporadic cases. The slower annual reduction of DAT ac- tivity might indicate the insidious degeneration of dopamine neurons and a benign prognosis. ■ Key words PINK1 gene · early-onset Parkinson’s disease · dopamine transporter · 99m Tc-TRODAT-1 SPECT
Transcript of PINK1 mutation in Taiwanese early-onset parkinsonism

J Neurol (2007) 254:1347–1355DOI 10.1007/s00415-007-0534-7 ORIGINAL COMMUNICATION
Yi-Hsin WengYah-Huei Wu ChouWen-Shiang WuKun-Ju LinHsiu-Chen ChangTzu-Chen YenRou-Shayn ChenShiaw-Pyng WeyChin-Song Lu
PINK1 mutation in Taiwanese early-onset
parkinsonism
Clinical, genetic, and dopamine transporter studies
JON
2534
Received: 27 April 2006Received in revised form: 30 October 2006Accepted: 17 January 2007Published online: 15 October 2007
Dr. Y.-H. Weng · Dr. R.-S. Chen · C.-S. Lu,MD (�) · W. S. Wu, Msc · H.-C. Chang, BSNeuroscience Research CenterSection of Movement DisordersDept. of NeurologyChang Gung Memorial Hospital and ChangGung University33305 5 Fu-Shin St.Kweishan, Taoyuan, TaiwanTel.: +886-3/328-1200 ext -8414Fax: +886-3/397-1504, +886-3/328-7226E-Mail: [email protected]
Dr. Y.-H. Wu ChouHuman Molecular Genetic LaboratoryChang Gung Memorial HospitalChang Gung University College of Medicine33305 5 Fu-Shin St.Kweishan, Taoyuan, Taiwan
Dr. K.-J. Lin · Dr. T.-C. YenDept. of Nuclear MedicineChang Gung Memorial HospitalChang Gung University College of Medicine.33305 5 Fu-Shin St.Kweishan, Taoyuan, Taiwan
Dr. S.-P. WeyDept. of Medical Imaging and RadiologicalSciencesChang Gung Memorial HospitalChang Gung University College of Medicine.33305 5 Fu-Shin St.Kweishan, Taoyuan, Taiwan
Dr. Y.-H. Weng · Dr. Y.-H. Wu Chou · Dr. K.-J. Lin · Dr. T.-C. Yen · Dr. R.-S. Chen ·Dr. C.-S. LuNeuroscience Research CenterChang Gung Memorial HospitalChang Gung University College of Medicine
■ Abstract The PINK1 gene muta-tion is probably the second mostcommon genetic cause of early-onset Parkinson’s disease (EOPD).The frequency and the characteris-tics of the PINK1 mutation in theTaiwanese population are un-known. This study was designed toinvestigate the genotype, pheno-type and dopaminergic function ofPINK1 in a cohort of EOPD pa-tients. The genetic settings were todetect the PINK1 gene mutations in138 EOPD patients and in 191 con-trols. Using the 99mTc-TRODAT-1(TRODAT) scan, we investigatedthe differences in the dopaminetransporter (DAT) activities be-tween the PINK1 patients, late-on-set Parkinson’s disease (LOPD) pa-tients and healthy controls. FourEOPD patients with 3 genotypicmutations in the PINK1 gene werefound: a compound heterozygousmutation (Q239X/R492X) in 2 sis-ters, a novel homozygous mutation(R492X) in a woman, and a novel
heterozygous mutation (G193R) ina man. The three PINK1 patientshad typical phenotype with juve-nile onset, benign course, and fre-quently with dyskinesias. The TRO-DAT scan showed a rather even andsymmetrical reduction of uptake inPINK1 patients, unlike the domi-nant decline in the putamen in theLOPD patients. The annual reduc-tion rate of uptake in the striatumwas much slower in PINK1 patientsthan that in the LOPD patients(1.7 % vs. 4.1 %; p < 0.005). In thepatient with a heterozygous muta-tion in the PINK1 gene, the reduc-tion ratio in the striatum, as well asthe annual reduction rate, werecloser to those in the LOPD group.We conclude that the incidence ofcarrying PINK1 mutations in thepresent cohort of Taiwanese EOPDpatients was low, accounting for2/39 (5.1 %) in familial cases, and2/99 (2 %) in sporadic cases. Theslower annual reduction of DAT ac-tivity might indicate the insidiousdegeneration of dopamine neuronsand a benign prognosis.
■ Key words PINK1 gene · early-onset Parkinson’s disease ·dopamine transporter · 99mTc-TRODAT-1 SPECT

1348
Introduction
Parkinson’s disease (PD) is a neurodegenerative disor-der caused by loss of the dopamine neurons in the sub-stantia nigra. Various etiologies have been proposedsuch as environmental factors, genetic factors, or a com-bination of both [1]. Genetic factors are responsible foronly a minor portion (10 %) of cases; however, they havecontributed greatly to the understanding of the patho-genesis of PD, especially in familial and/or sporadicEOPD patients whose disease begins before the age of 40years [1]. Several genes including SNCA, parkin, UCHL1,DJ1, LRRK2 have already been decoded for familialparkinsonism and/or sporadic EOPD [2–5]. PTEN-in-duced putative kinase 1 (PINK1) was recently identifiedto be responsible for some cases of familial parkinson-ism, and even sporadic PD patients mostly in the West-ern and Oriental countries [6–12]. It has been estimatedthat the mutation might be the second most commoncausative gene worldwide after parkin in autosomal re-cessive juvenile parkinsonism (ARJP) [10, 11]. However,the data on the prevalence of the PINK1 mutation in PDare still limited and controversial [6, 9, 12–14]. In Tai-wan, the genetic and clinical data in PINK1 patients withan age of onset under 40 years are lacking.
Parkinsonian patients with PINK1 mutations mightshow typical features of tremor, rigidity, bradykinesia,and sustained good levodopa responsiveness [15, 16].However, several clinical presentations, such as youngeronset (often ≤ 40 years), slower progression, diurnalfluctuation of symptoms, hyperreflexia, and early lev-odopa-induced dyskinesias,were different from those inpatients with late-onset Parkinson’s disease (LOPD).These clinical features are more closely related to thephenotypes of patients with parkin mutations [11], ex-cept for less focal dystonia [17, 18].
Dysfunction of the dopamine system has been docu-mented in several PINK1 patients by imaging ap-proaches [15, 19, 20]. However, the cases were extremelyrare, and the results of these studies were inconsistent.Using 99mTc-TRODAT-1 (TRODAT) single photon emis-sion computed tomography, we have measured thedopamine transporter (DAT) activity in various kinds ofparkinsonian disorders [21–24], particularly in PD, re-vealing a high sensitivity and specificity [25]. We havenow extended our experience to evaluate the DAT activ-ity in PINK1 patients.
Here we conducted a genetic screen on PINK1 muta-tions in a series of EOPD patients, and subsequently as-sessed the characteristics of clinical manifestation andTRODAT images in the PINK1 patients identified.
Materials and methods
■ Genetic analysis for the PINK1 mutation
Subjects
This research project was approved by the Institute of Review Boardof Chang Gung Memorial Hospital.Written informed consent was re-ceived from each participant.We have collected consecutively a seriesof 138 EOPD patients with an age at onset < 40 years (99 sporadic and39 familial from 28 families with unidentified causes) from the data-base of the Movement Disorders Unit. All of them were diagnosed asprobable PD according to the London Brain Bank criteria. The possi-bility of dopa-responsive dystonia, other secondary parkinsonismand particularly spinocerebellar ataxia type 2 and 3 were excluded[22, 23]. The detailed personal and familial histories, clinical assess-ments, activities of daily living, response to levodopa and adverse ef-fects were recorded. A group of 191 healthy individuals was includedas controls. The mean age at onset in EOPD patients (44 women, 94men) was 33.12 ± 7.38 years (range, 5–40), mean age at examination48.04 ± 9.93 years (20–70) and mean duration of illness 14.93 ± 7.78years (1–38). The mean age of controls (132 women, 59 men) was41.74 ± 14.08 years (range, 11–60).
Genetic analysis
Genomic DNA was extracted from leukocytes in peripheral blood us-ing standard methods. The exons of the PINK1 gene were first ampli-fied using PCR with primers and conditions similar to those previ-ously reported [8]. The 8 exons of the PINK1 gene were tested. Theamplicons were amplified in a final reaction volume of 25 μl contain-ing 100 ng of genomic DNA, 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 1.5mM MgCl2, 200 μM of each dNTP, 0.1 μM of each primer and 1.25 U ofTaq polymerase (Viogene). Subsequently the PCR products werescreened by single-strand conformation polymorphism analysis onGeneGel (Pharmacia Biotech) at 15 °C for 1.5 h and visualized by sil-ver staining. Then the results were further confirmed by direct se-quence using ABI prism 3100 automated DNA Sequence (AppliedBiosystems).
■ TRODAT scan
Three PINK1 patients and one patient with a heterozygous mutationwere further scanned with TRODAT imaging. Ten duration- andseverity-matched sporadic LOPD patients with an age at onset above50 years were studied as a disease-control group. All met the diag-nostic level of probable PD by London Brain Bank criteria. The clini-cal assessments including the Unified Parkinson’s Disease RatingScale (UPDRS),modified Hoehn and Yahr staging (H&Y),and Schwaband England scale of activity of daily living (SEADL) were all evalu-ated in the off state. The mean age in the LOPD group was signifi-cantly higher than that of PINK1 patients, where the duration of ill-ness was matched. To avoid the ageing effect on DAT activity whichdecreased with increasing age, we also enrolled 2 groups of healthycontrols; the first group (NC-1) consisted of 10 subjects, age-matchedto PINK1 patients and the second group (NC-2) also consisted of 10subjects, age-matched to the LOPD group.
The TRODAT scan was performed according to the imaging ac-quisition protocol described in our previous report [25].We have im-proved the analytic power by using the multi-modality software-PMOD (PMOD Technologies Ltd, Zurich, Switzerland), toautomatically co-register the TRODAT and MRI images in each indi-vidual for a precise anatomical template. Regions of interest (ROIs)corresponding to the caudate nucleus, putamen, and occipital cortexwere drawn on the co-registered images. No correction was made forpartial volume effect since all ROIs were restricted to regions com-pletely within the target to minimize this problem. Each ratio of spe-cific-to-nonspecific striatal 99mTc-TRODAT-1 binding was calculated

1349
as the specific uptake ratio (UR). UR equals (ROI counts – occipitalcortex counts)/occipital cortex counts. We also calculated the puta-men-to-caudate nucleus ratio (P/C), by (putamen counts – occipitalcortex counts)/(caudate nucleus counts – occipital cortex counts), toinvestigate the relative regional variability of DAT in the caudate nu-cleus and putamen.
To correct the aging effect, we used the reduction ratio instead ofthe UR for the statistical comparison of PINK1 and LOPD groups. TheUR in PINK1 patients was compared with the UR in the age-matchedhealthy subjects (NC-1), and the LOPD group with the NC-2, respec-tively. The reduction ratio then was calculated as: 100 % *(Mean URof NC – UR of disease group)/Mean UR of NC. The annual reductionrate was calculated as “(reduction ratio)/(duration of disease)”. Weused the nonparametric Mann-Whitney test to analyze the differ-ences of reduction ratios between the PINK1 and PD groups. The sig-nificance was defined as p < 0.05.
Results
■ Genetic studies
Table 1 summarizes the results of screening PINK1 mu-tations in the EOPD patients as well as in the controls.We identified 3 PINK1 mutations in 4 patients: com-pound heterozygous mutations (Q239X/R492X) in twosibs (Cases 1 and 2) have been reported previously [8], anovel homozygous mutation (Case 3,R492X) in one spo-radic patient, and a single heterozygous mutation (Case4, G193R) in the remaining patient that were not foundin the controls. None was born from a consanguineousmarriage. Mutations in the PINK1 gene were detected in4 out of 138 patients. Interestingly, we also detected 3novel variants (L286L, Y404Y, and Y454Y) in 5 sporadicpatients and 2 sites of single nucleotide polymorphismsin another 5 sporadic patients (G1018A in exon 5 in 2and A1562C in exon 8 in 3) in this EOPD series.
■ Clinical presentations
Table 2 summarizes the clinical demography of the fourcases with mutations in the PINK1 gene. The age at on-set in 3 women, 2 sibs (Cases 1 and 2) and one sporadic(Case 3), was the youngest which has ever been reported[8, 12]. The mean age at onset in the 3 patients was 18years, younger than that in the EOPD group (33 years)and mean duration of illness was 25 years, longer thanthat in EOPD group (15 years). These patients primarilypresented with a typical PD (unilateral onset of restingtremor, bradykinesia, and rigidity) with sleeping benefitand developed motor fluctuations and dyskinesias eventreated with a relatively low dose of levodopa. Particu-larly in Case 3, whose motor complications were partic-ularly severe, surgical intervention was required 31years after the onset. The severity of parkinsonian dis-ability and levodopa-induced dyskinesia seemed to becorrelated with the duration of illness in these com-pound heterozygous and homozygous mutated patients
Table 1 Screening of PINK1 mutations in EOPD and controls
Exon Nucleotide Amino acid EOPD Controlschange change, aa/codon (n = 138) (n = 191)
2 A577G hetero Gly193Arg 1 (Case 4) 0
3 C715T hetero Gln239Stop 2 (Case 1 & 2) 0
4 A804G hetero Leu268Leu 1 0
6 C1212T hetero Tyr404Tyr 1 0
7 C1362T hetero Tyr454Tyr 3 1
7 C1474T homo Arg492Stop 1 (Case 3) 0
7 C1474T hetero Arg492Stop 2 (Case 1 & 2) 0
Hetero heterozygous, homo homozygous, SNP single nucleotide polymorphism
Case 1 Case 2 Case 3 Case 4
Gender/Age, yrs F/38 F/42 F/50 M/55
Age at onset, yrs 18 17 19 37
Disease duration, yrs 20 25 31 18
Modified H&Y 3 2.5 5 2.5
SEADL, % 80 80 40 80
UPDRS (motor-III/whole) 24/42 33/46 61/98 12/23Initial symptoms Both legs tremor, R hand tremor Left hand tremor R hand clumsiness
right leg dystonia
Asymmetrical onset – + + +
Clinical features Sleeping benefit, Sleeping benefit, Sleeping benefit, Akinetic-rigid PDtypical PD typical PD typical PD
Levodopa response (mg/day) Excellent (500) Excellent (500) Excellent (900) Excellent (600)
LID + ++ +++ +
Motor fluctuations + + +++ +
Amino acid change Q239X/R492X Q239X/R492X R492X G193R
Modified H&Y modified Hoehn and Yahr Stage; SEADL Schwab and England scale of activity of daily living; UPDRS (III/whole)Unified Parkinson’s Disease Rating Scale (motor-III/whole); LID levodopa-induced dyskinesias
Table 2 Clinical demography of 4cases carrying PINK1 mutations
1350
with an onset under 20 years of age. Mild leg dystoniawas also found at the beginning in Case 1. Their parkin-sonian disability was mild,evidenced from the low mod-ified H&Y stage and UPDRS score even over 20 years af-ter onset of symptoms.Cases 1 and 2 still work full-time.The parkinsonism in all patients progressed very slowlywhich did not seem to have a correlation with the geno-type. The patient with a single heterozygous mutationmanifested similarly to those with homozygous andcompound heterozygous mutations, except an older on-set age, shorter duration and no tremor were found.
■ TRODAT scan
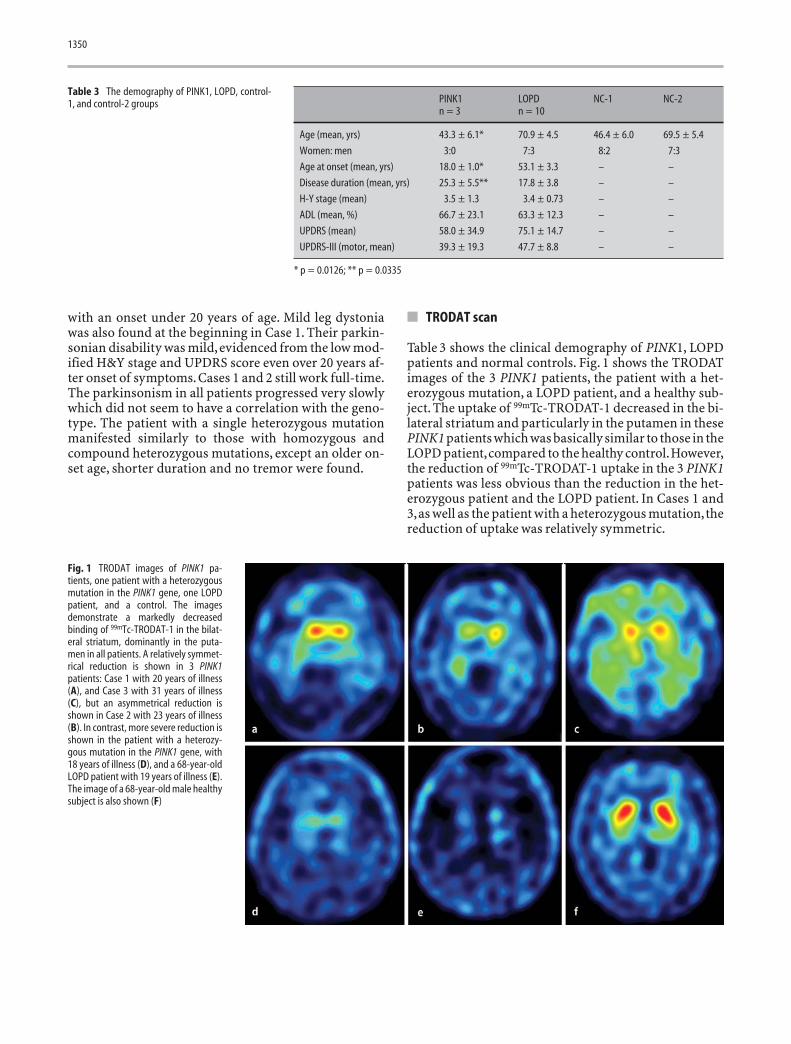
Table 3 shows the clinical demography of PINK1, LOPDpatients and normal controls. Fig. 1 shows the TRODATimages of the 3 PINK1 patients, the patient with a het-erozygous mutation, a LOPD patient, and a healthy sub-ject. The uptake of 99mTc-TRODAT-1 decreased in the bi-lateral striatum and particularly in the putamen in thesePINK1 patients which was basically similar to those in theLOPD patient,compared to the healthy control.However,the reduction of 99mTc-TRODAT-1 uptake in the 3 PINK1patients was less obvious than the reduction in the het-erozygous patient and the LOPD patient. In Cases 1 and3,as well as the patient with a heterozygous mutation,thereduction of uptake was relatively symmetric.
PINK1 LOPD NC-1 NC-2n = 3 n = 10
Age (mean, yrs) 43.3 ± 6.1* 70.9 ± 4.5 46.4 ± 6.0 69.5 ± 5.4
Women: men 3:0 7:3 8:2 7:3
Age at onset (mean, yrs) 18.0 ± 1.0* 53.1 ± 3.3 – –
Disease duration (mean, yrs) 25.3 ± 5.5** 17.8 ± 3.8 – –
H-Y stage (mean) 3.5 ± 1.3 3.4 ± 0.73 – –
ADL (mean, %) 66.7 ± 23.1 63.3 ± 12.3 – –
UPDRS (mean) 58.0 ± 34.9 75.1 ± 14.7 – –
UPDRS-III (motor, mean) 39.3 ± 19.3 47.7 ± 8.8 – –
* p = 0.0126; ** p = 0.0335
Table 3 The demography of PINK1, LOPD, control-1, and control-2 groups
Fig. 1 TRODAT images of PINK1 pa-tients, one patient with a heterozygousmutation in the PINK1 gene, one LOPDpatient, and a control. The imagesdemonstrate a markedly decreasedbinding of 99mTc-TRODAT-1 in the bilat-eral striatum, dominantly in the puta-men in all patients. A relatively symmet-rical reduction is shown in 3 PINK1patients: Case 1 with 20 years of illness(A), and Case 3 with 31 years of illness(C), but an asymmetrical reduction isshown in Case 2 with 23 years of illness(B). In contrast, more severe reduction isshown in the patient with a heterozy-gous mutation in the PINK1 gene, with18 years of illness (D), and a 68-year-oldLOPD patient with 19 years of illness (E).The image of a 68-year-old male healthysubject is also shown (F)

1351
The URs in the putamen, caudate nucleus and wholestriatum were significantly reduced in both PINK1 andLOPD patients compared to respective age-matchedcontrol groups (Table 4). The URs in all subregions weresignificantly higher in the PINK1 group than those in theLOPD group. The URs were also significantly differentbetween NC-1 and NC-2 (p < 0.0001), which might indi-cate the aging effect. To correct for the aging effect be-tween the PINK1 group and the LOPD group, we furthercalculated the reduction ratio of TRODAT uptake basedon the age-matched NC groups (Table 4). The reductionratios in the putamen were significantly lower in thePINK1 group than those in LOPD group. These resultsindicated a relatively benign degeneration of thedopamine neurons in the PINK1 patients.The URs in theputamen and caudate nucleus showed a negative trendwith the increased duration of illness in the three PINK1patients.A similar trend was also found in the reductionratios (data not shown).
The putamen/caudate (P/C) ratios that representedthe selectivity of dopaminergic degeneration were notdifferent between the NC-1, NC-2, and the PINK1 pa-tients, but were significantly lower in the LOPD group,particularly in the contralateral striatum (Table 5).These results disclosed that the loss of dopamine neu-rons in the PINK1 patients was even and symmetrical,which came close to a normal aging process.
Fig. 2 shows the annual reduction rate of URs in thecontralateral caudate nucleus and putamen in the PINK1patients, the patient with a heterozygous mutation, andthe LOPD patients. The three PINK1 patients had signif-icantly slower annual reduction rates than those in theLOPD patients in all subregions (PINK1/LOPD: con-tralateral striatum: 1.6 ± 0.5 %/4.3 ± 0.9 %; ipsilateralstriatum: 1.8 ± 0.2 %/3.80 ± 0.93 %; contralateral cau-date: 1.7 ± 0.6 %/3.0 ± 0.9 %; contralateral putamen:2.0 ± 0.6 %/5.1 ± 1.1 %, and ipsilateral putamen:2.1 ± 0.01 %/4.3 ± 1 %, respectively; all p = 0.0126), ex-cept the ipsilateral caudate (PINK1: 1.63 ± 0.57 %/,LOPD: 2.8 ± 0.98 %; p = 0.052). Within the PINK1 group,the annual reduction rates in the caudate nucleus andthe putamen were similar and symmetric, while within
Table 4 The specific uptake ratios in PINK1, LOPD, and control groups
Uptake Ratios (URs)(Mean ± SD)
C-Striatum I-Striatum C-Caudate nucleus I-Caudate nucleus C-Putamen I-Putamen
PINK1 (n = 3) 0.60 ± 0.21a, c 0.54 ± 0.15a, c 0.77 ± 0.31a 0.79 ± 0.34a 0.43 ± 0.18a, c 0.40 ± 0.10a, d
NC-1 (n = 10) 1.04 ± 0.13 1.02 ± 0.12 1.40 ± 0.13 1.40 ± 0.15 0.88 ± 0.12 0.86 ± 0.12URs reduction (%) 42.4 ± 19.8e 46.9 ± 14.3 44.5 ± 22.3 43.4 ± 24.2 50.6 ± 20e 53.2 ± 11.6f
LOPD (n = 10) 0.21 ± 0.06b 0.27 ± 0.07b 0.52 ± 0.13b 0.56 ± 0.12b 0.09 ± 0.06b 0.17 ± 0.11b
NC-2 (n = 10) 0.78 ± 0.08 0.78 ± 0.08 1.08 ± 0.13 1.06 ± 0.11 0.66 ± 0.08 0.66 ± 0.08URs reduction (%) 73.7 ± 7.0 64.9 ± 8.8 51.3 ± 11.9 47.5 ± 11.3 87.0 ± 8.9 74.2 ± 11.1
C contralateral and I ipsilateral to the dominant side, NC normal controla URs: PINK1 vs. NC-1, all P = 0.0112; b URs: LOPD vs. NC-2, all P < 0.0001; c URs: PINK1 vs. LOPD, P = 0.0126; d URs: PINK1 vs. LOPD, P = 0.0208; e Reduction (%): PINK1 vs.LOPD, P = 0.0126; f Reduction (%): PINK1 vs. LOPD, P = 0.0335
Table 5 The difference of P/C ratios between PINK1, LOPD and control groups
Groups Contralateral P/C ratios Ipsilateral P/C ratios(mean ± SD) (mean ± SD)
PINK1 (n = 3) 0.56 ± 0.07b 0.55 ± 0.15c
NC-1 (n = 10) 0.63 ± 0.05d 0.61 ± 0.06d
LOPD (n = 10) 0.17 ± 0.11a 0.32 ± 0.14a
NC-2 (n = 10) 0.62 ± 0.06 0.63 ± 0.06
P/C ratios putamen/caudate nucleus ratios; NC-1 normal control group 1; NC-2 nor-mal control group 2a LOPD vs. NC-2, all P < 0.0001; b PINK1 vs. LOPD, P = 0.0126; c PINK1 vs. LOPD,P = 0.04; d NC-1 vs. NC-2, all P = 0.4
Fig. 2 The annual reduction rates of uptake ratios in the contralateral caudate nu-cleus and putamen in 4 PINK1 patients and LOPD patients. In PINK1 patients, themean annual reduction rates were 1.69 ± 0.63 % in the contralateral caudate nu-cleus, and 1.96 ± 0.56 % in the contralateral putamen, respectively. In LOPD group,the mean annual reduction rates were 2.98 ± 0.92 % in the contralateral caudatenucleus, and 5.08 ± 1.06 % in the contralateral putamen. The annual reductionrates in the contralateral putamen and caudate nucleus were significantly slower inthe PINK1 patients than those in LOPD group (both p = 0.0126). In the patient witha heterozygous mutation in the PINK1 gene, the annual reduction rates were4.27 % and 4.39 % in the contralateral caudate nucleus and putamen, which werein the range of LOPD.
Contralateral caudate: Heterozygous
Contralateral putamen: LOPD
Contralateral caudate: LOPD
Contralateral putamen: PINK1
Contralateral putamen: HeterozygousContralateral caudate: PINK1
10
1
2
3
4
5
6
7
2 3 HeteroDiagnosis
Annu
al R
ate
of R
educ
tion
%
LOPD

1352
the LOPD group the annual reduction rates were signif-icantly higher in the putamen than in the caudate nu-cleus, and also higher in the contralateral striatum thanthe ipsilateral striatum (all p = 0.007).
In the patient with a heterozygous mutation in thePINK1 gene, the reduction ratios in the striatum weresimilar to those in the LOPD group as shown in Fig. 3.The annual reduction rate was also within the range ofthe LOPD group (Fig. 2). However, the contralateral andipsilateral P/C ratios in this patient were 0.57 and 0.63,which were significantly higher than those in the LOPDgroup, but close to the ratios in the PINK1 and NCgroups.
Discussion
We have identified four patients with 3 genotypic PINK1mutations in our cohort of EOPD with an age at onset≤40 years. Two of these were sisters in one family with atrait of autosomal recessive juvenile parkinsonism. In aseries of collaborative studies, Heaton et al. have docu-mented that PINK1 mutations account for eight of 90(8.9 %) parkin- and DJ-1-negative autosomal recessivePD families. They also suggest that PINK1 mutations arepossibly the second most common causative gene nextto parkin in ARJP,not only in Europe but also in Asia [26,27]. As indicated in our recent study, the genetic causesof familial parkinsonism consecutively identified wereparkin mutations in 7 families, PINK1 in 1, LRRK2 in 1,ataxin2 in 5, and ataxin3 in 1 [28]. In our study, the over-all incidence of PINK1 mutations in familial parkinson-ism (2/39) was less than those of parkin and ataxin2 mu-tations.
The two sisters were affected with pathogenic com-pound heterozygous mutations in the family [12, 26].Genetic testing in their parents and four brothers who
were not affected revealed only harboring of one het-erozygous mutation (either Q239X or R492X, data notshown). Therefore, the Mendelian autosomal recessiveinheritance was prevalent in this family. Another patho-genic mutation was a novel homozygous mutation(R492X), which did not occur within the kinase domainof the protein. Whether the mutation in the R492X is acommon site in ethnic Chinese is warranted for a furtherstudy.
The incidence of carrying PINK1 mutations in spo-radic EOPD patients is variable between several cohortstudies, which is low in Ireland (0/86) [15] and NorthAmerica (2/119) [6], but much higher in Italy (7/100)[10]. Valente et al. also reported a higher number of pa-tients (5 %) than controls (1 %) carrying a single het-erozygous mutation. We found a higher incidence ofPINK1 mutations in Taiwanese sporadic EOPD patients(2 in 99) than the incidence in Korea, where no PINK1mutation was identified in 94 EOPD patients [29].PINK1mutations are rarely detected in sporadic PD patients.The dosage on each amplified exon was also analyzed byusing ABI prism 7000 Sequence Detection System (Ap-plied Biosystems) [30]. However, no abnormal changesin the dosage of each exon or whole gene were detected.
The TRODAT scan showed a uniform binding patternamong our PINK1 patients: severe but relatively even re-duction in the putamen and caudate nucleus, and sym-metrical reduction bilaterally. The patterns of dopamin-ergic dysfunction in our PINK1 patients were similar tothose in four homozygous PINK1 patients who werestudied by Khan et al.using 18F-DOPA positron emissiontomography [19]. They also demonstrated an even re-duction of 18F-DOPA uptake in both the putamen andthe caudate nucleus in PINK1 patients, with a C/P indexof 1.8 that was equal to the P/C ratio in our study (0.55).Although the ligand is different from ours, both studiesshowed that PINK1 patients had an absence of an ante-rioposterior gradient in the striatal uptake, which is typ-ical in the PD patients.
Two previous reports also studying the DAT densityin homozygous PINK1 patients using SPECT and DATligands (123I-β-CIT and 123I-FP-CIT) have demonstrateda significant, but symmetrical decline of striatal bindingto the normal control [15, 20]. Interestingly, like our re-sults, one patient with younger age of onset (24 years)also had a high striatal 123I-FP-CIT binding as reportedby Kessler et al. [20]. These two studies concluded thatthe DAT binding in PINK1 patients was similar to that inthe PD patients. However, no duration- or severity-matched PD controls were available in those two studies.In our study,all PINK1 patients also had a significant de-cline of URs in comparison with the healthy subjects.Al-though our groups of PINK1 and idiopathic PD patientswere matched for disease duration and clinical diseaseseverity, they showed different patterns of nigrostriataldopaminergic dysfunction: First, the higher P/C ratios
Fig. 3 The percentage of URs reduction in the LOPD group and the patient with asingle heterozygous mutation in the PINK1 gene
% Contralateral striatum90
85
80
75
70
65
60
55
50Hetero LOPD
Diagnosis
Redu
ctio
n Ra
tios
% Ipsilateral striatum

1353
in PINK1 patients probably indicated a more uniformloss of the substantial dopaminergic neurons. The pref-erential degeneration of dopaminergic neurons in theventrolateral tier of substantia nigra pars compacta thatprojects to the dorsal putamen is a pathological markerof PD. However, to date there are no neuropathologicaldata regarding any patient with homozygous mutation.Our in vivo study suggested that PINK1 parkinsonismmight have different neurodegenerative processes in thesubstantia nigra compared to PD. Second, the reductionof TRODAT uptake in PINK1 patients did not correlatewith the severity and the duration of disease, which isoften observed in PD patients [25]. The result might beattributable to the small sample size; nevertheless, thediscrepancy between the clinical severity and thedopaminergic dysfunction had been reported by Khanet al. in PINK1 and parkin patients [19, 31]. The findingsmight suggest some uncertain compensatory mecha-nisms in such hereditary parkinsonism [19].
The annual reduction rate in the PINK1 patients wasabout 2 times lower than the rate in LOPD patients, butslightly higher than the normal aging rate [32]. Theslower rate of dopaminergic degeneration had been re-ported in four parkin patients from Khan et al. [31].They also found that the putamen and caudate nucleushad the same rates of deterioration in parkin patients.Therefore, we further compared the annual reductionrates in the PINK1 patients with those in five parkin pa-tients who were matched with age, duration and sever-ity of disease (mean age: 40.4 ± 7.4; onset age: 23.8 ± 6.0;duration: 16.6 ± 4.4; H&Y: 3.3 ± 0.6). The annual reduc-tion rates of the striatum in parkin patients (contralat-eral striatum: 2.0 ± 4.7 %, ipsilateral striatum:1.8 ± 0.6 %, respectively) were not significantly differentfrom the rates in PINK1 patients (all p > 0.05). The sim-ilar slower rate of dopaminergic degeneration in bothPINK1 and parkin patients might suggest the pathogen-esis mechanism in the hereditary parkinsonism was dif-ferent from sporadic PD.
The patient with a single heterozygous mutation hasthe typical presentations of the PINK1 disease, includingthe clinical characters and age of onset. The DAT activ-ity in the heterozygous PINK1 patient, including the up-take ratios and the annual reduction rate, were closer tothose in the LOPD patients. Heterozygous mutations ofparkin and PINK1 genes have been identified in cases ofsporadic PD [10, 31]. However, the contribution of thesingle heterozygous mutation to disease causation re-
mains debatable. This single heterozygous mutation wasrarely found in the normal controls. Several studies re-ported a higher number of sporadic PD patients thancontrols carrying a single heterozygous mutation [9,33].Our patient with a single heterozygous mutation showedsevere reduction of the striatal uptake 18 years after on-set. Other in vivo functional imaging studies of a clini-cally asymptomatic carrier have also shown a significantdecline of 18F-DOPA uptake, indicating preclinicaldopaminergic dysfunction [19]. Khan et al. also demon-strated a significant decrease of 18F-DOPA uptake in fourasymptomatic parkin carriers in three of whom subtleextrapyramidal symptoms had developed after a 10-year follow-up [31]. A recent post-mortem studydemonstrated that four PD patients with heterozygousPINK1 mutations were clinically and pathologicallyundistinguished from idiopathic PD [33]. The in vitropathological findings were compatible with the in vivoDAT imaging in our patient with a single heterozygousmutation, which was also undistinguished from theLOPD patient. However, the heterozygous mutations donot alter the expression or localization of the PINK1 pro-teins in the brain. The contribution of heterozygousPINK1 mutations in the pathogenesis of PD should befurther investigated.
In summary we have demonstrated three patientswith two genotypes of the PINK1 mutation and one pa-tient with a single heterozygous mutation in a series ofEOPD patients with an age at onset below 40 years. Thisis a pioneer cohort study in the Taiwanese population.The incidence of carrying PINK1 mutations in the pres-ent cohort of Taiwanese EOPD patients was low, ac-counting for 2/39 (5.1 %) in familial cases,and 2/99 (2 %)in sporadic cases. Phenotypic characteristics were simi-lar among different genotypes including early onset,sleeping benefit, prolonged course, and sustained effectto low-dose levodopa with dyskinesia. The DAT alter-ations were relatively uniform in the bilateral putamenand caudate nucleus. The slower annual reduction ofDAT activity might indicate the insidious degenerationof dopamine neurons and a benign prognosis.
■ Acknowledgement We gratefully acknowledge the supply of theTRODAT kit from the Institute of Nuclear Energy Research in Lontan,Taoyuan and the funding from the Chang Gung Medical ResearchCouncil (CMRPG 1015, CMRPG34007) and the National ScienceCouncil (NSC 93–2314-B-182A-209, NSC 95–2623–7–182A-001-NU).Dr. Lu had full access to all data in the study and takes responsibilityfor the integrity of the data and the accuracy of the data analysis.

1354
1. Tanner CM, Ottman R, Goldman SM,Ellenberg J, Chan P, Mayeux R,Langston JW (1999) Parkinson Diseasein Twins: An Etiologic Study. JAMA281:341–346
2. Bonifati V, Rizzu P, Squitieri F, KriegerE, Vanacore N, van Swieten JC, Brice A,van Duijn CM, Oostra B, Meco G,Heutink P (2003) DJ-1(PARK7), anovel gene for autosomal recessive,early onset parkinsonism. Neurol Sci24:159–160
3. Polymeropoulos MH, Lavedan C, LeroyE, Ide SE, Dehejia A, Dutra A, Pike B,Root H, Rubenstein J, Boyer R, Sten-roos ES, Chandrasekharappa S,Athanassiadou A, Papapetropoulos T,Johnson WG, Lazzarini AM, DuvoisinRC, Di Iorio G, Golbe LI, Nussbaum RL(1997) Mutation in the alpha-Synu-clein Gene Identified in Families withParkinson’s Disease. Science 276:2045–2047
4. Paisan-Ruiz C, Jain S, Evans EW, GilksWP, Simon J, van der Brug M, Lopez deMunain A, Aparicio S, Gil AM, Khan N,Johnson J, Martinez JR, Nicholl D,Carrera IM, Pena AS, de Silva R, LeesA, Marti-Masso JF, Perez-Tur J, WoodNW, Singleton AB. Cloning of the genecontaining mutations that causePARK8-linked Parkinson’s disease.Neuron 44:595–600
5. Kitada T, Asakawa S, Hattori N, Mat-sumine H, Yamamura Y, Minoshima S,Yokochi M, Mizuno Y, Shimizu N(1998) Mutations in the parkin genecause autosomal recessive juvenileparkinsonism. Nature 392:605–608
6. Rogaeva E, Johnson J, Lang AE, GulickC, Gwinn-Hardy K, Kawarai T, Sato C,Morgan A, Werner J, Nussbaum R, PetitA, Okun MS, McInerney A, Mandel R,Groen JL, Fernandez HH, Postuma R,Foote KD, Salehi-Rad S, Liang Y, Reim-snider S, Tandon A, Hardy J, George-Hyslop P, Singleton AB (2004) Analysisof the PINK1 gene in a large cohort ofcases with Parkinson disease. ArchNeurol 61:1898–1904
7. Valente EM, Brancati F, Caputo V, Gra-ham EA, Davis MB, Ferraris A, BretelerMM, Gasser T, Bonifati V, BentivoglioAR, De Michele G, Durr A, Cortelli P,Filla A, Meco G, Oostra BA, Brice A,Albanese A, Dallapiccola B, Wood NW,European Consortium on GeneticSusceptibility in Parkinson’s Disease(2002) PARK6 is a common cause offamilial parkinsonism. Neurol Sci 23:S117–118
8. Valente EM, Brancati F, Ferraris A,Graham EA, Davis MB, Breteler MM,Gasser T, Bonifati V, Bentivoglio AR,De Michele G, Durr A, Cortelli P,Wassilowsky D, Harhangi BS, Rawal N,Caputo V, Filla A, Meco G, Oostra BA,Brice A, Albanese A, Dallapiccola B,Wood NW, European Consortium onGenetic Susceptibility in Parkinson’sDisease (2002) PARK6-linked parkin-sonism occurs in several Europeanfamilies. Ann Neurol 51:14–18
9. Valente EM, Salvi S, Ialongo T,Marongiu R, Elia AE, Caputo V, RomitoL, Albanese A, Dallapiccola B, Ben-tivoglio AR (2004) PINK1 mutationsare associated with sporadic early-onset parkinsonism. Ann Neurol56:336–341
10. Valente EM, Abou-Sleiman PM, CaputoV, Muqit MM, Harvey K, Gispert S, AliZ, Del Turco D, Bentivoglio AR, HealyDG, Albanese A, Nussbaum R, Gonza-lez-Maldonado R, Deller T, Salvi S,Cortelli P, Gilks WP, Latchman DS,Harvey RJ, Dallapiccola B, Auburger G,Wood NW (2004) Hereditary early-onset Parkinson’s disease caused bymutations in PINK1. Science 304:1158–1160
11. Ibanez P, Lesage S, Lohmann E,Thobois S, De MG, Borg M, Agid Y,Durr A, Brice A, French Parkinson’sDisease Genetics Study Group (2006)Mutational analysis of the PINK1 genein early-onset parkinsonism in Europeand North Africa. Brain 129:686–694
12. Hatano Y, Li Y, Sato K, Asakawa S,Yamamura Y, Tomiyama H, Yoshino H,Asahina M, Kobayashi S, Hassin-BaerS, Lu CS, Ng AR, Rosales RL, ShimizuN, Toda T, Mizuno Y, Hattori N (2004)Novel PINK1 mutations in early-onsetparkinsonism. Ann Neurol 56:424–427
13. Healy DG, Abou-Sleiman PM, AhmadiKR, Muqit MM, Bhatia KP, Quinn NP,Lees AJ, Latchmann DS, Goldstein DB,Wood NW (2004) The gene responsi-ble for PARK6 Parkinson’s disease,PINK1, does not influence commonforms of parkinsonism. Ann Neurol56:329–335
14. Healy DGM, Abou-Sleiman PMP,Gibson JMM, Ross OAP, Jain SB,Gandhi SM, Gosal DM, Muqit MMK,Wood NWP, Lynch TM (2004) PINK1(PARK6) associated Parkinson diseasein Ireland. Neurology 63:1486–1488
15. Albanese A, Valente EM, Romito LM,Bellacchio E, Elia AE, Dallapiccola B(2005) The PINK1 phenotype can beindistinguishable from idiopathicParkinson disease. Neurology 64:1958–1960
16. Bentivoglio AR, Cortelli P, Valente EM,Ialongo T, Ferraris A, Elia A, MontagnaP, Albanese A (2001) Phenotypic char-acterisation of autosomal recessivePARK6-linked parkinsonism in threeunrelated Italian families. Mov Disord16:999–1006
17. Lohmann E, Periquet M, Bonifati V,Wood NW, De Michele G, Bonnet AM,Fraix V, Broussolle E, Horstink MW,Vidailhet M, Verpillat P, Gasser T,Nicholl D, Teive H, Raskin S, Rascol O,Destee A, Ruberg M, Gasparini F, MecoG, Agid Y, Durr A, Brice A (2003) Howmuch phenotypic variation can beattributed to parkin genotype? AnnNeurol 54:176–185
18. Saito M, Maruyama M, Ikeuchi K,Kondo H, Ishikawa A, Yuasa T, Tsuji S(2000) Autosomal recessive juvenileparkinsonism. Brain Develop 22:115–117
19. Khan NL, Valente EM, Bentivoglio AR,Wood NW, Albanese A, Brooks DJ,Piccini P (2002) Clinical and subclini-cal dopaminergic dysfunction inPARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol52:849–853
20. Kessler KR, Hamscho N, Morales B,Menzel C, Barrero F, Vives F, Gispert S,Auburger G (2005) Dopaminergicfunction in a family with the PARK6form of autosomal recessive Parkin-sons syndrome. J neural transm 112:1345–1353
21. Lu CS, Weng YH, Chen MC, Chen RS,Tzen KY, Wey SP, Ting G, Chang HC,Yen TC (2004) 99mTc-TRODAT-1 imag-ing of multiple system atrophy. J NuclMed 45:49–55
22. Lu CS, Chang HC, Kuo PC, Liu YL, WuWS, Weng YH, Yen TC, Chou YH (2004)The parkinsonian phenotype of spino-cerebellar ataxia type 3 in a Taiwanesefamily. Parkinsonism Related Disord10:369–373
23. Lu CS, Wu Chou YH, Kuo PC, ChangHC, Weng YH (2004) The parkinsonianphenotype of spinocerebellar ataxiatype 2. Arch Neurol 61:35–38
24. Yen TC, Lu CS, Tzen KY, Wey SP, ChouYH, Weng YH, Kao PF, Ting G (2000)Decreased dopamine transporterbinding in Machado-Joseph disease.J Nucl Med 41:994–998
25. Weng YH, Yen TC, Chen MC, Kao PF,Tzen KY, Chen RS, Wey SP, Ting G, LuCS (2004) Sensitivity and specificity of99mTc-TRODAT-1 SPECT imaging indifferentiating patients with idiopathicParkinson’s disease from healthy sub-jects. J Nucl Med 45:393–401
References

1355
26. Hatano Y, Sato K, Elibol B, Yoshino H,Yamamura Y, Tomiyama H, Bonifati V,Shinotoh H, Asahina M, Kobayashi S,Ng AR, Rosales RL, Hassin-Baer S,Shinar Y, Lu CS, Chang HC, Wu-ChouYH, Atac FB, Kobayashi T, Toda T,Mizuno Y, Hattori N (2004) PARK6-linked autosomal recessive early-onsetparkinsonism in Asia populations.Neurology 63:1482–1485
27. Li Y, Tomiyama H, Sato K, Hatano Y,Yoshino H, Atsumi M, Kitaguchi M,Sasaki S, Kawaguchi S, Miyajima H,Toda T, Mizuno Y, Hattori N (2006)Clinicogenetic study of PINK1 muta-tions in autosomal recessive early-onset parkinsonism. Neurology 64:1955–1957
28. Lu CS, Wu Chou YH, Weng YH, ChenRS (2006) Genetic and DAT imagingstudies of familial parkinsonism in aTaiwanese cohort. J Neural Transm 70(Suppl):235–240
29. Chung EJ, Ki CS, Lee WY, Kim IS, KimJY (2006) Clinical features and geneanalysis in Korea patients with early-onset Parkinson disease. Arch Neurol63:1170–1174
30. Hedrich K, Kann M, Lanthaler AJ,Dalski A, Eskelson C, Landt O,Schwinger E, Vieregge P, Lang AE,Breakefield XO, Ozelius LJ, PramstallerPP, Klein C (2001) The importance ofgene dosage studies: mutational analy-sis of the parkin gene in early-onsetparkinsonism. Hum Mol Genet 10:1649–1656
31. Khan NL, Brook DJ, Pavese, N, SweeneyMG, Wood NW, Lees AJ, Piccini P(2002) Progression of nigrostriataldysfunction in a parkin kindred: an[18F]dopa PET and clinical study. Brain125:2248–2256
32. Mozley PD, Acton PD, Barraclough ED,Plossl K, Gur RC, Alavi A, Mathur A,Saffer J, Kung HF (1999) Effects of ageon dopamine transporters in healthyhumans. J Nucl Med 40:1812–1817
33. Gandhi S, Muqit MMK, Stanyer L,Healy DG, Abou-Sleiman PM, Harg-reaves I, Heales S, Ganguly M, ParsonsL, Lees AJ, Latchman DS, Holton JL,Wood NW, Revesz T (2006) PINK1protein in normal human brain andParkinson’s disease. Brain 129:1720–1731




















